CHEMICAL BIOLOGY
Iron-Sulfur World, Origin of life in an
GUnter Wachtershauser, Chapel Hill, North Carolina
doi: 10.1002/9780470048672.wecb264
The theory of a chemo-autotrophic origin of life in an Iron-Sulfur World postulates quenched, aqueous flows of volcanic exhalations as sites for the emergence of the pioneer organism of life, which are characterized by a composite structure with an inorganic substructure and an organic superstructure. Iron, nickel, and other transition metal centers in the substructure are catalytic for the reductive formation of low-molecular-weight organic compounds by carbon fixation from volcanically derived carbonyl, cyano, sulfido, ammino, and other ligands. Some organic products become transition metal ligands in statu nascendi with the effect of ligand-accelerated autocatalysis as a chemical basis of pioneer reproduction, inheritance, and evolution. This unitary structure-function relationship of the pioneer organism constitutes the ''Anlage'' for two major parallel strands of evolution, genetization, and cellularization, and the ''mystery'' of an increase of complexity throughout evolution is simply caused by the primal synthetic drive of the autotrophic pioneer organism that may well be persisting to this day.
Conventional theories on the origin of life are characterized by a conjunction of several deep-seated misconceptions. Since the time of Nagel (1), the oldest and most persistent misconception is based on the fact that the bulk material of all extant organisms consists of polymers. It is, therefore, assumed widely that the first organisms of life were engaged already in the polycondensation of monomers to autocatalytic nucleic acids (“RNA World”) or to autocatalytic sets of polypeptides (“Protein World”). The origin of the monomers is relegated typically to an obscure chemical evolution in a “prebiotic broth” over thousands or millions of years. This view has been criticized severely, notably by Shapiro (see Further Reading). It suffers from a stark paradox. Liquid water is postulated as the medium of polycondensation; yet it is precisely this medium that tends to counteract polycondensation or polycondensation agents because of the hydrolytic mass effect of liquid water, notably under hot, non-neutral conditions. In the last 20 years, a new approach has been developed (2-5), which aims to avoid these pitfalls. The new theory is based not on a polymer metabolism with catalytic polymers, polycondensation, and replication, but rather on a small-molecule metabolism with catalytic transition metal precipitates and autocatalytic carbon fixation, which gives rise to small organic ligands. It is not restricted to main group chemistry, but it is critically dependent on transition metal coordination structures, organo-metal chemistry, and redox reactions, for which liquid water is a benefit rather than a detriment. In view of the dominance of iron among transition metals and the ubiquity of sulphur, the term “Iron-Sulfur World” has been coined for the setting of the origin of life (3), which is viewed not as a protracted affair of prebiotic chemistry but as a nearly instantaneous induction of an autocatalytic “pioneer organism.” The new theory is recognized in the field as one of the “two main theories on the origin of life” and as having generated a conflict over “hot, volcanic origin” versus “cold, oceanic origin” (6).
General Organization and Operation of the Pioneer Organism
The new theory places the pioneer organism in a locally and temporally coherent volcanic flow setting. Therefore, all its assumptions must be compatible with each other. Such a comprehensive theory cannot be conceived in one stroke. It has to evolve. Indeed, the theory presented here has been evolving over the past 20 years, its progress guided by chemical experiments and by the increase of its relative explanatory power (Popper). The partial aspects of the theory as it now stands shall be addressed one at a time in a manner that makes their coherence within the theory apparent.
Structural organization of the pioneer organism
An origin of life by autocatalytic carbon fixation requires a sufficient degree of localization and physical/compositional coherence for preventing decay of the chemical potentials by diffusion and dilution, while permitting access of the volcanic nutrients. These requirements are satisfied by a minimal structural organization that consists of an inorganic substructure of crustal origin and an organic superstructure of volcanic gas origin. Catalytic transition metal centers (e.g., Fe, Co, Ni, Cu, Mn, Mo, W, and V) in the inorganic substructure are exposed to volcanic gas nutrients (CO, HCN, CH3SH), which are quenched in the liquid water phase and bonded as ligands to the transition metal centers. The chemical potential of these inorganic ligands in conjunction with the catalytic properties of the transition metal centers leads to synthetic reaction cascades that generate low-molecular-weight organic compounds, which are functionalized sufficiently for bonding in statu nascendi as ligands to the transition metal centers (2). Thereby they become constituents of the organic superstructure attached to the transition metal centers in outer or inner surfaces of the inorganic substructure. The organic superstructure and the proximal regions of the inorganic substructure define the “pioneer organism.”
The organic superstructure constitutes a dynamic ligand sphere. Its constituents may undergo lateral transfer along the surfaces. At any given time the composition of the superstructure is a steady state between the rate of formation of organic constituents by carbon fixation and the rate of loss of organic constituents by detachment and diffusion into the vast expanses of the ocean (2).
The inorganic substructure should also be viewed as being dynamic. According to Ostwald’s rule of steps, it will change its constitution mainly by conversions of the structure-forming bridging ligands. It may begin as an extended polynuclear, polymodal, heteroleptic, and hydrated complex or as amorphous, hydrated hydroxy minerals or hydrogels [e.g., (Fe,Ni)(OH)2], which subsequently stabilize by crystallization. The additional development depends on the relative rates of introduction of polymerizing bidentate ligands (cyanidation, dehydration, or sulfidation) versus depolymerizing ligands (carbonylation or ammination). Under conditions of high sulfide activity, the substructure will convert to sulfides, which are at first amorphous or poorly crystalline, and finally to pyrites.
Inorganic starting conditions of the pioneer organism
The discovery that liquid water may well have existed on Earth 4.4 billion years ago has made room for the possibility that life originated deep in the Hadean, i.e., at a time when magma and crust of the Earth were much hotter than today and much more reducing. As a consequence, the water gas equilibrium of the volcanic gases
![]()
favors a high concentration of CO at the magmatic source. For example, under conditions of saturation with graphite, the molar ratio of CO/CO2 is about 1:1 at 1200 °C and 2 kbar or at 900° C and 0.1 kbar. If the rate of quenching along the flow path (by cold water or ice) is fast compared with the rate of equilibration, a significant disequilibrium concentration (chemical potential) of CO is the result (5). Quenched volcanic gases also contain H2, which increases in concentration by water gas equilibration (1), as well as the sulfur compounds H2S, CH3SH, and COS; the nitrogen compounds N2, NH3, and HCN (5); and the volatile phosphorus compound P4O10 (7).
The formation of low-molecular-weight organic compounds by carbon fixation from volcanic nutrients involves primarily electron transfer (redox) reactions. Redox reactions are creative chemically with thoroughgoing transformations of the electron configurations, whereas (poly)condensations are organizational, leaving the electron configurations of the monomers essentially unchanged. Redox reactions require catalysts that can act as electron shuttles. For the pioneer organism, only transition metals that can have different oxidation states are available as inorganic redox catalysts.
Reproduction of the pioneer organism
For the pioneer organism we postulate reproduction by synthetic reactions with autocatalytic product feedback. Now, it is well established that the catalytic activity of a transition metal center is dependent on its ligands and that organic ligands may increase the catalytic activity by a factor of up to 104. This ligand feedback is unpredictable theoretically and idiosyncratic (8). Therefore, it is suggested that certain products of the carbon fixation pathways may increase the catalytic activity of transition metal centers as ligands. This result may well be the simplest and earliest form of autocatalytic product feedback (ligand-accelerated autocatalysis). It constitutes “metabolic reproduction,” which gives rise to growth and inheritance by lateral spreading.
In broad terms, the proposed origin of life by autocatalytic ligand feedback is characterized by an extreme paucity of chemical possibilities because the starting materials are inorganic and the organic products have a low molecular weight. At the same time, the chemistry is highly selective because of mild chemical energy in combination with specific transition metal catalysis. As a consequence, only one possibility may exist for a chemo-autotrophic origin of life.
Flow setting of the pioneer organism
Precious little geological evidence exists concerning the geochemical conditions of the Hadean Earth. Cockell (see Further Reading) has proposed that heavy bombardment caused impact cratering, rock fracturing, and deposition of debris creating a bed with myriads of diverse flow ducts for volcanic, hydrothermal fluids. Such a flow bed would undergo several characteristic transformations.
The primal crust beneath the Hadean ocean may well have consisted of ultramafic rocks (e.g., komatiite), with orthosilicates. These rocks react with water, generating hydroxides according to the following simplified, notional formulas:

The alkaline (Mg, Ca)(OH)2 buffers the pH. After exhaustion of the buffer capacity by reaction with CO2 and with the acidic products of carbon fixation, the flow ducts undergo a pH development from alkaline toward neutral (pH zoning). (Fe, Ni)(OH)2 undergoes cyanidation, carbonylation, sulfidization, and pyritization. Specifically, cyanidation of Ni(OH)2 generates Ni(CN)2 having a layered crystal structure (9), and organic compounds may become trapped in the interspaces between the Ni(CN)2 layers to form clathrades.
As a consequence of the notions of surface catalysis and volcanic flow (2), the flow ducts would operate like a chromatographic reactor with interacting reactive and chromatographic processes. Organic constituents of the superstructure with differential ligand bonding strength would exhibit differential retention or residence times, with the best surface bonders being slowest travelers.
Toward the (Bio)Chemistry of the Pioneer Organism
We now turn to the (bio)chemistry of the pioneer organism and begin with reactions that provide reducing power and nutrients for the pioneer metabolism. The (bio)chemistry of the pioneer organism is testable experimentally with the aim of correlating extant metallo-enzymes with inorganic transition metal catalysts and with the ultimate goal of establishing a reproducing and evolving pioneer organism.
Sources of reducing power for the pioneer organism
CO is a source for reducing equivalents by reaction with H2O but also with H2S:
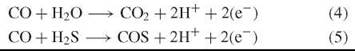
The reactions are precursors of the reactions catalyzed by the extant nickel-enzyme carbon monoxide dehydrogenase. They are catalyzed by nickel centers, and the reducing equivalents (e-) may appear primarily in the form of reduced nickel centers, Ni1+ or Ni°.
The reaction of volcanic dihydrogen (H2) is another electron source, which is correlated with the activity of extant Fe, Ni-hydrogenase:
![]()
Even more reducing equivalents may develop by established oxidative conversions of the inorganic substructure:
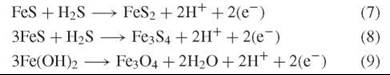
Inorganic nutrient interconversions in the surfaces of the inorganic substructure
Ammonia formation by nitrogen fixation under moderate temperature and pressure has been demonstrated unequivocally with 15N2 in conjunction with reaction (7) as an electron source (10). It has been suggested as a precursor of extant nitrogenase activity:
![]()
Hydrogen cyanide (HCN), the nitrogen analog of CO, forms by catalytic conversion of CO with NH3:
![]()
and it may be trapped as ligand by transition metals under conditions of moderate temperature and pressure (5). In the presence of catalytic NiS or FeS, COS, produced by reaction (5) (11), or from CO2by equilibration with H2S (12), respectively, is reduced via Ni-CH3 to methanethiol, which also becomes a ligand:
![]()
α-Hydroxy acids and α-amino acids
Amino acids are among the most important products of extant intermediary metabolism. Therefore, it is of importance that volcanic conditions have been found to generate a suite of α-amino acids and a corresponding suite of α-hydroxy acids at 1 bar CO and around 100° C with (Mg, Ca)(OH)2 as pH buffer and with an Ni-precipitate with CN-ligands. The CN-ligands serve as C-source and N-source. CO serves as reducing agent. The products satisfy the formula R-CHA-COOH, whereby A=NH2 or OH and R=H, HO-CH2, CH3, C2H5, n-C3H2, FC3H7 ..., which indicates a mechanism of chain extension (13).
The nickel precipitate provides not only catalytic nickel centers but also the carbon source and the source of electrons for the reactions. In this sense, the inorganic substructure reacts stoichiometrically and not catalytically. The transition from a stoichiometric reaction to a truly catalytic reaction is likely a result of evolution. It is remarkable that the a-amino acids and α-hydroxy acids are bidentate ligands for forming complexes with transition metals. Therefore, they are excellent candidates for ligand-accelerated autocatalysis.
The above experimental results suggest that among the most prominent pioneer amino acids of life were glycine (R=H), alanine (R=CH3), serine (R=HO-CH2), and valine (R=i-C3H7). Moreover, the facile formation of serine suggests that cysteine originated early on by sulfidization of serine.
α-keto acids and α-imino acids
In the above reactions under modest conditions, traces of pyruvate have been found (13), which indicates that α-keto or α-imino acids are intermediates in the pathways to α-hydroxy or α-amino acids. The reductive amination of α-keto acids has been demonstrated experimentally under alkaline conditions with FeS/H2S or Fe(OH)2 as catalyst and reducing agent (14). The formation of significant amounts of pyruvate from CO and FeS/nonylmercaptan at 250° C and 2000 bar has been reported (15). Remarkably, pyruvate is stable under these conditions and apparently not reduced to lactate.
Sugar alcohols and sugars
In the above reactions, significant amounts of ethylene glycol have been detected (13), which is the simplest sugar alcohol and may form by reduction of glycolaldehyde, the simplest sugar. It is well established that sugars and sugar alcohols are excellent ligands for transition metals, which suggests that we may trace the biosynthetic roots of nucleic acids down to the pioneer metabolism.
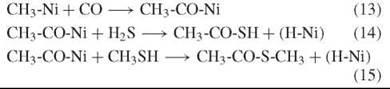
Acetyl-thioester
Experiments provide evidence that in the presence of Ni(OH)2, (Fe,Ni)S, NiS, or CoS, the volcanic reactants CO and H2S (or CO and CH3SH), are converted to activated forms of acetic acid (CH3-CO-Ni, CH3-CO-S-Ni, CH3-CO-S-H, or CH3-CO-S-CH3) as evolutionary precursor of acetyl-CoA (11):
Activated amino acids and peptides
The condensation of a-amino acids to peptides in dilute aqueous systems is endergonic and requires energy coupling. Under alkaline, volcanic conditions CO in the presence of H2S (or CH3SH) and (Ni,Fe)S is an efficient energy source for the formation of peptides, notably via COS (16). The latter operates as a short-lived coupling intermediate, and therefore, the hydrolysis problem associated with theories that require accumulation and/or transport of COS (17) is obviated. Interestingly, the addition of Na2HPO4 to the system (Fe,Ni)S/CO/CH3SH broadens the pH-range of efficient peptide synthesis (16), which indicates nucleophilic catalysis with a phosphorylated intermediate.
As a mechanism of peptide formation, it is suggested that a CO-derived activated intermediate (e.g., COS) is formed, which subsequently suffers a nucleophilic attack by the free amino group of an amino acid (aa) followed by ring closure to activate the carboxyl group of the amino acid in a five-membered cyclic intermediate, notably an amino acid N-carboxyanhydride (aa*) (16):
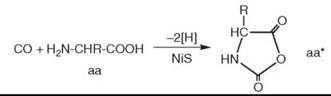
Subsequently the free amino group of another amino acid molecule (aa) reacts nucleophilically with the activated amino acid to generate a dipeptide (aa-aa):
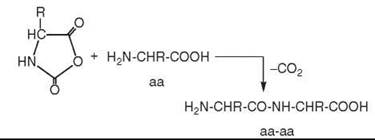
Alternatively the free amino group of a peptide may react with the activated amino acid (aa*), with the result that the peptide is extended by one amino acid unit at its N-terminal end. The redox energy of CO is converted into group activation energy, which then drives the endergonic peptide synthesis pathway. The synthetic pathway is controlled kinetically, and it runs as long as the energy source lasts (16).
Peptide cycle
Surprisingly, it was found that peptide-forming conditions actually support a peptide cycle (18), whereby the peptides react with CO to acquire an N-terminal hydantoin ring, which hydrolyzes to an N-terminal urea group. Finally, the urea group is hydrolyzed. The net result is a removal of the N-terminal amino acid unit. The following scheme shows the simplest case of a dipeptide cycle for glycine (Gly) through activated glycine (Gly*), glycyl-glycine (Gly-Gly), its hydantoin derivative (H-Gly), and its urea derivative (U-Gly):

Similar cycles pass through the tripeptide, tetrapeptide, and so on, which results in a concatenation of peptide cycles. The segment of the cycle up to the urea derivative is anabolic. The hydrolysis of the urea group constitutes a catabolic segment. Because of this anabolic-catabolic character, the peptide cycle generates a dynamic library of peptides, their hydantoin derivatives, and their urea derivatives (“peptide library”). The members of the peptide library come and go, and all transient members of this library are candidates for transition metal ligands. With an increase of the number of amino acids, the peptide library increases exponentially, and so does the likelihood of autocatalytic feedback.
It has been discovered that homochiral amino acids undergo slow racemization under the conditions of the peptide cycle, which may be attributed to the hydantoin stage. Racemic amino acids are actually an advantage from the point of view of ligand feedback, because racemic amino acids generate a greater variety of peptides than homochiral amino acids.
Remarkably, the dynamic peptide library is also self-selecting because differential bonding of its members as ligands to transition metal centers causes differential stabilization against hydrolysis. It means a self-selection of stable metallo-peptide structures.
The peptide cycle is driven by energy coupling from the redox energy of CO to group activation. The peptide cycle as a whole may be viewed as a catalytic cycle for the conversion of CO to CO2. It may therefore be viewed as a functional evolutionary precursor of extant carbon monoxide dehydrogenase. The Ni-catalyzed hydrolysis of the urea group is the functional evolutionary precursor of the extant Ni-enzyme urease. The hydantoin group is related structurally to the aminoacyl N-carboxyanhydride. It is also related to the imidazol ring of the purine bases, which suggests a synthetic as well as an evolutionary precursor relationship.
In the course of evolution of the metabolism by peptide feedback, the system will sooner or later become homochiral because of symmetry breaking by autocatalytic feedback. At this level it has been shown that α-amino acids undergo a facile enantioselective conversion to thermostable β-sheets (19).
Energy metabolism of the pioneer organism
In extant metabolism, it is expedient to distinguish an “energy metabolism” that generates a pool of energetic products, like ATP, for energy coupling with endergonic reactions. The initial chemical energy of a volcanic setting is redox energy, and it is surprising that the pioneer metabolism is engaged from the start in various forms of energy conversion and energy coupling.
A pool of redox energy originates, if the reducing equivalents of the oxidative half reactions (4) and (6)-(9) are transferred to the transition metal centers in the inorganic substructure, and subsequently, they may undergo lateral transfer through suitably spaced transition metal centers in the substructure. Such a redox pool would form the basis for redox-to-redox energy coupling between exergonic redox reactions and endergonic redox reactions, notably as the beginning of chemi-osmotic energy coupling.
Turning finally to redox-to-condensation energy coupling, the simplest case is shown in reaction (5). COS in turn will be the substrate for various forms of condensation-to-condensation energy couplings. For example, C-C-bond formation by carboxylation with COS will generate thioacids (3). Similarly, carboxylations of a-amino acids by COS generate thiocarbox-amides en route to aminoacyl N-carboxy-anhydrides:
![]()
Cascades of condensation-to-condensation energy coupling are among the hallmarks of extant metabolism. They proceed through compounds of decreasing hydrolytic energy and usually increasing reaction selectivity and constitute a form of chemical energy conservation.
Arguably the most important forms of energy coupling and energy conservation involve phosphorylation energy. Volatile phosphoric anhydride P4O10 in volcanic exhalations reacts hydrolytically on contact with liquid water through a cascade of oligophosphates (7). Pyrophosphate and other oligophosphates may have provided the group activation for the formation of phosphorylated organic compounds in early evolution (20).
It has been proposed by de Duve (see Further Reading) that primordial acetyl phosphate originates by reaction of acetyl-thioester with phosphate ions. According to the Iron-Sulfur World theory, phosphorylation energy may also result from a transfer of the redox energy of CO/H2S in the presence of amino acids (5), which received its first support by phosphate catalysis of peptide formation (16). Subsequently it was supported by the discovery of a formation of aminoacyl phosphate in reaction of phosphate with aminoacyl N-carboxyanhydride (21):
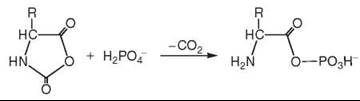
and by the discovery of a transfer of the condensation energy of COS to aminoacyl phosphate or aminoacyl adenylate and further to pyrophosphate (22).
The above-reported chemical reactions proceed under conditions that are compatible with an origin of life under the locally and temporally coherent conditions of a volcanic flow system. Therefore, the discovered reactions may well be components of the metabolic system of the pioneer organism. As additional components come into experimental view, the theory is expected to evolve. So far we have addressed the notions of growth and reproduction as aspects of one unitary chemical system. We now show that this unitary system is also the physical basis for the earliest mechanism of evolution and that it constitutes in fact the evolutionary “Anlage” for the emergence of the cellular and genetic features of extant forms of life.
Evolution from the Vantage Point of the Pioneer Organism
In extant forms of life, we distinguish a (fast) process of development over the lifetime of an organism from a (slow) process of evolution over many generations of reproduction. Going back in time we see these two processes fuse into one unitary process, which constitutes at the same time growth by the accumulation of carbon fixation products, reproduction by positive ligand feedback of some products, inheritance by rapid multiplication and lateral spreading of ligands with positive feedback, and metabolic evolution by variation of the set of ligands and by pathway extension. We now treat metabolic evolution in greater detail.
Ligand evolution
Let us assume that a member of the set of products of the pioneer metabolism binds as a ligand to a catalytic metal center and thereby increases the catalytic activity for a rate-determining step in the pioneer metabolism, which in turn increases the steady-state activities of all products downstream from said rate-determining step. This process induces additional ligand feedback effects and so forth, whereby a cascade of ligand feedback effects comes into play. It has the important consequence that the pioneer metabolism is self-expanding because of an avalanche of ligand feedback effects.
Under chromatographic flow conditions, strong bonding ligands have a long retention time and travel slowly in flow direction. Therefore, the strongest ligands (and the best candidates for ligand feedback) tend to become concentrated in upstream flow zones. These ligand zones are areas of ligand feedback concentration and thus constitute the spatial precondition for a localization of the pioneer organism. This catalyst evolution by ligand feedback is the most basic mechanism of evolution and the source mechanism for the emergence of a mechanism of pathway evolution to which we turn next.
Metabolic self-expansion
The evolutionary increase of catalytic activities and the concomitant widening of the spectrum of catalytic abilities elicit ever more complex expansions of the metabolism. These expansions may be from terminal extension or lateral branching of preexistent pathways. They lead to new synthetic products of the metabolism, which increase the set of potential ligands for the catalysts. Therefore, the catalysts evolve from strictly inorganic catalytic transition metal centers with inorganic ligands to hybrid inorganic-organic catalyst centers with a combination of inorganic and organic ligands. Within the latter category of catalysts, metallo-peptides become more and more dominant. Finally, with the emergence of the genetic machinery, primitive noncoded metallo-peptides are replaced by coded metallo-peptides and ultimately by folded metallo-proteins.
So far we have considered an evolving pioneer metabolism based on the full complement of primordial inorganic constituents of volcanic flow localities. With the expansion of the metabolism, new pathways come into play, which are independent of one or the other inorganic compound. This process allows the descendents of the pioneer organism to venture into spaces that are devoid of such inorganic compounds. For example, the coexistence of amino acid synthesis from CN-ligands and from CO/NH3-ligands allows the gradual colonization of spaces devoid of cyanide sources. Here the amino acid synthesis becomes restricted to CO-ligands as carbon source and indispensable CN-ligands (in hydrogenases) come to be biosynthesized. In subsequently conquered realms, the CO-ligands become replaced by carbon dioxide as carbon source and by FeS/H2S or H2 as reductant. In new chemical spaces, the catalysts that are dependent on unavailable or extremely depleted inorganic starting materials will be opportunistically lost. Therefore, newly conquered spaces turn from optional habitats into obligatory habitats and metabolic evolution leads by necessity to a biosphere with diverse variants in a diversity of habitats.
In the course of early evolution, the metabolism becomes more and more integrated and centralized, so much so that a chemical conversion of highly integrated constituents tends to weaken the metabolism. The more central the constituent, the more severe is this effect of metabolic decay by chemical conversions, which leads us to yet another strategy of metabolic evolution: the strategy of dual feedback.
Metabolic evolution by dual feedback
So far we have looked at early evolution from the point of view of ligand function for catalyst promotion. Now, we look at it from the point of view of ligand synthesis. Let us assume that a fluctuation in the chemical environment expands the metabolism by the synthesis of a new ligand with new positive feedback into the metabolism from a preexistent ligand. However, if the chemical environment returns to the original state, the new ligand will disappear again, unless it exhibits also a positive feedback into its own synthesis. More generally speaking, duality of feedback of new metabolic products (altruistically into the metabolism and egotistically into their own branch pathways) seems to be essential for metabolic evolution. Extant cellular organisms are replete with dual feedback catalysts. Ribosomes produce all proteins, including ribosomal proteins. Coenzymes catalyze many synthetic pathways, including coenzyme biosynthesis. Protein translocases transport many proteins, including translocase proteins. DNA codes for all genes, including DNA polymerase genes.
Autocatalytic metabolic cycles
According to a general rule of organic chemistry, reactions involving the smallest molecules are catalytically the most restrictive. This rule holds notably for the build-up of carbon skeletons with the arithmetic C1 + C1 = C2 (e.g., C2 = glycine or acetyl thioester). Therefore, it may not come as a surprise that in the course of metabolic evolution, these most “simple” carbon fixation reactions may fall by the wayside. Under these conditions, an autotrophic carbon fixation metabolism can only be maintained by a “metabolic cycle,” which multiplies the C2 unit autocatalytically in the absence of its de novo synthesis. A prominent example is the reductive citric acid cycle (C2 + C1 = C3; C3 + C1 = C4; C4 + C1 = C5; C5 + C1 = C6; C6 = C4 + C2) or variants thereof (2, 3). It is not clear at which level of catalyst evolution the first metabolic cycle (autotrophic and autocatalytic) would have emerged, but it surely allowed for a tremendous expansion of the spaces inhabitable by life.
Thermal evolution of life
By a reasoning that goes back to Thomas Brock, evolution can proceed only thermally downward from extremely thermophilic organisms to mesophiles by opportunistic losses of individual thermal stabilities. Such a view of evolution is compatible with an origin of metallo-peptide/protein folding structures with extremely thermostable covalent transition metal-sulfur cross-links, many of which were later replaced opportunistically and irreversibly by the cooperation of a multitude of weak, noncovalent group interactions in concert with an increase of fidelity of the emergent genetic machinery (4, 5).
Genetization and enzymatization
According to the RNA World theory, life begins with RNA replication, but the replicated RNA has no function other than its replication. According to the Iron-Sulfur World theory, the early novelties in the pioneer metabolism develop with the catalyst-promoting function of transition metal ligands. Therefore, early on, sugars, nucleosides, and (oligo)nucleotides must have evolved as ligands. Later oligonucleotides folded and were associated by base pairing, which ushered in a new type of catalysis: base pair-assisted positioning of RNA-bonded amino acids for the formation of peptides. This catalysis was of immediate benefit because the function of peptides as ligands was preestablished. It is the origin of the ur-ribosome, which is seen as preceding the origin of replication followed by a coevolution of translation fidelity, replication fidelity, and code expansion. In the process of the emergence of the genetic machinery, noncoded metallopeptides came to be replaced incrementally by coded metallo-enzymes. Thus, the earliest mechanism of evolution is a direct mechanism, in which new products of the metabolism feed back directly as ligands into the catalyst for their synthesis. It became first supplemented and later substituted by an indirect genetic mechanism of evolution, in which replication variations led indirectly to variations of pep- tide/protein ligands (5, 6). In the terminology of information metaphors, we may say that analog information of chemical feedback cycles precedes digital information of polymer sequences.
Cellularization
The emergence of any multicomponent genetic machinery depends on the existence of compartments. It has been suggested that the chemo-autotrophic origin of life occured in primordial compartments that were bounded by FeS-membranes. The proposal was based on experimental FeS-precipitations (23). However, the detected cell structures are likely an artifact of sample preparation by freeze-drying of an FeS-hydrogel. Freeze-drying usually produces a porous product, in which the rate of freezing determines the size of the ice crystals, which in turn determines the size of the pores (5, 6). Instead of such chemically unsupported FeS-cells, it is proposed that a cellularization of life occurred step by step with autotrophically generated lipids (2). The first lipids may have been long-chain a-hydroxy and a-amino acids (6) to be replaced later by products of Claissen condensation of acetyl thioester to fatty and isoprenoid acids (4). The first lipid function must have been lipophilization of the substructure by lipid accumulation. This process had the effect of lowering the H2O and H3O+ activities near the surface, thereby disfavoring hydrolytic reactions and favoring all kinds of condensation reactions. Because lipid syntheses from acetyl thioesters are condensation reactions, surface lipophilization is seen as collectively autocatalytic. The accumulating lipid molecules will inevitably organize into mineral-supported bilayer membrane patches, which ultimately will coalesce into an extended membrane that covers the mineral surface or surrounds a mineral particle (2).
Semicellular structures, which are bounded partly by a mineral surface and partly by a lipid membrane, form automatically for energetic reasons, when a lipid membrane spans over a small cavity in the mineral surface. The volcanic nutrients can pass freely through the membrane, whereas detached organic constituents are retained under the membrane. Now multicomponent systems can emerge within the incipient cytosol underneath the membrane along with the step-by-step enzymatization of the metabolism. Finally, self-supporting membrane envelopes appear after the emergence of wedge-shaped bis-acyl phospho-glycerol lipids. These lipids are chiral, but they must have appeared first as a racemate. The self-supporting lipid membrane formed the basis for chemi-osmosis by harnessing all kinds of environmental redox energy. Ultimately, the lipid racemate underwent chiral symmetry breaking by giving rise to the divergence of the domain Bacteria with one lipid enantiomer and the domain Archaea with the other lipid enantiomer (5, 6). Remarkably, up to the level of the origin of speciation, decisive events in the early evolution of life seem to have been preordained in the universal laws of physics and chemistry.
Concluding remarks
From the vantage point of a chemo-autotrophic origin of life, the overall mechanism of evolution is seen as a composite affair. It comprises a primal, direct, chemically deterministic, directional mechanism of evolution and a later, indirect, genetically stochastic mechanism of evolution. The latter is a derivative of and complementary to the primal mechanism of evolution. With the indirect mechanism of evolution appears the need for Darwinian selection, which gives rise to adaptations to the outer conditions of the environment and to the inner conditions of chemically restricted, preordained pathway possibilities. It is this association of chemical necessity and genetic chance that generates biochemical continuity between the origin of life and the extant biosphere. In fact the Iron-Sulfur World theory is not just a theory on the origin of life. It is also an explanatory theory of biochemistry.
Acknowledgments
Support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged. My gratitude goes to Claudia Huber and Wolfgang Eisenreich for seminal experiments and fruitful discussions, and to Adelbert Bacher for generously supplying laboratory facilities and material support.
References
1. Von Nageli C. Mechanisch-physiologische Theorie der Abstammungslehre. 1884. Verlag Oldenbourg, Munchen. pp. 83-101.
2. Wachtershauser G. Before enzymes and templates: theory of surface metabolism. Microbiol. Rev. 1988; 52:452-484.
3. Wachtershauser G. Groundworks for an evolutionary biochemistry — the iron-sulfur world. Prog. Biophys. Mol. Biol. 1992; 58:85-201.
4. Wachtershauser G. From volcanic origins of chemoautotrophic life to bacteria, archaea and eukarya. Philos. Trans. R. Soc. Ser. B 2006; 361:1787-1808.
5. Wachtershauser G. On the chemistry and evolution of the pioneer organism. Chem. Biodivers. 2007; 4:584-602.
6. Bada JL, Fegley B Jr, Miller SL, Lazcano A, Cleaves HJ, Hazen RM, Chalmers J. Debating evidence for the origin of life on Earth. Science 2007; 315:937-939.
7. Yamagata Y, Watanabe H, Saitoh M, Namba T. Volcanic production of polyphosphates and its relevance to prebiotic evolution. Nature 1991; 352:516-519.
8. Berrisford DJ, Bolm C, Sharpless KB. Ligand accelerated catalysis. Angew. Chem., Int. Ed. 1995; 34:1059-1070.
9. Ludi A, Huegi R. Preparation and structural properties of nickel(II) cyanide dihydrate and nickel(II) cyanide sesquihydrate. Helv. Chim. Acta 1967; 50:1283-1289.
10. 10. Dörr M, Käßbohrer J, Grunert R, Kreisel G, Brand WA, Werner RA, Geilmann H, Apfel C, Robl C, Weigand W. A possible prebiotic formation of ammonia from dinitrogen on iron sulfide surfaces. Angew. Chem. Int. Ed. 2003; 42:1540-1543.
11. Huber C, Wachtershauser G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 1997; 276:245-247.
12. Heinen W, Lauwers AM. Organic sulfur compounds resulting from the interaction of iron sulfide, hydrogen sulfide and carbon dioxide in an anaerobic aqueous environment. Orig. Life Evol. Biosph. 1996; 26:131-150.
13. Huber C, Wachtershauser G. α-Hydroxy and α-amino acids under possible hadean, volcanic origin-of-life conditions. Science 2006; 314:630-632.
14. Huber C, Wachtershauser G. Primordial reductive amination revisited. Tetrahedron Lett. 2003; 44:1695-1697.
15. Cody GD, Boctor NZ, Filley TR, Hazen RM, Scott JH, Sharma A, Yoder HS Jr. Primordial carbonylated iron-sulfur compounds and the synthesis of pyruvate. Science 2000; 289:1337-1340.
16. Huber C, Wachtershauser G. Peptides by activation of amino acids with CO on (Fe,Ni)S surfaces and implications for the origin of life. Science 1998; 281:670-682.
17. Leman LJ, Orgel LE, Ghadiri MR. Carbonyl sulfide-mediated prebiotic formation of peptides. Science 2004; 306:283-286.
18. Huber C, Eisenreich W, Hecht S, Wachtershauser G. A possible primordial peptide cycle. Science 2003; 301:938-940.
19. Brack A. From interstellar amino acids to prebiotic catalytic peptides. Chem. Biodivers. 2007; 4:665-679.
20. Baltscheffsky H. Inorganic pyrophosphate and the origin and evolution of biological energy transformation. In: Chemical Evolution and the Origin of Life. Buvet R, Ponnamperuma C, eds. 1971. North Holland, Amsterdam. pp. 466-470.
21. Biron J-P, Pascal R. Amino acid N-carboxyanhydrides: activated peptide monomers behaving as phosphate-activating agents in aqueous solution. J. Am. Chem. Soc. 2004; 126:9198-9199.
22. Leman LJ, Orgel LE, Ghadiri MR. Amino acid dependent formation of phosphate anhydrides in water mediated by carbonyl sulfide. J. Am. Chem. Soc. 2006; 128:20-21.
23. Martin W, Russell MJ. On the origin of cells: an hypothesis for the evolutionary transitions from abiotic geochemistry to chemoautotrophic prokaryotes, and from prokaryotes to nucleated cells. Philos. Trans. R. Soc. London, Ser. B 2003; 358:59-85.
Further Reading
Cockell CS. The origin and emergence of life under impact bombardment. Philos. Trans. R. Soc. London, Ser. B. 2006; 361:1845-1855.
Cody GD. Transition metal sulfides and the origin of metabolism. Annu. Rev. Earth Planet. Sci. 2004; 32:569-599.
DeDuve C. Blueprint for a Cell. 1991. Neil Patterson Publishers, Burlington, NC.
Fry I. The Emergence of Life on Earth. 2000. Rutgers Univerity Press, New Brunswick, NJ.
Schwartz AW. Intractable mixtures and the origin of life. Chem. Biodivers. 2007; 4:656-664.
Shapiro R. Small molecule interactions were central to the origin of life. Q. Rev. Biol. 2006; 81:105-125.
See Also
Origins of Life: Emergence of Amino Acids
Origins of Life: Emergence of an Rna World
Origins of Life: Emergence of Nucleic Acids
Prebiotic Membrane Formation
Transition Metals in Biology