Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
6. Homogeneous Reactions II: Photochemistry and Electrochemistry and Radiopharmaceutical Synthesis
Paul Watts and Charlotte Wiles
6.1. Photochemistry in Flow Reactors
Photochemistry can potentially provide an environmental-friendly and green approach to chemical synthesis; however, the ability to scale-up such photochemical processes is marred with problems, which are mainly associated with the power of light sources. The fact that a large number of microreactors are manufactured in glass, quartz, or transparent polymers is ideal for conducting photochemical processes, as the path length of such reactors is small meaning that it is very easy to irradiate the reaction mixture within the channel. Compared to other examples of chemical synthesis in flow reactors, the number of photochemical transformations performed under flow conditions has until recently been very limited. Early examples included benzopinacol formation [1], synthesis of cycloaddition products [2], and photosensitized diastereodifferentiation [3].
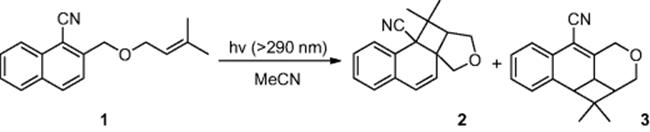
Mizuno and coworkers [4] reported an enhancement in reaction efficiency, as well as regioselectivity, by conducting the photocycloaddition of a naphthalene derivative (1) (Scheme 6.1) in a microreactor. The authors found that in batch, irradiation of cyanonaphthalene derivative (1), using a filtered xenon lamp (λ > 290 nm), afforded photocycloadducts (2) and (3) in 56% and 17% yield, respectively. In comparison, when conducting the reaction in a microreactor, employing an irradiation time of just 3.4 min, the desired compound (2) was obtained in an increased yield of 59%, while by-product (3) was reduced to 9%.
Scheme 6.1 Photocycloaddition of naphthalene derivative in a microreactor.
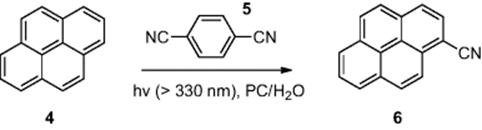
Kitamura [5] demonstrated the photocyanation of pyrene (4) (Scheme 6.2) across an oil–water interface within a microreactor [dimensions, 100 μm (wide) × 20 μm (deep) × 3.5 cm (length)]. To perform the reaction, an aqueous solution of sodium cyanide and pyrene (4) was added from one inlet and 1,4-dicyanobenzene (5) in propylene carbonate (PC) was introduced from the other inlet. Using a residence time of 3.5 min, while the reactor was irradiated using a high-pressure Hg lamp, the authors reported an optimized 73% conversion of pyrene (4) to 1-cyanopyrene (6).
Scheme 6.2 Photocyanation of pyrene an oil–water interface in a microreactor.
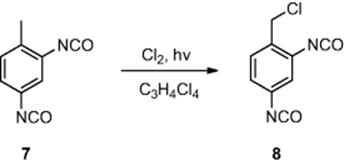
Another photochemical transformation conducted in a microreactor, involving a gaseous reagent, was the photochemical chlorination of toluene-2,4-diisocyanate (7) (Scheme 6.3) [6]. Employing a falling film microreactor [channel dimensions, 600 μm (wide) × 300 μm (deep) × 6.6 cm (length)] consisting of 32 parallel channels, the authors investigated the irradiation of gaseous chlorine, through a quartz window, generating chlorine radicals in the presence of toluene-2,4-diisocyanate (7) in tetrachloropropane as a solvent. The authors investigated the effect of varying the flow rate of chlorine and toluene-2,4-diisocyanate on the proportion of benzyl chloride-2,4-diisocyanate (8) produced. Maintaining the reactor at 130 °C, the authors identified the optimal residence time to be 9 s, affording benzyl chloride-2,4-diisocyanate (8) in 81% conversion.
Scheme 6.3 Photochemical chlorination of toluene-2,4-diisocyanate.
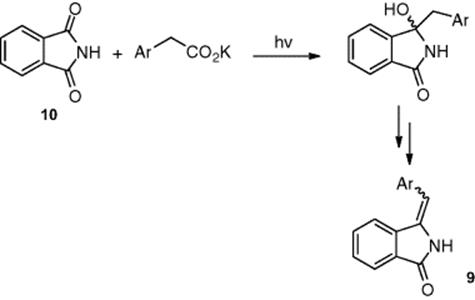
Oelgemöller and coworkers [7] focused on the photodecarboxylative benzylation of phthalimide (Scheme 6.4) as a means of providing access to 3-arylmethyleneisoindolin-1-ones (9) upon dehydration. With problems observed with this reaction using conventional photochemistry, including the formation of the product as a potassium salt and significant by-product formation upon dehydration, the authors investigated the reaction using a microreactor. Irradiating the phthalimide (10) solution, in a mixture of acetone and pH 7 buffer and in the presence of phenyl acetate, the authors obtained the target product (9) in 97% yield.
Scheme 6.4 Addition of phenylacetates to phthalimide (10) and dehydration to afford 3-arylmethyleneisoindolin-1-ones (9).
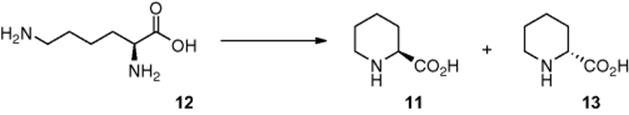
Takei and coworkers [8] demonstrated the synthesis of l-pipecolinic acid (11) from an aqueous solution of l-lysine (12) (Scheme 6.5). To achieve this photochemical transformation, the authors fabricated a Pyrex microreactor, in which the cover plate was coated in a 300 nm thick layer of TiO2, which was impregnated with platinum nanoparticles. The reactor was subsequently irradiated using a high-pressure Hg lamp and the selectivity to d-pipecolinic acid (13) and l-pipecolinic acid (11) was investigated. Employing a residence time of 50 s, the authors reported an 87% conversion and 22% selectivity for l-pipecolinic acid (11) synthesis.
Scheme 6.5 Photocatalytic synthesis of l-pipecolinic acid.
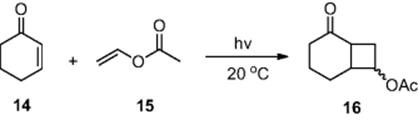
Using a series of UV-LEDs as the light source, Ryu and coworkers [9, 10] demonstrated increased reaction efficiency, compared to the Paternó–Büchi reaction performed using a 300 W Mercury lamp. Employing six UV-LED light sources, the authors performed the [2 + 2] cycloaddition of cyclohexen-2-one (14) with vinyl acetate (15) to afford the cycloadduct (16) (Scheme 6.6). Within a micro channel device [dimensions = 1000 μm (wide) × 200 μm (deep) × 56 cm (long)], the authors obtained a 200-fold increase in energy efficiency compared to a Mercury lamp and a 10-fold increase compared to black lights. With the developed protocol in hand, the authors also demonstrated the generality of the technique by varying the acetate used, affording an array of substituted cyclohexanone derivatives.
Scheme 6.6 Illustration of the Paternó–Büchi reaction performed in a photochemical microreactor.
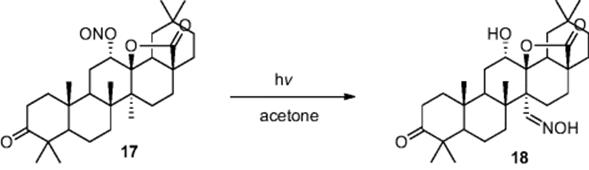
Other examples from the group include the black light promoted selective halogenation of cycloalkenes, reporting the efficient mono-bromination using Br2 and chlorinations using Cl2 and SOCl2 within a biphasic reaction system, and the Barton nitrite photolysis used for the conversion of nitrite derivative (17) to steroid (18) (Scheme 6.7) [11].
Scheme 6.7 Illustration of the Barton nitrite photolysis used in the synthesis of a complex saturated alcohol.
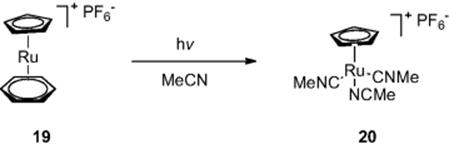
As well as organic synthesis, Jamison and colleagues [12] have demonstrated organometallic synthesis in flow reactors exploiting photochemistry. The group fabricated a flow reactor by wrapping PFA tubing (internal diameter, 762 μm (wide), volume = 1000 μl) around a standard 450 W medium-pressure mercury lamp. Reaction of complex (19) in acetonitrile at 0.02 M concentration afforded the desired CpRu(MeCN)3PF6 complex (20) in 99% yield (Scheme 6.8). While batch reactions commonly took 36 h to go to completion, the flow reactor gave the product in a residence time of just 5 min.
Scheme 6.8 Synthesis of CpRu(MeCN)3PF6 in a photochemical flow reactor.
With a view toward synthetic production using flow photochemistry, Freitag and coworkers [13] have reported the construction of multipass flow reactors as a means of increasing photochemical efficiency without the need for large irradiated areas. Incorporation of in-line IR enabled the authors to recirculate the reaction mixture until conversion had reached a preset level, at which point an automatic valve opened and diverted the reaction mixture to a collection vessel. In addition to single-phase photochemical transformations, continuous flow reactors have also been applied to heterogeneous photochemical reactions, using TiO2-coated channels to perform reductions [14], oxidations [15], and alkylations [16].