CHEMICAL BIOLOGY
Lipid Bilayers, Properties of
Winchil L. C. Vaz, Departamento de Quimica, Universidade de Coimbra, Coimbra, Portugal
doi: 10.1002/9780470048672.wecb281
Lipid bilayers are the structural basis of biological membranes; they provide excellent structures for microencapsulation and are interesting objects of study as thermotropic and lyotropic smectic liquid crystals. This article introduces the fundamental principles that lead to the formation of lipid bilayers and briefly lists some amphiphiles that form lipid bilayers in biological systems. The temperature-dependent phase behavior of bilayers formed from a single lipid species is discussed briefly followed by a discussion of bilayer dimensions and hydration. The article then introduces the reader to phase behavior of bilayers formed from mixtures of lipids and the consequences of phase coexistence for component compartmentalization and percolation in lipid bilayers. Special emphasis is placed on the phase behavior of bilayers that contain sterols. The article introduces the reader to some physical properties of lipid bilayers, which include the hydration force, elasto-mechanical properties, and the resistance of bilayers to lateral stretching and/or compression and bending. Brief discussions of molecular dynamics in lipid bilayers from trans/gauche isomerism and rotation of lipid chains in bilayers to rotational, wobbling, and translational diffusion of lipids in bilayers is followed by an introduction to the dynamics of insertion, desorption, and transverse translocation of lipids in bilayers. Lipid bilayers are then discussed in terms of their permeability to aqueous solutes and some models for bilayer permeation are mentioned briefly. Finally, the reader is introduced to the electrical properties of lipid bilayers with brief discussions of the dipolar potential, the surface electrostatic potential, and the transmembrane potential. The article ends with a brief discussion of lipid compositional asymmetry across the lipid bilayer in biological membranes and its possible causes. Key references are cited wherever relevant.
Lipid bilayers are formed by many amphiphilic molecules in the presence of water. Their interest derives not only from the fact that they are a major, if not the only, organizing principle of biological membranes (1), but also because they tend to form closed (usually) spherical structures (liposomes or lipid bilayer vesicles) in which inner and outer aqueous spaces are separated by the lipid bilayers (2) which thereby provides a means of encapsulation (3, 4).
Amphiphilic molecules (or amphiphiles), in general, tend to aggregate in aqueous solution above some critical concentration (the critical micellar concentration or CMC) because of the hydrophobic effect (5). The aggregate structure secludes the apolar portions of these molecules from the aqueous medium and exposes the polar portions to water at the surface. The form of the aggregate is dictated by packing constraints (6) given by the optimal area per amphiphile,1 a0; at the interface of the aggregate and the aqueous phase, the mean volume of the amphiphiles, V; and their critical length,2 lc. Lipid bilayers are formed when the packing parameter p = V/a0lc ≈ 1. Thus, a lipid bilayer is a self-aggregated sheet of amphiphiles (usually two molecules thick) in which the polar portions of the constituent molecules are exposed to water at the two surfaces of the sheet and the apolar portions are secluded from water in the volume between these two surfaces. Partial or total inter-digitation of the apolar portions of the amphiphiles from the two monolayers of the bilayer is possible under certain conditions. The surfaces of the bilayer in contact with the aqueous phase may be charged depending on the chemical identity of the polar portion of the amphiphiles and the conditions (pH, ionic strength) of the aqueous phase; the bilayer interior always is an apolar environment. The chemical nature of the apolar portions (usually long aliphatic chains) and the fact that these chains are anchored to the polar head groups of the amphiphiles located at the bilayer-water interface, make the lipid bilayer a highly anisotropic structure. Figure 1 (7, 8) shows some typical lipid bilayers.
This article discusses lipid bilayers formed by lipids of interest to biological membranes. The physics of these lipid bilayers have been the subject of excellent monographs (9, 10) and bilayers have been discussed in the context of their colloidal properties (11). Compilations of data on the physical properties of lipid bilayers may be found in References 12 and 13. Most generic physico-chemical properties are common to all bilayers. Much of our knowledge on the properties of lipid bilayers is the result of studies on bilayers formed in the laboratory by hydration of chemically defined diacylipids, phosphoglycerolipids, or sphingolipids. The aggregates formed by these lipids in water are lyotropic and thermotropic in character [i.e., the structure depends on the molar fraction of water and the temperature (14-16)]. These so-called “model” lipid bilayers form spontaneously on hydration of the amphiphiles. Usually, they are studied as oriented single bilayers or on stacked multibilayers on solid supports, multilamellar liposomes or vesicles (MLV), or unilamellar vesicles of different diameters: small unilamellar vesicles with a diameter of about 20 nm (17); large unilamellar vesicles with a diameter of about 100 nm (18); and giant unilamellar vesicles with a diameter in the pm range, usually 10-50 pm (19, 20). MLVs are composed of several concentric lipid bilayer vesicles each separated from the one inside and the one outside by a thin layer of water. This form of lipid bilayer is, therefore, a smectic liquid crystalline phase.

Figure 1. Different types of lipid bilayers. In the LC and LC, phase bilayers, the polar head groups are shown as squares with arrows to indicate that a crystalline order exists in the arrangement of the head groups. In the P' bilayers, the saw-tooth ripple is indicated by the parallel lines, and only a few lipids are drawn in this structure to show the way in which the lipids are arranged in the different domains of this structure. The reader is referred to References (7) and (8) for the structural details of the P' phase.
Chemical Composition
The main lipid constituents of bilayers in biological membranes are derivatives of sn-1,2- (or sn-2,3-) diacylglycerol (phosphatidic acids, phosphatidylethanolamines, phosphatidylcholines, phosphatidylserines, phosphatidylglycerols, phosphatidylinositols, and glycosylated diacylglycerols), derivatives of sphingosine (sphingomyelin, ceramide, and glycosylated derivatives of ceramides), and sterols (cholesterol in mammalian membranes, β-sitosterol, campesterol, and stigmasterol in plants, and ergosterol in eukaryotic microorganisms such as fungi). Cardiolipins (1,3-diphosphatidylglycerols) are important constituents of bacterial membranes and the membranes of mitochondria and chloroplasts. The aliphatic chains of glycerolipids may sometimes be linked to the glycerol by ether linkages as in the case of plasmalogens (important constituents of some mammalian membranes) and some bacterial glycerolipids. The membranes of extremophiles contain variable amounts of lipids in which long-chain (usually branched) α, ω-aliphatic diols are attached by ether linkage to two polyols (usually glycerol) to form bipolar lipid molecules that span the entire lipid bilayer. The aliphatic chains of naturally occurring lipids are usually from 14 to 24 carbon atoms long, and may be fully saturated or unsaturated with from 1 to 4 (usually) cis double bonds. The aliphatic chains of bacterial lipids are often branched and may include cyclic (3- to 6-membered ring) structures. Figure 2 shows the chemical structures of some lipids of biological importance.


Figure 2. Chemical structures of the more common lipids found in biological membranes: a) lipids derived from Diacylglycerols; b) lipids derived from sphingosine; c) the more common sterols; d) some unusual lipids.
Physical Properties
Phase behavior and phase transitions
At low temperatures, in lipid bilayers prepared from a single lipid species, the acyl/alkyl chains of the lipids in the bilayer are characterized by a high trans/gauche configurational ratio. The chains, viewed along their long axes, are packed parallel to each other in a more or less hexagonal lattice with a lattice spacing of about 0.4 nm, and the bilayers are characterized by a high degree of conformational, rotational, and translational order. These bilayers are said to be in the “gel” phase. In gel phases, the acyl/alkyl chain axes may be oriented perpendicular to the bilayer plane (the Lβphase) or be slightly tilted relative to this plane (the Lβ’ phase). The tilt is understood to be the result of the in-plane area of the head groups being slightly larger than the in-plane area of two acyl chains. Depending on the chemical identities of the lipids and the temperature, more than one ordered phase has been characterized in lipid bilayers. In the so-called “sub-gel” or LC (LC’ when the chains are tilted relative to the bilayer normal) phase, which is observed at very low temperatures, the acyl chain order is at least equal to but may be greater than in the Lβ (or Lβ’) phase and may include a crystalline ordering of the lipid molecules. The “rippled” or Pβ’ phase, which is observed in some ordered bilayers at higher temperatures, shows a periodic saw-tooth ripple in the bilayer plane. This rippled pattern has two types of domains, the two faces of the saw-tooth pattern, with distinctly different packing of the lipids, the transition from one domain to the next possibly including some disordered phase lipid (7, 8). The acyl chain order in the Pβ’ phase is lower than that in the Lβ (or Lβ’) and LC (or LC’) phases. Transitions between the various phases in ordered lipid bilayers occur at characteristic temperatures that depend on the chemical identity of the lipids and their acyl chain lengths. When raising the temperature above a characteristic temperature, Tm, which depends on the type of lipid that constitutes the bilayer, the ordered or gel phase is converted into a so-called “liquid-crystalline,” “fluid,” or Lα phase in which the acyl chain configuration is characterized by a low trans/gauche configurational ratio and the chains are still packed in a more or less hexagonal lattice but with a low coherence length and a lattice spacing of about 0.45 nm. Lipid bilayers in the fluid phase have low conformational, rotational, and translational order. The chain-melting transition at Tm is observed in all lipid bilayers regardless of the chemical identity of the lipid that constitutes the bilayer. The different transitions in the gel phase do not occur in all lipid bilayers, in which transitions depend on the lipid species of which the bilayer is constituted. When heating some lipid bilayers in the Lα phase even more (e.g., those formed from some phosphatidylethanolamines), the hydrated lipid aggregate may be converted into one of several other phases that include inverted hexagonal and cubic phases. In bilayers formed from a given class of lipids, the characteristic temperatures at which the various phase transitions occur are dependent on the acyl chain lengths of the lipids and their degree of saturation.
The dependence of the “chain-melting” or main phase transition temperature in a lipid bilayer, Tm, on the nature of the lipid head group as well as the length of the acyl chains and the degree and type (cis- or trans-) of unsaturation of the acyl chains has been studied exhaustively for some diacyl phospholipid classes (21). Bulkier head groups result in lower values of Tm for equivalent acyl chains. For head group homologs with two identical acyl chains, Tm increases monotonically, albeit in a nonlinear manner, with increasing acyl chain length. Usually, naturally occurring lipids have one saturated and one unsaturated acyl chain with one or more carbon-carbon double bonds, the latter usually is in the cis- configuration. When more than one double bond occurs in the same acyl chain, these are usually separated from each other by a methylene group. The unsaturated chain is usually attached to the sn-2 position of glycerol. Lipids with unsaturated acyl chains in the sn-2 position have Tmvalues that are considerably lower than their fully saturated homologs, the reduction depends on the position and the number of unsaturated bonds. Introduction of a single double bond results in a very large drop in Tm, the second unsaturated bond causes a smaller reduction in Tm, and little or no reduction occurs in Tm on further addition of double bonds in an acyl chain. In the case of lipids with a single double bond in the sn-2 acyl chain, the largest effect on Tm is observed when the position of the double bond is roughly in the middle of the acyl chain.
As might be expected, the lipid bilayer phase transitions from a more ordered to a less ordered state are endothermic in nature and can be followed by differential scanning calorimetry (15). Bilayers prepared from dipalmitoylphosphatidyl-choline (DPPC), for example, show at least three endothermic transitions: 1) the so-called subtransition (from the LC’ to the Lβ’ phase), which occurs at about 13°C with a transition enthalpy of ~23 kJ mol-1; 2) the so-called pre-transition (from the Lβ’ to the Pβ’ phase), which occurs at 35°C with a transition enthalpy of ~6.5 kJ mol-1; and 3) the so-called main (or chain-melting) transition, which occurs at 41.5° C with a transition enthalpy of 36.5 kJ mol-1. The enthalpies of the sub-transitions (when these occur) have been found to be ore or less the same for bilayers prepared from different lipids irrespective of acyl chain lengths or head group structure. The same seems to be true for the enthalpies of the pre-transition. The enthalpy of the main or chain-melting transition is, however, very dependent on the acyl chain lengths and the degree as well as the type (cis- or trans-) of unsaturation of the lipid that forms the bilayer. It is similar for similar chain lengths in bilayers formed from lipids with different head groups. The phase transitions are also accompanied by nonmonotonic changes in lipid bilayer volume. Figure 3 (22) shows a differential scanning calorimetric and a dilatometric scan for DPPC bilayers in excess water.

Figure 3. Comparison of a differential scanning calorimetric scan of fully hydrated DPPC multibilayer vesicles (continuous trace) and a dilatometric scan of the same vesicles (circles). The figure is taken from Reference 22.
Molecular and supramolecular dimensions in lipid bilayers
The exact dimensions of a phospholipid bilayer membrane in terms of the in-plane area and the height of the lipid molecules as well as the thickness of the water layer that is associated with them is dependent on the chemical identity of the phospholipid head group, the length and the degree of saturation of the acyl chains, and the degree of hydration. This information may be obtained from a combination of small-angle X-ray diffraction by MLV or oriented multi-bilayer samples of phospholipids in excess water, electron and/or neutron density profiles across lipid bilayers, and atomic level molecular dynamics simulations of hydrated lipid bilayers. 2H-NMR studies on selectively deuterated phospholipids have also been important in elucidating acyl chain and lipid head group conformations. Our current understanding of the structure of phospholipid bilayers in terms of atomic/molecular detail has been summarized critically in an excellent review (23). The structural details of lipid bilayers in ordered (or gel) phases have been easier to define than in fluid (or liquid crystalline) phases that are more relevant for biological membranes. The reasons for this difficulty have to do with the poor diffraction patterns that result from long wavelength fluctuations or undulations in liquid crystalline phase bilayers, which destroy crystalline long-range order in the multilayer and local molecular fluctuations within each bilayer of the liquid crystalline lipid bilayer stacks. In La phase phosphatidylcholine bilayers, the choline head group has a probability distribution function that places it very close to the probability distribution function for the phosphate group (i.e., the choline head group lies roughly parallel to the bilayer plane and only slightly above the level of the phosphate group). The glycerol backbone of the lipid is oriented almost perpendicular to the membrane plane and the sn-1 acyl chain proceeds vertically into the bilayer, whereas the sn-2 acyl chain is kinked at the second carbon. This structure makes the effective length of the sn-2 chain slightly (about 1.5 methylene groups) shorter than the sn-1 acyl chain. Table 1 lists some structural parameters for several phosphatidylcholine bilayers and one phosphatidyl-ethanolamine bilayer in the La phase. For comparison, the same structural parameters are listed for two of these bilayers [DPPC and dilauroylphosphatidyl- ethanolamine (DLPE)] in the gel phase as well. The most obvious structural differences among the La phase bilayer membranes listed in Table 1 can be attributed to the identity of the head group—the number of water molecules associated with the lipid bilayer is significantly different for the phosphatidylethanolamine lipid bilayers (DLPE) when compared with any of the bilayers prepared from phosphatidylcholines. Among the lipid bilayers prepared from phosphatidylcholines, the thickness of the hydrophobic region is slightly less and the area per lipid is slightly more when the acyl groups are unsaturated [1,2-dioleoylphosphatidylcholine (DOPC) and phosphatidylcholine fraction of egg-yolk phospholipids (EPC)] than when they are saturated (DPPC and DMPC). As might be expected, when gel phase bilayers are compared with La phase bilayers prepared from the same lipid chemical species, the hydrophobic core thickness is significantly larger and the area per lipid is significantly lower in the gel phase (fully extended lipid chains). The number of water molecules associated with the lipid bilayers is also significantly reduced in the gel phase compared with the La phase (13 compared with 30 in DPPC and 6 compared with 9 in DLPE).
Table 1. Structural parameters for some fully hydrated phospholipid bilayers (taken from Reference 23)
|
Lipid |
DPPC |
DPPC |
DMPC |
DOPC |
EPC |
DLPE |
DLPE |
|
Temperature |
20° C |
50° C |
30° C |
30° C |
30° C |
20° C |
35° C |
|
VL (nm3) |
1.144 |
1.232 |
1.101 |
1.303 |
1.261 |
0.863 |
0.907 |
|
D (nm) |
6.35 |
6.70 |
6.27 |
6.31 |
6.63 |
5.06 |
4.58 |
|
A (nm2) |
0.479 |
0.640 |
0.596 |
0.725 |
0.694 |
0.410 |
0.512 |
|
2DC (nm) |
3.44 |
2.85 |
2.62 |
2.71 |
2.71 |
3.00 |
2.58 |
|
DHH (nm) |
4.42 |
3.83 |
3.60 |
3.69 |
3.69 |
3.98 |
3.56 |
|
DB’ (nm) |
5.24 |
4.65 |
4.42 |
4.51 |
4.51 |
4.70 |
4.28 |
|
nW |
12.6 |
30.1 |
25.6 |
32.8 |
34.7 |
5.8 |
8.8 |
|
nW’ |
3.7 |
8.6 |
7.2 |
11.1 |
10.2 |
2.0 |
4.7 |
VL, lipid molecular volume; D, lamellar repeat spacing; A, average interfacial area per lipid; 2DC, thickness of the hydrocarbon core of the lipid bilayer; DHH, head group peak to head group peak distance in the electron density profile of a bilayer; DB’, steric thickness of the bilayer, this thickness includes some water intercalated in the polar head group of the lipid; nW, number of water molecules associated with each lipid in the bilayer; nW’, number of water molecules that are “bound” or intercalated in the polar region of the lipid bilayer.
Phase coexistence in lipid bilayers
The properties of lipid bilayers formed from mixtures of lipids are very relevant to the understanding of the lipid bilayers that form the basis of biological membranes. Detailed studies have been performed on bilayers formed from binary lipid mixtures, and some reports in the recent literature describe phase diagrams of lipid bilayers prepared from ternary mixtures that include cholesterol. Figure 4 (24-31) shows some phase diagrams of lipid bilayers formed from binary and ternary mixtures of lipids. The general observation is that lipids in a bilayer are not very miscible with each other in the gel phase. In binary mixtures of phosphatidylcholines, for example, differences in chemical identity of the head group and differences in chain length of four carbon atoms or more lead to gel phase immiscibility. Also, lipids with the capacity to form interdigitated gel phases do not mix well with lipids that do not have this capacity, even though their polar head groups and molecular masses are identical. Lipids generally seem to be mutually miscible in the fluid phase, although at least one report discusses immiscibility in the fluid phase of a bilayer prepared from a binary lipid mixture in which the polar head groups of the two chemical constituents of the bilayer were different.
Lipid bilayers that contain sterols are particularly interesting from the biological perspective because the plasma membranes of the cells of most eukaryotic organisms contain very large molar fractions of sterols (typically 30-50 mol%). In recent years, this aspect has received much attention because of the “raft” hypothesis (for a review on “rafts” see Reference 29). The condensing effect of cholesterol on fatty acids and phosphatidylcholine in monolayers has been known for a long time (32) and has been studied in detail. It was generally accepted that the condensing effect of cholesterol was a result of cholesterol-acyl chain interactions that forced the acyl chains in the liquid-expanded monolayer film to assume a conformation with a higher trans/gauche configurational ratio. Studies on bilayers that contain high cholesterol concentrations confirmed these conclusions for bilayers as well; they showed that cholesterol significantly increased the trans /gauche ratio (conformational order) in the acyl chains of the lipids to values expected for rotating all-trans chains and were significantly higher than those typical for the cholesterol-free La phase. Translational diffusion in the cholesterol-rich bilayers was also shown to be slower compared with cholesterol-free bilayers. The reduction of the long-range translational diffusion coefficient has been reported to be from about 2-3 fold (28, 33) up to about 10-fold (34). In 1987, Ipsen et al. (35) integrated this information into a thermodynamic and a microscopic model for the interaction of cholesterol with the phospholipid molecules. They proposed that cholesterol forms an La phase with lipids in which the lipid chains are ordered conformationally. This phase, which was denominated the liquid ordered (l0 or lo phase), is distinct from the La phase with conformationally disordered chains (which was denominated the ld or ld phase) observed in cholesterol-free lipid bilayers at temperatures above Tm. The formation of the La liquid-ordered phase is the consequence of the flat and rigid cholesterol structure that maximizes its interaction with the lipid acyl chains in an La phase bilayer by forcing these into a predominantly all-trans conformation but, at the same time, cholesterol cannot be incorporated into the in-plane crystalline lattice of a gel phase bilayer because of its size and shape. A compromise solution is to retain the all-trans configuration of the chains and simultaneously maintain the translational order of the fluid phase. Thus, in lipid bilayers that contain cholesterol, two fluid phases could possibly coexist in the lipid bilayer (see phase diagrams for ternary lipid mixtures in Fig. 4) depending on the molar fraction of cholesterol.
Phase coexistence in lipid bilayers may be an important physical property for membranes of cells. When two phases coexist in a bilayer, depending upon the relative mass fractions of the phases and the shapes of their domains, one of the phases is percolative (physically continuous) and the other is nonpercolative (physically discontinuous or dispersed as isolated domains). Changes in the physico-chemical properties of the membrane (lateral pressure, temperature, and chemical composition are the most relevant for biological membranes) result in interconversion between the two phases—one phase grows at the expense of the other. In phase-separated systems of this type, a critical mass ratio of phases called the percolation threshold, at which the previously continuous phase becomes discontinuous and the previously discontinuous phase becomes continuous, becomes an important physical property of the system (36). Phase separation in a membrane may lead to segregation of membrane components, which includes proteins (in-plane compartmentalization) based on their preferred solubility in one or the other of the coexisting phases; this segregation may have important consequences for in-plane bimolecular reactions that occur in these membranes (37). Crossing a percolation threshold (changes in the phase mass ratio may be induced by osmotic stress, temperature changes, or changes in chemical composition) can connect previously disconnected domains and their constituents. The previously continuous domain, and its constituents, becomes simultaneously discontinuous.

Figure 4. Some phase diagrams for lipid bilayers in excess water prepared from binary and ternary lipid mixtures. a) Multibilayer lipid vesicles prepared from binary mixtures of DMPC and DPPC (24); b) Multibilayer lipid vesicles prepared from binary mixtures of DMPC and DSPC [adapted by Reference (25); from data for perdeuterated lipids published by Knoll et al. (26)]; c) Multibilayer lipid vesicles prepared from binary mixtures of diC17:0PC and C22:0C12:0PC (27); d) Multibilayer lipid vesicles prepared from binary mixtures of DMPC and cholesterol (28); e) Multibilayer lipid vesicles prepared from ternary mixtures of palmitoyl sphingomyelin, POPC, and cholesterol [adapted by Reference (29), from data published by De Almeida et al. (30)]; Lipid bilayers prepared from ternary mixtures of DSPC, DOPC, and cholesterol (31).
In systems with phase coexistence, the free energy of the system is discontinuous at the interface between the phases. This discontinuity results in an interfacial tension that drives the system toward a minimization of the interface. In three-dimensional systems, the interfacial tension acts on the area (surface tension); in two-dimensional systems, it acts along the line (line tension) that separates the phases. In both cases, if the interfacial tension is large enough, then it may be expected that the coexisting phases separate into macroscopic domains with the smallest possible separating surface or line between them (a so-called bulk phase separation) at equilibrium. Interfacial tension may be reduced by surface-active agents, which are amphiphilic molecules that locate in the interfacial region and interact more favorably on one side with one of the phases and on the other side with the other phase. Such a reduction in interfacial tension is observed in thermodynamically stable emulsions and in metastable dispersions of immiscible phases. In the lipid bilayers of biological membranes, it is conceivable that some lipids and proteins may act as the amphiphiles that reduce interfacial line tension between domains of coexisting phases in the lipid bilayer.
Although the question of whether phase separations do occur in the lipid bilayers of biological membranes is hotly debated, it is generally accepted that these membranes do show spatial and temporal heterogeneities (domains) in lipid bilayer order. These heterogeneities may have several origins. First, membrane physiology involves processes of vesicle fusion with and budding from a given membrane. These processes will result in localized heterogeneities in chemical composition and physical properties in the lipid bilayer, which dissipate in a diffusion-limited manner within a short time. Second, protein aggregation by some force external to the lipid bilayer (reorganization of the cytoskeleton with its attached proteins; extra-membranal cross-linking of proteins, etc.) may cause a coalescence of the boundary lipid shells around the proteins, which results in a localized change in lipid order that is associated with the protein aggregate. Because the boundary layer has a coherence length of about two lipid shells around the protein surface, this heterogeneity may be expected to dissipate with the disintegration of the protein aggregate. Third, assuming that the system is in a steady state, the lipid bilayer of biological membranes may be a multiphase system because of its compositional complexity and the mutual immiscibility of its chemical constituents. Domains of these phases would be thermodynamically stable. As discussed above, if the line tension around the phase domains is small enough to be insufficient to drive domain growth, then phase coexistence in the bilayer does not necessarily imply a bulk phase separation or that the small dispersed domains of a given phase grow in time. They may, however, be dragged together by forces external to the membrane to form larger domains or platforms.
As discussed at the beginning of this article, the formation of lamellar aggregates such as bilayers by hydrated lipids is dependent on the shape of the lipid molecules, which are characterized by a critical packing parameter, p, whose value must be about equal to 1. When p ≈ 1/3, spherical micellar aggregates are formed, and for p > 1 the aggregate formed is an inverted micelle or an inverted hexagonal phase. Both aggregates are characterized by very large curvatures (curvature = 1/R, where R is the radius of curvature; see Fig. 5b for definitions) (38), the normal micellar aggregate has a positive and the inverted aggregate has a negative curvature. Some physiological events such as membrane fusion, membrane fission, and the drawing out of very narrow tubular membrane structures require the localized formation of membrane structures with high curvatures. Thus, to be responsive to these physiological events, the lipid bilayers in biological membranes must contain a certain amount of lipids that are readily available for and that can form these highly curved structures. At the same time, the presence of these lipids should not cause a significant perturbation of the bilayer membrane and its physical properties. This balance is maintained by biochemical mechanisms that change membrane lipid composition according to need (39).
The hydration force
The characteristics of the water associated with the polar lipid bilayer surfaces are of particular interest because they become very important in processes of physiological significance such as membrane fusion, and they may play a role in the mechanisms of association of proteins and small molecules with lipid bilayers and biological membranes. A very small fraction of the lipid bilayer-associated water molecules are actually immobilized, and a larger fraction (about 30% of the total bilayer-associated water in multilamellar PC bilayers in the La phase) has a probability distribution function that is more or less coincident with the probability distribution function for the polar head group of the lipids. Nevertheless, the pressure, P, which must be exerted to remove the bilayer-associated water, is large and varies (from ~0.5-500 N cm-2) with the thickness of the inter-bilayer water space as:
![]()
where dw and λ are the inter-bilayer water thickness in a multibilayer stack, and λ is a characteristic length (~0.2 nm in La phase PC bilayers). The resistance to removal of the inter-bilayer water has been called the “hydration force”; it is the consequence of contradictory interaction forces: A van der Waals interaction that brings bilayers together is counterpoised by repulsive interactions that result from long-range undulation fluctuations of the fluid lipid layers, steric repulsion of the head groups, and water dipoles with a preferential orientation at the bilayer-water interface (surface hydration). This phenomenon has been discussed by Parsegian and coworkers in excellent reviews (40, 41)
Elasto-mechanical properties of lipid bilayers
Lipid bilayers fall into the category of materials that have come to be known as “soft matter.” These materials are condensed phases that possess many characteristics of liquids but are simultaneously structured. The intermolecular interactions that cause lipid molecules to self-aggregate to fluid bilayers in the presence of water also impart a degree of “toughness” to these bilayers compared with conventional liquids. For detailed discussions of these properties of lipid bilayers, the reader is referred to References 10, 38, and 42. Toughness implies resistance to forces that shear, break, or bend the bilayer (Fig. 5). Bending without breaking and resistance to shear stresses, for instance, are essential properties of the membranes of cells (particularly erythrocytes and platelets) that are forced in the blood stream to squeeze through very narrow capillaries. This property is also essential to cell membranes (in particular the apical surface membranes of epithelial and endothelial cells) that line the lumen of tubes in the body through which relatively viscous fluids flow (for example, the endothelial cells of blood vessels, the epithelial cells of the gastrointestinal tract, the kidney and the urinary tract, the respiratory tract, etc.).
The lipid bilayer membrane in living cells is a fluid membrane and, therefore, has no shear rigidity. However, within the cell and subjacent to the membrane lies an intricate network of the cytoskeleton that is attached with some regularity to the lipid bilayer that constitutes the cell membrane via proteins that are anchored in the bilayer. The shear rigidity of cell membranes is thus provided in a large measure by the cell cytoskeleton.
Resistance of an infinitely thin sheet to stretching (or in-plane compression) and bending stresses are expressed in terms of the respective elasto-mechanical moduli. The area compressibility modulus, KA, is defined via the energy required per unit area EKA to produce an area increment ∆A in a sheet of reference area A0:
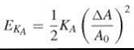
The resistance to bending is expressed in terms of two moduli: the mean curvature modulus, KC, and the Gaussian curvature modulus, KG, which are both defined via the energy, EK, required for bending the sheet (43):
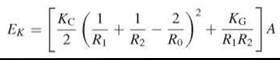
where A is the area of membrane, R1 and R2 are the principal radii of curvature, and R0 is the radius of spontaneous curvature of the sheet (see Fig. 5 for definitions). In the case of lipid bilayers, the spontaneous curvature is the result of the packing parameter, p, for the lipids that constitute the two monolayers of the lipid bilayer (see above for definition) not being exactly equal to 1. For bilaterally symmetrical bilayers, the spontaneous curvature (C0 = R-1) is zero because the two monolayers curve in opposite directions and the force resulting from the hydrophobic effect symmetrically forces them together. For asymmetric bilayers, however, this may not be the case. The mean curvature modulus of a bilayer, KC, and the area compressibility modulus, KA, are related to each other (KC = KAd2, for the simplest case of an infinitely thin sheet), the exact relationship is dependent on the coupling between the monolayers and whether the lateral pressure distribution is uniform across the bilayer (38). KC has a quadratic dependence on the thickness of the bilayer.
Marsh (38) has listed several measured values of the elastic moduli of lipid bilayers. Typically, the area compressibility moduli are in the range of 200-250 mN m-1 for fully hydrated symmetric bilayers in the La phase prepared from phosphatidylcholines, and they are not very dependent on the degree of saturation of the acyl chains. Cholesterol has a significant effect on the area compressibility modulus of a bilayer. Thus, KA for bilayers of 1-stearoyl-2-oleoylphosphatidylcholine (SOPC) increases from 235 mN m-1 in the absence of cholesterol (La liquid-disordered phase) to 640 mN m-1 in bilayers made from an equimolar mixture of SOPC and cholesterol (La liquid-ordered phase). The mean curvature elastic moduli have been reported to be in the range of 10-19 J for La phase phosphatidylcholine bilayers, but the exact value is dependent on the method used for their determination.

Figure 5. a) Schematic view of the mechanical stresses and consequent elasto-mechanical deformations that can occur in lipid bilayers. b) (left panel): Definition of the curvatures in a bent sheet (38); (right panel): Illustration of the spontaneous curvatures in a lipid monolayer.
Molecular dynamics, diffusion, trans-bilayer translocation, and lipid exchange in bilayers
The dynamics of molecules in a lipid bilayer is of fundamental importance in the role that the lipid bilayer plays in a biological membrane. Dynamics in a lipid bilayer spans a frequency scale from ~1014 s-1 (for the vibrational dynamics of single bonds in the lipid molecules) through ~10-5 s-1 for trans-membrane translocation (“flip-flop”) of a lipid molecule. From a functional perspective, the most important dynamic modes that have to be considered are the trans-gauche configurational isomerism in lipid molecules and the rotational and translational diffusional dynamics of these in the bilayer. These dynamic modes determine how fast the lipid bilayer responds to changes in volume and pressure (including localized changes resulting from conformational transitions of proteins associated with the bilayer of biological membranes) and the rate at which lipids exchange between different environments in a bilayer. Another important dynamic mode is the rate at which lipid molecules spontaneously desorb from and insert into lipid bilayers, because these rates determine the lower limit (noncatalyzed rates) at which lipids can exchange between the different (presumed noncontiguous) membrane environments in cells.
Changes in the configuration (trans-gauche isomerism) of single bonds in lipid molecules result in rotational motions in the lipid acyl chains and head groups; an increased configurational entropy is the cause of phase transitions in lipid bilayers. The global lipid conformation is a result of the combination of configurations about single bonds in the acyl chains and the head group. The headgroup and acyl chains may be viewed as anchored positionally at the glycerophosphate group at the aqueous interface, and they have the freedom to execute angular excursions relative to the bilayer normal. The headgroup is, therefore, capable of motion in a cone whose vertex lies at the carbon atom linked to the phosphate group and has some preferred orientation, which is known to be more or less parallel to the bilayer surface in bilayers prepared from phosphatidylcholines and from phosphatidylethanolamines. Each acyl chain is also capable of a conical excursion with the vertex close to the carbonyl group. The angular excursion executed by each carbon atom in the chain varies with the position of the carbon atom in the chain. In general, carbon atoms closer to the glycerophosphate anchor (approximately up to carbon atom 10) execute a smaller angular excursion than those farther away from it. The excursion angle is usually expressed in terms of the C-2H bond order parameters measured by 2H-NMR on lipid bilayers prepared from deuterated lipid molecules (44, 45). Frequencies for trans-gauche isomerization depend, as might be expected, on the physical state of the lipid bilayer, and are found to be 10-3 s-1 for the head group and ~104 s-1 in the chains in the crystalline (LC) phase and increase to ~109 s-1 in the fluid (La) phase (7).
In the fluid (La) phase, the constituents of a lipid bilayer have diffusional freedom that may be described in terms of the diffusion coefficients for wobbling motions of the lipid long axis (D⊥), rotation of the lipids around their long axis (Dll), and the translational diffusion in the plane of the lipid bilayer (Dt). Deterium NMR studies (46) and Molecular Dynamics simulations (47) of pure dimyristoylphosphatidylcholine (DMPC) bilayers at 40-45° C give values of Dll in the range of 108 to 109 s-1 and values of D⊥ of ~107 s-1. In La liquid-ordered phase bilayers prepared from equimolar binary mixtures of DMPC and cholesterol, Dll increases about threefold, whereas D⊥ decreases over 10-fold (48), which is consistent with a higher order and reduced chain entanglement in the liquid-ordered compared with the liquid-disordered phase. The translational diffusion coefficient (Dt) has been measured using a wide variety of techniques. In La, liquid-disordered phase lipid bilayers prepared from pure phosphatidylcholines Dt from 10-11 to 10-12m 2 s-1 depending on the lipid and the temperature (49), which corresponds to a hopping frequency (vh) of 107 to 108 s-1 assuming the area per lipid molecule in the bilayers to be ~0.5 nm2. In gel phase (Lβ’), phase phosphatidylcholine bilayer Dt has been reported to be from 3 to 9 orders of magnitude slower (10-1 s-1 ≤ vh ≤ 105 s-1) than in the fluid phase (for a review see Reference 50). In La liquid-ordered phase membranes prepared from binary mixtures of a phosphatidylcholine and cholesterol, the value of Dt has been reported to be from ~2-3 times lower to about 10 times lower than it is in La liquid-disordered phase membranes prepared from the phosphatidylcholine alone (28, 33, 34).
The theoretical description of translational diffusion in a lipid bilayer depends on the size of the diffusing particle. Theoretical descriptions based on fluid hydrodynamic theory (51, 52) have been shown to be applicable to particles whose radius in the plane of the bilayer is significantly larger than the radius of the lipid molecules that constitute the bilayer, in which case the diffusion coefficient may be given by:
![]()
where k is the Boltzmann constant, T is the temperature, h is the thickness of the bilayer sheet and height of the embedded diffusing particle considered to be a cylinder of radius RC, ηb and ηw are the viscosities of the bilayer fluid and the bounding water, respectively, with ηw « ηb, and γ = 0.5772 is Euler’s constant. The diffusion of lipids or particles with a radius that is equal to or smaller than that of the lipid molecules in a lipid bilayer is not described adequately by the continuum fluid hydrodynamic model; a theoretical model based on free volume theory has been proposed for this purpose according to which Dt may be given by (49, 50):
![]()
where Y is a factor that accounts for overlapping free volumes (0.5 ≤ γ ≤ 1), v0 is the van der Waals molecular volume of a lipid in the bilayer, v* is a critical free volume below which the diffusing particle does not move into an adjacent free volume, v0β is the free volume at the phase transition temperature, Tm, and α is the coefficient of volume expansion of the fluid lipid bilayer. In this model, particles that span the entire thickness of the bilayer feel the translational frictional drag, f, at both the bilayer water interfaces. Particles that span only a single monolayer feel the frictional drag at one of the bilayer-water interfaces and at the bilayer midplane. There are no dissipative interactions within the bilayer itself. The free volume, vf, of the fluid lipid bilayer is the difference between the volume per lipid at temperature T and the van der Waals volume of the lipid:
![]()
The viscous drag felt at the bilayer-water interface can be large when the bounding fluid viscosity approaches the viscosity of the membrane (53) or when the fluid lipid bilayer is associated with a rigid substrate (54).
The lipids in a lipid bilayer may translocate across the bilayer from one monolayer to the apposed monolayer. This transmembrane translocation process, which is also known as “flip-flop,” is slow for lipids with large polar head groups such as glycerolipids and sphingophospholipids but can be fast in the case of lipids with very small polar moieties such as cholesterol. Typical first-order rate constants for transmembrane translocation of a phospholipid-like molecule in La liquid-disordered phase bilayers prepared from 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) are ~10-4 s-1 and may be about 10-fold slower in La liquid-ordered phase bilayers prepared from binary mixtures of the same phosphatidylcholine and cholesterol but is much slower (~10-8 s-1) in La liquid-ordered phase membranes prepared from sphingomyelin and cholesterol (55). The activation free energy for the process, which corresponds to the energy necessary to put the translocating lipid molecule at the bilayer mid-plane, is ~100 kJ mol-1. In contrast, the rate constant for transmembrane translocation of cholesterol may be ~1 s-1 (56).
In any system that contains noncontiguous lipid bilayers, such as a living cell, the lipid components of these bilayers can exchange through a process that involves noncatalyzed desorption of single lipid molecules from one membrane and insertion into another. This lipid exchange involves three processes: 1) a first-order desorption of lipid molecules from the donor bilayer, 2) a diffusion-limited second-order encounter of the lipid monomers in the aqueous phase with the surface of the acceptor bilayer to form an encounter complex, and 3) a first-order insertion of the surface located lipid molecules into the acceptor bilayer. The rate constants for the first and last processes have been measured for lipid-derived probes (57). Desorption rate constants are on the order of 10-5 s-1 from Laliquid-disordered phase bilayers prepared from POPC and are about twofold slower when the bilayer is an La liquid-ordered phase prepared from sphingomyelin and cholesterol. Insertion rate constants are on the order of 1-10 s-1 in La phase bilayer membranes prepared from POPC, with or without cholesterol, and about 10-fold slower when the acceptor membrane is an La liquid-ordered phase prepared from a binary mixture of sphingomyelin and cholesterol. Thus, the noncatalyzed exchange of lipids between noncontiguous lipid bilayers in a cell is a slow process that is limited by the rate of desorption from the donor lipid bilayers.
Lipid bilayers as permeability barriers
Because of its apolar interior, the lipid bilayer is a barrier to diffusional equilibration of solutes between the two aqueous compartments that it separates. The ability of most small solute molecules (50 < molecular weight < 300) to cross the bilayer is directly proportional to their ability to partition into hexadecane or olive oil from an aqueous solution (58), which is an observation first made by Overton (59) and is often referred to as “Overton’s Law.” Permeation of lipid bilayers by small polar molecules and ions seems to occur via one or a combination of both of two mechanisms depending on the nature of the permeants and the nature of the bilayers. First, a “solubility-diffusion” mechanism treats the bilayer as a slab of liquid hydrocarbon sandwiched between two bulk aqueous compartments. The permeant must partition into the bilayer slab from one of the aqueous compartments, diffuse across it, and leave by dissolving into the second aqueous compartment. In this case, the permeability coefficient, P, is given by:
![]()
where KP is the partition coefficient of the permeant between the bilayer and aqueous phases, Dm is its translational diffusion coefficient in the bilayer phase and d is the bilayer thickness. Second, a pore mechanism assumes the formation of transient water-filled pores (often referred to as “water wires”) across the bilayer because of density fluctuations in it. The permeant (in a partially or fully hydrated state) is then assumed to diffuse through the bilayer via these transient pores. In this case, the permeation coefficient is proportional to the probability of formation of pores that span the entire bilayer, the mean pore radius, and the translational diffusion coefficient of the permeant in water. The expected dependence of the permeation coefficient on bilayer thickness is steeper in the case of the pore mechanism than it is in the solubility-diffusion mechanism, which thereby provides the basis for a critical test of which mechanism is applicable to the permeation of a given permeant particle (60). Uncharged, small polar molecules such as water, glycerol, and urea permeate bilayers, regardless of their thickness, via the solubility-diffusion mechanism. Protons seem to permeate bilayers predominantly via the pore mechanism for bilayers with an apolar layer thickness of up to ~3 nm (phosphatidylcholine bilayers with acyl chains up to about 16 to 18 carbon atoms); the solubility-diffusion mechanism becomes predominant for thicker bilayers. Potassium and halide ions permeate bilayers almost completely via a solubility-diffusion mechanism when the bilayers are thicker than ~2 nm (60, 61). Typical permeability coefficients, for permeation of bilayers prepared from phosphatidylcholines in the La liquid-disordered phase with a 3 nm hydrophobic thickness, are ~10-5 m s-1 for water permeation, ~10-6 ms-1 for the permeation of protons, ~3 x 10-14 ms-1 for potassium ions, and ~6 x 10-11 ms-1 for chloride ions.
Electrical properties of lipid bilayers
Lipid bilayers have electrical properties that are not just a result of the charges on the lipid molecules. The association of any charged particle with a lipid bilayer and/or the permeation of lipid bilayers by any charged particle are strongly conditioned by the electrical properties of the bilayer. Three types of electrical potentials are associated with lipid bilayers suspended in aqueous electrolyte solutions: 1) a dipole potential, 2) a surface potential, and 3) a transmembrane potential.
The dipole potential (discussed in Reference 62) results from the anisotropic orientation of the lipids in a bilayer and from orientation of water dipoles at the lipid bilayer-water interface. The dipole potential of an La liquid-disordered phase phosphatidylcholine bilayer is ~250 mV, which is positive inside the bilayer. The anisotropic orientation of the lipid molecules in a bilayer results in a “surface dipole moment” whose main contributions come from the dipole moments of the terminal methyl groups and the carbonyl groups of the lipid acyl chains, particularly the carbonyl group of the acyl chain attached to the sn-2 position of glycerol. The dipole moments of the lipid head groups and the chain methylene groups are assumed to contribute nothing because of their orientation parallel to the surface. A very significant, if not the major, contribution to the dipole potential may have its origin in the orientation of bound water dipoles at the membrane-water interface (63). As observed earlier, the orientation of water molecules at the bilayer surface is an important cause of the hydration force. The dipole potential of a lipid bilayer, which is dependent on lipid orientation, is different for different lipid phases. More ordered lipid bilayers might be generally expected to have a larger dipole potential. Differences in the dipole potential of different coexisting phases in a bilayer are responsible for determining the size, shape, and arrangement of domains in lipid monolayers (and possibly bilayers as well) and may make the coalescence of dispersed domains of the same phase to a macroscopically separated phase in the bilayer a very slow process (64). It may even determine the transverse superposition of domains of the same (or similar) phase across the lipid bilayer with phase coexistence (65).
The surface potential of a bilayer is a result of having charged lipids in this bilayer (for reviews see References 63 and 66). In fluid phase bilayers, rapid translational diffusion of the lipids allows the surface charge associated with the lipids to be considered a smeared charge and the electrostatic potential at the surface of the bilayer, ψ0, is well described by the Gouy-Chapman equation:
![]()
where z is the valence of the counter ion in the electrolyte solution, e is the electronic charge, k is the Boltzmann constant, T is the temperature, a is the surface charge density, c is the number of ions of a z:z electrolyte in the aqueous solution, A = (8ε0εwkT)-1/2 is a constant, ε0 is the permittivity of free space, and εw is the dielectric constant of water (~80). The electrostatic potential varies with distance, x, from the charged surface as
![]()
where ![]()
![]() is the Debye length. For small surface potentials, the Gouy-Chapman equation for the surface potential reduces to:
is the Debye length. For small surface potentials, the Gouy-Chapman equation for the surface potential reduces to:
![]()
and the expression for the distance-dependent potential reduces to:
![]()
The surface potential results in a concentration of the counter-ions and a depletion of the co-ions in the aqueous phase close to the charged surface, which results in the so-called electrical double layer. The ion concentrations in the aqueous phase at a given distance x from the charged surface are given by:
![]()
It is important to note that each monolayer that constitutes a lipid bilayer has its own surface potential, and whether the surface potential is equal on both sides of the lipid bilayer will depend on the transverse compositional symmetry of the bilayer. Lipid bilayers in biological membranes are usually compositionally asymmetric, which is a fact that could originate a potential difference (generally given as ∆Ф in the literature) between the two membrane surfaces that may be expected to play a role in ion permeation of the lipid bilayer and, therefore, be important in cell membrane physiology. One consequence of the surface potential is that protons are concentrated (or depleted in the case of a positively charged surface) in the electrical double layer with the resulting consequence that the apparent pKa of ionizable groups that suffer acid-base equilibria close to the surface of the bilayer is different from what it would be in bulk aqueous solution. The value of this apparent pKa is dependent on the ionic strength of the bulk aqueous electrolyte solution and approaches the value of the pKa in bulk solution as the ionic strength is raised.
The transmembrane potential is the difference in the electrical potentials of the two bulk aqueous phases separated by the bilayer (for a detailed treatment see Reference 67). The lipid bilayer may be viewed as an electrical capacitor in which the two conducting plates are the electrolyte solutions on either side of the bilayer and the apolar portion of the bilayer is the dielectric. Lipid bilayers are characterized by a very high specific capacitance (0.5-1 μF cm-2). The transmembrane potential is generated by the fact that some ions permeate lipid bilayers more rapidly than others do. An electrolyte concentration gradient (diffusion potential) across a lipid bilayer results in the more rapidly permeating ion crossing the bilayer much faster than its counter ion, which creates a charge separation and, therefore, an electrical potential across the membrane. The electrical potential works on the flux of the ions in a direction that is contrary to that of the diffusion potential. At equilibrium, the electrical potential and the diffusion potential are exactly balanced so that no net flux of ions occurs. The resulting transmembrane potential with respect to a given ion, δψN, is given by the Nernst equation:
![]()
where R is the gas constant, F is the Faraday constant, CN are the concentrations of the ion N, and the subscripts (1) and (2) represent the electrolyte solutions on the two sides of the bilayer. A biological membrane is bathed in electrolyte solutions that have different ions that permeate the bilayer, each at its own rate. Thus, if the bilayer is bathed in aqueous solutions of NaCl, then the concentrations of both Na+ and Cl- on both sides of the bilayer and the respective permeation coefficients, PNa+ and PCl- ,must be taken into account to account for the global transmembrane potential, which is done in the Goldman-Hodgkin-Katz equation:
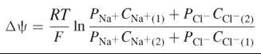
where the subscripts (1) and (2) refer, as before, to the electrolyte solutions on the two sides of the bilayer.
In living cells, ion channels and energy-dependent pumping mechanisms (with more or less specificity) facilitate or catalyze the permeation of certain ions in one direction or the other across the lipid bilayers that constitute the cell membranes. This mechanism makes the permeation of certain ions several orders of magnitude faster than the permeation of others; which thereby accentuates the transmembrane potentials discussed above. These processes are particularly important in the creation and the transmission of the action potential in nerves.
Lipid bilayer asymmetry in biological membranes and its possible causes
Most lipid bilayers prepared by hydration of lipids in the laboratory (the so-called model systems) are bilaterally symmetric at equilibrium (i.e., the chemical composition with regard to the lipids of each monolayer that constitutes these bilayers is identical).3 That is not the case in the lipid bilayers of biological membranes that are known to be compositionally asymmetric in all examined cases. An important contribution to the transverse asymmetry observed in biological membranes comes from the vectorial nature of lipid biosynthesis in cell organelles such as the endoplasmic reticulum and the Golgi compartments. Other contributions certainly come from the relatively slow transmembrane translocation (see above) of lipids in lipid bilayers, the asymmetric chemical composition of the aqueous compartments on the two sides of the membrane, and from the lipid shape-dependent (see discussion on the packing parameter above) spontaneous curvature of lipid monolayers. When all contributions are insufficient, the living cell seems to maintain lipid asymmetry in its membranes enzymatically using the so-called “flippases” that may or may not be dependent on metabolic energy (68, 69). Transverse translocation of most polar lipids across a lipid bilayer involves an intermediate “transition state,” in which the polar portion of the lipid molecule lies in the highly apolar bilayer midplane, with an activation free energy (∆G°) on the order of 100 kJ mol-1. It would seem, therefore, that transverse asymmetry is of paramount importance in cell membrane structure and physiology. It will be an important challenge from the perspective of Cell Biology and Biochemistry to understand why. From the perspective of lipid bilayer Physics and Chemistry it will be a nontrivial challenge to make relatively stable compositionally asymmetric lipid bilayers.
References
1. Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science 1972; 175:720-731.
2. Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface active agents as observed in the electron microscope. J. Mol. Biol. 1964; 8:660-668.
3. Gregoriadis G. Engineering liposomes for drug delivery: progress and problems. Trends Biotech. 1995; 13:527-537.
4. Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic aents to solid tumors. Pharm. Rev. 1999; 51:691-743.
5. Tanford C. The Hydrophobic Effect: Formation of Micelles and Biological Membranes. 1973. John Wiley and Sons, New York.
6. Israelachvili, JN, Mitchell DJ, Ninham BW. Theory of selfassembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans II 1976; 72:1525-1568.
7. Sun WJ, Tristram-Nagle S, Suter RM, Nagle JF. Structure of the ripple phase in lecithin bilayers. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:7008-7012.
8. De Vries AH, Yefimov S, Mark AE, Marrinck SJ. Molecular structure of the lecithin ripple phase. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:5392-5396.
9. Cevc G, Marsh D. Phospholipid Bilayers. 1987. John Wiley and Sons, New York. pp. 442.
10. Mouritsen OG. Life as a Matter of Fat: The Emerging Science of Lipidomics, 2005. Springer-Verlag, Berlin.
11. Evans DF, Wennerstrom H. The Colloidal Domain-Where Physics, Chemistry, Biology and Technology Meet, 2nd edition. 1994. Wiley-VCH, New York.
12. Marsh D. Handbook of Lipid Bilayers. 1990. CRC Press, Boca Raton, FL.
13. Koynova R, Caffrey M. An index of lipid phase diagrams. Chem. Phys. Lipids 2002; 115:107-219.
14. Chapman D. Liquid crystals and cell membranes. Ann. N.Y. Acad. Sci 1966; 137:745-754.
15. Chapman D, Williams RM, Ladbrooke BD. Physical studies of phospholipids. VI. Thermotropic and lyotropic mesomorphism of some 1,2-diacylphosphatidylcholines (lecithins). Chem. Phys. Lipids 1967; 1:445-475.
16. Luzzati V, Tardieu A. Lipid phases - structure and structural transitions. Annu. Rev. Phys. Chem. 1974; 25:79-94.
17. Huang CH. Studies on phosphatidylcholine vesicles. Formation and physical characteristics. Biochemistry 1969; 8:344-352.
18. Hope MJ, Bally MB, Webb G, Cullis PR. Production of large unilamellar vesicles by a rapid extrusion procedure - characterization of size distribution, trapped volume and ability to maintain a membrane potential. Biochim. Biophys. Acta 1985; 812:55-65.
19. Vaz WLC, Kapitza HG, Stilmpel J, Sackmann E, Jovin TM. Translational mobility of glycophorin in bilayer membranes of dimyristoylphosphatidylcholine. Biochemistry 1981; 20:1392-1396.
20. Angelova ML, Dimitrov DS. Liposome electroformation. Farady Disc. Chem. Soc. 1986; 81:303-311.
21. Huang CH, Li SS. Calorimetric and molecular mechanics studies of the thermotropic phase behavior of membrane phospholipids. Biochim. Biophys. Acta 1999; 1422:273-307.
22. Tristram-Nagle S, Nagle JF. Lipid bilayers: thermodynamics, structure, fluctuations, and interactions. Chem. Phys. Lipids 2004; 127:3-14.
23. Nagle JF, Tristram-Nagle S. Structure of lipid bilayers. Biochim. Biophys. Acta 2000; 1469:159-195.
24. Mabrey S, Sturtevant JM. Investigation of the phase transitions of lipids and lipid mixtures by high sensitivity differential scanning calorimetry. Proc. Natl. Acad. Sci. U.S.A. 1976; 73:3862-3866.
25. Vaz WLC, Melo ECC, Thompson TE. Translational diffusion and fluid domain connectivity in a two-component, two-phase phospholipid bilayer. Biophys. J. 1989; 56:869-875.
26. Knoll WK, Ibel K, Sackmann E. Small-angle neutron scattering study of lipid phase diagrams by the contrast variation method. Biochemistry 1981; 20:6379-6383.
27. Bultmann T, Vaz WLC, Melo ECC, Sisk RB, Thompson TE. Fluid-phase connectivity and translational diffusion in a eutectic two-component, two-phase phosphatidylcholine bilayer. Biochemistry 1991; 30:5573-5579.
28. Almeida PFF, Vaz WLC, Thompson TE. Lateral diffusion in the liquid phases of dimyriostoylphosphatidylcholine/cholesterol lipid bilayers: a free volume analysis. Biochemistry 1992; 31:6739- 6747.
29. Simons K, Vaz WLC. Model systems, lipid rafts, and cell membranes. Annu. Rev. Biophys. Biomol. Struct. 2004; 33:269-295.
30. De Almeida RFM, Fedorov A., Prieto M. Sphingomyelin/phosphatidyl choline/cholesterol phase diagram: boundaries and composition of lipid rafts. Biophys. J. 2003; 85:2406-2416.
31. Zhao J, Wu J, Heberle FA, Mills TT, Klawitter P, Huang G, Constanza G, Feigenson GW. Phase studies of model biomembranes: complex behavior of DSPC/DOPC/Cholesterol. Biochim. Biophys. Acta 2007; 1768:2764-2776.
32. Leathes JB. Croonian lectures on the role of fats in vital phenomena. Lancet 1925; 853-856.
33. Filippov A, Oradd G, Lindblom G. The effect of cholesterol on the lateral diffusion of phospholipids in oriented bilayers. Biophys. J. 2003; 84:3079-3086.
34. Korlach J, Schwille P, Webb WW, Feigenson GW. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8461-8466.
35. Ipsen JH, Karlstrom G, Mouritsen OG, Wennerstrom H, Zuckermann MJ. Phase equilibria in the phosphatidylcholine-cholesterol system. Biochim. Biophys. Acta 1987; 905:162-172.
36. Vaz WLC, Almeida PFF. Phase topology and percolation in multi-phase lipid bilayers: is the biological membrane a domain mosaic? Curr. Opin. Struct. Biol. 1993; 3:482-488.
37. Vaz WLC. Diffusion and chemical reactions in phase-separated membranes. Biophys. Chem. 1994; 50:139-145.
38. Marsh D. Elastic curvature constants of lipid monolayers and bilayers. Chem. Phys. Lipids 2006; 144:146-159.
39. Rilfors L, Lindblom G. Regulation of lipid composition in biological membranes - biophysical studies of lipids and lipid synthesizing enzymes. Colloids Surf. B 2002; 26:112-124.
40. Rand RP, Parsegian VA. Hydration forces between phospholipid bilayers. Biochim. Biophys. Acta 1989; 988:351-376.
41. Leikin S, Parsegian VA, Rau DC, Rand RP. Hydration forces. Annu. Rev. Phys. Chem. 1993; 44:369-395.
42. Evans EA, Skalak R. Mechanics and Thermodynamics of Biomembranes. 1980. CRC Press, Boca Raton, FL.
43. Helfrich W. Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch. 1973; 28:693-703.
44. Seelig J, Seelig A. Lipid conformation in model membranes and biological membranes. Quart. Rev. Biophys. 1980; 13:19-61.
45. Brown MF, Thurmond RL, Dodd SW, Otten D, Beyer K. Elastic deformation of membrane bilayers probed by deuterium NMR relaxation. J. Am. Chem. Soc. 2002; 124:8471-8484.
46. Nevzorov AA, Trouard TP, Brown MF. Lipid bilayer dynamics from simultaneous analysis of orientation and frequency dependence of deuterium spin-lattice and quadrupolar order relaxation. Phys. Rev. E 1998; 58:2259-2281.
47. Lindahl E, Edholm O. Molecular dynamics simulation of NMR relaxation rates and slow dynamics in lipid bilayers. J. Chem. Phys. 2001; 115:4938-4950.
48. Trouard TP, Nevzorov AA, Alam TM, Job C, Zajicek J, Brown MF. Influence of cholesterol on dynamics of dimyristoylphos- phatidylcholine bilayers as studied by deuterium NMR relaxation. J. Chem. Phys. 1999; 110:8802-8818.
49. Vaz WLC, Clegg RM, Hallmann D. Translational diffusion of lipids in liquid crystalline phase phosphatidylcholine multibilayers. A comparison of experiment with theory. Biochemistry 1985; 24:781-786.
50. Clegg RM, Vaz WLC. Translational diffusion of proteins and lipids in artificial lipid bilayer membranes. A comparison of experiment with theory. In: Progress in Protein-Lipid Interactions, Vol. I. Watts A, De Pont JJHHM, eds. 1985. Elsevier, Amsterdam, The Netherlands. pp. 173-229
51. Saffman PG. Brownian motion in thin sheets of viscous fluid. J. Fluid Mech. 1976; 73:593-602.
52. Hughes BD, Pailthorpe BA, White LR. The translational and rotation drag on a cylinder moving in a membrane. J. Fluid Mech. 1981; 110:349-372.
53. Vaz WLC, Stumpel J, Hallmann D, Gambacorta A, De Rosa M. Bounding fluid viscosity and translational diffusion in a fluid lipid bilayer. Eur. Biophys. J. 1987; 15:111-115.
54. Evans EA, Sackmann E. Translational and rotational drag coefficients for a disk moving in a liquid membrane associated with a rigid substrate. J. Fluid Mech. 1988; 194:553-561.
55. Moreno MJ, Estronca LMB, Vaz WLC. Translocation of phospholipids and dithionite permeability in liquid-ordered and liquid- disordered membranes. Biophys. J. 2006; 91:873-881.
56. Steck TL, Ye J, Lange Y. Probing red cell membrane cholesterol movement with cyclodextrin. Biophys. J. 2002; 83:2118-2125.
57. Estronca LMBB, Moreno MJ, Laranjinha JAN, Almeida LM, Vaz WLC. Kinetics and thermodynamics of lipid amphiphiles exchange between lipoproteins and albumin in serum. Biophys. J. 2005; 88:557-565.
58. Walter A, Gutknecht J. Permeability of small nonelectrolytes through lipid bilayer membranes. J. Membrane Biol. 1986; 90:207-217.
59. Overton E. Ueber die allgemeinen osmotischen Eigenschaften der Zelle, ihre vermutlichen Ursachen und ihre Bedeutung fur die Physiologie. Vierteljahrschr. Naturforsch. Ges. Zurich 1899; 44:88-135.
60. Paula S, Volkov AG, Van Hoek AN, Haines TH, Deamer DW. Permeation of protons, potassium ions, and small polar molecules through phospholipid bilayers as a function of membrane thickness. Biophys. J. 1996; 70:339-348.
61. Paula S, Volkov AN, Deamer DW. Permeation of halide anions through phospholipid bilayers occurs by the solubility-diffusion mechanism. Biophys. J. 1998; 74:319-327.
62. Brockman H. Dipole potential of lipid membranes. Chem. Phys. Lipids 1994; 73:57-79.
63. Cevc G. Membrane electrostatics. Biochim. Biophys. Acta 1990; 1031:311-382.
64. De Koker R, McConnell HM. Circle to dogbone: shapes and shape transitions of lipid monolayer domains. J. Phys. Chem. 1993; 97:13419-13424.
65. Smorodin V, Melo E. Shape and dimensions of gel domains in phospholipid bilayers: a theoretical study. J. Phys. Chem. 2001; 105:6010-6016.
66. McLaughlin S. The electrostatic properties of membranes. Annu. Rev. Biophys. Biophys. Chem. 1989; 18:113-136.
67. Hille B. Ion Channels of Excitable Membranes. 3rd edition. 2001. Sinauer Associates, Inc., Sunderland, MA.
68. Devaux PF. Phospholipid flippases. FEBS Lett. 1988; 234:8-12.
69. Daleke DL. Phospholipid flippases. J. Biol. Chem. 2007; 282: 821-825.
Further Reading
Cevc G, Marsh D. Phospholipid Bilayers: Physical Principles and Models. 1987. John Wiley and Sons, New York.
Gennis RB. Biomembranes: Molecular Structure and Function. 1989. Springer Verlag, Berlin.
Hanahan DJ. 1997. A Guide to Phospholipid Chemistry. Oxford University Press, New York.
Larsson K. Lipids: Molecular Organization, Physical Functions and Technical Applications. 1994. The Oily Press, Dundee.
Lipowsky R, Sackmann E, eds. Handbook of Biological Physics, Vol. 1A and B: Structure and Dynamics of Membranes, from Cells to Vesicles. 1996. Elsevier Science B.V., Amsterdam, The Netherlands. Mouritsen OG. 2005. Life - As a Matter of Fat: The Emerging Science of Lipidomics. Springer-Verlag, Berlin.
Yeagle PL, ed. The Structure of Biological Membranes. 2nd edition. 2005. CRC Press, Boca Raton, FL.
See Also
GENERAL/INTRODUCTION
Analytical Chemistry in Biology
Computational Chemistry in Biology
Lipid Micelle Organization
Membrane Architecture-function Relationships
Membrane Assembly in Living Systems
Membrane Fusion, Mechanisms of
Membranes: Topics in Chemical Biology
Modeling and Computation: Lipid Assemblies
Nature: A Model System for Chemists
Organic Chemistry in Biology
Physical Chemistry in Biology
Self Assembling Systems, Applications of
Self Assembling Systems, Synthetic, Applications of
Self Assembly, Bioenergetics of
Self Assembly, Computation and Modeling of
Therapeutics, Lipid Membrane Applications
Vesicle Formation, Dynamics of
Water, Properties of
CHEMICAL COMPOSITION
Chromatography of Lipids
Lipids from Whole Cells and Tissues, Extraction of
Fluorescence Techniques: Lipids
GC-MS of Lipids
Glycolipids, Synthesis of
Imaging Techniques: Lipids
Labeling Techniques: Lipids
Lipidomics
Lipids, Semi-synthetic
Steroid and Triterpene Biosynthesis
PHYSICAL PROPERTIES: PHASE BEHAVIOR AND PHASE TRANSITIONS
Calorimetry: Overview of Applications in Chemical Biology
Lipids, Phase Transitions of
PHYSICAL PROPERTIES: MOLECULAR AND SUPRAMOLECULAR DIMENSIONS IN LIPID BILAYERS
Lipid Orientation, Determination of
NMR of Lipids
NMR: Overview of Application in Chemical Biology
PHYSICAL PROPERTIES: PHASE COEXISTENCE IN LIPID BILAYERS
Lipid Domains, Chemistry of
Lipid Rafts
PHYSICAL PROPERTIES: ELASTO-MECHANICAL FORCES IN LIPID BILAYERS
Cell Membranes, Dynamics of
Membranes, Fluidity of
PHYSICAL PROPERTIES: MOLECULAR DYNAMICS, DIFFUSION, TRANS-BILAYER TRANSLOCATION, AND LIPID EXCHANGE IN BILAYERS
Cell Membranes, Dynamics of
Lipid Homeostasis, Chemistry of
Membrane Trafficking
PHYSICAL PROPERTIES: LIPID BILAYERS AS PERMEABILITY BARRIERS
Ion Transport
Water Channels
PHYSICAL PROPERTIES: ELECTRICAL PROPERTIES OF LIPID BILAYERS
Brain Development, Neurochemistry of
Electrochemical gradients
____________________
1The “optimal area per amphiphile” is defined as the mean area per amphiphile at the surface of the aggregate when the free energy per amphiphile in the aggregate is a minimum.
2The critical length is the mean length of the amphiphiles in an aggregate along the normal to the aggregate/aqueous interface.
3 This may not be the case when the model system is a small unilamellar vesicle in which the monolayer curvatures are significantly different for the inner and outer monolayer. If these vesicles are prepared from a mixture of lipids, the inner monolayer may be expected preferentially to contain that lipid species that has a more negative spontaneous curvature at the bilayer water interface.