CHEMICAL BIOLOGY
Membrane Proteins, Properties of
Linda Columbus, Department of Chemistry, University of Virginia, Charlottesville, Virginia
Robert K. Nakamoto, Department of Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, Virginia
David S. Cafiso, Department of Chemistry, University of Virginia, Charlottesville, Virginia
doi: 10.1002/9780470048672.wecb311
Membrane proteins constitute a significant fraction of the proteins that are encoded in the typical genome, and they are critical to many cellular processes, which include transport, cell signaling, and energy transduction. This importance is underscored by the fact that membrane proteins represent the major class of protein targets for pharmaceuticals that are currently in use. However, despite their importance, there are relatively few high-resolution models for membrane proteins, and little is known about their molecular function. The lack of information on membrane proteins in part reflects the difficulties in efficient expression of this class of proteins and difficulties with structural approaches, such as high-resolution nuclear magnetic resonance (NMR), that are not well suited to membrane proteins. In the field of structural biology, membrane proteins represent a significant challenge, and in recent years new structural and biochemical tools have been developed to characterize these systems. In this review, we discuss the functions and properties of membrane proteins, and we discuss the approaches and tools that yield new information on their structure and molecular function.
Membranes define the boundaries and compartments that make up the cellular matrix, and they are responsible for defining the chemical environment within the cell. Membranes also provide an interface that facilitates protein-protein interactions and other biochemical reactions necessary for cell signaling and trafficking. It is estimated that protein domains that reversibly associate with membrane interfaces represent the most abundant domains found in water-soluble proteins (1). Although the number of proteins that interact with or function at the membrane interface is large, this review will focus on membrane proteins that are sometimes termed integral membrane proteins. Integral membrane proteins are generally defined as proteins that cannot be isolated and purified without first dissolving the bilayer structure, typically with detergents. When compared with water-soluble proteins, membrane proteins have unique properties and present unique challenges in terms of their expression, isolation, and structural characterization. Inducing membrane protein expression in high yield can be more challenging than that of water-soluble proteins. Because the forces that dominate the fold of a membrane protein differ from those that stabilize water-soluble protein folds, purified membrane proteins must be studied in heterogeneous environments, such as membrane mimetic detergent micelles or reconstituted lipid bilayers. It is estimated that approximately 30% of the genome codes for membrane proteins (2), although the exact percentage is unknown. However, when compared with the total number of entries in the protein databank, membrane proteins represent less than 1% of the total number. Although X-ray crystallography remains the single most important method for generating structures of membrane proteins, several new spectroscopic methods are being employed to study membrane protein structure and dynamics.
Biological Functions and Distribution of Membrane Proteins
Classification of membrane proteins: nomenclature and architecture
Membrane proteins carry out a wide range of critical functions in cells, and they include passive and active transporters, ion channels, many classes of receptors, cellular toxins, proteins involved in membrane trafficking, and the enzymes that facilitate electron transport and oxidative phosphorylation. For example, the voltage-gated ion channels that facilitate the passive diffusion of sodium and potassium across the axonal membrane are responsible for the formation of an action potential. Active transport proteins establish ion gradients and are necessary for the uptake of nutrients into cells. Soluble hormones bind to membrane receptors, which then regulate the internal biochemistry of the cell.
At the present time, representative structures exist for approximately 21 unique β-barrel membrane protein families and 35 polytopic α-helical protein families. This sample is a small fraction of the predicted 300-500 α-helical folds and 700-1700 families (3). Although the structural biology of membrane proteins is in its infancy, it is clear that membrane proteins display a rich variety of structures that vary greatly in size and topology (Fig. 1). Of the structures observed thus far, all are based on two fundamental architectures: the α-helical bundle (4) and the β-barrel (5, 6).
The membrane β-barrel fold is unique to the outer membrane of mitochondria, chloroplasts, and Gram-negative bacteria, and they are found to be composed of an even number of β-strands that vary in number between 8 and 22. The inter-strand hydrogen-bonding pattern within the barrel allows structures to form, which do not have unsatisfied hydrogen bonds within the membrane interior. Frequently, these strands are configured so that amino acid side chains with an aliphatic composition reside on the barrel exterior facing the membrane hydrocarbon, and as observed in membrane proteins based on helical bundles, β-barrel proteins display more aromatic side chains at regions near the membrane bilayer interface.
Membrane proteins based on transmembrane a-helices (Fig. 1) are typically localized to the plasma membrane, organelle membranes, and inner membrane of mitochondria and bacteria. Thus, these proteins not only differ in secondary structure, but also they differ in localization. Helical membrane proteins can be formed from 1 to 19 transmembrane segments. When they possess a single transmembrane pass, they are sometimes referred to as either monotopic or bitopic. When these proteins have two or more transmembrane helices, they are referred to as polytopic. The transmembrane helices of a polytopic membrane protein associate into a bundle, and to maintain unsatisfied hydrogen bonds to a minimum, these helices are usually regular. Although helical membrane proteins may be quite flexible and dynamic, elements of helical structure within the bilayer are thought to be rigid.

Figure 1. Examples of several bacterial membrane proteins. The outer membrane (OM) of Gram-negative bacteria contains exclusively β-barrel proteins, and three examples are shown: BtuB (PDB ID: 1NQF), which is the 22 β-stranded βonB-dependent active transporter for vitamin B12; the LamB or maltoporin trimer (PDB ID: 1AF6), which is the 18 β-stranded passive sugar transporter; and OmpA (PDB ID: 1 BXW), which is an 8 P-stranded protein that provides structural support for the OM. Proteins in the cytoplasmic membrane (CM) are helical, and three examples are shown: the potassium channel KcsA (PDB ID: 1 BL8), which is a tetramer; Sec YEG (PDB ID: 1 RH5), which forms the protein transport channel in Methanococcus; and BtuCD (PDB ID: 1L7V), which is the ATP-driven transporter that imports vitamin B12 from the periplasm.
Membrane protein folding and membrane insertion
Integral membrane proteins must be stable and function in a unique and highly anisotropic environment. The aqueous facing domains of a membrane protein experience a very different environment than do the membrane facing regions of the protein, which must be stable in a low-dielectric region devoid of water (7). Because of the absence of a bulk aqueous phase, significant numbers of unsatisfied hydrogen bonds are highly unfavorable, and the folds of β-barrel or helical membrane proteins, as indicated above, must always be arranged to satisfy backbone hydrogen bonding.
In water-soluble proteins, the hydrophobic effect is believed to be a major force that drives the folding of proteins, dominating over van der Waals forces (8, 9). However, in membrane proteins, the hydrophobic effect is relatively unimportant in comparison to side-chain hydrogen bonding and van der Waals forces. Van der Waals forces in a helical bundle would be maximized by complementarity of the interacting surfaces on transmembrane helices; as expected, sequence motifs, such as GxxxG, have been identified that maximize the packing of membrane helices (10-12). Interhelix hydrogen bonding is also important in driving the association of helices, and it may be an important determinant of helix association during membrane protein synthesis (13). Although both interactions play important roles, a mutational study on one helical bundle suggests that van der Waals forces make the largest contribution to the stability of the bundle (14).
The arrangement of the transmembrane segments of a membrane protein and the orientation of the protein C and N-termini are determined by two main features of a membrane protein sequence: the stretch of hydrophobic residues that ultimately span the bilayer, and the position of positively charged segments (15, 16). The hydrophobic residues (Ala, Ile, Leu, Val) have the highest frequency in helical regions that lie in the center of the bilayer, whereas the two aromatic residues Tyr and Trp are most abundant near the bilayer interface (17). The balance of charge on either side of a transmembrane segment determines the orientation of the segment, so that the more positively charged portions of a membrane protein sequence are found on the cytoplasmic side of the bilayer. This structure is sometimes referred to as the “positive-inside rule” (18). By altering hydrophobicity and charge, differing topologies can be generated, and it is even possible to find homologous proteins that have naturally evolved different topologies by modifying these features (15). Biosynthetically, the insertion of the growing polypeptide chain into the bilayer is mediated by a translocon, which is a helical membrane protein that is believed to have a lateral gate that opens to the bilayer interior. In the rough endoplasmic reticulum, this protein is Sec61. It is generally thought that the growing polypeptide chain may sample the surrounding bilayer through this gate, so that a thermodynamic equilibrium is established with the surrounding lipid (16, 19).
The lipid bilayer is not passive in determining membrane protein activity and function, and an accumulating body of evidence indicates that there is a coupling of membrane proteins to lipid bilayer properties. These properties include the effect of bilayer curvature strain (20), the role of specific lipids such as phosphoinositides, (21) and the effect of thickness on membrane protein function (22). The lipid composition, as well as the bilayer properties that result from this composition, act as allosteric regulators of membrane protein function.
Molecular function of membrane proteins
Structural methods and other genetic and biochemical studies have provided clues to the molecular function of membrane proteins. For example, in the case of the bacterial K+ channel KcsA, the protein is designed to lower the energy for a potassium ion as it passes through the center of the channel. This function is facilitated both by a central cavity that is hydrated and by a helix that is positioned toward the channel pore so that its negatively charged end points toward this cavity (23). In the case of visual rhodopsin, a salt bridge that exists between Lys296 and Glu133 functions to maintain the protein in an inactive state. Disruption of this ionic interaction is responsible for the movement of helix 6, which activates the G-protein transducin (24), and mutations that disrupt this ionic interaction are responsible for the retinal diseases retinitis pigmentosa and congenital night blindness (25). Among outer membrane bacterial transporters, TonB-dependent transporters function to move rare nutrients, such as iron chelates, into the cell. They contain a large N-terminal domain of approximately 150 residues that is sometimes referred to as a “hatch.” The available high-resolution structures suggest that substantial rearrangements or an unfolding of this hatch domain takes place to allow the passage of substrates (26).
Expression, Isolation and Purification of Membrane Proteins
Difficulties in obtaining protein samples
In general, membrane proteins present challenges at every step on the way to structural determination. The synthesis and processing of these proteins is complex and often involves specific folding factors or chaperones (see References 27 and 28 for reviews). Expression in bacteria is favored because of the low cost to grow large culture volumes by fermentation, the potential for high yields (up to 10 to 100 mg of protein per liter of culture), the very fast growth rate, and the simplicity and flexibility of expression systems. However, intrinsic differences in how proteins are processed often prevent the expression of adequate amounts of protein with the proper fold, and it may not be possible to isolate and purify sufficient quantities of a membrane protein to a homogenous state. As a result, understanding the processing pathways for the specific protein of interest can be the important factor to achieve successful expression.
The biosynthetic membrane insertion of eukaryotic membrane proteins almost always takes place cotranslationally, whereas many prokaryotic proteins can be posttranslationally inserted, typically with the aid of chaperones. In eukaryotic cells, the proofreading mechanisms in the endoplasmic reticulum prevent misfolded proteins from leaving to the Golgi. Some of these mechanisms include glycosylation of the protein on its luminal domain (29). In addition, other protective mechanisms such as the yeast unfolded protein response may lead to increased degradation rates of improperly folded proteins (30).
Some proteins require specific types of processing. For example, β-barrel proteins in the outer membrane of Gram negative bacteria must pass through the cytoplasmic membrane in a linear fashion before being assembled in the outer membrane (for a review, see Reference 31). Processing of β-barrel proteins in the outer membranes of mitochondria and chloroplasts may also involve passage of the protein through the organelle outer membrane before insertion, but this process is not well understood (32). Several chaperones have been identified that help the assembly of mitochondrial and chloroplast membrane proteins. Some of these proteins are encoded in the nucleus and are imported to the proper compartment posttranslationally, whereas a few are encoded on the organelle’s own chromosome and processed from within. For these reasons, structural approaches for such proteins always rely on isolation from the native organelle.
Bacterial expression systems are often not an option for eukaryotic membrane proteins. Even if the protein is found embedded in the bacterial membrane, obtaining correctly folded protein is always a concern. Frequently, most or all expressed protein is found in an intracellular aggregate or inclusion body, and only a few examples have been refolded to their native state (33). In many cases, the apparent toxicity of the expressed membrane protein blocks expression as well as growth. Several reasons can explain protein toxicity (see Reference 34 for a review). For example, Miroux and Walker (35) suggest that the overproduction of a single mRNA in a typically used T7 polymerase-driven transcription system resulted in the uncoupling of transcription and translation. A strain, such as C43(DE3) (Lucigen, Middleton, WI) selected to avoid the effects of toxicity, demonstrated better yields of proteins despite a slower rate of synthesis. The requirement of a slower rate of synthesis suggests that cellular machinery, which is not overexpressed, may be required for proper folding. On the other hand, mixed results are observed in providing the cell with extra in- sertional machinery. Interestingly, one of the more productive approaches has been to provide extra chaperones or foldases in the expression strain (36).
Despite the severe limitations to membrane protein expression, successful approaches have been developed. Three general strategies are used by investigators to obtain sufficient amounts of protein for structural studies. First, protein can be obtained from a native source. Many membrane protein structures have been obtained from protein purified from tissue or organelles; for example, rhodopsin, the sarcoplasmic reticulum calcium pump, and the electron transport complexes from mitochondria. Obviously, this approach only works in situations where the protein of interest expresses at naturally high levels. Furthermore, genetic manipulation of the protein primary structure, for example to add a purification tag, is usually not possible. Second, large numbers of bacterial orthologs of the protein of interest can be screened. With the availability of chromosomal DNA or cDNA libraries from a wide range of species, investigators can clone several homologs and test for high level expression and folding in an Escherichia coli expression strain. The hope is that one or more of the genes will be amenable to stable expression, purification, and structural determination. This approach has been used successfully in many cases such as various transporters, aquaporins, mechanosensitive channels, and potassium channels (37). Finally, the eukaryotic protein can be produced in a eukaryotic cell. This approach has become increasingly feasible as synthetic media have brought the expense of large-scale growth into the realm of the academic laboratory. The cells of choice are insect cells, either those from the fall army worm, Spodoptera frugiperda (Sf9 or 21) or the cabbage looper Trichoplusia ni (High Five; Invitrogen, Carlsbad, CA). Mammalian cells, such as CHO or COS, have been used in a few cases, as have the yeast Saccharomyces cerevisiae or Pichia pastoris (from Invitrogen). This approach assumes that the eukaryotic cells can recognize the processing signals intrinsic to the protein sequence and that the proper chaperones and foldases will be present. Although this scenario is more likely, it is certainly not true in all cases. As mentioned above, other factors may be necessary for proper folding. Furthermore, a protein that is part of a complex may not express in a stable or properly folded manner in the absence of its partners or assembly factors.
Although the optimization of growth conditions for eukaryotic cells is limited, the growth conditions for bacterial systems can be varied widely, and they have a large influence on the expression of the protein target. Many “tricks” investigators use are anecdotal and may be specific to their protein of interest. Several publications detail the approaches and often many different conditions must be tested (see References 33 and 38 for extensive reviews). We will not attempt a thorough listing of such methods, but a few are worth noting. First, medium strength promoters are used to slow down synthesis rate. As mentioned above, the cell may respond to an overwhelming synthesis of a specific protein by activating degradation systems. Second, a lower culture temperature is used, as low as 15° C, with expression induced for many hours to days. Third, using a defined minimal medium may also result in accumulation of considerable amounts of protein, as the cells grow much more slowly. Development of expression protocols in defined medium is always advantageous for structural approaches because the investigator hopefully will need to generate seleno-methionine derivatives for MAD or SAD phasing crystallographic data, or isotopically 2H, 15N, 13C labeled samples for heteronuclear NMR.
Sample preparation
Without exception, isolation and purification of an integral membrane protein must involve detergents to solubilize the membrane and membrane-associated proteins. Although many detergents may be tested, the prudent investigator will generally carry out the initial membrane solubilization in a readily available and less expensive nonionic detergent. In our experience, almost all proteins will solubilize in one of the following: n-decyl-β-D-maltopyranoside, N,N-dimethyldodecylamine-N-oxide (LDAO or DAO-12), n-dodecylphosphocholine (Fos-choline 12 or DPC), n-octyl-β-D-glucopyanoside (octyl glucoside), or tetraethylene glycol monoctyl ether (C8E4). Solubilization is tested by adding the detergent solutions to a small amount of membrane preparation followed by an incubation period, usually at room temperature, and ultracentrifugation. The presence of the protein in the membrane pellet versus a detergent-protein micelle in solution is determined by Coomassie-stained SDS-PAGE gel or an immunoblot using a specific antibody or an antibody against the affinity tag.
Purification can usually follow the same approaches as soluble proteins except that all procedures are performed in the presence of the detergent. The function of commonly used affinity tags is usually not affected by detergent; however, membrane proteins are often at relatively low concentrations, and the affinity chromatography step should sometimes be followed by another form of chromatography, such as ion exchange and/or gel filtration. High-resolution gel filtration chromatography can also provide an indication of the oligomeric state of the protein. A general protocol for the purification of membrane proteins is shown in Fig. 2.
To characterize the protein preparation, the critical parameters are purity, which is estimated by SDS-PAGE, the fold and evaluated by secondary structure content via circular dichroism spectroscopy, as well as the oligomeric state determined by gel filtration, light scattering, or sedimentation velocity (39-41). Furthermore, the homogeneity of the protein can be evaluated by mass spectrometry. Detergents will normally interfere with mass spectrometry, but MALDI TOF approaches have been developed to avoid such problems (42). As discussed in the next section, a range of detergents must be explored for protein structural determination, and as a result, the characteristics of the protein in different detergents must be determined (see Fig. 2).
If possible, the function of the protein should be assessed. Unfortunately, detergent solublization can often make it difficult to assay protein activity, as in the case of an ion channel; however, other parameters may be assessed in such instances. Ligand binding affinity, enzymatic activity, or association with another protein may provide a convincing assessment of protein function. Other biochemical techniques will also indicate folding and homogeneity, such as limited protease digestion, reactivity with reagents such as sulfhydryl reactive compounds, or chemical cross-linking patterns. Spectroscopic approaches have also been used. The dynamics of specific regions on the protein can be evaluated by nitroxide spin labeling and electron paramagnetic spectroscopy. If the protein can be metabolically labeled with 2H, 15N, then a relatively quick heteronuclear single quantum coherence NMR experiment can provide critical information on the homogeneity and fold of the protein (39).

Figure 2. A flow chart that outlines the general strategies in preparing membrane proteins for structural studies.
Structural Characterization of Membrane Proteins
X-ray crystallography
High-resolution membrane protein structures are determined predominantly using X-ray crystallography. Although notoriously difficult to crystallize, several methods have been applied successfully to crystallize membrane proteins either by manipulating the detergent/lipid components or by altering the protein component. Most membrane protein structures have been determined using detergent solubilized protein in which the entire protein detergent complex is crystallized. Often, the best-quality crystals of a membrane protein may be obtained only in one or a few detergents, and extensive screening based on detergent properties is required (41).
Thirty-three detergents have been used to crystallize membrane proteins, and three of those detergents have been used to determine three NMR structures (43). Some detergents are fold-specific. For example, C8E4 is predominantly used for β-barrel proteins; whereas DDM has been mostly successful for α-helical membrane proteins. The four detergents that have been used to crystallize most membrane proteins are octyl-glucoside, lauryl dimethyl amine oxide, C8E4, and dodecyl maltoside. These four detergents vary in alkyl chain length and shape and size of the head group; however, they are all neutral. This commonality is very significant. For three-dimensional protein crystals to form, protein molecules need to contact each other to form crystal contacts that are essential to propagate the lattice. It is likely that charged or even zwitterionic repulsive forces would hinder the association of the protein detergent complexes, which is a process that must occur at early stages of crystal nucleation.
Membrane protein crystals have significantly more solvent (64%) content than soluble proteins [47% (44)] presumably because of the detergent in the crystal. The organization of the detergent in the membrane protein crystal has been investigated in a select few cases and is different in each case. In the LH2 crystal, the detergent forms a belt around the hydrophobic surface of the protein consistent with the dimension of the OG detergent molecule (45). Similar to LH2, the OG detergents form a belt around the hydrophobic surface of phospholipase A in the crystal; however, the belts fuse to form a continuous three-dimensional network throughout the crystal (46). The continuous density of detergent was also observed in crystals of porin and two photoreaction centers. Snijder et al. (46) suggest the possibility that organic amphiphilic additives in the crystal screen could facilitate this fusion; however, no experimental data correlate the detergent structure observed in the crystal and the physical properties of the detergent/amphiphile mixed micelle. In addition to the detergent used for solubilizing the membrane protein, the native (or synthetic) lipid concentration may have profound effects on diffraction quality as in the instance of the crystallization of LacY (47), cytocrome b6f (48), Ca2+-ATPase (49), and GlpT (50).
Lipid cubic (51) and sponge (52) phases, as well as bicelles (53), are alternatives to detergents that have been applied successfully to membrane protein crystallization. In these instances, the protein is embedded in a lipid bilayer environment, which is considered more natural compared with the detergents that form micellar phases. In the recent high-resolution crystal structure of the human β2 adrenergic G-protein-coupled receptor, lipid cubic phase was used with necessary cholesterol and 1,4-butandiol additives (54). The cholesterol and lipid molecules were important in facilitating protein-protein contacts in the crystal.
In addition to the influence of the detergent on crystallization, the introduction of additional protein components has proven to be successful. In several cases, an antibody fragment was cocrystallized with the membrane protein to provide essential crystal contacts for crystallization (55-57). The protein itself can be engineered with an insertion to provide crystal contacts as demonstrated by the recent success of human β2 adrenergic G-protein-coupled receptor structure, in which a lysozyme molecule was engineered into one of the loops (58).
Despite these advances, Oberai et al. (3) estimate that if no acceleration of membrane protein structure determination occurs, then it will take more than three decades to determine at least one structural representative of 90% of the α-helical membrane protein sequence families (3).
Solution nuclear magnetic resonance spectroscopy
Although solution NMR techniques do not require crystallization, a molecular weight limit does exist [33 kD is the largest to date (59)], and optimization of detergent conditions has proven to be difficult (60). Several NMR structures of β-barrel outer membrane proteins have been determined (59, 61-63); however, a solution structure of a polytopic α-helical membrane protein remains a challenge. Several research groups are making great strides with the first step to NMR structure determination, the sequential chemical shift assignment achieved for two polytopic membrane proteins diacylglycerol kinase (64) and the potassium channel KcsA (65). NMR structure determination of β-barrel proteins has been more successful than a-helical membrane proteins primarily because the nuclear Overhauser effects (NOEs) between amide protons are across strands (i.e., between secondary elements), rather than within the secondary element as in α-helices, which provides valuable structural constraints. The lack of NOE data is currently being overcome with measurements from paramagnetic relaxation (66) and residual dipolar coupling experiments (67).
In addition to the limited distance restraints, the preparation of membrane protein samples has proven to be a major challenge to high-resolution NMR. Similar to X-ray crystallography, the selection of detergent strongly influences the quality of NMR spectra. No single detergent is well suited for NMR studies of membrane proteins; solubility, dynamics, the hydrophobic surface area of the protein, and other physical properties differ for each protein detergent complex, and the proper combination still needs to be determined empirically through extensive screening (39, 68).
Beyond structure determination, solution NMR can be used to investigate backbone dynamics and protein-ligand interactions. Bax and colleagues (65, 69) have characterized backbone dynamics (69) and ion binding affinity of the tetrameric KcsA potassium channel (65). These studies added additional structural insights to the crystal structure. On the ps-ns time scale, the selectivity filter is not dynamic. In SDS micelles, the intracellular C-terminal α-helix is dynamic on the ns-ps timescale and does not associate into a tetrameric bundle. In addition to determining the PagP NMR solution structure (62), Hwang et al. (70) characterized a two state dynamic rearrangement in which the more flexible state facilitates the entry of the substrate into the central cavity of the β-barrel.
Site-directed spin labeling
Site-directed spin labeling (SDSL) is used to investigate membrane protein structure and dynamics in lipid bilayers as well as in detergents. In SDSL, a nitroxide probe is introduced to a unique site within a protein. In most cases, a cysteine residue is introduced and subsequently reacted with a sulfhydryl-reactive nitroxide reagent. The resulting nitroxide side chain is sensitive to the molecular environment, which allows the determination of secondary and tertiary structure (71), conformational dynamics (72), and site-specific dynamics (73). Unlike solution NMR, the technique does not have a molecular weight limit, and membrane proteins can be investigated in detergent solutions or lipid bilayers.
From the electron paramagnetic resonance (EPR) spectrum of the nitroxide side chain, four primary parameters are obtained: 1) solvent accessibility, 2) mobility of the R1 side chain, 3) a polarity index for its immediate environment, and 4) the distance between R1 and another paramagnetic center in the protein. Solvent accessibility of the side chain is determined from the collision frequency of the nitroxide with paramagnetic reagents in solution. The mobility, polarity, and distances are deduced from the EPR spectral line shape. For regular secondary structures, accessibility, mobility, and polarity are periodic functions of sequence position. The period and the phase of the function reveal the type of secondary structure and its orientation within the protein, respectively (71, 74). In the case of membrane proteins, the topography of the secondary structure with respect to the membrane surface can also be described (75, 76).
When a pair of spin labels is incorporated into a protein or a protein complex, the dipolar interactions between labels can be used to measure the distance and distance distribution between labels (77). If the labels are separated by 7 to 20 A, then the dipolar interactions are sufficiently strong that they can be observed and quantified by continuous wave (CW) EPR spectroscopy (78-81). If the labels are separated by a distance greater than 20 A, then the resulting weak dipolar interactions can be measured with newer pulse methods such as double electron-electron resonance (DEER) or double quantum coherence (82-84). These methods have been used to measure internitroxide distances and distance distributions out to 60 A or more. An advantage of the CW EPR measurements is that they can be performed at room temperature (78), whereas the pulse measurements, such as DEER, require that the samples be frozen, typically at liquid nitrogen temperatures or lower.
Changes in any of the SDSL parameters measured can be used to detect changes in protein conformations, and most importantly, the data can be interpreted in terms of helix rigid body motions, relative domain movement, and changes in secondary structure. Recently, SDSL distance measurements were used to map ligand-induced conformational changes in BtuB (85), LacY (86), and the NhaA antiporter (87). Figure 3 shows an example of the use of SDSL and DEER to determine structural changes that accompany ligand binding to the extracellular loops in the outer membrane transporter, BtuB (88).
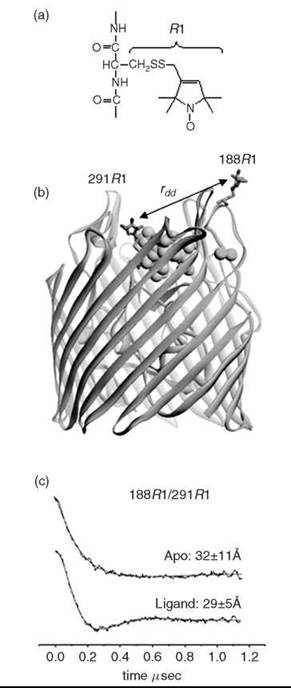
Figure 3. Site-directed spin labeling can be used to provide information on structural changes in membrane proteins (72). (a) A common nitroxide side chain R1, which is produced by cysteine mutagenesis and covalent modification. (b) The OM transporter, BtuB, showing the nitroxide side-chain, R1, at positions 188 and 291 in the extracellular loops of the transporter. The substrate, vitamin B12, is shown (CPK rendering), along with the co-ligand Ca2+. (c) DEER signals obtained from this spin pair in the absence and presence of the substrate (vitamin B12) and the Ca2+ co-ligand. The solid trace is a fit of the data to a single Gaussian distance distribution. Both the distance and distrance distribution (which is the standard deviation to the Gaussian) are shown. Ligand binding shortens the distance between spin pairs, which results in an ordering of the loop and narrowing of the distance distribution (88).
Solid-state nuclear magnetic spectroscopy
Solid-state NMR also can be applied to membrane proteins in lipid bilayers, and recent advancements in magic angle spinning solid-state NMR show promise for structure determination. Although the structures of small crystalline proteins (89) and membrane bound peptides (90) have been determined, the structure of a polytopic membrane protein has yet to be reported. The major necessity that is required to push the technique forward is the de novo sequential chemical shift assignment of the amino acid residues, and in the last few years, several groups have reported successful strategies (91, 92).
Beyond structure determination, solid-state NMR has been used to investigate the structures of membrane bound ligands. Several recent examples are a low-resolution structure of neurotensin bound to its G-protein coupled receptor (93), scorpion toxin bound to a chimeric potassium channel (94), retinal in both the rhodopsin and the metarhodopsin II intermediate (95), and acetylcholine bound to its receptor (96). These studies have great potential for designing and optimizing drugs-targeting membrane proteins (97).
Concluding Remarks
Several biophysical tools can provide information on the structure, dynamics, and conformational changes of membrane proteins. Many of these methods are beyond the scope of this review and were not mentioned here. However, each of these approaches has strengths and weaknesses; to investigate membrane protein structure and functional dynamics fully, a multitude of techniques is required. Static high-resolution structures are highly informative and provide a good starting point to generate hypotheses; however, they do not provide a complete understanding of protein molecular function.
References
1. DiNitto JP, Cronin TC, Lambright DG. Membrane recognition and targeting by lipid-binding domains. Sci. STKE 2003; 2003:re16.
2. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001; 305:567-580.
3. Oberai A, Ihm Y, Kim S, Bowie JU. A limited universe of membrane protein families and folds. Protein Sci. 2006; 15:1723-1734.
4. Engelman DM, Chen Y, Chin CN, Curran AR, Dixon AM, Dupuy AD, Lee AS, Lehnert U, Matthews EE, Reshetnyak YK, Senes A, Popot JL. Membrane protein folding: beyond the two stage model. FEBS Lett. 2003;555:122-125.
5. Tamm LK, Hong H, Liang B. Folding and assembly of beta-barrel membrane proteins. Biochim. Biophys. Acta 2004; 1666:250-263.
6. Wimley WC. The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 2003; 13:404-411.
7. von Heijne G. The membrane protein universe: what’s out there and why bother? J. Intern. Med. 2007; 261:543-557.
8. Dill KA. Polymer principles and protein folding. Protein Sci. 1999; 8:1166-1180.
9. Liang J, Dill KA. Are proteins well-packed? Biophys. J. 2001; 81:751-766.
10. Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000; 296:911-919.
11. Senes A, Engel DE, DeGrado WF. Folding of helical membrane proteins: the role of polar, GxxxG-like and proline motifs. Curr. Opin. Struct. Biol. 2004; 14:465-479.
12. North B, Cristian L, Fu Stowell X, Lear JD, Saven JG, Degrado WF. Characterization of a membrane protein folding motif, the Ser zipper, using designed peptides. J. Mol. Biol. 2006; 359:930-939.
13. Meindl-Beinker NM, Lundin C, Nilsson I, White SH, von Heijne G. Asn- and Asp-mediated interactions between transmembrane helices during translocon-mediated membrane protein assembly. EMBO Rep. 2006; 7:1111-1116.
14. Faham S, Yang D, Bare E, Yohannan S, Whitelegge JP, Bowie JU. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 2004; 335:297-305.
15. von Heijne G. Recent advances in the understanding of membrane protein assembly and structure. Q Rev. Biophys. 1999; 32:285-307.
16. von Heijne G. Membrane-protein topology. Nat. Rev. Mol. Cell. Biol. 2006; 7:909-918.
17. Ulmschneider MB, Sansom MS, Di Nola A. Properties of integral membrane protein structures: derivation of an implicit membrane potential. Proteins 2005; 59:252-265.
18. Nilsson J, Persson B, von Heijne G. Comparative analysis of amino acid distributions in integral membrane proteins from 107 genomes. Proteins 2005; 60:606-616.
19. White SH. Membrane protein insertion: the biology-physics nexus. J. Gen. Physiol. 2007; 129:363-369.
20. Epand RM. Membrane lipid polymorphism: relationship to bilayer properties and protein function. Methods Mol. Biol. 2007; 400:15-26.
21. Krauss M, Haucke V. Phosphoinositides: regulators of membrane traffic and protein function. FEBS Lett. 2007; 581:2105-2111.
22. Andersen OS, Koeppe RE 2nd. Bilayer thickness and membrane protein function: an energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 2007; 36:107-130.
23. Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 1998; 280:69-77.
24. Kim JM, Altenbach C, Kono M, Oprian DD, Hubbell WL, Khorana HG. Structural origins of constitutive activation in rhodopsin: role of the K296/E113 salt bridge. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:12508-12513.
25. Rao VR, Oprian DD. Activating mutations of rhodopsin and other G protein-coupled receptors. Annu. Rev. Biophys. Biomol. Struct. 1996; 25:287-314.
26. Wiener MC. TonB-dependent outer membrane transport: going for Baroque? Curr. Opin. Struct. Biol. 2005; 15:394-400.
27. Von Heijne G. Membrane protein assembly in vivo. Adv. Protein Chem. 2003; 63:1-18.
28. White SH, Von Heijne G. The machinery of membrane protein assembly. Curr. Opin. Struct. Biol. 2004; 14:397-404.
29. Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 2004; 73:1019-1049.
30. Griffith DA, Delipala C, Leadsham J, Jarvis SM, Oesterhelt D. A novel yeast expression system for the overproduction of quality-controlled membrane proteins. FEBS Lett. 2003; 553:45-50.
31. Ryan MT. Chaperones: inserting beta barrels into membranes. Curr. Biol. 2004; 14:R207-209.
32. Kutik S, Stojanovski D, Becker L, Becker T, Meinecke M, Kruger V, Prinz C, Meisinger C, Guiard B, Wagner R, Pfanner N, Wiedemann N. Dissecting membrane insertion of mitochondrial beta-barrel proteins. Cell 2008; 132:1011-1024.
33. Grisshammer R, Tate CG. Overexpression of integral membrane proteins for structural studies. Q. Rev. Biophys. 1995; 28:315-422.
34. Grisshammer R. Understanding recombinant expression of membrane proteins. Curr. Opin. Biotechnol. 2006; 17:337-340.
35. Miroux B, Walker JE. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996; 260:289-298.
36. Chen Y, Song J, Sui SF, Wang DN. DnaK and DnaJ facilitated the folding process and reduced inclusion body formation of magnesium transporter CorA overexpressed in Escherichia coli . Protein Expr. Purif. 2003; 32:221-231.
37. Locher KP, Bass RB, Rees DC. Structural biology. Breaching the barrier. Science 2003; 301:603-604.
38. Wang DN, Safferling M, Lemieux MJ, Griffith H, Chen Y, Li XD. Practical aspects of overexpressing bacterial secondary membrane transporters for structural studies. Biochim. Biophys. Acta 2003; 1610:23-36.
39. Columbus L, Lipfert J, Klock H, Millett I, Doniach S, Lesley SA. Expression, purification, and characterization of Thermotoga maritima membrane proteins for structure determination. Protein Sci. 2006; 15:961-975.
40. Savage DF, Anderson CL, Robles-Colmenares Y, Newby ZE, Stroud RM. Cell-free complements in vivo expression of the E. coli membrane proteome. Protein Sci. 2007; 16:966-976.
41. Wiener MC. A pedestrian guide to membrane protein crystallization. Methods 2004; 34:364-372.
42. Cadene M, Chait BT. A robust, detergent-friendly method for mass spectrometric analysis of integral membrane proteins. Anal. Chem. 2000; 72:5655-5658.
43. Raman P, Cherezov V, Caffrey M. The membrane protein data bank. Cell. Mol. Life Sci. 2006; 63:36-51.
44. Kantardjieff KA, Rupp B. Matthews coefficient probabilities: improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003; 12:1865-1871.
45. Prince SM, Howard TD, Myles DA, Wilkinson C, Papiz MZ, Freer AA, Cogdell RJ, Isaacs NW. Detergent structure in crystals of the integral membrane light-harvesting complex LH2 from Rhodopseudomonas acidophila strain 10050. J. Mol. Biol. 2003; 326:307-315.
46. Snijder HJ, Timmins PA, Kalk KH, Dijkstra BW. Detergent organisation in crystals of monomeric outer membrane phospholipase A. J. Struct. Biol. 2003; 141:122-131.
47. Guan L, Smirnova IN, Verner G, Nagamori S, Kaback HR. Manipulating phospholipids for crystallization of a membrane transport protein. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:1723-1726.
48. Zhang H, Kurisu G, Smith JL, Cramer WA. A defined protein-detergent-lipid complex for crystallization of integral membrane proteins: The cytochrome b6f complex of oxygenic photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:5160-5163.
49. Jidenko M, Nielsen RC, Sorensen TL, Moller JV, le Maire M, Nissen P, Jaxel C. Crystallization of a mammalian membrane protein overexpressed in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11687-11691.
50. Lemieux MJ, Song J, Kim MJ, Huang Y, Villa A, Auer M, Li XD, Wang DN. Three-dimensional crystallization of the Escherichia coli glycerol-3-phosphate transporter: a member of the major facilitator superfamily. Protein Sci. 2003; 12:2748-2756.
51. Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept 69. for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:14532-14535.
52. Wadsten P, Wohri AB, Snijder A, Katona G, Gardiner AT, Cogdell 70. RJ, Neutze R, Engstrom S. Lipidic sponge phase crystallization of membrane proteins. J. Mol. Biol. 2006; 364:44-53.
53. Faham S, Bowie JU. Bicelle crystallization: a new method for 71. crystallizing membrane proteins yields a monomeric bacterio-rhodopsin structure. J. Mol. Biol. 2002; 316:1-6.
54. Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, 72. Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007;318:1258-1265.
55. Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 1995; 376:660-669.
56. Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, Mac-Kinnon R. X-ray structure of a voltage-dependent K+ channel. Nature 2003; 423:33-41.
57. Hunte C, Michel H. Crystallisation of membrane proteins mediated by antibody fragments. Curr. Opin. Struct. Biol. 2002; 12:503-508.
58. Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, Schertler GF, Weis WI, Kobilka BK. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007; 450:383-387.
59. Liang B, Tamm LK. Structure of outer membrane protein G by solution NMR spectroscopy. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16140-16145.
60. Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn. Reson. Chem. 2006; 44:S24-S40.
61. Fernandez C, Hilty C, Wider G, Guntert P, Wuthrich K. NMR structure of the integral membrane protein OmpX. J. Mol. Biol. 2004; 336:1211-1221.
62. Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Prive GG, Bishop RE, Kay LE. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13560-13565.
63. Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001; 8:334-338.
64. Oxenoid K, Kim HJ, Jacob J, Sonnichsen FD, Sanders CR. NMR assignments for a helical 40 kDa membrane protein. J. Am. Chem. Soc. 2004; 126:5048-5049.
65. Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Sci. 2006; 15:684-689.
66. Liang B, Bushweller JH, Tamm LK. Site-directed parallel spinlabeling and paramagnetic relaxation enhancement in structure determination of membrane proteins by solution NMR spectroscopy.J. Am. Chem. Soc. 2006; 128:4389-4397.
67. Cierpicki T, Liang B, Tamm LK, Bushweller JH. Increasing the accuracy of solution NMR structures of membrane proteins by application of residual dipolar couplings. High-resolution structure of outer membrane protein A. J. Am. Chem. Soc. 2006; 128:6947-6951.
68. Vinogradova O, Sonnichsen F, Sanders CR 2nd. On choosing a detergent for solution NMR studies of membrane proteins. J. Biomol. NMR. 1998; 11:381-386.
69. Chill JH, Louis JM, Baber JL, Bax A. Measurement of 15N relaxation in the detergent-solubilized tetrameric KcsA potassium channel. J. Biomol. NMR 2006; 36:123-136.
70. Hwang PM, Bishop RE, Kay LE. The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:9618-9623.
71. Hubbell WL, Gross A, Langen R, Lietzow MA. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 1998; 8:649-656.
72. Hubbell WL, Cafiso D, Altenbach C. Identifying conformational changes with site-directed spin labeling. Nat. Struct. Biol. 2000; 7:735-739.
73. Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem. Sci. 2002; 27:288-295.
74. Hubbell WL, Altenbach C. Investigation of structure and dynamics in membrane proteins using site-directed spin labeling. Curr. Opin. Struct. Biol. 1994; 4:566-573.
75. Victor KG, Cafiso DS. Location and dynamics of basic peptides at the membrane interface: electron paramagnetic resonance spectroscopy of tetramethyl-piperidine-N-oxyl-4-amino-4-carboxylic acid-labeled peptides. Biophys. J. 2001; 81:2241-2250.
76. Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient-method to determine the immersion depth of nitrox- ides in lipid bilayers. Application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:1667-1671.
77. Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr. Opin. Struct. Biol. 2006; 16:644-653.
78. Altenbach C, Cai K, Klein-Seetharaman J, Khorana HG, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry 2001; 40:15471-15482.
79. Hustedt EJ, Smirnov AI, Laub CF, Cobb CE, Beth AH. Molecular distances from dipolar coupled spin-labels: the global analysis of multifrequency continuous wave electron paramagnetic resonance data. Biophys. J. 1997; 74:1861-1877.
80. Rabenstein MD, Shin Y-K. Determination of the distance between two spin labels attached to a macromolecule. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:8239-8243.
81. Steinhoff H-J, Radzwill N, Thevis W, Lenz V, Brandenburg D, Antson A, Dodson G, Wollmer A. Determination of interspin distances between spin labels attached to insulin: comparision of electron paramagnetic resonance data with the x-ray structure. Biophys. J. 1997; 73:3287-3298.
82. Borbat PP, McHaourab HS, Freed JH. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J. Am. Chem. Soc. 2002; 124: 5304-5314.
83. Jeschke G. Distance measurements in the nanometer range by pulse EPR. ChemPhysChem. 2002; 3:927-932.
84. Jeschke G, Polyhach Y. Distance measurements on spin-labelled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007; 9:1895-1910.
85. Xu Q, Ellena JF, Kim M, Cafiso DS. Substrate-dependent unfolding of the energy coupling motif of a membrane transport protein determined by double electron-electron resonance. Biochemistry 2006; 45:10847-10854.
86. Smirnova I, Kasho V, Choe JY, Altenbach C, Hubbell WL, Kaback HR. Sugar binding induces an outward facing conformation of LacY. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16504-16509.
87. Hilger D, Jung H, Padan E, Wegener C, Vogel KP, Steinhoff HJ, Jeschke G. Assessing oligomerization of membrane proteins by four-pulse DEER: pH-dependent dimerization of NhaA Na+/H+antiporter of E. coli. Biophys. J. 2005; 89:1328-1338.
88. Kim M, Xu Q, Murray D, Cafiso DS. Solutes alter the conformation of the ligand binding loops in outer membrane transporters. Biochemistry 2008; 47:670-679.
89. Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein, K Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature 2002; 420:98-102.
90. Hong M. Structure, topology, and dynamics of membrane peptides and proteins from solid-state NMR spectroscopy. J. Phys. Chem. B 2007; 111:10340-10351.
91. Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2007; 46:459-462.
92. Li Y, Berthold DA, Frericks HL, Gennis RB, Rienstra CM. Partial (13)C and (15)N chemical-shift assignments of the disulfide-bondforming enzyme DsbB by 3D magic-angle spinning NMR spectroscopy. ChemBioChem. 2007; 8:434-442.
93. Luca S, White JF, Sohal AK, Filippov DV, van Boom JH, Grisshammer R, Baldus M. The conformation of neurotensin bound to its G protein-coupled receptor. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:10706-10711.
94. Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, Becker S, Baldus M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature 2006; 440:959-962.
95. Patel AB, Crocker E, Eilers M, Hirshfeld A, Sheves M, Smith SO. Coupling of retinal isomerization to the activation of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10048-10053.
96. Williamson PT, Verhoeven A, Miller KW, Meier BH, Watts A. The conformation of acetylcholine at its target site in the membrane-embedded nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:18031-18036.
97. Watts A. Solid-state NMR in drug design and discovery for membrane-embedded targets. Nat. Rev. Drug Discov. 2005; 4:555-568.
Further Reading
Books
Luckey M, ed. Membrane Structural Biology: With Biochemical and Biophysical Foundations. 2008. Cambridge University Press, New York.
Lundstrom KH, ed. Structural Genomics on Membrane Proteins. 2006.
CRC Press, Taylor & Francis, Boca Raton, FL.
Pebay-Peyroula E, ed. Biophysical Analysis of Membrane Proteins: Investigating Structure and Function. 2008. Wiley-VCH, Weinheim, Germany.
Websites
http://pdbtm.enzim.hu/.
http://www.mpdb.ul.ie/.
http://blanco.biomol.uci.edU/Membrane_Proteins_xtal.html#Latest.
http://www.mpibp-frankfurt.mpg.de/michel/public/memprotstruct.html.
See Also
Ion Channels
Lipid Bilayers, Properties of
Membrane Assembly in Living Systems
Membrane Proteins, Properties of
Protein Targeting and Transport