March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 14. Substitution Reactions: Radical
This chapter discusses many types of radical reactions, including reactions in which radicals may be intermediates. Radicals are increasingly important in organic synthesis.1 The formation, fate, and properties of radicals were introduced in Section 5.C. Additional information concerning radicals may be found in Section 7.A, in the discussion of photochemical processes.
For the most part, this chapter discusses radical substitution reactions. Free radical additions to unsaturated compounds and rearrangements are discussed in Chapters 15 and 18, respectively. Fragmentation reactions are covered, in part, in Chapter 17. In addition, many of the oxidation–reduction reactions considered in Chapter 19 involve free radical mechanisms. Several important types of free radical reactions do not usually lead to reasonable yields of pure products and are not generally treated in this book.
14.A. Mechanisms
14.A.i. Radical Mechanisms in General2
A free radical process (or just a radical process) consists of at least two steps. Any radical reaction first step involve the formation of free radicals, usually by homolytic cleavage of a bond; that is, a cleavage in which each fragment retains one electron:
![]()
This is called an initiation step. It may happen spontaneously or may be induced by heat3 or light (see the discussion in Sec. 5.C.ii), depending on the type of bond.4 Peroxides, including hydrogen peroxide, dialkyl, diacyl, alkyl acyl peroxides, and peroxyacids are the most common source of free radicals. However, other organic compounds with low-energy bonds (e.g., azo compounds) are also used. Chlorine, bromine, and various ketones (see Chapter 7) are most commonly cleaved by light. Radicals can also be formed by a one-electron transfer (loss or gain) (e.g., A+ + e- → A•). One-electron transfers usually involve inorganic ions or electrochemical processes.5
Dialkyl peroxides (ROOR) or alkyl hydroperoxides (ROOH) decompose to hydroxy radicals (HO•) or alkoxy radicals (RO•) when heated.6 Cumene hydroperoxide (PhCMe2OOH), bi-tert-butylperoxide (Me3COOCMe3),7 and benzoyl peroxide [(PhCO)O2] undergo homolytic cleavage at temperatures compatible with many organic reactions. They are also reasonably soluble in organic solvents.8 In general, when a peroxide decomposes, the oxygen radical remains in a “cage” for ~ 10−11 s before diffusing away. The radical can
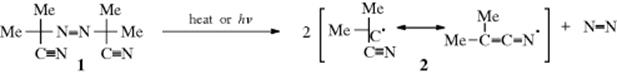
recombine (dimerize), or react with other molecules. Azo compounds, characterized by a –N=N– bond, are free radical precursors that liberate nitrogen gas (N≡N) upon decomposition. Azo bisisobutyronitrile (AIBN, 1) is a well-known example, which decomposes to give nitrogen gas and the cyano stabilized radical (2).9 Homolytic dissociation of symmetrical diazo compounds may be stepwise.10 A derivative has been developed that decomposes at room temperature: 2,2′-azobis(2,4-dimethyl-4-methoxyvaleronitrile), 3.11 Water soluble azo compounds are known, and can be used as radical initiators.12
Alkyl hypochlorites (R–O–Cl) generate chlorine radicals (Cl•) and alkoxy radicals (RO•) when heated.13 Heating N-alkoxydithiocarbamates is another useful source of alkoxy radicals (RO•).14 Alkoxy radicals, particularly those derived from cyclic compounds, may undergo β-scission reactions to give carbonyl derivatives.15
It has been known for many years that boron compounds participate in radical reactions.16 Trialkylboranes (R3B; see Reactions 15-27 and 12-27), such as triethylborane (Et3B), can be used to initiate radical reactions. Indeed, Et3B is widely used.17 In radical reactions, Et3B functions as both a radical initiator and also as a chain-propagation agent.18 Reactions are usually run in open vessels exposed to oxygen or under an oxygen atmosphere. It is known that O2is involved in the initiation step, as shown, in this case using an atom-transfer reaction with an alkyl iodide to give the alkyl radical, (R•). Trialkylborane/ water-mediated radical reactions are also known.19
![]()
In general, the order of reactivity is R3B > R2BOR > RB(OR)2 where R = alkyl.20 Boronic acids are less reactive, presumably due to π-bonding between B and O.17 However, B-alkycatecholboranes are very reactive, and highly useful for initiating radical reactions.21 Reaction conditions usually involve addition of catecholborane (4, abbreviated CatBH), and the B-alkyl derivative is presumably generated in situ by reaction with an alkene.22 Note that borole derivatives (the B analogue of pyrrole, 5) have been used to initiate radical reactions.23
Aldehydes can be a source of acyl radicals (•C=O) via reaction with transition metal salts (e.g., Mn(III) acetate or Fe(II) compounds).24 α,β-Unsaturated acyl radicals are subject to an isomerization that generates α-ketenyl radicals.25 Another useful variation employs imidoyl radicals as synthons for unstable aryl radicals.26
An important step in radical reactions involves the destruction of free radicals. This usually happens by a process opposite to the first, namely, a combination of two like or unlike radicals to form a new bond:27
![]()
This type of step is called termination because the product of the reaction is a neutral compound and not a radical.28 Note that this reaction constitutes a radical coupling process. The termination step rarely follows initiation because most radicals are very reactive, and there are several radical processes that occur faster than the termination step. In the usual situation, in which the concentration of radicals is low, a radical is more likely to react with a molecule rather than another radical (i.e., the radical coupling reaction is usually slower). When a radical, which has an odd number of electrons, reacts with a molecule, which has an even number, the total number of electrons in the products must be odd. In other words, the product is another radical. When a radical
![]()
reacts with a π bond, for example, the product is free radical (6). This reaction is called a radical addition. Another reaction constitutes an atom-transfer reaction. The abstraction of an atom (e.g., hydrogen atom) from an alkyl fragment gives two particles: R–H and the new radical (R′•), as shown. This type of atom-transfer reaction is called a hydrogen-transfer reaction.
![]()
Once again, the product is a free radical. This type of step is called propagation, since the newly formed radical can now react with another molecule and produce another radical, and so on, until two radicals undergo coupling and terminate the sequence. The process of initiation, propagation, and then termination constitutes what is called a chain reaction,29 and there may be hundreds or thousands of propagation steps between an initiation and a termination. Two other types of propagation reactions do not involve a molecule at all. These are (1) cleavage of a radical into, necessarily, a radical and a molecule and (2) rearrangement of one radical to another (see Chapter 18). When radicals are highly reactive (e.g., alkyl radicals), chains are long, since reactions occur with many molecules; but with radicals of low reactivity (e.g., aryl radicals), the radical may be unable to react with anything until it meets another radical, so that chains are short. Alternatively, the reaction may be a nonchain process. In any particular chain process, there is usually a wide variety of propagation and termination steps so there may be many products. Such reaction are often difficult to treat kinetically.30
![]()
Is it possible to terminate a radical reaction under controlled conditions? The answer is yes, using an atom-transfer reaction. When a carbon radical (R•) is generated in the presence of tributyltin hydride (n-Bu3SnH), a hydrogen atom is transferred to the radical to give R–H and a new radical (n-Bu3Sn•). The tin radical usually undergoes rapid coupling to another tin radical to give n-Bu3Sn–Sn–n-Bu3, which effectively terminates the chain-radical process. The carbon radical is reduced (R• → R–H) as a result of the hydrogen-atom transfer, and the tin dimer can be removed from the reaction. Again, hydrogen-atom transfer31 is simply a variation of the radical reaction known as atom transfer. Silanes (e.g., triethylsilane, Et3SiH), have also been used as an effective radical reducing agent.32 The rate constants for the reaction of both tributytin hydride and (Me3Si)3Si–H with acyl radical have been measured and the silane quenches the radical faster than the tin hydride.33 Thermolysis of bis(tri-n-butylstannyl)benzopinacolate has also been used as a source of n-Bu3Sn•, used to mediate radical reactions.34
The following are some general characteristics of free radical reactions35:
1. Reactions are fairly similar whether they are occurring in the vapor or liquid phase, but solvation of free radicals in solution does cause some differences.36
2. They are largely unaffected by the presence of acids or bases or by changes in the polarity of solvents, except that nonpolar solvents may suppress competing ionic reactions.
3. They are initiated or accelerated by typical free radical sources (e.g., the peroxides or diazo compounds) noted above, or by light. In the latter case, the concept of quantum yield applies (Sec. 7.A.viii). Quantum yields can be quite high (e.g., 1000), if each quantum generates a long chain, or low, in the case of nonchain processes.
4. Their rates are decreased or the reactions are suppressed entirely by substances that scavenge free radicals (e.g., nitric oxide, molecular oxygen, or benzoquinone). These substances are called inhibitors.37 Note that there are C-centered radicals in thermal equilibrium with their dimers that show poor reactivity with molecular oxygen, but good reactivity with peroxyl radicals.38
14.A.ii. Free Radical Substitution Mechanisms39
In a free radical substitution reaction
Step 1 ![]()
there must first be a cleavage of the substrate RX so that R• radicals are produced. This can happen by a spontaneous cleavage,
Step 2 ![]()
or it can be caused by light or heat, or, more often, there is no actual cleavage, but R• is produced by an abstraction of another atom, X by the radical W•.
Step 3 ![]()
The radical W• is produced by adding a compound (e.g., peroxide) that spontaneously forms free radicals. Such a compound is called an initiator (see above). Once R• is formed, it can go to product in this system, by another atom abstraction, such as the reaction with A–B to form R–A and a new radical B• (atom transfer).
Step 4 ![]()
Another reaction is coupling with another radical to form the neutral product R–Y.
Step 5 ![]()
In a reaction with a moderately long chain, much more of the product will be produced by abstraction (4) than by coupling (5). Cleavage steps like (2) have been called SH1 (H for homolytic), and abstraction steps like (3) and (4) have been called SH2; reactions can be classified as SH1 or SH2 on the basis of whether RX is converted to R by (2) or (3).40 Most chain substitution mechanisms follow the pattern (3), (4), (3), (4)...Chains are long and reactions go well where both (3) and (4) are energetically favored (no worse that slightly endothermic, see Sec. 14.B.i and 14.C.i). The IUPAC designation of a chain reaction that follows the pattern (3),(4)...is ArDR + ARDr (R stands for radical).
With certain radicals the transition state in an abstraction reaction has some polar character. Consider the abstraction of hydrogen from the methyl group of toluene by a bromine atom. Since bromine is more electronegative than carbon, it is reasonable to assume that there is a separation of charge in the transition state, with a partial negative charge on the halogen and a partial positive charge on the carbon:
![]()
Evidence for the polar character of the transition state is that electron-withdrawing groups in the para position of toluene, which would destabilize a positive charge, decrease the rate of hydrogen abstraction by bromine while electron-donating groups increase it.41 However, substituents have a smaller effect here (ρ ≈ −1.4) than they do in reactions where a completely ionic intermediate is involved (e.g., the SN1 mechanism, Sec. 10.A.ii). Other evidence for polar transition states in radical abstraction reactions is mentioned in Section 14.B.i, category 4. For abstraction by radicals such as methyl or phenyl, polar effects are very small or completely absent. For example, rates of hydrogen-atom abstraction from ring-substituted toluenes by methyl radical were relatively unaffected by the presence of electron-donating or electron-withdrawing substituents.42 Those radicals (e.g., Br•) that have a tendency to abstract electron-rich hydrogen atoms are called electrophilic radicals.
When the reaction step R–X → R• takes place at a stereogenic carbon, racemization is almost always observed because free radicals do not retain configuration. Exceptions to this rule are found with cyclopropyl substrates, where both inversion43 and retention44 of configuration have been reported, and in the reactions mentioned in Section 14.A.iv. Enantioselective radical processes have been reviewed.45
14.A.iii. Mechanisms at an Aromatic Substrate46
When R in reaction (1) is aromatic, the simple abstraction mechanism just discussed may be operating, especially in gas-phase reactions. However, mechanisms of this type cannot account for all reactions of aromatic substrates. In processes such as Reactions 13-27, 14-17, and 14-18:
Step 6 ![]()
which occur in solution, the simple coupling of two rings by abstraction of an entire group (e.g., phenyl) to generate H• by the free radical mechanism shown here is very unlikely (see Sec. 14.B.i).
Step 7 ![]()
The products can be explained by a mechanism similar to that of electrophilic and nucleophilic aromatic substitution. In the first step, the radical attacks the ring in much the same way as would an electrophile or a
Step 8 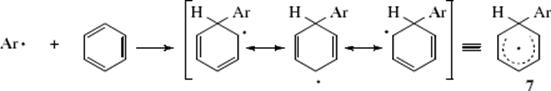
nucleophile to generate 7. The intermediate radical (7) is relatively stable because of the resonance. The reaction can terminate in three ways: by simple coupling to give 8, by disproportionation to give 9,
Step 9 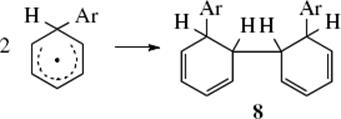
Step 10 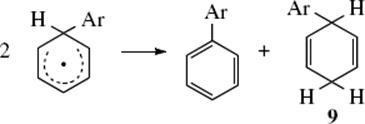
or, if a species (R′•) is present that abstracts hydrogen, by abstraction to give 10.47
Step 11 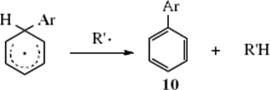
Coupling product 8 is a partially hydrogenated o-quaterphenyl (an o,o′-diphenylbiphenyl). Of course, the coupling need not be ortho–ortho, and other isomers can also be formed. Among the evidence (9) and (10) was isolation of compounds of types 8 and 9.48 However, under the reaction conditions dihydrobiphenyls like 9 are normally oxidized to the corresponding biphenyls. Other evidence for this mechanism is the detection of the intermediate 7 by CIDNP49 and the absence of isotope effects, expected if the rate-determining step were (7), which involves cleavage of the Ar–H bond. In the mechanism just given, the rate-determining step (8) does not involve loss of hydrogen. The reaction between aromatic rings and the HO• radical takes place by the same mechanism. Intramolecular hydrogen-transfer reactions of aryl radicals are known.50 A similar mechanism has been shown for substitution at some vinylic51 and acetylenic substrates, giving the substituted alkene (11).52 The kinetics of radical heterolysis reactions that form alkene radical cations has been studied.53
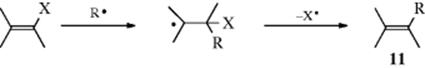
This is reminiscent of the nucleophilic tetrahedral mechanism at a vinylic carbon (Sec. 10.F).
There are many transition metal mediated coupling reactions of aromatic substrates that probably proceed by radical coupling. It is likely that many of these reactions do not proceed by free radicals, but rather by metal-mediated radicals or by ligand transfer at the metal. Reactions in these categories were presented in Chapter 13 for convenient correlation with other displacement reactions of aryl halides, aryl diazonium salts, and so on.
14.A.iv. Neighboring-Group Assistance in Free Radical Reactions
In a few cases, it has been shown that cleavage steps (2) and abstraction steps (3) were accelerated by the presence of neighboring groups. Photolytic halogenation (Reaction 14-1) is a process that normally leads to mixtures of many products. However, bromination of carbon chains containing a bromine atom occurs with high regioselectivity. Bromination of alkyl bromides gave 84–94% substitution at the carbon adjacent to the bromine already in the molecule.54 This result may seem surprising because, as will be seen (Sec. 14.B.i, category 3), positions close to a polar group (e.g., bromine) should actually be deactivated by the electron-withdrawing field effect of the bromine. However, the unusual regioselectivity is explained by a mechanism in which abstraction (3) is assisted by a neighboring bromine atom, as in 12.55 In the normal mechanism, Br• abstracts a hydrogen atom from RH, leaving R•. When a bromine is present in the proper position, it assists this process, giving a cyclic intermediate (a bridged free radical, 13).56 In the final step (very similar to R• + Br2 → RBr + Br•) the ring is broken. If this mechanism is correct, the configuration at the substituted carbon (marked ∗) should be retained. This has been shown to be the case: Optically active 1-bromo-2-methylbutane gave 1,2-dibromo-2-methylbutane with retention of configuration.55 Furthermore, when this reaction was carried out in
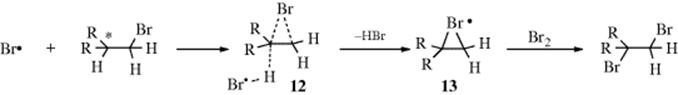
the presence of DBr, the “recovered” 1-bromo-2-methylbutane was found to be deuterated in the 2 position, and its configuration was retained.57 This is just what would be predicted if some of the 11 present abstracted D from DBR. There is evidence that Cl can form bridged radicals,58 though ESR spectra show that the bridging is not necessarily symmetrical.59 Still more evidence for bridging by Br has been found in isotope effects and other studies.60However, evidence from CIDNP shows that the methylene protons of the β-bromoethyl radical are not equivalent, at least while the radical is present in the radical pair [PhCOO• •CH2CH2Br] within a solvent cage.61 This evidence indicates that under these conditions BrCH2CH2• is not a symmetrically bridged radical, but it could be unsymmetrically bridged. A bridged intermediate has also been invoked, when a bromo group is in the proper position, in the Hunsdiecker reaction62 (14-30), and in abstraction of iodine atoms by the phenyl radical.63 Participation by other neighboring groups (e.g. SR, SiR3, SnR3) has also been reported.64
Note that a traditional explanation for the selectivity of bromine relative to chlorine is that abstraction of hydrogen by a bromine radical has a later transition state relative to abstraction of hydrogen by a chlorine radical. In a later transition state, the relative stability of the radical plays a greater role than the strength of the C–H bond. Since a tertiary radical is more stable than a secondary, which is more stable than a primary radical, bromination favors formation of the secondary radical over the primary, and a tertiary over secondary. This analysis is supported by the relative rates of hydrogen-atom abstraction by Br• when compared to Cl•, where there is a large difference between 1°, 2°, and 3° for Br• and a small difference for Cl•. This effect is discussed in more detail in Section 14.B.i, categories 1–5.