CHEMICAL BIOLOGY
Mass Spectrometry, Applications in Phosphoproteomics
Francesco Giorgianni, Charles B. Stout Neuroscience Mass Spectrometry Laboratory, The University of Tennessee Health Science Center, Memphis, Tennessee
Sarka Beranova-Giorgianni, Department of Pharmaceutical Sciences, The University of Tennessee Health Science Center, Memphis, Tennessee
doi: 10.1002/9780470048672.wecb304
Proteins are biological macromolecules whose structure and functions are essential to every biological process within cells. Protein phosphorylation is one of the most important post-translational modifications and it has a profound effect on protein function. Recently, concurrent advances in bioanalytical technologies and informatics enabled studies of proteins and phosphoproteins on a global scale. These large-scale approaches represent an integral component of systems biology, which is an area of scientific inquiry that focuses on a biological system as a whole. State-of-the art mass spectrometry is a key technology for global-scale protein and phosphoprotein analyses. Phosphorylated proteins from diverse biological systems can be probed with a combination of separation methods, tandem mass spectrometry, and bioinformatics, to reveal the identity of the phosphorylated protein and the exact localization of the site(s) of phosphorylation. Characterization of the phosphoproteomes in cells, tissues and biological fluids provides an excellent foundation on which to build new knowledge of living systems.
Proteins are the final products of gene expression, and while the genome provides the “blueprint” for the molecular components of a living cell, proteins are the essential molecules responsible for cellular structure and function. Decades of studies of proteins in a one-by-one fashion have generated a wealth of knowledge on proteins as individual parts of the cellular machinery. Recently, interrogation of proteins in biological systems on a global scale gave rise to a new area of scientific inquiry, termed proteomics. Proteomics focuses on the study of the proteome, which is defined as the array of proteins that are present in a cell, organ, or biological fluid at a specific time, under a specific set of conditions. The goals of proteomics are diverse and include elucidation of basic molecular mechanisms that regulate cell function in physiological and pathological state, discovery of novel targets for the development of improved drug treatments, discovery of biomarkers for early detection of a disease and for design of tailored therapies, and many other objectives.
From the analytical standpoint, large-scale, comprehensive analysis of proteins is an extremely challenging undertaking because of the enormous complexity of proteomes and their dynamic nature. In fact, the development of proteomics as a scientific discipline was made possible through the concurrent advances in separation sciences, mass spectrometry, and informatics. Mass spectrometry has been the essential technology that enabled interrogating proteins on a global scale, with a high degree of sensitivity and accuracy. The purpose of this chapter is to describe the basic principles of mass spectrometry in the context of proteomics. Specifically, the review focuses on the use of mass spectrometry for large-scale analysis of specific subsets of proteomes - the phosphoproteomes. The chapter includes discussion of the basics of gas-phase behavior of peptides and phosphopeptides, and shows the role of mass spectrometry as a component of a general analytical strategy for phosphoproteome analysis. Because of the diversity of the analytical platforms that are being used, this review is not intended as a comprehensive description of all approaches. Rather, this article includes an overview of selected methods, a sampling of relevant references, and an example of a mass spectrometry-based phosphoproteomics methodology used in the authors’ laboratories.
Biological Background
Proteins are high molecular weight organic molecules that are essential to every biological process within living systems. Proteins are structurally and functionally diverse. Some proteins are assembled in multi-unit complexes to form the cytoskeleton of cells or other mechanical structures, while others are enzymes that catalyze biochemical reactions, or they participate in signal transduction within a cell or in cell-to-cell communication. Post-translational modifications play a key role in regulatory cellular processes, and in particular, protein phosphorylation is central to most of the signaling events that ultimately determine the biological status of all eukaryotic cells. The intracellular regulation of protein phosphorylation within cells occurs via a very complex system of positive and negative feed-backs with the surrounding environment. Protein phosphorylation regulates critical protein functions such as protein-DNA, protein-RNA, and protein-protein interactions, enzyme activity, protein trafficking, protein intracellular localization, and protein degradation. Aberrations in protein phosphorylation can have deleterious consequences and have been linked to various diseases, including cancer. It is estimated that approximately 30% of all proteins in a mammalian cell are phosphorylated at any given time (1).
A proteome represents the complete repertoire of proteins present in a cell at any given time. The term phosphoproteome refers to a specific subset of the proteome that includes all the phosphorylated protein species. Phosphoproteomics focuses on the comprehensive characterization of phosphorylated proteins in biological systems, including identification of phosphorylated proteins, assignment of their exact sites of phosphorylation, and quantification of changes in protein phosphorylation. The expansion of proteomics and phosphoproteomics in recent years has been driven by technological developments. The greatest challenge for proteomics is the inherent complexity of cellular proteomes, which is due to the dynamic nature of the proteome, the large number and wide abundance range of cellular proteins, and their diverse physicochemical properties. It is recognized that the diversity and extent of proteome complexity cannot be solved by a single technology. Instead, the trend in proteomics is to develop an array of methodologies from which a method or a set of methods can be selected to tailor the analytical strategy to suit a specific study. Chromatography and electrophoresis are the central separation technologies for proteomics. High performance mass spectrometry in combination with bioinformatics tools are key components for protein identification and characterization.
Mass Spectrometry in Phosphoproteomics
Proteins are made of twenty “standard” amino acid residues joined together through peptide bonds. Each of the twenty amino acids has unique physico-chemical properties stemming from the size of the side chain, and the possible presence of an acidic or basic ionizable group. Protein phosphorylation most commonly occurs on serine, threonine, or tyrosine residues. The task to characterize phosphorylated proteins on a proteome-wide scale includes determination of protein identities and localization of the phosphorylated amino acid residues in these proteins. Mass spectrometry is the central technology for these tasks. Although there have been major advancements in the mass spectrometry analysis of intact proteins in proteomics (2), most approaches still focus on characterization of peptides and phosphopeptides from proteolytic digestion of proteins. Therefore, the discussion in this section will concentrate on these strategies.
Mass spectrometry has several inherent characteristics that make it an excellent choice for peptide analysis. The technique is rapid, versatile, highly amenable to automation, and it requires low-to-mid femtomole sample quantities to yield reliable information about the amino acid sequence of a peptide. (The field of mass spectrometry is continuously moving towards improved detection limits, and cutting-edge instruments provide sensitivity in the attomol range). For phosphoproteome analysis, the general analytical strategy (Fig. 1) includes isolation of the proteins from the biological system under study; protein or peptide fractionation and enrichment of phosphorylated proteins/peptides; mass spectrometry measurement of specific attributes of phosphopeptides, including their mass and fragmentation patterns; searches of protein sequence databases to identify the proteins and to assign phosphorylation sites.

Figure 1. Basic components of a general analytical strategy for phosphoproteome analysis.
Protein extraction
The first step in the analysis of proteomes and phosphoproteomes involves extraction of proteins from the biological system under study; the objective is to solubilize the proteins and to prepare them for subsequent analysis. Obviously, this step is critical for the overall success of the analysis, and choice of methods should be tailored to the characteristics of the biological system and to the goals of the study. Depending on the biological system, protein extraction may involve disruption of cells, removal of contaminants such as salts (e.g., by dialysis or ultrafiltration), and/or overabundant proteins (e.g., by immunoaffinity columns). For phosphoproteomics, particular care must be taken to preserve phosphorylation of the proteins, i.e., to prevent the action of protein phosphatases. This is achieved by controlling the temperature of the sample and by addition of phosphatase inhibitors to the extraction buffer.
Separation and enrichment
Complexity of the analyte mixtures is a critical issue for phosphoproteome analysis. The challenge is to probe a specific subset of molecules (phosphopeptides) among an enormous number of other peptides; diverse physicochemical properties and low abundance of many phosphoproteins/phosphopeptides further contribute to the challenge. To address this issue, multidimensional fractionations of analyte mixtures, often in conjunction with phosphoprotein/phosphopeptide enrichment, are necessary prior to the mass spectrometry analysis itself. Fractionation of the original analyte mixture produces multiple mixtures, each with reduced complexity than the starting material. The necessity to analyze all the fractions decreases the overall throughput of the analytical platform but results in an improved coverage of the phosphoproteome. The fractionation steps can be performed at the protein level or at the peptide level, using electrophoretic and/or chromatographic methods. At the protein level, the traditional approach for proteome fractionation involves two-dimensional gel electrophoresis (2D-PAGE) that combines isoelectric focusing (IEF) in the first dimension and SDS-PAGE in the second dimension. For phosphoproteomics, specific stains or immunoblotting may be used to selectively visualize phosphorylated proteins in 2D gels. One-dimensional electrophoretic separations using IEF or SDS-PAGE provide protein fractions of moderate complexity. Alternatively, separations may be performed at the peptide level after proteolytic digestion of the entire proteome. In the context of phosphoproteomics, the most widely used methodology involves strong cation exchange chromatography (3, 4).
The electrophoretic or chromatographic separation methods are often combined with enrichment strategies to specifically enrich for phosphorylated species. This enrichment can be employed at the protein level, preceding protein fractionation, and/or at the peptide level following protein fractionation and digestion. Enrichment is accomplished by various types of affinity chromatography. Affinity chromatography methods may involve immunoaffinity using antibodies specific for phosphorylated amino acid residues in proteins or peptides (5), immobilized metal ion affinity chromatography (IMAC) (6) or metal oxide affinity chromatography (MOAC) (7). Some analytical platforms include esterification to reduce non-specific binding of non-phosphorylated peptides to the IMAC column (8). Chemical derivatization methods have also been developed that replace the phosphate group with a chemically different moiety, e.g., to introduce a tag that allows subsequent capture of the modified peptides (reviewed in ref. 9).
Mass spectrometry of peptides and phosphopeptides
The basic goal of the mass spectrometry measurement in the context of peptide analysis in proteomics and phosphoproteomics is to determine specific attributes that are then used in subsequent database searches to provide: 1. the identity of the proteins present in the sample; 2. location of the site(s) of phosphorylation in these proteins. Both pieces of information are derived from the mass of the peptide and, most importantly, from the gas-phase dissociation patterns that are diagnostic of the peptide’s amino acid sequence and phosphosite location. The gas-phase dissociation patterns are obtained via tandem mass spectrometry (MS/MS). On a phosphoproteome-wide scale, the analysis includes measurement of the attributes for many thousands of individual peptides.
Basics of gas-phase behavior of peptides
Mass spectrometry deals with measurements of gas-phase ions. Therefore, the first step in peptide analysis by mass spectrometry is the conversion of the analytes into charged species in the gas phase. Peptides are relatively large, non-volatile biomolecules. In today’s world, the two ionization methods that are used in mass spectrometry of peptides and other biological macromolecules are matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI). MALDI and ESI are based on different principles (10, 11); however, both methods ionize peptides via protonation in an acidic environment. MALDI produces mainly singly-protonated peptide ions, while in ESI protonation occurs on all available basic sites in the peptide, thus yielding multiply-charged species. For tryptic peptides, which possess an amino group at the N-terminus, a basic amino acid (arginine or lysine) at the C-terminus, and possible internal basic amino acid residues, doubly- and triply-charged peptide ions usually predominate. Multiple charging in ESI has important implications for subsequent dissociations of the (activated) peptide ions. The “mobile” proton originally localized on the N-terminus migrates along the peptide backbone by internal solvation, producing a heterogeneous population of peptide ions that have the same sequence but different sites of protonation (12, 13). This heterogeneity of protonation sites drives the dissociations of peptide ions in MS/MS that are discussed below.
Mass analysis determines the mass-to-charge ratio (m/z) of ions derived from the analyte. For peptide ions, two characteristics can be obtained. The first characteristic is the molecular weight of the peptide, which can be calculated from the measured m/z of the source-generated intact peptide ion (the so-called molecular ion). The second characteristic is structural information that is obtained via an MS/MS analysis. An MS/MS experiment measures gas-phase dissociations of an activated molecular ion of the peptide to yield product-ion data that are diagnostic of the peptide sequence. The basic sequence of events in MS/MS includes: 1. mass selection of the peptide ion of interest (that is population of ions of a single m/z) as a so-called precursor ion; 2. activation of the precursor ion, most commonly through collisions with an inert gas, followed by dissociation of the activated precursor and formation of product ions; 3. Mass analysis of the product ions and recording of the MS/MS spectrum. The process of collisional activation and dissociation is termed collision-induced dissociation (CID). Depending on the instrument type, the MS/MS events can be separated in space (tandem-in-space) or in time (tandem-in-time).
In MS/MS, protonated peptide ions in the gas phase dissociate via cleavages along the peptide backbone; fragmentations can occur at any of the three types of bonds that make up the backbone of the peptide (Fig. 2). The nomenclature for peptide dissociations distinguishes six major series of sequence-determining product ions (14, 15). The N-terminal series encompass the a-, b-, and c-ion types; the C-terminal series include x-, y-, and z-ions. In addition to the six basic series, other types of product ions may also be observed (16) under certain conditions. The relative abundance of the different product ions depends on the amino acid sequence of the peptide, on the internal energy of the dissociating precursor ion, and on additional variables that affect the CID process (17). Under low-energy CID regime, used for example in ion trap mass spectrometers, the predominant types of product ions are the b-ions and y-ions that form by cleavages of the peptide bond. As shown in Fig. 2, adjacent (singly-charged) product ions from a series have a difference in mass that determines the amino acid present at that position of the peptide. For example, if the amino acid in position 3 of the tetrapeptide in Fig. 2 is a serine (R3=CH2OH), the mass difference between the y1 and y2 product ions will be 87 Da, corresponding to the mass of the serine residue -NH-CH(CH2OH)-CO-. Therefore, when an MS/MS spectrum of a peptide ion contains high quality data for one complete or several partial overlapping product-ion series, then the sequence of the peptide can be deduced from the MS/MS data.
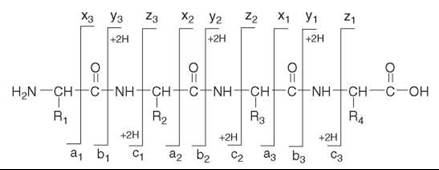
Figure 2. Nomenclature of peptide fragmentation. The possible product ion series that arise by cleavages along the peptide backbone are a-, b-, and c-series (N-terminal); and x-, y-, and z-series (C-terminal). (The designation +2H denotes addition of two hydrogens that are transferred onto the structures depicted in the figure to form the corresponding singly charged y- or c- product ions (21)). Under low-energy CID, y- and b-ions usually predominate. The mass differences between adjacent ions of the same series can be used to deduce portions of the peptide sequence.
Specifics of phosphopeptides
Phosphorylated peptides are modified peptides and therefore most of the basic concepts discussed above also apply to mass spectrometry of phosphopeptides.
In terms of ionization, the majority of large-scale phosphoproteomics strategies utilize ESI. The difficulty to effectively analyze phosphopeptides by ESI-based approaches is often attributed, among other factors, to selective suppression of phosphorylated peptides in the presence of unmodified peptides, and to decreased ionization efficiencies of phosphopeptides relative to their non-phosphorylated counterparts. However, this notion has been challenged in a recent study (18) underscoring the complexity of the phenomena associated with analyses of highly complex peptide/phosphopeptide mixtures.
Modification by phosphorylation adds 80 Da to the mass of the corresponding peptide. The principles of gas-phase dissociations of protonated phosphopeptide ions into sequencedetermining product ion series are analogous to those of non-phosphorylated peptides, with an additional issue that must be taken into consideration. Under CID conditions phosphorylated peptide ions undergo a facile neutral loss of H3PO4, corresponding to the loss of 98 Da. The mechanisms that underlie this dissociation behavior have been studied for phosphoserine-, phosphothreonine-, and phosphotyrosine-containing peptides (19, 20). The loss of phosphoric acid from the molecular ion produces a non-sequence specific product ion (M+nH-H3PO4)n+. This product ion can serve as a marker ion, indicating the presence of a phosphorylated peptide. An example of an MS/MS spectrum of a phosphorylated peptide is shown in Fig. 3. This spectrum illustrates the typical fragmentation behavior of protonated phosphopeptide ions in ion trap MS/MS. The spectrum is dominated by the intense (M+2H-H3PO4)2+ product ion; the spectrum further contains product ions of the y- and b- series that determine the amino acid sequence of the phosphopeptide and the location of the phosphorylation site. Often, the scenario is not so favorable. The loss of phosphoric acid dominates and not enough other product ions are observed for an unequivocal sequence determination. One way to remedy this unfavorable outcome is to perform an additional dissociation, an MS/MS/MS, where the primary product ion (M+nH-H3PO4)n+ is mass-selected and then dissociated via CID.

Figure 3. MS/MS spectrum of the phosphopeptide FNDS*EGDDTEETEDYR. The spectrum, which was acquired with an ion trap mass spectrometer, illustrates the typical behavior of phosphopeptide ions under low-energy CID. The molecular ion was (M+2H)2+, m/z 1001.9. The MS/MS spectrum is dominated by an intense product ion corresponding to the neutral loss of phosphoric acid from the activated precursor ion. In addition, the spectrum contains a number of product ions from the y- and b-series that determine the peptide sequence and the site of phosphorylation. (The product ions are singly charged unless noted otherwise). This phosphopeptide belongs to Bcl-2-associated transcription factor 1 (BCLF1_HUMAN) and it was identified in the analysis of the phosphoproteome in the LNCaP prostate cancer cell line.
The LC-MS/MS experiment
This section discusses a typical mass spectrometry experiment used for phosphopeptide analysis. Typically, after protein/peptide fractionation and enrichment, the peptide digest will contain phosphopeptides in mixture with nonphosphorylated peptides. This mixture will be of a high complexity. Commonly, the mass spectrometry measurement itself is preceded by a separation of this peptide digest by reversed-phase liquid chromatography, interfaced online to MS. This LC step, typically performed in nanoflow regime and using a shallow mobile phase gradient, will separate the peptides according to their hydrophobicities. The peptides eluting from the LC are ionized by nanoESI, and peptide mass and MS/MS data are measured. Two issues have to be addressed. First, because of the complexity of the starting mixture, even after LC separation, multiple peptides will coelute and therefore at any given time there will be more than one peptide ion present. Second, the characteristics, including the mass of the precursor ion that is needed to set the precursor selection in MS/MS are not known. These issues are dealt with through data-dependent acquisition mode, in which the mass spectrometer automatically cycles through a sequence of measurements of MS and MS/MS data. For peptide and phosphopeptide analysis, the instrument measures an MS spectrum to obtain masses of the analytes eluting from LC at that particular time. Based on the information from these MS data, subsequent MS/MS events are set - for example, 5 MS/MS measurements of 5 of the most intense ions from the MS spectrum, provided they are above a specified intensity threshold. The cycle is repeated many times during the LC-MS/MS analysis. To maximize information gained in the MS/MS steps, some strategies are incorporated such as a permanent exclusion from MS/MS of known contaminants throughout the entire analysis; and temporary exclusion of peptides whose MS/MS have already been measured for the duration of the expected time that it takes for the peptide to elute from the LC column. These strategies that decrease redundancy and maximize the number of peptides surveyed in the analysis are particularly important for phosphopeptides that are frequently minor components in a peptide digest. In a typical LC-MS/MS analysis, a large number of MS and MS/MS spectra are acquired, for example >10,000 MS/MS spectra on state-of-the-art ion trap instruments.
Bioinformatics
In proteomics and phosphoproteomics applications, search programs are used that utilize minimally processed MS/MS data without the need for manual interpretation (21). Development of these programs, for example SEQUEST, that allow the integration of mass spectrometry data with database searching has been one of the enabling developments in proteomics. In SEQUEST-based searches, the experimentally measured peptide mass is used to locate in the database peptide sequences whose masses match the measured mass, and then experimental product ion patterns are compared to theoretical patterns for each candidate peptide, and a correlation score is calculated. The highest scoring peptide sequences are reported. For phosphopeptide characterization, the search considers possible addition of 80 Da to serine, threonine, and tyrosine residues. After completion of the search, it is imperative that the spectra and the database search outputs are inspected before an ultimate decision about the correctness of the match is reached. For phosphopeptides, this examination includes verification that the correct amino acid sequence was retrieved, and verification of the assignment of the phosphorylation site. Once the phosphoproteins are identified and their sites are characterized, additional bioinformatics resources are available for in silico analysis and functional integration. For example, with the program Scan- site (scansite.mit.edu), sequences of the identified proteins are searched to locate motifs that would suggest phosphorylation by a specific kinase or a phospho-specific binding interaction. Information on protein phosphorylation is compiled in several databases, for example Phosphosite (www.phosphosite.org), Phosida (www.phosida.org), and others.
Additional approaches and current developments
Mass spectrometry-based phosphoproteomics is characterized by a great diversity of bioanalytical workflows and by continuous developments of new and improved strategies. Some of these new approaches are summarized in this section.
To address some of the issues associated with CID of phosphopeptides, new strategies for ion activation/dissociation have been introduced recently in the context of phosphoproteomics. In particular, electron transfer dissociation (ETD) is emerging as a promising new strategy for MS/MS-based phosphopeptide analysis (22, 23).
It should also be noted that besides the conventional production scanning, specialized MS/MS functions have been adopted for phosphopeptide analysis (9). These include precursor ion scanning for monitoring the precursors of the product ion PO3- (m/z 79) in the negative mode, or precursors of the phosphotyrosine-specific immonium ion at m/z 216.043 in the positive mode.
Finally, quantitative information in phosphoproteomics may be obtained through the use of stable isotope labeling. Stable isotope labeling is a proven approach for mass spectrometry- based quantification. Recent examples of stable isotope labeling methods that have been successfully adapted for large-scale quantification of protein phosphorylation include the iTRAQ methodology that involves chemical tagging at the peptide level (24), and the Stable Isotope Labeling of Amino Acids in Culture (SILAC) methodology that involves metabolic labeling (4).
Chemical Tools and Techniques
The study that serves to illustrate a possible bioanalytical stat- egy for characterization of a human phosphoproteome concerns characterization of protein phosphorylation in a human pituitary tissue (25). The analytical methodology involves in-gel IEF for protein separation, IMAC for enrichment of phospho- peptides after digestion, LC-MS/MS and database searches for phosphopeptide identification and localization of the phoshorylated amino acid residues.
The tissue sample is obtained from surgery or autopsy, and it must be frozen immediately to prevent protein degradation. Prior to analysis, the proteins including phosphoproteins are extracted via homogenization in the Trizol reagent that isolates proteins from RNA and DNA; a phosphatase inhibitor cocktail is added to minimize dephosphorylation. Proteins are obtained in the last step of the Trizol-based extraction in the form of a pellet that is then dissolved in a buffer suitable for isoelectric focusing. This buffer typically contains chaotropes such as urea and thiourea, CHAPS detergent, ampholytes that aid in the IEF, and dithiothreitol as a reducing agent. IEF that is performed in a commercially available immobilized pH gradient (IPG) strip separates the proteins based on their charge. After IEF, the strip is divided into sections. Each section still contains multiple proteins but the complexity of these mixtures is greatly reduced compared to that of the initial mixture. The proteins in each section of the IPG strip are digested with trypsin to produce mixtures of peptides that include phosphorylated and non-phosphorylated peptides. IMAC is used to enrich for phos- phorylated peptides. The steps in the IMAC procedure involve: 1. selective binding of the phosphopeptides via interaction of their phosphate groups with the immobilized metal ion (e.g., Ga3+) under carefully controlled acidic pH conditions; 2. washing of unbound and non-specifically bound material; 3. elution of phosphopeptides from the column under alkaline conditions. Following IMAC, desalting and volume reduction of the samples is performed with a C18 minicolumn, and the samples are analyzed by LC-MS/MS. The nano-LC setup includes a combined capillary column/spray needle packed with a C18 stationary phase. The i.d. of the column is 75 μm, the i.d. of the spray tip is 15 μm, and the flow-rate is on the order of 50-150 nL/min. Mobile phases typical for reversed-phase chromatography that are compatible with mass spectrometry are used, such as water/acetonitrile/formic acid or water/methanol/formic acid (26). Peptides and phosphopeptides eluting from the nanoLC are ionized by nanoelectrospray to produce multi-protonated ions in most cases (doubly or triply charged). MS and MS/MS spectra are acquired in the data-dependent mode. MS/MS/MS may be performed in non-data-dependent mode in a separate LC-MS/MS experiment. Alternatively, this step, where MS3 is triggered when a (M+nH-H3PO4)n+ product ion is present in the MS/MS spectrum, may be incorporated into data-dependent scanning (3). The set of data is used to search a protein sequence database such as the SWISSPROT or NCBInr. The search parameters include modifications on the amino acid residues where phosphorylation is expected to occur. The searches yield lists of phosphopeptide matches with scores indicating the quality of the match. The matches are evaluated manually. This evaluation has two objectives: confirmation of the correct amino acid sequence of the phosphopeptide which establishes the presence of the phosphorylated form of the corresponding protein in the pituitary; and assignment of the exact phosphorylation site(s) in the peptide. This validation includes inspection of the MS/MS data and the scores. Finally, the phosphorylated proteins are put into context of current scientific knowledge, using databases such as Phosphosite that extract and compile published information on protein phosphorylation.
Acknowledgments
The authors’ research activities are funded by the University of Tennessee College of Pharmacy, and by Chiesi Pharmaceuticals. Funds for mass spectrometry instrumentation have been provided in part by NIH grant 1S10 RR16679.
References
1. Cohen P. The regulation of protein function by multisite phosphorylation-a 25 year update. Trends Biochem. Sci. 2000; 25:596-601.
2. Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat. Methods 2007; 4:817-821.
3. Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U. S. A. 2004; 101:12130-12135.
4. Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006; 127:635-648.
5. Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005; 23:94-101.
6. Nuhse T, Yu K, Salomon A. Isolation of phosphopeptides by 26. immobilized metal ion affinity chromatography. Curr. Protoc. Mol. Biol. 2007; 18:18.13.
7. Thingholm TE, J0rgensen TJ, Jensen ON, Larsen MR. Highly selective enrichment of phosphorylated peptides using titanium dioxide. Nat Protoc. 2006; 1:1929-1935.
8. Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat Biotechnol. 2002; 20:301-305.
9. Collins MO, Yu L, Choudhary JS. Analysis of protein phosphorylation on a proteome-scale. Proteomics 2007; 7:2751-2768.
10. Dass C. Fundamentals of Contemporary Mass Spectrometry. 2007. Wiley, Hoboken, NJ, pp. 15-60.
11. Kinter M, Sherman NE. Protein Sequencing and Identification Using Tandem Mass Spectrometry. 2000. Wiley, New York, pp. 31-39.
12. Dongre AR, Jones JL, Somogyi A, Wysocki VH. Influence of peptide composition, gas-phase basicity, and chemical modifications on fragmentation efficiency: Evidence for the mobile proton model. J. Am. Chem. Soc. 1996; 118:8365-8374.
13. Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom. Rev. 2005; 24:508-548.
14. Roepstorff P, Fohlman J. Proposal for a common nomenclature for sequence ions in mass spectra of peptides. Biomed. Mass Spectrom. 1984; 11:601.
15. Biemann K. Contributions of mass spectrometry to peptide and protein structure. Biomed. Environ. Mass Spectrom. 1988; 16:99-111.
16. Dass C. Fundamentals of Contemporary Mass Spectrometry. 2007. Wiley, Hoboken, NJ, pp 317-322.
17. Wells JM, McLuckey SA. Collision-induced dissociation (CID) of peptides and proteins. Methods Enzymol. 2005; 402:148-185.
18. Steen H, Jebanathirajah JA, Rush J, Morrice N, Kirschner MW. Phosphorylation analysis by mass spectrometry: Myths, facts, and the consequences for qualitative and quantitative measurements. Mol. Cell. Proteomics 2006; 5:172-181.
19. DeGnore JP, Qin J. Fragmentation of phosphopeptides in an ion trap mass spectrometer. J. Am. Soc. Mass Spectrom. 1998; 9:1175-1188.
20. Palumbo AM, Tepe JJ, Reid GE. Mechanistic insights into the multistage gas-phase fragmentation behavior of phosphoserine- and phosphothreonine-containing peptides. J. Proteome Res. 2008; 7:771-779.
21. Hernandez P, Muller M, Appel RD. Automated protein identification by tandem mass spectrometry: issues and strategies. Mass Spectrom. Rev. 2006; 25:235-254.
22. Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF.
23. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U. S. A. 2004; 101:9528-9533.
24. Molina H, Horn DM, Tang N, Mathivanan S, Pandey A. Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U. S. A. 2007; 104:2199-2204.
25. Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc Natl Acad Sci U. S. A. 2007; 104:5860-5865.
26. Beranova-Giorgianni S, Zhao Y, Desiderio DM, Giorgianni F. Phosphoproteomic analysis of the human pituitary. Pituitary 2006; 9:109-120.
27. Giorgianni F, Cappiello A, Beranova-Giorgianni S, Palma P, Trufelli H, Desiderio DM. LC-MS/MS analysis of peptides with methanol as organic modifier: improved limits of detection. Anal. Chem. 2004; 76:7028-7038.
See Also
Post-Translational Modifications, Roles in Regulating Protein Function;
Proteins, Chemistry and Chemical Reactivity of