Liquid-State Physical Chemistry: Fundamentals, Modeling, and Applications (2013)
9. Modeling the Structure of Liquids: The Simulation Approach
9.4. An Example: Ammonia
In this section we provide, as an example of the results that can be obtained using simulation methods, a means of determining the structure of liquid NH3. This is achieved by discussing the results of combined quantum-mechanicalcalculations, pair potential fitting and MC simulations, as described by Hannongbuo [11]. In this investigation the interaction between NH3 molecules was obtained using HF-TZP calculations7) yielding a total energy for the molecule of −56.21603 Hartree and a dipole moment of 1.87 D. The latter value must be compared with other theoretical (quantum) calculations (Table 9.1), from which a dimer stabilization energy Estab = −2.53 kcal mol−1 was obtained with an optimum N–N distance of 3.4 Å and an optimum configuration α = β = 12°, where the angles are indicated in Figure 9.4. The interaction energy between two NH3 molecules was calculated for more than 1500 configurations, leading to a detailed energy landscape. For ease of further calculation, this energy landscape for the NH3–NH3 interaction was fitted with pair potentials to these configurations. This resulted in
![]()
![]()
![]()
where the units are ΔE [kcal mol–1] and r [Å].
Table 9.1 Dipole moments for NH3 calculated by various approximate Hartree–Fock (HF) calculations.
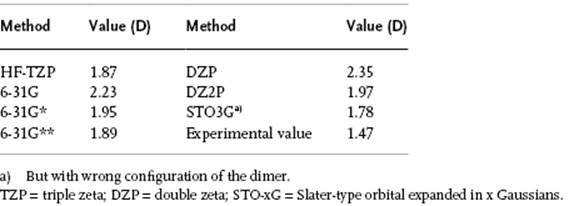
Figure 9.4 The general orientation for two NH3 molecules with optimal configuration α = β = 12°.
MC calculations were performed using 202 rigid NH3 molecules with the experimental structure as described by NH = 1.0124 Å and H–N–H = 106.67°. The simulations were made at an experimental density ρ = 0.633 g cm−3 at 277 K, which renders a box length of 20.99 Å.
These MC simulations were first made using ab initio HF-TZP calculations for each configuration. Although a logical choice for the cut-off value would be 10.45 Å, a value of 7.0 Å was chosen because the interaction almost disappears after 5–6 Å; the computing time was also reduced. Within a shell of 7 Å this implies a calculation for about 30 molecules. The MC simulations were also made with the fitted pair potential, but in this case an expected cut-off value of 10.45 Å was used.
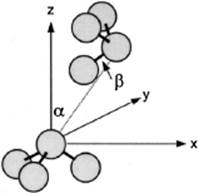
The N–N pair correlation function g(r) was calculated using 50 000 configurations for both (ab initio and pair potential fitted) energy landscapes, and compared with the experimental data of Narten [12] at 277 K and 1 atm. These results are shown in Figure 9.5, where the inset shows also the results for 25 000 configurations. The shape is typical for g(r). The first peak (∼3.4 Å) corresponds to the nearest-neighbor N–N distance. For the ab initio simulations a broad shoulder between 4.0 and 4.8 Å is observed, the absence of which for the pair potential simulations is attributed to a systematic error due to using simple analytical functions to represent the complicated energy surface. In particular, it was noted that for the ab initio simulations multiple minima were obtained with slightly different stabilization energies. The three most stable energies were for α = 0° with Estab = −2.40 kcal mol−1, α = 10° with Estab = −2.53 kcal mol−1, and α = 20° with Estab = −2.25 kcal mol−1 for the optimum N–N distance of 2.54 Å. For comparison, recall that RT ≅ 0.6 kcal mol−1. The pair potential fitting is unable to represent these minima.
Figure 9.5 The N–N pair correlation for NH3 as calculated from 50 000 configurations. The inset shows the results for 25 000 configurations.
The onset of the ab initio N–N g(r) at 2.3 Å, which starts earlier than the 2.7 Å X-ray value for the experimental N–N g(r), indicates less repulsion at short distances for the pair interaction derived from ab initio calculations during the simulation than in reality, where many-body effects are present. Another source of error which leads to discrepancy between the experimental and simulated results is the use of a fitted, nonpolarizable potential in the liquid phase simulations. It has been reported that variation of the pair potential, due to the addition of higher multipoles and of the polarization effects, influences the liquid structure by changing the dimer configurations corresponding to the local maxima of the correlation functions.
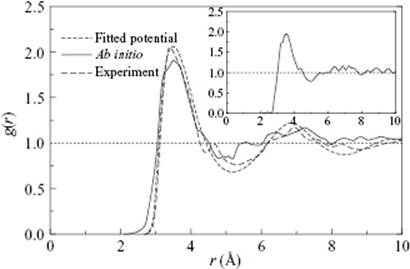
In order to investigate three-body interactions, calculations were made for the configurations as shown in Figure 9.6. The three-body contribution appeared to be important, and three configurations could be discerned within the range r1 ≤ 2 Å and r2 ≥ 10 Å. The configurations shown in Figure 9.6 can be described as donor–donor, donor–acceptor, and acceptor–acceptor, respectively. The donor–donor configuration appeared to be repulsive, while the donor–acceptor configuration was attractive, and the acceptor–acceptor configuration was strongly repulsive. At a N–N distance of 3.4 Å, the energy values for the donor–donor, acceptor–acceptor, and donor–acceptor configurations of 0.13, 0.65 and −0.22 kcal mol−1, respectively, led to a net effect for the E3bd value of 0.36 kcal mol−1, which equates to 14% of the global minimum of the pair interaction of 2.53 kcal mol−1. This influence is more pronounced for shorter distances because the three-body energy for the acceptor–acceptor increases much faster than for the other two terms.
Figure 9.6 (a) Donor–donor, (b) donor–acceptor, and (c) acceptor–acceptor configurations for three NH3 molecules.
The partial pair correlation function for H–H was rather featureless (Figure 9.7), whereas the N–H correlation showed a main peak at 3.751 Å and a wide hump from 2.3 to 3.11 Å, indicating very weak hydrogen bonding in liquid ammonia. The calculated H coordination number was 0.97, compared to 1.2 from previous simulation results and 1.2 from experimental data. It should be noted that the hydrogen bond distance in solid ammonia is 2.35 Å, with an average of three hydrogen bonds per nitrogen atom.
Figure 9.7 The H–H and N–H correlation functions for NH3.
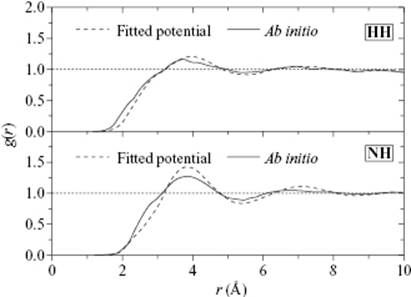
Overall, the remaining discrepancies between simulations and experimental can be attributed to statistical error, due mainly to the use of simple analytical functions to represent the complicated energy surface, together with many-body and the polarization effects.
Problem 9.4
Estimate the value for the average angle β from the N–N and N–H correlations functions given in Figures 9.5 and 9.7.
Notes
1) See footnote 4, Chapter 8.
2) Recall the notation introduced in Chapter 1: if a vector x is represented by a column matrix x, the inner product x·y is written as xTy and the set x1x2…xN (x1x2…xN) is represented as x (x).
3) Proper random generators are nowadays standard available for any computer.
4) See, for example, Frenkel and Smit [6].
5) Whilst for a small number of dimensions, a systematic sampling of space is more efficient, for a high number of dimensions random sampling is more efficient.
6) Whereas, in the MD method, a state means specified locations r1,r2,…,rN and specified momenta p1,p2,…,pN of the N particles, in the MC method a state means specified locations r1,r2,…,rN only.
7) These abbreviations and those used later in the comparison for the dipole moment indicate the type of basis set used in the molecular orbital calculations: HF = Hartree–Fock; TZP = triple zeta, indicating that for each occupied valence orbital in the atom three basis functions are used, plus polarization functions, indicating that nonoccupied valence orbitals in the atom are included, DZP = double zeta, similar to TZP but using two basis functions, STO-xG = Slater-type orbital expanded in x Gaussian functions. Essentially, this label indicates the quality of the basis set used.
References
1 Wood, W.W. and Parker, F.R. (1957) J. Chem. Phys., 27, 720.
2 See Berenden (2007).
3 See Friedman (1985).
4 See Sachs et al. (2006).
5 See Allen (1987).
6 See Frenkel and Smit (1987).
7 Verlet, L. (1967) Phys. Rev., A159, 98.
8 Berendsen, H.J.C., and van Gunsteren, W.F. (1986) in Molecular Dynamics Simulation of Statistical Mechanical Systems (eds G. Ciccotti and W. Hoover), North Holland, Amsterdam, p. 43.
9 Wainwright, T.E. and Alder, B.J. (1958) Nuovo Cimento, 9 (Suppl. 1), 116.
10 Metropolis, N., Rosenbluth, A.W., Rosenbluth, M.N., Teller, E., and Teller, A.H. (1953) J. Chem. Phys., 21, 1087.
11 Hannongbuo, S. (2000) J. Chem. Phys., 113, 4707.
12 Narten, A.H. (1977) J. Chem. Phys., 66, 3117.
Further Reading
Allen, M.P. and Tildesley, D.J. (1987) Computer Simulation of Liquids, Clarendon, Oxford.
Berendsen, H.J.C. (2007) Simulating the Physical World, Cambridge University Press, Cambridge.
Frenkel, D. and Smit, B. (2002) Understanding Molecular Simulation: From Algorithms to Applications, 2nd edn, Academic Press, London.
Friedman, H.L. (1985) A Course on Statistical Mechanics, Prentice-Hall, Englewood Cliffs, NJ.
Landau, D.P. and Binder, K. (2000) Monte Carlo Simulations in Statistical Physics, Cambridge University Press, Cambridge.
Leach, A.R. (2001) Molecular Modelling, Principles and Applications, 2nd edn, Pearson Prentice-Hall, Harlow, UK.
Sachs, I., Sen, S., and Sexton, J.C. (2006) Statistical Mechanics, Cambridge University Press, Cambridge.