CHEMICAL BIOLOGY
Metalloproteinases, Biophysics and Chemistry of
Ariel Solomon* and Tzvia Selzer*, The Weizmann Institute of Science, Israel
Marcos E. Milla, Group Leader, Department of Biochemical Pharmacology, Roche, Palo Alto, California
Irit Sagi, Associate Professor, Department of Structural Biology, The Weizmann Institute of Science, Israel
doi: 10.1002/9780470048672.wecb329
Zinc-dependent metalloproteases constitute a large family of enzymes, part of the proteinase superfamily. Fundamental to the structural integrity and catalytic activity of metalloproteases is the presence of both zinc and calcium ions in the structure of the protein. This article will focus on matrix metalloproteinases that are involved in extracellular matrix (ECM) catabolism and serve as important enzymes in many aspects of biology, ranging from cell proliferation, differentiation, and proliferation to cancer, tumor metastasis, inflammation, and other pathologic states. Despite their key role in many normal and pathologic processes, the molecular mechanisms by which zinc-dependent proteases hydrolyze their physiologic substrates are only known partly. Recent theoretical analyses have suggested reaction models for which limited and controversial experimental evidence exists. Here we will discuss the importance of quantifying the biophysical properties and the structural dynamic behavior of these enzymes to reveal their underlying molecular mechanisms. Such molecular knowledge holds promise in providing the basis for the novel design of specific antagonists as drug candidates for these important enzymes. In addition, we will discuss the use of real-time spectroscopic tools for studying the reactive metal sites in these enzymes.
______________________________
* These authors contributed equally to this article.
Introduction
Metalloproteinases catalyze the hydrolysis of the peptide bond, which is the most stable chemical bond in nature (1,2). Thermodynamic analysis of its hydrolysis reveals that although the free energy is relatively low, ~2.4 kcal/mol, and the spontaneous reaction is exothermic, the scissile bond cleavage is hindered by a high-activation energy barrier of ~20 kcal/mol (3). Thus, it may have taken 400 years to achieve the spontaneous bond cleavage in the absence of enzymes. Remarkably, metalloproteases accelerate peptide bond hydrolysis by a factor of 1016, which enables this process to be completed within milliseconds (4).
The zinc-dependent metalloproteases comprise a large family of enzymes with a wide variety of biologic roles. For example, important metalloproteinases for cell viability include the bacterial metalloendopeptidase thermolysin, the digestive exopeptidases, carboxypeptidase A or B, and the matrix metalloproteinases (MMPs) (Table 1). MMPs are members of the metzincin superfamily of greater than 770 zinc endopeptidases, which includes astacins, serralysins, adamalysins, leishmanolysins, and snapalysins. Metzincins are characterized by an absolutely conserved methionine residue C-terminal to the third histidine in the consensus sequence HEXXHXXGXXH/D, where the histidine residues chelate a catalytic zinc ion. Because of the extensive experimental and theoretical literature available, we will deliberately limit our discussion to MMPs that act within the extracellular matrix (ECM) milieu. Ample evidence exists on the role of MMPs in normal and pathologic processes, including embryogenesis, wound healing, inflammation, arthritis, connective tissue diseases, inflammation, cardiovascular and autoimmune diseases, and cancer (6-14). The MMP family consists of more than 25 enzymes, with differences in substrate preference, domain structure, and sequence homology. As depicted in Fig. 1, MMPs are multidomain proteins with a signal peptide, a “pro” domain that maintains enzyme latency until it is removed or disrupted, and a catalytic domain that is common to the entire family. Additional domains observed in different MMP structures include fibronectin-type
Table 1. Biologic roles of the zinc-dependent proteinases categorized into four subgroups based on the conserved structural motif of their active site (5)
|
Protein family |
Biologic distribution |
Main biologic function |
Preferred Substrate (cleavage site) |
References |
|
Thermolysins (Thermostable neutral proteinase ) |
Bacillus caldolyticus, sp., stearothermophilus, thermoproteolyticus |
Extracellular, thermostable, digestive bacterial proteases. Hydrolyze the N-terminal side of hydrophobic residues. |
Xaa---Leu > Xaa---Phe |
53-56 |
|
Carboxypeptidases |
Range from Yeast to Homo sapiens |
Degradation of a wide array of proteins. The well-studied pancreatic enzymes (carboxypeptidases A1, A2, and B) are involved in the digestion of food. Several members of the metallocarboxypeptidase gene family (carboxypeptidases D, E, M, and N) are more selective enzymes and are thought to play a role in the processing of intercellular peptide messengers |
Carboxyl terminal site |
37, 38, 57,58 |
|
Angiotensin-converting enzymes (ACEs) |
Eukaryotes |
ACEs are involved in blood chemistry. They mostly exist at the cell surface as ectoenzymes, where they hydrolyze circulating peptides. Gene-targeting studies in mice have established that the tissue-bound form of ACE controls both blood pressure and renal structure and function, although a soluble form of ACE, which is derived from the membrane form through the action of a secretase, is also present in serum and other body fluids. |
Nonspecific endopeptidase |
59, 60 |
|
Metzincins |
Procaryots-Eukaryotes |
Range from bacterial digestive proteins to human matrix metalloproteinases and ecto sheddses. The metzincins contain several sub-families. Astacins, bacterial serralysins, matrix metalloproteinases (MMPs), ADAMs (a disintegrating and metalloproteinase), ADAMTS (a disintegring and metalloproteinase with thrombospondin motifs). MMPs are involved in the catabolism of extracellular matrix (ECM) molecules and the activation of other MMPs; they also process several cell surfaces and serve as soluble regulators of cell behavior in bioactive molecules The ADAMs are major ectodomain sheddases; they release a variety of cell-surface proteins, including growth factors, cytokines, cell adhesion molecules, and receptors. ADAMs can also cleave and remodel components of the extracellular matrix. |
Nonspecific endopeptidase |
14, 22, 23, 61-67 |
Il-like, hemopexin-like, and transmembrane domains (15, 16). MMPs catalyze the hydrolysis of peptide bonds of a large range of biologic substrates, including collagen, gelatin, fibronectin, elastin, growth factors, cytokines, and chemokines. Notably, the broad association between MMP peptide hydrolysis activity and several serious diseases has made MMPs an attractive target for developing novel drugs aimed at inhibiting MMP activity (17).
In MMPs, the hydrolysis of a peptide bond is mediated by a catalytic zinc ion that resides in a structurally conserved catalytic cleft of the enzyme. Zinc is the second-most abundant transition metal in biology, after iron. It plays structural, chemical, and regulatory roles in biologic systems and is an essential ingredient at the active site in many enzymes (18, 19). Zinc plays a role in gene expression, stabilizes the structure of proteins and nucleic acids, preserves the integrity of subcellular organelles, participates in the transport process, and plays important roles in viral and immune phenomena (20). Moreover, zinc exhibits flexible coordination geometry and facilitates fast ligand exchange, and it is a Lewis acid with intermediate polarizability lacking redox activity (5). All the above characteristics give zinc its versatility, which enables it to form different types of chemical and bonding interactions. The families, motifs, en- zymology, and protein structures containing one or two zinc ions have been discussed (5, 18, 21).
The structural and chemical knowledge regarding enzymatic peptide cleavage in zinc-dependent metalloproteinases is limited, and experimental evidence is controversial, partly because the zinc atom is spectroscopically silent and hence difficult to study using conventional spectroscopic and analytical tools (20). In addition, most structural and biochemical studies carried out so far were limited to nondynamic structure/function characterization, thus preventing qualitative and quantitative analysis of the evolving intermediates formed during peptide cleavage mediated at the catalytic zinc-binding site. Most of our mechanistic knowledge comes from theoretical calculations derived from crystal structures. Unfortunately, this process results in controversial proposals as to how proteases hydrolyze their peptide substrates. Thus, in the absence of direct experimental tools that would enable us to characterize the reaction in real time, it will be difficult, not to say impossible, to elucidate the underlying reaction mechanisms.
Here we will discuss the progress achieved so far in our mechanistic understanding of peptide hydrolysis reactions by metalloproteinases. We will focus on the need for comparative structure-function analyses of individual metalloproteinases to reveal the reaction mechanisms. Finally, we will describe new frontiers in the biophysics and chemistry of metalloproteinases and the application of such studies to reveal directly reaction mechanisms and their relevance to drug discovery.
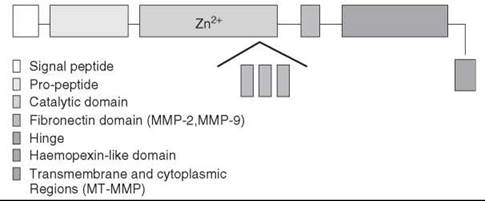
Figure 1. Schematics of the domain structures of the MMP family. The catalytic domain (represented by light purple) has an insertion of gelatin binding domain (fibronectin type-II-like domain) in MMP-2 and MMP-9. In all other MMPs, the catalytic domain is a continuous entity. Transmembrane and cytoplasmic regions (represented by yellow) are found in membrane-bound MMPs only.
Structures of Available Matrix Metalloproteinases
To date, the crystal structures of 12 different MMPs have been solved. Full structures were obtained for MMP-1 (2CLT), MMP-2 (1CK7), and MMP-7 (1MMP). As for the rest, only the catalytic domains in the presence of different inhibitors were determined. The hemopexin-like domains of MMP-2 (1RTG), MMP-9 (1ITV), and MMP-13 (1PEX) were crystallized, and structures were determined separately. Nuclear magnetic resonance (NMR) structures of the catalytic domains of MMP-1 (1AYK), MMP-2 (1HOV), MMP-3 (1UMS), MMP-12 (1YCM), and MMP-13 (1EUB) have also become available.
The overall structures of all MMP catalytic domains known so far are very similar (Fig. 2). These MMP catalytic domains are shaped like an oblate ellipsoid, with a small active-site cleft, harboring the catalytic zinc ion, notched into the flat ellipsoid surface. The active site cleft is defined by helix hB, which provides two histidine residues that coordinate to the catalytic zinc ion, and the catalytic Glu in between, all belonging to the zinc-binding consensus sequence HEXXHXXGXXH (5, 22, 23). The active-site helix ends at a Gly residue, where the peptide chain bends, presenting the third zinc-liganding His. The zinc ion also coordinates to a water molecule that is used to hydrolyze the peptide bond of the substrate (see the discussion in the next chapter). The water molecule is also held in place by the side chain of the active site Glu. Another basic feature of the MMP active site is the presence of three substrate-binding subsites. The surface of the protease that can accommodate a single side chain of a substrate residue is called the subsite. Subsites are numbered S1-Sn upward toward the N terminus of the substrate (nonprimed sites) and S1’-Sn’ toward the C terminus (primed sites), beginning from the sites on each side of the scissile bond. These subsites accommodate the side chains of the peptide to be cleaved, and the local structural characteristics and electrostatic environment of the individual subsites effectively determine the specificity of the substrate.

Figure 2. Ribbon structure of the MMP catalytic domain. The catalytic domain of MMP-8 (1ZP5) is superimposed with the catalytic domains of MMP-3 (green) (1HY7), MMP-12 (yellow) (1UTT), MT1-MMP (orange) (1BQQ), and MT3-MMP (pink) (1RM8); only the active site-conserved motif is shown for clarity. The catalytic and structural zinc (center and top) and the two calcium ions are displayed as red and blue spheres, respectively.
A detailed description of the fine structural differences between different MMPs can be found in recent reviews and publications (13, 15, 24). More relevant to the current article are the insights gained from integrating structural data obtained by different methods, more specifically by comparing X-ray structures (possibly more than one) with the solution structures obtained from NMR, to obtain a structural model that is beyond a single structural datum. Examples can be found in recent studies done by Rush et al. on MMP-1 and MMP-13 (25) and Bertini et al. on MMP-12 (26); both looked into the structure of these different MMPs complexed with various inhibitors by using both NMR and X-ray techniques. The backbone generalized order parameter (S2), a parameter related to the amplitude of fast (picosecond to nanosecond) movements of NH vectors, peak multiplicity, and weak or missing peaks, suggested active site mobility of inhibitor-free MMP-1 when compared with the structure of the inhibited enzyme. The data indicated that a slow conformational change in the active site results in a concerted motion of helix hB and the zinc-ligated histidines. Furthermore, the presence of an inhibitor that binds by chelating zinc effectively removes this motion, while maximizing the interaction of the inhibitor with the enzyme. The mobility of the random coil region in the vicinity of the active site was maintained even in the presence of a bound inhibitor. On the other hand, a comparison of the crystal structures of three MMP-12-inhibitor complexes indicated that the conformational heterogeneity observed is largely independent of the type of inhibitor. These studies have shown that flexibility/conformational heterogeneity in crucial parts of the catalytic domain is the rule rather than an exception in MMPs, and its extent may be underestimated by inspection of one X-ray structure.
Reaction Mechanisms of Metalloproteinase-Mediated Peptide Hydrolysis
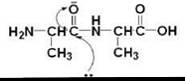
All proteinases achieve catalysis by providing a favorable electrostatic environment where the chemical reaction occurs. The bottleneck is the formation of the nucleophile, which in turn, attacks the carbonyl of the peptide that is properly located at the binding site and thus initiates bond cleavage (Scheme 1). All well-characterized proteinases are categorized, based on the nature of the most prominent functional group at the active site, into one of four families: serine, cysteine, aspartic, and metalloproteinase. Cysteine proteinases use a low pKa cysteine as a reactive nucleophile (27); serine proteases use the serine hydroxyl as the catalyst in conjugation with a hydrogen bond network that allows the general base catalysis (28, 29); a catalytically active diad of aspartates facilitates hydrolysis in aspartic proteinases; and metalloproteinases use zinc to promote catalysis.
The mechanism by which metalloproteinases execute catalysis has been of interest for many years. Most studies focused on carboxypeptidase A and thermolysin-like proteases for which extensive structural, chemical, and biochemical data are available. The first peptide hydrolysis mechanisms to be proposed were based on the available X-ray structures that have facilitated establishing structural models for the different reaction steps (30). Crystallographic analyses of enzyme-inhibitor complexes were used to reveal how enzymes interact with their substrates. Suitably chosen inhibitors were used to provide structural models for various stages in catalysis, including the Michaelis complex, transition states, and products. A classical example of a complex between an enzyme and a substrate analogue was described by Shoham et al. (31). In this study the X-ray crystal structure of the complex between carboxypeptidase A (CPA) and 5-amino-(N-t-butoxycarbonyl)-2-benzyl-4-oxo-6-phenylhexanoic acid (BBP), the ketomethylene substrate analogue of the peptide substrate N-(t-butoxycarbonyl)-L-phenylalanyl-L- phenylalanine, was resolved. It was shown that the enzyme specifically binds to the hydrated form of one of four stereoisomers of BBP that were present in the buffer solution in which the CPA crystals were soaked. This discovery was surprising at the time because the hydrated form of BBP was expected to be present in aqueous solution at a concentration of less than 0.2%. This result consequently led to the proposition that the enzyme-inhibitor complex is most stable in the presence of inhibitors whose structure resembles species along the reaction coordinate of a chemical reaction rather than species resembling a reactant or a product.
Biochemical studies and enzyme modification work, including mutagenesis of active site residues, zinc ion substitution, steady-state kinetics with different types of substrates, O18 exchange studies and others, together with the X-ray data, led to the proposition of two major types of hydrolysis mechanisms. The first mechanism to be proposed soon after the first crystal structure of carboxypeptidase A was published was the direct nucleophilic attack (also called the “Zinc-carbonyl mechanism” or “acyl pathway”) on the peptide carbonyl by the conserved active site Glu, resulting in the formation of an anhydride intermediate. Accumulating evidence from O18 exchange studies during the late 1960s and 1970s have ruled out the involvement of an anhydride intermediate in peptide hydrolysis (32, 33). A second type of mechanism proposed is the general-acid-general-base (GAGB) mechanism (the “Zinc-hydroxide mechanism” or the “promoted water pathway”) in which a water molecule initially attacks the carbonyl while, or after, losing a proton. This mechanism results in the formation of a gem-diol intermediate (34-36). Alternative paths for the GAGB mechanism were proposed and can be categorized into two groups. One alternative, following Christianson and Lipscomb (37), suggests that the substrate binds directly to the zinc ion while not replacing the water molecule that occupied the fourth coordination site of the zinc in the native structure. This water molecule is activated by the metal ion or by the conserved active site Glu (or both); it loses a proton and attacks the peptide carbonyl. According to this mechanism, the zinc ion has two roles: polarizing the carbonyl group of the substrate and facilitating the deprotonation of the water nucleophile. The second GAGB mechanism, following Mock and Zhang (38), suggests that the substrate carbonyl binds to the zinc ion and is activated by it. Nucleophilic attack is initiated by a water molecule that is deprotonated by the C-terminal carboxylate of the substrate itself, not by the conserved binding site Glu or by the zinc ion. In the latter, the role of zinc is minimized to the polarization of the carbonyl group of the substrate only, and the conserved active site Glu is not given any active role. Mock and Zhang’s (38) and Mock and Tsay’s (39) proposed mechanism was based on enzyme kinetics experiments that showed significant pH dependencies of the inhibition constants of transition-state analog inhibitors. However, several studies implied the importance of the active site Glu residue in achieving the enzymatic activity. Mutagenesis studies show that of all the active site residues, only the modification of active site Glu residues completely abolishes catalysis (40, 41).
As observed in other systems, the obvious difficulty in elucidating reaction mechanisms based on static structural snapshots subsequently initiated structural-dynamic theoretical studies of metalloproteinases. The active site chemistry of zinc-dependent enzymes has been studied using a variety of theoretical approaches. For example, mixed quantum mechanical/molecular calculations and classical molecular dynamic simulations have been employed, especially studies using density functional methods on redox-active metal centers (42).
Because of the availability of detailed structural information, the majority of theoretical studies focused on carboxypeptidase A and thermolysin-like proteases, and only one study investigated the mechanism of peptide hydrolysis by human MMP-3. Recently, Kilshtain-Vardi et al. compared the two GAGB alternative pathways for peptide cleavage by carboxypeptidase-A proposed by Lipscomb and Mock by performing semiempirical theoretical calculations (43). The proton transfer step to the nitrogen of the peptide, following the nucleophilic attack of the peptide carbonyl group by a hydroxide, was calculated to be rate limiting. It was shown that under kinetic control both reactions are feasible; however, a calculated thermodynamic enthalpy difference of ~20 kcal/mol indicated that the reaction path suggested by Lipscomb is more stable than the other.
Pelmenschikov and Siegbahn investigated the mechanism underlying peptide hydrolysis by human MMP-3 via quantum chemical methods (3) using the crystal structure of the inhibited enzyme with the transition state analogue piperidine sulfonamide inhibitor as the structural reference. The importance of the weakly bound water molecule as a potent electrophile for the zinc-coordinated substrate oxygen was revealed by reducing the activation barrier by about 5 kcal/mol. Furthermore, the conserved active site Glu residue was confirmed to play a key role by acting as a base during the reactant water deprotonation. Interestingly, the zinc ion was shown to retain pentacoordinated geometry during the reaction, with distorted trigonal bipyramidal coordination, whereas a tetrahedral coordination sphere was suggested for the final structure of the product (Fig. 3). Remarkably, the formation of a pentacoordinated zinc-protein complex is followed by distinct electronic transitions mediated by water and the conserved Glu residue. Essentially a single-step reaction mechanism has been obtained with activation energy of 13.1 kcal/mol. This work provided novel structural-dynamic insights into the reaction mechanism governing peptide hydrolysis. Naturally, such a theoretical model must be confirmed by experimental results, especially when the use of long-lived inhibitor-enzyme complexes (instead of enzyme-substrate complexes) may provide only structural models for different stages in catalysis. Such models are obviously not true intermediates; hence, in the absence of supporting experimental data, the obtained results must be interpreted with caution.

Figure 3. The catalytic cycle for the proteolytical GAGB mechanism of MMP-3 taken from Pelmenschikov et al. (3).
After more than 40 years of research on the structure/function of metalloproteases, tremendous advances in our understanding have been achieved. A consensus exists regarding the basics of peptide hydrolysis: 1) A water-formed hydroxyl nucle- ophilically attacks the peptide backbone carbonyl, 2) a carbonyl oxyanion is formed and coordinates the catalytic zinc ion, 3) the backbone amide is protonated, and 4) the zinc ion reduces the activation energy of this reaction by polarizing the oxygen carbonyl and by coordinating the negative charge of the intermediates evolving throughout the reaction (44). However, our understanding is far from complete, and several major questions remain unanswered: Is the catalytic water molecule bound to the zinc ion before proton abstraction by the conserved active site Glu base catalyst? Is the water-bound molecule pushed and replaced by the carbonyl oxygen of the peptide, or is a penta-coordinative complex consisting of both water and peptide formedFinally, although it is thought that protein function depends on protein flexibility, precisely how the molecule dynamics contribute to the catalytic mechanism remains unclear. Although internal protein dynamics are connected intimately to enzymatic catalysis, enzyme motions linked to substrate turnover remain largely unknown. Because of the scarcity of adequate experimental tools, the field is presented with a great challenge regarding quantification of protein conformational transitions during catalysis. In the next section we discuss the use of a real-time multidisciplinary structural-spectroscopic approach to study reactive sites in metalloenzymes during catalysis.
Time-Resolved, Structural Analysis-Correlation of Reaction Kinetics-Protein Conformations and Evolution of Reaction Intermediates
Enzymes are flexible moieties whose structures exhibit dynamic fluctuations on a wide range of timescales. This inherent mobility of a protein fold was shown to be manifested in the various steps constituting the catalytic cycle. The nature of this linkage between protein structure movement and function undoubtedly is complex and might involve the formation of a coupled network of interactions that bring the substrate closer, orient it properly, and provide a favorable electrostatic environment in which the chemical reaction can occur (45). However, the molecular details that link the catalytic chemistry to key kinetic, electronic, and structural events have remained elusive because of the difficulties associated with probing time-dependent, structure-function aspects of enzymatic reactions.
By using this argument, a single crystal structure generally is insufficient to enable the elucidation of enzymatic catalysis reaction mechanisms at an atomic level of detail. Typically, the catalytic cycle involves a series of intermediates and transition states, and for many of these states, no detailed structural information is available. Furthermore, determining the energies of the various stationary points in the cycle is highly nontrivial, from a theoretical or experimental point of view. For these reasons, as of today, a complete characterization of reactive enzymatic chemistry is unavailable.
Concentrating on metalloenzymes, we have developed a strategy based on stopped flow X-ray absorption spectroscopy (XAS) to elucidate in detail the molecular mechanisms at work during substrate turnover (Fig. 4). Importantly, XAS provides local structural and electronic information about the nearest coordination environment surrounding the catalytic metal ion within the active site of a metalloprotein in solution. When the X-rays hit a sample, the electromagnetic radiation interacts with the electrons bound in the metal atom. The radiation can be scattered by these electrons, or it can be absorbed, thereby exciting the electrons. At certain energies, the absorption increases drastically and gives rise to an absorption edge. Such edges occur when the energy of the X-ray beam is just sufficient to cause excitation of a core electron of the absorbing atom (in this case zinc) to a continuum state, for example, to produce a photoelectron. The energies of the absorbed radiation at these edges correspond to the binding energies of electrons in the K (or 1 s) shell of the zinc ion. When the photoelectron leaves the absorbing atom, its wave is backscattered by the neighboring atoms (in this case, the zinc-bound protein atoms). The constructive and destructive interference of these outgoing photoelectrons with the scattered waves from atoms surrounding the central metal atom gives rise to the extended X-ray absorption fine structure (EXAFS) oscillation pattern. Spectral analysis of the edge and EXAFS regions yields complementary electronic and structural information. Analysis of the edge region enables us to determine the oxidation state of the X-ray absorbing metal atom (in other words, the position of the absorption edge), whereas analysis of the EXAFS region provides precise information regarding distances between the absorbing metal atoms and the protein atoms that surround it. The high-resolution structural information that can be obtained by XAS studies makes XAS an advantageous tool for monitoring active site zinc coordination and electronics in metalloproteinases during different stages of the activation and inhibition processes, as demonstrated before (46-49). In addition, XAS is the only spectroscopic tool that can probe directly the otherwise spectroscopically silent zinc ion.
Time-resolved XAS (TRXAS) provides insight into the lifetimes and local atomic structures of metal-protein complexes during enzymatic reactions on millisecond-to-minute timescales (Fig. 4). This method is used to correlate the pre-steady-state kinetic behavior, with the structure of transient zinc-protein intermediates, and the local charge transitions that evolve during the initial peptide-protein interaction, as well as during the chemical step taking place at the catalytic site. Using this method, Kleifeld et al. detected the existence of two penta-coordinated intermediates during the oxidation of iso-propanol by alcohol dehydrogenase (49). The dehydrogenation reaction was shown to involve dynamic changes regarding zinc ion oxidation. In a recent study, Solomon et al. (50) have demonstrated that the catalytic zinc ion in TNF-α convertase (TACE), a zinc metalloproteinase (a close relative of MMPs), undergoes dynamic charge transitions before substrate binding to the metal ion, presumably during interaction of the substrate with distal protein side chains. Furthermore, TACE hydrolysis was shown to involve the formation of a penta-covalent complex at the catalytic zinc ion with a long lifetime of ~40 milliseconds during substrate binding to the catalytic zinc. Product release from the catalytic pocket resulted in restoration of a tetrahedral complex at the zinc ion. The most important point emerging from these studies is that by using TRXAS, the link between conformational changes in atomic resolution and the catalytic step in millisecond resolution can be followed directly as the enzyme loops through the catalytic cycle. Remarkably, these experiments revealed that peptide hydrolysis by TACE is governed by a process involving precursor charge transitions centered on the metal ion. In addition, they provided experimental evidence for the long-sought reaction mechanism proposed by Lipscomb et al. It is important to mention that the method described gives us atomic resolution structural snapshots of the nearest metal ion environment as the enzyme goes through catalysis. To build a detailed kinetic/mechanistic picture of the active site chemistry, the XAS analyses must be complemented with quantum mechanics/molecular mechanics calculations.

Figure 4. A schematic representation of the experimental approach for time-resolved XAS measurements. XAS provides local structural and electronic information about the nearest coordination environment surrounding the catalytic metal ion within the active site of a metalloprotein in solution. Spectral analysis of the various spectral regions yields complementary electronic and structural information, which allows the determination of the oxidation state of the X-ray absorbing metal atom and precise determination of distances between the absorbing metal atom and the protein atoms that surround it. Time-dependent XAS provides insight into the lifetimes and local atomic structures of metal-protein complexes during enzymatic reactions on millisecond to minute time scales. (a) The drawing describes a conventional stopped-flow machine that is used to rapidly mix the reaction components (e.g., enzyme and substrate) and derive kinetic traces as shown in (b). (b) The enzymatic reaction is studied by pre-steady-state kinetic analysis to dissect out the time frame of individual kinetic phases. (c) The stopped-flow apparatus is equipped with a freeze-quench device. Sample aliquots are collected after mixing and rapidly froze into X-ray sample holders by the freeze-quench device. (d) Frozen samples are subjected to X-ray data collection and analysis.
Comparative Structural-Dynamic Analysis and Its Relevance to Drug Discovery
Conducting real-time structural analysis on metalloproteinases was found to be a novel and effective approach for studying the highly structurally homologous catalytic sites residing in these enzymes via comparative structural-dynamic analysis. Remarkably, such high structural homology (Fig (2).) often hinders the design and production of highly selective inhibitors for this enzyme family. Thus, linking distinct protein conformational transitions with catalysis of individual enzymes is of great importance for revealing the molecular mechanisms underlying individual enzymes that may aid in the process of discovering new drugs. One may therefore inquire as to whether structurally homologous enzymes display similar dynamic profiles during binding and catalysis or whether functionally related enzymes that share similar structures can be distinguished based on their dynamic nature.
So far only a few reports have indicated distinct differences regarding the reaction chemistry and mechanisms among highly structural homologous MMP active sites. Studies by Fasciglione et al. demonstrate that the proton-linked behavior (in different pH environments) for kcat/Km, kcat, and Km is different among various MMPs, including MMP-9 and MMP-2 (51). In addition, the authors found that to have a fully consistent description of the enzymatic action of the various MMPs, they had to apply three protonating groups that are involved in the modulations of substrate interaction and catalysis. These results indicate that the details regarding active site modifications at the zinc ion are different for various MMPs and are of enormous importance for elucidating the mode of action of individual MMPs. Similar conclusions may be drawn from the recent study of Solomon et al. (52), which compares the inhibition mode of the mechanism-based inhibitor SB-3CT with the highly homologous catalytic sites of TACE and MMP-2. Importantly, it was shown that SB-3CT directly binds the metal ion of TACE, as observed before with MMP-2. However, in contrast to MMP-2, the binding mode of SB-3CT to the catalytic zinc ion of TACE is different regarding the length of the Zinc-S(SB-3CT) bond distance and the total effective charge of the catalytic zinc ion. In addition, SB-3CT inhibits TACE in a noncompetitive fashion by inducing marked conformational changes in the structure. For MMP-2, SB-3CT behaves as a competitive inhibitor, and no significant conformational changes are observed. Examination of the second shell amino acids surrounding the catalytic zinc ion of these enzymes indicated that the active site of TACE is more polar than that of MMP-2 and other MMPs. On the basis of these results, it was proposed that, although a seemingly high structural similarity exists between TACE and MMP-2, these enzymes are significantly diverse in the electronic and chemical properties within their active sites.
Overall, these reports raise the possibility that the highly structural analogous catalytic centers in MMPs exhibit different structural conformations and electronic behavior during catalysis and inhibition. Yet, an open question is as follows: How can such differences be quantifiedA comparative TRXAS analysis might provide the answer. By performing a comparative TRXAS analysis of functionally related enzymes that share similar structures, it might be possible to identify the intermediate states, the active site key amino acid residues, and the thermodynamic parameters of individual enzymes that are critical for efficient catalysis as well as for inhibition. Furthermore, the mobility so fundamental to protein structures is a major complicating factor to the general structure-based drug design approach. MMP’s active site dynamics were observed in the NMR structures of inhibitor-free MMP-1, MMP-3, and MMP-13 (25). The impact on drug design was clearly illustrated in MMP X-ray structures that demonstrated the ability of side chains in the active site to undergo conformational changes to accommodate a bound inhibitor. Thus, an inhibitor predicted to have poor inhibition activity against a specific MMP based on a poor fit in the specificity pocket (S1) may be accommodated in the binding site because of the mobility of the protein. By applying the time-resolved structural-dynamic approach to study catalysis as well as inhibition pathways, we can characterize unique reaction intermediates at the active site, as well as distinct conformational intermediates of the protein side chains, and can provide critical information about protein flexibility that additionally might be used for the design of potent and selective inhibitors. This approach departs from traditional drug design strategies used for metalloenzymes that target the catalytic metal with potent zinc-chelating peptidomimetic compounds.
Concluding Remarks
Here we reviewed the advances that have been made in the field of metalloproteinase chemistry over the past decades. Focusing on the critical need to understand better the chemical workings of these biologic machines at atomic detail, we noted the importance of quantifying the biophysical properties and the structural dynamic behavior of these enzymes to reveal their underlying molecular mechanisms.
A fundamental challenge for better understanding the function of proteins/enzymes is to characterize proteins as dynamic objects. In this article, we have presented novel experimental approaches that go beyond static structures, with the ultimate goal of characterizing macromolecules reacting at atomic resolution. More specifically, we have combined time-resolved X-ray spectroscopy with X-ray crystallography, enzymology, and computational mechanistic investigations to determine the structures and chemistry of transient intermediates as they evolve during the catalytic cycle. Our recent progress in the field of MMP reaction mechanisms demonstrates how a combined approach that uses a variety of biophysical techniques advances our fundamental understanding of complex biologic molecules.
References
1. Pauling L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry. 3rd. ed. 1960. Cornell University Press, Ithaca, NY.
2. Kozlov LV, Ginodman LM, Orekhovich VN, and Valueva TA. Biokhimiia 1966; 31:315-321.
3. Pelmenschikov V, Siegbahn PE. Inorg. Chem. 2002; 41:5659-5666.
4. Wolfenden R, Snider MJ. Acc. Chem. Res. 2001; 34:938-945.
5. Mott JD, Werb Z. Curr. Opin. Cell. Biol. 2004; 16:558-564.
6. Egeblad M, Werb Z. Nat. Rev. Cancer 2002; 2:161-174.
7. Coussens LM, Werb Z. Nature 2002; 420:860-867.
8. Sternlicht MD, Werb Z. Annu. Rev. Cell Dev. Biol. 2001; 17:463- 516.
9. Lee M, Fridman R, Mobashery S. Chem. Soc. Rev. 2004; 33:401- 409.
10. Overall CM, Lopez-Otin C. Nat. Rev. Cancer 2002; 2:657-672.
11. Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C. Internat. J. Devel. Biol. 2004; 48:411-424.
12. Bode W, Maskos K. Biol. Chem. 2003; 384:863-872.
13. Nagase H, Visse R, Murphy G. Cardiovasc. Res. 2006; 69:562-573.
14. Bode W, Fernandez-Catalan C, Tschesche H, Grams F, Nagase H, Maskos K. Cell. Mol. Life Sci. 1999; 55:639-652.
15. Massova I, Kotra LP, Fridman R, Mobashery S. FASEB J. 1998; 12:1075-1095.
16. Overall CM, Kleifeld O. Nat. Rev. Cancer 2006; 6:227-239.
17. Auld DS. BioMetals. 2001; 14:271-313.
18. Auld DS.In: Metal Sites in Proteins and ModelsVol. 89. 1997.
19. Thompson RB. Curr. Opin. Chem. Biol. 2005; 9:526-532.
20. Lipscomb WN, Strater N. Chem. Rev. 1996; 96:2375-2434.
21. Karlin S, Zhu Z-Y. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:14231-14236.
22. Stocker W, Bode W. Curr. Opin. Struct. Biol. 1995; 5:383-390.
23. Stocker W, Grams F, Baumann U, Reinemer P, Gomis-Ruth FX. McKay DB. Bode W. Protein Sci. 1995; 4:823-840.
24. Iyer S, Visse R, Nagase H, Acharya KR. J. Mol. Biol. 2006; 362:78-88.
25. Rush TS III, Powers R. Curr. Top. Med. Chem. 2004; 4:1311-1327.
26. Bertini I, Calderone V, Cosenza M, Fragai M, Lee Y, Luchinat C, Mangani S, Terni B, Turano P. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:5334-5339.
27. Pinitglang S, Watts AB, Patel M, Reid JD, Noble MA, Gul S, Bokth A, Naeem A, Patel H, Thomas EW, Sreedharan SK, Verma C, Brocklehurst K.) Biochemistry 1997; 36:9968-9982.
28. Elrod JP, Hogg JL, Quinn DM, Venkatasubban KS, Schowen RL. J. Am. Chem. Soc. 1980; 102:3917-3922.
29. Daggett V, Schroder S, Kollman P. J. Am. Chem. Soc. 1991; 113;8926-8935.
30. Kester WR, Matthews BW. Biochemistry 1977; 16:2506-2516.
31. Shoham G, Christianson DW, Oren DA. Proc. Natl. Acad. Sci. U.S.A. 1988; 85:684-688.
32. Breslow R, Wernick DL. Proc. Natl. Acad. Sci. U.S.A. 1977; 74:1303-1307.
33. Holmes MA, Tronrud DE, Matthews BW. Biochemistry 1983; 22:236-240.
34. Lipscomb WN, Hartsuck JA, Reeke GN, Quiocho FA, Bethge PH, Ludwig ML, Steitz TA, Muirhead H, Coppola JC. Brookhaven Symp Biol 1968; 21:24-90.
35. Mock WL. Bioorg. Chem. 1975; 4:270-278.
36. Christianson DW, Lipscomb WN. Proc. Natl. Acad. Sci. U.S.A. 1985; 82:6840-6844.
37. Christianson DW, Lipscomb WN. Acc. Chem. Res. 1989; 22:62-69.
38. Mock WL, Zhang JZ. J. Biol. Chem. 1991; 266:6393-6400.
39. Mock WL, Tsay J-T. Biochemistry 1986; 25:2920-2927.
40. Rozanov DV, Deryugina EI, Ratnikov BI, Monosov EZ, Marchenko GN, Quigley JP, Strongin AY. J. Biol. Chem. 2001; 276:25705-25714.
41. Homandberg GA, Minor ST, Peanasky RJ. Biochem. Biophys. Acta 1980; 612:384-394.
42. Friesner RA, Guallar V. Annu. Rev. Phys. Chem. 2005; 56:389-427.
43. Kilshtain-Vardi A, Shoham G, Goldblum A. Internat. J. Quant. Chem. 2002; 88:87-98.
44. Fersht A.In: Structure and Mechanisms in Protein Science, Vol. 1. 1999. W.H. Freedman.
45. Benkovic SJ, Hammes-Schiffer S. Science 2003; 301:1196-1202.
46. Kleifeld O, Van den Steen PE, Frenkel A, Cheng F, Jiang HL, Opdenakker G, Sagi I. J. Biol. Chem. 2000; 275:34335-34343.
47. Kleifeld O, Frenkel A, Bogin O, Eisenstein M, Bramfeld V, Burstein Y, Sagi I. Biochemistry. 2000; 39:7702-7711.
48. Rosenblum G, Meroueh SO, Kleifeld O, Brown S, Singson SP, Fridman R, Mobashery S, Sagi I. J. Biol. Chem. 2003; 301:139-200.
49. Kleifeld O, Frenkel A, Martin JM, Sagi I. Nat. Struct. Biol. 2003: 10:98-103.
50. Solomon A, Akabayov B, Frenkel A, Milla M, Sagi I. 2006.
51. Fasciglione GF, Marini S, D’Alessio S, Politi V, Coletta M. Biophys. J. 2000; 79:2138-2149.
52. Solomon A, Rosenblum G, Gonzales PE, Leonard JD, Mobashery S, Milla ME, Sagi I. J. Biol. Chem. 2004; 279:31646-31654.
53. Matsubara H, Singer A, Sasaki R, Jukes TH. Biochem. Biophys. Res. Commun. 1965; 21:242-247.
54. Latt SA, Holmquist B, Vallee BL. Biochem. Biophys. Res. Commun. 1969; 37:333-339.
55. Colman PM, Jansonius JN, Matthews BW. J. Mol. Biol. 1972; 70:701-724.
56. Matthews BJ, Colman PM, Schoenborn BP, Dupourque D. Nature (Lond) New Biol. 1972; 238:37-41.
57. Quiocho FA, Lipscomb WN. Adv. Protein Chem. 1971; 25:1-78.
58. Skidgel RA, Erdos EG. Immunol. Rev. 1998; 161:129-141.
59. Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi MR, Bernstein KE. J. Clin. Invest. 1997; 99:2375-2385.
60. Turner AJ, Hooper NM. Trends Pharmacol. Sci. 2002; 23:177-183.
61. Hooper NM. FEBS Lett. 1994; 354:1-6.
62. Bode W, Gomis-Ruth FX, Stockler W. FEBS Lett. 1993; 331:134-140.
63. Somerville RP, Oblander SA, Apte SS. Genome Biol. 2003; 4:216.
64. Schlondorff J, Blobel CP. J. Cell Sci. 1999; 112:3603-3617.
65. Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. J. Biol. Chem. 1997; 272:556-562.
66. White JM. Curr. Opin. Cell Biol. 2003; 15:598-606.
67. Black RA, White JM. Curr. Opin. Cell Biol. 1998; 10:654-659.