Microreactors in Organic Chemistry and Catalysis, Second Edition (2013)
11. Industrial Microreactor Process Development up to Production
11.2. Screening Studies in Laboratory
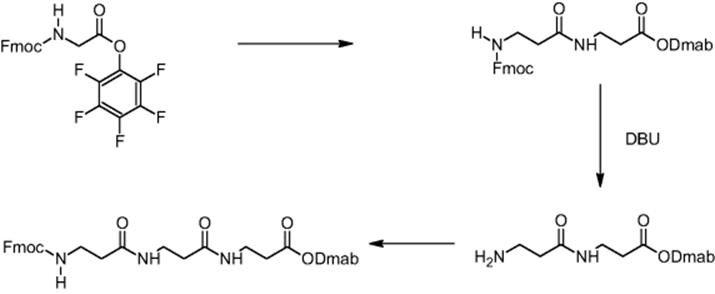
11.2.1 Peptide Synthesis
The Nervous System Research branch of Novartis Pharma Ltd in Basel, Switzerland, and the University of Hull investigated peptide synthesis in chip-based microreactors [12]. β-Amino acids were chosen for demonstrating feasibility of microreactor processing, as there are no chiral centers that may complicate analysis of the products [13].
Peptides are typically synthesized by solid-phase chemistry on polymer beads, a route discovered by and named after Merrifield [14, 15]. These polymer supports are expensive. Additional steps for linkage to and cleavage from the polymer are required. Hence, the motivation is to test solution chemistries as an alternative to the Merrifield approach.
The impact of mixing in a microreactor was demonstrated using the example of β-dipeptide synthesis by carbodiimide coupling using Dmab O-protection. Boc-β-alanine was O-protected (carboxylic moiety) by DMAP (4-dimethylaminopyridine) coupling with DmabOH (4-{N-[1-(4,4-dimethyl-2,6-dioxocyclohexyliden)-3-methylbutyl]amino}benzyl alcohol) yielding Dmab-β-alanine, whereas the Fmoc group was used for N-protection of β-alanine [16]. Thus, orthogonal protecting groups were established. During carbodiimide coupling, Dmab-β-alanine and Fmoc-β-alanine reacted and the synthesis of the corresponding β-dipeptide was realized.
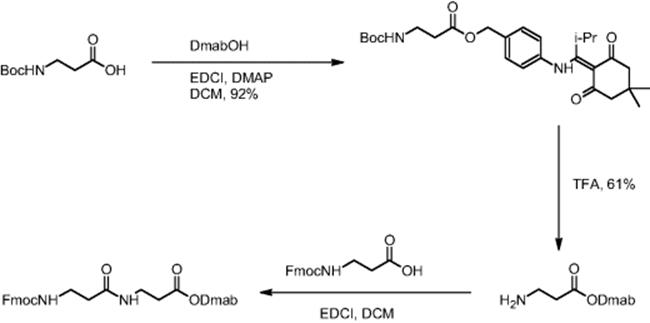
Electroosmotic flow (EOF) conditions were applied and yielded only 10% conversion with constant reactant movement [16]. Use of stopped-flow techniques, which periodically push and mix the flow, led to an increase in yield to 50%. A change in the coupling agent from 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) to dicyclohexylcarbodiimide (DCC) for reasons of limited solubility resulted in a 93% yield of the dipeptide. Batch β-dipeptide synthesis using EDCI gave a yield of 50% [6].
A reduction in reaction time by virtue of the improved transport properties in electroosmotically driven microreactors was demonstrated for the β-dipeptide synthesis using pentafluorophenyl O-activation. Fmoc-β-alanine was preactivated by introducing the pentafluorophenyl function as ester group [16]. Dmab-β-alanine and the pentafluorophenyl ester of Fmoc-β-alanine reacted and the synthesis of the corresponding β-dipeptide was realized.
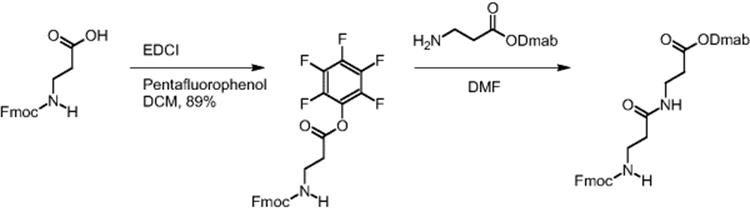
Quantitative yield of the dipeptide was achieved in only 20 min when using electroosmotically driven microreactors, while batch synthesis under the same conditions gave only 46% yield in 24 h [13, 16].
The same findings with respect to reaction time reduction were reported for the β-dipeptide synthesis using pentafluorophenyl O-activation. Boc-β-alanine was preactivated by introducing the pentafluorophenyl function as ester group [16]. Dmab-β-alanine and the pentafluorophenyl ester of Boc-β-alanine reacted and the synthesis of the corresponding β-dipeptide was realized.
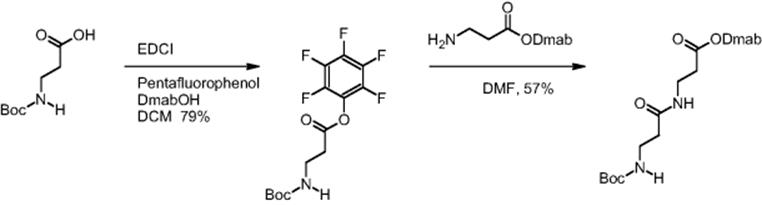
Electroosmotically driven microreactor processing gave quantitative yield of the dipeptide in only 20 min, while batch synthesis under the same conditions gave only 57% yield in 24 h [13, 16].
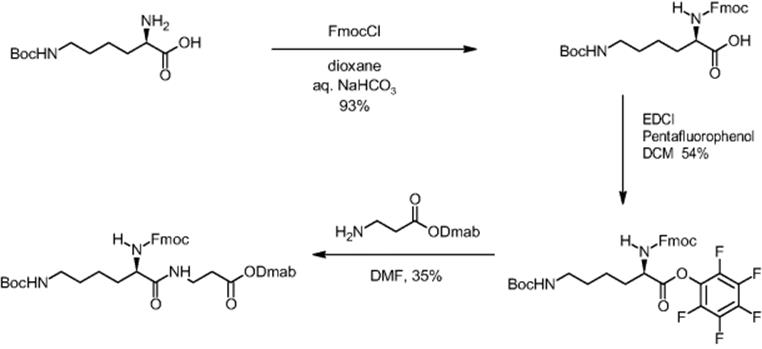
The formation of more complex dipeptides was also demonstrated by microchannel processing using the example of the amino acid N-ε-Boc-l-lysine with the additional amino function [5, 6]. While the batch yield was poor (9%), microreactor synthesis resulted in quantitative yield in 20 min.
In the next step, the preparation of longer chain peptides was done in electroosmotic microreactors. For this, deprotection and peptide bond forming reactions had to be developed resulting in a yield of 30% [13, 16]. In this way, the formation of tripeptide was achieved. Dmab-β-alanine and Fmoc-β-alanine were reacted in the first step to a dipeptide [13]. After cleavage of the Fmoc function, Fmoc-β-alanine was added to the dipeptide resulting in tripeptide formation with 30% yield.
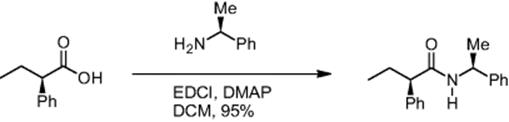
The degree of racemization was also monitored using the example of a simple carboxylic acid in peptide synthesis, 2-phenylbutyric acid. The pentafluorophenyl ester of (R)-2-phenylbutyric acid and (S)-1-phenylethylamine reacted to give the corresponding amino acid via an EDCI coupling [12]. In a control experiment, (R)-2-phenylbutyric acid and (R)-1-phenylethylamine were reacted as well. For the amino acid formation from (R)-2-phenylbutyric acid and (S)-1-phenylethylamine, a racemization of 4.2% was observed [6]. At higher concentration (0.5 M instead of 0.1 M), a higher degree of racemization was observed (7.8%).
11.2.2 Hantzsch Synthesis
GlaxoSmithKline Pharmaceuticals in Harlow, UK, performed the Hantzsch synthesis of 2-bromo-4′-methylacetophenone and 1-acetyl-2-thiourea in NMP (N-methyl-2-pyrrolidone) using a microchip reactor under EOF [10] (for EOF see Ref. [18]). This is claimed to be the first example of a heated organic reaction performed on a glass chip reactor under electroosmotic flow control, while earlier only room-temperature reactions were used. In a wider scope, the Hantzsch synthesis is a further example to evaluate the potential of microfluidic systems for high-throughput screening.
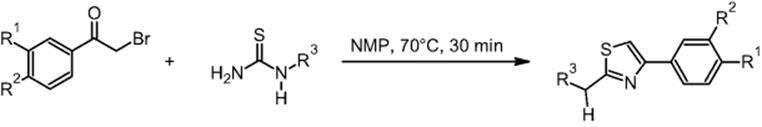
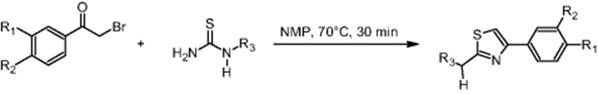
Yields from 42 to 99% were reported. Comparative and better yields were achieved when using a microchip reactor as compared to conventional laboratory batch technology. In case of improvement, the increase in yield amounted to about 10–20% [17, 19].
11.2.3 Knorr Synthesis
The Knorr synthesis of pyrazoles using a microchip reactor under electroosmotic flow conditions was developed by GlaxoSmithKline Pharmaceuticals in Harlow, UK [20]. The target was a much higher diversity campaign (7 × 32 libraries in the long run) by parallel operation of microchannels, after the feasibility of EOF processing had been demonstrated using the example of the Hantzsch synthesis. The Knorr synthesis is of interest for drug applications as products with a wide range of biological activity can be generated this way.
In the Knorr route, 1,3-dicarbonyl compounds react with hydrazines under ring closure to pyrazoles [20].
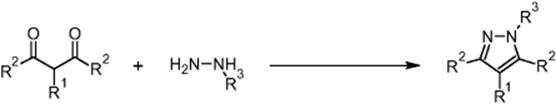
The following library consisting of seven (A1–A7) 1,3-dicarbonyl compounds and three hydrazines (B1–B3) was synthesized.
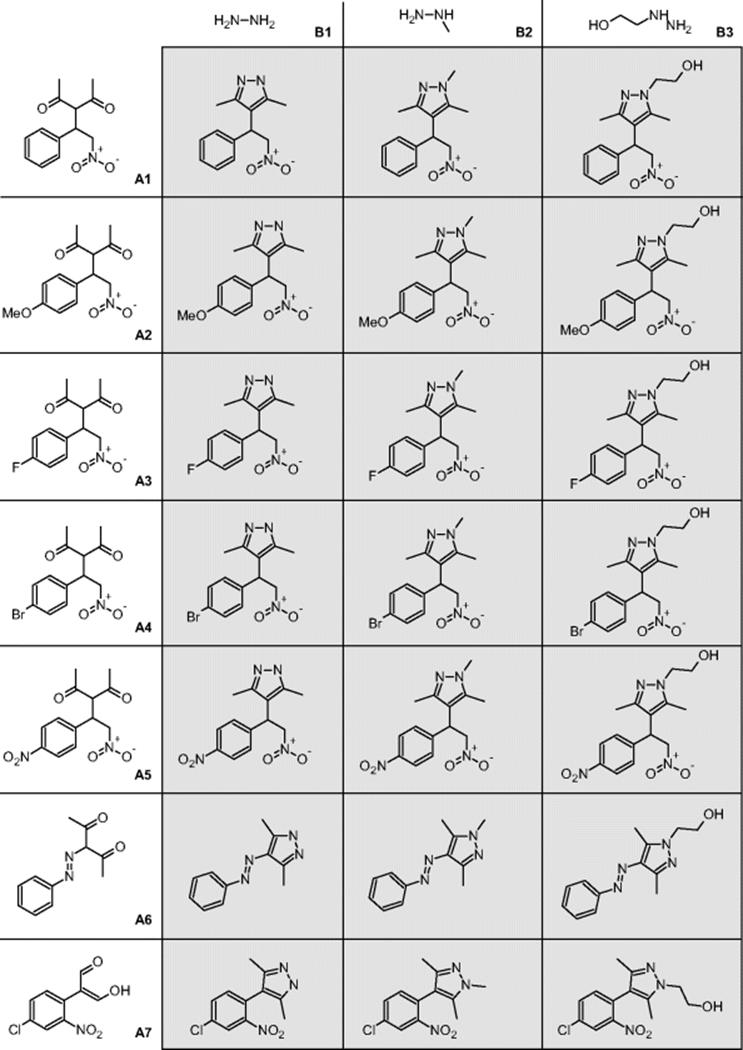
The 3 × 7 library was made in a sequential and automated way with conversions between 35 and 99% (quantitative; for 16 reactions). The corresponding chip is a commercial product of Caliper Technologies Company (110 Caliper chip™), originally designated for μTAS applications. The chips were constructed from two glass plates by means of standard photolithography. The etched microchannels have different widths for more stable flow, for example, to avoid dependence of capillary forces in the reservoirs. The glass chip is glued to a polymer caddy for interfacing with a multiport control device, the Caliper 42™ Workstation. This automated system consists of an autosampler (CTC-HTS Pal system) that introduces the reactant solutions in the chip via capillaries. A pumping system (μ-HPLC–CEC system) serves for fluid motion by hydrodynamically driven flow. A dilution system (Jasco PU-15(5)) is used for slug dilution on chip. A detection system (Jasco UV-1575) and an analysis system (LC–MS, Agilent 1100 series capLC-Waters micromass ZQ) are also used. All components were online and self-configured.
The results obtained were compared for consistency to single-reaction processing on the same chip. No cross-contamination was found during preparation of the library [20]. The products, by-products, or the hydrazine in excess were not intermixed.
11.2.4 Enamine Synthesis
GlaxoSmithKline Pharmaceuticals in Harlow, UK, performed an enamine synthesis using a microchip reactor under electroosmotic flow conditions [20]. The aim was a much higher research on enamine formation in microreactors focused on eliminating the need of using Lewis acid catalysts [21]. In addition, operation under mild conditions such as room-temperature processing was favored.
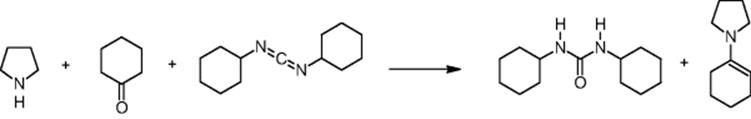
The microreactor yield (up to 42%) is comparable to batch Stork enamine reactions using p-toluenesulfonic acid in methanol under Dean and Stark conditions, that is, under water separation in a water trap (Dean–Stark apparatus).
11.2.5 Aldol Reaction
The Nervous Systems Research branch of Novartis Pharma Ltd in Basel, Switzerland, carried out the aldol reaction using a microchip reactor under electroosmotic flow conditions. Aldol reactions are well-established routes for CߝC bond formation in organic chemistry. The reaction requires the formation of enolates that themselves are one of the most profound species enabling CߝC bond formation [22]. Reducing processing time is a driver for microchannel processing of aldol reactions [22], which can be accomplished using reactive reactants such silyl enol ethers. For example, the reaction between 4-bromobenzaldehyde and the silyl enol ether of acetophenone was performed in a microreactor [22].
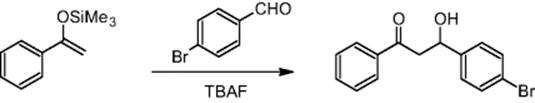
For this reaction, 100% conversion with respect to the silyl enol ether was achieved in 20 min [22]. The corresponding time for batch synthesis amounted to about 1 day.
11.2.6 Wittig Reaction
SmithKline Beecham Pharmaceuticals in Harlow, UK, carried out Wittig reactions using a microchip reactor under electroosmotic flow conditions. 2-Nitrobenzyltriphenylphosphonium bromide was reacted with methyl 4-formylbenzoate and four other aldehydes – 3-benzyloxybenzaldehyde, 2-naphthaldehyde, 5-nitrothiophene-2-carboxaldehyde, and 4-[3-dimethylamino)propoxy]benzaldehyde [23, 24].
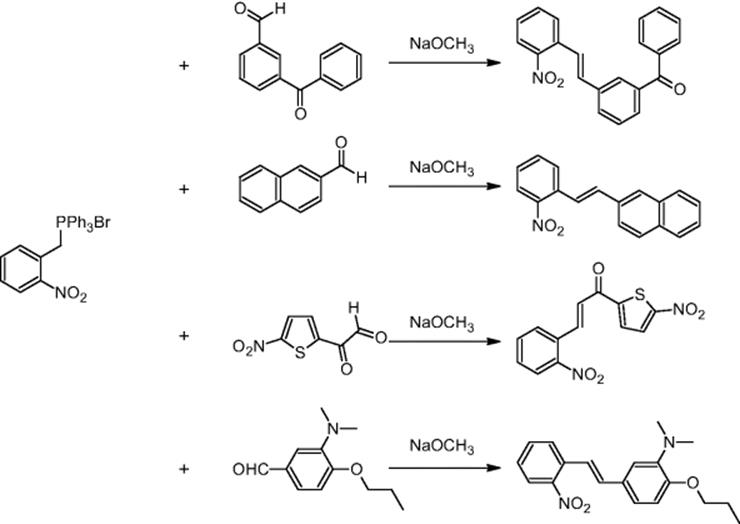
Using optimized reaction conditions, the Wittig reactions with four of the five aldehydes indeed resulted in improvement of their yields. The ratio of (E)- and (Z)-alkenes could be changed by simply adjusting the voltages in the electroosmotic flow-driven chip [25]. For a 1 : 1 ratio of the reactants, the Z/E ratio changed from 2.35–3.0 (premixed) to 0.82–1.09 (not premixed, separate movement) [24].
11.2.7 Polyethylene Formation
Dow in Midland, MI, USA, performed metallocene-catalyzed polymerization of ethylene using a home-built tube reactor setup with advanced, microflow tailored plant peripherals for heating, temperature monitoring, pressure control, and dosing via smart valves and injectors. Screening of process conditions was a driver [26]. Also, flexibility with regard to temperature and pressure at low sample consumption was an issue. Quality of the information is another motivation due to the advanced process control and sensing.
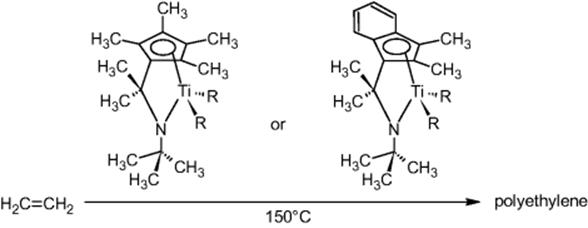
Ethylene is handled at 60 °C, well above the critical temperature [26]. Various combinations of precatalysts and activators were sampled and loaded by an autoinjector.
Temperature profiles versus time were taken for different positions at the reactor tube [26]. The maximum rise in temperature was about 23 °C. Improved pressure control was exerted by using advanced pressure control electronics [26]. In the regions of large temperature increase, pressure was slightly fluctuating; this effect diminished downstream. By deliberately changing pressure (in a loop), the temperature response followed immediately [26]. This proved that control of pressure is crucial for obtaining stable temperature baselines.
Catalyst plug-induced microchannel ethylene polymerization allows one to process about 10 runs per hour [26]. This is considerably more than achievable with conventional equipment (Parr reactors) processing only four to six runs per day.
11.2.8 Diastereoselective Alkylation
The Novartis Institute for BioMedical Research in Basel, Switzerland, and the University of Hull, UK, performed the diastereoselective alkylation of metal-stabilized enolates using a pressure-driven microreactor at −100 °C, whereby increased conversions and diastereoselectivity were observed compared to the batch mode [27].
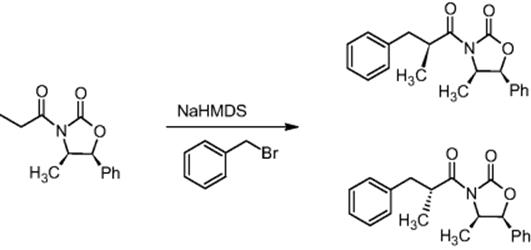
The diastereoselective synthesis of (2′S,4R,5S)-3-(2′-methyl-3′-phenylpropionyl)-4-methyl-5-phenyloxazolidin-2-one was demonstrated in a chip microreactor, whereby diastereoselectivities of >91 : 9 were obtained compared to 85 : 15 in the batch mode.
11.2.9 Multistep Synthesis of a Radiolabeled Imaging Probe
Several academic partners and Siemens Medical Solutions USA Inc. (Molecular Imaging) in Culver City, CA, USA, carried out the synthesis of an [18F]fluoride-radiolabeled molecular imaging probe, 2-deoxy-2-[18F]fluoro-d-glucose, in an integrated microfluidic device (see Figure 11.1) [21]. Five sequential processes were carried out: [18F]fluoride concentration, water evaporation, radiofluorination, solvent exchange, and hydrolytic deprotection. The half-life of [18F]fluorine (t1/2 = 110 min) makes rapid synthesis of doses essential. This is one of the first examples of an automated multistep synthesis in microflow fashion.
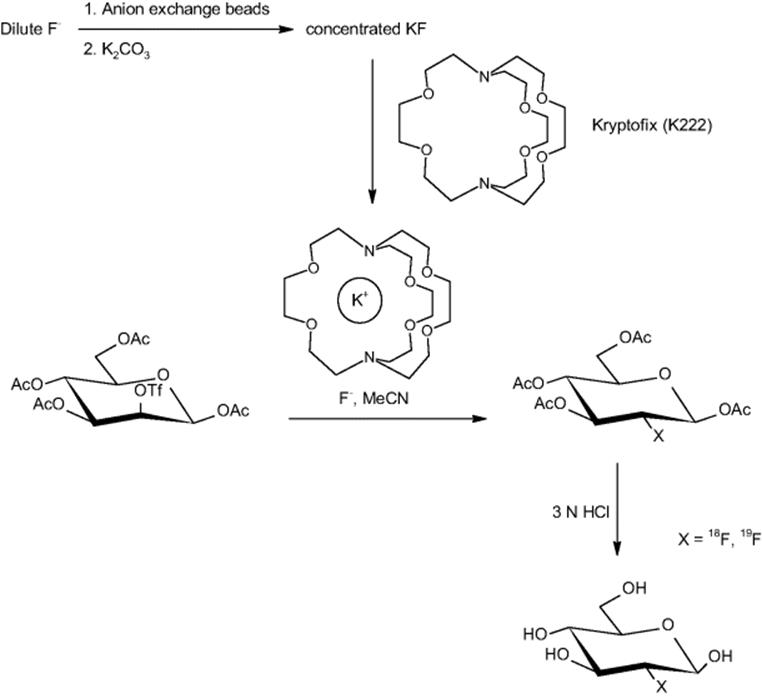
Figure 11.1 (a) Schematic of how to translate the chemical reaction steps of the synthesis of 2-deoxy-2-fluoro-d-glucose into a microfluidic design with microchannels and valves for consecutive fluid transport and manipulation. (b) Central area of the microfluidic circuit with channels filled with food dyes for visualization; same colors as in (a), plus green for fluidic microchannels. Inset: Photograph of the device. Source: Courtesy of AAAS [28].
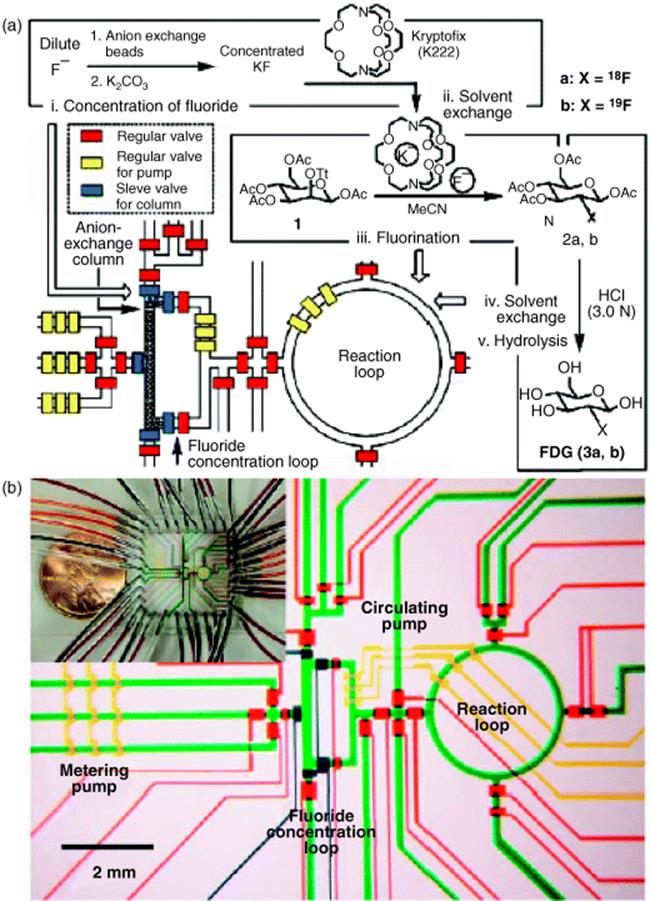
Compared to conventional automated synthesis, the radiochemical yield and purity of the compound obtained by microreactor processing were higher and also synthesis time was shorter [28]. Multiple doses of 2-deoxy-2-[18F]fluoro-d-glucose for positron emission tomography imaging studies in mice were prepared. Today, 2-deoxy-2-[18F]fluoro-d-glucose is routinely produced in about 50 min with the use of expensive commercial synthesizers and the radiolabeled compound for ~10–100 doses is produced in a single run.