CHEMICAL BIOLOGY
Bacterial Resistance to Antibiotics
Gerard D Wright, Antimicrobial Research Centre, Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, Ontario, Canada
doi: 10.1002/9780470048672.wecb034
Microbes have evolved a myriad of mechanisms to overcome the toxic effects of antibiotics, which include the production of enzymes that modify or degrade antibiotics, complex membrane-associated efflux systems that can pump antibiotics out of the cell, modification of antibiotic targets, and the production of immunity proteins. The biochemical logic of resistance is often intimately linked with the mode of action of the antibiotics. As a result, chemical biology approaches for understanding resistance not only have application in our understanding of this phenomenon, but also they can guide the generation and deployment of new antibiotics. In this review, a survey of the chemical strategies employed by bacteria to resist antibiotics is presented with an emphasis on the molecular mechanisms of resistance enzymes and proteins.
Modern anti-infective chemotherapy is founded on the deployment of an arsenal of potent and safe small-molecule inhibitors of microbial growth. As a result, it is not an understatement to say that antibiotics and antibacterial agents have revolutionized health care during the last century. These compounds are, respectively, natural product and synthetic small molecules that inhibit bacterial growth. For simplicity, in this review, the term “antibiotic” will be used to refer to both natural products and synthetic molecules.
Since the first articulation of the concept of the “magic bullet” by Paul Ehrlich to describe the discovery and use of small-molecule inhibitors of microbial growth (1), there has been a concerted effort to identify new antibiotics in chemical l.ibraries and from natural sources. Following Ehrlich’s efforts, the synthetic sulfa drugs that inhibit p-aminobenzoic acid biosynthesis dominated anti-infective therapy in the 1930s and early 1940s. The discovery of the highly potent fungal natural product penicillin, followed by peptide antibiotics such gramicidin from soil bacteria (2), ushered in a period of roughly 15 years of intense natural product drug discovery that revealed almost all the chemical classes of antibiotics in current clinical use.
As has been well documented elsewhere (3, 4), there was roughly a 25-year gap between the introduction of the quinolone antibiotics and the next new chemical class of antibiotics, the oxazolidinones. During this interval, innovation in antibiotic drug design and discovery was focused on the semisynthetic tailoring of natural product antibiotic scaffolds to improve pharmacologic properties and, most importantly, to overcome resistance to existing antibiotics.
Since the first use of antibiotics, bacterial insensitivity to these cytotoxic agents, both intrinsic and acquired, has been observed. Antibiotic resistance can be the result of an inability of the compound to enter the cell, active efflux from the cytosol, mutation or alteration of the primary molecular target, sequestration of the antibiotic, and enzymatic destruction or chemical modification of the compound (Table 1). Each of these mechanisms requires unique chemical strategies to achieve the same biological outcome: continued cell growth in the face of toxic compounds. This review will discuss the principal mechanisms of antibiotic resistance emphasizing the biochemical logic of the strategy to overcome the cytotoxic effects of antibiotics.
Biological Context of Antibiotic Resistance
Antibiotic resistance is a major clinical problem with great impact on the successful treatment of infectious disease (5). Resistance increases mortality and morbidity, and it lengthens hospital stays. Health-care costs associated with antibiotic-resistant infections range between $6000 and $30,000 USD per patient (6). Furthermore, the rate of resistance continues to rise in all important human pathogens year after year (7). Limits on the use of certain antibiotics and drug cycling can help in some cases to mitigate resistance; however, even when restricted-use protocols are in place, resistance continues to be a problem (8, 9). Therefore, once resistance emerges, it is a continuing clinical difficulty that must be managed.
Table 1 Molecular mechanisms of antibiotic resistance
|
Mechanism |
Example antibiotics |
|
Enzymatic |
|
|
Inactivation (e.g., Hydrolysis) |
β-Lactams, Fosfomycin |
|
Covalent Modification |
Aminoglycosides, Chloramphenicol |
|
Oxidation/Reduction |
Tetracycline |
|
Altered transport |
|
|
Efflux |
Tetracycline, Fluoroquinolones |
|
Reduced Uptake |
Aminoglycosides, Chloramphenicol |
|
Others |
|
|
Sequestration |
β-Lactams, Enediynes |
|
Immunity Proteins |
Bleomycin, Tetracycline |
|
Target Modification |
Fluoroquinolones, Macrolides |
|
Altered Metabolic Pathway |
Vancomycin |
|
Target Overexpression |
Trimethoprim |
Paradoxically, it is the remarkable potency of these drugs that is their Achilles heel. Resistant bacteria are selected by exposure to antibiotics, which nondiscriminately kill off susceptible organisms. The resultant and powerful “adapt or die” evolutionary pressure selects for microbes with the ability to evade the toxic effects of these agents. This antibiotic insensitivity can be intrinsic, i.e., a consequence of the genetic or physiologic makeup of microorganisms, or it can be acquired from other sources, generally through the aegis of mobile genetic elements.
Intrinsic antibiotic resistance
Microbial metabolism is largely contained within the cell envelope and the cell interior. The intracellular physiology and biochemistry (ribosomes and protein translation, nucleic acid replication and transcription machinery, metabolic pathways, etc.) are, for the most part, conserved throughout the bacterial kingdom. On the other hand, the bacterial cell envelope is more structurally diverse. In particular, the presence of a relatively impermeable outer membrane in Gram-negative bacteria is a barrier to many classes of antibiotics. The bacterial outer membrane comprises an asymmetric bilayer with a phospholipid interior and a lipopolysaccharide (LPS) exterior. This anionic LPS layer is bridged through cationic metals providing an additional physical diffusion barrier for small and large molecules. Access to the interior of the cell is provided by a series of membrane proteins (e.g., porins) that recognize and allow the transport of nutrients and other metabolites. Antibiotics such as the glycopeptides, macrolides, and others have difficulty penetrating the outer membrane. As a result, Gram-negative bacteria are consequently intrinsically resistant to these drugs; microbial physiology is therefore the resistance determinant.
The absence of a sensitive microbial target is also a form of genetic intrinsic resistance. The oxirane antibiotic fosfomycin covalently modifies the target protein MurA on a sensitive and catalytically relevant Cys residue (Fig. 1) (10). MurA catalyzes the formation of N-acetylmuramic acid via enoylpyruvyl transfer of phosphoenol pyruvate onto the acceptor sugar N-acetylglucosamine, providing the necessary carboxylate anchor for attachment of the pentapeptide required for bacterial peptidoglycan (cell wall) formation. Bacteria that encode a MurA orthologue where the susceptible Cys is replaced by an Asp (e.g., Mycobacteria) are intrinsically genetically resistant to fosfomycin (Fig. 1) (11, 12).
Another confounding physiologic barrier to antibiotic action is the bacterial growth state. Most bacteria can grow in planktonic form where cells are free living and are uniformly exposed to antibiotics. Most antibiotic susceptibility tests are in fact performed on this cell state to determine the minimal inhibitory concentration (MIC) of antibiotic required to arrest cell growth. However, many important pathogens such as Staphylococcus aureus and Pseudomonas aeruginosa can form biofilms on surfaces (e.g., on catheters or mucosal layers of the lung) where most cells are metabolically quiescent and often highly resistant to antibiotics. Furthermore, other organisms can enter eukaryotic cells and thus evade host defense systems. For pathogens such as Mycobacterium tuberculosis and Chlamydia, it is essential for pathogenesis, whereas for others such as the enteric pathogens Salmonella and Escherichia, it exacerbates infection. Once inside the cell, these organisms often shift metabolic activities and can enter a relatively inactive state, sometimes for long periods of time. Successful antibiotic therapy, therefore, requires not only penetration of the eukaryotic cell without associated toxicity, but also sufficient antimicrobial activity to kill the often slow-growing bacterial cells in this environment.
These passive obstacles to antibiotic action complicate treatment of infectious disease in the absence of highly accurate diagnostic tools to identify the infectious pathogen. Intrinsic resistance is problematic but is predictable and easily identified in the preclinical drug discovery stages. As a result, the spectrum of susceptible microbial species is well known before the compound enters the clinic.
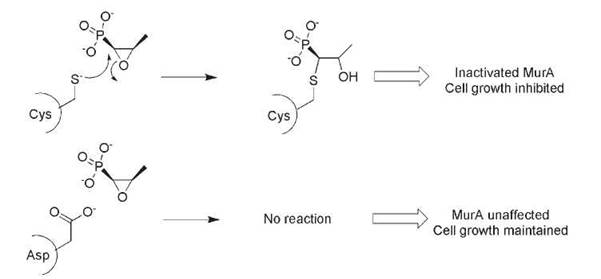
Figure 1. The action of the antibiotic fosfomycin requires an active-site Cys residue in the target MurA. Substitution of Asp for Cys in some bacterial species results in intrinsic fosfomycin resistance.
Acquired antibiotic resistance
In contrast to intrinsic resistance, which is genetically and physiologically “hard wired,” acquired antibiotic resistance occurs as a consequence of antibiotic use. Acquired resistance often requires the presence of the antibiotic for selection of resistant species within a susceptible genetic background. Resistance can emerge as a result of mutation of target genes on the chromosome (e.g., point mutations in DNA gyrase that confer fluoroquinone antibiotic resistance) (13) or by the presence of resistance genes that are captured on mobile and associated genetic elements such as plasmids, transposons, and integrons (14). The sequencing of entire bacterial genomes has revealed that these resistance-associated genetic elements can sometimes be components of the normal genetic makeup of a given bacterial species (especially transposons and integrons), but more often they are acquired as a result of selection in the face of antibiotic exposure. Conventional wisdom suggests that acquired resistance results in a genetic and physiologic burden that makes these organisms less fit in the absence of antibiotic selective pressure, although second site mutations can compensate (15).
One of the confounding issues with acquired resistance by way of mobile genetic elements is linkage of multiple resistance genes on a single element. R-Plasmids, integrons, and transposons often carry not only one resistance gene, but several (Fig. 2). As a result, selection of resistance to one antibiotic can inadvertently select for resistance to others, which can be especially problematic in clinical settings, can contribute to failure of antibiotic cycling countermeasure strategies within these institutions, and can add to the difficulty in eliminating antibiotic resistance once it has emerged.
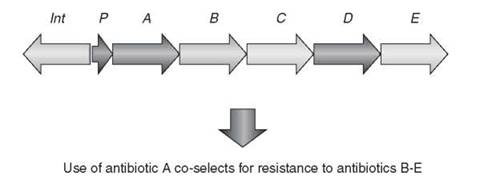
Figure 2. Integrons, transposons, and R-plasmids often collect multiple antibiotic resistance genes. As a result, selection of one resistance mechanism coselects for resistance to additional resistance genes. The scheme shows a typical arrangement for an integron with associated integrase-encoding gene (int), a promoter to drive gene transcription (P), and antibiotic resistance elements (A-E).
Chemical Biology of Antibiotic Resistance
Antibiotic resistance can be the result of several molecular mechanisms (Table 1). Some of the most important of these mechanisms include enzyme-catalyzed antibiotic inactivation or modification, altered transport such as efflux, and others such as metabolic bypass and sequestration. Each of these mechanisms requires the synthesis of associated proteins to mediate resistance. These are often highly specialized and efficient, and frequently the corresponding genes can be acquired on mobile genetic elements. In this fashion, the antibiotic resistome (16), which is the collection of all resistance genes within the bacterial kingdom, has the potential to be shared and can cross species and genus boundaries.
In a simplistic but useful view, antibiotic molecules comprised a target-specific “warhead” and a bioactive delivery vehicle or scaffold (Fig. 3). The warhead interacts specifically with the target (protein, RNA, DNA, membrane) and forms key molecular interactions with it. These interactions can be covalent in the case of electrophilic chemical fragments such as with β-lactams, β-lactones, and oxiranes, or noncovalent through hydrogen bonds, electrostatics, and so on. Consequently, alteration of the antibiotic at the warhead region or of the reciprocally vital antibiotic binding site on the target results in resistance. On the other hand, the delivery vehicle/scaffold portion of the antibiotic generally provides few essential interactions with the molecular target but instead contributes physical properties that enhance bioavailability (e.g., membrane permeability) or imparts structurally important features such as rigidity that ensure appropriate display and structure of the warhead. As a result, the scaffold portion of the antibiotic molecule tends to be more tolerant of chemical alteration. In fact, chemical modification of the molecular scaffold while preserving the antibiotic warhead has been the principal occupation of medicinal chemists since the first antibiotics were isolated from natural sources. The goal of these modifications is to improve pharmacology, evade resistance, and circumvent proprietary structure limitations. As a result, this region of the antibiotic molecule is generally not a principal target for resistance mechanisms; nonetheless, modification of key scaffold regions such as cyclizing bonds can result in resistance.
Next, various biochemical strategies employed by microorganisms to evade antibiotics are discussed with an emphasis on describing the chemical logic of the approach.

Figure 3. Antibiotics consist of an active warhead and associated chemical scaffold.
Antibiotic Resistance by Enzymatic Mechanisms
One of the most effective and potent strategies in which microbes have evolved to evade antibiotics is the chemical modification of these cytotoxic compounds. This approach is generally very selective to the specific antibiotic or class, thereby avoiding undesirable side reactions and minimally perturbing normal bacterial metabolism. The general strategy is to destroy or otherwise mask the warhead, which results in resistance. The biochemical logic of several examples is discussed below.
Antibiotic destruction
One of the most potent and physiologically irreversible mechanisms of antibiotic resistance is via chemical destruction of the antibiotic warhead or scaffold. This destruction can occur through several mechanisms, but hydrolytic approaches predominate (Fig. 4). Resistance to the highly important β-lactam antibiotics by β-lactamases is the archetype of this class. The β-lactams include the natural product penicillin and cephalos porin scaffolds as well as the synthetic mono-bactams such as azetronam. The highly strained β-lactam ring is the antibiotic warhead, covalently modifying target transpeptidase and carboxypeptidase enzymes required for the synthesis and maturation of peptidoglycan (Fig. 5). This molecular strategy is analogous to the modification of MurA by fosfomycin as discussed above (Fig. 1), but in this case, covalent enzyme inactivation affects late-stage peptidoglycan biosynthesis on the outside of the cell rather than early-stage intracellular steps. The β-lactamases hydrolyze the thermodynamically activated β-lactam ring irreversibly incapacitating the antibiotics.
Numerous β-lactamases have been cloned and characterized (recently reviewed in Reference 17). These enzymes are often secreted into the periplasmic region (the immediate extracellular space of the plasma membrane, bounded by the outer membrane in the case of Gram-negative bacteria) setting up a perimeter defense to intercept the antibiotics before they reach their molecular targets. The associated genes very frequently are on mobile genetic elements, but many bacteria also harbor orthologues within their genome (18).
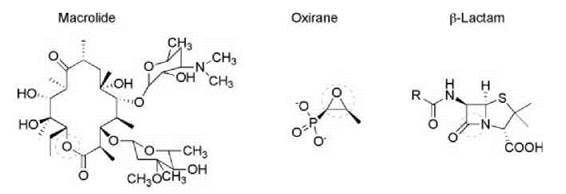
Figure 4. Hydrolytically sensitive antibiotics include the macrolides, oxirane, and β-lactams. The hydrolytically sensitive region is circled in each structural class.

Figure 5. Action of β-lactam antibiotics. Cell wall biosynthetic transpeptidases activate peptidoglycan peptides for cross-linking by formation of a covalent enzyme intermediate (A). The reactivity of the active-site Ser nucleophile is exploited by β-lactam antibiotics such as penicillin G (shown) to form a hydrolytically stable acylenzyme (B).
The β-lactamases have mirrored the chemical strategy of proteases to cleave the β-lactam bond. The main β-lactamase families emulate Ser or metallo-protease mechanisms to perform ring-opening hydrolysis of the β-lactam ring, destroying the antibiotic warhead in the process (Fig. 6). The Ser enzymes mimic the first step in peptidoglycan transpeptidase reaction by formation of an acyl-enzyme intermediate. However, unlike the transpeptidase, this intermediate is short lived and subsequent hydrolysis releases the inactive antibiotic (Fig. 6).

Figure 6. β-Lactamases hydrolytically cleave the susceptible β-lactam ring using Ser and metallo-protease mode chemistry.
Similarly, metallo-β-lactamases share with metallo-proteases activation of a hydrolytic water molecule by interaction with an active-site Zn2+ ion. These lactamases have gained clinical prominence in the past few years as a result of their association with carbapenem resistance. The carbapenems, such as meropenem and imipenem, are β-lactam antibiotics that have been introduced to circumvent Ser-β-lactamase activity. The trans stereochemistry across the 6-5 bond rather than the cis geometry found in most other β-lactams (Fig. 7) contributes to the relative stability of these antibiotics to Ser-β-lactamases and, as a result, are drugs of choice for treatment of infections caused by β-lactam-resistant bacteria. However, carbapenems are susceptible to metallo-β-lactamases such as IMP and VIM that are becoming widely distributed among Gram-negative bacteria and are a growing clinical concern (19).
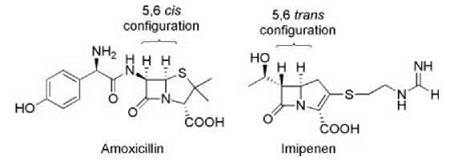
Figure 7. Comparison of the structures of amoxicillin and imipenem. The change in configuration across the 5-6 bond in imipenem maintains the ability to bind to penicillin binding proteins but provides insensitivity to many Ser-β-lactamases.
Hydrolytic enzymes are also known to inactivate the oxirane antibiotic fosfomycin and macrolides antibiotics such as erythromycin by ring-opening epoxidase and esterase activity, respectively (Fig. 4). The former mechanism is exemplified by the Mn2+-dependent enzyme FosX from the soil bacterium Mesorhizobium loti (20). The bulk of clinically important macrolide antibiotic resistance occurs through ribosomal RNA methylation or efflux mechanisms. As a result, macrolide esterases are presently not a major clinical problem. However, the genes encoding this mode of resistance have been found on mobile genetic elements (21-23) and, because they confer high level resistance to this important class of antibiotic, there is cause for concern that this mechanism could increase in frequency.
Conceptually similar, although mechanistically distinct, is Vgb, a class B streptogramin inactivating enzyme. Streptogramins consist of two distinct structural classes of antibiotics: polyketide-peptide hybrids (type A streptogramins) and cyclic hexa or hepta depsipeptides (type B streptogramins). The type B streptogramins are cyclized through an ester linkage between the C-terminal carboxyl group and the secondary hydroxyl group of an invariant Thr residue at position 2. Both classes of strep- togramin bind to the large subunit of the bacterial ribosome blocking the peptidyl transfer center (type A) and the peptide exit tunnel (type B). The warhead of the type B streptogramins consists primarily of the 3-hydroxypicolinc acid residue and the peptide backbone that blocks the entrance to the tunnel. Resistance to the type B streptogramins can occur through target modification or efflux, but also by the enzyme Vgb and orthologues (24). This enzyme linearizes the peptide backbone at the antibiotic’s thermodynamic weakest point, the ester linkage; however, unlike macrolide esterases, Vgb is not a hydrolase but a C-O lyase (25). The catalytic strategy likely involves acidification of the Thr α-proton by generation of the aci form of the amino acid followed by elimination of the carboxylate resulting in C-O bond cleavage and formation of the 2-amino-but-2-enoic acid amide product (Fig. 8).

Figure 8. Inactivation of type B streptogramin antibiotics by Vgb lyase. The type B streptogramins such as pristinamycin la (shown) are cyclic depsipeptides. Vgb lyase catalyzes the cleavage of the C-O depsipeptide bond.
Antibiotic modification
Chemical modification of key groups on the antibiotic can also result in resistance. Modification generally occurs on the warhead region of the molecule, blocking effective interaction with the target. Typical modifications include acylation, phosphorylation, and adenylylation. These add steric bulk to the antibiotic, obscure key target binding elements, and can alter charge of the compound. As the reactions require cosubstrates such as ATP and acetylCoA, antibiotic modifying enzymes are generally located within the cell interior where access to these high energy substrates is secure. This strategy impacts several classes of antibiotics and, in some cases, there is significant conservation of general protein fold among enzymes that modify chemically diverse antibiotics.
Acetylation of antibiotics is a common mechanism of modification. The acyltransferases generally use the metabolically abundant acetyl-CoA as the preferred acyl-donor. The aminoglycoside antibiotic acetyltransferases (AACs), for example, modify important amines that make contact with the target 16 S rRNA (Fig. 9), which results in a loss of positive charge and increased steric bulk that contribute to lowering the affinity of the antibiotics for the rRNA target by 600-fold (26). The AACs are members of the GCN5 superfamily of acyltransferases that include such members as the eukaryotic histone acetyltransferases and serotonin acetyltransferase (27).

Figure 9. Aminoglycoside antibiotic modifying enzymes. The aminoglycoside antibiotics such as kanamycin B (shown) bind to the 16 S rRNA of the bacterial ribosome impairing cognate codon-anticodon discrimination (A). Resistance occurs via acetylation (AAC), phosphorylation (APH), or adenylylation (ANT) of the antibiotic (B). A wide variety of enzymes are known with different regiospecificities of chemical modification, and the sites of some clinically important enzymes are shown in panel C.

Figure 10. Chloramphenicol modifying enzymes. Chloramphenicol binds to the 23 S rRNA of the large ribosomal subunit (A). Chemical modification of the essential hydroxyl groups by acetyltransferase or kinase enzymes confers resistance (B).
Strategically similar, but structurally distinct, are the chloramphenicol acetyltransferases (CATs). Chloramphenicol binds to the large ribosomal subunit at the nexus of the peptidyltransfer center and the peptide exit tunnel (28, 29). CATs are trimeric enzymes with active sites at the interface of the monomers (30) that catalyze acetylation of the hydroxyl group on position 3 (31), which is essential for interaction with the ribosome (Fig. 10). The well-characterized type I-III CATs are robust and efficient modifiers of the antibiotic, with kcatIKm values that approach diffusion-controlled reactions (32). These CATs are structurally distinct from a group of chloramphenicol acetyltransferases that have been classified as xenobiotic acetyltransferases (XAT) in view of their broader substrate specificity and relatively inferior affinity for chloramphenicol (31). The XATs belong to the large hexapeptide repeat family of proteins that incorporate a left-handed β-helix domain (33). Although they show no primary sequence or tertiary structure homology, both CATs and XATs are trimers with active-site residues at the interface of the monomer subunits and both use a catalyt- ically important His residue to assist in deprotonation of the antibiotic hydroxyl group undergoing acetylation. The streptogramin acetyltransferases (VATs) are also members of the XAT family (34) and are responsible for resistance to the type B streptogramins, notably the clinically used drug dalfopristin (Fig. 11) (35).

Figure 11. Chemical modification of Type B streptogramins. These antibiotics bind to the large ribosome adjacent to the chloramphenicol binding site (A). A key interaction with the 23 S rRNA is blocked by the action of VAT-dependent acetylation (B).
Phosphorylation is a common mechanism resulting in resistance to the aminoglycoside antibiotics. This chemical strategy also has been associated with resistance to the macrolides such as erythromycin, the tuberactinomycins such as viomycin, and chloramphenicol. The aminoglycoside kinases share 3D structural similarity with the SerlThrlTyr protein kinase family (36), and the conservation of kinase signature sequences in macrolide and tuberactinomycins kinases suggests that they will also share the family protein kinase 3D structure (37). Mirroring the effect of acetyl transfer, covalent modification of aminoglycosides alters charge and size, resulting in up to a 1300-fold decrease in affinity of the modified antibiotic for 16 S rRNA (Fig. 9) (26).
Like the aminoglycosides, the binding site of the macrolide antibiotics with the large ribosomal subunit has also been determined to atomic resolution by X-ray crystallography (29, 38, 39). Key interactions between the antibiotic and the 23 S rRNA occur and are mediated through the essential desosamine sugar: the antibiotic warhead (Fig. 12). Macrolide kinases (MPHs) phosphorylate position 2 of the desosamine sugar, impairing binding with the target. Genes encoding these enzymes have been identified in mobile genetic elements in various bacterial pathogens (40-43). Using an analogous evasive strategy, macrolide producing and other soil bacteria encode macrolide glycosyltransferases that catalyze glucosylation of the same essential 2'-position on the desosamine by UDP-glucose (44-46) (Fig. 12). The net result is the same with either approach: steric blockade of the appropriate target-antibiotic interaction resulting in resistance.

Figure 12. Macrolide modifying enzymes. Macrolide antibiotics such as erythromycin (shown) bind to the large ribosomal subunit through interactions with the 23 S rRNA (A). Chemical modification of the essential desosamine sugar blocks ribosome binding (B).
Chloramphenicol phosphotransferase from the producing bacterium Streptomyces venezuelae (47) is unrelated to the protein kinase family but rather shows more similarity to small- molecule kinases such as shikimate kinase (48). Analogous to the CAT strategy, phosphorylation occurs at the hydroxyl position 3, blocking this essential group from interacting with the ribosome (Fig. 10).
Aminoglycosides and lincosamides such as clarithromycin can be modified by O-adenylylation in an ATP-dependent fashion resulting in resistance. Position 2" of the aminoglycosides is the target for the clinically important ANT(2') enzymes that confer resistance to antibiotics such as tobramycin and gentamicin. In analogy to other aminoglycoside modifications that result in resistance discussed above, the addition of the bulky and negatively charged adenosine diphosphate to the antibiotic attenuates ribosome binding (Fig. 9). The lincosamide antibiotics are frequently modified by AMP at position 3 in a reaction catalyzed by the Lin proteins (49, 50). This modification blocks a key interaction of the antibiotic with the 23 S rRNA of the bacterial large subunit (Fig. 13) (29).

Figure 13. Adenylylation of lincosamides antibiotics. Lincosamides such as clindamycin (shown) interact with the large ribosomal subunit through a complex series of interactions (A). Modification of the antibiotic by adenylylation prevents binding to the ribosome (B).
Antibiotic Modification by Oxidation/Reduction Mechanisms
Antibiotic resistance via redox reactions is not as common as chemical modification; however, there are some examples of this strategy in nonpathogenic bacteria. The streptogramin antibiotic producer Streptomyces virginiae can reduce a vital ketone group of group A streptogramins in an NADH-dependent manner resulting in formation of the vicinal diol, which lacks antibiotic activity (51). Redox-dependent inactivation of rifampin has also been described by a gene product from Rhodococcus equi (52).
Tetracycline resistance occurs primarily by efflux and target protection mechanisms (53); however, inactivation by the redox enzyme TetX has been reported (54). TetX is a monooxygenase that selectively hydroxylates tetracycline antibiotics (55), including the latest generation of antibiotics of the class the glycylcyclines (56). Tetracycline binds to the A-site of bacterial ribosomes interfering with translation. Binding of the invariant β-diketone functional group characteristic of the class to the ribosome occurs through the mediation of a divalent cation (likely Mg2+) (Fig. 14). TetX catalyzes the mono-hydroxylation of the antibiotics at position 11a, effectively disrupting the Mg2+ binding site, resulting in resistance (Fig. 14). Furthermore, some tetracyclines undergo nonenzymatic degradation after mono-hydroxylation, resulting in irreversible destruction of the antibiotic.

Figure 14. Enzymatic modification of tetracycline. Tetracyclines bind to the ribosome in several areas, but the primary region is in the A site of the small subunit (shown) (A). The antibiotic binds divalent cations such as Mg2+, which is essential for ribosome binding. The enzyme TetX catalyzes the mono-hydroxylation of tetracycline at position 11a disrupting the Mg2+ binding site resulting in resistance.
Antibiotic Resistance Through Altered Transport
To be effective, antibiotics must accumulate to critical levels governed by their affinity for their molecular targets. Furthermore, these targets are frequently intracellular. As a result, antibiotics must efficiently permeate the bacterial cell envelope and achieve sufficiently effective local concentrations. In Gram-positive bacteria, the cell envelope consists of the cell wall and the phospholipid containing cell membrane. In contrast, Gram-negative bacteria deploy an additional outer membrane consisting of an asymmetric LPS/phospholipid bilayer to complement the peptidoglycan and phospholipid cell membrane components of the cell envelope. These architectures provide a substantial barrier to antibiotic access to the cell interior.
Transport-linked resistance to antibiotics can emerge as a result of poor penetration of the bacterial cell envelope or from the energy-dependent purging of antibiotic molecules that have managed to penetrate the envelope through the aegis of efflux proteins. Often, these two mechanisms, entry and efflux, combine to achieve high level resistance to many antibiotics. Like enzyme-based resistance, several of these transport-based resistance genes are encoded on mobile genetic elements; however, all bacteria encode an elaborate collection of transport proteins in their chromosomes, some of which have the potential to be recruited for antibiotic resistance. In particular, Gram-negative bacteria, such as those from the genera Pseudomonas and Burkholderia, are particularly adapted for transport-mediated resistance, and this mechanism contributes significantly to the high level of broad spectrum antibiotic resistance in these organisms.
Antibiotic efflux
Bacterial cells can express a variety of membrane proteins that mediate the energy-dependent efflux of toxic compounds (Table 2). Five structurally distinct classes of efflux proteins are recognized: ABC transporters, resistance-nodulation-division (RND) proteins, multidrug and toxin extrusion (MATE) proteins, small multidrug resistance (SMR) proteins, and major facilitator superfamily proteins (MFS). The latter four of these proteins promote exchange of a cation (H+ or Na+) for the antibiotic, whereas the ABC transporters use ATP hydrolysis to drive ejection of the compound from the cell. All of the protein classes have been associated with antibiotic resistance in various bacterial strains; nonetheless, some general rules have emerged from the past two decades of efflux research. First, with a few exceptions, plasmid- or integron-mediated resistance usually is associated with MFS and SMR proteins that have broad substrate specificity. Second, ABC transporters are analogous to the P-glycoprotein class of drug efflux systems in eukaryotes. They are not as prevalently associated with antibiotic resistance in pathogenic bacteria as the MFS class, but are very common in antibiotic biosynthesis gene clusters in producing bacteria. Third, the MFS proteins are common in Gram-positive and Gram-negative bacteria, whereas the RND class is generally found in Gram-negatives where they are chromosomally encoded. Fourth, sequencing of bacterial genomes has revealed that all microbial genomes have multiple predicted antibiotic efflux proteins (e.g., 25 in Staphylococcus aureus COL, 102 in Bacillus anthracis Ames).
Several excellent and comprehensive recent reviews cover this field in depth (57-61); therefore, the following description will briefly emphasize the proposed mechanisms rather than the comprehensive details of efflux systems and their individual components.
The MFS proteins are a large superfamily of transporters that are essential to the shuttling of numerous metabolites (sugars, amino acids, and anions) as well as cytotoxic agents such as antibiotics in exchange for cations. They consist of 12-14 membrane-spanning helices and can be found encoded on mobile genetic elements (e.g., QacA, which is clustered in many integrons) or on bacterial chromosomes (e.g., NorA, which is linked to fluoroquinolone resistance in S. aureus). Three-dimensional structures of members of the MFS superfamily have been reported, including the sugar transporters LacY (62) and GlpT (63) and, more recently, the multidrug transporter EmrD from E. coli (64). These structures reveal a collection of helices that form a pore-like structure to enable transport across the membrane. In EmrD, the interior of the pore is mostly hydrophobic, consistent with the substrate specificity of the protein that includes detergents such as sodium dodecyl-sulfate. This structure suggests a “rocker-switch” mechanism where the substrate enters the open pore from the cytoplasm or the inner leaflet of the cell membrane, the open pore then undergoes a conformational change to close around the substrate coupled with H+ transport, and finally the pore is reopened facing the exterior of the cell where the substrate can diffuse away (Fig. 15).

Figure 15. Proposed rocker-switch mechanism for MFS-mediated drug efflux. Antibiotic (circle) enters the open pore from the cytosolic face or inner membrane leaflet and is antiported to the cell exterior with an H+ ion coupled with conformational change within the protein.
The RND proteins have emerged as important mediators of multidrug resistance in Gram-negative bacteria, especially in the opportunistic pathogens P. aeruginosa, Stenotrophomonas maltophilia, Acinetobacter baumannii, and Burkholderia cepacia. These resistance modules combine with reduced uptake and a cadre of chromosomally encoded resistance enzymes to make these bacteria virtually impermeable to antibiotics. The groups of Nikaido and Poole have studied the RND efflux systems in detail from E. coli and P. aeruginosa, respectively, providing great insight into the components of individual RND transporters, their mechanisms of action, and substrate specificity. Consequently, the AcrAB/TolC system in E. coli and the MexAB/OprM triad in P. aeruginosa are the paradigms of the class. They are tripartite efflux systems consisting of an inner cell membrane transporter (AcrB, MexB), a periplasmic membrane fusion protein (AcrA, MexA), and an outer membrane pore (TolC, OprM) to seamlessly connect the inside of the cell across two membrane barriers to the external environment (Fig. 16). This structure provides an unimpeded conduit for toxic molecules from the cell interior to the exterior. Additionally, antibiotics such as β-lactams and other molecules are able to access the efflux system within the periplasmic space without entering the cell, thereby expanding the spectrum of the efflux system. The crystal structures of AcrB (65, 66), MexA (67), a fragment of AcrA (68), and TolC (69) have been determined. These structures confirm the predicted membrane spanning arrangement of the efflux system and serve to rationalize the broad substrate specificity of the complex as well as illustrate the challenge of overcoming efflux-mediated resistance.
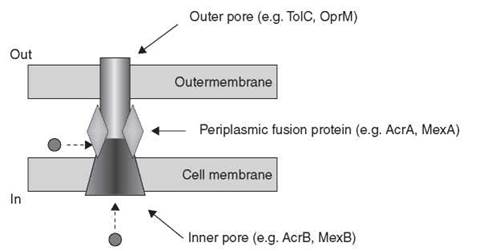
Figure 16. General structure of tripartite RND class of efflux proteins. Substrates (circle) can enter the pore from the cytosolic side or the periplasmic space for transport out of the cell.
Table 2. Selected antibiotic resistance efflux systems
|
Protein |
Class |
Found in... Mobile genetic Elementb Chromosome |
Organism |
Substratesa |
|
|
QuacA |
MFS |
√ |
|
many |
Quaternary ammonium compounds, dyes |
|
NorA |
MFS |
|
√ |
Staphylococcus aureus |
Dyes, fluoroquinolones |
|
Bmr |
MFS |
|
√ |
Bacillus subtilis |
Dyes, chloramphenicol, fluoroquinolones |
|
Smr (QuacC/D) |
SMR |
√ |
|
many |
Monovalent cations |
|
EmrE |
SMR |
|
√ |
Escherichia coli |
Monovalent cations, tetracycline |
|
MefE |
MSF |
|
√ |
Streptococcus pneumoniae |
Macrolides |
|
TetA |
MFS |
√ |
|
many |
Tetracycline |
|
Lsa |
ABC |
|
√ |
Enterococcus faecalis |
Type A streptogramins, lincosamides |
|
AcrA/AcrB/TolC |
RND |
|
√ |
Escherichia coli |
Dyes, bile salts, chloramphenicol, β-lactams |
|
MexA/MexB/OprM |
RND |
|
√ |
Pseudomonas aeruginosa |
Dyes, chloramphenicol, β-lactams, aminoglycosides, novobiocin |
|
MexC/MexD/OprJ |
RND |
|
√ |
Pseudomonas aeruginosa |
Chloramphenicol, fluoroquinolones, tetracycline, trimethroprim, triclosan |
|
AdeA/AdeB/AdeC |
RND |
|
√ |
Acinetobacter baumannii |
Aminoglycosides, chloramphenicol, fluoroquinolones, tetracycline, trimethroprim |
Partial list.
Includes plasmids, transposons, integrons.
Reduced uptake of antibiotics
Although efflux is a daunting obstacle for many antibiotics, it is the balance between influx and efflux that determines whether cells will be sensitive or resistant in the face of an antibiotic challenge. As a result, reduced uptake of antibiotics coupled with activation of efflux pumps is a potent combination that can overcome sensitivity to drugs in many pathogens. Many metabolites and compounds such as antibiotics penetrate cells via transport proteins and, as a result, mutation in these proteins or decrease in their expression can decrease antibiotic uptake. In Gram-negative bacteria, the outer membrane porins are required for entry of many antibiotics such as P-lactams and aminoglycosides, and antibiotic resistance can occur through modulation of these proteins. For example, imipenem resistance in P. aeruginosa is frequently associated with mutation in the OprD porin (70).
Other Mechanisms of Antibiotic Resistance
In addition to enzymatic modification or destruction and transport, bacteria can evade antibiotics through a variety of ways, including metabolic bypass, target overexpression or modification, and the production of various protective proteins.
Metabolic bypass: vancomycin resistance
The classic example of metabolic bypass resulting antibiotic resistance can be found in the glycopeptide antibiotics. Glycopep- tides such as vancomycin and teicoplanin are natural product antibiotics that bind to the ubiquitous acyl-D-Ala-D-Ala dipeptide termini of peptidoglycan (71), which results in sequestration of the substrate for cell wall biosynthetic enzymes including the transpeptidases and glycosyltransferases, resulting in impairment of cell wall synthesis and bacterial growth. Therefore, vancomycin binds to an essential small-molecule metabolite, which is the enzyme substrate, rather than to the protein itself. As a result, simple mutation of a single gene to overcome resistance is not possible.
Vancomycin and other glycopeptides bind to acyl-D-Ala-D- Ala through a series of five hydrogen bonds that includes a key interaction with the amide hydrogen of the D-Ala-D-Ala peptide bond (Fig. 17). The acyl-D-Ala-D-Ala glycopeptide-binding unit is synthesized in stepwise fashion through a series of metabolic steps involving first synthesis of D-Ala, ATP-dependent ligation of two D-Ala molecules to generate the D-Ala-D-Ala dipeptide, and a second ligation where the dipeptide is grafted onto the N-acetyl-muramic acid tripeptide acceptor for incorporation into the growing cell wall (Fig. 18). Vancomycin-resistant bacteria overcome the antibiotic by bypassing the normal cell wall biosynthesis. Instead of producing a cell wall terminating in the D-Ala-D-Ala dipeptide, a pep- tidoglycan is produced that terminates with the depsipeptide (ester) D-Ala-D-lactate (Fig. 17). This change of NH for O results in loss of a critical H-bond and electronic repulsion between the ester O and the carbonyl of vancomycin. The net effect is a 1000-fold decrease in KD resulting in vancomycin resistance (72). Incorporation of the D-Ala-D-lactate ester requires three enzymes: VanH, VanA, and VanX. VanH is a D-lactate dehydrogenase generating D-lactate from pyruvate. VanA is an ATP-dependent depsipeptide ligase that generates D-Ala-D-lactate. Finally, VanX is a highly specific D-Ala-D-Ala dipeptidase that clears the cell of D-Ala-D-Ala that continues to be synthesized by the chromosomally encoded D-Ala-D-Ala ligases. These three proteins work in concert to bypass the normal cellular cell wall biosynthetic metabolism and are each essential for resistance. Not surprisingly, they are colocated on resistance plasmids and transposons that have emerged in strains of enterococci and more recently in S. aureus.
In a variant of this strategy, vancomycin resistance can also emerge by incorporation of peptidoglycan terminating in D-Ala-D-Ser. Substitution of D-Ala for D-Ser has steric implications for tight vancomycin-peptidoglycan binding, which also can result in resistance.

Figure 17. Structure of the glycopeptide antibiotic vancomycin bound to the cell wall terminus D-Ala-D-Ala. Substitution of D-Ala-D-Ala for D-Ala-D-lactate eliminates an essential H bond resulting in resistance.

Figure 18. Synthesis of the D-Ala-D-Ala peptidoglycan terminus. D-Ala-D-Ala ligases (Ddl) synthesize the dipeptide, which is incorporated into the UDP-MurNAc-tripeptide by the D-Ala-D-Ala adding enzyme (MurF in E. coli).
Target modification
One of the most straightforward mechanisms of antibiotic resistance is mutation of the target to a form that has less affinity for the antibiotic. Spontaneous mutations that provide some benefit to the organism occur roughly 2 x 10-9 per replication (reviewed in Reference 73). Exposure to certain classes of antibiotics such as the rifamycins and fluoroquinolones can induce mutation, increasing the opportunity of developing resistance (74). Point mutations are associated with resistance to virtually all antibiotics, but this form of resistance is particularly important in the clinic for the fluoroquinolones, rifamycins such as rifampin, trimethroprim, and the sulfonamides.
Enzyme-mediated target modification can also be an important mechanism of antibiotic resistance. The Erm proteins are 23 S rRNA S-adenosylmethionine-dependentmethyltransferases that immunize the bacterial ribosome against the macrolide antibiotics such as erythromycin, the lincosamides, and type B streptogramins (75). The Erm proteins, specifically methylate A2058, provide a steric block for all three classes of antibiotics: a remarkable example of potent and strategic resistance parsimony. The erm genes are widespread in clinical isolates and can be constitutively expressed or strategically deployed through a complex induction mechanism (76).
By means of similar logic, methylation of the 16 S rRNA at position G1405 by Arm and related proteins results in high level aminoglycoside resistance (77).
Protective proteins
Expression of various proteins, which can collectively be called protective, can result in antibiotic resistance. These immunity proteins often work in stoichiometric fashion to sequester the compounds, or otherwise protect the cell against the toxic activity of the antibiotics.
Resistance to the peptide antibiotic/anticancer agent bleomycin can occur through the expression of the Blm family of proteins that sequester the antibiotic. Bleomycin includes a metal-binding region that complexes Fe2+ resulting in a potent generator of oxygen radicals (warhead) and a DNA intercalating portion (scaffold) that targets the complex to DNA. Bleomycin, therefore, is a remarkable molecule with the ability to produce toxic radicals in an unregulated fashion proximal to DNA. The Blm proteins, such as BlmA from the bleomycin producer Streptomyces verticillus and BlmT found on various resistance transposons such as TnJ, are small dimeric proteins (~14,000 Da/monomer), and high resolution crystal structures are available for each protein (78, 79). The structure of the BlmA protein in complex with a Cu(II)-bleomycin complex reveals that each monomer binds one antibiotic molecule and that binding of the second molecule of bleomycin is cooperative (80). The precise mechanism of resistance is likely through sequestration of both the warhead and scaffold portions of the antibiotic, thereby preventing optimal activation of the Fe(II) complex.
Similarly, resistance to the anticancer antibiotic mitomycin in the producing organism, Streptomyces lavendulae, occurs via sequestration of the antibiotic by the binding protein MRD (81, 82). This protein interacts with the mitomycin export system, thus additionally facilitating export of the antibiotic from the cell. The structure of MRD reveals similarity to the bleomycin resistance proteins and, in fact, the protein can also bind bleomycin and confer resistance to this antibiotic (83).
By similar means, resistance to the highly toxic enediyne antibiotics, which are active at ng/mL concentrations, occurs through binding proteins in the producing bacteria. These antibiotics include a conserved core warhead consisting of two acetylenic groups bridged by a carbon-carbon double bond. This warhead generates a diradical after thiol activation resulting in DNA cleavage (84). The class is divided into two structural groups, the 9-membered and 10-membered enediynes, referring to the size of the warhead-containing ring. The 9-membered enediynes are stabilized by coexpression and titration by self-protective binding proteins. The 3D NMR structure of one of these complexes, C-1027 with the binding protein CagA, has been reported (85). Resistance to the more chemically stable 10-membered enediynes (e.g., calicheamicin), however, involves the remarkable expression of the protective protein CalC that undergoes calicheamicin-dependent proteolytic self-sacrifice simultaneously inactivating the antibiotic (86).
Another example of protective protein expression is the tetracycline resistance TetO and TetM proteins and their analogues (87). These proteins structurally resemble the prokaryotic GT-Pase elongation factors such as EF-G and EF-Tu that bind to the ribosome and are essential for translation. TetO/M protection proteins do not replace elongation factors, but they do bind to the ribosome and displace bound tetracyclines in a GTP-dependent manner (88, 89).
Conclusions
The history of antibiotic use over the past 70 years is invariant: initial deployment to the clinic with little/no background resistance followed by emergence and global spread of resistance. Resistance to antibiotics is manifested through a number of different chemical and biophysical mechanisms, each with its own associated biochemical logic. A fundamental component of this logic is the sensitivity of the antibiotic warhead to chemical alteration. Although resistance is inevitable, understanding how resistance can occur, what the sensitive regions of the molecule are, and biochemical strategies that could overcome it such as the identification of specific inhibitors of resistance are essential to delay the emergence of resistance. Recently, it has been shown that penetration of antibiotic resistance in the microbial community at large is much deeper than previously appreciated (16). Understanding the origins and evolution of resistance will also provide drug discoverers with valuable information to circumvent established mechanisms. The concept of overcoming the inevitability of antibiotic resistance with small-molecule inhibitors of resistance mechanisms has been proven to be highly effective in the P-lactam class of antibiotics (90), and this approach should be broadly applicable (91). In fact, this area is one of intense interest, especially as applied to efflux mechanisms (92, 93). The routes forward in this important area of medicine require Chemical Biology approaches to understand antibiotic-target interactions and antibiotic resistance mechanisms. Chemical Biology and other multidisciplinary disciplines are therefore essential to prolong the utility of these essential therapeutic agents.
References
1. Winau F, Westphal O, Winau R. Paul Ehrlich—in search of the magic bullet. Microbes. Infect. 2004; 6:786-789.
2. Van Epps HL. Rene Dubos: unearthing antibiotics. J. Exp. Med. 2006; 203:259.
3. Nathan C. Antibiotics at the crossroads. Nature 2004; 431:899-902.
4. Norrby SR, Nord CE, Finch R. Lack of development of new antimicrobial drugs: a potential serious threat to public health. Lancet. Infect. Dis. 2005; 5:115-119.
5. Heymann DL. Resistance to anti-infective drugs and the threat to public health. Cell 2006; 124:671-675.
6. Cosgrove SE. The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs. Clin. Infect. Dis. 2006; 42:S82-S89.
7. Birnbaum D. Antimicrobial resistance: a deadly burden no country can afford to ignore. Can. Commun. Dis. Rep. 2003; 29:157-164.
8. Marshall DA, McGeer A, Gough J, Grootendorst P, Buitendyk M, Simonyi S, Green K, Jaszewski B, MacLeod SM, Low DE. Impact of antibiotic administrative restrictions on trends in antibiotic resistance. Can. J. Public Health 2006; 97:126-131.
9. Warren DK, Hill HA, Merz LR, Kollef MH, Hayden MK, Fraser VJ, Fridkin, SK. Cycling empirical antimicrobial agents to prevent emergence of antimicrobial-resistant Gram-negative bacteria among intensive care unit patients. Crit. Care Med. 2004; 32:2450-2456.
10. Skarzynski T, Mistry A, Wonacott A, Hutchinson SE, Kelly VA, Duncan K. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 1996; 4:1465-1474.
11. De Smet KA, Kempsell KE, Gallagher A, Duncan K, Young DB. Alteration of a single amino acid residue reverses fosfomycin resistance of recombinant MurA from Mycobacterium tuberculosis. Microbiology 1999; 145:3177-3184.
12. Kim DH, Lees WJ, Kempsell KE, Lane WS, Duncan K, Walsh CT. Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 1996; 35: 4923-4928.
13. Piddock LJ. Mechanisms of fluoroquinolone resistance: an update 1994-1998. Drugs 1999; 58:11-18.
14. Davies J. Inactivation of antibiotics and the dissemination of resistance genes. Science 1994; 264:375-382.
15. Bjorkman J, Nagaev I, Berg OG, Hughes D, Andersson DI. Effects of environment on compensatory mutations to ameliorate costs of antibiotic resistance. Science 2000; 287:1479-1482.
16. D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science 2006; 311:374-377.
17. Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to beta-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 2005; 105:395-424.
18. Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and beta-lactamases. Antimicrob. Agents Chemother. 1998; 42:1-17.
19. Walsh TR, Toleman MA, Poirel L, Nordmann P. Metallo-beta-lactamases: the quiet before the storm? Clin. Microbiol. Rev. 2005; 18:306-325.
20. Fillgrove KL, Pakhomova S, Newcomer ME, Armstrong RN. Mechanistic diversity of fosfomycin resistance in pathogenic microorganisms. J. Am. Chem. Soc. 2003; 125:15730-15731.
21. Nakamura A, Nakazawa K, Miyakozawa I, Mizukoshi S, Tsu- rubuchi K, Nakagawa M, O’Hara K, Sawai T. Macrolide esterase- producing Escherichia coli clinically isolated in Japan. J. Antibiot. (Tokyo) 2000; 53:516-524.
22. Plante I, Centron D, Roy PH. An integron cassette encoding erythromycin esterase, ere(A), from Providencia stuartii. J. Antimicrob. Chemother. 2003; 51:787-790.
23. Wondrack L, Massa M, Yang BV, Sutcliffe J. Clinical strain of Staphylococcus aureus inactivates and causes efflux of macrolides. Antimicrob. Agents Chemother. 1996; 40:992-998.
24. Mukhtar TA, Wright GD. Streptogramins, oxazolidinones, and other inhibitors of bacterial protein synthesis. Chem. Rev. 2005; 105:529-542.
25. Mukhtar TA, Koteva KP, Hughes DW, Wright GD. Vgb from Staphylococcus aureus inactivates streptogramin B antibiotics by an elimination mechanism not hydrolysis. Biochemistry 2001; 40:8877-8886.
26. Llano-Sotelo B, Azucena EF Jr., Kotra LP, Mobashery S, Chow CS. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2002; 9:455-463.
27. Vetting MW, S de Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 2005; 433:212-226.
28. Hansen JL, Moore PB, Steitz TA. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 2003; 330:1061-1075.
29. Schlunzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 2001; 413:814-821.
30. Leslie AG, Moody PC, Shaw WV. Structure of chloramphenicol acetyltransferase at 1.75-A resolution. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:4133-4137.
31. Murray IA, Shaw WV. O-Acetyltransferases for chloramphenicol and other natural products. Antimicrob. Agents Chemother. 1997; 41:1-6.
32. Shaw WV, Leslie AG. Chloramphenicol acetyltransferase. Annu. Rev. Biophys. Biophys. Chem. 1991; 20:363-386.
33. Beaman TW, Sugantino M, Roderick SL. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa. Biochemistry 1998; 37:6689-6696.
34. Sugantino M, Roderick SL. Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry 2002; 41:2209-2216.
35. Kehoe LE, Snidwongse J, Courvalin P, Rafferty JB, Murray IA. Structural basis of Synercid (quinupristin-dalfopristin) resistance in Gram-positive bacterial pathogens. J. Biol. Chem. 2003; 278:29963-29970.
36. Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DS, Wright GD, Berghuis AM. Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell 1997; 89:887-895.
37. Wright GD, Thompson PR. Aminoglycoside phosphotransferases: proteins, structure, and mechanism. Front. Biosci. 1999; 4:D9-D21.
38. Tu D, Blaha G, Moore PB, Steitz TA. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell 2005; 121:257-270.
39. Hansen JL, Ippolito JA, Ban N, Nissen P, Moore PB, Steitz TA. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell 2002; 10:117-128.
40. Chen CY, Nace GW, Solow B, Fratamico P. Complete nucleotide sequences of 84.5- and 3.2-kb plasmids in the multi-antibiotic resistant Salmonella enterica serovar Typhimurium U302 strain G8430. Plasmid 2006; 57:29-43.
41. Matsuoka M, Endou K, Kobayashi H, Inoue M, Nakajima Y. A plasmid that encodes three genes for resistance to macrolide antibiotics in Staphylococcus aureus. FEMS Microbiol. Lett. 1998; 167:221-227.
42. Nakamura A, Miyakozawa I, Nakazawa K, O’Hara K, Sawai T. Detection and characterization of a macrolide Tl-phosphotransferase from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 2000; 44:3241-3242.
43. Noguchi N, Emura A, Matsuyama H, O’Hara K, Sasatsu M, Kono M. Nucleotide sequence and characterization of erythromycin resistance determinant that encodes macrolide 2;-phosphotransferase I in Escherichia coli. Antimicrob. Agents Chemother. 1995; 39:2359-2363.
44. Quiros LM, Carbajo RJ, Brana AF, Salas JA. Glycosylation of macrolide antibiotics. Purification and kinetic studies of a macrolide glycosyltransferase from Streptomyces antibioticus. J. Biol. Chem. 2000; 275:11713-11720.
45. Gourmelen A, Blondelet-Rouault MH, Pernodet JL. Characterization of a glycosyl transferase inactivating macrolides, encoded by gimA from Streptomyces ambofaciens. Antimicrob. Agents Chemother. 1998; 42(10):2612-2619.
46. Cundliffe E. Glycosylation of macrolide antibiotics in extracts of Streptomyces lividans. Antimicrob. Agents Chemother. 1992; 36(2):348-352.
47. Mosher RH, Camp DJ, Yang K, Brown MP, Shaw WV, Vining LC. Inactivation of chloramphenicol by O -phosphorylation. A novel resistance mechanism in Streptomyces venezuelae ISP5230, a chloramphenicol producer. J. Biol. Chem. 1995; 270(45):27000-27006.
48. Izard T, Ellis J. The crystal structures of chloramphenicol phosphotransferase reveal a novel inactivation mechanism. EMBO J. 2000; 19(11):2690-2700.
49. Bozdogan B, Berrezouga L, Kuo MS, Yurek DA, Farley KA, Stockman BJ, Leclercq R. A new resistance gene, linB, conferring resistance to lincosamides by nucleotidylation in Enterococcus faecium HM1025. Antimicrob. Agents Chemother. 1999; 43(4):925-929.
50. Leclercq R, Brisson-Noel A, Duval J, Courvalin P. Phenotypic expression and genetic heterogeneity of lincosamide inactivation in Staphylococcus spp. Antimicrob. Agents Chemother. 1987; 31(12):1887-1891.
51. Lee CK, Minami M, Sakuda S, Nihira T, Yamada Y. Stereospecific reduction of virginiamycin M1 as the virginiamycin resistance pathway in Streptomyces virginiae. Antimicrob. Agents Chemother. 1996; 40(3):595-601.
52. Andersen SJ, Quan S, Gowan B, Dabbs ER. Monooxygenase-like sequence of a Rhodococcus equi gene conferring increased resistance to rifampin by inactivating this antibiotic. Antimicrob. Agents Chemother. 1997; 41(1):218-221.
53. Roberts MC. Update on acquired tetracycline resistance genes. FEMS Microbiol. Lett. 2005; 245(2):195-203.
54. Speer BS, Salyers AA. Novel aerobic tetracycline resistance gene that chemically modifies tetracycline. J. Bacteriol. 1989; 171(1):148-153.
55. Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD. TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J. Biol. Chem. 2004; 279(50): 52346-52352.
56. Moore IF, Hughes DW, Wright GD. Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry 2005; 44(35):11829-11835.
57. Kumar A, Schweizer HP. Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv. Drug Deliv. Rev. 2005; 57(10): 1486-1513.
58. Li XZ, Nikaido H. Efflux-mediated drug resistance in bacteria. Drugs 2004; 64(2):159-204.
59. Piddock LJ. Multidrug-resistance efflux pumps—not just for resistance. Nat. Rev. Microbiol. 2006; 4(8):629-636.
60. Piddock LJ. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin. Microbiol. Rev. 2006; 19(2):382-402.
61. Poole K. Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 2005; 56(1):20-51.
62. Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003; 301(5633):610-615.
63. Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 2003; 301(5633):616-620.
64. Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science 2006; 312(5774):741-744.
65. Yu EW, Aires JR, McDermott G, Nikaido H. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J. Bacteriol. 2005; 187(19):6804-6815.
66. Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE Jr. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 2003; 300(5621):976-980.
67. Higgins MK, Bokma E, Koronakis E, Hughes C, Koronakis V. Structure of the periplasmic component of a bacterial drug efflux pump. Proc. Natl. Acad. Sci. U.S.A. 2004; 101(27):9994-9999.
68. Mikolosko J, Bobyk K, Zgurskaya HI, Ghosh P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure 2006; 14(3):577-587.
69. Koronakis V, Li J, Koronakis E, Stauffer K. Structure of TolC, the outer membrane component of the bacterial type I efflux system, derived from two-dimensional crystals. Mol. Microbiol. 1997; 23(3):617-626.
70. Wolter DJ, Hanson ND, Lister PD. Insertional inactivation of oprD in clinical isolates of Pseudomonas aeruginosa leading to car- bapenem resistance. FEMS Microbiol. Lett. 2004; 236(1):137-143.
71. Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005; 105(2):425-448.
72. Bugg TDH, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide pepti- doglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry 1991; 30:10408-10415.
73. Denamur E, Matic I. Evolution of mutation rates in bacteria. Mol. Microbiol. 2006; 60(4):820-827.
74. Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005; 3(6):e176.
75. Maravic G. Macrolide resistance based on the Erm-mediated rRNA methylation. Curr. Drug Targets Infect. Disord. 2004; 4(3):193-202.
76. Weisblum B. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother. 1995; 39(3):577-585.
77. Liou GF, Yoshizawa S, Courvalin P, Galimand M. Aminoglycoside resistance by ArmA-mediated ribosomal 16S methylation in human bacterial pathogens. J. Mol. Biol. 2006; 359(2):358-364.
78. Kawano Y, Kumagai T, Muta K, Matoba Y, Davies J, Sugiyama M. The 1.5 A crystal structure of a bleomycin resistance determinant from bleomycin-producing Streptomyces verticillus. J. Mol. Biol. 2000; 295(4):915-925.
79. Maruyama M, Kumagai T, Matoba Y, Hayashida M, Fujii T, Hata Y, Sugiyama M. Crystal structures of the transposon Tn5-carried bleomycin resistance determinant uncomplexed and complexed with bleomycin. J. Biol. Chem. 2001; 276(13):9992-9999.
80. Sugiyama M, Kumagai T, Hayashida M, Maruyama M, Matoba Y. The 1.6-A crystal structure of the copper(II)-bound bleomycin complexed with the bleomycin-binding protein from bleomycin-producing Streptomyces verticillus. J. Biol. Chem. 2002; 277(3):2311-2320.
81. Martin TW, Dauter Z, Devedjiev Y, Sheffield P, Jelen F, He M, Sherman DH, Otlewski J, Derewenda ZS, Derewenda U. Molecular basis of mitomycin C resistance in streptomyces: structure and function of the MRD protein. Structure 2002; 10(7):933-942.
82. Sheldon PJ, Johnson DA, August PR, Liu HW, Sherman DH. Characterization of a mitomycin-binding drug resistance mechanism from the producing organism, Streptomyces lavendulae. J. Bacteriol. 1997; 179(5):1796-1804.
83. Danshiitsoodol N, de Pinho CA, Matoba Y, Kumagai T, Sugiyama M. The mitomycin C (MMC)-binding protein from MMC- producing microorganisms protects from the lethal effect of bleomycin: crystallographic analysis to elucidate the binding mode of the antibiotic to the protein. J. Mol. Biol. 2006; 360(2):398-408.
84. Galm U, Hager MH, Van Lanen SG, Ju J, Thorson JS, Shen B. Antitumor antibiotics: bleomycin, enediynes, and mitomycin. Chem. Rev. 2005; 105(2):739-758.
85. Tanaka T, Fukuda-Ishisaka S, Hirama M, Otani T. Solution structures of C-1027 apoprotein and its complex with the aromatized chromophore. J. Mol. Biol. 2001; 309(1):267-283.
86. Biggins JB, Onwueme KC, Thorson JS. Resistance to enediyne antitumor antibiotics byCalC self-sacrifice. Science 2003; 301(5639): 1537-1541.
87. Connell SR, Tracz DM, Nierhaus KH, Taylor DE. Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob. Agents Chemother. 2003; 47(12):3675-3681.
88. Burdett V. Tet(M)-promoted release of tetracycline from ribosomes is GTP dependent. J. Bacteriol. 1996; 178(11):3246-3251.
89. Trieber CA, Burkhardt N, Nierhaus KH, Taylor DE. Ribosomal protection from tetracycline mediated by Tet(O): Tet(O) interaction with ribosomes is GTP-dependent. Biol. Chem. 1998; 379(7):847-855.
90. Maiti SN, Phillips OA, Micetich RG, Livermore DM. beta-lactamase inhibitors: agents to overcome bacterial resistance. Curr. Med. Chem. 1998; 5(6):441-456.
91. Wright GD. Resisting resistance: new chemical strategies for battling superbugs. Chem. Biol. 2000; 7(6):R127-R132.
92. Lomovskaya O, Bostian KA. Practical applications and feasibility of efflux pump inhibitors in the clinic—a vision for applied use. Biochem. Pharmacol. 2006; 71(7):910-918.
93. Pages JM, Masi M, Barbe J. Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med. 2005; 11(8):382-389.
Further Reading
Walsh CT. Antibiotics: Actions, Origins, Resistance. 2003. ASM Press, Washington, D.C.
White DG, Alekshun MN, McDermott PF, eds. Frontiers in Antimicrobial Resistance, A Tribute to Stuart Levy. 2005. ASM Press, Washington, D.C.
See Also
Antibiotics, Mechanisms of
Antibiotics, Biosynthesis of
Enzyme Catalysis, Chemistry of
Secondary Metabolite, Chemical Diversity of