CHEMICAL BIOLOGY
The Chemistry of Metallo-Enzymes and Metallo-Proteins
Robert J.P. Williams, Oxford University, Oxford, United Kingdom
doi: 10.1002/9780470048672.wecb327
Metallo-enzymes and metallo-proteins are a combination of a metal ion or several metal ions in which the metal ion is strongly held in a protein. Hence, before describing their properties and functions, it is necessary to appreciate which metal ions bind strongly to proteins and exchange slowly. The biological significance of metallo-enzymes is that they are essential catalysts, especially for the metabolism of small molecules such as H2, CH4, CO, N2, and O2, as well as for long-range electron transfer. The control of uptake and the synthesis of their proteins rest with feedback in a series of metallo-proteins.
Before describing these enzymes (see the examples in Table 1), it is necessary to make a broad division of metal interactions in enzymes. In one case, the metal ion is firmly attached to the protein so that like a nonmetal of an amino acid it does not exchange within days. In such a situation, the isolation of the metallo-protein, correctly named, can be followed through all steps of purification until further purification procedures fail to alter the stoichiometry of the metal/protein, and this is a whole number. At the same time, if the metallo-protein is an enzyme, activity will become optimal. This approach to metallo-enzymes was first developed by Professor B. L. Vallee of Harvard. A different extreme is one in which the metal ion is loosely attached to the protein when isolation procedures may well result in an apoprotein, and if it was an enzyme, it would then be devoid of activity. The problem in such a case is to know which metal ion was intrinsically involved in activity in vivo. Now a great number of metal ion/protein interactions are intermediate in character between extremes so that purification results in the discovery of “fractional stoichiometry” and can be beset by contamination. Confusion is increased because many enzymes have several metal ion centers when it may require astute experimentation to reveal the nature of the original metal ion complement. We must be aware of both binding constants of metal ions to proteins of different kinds and of their rates of exchange so as to establish their nature correctly as it is related to function (1-4).
Table 1. Examples of metallo-enzymes
|
Class of catalysis |
Example of active site Mn+ |
Enzyme |
|
Acid/Base Hydrolysis Electron Transfer Oxidation (O2) Oxidation (H2O) Oxidation (H2O2, RO2H) Hydrogenation Group Transfer (-CH3) Group Transfer (-OPO32-) Group Transfer (CO) |
Zn (Mg, Co, Ni, Ca) Fe, heme0 (Fe), Cu Fe, heme0 (Fe), Cu Mo0, W0, Mn Se, Fe, heme0 (Fe) Ni, Fe Co (B12)0 Mg (weakly bound) Ni (F-430)0 |
Carbonic Anhydrase Cytochromes Cytochrome P-450 Aldehyde Oxidase Catalase, Peroxidase Hydrogenase Methylmalonyl Isomerase Kinase Acetyl Synthetase |
NOTES: Organic side chains and metal ions can be substituted. The organic side-chain substitution is usually done by gene mutation, but metal ion substitution is done by direct exchange and, in fact, can be done for S and Se.
0In these cases, the metal ion is in a metal complex.
Binding Constants
Over many years orders of binding strengths of metal ions have been established. Some general features are that the ions of sodium and potassium bind very weakly if at all, those of magnesium and calcium bind more strongly, and certain transition metal ions bind with very considerable strength. In more detail, the binding of divalent ions to virtually all ligands follows the Irving-Williams order and the metal ions toward the end of the series are those that form stable metallo-enzymes.
Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+
Binding strength of the monovalent Cu+ is large like that of Cu2+, but binding of trivalent ions may also be very strong, especially Fe3+ and Mn3+. (Changes of oxidation state can occur during isolation when the properties of interest may be hidden.) A further specific difficulty is that the binding of Ca2+ is extremely variable in magnitude relative to that of Mg2+. Although the above sequence is roughly that of increasing electron affinity and hence of binding constant strength, as the ions do not differ greatly in size, the size of Ca2+ is very different so ligands can be devised for it that are of greater binding strength than Mg2+ by using structural constraints on ligand conformation, creating a disadvantage for Mg2+ binding.
The fact that so many metal ions are available and that they vary in availability in environmental waters means that specifically separating each one with a given ligand, assuming at first that equilibrium is attained (i.e., that is, binding constant orders are obeyed), make for a problem of selection. It is largely solved in cells as follows; see Fig. 1.
A cell can limit the free concentration of a metal ion in a given compartment by using energized pumps either inwardly or outwardly directed in the containing membrane of the compartment (Fig. 1). It can also use selective reagents outside the cell, siderophores, or proteins to scavenge for metal ions, particularly iron, bringing them into the cell where they may be reduced (5). In this manner, the concentration of ions in a cell can be fixed; for example, Mg2+ at about 10-3 M in the cytoplasm greatly exceeds that of all other ions. The cell has certain binding agents, potential ligands (L) that, although they bind in a given order of strength, Mg2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+, show only a modest change from Mg2+ to Cu2+ (e.g., the pyrophosphate of ATP). Given that concentration of the ion bound depends on the product K[M2+][L] = [ML], the high value of [M2+] for Mg2+ makes it the only ion able to bind to a very weak-binding ligand L, which has a general binding constant between 103 M-1 and 104 M-1. In fact, only Mg2+ is found in many enzymes using ATP, where Mg2+ ATP is the substrate. No other metal ion is observed in these metal/ligand/protein complexes (e.g., the kinases).

Figure 1. An example of the way metallo-enzymes are under controlled formation through both controlled uptake (rejection) of a metal ion and controlled synthesis of all the proteins connected to its metabolism and functions. The example is that of iron. Iron is taken up via a molecular carrier by bacteria but by a carrier protein, transferrin, in higher organisms. Pumps transfer either free iron or transferrin into the cell where Fe3+ ions are reduced to Fe2+ ions. The Fe2+ ions form heme, aided by cobalamin (cobalt B12 controls) and a zinc enzyme for α-laevulinic acid (ALA) synthesis. Heme or free iron then goes into several metallo-enzymes. Free Fe2+also forms a metallo-protein transcription factor, which sees to it that synthesis of all iron carriers, storage systems, metallo-proteins, and metallo-enzymes are in fixed amounts (homeostasis). There are also iron metallo-enzymes for protection including Fe SOD (superoxide dismutase). Adenosine triphosphate (ATP) and H+ gradients supply energy for all processes. See References 1-3.
Now consider the opposite extreme of binding strength Cu2+(Cu+). Its concentration in a cell is limited to around 10-15 M by pumps, so a very strong binding ligand is required to retain copper. As Cu2+(Cu+) has such a strong binding constant, it alone can still form its ML complex so long as the free concentrations of the other metal ions in the product K[M][L] are weaker than for Cu2+, which then links Cu to a given L, but if excess [L] over [Cu] were allowed, then the excess of [L] above that of [CuL] would let this ligand, L, bind other metal ions. This possibility is prevented as the free [Cu] binds to a transcription factor for the synthesis of its L (see Fig. 1) (6), with equal strength such that when free [Cu] is at a specific value, say 10-15 M, the production of L ceases; hence, [CuL] alone is formed. No other metal ion has a sufficient K[M][L] to form such complexes. The transcription factors bind to promoter regions of DNA. By a subtle combination over such controls of both free [M] and free [L], specific combinations can be obtained for all divalent ions. In fact, very little cross contamination of M and L is observed in vivo (see the cases in Table 1). (In all these considerations we must remember that K is an effective binding constant in cells dependent on pH and the presence of other interacting ions or molecules. It is the size of the effective constant K that makes certain that binding is selective.) We estimate the free ion concentration, molarity, in cells to be in the inverse order to binding strength
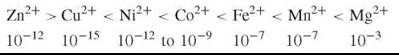
which is independent of many cell types in their cytoplasm. The concentration profile could be called a free metal ion signature of all cell cytoplasms.
We can now turn to the rate of exchange of metal ions from proteins.
Exchange Rates
The exchange rate of a metal ion (7) bound to a protein is important if the ion 1) acts as a regulator or transcription factor when it needs to exchange or 2) needs to be transported, when the ion is not necessarily open to simple exchange with its aquated state or is only present at very high dilution. We can start the consideration of exchange rates with the general observation that an equilibrium binding constant K can be looked upon as a simple ratio of the rate of ligand, L, binding, kon, relative to the rate of leaving koff (i.e., K = kon/koff). As kon is limited by loss of water from around the ion, which cannot exceed 109 s-1, koff is always less than 1 second for K > 109 M-1, and for K > 1012 M-1 the off-rate is close to or more than 1 hour. For slower on-rates, the off-rate for a given K is correspondingly slower. It is these rates, often related to equilibrium binding constants, that decide whether an enzyme containing a metal can be separated in pure bound form, a true metallo-enzyme, that is in slow exchange, (8), or will need a metal ion to be added to an isolated apo-enzyme to form a metal ion protein complex that is a fast exchange (9-12). Now certain ions always have relatively fast exchange rates. Examples are Na+, K+, Mg2+, Mn2+, and sometimes Ca2+. Examples of metal ions that have slower exchange rates and readily form metallo-enzymes are Cu+, Cu2+, Zn2+, Ni2+, Mo6+, Fe3+, Mn3+, and possibly Co2+. In an intermediate group is Fe2+, the enzymes of which are usually included in the class of metallo-enzymes. We must also remember that some metal ions are trapped not bound directly to protein side chains but to complex ligands such as porphyrins, for example, Fe, Mg, Co, and Ni (see Fig. 2) or special dithiols such as Mo (see Fig. 2) and W from which exchange is slow and the metal ions are frequently in low-spin states. The bindings of these complexes is often very tight and in slow exchange, and we include their enzymes in the class of metallo-enzymes. It is generally found that these ions are inserted into the complex ligand by selective processes. Slow exchange is also observed for Fe2S2 centers, but certainly some Fe4S4 centers exchange at intermediate rates.

Figure 2. Some examples of active sites in metallo-enzymes. Top left: cobalamin co-enzyme of isomerase; top right in succession: the molybdenum cofactor (Moco) of aldehyde oxidase; the manganese/calcium center of photochemical oxygen production; the complex iron site of hydrogenase.
To transfer or insert the cations that are in slow exchange with aqueous cations or are very dilute in solution demands special transporting proteins (Fig. 1). The first of these to be discovered was the protein for calcium transport across cells, calbindin, a member of the S-1 class of proteins. More recently, transporters have been discovered for Ni, Cu, Mo, and for molecules such as vitamin B12(Co). In essence, a transport metallo-protein, T, with only very slow loss of the metal ion, carries the ion and delivers it from a pump to an apoprotein, A, which becomes an enzyme and implies that the transporter, sometimes called a chaperone, and the apoprotein form a complex TM.A, which by exchange becomes T.MA. TM must also exchange with a pump, P, in the same way PM.T becomes P.MT. It is interesting that the genes for T, P, and A often lie close together under one promotor and transcription factor F (Fig. 1), which senses either the CM or that of the free M concentration giving F.M, and all these metallo-proteins have rather similar binding constants and selective functional groups.
We have now described the intracellular movement leading to the synthesis of a metallo-enzyme and we see that the control of synthesis rests with the supply of M to the cell from the environment, with its genetic system, which controls synthesis of the apoprotein, with its carrier and its pumps. All must be in a tight feedback circuit so that each metal has its selected proteins and its free concentration in homeostasis, which implies that all their binding constants are closely related to the Irving-Williams series and that to obtain selectivity the binding groups of the proteins are such that thiols are common for binding Cu+, Zn2+, Mo6+, W6+, and Fe3+; N-donors are more common for Fe2+ and Mn2+; whereas O-donors are observed to bind Mg2+, Ca2+, and K+. We need to go forward to the properties of these proteins as isolated and then return to their cellular functions. We shall take it that clear-cut metallo-proteins are those containing simple Cu, Zn, Ni, and Mo (W) ions including the cases of heme (Fe), some FenSn, BI2 (Co) and F-430 (Ni), as well as some mixed metal ion proteins of Fe2+/Fe3+, of Mn3+, or of Ni/Fe and Mo/Fe.
Properties Of Isolated Metallo-Proteins
Metallo-proteins have been studied by a great range of physical methods, which have revealed that frequently the metal ion is held in a constrained (sometimes called entatic) state (Figs. 3 and 4) (8-28). The simplest explanation of this state is that the binding energy of the metal ion in the resting state of the enzyme is such as to lower the activation energy required for it to function in a catalytic act relative to that energy required for free ion activity, which is most easily understood if we refer to a free energy diagram for a reaction pathway. In the course of the reaction the substrate is transformed while the catalyst cycles. Referring to the discussion of substrate activation in a catalyst, Pauling proposed that its binding energy was used in part to distort its structure to match as far as possible the transition state for the reaction presuming the structure of the catalyst was fixed. Turning now to the metal ion, or any other part of a catalyst that acts in the catalytic act, we can examine its bonding relative to that normally observed in the ground state of its simple compounds where its bonds are relaxed to optimal free energy binding conditions. As examples, the bond angles and bond lengths of amino acids and of metal ions in simple aqueous complexes (no steric hindrance or other constraint) are well known and the corresponding physical-chemical properties are understood. It is now known that in a protein complex the binding of any group can be strained by the need for the overall structure to have as great an activity as possible (1). It is usually found that a metal ion in a metallo-protein has distinctly unusual bond properties (8). The observed structural consequence is that the bonding geometry of any single unit, here a metal ion, can be seen to match the catalytic requirement. Some ways in which catalytic properties are achieved are for the metal ion 1) to have a small number of ligating atoms giving the metal ion an enhanced electron affinity for acid attack (Fig. 4) or 2) to have a ligand geometry matching its redox properties with those required for redox catalysis (Fig. 3). In addition, if during the catalytic cycle the metal ion has to change coordination number or oxidation state, then the constrained condition of the metal ion ensures that the relaxation energies between states in the cycle remain small. Note immediately that strong binding and non-exchanging ions are most likely to show these properties. In addition, it may be helpful if the metal ion has an open-sided structure and has a disposition of ligands to repel product while accepting substrate. Examples illustrate these generalities (see Table 2).
Table 2. Examples off constraints in metallo-enzymes
|
Metal |
Constraint |
|
Cu |
Cu+/Cu2+ held in fixed coordination allowing fast electron transfer (Fig. 3) |
|
Zn |
Zn2+ held in 5-coordination (one water molecule) which is readily adjustable (Fig. 4) |
|
Fe |
Heme enzymes have open-sided Fe2+ to give easy access to O2 |
|
Co |
Cobalt in coenzyme B12 (Fig. 2) has a Co-carbon bond that breaks on substrate binding |
|
Ni |
Ni2+ in close to tetrahedral geometry in hydrogenase |
|
Mn |
Mn3+ held in a peculiar cluster with calcium in O2-production (Fig. 2) |
NOTES: The constraints have been shown to be related to function in many cases.

Figure 3. A typical metallo-enzyme, azurin. The copper ion in it is a constrained (entatic) state matching its function. The copper in the enzyme is not open to any substrate—it is an electron-transfer protein. See Reference 8.

Figure 4. A zinc metallo-enzyme carbonic anhydrase for the very fast reversible formation of carbonic acid, H2CO3 from CO2 and H2O. Note the complexity of the protein required apparently to secure selectivity and the constrained, 5-coordinate, state of the zinc. There is a channel for substrates to the zinc in contrast to the copper site shown in Figure 3.
The case of Lewis acid function of metallo-enzymes is given by many a zinc enzyme (Fig. 4). Taking the case of zinc in carbonic anhydrase, the ground state structure shows it to be open sided with easy access for H2O and CO2 but not larger substrates. Moreover, these neutral molecules bind more readily than the product HCO3-. The zinc ion is known to be open sided and to go readily between 4- and 5-coordination, which is extremely useful in the required catalytic cycle. Moreover, the pKa of attached water is close to 7.0, an exceedingly low value for Zn2+(H2O) complexes. The reason for the selection of zinc as a biological Lewis acid catalyst is now clear. Functionally, it has optimum properties by being readily constrained in the ground state and it easily passes through different coordination states all the time in fast exchange internally with substrate intermediates in the site. If we look at other available Lewis acid cations, we observe that of the metal ions with possible similar advantages are 1) Mg2+, but it binds weakly and is a weak acid; 2) Ni2+, but it does not change geometry easily; and 3) Cu2+, but it would readily react with oxygen that can access the site. Only Co2+ is an ideal substitute, but it is a somewhat weaker acid than Zn2+. However, in confirmation that Co2+ is the only obvious substitute, it is found that the catalytic power of the M2+ ions in carbonic anhydrase follows the order
![]()
Notice that this order is not the relative Lewis acid strength or binding strength to the site as demonstrated in simple aqueous complexes that follow the order
Cu2+ > Zn2+ = Ni2+ > Co2+ > Fe2+ > Mn2+ > Mg2+
but is a result of the similar coordination geometries of Zn2+ and Co2+. In addition, Fe2+, Mn2+, and Mg2+ are used in enzymes for certain acid catalyses; however, these enzymes are not true metallo-enzymes but are metal/enzyme complexes from which the metal ion dissociates relatively easily, and hence these ions function both as a catalyst and as a free unbound regulatory ion (Fig. 1). Probably the tightly bound ions Zn2+, Cu+, and Ni2+ only act as regulators while bound to proteins.
We turn next to a simple example of a redox metallo-enzyme—azurin, which is a copper electron-transfer protein and, as the electron has no volume, it is best if the metal ion is enclosed for protection from all other agents. The structure (Fig. 3) shows the copper to be 4-coordinate in an unusual stereochemistry for Cu2+ or Cu+, a compromise between the demands of these two oxidation states. The redox potential is now fixed both by the degree to which the copper is structured and exposed to internal protein groups and to water at a distance. Now the electron-transfer rate of free Cu2+/Cu+ in water is slow because of the large relaxation energy between the normal different stereochemistries of the two ions. In this protein, however, the compromised constrained structure at Cu changes little with oxidation state, making electron transfer fast. If the favorable redox potential of the reaction matches the unfavorable relaxation energy, then following Marcus theory electron transfer can occur not by thermal excitation but by tunneling, which is a very clear-cut case of a constrained state of a metal ion (8).
We can compare this protein with a Cu metallo-enzyme for oxidation using molecular oxygen (e.g., laccase). The reaction is the generation of a free radical from a substrate X, say ascorbic acid, by removal of electrons at a single-copper site and transfer of the electron to a distant three-copper site where oxygen is reduced to water by four successive additions of single electrons. Now the three-copper site must be open sided to give access to O2, but the substrate to be oxidized by one of the electron-transfer steps can be as far away as a reasonable electron-transfer rate will permit, 10-15A, from the second, single-copper site. The second site is very much like that of azurin. Several copper ions work in a fast one-electron relay: substrate electrons to Cu (azurin-like), then electrons from this Cu site to the copper of oxygen-binding capability and finally electrons to O2. The O2 is retained as it goes through four reductive steps to 2H2O. Note that all the copper is firmly bound and does not exchange with external aqueous copper.
Similar relays of electrons are found in many iron metallo-enzymes, but here a difficulty had to be overcome in evolution. It is possible but not easy to retain a single Fe2+ in a site as it’s binding is weak, hence it tends to give metal/protein complexes. It is, however, possible to retain Fe3+ so that in one type of electron transfer-protein Fe is held in the Fe3+ state, rubredoxin. More usually, iron electron-transfer proteins are based on mixed valent Fe3+.Fe2+(S2—) clusters where the numbers of metal ions and sulfides may be anything up to eight. These electron-transfer clusters are shut off from the surrounding media and both Fe2+ and Fe3+ are held in a tetrahedral geometry of S2 and —RS. The problem of a large relaxation energy does not develop, and in some cases electron transfer in the cluster is so fast that the metal ions are effectively in the state Fe2 5+. Once again, we can contrast these clusters with those that are valuable in direct reaction with substrates such as H2, CO, and N2. Here, the clusters may contain other metal ions such as of Ni, Mo, W, or V, and at least one of these ions must have an open-sided structure. Products (e.g., H+, CH3CO-, and NH3) are now very different from reactants (e.g., H2, CO, CH4, and N2, respectively). Notice that all the active metal ions here (Ni2+, MoS22+, WS22+, and V) bind strongly. The selection of metal ions for particular reactions often reflects the intrinsic value of the metal electronic structure.
As mentioned, there is another way to avoid fast exchange of a metal ion in a metallo-enzyme: to enclose it in a chelate from which it cannot escape (Fig. 2). Very well-known examples of such very strong chelating agents are provided by porphyrin and its derivatives as in Fe2+.heme, Co2+.Bi2, Ni.F-430, and even Mg2+.chlorin, although the last is only found in electron-transfer enzymes of photo-centers. For the three transition metal ions, the placing of them in the strong ligands of the porphyrin series generates low spin states in contrast with all the examples we have given so far where metal ions are high spin. Low spin states are more common among second and third rows of transition metal ions such as of elements Rh, Pd, and Pt. Man has made great use of these ions as catalysts, but they are not environmentally available to organisms, hence the resort to porphyrins. A big advantage of porphyrin is that it induces low spin states in its Fe, Co, and Ni complexes where redox change is usually of small relaxation energy so that relaxation is usually easy as various oxidation states can be held in or closely in the porphyrin plane. Of course the binding of additional ligands above and below the plane or just below it tunes the function and once again the protein can create constrained advantageous conditions. With two ligands bound, the heme iron is an excellent electron-transfer metallo-enzyme known in the series of cytochromes a, b, and c. The metal ions Co2+(Bi2) and Ni2+(F-430) by contrast are naturally open sided. They, together with open-sided Fe(heme), readily bind ligands but of very different kinds. Open-sided Fe2+.heme is electron excessive and acts as a binding agent for substrates such as O2 (in enzymes like cytochromes a3 and P-430) as well as for NO2- and SO32-when it becomes Fe3+. Co2+.B12 has a single electron occupying the sixth (open) side (Fig. 2). It acts as a radical catalyst as it cycles through the condition Co3+-CH2R to Co2+...●CH2R with homolytic bond breaking. Ni3+ can behave similarly or can act in P-430 as a base as it has a lone pair in the Ni2+ low spin state; but even if it has no ligands above and below the porphyrin plane it becomes low spin. It is well known that low spin d6 to d8 electron shell configurations are powerful redox catalysts. B12 coenzymes can also catalyze alkyl-transfer where Co+ acts as a strong nucleophile, for example, in methionine synthatase.
Now, as in the cases of the copper oxidases, it is found that the catalyst centers can form chains linked by electron-transfer proteins. They are known in complex enzymes such as nitroge- nases (Fig. 5), but outstanding are the long chains of catalysts in the bioenergetic devices of photosynthesis and oxidative phosphorylation.
Overall, the metallo-enzymes are excellent catalysts for reactions of especially small inert small molecules for which the organic side chains of proteins cannot be used (e.g., for H2, CH4, N2, O2, and CO). These proteins are the basic starting compounds of biological synthesis. Hence, metallo-enzymes have always been the basic catalysts of organisms. In fact, they have increased in significance with evolution.

Figure 5. Some metallo-enzymes are formed by a set of subunits. Shown here is the structure of nitrogenase that has an energized supply of electrons (energy from ATP) via two iron/sulfur clusters and an active site FeMoco shown in the inset. There are two distinct protein units linked to one supply line of a separate unit. Note there is a light non-metal in a hole in the structure of Fe7MoS9 but, as it is of uncertain nature, it is not shown.
References
1. Frausto da Silva JJR, Williams RJP. The Biological Chemistry of The Elements. 2nd edition. 2001. Oxford University Press, Oxford, UK.
2. Valentine JS, Bertini I, Gray HB. Biological Inorganic Chemistry: Structure and Reactivity. 2000. Univ. Science Books, New York.
3. Kaim W, Schwederski B. Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life. 1994. Wiley, Chichester, UK.
4. Ledin M. Accumulation of metals by micro-organisms. Earth Sci. Rev. 2000; 51:1-31.
5. Nelson N. Metal ion transporters and homeostasis. EMBO J. 1999; 18:4361-4371.
6. Sarkar B, ed. Metals and Genetics. 1998. Plenum Press, New York.
7. Connors KA. Chemical Reactions in Solution. 1992. VCH, New York.
8. Gray HB, Malmstrom BG, Williams RJP. Copper coordination in blue proteins. J. Biolog. Inorg. Chem. 2000; 5:551-559.
The following references are arranged in sequence to give quick access to different metal ion/protein complexes. Na, K, Mg, Ca (9)-(14), Zn (13,14), Fe (15)-(18), Mn (19), Cu (20,21), Co (22,23), Ni(24), V(25), W(26), Mo (27), Se (28).
9. Williams RJP. The biochemistry of sodium, potassium, magnesium and calcium. Quart. Revs. Chem. Soc. 1970; 24:331-360.
10. Cowan JA, ed. Biological Chemistry of Magnesium. 1995. VCH, New York.
11. Carafoli E, Klee C, eds. Calcium as a Cell Regulator. 1998. Oxford University Press, Oxford, UK.
12. Pochet R, ed. Calcium: The Molecular Basis of Calcium Action. 2000. Kluwer Academic, Dordrecht, the Netherlands.
13. Coleman JE. Zinc enzymes. Curr. Opin. Chem. Biochem. 1998; 2: 222-234.
14. Klug A. Zinc finger peptides for the regulation of gene expression. J. Mol. Biol. 1999; 293:215-218.
15. Beinert H. Iron-sulfur proteins: ancient structures still full of surprises. J. Biol. Inorg. Chem. 2000; 5:2-15.
16. Moore GR, Pettigrew GW. Cytochrome c biological aspects. 1990. Springer-Verlag, Berlin.
17. Isaac IF, Dawson JH. Haem-containing peroxidases. Essays Biochem. 1999; 34:51-69.
18. Michel H, Behr J, Harrenga A, Kanut A. Cytochrome c oxidase structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 1998; 27:329-356.
19. Siegel H, Siegel A, eds. Metal Ions in Biological Systems. Vol. 37. 2000. Marcel Dekker, New York.
20. Malmstrom BG, Leckner R. The chemical biology of copper. Curr. Opin. Chem. Biol. 1998; 2:286-292.
21. Huber R. Copper clusters in oxidases. Angewandte Chemie (Int. Ed.) 1989; 28:848-869.
22. Lowther WT, Matthews BW. Structure based perspectives on Bi2-dependent enzymes. Ann. Rev. Biochem. 1997; 66:269-313.
23. Krantler B (ed). Vitamin B12 and B12-proteins. 1998. Wiley-VCH, Weinheim, Germany.
24. Fontecilla-Camps J-C, Ragland SW. Nickel-iron-sulfur sites. In: Advances in Inorganic Chemistry. Cammack R, Sykes AG, eds. 1999. Academic Press, San Diego, pp. 283-332.
25. J. Inorg. Biochem. Articles devoted to vanadium biochemistry. 2000; 30.
26. Kletzin A, Adams MWW. Tungsten in biological systems. FEMS Microbiol. Rev. 1996; 18:5-63.
27. Hille R. Molybdenum enzymes. Essay Biochem. 1990; 34:125-137.
28. Stadtman TC. Selenium biochemistry. Ann Rev. Biochem. 1990; 65:83-100.
Further Reading
The following volumes contain detailed structures and properties of virtually all metallo-enzymes and metallo-proteins.
Messerschmidt A, Huber R, Poulos T, Weighhardt K. Handbook of Metalloproteins. Vol. I and II. 2001. Wiley, New York.
Messerschmidt A, Bode W, Cyglar M. Handbook of Metalloproteins. Vol. III. 2004. Wiley, New York.