March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 15. Addition to Carbon–Carbon Multiple Bonds
15.C. Reactions
15.C.i. Isomerization of Double and Triple Bonds
15-1 Isomerization
![]()
Without a transition metal catalyst, there is usually a rather high-energy barrier for the excited state required for (E/Z) isomerization.146 The transition metal catalyzed isomerization of an alkene from (E) to (Z) or (Z) to (E) is a well-studied reaction.147 Among the metals used, Pt is widely used, and rather selective.148 The Pd catalyzed isomerization of (Z)-alkenes to (E)-alkenes required the presence of Bu3SnH.149 However, a 1:1 mixture of cis/trans-styrene derivatives was isomerized to a 90% yield of the trans-styrene derivatives reported using a Pd catalyst.150 Isomerization of cyclic alkenes is difficult for rings of seven members and less, but cis/trans isomerization of cyclooctene is induced photochemically.151 Radical-induced (E/Z) isomerization is known.152 Isomerization of the C=C units in dienes is also induced photochemically.153
In a different type of reaction, isomerization of alkynes to 1,3-dienes is possible using Rh or Pd catalysts.154
There are several reagents that lead to isomerization of a double bond to form a new alkene. In general, there is an energetic preference of an α,β- versus β,γ-double bond.155 Allylic arenes (Ar–CH2CH=CH2) have been converted to the corresponding (Z)-1-propenyl arene (Ar–CH=CHMe using an Ru catalyst156 or a polymer-supported Ir catalyst.157 In the presence of a Rh catalyst, certain allylic amines are converted to an enamine with high selectivity for the (Z) isomer.158 Double-bond migration has been observed in sulfide photoirradiation, induced by singlet oxygen.159 Many of these reactions were discussed in Reaction 12-2.
For conjugated carbonyl compounds that have a hydrogen atom at the γ-position (C-4), it is possible to move a double bond out of conjugation. Photolysis of conjugated esters, at −40 °C in the presence of N,N-dimethylaminoethanol, gave the nonconjugated ester.160 Heating an N-allylic amide (N–C–C=C) with Fe(CO)5, neat, gave the enamide (N–C=C–C).161 Conjugated aldehydes have been isomerized using thiourea in DMF.162
Double bonds of atoms other than carbon are subject to isomerization. Azobenzenes (Ar–N=N–Ar) exist as (E) and (Z) isomers, and photochemical isomerization is possible.163
15.C.ii. Reactions in Which Hydrogen Adds to One Side
A. Halogen on the Other Side
15-2 Addition of Hydrogen Halides
Hydro-halo-addition
![]()
Any of the four hydrogen halides can be added to double bonds.164 Alkenes react as Br![]() nsted–Lowry bases with HI, HBr, and HF165 at room temperature, but reaction with HCl is more difficult and usually requires heat.24Hydrogen Chloride adds easily in the presence of silica gel. However,166 HF is difficult to handle, but a convenient method for the addition of HF involves the use of a polyhydrogen fluoride–pyridine solution in THF.167
nsted–Lowry bases with HI, HBr, and HF165 at room temperature, but reaction with HCl is more difficult and usually requires heat.24Hydrogen Chloride adds easily in the presence of silica gel. However,166 HF is difficult to handle, but a convenient method for the addition of HF involves the use of a polyhydrogen fluoride–pyridine solution in THF.167
The addition of hydrogen halides to simple alkenes, in the absence of peroxides, takes place by an electrophilic mechanism, and the orientation is in accord with Markovnikov's rule.168 In other words, the π bond of the alkenes donates two electrons to the acidic proton of H–X. The addition follows second-order kinetics.169 When peroxides are added, the addition of HBr occurs by a free-radical mechanism and the orientation is anti-Markovnikov (Sec. 15.B.i).170 It must be emphasized that this is true only for HBr. Free radical addition of HF and HI has never been observed, even in the presence of peroxides; free radical addition of HCl has been observed only rarely. In the rare cases where free radical addition of HCl was noted, the orientation was still Markovnikov, presumably because the more stable product was formed.171 Free radical addition of HF, HI, and HCl is energetically unfavorable (see the discussions in Sec. 14.B.i and 14.C.i). It is known that under some conditions anti-Markovnikov addition of HBr takes place even when peroxides have not been added. This happens because the substrate alkenes absorb oxygen from the air, forming small amounts of peroxides (Reaction 14-7). Markovnikov addition can be ensured by rigorous purification of the substrate, but in practice this is not easy to achieve. It is more common to add inhibitors (e.g., phenols or quinones), which suppress the free radical pathway. The presence of free radical precursors (e.g., peroxides) does not inhibit the ionic mechanism, but does inhibit the more rapid radical reaction, which is a chain process. In most cases, it is possible to control the mechanism (and hence the orientation) by adding peroxides to achieve complete free radical addition, or inhibitors to achieve complete electrophilic addition, although there are some cases where the ionic mechanism is fast enough to compete with the free radical mechanism and complete control cannot be attained. Markovnikov addition of HBr, HCl, and HI has also been accomplished, in high yields, by the use of phase transfer catalysis.172 For alternative methods of adding HBr (or HI) with anti-Markovnikov orientation, see Reaction 12-31.
Alkynes also react as bases with acids (e.g., HX). It is possible to add 1173 or 2 equiv of any of the four hydrogen halides to triple bonds. Markovnikov's rule ensures that gem-dihalides and not vic-dihalides are the products of the addition of 2 equiv.
![]()
Chlorotrimethylsilane can be added to alkenes to give alkyl chlorides. 1-Hexene reacts with Me3SiCl in water, for example, to give 2-chlorohexane.174 Treatment of an alkene with KHF2 and SiF4 leads to the alkyl fluoride,175 and bromotrimethylsilane adds to alkynes to give the vinyl bromide.176
Br![]() nsted–Lowry acids (e.g., HX) are electrophilic reagents, and many polyhalo or polycyano alkenes do not react with them in the absence of free radical conditions. Vinylcyclopropanes, however, react with opening of the cyclopropane ring to give a homoallylic chloride.177 When such reactions do occur, however, they take place by a nucleophilic addition mechanism, that is, initial attack is by X−. This type of mechanism also occurs with Michael-type substrates (C=C–Z),178 where the orientation is always such that the halogen goes to the carbon that does not bear the Z, so the product is of the form X–C–CH–Z, even in the presence of free radical initiators.
nsted–Lowry acids (e.g., HX) are electrophilic reagents, and many polyhalo or polycyano alkenes do not react with them in the absence of free radical conditions. Vinylcyclopropanes, however, react with opening of the cyclopropane ring to give a homoallylic chloride.177 When such reactions do occur, however, they take place by a nucleophilic addition mechanism, that is, initial attack is by X−. This type of mechanism also occurs with Michael-type substrates (C=C–Z),178 where the orientation is always such that the halogen goes to the carbon that does not bear the Z, so the product is of the form X–C–CH–Z, even in the presence of free radical initiators.
The reaction has been carried out with conjugated dienes, where both 1,2- and 1,4-addition are possible. Hydrogen iodide adds 1,4 to conjugated dienes in the gas phase by a pericyclic mechanism:179
![]()
In a related reaction, HX can be added to ketenes180 to give acyl halides:
![]()
OS I, 166; II, 137, 336; III, 576; IV, 238, 543; VI, 273; VII, 59; 80, 129.
B. Oxygen on the Other Side
15-3 Hydration of Double bonds
Hydro-hydroxy-addition
![]()
Double bonds can be hydrated by treatment with water and an acid catalyst. Sulfuric acid is a common catalyst, but other acids that have relatively non-nucleophilic counterions, such as nitric, perchloric, or more commonly sulfonic acids (p-toluenesulfonic acid, methanesulfonic acid, etc.) can also be used. The mechanism is electrophilic and begins with attack of the π bond on an acidic proton (Sec. 15.A.i). The resulting carbocation is then attacked by negative species (e.g., HSO4−, or a similar counterion in the case of other acids), to give the initial product (32), which can be isolated in some cases. However, such compounds are rather unstable, and
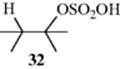
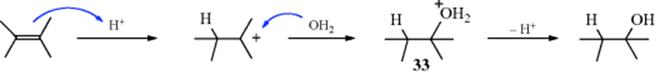
under the conditions of the reaction are usually hydrolyzed to the alcohol (Reaction 10-4). Under some reaction conditions, other nucleophiles are present in the reaction, either from the solvent or from added compounds. In an aqueous medium, water is a competitive nucleophile, and attack by water forms oxonium ion 33. Products, such as 32, are not involved when other nucleophiles react with the carbocation, and the mechanism is exactly (by the principle of microscopic reversibility) the reverse of El elimination of alcohols (Reaction 17-1).181 The initial carbocation occasionally rearranges to a more stable one. For example, hydration of CH2=CHCH(CH3)2 gives CH3CH2COH(CH3)2. Hydration of simple alkenes leads to alcohols predicted by Markovnikov's rule.
Oxymercuration182 (addition of oxygen and mercury) of alkenes followed by in situ treatment with sodium borohydride183 (Reaction 12-24), gives an alcohol (see example) under mild conditions, in high yields, and without rearrangement products. For example, treatment of 2-methyl-1-butene with mercuric acetate,184 followed by NaBH4, gave 2-methyl-2-butanol. Oxymercuration of alkenes has been reported in water, using cyclodextrins as phase-transfer catalysts.185
![]()
This method, which is applicable to mono-, di-, tri-, and tetraalkyl, as well as phenyl-substituted alkenes, gives almost complete Markovnikov addition. Hydroxy, methoxy, acetoxy, halo, and other groups may be present in the substrate, and generally do not cause difficulties.186 When two double bonds are present in the same molecule, changing the carboxylic acid ligand of the mercuric salt allows oxymercuration of the less-substituted one without affecting the other, with ultrasound.187 A related reaction treats an alkene with zinc borohydride on silica gel to give a 35:65 mixture of secondary/primary alcohols.188
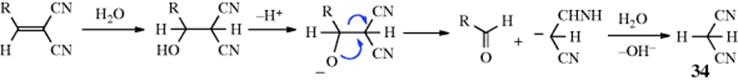
With substrates of the type C=C–Z (Z is as defined in Sec.15.A.ii) the product is almost always HO–C–CH–Z and the mechanism is usually nucleophilic,189 although electrophilic addition gives the same product190 since a cation CH–C–Z would be destabilized by the positive charges (full or partial) on two adjacent atoms. However, the α-hydroxy compound HC–CH(OH)Z, was obtained by treatment of the substrate with O2, PhSiH3, and a manganese-complex catalyst.191 Addition of water to RCH=CZZ′ substrates may result in cleavage of the adduct to give an aldehyde and CH2ZZ′ (34).192 The cleavage step is an example of Reaction 12-41.
For another method of anti-Markovnikov hydration, see hydroboration (Reaction 15-16).
Indirect hydration, with anti-Markovnikov orientation, was achieved by treatment of the alkene with a 1:1 mixture of PhCH2NEt3+ BH4− and Me3SiCl, followed by addition of an aqueous solution of K2CO3.193 Reaction of alkenes with Ti(BH4)3, and then aq K2CO3 also leads to the anti-Markovnikov alcohol.194 Alkenes react with PhO2BH and a Nb catalyst, followed by oxidation with NaOO−, to give the alcohol,195 and Cp2TiCl4 can also be used.196Conjugated alkenes also react with PhSiH2 and oxygen, with a Mn catalyst, to give an α-hydroxy ketone.197 Alkenes react with molecular oxygen in the presence of a Co porphyrin catalyst, and reduction with P(OMe)3 leads to the secondary alcohol.198 This procedure has also been used to hydrate conjugated dienes,199 although conjugated dienes are seldom hydrated.
The addition of water to enol ethers causes hydrolysis to aldehydes or ketones (Reaction 10-6). Ketenes add water to give carboxylic acids (R2C=C=O → R2CO2H) in a reaction catalyzed by acids200:
OS IV, 555, 560; VI, 766. Also see, OS V, 818.
15-4 Hydration of Triple Bonds
Dihydro-oxo-biaddition
![]()
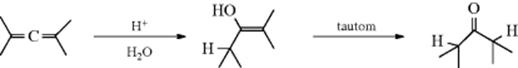
The hydration of triple bonds is generally carried out with mercuric ion salts (often the sulfate or acetate or even mercuric oxide) as catalysts.201 In contrast to oxymercuration of alkenes, the organomercury intermediate from this reaction is unstable and loss of mercury in situ leads to an enol product. The enol tautomerizes to the ketone (see Sec. 2.N.i), so the isolated product is the ketone from either an internal or terminal alkyne (the OH unit will always be on the more substituted carbon via the more stable secondary vinyl carbocation). Only acetylene gives an aldehyde. With alkynes of the form RC![]() CR′ both possible ketone products are usually obtained. The reaction can be conveniently carried out with a catalyst prepared by impregnating mercuric oxide onto Nafion-H (a superacidic perfluorinated resinsulfonic acid; see Sec. 5.A.ii).202 Gold,203 In,204 and Ru205 catalysts have been used to convert alkynes to the ketone. Gold(I) catalysts have also been used in the hydration of allenes.206 Internal alkynes were treated with 2-aminophenol in refluxing dioxane using a Pd catalyst to produce the corresponding ketone.207 Lactones have been prepared from trimethylsilyl alkenes containing a hydroxyl unit elsewhere in the molecule, when reacted with molecular oxygen, CuCl2, and a Pd catalyst.208 When a carboxylic acid that contains a double bond in the chain is treated with a strong acid, the intramolecular hydration reaction gives a γ- and/or a δ-lactone, regardless of the original position of the double bond in the chain, since strong acids catalyze double-bond shifts (Reaction 15-1; and see 12-2).209 The double bond always migrates to a position favorable for the reaction, whether this has to be toward or away from the carboxyl group. The use of a chiral Cinchonidine alkaloid additive leads to lactone formation with modest enantioselectivity.210
CR′ both possible ketone products are usually obtained. The reaction can be conveniently carried out with a catalyst prepared by impregnating mercuric oxide onto Nafion-H (a superacidic perfluorinated resinsulfonic acid; see Sec. 5.A.ii).202 Gold,203 In,204 and Ru205 catalysts have been used to convert alkynes to the ketone. Gold(I) catalysts have also been used in the hydration of allenes.206 Internal alkynes were treated with 2-aminophenol in refluxing dioxane using a Pd catalyst to produce the corresponding ketone.207 Lactones have been prepared from trimethylsilyl alkenes containing a hydroxyl unit elsewhere in the molecule, when reacted with molecular oxygen, CuCl2, and a Pd catalyst.208 When a carboxylic acid that contains a double bond in the chain is treated with a strong acid, the intramolecular hydration reaction gives a γ- and/or a δ-lactone, regardless of the original position of the double bond in the chain, since strong acids catalyze double-bond shifts (Reaction 15-1; and see 12-2).209 The double bond always migrates to a position favorable for the reaction, whether this has to be toward or away from the carboxyl group. The use of a chiral Cinchonidine alkaloid additive leads to lactone formation with modest enantioselectivity.210
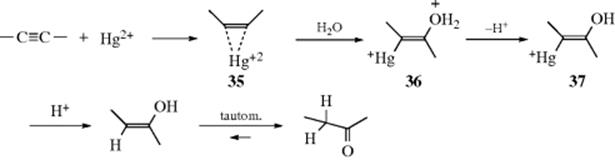
The first step of the mercury-mediated mechanism is formation of a complex (35). It is known that ions like Hg2+ form complexes with alkynes (Sec. 3.C.i). Water then attacks in an SN2 type process to give the intermediate 36, which loses a proton to give 37. Hydrolysis of 37 (an example of Reaction 12-34) gives the enol, which tautomerizes to the product. A spectrum of the enol was detected by flash photolysis when phenylacetylene was hydrated photolytically.211 Note that another possibility for 35 is a mercury-stabilized carbocation rather than a formal three-membered ring complex. In such a carbocation, the carbocation is stabilized by back-donation from the metal, and nucleophilic attack is more like a SN1 type process.
Metal-free reactions are known, often using strong acids. Phenyl acetylene was converted to acetophenone [e.g., in water at 100 °C with a catalytic amount of Tf2NH (trifluoromethanesulfonimide)], which is a very powerful acid.212 Simple alkynes can also be converted to ketones by heating with formic acid, without a catalyst.213 Metal-free hydration of terminal alkynes occurs by reacting water, heated with microwave irradiation, to give the corresponding methyl ketone.214 1-Selenoalkynes (e.g., PhSe-C![]() C-Ph) react with tosic acid in dichloromethane to give a seleno ester [PhSeC(=O)SH2Ph] after treatment with water.215 Allenes can be hydrolyzed to ketones using an acid catalyst.216
C-Ph) react with tosic acid in dichloromethane to give a seleno ester [PhSeC(=O)SH2Ph] after treatment with water.215 Allenes can be hydrolyzed to ketones using an acid catalyst.216
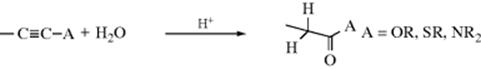
Carboxylic esters, thiol esters, and amides can be made, respectively, by acid-catalyzed hydration of acetylenic ethers, thioethers,217 and ynamines, without a mercuric catalyst.218
This is ordinary electrophilic addition, with rate-determining protonation as the first step.219 Certain other alkynes have also been hydrated to ketones with strong acids in the absence of mercuric salts.220
Catalysts have been developed for the anti-Markovnikov hydration of alkynes.221 When 1-octyne was heated with water, isopropyl alcohol and a Ru catalyst, for example, the product was octanal.222 The presence of certain functionality can influence the regioselectivity of hydration.
A Ni catalyzed reaction has been reported between alkynes and allyl phenyl sulfides to give thioallylation.223
OS III, 22; IV, 13; V, 1024.
15-5 Addition of Alcohols and Phenols
Hydro-alkoxy-addition
![]()
Just as water adds to an alkene via hydration to form an alcohol, alcohols can also add to form an ether. Addition of alcohols and phenols to double bonds is catalyzed by acids or bases. When the reactions are acid catalyzed, the mechanism is electrophilic, where H+ of the acid catalyst, is attacked by the π bond. The more stable carbocation is formed and subsequently attacked by a molecule of alcohol to give an oxonium ion (38).
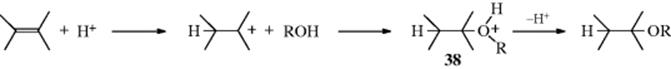
The addition, therefore, follows Markovnikov's rule. Primary alcohols give better results than secondary, and tertiary alcohols are relatively inactive. This method is convenient for the preparation of tertiary ethers by the use of a suitable alkene (e.g., Me2C=CH2). Addition of alcohols to allylic systems can proceed with rearrangement, and the use of chiral additive can lead to asymmetric induction.224 The uncatalyzed addition of alcohols occurs in supercritical alcohols.225
Metal-catalyzed addition to alkenes is a useful variation. The Pd catalyzed addition of alcohols to aryl alkenes gives the ether.226 The Au(III)–CuCl2 catalyzed reaction of alcohols and alkenes gives the ether.227 Gold(I) catalyzed intermolecular addition of phenols leads to aryl ethers.228
Alcohols add intramolecularly to alkenes to generate cyclic ethers, and the product often bears a hydroxyl unit,229 but not always.230 Cyclization is facilitated by Re,231 Ti,232 or Pt compounds,233 forming functionalized tetrahydrofurans or tetrahydropyans. Intramolecular addition of alcohols to alkenes can be promoted by a Pd catalyst, but migration of the double bond in the final product is sometimes a problem.234 Furan derivatives are available from alkene–ketones using CuCl2 and a Pd235 Cr,236 Ag(I),237 or lanthanide catalyst.238 A gold catalyst was used with conjugated ketones bearing an alkyne substituent to give fused-ring furans.239 Note that the reaction of an alkene–alcohol and NIS with a chiral Ti catalyst leads to a THF with a pendant iodoalkyl group, with modest enantioselectivity.240
Alcohols add to alkynes under certain conditions to give vinyl ethers. In an excess of alcohol, and in the presence of a Pt241 or a Au catalyst,242 internal alkynes are converted to ketals. The alcohol to alkyne addition reaction is quite useful for the preparation of heterocycles. Dihydrofurans,243 furans,244 benzofurans,245 and pyran derivatives246 have been prepared using this approach. Tetrahydrofurans bearing an exocyclic double bond (vinylidene tetrahydrofurans) were prepared from alkynyl alcohols and a silver carbonate catalyst.247
Allenes that react with alcohols and allenic alcohols have been converted to THF derivatives bearing a vinyl group at the α-position, using diphenyliodonium salts.248 In the presence of allylic bromide and a Pd catalyst, allenic alcohols lead to allylically substituted dihydrofurans.249 The intramolecular Au(I) catalyzed reaction of alcohols and allenes has been reported.250 Intramolecular addition of alcohols to allenes leads to cyclic vinyl ethers.251
Functionalized ethers can be formed in the presence of other reagents. In methanol with a R–Se–Br reagent, alkenes are converted to selenoalkyl ethers (MeO–C–C–SeR).252
Base-catalyzed reactions are known. For those substrates more susceptible to nucleophilic attack, for example, polyhalo alkenes and alkenes of the type C=C–Z, it is better to carry out the reaction in basic solution, where the attacking species is RO−.253 The reactions with C=C–Z are of the Michael type, and OR goes to the side away from the Z.254
Since triple bonds are more susceptible to nucleophilic attack than double bonds, it might be expected that bases would catalyze addition to triple bonds particularly well. This is the case, and enol ethers and acetals can be produced by this reaction.255 Because enol ethers are more susceptible than triple bonds to electrophilic attack, the addition of alcohols to enol ethers can also be catalyzed by acids.256 One utilization of this reaction
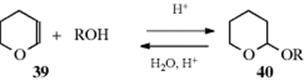
involves the compound dihydropyran (39), which is often used to protect the OH groups of primary and secondary alcohols257 and phenols.258 When the desired reactions are completed, 40 is easily cleaved by treatment with dilute acids (Reaction 10-6). In base-catalyzed addition to triple bonds, the rate falls in going from a primary to a tertiary alcohol, and phenols require more severe conditions.
Photochemical addition of alcohols to certain double-bond compounds (cyclohexenes, cycloheptenes) is possible259 in the presence of a photosensitizer (e.g., benzene). The mechanism is electrophilic and Markovnikov orientation is found. The alkenes react in their first excited triplet states.260
The oxymercuration–demercuration procedure mentioned in Reaction 15-3 can be adapted to the preparation of ethers in what is known as alkoxymercuration–demercuration (Markovnikov orientation), if the reaction is carried out in an alcohol (ROH) solvent.261 For example, oxymercuration of 2-methyl-1-butene in ethanol gives EtMe2COEt.262 Primary alcohols give good yields when mercuric acetate is used, but for secondary and tertiary alcohols, it is necessary to use mercuric trifluoroacetate.263 However, even this reagent fails where the product would be a ditertiary ether. It is possible to combine the alcohol reactant with another reagent. The reaction of an alkene with iodine and allyl alcohol, in the presence of HgO, gave the vic-iodo ether.264 Alkene-alcohols react with mercuric trifluoroacetate and the aq KBr (with LiBH4/BEt3) to give a THF derivative bearing an iodoalkyl substituent [–O–C–CH(I)R].265 Alkynes generally react under the same conditions to give acetals. If the oxymercuration is carried out in the presence of a hydroperoxide instead of an alcohol, the product (after demercuration with NaBH4) is an alkyl peroxide (peroxy-mercuration).266 This can be done intramolecularly.267
Both alcohols and phenols add to ketenes to give carboxylic esters [R2C=C=O + ROH → R2CHCO2R].268 This has been done intramolecularly (with the ketene end of the molecule generated and used in situ) to form medium- and large-ring lactones.269 In the presence of a strong acid, ketene reacts with aldehydes or ketones (in their enol forms) to give enol acetates. 1,4-Asymmetric induction is possible when chiral alcohols add to ketenes.270
OS III, 371, 774, 813; IV, 184, 558; VI, 916; VII, 66, 160, 304, 334, 381; VIII, 204, 254; IX, 472.
15-6 Addition of Carboxylic Acids to Form Esters
Hydro-acyloxy-addition
![]()
Carboxylic esters are produced by the addition of carboxylic acids to alkenes, a reaction that is usually acid catalyzed (by Br![]() nsted–Lowry or Lewis acids271) and similar in mechanism to Reaction 15-5. Since Markovnikov's ruleis followed, hard-to-get esters of tertiary alcohols can be prepared from alkenes of the form R2C=CHR.272 Carboxylic esters have also been prepared by the acyloxymercuration–demercuration of alkenes (similar to the procedures mentioned in Reactions 15-3 and 15-4).273 Addition of carboxylic acids to alkenes to form esters or lactones is catalyzed by Pd compounds.274 Thallium acetate also promotes this cyclization reaction.275 Diene carboxylic acids have been cyclized using acetic acid and a Pd catalyst to form lactones that have an allylic acetate moiety elsewhere in the molecule.276
nsted–Lowry or Lewis acids271) and similar in mechanism to Reaction 15-5. Since Markovnikov's ruleis followed, hard-to-get esters of tertiary alcohols can be prepared from alkenes of the form R2C=CHR.272 Carboxylic esters have also been prepared by the acyloxymercuration–demercuration of alkenes (similar to the procedures mentioned in Reactions 15-3 and 15-4).273 Addition of carboxylic acids to alkenes to form esters or lactones is catalyzed by Pd compounds.274 Thallium acetate also promotes this cyclization reaction.275 Diene carboxylic acids have been cyclized using acetic acid and a Pd catalyst to form lactones that have an allylic acetate moiety elsewhere in the molecule.276
Triple bonds can give enol esters277 or acylals when treated with carboxylic acids. Mercuric salts are usually catalysts,278 and vinylmercury compounds (RO2C–C=C–HgX) are intermediates,279 but Ru complexes have also been used.280 Terminal alkynes (RC![]() CH) react with CO2, a secondary amine (R′2NH), and a Ru complex catalyst, to give enol carbamates [RCH=CHOC(=O)NR].281 This reaction has also been performed intramolecularly, to produce unsaturated lactones.282 Cyclic unsaturated lactones (internal vinyl esters) have been generated from alkyne-carboxylic acids using a Pd283 or a Ru catalyst.284 Carboxylic esters can also be obtained by the addition to alkenes of diacyl peroxides.285 These reactions are catalyzed by Cu and are free radical processes.
CH) react with CO2, a secondary amine (R′2NH), and a Ru complex catalyst, to give enol carbamates [RCH=CHOC(=O)NR].281 This reaction has also been performed intramolecularly, to produce unsaturated lactones.282 Cyclic unsaturated lactones (internal vinyl esters) have been generated from alkyne-carboxylic acids using a Pd283 or a Ru catalyst.284 Carboxylic esters can also be obtained by the addition to alkenes of diacyl peroxides.285 These reactions are catalyzed by Cu and are free radical processes.
Allene carboxylic acids have been cyclized to butenolides with copper(II) chloride.286 Allene esters were converted to butenolides by treatment with acetic acid and LiBr.287 Cyclic carbonates can be prepared from allene alcohols using carbon dioxide and a Pd catalyst.288 Carboxylic acids react with ketenes to give anhydrides289 and acetic anhydride is prepared industrially in this manner [CH2=C=O + MeCO2H → (MeC=O)2O].
Sulfonic acids add to alkenes and alkynes. The reaction of an alkyne with p-toluenesulfonic acid and treatment with silica gives the vinyl sulfonate (C=C–OSO2Tol).290 Cyclic sulfonates can be generated by the reaction of an allylic sulfonate salt (C=C–C–OSO3−) with silver nitrate in acetonitrile containing an excess of bromine and a catalytic amount of water.291 Sultones are formed when alkenes react with PhIO and 2 equiv of Me2SiSO3Cl.292
OS III, 853; IV, 261, 417, 444; V, 852, 863; VII, 30, 411. Also see, OS I, 317.
C. Sulfur on the Other Side
15-7 Addition of H2S and Thiols
Hydro-alkylthio-addition
![]()
Hydrogen sulfide (H2S) and thiols add to alkenes to give alkyl thiols or sulfides by electrophilic, nucleophilic, or free radical mechanisms.293 In the absence of initiators, the addition to simple alkenes is by an electrophilic mechanism, similar to that in Reaction 15-5, and Markovnikov's rule is followed. However, this reaction is usually very slow and often cannot be done or requires very severe conditions unless a Br![]() nsted–Lowry or Lewis acid catalyst is used. For example, the reaction can be performed in concentrated H2SO4294 or with the addition of AlCl3.295 In the presence of free radical initiators, H2S and thiols add to double and triple bonds by a free radical mechanism and the orientation is anti-Markovnikov.296 Anti-Markovnikov addition of thiols to vinyl ethers occurs under solvent- and catalyst-free conditions,297 and is promoted by water.298
nsted–Lowry or Lewis acid catalyst is used. For example, the reaction can be performed in concentrated H2SO4294 or with the addition of AlCl3.295 In the presence of free radical initiators, H2S and thiols add to double and triple bonds by a free radical mechanism and the orientation is anti-Markovnikov.296 Anti-Markovnikov addition of thiols to vinyl ethers occurs under solvent- and catalyst-free conditions,297 and is promoted by water.298
Additives can influence the regioselectivity. Styrene reacts with thiophenol to give primarily the anti-Markovnikov product, whereas addition of thiophenol in the presence of Montmorillonite K-10 clay gives primarily the Markovnikov addition product.299 The addition of thiophenol to an alkene with a zeolite, however, leads to the anti-Markovnikov sulfide.300 In fact, the orientation can be used as a diagnostic tool to indicate which mechanism is operating. Free radical addition can be done with H2S, RSH (R may be primary, secondary, or tertiary), ArSH, or RCOSH.301 The R group may contain various functional groups. The alkenes may be terminal, internal, contain branching, be cyclic, and have various functional groups including OH, CO2H, CO2R, NO2, RSO2, and so on. Addition of Ph3SiSH to terminal alkenes under radical conditions also leads to the primary thiol.302
Alkynes react with thiols to give vinyl sulfides. With alkynes it is possible to add 1 or 2 molar equivalents of RSH, giving a vinyl sulfide303 or a dithioketal, respectively. Thiols also add to alkynes with a Pd catalyst to give vinyl sulfides.304 Thiols add to alkenes under photochemical conditions to form thioethers, and the reaction can be done intramolecularly to give cyclic thioethers.305 Thiocarbonates function as thiol surrogates, converting alkenes to alkyl thiols in the presence of TiCl4 and CuO.306 Sulfonic acids add to alkynes to give vinyl sulfonates in the presence of a Au catalyst.307 A cesium carbonate catalyzed reaction gives the vinyl sulfide with good (Z)-selectivity.308 The Rh−309 In−,310 organoactinide−,311 organozirconium-,312 or Pt catalyzed313 reaction of alkynes with thiols gives the corresponding vinyl sulfide. Similar results were obtained under solvent-free conditions using an alumina–KF system.314Alternative preparations are available, as in the reaction of a terminal alkyne with Cp2Zr(H)Cl followed by PhSCl to give the vinyl sulfide with the SPh unit at the less substituted position (PhCH=CHSPh).315 The intramolecular addition of a thiol to an ene–yne, with a Pd catalyst, leads to substituted thiophene derivatives.316 Alkenes react with diphenyl disulfide in the presence of GaCl3 to give the product with two phenylthio units (PhS–C–C–SPh).317 The reaction of an alkyne with diphenyl disulfide and a Pd catalyst leads to the bis(vinyl) sulfide (PhS–C=C–SPh).318
When thiols are added to substrates susceptible to nucleophilic attack, bases catalyze the reaction and the mechanism is nucleophilic. These substrates may be of the Michael type319 or may be polyhalo alkenes or alkynes.255 As with the free-radical mechanism, alkynes can give either vinylic thioethers or dithioacetals:
![]()
By any mechanism, the initial product of addition of H2S to a double bond is a thiol, which is capable of adding to a second molecule of alkene, so that sulfides are often produced: C=C → H–C–C–S–C–C–H. As with alcohols, ketenes add thiols to give thiol esters [R2C=C=O + RSH → R2CHCOSR].320
Selenium compounds (RSeH) add in a similar manner to thiols.321 Vinyl selenides can be prepared from alkynes using diphenyl diselenide and sodium borohydride.322 A Pd(II) catalyzed reaction of PhSeH with alkynes, in pyridine, also gives the corresponding vinyl selenide.323
The conjugate addition of thiols to α,β-unsaturated carbonyl derivatives is discussed in Reaction 15-31.
OS III, 458; IV, 669; VIII, 302. See also, OS VIII, 458.
D. Nitrogen or Phosphorus on the Other Side
15-8 Addition of Ammonia and Amines, Phosphines, and Related Compounds324
Hydro-amino-addition
Hydro-phosphino-addition
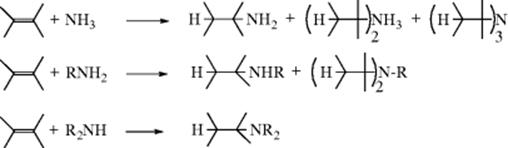
Ammonia and primary and secondary amines add to alkenes in some cases.325 Ammonia and amines are much weaker acids than water, alcohols, and thiols (see Reactions 15-3, 15-5, and 15-7) and since acids turn NH3 into the weak acid, the ammonium ion (NH4+), this reaction does not occur by an electrophilic mechanism. The reaction tends to give very low yields, if any, with ordinary alkenes, unless extreme conditions are used (e.g., 178–200 °C, 800–1000 atm, and the presence of metallic Na, for the reaction between NH3 and ethylene326). There is, however, a proton-catalyzed hydroamination reaction in which aniline derivatives add to alkenes in the presence of anilinium salts, in 20–90% yield depending on the alkene.327
There are many examples of transition-catalyzed addition of nitrogen compounds to alkenes, alkynes,328 and so on. Amines can be added to certain nonactivated alkenes using Pd,329 Rh,330 In,331 Ti,332 Fe,333 Ta,334 Au,335 Y,336Mo,337 and various lanthanide catalysts.338 1,3-Dienes,339 and also allenes,340 undergo hydroamination in the presence of an Au catalyst. Complexation with the metal lowers the electron density of the double bond, facilitating nucleophilic attack.341 Markovnikov orientation is observed and the addition is anti.342 Aniline reacts with dienes and a Pd catalyst to give allylic amines.343 Diene amines react with Sm catalysts to give 2-alkenyl pyrrolidines.344 The mechanism of the Au(I) catalyzed hydroamination reaction of alkenes has been studied.345 It is believed to involve a ligand substitution reaction in the active Au species followed by nucleophile attack of the N-nucleophile on the activated double bond, which is followed by proton transfer from the NH2 group to the unsaturated carbon atom.
Cyclization reactions are useful variations of this reaction. An intramolecular addition of an amine unit to an alkene to form a pyrrolidine was reported using a Pd346 Rh,347 Sc,348 Sm,349 Ti,350 Zr,351 or a Lu catalyst.352 as well as a lanthanide reagent,353 or a Y reagent.354 An intramolecular Ca mediated reaction of amino alkenes leads to cyclic amines.355 Alkenyl amines give cyclic amines as the major product, in good yield, when treated with n-butyllithium.356 Reaction of a secondary amine with butyllithium generates an amide base, which reacts with alkenes to give alkyl amines,357 and can add intramolecularly to an alkene to form a pyrrolidine.358 Pyrroles can be generated in this manner.359
Other nitrogen compounds, among them hydroxylamine and hydroxylamines,360 hydrazines, and amides (Reaction 15-9), also add to alkenes. Tertiary amines (except those that are too bulky) add to Michael-type substrates (C=C–Z) in a reaction that is catalyzed by acids like HCl or HNO3 to give the corresponding quaternary ammonium salts (R3N+–C–C–Z).361 The tertiary amine can be aliphatic, cycloalkyl, or heterocyclic (including pyridine). The reaction of NaOH with an amine containing two distal alkene units, followed by addition of a neodymium catalyst, leads to a bicyclic amine.362
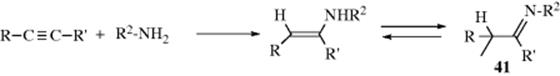
Primary amines add to triple bonds363 to give enamines that have a hydrogen on the nitrogen and (analogously to enols) tautomerize (Sec. 2.N.ii, category 4) to the more stable imines (41).364 The reaction has been done with a Pd,365 a Ti,366 a Ta,367 a Cu,368 or an Au catalyst.369 An intramolecular addition of amines to an alkyne unit in the presence of a Pd catalyst generated heterocyclic or cyclic amine compounds.370 A variation treats an alkynyl imine with CuI to form pyrroles.371 N,N-Diphenylhydrazine reacts with diphenyl acetylene and a Ti catalyst to give indole derivatives.372 Treatment of an imine of 2-alkynyl benzaldehyde with iodide gave a functionalized isoquinoline.373When ammonia is used instead of a primary amine, the corresponding R2C=NH imine product is not stable enough for isolation, but polymerizes. Ammonia and primary amines (aliphatic and aromatic) add to conjugated diynes to give pyrroles (42).374 Anti-Markovnikov addition of alkynes is possible using a Cu catalyst.375
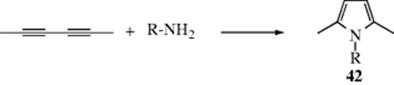
A related reaction of amines and alkynes, in supercritical CO2, leads to amides.376
Allenes are reaction partners,377 and amines add to allenes in the presence of a catalytic amount of CuBr,378 Au,379 or Pd compounds.380 Intramolecular reaction of allene amines leads to dihydropyrroles, using a Au catalyst.381Cyclic imines can be prepared from allene amines using a Ti catalyst.382
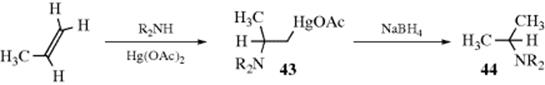
A NH2 or NR2 unit can be added to double bonds (even ordinary double bonds) in an indirect manner by the use of hydroboration (Reaction 15-16) followed by treatment with NH2Cl or NH2OSO2OH (Reaction 12-32). This produces a primary amine with anti-Markovnikov orientation. An indirect way of adding a primary or secondary amine to a double bond consists of aminomercuration to give 43, followed by reduction (see Reaction 15-3 for the analogous oxymercuration–demercuration procedure) to give amine 44.383 The addition of a secondary amine produces a tertiary amine, while addition of a primary amine gives a secondary amine. The overall orientation follows Markovnikov's rule. For conversion of 43 to other products, see Reaction 15-53.
![]()
Phosphines add to alkenes to give alkyl phosphines (45) and to alkynes to give vinyl phosphines. A Pd catalyzed reaction of alkenes and triarylphosphines gives alkyltriarylphosphonium salts.384 Alkenes also react with diarylphosphines and a Ni catalyst to give the alkyl phosphine.385 Silylphosphines (R3Si–PAr2) react with alkenes and Bu4NF to give the anti-Markovnikov allyl phosphine.386 Phosphine oxides can be prepared by the reaction of an aryl substituted alkene and diphenylphosphine oxide, Ph2P(=O)H.387 Phosphonate esters were similarly prepared from alkenes and diethyl phosphite [(EtO)2P(=O)H] and an Mn catalyst in a reaction exposed to oxygen.388 Similar addition was observed in the reaction of an alkene with NaH2PO2 to give the phosphinate [RCH=CH2 → RCH2CH2PH(=O)ONa].389 Palladium catalysts were used for the preparation of similar compounds from alkenes390 and the reaction of terminal alkynes with dimethyl phosphite and a Ni catalyst gave the Markovnikov vinyl phosphonate ester.391
In the presence of an Yb catalyst, diphenylphosphine added to diphenyl acetylene to give the corresponding vinyl phosphine.392 A Pd catalyst was used for the addition of diphenylphosphine to terminal alkynes, giving the anti-Markovnikov vinyl phosphine, but a Ni catalyst led to the Markovnikov vinyl phosphine.393 A Co catalyst has also been used.394 Diphenylphosphine oxide also reacted with terminal alkynes to give the anti-Markovnikov vinyl phosphine oxide using an Rh catalyst.395 Other phosphites were added to dienes to give an allylic phosphonate ester using a Pd catalyst.396 Diarylphosphines react with vinyl ethers and a Ni catalyst to give α-alkoxy phosphonate esters.397
OS I, 196; III, 91, 93, 244, 258; IV, 146, 205; V, 39, 575, 929; VI, 75, 943; VIII,188, 190, 536; 80, 75. See also, OS VI, 932.
15-9 Addition of Amides
Hydro-amido-addition
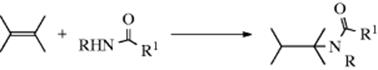
Under certain conditions, primary and secondary amides can add directly to alkenes to form N-alkylated amides. Sulfonamides react in a similar manner. Alkenes react with amides and related compounds in the presence of certain transition metals. The Ti catalyzed reaction of alkenyl N-tosylamines gives N-tosyl cyclic amines.398
The reaction can be done intramolecularly. 3-Pentenamide cyclized to 5-methyl-2-pyrrolidinone by treatment with trifluorosulfonic acid.399 N-Benzyl pent-4-ynamide reacted with tetrabutylammonium fluoride to an alkylidene lactam.400 Acyl hydrazine derivatives also cyclize in the presence of hypervalent iodine reagents to give lactams.401 Treatment of triflamide alkenes with triflic acid gives the corresponding N-triflyl cyclic amine.402 Intramolecular cyclization of sulfamate esters, catalyzed by a Rh complex, leads to cyclic sulfamates.403
Alkynes and allenes also react with amides. A Ru/In catalyzed addition of sulfonamides to alkynes leads to cyclic N-sulfonyl derivatives.404 Palladium-catalyzed reactions give similar results,405 and Bi and Hf have been used as catalysts.406 Phenylthiomethyl alkynes were converted to N-Boc-N-phenylthio allenes with Boc azide and an iron catalyst.407 Enamides are prepared by the Re408 or Ru catalyzed409 hydroamidation of terminal alkynes with amides. The Pd catalyzed reaction of an allene amide, with iodobenzene, leads to N-sulfonyl aziridines having an allylic group at C-1.410 Other allene N-tosylamines similarly give N-tosyl tetrahydropyridines.411
N-Bromocarbamates also add to alkenes, in the presence of BF3·OEt2 to give a vic-bromo N-Boc amine.412 When a carbamate was treated with Bu3SnH, and AIBN, addition to an alkene led to a bicyclic lactam.413 Alkenyl amides and carbamates react with transition metal catalysts to form lactams or cyclic carbamates. Similar addition of a tosylamide–alkene, with a Pd catalyst, led to a vinyl N-tosyl pyrrolidine.414 Both Pd415 and Au416 catalyzed cyclization reactions of carbamates are known, and Os compounds have been used.417 Ionic liquids have been used to catalyze these reactions.418
Imides can also add to alkenes or alkynes. Phthalimide reacts with an alkene in the presence of a Pd catalyst.419 Ethyl 2-propynoate reacted with phthalimide, in the presence of a Pd catalyst, to give ethyl 2-phthalimido-2-propenoate.420
Nickel-catalyzed hydrophosphinylation reactions are known, such as the reaction of an alkyne with an alkyl phosphinate to give a vinyl phosphinate ester.421 Both H-phosphinates and secondary phosphine oxides add to alkenes in an anti-Markovnikov manner, induced by air in what is likely a radical reaction.422
15-10 Addition of Hydrazoic Acid
Hydro-azido-addition
![]()
Hydrazoic acid (HN3) can be added to certain Michael-type substrates (Z is as defined in Sec. 15.A.ii) to give β-azido compounds.423 The reaction apparently fails if R is phenyl. Hydrazoic acid also adds to enol ethers CH2=CHOR to give CH3 –CH(OR)N3, and to silyl enol ethers,424 but it does not add to ordinary alkenes unless a Lewis acid catalyst (e.g., TiCl4) is used, in which case good yields of azide can be obtained.424 Hydrazoic acid can also be added indirectly to ordinary alkenes by azidomercuration, followed by demercuration,425 analogous to the
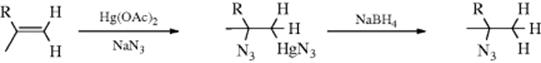
similar procedures mentioned in Reactions 15-3, 15-5, 15-6, and 15-8. The method can be applied to terminal alkenes or strained cycloalkenes (e.g., norbornene), but fails for unstrained internal alkenes. A variation is the hydroazidation reaction of alkenes using a Co catalyst and tert-BuOOH to give the alkyl azide.426
E. Hydrogen on Both Sides
15-11 Hydrogenation of Double and Triple Bonds427
Dihydro-addition
![]()
Most carbon–carbon double bonds, whether substituted by electron-donating or electron-withdrawing substituents, can be catalytically hydrogenated, usually in quantitative or near-quantitative yields.428 However, a transition metal catalyst is required to break apart H2 into metal-bound hydrogen atoms before reaction can occur with the alkene. Almost all known alkenes added hydrogen at temperatures between 0 and 275 °C. The catalysts used can be divided into two broad classes, both of which mainly consist of transition metals and their compounds: (1) Catalysts insoluble in the reaction medium (heterogeneous catalysts). Among the most effective are Raney nickel,429 Pd-on-charcoal (perhaps the most common),430 NaBH4 reduced nickel431 (also called nickel boride), Pt metal or its oxide, Rh, Ru, and zinc oxide,432 (2) Catalysts soluble in the reaction medium (homogeneous catalysts).433 An important example is chlorotris(triphenylphosphine)rhodium [RhCl(Ph3P)3,434 46, Wilkinson's catalyst),435 which catalyzes the hydrogenation of many alkenyl compounds without disturbing such groups as CO2R, NO2, CN, or COR present in the same molecule.436 Even unsaturated aldehydes can be reduced to saturated aldehydes,437 although in this case decarbonylation (Reaction 14-32) may be a side reaction. In general, for catalytic hydrogenation many functional groups may be present in the molecule, for example, OH, COOH, NR2 (including NH2), and N(R)COR′ (including carbamates),438 CHO, COR, CO2R, or CN. Vinyl esters can be hydrogenated using homogeneous Rh catalyst.439Some of these groups are also susceptible to catalytic reduction, but it is usually possible to find conditions under which double bonds can be reduced selectively440 (see Table 19.2). Controlling the solvent allows catalytic hydrogenation of an alkene in the presence of an aromatic nitro group.441
Modification of the catalyst includes a polymer bound Ru catalyst,442 and a polymer incarcerated Pd catalyst.443 A nanoparticulate Pd catalyst in an ionic liquid has been used for the hydrogenation of alkenes.444
Homogeneous catalysts have the advantages of better catalyst reproducibility and better selectivity. Apart from Wilkinson's catalyst (46), chlorotris(triphenylphosphine)hydridoruthenium(II) [(Ph3P)3RuClH]445 is an important homogeneous catalyst that is specific for terminal double bonds (other double bonds are hydrogenated slowly or not at all). Homogeneous catalysts are also less susceptible to catalyst poisoning.446 Heterogeneous catalysts are usually poisoned by small amounts of sulfur, often found in rubber stoppers, or by thiols and sulfides.447 Note that heterogeneous catalysts are usually easier to separate from a reaction mixture.
Using soluble, homogeneous catalysts, unfunctionalized alkenes are hydrogenated with good diastereoselectivity and enantioselectivity448 using various metal catalysts (e.g., Ir,449 Pd,450 or Zr,451 and chiral ligands).452 The chiral transition metal catalyst (Rh and Ru are probably the most common) is usually prepared with suitable chiral ligands prior to addition to the reaction. Alternatively, an achiral catalyst (e.g., Wilkinson's catalyst, 46), is simply added along with a chiral ligand. With monophosphine chiral ligands, 453 the phosphorous may be chiral, as in 47 (called R-camp)454 or bis(phosphine) 48 (called dipamp),455 but pyramidal inversion at elevated temperatures (see Sec. 4.C) limits the utility of such ligands. The alternative is to prepare phosphines that contain a chiral carbon, as in 49 (known as Chiraphos),456 but there are many variations of chiral bis(phosphine) ligands.457 Chiral poisoning has been used as a strategy for asymmetric catalysis.458
Hydrogenations are carried out at room temperature and just above atmospheric pressure, in most cases, but some double bonds are more resistant and require higher temperatures and pressures. The poor reactivity is usually a function of increasing substitution and is presumably caused by steric factors. Trisubstituted double bonds require, say, 25 °C and 100 atm, while tetrasubstituted double bonds may require 275 °C and 1000 atm. Among the double bonds most difficult to hydrogenate or which cannot be hydrogenated at all are those common to two rings, as in steroid 49. Hydrogenations, even at about atmospheric pressure, are often performed in a special hydrogenator, but this is not always necessary. Indeed, placing a hydrogen-filled balloon over the reaction flask is common for small-scale hydrogenations that do not require heat or pressure. The great variety of catalysts available often allows an investigator to find one that is highly selective. For example, the catalyst Pd(salen) encapsulated in zeolites permitted the catalytic hydrogenation of 1-hexene in the presence of cyclohexene.459 It has been shown that the pressure of the reaction can influence enantioselectivity in asymmetric catalytic hydrogenations.460
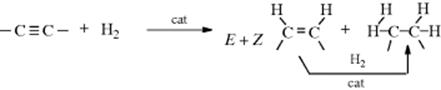
Triple bonds can be reduced, either by catalytic hydrogenation or by the other methods mentioned in the following two sections. The comparative reactivity of triple and double bonds depends on the catalyst. With most catalysts (e.g., Pd), triple bonds are hydrogenated more easily, and therefore it is usually possible to add just 1 equiv of hydrogen and reduce a triple to a double bond or to reduce a triple bond without affecting a double bond present in the same molecule.461 A particularly good catalyst for this purpose is the Lindlar catalyst (Pd–CaCO2–PbO),462 which gives rather lean syn addition, and a (Z)-alkene. An alternative catalyst used for selective hydrogenation to cis-alkenes is the Pd on a barium sulfate (BaSO4) catalyst, poisoned with quinoline463 (sometimes called the Rosenmund catalyst). Palladium on calcium carbonate in PEG has also been used as a recyclable catalyst system.464 Catalytic hydrogenation of alkynes leads to cis-alkenes with a Pd catalyst and DMF/KOH was found to be an efficient transfer-hydrogen source.465 Hydrogenation of a C![]() C unit occurs in the presence of other functional groups, including NR2 including NH2,466 and sulfonyl.467
C unit occurs in the presence of other functional groups, including NR2 including NH2,466 and sulfonyl.467
Conjugated dienes can add hydrogen by 1,2- or 1,4-addition. Selective 1,4-addition can be achieved by hydrogenation in the presence of CO, with bis(cyclopentadienyl)chromium as catalyst.468 With allenes,469 catalytic hydrogenation usually reduces both double bonds. Hydrogenation of functionalized alkenes is possible. The Rh catalyzed hydrogenation of enamines leads to amines,470 for example. Hydrogenation of fluorinated alkenes has been reported using an Ir catalyst.471 The hydrogenation of conjugated alkenes is discussed in Reaction 15-14.
Most catalytic reductions of double or triple bonds, whether heterogeneous or homogeneous, have been shown to be mostly syn, with the hydrogen atoms incorporated from the less-hindered side of the molecule.472 This selectivity depends in large part of how well the reactive intermediates are bound to the metal, and isomerization of the double bond is possible. Stereospecificity can be investigated only for tetrasubstituted alkenes (except when the reagent is D2), which are the hardest to hydrogenate, but the results of these investigations show that the addition is usually 80–100% syn, although some of the anti-addition product is normally also found and in some cases predominates. Catalytic hydrogenation of alkynes is nearly always stereoselective, giving the cis-alkene (usually at least 80%), even when it is thermodynamically less stable. For example, 50 gave 51, even though the steric hindrance is such that a planar molecule is impossible.473 This is
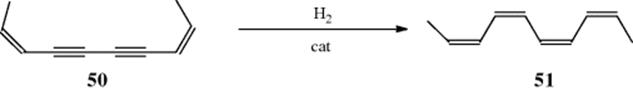
thus a useful method for preparing cis-alkenes.474 However, when steric hindrance is too great, the trans-alkene may be formed. One factor that complicates the study of the stereochemistry of heterogeneous catalytic hydrogenation is that exchange of hydrogen atoms takes place, as can be shown by hydrogenation with deuterium.475 Thus deuterogenation of ethylene produced all the possible deuterated ethylenes and ethanes (even C2H6), as well as HD.476 With 2-butene, it was found that double-bond migration, cis–trans isomerization, and even exchange of hydrogen with groups not on the double bond could occur (e.g., C4H2D8 and C4HD9 were detected on treatment of cis-2-butene with deuterium and a catalyst).477 Indeed, alkanes have been found to exchange with deuterium over a catalyst,478 and even without deuterium (e.g., CH4 + CD4 → CHD3 + CH3D in the gas phase), with a catalyst. All this makes it difficult to investigate the stereochemistry of heterogeneous catalytic hydrogenation.
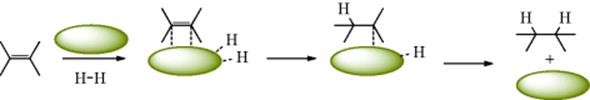
The mechanism of the heterogeneous catalytic hydrogenation of double bonds is not thoroughly understood because it is a very difficult reaction to study.479 Because the reaction is heterogeneous, kinetic data, although easy to obtain (measurement of decreasing hydrogen pressure), are difficult to interpret. Furthermore, there are the difficulties caused by the aforementioned hydrogen exchange. The currently accepted mechanism for the common two-phase reaction was originally proposed in 1934.480 According to this, the alkene is adsorbed onto the surface of the metal, although the nature of the actual bonding is unknown,481 despite many attempts to elucidate it.482 In the 1934 work, the metallic site was indicated by an asterisk, but here ![]() is used. For steric reasons it is apparent that adsorption of the alkene takes place with its less-hindered side attached to the catalyst surface, probably as an η2complex (Sec. 3.C.i). The fact that addition of hydrogen is generally also from the less-hindered side indicates that the hydrogen too is probably adsorbed on the catalyst surface before it reacts with the alkene. It is likely that as the H2 molecule is adsorbed on (coordinated to) the metal catalyst, cleavage occurs to give η1-coordinated hydrogen atoms (Sec. 3.C.i). Note that this model suggests a single metal particle for coordination of the alkene and the hydrogen atoms, but the hydrogen atoms and the alkene could be coordinated to different metal particles. It has been shown that Pt catalyzes homolytic cleavage of hydrogen molecules.483 In the second step, one of the adsorbed (η1-coordinated) hydrogen atoms becomes attached to a carbon atom, creating in effect, an alkyl radical, which is still bound to the catalyst although only by one bond, probably η1-coordination. Transfer of a hydrogen atom to carbon opens a site on the metal catalyst for coordination to additional hydrogen atoms. Finally, another hydrogen atom (not necessarily the one originally connected to the first hydrogen) combines with the radical to give the reaction product, freed from the catalyst surface, and the metal catalyst that is now available for coordination of additional hydrogen atoms and/or alkenes. All the various side reactions, including hydrogen exchange and isomerism, can be explained by this type of process.484 Although this mechanism is satisfactory as far as it goes,485 there are still questions it does not answer, among them questions486 that involve the nature of the asterisk, the nature of the bonding, and the differences caused by the differing nature of each catalyst.487
is used. For steric reasons it is apparent that adsorption of the alkene takes place with its less-hindered side attached to the catalyst surface, probably as an η2complex (Sec. 3.C.i). The fact that addition of hydrogen is generally also from the less-hindered side indicates that the hydrogen too is probably adsorbed on the catalyst surface before it reacts with the alkene. It is likely that as the H2 molecule is adsorbed on (coordinated to) the metal catalyst, cleavage occurs to give η1-coordinated hydrogen atoms (Sec. 3.C.i). Note that this model suggests a single metal particle for coordination of the alkene and the hydrogen atoms, but the hydrogen atoms and the alkene could be coordinated to different metal particles. It has been shown that Pt catalyzes homolytic cleavage of hydrogen molecules.483 In the second step, one of the adsorbed (η1-coordinated) hydrogen atoms becomes attached to a carbon atom, creating in effect, an alkyl radical, which is still bound to the catalyst although only by one bond, probably η1-coordination. Transfer of a hydrogen atom to carbon opens a site on the metal catalyst for coordination to additional hydrogen atoms. Finally, another hydrogen atom (not necessarily the one originally connected to the first hydrogen) combines with the radical to give the reaction product, freed from the catalyst surface, and the metal catalyst that is now available for coordination of additional hydrogen atoms and/or alkenes. All the various side reactions, including hydrogen exchange and isomerism, can be explained by this type of process.484 Although this mechanism is satisfactory as far as it goes,485 there are still questions it does not answer, among them questions486 that involve the nature of the asterisk, the nature of the bonding, and the differences caused by the differing nature of each catalyst.487
Another problem with any study of heterogeneous catalysis is that it occurs at the surface, and different types of metal particles are exposed to the medium and reactants. Maier488 suggested the presence of terrace-, step-, and kink-type atoms (in Fig. 15.1)488 on the surface of a heterogeneous catalyst. These terms refer to different atom types, characterized by the number of nearest neighbors, which correspond to different transition metal fragments, as well as to different coordination states of that metal.489 A terrace-type atom (A in Fig. 15.1) typically has eight or nine neighbors and corresponds to a geometry shown for the ML5 particle. The step type of atom (B) usually has seven neighbors and can be correlated with the geometry shown for the ML4 particle. Finally, the kink-type atom (C) has six neighbors and corresponds to geometry shown for the ML3 particle. In general, as the particle size increases, the relative concentration of terrace atoms will increase, whereas small particle size favors the kink type of surface atoms.
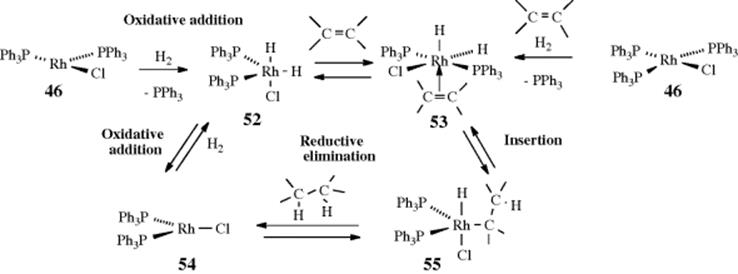
Fig. 15.1 The principal surface and particle sites for heterogeneous catalysts. [Reprinted with permission from Maier, W.F. Angew. Chem. Int. Ed. 1989, 135, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 1989by Wiley-VCH Verlag]
The mechanism of homogeneous hydrogenation490 catalyzed by RhCl(Ph3P)3 (46, Wilkinson's catalyst)491 involves reaction of the catalyst with hydrogen to form a metal hydride (PPh3)2RhH2Cl (52).492 Replacement of a triphenylphosphine ligand with two toms of hydrogen constitutes an oxidative addition. After coordination of the alkene to form 53, transfer of hydrogen to carbon is an insertion process, presumably generating 55, and a second insertion liberates the hydrogenated compound, and Rh species 54, which adds hydrogen by oxidative addition to give 55. In a different study of Pd catalyzed hydrogenations, a palladium hydride species was detected.493Alternatively, replacement of triphenylphosphine can lead to 53, with two hydrogen atoms and a η2 alkene complex. If a mixture of H2 and D2 is used, the product contains only dideuterated and nondeuterated compounds; no monodeuterated products are found, indicating that (unlike the case of heterogeneous catalysis) H2 or D2 has been added to one alkene molecule and that no exchange takes place.491 Although conversion of 53 to the products takes place in two steps,494 the addition of H2 to the double bond is syn, although bond rotation in 55 can lead to stereochemical mixtures.
The occurrence of hydrogen exchange and double-bond migration in heterogeneous catalytic hydrogenation means that the hydrogenation does not necessarily take place by straightforward addition of two hydrogen atoms at the site of the original double bond. Consequently, this method is not synthetically useful for adding D2 to a double or triple bond in a regioselective or stereospecific manner. However, this objective can be achieved (with syn addition) by a homogeneous catalytic hydrogenation, which usually adds D2 without scrambling495 or by the use of one of the diimide methods (Reaction 15-12). Deuterium can also be regioselectively added by the hydroboration–reduction procedure previously mentioned.
Reductions of double and triple bonds are found at OS I, 101, 311; II, 191, 491; III, 385, 794; IV, 298, 304, 408; V, 16, 96, 277; VI, 68, 459; VII, 226, 287; VIII, 420. 609; IX, 169, 533.
Catalysts and apparatus for hydrogenation are found at OS I, 61, 463; II, 142; III, 176, 181, 685; V, 880; VI, 1007.
15-12 Other Reductions of Double and Triple Bonds
![]()
Although catalytic hydrogenation is the method most often used, double or triple bonds can be reduced by other reagents as well. These include sodium in ethanol, sodium and tert-butyl alcohol in HMPA,496 lithium in aliphatic amines497 (see also, Reaction 15-13), zinc and acids, and (EtO)3SiH–Pd(OAc)2.498 Trialkylsilanes (R3SiH) in conjunction with an acid will reduce double bonds.499 When double bonds are reduced by lithium in ammonia or amines, the mechanism is similar to that of the Birch reduction (Reaction 15-13).500 The reduction with trifluoroacetic acid and Et3SiH has an ionic mechanism, with H+ coming in from the acid and H− from the silane.289 In accord with this mechanism, the reaction can be applied only to those alkenes that when protonated can form a tertiary carbocation or one stabilized in some other way (e.g., by a OR substitution).501 It has been shown, by the detection of CIDNP, that reduction of α-methylstyrene by hydridopentacarbonylmanganese(I), [HMn(CO)5], involves free radical addition.502
Triethylamine reduces alkynes in the presence of a Pd catalyst.503 Samarium iodide in water and a triamine additive led to reduction of alkenes.504 Similar reduction was reported using Co2(CO)8 and an excess of water in dimethoxyethane.505
Another hydrogenation method is called transfer hydrogenation.506 In this method, the hydrogen atom comes from another organic molecule, and that molecule is oxidized. A transition metal catalyst, heterogeneous or homogeneous, is frequently employed. A common reducing agent is cyclohexene, which, when a Pd catalyst is used, is oxidized to benzene, losing 2 molar equivalents of hydrogen. Nickel nanoparticles reduce alkenes by transfer hydrogenation using 2-propanol.507
Diimide (NH=NH) is a reducing agent for simple alkenes, formed in situ from N2H4 from the reaction of a mixture of hydrazine and hydroxylamine.508 The rate of this reaction has been studied.509 Diimide is also generated from hydrazine using a flavin catalyst in an oxygen atmosphere.510 Although both the syn and anti forms of diimide are produced, only the syn form reduces the double bond,511 at least in part by a cyclic mechanism:512

The addition is stereospecifically syn513 and, like catalytic hydrogenation, generally takes place from the less-hindered side of a double bond, although not much discrimination in this respect is observed where the difference in bulk effects is small.514 Diimide reductions are most successful with symmetrical multiple bonds (C=C, C![]() C, N=N) and are not useful for those inherently polar (C
C, N=N) and are not useful for those inherently polar (C![]() N, C=N, C=O, etc.). Diimide is not stable enough for isolation at ordinary temperatures, although it has been prepared515 as a yellow solid at −196°C.
N, C=N, C=O, etc.). Diimide is not stable enough for isolation at ordinary temperatures, although it has been prepared515 as a yellow solid at −196°C.
An indirect method516 of double-bond reduction involves formation of an alkylborane from an alkene, followed by hydrolysis of the borane (prepared by Reaction 15-16). Trialkylboranes can be hydrolyzed by heating to reflux with carboxylic acids,517 while monoalkylboranes (RBH2) can be hydrolyzed with base.518 A mild procedure involves treatment of the alkene with 2 molar equivalents of catecholborane, a catalytic amount of MeCONMe2, followed by reduction of the organoborane with 4 molar equivalents of MeOH and then treatment with air.519 Triple bonds can be similarly reduced to cis-alkenes.520 Further reduction is also possible. When an alkyne was treated with decaborane and Pd/C in methanol, 2 equiv of hydrogen are transferred to give the alkane.521 Reduction of alkenes occurs with tert-butylamine·borane complex in methanol with 10% Pd/C.522 In a related reaction, reduction occurs in situ when an alkene is treated with NaBH4, NiCl2·6 H2O with moist alumina.523 Hydrogenation with Ni2B on borohydride-exchange resin (BER) has also been used.524
Metallic hydrides (e.g., lithium aluminum hydride and sodium borohydride) do not typically reduce carbon–carbon double bonds, although in special cases where the double bond is polar, as in 1,1-diarylethenes525 and in enamines,526 reduction can occur. Note that both LiAlH4 and NaBH4, as well as NaH, reduce ordinary alkenes and alkynes when complexed with transition metal salts (e.g., FeCl2 or CoBr2).527 Lithium aluminum hydride reduces cyclopropenes with a pendant alcohol in the allylic position to the corresponding cyclopropane.528 Transition metals catalyze the reduction of alkenes using NaBH4. Among the metals used for this purpose are Pd,529 and Ru.530 A mixture of NaBH4 and BiCl3 also reduced certain alkenes.531 Zinc metal catalyzes the reduction of alkenes in water in the presence of a Rh complex.532
The fact that ordinary double bonds are inert toward metallic hydrides is quite useful, since it permits reduction of, say, a carbonyl or nitro group, without disturbing a double bond in the same molecule (see Chap 19 for a discussion of selectivity in reduction reactions). Sodium in liquid ammonia also does not reduce ordinary double bonds,533 although it does reduce alkynes, allenes, conjugated dienes,534 and aromatic rings (Reaction 15-13).
Enantioselective reduction of certain alkenes has also been achieved by reducing with baker's yeast.535
Catalytic hydrogenation of triple bonds and the reaction with Dibal-H (diisobutylaluminum hydride) usually give the cis-alkene (Reaction 15-11). Most of the other methods of triple-bond reduction lead to the more thermodynamically stable trans-alkene. However, this is not the case with the method involving hydrolysis of boranes or with the reductions with activated zinc, hydrazine, or NH2OSO3H, which also give the cis products.
Triple bonds can also be selectively reduced to double bonds with Dibal-H,536 with activated zinc (see Reaction 12-38),537 or (internal triple bonds only) with alkali metals (Na, Li) in liquid ammonia or a low-molecular-weight amine.538 Terminal alkynes are not reduced by the Na–NH3 procedure because they are converted to acetylide ions under these conditions. However, terminal triple bonds can be reduced to double bonds by the addition to the Na–NH2 solution of (NH4)2SO4, which liberates the free ethynyl group.539 The reaction of a terminal alkyne with lithium naphthalenide and NiCl2 effectively reduced the alkyne unit.540 This reagent is also effective for the reduction of simple alkenes.541
Alkynes are converted to trans-alkenes with siloxanes [(RO)3SiH] 542 and a Ru catalyst, followed by treatment with AgF, or silanes543 and a Ru catalyst, followed by treatment with CuI and Bu4NF. Reduction of an alkyne to an alkene can be done via an organometallic, by heating the alkyne with In metal in aq ethanol.544 Alkynes are reduced with palladium acetate and sodium ethoxide. In methanol, the product is the alkane, whereas in THF the product is the cis-alkene.545
Reduction of just one double bond of an allene, to give an alkene, has been accomplished by treatment with Na–NH3546 or with Dibal-H,547 and by hydrogenation with RhCl(PPh3)3 as catalyst.548
Reductions of double and triple bonds are found at OS III, 586, 742; IV, 136, 302, 887; V, 281, 993; VII, 524; 80, 120.
15-13 Hydrogenation of Aromatic Rings
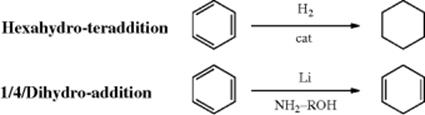
Aromatic rings can be reduced by catalytic hydrogenation,549 but higher temperatures (100–200 °C) are required than for double bonds in alkenes.550 Although the reaction is usually carried out with heterogeneous catalysts, homogeneous catalysts have also been used; conditions are much milder with these.551 Hydrogenations using phase transfer catalysts often proceed under mild conditions.552 Hydrogenation in ionic liquids is known,553 and also hydrogenation in supercritical ethane containing water.554 Many functional groups (e.g., OH, O−, CO2H, CO2R, NH2) do not interfere with the reaction, but some groups may be preferentially reduced. Among these are CH2OH groups, which undergo hydrogenolysis to CH3 (Reaction 19-54). Phenols may be reduced to cyclohexanones, presumably through the enol. A computational study of the mechanism of hydrogenation of aromatic compounds has been reported, and it was shown that the barrier for uncatalyzed 1,4-hydrogenation is substantially lower than that for 1,2-hydrogenation, despite similar reaction enthalpies.555
It is usually impossible to stop the reduction of benzene rings after only one or two bonds have been reduced, since alkenes are more easily reduced than aromatic rings.556 Thus, 1 molar equivalent of benzene, treated with 1 molar equivalent of hydrogen, gives no cyclohexadiene or cyclohexene, but one-third equivalent of cyclohexane and two-thirds equivalent of recovered benzene. This is not true for all aromatic systems. With anthracene, for example, it is easy to stop after only the 9,10-bond has been reduced (Sec. 2.I.i). Hydrogenation of phenol derivatives can lead to conjugated cyclohexenones.557 Hydrogenation of toluene in an ionic liquid using a Ru catalyst gave methylcyclohexane.558
Heterocyclic compounds are often reduced by hydrogenation.559 Furan gives THF, pyrroles560 give pyrrolidines, and pyridines561 give piperidines. The nitrogen-containing ring of quinolines is reduced by hydrogenation using iodine and an Ir catalyst.562 Catalytic hydrogenation of the five-membered ring in indole derivatives using a chiral Rh catalyst gave hydroindoles with excellent enantioselectivity.563
When aromatic rings are reduced by Li (or K or Na) in liquid ammonia (such reductions are known as dissolving metal reductions), usually in the presence of an alcohol (often ethyl, isopropyl, or tert-butyl alcohol), 1,4-addition of hydrogen takes place and nonconjugated cyclohexadienes are produced.564 This reaction is called the Birch reduction.565 Heterocycles (e.g., pyrroles,566 furans,567 pyridines,568 and indolones569) can be reduced using Birch reduction. Ammonia obtained commercially often has iron salts as impurities that lower the yield in the Birch reduction. Therefore it is often necessary to distill the ammonia. When substituted aromatic compounds are subjected to the Birch reduction, electron-donating groups (e.g., alkyl or alkoxyl) decrease the rate of the reaction and are generally found on the nonreduced positions of the product. For example, anisole gives 1-methoxy-1,4-cyclohexadiene, not 3-methoxy-1,4-cyclohexadiene. On the other hand, electron-withdrawing groups (e.g., COOH or CONH2) increase the reaction rate and are found on the reduced positions of the product.570 The regioselectivity of the reaction has been examined.571 The mechanism involves solvated electrons,572 which are transferred from the metal to the solvent, and hence to the ring:573
The sodium becomes oxidized to Na+ and creates a radical ion (56).574 There is a great deal of evidence from ESR spectra for these species.575 The radical ion accepts a proton from the alcohol to give a radical, which is reduced to a carbanion by another sodium atom. Finally, 57 accepts another proton. Thus the function of the alcohol is to supply protons, since with most substrates ammonia is not acidic enough for this purpose. In the absence of the alcohol, products arising from dimerization of 56 are frequently obtained. There is evidence576 at least with some substrates (e.g., biphenyl) that the radical ion corresponding to 56 is converted to the carbanion corresponding to 56 by a different pathway, in which the order of the steps is reversed: First a second electron is gained to give a dianion,573 which then acquires a proton, producing the intermediate (e.g., 56).
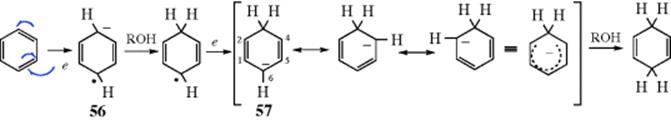
Ordinary alkenes are usually unaffected by Birch-reduction conditions, and double bonds may be present in the molecule if they are not conjugated with the ring. However, phenylated alkenes, internal alkynes (Reaction 15-12),577and conjugated alkenes (with C=C or C=O) are reduced under these conditions.
Note that 56 is a resonance hybrid; that is, two additional canonical forms can be written. The question therefore arises: Why does the carbanion pick up a proton at the 6 position to give the 1,4-diene? Why not at the 2 position to give the 1,3-diene?578 An answer to this question has been proposed by Hine,579 who suggested that this case is an illustration of the operation of the principle of least motion. According to this principle, “those elementary reactions will be favored that involve the least change in atomic position and electronic configuration.”578 The principle can be applied to the case at hand in the following manner (simplified): The valence bond bond orders (Sec. 2.A) for the six carbon–carbon bonds (on the assumption that each of the three forms contributes equally) are (going around the ring) 1![]() , 1, 1, 1
, 1, 1, 1![]() , 1
, 1![]() , and 1
, and 1![]() . When the carbanion is converted to the diene, these bond orders change as follows:
. When the carbanion is converted to the diene, these bond orders change as follows:
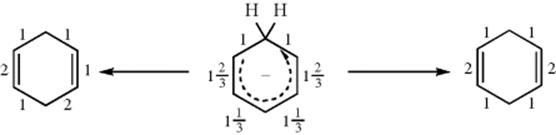
It can be seen that the two bonds whose bond order is 1 are unchanged in the two products, but for the other four bonds there is a change. If the 1,4-diene is formed, the change is ![]() , while formation of the 1,3-diene requires a change of
, while formation of the 1,3-diene requires a change of ![]() . Since a greater change is required to form the 1,3-diene, the principle of least motion predicts formation of the 1,4-diene. This may not be the only factor, because the 13C NMR spectrum of 111shows that the 6 position has a somewhat greater electron density than the 2 position, which presumably would make the former more attractive to a proton.580
. Since a greater change is required to form the 1,3-diene, the principle of least motion predicts formation of the 1,4-diene. This may not be the only factor, because the 13C NMR spectrum of 111shows that the 6 position has a somewhat greater electron density than the 2 position, which presumably would make the former more attractive to a proton.580
Reduction of aromatic rings with Li581 or Ca582 in amines (instead of ammonia: called Benkeser reduction) proceeds further and cyclohexenes are obtained. It is thus possible to reduce a benzene ring, by proper choice of reagent, so that one, two, or all three double bonds are reduced.583 Lithium triethylborohydride (LiBEt3H) has also been used, to reduce pyridine derivatives to piperidine derivatives.584
Transition metals and metal compounds can reduce aromatic rings in the proper medium. Indium metal reduces the pyridine ring in quinoline in aq ethanol solution,585 as well as the C=C unit in the five-membered ring of indole derivatives.586 Samarium iodide (SmI2) reduces pyridine in aq THF587 and phenol in MeOH/KOH.588 Ammonium formate and a Pd–C catalyst reduces pyridine N-oxide to piperidine in methanol.589 The nitrogen-containing ring of quinolines is reduced with an In catalyst in isopropyl alcohol.590
OS I, 99, 499; II, 566; III, 278, 742; IV, 313, 887, 903; V, 398, 400, 467, 591, 670, 743, 989; VI, 371, 395, 461, 731, 852, 856, 996; VII, 249.
15-14 Reduction of the Double or Triple Bonds Conjugated to Carbonyls, Cyano, Nitro, and so on
Reduction of only the C=C bond of conjugated C=C–C=O and C=C–C![]() N systems591 has been achieved by many reducing agents,592 including catalytic hydrogenation with a Rh,593 a Ru,594 a Pd,595 or an Ir catalyst,596 and with Raney nickel alone.597 Reagents, such as SmI2598 and catecholborane,599 are effective. Conjugated ketones react with 2 equiv of Cp2TiCl in THF/MeOH to give the corresponding saturated ketone.600 Zinc and acetic acid has been used for the conjugate reduction of dihydropyridin-4-ones.601 Formic acid with a Pd catalyst reduced conjugated carboxylic acids.602 A zinc–titanocene protocol has been developed for conjugate reductions.603 Both NaBH4 in MeOH–THF604 and NaCNBH3 on a zeolite605 reduce α,β-unsaturated nitro compounds to nitroalkanes.
N systems591 has been achieved by many reducing agents,592 including catalytic hydrogenation with a Rh,593 a Ru,594 a Pd,595 or an Ir catalyst,596 and with Raney nickel alone.597 Reagents, such as SmI2598 and catecholborane,599 are effective. Conjugated ketones react with 2 equiv of Cp2TiCl in THF/MeOH to give the corresponding saturated ketone.600 Zinc and acetic acid has been used for the conjugate reduction of dihydropyridin-4-ones.601 Formic acid with a Pd catalyst reduced conjugated carboxylic acids.602 A zinc–titanocene protocol has been developed for conjugate reductions.603 Both NaBH4 in MeOH–THF604 and NaCNBH3 on a zeolite605 reduce α,β-unsaturated nitro compounds to nitroalkanes.
In certain cases,606 metallic hydride reagents may selectively reduce double bonds that are in conjugation with C=O bonds,607 although the C=O bonds are also reduced in many cases, as in the conversion of cyclopentenone to cyclopentanol.608 The reagent NaBH4 has a greater tendency than LiAlH4 to effect this double reduction, although even with NaBH4 the product of 1,2-reduction (of the C=O bond) is usually formed in larger amount than the doubly reduced product. The C=C unit proximal to the carbonyl in dienyl amides is selectively reduced with NaBH4/I2.609 Mixed hydride reducing agents (e.g., NaBH4–BiCl3,610 NaBH4–InCl3,611 and Dibal-H–Co(acac)2)612 have been used. The InCl3–NaBH4 reagent was used to covert conjugated diene ketones (C=C–C=C–C=O) selectively to the nonconjugated alkenyl ketone (C=C–CH2CH2–C=O).613 Lithium aluminum hydride also reduces the double bonds of allylic alcohols.614
Transfer hydrogenation can be applied to the reduction of conjugated alkenes. Reduction of the C=C unit of conjugated aldehydes is accomplished with an imidazolidinone catalyst615 or an amino ester616 in the presence of a Hantzch ester (e.g., 58). Nitroalkenes are hydrogenated using a thiourea catalyst in the presence of 58.617 Palladium/carbon with microwave heating is used for the transfer hydrogenation of conjugated carboxylic acids, using 1,4-cyclohexadiene as the hydrogen-transfer agent.618 Solvent-free transfer hydrogenation is possible using a Ru complex, with formic acid or water619 as the hydrogen donor.620 Palladium–P(t-Bu)3 has been used as a mild catalyst for transfer hydrogenation.621 Titanium-catalyzed reductions are also known.622
Silanes reduce the C=C unit in conjugated systems in the presence of Cu species.623 An asymmetric CuH catalyzed hydrosilation reaction is known.624 Phenylsilane (PhSiH3) and a Ni catalyst,625 CuCl,626 a Mn catalyst627 or a Mo628 catalyst have been used for hydrosilation reactions. Triphenylsilane was also used for the asymmetric reduction of nitro alkenes (C=C–NO2).629 Poly(methylhydrosiloxane) with a chiral Cu catalyst gave conjugate reduction of conjugated esters to give the saturated derivative with high enantioselectivity.630 Polymethylhydrosiloxane, in the presence of a Co-catalyst reduces conjugated nitriles.631 A β-bromo conjugated lactone was reduced to the β-bromolactone with modest enantioselectivity using an excess of Ph3SiH and a CuCl catalyst with a chiral ligand.632 Tributyltin hydride, in the presence of MgBr2·OEt2 gave 1,4-reduction of conjugated esters.633
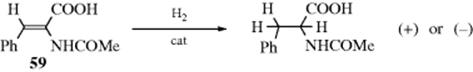
Optically active homogeneous hydrogenation catalysts have been used to achieve the enantioselective hydrogenation634 of many prochiral conjugated substrates.635 For example,636 hydrogenation of 59 with a suitable catalyst gives the (+) or (−) amino ester (depending on which enantiomer of the catalyst is used) with an ee as high as 96%.637 Prochiral substrates that give such high optical yields generally contain functional groups (e.g., a carbonyl group,638 amide groups, cyano groups) or combinations of such groups as in 58.639 The catalyst in such cases640 is usually a Ru641 or Rh complex642with chiral phosphine ligands.643 Good asymmetric induction644 has been achieved using chiral Rh complexes with other chiral additives.645 Iridium complexes have been used with excellent enantioselectivity.646 The role of solvent has been examined.647 A pressure-dependent enantioselective hydrogenation has been reported.648 Asymmetric catalytic hydrogenation has been reported for conjugated carboxylic acids649 and conjugated ketones.650 Asymmetric hydrogenation of conjugated carboxylic acids in an ionic liquid is known using a chiral Ru complex651
See Reaction 19-36 for methods of reducing C=O bonds in the presence of conjugated C=C bonds.
The C=C unit of conjugated aldehydes has been reduced using AlMe3 with a catalytic amount of CuBr652 and with ammonium formate/Pd–C.653 Polymer-supported formate has been used for the 1,4-reduction of conjugated ketones654 and for conjugated acids using an Rh catalyst and microwave irradiation.655 Isopropyl alcohol and an Ir catalyst gives conjugate reduction of conjugated ketones.656 The reaction of conjugated ketones with aluminum chlorides, followed by treatment with water generates the saturated ketone.657
Enzymatic reduction of conjugated systems requires the reactivity with certain purified or whole cell enzymes. Baker's yeast reduces conjugated nitro compounds to nitroalkanes658 and also the C=C unit of conjugated ketones.659Other enzymatic reductions are possible. A reductase from Nicotiana tabacum reduced a conjugated ketone to the saturated ketone, with excellent enantioselectivity.660 Enzyme YNAR-I and NADP-H reduces conjugated nitro compounds to nitroalkanes.661 Conjugated nitro compounds are reduced in the presence of Clostridium sporogenes.662
15-15 Reductive Cleavage of Cyclopropanes
![]()
Cyclopropanes can be cleaved by catalytic hydrogenolysis.663 Among the catalysts used have been Ni, Pd, Rh,664 and Pt. The reaction can often be run under mild conditions.665 Certain cyclopropane rings, especially cyclopropyl ketones and aryl-substituted cyclopropanes,666 can be reductively cleaved by an alkali metal (generally Na or Li) in liquid ammonia.667 Similar reduction has been accomplished photochemically in the presence of LiClO4.668 This reaction is an excellent way to introduce a gem-dimethyl unit into a molecule. Hydrogenation of the cyclopropane ring in 60, for example, gave the gem-dimethyl unit in 61 using PtO2 (Adam's catalyst).669
F. A Metal on the Other Side
15-16 Hydroboration
![]()
When alkenes are treated with borane670 in ether solvents, BH3 adds across the double bond.671 The alkene reacts as a base with the boron essentially reacting as a Lewis acid. Borane cannot be prepared as a stable pure compound672 (it dimerizes to diborane, B2H6), but it is commercially available in the form of “ate” complexes with THF, Me2S,673 phosphines, or tertiary amines. The alkenes can be treated with a solution of one of these complexes. Tetrahydrofuran–BH3 reacts at 0 °C and is the most convenient to use and R3N–BH3 generally require temperatures of ~100 °C. The latter can be prepared as air-stable liquids or solids, while the former can only be used as relatively dilute solutions in THF and are decomposed by moisture in air) or with a mixture of NaBH4 and BF3 etherate, which generates borane in situ.674 Pyridine–borane can be used for the hydroboration of alkenes at room temperature.675With relatively unhindered alkenes, the process cannot be stopped with the addition of one molecule of BH3 because the resulting RBH2 adds to another molecule of alkene to give R2BH, which in turn adds to a third alkene molecule, so that the isolated product is a trialkylborane (R3B). The reaction can be performed on alkenes with one to four substituents, including cyclic alkenes, but when the alkene is moderately hindered, the product is the dialkylborane (R2BH) or even the monoalkylborane (RBH2).676 For example, 62 (disiamylborane) and 63 (thexylborane)677 have been prepared in this manner. Monoalkylboranes (RBH2), which can be prepared from hindered alkenes, (as above) and dialkylboranes (R2BH) also add to alkenes, to give the mixed trialkylboranes (RR′2B and R2R′B), respectively. Surprisingly, when methylborane (MeBH2),678 which is not a bulky molecule, adds to alkenes in the solvent THF, the reaction can be stopped with one addition to give dialkylboranes (RMeBH).679 Reaction of this with a second alkene produces the trialkylborane (RR′MeB).680 Other monoalkylboranes (iPrBH2, n-BuBH2, s-BuBH2, and t-BuBH2), behave similarly with internal alkenes, but not with alkenes of the type RCH=CH2.681
In all cases, the boron goes to the side of the double bond that has more hydrogen atoms (less substituted), whether the substituents are aryl or alkyl.682 Technically, this follows Markovnikov's rule, since boron is more positive than hydrogen. The regioselectivity is caused mostly by steric factors, although electronic factors also play a part. Studies of the effect of ring substituents on rates and on the direction of attack in hydroboration of substituted styrenes showed that the reaction with boron and the alkene has electrophilic character.683 When both sides of the double bond are monosubstituted or disubstituted, about equal amounts of each isomer are obtained. However, it is possible in such cases to make the addition regioselective by the use of a large borane molecule. For example, treatment of iPrCH=CHMe with borane gave 57% of product with boron on the methyl-bearing carbon and 43% of the other, while treatment with 62 gave 95% 64 and only 5% of the other isomer.684
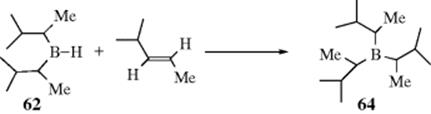
Another reagent with high regioselectivity is 9-borabicyclo[3.3.1]nonane (9-BBN, 65), which is prepared by hydroboration of 1,5-cyclooctadiene,685 and has the advantage that it is stable in air. Borane is quite unselective and attacks all sorts of double bonds. Disiamylborane, 9-BBN, and similar molecules are far more selective and preferentially react at the less-hindered bonds, so it is often possible to hydroborate one double bond in a molecule and leave others unaffected or to hydroborate one alkene in the presence of a less reactive alkene.686 For example, 1-pentene can be removed from a mixture of 1- and 2-pentenes, and a cis-alkene can be selectively hydroborated in a mixture of the cis and trans isomers.
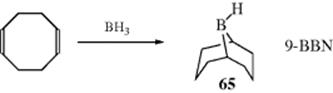
For most substrates, the addition in hydroboration is stereospecific and syn, with attack taking place from the less-hindered side.687 Note that organoboranes can be analyzed using 11B NMR.688 The mechanism689 may be a cyclic four-center one:690
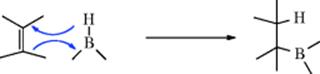
When the substrate is an allylic alcohol or amine, the addition is generally anti,691 although the stereoselectivity can be changed to syn by the use of catecholborane and Rh complexes.692 Because the mechanism is different, use of this procedure can result in a change in regioselectivity as well (e.g., styrene, PhCH=CH2, gave PhCH(OH)CH3).693
Monochloroborane694 (BH2Cl) coordinated with DMS shows greater regioselectivity than BH3 for terminal alkenes or those of the form R2C=CHR, and the hydroboration product is a dialkylchloroborane (R2BCl).695 For example, 1-hexene gave 94% of the anti-Markovnikov product (the boron is on the less substituted carbon) with BH3–THF, but 99.2% with BH2Cl−SMe2. Treatment of alkenes with dichloroborane–DMS (BHCl2–SMe2) in the presence of BF3696 or with BCl3 and Me3SiH697 gives alkyldichloroboranes (RBCl2). Extensions of this basic approach are possible with dihalo alkylboranes. The reaction of an alkene with allyl dibromoborane, incorporated an allyl group and the boron on adjacent carbons.698
An important use of the hydroboration reaction is oxidation of an organoborane to alcohols with H2O2 and NaOH (see Reaction 12-27). The synthetic result is an indirect way of adding H2O across a double bond in an anti-Markovnikov manner. However, boranes undergo many other reactions as well. Among other things, they react with α-halo carbonyl compounds to give alkylated products (Reaction 10-73), with α,β-unsaturated carbonyl compounds to give Michael-type addition of R and H (Reaction 15-27), with CO to give alcohols and ketones (Reactions 18-23–18-24); they can be reduced with carboxylic acids (Reaction 15-11), or they can be oxidized with chromic acid or pyridinium chlorochromate to give ketones699 or aldehydes (from terminal alkenes),700 dimerized with silver nitrate and NaOH (Reaction 14-26), isomerized (Reaction 18-11), or converted to amines (Reaction 12-32), halides (Reaction 12-31), or carboxylic acids.701 They are thus useful intermediates for the preparation of a wide variety of compounds. Intramolecular hydroboration reactions are possible.702
Such functional groups as OR, OH, NH2, SMe, halogen, and CO2R may be present in the molecule,703 but not groups that are reducible by borane (e.g., COOH). Hydroboration of enamines with 9-BBN provides an indirect method for reducing an aldehyde or ketone to an alkene, for example,704
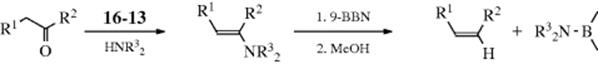
The presence of certain functional groups has directing effects on hydroboration reactions. Amides direct hydroboration reactions in alkenyl amides, for example.705 Intramolecular hydroboration is directed by amine groups in alkenyl amines.706 Alkenyl alcohols or ethers also undergo hydroboration, where delivery of boron is directed by the oxygen.707
Use of the reagent diisopinocampheylborane (66, prepared by treating optically active α-pinene with BH3) results in enantioselective hydroboration–oxidation.708 Since both (+) and (−) α-pinene are readily available, both enantiomers can be prepared. Alcohols with moderate-to-excellent enantioselectivities have been obtained
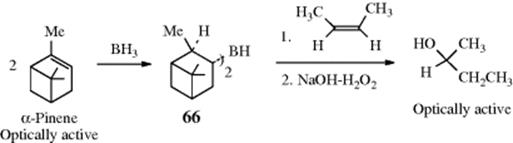
in this way.709 However, 66 does not give good results with even moderately hindered alkenes; a better reagent for these compounds is isopinocampheylborane710 although optical yields are lower. Limonylborane,711 2- and 4-dicaranylboranes,712 a myrtanylborane,713 and dilongifolylborane714 have also been used. Other new asymmetric boranes have also been developed. The chiral cyclic boranes trans-2,15-dimethylborolanes (67 and 68) also add enantioselectively to alkenes (except alkenes of the form RR′C=CH2) to give boranes of high optical purity.715 When chiral boranes are added to trisubstituted alkenes of the form RR′C=CHR″, two new chiral centers are created, and, with 67 or 68, only one of the four possible diastereomers is predominantly produced, in yields >90%.715 This has been called double-asymmetric synthesis.716 An alternative asymmetric synthesis of alcohols involves the reaction of catechol borane with an alkene in the presence of a chiral Rh catalyst, giving the alcohol enantioselectivity after the usual oxidation.717
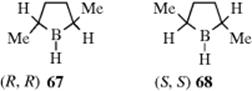
The double bonds in a conjugated diene are hydroborated separately; that is, there is no 1,4-addition. However, it is not easy to hydroborate just one of a conjugated system, since conjugated double bonds are less reactive than isolated ones. Thexylborane677 (63) is particularly useful for achieving the cyclic hydroboration of dienes, conjugated or nonconjugated, as in the formation of 69.718
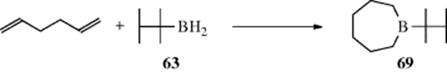
Rings of five, six, or seven members can be formed in this way. Similar cyclization can also be accomplished with other monoalkylboranes and, in some instances, with BH3 itself.719 One example is the formation of 9-BBN, shown above. Another is conversion of 1,5,9-cyclododecatriene to perhydro-9b-boraphenalene (70).720
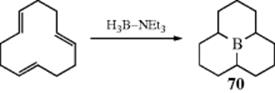
Boronate esters are prepared from alkenes. The reaction of an alkene with pyridine iodoborane, followed by treatment with pinacol and NaOH, for example, leads to the pinacol boronate ester.721
Triple bonds722 can be monohydroborated to give vinylic boranes, which can be reduced with carboxylic acids to cis-alkenes or oxidized and hydrolyzed to aldehydes or ketones. Terminal alkynes give aldehydes by this method, in contrast to the mercuric or acid-catalyzed addition of water discussed in Reaction 15-4. However, terminal alkynes give vinylic boranes723 (and hence aldehydes) only when treated with a hindered borane (e.g., 62, 63, or catecholborane, Reaction 12-31 and Sec. 14.A.i),724 or with BHBr2–SMe2.725 The reaction between terminal alkynes and BH3 produces 1,1-dibora compounds, which can be oxidized either to primary alcohols (with NaOH–H2O2) or to carboxylic acids (with m-chloroperoxybenzoic acid).726 Double bonds can be hydroborated in the presence of triple bonds if the reagent is 9-BBN.727 On the other hand, dimesitylborane selectively hydroborates triple bonds in the presence of double bonds.728 Furthermore, it is often possible to hydroborate selectively one particular double bond of a nonconjugated diene.729 A triple bond can be hydroborated in the presence of a ketone, and treatment with acetic acid reduces the C![]() C unit to a cis- alkene (see Reaction 15-12).730 When the reagent is catecholborane, hydroboration is catalyzed by Rh complexes,731 (e.g., Wilkinson's catalyst, 46,732 by SmI2,733 or lanthanide reagents).734Enantioselective hydroboration–oxidation has been achieved by the use of optically active Rh complexes.735
C unit to a cis- alkene (see Reaction 15-12).730 When the reagent is catecholborane, hydroboration is catalyzed by Rh complexes,731 (e.g., Wilkinson's catalyst, 46,732 by SmI2,733 or lanthanide reagents).734Enantioselective hydroboration–oxidation has been achieved by the use of optically active Rh complexes.735
A chain extension variation involved the reaction of styrene with catecholborane and then Me3SiCHN2.736 Subsequent oxidation with NaOH/H2O2 and the reaction with Bu4NF gave 3-phenyl-1-propanol.
OS VI, 719, 852, 919, 943; VII, 164, 339, 402, 427; VIII, 532.
15-17 Other Hydrometalation
Hydro-metallo-addition
![]()
Metal hydrides of Groups 13 and 14 of the periodic table (e.g., AlH3, GaH3), as well as many of their alkyl and aryl derivatives (e.g., R2AlH and Ar3SnH) add to double bonds to give organometallic compounds.737 In each case, the alkene reacts with the Lewis acid. The hydroboration reaction (15-16) is the most important example but, as mentioned, other important metals in this reaction are Al,738 Sn,739 and Zr740 (a Group 4 metal). Some of these reactions are uncatalyzed, but in other cases various types of catalyst have been used.741 Hydrozirconation is most commonly carried out with Cp2ZrHCl (Cp = cyclopentadienyl),742 known as Schwartz's reagent. The mechanism with Group 13 hydrides seems to be electrophilic (or four-centered pericyclic with some electrophilic characteristics) while with Group 14 hydrides a mechanism involving free radicals seems more likely. Dialkylmagnesium reagents have been obtained by adding MgH2 to double bonds.743 With some reagents triple bonds744 can add 1 or 2 equiv.745 When 2 molar equivalents are added, electrophilic addition generally gives 1,1-dimetallic products (as with hydroboration), while free radical addition usually gives the 1,2-dimetallic products.
OS VII, 456; VIII, 268, 295, 507; 80, 104. See also, OS VIII, 277, 381.
G. Carbon or Silicon on the Other Side
15-18 Addition of Alkanes
Hydro-alkyl-addition
![]()
There are two important ways of adding alkanes to alkenes: direct heating, and acid catalysis.746 Both give mixtures, and neither is useful for the preparation of relatively pure compounds in reasonable yields. However, both are useful industrially. In the thermal method, the reactants are heated to high temperatures (~500 °C) at high pressures (150–300 atm) without a catalyst. As an example, propane and ethylene gave 55.5% isopentane, 7.3% hexanes, 10.1% heptanes, and 7.4% alkenes.747 The mechanism is undoubtedly of a free radical type and can be illustrated by one possible sequence in the reaction between propane and ethylene:
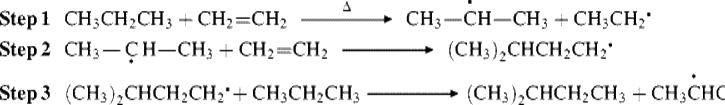
There is kinetic evidence that the initiation takes place primarily by steps like 1, which are called symproportionation steps748 (the opposite of disproportionation, see Sec. 5.C.ii).
In the acid-catalysis method, a protonic or Lewis acid is used as the catalyst and the reaction is carried out at temperatures between –30 and 100 °C. This is a Friedel–Crafts process that proceeds via a carbocation mechanism749(illustrated for a proton acid catalyst):
Carbocation 72 often rearranges before a hydride is transferred, explaining, for example, why the principal product from the reaction between isobutane and ethylene is 2,3-dimethylbutane. Instead of abstracting a hydride, it is also possible for 71 (or 72) to add to another molar equivalent of alkene, so that not only rearrangement products, but also dimeric and polymeric products, are frequent. If the tri- or tetrasubstituted alkenes are treated with Me4Si, HCl, and AlCl3, protonation gives a tertiary carbocation, which reacts with the Me4Si to give a product that is the result of addition of H and Me to the original alkene.750 (For a free radical hydro-methyl addition, see Reaction 15-28.) An intramolecular cyclization of 1-dodecene to cyclododecane was reported using aluminum chloride in an ionic liquid.751
Alkanes add to alkynes under photolysis conditions to give an alkene.752 Tetrahydrofuran adds to alkynes to give the alkene with microwave irradiation.753
The reaction can also be base catalyzed, in which case there is nucleophilic addition and a carbanion mechanism.754 Carbanions most often used are those stabilized by one or more α-aryl groups. For example, toluene adds to styrene in the presence of sodium to give 1,3-diphenylpropane:755
![]()
Conjugated dienes give 1,4-addition.756 This reaction has also been performed with salts of carboxylic acids in what amounts to a method of alkylation of carboxylic acids757 (see also, Reaction 10-59).
![]()
There are transition metal catalyzed addition reactions of alkyl units to alkenes,758 often proceeding with metal hydride elimination to form an alkene. An intramolecular cyclization reaction of an N-pyrrolidino amide alkene was reported using an iridium catalyst for addition of the carbon α to nitrogen at the alkene unit.759
OS I, 229; IV, 665; VII, 479.
15-19 Addition of Silanes (Hydrosilation)
Silyl-hydro-addition
![]()
Although silanes bearing at least one Si–H unit do not generally react with alkenes or alkynes, addition occurs to give the corresponding alkyl or vinyl silane in the presence of transition metal catalysts.760 This reaction is known as hydrosilation. The reaction of an alkylsilane and an alkene with a Ru,761 Rh,762 Pd,763 Re,764 La,765 Y,766 Pt767 Cu,768 or Sm769 catalyst leads to addition with high anti-Markovnikov selectivity. Silanes add to dienes with a Pd catalyst, and asymmetric induction is achieved by using a chiral binapthyl additive.770 Alkenes react with Li metal and t-Bu2SiCl2 to give a three-membered ring silane.771
Dienes react with zirconium compounds and silanes to produce cyclic compounds in which the silyl group has also added to one C=C unit.772 With an Y catalyst, PhSiH3 reacts with nonconjugated dienes to give cyclic alkenes with a pendant CH2SiH2Ph group.773 Rhodium compounds allow silanes to add to enamides to give the α-silylamide.774 Formation of silanes via reaction with alkenes can be followed by reaction with fluoride ion and then oxidation to give an alcohol775 (see Reaction 10-16; Tamao–Fleming oxidation). A variation of this reaction generated allylic silanes from terminal alkenes and a dianion-type zincate using a Ti catalyst.776
In the presence of BEt3, silanes add to alkenes to give the alkylsilane with anti-Markovnikov selectivity,777 or to alkynes to give the corresponding vinyl silane.778 Similar selectivity was observed when a silylated zinc reagent was added to a terminal alkyne.779 Hydrosilation of alkynes is accomplished using transition metal catalysts (e.g., Ru780 Pt,781 Ti,782 or Ir).783 Organocatalyts have been used as well.784 Siloxanes [e.g., (RO)3SiH] add to alkynes with a Ru catalyst to give the corresponding vinyl silane.785 The reaction of Cl2MeSiH and terminal alkynes, in ethanol–triethylamine with a Ru catalyst, to give primarily the Markovnikov vinyl silane.786 However, Et3SiH adds to terminal alkynes with a Rh787 or a Pt788 catalyst to give the anti-Markovnikov vinyl silane. Using a 0.5 molar equivalent of HfClO4 with alkynes bearing a dimethylphenylsilyl unit gave a cyclic vinyl silane with transfer of the phenyl group to carbon (see 73).789
Silanes add to alkenes under radical conditions (using AIBN) with high anti-Markovnikov selectivity.790 An alternative route to alkylsilanes reacted an alkene with Li metal in the presence of 3 equiv of chlorotrimethylsilane, giving bis-1,2-trimethylsilyl compounds after treatment with water.791 Silanes also add to alkenes to form the anti-Markovnikov alkylsilane (R3Si–C–C–R′) in the presence of a hyponitrite.792
In a reaction more related to those in Reaction 15-24, vinyl silanes add to conjugated carbonyl compounds in the presence of a Ru catalyst,793 or to acrylonitriles with a Co catalyst.794 Silyl phosphines react with conjugated ynones directly to give an enone with an α-trimethylsilyl and a β-phosphine group.795 Siloxanes of the type (RO)3SiH add to the α-carbon of enamines in the presence of a dirhodium catalyst.796 The uncatalyzed reaction of trimethylsilyl cyanide and ynamines, however, gave an enamine with a β-trimethylsilyl and an α-cyano group.797
bis(Silanes) add to alkylidene malonate derivatives (see Reaction 15-24) in the presence of a Cu catalyst to give β-silyl malonates [RCH(SiR3)CH(CO2Me)2].798 Alkylsilane units add using bis(trialkylsilyl)zinc reagents with a CuCN catalyst.799 Trimethylsilyl cyanide (Me3SiCN) adds a cyano group to α,β-unsaturated amines with a specialized Al(salen)-Y catalyst.800
15-20 Addition of Alkenes and/or Alkynes to Alkenes and/or Alkynes
Hydro-alkenyl-addition
![]()
With certain substrates, alkenes can be dimerized by acid catalysts, so that the product is a dimer that contains one double bond.801 A catalyst comprised of a combination of Zn and a CoCl2 accomplished this type of coupling.802One alkene adds to another in the presence of a Ni catalyst.803 α-Alkenes add to dienes in a 1,4-manner in the presence of an Fe catalyst.804
This reaction is more often carried out internally, as in the formation of cyclohexene (74). A Pd catalyzed cyclization is known, in which dienes are converted to cyclopentene derivatives (e.g., 75).805
Ring-forming reactions are possible. A Ru catalyzed version of this reaction gave the five-membered ring with an exocyclic double bond.806 Carbocyclization of an alkene unit to another alkene unit was reported using a Y,807 or a Ti catalyst.808 In some cases, internal coupling of two alkenes can form larger rings.809 Carbocyclization was reported using Pd,810 Rh,811 Ru,812 Ir,813 or a Zr catalyst.814 Alkene allene substrates were cyclized to form cyclic products with an exocyclic double bond using a Pd catalyst.815 An interesting variation adds a silyl enol ether to an alkyne using GaCl3 to give an unconjugated ketone (O=C–C–C=C).816 Alkenes and alkynes can also add to each other to give cyclic products in other ways (see Reaction 15-63 and 15-65).
Processes of this kind are important in the biosynthesis of steroids and tetra- and pentacyclic terpenes. For example, squalene 2,3-oxide is converted by enzymatic catalysis to dammaradienol. The squalene → lanosterol biosynthesis, which is a key step in the biosynthesis of cholesterol, is similar. The idea that the biosynthesis of
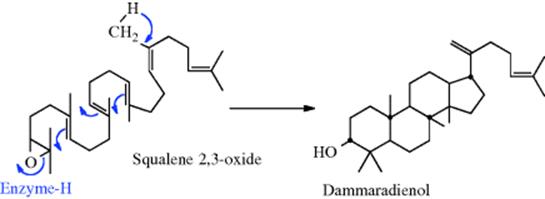
such compounds involves this type of multiple ring closing was proposed in 1955 and is known as the Stork–Eschenmoser hypothesis.817 Such reactions can also be carried out in the laboratory, without enzymes.818 By putting cation-stabilizing groups at positions at which positive charges develop, Johnson and co-workers819 have been able to close as many as four rings stereoselectively and in high yield, in one operation. An example is formation of 76,820 by what is known as the Johnson polyene cyclization.821 Lewis acids can be used to initiate this cyclization,822 including EtAlCl2 used for the coupling of an alkyne and an alkene.823 A Pd catalyst has been used for a similar cyclization reaction.824 A radical cyclization approach (Reaction 15-30) to polyene cyclization using a seleno–ester anchor gave a tetracyclic system.825
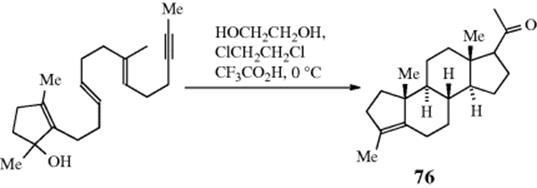
The addition of alkenes to alkenes826 can also be mediated by bases.827 Coupling reactions can occur using transition metal catalyst systems828 (e.g., alkylaluminum compounds, known as Ziegler catalysts),829 Rh,830 Fe,831 or Ni832catalysts. The 1,4-addition of alkenes to conjugated dienes to give nonconjugated dienes833 occurs with various transition metal catalysts, as does the dimerization of 1,3-butadienes to octatrienes.834 A
![]()
molecule containing two distal conjugated diene units was cyclized to give a bicyclic molecule with an exocyclic double bond using a Pd catalyst.835 A Ni catalyst converted a similar system to a saturated five-membered ring containing an allylic and a vinyl group.836 Ethylene adds to alkenes to form a new alkene in the presence of a Ni837 or a Zr catalyst,838 and to alkynes in the presence of a Ru catalyst839 to form a diene. Allenes add to alkynes to give a diene with a Ti catalyst.840
In the presence of cuprous chloride and ammonium chloride, acetylene undergoes self-coupling to give vinylacetylene. Alkynes are coupled to dienes to give enynes in the presence of a Ni catalyst.841
![]()
Another type of alkyne dimerization is the reductive coupling in which two molecules of alkyne, the same or different, give a 1,3-diene, as shown.842 In this method, one alkyne is treated with Schwartz's reagent (see Reaction 15-17) to produce a vinylic zirconium intermediate. Addition of MeLi or MeMgBr, followed by the second alkyne, gives another intermediate, which, when treated with aq acid, gives the diene in moderate-to-good yields.843If the second intermediate is treated with I2 instead of aq acid, the 1,4-diiodo-1,3-diene is obtained, in comparable yield and isomeric purity. The reaction of an alkyne with a Grignard reagent, followed by an Fe complex and then an alkene leads to enynes.844 A Rh catalyzed coupling reaction of alkenes and electron-deficient internal alkynes leads to 1,3-dienes.845 A combination of Zr and Cr reagents allows the coupling of alkynes to form linear polyenes.846Alkynes can also be coupled to allylic silyl ethers with a Ru catalyst to give dienes.847 Other alkyne–allylic coupling reactions are known to give dienes.848
This reaction can also be done intramolecularly, as in the cyclization of diyne 77 to (E,E)-exocyclic dienes (78) by treatment with a Zr,849 Rh,850 Ru,851 Au,852 or Pt complex.853 A similar reaction was reported using a Ti catalyst from a diyne amide.854 Rings of four, five, and six members were obtained in high yield; seven-membered rings in lower yield. When the reaction is applied to enynes, compounds similar to 78 are formed using various catalysts, but with only one double bond855 Larger rings can be formed from the appropriate enyne, including forming cyclohexadiene compounds.856 Spirocyclic compounds can be prepared from enynes in this manner using formic acid and a Pd catalyst.857
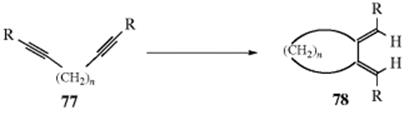
The Rh catalyzed cyclization of 1,6-enynes, triggered by arylboronic acids, leads to rings with an exocyclic alkylidene group.858 Alkynes are coupled to give enynes using Ni,859 Pd,860 Lu,861 and Ru862 catalysts. Similar products are obtained by cross-coupling terminal alkynes with an allene, using a combination of Pd and CuI catalysts.863 The reaction has been carried out internally to convert diynes to large-ring cycloalkynes with an exocyclic double bond.864 Diynes have also been cyclized to form cyclic enynes (an endocyclic double bond) using a diruthenium catalyst with ammonium tetrafluoroborate in methanol.865 Enynes are similarly cyclized to cyclic alkenes with an endocyclic C=C unit using a dicobalt catalyst.866
![]()
Enynes can also be converted to cyclic and bicyclic compounds using a Au867 Rh,868 Fe,869 or Pd870 catalyst. Enynes having a conjugated alkene unit also undergo this reaction in the presence of ZnBr2.871 Using mercury(II) triflate in water, cyclization leads to five-membered rings having an exocyclic double bond, and a pendant alcohol group.872 Enynes give cyclic compounds with an endocyclic double bond conjugated to another alkene unit (a conjugated diene) when treated with GaCl3873 or a Pt catalyst in an ionic liquid.874 Allene–alkenes give a similar product with a Pd875 or a Ru catalyst,876 as do alkyne–allenes with a dirhodium catalyst.877
There are many useful variations. Internal coupling of an alkyne with a vinyl halide, using triethylsilane and a Pd catalyst, gave the saturated cyclic compound with two adjacent exocyclic double bonds (a 2,3-disubstituted diene).878 Alkynes are added to propargyl acetates using Pd catalyst to give an alkyne–allene.879 Alkyne–alkenes were formed by coupling terminal alkynes and allenes in the presence of a Pd catalyst.880 An alkyne was coupled internally to give an allene using a Pd catalyst, to give an product that has an exocyclic methylene group and a vinyltin derivative.881 A similar process occurred with RhCl(PPh3)3 to incorporate a vinyl chloride.882 Allene–allylic halide systems reacted with phenylboronic acid and a Pd catalyst to give cyclopentane rings with two pendant vinyl groups, one of which contained a phenyl group.883
OS VIII, 190, 381, 505; IX, 310.
15-21 Addition of Organometallics to Double and Triple Bonds Not Conjugated to Carbonyls
Hydro-alkyl-addition
Neither Grignard reagents nor lithium dialkylcopper reagents generally add to ordinary C=C double bonds in the absence of a transition metal catalyst.884 Grignard reagents usually add only to double bonds susceptible to nucleophilic attack (e.g., fluoroalkenes and tetracyanoethylene).885 However, active Grignard reagents (benzylic, allylic) also add to the double bonds of allylic amines,886 and of allylic and homoallylic alcohols,887 as well as to the triple bonds of propargyl alcohols and certain other alkynols.888 Transition metal complexes facilitate the addition of Grignard reagents to alkenes. Examples include Ti,889 Mn,890 Zr,891 Ni,892 Fe,893 and Cu compounds.894Cyclopropenes are an exception, and an excess of a Grignard reagent will add at low temperatures.895 Cyclopropene derivatives also react with CuI and then allyl bromide.896
Benzylic alkenes react with silylmethyl Grignard reagents in the presence of oxygen.897 It is likely that cyclic intermediates are involved in these cases, in which the Mg coordinates with the heteroatom. Allylic, benzylic, and tertiary alkyl Grignard reagents also add to 1-alkenes and strained internal alkenes (e.g., norbornene), if the reaction is carried out in a hydrocarbon solvent (e.g., pentane) rather than ether, or in the alkene itself as solvent, heated, under pressure if necessary, to 60–130 °C.898 Yields are variable.
Intramolecular addition of RMgX to completely unactivated double and triple bonds has been demonstrated.899 The reaction of tosylates bearing a remote alkene unit and a Grignard reagent leads to cyclization when a Zr catalyst is used.900 The intramolecular addition of a CH2Br unit to the C=C unit of an allylic ether was accomplished using PhMgBr and a Co catalyst, give a functionalized THF and incorporation of the phenyl group on the C=C unit as well.901
In a useful variation, vinyl epoxides react with Grignard reagents and CuBr to give an allylic alcohol via reaction at the C=C unit and concomitant opening of the epoxide.902 Conjugated dienes react with arylmagnesium halides, Ph3SiCl, and a Pd catalyst to give a coupling product involving the reaction of 2 equiv of the diene and incorporation of two SiPh3 units.903 The Cr catalyzed formation of arylmagnesium compounds904 and the Rh catalyzed hydroarylation905 of alkynes are also known. The Pd catalyzed hydroarylation906 of alkynes is possible using arenediazonium salts907 as substrates or hydroarylation of 1,3-dienes with boronic acids.908
Organolithium reagents (primary, secondary, and tertiary alkyl and in some cases aryl) add to the double and triple bonds of allylic and propargylic alcohols,909 (tetramethylethylenediamine is a catalyst) and also to certain other alkenes containing hetero groups (e.g., OR, NR2, or SR). Mixing an organolithium reagent with transition metal compounds [e.g., CeCl3910 or Fe(acac)3]911 leads to addition of the alkyl group. Cyclopropane derivatives have been formed in this manner.912 The organolithium reagents can contain heteroatoms (e.g., nitrogen) elsewhere in the molecule, and the organolithium species can be generated from an intermediate organotin derivative.913 Organolithium reagents add to the less substituted C=C unit of conjugated dienes.914 Addition of butyllithium to alkenes has been observed with good enantioselectivity when sparteine was added.915
The intramolecular addition of RLi and R2CuLi has been reported.916 Organolithium reagents containing an alkene917 or alkyne918 unit cyclize919 at low temperatures and quenching with methanol replaces the new C–Li bond with C–H. Tandem cyclization is possible with dienes and enynes to form more than one ring,920 including bicyclic compounds.921 Tandem cyclization is possible with alkyne iodides922 or alkynes with a homoallylic CH2Li unit.923 The organolithium compound can be generated in situ by reaction of an organotin compound with butyllithium, allowing cyclization to occur upon treatment with an excess of LiCl.924
Unactivated alkenes or alkynes925 react with other organometallic compounds under certain conditions. The Pt catalyzed addition to alkynes leads to functionalized alkenes.926 Phenylboronic acids add to alkynes in the presence of a Co927 or a Pd catalyst.928 Trimethylaluminum reacts with 4-methyl-1-pentene, in the presence of Cl2ZrCp2, for example, and subsequent reaction with molecular oxygen leads to (2R),4-dimethyl-1-pentanol in good yield and 74% ee.929 These reagents also add to alkynes.930 Ruthenium catalysts have been used to mediate the addition of allylic alcohols to alkynes.931 Samarium iodide (SmI2) induces cyclization of a halide moiety to an alkyne unit932 or an alkene unit933 to form cyclized products. Copper complexes can catalyze similar cyclization to alkenes, even when an ester unit is present in the molecule.934 Allyl manganese compounds add to allenes to give nonconjugated dienes.935
Organomanganese reagents add to alkenes.936 Manganese triacetate [Mn(OAc)3], in the presence of cupric acetate, facilitates intramolecular cyclization of a halide unit to an alkene.937 Alkynes react with In reagents [e.g., (allyl)3In2I3] to form dienes (allyl substituted alkenes from the alkyne).938 Allylic halides add to propargyl alcohols using In metal to form the aryl organometallic in situ.939 Allyltin reagents add to alkynes in a similar manner in the presence of ZrCl4.940 Alkylzinc reagents add to alkynes to give substituted alkenes in the presence of a Pd catalyst.941 Allylzinc reagents add to alkynes in the presence of a Co catalyst.942 A vinyltellurium reagent adds to alkynes in the presence of CuI/PdCl2.943
Ketones with an α-hydrogen add to alkenes intramolecularly when heated in a sealed tube with CuCl2 and a Pd catalyst.944 A similar reaction was reported using Yb(OTf)3 and a Pd catalyst945 or an In catalyst.946 Keto esters add to alkynes using 10% benzoic acid and a Pd catalyst,947 or an In catalyst.948 1,3-Diketones add to dienes (1,4-addition) using a Pd catalyst,949 a AuCl3/AgOTf catalyst,950 and this addition has been done intramolecularly using 2.4 molar equivalents of CuCl2 and a Pd catalyst.951 The intermolecular addition of diesters (e.g., malonates) to alkynes was accomplished in acetic acid and a Pd catalyst under microwave irradiation.952 The enolate anion derived from the reaction of a nitrile with potassium tert-butoxide added to the less substituted carbon of the C=C unit of styrene in DMSO.953 Silyl enol ethers add to alkynes using a W catalyst.954 Malonate derivatives add to alkenes in the presence of an Al(OR)3 catalyst.955 1,3-Dicarbonyl compounds add to allenes in the presence of a Pd catalyst.956
Aryl iodides add to alkynes using a Pt complex in conjunction with a Pd catalyst.957 A Pd catalyst has been used alone for the same purpose,958 and the intramolecular addition of a arene to an alkene was accomplished with a Pd959or a GaCl3 catalyst.960 Alkyl iodides add intramolecularly to alkenes with a Ti catalyst,961 or to alkynes using In metal and additives.962 The latter cyclization of aryl iodides to alkenes was accomplished with In and I2963 or with SmI2.964
Aromatic hydrocarbons (e.g., benzene) add to alkenes using a Ru catalyst965 a catalytic mixture of AuCl3/AgSbF6,966 or a Rh catalyst,967 and Ru complexes catalyze the addition of heteroaromatic compounds (e.g., pyridine) to alkynes.968 Such alkylation reactions are clearly reminiscent of the Friedel–Crafts reaction (11-11). Palladium catalysts can also be used for the addition of aromatic compounds to alkynes,969 and Rh catalysts for addition to alkenes (with microwave irradiation).970 Note that vinylidene cyclopropanes react with furans and a Pd catalyst to give allylically substituted furans.971
Arylboronic acids (see Reaction 13-13) add to alkynes to give the substituted alkene using a Rh catalyst.972 Allenes react with phenylboronic acid and an aryl iodide, in the presence of a Pd catalyst, to give a substituted alkene.9732-Bromo-1,6-dienes react with phenylboronic acid with a Pd catalyst to give a cyclopentane with an exocyclic double bond and a benzyl substituent.974
An indirect addition converts alkynes to an organozinc compound using a Pd catalyst, which then reacts with allylic halides.975 Similarly, the reaction of an alkyne with Ti(OiPr)4/2 iPrMgCl followed by addition of an alkyne leads to a conjugated diene.976
OS 81, 121.
15-22 The Addition of Two Alkyl Groups to an Alkyne
Dialkyl-addition
![]()
Two different alkyl groups can be added to a terminal alkyne977 in one laboratory step by treatment with an alkylcopper–magnesium bromide reagent (called Normant reagents)978 and an alkyl iodide in ether–HMPA containing triethylphosphite.979 The groups add stereoselectively syn. The reaction, which has been applied to primary980 R′ and to primary, allylic, benzylic, vinylic, and α-alkoxyalkyl R′, involves initial addition of an intermediate alkylcopper reagent,981 followed by a coupling reaction (10-57):
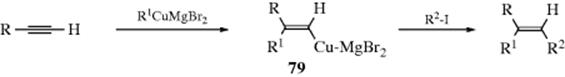
Acetylene itself (R = H) undergoes the reaction with R2CuLi instead of the Normant reagent.982 The use of R′ containing functional groups has been reported.983 If the alkyl iodide is omitted, the vinylic copper intermediate (79) can be converted to a carboxylic acid by the addition of CO2 (see Reaction 16-30) or to an amide by the addition of an isocyanate, in either case in the presence of HMPA and a catalytic amount of triethyl phosphite.984 The use of I2results in a vinylic iodide.985
![]()
Similar reactions, in which two alkyl groups are added to a triple bond, have been carried out with trialkylalanes (R3Al) and zirconium complexes as catalysts.986 Internal alkynes undergo bis(allylation) using a Ni catalysts and triallylindium.987 Allyl ethers and iodobenzene have also been added using a Zr complex.988 Similarly, allyl ethers and allyl chlorides have been added.989
Arylboronic acids (see Reaction 13-13) react with alkynes and 1 equiv of an aryl iodide, with a Pd catalyst, to add two aryl groups across the triple bond.990
OS VII, 236, 245, 290.
15-23 The Ene Reaction
Hydro-allyl-addition
An interesting addition of RH to a double bond involves the reaction of alkenes with an alkene having an allylic hydrogen (80), and is called the ene reaction or the ene synthesis.991 The reaction proceeds without a catalyst, but one of the components must be a reactive dienophile (reacts with a diene; see Reaction 15-60 for a definition of this word), such as maleic anhydride, but the other (which supplies the hydrogen) may be a simple alkene (e.g., propene) as long as there is an allylic hydrogen atom. Rather high reaction temperatures (250–450 °C) are common unless the substrates are very activated, but steric acceleration of the uncatalyzed ene reaction is known.992 The reaction is compatible with a variety of functional groups that can be appended to the ene and dienophile. N,N-Diallyl amides give an ene cyclization, for example.993 There has been much discussion of the mechanism of this reaction, and both concerted pericyclic (as shown above) and stepwise mechanisms have been suggested. The mechanism of the ene reaction of singlet (![]() ) oxygen with simple alkenes was found to involve two steps, with no intermediate.994 A retro-ene reaction is known with allylic dithiocarbonate.995 An intramolecular ene reaction between oxazolones and enol ethers leads to functionalized oxazolones.996 The reaction between maleic anhydride and optically active PhCHMeCH=CH2 gave an optically active product (81),997 which is strong evidence for a concerted rather than a stepwise mechanism.998 The reaction can be highly stereoselective.999
) oxygen with simple alkenes was found to involve two steps, with no intermediate.994 A retro-ene reaction is known with allylic dithiocarbonate.995 An intramolecular ene reaction between oxazolones and enol ethers leads to functionalized oxazolones.996 The reaction between maleic anhydride and optically active PhCHMeCH=CH2 gave an optically active product (81),997 which is strong evidence for a concerted rather than a stepwise mechanism.998 The reaction can be highly stereoselective.999
The reaction can be extended to less-reactive enophiles by the use of Lewis acid catalysts, especially alkylaluminum halides.1000 Titanium catalysts,1001 Sc,1002 LiClO41003 Y,1004 In,1005 Pd,1006 Co,1007 Ni catalysts,1008 as well as a combination of Ag and Au catalysts1009 have also been used. A magnesium–ene cyclization stereochemically directed by an allylic oxyanionic group has been reported.1010 The Lewis acid catalyzed reaction probably has a stepwise mechanism.1011 An Ir catalyzed ene reaction has been done in an ionic liquid.1012 An ene reaction of arynes with alkynes leads to aryl allenes.1013 An aza–ene reaction1014 has been used in a synthesis of enantioenriched piperidines, using two different imines as starting materials.1015 Ene reactions of imines are sometimes called imino–ene reactions.1016
The carbonyl–ene reaction1017 is also very useful, and often gives synthetically useful yields of products when catalyzed by Lewis acids.1018 Scandium triflate1019 and chromium complexes1020 are useful Lewis acids in this reaction. Asymmetric catalysts1021 for enantioselective carbonyl ene reactions have been reported using chiral Sc catalysts1022 In,1023 or chiral Ni catalysts.1024 Ketoester ene reactions with silyl enol ethers occurs in the presence of Pd and Ag catalysts.1025 Carbonyl–ene cyclization has been reported on silica gel at high pressure (15 kbar).1026 Ene reactions with imines,1027 nitrile oxides,1028 as well as nitroso–ene reactions are known.1029 Vinyl boronates have been prepared via a Ru catalyzed ene reaction.1030
OS IV, 766; V, 459. See also, OS VIII, 427.
15-24 The Michael Reaction
Hydro-bis(ethoxycarbonyl)methyl-addition, and so on
Compounds containing electron-withdrawing groups (Z is defined in Sec. 15.A.ii as an electron-withdrawing group) add to alkenes of the form C=C–Z in the presence of bases.1031 This is called the Michael reaction and is formally a conjugate addition.1032 The product formed, RCH2Z or RCHZZ′, can include aldehydes,1033 ketones,1034 esters,1035 diesters,1036 diketones,1037 keto-esters,1038 carboxylic acids, dicarboxylic acids,1039 nitriles,1040 vinyl sulfones,1041 nitro compounds (see below),1042 and others of the form ZCH3, ZCH2R, ZCHR2, and ZCHRZ′. 1043 In the most common examples, a base removes the acidic proton from the substrate adding to C=C–Z and the mechanism is as outlined in Section 15.A.ii. Michael addition is known to be catalyzed by phosphines,1044 as well as other organocatalysts.1045 Catalysts are known that are compatible with an aqueous medium.1046 A double Michaelprocess is possible, where conjugate addition to an alkynyl ketone is followed by an intramolecular Michael reaction to form a functionalized ring.1047 Vinylogous Michael reactions are well known, using a variety of nucleophilic species1048 (see Sec. 6.B for vinylogy). 1,6-Additions are also known.1049 Arylsilanes add to conjugated esters and amides in the presence of a Rh catalyst.1050 Nitro compounds add to conjugated ketones via Michael addition.1051Niroalkenes are Michael acceptors1052 for the enolate anions of β-keto esters1053 or malonate derivatives with a Ni catalyst1054 or an organocatalyst.1055 Other substrates have been added to nitroalkenes via Michael addition.1056Malonate derivatives also add to conjugated ketones,1057 and keto esters add to conjugated esters.1058 Vinyl sulfones undergo Michael addition.1059
It is known that 1,2-addition (to the C=O or C![]() N group) often competes and sometimes predominates (Reaction 16-38).1060 In particular, α,β-unsaturated aldehydes seldom give 1,4-addition.1061 The Michael reaction generally gives better yields with fewer side reactions by conversion of the nucleophile to its enolate form (a preformed enolate).1062 Phase-transfer catalysts have been used,1063 and ionic liquids have been used in conjunction with phase-transfer catalysis.1064 Transition metal compounds Ce,1065 Yb,1066 Bi,1067 Fe,1068 Ni,1069 Cu,1070 La,1071 Ru,1072 or Sc1073 also induce the reaction Conjugate addition has also been promoted by Y-zeolite,1074 and water-promoted Michael additions have also been reported.1075 Acylsilanes, when treated with fluoride ion, add to conjugated amides under certain conditions.1076 Addition to the meta-position of phenolic compounds leads to bicyclic ketones.1077Conjugate addition of nitrones using SmI2 has been reported.1078 Cyanide ion adds to Michael acceptors, and TMSCN also adds in a Michael reaction.1079
N group) often competes and sometimes predominates (Reaction 16-38).1060 In particular, α,β-unsaturated aldehydes seldom give 1,4-addition.1061 The Michael reaction generally gives better yields with fewer side reactions by conversion of the nucleophile to its enolate form (a preformed enolate).1062 Phase-transfer catalysts have been used,1063 and ionic liquids have been used in conjunction with phase-transfer catalysis.1064 Transition metal compounds Ce,1065 Yb,1066 Bi,1067 Fe,1068 Ni,1069 Cu,1070 La,1071 Ru,1072 or Sc1073 also induce the reaction Conjugate addition has also been promoted by Y-zeolite,1074 and water-promoted Michael additions have also been reported.1075 Acylsilanes, when treated with fluoride ion, add to conjugated amides under certain conditions.1076 Addition to the meta-position of phenolic compounds leads to bicyclic ketones.1077Conjugate addition of nitrones using SmI2 has been reported.1078 Cyanide ion adds to Michael acceptors, and TMSCN also adds in a Michael reaction.1079
Intramolecular versions of Michael addition are known.1080 The intramolecular Michael addition of an α-chloro ketone enolate anion, formed in situ using DABCO, leads to formation of a bicyclo[4.1.0] diketone.1081
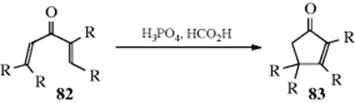
An important cyclization procedure involves the acid-catalyzed addition of diene-ketones (e.g., 82), where one conjugated alkene adds to the other conjugated alkene to form cyclopentenones (83). This is called the Nazarov cyclization.1082 While it may be categorized in Reaction 15-20 because one alkene unit adds to another, the addition is formally a Michael addition, and so is placed here. Structural variations are possible that prepare a variety of cyclopentenones. The steric influence of substituents has been discussed,1083 and the rate-accelerating influence observed in N- and S-heterocycles.1084 Substituents on the C=C units lead to cyclopentenones that bear those substituents. Gold1085 and V catalyzed1086 cyclizations have been reported, as well as a Sc catalyzed cyclization in water.1087 Heating dienones in DME or an ionic liquid has been shown to give the Nazarov product without addition of a Lewis acid.1088 Allenes participate in Nazarov cyclization reactions.1089 A vinylogous Nazarov reaction is involved in the cyclization of cross-conjugated trienes1090 (see Sec. 6.B for vinylogy). The use of a chiral ligand gave the cyclopentenone with modest enantioselectivity.1091 Reductive cyclization can also give the nonconjugated five-membered ring.1092 Note that a retro-Nazarov is possible with α-bromocyclopentanones.1093 In one variation using an Al complex, a cyclohexenone was formed.1094 There is a so-called interrupted Nazarov, in which an amine is trapped after cyclization to give an α-amino conjugated cyclopentenone.1095 Spirocycles have been prepared via a tandem Nazarov–Wagner–Meerwein sequence.1096

In a Michael reaction with suitably different R groups, two new stereogenic centers may be created (see 84). In a diastereoselective process, one of the two pairs is formed exclusively or predominantly as a racemic mixture.1097When either or both of the reaction components have a chiral substituent, the reaction can be enantioselective (only one of the four diastereomers formed predominantly).1098 There are many examples of catalytic enantioselective Michael additions,1099 often by the use of a chiral catalyst1100 or by using optically active enamines instead of enolates.1101 Common chiral catalysts used with carbonyl substrates include Ru,1102 Ni,1103 Sr,1104 and Al(salen) complexes.1105 Chiral organocatalysts1106 (e.g., oxazolidinones)1107 have been developed and chiral imines have also been used.1108 Certain antibodies have been used to facilitate chiral, intramolecular Michael addition reactions.1109Enzymes have been used as for asymmetric Michael reactions.1110 Addition of chiral additives to the reaction (e.g., metal–salen complexes,1111 proline derivatives,1112 or (−)-sparteine1113) lead to product formation with good-to-excellent asymmetric induction. Ultrasound has been used to promote asymmetric Michael reactions.1114 In reactions of enolate anions, both the enolate anion and substrate can exist as (Z) or (E) isomers. With enolates derived from ketones or carboxylic esters, the (E) enolates gave the syn pair of enantiomers (Sec. 4.G), while (Z) enolates gave the anti pair.1115
When the substrate contains gem-Z groups, (e.g., 86), bulky groups can be added, if the reaction is carried out under aprotic conditions. For example, addition of enolate 85 to 86 gave 87 in which two adjacent quaternary centers have been formed.1116
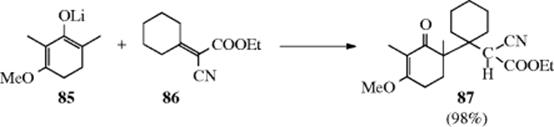
In certain cases, Michael reactions can take place under acidic conditions.1117 Michael-type addition of radicals to conjugated carbonyl compounds is also known.1118 Radical addition can be catalyzed by Yb(OTf)3,1119 but radicals add under standard conditions as well, even intramolecularly.1120 Electrochemical-initiated Michael additions are known.
Alkynes are reactive, and Michael reactions are sometimes applied to substrates of the type C![]() C–Z, where the coproducts are conjugated systems of the type C=C–Z.1121 Terminal alkynes add to conjugated systems.1122 Due to the greater susceptibility of triple bonds to nucleophilic attack, it is even possible for nonactivated alkynes (e.g., acetylene), to be substrates in this reaction.1123
C–Z, where the coproducts are conjugated systems of the type C=C–Z.1121 Terminal alkynes add to conjugated systems.1122 Due to the greater susceptibility of triple bonds to nucleophilic attack, it is even possible for nonactivated alkynes (e.g., acetylene), to be substrates in this reaction.1123
Silyl enol ethers (e.g., 88) add to α,β-unsaturated ketones and esters when catalyzed1124 by TiCl41125 or InCl3.1126 Aluminum compounds also catalyze this reaction1127 and the reaction has been done in neat tri-n-propylaluminum.1128 A solid-state version of the reaction used alumina·ZnCl2.1129 Tin-enolates have been used.1130 This reaction has been performed with good diastereoselectivity,1131 and silyl enol ethers have been used in conjunction with chiral additives.1132
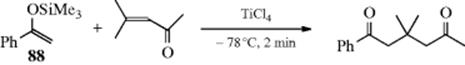
The reaction of C=C–Z compounds with enamines (10-69) can also be considered a Michael reaction.
OS I, 272; II, 200; III, 286; IV, 630, 652, 662, 776; V, 486, 1135; VI, 31, 648, 666, 940; VII, 50, 363, 368, 414, 443; VIII, 87, 210, 219, 444, 467; IX, 526. See also, OS VIII, 148.
15-25 1,4-Addition of Organocuprates and Other Organometallic Compounds to Activated Double Bonds
Hydro-alkyl-addition
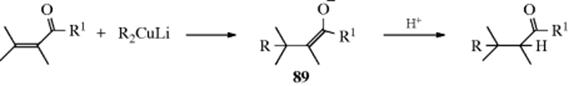
Lithium dialkylcopper reagents (R2CuLi reagents,1133 also known as Gilman reagents, see Reaction 10-57) add to α,β-unsaturated aldehydes1134 and ketones (R′ = H, R, Ar) and other systems of the form C![]() C–C=O1135 to give conjugate addition products1136 in a reaction closely related to the Michael reaction. α,β-Unsaturated esters are less reactive,1137 and the corresponding acids do not react at all. The R group can be primary alkyl, vinylic,1138 or aryl. If Me3SiCl is present, the reaction takes place much faster and in higher yield; in the example given, the product is the silyl enol ether of 89 (see Reaction 12-17).1139 The use of Me3SiCl also permits good yields with allylic R groups.1140 Conjugated alkynyl-ketones also react via 1,4-addition to give substituted alkenyl-ketones.1141 Solvent effects are important for the reactivity of organocuprates,1142 which influence aggregation and aggregation state of the dialkyl cuprate.1143 Organocuprate reactions have been shown to undergo oscillations in complexation.1144
C–C=O1135 to give conjugate addition products1136 in a reaction closely related to the Michael reaction. α,β-Unsaturated esters are less reactive,1137 and the corresponding acids do not react at all. The R group can be primary alkyl, vinylic,1138 or aryl. If Me3SiCl is present, the reaction takes place much faster and in higher yield; in the example given, the product is the silyl enol ether of 89 (see Reaction 12-17).1139 The use of Me3SiCl also permits good yields with allylic R groups.1140 Conjugated alkynyl-ketones also react via 1,4-addition to give substituted alkenyl-ketones.1141 Solvent effects are important for the reactivity of organocuprates,1142 which influence aggregation and aggregation state of the dialkyl cuprate.1143 Organocuprate reactions have been shown to undergo oscillations in complexation.1144
An excess of the cuprate reagent relative to the conjugated substrate is often required. In general, only one of the R groups of R2CuLi adds to the substrate; the other is wasted with respect to the conjugated substrate. This can be a limitation where the precursor (RLi or RCu, see Reaction 12-36) is expensive or available in limited amounts, particularly if an excess of the reagent is required. The difficulty of group transfer can be overcome by using one of the mixed reagents [R(R′C![]() C)CuLi,1145 R(O-t-Bu)CuLi,1146 R(PhS)CuLi1147 each of which transfers only the R group. Mixed reagents are easily prepared by the reaction of RLi with R′C
C)CuLi,1145 R(O-t-Bu)CuLi,1146 R(PhS)CuLi1147 each of which transfers only the R group. Mixed reagents are easily prepared by the reaction of RLi with R′C![]() CCu (R′ = n-Pr or t-Bu), t-BuOCu, or PhSCu, respectively. A further advantage of the mixed reagents is that good yields of addition product are achieved when R is tertiary, so that use of one of them permits the introduction of a tertiary alkyl group. The mixed reagents R(CN)CuLi1148 (prepared from RLi and CuCN) and R2Cu(CN)Li21149 also selectively transfer the R group.1150 With mixed cuprates, one of the ligands may be less prone to transfer than the other, as R(R′Se)Cu(CN)Li2, leading to selective transfer of the R group.1151 This less transferable ligand is sometimes referred to as a “dummy ligand”. The selectivity of ligand transfer depends on two factors, thermodynamics of groups (e.g., alkyl or thioalkyl) and kinetic reactivity of groups (e.g., silylalkyl or vinyl).1152 The selectivity arises in the Cu(III) intermediate formed by complexation of the cuprate and the unsaturated carbonyl compound.1150,1153 A Cu(III) complex has been detected using rapid injection NMR.1154
CCu (R′ = n-Pr or t-Bu), t-BuOCu, or PhSCu, respectively. A further advantage of the mixed reagents is that good yields of addition product are achieved when R is tertiary, so that use of one of them permits the introduction of a tertiary alkyl group. The mixed reagents R(CN)CuLi1148 (prepared from RLi and CuCN) and R2Cu(CN)Li21149 also selectively transfer the R group.1150 With mixed cuprates, one of the ligands may be less prone to transfer than the other, as R(R′Se)Cu(CN)Li2, leading to selective transfer of the R group.1151 This less transferable ligand is sometimes referred to as a “dummy ligand”. The selectivity of ligand transfer depends on two factors, thermodynamics of groups (e.g., alkyl or thioalkyl) and kinetic reactivity of groups (e.g., silylalkyl or vinyl).1152 The selectivity arises in the Cu(III) intermediate formed by complexation of the cuprate and the unsaturated carbonyl compound.1150,1153 A Cu(III) complex has been detected using rapid injection NMR.1154
Various functional groups (e.g., OH and unconjugated C=O groups) may be present in the substrate when organocuprates are employed.1155 There is generally little or no competition from 1,2-addition (to the C=O). However, when R is allylic, 1,4-addition is observed with some substrates and 1,2-addition with others.1156 The R2CuLi group also adds to α,β-unsaturated sulfones,1157 but not to simple α,β-unsaturated nitriles.1158 Organocopper reagents (RCu) as well as certain R2CuLi add to α,β-unsaturated and acetylenic sulfoxides.1159 The reaction has been carried out1160 with α,β-acetylenic ketones,1161 esters, and nitriles. Conjugate addition to α,β-unsaturated and acetylenic acids and esters, as well as ketones, can be achieved by the use of the coordinated reagents RCu·BF3 (R = primary).1162 Amine units have been transferred using α-lithio amides, CuCN, and various additives, which gave conjugate addition of an amidomethyl unit [–CH2N(Me)Boc].1163 Other amino-cuprates are known to give conjugate addition reactions.1164
Conjugate addition of the cuprate to the α,β-unsaturated ketone leads to an enolate ion (89), as noted above. It is possible for this enolate anion to react with an electrophilic species (tandem vicinal difunctionalization), in some cases at the O and in other cases at the C.1165 For example, if an alkyl halide R2X is present (R2 = primary alkyl or allylic), the enolate (89) can be alkylated directly to give 90.1166 Thus, by this method, both the α and β positions of a ketone are alkylated in one synthetic operation (see also, Reaction 15-22).
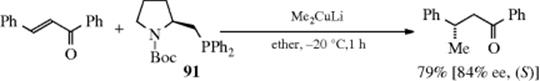
As with the Michael reaction (15-24) the 1,4-addition of organometallic compounds has been performed diastereoselectivity1167 and enantioselectively.1168 The influence of solvent and additives on yield and selectivity has been examined.1169 Addition of chiral ligands to the organocuprate conjugate addition reaction leads to alkylation with good-to-excellent enantioselectivity.1170 The conjugate addition of dimethyl cuprate in the presence of a chiral ligand (e.g., 91) is an example.1171 Chiral bis(oxazoline) copper catalysts have been used for the conjugate addition of indoles to α,β-unsaturated esters.1172 In the presence of a chiral additive and a Cu catalyst, conjugate addition to trisubstituted cyclohexeneones leads to the formation of stereogenic quaternary centers.1173 Enantioselectivity was improved by the addition of styrene in the conjugate addition reactions of α-halo enones.1174 Enantioselectivity is effectively controlled by the choice of ligand and its interaction with the copper compound, where different ligands on the metal may lead to differences in selectivity.1175 Chiral templates have also been used with Grignard reagents, directly1176 and in the presence of AlMe2Cl.1177
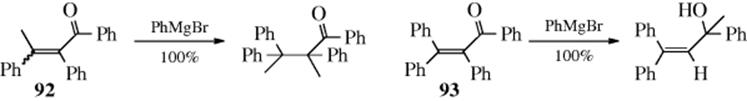
Other organometallic compounds add to conjugated systems. Grignard reagents add to conjugated substrates (e.g., α,β-unsaturated ketones, cyano-ketones,1178 esters, and nitriles),1179 but 1,2-addition may seriously compete:1180The extent of 1,4-addition of Grignard reagents can be increased by the use of a Cu catalyst [e.g., CuCl and Cu(OAc)2],1181 forming a magnesium cuprate in situ. Formation of the conjugate addition product is often controlled by steric factors. Thus 92 with phenylmagnesium bromide gives 100% 1,4-addition, while 93 gives 100% 1,2-addition. In general, substitution at the carbonyl group increases 1,4-addition, while substitution at the double bond increases 1,2-addition. In most cases, both products are obtained, but α,β-unsaturated aldehydes nearly always give exclusive 1,2-addition when treated with Grignard reagents. Grignard reagents mixed with CeCl3 generates a reactive species that gives primarily 1,4-addition.1182 It is likely that alkylcopper reagents, formed from RMgX and Cu+ (cupric acetate is reduced to cuprous ion by excess RMgX), are the actual reactive species in these cases.1134 Alkylidene malonic ester derivatives [C=C(CO2R)] increase the facility of 1,4-addition with the two electron-withdrawing groups.1183
A dialkyl copper–magnesium iodide complex (R2Cu·MgI) has been used for conjugate addition to chiral α,β-unsaturated amides.1184 Catalytic enantioselective conjugate addition has been reported with Grignard reagents.1185
Organolithium reagents1186 generally react with conjugated aldehydes, ketones, and esters by 1,2-addition,1187 but 1,4-addition was achieved with esters of the form C=C–CO2Ar, where Ar was a bulky group (e.g., 2,6-di-tert-butyl-4-methoxyphenyl).1188 Alkyllithium reagents can be made to give 1,4-addition with α,β-unsaturated ketones1189 and aldehydes,1190 if the reactions are conducted in the presence of HMPA.1191 Among organolithium reagents that have been found to add 1,4 in this manner are 2-lithio-1,3-dithianes (see Reaction 10-71),1192 vinyllithium reagents,1193 and α-lithio allylic amides.1194 Lithium–halogen exchange (12-22) generates an organolithium species that adds intramolecularly to conjugated esters to give cyclic and bicyclic products.1195 A reagent based on RMgX–3 MeLi gave conjugate addition with α,β-unsaturated amides and carboxylic acid derivatives.1196 1,4-Addition of alkyllithium reagents to α,β-unsaturated aldehydes can also be achieved by converting the aldehyde to a benzothiazole derivative (masking the aldehyde function),1197 from which the aldehyde group can be regenerated. α,β-Unsaturated nitro compounds undergo conjugate addition with aryllithium reagents, and subsequent treatment with acetic acid gives the α-aryl ketone.1198
If the organolithium reagent is complexed, 1,4-addition is more successful. The reaction of an aryllithium reagent with B(OMe)3, for example, led to a Rh catalyzed conjugate addition with excellent enantioselectivity when a chiral ligand was employed.1199 Allylic Te reagents that are treated with lithium diisopropyl amide and then conjugated esters give the 1,4-addition product, which cyclizes to form the corresponding cyclopropane derivative.1200
Organozinc compounds add to conjugated systems, especially dialkyl zinc compounds (R2Zn). Many dialkylzinc compounds can be used, including vinylzinc compounds.1201 The use of chiral ligands is effective for conjugate addition of dialkylzinc compounds to α,β-unsaturated ketones, esters, and so on,1202 including conjugated lactones.1203 The addition of a chiral complex to dialkylzinc compounds leads to enantioselective conjugate addition in conjunction with Cu(OTf)21204 CuCN,1205 or other copper compounds.1206 Chiral ionic liquids have also been employed.1207 Diethylzinc adds to conjugated nitro compounds in the presence of a catalytic amount of Cu(OTf)2 to give the conjugate addition product.1208 1,6-Addition of dialkylzinc compounds has been reported, in the presence of a Rh catalyst.1209 Other transition metal compounds can be used in conjunction with dialkylzinc compounds1210 or with arylzinc halides (ArZnCl).1211 Reaction of alkyl iodides with Zn/CuI and ultrasound generates an organometallic that adds to conjugated esters.1212 Diarylzinc compounds (prepared with the aid of ultrasound) in the presence of nickel acetylacetonate, undergo 1,4-addition not only to α,β-unsaturated ketones, but also to α,β-unsaturated aldehydes.1213 Mixed-alkylzinc compounds also add to conjugated systems.1214 Functionalized allylic groups can be added to terminal alkynes with allylic halides, zinc, and ultrasound, to give 1,4-dienes.1215 Internal alkynes undergo 1,4-addition to conjugated esters using a combination of zinc metal and a Co complex as catalysts.1216
Trialkylalanes (R3Al) add 1,4 to α,β-unsaturated carbonyl compounds in the presence of nickel acetylacetonate1217 or Cu(OTf)2.1218 In the presence of aluminum chloride, benzene reacts with conjugated amides to add a phenyl group to C-4.1219 Alkyl halides react via conjugate addition using BEt3 or AlEt3.1220 Other metals are known to catalyzed conjugate addition of alkyl or aryl groups, including Co.1221 An In/Cu mediated conjugate addition reaction is known using unactivated alkyl iodides.1222
Terminal alkynes add to conjugated systems when using a Ru,1223 Pd,1224 Ni,1225 or a Rh catalyst.1226 Intramolecular addition of terminal alkynes, in the presence of phenylboronic acid and a Rh catalyst, leads to cyclic compounds.1227 Lithium tetraalkylgallium reagents give 1,4-addition.1228 Trimethyl(phenyl)tin and a Rh catalyst gives conjugate addition of a methyl group1229 and tetraphenyltin and a Pd catalyst adds a phenyl group.1230Triphenylbismuth (Ph3Bi) and a Rh catalyst give conjugate addition of the phenyl group upon exposure to air.1231 Similar reactivity is observed with a Pd catalyst in aqueous media.1232 Allyltin compounds add an allyl group in the presence of a Sc catalyst.1233 Benzylic bromides add to conjugated nitriles using a 2:1 mixture of CrCl3 and Mn metal.1234 Aryl halides add in the presence of NiBr2.1235 Vinyl Zr complexes undergo conjugate addition when using a Rh catalyst.1236
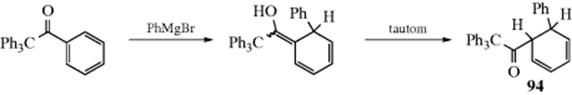
In certain cases, Grignard reagents add 1,4 to aromatic systems to give 94 after tautomerization (Sec. 2.N.i) of the initial formed enol.1237 Such cyclohexadienes are easily oxidizable to benzenes (often by atmospheric oxygen), so this reaction becomes a method of alkylating and arylating suitably substituted (usually hindered) aryl ketones. A similar reaction has been reported for aromatic nitro compounds where 1,3,5-trinitrobenzene reacts with excess methylmagnesium halide to give 2,4,6-trinitro-1,3,5-trimethylcyclohexane.1238

The mechanisms of most of these reactions are not well known. The 1,4-uncatalyzed Grignard reaction has been postulated to proceed by the cyclic mechanism shown, but there is evidence against it.1239 The R2CuLi1240 and copper-catalyzed Grignard additions may involve a number of mechanisms, since the actual attacking species and substrates are so diverse.1241 A free radical mechanism of some type (perhaps SET) has been suggested1242 although the fact that retention of configuration at R has been demonstrated in several cases completely rules out a free R• radical.1243 For simple α,β-unsaturated ketones (e.g., 2-cyclohexenone and Me2CuLi), there is evidence1244 for this mechanism:
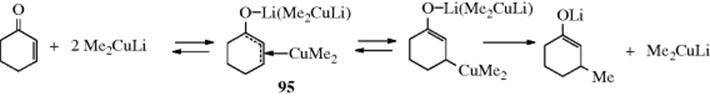
95 is a d, π∗ complex, with bonding between copper, as a base supplying a pair of d electrons, and the enone as a Lewis acid using the π∗ orbital of the allylic system.1242 The ![]() NMR spectrum of an intermediate similar to 95 has been reported.1245
NMR spectrum of an intermediate similar to 95 has been reported.1245
For the addition of organocopper reagents to alkynes and conjugated dienes, see Reaction 15-22.
OS IV, 93; V, 762; VI, 442, 666, 762, 786; VIII, 112, 257, 277, 479; IX, 328, 350, 640.
15-26 The Sakurai Reaction
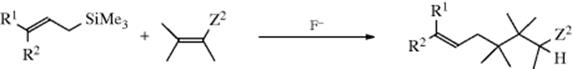
Allylic silanes (R2C=CHCH2SiMe3) can be added to conjugated systems rather than silyl enol ethers in what is known as the Sakurai reaction.1246 For example, an allyl group can be added to α,β-unsaturated carboxylic esters, amides and nitriles, with CH2=CHCH2SiMe3 and F− ion (see Reaction 15-47).1247 This reagent gave better results than lithium diallylcuprate (Reaction 15-25). Catalytic Sakurai reactions are known.1248 The Pd catalyzed reaction of conjugated ketones with PhSi(OEt)3 and SbCl3 and Bu4NF in acetic acid gave the 1,4-addition product.1249 A similar reaction was reported using PhSi(OMe)3 with a Rh catalyst.1250 Silver fluoride was used to catalyze the reaction with allyl(trimethoxy)silane.1251 In a related reaction, Ph2SiCl2, NaF, and a Rh catalyst gives conjugate addition of a phenyl group to α,β-unsaturated ketones.1252 An interesting Rh catalyzed, conjugate addition of a phenyl group was reported using a siloxane polymer bearing Si–Ph units.1253 The Sakurai reaction has been used in multi-component reactions.1254
15-27 Conjugate Addition of Boranes to Activated Double Bonds
Hydro-alkyl-addition (overall transformation)
![]()
Just as trialkylboranes add to simple alkenes (Reaction 15-16), they rapidly add to the double bonds of acrolein, methyl vinyl ketone, and certain related derivatives in THF to give enol borinates (also see Reaction 10-68), which can be hydrolyzed to aldehydes or ketones.1255 If water is present in the reaction medium from the beginning, the reaction can be run in one laboratory step. Since the boranes can be prepared from alkenes (Reaction 15-16), this reaction provides a means of lengthening a carbon chain by three or four carbons, respectively. Compounds containing a terminal alkyl group [e.g., such as crotonaldehyde (CH3CH=CHCHO) and 3-penten-2-one], fail to react under these conditions, as does acrylonitrile, but these compounds can be induced to react by the slow and controlled addition of O2 or by initiation with peroxides or UV light.1256 A disadvantage is that only one of the
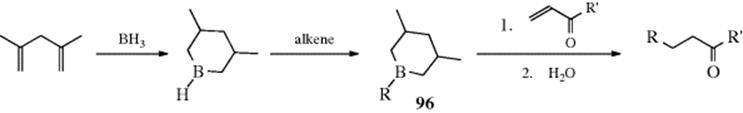
three R groups of R3B adds to the substrate, so that the other two are wasted. This difficulty is overcome by the use of a β-alkyl borinate (e.g., 96),1257 which can be prepared as shown. Borinate (96, R = tert-butyl) can be made by treatment of 96 (R = OMe) with t-BuLi. The use of this reagent permits tert-butyl groups to be added. β-1-Alkenyl-9-BBN compounds β-RCH=CR′-9-BBN (prepared by treatment of alkynes with 9-BBN or of RCH=CR′Li with β-methoxy-9-BBN1258) add to methyl vinyl ketones to give, after hydrolysis, γ,δ-unsaturated ketones,1259 although β-R-9-BBN, where R = a saturated group, are not useful here, because the R group of these reagents does not preferentially add to the substrate.1253 Transition metals catalyze the addition of trialkylboranes to conjugated systems (e.g., the addition of allylboranes in the presence of a Ni catalyst).1260 The Ni catalyzed addition was enhanced by the addition of methanol.1261
The corresponding β-1-alkynyl-9-BBN compounds also give the reaction.1262 Since the product 97 is an α,β-unsaturated ketone, it can be made to react with another BR3, the same or different, to produce a wide variety of ketones (98).
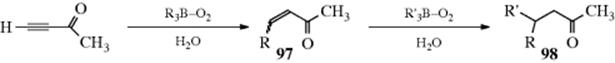
Vinyl boranes add to conjugated ketones in the presence of a Rh catalyst (with high asymmetric induction in the presence of BINAP).1263 Alkynyl-boranes also add to conjugated ketones, in the presence of BF3.1264
Other boron reagents add to conjugated carbonyl compounds.1265 Tetraphenylborates add to conjugated alkynes in the presence of a Pd catalyst in a reaction known as hydrophenylation.1266 Alkynyl boronate esters (Reaction 12-28) give conjugate addition1267 in the presence of boron trifluoride etherate,1268 as do arylboronic acids (Reaction 12-28) with a Rh,1269 Pd,1270 or a Bi catalyst.1271 Diethylzinc has also been used.1272 Aryl boronic acids add to the double bond of vinyl sulfones in the presence of a Rh catalyst.1273 Vinylboronic acids add directly to conjugated ketones.1274 An Ir catalyzed 1,6-addition of arylboronic acids is known.1275 Conjugated alkynes undergo conjugate addition with arylboronic acids in the presence of a Cu catalyst.1276 Organocatalysts have also been used for the conjugate addition of arylboronic acids to conjugated systems.1277 Potassium vinyltrifluoroborates (see Reactions 10-59, 13-10, and 13-13) give 1,4-addition with a Rh catalyst,1278 as do aryltrifluoroborates.1279 The Rh catalyzed addition of vinyl tetrafluoroborates has been reported.1280
In the presence of a Rh catalyst, LiBPh(OMe)3 gave conjugate addition of the phenyl group to α,β-unsaturated esters.1281 The fact that these reactions are catalyzed by free radical initiators and inhibited by galvinoxyl1282 (a free radical inhibitor) indicates that free-radical mechanisms are involved.
15-28 Radical Addition to Activated Double Bonds
Hydro-alkyl-addition
![]()
In a reaction similar to 15-25, alkyl groups can be added to alkenes activated by such groups as COR′, CO2R′, CN, and even Ph.1283 This is a radical addition reaction.1284 In the method illustrated above, the R group comes from an alkyl halide (R = primary, secondary, or tertiary alkyl; X = Br or I) and the hydrogen from the tin hydride (H atom transfer agents). The reaction of tert-butyl bromide, Bu3SnH and AIBN (Sec. 14.A.i), for example, adds a tert-butyl group to a conjugated ester via 1,4-addition.1285 An alkene is converted to an alkylborane with catecholborane (Reaction 12-28) and when treated with a conjugated ketone and O2, radical conjugate addition leads to the β-substituted ketone.1286 The Bu3SnH can also be generated in situ, from R3SnX and NaBH4. Like Reaction 15-27, these additions have free radical mechanisms. The reaction has been used for free radical cyclizations of the type discussed in Reaction 15-30.1287 Such cyclizations normally give predominant formation of five-membered rings, but large rings (11–20 members) have also been synthesized by this reaction.1288
A BEt3 (see Sec. 14.A.i) initiated reaction of conjugated amides with an alkyl iodide, in the presence of Bu3SnH and O2, leads to conjugate addition of the alkyl group.1289 Enantioselective radical addition has been reported.1290
Conjugate addition is possible using photolysis. The photoinduced 1,4-addition of indoles to enones proceeds when irradiated at 350 nm.1291
OS VII, 105.
15-29 Radical Addition to Unactivated Double Bonds1292
Alkyl-hydro-addition
![]()
Radical addition to alkenes is usually difficult, except when addition occurs to conjugated carbonyl compounds (Reaction 15-24). An important exception involves radicals bearing a heteroatom α to the carbon bearing the radical center. Such radicals are much more stable and can add to alkenes, usually with anti-Markovnikov orientation, as in the radical induced addition of HBr to alkenes (Reaction 15-2).1293
Examples of this type of reaction include the use of alcohol-, ester-,1294 amino-, and aldehyde-stabilized radicals.483 The alkyl group of alkyl iodides adds to alkenes with BEt3/O2 as the initiator and in the presence of a tetraalkylammonium hypophosphite.1295 The radical generated from (EtO)2POCH2Br adds to alkenes to generate a new phosphonate ester.1296 α-Bromo esters add to alkenes in the presence of BEt3/air to give a γ-bromo ester.1297 α-Bromo amides add the Br and the acyl carbon to an alkene using Yb(OTf)3 with BEt3/O2 as the radical initiator.1298 α-Iodo amides add to alkenes using a water-soluble azobis initiator (see Sec. 14.A.i) to give the iodo ester, which cyclizes under the reaction conditions to give a lactone.1299 β-Keto dithiocarbonates [RC(=O)–C–SC(=S)OEt] generate the radical in the presence of a peroxide and add to alkenes.1300 2-Fluoropyridyl derivatives of allylic alcohols react with xanthates in the presence of lauroyl peroxide to give alkenes.1301 Malonate derivatives add to alkenes in the presence of a mixture of Mn/Co catalyst, in oxygenated acetic acid.1302
Other radicals can add to alkenes, and the rate constant for the addition of methyl radicals to alkenes has been studied.1303 The rate of radical additions to alkenes in general has also been studied.1304 The kinetic and thermodynamic control of a radical addition regiochemistry has also been studied.1305 Alkynes are generally less reactive than alkenes in radical coupling reactions.1306 Nonradical nucleophiles usually react faster with alkynes than with alkenes, however.1307
15-30 Radical Cyclization1308
Alkyl-hydro-addition

ω-Haloalkenes generate radicals upon treatment with radical initiator reagents (e.g., AIBN) or under photolysis conditions,1309 and the radical carbon adds to the alkene to form cyclic compounds.1310 This intramolecular addition of a radical to an alkene is called radical cyclization. In a typical example, haloalkene (101) reacts with the radical produced by AIBN to give radical 100. The radical can add to the more substituted carbon to give 102 via a 5-exo-trig reaction (Sec. 6.E).1311 If the radical adds to the less substituted carbon, 103 is formed via a 6-endo-trig reaction.1312 In both cases, the product is another radical, which must be converted to an unreactive product. This is generally accomplished by adding a hydrogen-transfer agent1313 [e.g., tributyltin hydride (Bu3SnH)], which reacts with 102 to form methylcyclopentane and Bu3Sn•, or with 103 to give cyclohexane. The Bu3Sn• formed in both cases usually dimerizes to form Bu3SnSnBu3. Cyclization can compete with hydrogen transfer1314 from Bu3SnH to 100 to give 99, the reduction product. Atom-transfer cyclization is possible with other atoms (e.g., halogen), catalyzed by InCl31315 or CuBr.1316 Tin-free radical cyclizations are known using peroxyacids.1317
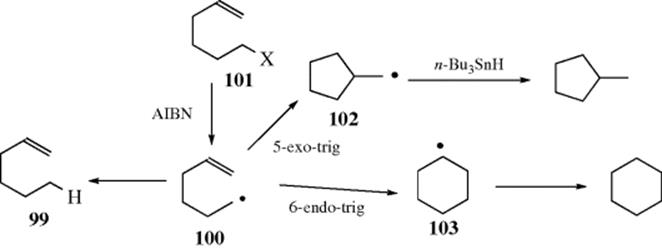
In general, formation of the five-membered ring dominates the cyclization, but if addition to the C=C unit is relatively slow, the reduction product is formed preferentially. Radical rearrangements can also diminish the yield of the desired product.1318 Given a choice between a larger and a smaller ring, radical cyclization generally gives the smaller ring,1319 but not always.1320 Formation of other size rings is possible of course. A 4-exo-trig radical cyclization has been studied,1321 selectivity in a 7-endo versus 6-exo cyclization,1322 and also an 8-endo-trig reaction.1323 In radical cyclization to form large rings, 1,5- and 1,9-hydrogen atom abstractions can pose a problem1324 Ring expansion during radical cyclization is possible when the terminal intermediate is a cyclobutylcarbinyl radical.1325
The mechanism of this reaction has been discussed.1326 Cyclization via 5-endo-dig transition states require reorientation of the radical orbital needed to reach the in-plane acetylene π orbital in the bond-forming step, with accompanying loss of conjugative stabilization, and an increase in the activation energy. Therefore, many 5-endo cyclizations undergo H abstraction or equilibration with an isomeric radical.1327
In cases where hydrogen atom transfer gives primarily reduced products, one solution to promote cyclization generates the radical by photochemical cleavage of Bu3Sn–SnBu3 and the resulting carbon radical can cyclize (see Reaction 15-46).1328 A halogen atom transfer agent (e.g., iodoethane) is used rather than a hydrogen-transfer agent, so the final product is an alkyl iodide.
A mixture of a Grignard reagent and CoCl2 has been used to initiate aryl radical cyclizations.1329 Titanium(III)-mediated radical cyclizations are known,1330 and SmI2 mediated reactions are possible in the presence of a Ni catalyst.1331 Organoborane-mediated radical cyclizations are known (see Sec. 14.A.i).1332 The influence of the halogen atom on radical cyclization has been studied.1333
Both phenylthio1334 and phenylseleno groups1335 can be used as “leaving groups” for radical cyclization, where S or Se atom transfer leads to formation of the radical. A seleno ester (R2N–CH2C(–O)SeMe) has also been used with (Me3Si)3SiH (tristrimethylsilylane, TTMSS) and AIBN to generate R2NCH2•.1336 O-Phosphonate esters have also served as the leaving group.1337 N-(2-bromophenylbenzyl)methylamino have been used as leaving groups for formation of a radical.1338
Radical cyclization reaction often proceeds with high diastereoselectivity1339 and high asymmetric induction when chiral precursors are used. Internal alkynes are good substrates for radical cyclization,1340 but terminal alkynes tend to give mixtures of exo/endo-dig products (Sec. 6.E).1341 Radical cyclization has been used to transfer asymmetry from transient atropisomers to form lactams.1342
Radical cyclization is compatible with the presence of other functional groups, and heterocyclic rings may be formed via radical cyclization.1343 Aryl radicals participate in radical cyclization reactions when the aromatic ring has an alkene or alkyne substituent. o-Iodo aryl allyl ethers cyclize to benzofuran derivatives, for example, when treated with AIBN, aq H3PO2 and NaHCO3 in ethanol.1344 Cyclization of vinyl radicals1345 and allenyl radicals1346 are also well known. Treatment of XCH2CON(R)–C(R1)=CH2 derivatives (X = Cl, Br, I) with Ph3SnH and AIBN led to formation of a lactam via radical cyclization.1347 Cyclization of N-iodoethyl-5-vinyl-2-pyrrolidinone led to the corresponding bicyclic lactam,1348 and there are other examples of radical cyclization with molecules containing a lactam unit1349 or an amide unit.1350 β-Lactams can be produced by radical cyclization, using Mn(OAc)3.1351 Radical cyclization occurs with enamines as well.1352 Radical cyclization occurs with oximes to form the corresponding heterocyclic ring.1353 Phenylseleno N-allylamines led to cyclic amines.1354 ω-Iodo acrylate esters cyclize to form lactones,1355 and allylic acetoxy compounds of the type C=C–C–O2C–CH2I cyclize in a similar manner to give lactones.1356 Iodolactonization (see Reaction 15-41) occurs under standard radical cyclization conditions using allylic acetoxy compounds1357 and HGaCl2/BEt3 has been used to initiate the radical process.1358 α-Bromo mixed acetals give α-alkoxy THF derivatives1359 and α-iodoacetals cyclize to give similar products.1360 The reaction of an o-alkynyl aryl isonitrile with AIBN and 2.2 equiv of Bu3SnH gave an indole via 5-exo-dig cyclization.1361 Indole derivatives have also been prepared from o-iodoaniline derivatives, using AIBN and TTMSS.1362 Samarium(II) has been used to initial 5-exo-trig ketyl-alkene coupling, and the mechanism of the reaction has been examined.1363
Acyl radicals can be generated and they cyclize in the usual manner.1364 Molecular orbital calculations have shown that acyl, as well as silyl radicals, simultaneously use SOMO–LUMO (SOMO = singly occupied molecular orbital and LUMO = lowest unoccupied molecular orbital) and LUMO–HOMO interactions in reactions with alkenes.1365 A polyene-cyclization reaction generated four rings, initiating the sequence by treatment of a phenylseleno ester with Bu3SnH/AIBN to form the acyl radical, which added to the first alkene unit.1366 The newly formed carbon radical added to the next alkene, and so on. Acyl radicals generated from Ts(R)NCOSePh derivatives cyclize to form lactams.1367
Radical cyclization of iodo aldehydes or ketones, at the carbon of the carbonyl, is effectively an acyl addition reaction (16-24 and 16-25). This cyclization is often reversible, and there are many fewer examples of addition to an alkene or alkyne. In one example, a δ-iodo aldehyde was treated with BEt3/O2 to initiate formation of the radical, and in the presence of Bu3SnH cyclization gave a cyclopentanol.1368 The reaction of an aldehyde-alkene with AIBN, 0.5 PhSiH3 and 0.1 Bu3SnH generated a radical from the alkene, which cyclized at the aldehyde to give cyclopentanol derivatives.1369 An aldehyde–O-methyloxime generated a radical adjacent to nitrogen under standard conditions, which cyclized at the carbonyl to give a cyclic α-hydroxy N-methoxyamine.1370 Alternatively, an α-bromoacetal–O-methyl oxime cyclized at the C=NOMe unit under electrolytic conditions in the presence of cobaloxime.1371Alkynyl-imines are cyclized to the imino carbon to form alkylidene lactams under radical conditions in the presence of CO.1372
The attacking radical in radical cyclization reactions is not limited to a carbon, and a number of heterocycles can be prepared.1373 Amidyl radicals are known and give cyclization reactions.1374 Aminyl radical cyclizations have been reported.1375 N-Chloroamine-alkenes give an aminyl radical when treated with TiCl3·BF3, and cyclization gave a pyrrolidine derivative with a pendant chloromethyl group.1376 N-(S-substituted) amines give similar results using AIBN/Bu3SnH.1377 Oxime-alkenes cyclize to imines when treated with PhSSPh and TEMPO (Sec. 5.C.i).1378 An oxygen radical can be generated under photochemical conditions, and they add to alkenes in a normal manner.1379Note that radical substitution occurs, and reaction of Ph3SnH/AIBN and an O-amidyl compound having a phosphonate ester elsewhere in the molecule gave cyclization to a THF derivative.1380
15-31 Conjugate Addition with Heteroatom Nucleophiles
![]()
Heteroatom nucleophiles add to conjugated systems to give Michael-type products. Conjugated carbonyl compounds react via conjugate addition with amines to give β-amino derivatives (See Reaction 15-31)1381 Conjugate addition of nitrogen-containing compounds is often called the aza-Michael reaction.1382 Amines add to conjugated systems in the presence of In,1383 Pd,1384 Sm,1385 Bi,1386 Cu,1387 Ce,1388 La,1389 or Yb compounds1390 to give β-amino derivatives. This reaction can be initiated photochemically1391 or with microwave irradiation.1392 Aniline derivatives add to conjugated aldehydes in the presence of a catalytic amount of DBU,1393 and indeed, DBU promotes the aza-Michael reaction.1394 Lithium amides add to conjugated esters to give the β-amino ester.1395 Amidocuprates add to conjugated systems to give β-nitrogen compounds, and a β-silyl group has an activating effect of the amidocuprate.1396 A solvent-free conjugate addition of amines occurs on alumina in the presence of a Ce catalyst.1397 Boric acid has been used as a catalyst of aza-Michael reactions in water.1398 An intramolecular addition of an amine unit to a conjugated ketone in the presence of a Pd catalyst, or photochemically, led to cyclic amines.1399 Amines add to conjugated thiolactams.1400
There are asymmetric versions of the aza-Michael reaction,1401 and high enantioselectivity is possible using an organocatalyst.1402 Chiral catalysts lead to enantioselective reactions.1403 Chiral additives (e.g., chiral Cinchona alkaloids1404 or chiral naphthol derivatives)1405 have also been used. Chiral imines add in a highly stereoselective manner.1406 Chiral catalysts have been used for the conjugated addition of carbamates.1407 Indoles add to nitro alkenes in the presence of an organocatalyst.1408 Other N-heterocycles add with good enantioselectivity in the presence of an organocatalyst.1409
Lactams have been shown to add to conjugated esters in the presence of Si(OEt)4 and CsF.1410 Phthalimide adds to alkylidene malononitriles via 1,4-addition with a Pd catalyst, and the resulting anion can be alkylated with an added allylic halide.1411 Alkylidene amido-amides, C=C(NHAc)CONHR, react with secondary amines in water to give the β-amino amido amide.1412 Amines also add in a conjugate manner to alkynyl phosphonate esters, C![]() C–PO(OEt)2, using a CuI catalyst.1413 Hydroxylamines add to conjugated nitro compounds to give 2-nitro hydroxylamines.1414 N,O-Trimethylsilyl hydroxylamines add to conjugated esters, via nitrogen, using a Cu catalyst.1415Trimethylsilyl azide with acetic acid reacts with conjugated ketones to give the β-azido ketone.1416 Sodium azide adds to conjugated ketones in aq acetic acid and 20% PBu3.1417 An interesting variation involves a double Michael addition of amido amines, amido alcohols or amido thiols to conjugated alkynes, forming pyrrolidine, oxazolidine, or thiazolidine derivatives.1418
C–PO(OEt)2, using a CuI catalyst.1413 Hydroxylamines add to conjugated nitro compounds to give 2-nitro hydroxylamines.1414 N,O-Trimethylsilyl hydroxylamines add to conjugated esters, via nitrogen, using a Cu catalyst.1415Trimethylsilyl azide with acetic acid reacts with conjugated ketones to give the β-azido ketone.1416 Sodium azide adds to conjugated ketones in aq acetic acid and 20% PBu3.1417 An interesting variation involves a double Michael addition of amido amines, amido alcohols or amido thiols to conjugated alkynes, forming pyrrolidine, oxazolidine, or thiazolidine derivatives.1418
The nitrogen of carbamates adds to conjugated ketones with a Pt,1419 Pd,1420 Cu,1421 or with a bis(triflamide) organocatalyst.1422 The amine moiety of a carbamate adds to conjugated ketones with a polymer-supported acid catalyst,1423 or with BF3·OEt2.1424 The reaction of ammonium formate with 1,4-diphenylbut-2-en-1,4-dione, in PEG-200 and a Pd catalyst under microwave irradiation, gave 2,5-diphenylpyrrole.1425
Phosphines react similarly to amines under certain conditions. Conjugate addition of R2PH and a Ni catalyst give conjugate addition to α,β-unsaturated nitriles.1426 A Pd catalyzed addition of diarylphosphines proceeds with good enantioselectivity to give chiral phosphines.1427 Phosphites add to nitroalkenes in the presence of a chiral organocatalyst to give the corresponding nitro phosphite compound.1428
Alcohols add to conjugated ketones with a PMe3 catalyst to give the β-alkoxy ketone.1429 This reaction is called an oxy-Michael reaction.1430 Alcohol addition is catalyzed by N-heterocyclic carbenes1431 and other organocatalysts,1432 often with enantioselectivity.1433 The conjugate addition of peroxide anions (HOO− and ROO−) to α,β-unsaturated carbonyl compounds is discussed in Reaction 15-48. An intramolecular variation is known that produces dihydropyrones.1434
Thiophenol and butyllithium (lithium phenylthiolate) adds to conjugated esters.1435 Similar addition is observed with selenium compounds (RSeLi).1436 Thiols react with conjugated amides via 1,4-addition with the addition of 10% Hf(OTf)4 or other lanthanide triflates1437 or to conjugated ketones in ionic solvents.1438 Alkyl thiols add to conjugated carbonyl compounds with high enantioselectivity using an organocatalyst.1439 Iron(III)-catalyzed addition of thiols occurs under solvent-free conditions.1440 Thiols add without a catalyst in water1441 in PEG,1442 or in ionic liquids.1443 Thiol addition is also catalyzed by iodine under solvent-free conditions.1444 Ceric ammonium nitrate promotes the conjugate addition of thiols.1445 Thioaryl moieties can be added in the presence of Yb1446 or a catalytic amount of (DHQD)2PYR (a dihydroquinidine, see Reaction 15-48).1447 Thioalkyl units (e.g., BuS–) add to conjugated ketones using BuS–SnBu and In–I.1448 Addition of conjugated lactones is possible to produce β-arylthiolated lactones.1449 Dithiocarbamates are prepared by the reaction of an amine, CS2, and a conjugated carbonyl compound.1450 α,β-Unsaturated sulfones undergo conjugate addition of a cyano group using Et2AlCN.1451
15-32 Acylation of Activated Double Bonds and of Triple Bonds
Hydro-acyl-addition

Under some conditions, acid derivatives add directly to activated double bonds. Acetic anhydride, Mg metal, and Me3SiCl react with conjugated esters to give a γ-keto ester.1452 Similar reaction with vinyl phosphonate esters leads to a γ-keto phosphonate ester.1453 Thioesters undergo conjugate addition to α,β-unsaturated ketones in the presence of SmI2.1454 Using DBU and a thioimidazolium salt, acyl silanes, Ar(C=O)SiMe3, add in a similar manner.1455 Under microwave irradiation, aldehydes add to conjugated ketones using DBU/Al2O3 and a thiazolium salt.1456 The conjugate addition of acyl zirconium complexes in the presence of BF3·OEt2 is catalyzed by palladium acetate.1457
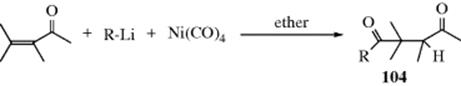
An acyl group can be introduced into the 4 position of an α,β-unsaturated ketone by treatment with an organolithium compound and nickel carbonyl1458 to give a 1,4-diketone (104). The R group may be aryl or primary alkyl. The reaction can also be applied to alkynes, which need not be activated, in which case 2 molar equivalents add and the product is also a 1,4-diketone (e.g., R′C![]() CH → RCOCHR′CH2COR).1459 In a different procedure, α,β-unsaturated ketones and aldehydes are acylated by treatment at −110°C with R2(CN)CuLi2 and CO. This method is successful for R = primary, secondary, and tertiary alkyl.1460 For secondary and tertiary groups, R(CN)CuLi, which does not waste an R group, can be used instead.1461
CH → RCOCHR′CH2COR).1459 In a different procedure, α,β-unsaturated ketones and aldehydes are acylated by treatment at −110°C with R2(CN)CuLi2 and CO. This method is successful for R = primary, secondary, and tertiary alkyl.1460 For secondary and tertiary groups, R(CN)CuLi, which does not waste an R group, can be used instead.1461
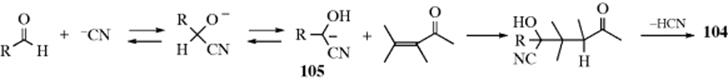
The reaction of an aldehyde and cyanide ion (See Reaction 16-52) in a polar aprotic solvent (e.g., DMF or DMSO) leads to a cyanohydrin, which generates a diketone via loss of HCN.1462 This method has been applied to α,β-unsaturated ketones, esters, and nitriles to give the corresponding 1,4-diketones, γ-keto esters, and γ-keto nitriles, respectively (see also, Reaction 16-55). The initial product of this reaction is ion 105, which is a synthon for the unavailable R−C=O anion (see Reaction 10-68). It is a masked R−C=O anion that upon reaction with the conjugated carbonyl gives 104 after loss of HCN from the cyanohydrin addition product. Other masked carbanions that have been used in this reaction are the RC−(CN)NR ion,1463 the EtSCRSOEt ion,1464 the CH2=C−OEt ion,1465 CH2=C(OEt)Cu2Li,1466 CH2=CMe(SiMe3),1466 and the RC−(OCHMeOEt)CN ion.1467 In the last case, best results are obtained when R is a vinylic group. Anions of 1,3-dithianes (Reaction 10-71) do not give 1,4-addition to these substrates (except in the presence of HMPA, see Reaction 15-25), but add 1,2 to the C=O group instead (Reaction 16-38).
Interestingly, acylation occurs at the α-position of an enone when and α,β-unsaturated ketone is treated with an acid chloride and Et2Zn in the presence of a Rh catalyst.1468
In another procedure, acyl radicals derived from phenyl selenoesters (ArCOSePh) (by treatment with Bu3SnH) add to α,β-unsaturated esters and nitriles to give γ-keto esters and γ-keto nitriles, respectively.1469
OS VI, 866; VIII, 620.
15-33 Addition of Alcohols, Amines, Carboxylic Esters, Aldehydes, and so on
Hydro-acyl-addition, and so on
Formates, primary and secondary alcohols, amines, ethers, alkyl halides, compounds of the type Z–CH2–Z′, and a few other compounds add to double bonds in the presence of free radical initiators.1470 This is formally the addition of RH to a double bond, but the “R” is not just any carbon, but one connected to an oxygen or a nitrogen, a halogen, or to two Z groups (defined as in Sec. 15.A.ii). Formates and formamides1471 add similarly:
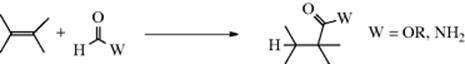
Alcohols, ethers, amines, and alkyl halides add as follows (shown for alcohols):

The ZCH2Z′ compounds react at the carbon bearing the active hydrogen1472:
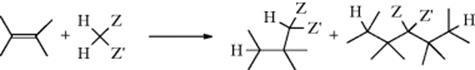
Similar additions have been successfully carried out with carboxylic acids, anhydrides,1473 acyl halides, carboxylic esters, nitriles, and other types of compounds.1474
Similar reactions have been carried out on acetylene.1475 In an interesting variation, thiocarbonates add to alkynes in the presence of a Pd catalyst to give a β-phenylthio α,β-unsaturated ester.1476 Aldehydes add to alkynes in the presence of a Rh catalyst to give conjugated ketones.1477 In a cyclic version of the addition of aldehydes, 4-pentenal was converted to cyclopentanone with a Rh complex catalyst.1478 An intramolecular acyl addition to an alkyne was reported using silyl ketones, acetic acid, and a Rh catalyst.1479 Formamides add to alkynes in the presence of a Pd catalyst to form conjugated amides.1480
OS IV, 430; V, 93; VI, 587, 615.
15-34 Addition of Aldehydes
Alkyl-carbonyl-addition
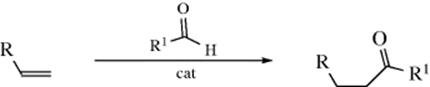
In the presence of metal catalysts (e.g., Rh1481 or Yb1482), aldehydes can add directly to alkenes to form ketones. Additives play an important role in such reactions.1483 The reaction of ω-alkenyl aldehydes with a Rh catalyst leads to cyclic ketones,1484 with high enantioselectivity if chiral ligands are employed. A carbene organocatalyst was used for an enantioselective intramolecular reaction.1485 β,γ-Unsaturated ketones are prepared by the Rh catalyzed addition of aldehydes to dienes.1486 The addition of aldehydes to activated double bonds, mediated by a catalytic amount of thiazolium salt in the presence of a weak base, is called the Stetter reaction,1487 An internal addition of an alkynyl aldehyde, catalyzed by a Rh complex, led to a cyclopentenone derivative.1488 These reactions are not successful when the alkene contains electron-withdrawing groups (e.g., halo or carbonyl groups). A free radical initiator is required,1489 usually peroxides or UV light. The mechanism is illustrated for aldehydes, but is similar for the other compounds:
In the presence of BF3 and a Ag salt, aldehydes add to alkynes to give the corresponding conjugated ketone.1490 Polymers are often side products. Photochemical addition of aldehyde to conjugated C=C units can be efficient when a triplet sensitizer (Sec. 7.A.vi, category 5, e.g., benzophenone) is used.1491
A variation that is more of an acyl addition (Reaction 16-25) involves the reaction of an allylic alcohol with benzaldehyde. With a Ru catalyst and in an ionic liquid, the C=C unit reacts with the aldehyde, with concomitant oxidation of the allylic alcohol unit, to give a β-hydroxy ketone, PhCHO + C=C–CH(OH)R → PhCH(OH)–CH(Me)COR.1492 In another variation, formate esters add to alkenes using a Ru catalyst to give an alkyl ester via a formylation process.1493
15-35 Hydrocarboxylation
Hydro-carboxy-addition
![]()
The acid-catalyzed hydrocarboxylation of alkenes (the Koch reaction) can be performed in a number of ways.1494 In one method, the alkene is treated with CO and water at 100–350°C and 500–1000-atm pressure with a mineral acid catalyst. However, the reaction can also be performed under milder conditions. If the alkene is first treated with CO and catalyst and then water added, the reaction can be accomplished at 0–50°C and 1–100 atm. If formic acid is used as the source of both the CO and the water, the reaction can be carried out at room temperature and atmospheric pressure.1495 The formic acid procedure is called the Koch–Haaf reaction (the Koch–Haaf reaction can also be applied to alcohols, see Reaction 10-77). Nearly all alkenes can be hydrocarboxylated by one of these procedures. However, conjugated dienes are polymerized under these conditions. Hydrocarboxylation can also be accomplished under mild conditions (160°C and 50 atm) by the use of nickel carbonyl as catalyst. Acid catalysts are used along with the nickel carbonyl, but basic catalysts can also be employed.1496 The Ni(CO)4 catalyzed oxidative carbonylation with CO and H2O as a nucleophile is often called Reppe carbonylation.1497 The toxic nature of nickel tetracarbonyl has led to development of other catalysts.1498 Indeed, variations in the reaction procedure include the use of Pd,1499Pt,1500 and Rh1501 catalysts. This reaction converts alkenes, alkynes, and dienes and is tolerant of a wide variety of functional groups. When the additive is alcohol or acid, saturated or unsaturated acids, esters, or anhydrides are produced (see Reaction 15-36). The transition metal catalyzed carbonylation has been done enantioselectively, with moderate-to-high optical yields, by the use of an optically active palladium-complex catalyst.1502 Alkenes also react with Fe(CO)5 and CO to give carboxylic acids.1503 Electrochemical carboxylation procedures have been developed, including the conversion of alkenes to 1,4-butanedicarboxylic acids.1504 A reductive carboxylation of alkenes with CO and cesium carbonate has been reported.1505
When applied to triple bonds, hydrocarboxylation gives α,β-unsaturated acids under very mild conditions. Triple bonds give unsaturated acids and saturated dicarboxylic acids when treated with CO2 and an electrically reduced Ni complex catalyst.1506 Alkynes also react with NaHFe(CO)4, followed by CuCl2·2 H2O, to give alkenyl acid derivatives.1507 A related reaction with CO and Pd catalysts in the presence of SnCl2 leads to conjugated acid derivatives.1508 Terminal alkynes react with CO2 and Ni(cod)2 (cod = 1,5-cycloctadiene), and subsequent treatment with DBU gives the α,β-unsaturated carboxylic acid.1509
When acid catalysts are employed, in the absence of nickel carbonyl, the mechanism1510 involves initial attack on a proton, followed by attack by CO on the resulting carbocation to give an acyl cation, and subsequent reaction with water gives the product 107. Markovnikov's rule is followed, and carbon skeleton rearrangements and double-bond isomerizations (prior to attack by CO) are frequent.
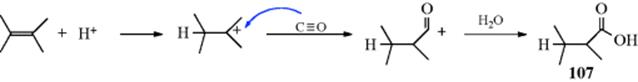
For the transition metal catalyzed reactions, the nickel carbonyl reaction has been well studied and the addition is syn for both alkenes and alkynes.1511 The following is the accepted mechanism:1511
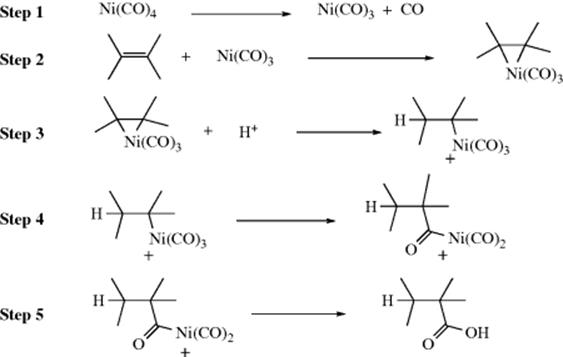
Step 3 is an electrophilic substitution. The principal step of the mechanism, step 4, is a rearrangement.
An indirect method for hydrocarboxylation involves the reaction of an alkene with a borate [(RO)2BH] and a Rh catalyst. Subsequent reaction with LiCHCl2, and then NaClO2, gives the Markovnikov carboxylic acid (RC=C → RC(CO2H)CH3.1512 When a chiral ligand is used, the reaction proceeds with good enantioselectivity.
15-36 Carbonylation, Alkoxycarbonylation, and Aminocarbonylation of Double and Triple Bonds
Alkyl, Alkoxy, or Amino-carbonyl-addition
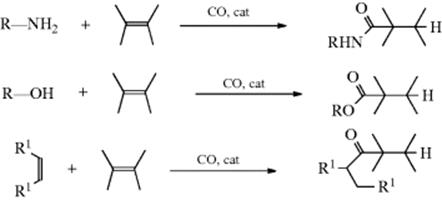
In the presence of certain metal catalysts, alkenes and alkynes can be carbonylated or converted to give an amide or an ester.1513 There are several variations. The reaction of an alkyl iodide and a conjugated ester with CO, (Me3Si)3SiH, and AIBN in supercritical CO2 (Sec. 9.D.ii) gave a γ-keto ester.1514 Terminal alkynes react with CO and methanol in the presence of CuCl2 and PdCl2 to give a β-chloro-α,β-unsaturated methyl ester.1515 Conjugated dienes react with thiophenol, CO and Pd(OAc)2 to give the β,γ-unsaturated thioester.1516 Allene reacts with CO, CH3OH, and a Ru catalyst to give methacrylic acid.1517 Alkynes react with thiophenol and CO with a Pd1518 or Pt1519catalyst to give a conjugated thioester. Terminal alkynes react with CO and CH3OH, using a combination of a palladium(II) halide and a copper(II) halide, to give a conjugated diester, MeO2C–C=C–CO2Me.1520 A similar reaction with alkenes using a combination of a Pd and a Mo catalyst led to a saturated diester (MeO2C–C–C–CO2Me).1521 Alkenes were converted to the dimethyl ester of 1,4-butanedioic acid derivatives with CO/O2 and a combination of PdCl2 and CuCl catalysts.1522 Note that alkenes primarily are converted to the anti-Markovnikov ester upon treatment with arylmethyl formate esters (ArCH2OCHO) and a Ru catalyst.1523 Terminal alkynes react with tosyl azide, water, and a catalytic amount of CuI to give an N-tosyl amide.1524
A bicyclic ketone was generated when 1,2-diphenylethyne was heated with carbon monoxide, methanol and a dirhodium catalyst.1525 2-Iodostyrene reacted at 100 °C with CO and a Pd catalyst to give the bicyclic ketone 1-indanone.1526 Another variation reacted a conjugated allene–alkene with 5 atm of CO and a Rh catalyst to give a bicyclic ketone.1527 An intermolecular version of this reaction is known using a Co catalyst, giving a cyclopentenone1528 in a reaction related to the Pauson–Khand reaction (see below). The reaction of a conjugated diene having a distal alkene unit and CO with a Rh catalyst led to a bicyclic conjugated ketone.1529 When a Stille coupling (Reaction 12-15) is done in a CO atmosphere, conjugated ketones of the type C=C–CO–C=C are formed,1530 suitable for a Nazarov cyclization (Reaction 15-20). Alkynes were converted to cyclobutenones using Fe3(CO)12to form an initial complex, followed by reaction with copper(II) chloride.1531 An interesting variation treated cyclohexene with 5 molar equivalents of Oxone and a RuCl3 catalyst to give 2-hydroxycyclohexanone.1532
The reaction of dienes, diynes, or enynes with transition metals1533 (usually Co)1534 forms organometallic coordination complexes. Rhodium,1535 Ti,1536 Mo,1537 and W1538 complexes have been used for this reaction. In the presence of CO, the metal complexes derived primarily from enynes (alkene–alkynes) generate cyclopentenone derivatives in what is known as the Pauson–Khand reaction.1539 This reaction involves (1) formation of a hexacarbonyldicobalt–alkyne complex and (2) decomposition of the complex in the presence of an alkene.1540 A typical example is the preparation of 108.1541 Cyclopentenones can be prepared by an intermolecular reaction of a vinyl silane and an alkyne using CO and a Ru catalyst.1542 Carbonylation of an alkene–diene using a Rh catalyst leads to cyclization to an α-vinyl cyclopentanone.1543 An yne–diene can also be used for the Pauson–Khand reaction.1544
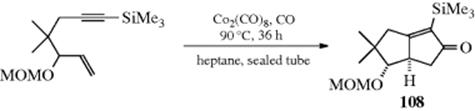
The reaction can be promoted photochemically1545 and the rate is enhanced by the presence of primary amines.1546 Coordinating ligands also accelerate the reaction,1547 polymer-supported promoters have been developed1548 and there are many possible variations in reaction conditions.1549 The Pauson–Khand reaction has been done under heterogeneous reaction conditions,1550 with Co nanoparticles,1551 and in water.1552 A dendritic Co catalyst has been used.1553 Ultrasound promoted1554 and microwave promoted1555 reactions have been developed. Polycyclic compounds (tricyclic and higher) are prepared in a relatively straightforward manner using this reaction.1556 Asymmetric Pauson–Khand reactions are known.1557
The Pauson–Khand reaction is compatible with other groups or heteroatoms elsewhere in the molecule. These include ethers and aryl halides,1558 esters,1559 amides,1560 alcohols,1561 diols,1562 and an indole unit.1563 A silicon-tethered Pauson–Khand reaction is known.1564 Allenes are reaction partners in the Pauson–Khand reaction.1565 This type of reaction can be extended to form six-membered rings using a Ru catalyst.1566 A double-Pauson–Khand process was reported.1567 In some cases, an aldehyde can serve as the source of the carbonyl for carbonylation.1568
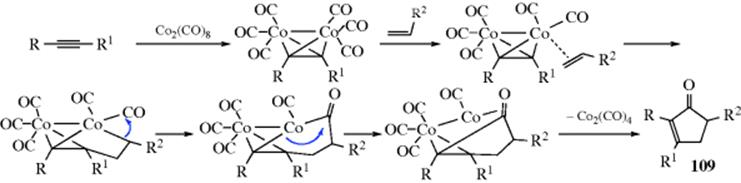
The accepted mechanism was proposed by Magnus and Principe,1569 shown for the formation of 109,1570 and supported by Krafft's work.1571 It has been shown that CO is lost from the Pauson–Khand complex prior to alkene coordination and insertion.1572 Calculations concluded that the LUMO of the coordinated alkene plays a crucial role in alkene reactivity by determining the degree of back-donation in the complex.1573
Other carbonylation methods are available. Carbonylation occurs with conjugated ketones to give 1.4-diketones, using phenylboronic acid (see Reaction 13-12), CO and a Rh catalyst.1574 A noncarbonylation route treated a conjugated diene with an excess of tert-butyllithium, and quenching with CO2 led to a cyclopentadienone.1575 When quenched with CO rather than CO2, a nonconjugated cyclopentenone was formed.1576 Note that a carbonylation reaction with CO, a diyne, and an Ir1577 or a Co catalyst1578 provided similar molecules.
With any method, if the alkene contains a functional group (e.g., OH, NH2, or CONH2), the corresponding lactone (Reaction 16-63),1579 lactam (Reaction 16-74), or cyclic imide may be the product.1580 Titanium,1581 Pd,1582Ru,1583 and Rh1584 catalysts have been used to generate lactones. Allenic alcohols are converted to butenolides with 10 atm of CO and a Ru catalyst.1585 Larger ring conjugated lactones can also be formed by this route using the appropriate allenic alcohol.1586 Propargylic alcohols lead to β-lactones1587 or to butenolides with CO/H2O and a Rh catalyst.1588 Allenic tosyl-amides are converted to N-tosyl α,β-unsaturated pyrrolidinones using 20 atm of CO and a Ru catalyst.1589 Conjugated imines are converted to similar products with CO, ethylene, and a Ru catalyst.1590 Propargyl alcohols generate lactones when treated with a chromium pentacarbonyl carbene complex.1591 Amines add to allenes, in the presence of CO and a Pd catalyst, to form conjugated amides.1592
The reaction of a secondary amine, CO, a terminal alkyne, and t-BuMe2SiH with a Rh catalyst led to a conjugated amide bearing the silyl group of the C=C unit.1593 Reaction of a molecule containing an amine and an alkene unit was carboxylated with CO in the presence of a Pd catalyst to give a lactam.1594 A similar reaction with a molecule containing an amine and an alkyne also generated a lactam, in the presence of CO and a Rh catalyst.1595 An intramolecular carbonylation reaction of a conjugated imine, with CO, ethylene and a Ru catalyst, led to a highly substituted β,γ-unsaturated lactam.1596
15-37 Hydroformylation
Hydro-formyl-addition
![]()
Alkenes can be hydroformylated1597 by treatment with CO and hydrogen over a catalyst, usually a Co carbonyl (see below for a description of the mechanism) or a Rh complex,1598 but other transition metal compounds have also been used. Cobalt catalysts are less active than the Rh type, and catalysts of other metals are generally less active.1599 Commercially, this is called the oxo process, but it can be carried out in the laboratory in an ordinary hydrogenation apparatus. The order of reactivity is straight-chain terminal alkenes > straight-chain internal alkenes > branched-chain alkenes. With terminal alkenes, for example, the aldehyde unit is formed on both the primary and secondary carbon, but proper choice of catalyst and additive leads to selectivity for the secondary1600 or primary product.1601 Alkylidenecyclopropane derivatives undergo hydroformylation to give aldehydes with a quaternary center.1602
Good yields for hydroformylation have been reported using Rh catalysts in the presence of certain other additives.1603 Among the side reactions are the aldol Reaction (16-34), acetal formation, the Tischenko Reaction (19-82), and polymerization. In one case using a Rh catalyst, 2-octene gave nonanal, presumably via a η3-allyl complex (Sec. 3.C).1604 Conjugated dienes give dialdehydes when Rh catalysts are used1605 but saturated monoaldehydes (the second double bond is reduced) with cobalt carbonyls. Both 1,4- and 1,5-dienes may give cyclic ketones.1606
Hydroformylation of triple bonds proceeds very slowly, and few examples have been reported.1607 However, in the presence of a Rh catalyst, the triple bond of a conjugated enyne is formylated.1608 The Rh catalyzed reaction can be regioselective.1609 Many functional groups (e.g., OH, CHO, CO2R,1610 CN), can be present in the molecule, although halogens usually interfere. Stereoselective syn addition has been reported,1611 and also stereoselective anti addition.1612
Asymmetric hydroformylation of alkenes has been accomplished with a chiral catalyst,1613 and in the presence of chiral additives.1614 The choice of ligand is important in such reactions.1615 Cyclization to prolinal derivatives has been reported with allylic amines.1616
When dicobalt octacarbonyl [Co(CO)4]2 is the catalyst, the species that actually adds to the double bond is tricarbonylhydrocobalt [HCo(CO)3].1617 Carbonylation [RCo(CO)3 + CO → RCo(CO)4] takes place followed by a rearrangement and a reduction of the C–Co bond, similar to steps 4 and 5 of the nickel carbonyl mechanism shown in Reaction 15-35. The reducing agent in the reduction step is tetracarbonylhydrocobalt [HCo(CO)4],1618 or, under some conditions, H2.1619 When HCo(CO)4 was the agent used to hydroformylate styrene, the observation of CIDNP (Sec. 5.C.i) indicated that the mechanism is different, and involves free radicals.1620 Key intermediates have been detected in the Co catalyzed hydroformylation reaction.1621 Alcohols can be obtained by allowing the reduction to continue after all the CO is used up. It has been shown1622 that the formation of alcohols is a second step, occurring after the formation of aldehydes, and that HCo(CO)3 is the reducing agent.
OS VI, 338.
15-38 Addition of HCN
Hydro-cyano-addition
![]()
Ordinary alkenes do not react with HCN, but polyhalo alkenes and alkenes of the form C=C–Z add HCN to give nitriles.1623 The reaction is therefore a nucleophilic addition and is base catalyzed. Hydrogen cyanide can be added to ordinary alkenes in the presence of dicobalt octacarbonyl1624 or certain other transition metal compounds.1625 When Z is COR or, more especially, CHO, 1,2-addition (Reaction 16-53) is an important competing reaction and may be the only reaction. An acid-catalyzed hydrocyanation is also known.1626 Triple bonds react very well when catalyzed by an aqueous solution of CuCl, NH4Cl, and HCl or by Ni or Pd compounds.1627 The HCN can be generated in situ from acetone cyanohydrin (see Reaction 16-52), avoiding the use of the poisonous HCN.1628 Alkenes react with HCN via this procedure to give a nitrile in the presence of a Ni complex.1629
One or 2 molar equivalents of HCN can be added to a triple bond, since the initial product is a Michael-type substrate. Acrylonitrile is commercially prepared this way, by the addition of HCN to acetylene. Alkylaluminum cyanides (e.g., Et2AlCN), or mixtures of HCN and trialkylalanes (R3Al) are especially good reagents for conjugate addition of HCN1630 to α,β-unsaturated ketones and α,β-unsaturated acyl halides. An indirect method for the addition of HCN to ordinary alkenes uses an isocyanide (RNC) and Schwartz's reagent (see Reaction 15-17); this method gives anti-Markovnikov addition.1631 tert-Butyl isocyanide and TiCl4 have been used to add HCN to C=C–Z alkenes.1632 Pretreatment with NaI/Me3SiCl followed by CuCN converts alkynes to vinyl nitriles.1633
When an alkene is treated with Me3SiCN and AgClO4, followed by aq NaHCO3, the product is the isonitrile (RNC) formed with Markovnikov selectivity.1634 Enantioselective cyanation using TMSCN and HCN, and a Gd catalyst, leads to β-cyano amides.1635
OS I, 451; II, 498; III, 615; IV, 392, 393, 804; V, 239, 572; VI, 14.
For addition of ArH, see Reaction 11-12 (Friedel–Crafts alkylation).
15.C.iii Reactions in Which Hydrogen Adds to Neither Side
Some of these reactions are cycloadditions (Reactions 15-50, 15-62, 15-54, and 15-57–15-66). In such cases, addition to the multiple bond closes a ring:

A. Halogen on One or Both Sides
15-39 Halogenation of Double and Triple Bonds (Addition of Halogen, Halogen)
Dihalo-addition
![]()
Most double bonds are easily halogenated1636 with bromine, chlorine, or inter-halogen compounds.1637 Substitution can compete with addition in some cases.1638 Iodination has also been accomplished, but the reaction is slower.1639 Under free radical conditions, iodination proceeds more easily.1640 However, vic-diiodides are generally unstable and tend to revert to iodine and the alkene.
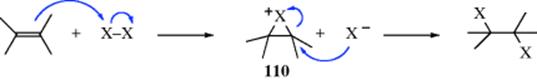
The mechanism is usually electrophilic (see Sec. 15.A.i), involving formation of an halonium ion (Reaction 110),1641 followed by nucleophilic ring opening to give the vic-dihalide. Nucleophilic attack occurs with selectivity for the less substituted carbon. When free radical initiators (or UV light) are present, addition can occur by a free radical mechanism.1642 Once Br• or Cl• radicals are formed, however, substitution may compete (Reactions 14-1 and 14-3). This is especially important when the alkene has allylic or benzylic hydrogen atoms. Under free radical conditions (UV light) bromine or chlorine adds to a benzene substituent to give, respectively, hexabromo- and hexachlorocyclohexane. These are mixtures of stereoisomers (see Sec. 4.K.ii).1643
Under ordinary conditions fluorine itself is too reactive to give simple addition, and mixtures are obtained.1644 However, F2 has been successfully added to certain double bonds in an inert solvent at low temperatures (−78 °C), usually by diluting the F2 gas with Ar or N2.1645 Addition of fluorine has also been accomplished with other reagents (e.g., p-Tol-IF2/Et3N·5 HF),1646 and a mixture of PbO2 and SF4.1647 The Au catalyzed reaction of Et3N–HF with alkynes gives vinyl fluorides.1648
The reaction with bromine is very rapid and is easily carried out at room temperature,1649 although the reaction is reversible under some conditions.1650 In the case of bromine, an alkene·Br2 complex has been detected in at least one case.1651 Bromine is often used as a qualitative or quantitative test for unsaturation1652 because the vast majority of double bonds can be successfully brominated. Even when functions (aldehyde, ketone, amine, etc.) are present in the molecule, they do not interfere, since the reaction with double bonds is faster. Bromination has been carried out in an ionic liquid.1653
Several reagents other than chlorine gas add Cl2 to double bonds, among them Me3SiCl−MnO2,1654 BnNEt3MnO4/Me3SiCl,1655 and KMnO4–oxalyl chloride.1656 A convenient reagent for the addition of Br2 to a double bond on a small scale is the commercially available pyridinium bromide perbromide (C5H5NH+ Br3−).1657 Potassium bromide with ceric ammonium nitrate, in water/dichloromethane, gives the dibromide.1658 A combination of KBr and Selectfluor also give the dibromide.1659 A combination of CuBr2 in aq THF and a chiral ligand led to the dibromide with good enantioselectivity.1660 Either Br2 or Cl2 can also be added using CuBr2 or CuCl2 in the presence of acetonitrile, methanol, or triphenylphosphine.1661 Alkenes are brominated using KBr and diacetoxyiodobenzene.1662 Note that theoretical and experimental studies have shown that in nonpolar solvents the bromination of acetylene via a covalent tribromide adduct is strongly favored over the textbook mechanism via a bridged bromonium ion.
Mixed halogenations have also been achieved, and the order of activity for some of the reagents is BrCl > ICl1663 > Br2 > IBr > I2.1664 Mixtures of Br2 and Cl2 have been used to give bromochlorination,1665 as has tetrabutylammonium dichlorobromate (Bu4NBrCl2).1666 Iodochlorination has been achieved with KICl2,1667 CuCl2, and either I2, HI, or CdI2; iodofluorination1668 with mixtures of AgF and I2;1669 and mixtures of N-bromo amides in anhydrous HF give bromofluorination.1670 Bromo-, iodo-, and chlorofluorination have also been achieved by treatment of the substrate with a solution of Br2, I2, or an N-halo amide in polyhydrogen fluoride–pyridine;1671 while addition of I along with Br, Cl, or F has been accomplished with the reagent bis(pyridine)iodo(I) tetrafluoroborate [I(Py)2BF4] and Br−, Cl−, or F−, respectively.1672 This reaction, which is also successful for triple bonds,1673 can be extended to addition of I and other nucleophiles (e.g., NCO, OH, OAc, and NO2).1673
Conjugated systems give both 1,2- and 1,4-addition.1644 Triple bonds add bromine, although generally more slowly than double bonds (see Sec. 15.B.i). Molecules that contain both double and triple bonds are preferentially attacked at the double bond. Addition of 2 molar equivalents of bromine to triple bonds gives tetrabromo products. There is evidence that the addition of the first molar equivalent of bromine to a triple bond may take place by a nucleophilic mechanism.1674 Molecular diiodine on Al2O3 adds to triple bonds to give good yields of 1,2-diiodoalkenes.1675 Interestingly, 1,1-diiodo alkenes are prepared from an alkynyltin compound, via initial treatment with Cp2Zr(H)Cl, and then 2.15 equiv of iodine.1676 A mixture of NaBO3 and NaBr adds two bromine atoms across a triple bond.1677 With allenes it is easy to stop the reaction after only 1 equiv has added, to give X–C–CX=C.1678Addition of halogen to ketenes gives α-halo acyl halides, but the yields are not good.
OS I, 205, 521; II, 171, 177, 270, 408; III, 105, 123, 127, 209, 350, 526, 531, 731, 785; IV, 130, 195, 748, 851, 969; V, 136, 370, 403, 467; VI, 210, 422, 675, 862, 954; IX, 117; 76, 159.
15-40 Addition of Hypohalous Acids and Hypohalites (Addition of Halogen, Oxygen)
Hydroxy-chloro-addition, and so on.1679
Alkoxy-chloro-addition, and so on
Hypohalous acids (HOCl, HOBr, and HOI) react with alkenes1680 to produce halohydrins.1681 Both HOBr and HOCl can be generated in situ by the reaction between water and Br2 or Cl2, respectively. The compound HOI, generated from I2 and H2O, also adds to double bonds, if the reaction is carried out in tetramethylene sulfone–CHCl31682 or if an oxidizing agent (e.g., HIO3) is present.1683 Iodine and cerium sulfate in aq acetonitrile generates iodohydrins,1684 as do iodine and ammonium acetate in acetic acid,1685 or NaIO4 with sodium bisulfite.1686
The HOBr can also be conveniently added by the use of a reagent consisting of an N-bromo amide (e.g., NBS or N-bromoacetamide) and a small amount of water in a solvent (e.g., DMSO or dioxane).1687 N-Iodosuccinimide in aq dimethoxyethane leads to the iodohydrin.1688 An especially powerful reagent for HOCl addition is tert-butyl hydroperoxide (or di-tert-butyl peroxide) along with TiCl4.1689 Chlorohydrins can be conveniently prepared by treatment of the alkene with Chloramine T (TsNCl− Na+)1690 in acetone–water.1691 The compound HOI can be added by treatment of alkenes with periodic acid and NaHSO3.1692 There are Se catalyzed iodohydrin forming reactions.1693 The reaction of an alkene with polymeric (SnO)n, and then HCl with Me3SiOOSiMe3, leads to the chlorohydrin.1694 Hypervalent iodine compounds react with an alkene and iodine in aqueous media to give the iodohydrin.1695Halohydrins are produced in ionic liquids.1696 N-Bromo and N-iodosaccharin have been used to prepare the corresponding halohydrins.1697
The compound HOF has also been added, but this reagent is difficult to prepare in a pure state and explosions have occurred.1698
The mechanism of HOX addition is electrophilic, with initial attack by the alkene on the positive halogen end of the HOX dipole. Following Markovnikov's rule, the positive halogen goes to the side of the double bond that has more hydrogen atoms (forming a more stable carbocation). This carbocation (or bromonium or iodonium ion in the absence of an aqueous solvent) reacts with ![]() or H2O to give the product. If the substrate is treated with Br2 or Cl2 (or another source of positive halogen, e.g., NBS) in an alcohol or a carboxylic acid solvent, it is possible to obtain, directly C–C–C–OR or X–C–C–OCOR, respectively (see also, Reaction 15-48).1699 Even the weak nucleophile CF3SO2O− can participate in the second step. The addition of Cl2 or Br2 to alkenes in the presence of this ion resulted in the formation of some β-haloalkyl triflates.1700 There is evidence that the mechanism with Cl2 and H2O is different from that with HOCl.1701 Both HOCl and HOBr can be added to triple bonds to give dihalo carbonyl compounds (–CX2–CO–).
or H2O to give the product. If the substrate is treated with Br2 or Cl2 (or another source of positive halogen, e.g., NBS) in an alcohol or a carboxylic acid solvent, it is possible to obtain, directly C–C–C–OR or X–C–C–OCOR, respectively (see also, Reaction 15-48).1699 Even the weak nucleophile CF3SO2O− can participate in the second step. The addition of Cl2 or Br2 to alkenes in the presence of this ion resulted in the formation of some β-haloalkyl triflates.1700 There is evidence that the mechanism with Cl2 and H2O is different from that with HOCl.1701 Both HOCl and HOBr can be added to triple bonds to give dihalo carbonyl compounds (–CX2–CO–).
Alcohols and halogens react with alkenes to form halo ethers. When a homoallylic alcohol is treated with bromine, cyclization occurs to give a 3-bromotetrahydrofuran derivative.1702 tert-Butyl hypochlorite (Me3COCl), hypobromite, and hypoiodite1703 add to double bonds to give halogenated tert-butyl ethers (X–C–C–OCMe3). This is a convenient method for the preparation of tertiary ethers. Iodine and ethanol convert some alkenes to iodo-ethers.1704 Iodine, alcohol, and a Ce(OTf)2 catalyst also generates the iodo-ether.1705 When Me3COCl or Me3COBr is added to alkenes in the presence of excess ROH, the ether X–C–C–OR is produced.1706 Vinylic ethers give β-halo acetals.1707 Chlorine acetate [solutions of which are prepared by treating Cl2 with Hg(OAc)2 in an appropriate solvent] adds to alkenes to give acetoxy chlorides.1708 Acetoxy fluorides have been obtained by treatment of alkenes with CH3COOF.1709
For a method of iodoacetyl addition, see Reaction 15-48.
OS I, 158; IV, 130, 157; VI, 184, 361, 560; VII, 164; VIII, 5, 9.
15-41 Halolactonization and Halolactamization
Halo-alkoxylation
Halo esters can be formed by addition of halogen atoms and ester groups to an alkene. Alkene carboxylic acids give a tandem reaction of formation of a halonium ion followed by intramolecular displacement of the carboxylic group to give a halo lactone. This tandem addition of X and OCOR is called halolactonization.1710
The most common version of this reaction is known as iodolactonization,1711 and a typical example is the conversion of 111 to 112.1712 Bromo lactones and, to a lesser extent, chloro lactones have also been prepared. In general, addition of the halogen to an alkenyl acid, as shown, leads to the halo-lactone. Other reagents include I+(collidine)2PF6−,1713 KI/sodium persulfate.1714 The Tl1715 and Y1716 reagents, along with the halogen, have also been used. An enantioselective 5-endo-halolactonization procedure has been reported using systems, such as iodobis(collidine) hexafluorophosphate or AgSbF6, followed by iodine.1717 When done in the presence of a chiral Ti reagent, I2, and CuO, lactones are formed with good enantioselectivity.1718 Iodine monochloride (ICl) has been used, with formation of a quaternary center at the oxygen-bearing carbon of the lactone.1719 Organocatalysts have also been used to mediate asymmetric halolactonization reactions.1720 Enantioselective iodolactonization occurs with pentenoic acid derivatives in the presence of a chiral Co(salen) complex.1721
In the case of γ,δ-unsaturated acids, five-membered rings (γ-lactones) are predominantly formed (as shown above; note that Markovnikov's rule is followed), but six-membered and even four-membered lactones have also been made by this procedure. There is a gem-dimethyl effect that favors formation of 7–11 membered ring lactones by this procedure.1722
Formation of halo-lactams (Reaction 15-43) by a procedure similar to halolactonization is difficult, but the problems have been overcome. Formation of a triflate from 113 followed by treatment with iodine leads to the iodolactam (114).1723 A related cyclization of N-sulfonyl-amino alkenes and NBS gave the bromo-lactam,1724 and a dichloro-N,N-bis(allylamide) was converted to a dichloro-lactam with FeCl2.1725 Note that lactone formation is possible from unsaturated amides
OS IX, 516
15-42 Addition of Sulfur Compounds (Addition of Halogen, Sulfur)
Alkylsulfonyl-chloro-addition, and so on1726
![]()
Sulfonyl halides add to double bonds to give β-halo sulfones, in the presence of free radical initiators or UV light. A particularly good catalyst is cuprous chloride.1727 In the presence of TsCl, AIBN and a Ru catalyst, β-chloro sulfones are generated from alkenes.1728 A combination of the anion ArSO2Na, NaI, and ceric ammonium nitrate converts alkenes to vinyl sulfones.1729 Triple bonds behave similarly, to give β-halo-α,β-unsaturated sulfones.1730 In a similar reaction, sulfenyl chlorides, (RSCl) give β-halo thioethers.1731 The latter may be free radical or electrophilic additions, depending on conditions. The addition of MeS and Cl has also been accomplished by treating the alkene with Me3SiCl and Me2SO.1732 The use of Me3SiBr and Me2SO does not give this result; dibromides (Reaction 15-39) are formed instead.
β-Iodothiocyanates can be prepared from alkenes by treatment with I2 and isothiocyanatotributylstannane (Bu3SnNCS).1733 Bromothiocyanation can be accomplished with Br2 and thallium(I) thiocyanate.1734 Lead(II) thiocyanate reacts with terminal alkynes in the presence of PhICl2 to give the bis(thiocyanato) alkene [ArC(SCN)–CHSCN].1735 Such compounds were also prepared from alkenes using KSCN and FeCl31736 or iodine thiocyanate.1737 β-Halo disulfides, formed by addition of arenethiosulfenyl chlorides to double-bond compounds, are easily converted to thiiranes by treatment with sodium amide or sodium sulfide.1738
OS VIII, 212. See also, OS VII, 251.
15-43 Addition of Halogen and a Nitrogen Group (Addition of Halogen, Nitrogen)
Dialkylamino-chloro-addition
![]()
The groups R2N and Cl can be added directly to alkenes, allenes, conjugated dienes, and alkynes, by treatment with dialkyl-N-chloroamines and acids.1739 N-Halo amides (RCONHX) add RCONH and X to double bonds under the influence of UV light or chromous chloride.1740 N-Bromoamides add to alkenes in the presence of a transition metal catalyst (e.g., SnCl4) to give the corresponding β-bromo amide.1741 The reaction of TsNCl2 and a ZnCl2 catalyst gave the chloro tosylamine.1742 Aminochlorination of alkenes occurs in a CO2 promoted reaction with Chloramine-T (TolSO2N−–Cl).1743 These are free radical additions, with initial attack by the R2NH•+ radical ion.1744 Amines add to allenes in the presence of a Pd catalyst.1745 A mixture of N-(2-nosyl)NCl2 and sodium N-(2-nosyl)NH− with a CuOTf catalyst reacted with conjugated esters to give the vicinal (E)-3-chloro-2-amino ester.1746 A variation of this latter reaction was done in an ionic liquid.1747
15-44 Addition of NOX and NO2X (Addition of Halogen, Nitrogen)
Nitroso-chloro-addition
![]()
There are three possible products when NOCl is added to alkenes, a β-halo nitroso compound, an oxime, or a β-halo nitro compound.1748 The initial product is always the β-halo nitroso compound,1749 but these are stable only if the carbon bearing the nitrogen has no hydrogen. If it has, the nitroso compound tautomerizes to the oxime, H–C–N=O → C=N–OH. With some alkenes, the initial β-halo nitroso compound is oxidized by the NOCl to a β-halo nitro compound.1750 Many functional groups may be present without interference (e.g., CO2H, CO2R, CN, OR). The mechanism in most cases is probably simple electrophilic addition, and the addition is usually anti, although syn addition has been reported in some cases.1751 Markovnikov's rule is followed, the positive NO going to the carbon that has more hydrogen atoms.
Nitryl chloride (NO2Cl) also adds to alkenes, to give β-halo nitro compounds, but this is a free radical process. The NO2 goes to the less-substituted carbon.1752 Nitryl chloride also adds to triple bonds to give the expected 1-nitro-2-chloro alkenes.1753 The compound FNO2 can be added to alkenes1754 by treatment with HF in HNO31755 or by addition of the alkene to a solution of nitronium tetrafluoroborate (NO2+ BF4−; see Reaction 11-2) in 70% polyhydrogen fluoride–pyridine solution1756 (see also, Reaction 15-37).
OS IV, 711; V, 266, 863.
15-45 Addition of XN3 (Addition of Halogen, Nitrogen)
Azido-iodo-addition
![]()
The addition of iodine azide to double bonds gives β-iodo azides.1757 The reagent can be prepared in situ from KI–NaN3 in the presence of Oxone–wet alumina.1758 The addition is stereospecific and anti, suggesting that the mechanism involves a cyclic iodonium ion intermediate.1759 The reaction has been performed on many double-bond compounds, including allenes1760 and α,β-unsaturated ketones. Similar reactions can be performed with BrN31761and ClN3. 1,4-Addition has been found with acyclic conjugated dienes.1762 In the case of BrN3, both electrophilic and free radical mechanisms are important,1763 while with ClN3 the additions are chiefly free radical.1764 Iodine monoazide (IN3) also adds to triple bonds to give β-iodo-α,β-unsaturated azides.1765
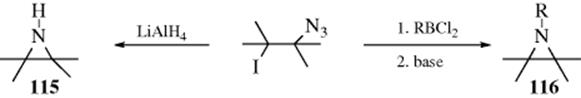
β-Iodo azides can be reduced to aziridines (115) with LiAlH41766 or converted to N-alkyl- or N-arylaziridines (116) by treatment with an alkyl- or aryldichloroborane followed by a base.1767 In both cases the azide is first reduced to the corresponding amine (primary or secondary, respectively) and ring closure (Reaction 10-31) follows. With Chloramine T (TsNCl− Na+) and 10% of pyridinium bromide perbromide, however, the reaction with alkenes give an N-tosyl aziridine directly.1768
OS VI, 893.
15-46 Addition of Alkyl Halides (Addition of Halogen, Carbon)
Alkyl-halo-addition1135
![]()
Alkyl halides can be added to alkenes in the presence of a Friedel–Crafts catalyst, most often AlCl3.1769 The yields are best for tertiary R. Secondary R can also be used, but primary R give rearrangement products (as with Reaction 11-11). The reactive species is the carbocation formed from the alkyl halide and the catalyst (see Reaction 11-11).1770 The reaction with an alkene follows Markovnikov's rule, and generates the more stable carbocation from the alkene after reaction with the carbocation. Methyl and ethyl halides, which cannot rearrange to a more stable secondary or tertiary carbocation, give no reaction at all. Substitution is a side reaction, arising from loss of hydrogen from the carbocation (117). Conjugated dienes give 1,4-addition.1771 Triple bonds also undergo the reaction, to give vinylic halides.1772
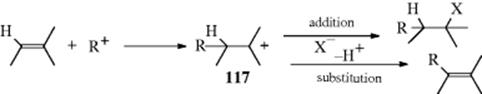
Simple polyhalo alkanes (e.g., CCl4, BrCCl3, ICF3 and related molecules) add to alkenes in good yield.1773 These are free radical additions and require initiation, for example,1774 by peroxides, metal halides (e.g., FeCl2, CuCl),1775Ru catalysts,1776 or UV light. The initial reaction generates the more stable radical intermediate, as in most free radical reactions with alkenes:
![]()
Polyhalo alkanes add to halogenated alkenes in the presence of AlCl3 by an electrophilic mechanism. This has been called the Prins reaction (not to be confused with the other Prins Reaction, 16-54).1777
α-Iodolactones add to alkenes in the presence of BEt3/O2 to give the addition product.1778 Other α-iodoesters add under similar conditions to give the lactone.1779 Iodoesters also add to alkenes in the presence of BEt3 to give iodo-esters that have not cyclized.1780
A variant of the free radical addition method has been used for ring closure (see Reaction 15-30).
For another method of adding R and I to a triple bond, see Reaction 15-23.
OS II, 312; IV, 727; V, 1076; VI, 21; VII, 290.
15-47 Addition of Acyl Halides (Addition of Halogen, Carbon)
Acyl-halo-addition

Acyl halides add to many alkenes using Friedel–Crafts catalysts, although polymerization is a problem. The reaction has been applied to straight-chain, branched, and cyclic alkenes, but to very few containing functional groups, other than halogen.1781 The mechanism is similar to that of Reaction 15-46, and, as in that case, substitution competes (Reaction 12-16). Increasing temperature favors substitution,1782 but good yields of addition products can be achieved if the temperature is kept under 0°C. The reaction usually fails with conjugated dienes, since polymerization predominates.1783 Iodo acetates have been formed from alkenes using iodine and Pb(OAc)2 in acetic acid.1784Rhodium-catalyzed variations are known.1785 The reaction can be performed on triple-bond compounds, producing compounds of the form RCO–C=C–Cl.1786 A formyl group and a halogen can be added to triple bonds by treatment with N,N-disubstituted formamides and POCl3 (Vilsmeier conditions, Reaction 11-18).1787 Chloroformates add to allenes in the presence of a Rh catalyst to give a β-chloro, β,γ-unsaturated ester.1788
OS IV, 186; VI, 883; VIII, 254.
B. Oxygen, Nitrogen, or Sulfur on One or Both Sides
15-48 Dihydroxylation and Dialkoxylation (Addition of Oxygen, Oxygen)
Dihydroxy-addition, Dialkoxy-addition
![]()
There are many reagents that add two OH groups to a double bond (dihydroxylation).1789 The most common are OsO4,1790 first used by Criegee in 1936,1791 and alkaline KMnO4.1792 Both give syn addition from the less-hindered side of the double bond. Less substituted double bonds are oxidized more rapidly than more substituted alkenes.1793 Permanganate adds to alkenes to form an intermediate manganate ester (Reaction 118), which is decomposed under alkaline conditions. Transition state structures and the energetics of the permanganate oxidation of alkenes has been studied using molecular mechanics.1794 Bases catalyze the decomposition of 118 by coordinating with the ester. Note that there are alternative Mn complexes that may be used for cis-dihydroxylation of alkenes.1795 Osmium tetroxide adds rather slowly but almost quantitatively to form a cyclic osmate ester (e.g., 119) as an intermediate,1796which may be isolated in some cases, but is usually decomposed in solution with sodium sulfite (Na2SO3) in ethanol or other reagents.1797
The chief drawbacks to the use of OsO4 are the facts that it is expensive and toxic, but the reaction is made catalytic in OsO4 by using N-methylmorpholine-N-oxide (NMO),1798 tert-butyl hydroperoxide in alkaline solution,1799H2O2,1800 peroxyacid,1801 K3Fe(CN)6,1802 and non-heme iron catalysts.1803 Polymer-bound OsO4,1804 and encapsulated OsO4 have been shown to give the diol in the presence of NMO,1805 as well as OsO42− on an ion exchange resin.1806 Dihydroxylation has also been reported in ionic liquids.1807 Other metals have been used to catalyze dihydroxylation, including Fe1808 or Ru catalyzed1809 reactions with H2O2. A catalytic amount of K2OsO4 with a Cinchona alkaloid on a ordered inorganic support, in the presence of K3Fe(CN)6, gives the cis-diol.1810
The end product of the reaction is a 1,2-diol. Potassium permanganate is a strong oxidizing agent and can oxidize the glycol product1811 (see Reaction 19-7 and 19-10). In acidic and neutral solution, it always does so; hence glycols must be prepared with alkaline1812 permanganate, but the conditions must be mild. Even so, yields are seldom >50%, although they can be improved with phase-transfer catalysis1813 or increased stirring.1814 The use of ultrasound with permanganate has resulted in good yields of the diol.1815 This reaction is the basis of the Baeyer test for the presence of double bonds. The oxidation is compatible with a number of functional groups, including trichloroacetamides.1816
Anti-hydroxylation can be achieved by treatment with H2O2 and formic acid. In this case, epoxidation (Reaction 15-50) occurs first, followed by an SN2 reaction, which results in overall anti addition:
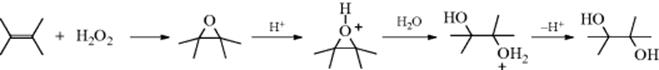
The same result can be achieved in one step with m-chloroperoxybenzoic acid and water.1817 Overall anti addition can also be achieved by the method of Prévost (the Prévost reaction). In this method, the alkene is treated with iodine and silver benzoate in a 1:2 molar ratio. The initial addition is anti and results in a β-halo benzoate, as shown. These can be isolated, and this represents a method of addition of IOCOPh. However, under normal reaction conditions, the iodine is replaced by a second PhCOO group. This is a nucleophilic substitution reaction via the neighboring-group mechanism (Sec. 10.C), so the groups are still anti:
![]()
Hydrolysis of the ester does not change the configuration. The Woodward modification of the Prévost reaction is similar, but results in overall syn hydroxylation.1818 In this procedure, the alkene is treated with iodine and silver acetate in a 1:1 molar ratio in acetic acid containing water. Here again, the initial product is a β-halo ester; the addition is anti and a nucleophilic replacement of the iodine occurs. However, in the presence of water, neighboring-group participation is prevented or greatly decreased by solvation of the ester function, and the mechanism is the normal SN2 process,1819 so the monoacetate is syn and hydrolysis gives the diol as the product, with overall syn addition. Although the Woodward method results in overall syn addition, the product may be different from that with OsO4 or KMnO4, since the overall syn process is from the more hindered side of the alkene.1820 Both the Prévostand the Woodward methods1821 have been carried out in high yields with thallium(I) acetate and thallium(I) benzoate instead of the silver carboxylates.1822 Note that cyclic sulfates can be prepared from alkenes by reaction with PhIO and SO3·DMF.1823 Diacetates have been prepared from alkenes using a Cu catalyzed reaction with PhI(OAc)2 as the oxidizing agent.1824 A similar Pd/Cu catalyzed reaction is known using O2 as the oxidant.1825
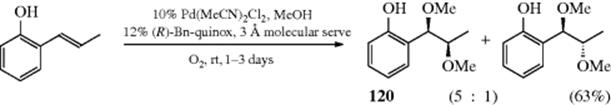
Dialkoxylation reactions are possible. The reaction of an aryl alkene with CH3OH, O2, and a Pd catalyst leads to the dimethoxy compound (see Reaction 120), with moderate enantioselectivity if a chiral ligand is used.1826Dihydroxylation to alkenes of the form RCH=CH2 has been made enantioselective, and addition to RCH=CHR′ both diastereoselective1827 and enantioselective,1828 using chiral additives or chiral catalysts1829 (e.g., 121 or 122, derivatives of the naturally occurring quinine and quinuclidine),1830 along with OsO4, in what is called
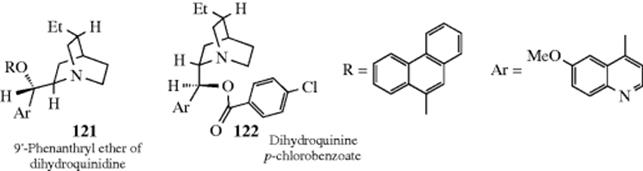
Sharpless asymmetric dihydroxylation.1831 Other chiral ligands1832 have also been used, as well as polymer1833 and silica-bound1834 Cinchona alkaloids. These amines bind to the OsO4 in situ as chiral ligands, causing it to add asymmetrically.1835 This has been done both with the stoichiometric and with the catalytic method.1836 The catalytic method has been extended to conjugated ketones1837 and to conjugated dienes, which give tetrahydroxy products diastereoselectively.1838 Asymmetric dihydroxylation has also been reported with chiral alkenes.1839 Ligands 121 and 122 not only cause enantioselective addition, but also accelerate the reaction, so that they may be useful even where enantioselective addition is not required.1840 Although 121 and 122 are not enantiomers, they give enantioselective addition to a given alkene in the opposite sense; for example, styrene predominantly gave the (R) diol with 121, and the (S) diol with 122.1841 Note that ionic liquids have been used in asymmetric dihydroxylation.1842
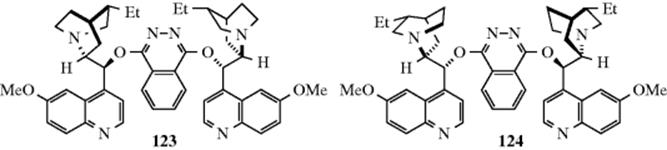
Two phthalazine derivatives,1843 (DHQD)2PHAL (123) and (DHQ)2PHAL (124) are used in conjunction with an Os reagent to improve the efficiency and ease of use, and are commercial available as AD-mix-β (using 123) and AD-mix-α (using 124). Catalyst 123 is prepared from dihydroquinidine (DHQD) and 1,4-dichlorophthalazine (PHAL), and 124 is prepared from dihydroquinine (DHQ) and PHAL. The actual oxidation using AD-mix α or β- uses 124 or 123, respectively, mixed with potassium osmate [K2OsO2(OH)6], powdered K3Fe(CN)6, and powdered K2CO3 in an aqueous solvent mixture.1843 One study showed that osymylation does not always occur preferentially on the most electron-rich double bond. There are examples of the less-rich double bond reacting preferentially, and such preferences may be amplified using AD type reagents, which adds significant steric hindrance to the overall system.1844
These additives have been used in conjunction with microencapsulated OsO4,1845 and polymer bound 123 has been used.1846 An asymmetric dihydroxylation was reported catalyzed by ionic polymer-supported OsO4.1847 A catalytic amount of flavin has been used.1848 Both 1231849 and 1241850 have been used to generate diols with high enantioselectivity. Oxidation of a terminal alkene with AD-mix and then oxidation with TEMPO/NaOCl/NaOCl2leads to α-hydroxyl carboxylic acids with high enantioselectivity.1851
Enantioselective and diastereoselective addition have also been achieved by using preformed derivatives of OsO4, already containing chiral ligands,1852 and by the use of OsO4 on alkenes that have a chiral group elsewhere in the molecule.1853 A Rh catalyzed diboration of alkenes in the presence of a chiral ligand, leads to the corresponding diol with good enantioselectivity after oxidation.1854
Alkenes can also be oxidized with metallic acetates [e.g., lead tetraacetate1855 or thallium(III) acetate]1856 to give bis(acetates) of glycols.1857 Oxidizing agents (e.g., benzoquinone, MnO2, or O2), along with palladium acetate, have been used to convert conjugated dienes to 1,4-diacetoxy-2-alkenes (1,4-addition).1858
1,2-Diols are also generated from terminal alkynes by two sequential reactions with a Pt catalyst and then a Pd catalyst, both with HSiCl3, and a final oxidation with H2O2–KF.1859 The dihydroxylation of a vinyl ether, derived from an alkyne, leads to α-hydroxy aldehydes.1860 Dihydroxylation of alkenes has been reported using a lipase and hydrogen peroxide, under microwave irradiation.1861 A Pd catalyzed diacetoxylation is also known.1862
1,2-Dithiols can be prepared from alkenes by largely indirect methods.1863
OS II, 307; III, 217; IV, 317; V, 647; VI, 196, 342, 348; IX, 251, 383.
15-49 Dihydroxylation of Aromatic Rings
Dihydroxy-addition

One π bond of an aromatic ring can be converted to a cyclohexadiene 1,2-diol by reaction with enzymes associated with P. putida.1864 A variety of substituted aromatic compounds can be oxidized, including bromobenzene, chlorobenzene,1865 and toluene.1866 In these latter cases, introduction of the hydroxyl groups generates a chiral molecule that can be used as a template for asymmetric syntheses.1867
OS X, 217.
15-50 Epoxidation (Addition of Oxygen, Oxygen)
epi-Oxy-addition
![]()
Alkenes are converted to epoxides (oxiranes) by reaction with many peroxyacids.1868 The reaction, called the Prilezhaev reaction, has wide utility.1869 The most common is probably m-chloroperoxybenzoic acid, but peroxyacetic and peroxybenzoic are available, and trifluoroperoxyacetic acid1870 and 3,5-dinitroperoxybenzoic acid1871 are particularly reactive. The limiting factor concerning choice of the peroxyacid is usually whether or not it is commercially available because an in-lab preparation is potentially rather dangerous. Magnesium monoperoxyphthalate (MMPP)1872 is commercially available, and has been shown to be a good substitute for m-chloroperoxybenzoic acid in a number of reactions. Alkyl, aryl, hydroxyl, ester, and other groups may be present, but not amino groups since they are oxidized by the reagent. The presence of electron-donating groups increases the rate, and the reaction is particularly rapid with tetraalkyl alkenes. Conditions are mild and yields are high. Transition metal catalysts can facilitate epoxidation of alkenes at low temperatures or with alkenes that may otherwise react sluggishly.1873
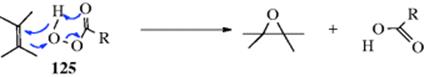
The one-step mechanism involving a transition state (e.g., 125)1874 was proposed by Bartlett.1875 Evidence for this concerted mechanism is as follows1876: (1) The reaction is second order. If ionization were the rate-determining step, it would be first order in peroxyacid. (2) The reaction readily takes place in nonpolar solvents, where formation of ions is inhibited.1877 (3) Measurements of the effect on the reaction rate of changes in the substrate structure show that there is no carbocation character in the transition state.1878 (4) The addition is stereospecific (i.e., a trans-alkene gives a trans-epoxide and a cis-alkene gives a cis-epoxide) even in cases where electron-donating substituents would stabilize a hypothetical carbocation intermediate.1879 However, where there is an OH group in the allylic or homoallylic position, the stereospecificity diminishes or disappears, with both cis and trans isomers giving predominantly and exclusively the product where the incoming oxygen is syn to the OH group. This probably indicates a transition state in which there is hydrogen bonding between the OH group and the peroxyacid.1880
In general, peroxides (HOOH1881 and ROOH) are poor reagents for epoxidation of simple alkenes since OH and OR are poor leaving groups in the concerted mechanism shown above.1882 Transition metal catalysts1883 have been used with alkyl hydroperoxides,1884 however. Epoxidation occurs with Fe,1885 and with Ti1886 or V catalysts.1887 In the presence of some other reagents,1888 peroxides give good yields of the epoxide. These coreagents include DCC,1889 magnesium aluminates,1890 metalloporphyrins,1891 hydrotalcite1892 with microwave irradiation,1893 and arsines in fluorous solvents.1894 The catalyst MeReO31895 has been used for epoxidation using sodium percarbonate and pyrazole,1896 or hydrogen peroxide,1897 or urea–H2O2.1898
Epoxidation has been done in ionic liquids using 10% H2O2 with MnSO41899 or an Fe catalyst.1900 Hypervalent iodine compounds [e.g., PhI(OAc)2], in conjunction with a Ru catalyst in aqueous media, converts alkenes to epoxides.1901 This reagent has been used in an ionic liquid with a Mn catalyst.1902 Sodium chlorite (NaClO2) in water gives epoxidation from alkenes.1903 Microwave assisted epoxidations are known using H2O2.1904 Epoxidation of vinyl ethers has been studied.1905
Several homogeneous and heterogeneous asymmetric epoxidation protocols have been developed.1906 Enzymatic epoxidation1907 and epoxidation with catalytic antibodies1908 have been reported. Organocatalysts (e.g., chiral iminium salts) have been used.1909 Asymmetric Weitz–Scheffer epoxidation1910 (epoxidation of electron-deficient alkenes using H2O2 in a strong alkaline solution) is common. Cinchona-derived phase-transfer catalysts, initially used by Wynberg, are now common.1911 Enantioselectivities can be significantly improved by changes of the catalyst structure, as well as the type of oxidant.1912 A Yb–BINOL complex, with t-BuOOH led to epoxidation of conjugated ketones with high asymmetric induction,1913 as did a mixture of NaOCl and a Cinchona alkaloid.1914 Treatment with aq NaOCl1915 or with an alkyl hydroperoxide1916 and a chiral phase-transfer agent leads to chiral nonracemic epoxy-ketones. Epoxides can also be prepared by treating alkenes with oxygen or with an alkyl peroxide1917 catalyzed by a complex of a transition metal (e.g., V, Mo, Ti, La,1918 Y,1919 or Co).1920 The use of chiral additive leads to enantioselective epoxidation,1921 and organocatalysts have been used as well.1922 Chiral hydroperoxides have been used for enantioselective epoxidation.1923
Other epoxidation methods are available. Dioxiranes,1924 (e.g., dimethyl dioxirane, 126),1925 either isolated or generated in situ,1926 are important epoxidation reagents. With dimethyloxirane, C–H insertion reactions can occur preferentially.1927 The reaction with alkenes is rapid, mild, safe, and a variety of methods have been developed using an oxidant as a coreagent. Substituent effects in such reactions have been studied1928 and also substrate variations.1929 The most commonly used coreagent is probably potassium peroxomonosulfate (KHSO5). Oxone (2KHSO5·KHSO4·K2SO4) is a common source of KHSO5. Oxone reacts with ketones1930 and sodium bicarbonate to convert an alkene to an epoxide. Oxone converts alkenes to epoxides in the presence of certain additives (e.g., N,N-dialkylalloxans).1931 Oxone, with hydrogen peroxide or a similar oxidant, can be used with chiral ketones1932 or aldehydes to convert alkenes to chiral, nonracemic epoxides.1933 This reaction probably converts alkenes to epoxides with good enantioselectivity by in situ generation of dioxirane.1934 Chiral dioxiranes have reportedly given nonracemic epoxides.1935 This transformation with chiral carbohydrates is sometimes called Shi epoxidation.1936 Epoxidation does not occur in good yields with these reagents in most other solvents, and it is suggested that the active agent that generates dioxirane is peroxyimidic acid [MeC(=NH)OOH].1937 Note that benzaldehyde with Chloramine-M1938 will convert alkenes to epoxides.1939
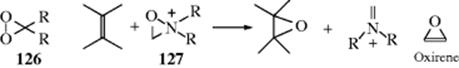
Oxone oxidizes iminium salts to an oxaziridinium intermediate (127), which can transfer oxygen to an alkene to form an epoxide and regenerate the iminium salt.1940 This variation has been applied to asymmetric1941 epoxidations using chiral iminium salt precursors.1942 Other asymmetric epoxidation reactions of alkenes use chiral ketones and iminium salts with an organocatalyst.1943 Direct epoxidation of alkenes has been done using oxaziridinium salts.1944
Although cis–trans isomerization of epoxides is not formally associated with this section, it is a potential issue in the conversion of an alkene to an epoxide. There are several catalysts for this process.1945
It would be useful if triple bonds could be similarly epoxidized to give oxirenes (see oxirene, above), but they are not stable compounds.1946 Two oxirenes have been trapped in solid argon matrices at very low temperatures, but they decayed upon warming to 35 K.1947 Oxirenes probably form in the reaction,1948 but react further before they can be isolated. Note that oxirenes bear the same relationship to cyclobutadiene that furan does to benzene and may therefore be expected to be antiaromatic (Sec. 2.B and 2.K.ii).
Conjugated dienes can be epoxidized (1,2-addition), although the reaction is slower than for corresponding alkenes, but α,β-unsaturated ketones do not generally give epoxides when treated with peroxyacids.1949 The epoxidation of α,β-unsaturated ketones with H2O2 under basic conditions is known as the Waits–Scheffer epoxidation, discovered in 1921.1950 This fundamental reaction has been extended to α,β-unsaturated ketones (including quinones), aldehydes, and sulfones.1951 This is a nucleophilic addition by a Michael-type mechanism, involving attack by HO2−1952: This reaction is another example of 1,4-addition of a heteroatom-containing species, as discussed in Reaction 15-31.
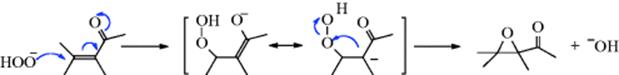
α,β-Unsaturated compounds can be epoxidized alkyl hydroperoxides and a base,1953 or with H2O2 and a base1954or heteropoly acids.1955 The reaction has been done with LiOH and polymer-bound quaternary ammonium salts.1956
Another important asymmetric epoxidation of a conjugated system is the reaction of alkenes with polyleucine,1957 DBU, and urea–H2O2, giving an epoxy–carbonyl compound with good enantioselectivity.1958 The hydroperoxide anion epoxidation of conjugated carbonyl compounds with a polyamino acid (e.g., poly-l-alanine or poly-l-leucine is known as the Juliá–Colonna epoxidation.1959 Epoxidation of conjugated ketones to give nonracemic epoxy-ketones was done with aq NaOCl and a Cinchona alkaloid derivative as catalyst.1960 A triphasic phase-transfer catalysis protocol has also been developed.1961 β-Peptides have been used as catalysts in this reaction.1962
When a carbonyl group is elsewhere in the molecule, but not conjugated with the double bond, the Baeyer–Villiger Reaction (18-19) may compete. Allenes1963 are converted by peroxyacids to allene oxides1964 or spiro dioxides, both of which species can in certain cases be isolated,1965 but more often are unstable under the reaction conditions and react further to give other products.1966
Allylic alcohols can be converted to epoxy-alcohols with tert-butylhydroperoxide on molecular sieves,1967 or with peroxyacids.1968 The addition of an appropriate chiral ligand to the metal-catalyzed hydroperoxide epoxidation of allylic alcohols leads to high enantioselectivity. This important modification is known as the Sharpless asymmetric epoxidation,1969 where allylic alcohols are converted to optically active epoxides with excellent enantioselectivity by treatment with t-BuOOH, titanium tetraisopropoxide, and optically active diethyl tartrate.1970 The Ti(OCHMe2)4 and diethyl tartrate can be present in catalytic amounts (15–10 mol%) if molecular sieves are present.1971 Polymer-supported catalysts have also been reported.1972 The use of a tartrate–PEG reagent (PEG350 or PEG750) allows generation of both enantiomers.1973 Both (+) and (−) diethyl tartrate are readily available, so either enantiomer of the product can be prepared. The method has been successful for a wide range of primary allylic alcohols, including substrates where the double bond is mono-, di, tri-, and tetrasubstituted,1974 and is highly useful in natural product synthesis. The mechanism of the Sharpless epoxidation is believed to involve attack on the substrate by a compound1975 formed from the titanium alkoxide and the diethyl tartrate to produce a complex that also contains the substrate and the t-BuOOH.1976
Ordinary alkenes (without an allylic OH group) do not give optically active alcohols by the Sharpless protocol because binding to the catalyst is necessary for enantioselectivity. Homoallylic alcohols have been converted to the epoxide, however, using a V catalyst in the presence of a chiral bis(hydroxyamide).1977 Simple alkenes can be epoxidized enantioselectively with sodium hypochlorite (NaOCl, commercial bleach) and an optically active manganese complex catalyst.1978 Apart from the commonly used NaOCl, urea–H2O2 has been used.1979
The use of a manganese–salen complex1980 with various oxidizing agents, in what is called the Jacobsen–Katsuki reaction.1981 Simple alkenes can be epoxidized with high enantioselectivity.1982 In addition to Mn, Cr–salen,1983 Ti–salen,1984 and Ru–salen complexes1985 have been used for epoxidation.1986 Note that salen ligands are based on salen. The mechanism of this reaction has been examined.1987 Radical intermediates have been suggested for this reaction,1988 A polymer-bound Mn(III)–salen complex, in conjunction with NaOCl, has been used for asymmetric epoxidation,1989 and manganese porphyrin complexes have also been used.1990 Cobalt complexes give similar results.1991 A related epoxidation reaction used an iron complex with molecular oxygen and isopropanal.1992 Nonracemic epoxides can be prepared from racemic epoxides with (salen) cobalt(II) catalysts following a modified procedure for kinetic resolution.1993
In a different type of reaction, alkenes are photooxygenated (with singlet O2, see Reaction 14-7) in the presence of a Ti, V, or Mo complex to give epoxy alcohols (e.g., 128), formally derived from allylic hydroxylation followed by epoxidation.1994 In other cases, modification of the procedure gives simple epoxidation.1995 Alkenes react with aldehydes and oxygen, with Pd-on-silica1996 or a Ru catalyst,1997 to give the epoxide.
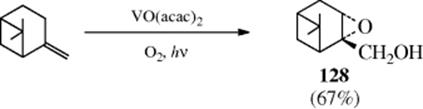
Thiiranes can be prepared directly from alkenes using specialized reagents.1998 Thiourea with a tin catalyst gives the thiirane, for example.1999 Interestingly, internal alkynes were converted to 1,2-dichorothiiranes by reaction with S2Cl2 (sulfur monochloride).2000 Note that epoxides are converted to thiiranes with ammonium thiocyanate and a cerium complex.2001 A trans-thiiration reaction occurs with a Mo catalyst, in which an alkene reacts with styrene thiirane to give the new thiirane.2002
OS I, 494; IV, 552, 860; V, 191, 414, 467, 1007; VI, 39, 320, 679, 862; VII, 121, 126, 461; VIII, 546; IX, 288; X, 29; 80, 9.
15-51 Hydroxysulfenylation (Addition of Oxygen, Sulfur)
Hydroxy-arylthio-addition (overall transformation)
![]()
Both hydroxy and an arylthio group are added to a double bond by treatment with an aryl disulfide and lead tetraacetate in the presence of trifluoroacetic acid.2003 Manganese and copper acetates have been used instead of Pb(OAc)4.2004 Addition of the groups OH and RSO has been achieved by treatment of alkenes with O2 and a thiol (RSH).2005 Addition to RS groups to give vic-dithiols was observed by treatment of the alkene with a disulfide (RSSR) and BF3–etherate.2006 This reaction has been carried out intramolecularly.2007 In a similar manner, the reaction of alkenes with ceric ammonium nitrate and diphenyl diselenide in methanol leads to vicinally substituted phenylselenyl methyl ethers.2008 Dimethyl diselenide adds to alkenes to form vicinal bis(methylselenyl) compounds, in the presence of tin tetrachloride.2009
Halo-ethers can be formed by the reaction of alkenyl alcohols with various reagents. Hept-6-en-1-ol reacts with (collidine)2I+PF6−, for example, to form 2-iodomethyl-1-oxacycloheptane.2010
15-52 Oxyamination (Addition of Oxygen, Nitrogen)
Tosylamino-hydroxy-addition
![]()
N-Tosylated β-hydroxy alkylamines, which can be easily hydrolyzed to β-hydroxyamines2011, can be prepared2012 by treatment of alkenes with the trihydrate of Chloramine-T (N-chloro-p-toluenesulfonamide sodium salt)1690 and a catalytic amount of OsO4.2013 In some cases, yields can be improved by the use of phase-transfer catalysis.2014 The reaction has been carried out enantioselectively.2015 Alkenes can be converted to amido alcohols enantioselectivity by modification of this basic scheme. The Sharpless asymmetric aminohydroxylation employs a catalyst consisting of Cinchona alkaloid derived ligands and an osmium species in combination with a stoichiometric nitrogen source that also functions as the oxidant.2016 The Cu catalyzed reaction of an alkene with a N-sulfone oxaziridine leads to an oxazolidine.2017 N-Chlorosulfonyl isocyanate has been used to prepare 1,2-amino alcohols.2018 The Cu catalyzed hydroxyamination of alkenes was reported using Boc-hydroxylamine.2019
The reaction of a carbamate with (DHQ)2PHAL (124) and the osmium compound, with NaOH and tert-butyl hypochlorite, leads to a diastereomeric mixture of amido alcohols 129 and 130, each formed with high enantioselectivity.2020 An enantioselective aminohydroxylation of acrylamides has been reported.2021
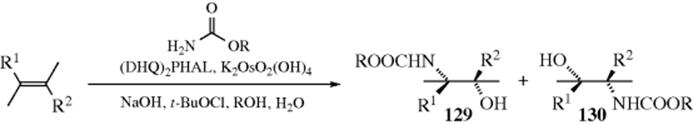
In general, the nitrogen adds to the less sterically hindered carbon of the alkene to give the major product. N-Bromoamides, in the presence of a catalytic amount of (DHQ)2PHAL and LiOH converts conjugated esters to β-amido-α-hydroxy esters with good enantioselectivity.2022 Another oxyamination reaction involves treatment of a Pd complex of the alkene with a secondary or primary amine, followed by lead tetraacetate or another oxidant.2023
The organolanthanide-catalyzed alkene hydroamination has been reported.2024 With this approach, amino alkenes (not enamines) can be cyclized to form cyclic amines,2025 and amino alkynes lead to cyclic imine.2026 The use of synthesized C-12027 and C-2 symmetric2028 chiral organolanthanide complexes give the amino alcohol with good enantioselectivity.
β-Amino alcohols can be prepared by treatment of an alkene with a reagent prepared from HgO and HBF4 along with aniline to give an aminomercurial compound (PhHN–C–C–HgBF4, aminomercuration; see Reaction 15-7), which is hydrolyzed to PhHN–C–C–OH.2029 The use of an alcohol instead of water gives the corresponding amino ether. β-Azido alcohols are prepared by the reaction of an alkene with Me3SiOOSiMe3, Me3SiN3, and 20% (Cl2SnO)n, followed by treatment with aqueous acetic acid.2030
OS VII, 223, 375.
15-53 Diamination (Addition of Nitrogen, Nitrogen)
Di(alkylarylamino)-addition
![]()
Primary (R = H) and secondary aromatic amines react with alkenes in the presence of thallium(III) acetate to give vic-diamines in good yields.2031 The reaction is not successful for primary aliphatic amines. In another procedure, alkenes can be diaminated by treatment with the Os compounds R3NOsO (R = t-Bu) and R2NOsO2,2032 analogous to the Os compound mentioned at Reaction 15-52.2033 The Pd promoted method of Reaction 15-52 has also been extended to diamination.2034 Alkenes can also be diaminated2035 indirectly by treatment of the aminomercurial compound mentioned in Reaction 15-52 with a primary or secondary aromatic amine.2036 The reaction of an alkene with N-arylsulfonyl dichloroamines (ArSO2NCl2) followed by reaction with aq Na2SO3, gives the anti-vic-diacetamde.2037 The Pd catalyzed addition of saccharin and H(NTs)2 with an alkene, in the presence of a hypervalent iodine oxidant leads to a precursor that can be converted to a 1,2-diamine.2038
Two azido groups can be added to double bonds by treatment with sodium azide and iodosobenzene in acetic acid, C=C + NaN3 + PhIO → N3–C–C–N3.2039
Dienes react with ureas in the presence of a Pd catalyst, to give an oxazolidinone.2040 A Pd catalyzed reaction of dienes with di-tert-butyldiaziridinone also leads to an oxazolidinone.2041
Alkynes react with the bis(tosylate) of ethylenediamine, in the presence of a CuI catalyst, to give a dihydropiperazine.2042
15-54 Formation of Aziridines (Addition of Nitrogen, Nitrogen)
epi-Arylimino-addition, and so on
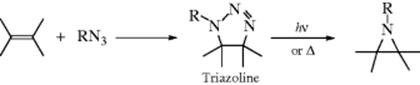
Aziridines can be prepared directly from double-bond compounds by photolysis or thermolysis of a mixture of the substrate and an azide.2043 The reaction has been carried out with R = aryl, cyano, EtOOC, and RSO2, as well as other groups. The reaction can take place by at least two pathways.
In one pathway, a 1,3-dipolar addition (Reaction 15-58) takes place to give a triazoline, which can be isolated, followed thermal by extrusion of nitrogen (Reaction 17-34). Evidence for the nitrene pathway is most compelling for R = acyl groups. In the other, the azide is converted to a nitrene, which adds to the double bond in a manner analogous to that of carbene addition (Reaction 15-64). Sulfonyloxy amines (e.g., ArSO2ONHCO2Et) form an aziridine when treated with CaO in the presence of a conjugated carbonyl compound.2044 In the presence of Cu,2045 Co,2046 or Rh complexes,2047 ethyl diazoacetate adds to imines to give aziridines. Diazirenes (Sec. 5.D.ii) with n-butyllithium converted conjugated amides to the α,β-aziridino amide.2048 Calcium oxide has also been used to generate the nitrene,2049 including nitrene precursors that have an attached chiral ester.2050 Other specialized reagents have also been used.2051 As discussed in Section 5.E, singlet nitrenes add stereospecifically while triplet nitrenes do not. Aminonitrenes (R2NN:) have been shown to add to alkenes2052 to give N-substituted aziridines and to triple bonds to give 1-azirines, which arise from rearrangement of the initially formed 2-azirines.2053 N-Aminophthalimide generates a nitrene in the presence of a Pd catalyst, giving an N-phthalimido aziridine upon reaction with electron-deficient alkenes.2054 Alkyl azides add to conjugated alkenes in the presence of an acid.2055 Intramolecular aziridination reactions are known (e.g., the Pd catalyzed addition of N-tosyloxycarbamates to alkenes to form the bicyclic oxaziridinone).2056 Tosylamines react with alkenes in the presence of a Rh catalyst2057 or with iodine/PhI(OAc)2.2058 Trichloroethylsulfamate esters react with PhI(OAc)2 and a Rh catalyst to give the corresponding N-sulfonyl aziridine.2059
Like oxirenes (see Reaction 15-50), 2-azirines are unstable. 1-Azirines can be reduced to give chiral aziridines.2060
An alternative preparation of aziridines reacts an alkene with iodine and Chloramine-T, generating the corresponding N-tosyl aziridine.2061 Chloramine T and NBS also give the N-tosyl aziridine,2062 and Bromamine-T (TsNBr−Na+) or TsNIK and have also been used in a similar manner,2063,2064 Diazoalkanes react with imines to give aziridines.2065 Another useful reagent is NsN=IPh, which reacts with alkenes in the presence of Rh compounds2066 or Cu complexes2067 to give N-Ns aziridines. Other sulfonamide reagents can be used,2068 including PhI=NTs.2069 Enantioselective aziridination is possible using this reaction with chiral ligands.2070 This reagent has been used in ionic liquids with a Cu catalyst.2071 Palladium catalyzes such reactions2072 and we can also use methyl trioxorhenium (MeReO3).2073 Manganese(salen) catalysts have also been used with this reagent.2074 A nitrido Mn(salen) complex was used with ditosyl anhydride, converting a conjugated diene to an allylic N-tosylaziridine.2075 Arylsulfonamides react with alkenes via the nitrene using an Au2076 or a Cu catalyst.2077
Organocatalysts have been used for the enantioselective aziridination of the C=C unit in conjugated aldehydes.2078
Nitrenes can add to aromatic rings to give ring-expansion products analogous to those mentioned in Reaction 15-62.2079
OS VI, 56.
15-55 Aminosulfenylation (Addition of Nitrogen, Sulfur)
Arylamino-arylthio-addition
![]()
An amino and an arylthio group can be added to a double bond by treatment with a sulfenanilide (PhSNHAr) in the presence of BF3–etherate.2080 The addition is anti, and the mechanism probably involves a thiiranium ion.2081 In another aminosulfenylation procedure, the substrate is treated with dimethyl(methylthio)sulfonium fluoroborate (MeSSMe2 BF4−) and ammonia or an amine,2082 the latter acting as a nucleophile. This reaction was extended to other nucleophiles:2083 N3−,2084 NO2−, CN−, ![]() , and
, and ![]() to give MeS–C–C–A, where A = N3, NO2, CN, OH, and OAc, respectively. An RS (R = alkyl or aryl) and an NHCOMe group have been added in an electrochemical procedure.2085
to give MeS–C–C–A, where A = N3, NO2, CN, OH, and OAc, respectively. An RS (R = alkyl or aryl) and an NHCOMe group have been added in an electrochemical procedure.2085
15-56 Acylacyloxylation and Acylamidation (Addition of Oxygen, Carbon, or Nitrogen, Carbon)
Acyl-acyloxy-addition
An acyl and an acyloxy group can be added to a double bond by treatment with an acyl fluoroborate and acetic anhydride.2086 As expected, the addition follows Markovnikov's rule, with the electrophile Ac+ going to the carbon with more hydrogen atoms. In an analogous reaction, an acyl and an amido group can be added to give 131, if a nitrile is used in place of the anhydride. Similarly, halo-acetoxylation is known.2087 This reaction has also been carried out on triple bonds, to give the unsaturated analogues of 131 (syn addition).2088
15-57 The Conversion of Alkenes or Alkynes to Lactones (Addition of Oxygen, Carbon)
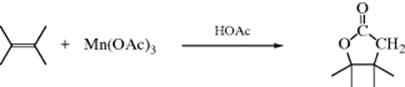
This reaction is clearly related to forming esters and lactones by reaction of carboxylic acids with alkenes (Reaction 15-6), but the Mn reagent leads to differences. Alkenes react with manganese(III) acetate to give γ-lactones.2089The mechanism is probably free radical, involving addition of •CH2COOH to the double bond. Ultrasound improves the efficiency of the reaction.2090 In a related reaction, cyclohexene reacted with MeO2CCH2CO2K and Mn(OAc)3to give an α-carbomethoxy bicyclic lactone.2091 The use of dimethyl malonate and ultrasound in this reaction gave the same type of product.2092 Lactone formation has also been accomplished by treatment of alkenes with α-bromo carboxylic acids in the presence of benzoyl peroxide as catalyst,2093 and with alkylidene Cr(CO)5 complexes.2094 Alkenes can also be converted to γ-lactones by indirect routes.2095 Chromium carbene complexes add to alkenes to give β-lactones using ultrasound.2096
Cyclic dienes react with β-keto esters, in the presence of a Ga2097 catalyst and water, to give an α-acyl bicyclic lactone.
Alkenyl acids cyclize to the corresponding lactone upon treatment with sodium hypochlorite and a Lewis acid.2098 Alkynyl acids cyclize upon treatment with PIFA [phenyliodine(III)-bis(trifluoroacetate)] to give ω-acyl lactones.2099 A variation of this reaction also employs a diselenide.2100 Treatment of alkynyl acids with a Au catalyst2101 leads to an alkylidene lactone.
An intramolecular variation of this reaction is known, involving amides, which generate a lactam.2102
OS VII, 400.
Note that the related halolacctonization reaction, including iodolactonization, is discussed in Reaction 15-41.
For addition of aldehydes and ketones, see the Prins reaction (16-54), and Reactions 16-95 and 16-96.
15.C.iv. Cycloaddition Reactions
15-58 1,3-Dipolar Addition (Addition of Oxygen, Nitrogen, Carbon)
There are a large group of reactions ([3 + 2]-cycloadditions) in which five-membered heterocyclic compounds are prepared by addition of 1,3-dipolar compounds to double bonds. This reaction is quite useful in the synthesis of alkaloids,2103 including asymmetric syntheses.2104 These dipolar compounds have a sequence of three atoms a–b–c, of which a has a sextet of electrons in the outer shell and c has an octet with at least one unshared pair (see Table 15.3).2105 The reaction can then be formulated as shown to generate 132. Note that the initial reaction of potassium permanganate (Reaction 15-48) occurs by [3 + 2]-cycloaddition to give a manganate ester (119).2106 [3 + 2]-Cycloaddition occurs with other metal oxides.2107 Hydrazones have also been reported to give [3 + 2]-cycloadditions.2108
Table 15.3 Some Common 1,3-Dipolar Compounds.
1,3-Dipoles of the type shown in Table 15.3 have an atom with six electrons in the outer shell, which is usually unstable. Such compounds will delocalize the change to alleviate this electronic arrangement (they are resonance stabilized). 1,3-Dipolar compounds can be divided into two main types:
1. Those in which the dipolar canonical form has a double bond on the sextet atom and the other canonical form has a triple bond on that atom:
![]()
If the discussion is limited to the first row of the periodic table, b can only be nitrogen, c can be carbon or nitrogen, and a can be carbon, oxygen, or nitrogen; hence there are six types. Among these are azides (a = b = c = N) and diazoalkanes.
2. Those in which the dipolar canonical form has a single bond on the sextet atom and the other form has a double bond:
![]()
Here b can be nitrogen or oxygen, and a and c can be nitrogen, oxygen, or carbon, but there are only 12 types, since, for example, N–N–C is only another form of C–N–N. Examples are shown in Table 15.3.
Of the 18 systems, some of which are unstable and must be generated in situ,2117 the reaction has been accomplished for at least 15, but not in all cases with a carbon–carbon double bond (the reaction also can be carried out with other double bonds2118). Not all alkenes undergo 1,3-dipolar addition equally well. The reaction is most successful for those that are good dienophiles in the Diels–Alder Reaction (15-60).
The addition is stereospecific and syn, and the mechanism is probably a one-step concerted process,2119 as illustrated above,2120 largely controlled by Frontier Molecular Orbital considerations.2121 Reactivity has been shown to correlate with the energy required to distort 1,3-dipole and dipolarophiles to the transition state.2122 In-plane aromaticity has been invoked for these dipolar cycloadditions.2123 As expected for this type of mechanism, the rates do not vary much with changes in solvent,2124 although rate acceleration has been observed in ionic liquids.2125 Nitrile oxide cycloadditions have also been done in supercritical carbon dioxide.2126 There are no simple rules covering orientation in 1,3-dipolar additions. The regioselectivity has been explained by MO treatments,2127 where overlap of the largest orbital coefficients of the atoms forming the new bonds leads to the major regioisomer. When the 1,3-dipolar compound is a thiocarbonyl ylid (R2C=S+–CH2−) the addition has been shown to be nonstereospecific with certain substrates, but stereospecific with others, indicating a nonsynchronous mechanism in these cases, and in fact, a diionic intermediate (see mechanism c in Reaction 15-63, category 4) has been trapped in one such case.2128 In a theoretical study of the 1,3-dipolar cycloadditions (diazomethane and ethene; fulminic acid [H–C![]() N–O] and ethyne),2129 calculations based on valence bond descriptions suggest that many concerted 1,3-dipolar cycloaddition reactions follow an electronic heterolytic mechanism where the movement of well-identifiable orbital pairs is retained along the entire reaction path from reactants to product.2130
N–O] and ethyne),2129 calculations based on valence bond descriptions suggest that many concerted 1,3-dipolar cycloaddition reactions follow an electronic heterolytic mechanism where the movement of well-identifiable orbital pairs is retained along the entire reaction path from reactants to product.2130
An antibody-catalyzed [3 + 2]-cycloaddition has been reported.2131 Metal-assisted dipolar additions are also known.2132 In a different metal-mediated reaction, alkenyl Fischer carbene complexes react with alkynes, in the presence of a Ni catalyst, to give cyclopentenones.2133 Fischer carbene complexes take the form R2C=M(CO)x,2134 and the metals include those of low oxidation state, and Fe, Mo, Cr, or W. Ligands include π-electron acceptors and π-donor substituents on methylene groups (e.g., alkoxy and amino groups).
Many of the cycloadducts formed from the dipoles in Table 15.3 are unstable, which lead to other products. The reaction of alkyl azides with alkenes generates triazolines (Reaction 15-54), which extrude nitrogen (N![]() N) upon heating or photolysis to give an aziridine.2135 With a transition metal catalyst, alkyl azides add to alkynes to give triazoles.2136 Retro [3 + 2]-cycloaddition reactions are also known.2137 Cycloaddition of azides to allenes leads to pyrrolidines.2138
N) upon heating or photolysis to give an aziridine.2135 With a transition metal catalyst, alkyl azides add to alkynes to give triazoles.2136 Retro [3 + 2]-cycloaddition reactions are also known.2137 Cycloaddition of azides to allenes leads to pyrrolidines.2138
[3 + 2]-Cycloaddition reactions occur intramolecularly to generate bicyclic and polycyclic compounds.2139 The intramolecular cycloaddition of azomethine imines give bicyclic pyrazolidines, for example.2140 When diazoalkanes (including diazo acetates, e.g., ethyl diazoacetate, N2CHCO2Et) react with an alkene and a Cr catalyst the initially formed product is a five-membered ring, a pyrazoline.2141 Pyrazolines are generally unstable and extrusion of nitrogen leads to a cyclopropane.2142 Rhodium-catalyzed cycloaddition using chiral ligands leads to formation of cyclopropanes with good enantioselectivity.2143
There are many cases where the [3 + 2]-cycloaddition leads to cycloadducts with high enantioselectivity.2144 Cycloaddition of diazo esters with a Co catalyst having a chiral ligand leads to cyclopropane derivatives with good enantioselectivity.2145 Cycloaddition of nitrones and pyrazolinones with a Cu catalyst and a chiral ligand leads to pyrrolidine derivatives with good enantioselectivity.2146 In the presence of a Ni catalyst and a chiral ligand, nitrones react with activated cyclopropanes to give a tetrahydro-1,2-oxazine, with high enantioselectivity.2147 Nitrones react with conjugated carbonyl compounds, with a transition metal catalyst (e.g., a Ti complex) to give an 1,2-oxazoline.2148
Conjugated dienes generally give exclusive 1,2-addition, although 1,4-addition (a 3 + 4 cycloaddition) has been reported.2149 Carbon–carbon triple bonds can also undergo 1,3-dipolar addition.2150 For example, azides react to give triazoles, (133).
The 1,3-dipolar reagent can in some cases be generated by the in situ opening of a suitable three-membered ring system. For example, aziridines open to give a zwitterion (e.g., 134), which can add to activated double bonds to give pyrrolidines.2151
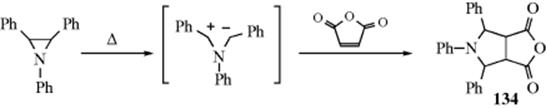
Aziridines also add to C![]() C triple bonds, as well as to other unsaturated linkages, including C=O, C=N, and C
C triple bonds, as well as to other unsaturated linkages, including C=O, C=N, and C![]() N.2152 In some of these reactions, it is a C–N bond of the aziridine that opens rather than the C–C bond.
N.2152 In some of these reactions, it is a C–N bond of the aziridine that opens rather than the C–C bond.
For other [3 + 2]-cycloadditions, see Reaction 15-59.
OS V, 957, 1124; VI, 592, 670; VIII, 231. Also see, OS IV, 380.
C. Carbon on Both Sides
Reactions 15-58–15-64 are cycloaddition reactions.2153
15-59 All-Carbon [3 + 2]-Cycloadditions2154
Several methods have been reported for the formation of cyclopentanes by [3 + 2]-cycloadditions.2155 Heating conjugated ketones with trialkylphosphines generates an intermediate that adds to conjugated alkynes.2156 One type involves reagents that produce intermediates 135 or 136.2157 A synthetically useful example2158 uses 2-[(trimethylsilyl)methyl]-2-propen-1-yl acetate (139), which is commercially available, and a Pd or other transition metal catalyst to generate 135 or 136, which adds to double bonds to give cyclopentanes
with an exocyclic double bond. The reaction occurs with 139 to generate trimethylenemethane in situ, which reacts with alkenes to give methylenecyclopentane derivatives.2159 A similar reaction occurs with imines to give methylene pyrrolidines.2160 The Pd catalyzed reaction with CO2 leads to butenolides.2161
Note that 136 also reacts with N-tosyl aziridines, with 20% n-butyllithium and 10% of Pd(OAc)2, to give a vinylidene piperidine derivative.2162 Similar or identical intermediates generated from bicyclic azo compounds 137 (see Reaction 17-34) or methylenecyclopropane (138)2163 also add to activated double bonds. With suitable substrates the addition can be enantioselective.2164
In a different type of procedure, [3 + 2]-cycloadditions are performed with allylic anions. Such reactions are called 1,3-anionic cycloadditions.2165 For example, α-methylstyrene adds to stilbene on treatment with the strong base LDA.2166
The mechanism can be outlined as:
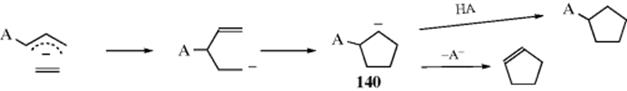
In the case above, 140 is protonated in the last step by the acid HA, but if the acid is omitted and a suitable nucleofuge is present, it may leave, resulting in a cyclopentene.2167 In these cases, the reagent is an allylic anion, but similar [3 + 2]-cycloadditions involving allylic cations have also been reported.2168
OS VIII,173, 347.
15-60 The Diels–Alder Reaction
(4 + 2) cyclo- Ethylene-1/4/addition or (4 + 2)cyclo-[Bu t-2-ene-1,4-diyl]-1/2/addition, and so on
![]()
In the prototype Diels–Alder reaction, the double bond of an alkene adds 1,4- to a conjugated diene (a [4 + 2]-cycloaddition),2169 so the product is always a cyclohexene. The cycloaddition is not limited to alkenes or to dienes (see Reaction 15-61), but the substrate that reacts with the diene is called a dienophile. The reaction is of very broad scope2170 and reactivity of dienes and dienophiles can be predicted based on analysis of the HOMOs2171 and LUMOs of these species (FMO theory).2172 Ethylene and simple alkenes make poor dienophiles, unless high temperatures and/or pressures are used. Most dienophiles are of the form –C=C–Z or Z–C=C–Z′, where Z and Z′ are electron-withdrawing groups2173 (e.g., CHO, COR,2174 CO2H, CO2R, COCl, COAr, CN,2175 NO2,2176 Ar, CH2OH, CH2Cl, CH2NH2, CH2CN, CH2CO2H, halogen, PO(OEt)2,2177 or C=C). In the last case, the dienophile is itself a diene.2178
The low reactivity of simple alkenes can be overcome by incorporating an electron-withdrawing group to facilitate the cycloaddition, as indicated above. Electron-withdrawing groups may be incorporated to facilitate the Diels–Alder reaction and then removed after cycloaddition. An example is phenyl vinyl sulfone (PhSO2CH=CH2),2179 and the PhSO2 group can be easily removed with Na–Hg after the ring-closure reaction. Similarly, phenyl vinyl sulfoxide (PhSOCH=CH2) can be used as a synthon for acetylene.2180 In this case, PhSOH is lost from the sulfoxide product (Reaction 17-12).
Electron-donating substituents in the diene accelerate the reaction; electron-withdrawing groups retard it.2181 For the dienophile, it is just the reverse: donating groups decrease the rate, and withdrawing groups increase it. The s-cis (cisoid) conformation is required for the cycloaddition,2182 and acyclic dienes are conformationally mobile so the s-cis conformation will be available.2183 Cyclic dienes, in which the s-cis conformation is built in, usually react faster than the corresponding open-chain compounds, which have to achieve the s-cis conformation by rotation.2184 Dienes can be open-chain, inner-ring (e.g., 141), outer-ring2185 (e.g., 142), across rings (e.g., 143), or inner–outer (e.g., 145), except that they may not be frozen into a s-trans (transoid) conformation (see category 3). They need no special activating groups, and nearly all conjugated dienes undergo the reaction with suitable dienophiles.2186
While Diels–Alder reactions generally require no catalyst, Lewis acids are effective catalysts,2187 particularly those in which Z in the dienophile is a C=O or C=N group.2188 Chemoselectivity is related to the choice of Lewis acid or Br![]() nsted–Lowry acid catalyst.2189 A Lewis acid catalyst usually increases both the regioselectivity of the reaction (in the sense given above) and the extent of endo addition,2190 and, in the case of enantioselective reactions, the extent of enantioselectivity. Copper catalysts have been used.2191 Br
nsted–Lowry acid catalyst.2189 A Lewis acid catalyst usually increases both the regioselectivity of the reaction (in the sense given above) and the extent of endo addition,2190 and, in the case of enantioselective reactions, the extent of enantioselectivity. Copper catalysts have been used.2191 Br![]() nsted acids have also been used to accelerate the rate of the Diels–Alder reaction.2192 Diels–Alder reactions have been done in ionic liquids (see Sec. 9.D.iii).2193Lanthanum triflate [La(OTf)3] has been reported as a reusable catalyst2194 and Me3SiNTf2 has been used as a green Lewis acid catalyst.2195 Cationic Diels–Alder catalysts have been developed (e.g., oxazaborolidine catalysts).2196Some Diels–Alder reactions can also be catalyzed by the addition of a stable cation radical,2197 for example, tris(4-bromophenyl)aminium hexachloroantimonate (Ar3N•+ SbCl6−).2198 Zirconocene-catalyzed cationic Diels–Alder reactions are known.2199 Certain antibodies have been developed that catalyze Diels–Alder reactions.2200 Photochemically induced Diels–Alder reactions are also known.2201
nsted acids have also been used to accelerate the rate of the Diels–Alder reaction.2192 Diels–Alder reactions have been done in ionic liquids (see Sec. 9.D.iii).2193Lanthanum triflate [La(OTf)3] has been reported as a reusable catalyst2194 and Me3SiNTf2 has been used as a green Lewis acid catalyst.2195 Cationic Diels–Alder catalysts have been developed (e.g., oxazaborolidine catalysts).2196Some Diels–Alder reactions can also be catalyzed by the addition of a stable cation radical,2197 for example, tris(4-bromophenyl)aminium hexachloroantimonate (Ar3N•+ SbCl6−).2198 Zirconocene-catalyzed cationic Diels–Alder reactions are known.2199 Certain antibodies have been developed that catalyze Diels–Alder reactions.2200 Photochemically induced Diels–Alder reactions are also known.2201
A number of other methods have been reported for the acceleration of Diels–Alder reactions,2202 including the use of microwave irradiation,2203 ultrasound,2204 absorption of the reactants on chromatographic absorbents,2205 via encapsulation techniques,2206 and the use of an ultracentrifuge2207 (one of several ways to achieve reaction at high pressures).2208 Solid-state Diels–Alder reactions are known.2209 One of the most common methods is to use water as a solvent or a cosolvent (a hydrophobic effect).2210 Catalysts have been developed for aq Diels–Alder reactions2211 that are suitable for ionic Diels–Alder reactions.2212 There are cases of hydrogen-bonding acceleration.2213 The influence of the hydrophobicity of reactants on the reaction has been examined,2214 as has micellular effects.2215 Another alternative reaction medium is the use of 5M LiClO4 in Et2O as solvent.2216 An alternative to lithium perchlorate in ether is lithium triflate in acetonitrile.2217 The addition of HPO4− to an aq ethanol solution has also been shown to give a small rate enhancement.2218 This appears to be the only case where an anion is responsible for a rate enhancement. The retro-Diels–Alder reaction has also been done in water.2219
Note that Diels–Alder reactions have been done with supercritical carbon monoxide2220 and with supercritical water2221 as solvents. Diels–Alder reactions on solid supports also have been reported,2222 and zeolites have been used in conjunction with catalytic agents.2223 Alumina has been used to promote Diels–Alder reactions.2224 Diels–Alder reactions can be done in ionic liquids,2225 including asymmetric Diels–Alder reactions.2226 Note that the rate of Diels–Alder reactions is faster in water than in ionic liquids.2227
When an unsymmetrical diene adds to an unsymmetrical dienophile, regioisomeric products (not counting stereoisomers) are possible. Rearrangements have been encountered in some cases.2228 In simple cases, 1-substituted dienes give cyclohexenes with a 1,2- and a 1,3- substitution pattern. 2-Substituted dienes lead to 1,4- and 1,3-disubstituted products.
Although mixtures are often obtained, one usually predominates (the one shown lowed major above), but selectivity depends on the nature of the substituents on both diene and alkene. This regioselectivity, in which the “ortho” or “para” product is favored over the “meta”, has been explained by molecular orbital considerations.2229 When X = NO2, regioselectivity to give the “ortho” or “para” product was very high at room temperature. This method, combined with subsequent removal of the NO2 (see Reaction 19-67), has been used to perform regioselective Diels–Alder reactions.2230 Competing reactions are polymerization of the diene or dienophile, or both, and [1,2]-cycloaddition (15-63).
The stereochemistry of the Diels–Alder reaction can be considered from several aspects:2231
1. With respect to the dienophile, the addition is stereospecifically syn, with very few exceptions.2232 This means that groups that are cis in the alkene will be cis in the cyclohexene ring (A–B and C–D) and groups that are trans in the alkene will be trans in the cyclohexene ring (A–D and C–B).

2. With respect to 1,4-disubstituted dienes, fewer cases have been investigated, but here too the reaction is stereospecific and syn. Thus, trans,trans-1,4-diphenylbutadiene gives cis-1,4-diphenylcyclohexene derivatives. This selectivity is predicted by disrotatory motion of the substituent in the transition state2233 of the reaction (see 18-27).
3. The diene must be in the s-cis conformation. If it is frozen into the s-trans conformation, as in 144, the reaction does not take place. The diene either must be frozen into the s-cis conformation or must be able to achieve it during the reaction.
4. There are two possible ways in which addition can occur to a cyclic diene, if the dienophile is not symmetrical: say a monosubstituted alkene. The substituent on the dienophile (usually an electron-withdrawing substituent) may approach under the ring (endo addition), or away from the ring (exo addition):
Most of the time, the addition is predominantly endo; that is, the more bulky side of the alkene is under the ring, and this is probably true for open-chain dienes also.2234 However, exceptions are known, and in many cases mixtures of exo and endo addition products are found.2235 An imidazolidone catalyst was used to give a 1:1.3 mixture favoring the exo isomer in a reaction of conjugated aldehydes and cyclopentadiene.2236 Secondary orbital interactions.2237 have been invoked, but this approach has been called into question.2238 There has been a direct evaluation of such interactions, however.2239 It has been argued that facial selectivity is not due to torsional angle decompression.2240 The endo/exo ratio can be influenced by the nature of the solvent.2241
5. As seen previously, the Diels–Alder reaction can be both stereoselective and regioselective.2242 In some cases, the Diels–Alder reaction can be made enantioselective,2243 as described above. Solvent effects are important in such reactions.2244 The role of reactant polarity on the course of the reaction has been examined.2245 Most enantioselective Diels–Alder reactions have used a chiral dienophile (e.g., 145) and an achiral diene,2246 along with a Lewis acid catalyst (see below). In such cases, addition of the diene to the two
faces2247 of 145 takes place at different rates, and 146 and 147 are formed in different amounts.2248 An achiral compound can be converted to a chiral compound by a chemical reaction with a compound that is enantiopure. After the reaction, the resulting diastereomers can be separated, providing enantiopure compounds, each with a bond between the molecule of interest and chiral compound (a chiral auxiliary). Common chiral auxiliaries include chiral carboxylic acids, alcohols, or sultams. In the case illustrated, hydrolysis of the product removes the chiral R group, making it a chiral auxiliary in this reaction. Asymmetric Diels–Alder reactions have also been carried out with achiral dienes and dienophiles, but with an optically active catalyst.2249 Many chiral catalysts have been developed.2250 In many cases, asymmetric Lewis acids form a chiral complex with the dienophile.2251 Chiral organocatalysts are increasingly important.2252
Triple bond compounds (–C![]() C–Z or Z–C
C–Z or Z–C![]() C–Z′) may be dienophiles,2253 generating nonconjugated cyclohexadienes (148). This reaction can be catalyzed by transition metal compounds.2254 Aromatic rings can be generated by cycloaddition of aryl alkynes.2255 Allenes react as dienophiles, but without activating groups are very poor dienophiles.2256 Ketenes, however, do not undergo Diels–Alder reactions.2257
C–Z′) may be dienophiles,2253 generating nonconjugated cyclohexadienes (148). This reaction can be catalyzed by transition metal compounds.2254 Aromatic rings can be generated by cycloaddition of aryl alkynes.2255 Allenes react as dienophiles, but without activating groups are very poor dienophiles.2256 Ketenes, however, do not undergo Diels–Alder reactions.2257
Many interesting compounds can be prepared by the Diels–Alder reaction,2258 some of which would be hard to make in any other way. Some aromatic compounds can behave as dienes,2259 but benzene is very unreactive toward dienophiles,2260 and very few dienophiles (one of them is benzyne) have been reported to give Diels–Alder adducts with it.2261 Benzynes (e.g., 149), although not isolable, act as dienophiles and can be trapped with dienes.2262 The interesting compound triptycene can be prepared by a Diels–Alder reaction between benzyne and anthracene.2263 Naphthalene and phenanthrene are poor reaction partners, although naphthalene has given Diels–Alder addition at high pressures.2264 Anthracene and other compounds with at least three linear benzene rings give Diels–Alder reactions readily.
For both all-carbon and hetero systems, the “diene” can be a conjugated enyne. If the geometry of the molecule is suitable, the diene can even be nonconjugated, for example,2265

This last reaction is known as the homo-Diels–Alder reaction. A similar reaction has been reported with alkynes, using a mixture of a Co complex, ZnI2, and tetrabutylammonium borohydride.2266
Intramolecular versions of the Diels–Alder reaction are well known,2267and this is a powerful method for the synthesis of mono- and polycyclic compounds.2268 There are many examples and variations, including Lewis acid catalysis.2269 The origin of cis/trans stereoselectivity has been examined using density functional theory.2270
Internal Diels–Alder reactions can be viewed as linking the diene and alkene by a tether, usually of carbon atoms. Dienophile twisting and substituent effects influence the rate of cycloaddition.2271 If the tether is replaced by functional groups that allow the selectivity inherent to the intramolecular cycloaddition, but can be cleaved afterward, a powerful modification is available. Indeed, such tethered cycloaddition reactions are increasingly common. After cycloaddition, the tether can be cleaved to give a functionalized cyclohexene derivative. Such tethered reactions allow enhancement of stereoselectivity2272 and sometimes reactivity, relative to an untethered reaction, giving an indirect method for enhancing those parameters. Tethers or linkages include C–O–SiR2–C2273 or a C–O–SiR2–O–C,2274 or hydroxamides.2275 The nature of the tether plays a role in cis/trans selectivity for the intramolecular reaction.2276 Transient tethers can be used, as in the reaction of a diene having an allylic alcohol unit in a reaction with allyl alcohol, with AlMe3 to give the cycloadduct with good selectivity.2277
The Diels–Alder reaction is usually reversible, although the retro reaction typically occurs at significantly higher temperatures than the forward reaction. However, the reaction is reversible2278 and this fact has been used. A convenient substitute for butadiene in the Diels–Alder reaction is the compound 3-sulfolene, since the latter is a solid, which is easy to handle while the former is a gas.2279 Butadiene is generated in situ by a reverse Diels–Alder reaction (see 17-20).
There are, broadly speaking, three possible mechanisms that have been considered for the uncatalyzed Diels–Alder reaction.2280 In mechanism a, there is a cyclic six-centered transition state and no intermediate. The reaction is concerted and occurs in one step. In mechanism b, one end of the diene fastens to one end of the dienophile first to give a diradical, and then, in a second step, the other ends become fastened.2281 A diradical formed in this manner must be a singlet; that is, the two unpaired electrons must have opposite spins, by an argument similar to that outlined in Section 5.C.i. The third mechanism (c, not shown) is similar to mechanism b, but the initial bond and the subsequent bond are formed by movements of electron pairs and the intermediate is a diion. Electrophilicity–nucleophilicity indices have been analyzed to understand the mechanism of polar Diels–Alder reactions.2282 There have been many mechanistic investigations of the Diels–Alder reaction. The bulk of the evidence suggests that most Diels–Alder reactions take place by the one-step cyclic mechanism a,2283 although it is possible that a diradical2284 or even a diion2285 mechanism may be taking place in some cases. Radical cation Diels–Alder reactions have been considered.2286 The main evidence in support of mechanism a is as follows: (1) the reaction is stereospecific in both the diene and dienophile. A completely free diradical or diion probably would not be able to retain its configuration. (2) In general, the rates of Diels–Alder reactions depend very little on the nature of the solvent. This would rule out a diion intermediate because polar solvents increase the rates of reactions that develop charges in the transition state. (3) It was shown that, in the decomposition of 150, the isotope effect kI/kII was equal to 1.00 within experimental error.2287 If bond x were
to break before bond y, there should surely be a secondary isotope effect. This result strongly indicates that the bond breaking of x and y is simultaneous. This is the reverse of a Diels–Alder reaction, and by the principle of microscopic reversibility, the mechanism of the forward reaction should involve simultaneous formation of bonds x and y. Subsequently, a similar experiment was carried out on the forward reaction2288 and the result was the same. There is other evidence for mechanism a.2289 However, the fact that the mechanism is concerted does not necessarily mean that it is synchronous.2290 In the transition state of a synchronous reaction both new σ bonds would be formed to the same extent, but a Diels–Alder reaction with nonsymmetrical components might very well be nonsynchronous;2291 that is, it could have a transition state in which one bond has been formed to a greater degree than the other.2291,2292 A biradical mechanism has been proposed for some Diels–Alder reactions.2293
In another aspect of the mechanism, the effects of electron-donating and electron-withdrawing substituents (see above) indicate that the diene behaves as a nucleophile and the dienophile as an electrophile. However, this can be reversed. Perchlorocyclopentadiene reacts better with cyclopentene than with maleic anhydride and not at all with tetracyanoethylene, although the latter is normally the most reactive dienophile known. This diene is said to be the electrophile in its Diels–Alder reactions.2294 Reactions of this type are said to proceed with inverse electron demand.2295 It is known that alkynylboronates participate in inverse electron demand cyclization.2296
The Diels–Alder reaction generally takes place rapidly and conveniently. In sharp contrast, the apparently similar dimerization of alkenes to cyclobutanes (Reaction 15-63) gives very poor results in most cases, except when photochemically induced. Woodward and Hoffmann2297 showed that these contrasting results can be explained by the principle of conservation of orbital symmetry, which predicts that certain reactions are allowed and others are forbidden. The orbital-symmetry rules (also called the Woodward–Hoffmann rules)2298 apply only to concerted reactions (e.g., mechanism a) and are based on the principle that reactions take place in such a way as to maintain maximum bonding throughout the course of the reaction. In a separate work, Fukui used MO arguments to explain these reactions. There are several ways of applying the orbital-symmetry principle to cycloaddition reactions, three of which are used more frequently than others.2299 Of these three, two will be discussed: the FMO and the Möbius–Hückel method. The third, called the correlation-diagram method,2300 is less convenient to apply than the other two.
The Frontier-Orbital Method2301
As applied to cycloaddition reactions, the rule is that reactions are allowed only when all overlaps between the HOMO of one reactant and the LUMO of the other are such that a positive lobe overlaps only with another positive lobe and a negative lobe only with another negative lobe.2302 Recall that monoalkenes have two π molecular orbitals (Sec. 1.D) and that conjugated dienes have four (Sec. 2.C), as shown in Fig. 15.2. A concerted cyclization of two monoalkenes (a [2 + 2] reaction) is not allowed because it would require that a positive lobe overlap with a negative lobe (Fig. 15.3). On the other hand, the Diels–Alder reaction (a [4 + 2] reaction) is allowed, when considered from either direction (Fig. 15.4).
Fig. 15.2 Schematic drawings of the π orbitals of an isolated C=C bond and a conjugated diene
Fig. 15.3 Overlap of orbitals in a thermal [2 + 2]-cycloaddition
Fig. 15.4 Two ways for orbitals to overlap in a thermal [4 + 2]-cycloaddition
These considerations are reversed when the ring closures are photochemically induced, since in such cases an electron is promoted to a vacant orbital before the reaction occurs. Obviously, the [2 + 2] reaction is now allowed (Fig. 15.5) and the [4 + 2]-reaction is disallowed. The reverse reactions follow the same rules, by the principle of microscopic reversibility. In fact, Diels–Alder adducts are usually cleaved quite readily, while cyclobutanes, despite the additional strain, require more strenuous conditions.
Fig. 15.5 Overlap of orbitals in a photochemical [2 + 2]-cycloaddition
The Möbius–Hückel Method2303
In this method, the orbital symmetry rules are related to the Hückel aromaticity rule discussed in Chapter 2.2304 Hückel's rule, which states that a cyclic system of electrons is aromatic (hence, stable) when it consists of 4n + 2 electrons, applies of course to molecules in their ground states. In applying the orbital symmetry principle, we are not concerned with ground states, but with transition states. In the present method, do not examine the molecular orbitals themselves, but rather the p orbitals before they overlap to form the molecular orbitals. Such a set of p orbitals is called a basis set (Fig. 15.6). In investigating the possibility of a concerted reaction, the basis sets are put into the position they would occupy in the transition state. Figure 15.7 shows this for both the [2 + 2] and the [4 + 2] ring closures, looking for sign inversions. In Fig. 15.7, there are no sign inversions in either case. That is, the dashed line connects only lobes with a minus sign. Systems with zero or an even number of sign inversions are called Hückel systems. Because they have no sign inversions, both of these systems are Hückel systems. Systems with an odd number of sign inversions are called Möbius systems (because of the similarity to the Möbius strip, which is a mathematical surface, shown in Fig. 15.8).2305 Möbius systems do not enter into either of these reactions, but an example of such a system is shown in Reaction 18.-28, B. Double-twist Möbius aromaticity has been invoked in the Diels–Alder transition state for the reaction of a 5,6-di-tert-butyl substituted decapentaene.2306
Fig. 15.6 Some basis sets
Fig. 15.7 Transition states illustrating Hückel–Möbius rules for cycloaddition reactions
Fig. 15.8 A Möbius strip. Such a strip is easily constructed by twisting a thin strip of paper 180° and fastening the ends together
The rule may then be stated: A thermal pericyclic reaction involving a Hückel system is allowed only if the total number of electrons is 4n + 2. A thermal pericyclic reaction involving a Möbius system is allowed only if the total number of electrons is 4n. For photochemical reactions, these rules are reversed. Since both the [4 + 2]- and [2 + 2]-cycloadditions are Hückel systems, the Möbius–Hückel method predicts that the [4 + 2]-reaction, with 6 electrons, is thermally allowed, but the [2 + 2]-reaction is not. One the other hand, the [2 + 2] -reaction is allowed photochemically, while the [4 + 2]-reaction is forbidden.
Note that both the [2 + 2] and [4 + 2] transition states are Hückel systems no matter what basis sets were chosen. For example, Fig. 15.9 shows other basis sets we might have chosen. In every case there will be zero or an even number of sign inversions.
Fig. 15.9 Transition states of {2 + 2] - and [4 + 2]-cyclizations involving other basis sets
Thus, the FMO and Hückel–Möbius methods (and the correlation-diagram method as well) lead to the same conclusions: Thermal [4 + 2]-cycloadditions and photochemical [2 + 2]-cycloadditions (and the reverse ring openings) are allowed, while photochemical [4 + 2]- and thermal [2 + 2]-ring closings (and openings) are forbidden.
Application of the same procedures to other ring closures shows that [4 + 4]- and [2 + 6]-ring closures and openings require photochemical induction while the [4 + 6]- and [2 + 8]-reactions can take place only thermally (see Reaction 15-53). In general, cycloaddition reactions allowed thermally are those with 4n + 2 electrons, while those allowed photochemically have 4n electrons.
It must be emphasized once again that the rules apply only to cycloaddition reactions that take place by cyclic mechanisms, which is where two σ bonds are formed (or broken) at about the same time.2307 The rule does not apply to cases where one bond is clearly formed (or broken) before the other. It must further be emphasized that the fact that the thermal Diels–Alder reaction (mechanism a) is allowed by the principle of conservation of orbital symmetry does not constitute proof that any given Diels–Alder reaction proceeds by this mechanism. The principle merely says the mechanism is allowed, not that it must go by this pathway. However, the principle does say that thermal [2 + 2]-cycloadditions in which the molecules assume a face-to-face geometry cannot2308 take place by a cyclic mechanism because their activation energies would be too high (however, see below). In Reaction 15-62 it will be seen that such reactions largely occur by two-step mechanisms. Similarly, [4 + 2]-photochemical cycloadditions are also known, but the fact that they are not stereospecific indicates that they also take place by the two-step diradical mechanism (mechanism b).2309
In all of the above discussions, it has been assumed that a given molecule forms both the new σ bonds from the same face of the π system. This manner of bond formation, called suprafacial, is certainly most reasonable and almost always takes place. The subscript s is used to designate this geometry, and a normal Diels–Alder reaction would be called a [π2s + π4s]-cycloaddition (the subscript π indicates that π electrons are involved in the cycloaddition). However, there is another approach in which the newly forming bonds of the diene lie on opposite faces of the π system, that is, they point in opposite directions. This type of orientation of the newly formed bonds is called antarafacial, and the reaction would be a [π2s + π4s]-cycloaddition (a stands for antarafacial). The FMO method shows that this reaction (and consequently the reverse ring-opening reactions) are thermally forbidden and photochemically allowed. Thus in order for a [π2s + π4a] reaction to proceed, overlap between the highest-occupied π orbital of the alkene and the lowest-unoccupied π orbital of the diene would have to occur as shown in Fig. 15.10, with a + lobe overlapping a −lobe. Since like signs are no
longer overlapping, the thermal reaction is now forbidden. Similarly, thermal [[π4s + π2a] and [π2s + π4a]-cyclizations are forbidden, while thermal [π4a + π2a]- and [π2s + π2a]-cyclizations are allowed. These considerations are reversed for the corresponding photochemical processes. Of course, an antarafacial approach is highly unlikely in a [4 + 2]-cyclization,2310 but larger ring closures could take place by such a pathway, and [2 + 2] -thermal cyclizations, where the [[π2s + π2s]-pathway is forbidden, can also do so in certain cases (see Reaction 15-63). Whether a given cycloaddition is allowed or forbidden depends on the geometry of approach of the two molecules involved.
Fig. 15.10 Overlap of orbitals in an antarafacial thermal [4 + 2]-cycloaddition
Symmetry considerations have also been advanced to explain predominant endo addition.2311 In the case of [4 + 2]-addition of butadiene to acrolein, the approach can be exo or endo. It can be seen (Fig. 15.11) that whether the HOMO of the diene overlaps with the LUMO of acrolein or vice versa, the endo orientation is stabilized by additional secondary overlap of orbitals2312 of like sign (dashed lines between heavy dots). Addition from the exo direction has no such stabilization since the sign of the orbitals do not match. Evidence for secondary orbital overlap as the cause of predominant endo orientation, at least in some cases, is that [4 + 6]-cycloaddition is predicted by similar considerations to proceed with predominant exo orientation, and that is what is found.2313 However, this explanation does not account for endo orientation in cases where the dienophile does not possess additional π orbitals, and a number of alternative explanations have been offered.2314
Fig. 15.11 Overlap of orbitals in [4 + 2]-cycloaddition of 1,3-butadiene with acrolein
OS II, 102; III, 310, 807; IV, 238, 738, 890, 964; V, 414, 424, 604, 985, 1037; VI, 82, 196, 422, 427, 445, 454; VII, 4, 312, 485; VIII, 31, 38, 298, 353, 444, 597; IX, 186, 722; 75, 201; 81, 171. For a reverse Diels–Alder reaction, see OS VII, 339.
15-61 Heteroatom Diels–Alder Reactions
![]()
Alkenes, alkynes, and dienes are not the only units that can participate in Diels–Alder reactions. Other double- and triple-bond compounds can be dienophiles and they give rise to heterocyclic compounds.2315 Among these are N![]() C–, –N=C–,2316 iminium salts,2317 –N=N–, O=N–,2318 and –C=O compounds,2319 and even molecular oxygen (Reaction 15-62). Several catalysts can be used, depending on the nature of the heteroatoms incorporated into the alkene or diene.2320 Intramolecular cycloaddition with a diene–imine substrate leads to pyrrolidines.2321
C–, –N=C–,2316 iminium salts,2317 –N=N–, O=N–,2318 and –C=O compounds,2319 and even molecular oxygen (Reaction 15-62). Several catalysts can be used, depending on the nature of the heteroatoms incorporated into the alkene or diene.2320 Intramolecular cycloaddition with a diene–imine substrate leads to pyrrolidines.2321
Aldehydes react with suitably functionalized dienes (e.g., 151), known as Danishefsky's diene,2322 and the reaction usually requires a Lewis acid catalyst (e.g., lanthanide compounds).2323 The Diels–Alder reaction of aldehydes with dienes can be catalyzed by many transition metal compounds, including Co2324 and In2325 catalysts. Aldehydes react using a chiral Ti2326 or a Zr2327 catalyst to give the dihydropyran with good enantioselectivity. Copper(I) catalysts have been used as well.2328 Note that the reaction of Danishefsky's diene with an imine,2329 formed in situ by reaction of an aryl aldehyde and an aniline derivative, proceeds without a Lewis acid.2330 Ketones also react with suitably functionalized dienes.2331 Indium trichloride (InCl3) is a good catalyst for imino-Diels–Alder reactions.2332 Hetero-Diels–Alder reactions involving carbonyls have been done in water.2333 Ultrasound has been used to promote the Diels–Alder reactions of 1-azadienes.2334 Polymer-supported dienes have been used.2335
Hetero-Diels–Alder reactions that proceed with good-to-excellent asymmetric induction are well known.2336 Asymmetric Diels–Alder reactions of carbonyl compounds are well known.2337 Chiral 1-aza-dienes have been developed as substrates, for example.2338 Azadienes also react with chiral dienophiles.2339 Chiral catalysts have been developed.2340
Dienes related to 151 are known, and their reactivity has been examined. Amino-substituted dienes undergo what is known as hydrogen-bonding catalyzed reactions.2341 Imines react with other substrates (e.g., allenes), to give tetrahydropyridine derivatives, with good enantioselectivity in the presence of a chiral ligand.2342 Azo compounds (–N=N–) react as dienophiles in the presence of an Ag catalyst.2343 Iminium ions undergo Diels–Alder cycloaddition.2344
Azadienes undergo Diels–Alder reactions to form pyridine, dihydro- and tetrahydropyridine derivatives.2345 Aza-Diels–Alder reactions have been done in ionic liquids.2346 Similarly, acyl iminium salts (C=N(R)–C=O) react with alkenes via cycloaddition.2347 N-Vinyl lactim ethers undergo Diels–Alder reactions with a limited set of dienophiles.2348 Br![]() nsted acids can catalyze inverse electron demand aza-Diels–Alder reactions.2349
nsted acids can catalyze inverse electron demand aza-Diels–Alder reactions.2349
Thioketones react with dienes to give Diels–Alder cycloadducts.2350 The carbonyl group of lactams has also been shown to be a dienophile.2351 Certain heterocyclic aromatic rings (among them furans)2352 can also behave as dienes in the Diels–Alder reaction. Some hetero dienes that give the reaction are –C=C–C=O, O=C–C=O, and N=C–C=N.2352 Nitroso compounds of the type t-BuO2C-N=O react with dienes to give the corresponding 2-azadihydropyran.2353 Conjugated aldehydes react with vinyl ethers, with a chiral Cr catalyst, in an inverse electron demand cycloaddition that give a dihydropyran with good enantioselectivity.2354 Vinyl sulfilimines have been used in chiral Diels–Alder reactions.2355
OS IV, 311; V, 60, 96; 80, 133. See also, OS VII, 326.
15-62 Photooxidation of Dienes (Addition of Oxygen, Oxygen)
[4 + 2] OC,OC-cyclo-Peroxy-1/4/addition
Conjugated dienes react with oxygen under the influence of light to give cyclic peroxides (152).2356 The reaction has mostly2357 been applied to cyclic dienes.2358 Cycloaddition of furan has been reported using singlet oxygen.2359The scope extends to certain aromatic compounds (e.g., phenanthrene).2360 Besides those dienes and aromatic rings that can be photooxidized directly, there is a larger group that gives the reaction in the presence of a photosensitizer (see Sec. 7.A.vi, category 5; e.g., eosin, a red xanthene dye). Among these is α-terpinene, which is converted to ascaridole:
![]()
As in Reaction 14-7, it is not the ground-state oxygen (the triplet), that reacts, but the excited singlet state,2361,2362 so the reaction is actually a Diels–Alder reaction (see Reaction 15-60) with singlet oxygen as dienophile:2363

Like Reaction 15-60, this reaction is reversible.
As previously discussed, the reaction of singlet oxygen with double-bond compounds gives hydroperoxides (Reaction 14-7), but singlet oxygen can also react with double bonds in another way to give a dioxetane intermediate2364(Reaction 153), which usually cleaves to aldehydes or ketones,2365 but has been isolated.2366 Both the six-membered cyclic peroxides2367 and the four-membered 2052368 have been formed from oxygenation reactions that do not involve singlet oxygen. If cyclic peroxides (e.g., 205) are desired, better reagents2369 are triphenyl phosphite ozonide [(PhO)3PO3] and triethylsilyl hydrotrioxide [(Et3SiOOOH)], but yields are not high.2370
15-63 [2 + 2]-Cycloadditions
[2 + 2]cyclo-Ethylene-1/2/addition
![]()
Two alkene molecules react under thermal conditions to give cyclobutane derivatives in what is known as a [2 + 2]-cycloaddition. The cycloaddition occurs when the alkenes are the same or different, but the reaction is not general for all alkenes.2371 Certain transition metal complexes can catalyze the cycloaddition.2372 Benzynes undergo cycloaddition to give biphenylene derivatives (154),2373 activated alkenes (e.g., styrene, acrylonitrile, butadiene), and certain methylenecyclopropanes.2374 Alkenes react with alkynes2375 or with activated alkynes, with a Ru catalyst, to give cyclobutenes.2376 Dimerization of allenes leads to bis(alkylidene) cyclobutanes.2377 Substituted ketenes can dimerize to give cyclobutenone derivatives, although ketene itself dimerizes in a different manner, to give an unsaturated β-lactone (Reaction 16-95).2378
Intramolecular [2 + 2]-cycloadditions are common in which a diene is converted to a bicyclic compound with a four-membered ring fused to another ring. Transition metal catalyzed reactions have been reported, including the use of Fe complexes.2379 Heating N-vinyl imines, where the vinyl moiety is a silyl enol, gives β-lactams.2380 Apart from photochemical initiation of such reactions (see below), intramolecular cycloaddition of two conjugated ketone units, in the presence of PhMeSiH2 and catalyzed by Co compounds, leads to the bicyclic compound with two ketone substituents.2381 In a variation of this reaction, a diyne was treated with Ti(OiPr)4/2 iPrMgCl to generate a bicyclic cyclobutene with two vinylidene units.2382
Ketenes react with many alkenes to give cyclobutanone derivatives2383 and intermolecular cycloadditions are well known.2384 A typical reaction of dimethylketene and ethene gives 2,2-dimethylcyclobutanone, as shown.2385Ketenes react with imines via [2 + 2]-cycloaddition to produce β-lactams.2386 Cycloaddition of an imine with a conjugated ester in the presence of Et3MeSiH and an Ir catalyst also gives a β-lactam.2387 See Reaction 19-66 for a discussion of reactions that give β-lactams.

Different alkenes combine as follows:
1. F2C=CX2 (X = F or Cl), especially F2C=CF2, form cyclobutanes with many alkenes. Compounds of this type even react with conjugated dienes to give four-membered rings rather than undergoing normal Diels–Alder reactions.2388
2. Allenes2389 and ketenes2390 react with activated alkenes and alkynes. Ketenes give 1,2-addition, even with conjugated dienes.2391 Ketenes also add to unactivated alkenes if sufficiently long reaction times are used.2392 Allenes and ketenes also add to each other.2393
3. Enamines2394 form four-membered rings with Michael-type alkenes2395 and ketenes.2396 In both cases, only enamines from aldehydes give stable four-membered rings:
Generating the ketene in situ from an acyl halide and a tertiary amine is a convenient way to carry out the reaction of enamines with ketenes.
4. Alkenes with electron-withdrawing groups may form cyclobutanes with alkenes containing electron-donating groups.2397 The enamine reactions, mentioned above, are examples of this, but it has also been accomplished with tetracyanoethylene and similar molecules, which give substituted cyclobutanes when treated with alkenes of the form C=C–A, where A may be OR,2398 SR (enol and thioenol ethers),2399 cyclopropyl,2400 or certain aryl groups.2401
Enantioselective [2 + 2]-cycloaddition reactions are known. Chiral organocatalysts lead to chiral cyclobutane derivatives.2402
Solvents are not necessary for [2 + 2]-cycloadditions. They can be carried out at 100–225 °C under pressure, although the reactions in Group 4 occur under milder conditions. However, the choice of solvent can control the distribution of products in photochemical [2 + 2]-cycloaddition.2403
It has been found that certain [2 + 2]-cycloadditions that do not occur thermally can be made to take place without photochemical initiation using certain catalysts, usually transition metal compounds.2404 Among the catalysts used are Lewis acids2405 and phosphine–Ni complexes.2406 The role of the catalyst is not certain and may be different in each case. One possibility is that the presence of the catalyst causes a forbidden reaction to become allowed, through coordination of the catalyst to the π or s bonds of the substrate.2407 In such a case, the reaction would of course be a concerted [2s + 2s] process.2408 However, the available evidence is more consistent with nonconcerted mechanisms involving metal–carbon σ-bonded intermediates, at least in most cases.2409 For example, such an intermediate was isolated in the dimerization of norbornadiene, catalyzed by Ir complexes.2410 Photochemical2411 [π2 + s2]-cycloadditions have also been reported. Visible light mediates cycloaddition in the presence of a Rh catalyst.2412 Some reverse cyclobutane ring openings can also be catalytically induced (Reaction 18-38).
Thermal cycloadditions leading to four-membered rings can also take place between a cyclopropane ring and an alkene or alkyne2413 bearing electron-withdrawing groups.2414 These reactions are [π2 + s2]-cycloadditions. Ordinary cyclopropanes do not undergo the reaction, but it has been accomplished with strained systems (e.g., bicyclo[1.1.0]butanes2415 and bicyclo[2.1.0]pentanes). For example, bicyclo[2.1.0]pentane reacts with maleonitrile (or fumaronitrile) to give all three isomers of 2,3-dicyanonorbornane, as well as four other products.2416 The lack of stereospecificity and the negligible effect of solvent on the rate indicate a diradical mechanism.
If dienes are involved in the reaction, the Diels–Alder reaction may compete, although most alkenes react with a diene either entirely by 1,2- or entirely by 1,4-addition. Three mechanisms have been proposed for [2 + 2]-cycloaddition.2417
Mechanism a is a concerted pericyclic process, and mechanisms b and c are two-step reactions involving, respectively, a diradical (155) and a diion (156) intermediate. As in Reaction 15-60, a diradical intermediate must be a singlet. In searching for ways to tell which mechanism is operating in a given case, mechanism c is expected to be sensitive to changes in solvent polarity, while mechanisms a and b should be insensitive. Mechanism a is expected to be stereospecific, while mechanisms b and c probably would not be stereospecific. However, if the second step of these processes takes place very rapidly, before 155 or 156 have a chance to rotate about the newly formed single bond, stereospecificity might be observed. Because of entropy considerations, such rapid ring closure might be more likely here than in a [4 + 2]-cycloaddition.
There is evidence that the reactions can take place by all three mechanisms, depending on the structure of the reactants. A thermal [π2s + π2s] mechanism is ruled out for most of these substrates by the orbital symmetry rules, but a [π2s + π2a] mechanism is allowed (see above), and there is much evidence that ketenes and certain other linear molecules2418 in which the steric hindrance to such an approach is minimal can and often do react by this mechanism. In a [π2s + π2a] cycloaddition, the molecules must approach each other in such a way (Fig. 15.12a) that the HOMO–LUMO overlap requires that the groups of one molecule project into the plane of the other. This does not happen with ordinary alkenes,2419 but if one molecule is a ketene (Fig. 15.12b), a group on the carbon of the C=C unit is missing (relative to an alkene) and the [π2s + π2a]-reaction can take place. Among the evidence2420 for this mechanism2421 is the following: (1) the reactions are stereospecific.2422 (2) The isomer that forms is the more hindered one. Thus the reaction of methylketene plus cyclopentadiene gave only
the endo product (157, A = H, R = CH3).2423 Even more remarkably, when haloalkyl ketenes RXC=C=O were treated with cyclopentadiene, the endo/exo ratio of the product (157, 158, A = halogen) actually increased substantially when R was changed from Me to iPr to t-Bu!2424 One would expect preferential formation of the exo products (158) from [π2s + π2s]-cycloadditions where the molecules approach each other face-to-face. However, a [π2s + π2a]-process leads to endo products because the ketene molecule (which for steric reasons would approach with its smaller group, methyl in the figure, directed toward the alkene) must twist as shown in Fig. 15.13 (tert-butyl = larger; methyl = smaller group) in order for the + lobes to interact. This process swings the larger group (tert-butyl) into the endo position.2425 The experimental results in which the amount of endo isomer increases with the increasing size of the R group appears to be contrary to what would be expected from steric hindrance considerations (called masochistic steric effects), but they are just what is predicted for a [π2s + π2a] reaction. (3) There is only moderate polar solvent acceleration.2426 (4) The rate of the reaction is not very sensitive to the presence of electron-withdrawing or electron-donating substituents.2427 Because cycloadditions involving allenes are often stereospecific, it has been suggested that these also take place by the [π2s + π2a] mechanism,2428 but the evidence in these cases is more consistent with the diradical mechanism b.2429
Fig. 15.12 Steric interactions in [π2s + π2s]-cycloaddition between (a) two alkene molecules and (b) a ketene and an alkene
Fig. 15.13 Orbital overlap in the reaction of a ketene with cyclopentadiene. Here S and L represent small and large. [Reproduced from Brook, P.R.; Harrison, J.M.; Duke, A.J. Chem. Commun. 1970, 589 with permission from the Royal Society of Chemistry]
The diradical mechanism b is most prominent in the reactions involving fluorinated alkenes.2430 These reactions are generally not stereospecific2431 and are insensitive to solvent effects. Further evidence that a diion is not involved is that head-to-head coupling is found when an unsymmetrical molecule is dimerized. Thus dimerization of F2C=CFCl gives 159, not 160. If one pair of electrons moved before the other, the positive end of one molecule would be expected to attack the negative end of the other.2432
The diion mechanism2433 c has been reported for at least some of the reactions2434 in categories 3 and 4,2435 as well as some ketene dimerizations.2436 The rate of the reaction between 1,2-bis(trifluoromethyl)-1,2-dicyanoethene and ethyl vinyl ether, for example, was strongly influenced by changes in solvent polarity.2437 Some of these reactions are nonstereospecific, but others are stereospecific.2438 As previously indicated, it is likely that in the latter cases the diionic intermediate closes before rotation can take place. Such rapid ring closure is more likely for a diion than for a diradical because of the attraction between the opposite charges. Other evidence for the diion mechanism in these cases is that reaction rates are greatly dependent on the presence of electron-donating and electron-withdrawing groups and that it is possible to trap the diionic intermediates.
Whether a given alkene reacts by the diradical or diion mechanism depends, among other things, on the groups attached to it. For example, phenyl and vinyl groups at the α positions of 155 or 156 help to stabilize a diradical, while donors (e.g., oxygen and nitrogen) favor a diion (they stabilize the positively charged end).2439 A table in Ref. 2439 (see p. 451) shows which mechanism is more likely for [2 + 2]-cycloadditions of various pairs of alkenes.
Thermal cleavage of cyclobutanes2440 to give two alkene molecules (cycloreversion,2441 the reverse of [2 + 2]-cycloaddition) operates by the diradical mechanism, and the [σ2s + σ2a]-pathway has not been found2442 (the subscripts σ indicate that σ bonds are involved in this reaction).
In some cases, double bonds add to triple bonds to give cyclobutenes, apparently at about the same rate that they add to double bonds. The addition of triple bonds to triple bonds would give cyclobutadienes, and this has not been observed, except where these rearrange before they can be isolated (see Reaction 15-65)2443 or in the presence of a suitable coordination compound, so that the cyclobutadiene is produced in the form of a complex (Sec. 2.K.ii).2444
Although thermal [2 + 2]-cycloaddition reactions are essentially limited to the cases described above, many (although by no means all) double-bond compounds react when photochemically excited (either directly or by a photosensitizer, see Sec. 7.A.vi, category 5), even if they are not in the above categories.2445 Simple alkenes absorb in the far-UV (Sec. 7.A.iii), which is difficult to reach experimentally, although this problem can sometimes be overcome by the use of suitable photosensitizers. The reaction has been applied to simple alkenes2446 (especially to strained compounds, e.g., cyclopropenes and cyclobutenes), but more often the double-bond compounds involved are conjugated dienes,2447 α,β-unsaturated ketones,2448 conjugated acids or acid derivatives, and quinones. Since these compounds, are conjugated, they absorb at longer wavelengths (Sec 7.A.iii). Both dimerizations and mixed additions are common. Some examples follow:
Ref. 2449
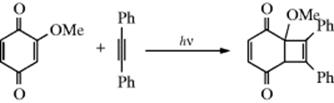
Ref. 2450
See also, Section 7.A.vii (Reaction 7-9). Photochemical [2 + 2]-cycloadditions can also take place intramolecularly if a molecule has two double bonds that are properly oriented.2451 The cyclization of the quinone dimer shown above is one example. Other examples are
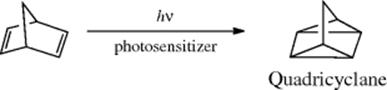
Ref. 2452
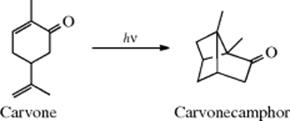
Ref. 2453
It is obvious that many molecules can be constructed in this way that would be difficult to make by other procedures. However, attempted cyclizations of this kind are not always successful. In many cases, polymeric or other side products are obtained instead of the desired product.
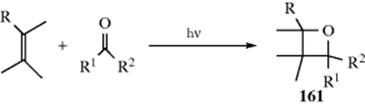
The photochemical cycloaddition of a carbonyl, generally from an aldehyde or ketone and an alkene, is called the Paternò–Büchi reaction.2454 This [2 + 2]-cycloaddition gives an oxetane (161) and the reaction is believed to proceed via a diradical intermediate. Silyl enol ethers react with aldehydes under nonphotochemical conditions using ZnCl2 at 25°C or SnCl4 at −78°C.2455
It is possible that some of these photochemical cycloadditions take place by a [π2s + π2s]-mechanism, which is of course allowed by orbital symmetry; when and if they do, one of the molecules must be in the excited singlet state (S1) and the other in the ground state.2456 The nonphotosensitized dimerizations of cis- and trans-2-butene are stereospecific,2457 making it likely that the [π2s + π2s]-mechanism is operating in these reactions. However, in most cases it is a triplet excited state that reacts with the ground-state molecule; in these cases the diradical (or in certain cases, the diionic) mechanism is taking place.2458 In one intramolecular case, the intermediate diradical has been trapped.2459 Photosensitized [2π + 2π]-cycloadditions almost always involve the triplet state, and hence a diradical (or diionic) mechanism.
The photochemical diradical mechanism is not quite the same as the thermal diradical mechanism. In the thermal mechanism, the initially formed diradical must be a singlet, but in the photochemical process a triplet excited state is adding to a ground state, which is of course a singlet. Thus, in order to conserve spin,2460 the initially formed diradical must be a triplet; that is, the two electrons must have the same spin. Consequently, the second, or ring closing, step of the mechanism cannot take place at once, because a new bond cannot form from a combination of two electrons with the same spin, and the diradical has a reasonably long lifetime before collisions with molecules in the environment allow a spin inversion to take place and the diradical to cyclize. The prediction is nonstereospecificity, and that is what is found.2461 It has been believed that at least some [2 + 2] photocycloadditions take place by way of exciplex intermediates2462 [an exciplex2463 is an excited EDA complex (Sec. 7.A.vii) that is dissociated in the ground state; in this case one double bond is the donor and the other is the acceptor], but there is evidence against this.2464
In Reaction 15-60, the principle of conservation of orbital symmetry was used to explain why certain reactions take place readily and others do not. The orbital-symmetry principle can also explain why certain molecules are stable although highly strained. Quadricyclane and hexamethylprismane2465 are thermodynamically much less stable (because they are much more strained), for example, than their corresponding isomeric dienes, norbornadiene and
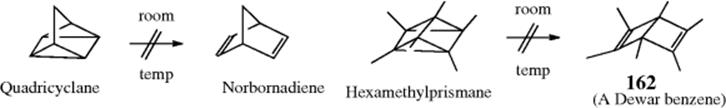
hexamethylbicyclo[2.2.0]hexadiene (162).2466 Yet the former two compounds can be kept indefinitely at room temperature, although in the absence of orbital-symmetry considerations it is not easy to understand why the electrons simply do not move over to give the more stable diene isomers. The reason is that both these reactions involve the conversion of a cyclobutane ring to a pair of double bonds (a s2 + s2 process) and, as seen previously, a thermal process of this sort is forbidden by the Woodward–Hoffmann rules. The process is allowed photochemically, so both quadricyclane and hexamethylprismane are photochemically converted to the respective dienes at room temperature or below.2467 It is also possible to conceive of simple bond rearrangements whereby hexamethylprismane is converted to hexamethylbenzene (as shown below), which of course is far more stable than either hexamethylprismane or 162. It has been calculated that hexamethylbenzene is at least 90 kcal mol−1 (380 kJ mol−1) more stable than hexamethylprismane. A correlation diagram for this reaction2468 discloses that it too is a symmetry-forbidden process. All three of these “forbidden” reactions do take place when the compounds are heated, but the diradical mechanism is likely under these conditions.2468
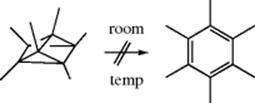
Bicyclo[2.2.0]hexadienes and prismanes are valence isomers of benzenes.2469 These compounds actually have the structures that were proposed for benzenes in the nineteenth century. Prismanes have the Ladenburg formula, and bicyclo[2.2.0]hexadienes have the Dewar formula. Because of this, bicyclo[2.2.0]hexadiene is often called Dewar benzene. In the paragraph prior to Section 2.A, it was mentioned that Dewar formulas are canonical forms (although not very important) of benzenes. Yet they also exist as separate compounds in which the positions of the nuclei are different from those of benzenes.
OS V, 54, 235, 277, 297, 370, 393, 424, 459, 528; VI, 378, 571, 962, 1002, 1024, 1037; VII, 177, 256, 315; VIII, 82, 116, 306, 377; IX, 28, 275; 80, 160. For the reverse reaction, see OS V, 734.
15-64 The Addition of Carbenes and Carbenoids to Double and Triple Bonds
epi-Methylene-addition

Carbenes and substituted carbenes add to double bonds to give cyclopropane derivatives by what can be considered as a formal [1 + 2]-cycloaddition.2470 Many carbene derivatives [e.g., PhCH, ROCH,2471 Me2C=C, and C(CN)2] have been added to double bonds, but the reaction is often performed with CH2 itself, with halo and dihalocarbenes,2472 and with carbalkoxycarbenes2473 (generated from diazoacetic esters). Alkylcarbenes (HCR) have been added to alkenes,2474 but more often these rearrange to give alkenes (Sec. 5.D.ii, category 4). The carbene can be generated in any of the ways normally used (Sec. 5.D.ii). However, most reactions in which a cyclopropane is formed by treatment of an alkene with a carbene “precursor” do not actually involve free carbene intermediates. In some cases, it is certain that free carbenes are not involved, and in other cases there is doubt. Because of this, the term carbene transfer is often used to cover all reactions in which a double bond is converted to a cyclopropane, whether a carbene or a carbenoid (Sec. 5.D.ii) is actually involved.
Carbene itself (:CH2) is extremely reactive and gives many side reactions, especially insertion reactions (12-21), which greatly reduce yields. This competition is also true with Rh catalyzed diazoalkane cyclopropanations2475 (see below). When: CH2 must be added for preparative purposes, a free carbene is not used, but the Simmons–Smith procedure (see 167) or some other method that does not involve free carbenes is employed instead. Halocarbenes are less active than carbenes, and this reaction proceeds quite well, since insertion reactions do not interfere.2476 Vinyldiazolactone is a vinylcarbene precursor for a reaction with alkenes to give spirolactones.2477
The absolute rate constant for addition of selected alkoxychlorocarbene to butenes has been measured to range from 330 to 1 × 104 M−1 s−1.2478 Both entropy and enthalpy play a role in addition of some carbenes.2479 Shown are a few of the many ways2480 in which halocarbenes or carbenoids are generated,2481 and most involve formal elimination (for the first two steps of the SN1cB mechanism, see Sec. 10.G.iii):
![]()
![]()
![]()
![]()
Ref. 2482
![]()
Ref. 2483
![]()
Ref. 2484
The reaction between CHCl3 and HO− is often carried out under phase-transfer conditions.2485 It has been shown that the reaction between PhCHCl2 and t-BuOK produces a carbenoid, but when the reaction is run in the presence of a crown ether, the carbene [Ph(Cl)C:] is formed instead.2486 The reaction of iodoform and CrCl2 leads to iodocyclopropanes upon reaction with alkenes.2487 Dihalocyclopropanes are very useful compounds2488 that can be reduced to cyclopropanes, treated with Mg or Na to give allenes (Reaction 18-3), or converted to a number of other products.
Alkenes of all types can be converted to cyclopropane derivatives by this reaction, but difficulty may be encountered with sterically hindered ones.2489 Even tetracyanoethylene, which responds very poorly to electrophilic attack, gives cyclopropane derivatives with carbenes.2490 Conjugated dienes give 1,2-addition to give a vinylcyclopropane.2491 Addition of a second molar equivalent gives bicyclopropyl derivatives.2492 1,4-Addition is rare but has been reported in certain cases.2493 Carbene adds to ketene to give cyclopropanone.2494 Allenes react with carbenes to give cyclopropanes with exocyclic unsaturation:2495
![]()
A second equivalent gives spiropentanes. In fact, any size ring with an exocyclic double bond can be converted by a carbene to a spiro compound.2496
Free carbenes can also be avoided by using transition metal–carbene complexes (LnM = CRR′, L = a ligand, M = a metal),2497 which add the group CRR′ to double bonds.2498 An example is the reaction of iron carbene (163).2499These complexes can be isolated in some cases; in others they are generated in situ from appropriate precursors, of which diazo compounds are among the most important. Chromium complexes have been used for the cyclopropanation of alkenes.2500
Diazo compounds, including CH2N2 and other diazoalkanes, react with metals or metal salts (Cu, Pd,2501 Ag,2502 La,2503 and Rh2504 are commonly used) to give the carbene complexes that add:CRR′ to double bonds.2505 Polymer-supported benzenesulfonyl azides have been developed as a safe diazo-transfer reagent.2506 Diazoketones and diazoesters react with alkenes to give the cyclopropane derivative, usually with a transition metal catalyst (e.g., a Cu complex).2507 The Ru catalyst reaction of diazoesters with an alkyne gives a cyclopropene.2508 An X-ray structure of an Os catalyst intermediate has been determined.2509 Electron-rich alkenes react faster than simple alkenes.2510
Asymmetric cyclopropanation reactions are a growing area of interest,2511 and chiral complexes have been used for enantioselective cyclopropane synthesis.2512 Decomposition of diazoalkanes in the presence of chiral Rh2513Cu,2514 Ir,2515 Co,2516 Au,2517 or Ru2518 complexes leads to optically active cyclopropanes. Diazosulfonate esters have been used in asymmetric cyclopropanations.2519 The use of chiral additives or auxiliaries with a metal complex also leads to cyclopropanes enantioselectively.2520 An important chiral species is Rh2(S-DOSP)4,2521 which leads to cyclopropanes with excellent enantioselectivity in carbene cyclopropanation reactions.2522 Chiral organocatalysts have been used.2523 The Cu catalyzed diazoester cyclopropanation was reported in an ionic liquid.2524 Phosphonate esters have been incorporated into the diazo compound.2525 Fischer carbene compounds (see Reaction 15-58) react with enolate anions to give cyclopropane derivatives.2526 A Cr promoted cyclopropanation of conjugated amides has been reported.2527
Asymmetric, intramolecular cyclopropanation reactions have been reported.2528 Note that the reaction of a diazoester with a chiral dirhodium catalyst leads to β-lactones with modest enantioselectivity.2529
Triple-bond compounds2530 react with carbenes to give cyclopropenes, except that in the case of acetylene itself, the cyclopropenes first formed cannot be isolated because they rearrange to allenes.2531 Cyclopropenones (Sec. 2.K.i) are obtained by hydrolysis of dihalocyclopropenes.2532
Most carbenes are electrophilic, and, in accord with this, electron-donating substituents on the alkene increase the rate of the reaction, and electron-withdrawing groups decrease it,2533 although the range of relative rates is not very great.2534 As discussed in Section 5.D.i, carbenes in the singlet state, which is the most common state, react stereospecifically and syn,2535 probably by a one-step mechanism,2536 similar to mechanism a of Reactions 15-60 and 15-63:
Infrared spectra of a carbene and the cyclopropane product have been observed in an Ar matrix at 12–45 K.2537 Carbenes in the triplet state react nonstereospecifically,2538 probably by a diradical mechanism, similar to mechanism bof Reactions 15-49 and 15-63:
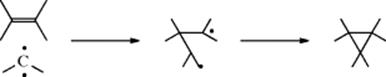
For carbenes or carbenoids of the type R–C–R′ there is another aspect of stereochemistry.2539 When these species are added to all but symmetrical alkenes, two isomers are possible, even if the four groups originally on the double-bond carbons maintain their configurations:
Which isomer is predominantly formed depends on R, R′, and on the method by which the carbene or carbenoid is generated. Most studies have been carried out on monosubstituted species (R′ = H), and in these studies it is found that aryl groups generally prefer the more substituted side (syn addition) while carbethoxy groups usually show antistereoselectivity. When R = halogen, free halocarbenes show little or no stereochemical preference, while halocarbenoids exhibit a preference for syn addition. Beyond this, it is difficult to make simple generalizations.
Carbenes are so reactive that they add to the “double bonds” of aromatic rings.2540 The products are usually unstable and rearrange to give ring expansion. Carbene reacts with benzene to give cycloheptatriene (164),2541 but not all carbenes are reactive enough to add to benzene. The norcaradiene intermediate cannot be isolated in this case2542 (it undergoes an electrocyclic rearrangement, Reaction 18-27), although certain substituted norcaradienes, [e.g., the product of addition of:C(CN)2 to benzene],2543 have been isolated.2544 With:CH2, insertion is a major side reaction, and, for example, benzene gives toluene as well as cycloheptatriene. A method of adding:CH2 to benzene rings without the use of free carbene is the catalytic decomposition of diazomethane (CH2N2) in the aromatic compound as a solvent with CuCl or CuBr.2545 By this method, better yields of cycloheptatrienes are obtained without insertion of side products. Picosecond optical grating calorimetry has been used to investigate the photochemical decomposition of diazomethane in benzene, and it appears that a transient is formed that is consistent with a weak complex between singlet methylene and benzene.2546 Chlorocarbene (:CHCl) is active enough to add to benzene, but dihalocarbenes do not add to benzene or toluene, only to rings with greater electron density. Pyrroles and indoles can be expanded, respectively, to pyridines and quinolines by treatment with halocarbenes2547 via the initially formed adduct 165 in the case of the indole. In such cases, a side reaction that sometimes occurs is expansion of the six-membered ring. Ring expansion can occur even with nonaromatic compounds, when the driving force is supplied by relief of strain (see Reaction 166).2548
As previously mentioned, free carbene is not very useful for additions to double bonds since it gives too many side products. The Simmons–Smith procedure accomplishes the same result without a free carbene intermediate and without insertion of side products.2549 This is known as a carbenoid reaction. Intramolecular variations are known.2550 This procedure involves treatment of the double-bond compound with CH2I2 and a Zn–Cu couple and leads to cyclopropane derivatives in good yields.2551 The Zn–Cu couple can be prepared in several ways,2552 and heating Zn dust with CuCl in ether under nitrogen2553 is particularly convenient. The reaction has also been done with unactivated Zn and ultrasound.2554 When TiCl4 is used along with Zn and CuCl, CH2I2 can be replaced by the cheaper CH2Br2.2555
The actual attacking species is an organozinc intermediate, probably (ICH2)2Zn·ZnI2, which is stable enough for isolable solutions.2556 An X-ray crystallographic investigation of the intermediate, complexed with a diether, has been reported.2557 The addition is stereospecifically syn, and a concerted mechanism2558 is likely, perhaps involving 167.2559 An iodomethylzinc phosphate has also been used for cyclopropanation reactions.2560 Diiodomethane gives cyclopropanes in a reaction mediated by indium.2561
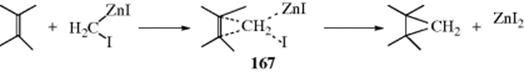
Asymmetric induction is possible when chiral additives are used.2562 Chiral complexes also lead to enantioselectivity in the cyclopropanation reaction.2563 Organocatalysts have been used.2564
With the Simmons–Smith procedure, as with free carbenes, conjugated dienes give 1,2-addition,2565 and allenes give methylenecyclopropanes or spiropentanes.2566
An alternative way of carrying out the Simmons–Smith reaction is by treatment of the substrate with CH2I2 or another dihalomethane and Et2Zn in ether.2567 This method can be adapted to the introduction of RCH and ArCH by the use of RCHI2 or ArCHI2 instead of the dihalomethane.2568 The reaction is compatible with other functionality in the carbenoid complex. The reaction of RCO2CH2I with diethyl zinc and an alkene under photolysis conditions give a cyclopropane.2569 In another method, CH2I2 or MeCHI2 is used along with an alane (R3Al) to transfer CH2 or CHMe.2570 Titanium complexes have been used similarly.2571 Samarium and CH2I2 has been used for the cyclopropanation of conjugated amides.2572 For the conversion of enolate anions to cyclopropanols, CH2I2 has been used along with SmI2.2573 Diodomethane in the presence of isopropylmagnesium chloride has been used to cyclopropanate allyl alcohols.2574
The Simmons–Smith reaction is the basis of a method for the indirect α methylation of a ketone.2575 The ketone (illustrated for cyclohexanone) is first converted to an enol ether, an enamine (Reaction 16-13) or silyl enol ether2576(Reaction 12-17), and cyclopropanation via the Simmons–Smith reaction is followed by hydrolysis to give the α methylated ketone. A related procedure using diethylzinc and diiodomethane allows ketones to be chain-extended by one carbon.2577 In another variation, phenols can be ortho methylated in one laboratory step, by treatment with Et2Zn and CH2I2.2578
Diazoesters react with amines with a Rh catalyst to give α-amino esters.2579 Diazoesters also react with aldehydes and a Rh catalyst. The product is an α,β-epoxy ester.2580 Diazoalkanes react similarly with aldehydes to give an alkene (Me3SiCH=N2 + ArCHO → ArCH=CHOSiMe3).2581
OS V, 306, 855, 859, 874; VI, 87, 142, 187, 327, 731, 913, 974; VII, 12, 200, 203; VIII, 124, 196, 321, 467; IX, 422; 76, 86.
15-65 Trimerization and Tetramerization of Alkynes
![]()
Aromatic compounds can be prepared by cyclotrimerization of alkynes2582 or triynes. Cyclotrimerization is possible by heating to 450–600 °C with no catalyst.2583 The spontaneous (no catalyst) trimerization of t-BuC![]() CF gave 1,2,3-tri-tert-butyl-4,5,6-trifluorobenzene (169), which the first time was three adjacent tert-butyl groups put onto a benzene ring.2584 The fact that this is a head-to-head joining allows formation of 169 from two alkynes. The fact that 168 (a Dewar benzene) was also isolated lends support to this scheme.2585
CF gave 1,2,3-tri-tert-butyl-4,5,6-trifluorobenzene (169), which the first time was three adjacent tert-butyl groups put onto a benzene ring.2584 The fact that this is a head-to-head joining allows formation of 169 from two alkynes. The fact that 168 (a Dewar benzene) was also isolated lends support to this scheme.2585
When acetylene is heated with nickel cyanide, other Ni(II) or Ni(0) compounds, or similar catalysts, it gives benzene and cyclooctatetraene.2586 It is possible to get more of either product by a proper choice of catalyst. Substituted acetylenes give substituted benzenes,2587 and this reaction has been used to prepare very crowded molecules. Dialkylalkynes were trimerized over CO2(CO)82588 and over Hg[Co(CO)4]2 to give hexaisopropylbenzene.2589 The six isopropyl groups are not free to rotate, but are lined up perpendicular to the plane of the benzene ring. Highly substituted benzene derivatives have also been prepared via cyclotrimeriztion using a Rh,2590 Ni,2591 Ti,2592 Mo,2593Ru,2594 Co,2595 or a Pd2596 catalyst. Alkynes react with allenes and a Ni catalyst to give highly substituted benzene derivatives.2597 Conjugated ketones react with internal alkynes with Me3Al and a Ni catalyst2598 to give an aromatic ring fused to a cyclic ketone after reaction with DBU and air.2599 N-Aryl chloroimines react with alkynes and a Rh catalyst to give quinolines,2600 as do N-aryl alkynyl imines with a W complex.2601
Intramolecular cyclotrimerizations have been reported by condensation of a diyne2602 with an alkyne in the presence of a Pd,2603 Mo,2604 Ni,2605 Rh,2606 Ir,2607 Ag,2608 Co,2609 or Ru catalyst.2610 Triynes have been similarly condensed with a Rh catalyst.2611 Note that this type of cyclization has been labeled as a [2 + 2 + 2]-cycloaddition reaction, which is discussed in Reaction 15-66. The internal cyclotrimerization of a triyne, utilizing a siloxy tether and a Co catalyst has been reported.2612 Fused-ring aromatic compounds are prepared by this method. Similar results were obtained from diynes and allenes with a Ni catalyst.2613 Solid-supported cyclotrimerizations have been reported.2614 Endiynes are cyclized to bicyclic arenes using a Pd2615 or Ru2616 catalyst, as are yndienes with a Ru catalyst.2617 Alkynyl biaryls are cyclized to phenanthrene derivatives using ICl.2618 In the presence of PhMe2SiH, CO, and a Rh catalyst, a nonconjugated triyne leads to a tricyclic compound in which a benzene ring is fused to two carbocyclic rings.2619 Internal cyclotrimerization of an aryl alkynyl ketone where the aryl group has an orthotrimethylsiylalkyne substituent gives a tetracyclic naphthalene derivative with a fused cyclopentanone unit.2620 Benzene derivatives with ortho alkyne units can be converted to naphthalene derivatives in aq NaOH with hydrazine, Te, NaBH4 and sonication.2621 Vinyl and alkyne substituents with a Ru catalyst lead to naphthalene derivatives.2622 Cyclotrimerization occurs with alkynyl boronic esters.2623
Imino and iodo substituents with a silyl alkyne and a Pd catalyst leads to an isoquinoline.2624 Benzene derivatives having ortho imine and alkyne substituents give an isoquinoline when treated with iodine2625 or with a Pd catalyst.2626 Diynes with nitriles and a Ru catalyst lead to isoquinolines.2627 Pyridines fused to carboxylic rings can be prepared by similar methodology using a cyanoamine and a Co catalyst.2628 An isocyanate (Ar–N=C=O) reacts with a diyne and a Ru catalyst to give a bicyclic pyridone.2629 Isocyanides and alkynes also react with a phosphine catalyst to give pyrroles.2630 Ortho alkynyl and epoxy substituents leads to β-naphthols using a Ru catalyst.2631
Nitriles react with 2 molar equivalents of acetylene, in the presence of a Co catalyst, to give 2-substituted pyridines.2632 Propargyl amines react with cyclohexanone derivatives and a Au complex to give tetrahydroquinolines.2633Treatment of alkynes with Cp2ZrEt2 followed by reaction with acetonitrile and then a second alkyne with a Ni catalyst gives a highly substituted pyridine.2634 This reaction can be done intramolecularly using a photochemically induced reaction with a Co catalyst and p-TolCN to give pyridines incorporated into macrocycles.2635 Diynes react with N-heterocyclic carbenes in the presence of a Ni catalyst to give pyridines.2636 Alkynyl esters react with enamino esters with a ZnBr2 catalyst to give substituted pyridines.2637 α-Halo oxime ethers react with alkynes and Grignard reagents, with a mixture of Pd and Cu catalysts, to give pyrimidines.2638 Triketones fix nitrogen gas in the presence of TiCl4 and Li metal to form bicyclic pyrrole derivatives.2639
In contrast to the spontaneous reaction, the catalyzed process seldom gives the 1,2,3-trisubstituted benzene isomer from an acetylene (RC![]() CH). The chief product is usually the 1,2,4-isomer,2640 with lesser amounts of the 1,3,15-isomer also generally obtained, but little if any of the 1,2,3-isomer. The mechanism of the catalyzed
CH). The chief product is usually the 1,2,4-isomer,2640 with lesser amounts of the 1,3,15-isomer also generally obtained, but little if any of the 1,2,3-isomer. The mechanism of the catalyzed
reaction to form benzenes2641 is believed to go through a species 170 in which two molecules of alkyne coordinate with the metal, and another species (171), a five-membered heterocyclic intermediate.2642 Such intermediates (where M = Rh, Ir, Zr,2643 or Ni) have been isolated and are shown to give benzenes (172) when treated with alkynes.2644 Note that this pathway accounts for the predominant formation of the 1,2,4-isomer. Two possibilities for the last step are a Diels–Alder reaction, and a ring expansion, each followed by extrusion of the metal:2645
In at least one case, the mechanism is different, going through a cyclobutadiene–nickel complex (see Sec. 2.K.ii), which has been isolated.2646 Similar results were obtained with a Ti complex.2647 Using a mixture of PdCl2 and CuCl2, however, aliphatic alkynes are converted to the 1,3,5-trialkyl benzene derivative.2648
Alkoxy chromium carbenes (Fischer carbene complexes, see Reaction 15-58) react with phenylalkynes to give naphthalene derivatives.2649 These Cr carbenes react with alkynyl boronates, cerium(IV) compounds, and then PhBr and a Pd catalyst to give a naphthoquinone.2650 Diynes react to give cyclotrimerization.2651 Note that vinyl Chromium carbenes react directly with alkynes to give spirocyclic compounds (spiro[4.4]nona-1,3,6-trienes).2652Benzofurans can be prepared using methoxy carbenes.2653 Amino-substituted chromium carbenes react with alkynes and then silica to give substituted benzene derivatives that have an aminoalkyl (–NR2) substituent.2654 Imino-substituted Chromium carbenes react with alkynes to give pyrrole derivatives.2655 Fischer carbene complexes react with alkynes to give the Dötz benzannulation,2656 giving p-alkoxylphenol derivatives. Modification of this basic technique can lead to eight-membered ring carbocycles (see Reaction 15-66).2657
When benzene, in the gas phase, was adsorbed onto a surface of 10% rhodium-on-alumina, the reverse reaction took place, and acetylene was formed.2658
Heating ketones in the presence of TlCl3OTf leads to 1,3,5-trisubstituted arenes.2659 Heating acetophenone with TiCl4 gives 1,3,5-triphenylbenzene.2660
OS VII, 256; IX, 1; 80, 93.
15-66 Other Cycloaddition Reactions
cyclo-[But-2-en-1,4-diyl]-1/4/addition, and so on
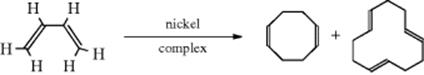
Cycloaddition reactions other than [4 + 2], [3 + 2], or [2 + 2] are possible, often providing synthetically useful routes to cyclic compounds. Conjugated dienes can be dimerized or trimerized at the 1,4-positions (formally, [4 + 4] and [4 + 4 + 4] cycloadditions) by treatment with certain complexes or other transition metal compounds.2661 Thus butadiene gives 1,5-cyclooctadiene and 1,5,9-cyclododecatriene.2662 The relative amount of each product can be controlled by use of the proper catalyst. For example, Ni:P(OC6H4-o-Ph)3 gives predominant dimerization, while Ni(cyclooctadiene)2 gives mostly trimerization. The products arise, not by direct 1,4 to 1,4 attack, but by stepwise mechanisms involving metal–alkene complexes.2663 The Rh catalyzed intramolecular cycloaddition of a furan with a conjugated diazoester gives a [3 + 4]-cycloadduct.2664 The suprafacial thermal addition of an allylic cation to a diene (a [4 + 3] cycloaddition) is allowed by the Woodward–Hoffmann rules (this reaction would be expected to follow the same rules as the Diels–Alder reaction2665). Pyrroles react with allylic diazo compounds, in the presence of a Rh catalyst, to give bicyclic amines in a [4 + 3]-cycloaddition.2666 A different [4 + 3]-cycloaddition involves the intramolecular reaction of a diene with an alkylidenecyclopropane unit, in the presence of a Pd catalyst, to give a seven-membered ring as part of a bicyclic system.2667 A [3 + 2 + 2]-cycloaddition was reported with an alkyne and an alkene–alkylidenecyclopropane substrate, in the presence of a Rh catalyst.2668 Reaction with a chiral Rh catalyst converts dienes and diazo compounds to cycloheptadienes.2669 Chiral cations have been used in [4 + 3] cycloadditions.2670 A [5 + 2]-cycloaddition of vinylcyclopropanes and alkenes, in the presence of a Rh catalyst, leads to seven-membered rings.2671 The reaction of a conjugated carbonyl compound with a diazo ester, in the presence of a Cu catalyst, leads to a dihydropyran in what is labeled a [4 + 1]-cycloaddition.2672 Dienes react with nitriles in a Ti mediated [4 + 1]-cycloaddition.2673 Cycloheptatriene reacts with terminal alkynes, using a complex catalyst involves Co and Zn compounds, to give a bicyclic triene via a [6 + 2]-cycloaddition.2674
As seen in Reaction 15-60, the Woodward–Hoffmann rules allow suprafacial concerted cycloadditions to take place thermally if the total number of electrons is 4n + 2 and photochemically if the number is 4n. Furthermore, forbidden reactions become allowed if one molecule reacts antarafacially. It would thus seem that syntheses of many large rings could easily be achieved. However, when the newly formed ring is eight membered or greater, concerted mechanisms, although allowed by orbital symmetry for the cases stated, become difficult to achieve. Due to the entropy factor the two ends of one system must simultaneously encounter the two ends of the other, unless one or both components are cyclic, in which case the molecule has many fewer possible conformations. There have been a number of reports of cycloaddition reactions leading to eight membered and larger rings, some thermally and some photochemically induced, but (apart from the dimerization and trimerization of butadienes mentioned above, which are known not to involve direct [4 + 4]- or [4 + 4 + 4]-cycloaddition) in most cases evidence is lacking to indicate whether they are concerted or stepwise processes. Some examples follows:
Ref.2675
Ref.2676
Ref.2677
Ref.2678
Benzene rings can undergo photochemical cycloaddition with alkenes.2679 The major product is usually the 1,3 addition product (173, in which a three-membered ring has also been formed), although some of the 1,2-product (174Reaction 15-63) is sometimes formed as well. Compound 174 is usually the main product where the alkene bears electron-withdrawing groups and the aromatic compound electron-donating groups, or vice versa. The 1,4-product (175) is rarely formed. The reaction has also been run with benzenes substituted with alkyl, halo, OR, CN, and other groups, and with acyclic and cyclic alkenes bearing various groups.2680
[2 + 2 + 2]-Cycloaddition reactions are known2681 (also see Reaction 15-65), usually with diynes, enynes, or intermolecular reactions of alkynes or alkenes with an alkyne, and facilitated by Ni2682 Ru,2683 or a Co catalyst.2684 A mechanistic density functional study has been reported for this reaction.2685 With a Co catalyst, an intramolecular [2 + 2 + 2]-cycloaddition of diynes with nitriles leads to bicyclic pyridines.2686 Alkenyl isocyanates and alkynes react via [2 + 2 + 2]-cycloaddition, in the presence of a Ru catalyst, to form bicyclic conjugated lactams.2687 Pyridines can also be prepared by metal-catalyzed [2 + 2 + 2]-cycloaddition.2688 Alkynes and isocyanates react with CO in the presence of a Ru catalyst to give imides,2689 and other [2 + 2 + 1]-cycloadditions are known,2690 A Co catalyst is used for a [4 + 2 + 2]-cycloaddition of 1,3-butadiene and bicyclo[2.2.2]octa-2,5-diene.2691 Eight-membered rings are produced by a Rh catalyzed [4 + 2 + 2] cycloaddition.2692 Yne-dienes undergo [4 + 2 + 1]-cycloaddition in the presence of a Ni catalyst.2693 Chromium catalysts are available for [6 + 4]-cycloadditions.2694 Ene-diynes undergo [2 + 2 + 2 + 1]-cycloaddition to form seven-membered ring ketones, in the presence of CO and a Rh catalyst.2695 Nickel catalyzed [2 + 2 + 2 + 2]-cycloadditions of alkynes lead to eight-membered rings.2696
Allenes and vinylcyclopropanes undergo [5 + 2]- and [5 + 2 + 1]-cycloadditions in the presence of a Rh catalyst.2697 A reductive elimination step determines the selectivity for various substrates in Rh catalyzed catalyzed [5 + 2]-cycloadditions.2698
OS VI, 512; VII, 485; X, 1, 336.
Notes
1. See de la Mare, P.B.D.; Bolton, R. Electrophilic Additions to Unsaturated Systems, 2nd ed., Elsevier, NY, 1982. For reviews, see Schmid, G.H. in Patai, S. Supplement A: The Chemistry of Double-bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 679–731; Schmid, G.H.; Garratt, D.G. in Patai, S. Supplement A: The Chemistry of Double-bonded Functional Groups, Vol. 1, pt. 2, Wiley, NY, 1977, pp. 725–912; Freeman, F. Chem. Rev. 1975, 75, 439.
2. See Mayr, H.; Kempf, B.; Ofial, A.R. Acc. Chem. Res. 2003, 36, 66.
3. See Fahey, R.C. Top. Stereochem. 1968, 3, 237; Bartlett, P.A. Tetrahedron 1980, 36, 2, pp. 3–15.
4. Heasley, G.E.; Bower, T.R.; Dougharty, K.W.; Easdon, J.C.; Heasley, V.L.; Arnold, S.; Carter, T.L.; Yaeger, D.B.; Gipe, B.T.; Shellhamer, D.F. J. Org. Chem. 1980, 45, 5150.
5. See Roberts, R.M.G. J. Chem. Soc. Perkin Trans. 2,1976, 1374; Pasto, D.J.; Gadberry, J.F. J. Am. Chem. Soc. 1978, 100, 1469; Naab, P.; Staab, H.A. Chem. Ber. 1978, 111, 2982.
6. Slebocka-Tilk, H.; Ball, R.G.; Brown, R.S. J. Am. Chem. Soc. 1985, 107, 4504.
7. Fischer, E. Liebigs Ann. Chem. 1911, 386, 374; McKenzie, A. Proc. Chem. Soc. 1911, 150; J. Chem. Soc. 1912, 101, 1196.
8. Michael, A. J. Prakt. Chem. 1892, 46, 209.
9. Francis, A.W. J. Am. Chem. Soc. 1925, 47, 2340.
10. See Zefirov, N.S.; Koz'min, A.S.; Dan'kov, Yu.V.; Zhdankin, V.V.; Kirin, V.N. J. Org. Chem. USSR 1984, 20, 205.
11. Hamilton, T.P.; Schaefer, III, H.F. J. Am. Chem. Soc. 1990, 112, 8260.
12. Ruasse, M.; Motallebi, S.; Galland, B. J. Am. Chem. Soc. 1991, 113, 3440; Bellucci, G.; Bianchini, R.; Chiappe, C.; Brown, R.S.; Slebocka-Tilk, H. J. Am. Chem. Soc. 1991, 113, 8012; Bennet, A.J.; Brown, R.S.; McClung, R.E.D.; Klobukowski, M.; Aarts, G.H.M.; Santarsiero, B.D.; Bellucci, G.; Bianchini, R. J. Am. Chem. Soc. 1991, 113, 8532.
13. Fahey, R.C.; Schneider, H. J. Am. Chem. Soc. 1968, 90, 4429. See also, Rolston, J.H.; Yates, K. J. Am. Chem. Soc. 1969, 91, 1469, 1477, 1483.
14. Ruasse, M.; Dubois, J.E. J. Am. Chem. Soc. 1975, 97, 1977; Bellucci, G.; Bianchini, R.; Chiappe, C.; Marioni, F. J. Org. Chem. 1990, 55, 4094.
15. Pincock, J.A.; Yates, K. Can. J. Chem. 1970, 48, 3332.
16. Cadogan, J.I.G.; Cameron D.K.; Gosney, I.; Highcock, R.M.; Newlands, S.F. J. Chem. Soc., Chem. Commun. 1985, 1751. For a review, see Ruasse, M. Acc. Chem. Res. 1990, 23, 87.
17. See Naae, D.G. J. Org. Chem. 1980, 45, 1394.
18. Kokil, P.B.; Fry, A. Tetrahedron Lett. 1986, 27, 5051.
19. Fahey, R.C. Top. Stereochem. 1968, 3, 237, pp. 273–277.
20. Hassner, A.; Boerwinkle, F.; Levy, A.B. J. Am. Chem. Soc. 1970, 92, 4879.
21. Capozzi, G.; Modena, G. in Bernardi, F.; Csizmadia, I.G.; Mangini, A. Organic Sulfur Chemistry, Elsevier, NY, 1985, pp. 246–298; Dittmer, D.C.; Patwardhan, B.H. in Stirling, C.J.M. The Chemistry of the Sulphonium Group, pt. 1, Wiley, NY, 1981, pp. 387–412; Capozzi, G.; Lucchini, V.; Modena, G.; Rev. Chem. Intermed. 1979, 2, 347; Schmid, G.H. Top. Sulfur Chem. 1977, 3, 102; Mueller, W.H. Angew. Chem. Int. Ed. 1969, 8, 482. The specific nature of the three-membered sulfur-containing ring is in dispute; see Smit, W.A.; Zefirov, N.S.; Bodrikov, I.V.; Krimer, M.Z. Acc. Chem. Res. 1979, 12, 282; Schmid, G.H.; Garratt, D.G.; Dean, C.L. Can. J. Chem. 1987, 65, 1172; Schmid, G.H.; Strukelj, M.; Dalipi, S. Can. J. Chem. 1987, 65, 1945.
22. See Bellucci, G.; Bianchini, R.; Chiappe, C.; Marioni, F.; Ambrosetti, R.; Brown, R.S.; Slebocka-Tilk, H. J. Am. Chem. Soc. 1989, 111, 2640.
23. Ohta, B.K.; Hough, R.E.; Jeffrey W.; Schubert, J.W. Org. Lett. 2007, 9, 2317.
24. See Sergeev, G.B.; Smirnov, V.V.; Rostovshchikova, T.N. Russ. Chem. Rev. 1983, 52, 259.
25. Also see Hampel, M.; Just, G.; Pisanenko, D.A.; Pritzkow, W. J. Prakt. Chem. 1976, 318, 930; Allen, A.D.; Tidwell, T.T. J. Am. Chem. Soc. 1983, 104, 3145.
26. Schubert, W.M.; Keeffe, J.R. J. Am. Chem. Soc. 1972, 94, 559; Chiang, Y.; Kresge, A.J. J. Am. Chem. Soc. 1985, 107, 6363.
27. Schmid, G.H.; Garratt, D.G. Can. J. Chem. 1973, 51, 2463.
28. See Anantakrishnan, S.V.; Ingold, C.K. J. Chem. Soc. 1935, 1396; Swern, D. in Swern, D. Organic Peroxides, Vol. 2, Wiley, NY, 1971, pp. 451–454; Nowlan, V.J.; Tidwell, T.T. Acc. Chem. Res. 1977, 10, 252.
29. See Bartlett, P.D.; Sargent, G.D. J. Am. Chem. Soc. 1965, 87, 1297 and are references cited therein.
30. See Mayr, H.; Pock, R. Chem. Ber. 1986, 119, 2473.
31. See Stammann, G.; Griesbaum, K. Chem. Ber. 1980, 113, 598.
32. Hammond, G.S.; Nevitt, T.D. J. Am. Chem. Soc. 1954, 76, 4121; See also, Pasto, D.J.; Meyer, G.R.; Lepeska, B. J. Am. Chem. Soc. 1974, 96, 1858.
33. Collins, C.H.; Hammond, G.S. J. Org. Chem. 1960, 25, 911.
34. See Heasley, G.E.; Bower, T.R.; Dougharty, K.W.; Easdon, J.C.; Heasley, V.L.; Arnold, S.; Carter, T.L.; Yaeger, D.B.; Gipe, B.T.; Shellhamer, D.F. J. Org. Chem. 1980, 45, 5150.
35. Becker, K.B.; Grob, C.A. Synthesis 1973, 789. See also, Marcuzzi, F.; Melloni, G.; Modena, G. Tetrahedron Lett. 1974, 413; Naab, P.; Staab, H.A. Chem. Ber. 1978, 111, 2982.
36. See Rappoport, Z. React. Intermed. (Plenum) 1983, 3, 427, pp. 428–440; Stang, P.J.; Rappoport, Z.; Hanack, M.; Subramanian, L.R. Vinyl Cations, Academic Press, NY, 1979, pp. 24–151; Stang, P.J. Prog. Phys. Org. Chem.1973, 10, 205; Modena, G.; Tonellato, U. Adv. Phys. Org. Chem. 1971, 9, 185, pp. 187–231.
37. See Bellucci, G.; Berti, G.; Ingrosso, G.; Mastrorilli, E. Tetrahedron Lett. 1973, 3911.
38. Patai, S.; Rappoport, Z. in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, pp. 469–584.
39. See in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 1, Wiley, NY, 1989, the articles by Boyd, G.V. pp. 281–315; Duval, D.; Géribaldi, S. pp. 355–469.
40. See Kutyrev, A.A.; Moskva, V.V. Russ. Chem. Rev. 1991, 60, 72; Finley, K.T. in Patai, S.; Rappoport, Z. The Chemistry of the Quinonoid Compounds, Vol. 2, pt. 1, Wiley, NY, 1988, pp. 537–717, see pp. 539–589; Finley, K.T. in Patai, S. The Chemistry of the Quinonoid Compounds, pt. 2, Wiley, NY, 1974, pp. 877–1144.
41. See Simpkins, N.S. Tetrahedron 1990, 46, 6951; Fuchs, P.L.; Braish, T.F. Chem. Rev. 1986, 86, 903.
42. See Bernasconi, C.F. Tetrahedron 1989, 45, 4017.
43. See Barbot, F.; Kadib-Elban, A.; Miginiac, P. J. Organomet. Chem. 1988, 345, 239.
44. See, however, Klumpp, G.W.; Mierop, A.J.C.; Vrielink, J.J.; Brugman, A.; Schakel, M. J. Am. Chem. Soc. 1985, 107, 6740.
45. Truce, W.E.; Levy, A.J. J. Org. Chem. 1963, 28, 679.
46. Truce, W.E.; Levy, A.J. J. Am. Chem. Soc. 1961, 83, 4641; Zefirov, N.S.; Yur'ev, Yu.K.; Prikazchikova, L.P.; Bykhovskaya, M.Sh. J. Gen. Chem. USSR 1963, 33, 2100.
47. Mohrig, J.R.; Fu, S.S.; King, R.W.; Warnet, R.; Gustafson, G. J. Am. Chem. Soc. 1990, 112, 3665.
48. See Truce, W.E.; Tichenor, G.J.W. J. Org. Chem. 1972, 37, 2391.
49. Hayakawa, K.; Kamikawaji, Y.; Wakita, A.; Kanematsu, K. J. Org. Chem. 1984, 49, 1985.
50. Truce, W.E.; Brady, D.G. J. Org. Chem. 1966, 31, 3543; Prilezhaeva, E.N.; Vasil'ev, G.S.; Mikhaleshvili, I.L.; Bogdanov, V.S. Bull. Acad. Sci. USSR Div. Chem. Sci. 1970, 1820.
51. Huyser, E.S. Free-Radical Chain Reactions, Wiley, NY, 1970; Nonhebel, D.C.; Walton, J.C. Free-Radical Chemistry Cambridge University Press, London, 1974; Pyor, W.A. Free Radicals McGraw-Hill, NY, 1965. See Giese, B. Rev. Chem. Intermed. 1986, 7, 3; Angew. Chem. Int. Ed. 1983, 22, 753; Abell, P.I. in Kochi, J.K. Free Radicals, Vol. 2, Wiley, NY, 1973, pp. 63–112; Minisci, F. Acc. Chem. Res. 1975, 8, 165.
52. Héberger, K.; Lopata, A. J. Chem. Soc. Perkin Trans. 2,1995, 91.
53. RajanBabu, T.V. Acc. Chem. Res. 1991, 24, 139; Beckwith, A.L.J. Rev. Chem. Intermed. 1986, 7, 143; Giese, B. Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Elmsford, NY, 1986, pp. 141–209; Surzur, J. React. Intermed. (Plenum) 1982, 2, 121–295; Julia, M. Acc. Chem. Res. 1972, 4, 386; Pure Appl. Chem. 1974, 40, 553; Thebtaranonth, C.; Thebtaranonth, Y. Tetrahedron 1990, 46, 1385.
54. Denis, R.C.; Rancourt, J.; Ghiro, E.; Boutonnet, F.; Gravel, D. Tetrahedron Lett. 1993, 34, 2091.
55. Goering, H.L.; Abell, P.I.; Aycock, B.F. J. Am. Chem. Soc. 1952, 74, 3588. See also, LeBel, N.A.; Czaja, R.F.; DeBoer, A. J. Org. Chem. 1969, 34, 3112.
56. Skell, P.S.; Allen, R.G. J. Am. Chem. Soc. 1958, 80, 5997.
57. Ogura, K.; Kayano, A.; Fujino, T.; Sumitani, N.; Fujita, M. Tetrahedron Lett. 1993, 34, 8313.
58. Goering, H.L.; Larsen, D.W. J. Am. Chem. Soc. 1959, 81, 5937. Also see, Skell, P.S.; Freeman, P.K. J. Org. Chem. 1964, 29, 2524.
59. Abell, P.I.; Piette, L.H. J. Am. Chem. Soc. 1962, 84, 916. See also, Leggett, T.L.; Kennerly, R.E.; Kohl, D.A. J. Chem. Phys. 1974, 60, 3264.
60. Golden, D.M.; Furuyama, S.; Benson, S.W. Int. J. Chem. Kinet. 1969, 1, 57.
61. Khristov, V.Kh.; Angelov, Kh.M.; Petrov, A.A. Russ. Chem. Rev. 1991, 60, 39.
62. Mislow, K. J. Am. Chem. Soc. 1953, 75, 2512.
63. Kharasch, M.S.; Kritchevsky, J.; Mayo, F.R. J. Org. Chem. 1938, 2, 489.
64. Nordlander, J.E.; Owuor, P.O.; Haky, J.E. J. Am. Chem. Soc. 1979, 101, 1288.
65. Afanas'ev, I.B.; Samokhvalov, G.I. Russ. Chem. Rev. 1969, 38, 318.
66. Curran, D.P.; Qi, H.; Porter, N.A.; Su, Q.; Wu, W.-X. Tetrahedron Lett. 1993, 34, 4489.
67. Giese, B.; Damm, W.; Roth, M.; Zehnder, M. Synlett 1992, 441.
68. Ferreri, C.; Ballestri, M.; Chatgilialoglu, C. Tetrahedron Lett. 1993, 34, 5147.
69. Table 15.1 is from de la Mare, P.B.D. Q. Rev. Chem. Soc. 1949, 3, 126, p. 145. Table 15.2 is from Dubois, J.E.; Mouvier, G. Tetrahedron Lett. 1963, 1325. See also, Grosjean, D.; Mouvier, G.; Dubois, J.E. J. Org. Chem. 1976, 41, 3869, 3872.
70. Shelton, J.R.; Lee, L. J. Org. Chem. 1960, 25, 428.
71. See Chambers, R.D.; Mobbs, R.H. Adv. Fluorine Chem. 1965, 4, 51.
72. See Fatiadi, A.J. Synthesis 1987, 249, 749; Dhar, D.N. Chem. Rev. 1967, 67, 611.
73. See Olah, G.A.; Mo, Y.K.J. Org. Chem. 1972, 37, 1028; Belen'kii, G.G.; German, L.S. Sov. Sci. Rev. Sect. B 1984, 5, 183; Dyatkin, B.L.; Mochalina, E.P.; Knunyants, I.L. Fluorine Chem. Rev. 1969, 3, 45.
74. Dickinson, C.L.; Wiley, D.W.; McKusick, B.C. J. Am. Chem. Soc. 1960, 82, 6132. For another example, see Atkinson, R.C.; de la Mare, P.B.D.; Larsen, D.S. J. Chem. Soc. Perkin Trans. 2,1983, 271.
75. Miller, Jr., W.T.; Fried, J.H.; Goldwhite, H. J. Am. Chem. Soc. 1960, 82, 3091.
76. Müllen, K.; Wolf, P. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 1, Wiley, NY, 1989, pp. 513–558.
77. See Ring, R.N.; Tesoro, G.C.; Moore, D.R. J. Org. Chem. 1967, 32, 1091.
78. Shenhav, H.; Rappoport, Z.; Patai, S. J. Chem. Soc. B 1970, 469.
79. Curran, D.P.; Ko, S.-B. Tetrahedron Lett. 1998, 39, 6629.
80. See in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, Wiley, NY, 1978, the articles by Schmid, G.H. pt. 1, pp. 275–341, and by Dickstein, J.I.; Miller, S.I. pt. 2, pp. 813–955; Miller, S.I.; Winterfeldt, E. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 267–334. For comparisons of double and triple bond reactivity, see Melloni, G.; Modena, G.; Tonellato, U. Acc. Chem. Res. 1981, 14, 227; Allen, A.D.; Chiang, Y.; Kresge, A.J.; Tidwell, T.T. J. Org. Chem. 1982, 47, 775.
81. See Strozier, R.W.; Caramella, P.; Houk, K.N. J. Am. Chem. Soc. 1979, 101, 1340.
82. Petrov, A.A. Russ. Chem. Rev. 1960, 29, 489.
83. Melloni, G.; Modena, G.; Tonellato, U. Acc. Chem. Res. 1981, 14, 227, p. 228.
84. Robertson, P.W.; Dasent, W.E.; Milburn, R.M.; Oliver, W.H. J. Chem. Soc. 1950, 1628.
85. Wolf, S.A.; Ganguly, S.; Berliner, E. J. Am. Chem. Soc. 1985, 50, 1053.
86. Walsh, A.D. Q. Rev. Chem. Soc. 1948, 2, 73.
87. Ng, L.; Jordan, K.D.; Krebs, A.; Rüger, W. J. Am. Chem. Soc. 1982, 104, 7414.
88. See Schmid, G.H.; Modro, A.; Lenz, F.; Garratt, D.G.; Yates, K. J. Org. Chem. 1976, 41, 2331.
89. See Winterfeldt, E. Angew. Chem. Int. Ed. 1967, 6, 423; Newer Methods Prep. Org. Chem. 1971, 6, 243.
90. Nelson, D.J.; Cooper, P.J.; Soundararajan, R. J. Am. Chem. Soc. 1989, 111, 1414.
91. See Tedder, J.M. Angew. Chem. Int. Ed. 1982, 21, 401.
92. Giese, B.; Lachhein, S. Angew. Chem. Int. Ed. 1982, 21, 768.
93. See Volovik, S.V.; Dyadyusha, G.G.; Staninets, V.I. J. Org. Chem. USSR 1986, 22, 1224.
94. Butler, D.N.; Gupt, I.; Ng, W.W.; Nyburg, S.C. J. Chem. Soc., Chem. Commun. 1980, 596.
95. Bolze, R.; Eierdanz, H.; Schlüter, K.; Massa, W.; Grahn, W.; Berndt, A. Angew. Chem. Int. Ed. 1982, 21, 924.
96. See Isenberg, N.; Grdinic, M. J. Chem. Educ. 1969, 46, 601; Grdinic, M.; Isenberg, N. Intra-Sci. Chem. Rep.,1970, 4, 145–162.
97. Sæthre, L.J.; Thomas, T.D.; Svensson, S. J. Chem. Soc. Perkin Trans. 2,1997, 749.
98. See Dubois, J.E.; Chrétien, J.R. J. Am. Chem. Soc. 1978, 100, 3506.
99. Myhre, P.C.; Andrews, G.D. J. Am. Chem. Soc. 1970, 92, 7595, 7596. See also, Newton, T.A. J. Chem. Educ. 1987, 64, 531.
100. Suresh, C.H.; Koga, N.; Gadre, S.R. J. Org. Chem. 2001, 66, 6883.
101. Tedder, J.M.; Walton, J.C. Tetrahedron 1980, 36, 701; Acc. Chem. Res. 1976, 9, 183. See also, Giese, B. Rev. Chem. Intermed. 1986, 7, 3; Tedder, J.M. J. Chem. Educ. 1984, 61, 237.
102. See, however, Gleicher, G.J.; Mahiou, B.; Aretakis, A.J. J. Org. Chem. 1989, 54, 308.
103. For an exception, see Wilt, J.W. Tetrahedron 1985, 41, 3979.
104. See Beckwith, A.L.J.; Schiesser, C.H. Tetrahedron 1985, 41, 3925; Spellmeyer, D.C.; Houk, K.N. J. Org. Chem. 1987, 52, 959.
105. See Beckwith, A.L.J.; Easton, C.J.; Serelis, A.K. J. Chem. Soc., Chem. Commun. 1980, 482.
106. See Chuang, C.; Gallucci, J.C.; Hart, D.J.; Hoffman, C. J. Org. Chem. 1988, 53, 3218.
107. See Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis Wiley, NY, 1984; Pasto, D.J. Tetrahedron 1984, 40, 2805; Smadja, W. Chem. Rev. 1983, 83, 263; in Landor, S.R. The Chemistry of Allenes, Vol. 2, Academic Press, NY, 1982, articles by Landor, S.R., Jacobs, T.L.; Hopf, H. pp. 351–577; Stang, P.J.; Rappoport, Z.; Hanack, M.; Subramanian, L.R. Vinyl Cations, Academic Press, NY, 1979, pp. 152–167; Richey, Jr., H.G.; Richey, J.M. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 917–922; Taylor, D.R. Chem. Rev. 1967, 67, 317, pp. 338–346; Griesbaum, K. Angew. Chem. Int. Ed. 1966, 5, 933. See Ma, S. Pure Appl. Chem. 2006, 78,197.
108. See Okuyama, T.; Izawa, K.; Fueno, T. J. Am. Chem. Soc. 1973, 95, 6749.
109. Also see Poutsma, M.L.; Ibarbia, P.A. J. Am. Chem. Soc. 1971, 93, 440.
110. Jacobs, T.L. in Landor, S.R. The Chemistry of Allenes, Vol. 2, Academic Press, NY, 1982, pp. 399–415.
111. Griesbaum, K.; Oswald, A.A.; Quiram, E.R.; Naegele, W. J. Org. Chem. 1963, 28, 1952.
112. See Pasto, D.J.; L'Hermine, G. J. Org. Chem. 1990, 55, 685.
113. See Pasto, D.J.; Warren, S.E.; Morrison, M.A. J. Org. Chem. 1981, 46, 2837. See, however, Bartels, H.M.; Boldt, P. Liebigs Ann. Chem. 1981, 40.
114. Henbest, H.B.; McCullough, J.J. Proc. Chem. Soc. 1962, 74.
115. See Traylor, T.G. Acc. Chem. Res. 1969, 2, 152.
116. Koga, N.; Ozawa, T.; Morokuma, K. J. Phys. Org. Chem. 1990, 3, 519.
117. Brown, H.C.; Kawakami, J.H.; Liu, K. J. Am. Chem. Soc. 1973, 95, 2209.
118. Brown, H.C.; Liu, K. J. Am. Chem. Soc. 1975, 97, 600, 2469.
119. See Azovskaya, V.A.; Prilezhaeva, E.N. Russ. Chem. Rev. 1972, 41, 516.
120. Srivastava, S.; le Noble, W.J. J. Am. Chem. Soc. 1987, 109, 5874. See also, Bodepudi, V.R.; le Noble, W.J. J. Org. Chem. 1991, 56, 2001.
121. Cieplak, A.S.; Tait, B.D.; Johnson, C.R. J. Am. Chem. Soc. 1989, 111, 8447.
122. Cieplak, A.S. J. Am. Chem. Soc. 1981, 103, 4540. See also, Jorgensen, W.L. Chemtracts: Org. Chem. 1988, 1, 71.
123. Senda, Y.; Nakano, S.; Kunii, H.; Itoh, H. J. Chem. Soc. Perkin Trans. 2,1993, 1009.
124. Coxon, J.M.; McDonald, D.Q. Tetrahedron 1992, 48, 3353.
125. Readio, P.D.; Skell, P.S. J. Org. Chem. 1966, 31, 753, 759.
126. See Anselmi, C.; Berti, G.; Catelani, G.; Lecce, L.; Monti, L. Tetrahedron 1977, 33, 2771.
127. LeBel, N.A.; Czaja, R.F.; DeBoer, A. J. Org. Chem. 1969, 34, 3112
128. See Giese, B. Angew. Chem. Int. Ed. 1989, 28, 969.
129. Charton, M. in Zabicky, J. The Chemistry of Alkenes, vol 2., Wiley, NY, 1970, pp. 569–592; Reissig, H. Top. Curr. Chem. 1988, 144, 73; Wong, H.N.C.; Hon, M.; Tse, C.; Yip, Y.; Tanko, J.; Hudlicky, T. Chem. Rev. 1989, 89, 165.
130. See, however, Gordon, A.J. J. Chem. Educ. 1967, 44, 461.
131. Moon, S. J. Org. Chem. 1964, 39, 3456.
132. See DePuy, C.H. Top. Curr. Chem. 1973, 40, 73–101. For a list of references to pertinent mechanistic studies, see Wiberg, K.B.; Kass, S.R. J. Am. Chem. Soc. 1985, 107, 988.
133. Kramer, G.M. J. Am. Chem. Soc. 1970, 92, 4344.
134. See Hendrickson, J.B.; Boeckman, Jr., R.K. J. Am. Chem. Soc. 1969, 91, 3269.
135. See Hogeveen, H.; Roobeek, C.F.; Volger, H.C. Tetrahedron Lett. 1972, 221; Battiste, M.A.; Mackiernan, J. Tetrahedron Lett. 1972, 4095. See also, Coxon, J.M.; Steel, P.J.; Whittington, B.I. J. Org. Chem. 1990, 55, 4136.
136. DePuy, C.H.; Fünfschilling, P.C.; Andrist, A.H.; Olson, J.M. J. Am. Chem. Soc. 1977, 99, 6297.
137. Hendrickson, J.B.; Boeckman, Jr., R.K. J. Am. Chem. Soc. 1971, 93, 4491.
138. Collins, C.J. Chem. Rev. 1969, 69, 543; Lee, C.C. Prog. Phys. Org. Chem. 1970, 7, 129.
139. Coxon, J.M.; Steel, P.J.; Whittington, B.I.; Battiste, M.A. J. Org. Chem. 1989, 54, 1383; Coxon, J.M.; Steel, P.J.; Whittington, B.I. J. Org. Chem. 1989, 54, 3702.
140. Lambert, J.B.; Chelius, E.C.; Bible, Jr., R.H.; Hadju, E. J. Am. Chem. Soc. 1991, 113, 1331.
141. Koch, W.; Liu, B.; Schleyer, P.v.R. J. Am. Chem. Soc. 1989, 111, 3479, and references cited therein.
142. Wiberg, K.B.; Kass, S.R. J. Am. Chem. Soc. 1985, 107, 988.
143. Maynes, G.G.; Applequist, D.E. J. Am. Chem. Soc. 1973, 95, 856; Incremona, J.H.; Shea, K.J.; Skell, P.S. J. Am. Chem. Soc. 1973, 95, 6728; Upton, C.J.; Incremona, J.H. J. Org. Chem. 1976, 41, 523.
144. See Wiberg, K.B.; Waddell, S.T.; Laidig, K. Tetrahedron Lett. 1986, 27, 1553.
145. Sarel, S.; Ben-Shoshan, B. Tetrahedron Lett. 1965, 1053. See also, Danishefsky, S. Acc. Chem. Res. 1979, 12, 66.
146. Arai, T.; Takahashi, O. J. Chem. Soc., Chem. Commun. 1995, 1837.
147. See Dugave, C.; Demange, L. Chem. Rev. 2003, 103, 2475. See Bond, G.C.; Wells, P.B. Adv. Catal. 1964, 15, 91; Anderson, J.R.; Baker, B.G. in Chemisorption and Reactions on Metallic Films, Vol. 2 Anderson, J.R., Ed. Academic Press, London, 1971, p. 63; Zaera, F. Langmuir 1996, 12, 88.
148. Lee, I.; Zaera, F. J. Am. Chem. Soc. 2005, 127, 12174.
149. Kim, I.S.; Dong, G.R.; Jung, Y.H. J. Org. Chem. 2007, 72, 5424.
150. Yu, J.; Gaunt, M.J.; Spencer, J.B. J. Org. Chem. 2002, 67, 4627.
151. Royzen, M.; Yap, G.P.A.; Fox, J.M. J. Am. Chem. Soc. 2008, 130, 3760.
152. Baag, Md.M.; Kar, A.; Argade, N.P. Tetrahedron 2003, 59, 6489.
153. Wakamatsu, K.; Takahashi, Y.; Kikuchi, K.; Miyashi, T. J. Chem. Soc. Perkin Trans. 2,1996, 2105.
154. Yasui, H.; Yorimitsu, H.; Oshima, K. Synlett 2006, 1783. For a review, see Kwong, C.K.-W.; Fu, M.Y.; Lam, C.S.-L.; Toy, P.H. Synthesis 2008, 2307.
155. Lee, P.S.; Du, W.; Boger, D.L.; Jorgensen, W.L. J. Org. Chem. 2004, 69, 5448.
156. Sato, T.; Komine, N.; Hirano, M.; Komiya, S. Chem. Lett. 1999, 441.
157. Baxendale, I.R.; Lee, A.-L.; Ley, S.V. Synlett 2002, 516.
158. Alphonse, F.-A.; Yudin, A.K. J. Am. Chem. Soc. 2006, 128, 11754. See Krompiec, S.; Pigulla, M.; Krompiec, M.; Baj, S.; Mrowiec-Bialon, J.; Kasperczyk, J. Tetrahedron Lett. 2004, 45, 5257.
159. Clennan, E.L.; Aebisher, D. J. Org. Chem. 2002, 67, 1036.
160. Bargiggia, F.; Piva, O. Tetrahedron Asymmetry 2001, 12, 1389.
161. Sergeyev, S.; Hesse, M. Synlett 2002, 1313.
162. Phillips, O.A.; Eby, P.; Maiti, S.N. Synth. Commun. 1995, 25, 87.
163. Carreño, M.C.; García, I.; Ribagorda, M.; Merino, E.; Pieraccini, S.; Spada, G.P. Org. Lett. 2005, 7, 2869.
164. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 633–636.
165. See Sharts, C.M.; Sheppard, W.A. Org. React. 1974, 21, 125, pp. 192–198, 212–214; Hudlicky, M. The Chemistry of Organic Fluorine Compounds, 2nd ed., Ellis Horwood, Chichester, 1976, pp. 36–41.
166. Kropp, P.J.; Daus, K.A.; Tubergen, M.W.; Kepler, K.D.; Wilson, V.P.; Craig, S.L.; Baillargeon, M.M.; Breton, G.W. J. Am. Chem. Soc. 1993, 115, 3071.
167. Olah, G.A.; Welch, J.T.; Vankar, Y.D.; Nojima, M.; Kerekes, I.; Olah, J.A. J. Org. Chem. 1979, 44, 3872. For a related method, see Olah, G.A.; Li, X. Synlett 1990, 267.
168. See Sergeev, G.B.; Smirnov, V.V.; Rostovshchikova, T.N.; Russ. Chem. Rev. 1983, 52, 259; Dewar, M.J.S. Angew. Chem. Int. Ed. 1964, 3, 245.
169. Boregeaud, R.; Newman, H.; Schelpe, A.; Vasco, V.; Hughes, D.E.P. J. Chem. Soc., Perkin Trans. 2,2002, 810.
170. See Thaler, W.A. Methods Free-Radical Chem. 1969, 2, 121, see pp. 182–195.
171. Mayo, F.R. J. Am. Chem. Soc. 1962, 84, 3964.
172. Landini, D.; Rolla, F. J. Org. Chem. 1980, 45, 3527.
173. See Cousseau, J. Synthesis 1980, 805; Kamiya, N.; Chikami, Y.; Ishii, Y. Synlett 1990, 675.
174. Boudjouk, P.; Kim, B.-K.; Han, B.-H. Synth. Commun. 1996, 26, 3479.
175. Tamura, M.; Shibakami, M.; Kurosawa, S.; Arimura, T.; Sekiya, A. J. Chem. Soc., Chem. Commun. 1995, 1891.
176. Su, M.; Yu, W.; Jin, Z. Tetrahedron Lett. 2001, 42, 3771.
177. Siriwardana, A.I.; Nakamura, I.; Yamamoto, Y. Tetrahedron Lett. 2003, 44, 985.
178. For an example, see Marx, J.N. Tetrahedron 1983, 39, 1529.
179. Gorton, P.J.; Walsh, R. J. Chem. Soc., Chem. Commun. 1972, 782. For evidence that a pericyclic mechanism may be possible, even for an isolated double bond, see Sergeev, G.B.; Stepanov, N.F.; Leenson, I.A.; Smirnov, V.V.; Pupyshev, V.I.; Tyurina, L.A.; Mashyanov, M.N. Tetrahedron 1982, 38, 2585.
180. Tidwell, T.T. Acc. Chem. Res. 1990, 23, 273; Seikaly, H.R.; Tidwell, T.T. Tetrahedron 1986, 42, 2587; Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev. 1975, 4, 231.
181. See Vinnik, M.I.; Obraztsov, P.A. Russ. Chem. Rev. 1990, 59, 63; Liler, M. Reaction Mechanisms in Sulphuric Acid Academic Press, NY, 1971, pp. 210–225.
182. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986; Kitching, W. Organomet. React. 1972, 3, 319; Organomet. Chem. Rev. 1968, 3, 61; Oullette, R.J. in Trahanovsky, W.S. Oxidation in Organic Chemistry pt. B, Academic Press, NY, 1973, pp. 140–166; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 387–396.
183. Brown, H.C.; Geoghegan, Jr., P.J. J. Org. Chem. 1972, 37, 1937; Brown, H.C.; Geoghegan Jr., P.J.; Lynch, G.J.; Kurek, J.T. J. Org. Chem. 1972, 37, 1941; Barrelle, M.; Apparu, M. Bull. Soc. Chim. Fr. 1972, 2016.
184. See Butler, R.N. in Pizey, J.S. Synthetic Reagents, Vol. 4, Wiley, NY, 1981, pp. 1–145.
185. Abreu, A.R.; Costa, I.; Rosa, C.; Ferreira, L.M.; Lourenço, A.; Santos, P.P. Tetrahedron 2005, 61, 11986.
186. See the extensive tables in Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 4–71.
187. Einhorn, J.; Einhorn, C.; Luche, J.L. J. Org. Chem. 1989, 54, 4479.
188. Campelo, J.M.; Chakraborty, R.; Marinas, J.M. Synth. Commun. 1996, 26, 1639.
189. See Bernasconi, C.F.; Leonarduzzi, G.D. J. Am. Chem. Soc. 1982, 104, 5133, 5143.
190. See Noyce, D.S.; DeBruin, K.E. J. Am. Chem. Soc. 1968, 90, 372.
191. Magnus, P.; Scott, D.A.; Fielding, M.R. Tetrahedron Lett. 2001, 42, 4127.
192. Bernasconi, C.F.; Paschalis, P. J. Am. Chem. Soc. 1989, 111, 5893, and other papers in this series.
193. Baskaran, S.; Gupta, V.; Chidambaram, N.; Chandrasekaran, S. J. Chem. Soc., Chem. Commun. 1989, 903.
194. Kumar, K.S.R.; Baskaran, S.; Chandrasekaran, S. Tetrahedron Lett. 1993, 34, 171.
195. Burgess, K.; Jaspars, M. Tetrahedron Lett. 1993, 34, 6813.
196. Burgess, K.; van der Donk, W.A. Tetrahedron Lett. 1993, 34, 6817.
197. Magnus, P.; Payne, A.H.; Waring, M.J.; Scott, D.A.; Lynch, V. Tetrahedron Lett. 2000, 41, 9725.
198. Matsushita, Y.; Sugamoto, K.; Matsui, T. Chem. Lett. 1993, 925.
199. Matshshita, Y.; Sugamoto, K.; Nakama, T.; Sakamoto, T.; Matsui, T.; Nakayama, M. Tetrahedron Lett. 1995, 36, 1879.
200. See Poon, N.L.; Satchell, D.P.N. J. Chem. Soc. Perkin Trans. 2 1986, 1485; Tidwell, T.T. Acc. Chem. Res. 1990, 23, 273; Seikaly, H.R.; Tidwell, T.T. Tetrahedron 1986, 42, 2587; Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev. 1975, 4, 231.
201. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 123–148; Khan, M.M.T.; Martell, A.E. Homogeneous Catalysis by Metal Complexes, Vol. 2, Academic Press, NY, 1974, pp. 91–95. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1217–1219.
202. Olah, G.A.; Meidar, D. Synthesis 1978, 671.
203. Marion, N.; Ramn, R.S.; Nolan, S.P. J. Am. Chem. Soc. 2009, 131, 448; Leyva, A.; Corma, A. J. Org. Chem. 2009, 74, 2067.
204. Hirabayashi, T.; Okimoto, Y.; Saito, A.; Morita, M.; Sakaguchi, S.; Ishii, Y. Tetrahedron 2006, 62, 2231.
205. Alvarez, P.; Basetti, M.; Gimeno, J.; Mancini, G. Tetrahedron Lett. 2001, 42, 8467.
206. Zhang, Z.; Lee, S.D.; Fisher, A.S.; Widenhoefer, R.A. Tetrahedron 2009, 65, 1794.
207. Shimada, T.; Yamamoto, Y. J. Am. Chem. Soc. 2002, 124, 12670.
208. Compain, P.; Goré, J.; Vatèle, J.-M. Tetrahedron 1996, 52, 10405.
209. See Ansell, M.F.; Palmer, M.H. Q. Rev. Chem. Soc. 1964, 18, 211. For a Rh(I)-catalyzed reaction in ionic liquids, see Oonishi, Y.; Ogura, J.; Sato, Y. Tetrahedron Lett. 2007, 48, 7505.
210. Wang, M.; Gao, L.X.; Mai, W.P.; Xia, A.X.; Wang, F.; Zhang, S.B. J. Org. Chem. 2004, 69, 2874.
211. Chiang, Y.; Kresge, A.J.; Capponi, M.; Wirz, J. Helv. Chim. Acta 1986, 69, 1331.
212. Tsuchimoto, T.; Joya, T.; Shirakawa, E.; Kawakami, Y. Synlett 2000, 1777.
213. Menashe, N.; Reshef, D.; Shvo, Y. J. Org. Chem. 1991, 56, 2912.
214. Vasudevan, A.; Verzas, M.K. Synlett 2004, 631. An acid catalyst may be added, see Le Bras, G.; Provot, O.; Peyrat, J.-F.; Alami, M.; Brion, J.-D. Tetrahedron Lett. 2006, 47, 5497.
215. Sheng, S.; Liu, X. Org. Prep. Proceed. Int. 2002, 34, 499.
216. See Cramer, P.; Tidwell, T.T. J. Org. Chem. 1981, 46, 2683.
217. Braga, A.L.; Martins, T.L.C.; Silveira, C.C.; Rodrigues, O.E.D. Tetrahedron 2001, 57, 3297. Also see Brandsma, L.; Bos, H.J.T.; Arens, J.F. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 751–860.
218. Arens, J.F. Adv. Org. Chem. 1960, 2, 163; Brandsma, L.; Bos, H.J.T.; Arens, J.F. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 774–775.
219. Banait, N.; Hojatti, M.; Findlay, P.; Kresge, A.J. Can. J. Chem. 1987, 65, 441.
220. Noyce, D.S.; Schiavelli, M.D. J. Org. Chem. 1968, 33, 845; J. Am. Chem. Soc. 1968, 90, 1020, 1023.
221. Labonne, A.; Kribber, T.; Hintermann, L. Org. Lett. 2006, 8, 5853.
222. Suzuki, T.; Tokunaga, M.; Wakatsuki, Y. Org. Lett. 2001, 3, 735. Also see Grotjahn, D.B.; Lev, D.A. J. Am. Chem. Soc. 2004, 126, 12232.
223. Hua, R.; Takeda, H.; Onozawa, S.-y.; Abe, Y.; Tanaka, M. Org. Lett. 2007, 9, 263.
224. See Nakamura, H.; Ishihara, K.; Yamamoto, H. J. Org. Chem. 2002, 67, 5124.
225. Kamitanaka, T.; Hikida, T.; Hayashi, S.; Kishida, N.; Matsuda, T.; Harada, T. Tetrahedron Lett. 2007, 48, 8460.
226. Gligorich, K.M.; Schultz, M..J.; Sigman, M.S. J. Am. Chem. Soc. 2006, 128, 2794.
227. Zhang, X.; Corma, A. Chem. Commun. 2007, 3080.
228. Yang, C.-G.; He, C. J. Am. Chem. Soc. 2005, 127, 6966.
229. Gruttadauric, M.; Aprile, C.; Riela, S.; Noto, R. Tetrahedron Lett. 2001, 42, 2213.
230. Miura, K.; Hondo, T.; Okajima, S.; Nakagawa, T.; Takahashi, T.; Hosomi, A. J. Org. Chem. 2002, 67, 6082; Marotta, E.; Foresti, E.; Marcelli, T.; Peri, F.; Righi, P.; Scardovi, N.; Rosini, G. Org. Lett. 2002, 4, 4451.
231. McDonald, F.E.; Towne, T.B. J. Org. Chem. 1995, 60, 5750.
232. Lattanzi, A.; Della Sala, G.D.; Russo, M.; Screttri, A. Synlett 2001, 1479.
233. Qian, H.; Han, X.; Widenhoefer, R.A. J. Am. Chem. Soc. 2004, 126, 9536.
234. Rönn, M.; Bäckvall, J.-E.; Andersson, P.G. Tetrahedron Lett. 1995, 36, 7749. See Tiecco, M.; Testaferri, L.; Santi, C. Eur. J. Org. Chem. 1999, 797.
235. Han, X.; Widenhoefer, R.A. J. Org. Chem. 2004, 69, 1738.
236. Miki, K.; Nishino, F.; Ohe, K.; Uemura, S. J. Am. Chem. Soc. 2002, 124, 5260.
237. Yang, C.-G.; Reich, N.W.; Shi, Z.; He, C. Org. Lett. 2005, 7, 4553.
238. Yu, X.; Seo, S.Y.; Marks, T.J. J. Am. Chem. Soc. 2007, 129, 7244.
239. Yao, T.; Zhang, X.; Larock, R.C. J. Am. Chem. Soc. 2004, 126, 11164.
240. Kang, S.H.; Park, C.M.; Lee, S.B.; Kim, M. Synlett 2004, 1279.
241. Hartman, J.W.; Sperry, L. Tetrahedron Lett. 2004, 45, 3787.
242. Antoniotti, S.; Genin, E.; Michelet, V.; Genêt, J.-P. J. Am. Chem. Soc. 2005, 127, 9976; Santos, L.L.; Ruiz, V.R.; Sabater, M.J.; Corma, A. Tetrahedron 2008, 64, 7902.
243. Gabriele, B.; Salerno, G.; Lauria, E. J. Org. Chem. 1999, 64, 7687.
244. Qing, F.L.; Gao, W.-Z.; Ying, J. J. Org. Chem. 2000, 65, 2003. See Kel'in, A.V.; Gevorgyan, V. J. Org. Chem. 2002, 67, 95.
245. Nan, Y.; Miao, H.; Yang, Z. Org. Lett. 2000, 2, 297. See also, Arcadi, A.; Cacchi, S.; DiGiuseppe, S.; Fabrizi, G.; Marinelli, F. Synlett 2002, 453.
246. Davidson, M.H.; McDonald, F.E. Org. Lett. 2004, 6, 1601.
247. Pale, P.; Chuche, J. Eur. J. Org. Chem. 2000, 1019.
248. The phenyl group also added to the allene. Kang, S.-K.; Baik, T.-G.; Kulak, A.N. Synlett 1999, 324.
249. Ma, S.; Gao, W. J. Org. Chem. 2002, 67, 6104.
250. Zhang, Z.; Widenhoefer, R.A. Angew. Chem. Int. Ed. 2007, 46, 283.
251. Mukai, C.; Ohta, M.; Yamashita, H.; Kitagaki, S. J. Org. Chem. 2004, 69, 6867.
252. Back, T.G.; Moussa, Z.; Parvez, M. J. Org. Chem. 2002, 67, 499.
253. See Chambers, R.D.; Mobbs, R.H. Adv. Fluorine Chem. 1965, 4, 51, pp. 53–61.
254. See Farnsworth, M.V.; Cross, M.J.; Louie, J. Tetrahedron Lett. 2004, 45, 7441.
255. Shostakovskii, M.F.; Trofimov, B.A.; Atavin, A.S.; Lavrov, V.I. Russ. Chem. Rev. 1968, 37, 907.
256. See Kresge, A.J.; Yin, Y. J. Phys. Org. Chem. 1989, 2, 43.
257. See Bolitt, V.; Mioskowski, C.; Shin, D.; Falck, J.R. Tetrahedron Lett. 1988, 29, 4583.
258. See Johnston, R.D.; Marston, C.R.; Krieger, P.E.; Goem G.L. Synthesis 1988, 393.
259. See Wan, P.; Yates, K. Rev. Chem. Intermed. 1984, 5, 157.
260. Marshall, J.A. Acc. Chem. Res. 1969, 2, 33.
261. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 162–345.
262. Brown, H.C.; Rei, M. J. Am. Chem. Soc. 1969, 91, 5646.
263. Brown, H.C.; Kurek, J.T.; Rei, M.; Thompson, K.L. J. Org. Chem. 1984, 49, 2551; 1985, 50, 1171.
264. Talybov, G.M.; Mekhtieva, V.Z.; Karaev, S.F. Russ. J. Org. Chem. 2001, 37, 600.
265. Kang, S.H.; Kim, M. J. Am. Chem. Soc. 2003, 125, 4684. For an enantioselective example, see Kang, S.H.; Lee, S.B.; Park, C.M. J. Am. Chem. Soc. 2003, 125, 15748.
266. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 346–366.
267. Garavelas, A.; Mavropoulos, I.; Perlmutter, P.; Westman, F. Tetrahedron Lett. 1995, 36, 463.
268. Quadbeck, G. Newer Methods Prep. Org. Chem. 1963, 2, 133–161. See Tidwell, T.T. Acc. Chem. Res. 1990, 23, 273; Seikaly, H.R.; Tidwell, T.T. Tetrahedron 1986, 42, 2587; Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev.1975, 4, 231.
269. Boeckman, Jr., R.K.; Pruitt, J.R. J. Am. Chem. Soc. 1989, 111, 8286.
270. Cannizzaro, C.E.; Strassner, T.; Houk, K.N. J. Am. Chem. Soc. 2001, 123, 2668.
271. See Ballantine, J.A.; Davies, M.; Purnell, H.; Rayanakorn, M.; Thomas, J.M.; Williams, K.J. J. Chem. Soc., Chem. Commun. 1981, 8.
272. See Peterson, P.E.; Tao, E.V.P. J. Org. Chem. 1964, 29, 2322.
273. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis Springer, NY, 1986, pp. 367–442.
274. Larock, R.C.; Hightower, T.R. J. Org. Chem. 1993, 58, 5298; Annby, U.; Stenkula, M.; Andersson, C.-M. Tetrahedron Lett. 1993, 34, 8545.
275. Ferraz, H.M.C.; Ribeiro, C.M.R. Synth. Commun. 1992, 22, 399.
276. Verboom, R.C.; Persson, B.A.; Bäckvall, J.-E. J. Org. Chem. 2004, 69, 3102.
277. Goossen, L.J.; Paetzold, J.; Koley, D. Chem. Commun. 2003, 706. See Hua, R.; Tian, X. J. Org. Chem. 2004, 69, 5782.
278. See Bianchini, C.; Meli, A.; Peruzzini, M.; Zanobini, F.; Bruneau, C.; Dixneuf, P.H. Organometallics 1990, 9, 1155.
279. Bassetti, M.; Floris, B. J. Chem. Soc. Perkin Trans. 2,1988, 227; Grishin, Yu.K.; Bazhenov, D.V.; Ustynyuk, Yu.A.; Zefirov, N.S.; Kartashov, V.R.; Sokolova, T.N.; Skorobogatova, E.V.; Chernov, A.N. Tetrahedron Lett. 1988, 29, 4631.
280. Mitsudo, T.; Hori, Y.; Yamakawa, Y.; Watanabe, Y. J. Org. Chem. 1987, 52, 2230
281. Mahé, R.; Sasaki, Y.; Bruneau, C.; Dixneuf, P.H. J. Org. Chem. 1989, 54, 1518.
282. See Sofia, M.J.; Katzenellenbogen, J.A. J. Org. Chem. 1985, 50, 2331. For a list of other examples, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 1895.
283. Liao, H.-Y.; Cheng, C.-H. J. Org. Chem. 1995, 60, 3711
284. Jiménez-Tenorio, M.; Puerta, M.C.; Valerga, P.; Moreno-Dorado, F.J.; Guerra, F.M.; Massanet, G.M. Chem. Commun. 2001, 2324.
285. Kharasch, M.S.; Fono, A. J. Org. Chem. 1959, 24, 606; Kochi, J.K. J. Am. Chem. Soc. 1962, 84, 1572.
286. Ma, S.; Wu, S. J. Org. Chem. 1999, 64, 9314.
287. Ma, S.; Li, L.; Wei, Q.; Xie, H.; Wang, G.; Shi, Z.; Zhang, J. Pure. Appl. Chem. 2000, 72, 1739.
288. Uemura, K.; Shiraishi, D.; Noziri, M.; Inoue, Y. Bull. Chem. Soc. Jpn. 1999, 72, 1063.
289. See Blake, P.G.; Vayjooee, M.H.B. J. Chem. Soc. Perkin Trans. 2,1976, 1533.
290. Braga, A.L.; Emmerich, D.J.; Silveira, C.C.; Martins, T.L.C.; Rodrigues, O.E.D. Synlett 2001, 371.
291. Steinmann, J.E.; Phillips, J.H.; Sanders, W.J.; Kiessling, L.L. Org. Lett. 2001, 3, 3557.
292. Bassindale, A.R.; Katampe, I.; Maesano, M.G.; Patel, P.; Taylor, P.G. Tetrahedron Lett. 199, 40, 7417.
293. See Wardell, J.L. in Patai, S. The Chemistry of the Thiol Group, pt. 1, Wiley, NY, 1974, pp. 169–178.
294. Shostakovskii, M.F.; Kul'bovskaya, N.K.; Gracheva, E.P.; Laba, V.I.; Yakushina, L.M. J. Gen. Chem. USSR 1962, 32, 707.
295. Belley, M.; Zamboni, R. J. Org. Chem. 1989, 54, 1230.
296. See Voronkov, M.G.; Martynov, A.V.; Mirskova, A.N. Sulfur Rep.,1986, 6, 77; Griesbaum, K. Angew. Chem. Int. Ed. 1970, 9, 273; Oswald, A.A.; Griesbaum, K. in Kharasch, N.; Meyers, C.Y. Organic Sulfur Compounds, Vol. 2, Pergamon, Elmsford, NY, 1966, pp. 233–256; Stacey, F.W.; Harris, Jr., J.F. Org. React. 1963, 13, 150, pp. 165–196, 247–324.
297. Lou, F.-W.; Xu, J.-M.; Liu, B.-K.; Wu, Q.; Pan, Q.; Lin, X.-F. Tetrahedron Lett. 2007, 48, 8815. For a solvent free, Ce(III)-promoted reaction, see Silveira, C.C.; Mendes, S.R.; Líbero, F.M. Synlett 2010, 790.
298. Ranu, B.C.; Mandal, T. Synlett 2007, 925; Ranu, B.C. Can. J. Chem. 2009, 87, 1605.
299. Kanagasabapathy, S.; Sudalai, A.; Benicewicz, B.C. Tetrahedron Lett. 2001, 42, 3791.
300. Kumar, P.; Pandey, R.K.; Hegde, V.R. Synlett 1999, 1921.
301. Janssen, M.J. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 720–723.
302. Haché, B.; Gareau, Y. Tetrahedron Lett. 1994, 35, 1837.
303. See Arjona, O.; Medel, R.; Rojas, J.; Costa, A.M.; Vilarrasa, J. Tetrahedron Lett. 2003, 44, 6369.
304. Kuniyasu, H.; Ogawa, A.; Sato, K.-I.; Ryu, I.; Kambe, N.; Sonoda, N. J. Am. Chem. Soc. 1992, 114, 5902.
305. Kirpichenko, S.V.; Tolstikova, L.L.; Suslova, E.N.; Voronkov, M.G. Tetrahedron Lett. 1993, 34, 3889.
306. Mukaiyama, T.; Saitoh, T.; Jona, H. Chem. Lett. 2001, 638.
307. Cui, D.-M.; Meng, Q.; Zheng, J.-Z.; Zhang, C. Chem. Commun. 2009, 1577.
308. Kondoh, A.; Takami, K.; Yorimitsu, H.; Oshima, K. J. Org. Chem. 2005, 70, 6468.
309. Cao, C.; Fraser, L.R.; Love, J.A. J. Am. Chem. Soc. 2005, 127, 17614; Yang, J.; Sabarre, A.; Fraser, L.R.; Patrick, B.O.; Love, J.A. J. Org. Chem. 2009, 74, 182.
310. Yadav, J.S.; Subba Reddy, B.V.; Raju, A.; Ravindar, K.; Baishya, G. Chem. Lett. 2007, 36, 1474
311. Weiss, C.J.; Wobser, S.D.; Marks, T.J. J. Am. Chem. Soc. 2009, 131, 2062.
312. Weiss, C.J.; Marks, T.J. J. Am. Chem. Soc. 2010, 132, 10533.
313. Yamashita, F.; Kuniyasu, H.; Terao, J.; Kambe, N. Org. Lett. 2008, 10, 101.
314. Silva, M.S.; Lara, R.G.; Marczewski, J.M.; Jacob, R.G.; Lenardão, E.J.; Perin, G. Tetrahedron Lett. 2008, 49, 1927.
315. Huang, X.; Zhong, P.; Guo, W.-r. Org. Prep. Proceed. Int. 1999, 31, 201.
316. Gabriele, B.; Salerno, G.; Fazio, A. Org. Lett. 2000, 2, 351.
317. Usugi, S.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2004, 6, 601.
318. Ananikov, V.P.; Beletskaya, I.P. Org. Biomol. Chem. 2004, 2, 284.
319. See Gassman, P.G.; Gilbert, D.P.; Cole, S.M. J. Org. Chem. 1977, 42, 3233.
320. See Blake, A.J.; Friend, C.L.; Outram, R.J.; Simpkins, N.S.; Whitehead, A.J. Tetrahedron Lett. 2001, 42, 2877.
321. Kuniyasu, H.; Ogawa, A.; Sato, K.-I.; Ryu, I.; Sonoda, N. Tetrahedron Lett. 1992, 33, 5525.
322. Dabdoub, M.J.; Baroni, A.C.M.; Lenardão, E.J.; Gianeti, T.R.; Hurtado, G.R. Tetrahedron 2001, 57, 4271.
323. Kamiya, I.; Nishinaka, E.; Ogawa, A. J. Org. Chem. 2005, 70, 696.
324. Müller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Chem. Rev. 2008, 108, 3795.
325. See Gasc, M.B.; Lattes, A.; Périé, J.J. Tetrahedron 1983, 39, 703; Pines, H.; Stalick, W.M. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds Academic Press, NY, 1977, pp. 423–454; Beller, M.; Breindl, C.; Eichberger, M.; Hartung, C.G.; Seayad, J.; Thiel, O.R.; Tillack, A.; Trauthwein, H. Synlett 2002, 1579. For selectivity, see Tillack, A.; Khedkar, V.; Beller, M. Tetrahedron Lett. 2004, 45, 8875.
326. Howk, B.W.; Little, E.L.; Scott, S.L.; Whitman, G.M. J. Am. Chem. Soc. 1954, 76, 1899.
327. Anderson, L.L.; Arnold, J.; Bergman, R.G. J. Am. Chem. Soc. 2005, 127, 14542.
328. For a review, see Doye, S. Synlett 2004, 1653.
329. See Gasc, M.B.; Lattes, A.; Périé, J.J. Tetrahedron 1983, 39, 703; Bäckvall, J. Adv. Met.-Org. Chem. 1989, 1, 135. Also see Rogers, M.M.; Kotov, V.; Chatwichien, J.; Stahl, S.S. Org. Lett. 2007, 9, 4331. See also Singer, R.A.; Doré, M.; Sieser, J.E.; Berliner, M.A. Tetrahedron Lett. 2006, 47, 3727.
330. The anti-Markovkinov amine is produced: Utsonomiya, M.; Hartwig, J.F. J. Am. Chem. Soc. 2004, 126, 2702; Routaboul, L.; Buch, C.; Klein, H.; Jackstell, R.; Beller, M. Tetrahedron Lett. 2005, 46, 7401. For mechansitic studies, see Takaya, J.; Hartwig, J.F. J. Am. Chem. Soc. 2005, 127, 5756.
331. Huang, J.-M.; Wong, C.-M.; Xu, F.-X.; Loh, T.-P. Tetrahedron Lett 2007, 48, 3375.
332. Kaspar, L.T.; Fingerhut, B.; Ackermann, L. Angew. Chem. Int. Ed. 2005, 44, 5972.
333. Michaux, J.; Terrasson, V.; Marque, S.; Wehbe, J.; Prim, D.; Campagne, J.-M. Eur. J. Org. Chem. 2007, 2601.
334. Herzon, S.B.; Hartwig, J.F. J. Am. Chem. Soc. 2007, 129, 6690.
335. Hesp, K.D.; Stradiotto, M. J. Am. Chem. Soc. 2010, 132, 18026.
336. O'Shaughnessy, P.N.; Scott, P. Tetrahedron Asymmetry 2003, 14, 1979.
337. Srivastava, R.S.; Nicholas, K.M. Chem. Commun. 1996, 2335.
338. Hultzsch, K.C. Org. Biomol. Chem. 2005, 3, 1819; Yin, P.; Loh, T.-P. Org. Lett. 2009, 11, 3791. See Reznichenko, A.L.; Nguyen, H.N.; Hultzsch, K.C. Angew. Chem. Int. Ed. 2010, 49, 8984.
339. Brouwer, C.; He, C. Angew. Chem. Int. Ed. 2006, 45, 1744.
340. Nishina, N.; Yamamoto, Y. Angew. Chem. Int. Ed. 2006, 45, 3314.
341. See Hegedus, L.S.; Åkermark, B.; Zetterberg, K.; Olsson, L.F. J. Am. Chem. Soc. 1984, 106, 7122.
342. See Utsunomiya, M.; Hartwig, J.F. J. Am. Chem. Soc. 2003, 125, 14286.
343. Minami, T.; Okamoto, H.; Ikeda, S.; Tanaka, R.; Ozawa, F.; Yoshifuji, M. Angew. Chem. Int. Ed. 2001, 40, 4501.
344. Hong, S.; Marks, T.J. J. Am. Chem. Soc. 2002, 124, 7886.
345. Kovács, G.; Ujaque, G.; Lledós, A. J. Am. Chem. Soc. 2008, 130, 853.
346. Ney, J.E.; Wolfe, J.P. J. Am. Chem. Soc. 2005, 127, 8644. See also, Sakai, N.; Ridder, A.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 8134; Rogers, M.M.; Wendlandt, J.E.; Guzei, I.A.; Stahl, S.S. Org. Lett. 2006, 8, 2257.
347. Takemiya, A.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 6042; Liu, Z.; Hartwig, J.F. J. Am. Chem. Soc. 2008, 130, 1570.
348. Kim, J.Y.; Livinghouse, T. Org. Lett. 2005, 7, 4391.
349. Quinet, C.; Ates, A.; Markó, I.E. Tetrahedron Lett. 2008, 49, 5032.
350. Müller, C.; Loos, C.; Schulenberg, N.; Doye, S. Eur. J. Org. Chem. 2006, 2499.
351. Kim, H.; Lee, P.H.; Livinghouse, T. Chem. Commun. 2005, 5205; Yang, L.; Xu, L.-W.; Zhou, W.; Gao, Y.-H.; Sun, W.; Xia, C.-G. Synlett 2009, 1167.
352. Gribkov, D.V.; Hultzsch, K.C.; Hampel, F. J. Am. Chem. Soc. 2006, 128, 3748.
353. Ryu, J.-S.; Marks, T.J.; McDonald, F.E. Org. Lett. 2001, 3, 3091. See Hong, S.; Tian, S.; Metz, M.V.; Marks, T.J. J. Am. Chem. Soc. 2003, 125, 14768.
354. Kim, Y.K.; Livinghouse, T.; Bercaw, J.E. Tetrahedron Lett. 2001, 42, 2933.
355. Crimmin, M.R.; Casely, I.J.; Hill, M.S. J. Am. Chem. Soc. 2005, 127, 2042.
356. Ates, A.; Quinet, C. Eur. J. Org. Chem. 2003, 1623.
357. Hartung, C.G.; Breindl, C.; Tillack, A.; Beller, M. Tetrahedron 2000, 56, 5157.
358. Fujita, H.; Tokuda, M.; Nitta, M.; Suginome, H. Tetrahedron Lett. 1992, 33, 6359.
359. Dieter, R.K.; Yu, H. Org. Lett. 2000, 2, 2283.
360. Singh, S.; Nicholas, K.M. Synth. Commun. 2001, 31, 3087.
361. Le Berre, A.; Delacroix, A. Bull. Soc. Chim. Fr. 1973, 640, 647. See also, Vogel, D.E.; Büchi, G. Org. Synth., 66, 29.
362. Molander, G.A.; Pack, S.K. J. Org. Chem. 2003, 68, 9214.
363. See Chekulaeva, I.A.; Kondrat'eva, L.V. Russ. Chem. Rev. 1965, 34, 669; Hartung, C.G.; Tillack, A.; Trauthwein, H.; Beller, M. J. Org. Chem. 2001, 66, 6339.
364. See Kruse, C.W.; Kleinschmidt, R.F. J. Am. Chem. Soc. 1961, 83, 213, 216.
365. Patil, N.T.; Lutete, L.M.; Wu, H.; Pahadi, N.K.; Gridnev, I.D.; Yamamoto, Y. J. Org. Chem. 2006, 71, 4270.
366. Khedkar, V.; Tillack, A.; Beller, M. Org. Lett. 2003, 5, 4767; Tillack, A.; Castro, I.G.; Hartung, C.G.; Beller, M. Angew. Chem. Int. Ed. 2002, 41, 2541; Bytschkov, I.; Doye, S. Eur. J. Org. Chem. 2001, 4411.
367. Anderson, L.L.; Arnold, J.; Bergman R.G. Org. Lett. 2004, 6, 2519; Cao, C.; Li, Y.; Shi, Y.; Odom, A.L. Chem. Commun.,2004, 2002.
368. Shanbhag, G.V.; Kumbar, S.M.; Joseph, T.; Halligudi, S.B. Tetrahedron Lett. 2006, 47, 141.
369. Mizushima, E.; Hayashi, T.; Tanaka, M. Org. Lett. 2003, 5, 3349.
370. MülHiroya, K.; Matsumoto, S.; Sakamoto, T. Org. Lett. 2004, 6, 2953; Lutete, L.M.; Kadota, I.; Yamamoto, Y. J. Am. Chem. Soc. 2004, 126, 1622. See also, Karur, S.; Kotti, S.R.S.S.; Xu, X.; Cannon, J.F.; Headley, A.; Li, G. J. Am. Chem. Soc. 2003, 125, 13340.
371. Kel'in, A.; Sromek, A.W.; Gevorgyan, V. J. Am. Chem. Soc. 2001, 123, 2074.
372. Ackermann, L.; Born, R. Tetrahedron Lett. 2004, 45, 9541. For a different approach using hypervalent iodine, see Barluenga, J.; Trincado, M.; Rubio, E.; González, J.M. Angew. Chem. Int. Ed. 2003, 42, 2406.
373. Huang, Q.; Hunter, J.A.; Larock, R.C J. Org. Chem. 2002, 67, 3437.
374. Schult, K.E.; Reisch, J.; Walker, H. Chem. Ber. 1965, 98, 98.
375. Zhou, L.; Bohle, D.S.; Jiang, H.-F.; Li, C.-J. Synlett 2009, 937.
376. Mak, X.Y.; Ciccolini, R.P.; Robinson, J.M.; Tester, J.W.; Danheiser, R.L. J. Org. Chem. 2009, 74, 9381.
377. Meguro, M.; Yamamoto, Y. Tetrahedron Lett. 1998, 39, 5421.
378. Geri, R.; Polizzi, C.; Lardicci, L.; Caporusso, A.M. Gazz. Chim. Ital.,1994, 124, 241.
379. Zeng, X.; Soleilhavoup, M.; Bertrand, G. Org. Lett. 2009, 11, 3166.
380. Davies, I.W.; Scopes, D.I.C.; Gallagher, T. Synlett 1993, 85.
381. Morita, N.; Krause, N. Org. Lett. 2004, 6, 4121.
382. Ackermann, L.; Bergman, R.G.; Loy, R.N. J. Am. Chem. Soc. 2003, 125, 11956.
383. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 443–504. See also, Barluenga, J.; Perez-Prieto, J.; Asensio, G. Tetrahedron 1990, 46, 2453.
384. Arisawa, M.; Yamaguchi, M. J. Am. Chem. Soc. 2006, 128, 50. See Kawaguchi, S.-i.; Nagata, S.; Nomoto, A.; Sonoda, M.; Ogawa, A. J. Org. Chem. 2008, 73, 7928.
385. Shulyupin, M.O.; Kazankova, M.A.; Beletskaya, I.P. Org. Lett.. 2002, 4, 761.
386. Hayashi, M.; Matsuura, Y.; Watanabe, Y. Tetrahedron Lett. 2004, 45, 9167.
387. Bunlaksananusorn, T.; Knochel, P. J. Org. Chem. 2004, 69, 4595.
388. Tayama, O.; Nakano, A.; Iwahama, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2004, 69, 5494.
389. Deprèle, S.; Montchamp, J.-L. J. Org. Chem. 2001, 66, 6745.
390. Deprèle, S.; Montchamp, J.-L. J. Am. Chem. Soc. 2002, 124, 9386.
391. Han, L.-B.; Zhang, C.; Yazawa, H.; Shimada, S. J. Am. Chem. Soc. 2004, 126, 5080.
392. Takaki, K.; Koshoji, G.; Komeyama, K.; Takeda, M.; Shishido, T.; Kitani, A.; Takehira, K. J. Org. Chem. 2003, 68, 6554.
393. Kazankova, M.A.; Efimova, I.V.; Kochetkov, A.N.; Atanas'ev, V.V.; Beletskaya, I.P.; Dixneuf, P.H. Synlett 2001, 497.
394. Ohmiya, H.; Yorimitsu, H.; Oshima, K. Angew. Chem. Int. Ed. 2005, 44, 2368.
395. Han, L.-B.; Zhao, C.-Q.; Tanaka, M. J. Org. Chem. 2001, 66, 5929.
396. Mirzaei, F.; Han, L.-B.; Tanaka, M. Tetrahedron Lett. 2001, 42, 297.
397. Kazankova, M.A.; Shulyupin, M.O.; Beletskaya, I.P. Synlett 2003, 2155.
398. Miura, K.; Hondo, T.; Nakagawa, T.; Takahashi, T.; Hosomi, A. Org. Lett. 2000, 2, 385.
399. Marson, C.M.; Fallah, A. Tetrahedron Lett. 1994, 35, 293.
400. Jacobi, P.A.; Brielmann, H.L.; Hauck, S.I. J. Org. Chem. 1996, 61, 5013.
401. Scartozzi, M.; Grondin, R.; Leblanc, Y. Tetrahedron Lett. 1992, 33, 5717.
402. Schlummer, B.; Hartwig, J.F. Org. Lett. 2002, 4, 1471; Haskins, C.M.; Knight, D.W. Chem. Commun. 2002, 2724.
403. Zalatan, D.N.; Du Bois, J. J. Am. Chem. Soc. 2008, 130, 9220.
404. Trost, B.M.; Maulide, N.; Livingston, R.C. J. Am. Chem. Soc. 2008, 130, 16502.
405. Bajracharya, G.B.; Huo, Z.; Yamamoto, Y. J. Org. Chem. 2005, 70, 4883; Patil, N.T.; Huo, Z.; Bajracharya, G.B.; Yamamoto, Y. J. Org. Chem. 2006, 71, 3612; Narsireddy, M.; Yamamoto, Y. J. Org. Chem. 2008, 73, 9698.
406. Qin, H.; Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. Chemistry: Asian J. 2007, 2, 150.
407. Bacci, J.P.; Greenman, K.L.; van Vranken, D.L. J. Org. Chem. 200368, 4955.
408. Yudha S., S.; Kuninobu, Y.; Takai, K. Org. Lett. 2007, 9, 5609.
409. Gooßen, L.J.; Salih, K.S.M.; Blanchot, M. Angew. Chem. Int. Ed. 2008, 47, 8492.
410. Ohno, H.; Toda, A.; Miwa, Y.; Taga, T.; Osawa, E.; Yamaoka, Y.; Fujii, N.; Ibuka, T. J. Org. Chem. 1999, 64, 2992.
411. Na, S.; Yu, F.; Gao, W. J. Org. Chem. 2003, 68, 5943; Ma, S.; Gao, W. Org. Lett. 2002, 4, 2989.
412. Sliwnska, A.; Zwierzak, A. Tetrahedron 2003, 59, 5927.
413. Callier, A.-C.; Quiclet-Sire, B.; Zard, S.Z. Tetrahedron Lett. 1994, 35, 6109.
414. Larock, R.C.; Hightower, T.R.; Hasvold, L.A.; Peterson, K.P. J. Org. Chem. 1996, 61, 3584. See also, Pinho, P.; Minnaard, A.J.; Feringa, B.L. Org. Lett. 2003, 5, 259. For an electrochemical variation, see Xu, H.-C; Moeller, K.D. J. Am. Chem. Soc. 2008, 130, 13542.
415. Alexanian, E.J.; Lee, C.; Sorensen, E.J. J. Am. Chem. Soc. 2005, 127, 7690; Michael, F.E.; Cochran, B.M. J. Am. Chem. Soc. 2006, 128, 4246. For a related reaction, see Zabawa, T.P.; Kasi, D.; Chemler, S.R. J. Am. Chem. Soc. 2005, 127, 11250.
416. Zhang, Z.; Liu, C.; Kinder, R.E.; Han, X.; Qian, H.; Widenhoefer, R.A. J. Am. Chem. Soc. 2006, 128, 9066; LaLonde, R.L.; Sherry, B.D.; Kang, E.J.; Toste, F.D. J. Am. Chem. Soc. 2007, 129, 2452; Han, X.; Widenhoefer, R.A. Angew. Chem. Int. Ed. 2006, 45, 1747.
417. Donohoe, T.J.; Chughtai, M.J.; Klauber, D.J.; Griffin, D.; Campbell, A.D. J. Am. Chem. Soc. 2006, 128, 2514.
418. Yang, L.; Xu, L.-W.; Xia, C.-G. Synthesis 2009, 1969.
419. Brice, J.L.; Harang, J.E.; Timokhin, V.I.; Anastasi, N.R.; Stahl, S.S. J. Am. Chem. Soc. 2005, 127, 2868; Liu, G.; Stahl, S.S. J. Am. Chem. Soc. 2006, 128, 7179; Qian, H.; Widenhoefer, R.A. Org. Lett. 2005, 7, 2635.
420. Trost, B.M.; Dake, G.R. J. Am. Chem. Soc. 1997, 119, 7595.
421. Ribière, P,; Bravo-Altamirano, K.; Antczak, M.I.; Hawkins, J.D.; Montchamp, J.-L. J. Org. Chem. 2005, 70, 4064.
422. Hirai, T.; Han, L.-B. Org. Lett. 2007, 9, 53.
423. Harvey, G.R.; Ratts, K.W. J. Org. Chem. 1966, 31, 3907. See Biffin, M.E.C.; Miller, J.; Paul, D.B. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 120–136.
424. Hassner, A.; Fibiger, R.; Andisik, D. J. Org. Chem. 1984, 49, 4237.
425. Heathcock, C.H. Angew. Chem. Int. Ed. 1969, 8, 134. See Larock, R.C. Solvation/Demercuration Reactions in Organic Synthesis, Springer, NY, 1986, pp. 522–527.
426. Waser, J.; Nambu, H.; Carreira, E.M. J. Am. Chem. Soc. 2005, 127, 8294.
427. See Mitsui, S.; Kasahara, A. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 175–214. Also see, Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 422–438.
428. See Rylander, P.N. Hydrogenation Methods Academic Press, NY, 1985; Catalytic Hydrogenation in Organic Synthesis Academic Press, NY, 1979; Freifelder, M. Catalytic Hydrogenation in Organic Synthesis Wiley, NY, 1978; Practical Catalytic Hydrogenation Wiley, NY, 1971; Augustine, R.L. Catalytic Hydrogenation Marcel Dekker, NY, 1965; Parker, D. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 979–1047.
429. Pizey, J.S. Synthetic Reagents, Vol. 2, Wiley, NY, 1974, pp. 175–311; Pojer, P.M. Chem. Ind. (London) 1986, 177.
430. See Chandrasekhar, S.; Narsihmulu, Ch.; Chandrashekar, G.; Shyamsunder, T. Tetrahedron Lett. 2004, 45, 2421.
431. See Ganem, B.; Osby, J.O. Chem. Rev. 1986, 86, 763.
432. See Minachev, Kh.M.; Khodakov, Yu.S.; Nakhshunov, V.S. Russ. Chem. Rev. 1976, 45, 142.
433. James, B.R. Homogeneous Hydrogenation Wiley, NY, 1973; Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry University Science Books, Mill Valley, CA 1987, pp. 523–564; Birch, A.J.; Williamson, D.H. Org. React. 1976, 24, 1; James, B.R. Adv. Organomet. Chem. 1979, 17, 319; Harmon, R.E.; Gupta, S.K.; Brown, D.J. Chem. Rev. 1973, 73, 21; Rylander, P.N. Organic Syntheses with Noble Metal Catalysts Academic Press, NY, 1973, pp. 60–76.
434. See van Bekkum, H.; van Rantwijk, F.; van de Putte, T. Tetrahedron Lett. 1969, 1.
435. See Jardine, F.H. Prog. Inorg. Chem. 1981, 28, 63–202.
436. Harmon, R.E.; Parsons, J.L.; Cooke, D.W.; Gupta, S.K.; Schoolenberg, J. J. Org. Chem. 1969, 34, 3684. See also, Mohrig, J.R.; Dabora, S.L.; Foster, T.F.; Schultz, S.C. J. Org. Chem. 1984, 49, 5179.
437. Jardine, F.H.; Wilkinson, G. J. Chem. Soc. C 1967, 270.
438. Hattori, K.; Sajiki, H.; Hirota, K. Tetrahedron 2000, 56, 8433.
439. Tang, W.; Liu, D.; Zhang, X Org. Lett. 2003, 5, 205.
440. See Rylander, P.N. Catalytic Hydrogenation over Platinum Metals, Academic Press, NY, 1967, pp. 59–120. Also see, Hudlicky, M. Reductions in Organic Chemistry Ellis Horwood Ltd., Chichester 1984.
441. Jourdant, A.; González-Zamora, E.; Zhu, J. J. Org. Chem. 2002, 67, 3163.
442. Taylor, R.A.; Santora, B.P.; Gagné, M.R. Org. Lett. 2000, 2, 1781.
443. Okamoto, K.; Akiyama, R.; Kobayashi, S. J. Org. Chem. 2004, 69, 2871. See also, Bremeyer, N.; Ley, S.V.; Ramarao, C.; Shirley, I.M.; Smith, S.C. Synlett 2002, 1843.
444. Huang, J.; Jiang, T.; Han, B.; Gao, H.; Chang, Y.; Zhao, G.; Wu, W. Chem. Commun. 2003, 1654.
445. Hallman, P.S.; McGarvey, B.R.; Wilkinson, G. J. Chem. Soc. A 1968, 3143; Jardine, F.H.; McQuillin, F.J. Tetrahedron Lett. 1968, 5189.
446. Birch, A.J.; Walker, K.A.M. Tetrahedron Lett. 1967, 1935.
447. Barbier, J.; Lamy-Pitara, E.; Marecot, P.; Boitiaux, J.P.; Cosyns, J.; Verna, F. Adv. Catal. 1990, 37, 279–318.
448. Cui, X.; Burgess, K. Chem. Rev. 2005, 105, 3272.
449. Roseblade, S.J.; Pfaltz, A. Acc. Chem. Res. 2007, 40, 1402; Källström, K.; Andersson, P.G. Tetrahedron Lett. 2006, 47, 7477; Li, X.; Kong, L.; Gao, Y.; Wang, X. Tetrahedron Lett. 2007, 48, 3915; Özkar, S.; Finke, R.G. J. Am. Chem. Soc. 2005, 127, 4800; Schrems, M.G.; Neumann, E.; Pfaltz, A. Angew. Chem. Int. Ed. 2007, 46, 8274; Källström, K.; Munslow, I.; Andersson, P.G. Chemistry: European J. 2006, 12, 3194.
450. Callis, N.M.; Thiery, E.; Le Bras, J.; Muzart, J. Tetrahedron Lett. 2007, 48, 8128. See Brunel, J.M. Tetrahedron 2007, 63, 3899.
451. Troutman, M.V.; Appella, D.H.; Buchwald, S.L. J. Am. Chem. Soc. 1999, 121, 4916.
452. Imamoto, T.; Sugita, K.; Yoshida, K. J. Am. Chem. Soc. 2005, 127, 11934; Hedberg, C.; Källström, K.; Brandt, P.; Hansen, L.K.; Andersson, P.G. J. Am. Chem. Soc. 2006, 128, 2995. See McIntosh, A.I.; Watson, D.J.; Burton, J.W.; Lambert, R.M. J. Am. Chem. Soc. 2006, 128, 7329; Diguez, M.; Mazuela, J.; Pmies, O.; Verendel, J.J.; Andersson, P.G. J. Am. Chem. Soc. 2008, 130, 7208; Chen, W.; Roberts, S.M.; Whittall, J. Tetrahedron Lett. 2006, 47, 4263; Tang, W.-J.; Huang, Y.-Y.; He, Y.-M.; Fan, Q.-H. Tetrahedron Asymmetry 2006, 17, 536; Tang, W.; Qu, B.; Capacci, A.G.; Rodriguez, S.; Wei, X.; Haddad, N.; Narayanan, B.; Ma, S.; Grinberg, N.; Yee, N.K.; Krishnamurthy, D.; Chris H.; Senanayake, C.H. Org. Lett. 2010, 12, 176; Zhang, X.; Huang, K.; Hou, G.; Cao, B.; Zhang, X. Angew. Chem. Int. Ed. 2010, 49, 6421.
453. Huang, H.; Zheng, Z.; Luo, H.; Bai, C.; Hu, X.; Chen, H. J. Org. Chem. 2004, 69, 2355; Hua, Z.; Vassar, V.C.; Ojima, I. Org. Lett. 2003, 5, 3831. For a review, see Jerphagnon, T.; Renaud, J.-L.; Bruneau, C. Tetrahedron Asymmetry 2004, 15, 2101.
454. Knowles, W.S.; Sabacky, M.J.; Vineyard, B.D. Adv. Chem. Ser 1974, 132, 274.
455. Brown, J.M.; Chaloner, P.A. J. Chem. Soc., Chem. Commun. 1980, 344; 1978, 321; J. Am. Chem. Soc. 1980, 102, 3040.
456. See Chan. A.S.C.; Halpern, J. J. Am. Chem. Soc. 1980, 102, 838; Chua, P.S.; Roberts, N.K.; Bosnich, B.; Okrasinski, S.J.; Halpern, J. J. Chem. Soc. Chem. Commun. 1981, 1278.
457. See Tang, W.; Zhang, X. Chem. Rev. 2003, 103, 3029.
458. Faller, J.W.; Parr, J. J. Am. Chem. Soc. 1993, 115, 804.
459. Kowalak, S.; Weiss, R.C.; Balkus Jr., K.J. J. Chem. Soc., Chem. Commun. 1991, 57.
460. Sun, Y.; Landau, R.N.; Wang, J.; LeBlond, C.; Blackmond, D.G. J. Am. Chem. Soc. 1996, 118, 1348.
461. See Hutchins, R.O.; Hutchins, M.G.K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 1, Wley, NY, 1983, pp. 571–601; Marvell, E.N.; Li, T. Synthesis 1973, 457; Gutmann, H.; Lindlar, H. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 355–363.
462. Lindlar, H.; Dubuis, R. Org. Synth. V, 880. See also, Rajaram, J.; Narula, A.P.S.; Chawla, H.P.S.; Dev, S. Tetrahedron 1983, 39, 2315; McEwen, A.B.; Guttieri, M.J.; Maier, W.F.; Laine, R.M.; Shvo, Y. J. Org. Chem. 1983, 48, 4436.
463. Cram, D.J.; Allinger, N.L. J. Am. Chem. Soc. 1956, 78, 2518; Rosenmund, K.W. Ber. 1918, 51, 585; Mosettig, E.; Mozingo, R. Org. React. 1948, 4, 362.
464. Chandrasekhar, S.; Narsihmulu, Ch.; Chandrashekar, G.; Shyamsunder, T. Tetrahedron Lett. 2004, 45, 2421.
465. Li, J.; Hua, R.; Liu, T. J. Org. Chem. 2010, 75, 2966.
466. Campos, K.R.; Cai, D.; Journet, M.; Kowal, J.J.; Larsen, R.D.; Reider, P.J. J. Org. Chem. 2001, 66, 3634.
467. Zhong, P.; Huang, X.; Ping-Guo, M. Tetrahedron 2000, 56, 8921.
468. Miyake, A.; Kondo, H. Angew. Chem. Int. Ed. 1968, 7, 631. For other methods, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 403–404.
469. See Schuster, H.F.; Coppola, G.M. Allenes in Organoic Synthesis Wiley, NY, 1984, pp. 57–61.
470. Hou, G.-H.; Xie, J.-H.; Wang, L.-X.; Zhou, Q.-L. J. Am. Chem. Soc. 2006, 128, 11774.
471. Engman, M.; Diesen, J.S.; Paptchikhine, A.; Andersson, P.G. J. Am. Chem. Soc. 2007, 129, 4536.
472. See Brown, J.M. Angew. Chem. Int. Ed. 1987, 26, 190.
473. Holme, D.; Jones, E.R.H.; Whiting, M.C. Chem. Ind. (London) 1956, 928.
474. See Burch, R.R.; Muetterties, E.L.; Teller, R.G.; Williams, J.M. J. Am. Chem. Soc. 1982, 104, 4257.
475. See Gudkov, B.S. Russ. Chem. Rev. 1986, 55, 259.
476. Turkevich, J.; Schissler, D.O.; Irsa, P. J. Phys. Chem. 1951, 55, 1078.
477. Wilson, J.N.; Otvos, J.W.; Stevenson, D.P.; Wagner, C.D. Ind. Eng. Chem. 1953, 45, 1480.
478. See Gudkov, B.S.; Balandin, A.A. Russ. Chem. Rev. 1966, 35, 756. For an intramolecular exchange, see Lebrilla, C.B.; Maier, W.F. Tetrahedron Lett. 1983, 24, 1119. See also, Poretti, M.; Gäumann, T. Helv. Chim. Acta 1985, 68, 1160.
479. See Webb, G. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 20; Elsevier, NY, 1978, pp. 1–121; Clarke, J.K.A.; Rooney, J.J. Adv. Catal. 1976, 25, 125–183.
480. Horiuti, I.; Polanyi, M. Trans. Faraday Soc. 1934, 30, 1164.
481. See Burwell, Jr., R.L.; Schrage, K. J. Am. Chem. Soc. 1965, 87, 5234.
482. See Bautista, F.M.; Campelo, J.M.; Garcia, A.; Guardeño, R.; Luna, D.; Marinas, J.M. J. Chem. Soc. Perkin Trans. 2,1989, 493.
483. Krasna, A.I. J. Am. Chem. Soc. 1961, 83, 289.
484. Smith, G.V.; Burwell, Jr., R.L. J. Am. Chem. Soc. 1962, 84, 925.
485. A different mechanism has been proposed by Zaera, F.; Somorjai, G.A. J. Am. Chem. Soc. 1984, 106, 2288, but there is evidence against it: Beebe, Jr., T.P.; Yates, Jr., J.T. J. Am. Chem. Soc. 1986, 108, 663. See also, Thomson, S.J.; Webb, G. J. Chem. Soc., Chem. Commun. 1976, 526.
486. See Augustine, R.L.; Yaghmaie, F.; Van Peppen, J.F. J. Org. Chem. 1984, 49, 1865; Maier, W.F. Angew. Chem. Int. Ed. 1989, 28, 135.
487. See Schlögl, R.; Noack, K.; Zbinden, H.; Reller, A. Helv. Chim. Acta 1987, 70, 627.
488. Maier, W.F. Angew. Chem. Int. Ed. 1989, 28, 135.
489. Maier, W.F. in Rylander, P.N.; Greenfield, H.; Augustine, R.L. Catalysis of Organic Reactions, Marcel Dekker, NY, 1988, pp 211–231, Cf. p. 220.
490. See Crabtree, R.H. Organometallic Chemistry of the Transition Metals, Wiley, NY, 1988, pp. 190–200; Jardine, F.H. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 1049–1071.
491. Koga, N.; Daniel, C.; Han, J.; Fu, X.Y.; Morokuma, K. J. Am. Chem. Soc. 1987, 109, 3455.
492. Tolman, C.A.; Meakin, P.Z.; Lindner, D.L.; Jesson, J.P. J. Am. Chem. Soc. 1976, 96, 2762.
493. López-Serrano, J.; Duckett, S.B.; Lledós, A. J. Am. Chem. Soc. 2006, 128, 9596.
494. Smith, G.V.; Shuford, R.J. Tetrahedron Lett. 1970, 525; Atkinson, J.G.; Luke, M.O. Can. J. Chem. 1970, 48, 3580.
495. Morandi, J.R.; Jensen, H.B. J. Org. Chem. 1969, 34, 1889. See, however, Atkinson, J.G.; Luke, M.O. Can. J. Chem. 1970, 48, 3580.
496. Whitesides, G.M.; Ehmann, W.J. J. Org. Chem. 1970, 35, 3565.
497. Benkeser, R.A.; Schroll, G.; Sauve, D.M. J. Am. Chem. Soc. 1955, 77, 3378.
498. Tour, J.M.; Pendalwar, S.L. Tetrahedron Lett. 1990, 31, 4719.
499. Masuno, M.N.; Molinski, T.F. Tetrahedron Lett. 2001, 42, 8263; Kursanov, D.N.; Parnes, Z.N.; Kalinkin, M.I.; Loim, N.M. Ionic Hydrogenation and Related Reactions Harwood Academic Publishers, Chur, Switzerland, 1985.
500. See Toromanoff, E. Bull. Soc. Chim. Fr. 1987, 893–901; Russell, G.A. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 2, Wiley, NY, 1989, pp. 471–512.
501. Parnes, Z.N.; Bolestova, G.I.; Kursanov, D.N. Bull. Acad. Sci. USSR Div. Chem. Sci. 1972, 21, 1927.
502. Sweany, R.L.; Halpern, J. J. Am. Chem. Soc. 1977, 99, 8335. See also, Bullock, R.M.; Samsel, E.G. J. Am. Chem. Soc. 1987, 109, 6542.
503. Luo, F.; Pan, C.; Wang, W.; Ye, Z.; Cheng, J. Tetrahedron 2010, 66, 1399. See Han, J.W.; Hayashi, T. Tetrahedron Asymm. 2010, 21, 2193.
504. Dahlén, A.; Hilmersson, G. Tetrahedron Lett. 2003, 44, 2661.
505. Lee, H.-Y.; An, M. Tetrahedron Lett. 2003, 44, 2775.
506. Johnstone, R.A.W.; Wilby, A.H.; Entwistle, I.D. Chem. Rev. 1985, 85, 129; Brieger, G.; Nestrick, T.J. Chem. Rev. 1974, 74, 567.
507. Alonso, F.; Riente, P.; Yus, M. Tetrahedron 2009, 65, 10637.
508. See Pasto, D.J.; Taylor, R.T. Org. React. 1991, 40, 91; Hünig, S.; Müller, H.R.; Thier, W. Angew. Chem. Int. Ed. 1965, 4, 271.
509. Nelson, D.J.; Henley, R.L.; Yao, Z.; Smith, T.D. Tetrahedron Lett. 1993, 34, 5835.
510. Imada, Y.; Iida, H.; Naota, T. J. Am. Chem. Soc. 2005, 127, 14544.
511. Aylward, F.; Sawistowska, M.H. J. Chem. Soc. 1964, 1435.
512. Willis, C.; Back, R.A.; Parsons, J.A.; Purdon, J.G. J. Am. Chem. Soc. 1977, 99, 4451.
513. Corey, E.J.; Pasto, D.J.; Mock, W.L. J. Am. Chem. Soc. 1961, 83, 2957.
514. van Tamelen, E.E.; Timmons, R.J. J. Am. Chem. Soc. 1962, 84, 1067.
515. Wiberg, N.; Fischer, G.; Bachhuber, H. Angew. Chem. Int. Ed. 1977, 16, 780. See also, Craig, N.C.; Kliewer, M.A.; Shih, N.C. J. Am. Chem. Soc. 1979, 101, 2480.
516. See Zweifel, G. Intra-Sci. Chem. Rep. 1973, 7(2), 181–189.
517. Brown, H.C.; Murray, K.J. Tetrahedron 1986, 42, 5497; Kabalka, G.W.; Newton Jr., R.J.; Jacobus, J. J. Org. Chem. 1979, 44, 4185.
518. Weinheimer, A.J.; Marisco, W.E. J. Org. Chem. 1962, 27, 1926.
519. Pozzi, D.; Scanlan, E.M.; Renaud, P. J. Am. Chem. Soc. 2005, 127, 14204.
520. Brown, H.C.; Zweifel, G. J. Am. Chem. Soc. 1959, 81, 1512.
521. Lee, S.H.; Park, Y.J.; Yoon, C.M. Tetrahedron Lett. 2000, 41, 887.
522. Couturier, M.; Andresen, B.M.; Tucker, J.L.; Dubé, P.; Brenek, S.J.; Negri, J.J. Tetrahedron Lett. 2001, 42, 2763.
523. Yakabe, S.; Hirano, M.; Morimoto, T. Tetrahedron Lett. 2000, 41, 6795.
524. Choi, J.; Yoon, N.M. Synthesis 1996, 597.
525. See Granoth, I.; Segall, Y.; Leader, H.; Alkabets, R. J. Org. Chem. 1976, 41, 3682.
526. Gribble, G.W.; Nutaitis, C.F. Org. Prep. Proced. Int. 1985, 17, 317; Nilsson, A.; Carlson, R. Acta Chem. Scand. Sect. B 1985, 39, 187.
527. See Ashby, E.C.; Lin, J.J. J. Org. Chem. 1978, 43, 2567; Chung, S. J. Org. Chem. 1979, 44, 1014. See also, Osby, J.O.; Heinzman, S.W.; Ganem, B. J. Am. Chem. Soc. 1986, 108, 67.
528. Zohar, E.; Marek, I. Org. Lett. 2004, 6, 341.
529. Tran, A.T.; Huynh, V.A.; Friz, E.M.; Whitney, S.K.; Cordes, D.B. Tetrahedron Lett. 2009, 50, 1817.
530. Adair, G.R.A.; Kapoor, K.K.; Scolan, A.L.B.; Williams, J.M.J. Tetrahedron Lett. 2006, 47, 8943.
531. Ren, P.-D.; Pan, S.-F.; Dong, T.-W.; Wu, S.-H. Synth. Commun. 1996, 26, 763.
532. Sato, T.; Watanabe, S.; Kiuchi, H.; Oi, S.; Inoue, Y. Tetrahedron Lett. 2006, 47, 7703.
533. There are some exceptions. Butler, D.N. Synth. Commun. 1977, 7, 441, and references cited therein.
534. See Caine, D. Org. React. 1976, 23, 1–258.
535. See Ferraboschi, P.; Reza-Elahi, S.; Verza, E.; Santaniello, E. Tetrahedron Asymmetry 1999, 10, 2639. For reviews of baker's yeast, see Csuk, R.; Glänzer, B.I. Chem. Rev. 1991, 91, 49; Servi, S. Synthesis 1990, 1.
536. Ulan, J.G.; Maier, W.F.; Smith, D.A. J. Org. Chem. 1987, 52, 3132.
537. Chou, W.; Clark, D.L.; White, J.B. Tetrahedron Lett. 1991, 32, 299. See Kaufman, D.; Johnson, E.; Mosher, M.D. Tetrahedron Lett. 2005, 46, 5613.
538. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 405–410.
539. Henne, A.L.; Greenlee, K.W. J. Am. Chem. Soc. 1943, 65, 2020.
540. Alonso, F.; Yus, M. Tetrahedron Lett. 1997, 38, 149.
541. Alonso, F.; Yus, M. Tetrahedron Lett. 1996, 37, 6925.
542. Fürstner, A.; Radkowski, K. Chem. Commun. 2002, 2182.
543. Trost, B.M.; Ball, Z.T.; Jöge, T. J. Am. Chem. Soc. 2002, 124, 7922.
544. Ranu, B.C.; Dutta, J.; Guchhait, S.K. J. Org. Chem. 2001, 66, 5624.
545. Wei, L.-L.; Wei, L.-M.; Pan, W.-B.; Leou, S.-P.; Wu, M.-J. Tetrahedron Lett. 2003, 44, 1979.
546. Vaidyanathaswamy, R.; Joshi, G.C.; Devaprabhakara, D. Tetrahedron Lett. 1971, 2075.
547. Montury, M.; Goré, J. Tetrahedron Lett. 1980, 21, 51.
548. Bhagwat, M.M.; Devaprabhakara, D. Tetrahedron Lett. 1972, 1391.
549. See Karakhanov, E.A.; Dedov, A.G.; Loktev, A.S. Russ. Chem. Rev. 1985, 54, 171.
550. See Timmer, K.; Thewissen, D.H.M.W.; Meinema, H.A.; Bulten, E.J. Recl. Trav. Chim. Pays-Bas 1990, 109, 87.
551. Muetterties, E.L.; Bleeke, J.R. Acc. Chem. Res. 1979, 12, 324. See also, Tsukinoki, T.; Kanda, T.; Liu, G.-B.; Tsuzuki, H.; Tashiro, M. Tetrahedron Lett. 2000, 41, 5865.
552. Januszkiewicz, K.R.; Alper, H. Organometallics 1983, 2, 1055.
553. Dyson, P.J.; Ellis, D.J.; Parker, D.G.; Welton, T. Chem. Commun. 1999, 25. Rhodium nanoparticles are used as a catalyst: Mu, X.-d.; Meng, J.-q.; Li, Z.-C.; Kou, Y. J. Am. Chem. Soc. 2005, 127, 9694.
554. Bonilla, R.J.; James, B.R.; Jessop, P.G. Chem. Commun. 2000, 941.
555. Zhong, G.; Chan, B.; Radom, L. J. Am. Chem. Soc. 2007, 129, 924.
556. For an indirect method of hydrogenating benzene to cyclohexene, see Harman, W.D.; Taube, H. J. Am. Chem. Soc. 1988, 110, 7906.
557. Higashijima, M.; Nishimura, S. Bull. Chem. Soc. Jpn. 1992, 65, 824.
558. Boxwell, C.J.; Dyson, P.J.; Ellis, D.J.; Welton, T. J. Am. Chem. Soc. 2002, 124, 9334.
559. Zhou, Y.-G. Acc. Chem. Res. 2007, 40, 1357.
560. Kuwano, R.; Kashiwabara, M.; Ohsumi, M.; Kusano, H. J. Am. Chem. Soc. 2008, 130, 808.
561. Piras, L.; Genesio, E.; Ghiron, C.; Taddei, M. Synlett 2008, 1125.
562. Wang, W.-B.; Lu, S.-M.; Yang, P.-Y.; Han, X.-W.; Zhou, Y.-G. J. Am. Chem. Soc. 2003, 125, 10536.
563. Kuwano, R.; Kaneda, K.; Ito, T.; Sato, K.; Kurokawa, T.; Ito, Y. Org. Lett. 2004, 6, 2213. See Kim, J.T.; Gevorgyan, V. J. Org. Chem. 2005, 70, 2054.
564. See Brandsma, L.; van Soolingen, J.; Andringa, H. Synth. Commun. 1990, 20, 2165. Also see, Weitz, I.S.; Rabinovitz, M. J. Chem. Soc. Perkin Trans. 1,1993, 117.
565. Akhrem, A.A.; Reshotova, I.G.; Titov, Yu.A. Birch Reduction of Aromatic Compounds Plenum, NY, 1972; Birch, A.J. Pure Appl. Chem. 1996, 68, 553; Rabideau, P.W. Tetrahedron 1989, 45, 1579; Birch, A.J.; Subba Rao, G. Adv. Org. Chem. 1972, 8, 1; Kaiser, E.M. Synthesis 1972, 391.
566. Donohoe, T.J.; House, D. J. Org. Chem. 2002, 67, 5015.
567. Kinoshita, T.; Ichinari, D.; Sinya, J. J. Heterocyclic Chem. 1996, 33, 1313.
568. Donohoe, T.J.; McRiner, A.J.; Helliwell, M.; Sheldrake, P. J. Chem. Soc., Perkin Trans. 1 2001, 1435.
569. Guo, Z.; Schultz, A.G. J. Org. Chem. 2001, 66, 2154.
570. See Zimmerman, H.E.; Wang, P.A. J. Am. Chem. Soc. 1990, 112, 1280; Rabideau, P.W.; Karrick, G.L. Tetrahedron Lett. 1987, 28, 2481.
571. Zimmerman, H.E.; Wang, P.A. J. Am. Chem. Soc. 1993, 115, 2205.
572. For reviews of solvated electrons and related topics, see Dye, J.L. Prog. Inorg. Chem. 1984, 32, 327–441; Alpatova, N.M.; Krishtalik, L.I.; Pleskov, Y.V. Top. Curr. Chem. 1987, 138, 149–219.
573. Birch, A.J.; Nasipuri, D. Tetrahedron 1959, 6, 148.
574. See Holy, N.L. Chem. Rev. 1974, 74, 243.
575. See Jones, M.T. in Kaiser, E.T.; Kevan, L. Radical Ions, Wiley, NY, 1968, pp. 245–274; Bowers, K.W. Adv. Magn. Reson.,1965, 1, 317; Carrington, A. Q. Rev. Chem. Soc. 1963, 17, 67.
576. Rabideau, P.W.; Peters, N.K.; Huser, D.L. J. Org. Chem. 1981, 46, 1593.
577. See Brandsma, L.; Nieuwenhuizen, W.F.; Zwikker, J.W.; Mäeorg, U. Eur. J. Org. Chem. 1999, 775.
578. See Rabideau, P.W.; Huser, D.L. J. Org. Chem. 1983, 48, 4266.
579. Hine, J. J. Org. Chem. 1966, 31, 1236; Hine, J. Adv. Phys. Org. Chem. 1977, 15, 1. See also, Jochum, C.; Gasteiger, J.; Ugi, I. Angew. Chem. Int. Ed. 1980, 19, 495.
580. Bates, R.B.; Brenner, S.; Cole, C.M.; Davidson, E.W.; Forsythe, G.D.; McCombs, D.A.; Roth, A.S. J. Am. Chem. Soc. 1973, 95, 926.
581. Benkeser, R.A.; Agnihotri, R.K.; Burrous, M.L.; Kaiser, E.M.; Mallan, J.M.; Ryan, P.W. J. Org. Chem. 1964, 29, 1313; Kwart, H.; Conley, R.A. J. Org. Chem. 1973, 38, 2011.
582. Benkeser, R.A.; Belmonte, F.G.; Kang, J. J. Org. Chem. 1983, 48, 2796. See also, Benkeser, R.A.; Laugal, J.A.; Rappa, A. Tetrahedron Lett. 1984, 25, 2089.
583. See Keay, J.G. Adv. Heterocycl. Chem. 1986, 39, 1.
584. Blough, B.E.; Carroll, F.I. Tetrahedron Lett. 1993, 34, 7239.
585. Moody, C.J.; Pitts, M.R. Synlett 1998, 1029.
586. Pitts, M.R.; Harrison, J.R.; Moody, C.J. J. Chem. Soc., Perkin Trans. 1, 2001, 955.
587. Kamochi, Y.; Kudo, T. Heterocycles 1993, 36, 2383.
588. Kamochi, Y.; Kudo, T. Tetrahedron Lett. 1994, 35, 4169.
589. Zacharie, B.; Moreau, N.; Dockendorff, C. J. Org. Chem. 2001, 66, 5264.
590. Fujita, K.; Kitatsuji, C.; Furukawa, S.; Yamaguchi, R. Tetrahedron Lett. 2004, 45, 3215.
591. See Keinan, E.; Greenspoon, N. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 2, Wiley, NY, 1989, pp. 923–1022.; Augustine, R.L. Adv. Catal. 1976, 25, 56.
592. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 13–27.
593. Cabello, J.A.; Campelo, J.M.; Garcia, A.; Luna, D.; Marinas, J.M. J. Org. Chem. 1986, 51, 1786.
594. Wang, C.-J.; Tao, H.; Zhang, X. Tetrahedron Lett. 2006, 47, 1901.
595. Nagano, H.; Yokota, M.; Iwazaki, Y. Tetrahedron Lett. 2004, 45, 3035.
596. Yue, T.-Y.; Nugent, W.A. J. Am. Chem. Soc. 2002, 124, 13692;
597. Barrero, A.F.; Alvarez-Manzaneda, E.J.; Chahboun, R.; Meneses, R. Synlett 1999, 1663; Wang, H.; Lian, H.; Chen, J.; Pan, Y.; Shi, Y. Synth. Commun. 1999, 29, 129.
598. Cabrera, A.; Alper, H. Tetrahedron Lett. 1992, 33, 5007. See also, Tarnopolsky, A.; Hoz, S. J. Am. Chem. Soc. 2007, 129, 3402.
599. Evans, D.A.; Fu, G.C. J. Org. Chem. 1990, 55, 5678.
600. Moisan, L.; Hardouin, C.; Rousseau, B.; Doris, E. Tetrahedron Lett. 2002, 43, 2013.
601. Comins, D.L.; Brooks, C.A.; Ingalls, C.L. J. Org. Chem. 2001, 66, 2181.
602. Arterburn, J.B.; Pannala, M.; Gonzlez, A.M.; Chamberlin, R.M. Tetrahedron Lett. 2000, 41, 7847.
603. Kosal, A.D.; Ashfeld, B.L. Org. Lett. 2010, 12, 44.
604. Varma, R.S.; Kabalka, G.W. Synth. Commun. 1985, 15, 151.
605. Gupta, A.; Haque, A.; Vankar, Y.D. Chem. Commun. 1996, 1653.
606. See Meyer, G.R. J. Chem. Educ. 1981, 58, 628.
607. For a discussion of hydride affinity with respect to hydride reducing agents, see Zhu, X.-Q.; Zhang, M.; Liu, Q.-Y.; Wang, X.-X.; Zhang, J.-Y.; Cheng, J.-P. Angew. Chem. Int. Ed. 2006, 45, 3954; Vianello, R.; Peran, N.; Maksi![]() , Z.B. Eur. J. Org. Chem. 2007, 5296.
, Z.B. Eur. J. Org. Chem. 2007, 5296.
608. Brown, H.C.; Hess, H.M. J. Org. Chem. 1969, 34, 2206. For other methods of reducing both double bonds, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 1096.
609. Das, B.; Kashinatham, A.; Madhusudhan, P. Tetrahedron Lett. 1998, 39, 677.
610. Ren, P.-D.; Pan, S.-F.; Dong, T.-W.; Wu, S.-H. Synth. Commun. 1995, 25, 3395.
611. Ranu, B.C.; Samanta, S. Tetrahedron Lett. 2002, 43, 7405.
612. Ikeno, T.; Kimura, T.; Ohtsuka, Y.; Yamada, T. Synlett 1999, 96.
613. Ranu, B.C.; Samanta, S. J. Org. Chem. 2003, 68, 7130.
614. See Blunt, J.W.; Hartshorn, M.P.; Soong, L.T.; Munro, M.H.G. Aust. J. Chem. 1982, 35, 2519; Vincens, M.; Fadel, R.; Vidal, M. Bull. Soc. Chim. Fr. 1987, 462.
615. Ouellet, S.G.; Tuttle, J.B.; MacMillan, D.W.C. J. Am. Chem. Soc. 2005, 127, 32; Tuttle, J.B.; Ouellet, S.G.; MacMillan, D.W.C. J. Am. Chem. Soc. 2006, 128, 12662; Adolfsson, H. Angew. Chem. Int. Ed. 2005, 44, 3340.
616. Martin, N.J.A.; List, B. J. Am. Chem. Soc. 2006, 128, 13368.
617. Martin, N.J.A.; Ozores, L.; List, B. J. Am. Chem. Soc. 2007, 129, 8976.
618. Quinn, J.F.; Razzano, D.A.; Golden, K.C.; Gregg, B.T. Tetrahedron Lett. 2008, 49, 6137.
619. Li, X.; Li, L.; Tang, Y.; Zhong, L.; Cun, L.; Zhu, J.; Liao, J.; Deng, J. J. Org. Chem. 2010, 75, 2981.
620. Naskar, S.; Bhattacharjee, M. Tetrahedron Lett. 2007, 48, 465.
621. Brunel, J.M. Synlett 2007, 330.
622. Che, J.; Lam, Y. Synlett 2010, 2415.
623. Mori, A.; Fujita, A.; Nishihara, Y.; Hiyama, R. Chem. Commun. 1997, 2159; Mori, A.; Fujita, A.; Kajiro, H.; Nishihara, Y.; Hiyama, T. Tetrahedron 1999, 55, 4573.
624. Huang, S.; Voigtritter, K.R.; Unger, J.B.; Lipshutz, B.H. Synlett 2010, 2041.
625. Boudjouk, P.; Choi, S.-B.; Hauck, B.J.; Rajkumar, A.B. Tetrahedron Lett. 1998, 39, 3951.
626. Ito, H.; Ishizuka, T.; Arimoto, K.; Miura, K.; Hosomi, A. Tetrahedron Lett. 1997, 38, 8887.
627. Magnus, P.; Waring, M.J.; Scott, D.A. Tetrahedron Lett. 2000, 41, 9731.
628. Keinan, E.; Perez, D. J. Org. Chem. 1987, 52, 2576.
629. Czekelius, C.; Carreira, E.M. Org. Lett. 2004, 6, 4575.
630. See Jurkauskas, V.; Buchwald, S.L. J. Am. Chem. Soc. 2002, 124, 2892; Lipshutz, B.H.; Servesko, J.M.; Taft, B.R. J. Am. Chem. Soc. 2004, 126, 8352.
631. Kim, D.; Park, B.-M.; Yun, J. Chem. Commun. 2005, 1755.
632. Hughes, G.; Kimura, M.; Buchwald, S.L. J. Am. Chem. Soc. 2003, 125, 11253.
633. Hirasawa, S.; Nagano, H.; Kameda, Y. Tetrahedron Lett. 2004, 45, 2207.
634. See Gridnev, I.D.; Imamoto, T. Acc. Chem. Res. 2004, 37, 633.
635. Ojima, I.; Clos, N.; Bastos, C. Tetrahedron 1989, 45, 6901, pp. 6902–6916; Jardine, F.H. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 751–775; Nógrádi, M. Stereoselective SynthesisVCH, NY, 1986, pp. 53–87; Knowles, W.S. Acc. Chem. Res. 1983, 16, 106; Brunner, H. Angew. Chem. Int. Ed. 1983, 22, 897; Caplar, V.; Comisso, G.; Sunjic, V. Synthesis 1981, 85. See also, Wroblewski, A.E.; Applequist, J.; Takaya, A.; Honzatko, R.; Kim, S.; Jacobson, R.A.; Reitsma, B.H.; Yeung, E.S.; Verkade, J.G. J. Am. Chem. Soc. 1988, 110, 4144; Knowles, W.S. Angew. Chem. Int. Ed. 2002, 41, 1999.
636. See Ashby, M.T.; Halpern, J. J. Am. Chem. Soc. 1991, 113, 589; Heiser, B.; Broger, E.A.; Crameri, Y. Tetrahedron: Asymmetry 1991, 2, 51; Burk, M.J. J. Am. Chem. Soc. 1991, 113, 8518.
637. Koenig, K.E. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, p. 74.
638. Reetz, M.T.; Mehler, G. Angew. Chem. Int. Ed. 2000, 39, 3889.
639. Koenig, K.E. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, pp. 83–101.
640. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 8–12. See Izumi, Y. Adv. Catal. 1983, 32, 215; Mortreux, A.; Petit, F.; Buono, G.; Peiffer, G. Bull. Soc. Chim. Fr. 1987, 631.
641. Wu, H.-P.; Hoge, G. Org. Lett. 2004, 6, 3645; Tang, W.; Wu, S.; Zhang, X. J. Am. Chem. Soc. 2003, 125, 9570.
642. Kanazawa, Y.; Tsuchiya, Y.; Kobayashi, K.; Shiomi, T.; Itoh, J.-i.; Kikuchi, M.; Yamamoto, Y.; Nishiyama, H. Chemistry: European J. 2006, 12, 63.
643. Pagenkopf, B.L. J. Org. Chem. 2004, 69, 4177; Fu, Y.; Guo, X.-X.; Zhu, S.-F.; Hu, A.-G.; Xie, J.-H.; Zhou, Q.-L. J. Org. Chem. 2004, 69, 4648; Yi, B.; Fan, Q.-H.; Deng, G.-J.; Li, Y.-M.; Qiu, L.-Q.; Chan, A.S.C. Org. Lett.2004, 6, 1361.; Hoen, R.; van den Berg, M.; Bernsmann, H.; Minnaard, A.J.; de Vries, J.G.; Feringa, B.L. Org. Lett. 2004, 6, 1433; Fu, Y.; Hou, G.-H.; Xie, J.-H.; Xing, L.; Wang, L.-X.; Zhou, Q.-L. J. Org. Chem. 2004, 69, 8157; Hoge, G.; Wu, H.-P.; Kissel, W.S.; Pflum, D.A.; Greene, D.J.; Bao, J. J. Am. Chem. Soc. 2004, 126, 5966; Ikeda, S.-i.; Sanuki, R.; Miyachi, H.; Miyashita, H.; Taniguchi, M.; Odashima, K. J. Am. Chem. Soc. 2004, 126, 10331; Huang, H.; Liu, X.; Chen, S.; Chen, H.; Zheng, Z. Tetrahedron Asymmetry 2004, 15, 2011; Hattori, G.; Hori, T.; Miyake, Y.; Nishibayashi, Y. J. Am. Chem. Soc. 2007, 129, 12930.
644. Zhu, G.; Zhang, X. J. Org. Chem. 1998, 63, 9590; Burk, M.J.; Casy, G.; Johnson, N.B. J. Org. Chem. 1998, 63, 6084; Burk, M.J.; Allen, J.G.; Kiesman, W.F. J. Am. Chem. Soc. 1998, 120, 657.
645. Noyori, R.; Hashiguchi, S. Accts. Chem. Res. 1997, 30, 97.
646. Li, S.; Zhu, S.-F.; Zhang, C.-M.; Song, S.; Zhou, Q.-L. J. Am. Chem. Soc. 2008, 130, 8584; Lu, W.-J.; Chen, Y.-W.; Hou, X.-L. Angew. Chem. Int. Ed. 2008, 47, 10133.
647. Heller, D.; Drexler, H.-J.; Spannenberg, A.; Heller, B.; You, J.; Baumann, W. Angew. Chem. Int. Ed. 2002, 41, 777.
648. Heller, D.; Holz, J.; Drexler, H.-J.; Lang, J.; Drauz, K.; Krimmer, H.-P.; Börner, A. J. Org. Chem. 2001, 66, 6816.
649. Suárez, A.; Pizzano, A. Tetrahedron Asymmetry 2001, 12, 2501. See Okano, T.; Kaji, M.; Isotani, S.; Kiji, J. Tetrahedron Lett. 1992, 33, 5547 for the influence of water on the regioselectivity of this reduction.
650. Yamaguchi, M.; Nitta, A.; Reddy, R.S.; Hirama, M. Synlett 1997, 117.
651. Brown, R.A.; Pollet, P.; McKoon, E.; Eckert, C.A.; Liotta, C.L.; Jessop, P.G. J. Am. Chem. Soc. 2001, 123, 1254.
652. Kabbara, J.; Flemming, S.; Nickisch, K.; Neh, H.; Westermann, J. Synlett 1994, 679.
653. Ranu, B.C.; Sarkar, A. Tetrahedron Lett. 1994, 35, 8649.
654. Basu, B.; Bhuiyan, Md.M.H.; Das, P.; Hossain, I. Tetrahedron Lett. 2003, 44, 8931.
655. Desai, B.; Danks, T.N. Tetrahedron Lett. 2001, 42, 5963.
656. Sakaguchi, S.; Yamaga, T.; Ishii, Y. J. Org. Chem. 2001, 66, 4710.
657. Koltunov, K.Yu.; Repinskaya, I.B.; Borodkin, G.I. Russ. J. Org. Chem. 2001, 37, 1534.
658. Kawai, Y.; Inaba, Y.; Tokitoh, N. Tetrahedron Asymmetry 2001, 12, 309.
659. Filho, E.P.S.; Rodrigues, J.A.R.; Moran, P.J.S. Tetrahedron Asymmetry 2001, 12, 847; Kawai, Y.; Hayashi, M.; Tokitoh, N. Tetrahedron Asymmetry 2001, 12, 3007.
660. Shimoda, K.; Kubota, N.; Hamada, H. Tetrahedron Asymmetry 2004, 15, 2443.
661. Kawai, Y.; Inaba, Y.; Hayashi, M.; Tokitoh, N. Tetrahedron Lett. 2001, 42, 3367.
662. Fryszkowska, A.; Fisher, K.; Gardiner, J.M.; Stephens, G.M. J. Org. Chem. 2008, 73, 4295.
663. See Charton, M. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 588–592; Newham, J. Chem. Rev. 1963, 63, 123; Rylander, P.N. Catalytic Hydrogenation over Platinum Metals Academic Press, NY,1967, pp. 469–474.
664. Bart, S.C.; Chirik, P.J. J. Am. Chem. Soc. 2003, 125, 886.
665. See Woodworth, C.W.; Buss, V.; Schleyer, P.v.R. Chem. Commun. 1968, 569.
666. See Walborsky, H.M.; Aronoff, M.S.; Schulman, M.F. J. Org. Chem. 1970, 36, 1036.
667. For a review, see Staley, S.W. Sel. Org. Transform. 1972, 2, 309.
668. Cossy, J.; Furet, N. Tetrahedron Lett. 1993, 34, 8107.
669. Karimi, S.; Tavares, P. J. Nat. Prod. 2003, 66, 520.
670. See Lane, C.F. in Pizey, J.S. Synthetic Reagents, Vol. 3, Wiley, NY, 1977, pp. 1–191.
671. See Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, Organic Syntheses Via Boranes Wiley, NY, 1975; Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973; Matteson, D.S. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 307–409, pp. 315–337; Suzuki, A.; Dhillon, R.S. Top. Curr. Chem. 1986, 130, 23.
672. Fehlner, T.P. J. Am. Chem. Soc. 1971, 93, 6366.
673. See Hutchins, R.O.; Cistone, F. Org. Prep. Proced. Int. 1981, 13, 225; Cadot, C.; Dalko, P.I.; Cossy, J. Tetrahedron Lett. 2001, 42, 1661.
674. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1005–1009.
675. Clay, J.M.; Vedejs, E. J. Am. Chem. Soc. 2005, 127, 5766.
676. Unless coordinated with a strong Lewis base, such as a tertiary amine, mono- and dialkylboranes actually exist as hydrogen-bridged dimers: Brown, H.C.; Klender, G.J. Inorg. Chem. 1962, 1, 204.
677. See Negishi, E.; Brown, H.C. Synthesis 1974, 77.
678. See Brown, H.C.; Cole, T.E.; Srebnik, M.; Kim, K. J. Org. Chem. 1986, 51, 4925.
679. Srebnik, M.; Cole, T.E.; Brown, H.C. J. Org. Chem. 1990, 55, 5051.
680. See Kulkarni, S.U.; Basavaiah, D.; Zaidlewicz, M.; Brown, H.C. Organometallics 1982, 1, 212.
681. Srebnik, M.; Cole, T.E.; Ramachandran, P.V.; Brown, H.C. J. Org. Chem. 1989, 54, 6085.
682. Cragg, G.M.L. Organoboranes in Organic Synthesis Marcel Dekker, NY, 1973, pp. 63–84, 137–197; Brown, H.C.; Vara Prasad, J.V.N.; Zee, S. J. Org. Chem. 1986, 51, 439.
683. Brown, H.C.; Sharp, R.L. J. Am. Chem. Soc. 1966, 88, 5851; Klein, J.; Dunkelblum, E.; Wolff, M.A. J. Organomet. Chem. 1967, 7, 377. See also, Marshall, P.A.; Prager, R.H. Aust. J. Chem. 1979, 32, 1251; Mo, Y.; Jiao, H.; Schleyer, P.v.R. J. Org. Chem. 2004, 69, 3493.
684. Brown, H.C.; Zweifel, G. J. Am. Chem. Soc. 1961, 83, 1241.
685. See Brown, H.C.; Chen, J.C. J. Org. Chem. 1981, 46, 3978; Soderquist, J.A.; Brown, H.C. J. Org. Chem. 1981, 46, 4599.
686. Brown, H.C.; Sharp, R.L. J. Am. Chem. Soc. 1966, 88, 5851; Klein, J.; Dunkelblum, E.; Wolff, M.A. J. Organomet. Chem. 1967, 7, 377.
687. Kabalka, G.W.; Newton Jr., R.J.; Jacobus, J. J. Org. Chem. 1978, 43, 1567.
688. Medina, J.R.; Cruz, G.; Cabrera, C.R.; Soderquist, J.A. J. Org. Chem. 2003, 68, 4631.
689. See Nelson, D.J.; Cooper, P.J. Tetrahedron Lett. 1986, 27, 4693; Brown, H.C.; Chandrasekharan, J. J. Org. Chem. 1988, 53, 4811.
690. Narayana, C.; Periasamy, M. J. Chem. Soc., Chem. Commun. 1987, 1857. See, however, Jones, P.R. J. Org. Chem. 1972, 37, 1886.
691. See Still, W.C.; Barrish, J.C. J. Am. Chem. Soc. 1983, 105, 2487.
692. See Burgess, K.; Cassidy, J.; Ohlmeyer, M.J. J. Org. Chem. 1991, 56, 1020; Burgess, K.; Ohlmeyer, M.J. J. Org. Chem. 1991, 56, 1027.
693. Zhang, J.; Lou, B.; Guo, G.; Dai, L. J. Org. Chem. 1991, 56, 1670.
694. See Brown, H.C.; Kulkarni, S.U. J. Organomet. Chem. 1982, 239, 23.
695. Brown, H.C.; Ravindran, N.; Kulkarni, S.U. J. Org. Chem. 1979, 44, 2417.
696. Brown, H.C.; Racherla, U.S. J. Org. Chem. 1986, 51, 895.
697. Soundararajan, R.; Matteson, D.S. J. Org. Chem. 1990, 55, 2274.
698. Frantz, D.E.; Singleton, D.A. Org. Lett. 1999, 1, 485.
699. Parish, E.J.; Parish, S.; Honda, H. Synth. Commun. 1990, 20, 3265.
700. Brown, H.C.; Kulkarni, S.U.; Rao, C.G.; Patil, V.D. Tetrahedron 1986, 42, 5515.
701. Soderquist, J.A.; Martinez, J.; Oyola, Y.; Kock, I. Tetrahedron Lett. 2004, 45, 5541.
702. See Shapland, P.; Vedejs, E. J. Org. Chem. 2004, 69, 4094.
703. See Brown, H.C.; Unni, M.K. J. Am. Chem. Soc. 1968, 90, 2902; Brown, H.C.; Gallivan, Jr., R.M. J. Am. Chem. Soc. 1968, 90, 2906; Brown, H.C.; Sharp, R.L. J. Am. Chem. Soc. 1968, 90, 2915.
704. Singaram, B.; Rangaishenvi, M.V.; Brown, H.C.; Goralski, C.T.; Hasha, D.L. J. Org. Chem. 1991, 56, 1543.
705. Smith, S.M.; Thacker, N.C.; Takacs, J.M. J. Am. Chem. Soc. 2008, 130, 3734.
706. Scheideman, M.; Wang, G.; Vedejs, E. J. Am. Chem. Soc. 2008, 130, 8669.
707. Rarig, R.-A.F.; Scheideman, M.; Vedejs, E. J. Am. Chem. Soc. 2008, 130, 9182.
708. Brown, H.C.; Vara Prasad, J.V.N. J. Am. Chem. Soc. 1986, 108, 2049.
709. Brown, H.C.; Singaram, B. Acc. Chem. Res. 1988, 21, 287; Srebnik, M.; Ramachandran, P.V. Aldrichimica Acta 1987, 20, 9; Brown, H.C.; Jadhav, P.K. in Morrison, J.D. Asymmetric Synthesis Vol. 2, Academic Press, NY, 1983, pp. 1–43. For a study of electronic effects, see Garner, C.M.; Chiang, S.; Nething, M.; Monestel, R. Tetrahedron Lett. 2002, 43, 8339.
710. Brown, H.C.; Jadhav, P.K.; Mandal, A.K. J. Org. Chem. 1982, 47, 5074. See also, Brown, H.C.; Weissman, S.A.; Perumal, P.T.; Dhokte, U.P. J. Org. Chem. 1990, 55, 1217. For the crystal structure of this adduct, see Soderquist, J.A.; Hwang-Lee, S.; Barnes, C.L. Tetrahedron Lett. 1988, 29, 3385.
711. Jadhav, P.K.; Kulkarni, S.U. Heterocycles 1982, 18, 169.
712. Brown, H.C.; Vara Prasad, J.V.N.; Zaidlewicz, M. J. Org. Chem. 1988, 53, 2911.
713. Kiesgen de Richter, R.; Bonato, M.; Follet, M.; Kamenka, J. J. Org. Chem. 1990, 55, 2855.
714. Jadhav, P.K.; Brown, H.C. J. Org. Chem. 1981, 46, 2988.
715. Masamune, S.; Kim, B.M.; Petersen, J.S.; Sato, T.; Veenstra, J.S.; Imai, T. J. Am. Chem. Soc. 1985, 107, 4549. See Thomas, S.P.; Aggarwal, V.K. Angew. Chem. Int. Ed. 2009, 48, 1896.
716. For another enantioselective hydroboration method, see Reaction 15-16.
717. Demay, S.; Volant, F.; Knochel, P. Angew. Chem. Int. Ed. 2001, 40, 1235.
718. Brown, H.C.; Negishi, E. J. Am. Chem. Soc. 1972, 94, 3567.
719. Cyclic hydroboration: see Brown, H.C.; Negishi, E. Tetrahedron 1977, 33, 2331. See also, Brown, H.C.; Pai, G.G.; Naik, R.G. J. Org. Chem. 1984, 49, 1072.
720. Brown, H.C.; Negishi, E.; Dickason, W.C. J. Org. Chem. 1985, 50, 520.
721. Karatjas, A.G.; Vedejs, E. J. Org. Chem. 2008, 73, 9508.
722. See Hudrlik, P.F.; Hudrlik, A.M. in Patai, S. The Chemistry of the Carbn–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 203–219.
723. Brown, H.C.; Campbell, Jr., J.B. Aldrichimica Acta 1981, 14, 1.
724. Brown, H.C.; Gupta, S.K. J. Am. Chem. Soc. 1975, 97, 5249. See Garrett, C.E.; Fu, G.C. J. Org. Chem. 1996, 61, 3224.
725. Brown, H.C.; Campbell Jr., J.B. J. Org. Chem. 1980, 45, 389.
726. Zweifel, G.; Arzoumanian, H. J. Am. Chem. Soc. 1967, 89, 291.
727. Brown, H.C.; Coleman, R.A. J. Org. Chem. 1979, 44, 2328.
728. Pelter, A.; Singaram, S.; Brown, H.C. Tetrahedron Lett. 1983, 24, 1433.
729. See Gautam, V.K.; Singh, J.; Dhillon, R.S. J. Org. Chem. 1988, 53, 187. See also, Suzuki, A.; Dhillon, R.S. Top. Curr. Chem. 1986, 130, 23.
730. Kabalka, G.W.; Yu, S.; Li, N.-S. Tetrahedron Lett. 1997, 38, 7681.
731. Burgess, K.; van der Donk, W.A.; Westcott, S.A.; Marder, T.B.; Baker, R.T.; Calabrese, J.C. J. Am. Chem. Soc. 1992, 114, 9350; Evans, D.A.; Fu, G.C.; Hoveyda, A.H. J. Am. Chem. Soc. 1992, 114, 6671.
732. Burgess, K.; Ohlmeyer, M.J. Chem. Rev. 1991, 91, 1179.
733. Evans, D.A.; Muci, A.R.; Stürmer, R. J. Org. Chem. 1993, 58, 5307.
734. Harrison, K.N.; Marks, T.J. J. Am. Chem. Soc. 1992, 114, 9220.
735. Sato, M.; Miyaura, N.; Suzuki, A. Tetrahedron Lett. 1990, 31, 231; Brown, J.M.; Lloyd-Jones, G.C. Tetrahedron: Asymmetry 1990, 1, 869.
736. Goddard, J.-P.; LeGall, T.; Mioskowski, C. Org. Lett. 2000, 2, 1455.
737. Negishi, E. Adv. Met.-Org. Chem. 1989, 1, 177; Eisch, J.J. The Chemistry of Organometallic Compounds; Macmillan, NY, 1967, pp. 107–111; Eisch, J.J.; Fichter, K.C. J. Organomet. Chem. 1983, 250, 63.
738. See Dzhemilev, U.M.; Vostrikova, O.S.; Tolstikov, G.A. Russ. Chem. Rev. 1990, 59, 1157; Maruoka, K.; Yamamoto, H. Tetrahedron 1988, 44, 5001.
739. See Negishi, E. Organometallics in Organic Synthesis Vol. 1, Wiley, NY, 1980, pp. 45–48, 357–363, 406–412; Speier, J.L. Adv. Organomet. Chem. 1979, 17, 407; Andrianov, K.A.; Soucek, J.; Khananashvili, L.M. Russ. Chem. Rev. 1979, 48, 657.
740. Negishi, E.; Takahashi, T. Synthesis 1988, 1; Dzhemilev, U.M.; Vostrikova, O.S.; Tolstikov, G.A. J. Organomet. Chem. 1986, 304, 17. Also see, Hoveyda, A.H.; Morken, J.P. J. Org. Chem. 1993, 58, 4237.
741. Doyle, M.P.; High, K.G.; Nesloney, C.L.; Clayton, Jr., T.W.; Lin, J. Organometallics 1991, 10, 1225.
742. Lipshutz, B.H.; Keil, R.; Ellsworth, E.L. Tetrahedron Lett. 1990, 31, 7257.
743. See Bogdanovic, B. Angew. Chem. Int. Ed. 1985, 24, 262.
744. See Hudrlik, P.F.; Hudrlik, A.M. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 219–232.
745. Eisch, J.J.; Kaska, W.C. J. Am. Chem. Soc. 1966, 88, 2213; Eisch, J.J.; Rhee, S. Liebigs Ann. Chem. 1975, 565.
746. See Shuikin, N.I.; Lebedev, B.L. Russ. Chem. Rev. 1966, 35, 448; Schmerling, L. in Olah, G.A. Friedel–Crafts and Related Reactions, Vol. 2, Wiley, NY, 1964, pp. 1075–1111, 1121–1122.
747. Frey, E.J.; Hepp, H.J. Ind. Eng. Chem. 1936, 28, 1439.
748. Metzger, J.O. Angew. Chem. Int. Ed. 1983, 22, 889; Hartmanns, J.; Klenke, K.; Metzger, J.O. Chem. Ber. 1986, 119, 488.
749. See Mayr, H. Angew. Chem. Int. Ed. 1990, 29, 1371.
750. Bolestova, G.I.; Parnes, Z.N.; Kursanov, D.N. J. Org. Chem. USSR 1983, 19, 2175.
751. Qiao, K.; Deng, Y. Tetrahedron Lett. 2003, 44, 2191.
752. Geraghty, N.W.A.; Hannan, J.J. Tetrahedron Lett. 2001, 42, 3211.
753. Zhang, Y.; Li, C.-J. Tetrahedron Lett. 2004,45, 7581.
754. See Pines, H.; Stalick, W.M. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds, Academic Press, NY, 1977, pp. 240–422; Pines, H. Acc. Chem. Res. 1974, 7, 155.
755. Pines, H.; Wunderlich, D. J. Am. Chem. Soc. 1958, 80, 6001.
756. Eberhardt, G.G.; Peterson, H.J. J. Org. Chem. 1965, 30, 82; Pines, H.; Stalick, W.M. Tetrahedron Lett. 1968, 3723.
757. Schmerling, L.; Toekelt, W.G. J. Am. Chem. Soc. 1962, 84, 3694.
758. Kakiuchi, F.; Murai, S. Acc. Chem. Res. 2002, 35, 826.
759. DeBoef, B.; Pastine, S.J.; Sames, D. J. Am. Chem. Soc. 2004, 126, 6556.
760. Buch, F.; Brettar, J.; Harder, S. Angew. Chem. Int. Ed. 2006, 45, 2741.
761. Glaser, P.B.; Tilley, T.D. J. Am. Chem.Soc. 2003, 125, 13640.
762. See Tsuchiya, Y.; Uchimura, H.; Kobayashi, K.; Nishiyama, H. Synlett 2004, 2099.
763. Motoda, D.; Shinokubo, H.; Oshima, K. Synlett 2002, 1529.
764. Zhao, W.-G.; Hua, R. Eur. J. Org. Chem. 2006, 5495.
765. Takaki, K.; Sonoda, K.; Kousaka, T.; Koshoji, G.; Shishido, T.; Takehira, K. Tetrahedron Lett. 2001, 42, 9211.
766. Molander, G.A.; Julius, M. J. Org. Chem. 1992, 57, 6347.
767. See Sabourault, N.; Mignani, G.; Wagner, A.; Mioskowski, C. Org. Lett. 2002, 4, 2117.
768. Nakamura, S.; Uchiyama, M. J. Am. Chem. Soc. 2007, 129, 28; Lipshutz, B.H.; Lower, A.; Kucejko, R.J.; Noson, K. Org. Lett. 2006, 8, 2969.
769. Hou, Z.; Zhang, Y.; Tardif, O.; Wakatsuki, Y. J. Am. Chem. Soc. 2001, 123, 9216.
770. Hatanaka, Y.; Goda, K.; Yamashita, F.; Hiyama, T. Tetrahedron Lett. 1994, 35, 7981.
771. Driver, T.G.; Franz, A.K.; Woerpel, K.A. J. Am. Chem. Soc. 2002, 124, 6524.
772. Molander, G.A.; Corrette, C.P. Tetrahedron Lett. 1998, 39, 5011.
773. Muci, A.R.; Bercaw, J.E. Tetrahedron Lett. 2000, 41, 7609.
774. Murai, T.; Oda, T.; Kimura, F.; Onishi, H.; Kanda, T.; Kato, S. J. Chem. Soc., Chem. Commun. 1994, 2143.
775. Jensen, J.F.; Svendsen, B.H.; la Cour, T.V.; Pedersen, H.L.; Johannsen, M. J. Am. Chem. Soc. 2002, 124, 4558.
776. Nakamura, S.; Uchiyama, M.; Ohwada, T. J. Am. Chem. Soc. 2005, 127, 13116.
777. Rubin, M.; Schwier, T.; Gevorgyan, V. J. Org. Chem. 2002, 67, 1936.
778. Miura, K.; Oshima, K.; Utimoto, K. Bull. Chem. Soc. Jpn. 1993, 66, 2356.
779. Nakamura, S.; Uchiyama, M.; Ohwada, T. J. Am. Chem. Soc. 2004, 126, 11146.
780. Trost, B.M.; Ball, Z.T. J. Am. Chem. Soc. 2005, 127, 17644; Maifeld, S.V.; Tran, M.N.; Lee, D. Tetrahedron Lett. 2005, 46, 105.
781. Hamze, A.; Provot, O.; Brion, J.-D.; Alami, M. Synthesis 2007, 2025.
782. Takahashi, T.; Bao, F.; Gao, G.; Ogasawara, M. Org. Lett. 2003, 5, 3479.
783. Miyake, Y.; Isomura, E.; Iyoda, M. Chem. Lett. 2006, 35, 836.
784. Berthon-Gelloz, G.; Schumers, J.-M.; De Bo, G.; Markó, I.E. J. Org. Chem. 2008, 73, 4190.
785. Trost, B.M.; Ball, Z.T. J. Am. Chem. Soc. 2001, 123, 12726.
786. Kawanami, Y.; Sonoda, Y.; Mori, T.; Yamamoto, K. Org. Lett. 2002, 4, 2825.
787. Sato, A.; Kinoshita, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2004, 6, 2217.
788. Wu, W.; Li, C.-J. Chem. Commun. 2003, 1668.
789. Asao, N.; Shimada, T.; Shimada, T.; Yamamoto, Y. J. Am. Chem. Soc. 2001, 123, 10899. See also, Sudo, T.; Asao, N.; Yamamoto, Y. J. Org. Chem. 2000, 65, 8919.
790. Kopping, B.; Chatgilialoglu, C.; Zehnder, M.; Giese, B. J. Org. Chem. 1992, 57, 3994.
791. Yus, M.; Martínez, P.; Guijarro, D. Tetrahedron 2001, 57, 10119.
792. Dang, H.-S.; Roberts, B.P. Tetrahedron Lett. 1995, 36, 2875.
793. Kakiuchi, F.; Tanaka, Y.; Sato, T.; Chatani, N.; Murai, S. Chem. Lett. 1995, 679; Trost, B.M.; Imi, K.; Davies, I.W. J. Am. Chem. Soc. 1995, 117, 5371.
794. Tayama, O.; Iwahama, T.; Sakaguchi, S.; Ishii, Y. Eur. J. Org. Chem. 2003, 2286.
795. Reisser, M.; Maier, A.; Maas, G. Synlett 2002, 1459.
796. Hewitt, G.W.; Somers, J.J.; Sieburth, S.Mc.N. Tetrahedron Lett. 2000, 41, 10175.
797. Lukashev, N.V.; Kazantsev, A.V.; Borisenko, A.A.; Beletskaya, I.P. Tetrahedron 2001, 57, 10309.
798. Clark, C.T.; Lake, J.F.; Scheidt, K.A. J. Am. Chem. Soc. 2004, 126, 84.
799. Oestreich, M.; Weiner, B. Synlett 2004, 2139.
800. See Sammis, G.M.; Danjo, H.; Jacobsen, E.N. J. Am. Chem. Soc. 2004, 126, 9928.
801. See Onsager, O.; Johansen, J.E. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 3, Wiley, NY, 1985, pp. 205–257.
802. Wang, C.-C.; Lin, P.-S.; Cheng, C.-H. Tetrahedron Lett. 2004, 45, 6203.
803. Ng, S.-S.; Ho, C.-Y.; Schleicher, K.D.; Jamison, T.F. Pure Appl. Chem. 2008, 80, 929.
804. Moreau, B.; Wu, J.Y.; Ritter, T. Org. Lett. 2009, 11, 337.
805. Kisanga, P.; Goj, L.A.; Widenhoefer, R.A. J. Org. Chem. 2001, 66, 635.
806. Mori, M.; Saito, N.; Tanaka, D.; Takimoto, M.; Sato, Y. J. Am. Chem. Soc. 2003, 125, 5606; Michaut, M.; Santelli, M.; Parrain, J.-L. Tetrahedron Lett. 2003, 44, 2157.
807. Molander, G.A.; Dowdy, E.D.; Schumann, H. J. Org. Chem. 1998, 63, 3386.
808. Okamoto, S.; Livinghouse, T. J. Am. Chem. Soc. 2000, 122, 1223. See Hart, D.J.; Bennett, C.E. Org. Lett. 2003, 5, 1499.
809. Toyota, M.; Majo, V.J.; Ihara, M. Tetrahedron Lett. 2001, 42, 1555.
810. Kende, A.S.; Mota Nelson, C.E.; Fuchs, S. Tetrahedron Lett. 2005, 46, 8149.
811. Wender, P.A.; Dyckman, A.J. Org. Lett. 1999, 1, 2089; Cao, P.; Wang, B.; Zhang, X. J. Am. Chem. Soc. 2000, 122, 6490; Cao, P.; Zhang, X. Angew. Chem. Int. Ed. 2000, 39, 4104.
812. Fernández-Rivas, C.; Méndez, M.; Echavarren, A.M. J. Am. Chem. Soc. 2000, 122, 1221.
813. Chatani, N.; Inoue, H.; Morimoto, T.; Muto, T.; Murai, S. J. Org. Chem. 2001, 66, 4433.
814. Miura, K.; Funatsu, M.; Saito, H.; Ito, H.; Hosomi, A. Tetrahedron Lett. 1996, 37, 9059. Also see, Maye, J.P.; Negishi, E. Tetrahedron Lett. 1993, 34, 3359.
815. See Ohno, H.; Takeoka, Y.; Kadoh, Y.; Miyamura, K.; Tanaka, T. J. Org. Chem. 2004, 69, 4541.
816. Yamaguchi, M.; Tsukagoshi, T.; Arisawa, M. J. Am. Chem. Soc. 1999, 121, 4074.
817. Stork, G.; Burgstahler, A.W. J. Am. Chem. Soc. 1955, 77, 5068; Eschenmoser, A.; Ruzicka, L.; Jeger, O.; Arigoni, D. Helv. Chim. Acta 1955, 38, 1890.
818. Gnonlonfoun, N. Bull. Soc. Chim. Fr. 1988, 862; Johnson, W.S. Angew. Chem. Int. Ed. 1976, 15, 9; Acc. Chem. Res. 1968, 1, 1; van Tamelen, E.E. Acc. Chem. Res. 1975, 8, 152. See Bartlett, P.A. in Morrison, J.D. Asymmetric Synthesis Vol. 3, Academic Press, NY, 1985, pp. 341–409.
819. Guay, D.; Johnson, W.S.; Schubert, U. J. Org. Chem. 1989, 54, 4731 and references cited therein.
820. Johnson, W.S.; Gravestock, M.B.; McCarry, B.E. J. Am. Chem. Soc. 1971, 93, 4332.
821. Johnson, W.S. Acc. Chem. Res. 1968, 1, 1; Kametani T.; Fukumoto, K. Synthesis 1972, 657.
822. Ishihara, K.; Nakamura, S.; Yamamoto, H. J. Am. Chem. Soc. 1999, 121, 4906.
823. Asao, N.; Shimada, T.; Yamamoto, Y. J. Am. Chem. Soc. 1999, 121, 3797.
824. Mullen, C.A.; Gagné, M.R. J. Am. Chem. Soc. 2007, 129, 11880.
825. Chen, L.; Gill, G.B.; Pattenden, G. Tetrahedron Lett. 1994, 35, 2593.
826. Fel'dblyum, V.Sh.; Obeshchalova, N.V. Russ. Chem. Rev. 1968, 37, 789.
827. For a review, see Pines, H. Synthesis 1974, 309.
828. Pillai, S.M.; Ravindranathan, M.; Sivaram, S. Chem. Rev. 1986, 86, 353; Jira, R.; Freiesleben, W. Organomet. React. 1972, 3, 1, p. 117; Heck, R.F. Organotransition Metal Chemistry Academic Press, NY, 1974, pp. 84–94, 150–157. Also see, Kaur, G.; Manju, K.; Trehan, S. Chem. Commun. 1996, 581.
829. See Fischer, K.; Jonas, K.; Misbach, P.; Stabba, R.; Wilke, G. Angew. Chem. Int. Ed. 1973, 12, 943.
830. Takahashi, N.; Okura, I.; Keii, T. J. Am. Chem. Soc. 1975, 97, 7489.
831. Takacs, J.M.; Myoung, Y.C. Tetrahedron Lett. 1992, 33, 317.
832. Zhang, A.; RajanBabu, T.V. J. Am. Chem. Soc. 2006, 128, 54.
833. Hilt, G.; du Mesnil, F.-X.; Lüers, S. Angew. Chem. Int. Ed. 2001, 40, 387. For a review see Su, A.C.L. Adv. Organomet. Chem. 1979, 17, 269.
834. See Denis, P.; Jean, A.; Croizy, J.F.; Mortreux, A.; Petit, F. J. Am. Chem. Soc. 1990, 112, 1292.
835. Takacs, J.M.; Leonov, A.P. Org. Lett. 2003, 5, 4317.
836. Takimoto, M.; Nakamura, Y.; Kimura, K.; Mori, M. J. Am. Chem. Soc. 2004, 126, 5956.
837. Nomura, N.; Jin, J.; Park, H.; RajanBabu, T.V. J. Am. Chem. Soc. 1998, 120, 459.
838. Takahashi, T.; Xi, Z.; Fischer, R.; Huo, S.; Xi, C.; Nakajima, K. J. Am. Chem. Soc. 1997, 119, 4561.
839. Kinoshita, A.; Sakakibara, N.; Mori, M. J. Am. Chem. Soc. 1997, 119, 12388.
840. Urabe, H.; Takeda, T.; Hideura, D.; Sato, F. J. Am. Chem. Soc. 1997, 119, 11295.
841. Shirakura, M.; Suginome, M. J. Am. Chem. Soc. 2008, 130, 5410.
842. Buchwald, S.L.; Nielsen, R.B. J. Am. Chem. Soc. 1989, 111, 2870.
843. Ryan, J.; Micalizio, G.C. J. Am. Chem. Soc. 2006, 128, 2764; Kanno, K.-i.; Igarashi, E.; Zhou, L.; Nakajima, K.; Takahashi, T. J. Am. Chem. Soc. 2008, 130, 5624.
844. Hatakeyama, T.; Yoshimoto, Y.; Gabriel, T.; Nakamura, M. Org. Lett. 2008, 10, 5341.
845. Shibata, Y.; Hirano, M.; Tanaka, K. Org. Lett. 2008, 10, 2829; Katagiri, T.; Tsurugi, H.; Satoh, T.; Miura, M. Chem. Commun. 2008, 3405.
846. Takahashi, T.; Liu, Y.; Iesato, A.; Chaki, S.; Nakajima, K.; Kanno, K.-i. J. Am. Chem. Soc. 2005, 127, 11928.
847. Trost, B.M.; Surivet, J.-P.; Toste, F.D. J. Am. Chem. Soc. 2001, 123, 2897.
848. Giessert, A.J.; Snyder, L.; Markham, J.; Diver, S.T. Org. Lett. 2003, 5, 1793.
849. Nugent, W.A.; Thorn, D.L.; Harlow, R.L. J. Am. Chem. Soc. 1987, 109, 2788. See Trost, B.M.; Lee, D.C. J. Am. Chem. Soc. 1988, 110, 7255; Tamao, K.; Kobayashi, K.; Ito, Y. J. Am. Chem. Soc. 1989, 111, 6478.
850. Jang, H.-Y.; Krische, M.J. J. Am. Chem. Soc. 2004, 126, 7875.
851. Varela, J.A.; Rubín, S.G.; González-Rodríguez, C.; Castedo, L.; Saá, C. J. Am. Chem. Soc. 2006, 128, 9263.
852. Zhang, C.; Cui, D.-M.; Yao, L.-Y.; Wang, B.-S.; Hu, Y.-Z.; Hayashi, T. J. Org. Chem. 2008, 73, 7811.
853. Méndez, M.; Muñoz, M.P.; Nevado, C.; Cárdenas, D.J.; Echavarren, A.M. J. Am. Chem. Soc. 2001, 123, 10511; Wang, X.; Chakrapani, H.; Madine, J.W.; Keyerleber, M.A.; Widenhoefer, R.A. J. Org. Chem. 2002, 67, 2778. See also, Fürstner, A.; Stelzer, F.; Szillat, H. J. Am. Chem. Soc. 2001, 123, 11863.
854. Urabe, H.; Nakajima, R.; Sato, F. Org. Lett. 2000, 2, 3481.
855. Chakrapani, H.; Liu, C.; Widenhoefer, R.A. Org. Lett. 2003, 5, 157; Lee, P.H.; Kim, S.; Lee, K.; Seomoon, D.; Kim, H.; Lee, S.; Kim, M.; Han, M.; Noh, K.; Livinghouse, T. Org. Lett. 2004, 6, 4825.
856. Yamamoto, Y.; Kuwabara, S.; Ando, Y.; Nagata, H.; Nishiyama, H.; Itoh, K. J. Org. Chem. 2004, 69, 6697.
857. Hatano, M.; Mikami, K. J. Am. Chem. Soc. 2003, 125, 4704.
858. Miura, T.; Shimada, M.; Murakami, M. J. Am. Chem. Soc. 2005, 127, 1094.
859. Ogoshi, S.; Ueta, M.; Oka, M.-a.; Kurosawa, H. Chem. Commun. 2004, 2732.
860. Rubina, M.; Gevergyan, V. J. Am. Chem. Soc. 2001, 123, 11107; Yang, C.; Nolan, S.P. J. Org. Chem. 2002, 67, 591.
861. Nishiura, M.; Hou, Z.; Wakatsuki, Y.; Yamaki, T.; Miyamoto, T. J. Am. Chem. Soc. 2003, 125, 1184.
862. Bassetti, M.; Pasquini, C.; Raneri, A.; Rosato, D. J. Org. Chem. 2007, 72, 4558.
863. Bruyere, D.; Grigg, R.; Hinsley, J.; Hussain, R.K.; Korn, S.; Del Cierva, C.O.; Sridharan, V.; Wang, J. Tetrahedron Lett. 2003, 44, 8669.
864. Trost, B.M.; Matusbara, S.; Carninji, J.J. J. Am. Chem. Soc. 1989, 111, 8745.
865. Nishibayashi, Y.; Yamanashi, M.; Wakiji, I.; Hidai, M. Angew. Chem. Int. Ed. 2000, 39, 2909.
866. Ajamian, A.; Gleason, J.L. Org. Lett. 2003, 5, 2409.
867. Luzung, M.R.; Markham, J.P.; Toste, F.D. J. Am. Chem. Soc. 2004, 126, 10858; Zhang, L.; Kozmin, S.A. J. Am. Chem. Soc. 2005, 127, 6962.
868. Kim, H.; Lee, C. J. Am. Chem. Soc. 2005, 127, 10180. With AgBF4, Evans, P.A.; Lai, K.W.; Sawyer, J.R. J. Am. Chem. Soc. 2005, 127, 12466; Kim, H.; Lee, C. J. Am. Chem. Soc. 2006, 128, 6336. See Denmark, S.E.; Liu, J.H.-C. J. Am. Chem. Soc. 2007, 129, 3737.
869. Fürstner, A.; Martin, R.; Majima, K. J. Am. Chem. Soc. 2005, 127, 12236.
870. Ma, S.; Gu, Z. J. Am. Chem. Soc. 2005, 127, 6182; Corkey, B.K.; Toste, F.D. J. Am. Chem. Soc. 2007, 129, 2764.
871. Yamazaki, S.; Yamada, K.; Yamabe, S.; Yamamoto, K. J. Org. Chem. 2002, 67, 2889.
872. Nishizawa, M.; Yadav, V.K.; Skwarczynski, M.; Takao, H.; Imagawa, H.; Sugihara, T. Org. Lett. 2003, 5, 1609.
873. Chatani, N.; Inoue, H.; Kotsuma, T.; Morai, S. J. Am. Chem. Soc. 2002, 124, 10294.
874. Miyanohana, Y.; Inoue, H.; Chatani, N. J. Org. Chem. 2004, 69, 8541.
875. Frazén, J.; Löfstedt, J.; Dorange, I.; Bäckvall, J.-E. J. Am. Chem. Soc. 2002, 124, 11246.
876. Kang, S.-K.; Ko, B.-S.; Lee, D.-M. Tetrahedron Lett. 2002, 43, 6693.
877. Brummond, K.M.; Chen, H.; Sill, P.; You, L. J. Am. Chem. Soc. 2002, 124, 15186. See also, Candran, N.; Cariou, K.; Hervé, G.; Aubert, C.; Fensterbank, L.; Malacria, M.; Marco-Contelles, J. J. Am. Chem. Soc. 2004, 126, 3408.
878. Oh, C.H.; Park, S.J. Tetrahedron Lett. 2003, 44, 3785.
879. Condon-Gueugnot, S.; Linstrumelle, G. Tetrahedron 2000, 56, 1851.
880. Rubin, M.; Markov, J.; Chuprakov, S.; Wink, D.J.; Gevorgyan, V. J. Org. Chem. 2003, 68, 6251.
881. Shin, S.; RajanBabu, T.V. J. Am. Chem. Soc. 2001, 123, 8416.
882. Tong, X.; Zhang, Z.; Zhang, X. J. Am. Chem. Soc. 2003, 125, 6370.
883. Zhu, G.; Zhang, Z. Org. Lett. 2004, 6, 4041.
884. Wardell, J.L.; Paterson, E.S. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 219–338, pp. 268–296.
885. Gardner, H.C.; Kochi, J.K. J. Am. Chem. Soc. 1976, 98, 558.
886. Richey Jr., H.G.; Moses, L.M.; Domalski, M.S.; Erickson, W.F.; Heyn, A.S. J. Org. Chem. 1981, 46, 3773.
887. Kang, J. Organometallics 1984, 3, 525.
888. Eisch, J.J.; Merkley, J.H. J. Am. Chem. Soc. 1979, 101, 1148; Miller, R.B.; Reichenbach, T. Synth. Commun. 1976, 6, 319. See also, Duboudin, J.G.; Jousseaume, B. J. Organomet. Chem. 1979, 168, 1; Synth. Commun. 1979, 9, 53.
889. See Sato, F. J. Organomet. Chem. 1985, 285, 53–64.; Dzhemilev, U.M.; D'yakonov, V.A.; Khafizova, L.O.; Ibragimov, A.G. Russ. J. Org. Chem. 2005, 41, 352.
890. Yorimitsu, H.; Tang, J.; Okada, K.; Shinokubo, H.; Oshima, K. Chem. Lett. 1998, 11.
891. de Armas, J.; Hoveyda, A.H. Org. Lett. 2001, 3, 2097.
892. Pellet-Rostaing, S.; Saluzzo, C.; Ter Halle, R.; Breuzard, J.; Vial, L.; LeGuyader, F.; Lemaire, M. Tetrahedron Asymmetry 2001, 12, 1983.
893. Zhang, D.; Ready, J.M. J. Am. Chem. Soc. 2006, 128, 15050.
894. Shirakawa, E.; Yamagami, T.; Kimura, T.; Yamaguchi, S.; Hayashi, T. J. Am. Chem. Soc. 2005, 127, 17164.
895. Liu, X.; Fox, J.M. J. Am. Chem. Soc. 2006, 128, 5600,
896. Yan, N.; Liu, X.; Fox, J.M. J. Org. Chem. 2008, 73, 563.
897. Nobe, Y.; Arayama, K.; Urabe, H. J. Am. Chem. Soc. 2005, 127, 18006.
898. Lehmkuhl, H.; Janssen, E. Liebigs Ann. Chem. 1978, 1854. This is actually a type of ene reaction, see Oppolzer, W. Angew. Chem. Int. Ed. 1989, 28, 38.
899. Felkin, H.; Umpleby, J.D.; Hagaman, E.; Wenkert, E. Tetrahedron Lett. 1972, 2285; Hill, E.A.; Myers, M.M. J. Organomet. Chem. 1979, 173, 1. See also, Yang, D.; Gu, S.; Yan, Y.-L.; Zhu, N.-Y.; Cheung, K.-K. J. Am. Chem. Soc. 2001, 123, 8612.
900. Cesati III, R.R.; de Armas, J.; Hoveyda, A.H. Org. Lett. 2002, 4, 395.
901. Wakabayashi, K.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2001, 123, 5374.
902. Taber, D.F.; Mitten, J.V. J. Org. Chem. 2002, 67, 3847.
903. Terao, J.; Oda, A.; Kambe, N. Org. Lett. 2004, 6, 3341.
904. Murakami, K.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. Org. Lett. 2007, 9, 1569.
905. Schipper, D.J.; Hutchinson, M.; Fagnou, K. J. Am. Chem. Soc. 2010, 132, 6910.
906. The mechanism of the phosphine free Pd catalyzed reaction has been studied. See Ahlquist M.; Fabrizi, G.; Sandro Cacchi,, S.; Norrby, P.-O. J. Am. Chem. Soc. 2006, 128, 12785.
907. Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Persiani, D. Org. Lett. 2008, 10, 1597.
908. Liao, L.; Sigman, M.S. J. Am. Chem. Soc. 2010, 132, 10209.
909. See Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11; VCH, NY, 1988, pp. 129–142; García, G.V.; Budelman, N.S. Org. Prep. Proceed. Int. 2003, 35, 445.
910. Bartoli, G.; Dalpozzo, R.; DeNino, A.; Procopio, A.; Sanbri, L.; Tagarelli, A. Tetrahedron Lett. 2001, 42, 8823.
911. Hojo, M.; Murakami, Y.; Aihara, H.; Sakuragi, R.; Baba, Y.; Hosomi, A. Angew. Chem. Int. Ed. 2001, 40, 621.
912. Gandon, V.; Laroche, C.; Szymoniak, J. Tetrahedron Lett. 2003, 44, 4827.
913. Coldham, I.; Hufton, R.; Rathmell, R.E. Tetrahedron Lett. 1997, 38, 7617; Coldham, I.; Lang-Anderson, M.M.S.; Rathmell, R.E.; Snowden, D.J. Tetrahedron Lett. 1997, 38, 7621.
914. Norsikian, S.; Baudry, M.; Normant, J.F. Tetrahedron Lett. 2000, 41, 6575.
915. Norsikian, S.; Marek, I.; Poisson, J.-F.; Normant, J.F. J. Org. Chem. 1997, 62, 4898.
916. Wender, P.A.; White, A.W. J. Am. Chem. Soc. 1988, 110, 2218; Bailey, W.F.; Nurmi, T.T.; Patricia, J.J.; Wang, W. J. Am. Chem. Soc. 1987, 109, 2442.
917. Bailey, W.F.; Khanolkar, A.D. J. Org. Chem. 1990, 55, 6058 and references cited therein; Bailey, W.F.; Daskapan, T.; Rampalli, S. J. Org. Chem. 2003, 68, 1334. Also see, Fretwell, P.; Grigg, R.; Sansano, J.M.; Sridharan, V.; Sukirthalingam, S.; Wilson, D.; Redpath, J. Tetrahedron 2000, 56, 7525.
918. Bailey, W.F.; Ovaska, T.V. J. Am. Chem. Soc. 1993, 115, 3080, and references cited therein. See Funk, R.L.; Bolton, G.L.; Brummond, K.M.; Ellestad, K.E.; Stallman, J.B. J. Am. Chem. Soc. 1993, 115, 7023.
919. A conformational radical clock has been developed to evaluate organolithium-mediated cyclization reactions. See Rychnovsky, S.D.; Hata, T.; Kim, A.I.; Buckmelter, A.J. Org. Lett. 2001, 3, 807.
920. Bailey, W.F.; Ovaska, T.V. Chem. Lett. 1993, 819.
921. Bailey, W.F.; Khanolkar, A.D.; Gavaskar, K.V. J. Am. Chem. Soc. 1992, 114, 8053.
922. Harada, T.; Fujiwara, T.; Iwazaki, K.; Oku, A. Org. Lett. 2000, 2, 1855.
923. Wei, X.; Taylor, R.J.K. Angew. Chem. Int. Ed. 2000, 39, 409.
924. Komine, N.; Tomooka, K.; Nakai, T. Heterocycles 2000, 52, 1071.
925. See Trost, B.M.; Ball, Z.T. Synthesis 2005, 853.
926. Nakamura, I.; Mizushima, Y.; Yamamoto, Y. J. Am. Chem. Soc. 2005, 127, 15022; Fürstner, A.; Davies, P.W. J. Am. Chem. Soc. 2005, 127, 15024.
927. Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chemistry: European J. 2008, 14, 11296;
928. Gupta, A.K.; Kim, K.S.; Oh, C.H. Synlett 2005, 457.
929. Kondakov, D.Y.; Negishi, E. J. Am. Chem. Soc. 1995, 117, 10771. Also see, Shibata, K.; Aida, T.; Inoue, S. Tetrahedron Lett. 1993, 33, 1077.
930. Wipf, P.; Lim, S. Angew. Chem. Int. Ed. 1993, 32, 1068.
931. Trost, B.M.; Indolese, A.F.; Müller, T.J.J.; Treptow, B. J. Am. Chem. Soc. 1995, 117, 615.
932. Zhou, Z.; Larouche, D.; Bennett, S.M. Tetrahedron 1995, 51, 11623.
933. Fukuzawa, S.; Tsuchimoto, T. Synlett 1993, 803.
934. Pirrung, F.O.H.; Hiemstra, H.; Speckamp, W.N.; Kaptein, B.; Schoemaker, H.E. Tetrahedron 1994, 50, 12415.
935. Nishikawa, T.; Shinokubo, H.; Oshima, K. Org. Lett. 2003, 5, 4623.
936. Nakao, J.; Inoue, R.; Shinokubo, H.; Oshima, K. J. Org. Chem. 1997, 62, 1910.
937. Snider, B.B.; Merritt, J.E. Tetrahedron 1991, 47, 8663.
938. Fujiwara, N.; Yamamoto, Y. J. Org. Chem. 1999, 64, 4095.
939. Klaps, E.; Schmid, W. J. Org. Chem. 1999, 64, 7537.
940. Asao, N.; Matsukawa, Y.; Yamamoto, Y. Chem. Commun. 1996, 1513.
941. Luo, F.-T.; Fwu, S.-L.; Huang, W.-S. Tetrahedron Lett. 1992, 33, 6839.
942. Nishikawa, T.; Yorimitsu, H.; Oshima, K. Synlett 2004, 1573.
943. Zeni, G.; Nogueira, C.W.; Pena, J.M.; Pialssa, C.; Menezes, P.H.; Braga, A.L.; Rocha, J.B.T. Synlett 2003, 579.
944. Wang, X.; Pei, T.; Han, X.; Widenhoefer, R.A. Org. Lett. 2003, 5, 2699.
945. Yang, D.; Li, J.-H.; Gao, Q.; Yan, Y.-L. Org. Lett. 2003, 5, 2869.
946. Itoh, Y.; Tsuji, H.; Yamagata, K.-i.; Endo, K.; Tanaka, I.; Nakamura, M.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 17161.
947. Patil, N.T.; Yamamoto, Y. J. Org. Chem. 2004, 69, 6478.
948. Nakamura, M.; Endo, K.; Nakamura, E. J. Am. Chem. Soc. 2003, 125, 13002.
949. Leitner, A.; Larsen, J.; Steffens, C.; Hartwig, J.F. J. Org. Chem. 2004, 69, 7552.
950. Yao, X.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 6884.
951. Pei, T.; Wang, X.; Widenhoefer, R.A. J. Am. Chem. Soc. 2003, 125, 648.
952. Patil, N.T.; Khan, F.N.; Yamamoto, Y. Tetrahedron Lett. 2004, 45, 8497.
953. Rodriguez, A.L.; Bunlaksananusorn, T.; Knochel, P. Org. Lett. 2000, 2, 3285.
954. Iwasawa, N.; Miura, T.; Kiyota, K.; Kusama, H.; Lee, K.; Lee, P.H. Org. Lett. 2002, 4, 4463.
955. Black, P.J.; Harris, W.; Williams, J.M.J. Angew. Chem. Int. Ed. 2001, 40, 4475.
956. Trost, B.M.; Simas, A.B.C.; Plietker, B.; Jäkel, C.; Xie, J. Chemistry: European J. 2005, 11, 7075.
957. Denmark, S.E.; Wang, Z. Org. Lett. 2001, 3, 1073.
958. Havránek, M.; Dvorák, D. J. Org. Chem. 2002, 67, 2125. See also, Lee, K.; Seomoon, D.; Lee, P.H. Angew. Chem. Int. Ed. 2002, 41, 3901.
959. Huang, Q.; Fazio, A.; Dai, G.; Campo, M.A.; Larock, R.C. J. Am. Chem. Soc. 2004, 126, 7460.
960. Inoue, H.; Chatani, N.; Murai, S. J. Org. Chem. 2002, 67, 1414.
961. Zhou, L.; Hirao, T. J. Org. Chem. 2003, 68, 1633.
962. Yanada, R.; Koh, Y.; Nishimori, N.; Matsumura, A.; Obika, S.; Mitsuya, H.; Fujii, N.; Takemoto, Y. J. Org. Chem. 2004, 69, 2417; Yanada, R.; Obika, S.; Oyama, M.; Takemoto, Y. Org. Lett. 2004, 6, 2825.
963. Yanada, R.; Obika, S.; Nishimori, N.; Yamauchi, M.; Takemoto, Y. Tetrahedron Lett. 2004, 45, 2331.
964. Dahlén, A.; Petersson, A.; Hilmersson, G. Org. Biomol. Chem. 2003, 1, 2423.
965. Lail, M.; Arrowood, B.N.; Gunnoe, T.B. J. Am. Chem. Soc. 2003, 125, 7506.
966. Reetz, M.T.; Sommer, K. Eur. J. Org. Chem. 2003, 3485.
967. Thalji, R.K.; Ellman, J.A.; Bergman, R.G. J. Am. Chem. Soc. 2004, 126, 7192.
968. See Alonso, F.; Beletskaya, I.P.; Yus, M. Chem. Rev. 2004, 104, 3079.
969. Tsukada, N.; Mitsuboshi, T.; Setoguchi, H.; Inoue, Y. J. Am. Chem. Soc. 2003, 125, 12102.
970. Vo-Thanh, G.; Lahrache, H.; Loupy, A.; Kim, I.-J.; Chang, D.-H.; Jun, C.-H. Tetrahedron 2004, 60, 5539.
971. Nakamura, I.; Siriwardana, A.I.; Saito, S.; Yamamoto, Y. J. Org. Chem. 2002, 67, 3445.
972. Lautens, M.; Yoshida, M. J. Org. Chem. 2003, 68, 762; Genin, E.; Michelet, V.; Genêt, J.-P. Tetrahedron Lett. 2004, 45, 4157.
973. Yoshida, M.; Gotou, T.; Ihara, M. Chem. Commun. 2004, 1124.
974. Oh, C.H.; Sung, H.R.; Park, S.J.; Ahn, K.H. J. Org. Chem. 2002, 67, 7155.
975. Matsubara, S.; Ukai, K.; Toda, N.; Utimoto, K.; Oshima, K. Synlett 2000, 995.
976. Tanaka, R.; Hirano, S.; Urabe, H.; Sato, F. Org. Lett. 2003, 5, 67.
977. Raston, C.L.; Salem, G. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 159–306, 233–248; Normant, J.F.; Alexakis, A. Synthesis 1981, 841; Hudrlik, P.F.; Hudrlik, A.M. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 233–238. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 452–460.
978. See Ashby, E.C.; Goel, A.B. J. Org. Chem. 1983, 48, 2125.
979. Gardette, M.; Alexakis, A.; Normant, J.F. Tetrahedron 1985, 41, 5887 and references cited therein. For an extensive list of references, see Marfat, A.; McGuirk, P.R.; Helquist, P. J. Org. Chem. 1979, 44, 3888.
980. See Rao, S.A.; Periasamy, M. Tetrahedron Lett. 1988, 29, 4313.
981. See Westmijze, H.; Kleijn, H.; Meijer, J.; Vermeer, P. Recl. Trav. Chim. Pays-Bas 1981, 100, 98, and references cited therein.
982. Furber, M.; Taylor, R.J.K.; Burford, S.C. J. Chem. Soc. Perkin Trans. 1,1986, 1809.
983. Rao, S.A.; Knochel, P. J. Am. Chem. Soc. 1991, 113, 5735.
984. Normant, J.F.; Cahiez, G.; Chuit, C.; Villieras, J. J. Organomet. Chem. 1973, 54, C53.
985. Alexakis, A.; Cahiez, G.; Normant, J.F. Org. Synth. VII, 290.
986. See Negishi, E. Acc. Chem. Res. 1987, 20, 65; Pure Appl. Chem. 1981, 53, 2333; Negishi, E.; Takahashi, T. Aldrichimica Acta 1985, 18, 31.
987. Hirashita, T.; Akutagawa, K.; Kamei, T.; Araki, S. Chem. Commun. 2006, 2598.
988. Hara, R.; Nishihara, Y.; Landré, P.D.; Takahashi, T. Tetrahedron Lett. 1997, 38, 447.
989. Takahashi, T.; Kotora, M.; Kasai, K.; Suzuki, N. Tetrahedron Lett. 1994, 35, 5685.
990. Zhou, C.; Emrich, D.E.; Larock, R.C. Org. Lett. 2003, 5, 1579; Zhou, C.; Larock, R.C. J. Org. Chem. 2005, 70, 3765; Zhou, C.; Larock, R.C. Org. Lett. 2005, 7, 259.
991. Carruthers, W. Cycloaddition Reactions in Organic Synthesis Pergamon, Elmsford, NY, 1990; Boyd, G.V. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 477–525; Hoffmann, H.M.R. Angew. Chem. Int. Ed. 1969, 8, 556. For reviews of intramolecular ene reactions see Taber, D.F. Intramolecular Diels–Alder and Alder Ene Reactions Springer, NY, 1984; pp. 61–94; Oppolzer, W.; Snieckus, V. Angew. Chem. Int. Ed. 1978, 17, 476–486; Conia, J.M.; Le Perchec, P. Synthesis 1975, 1. See Desimoni, G.; Faita, G.; Righetti, P.P.; Sfulcini, A.; Tsyganov, D. Tetrahedron 1994, 50, 1821 for solvent effects in the ene reaction.
992. Choony, N.; Kuhnert, N.; Sammes, P.G.; Smith, G.; Ward, R.W. J. Chem. Soc., Perkin Trans. 1 2002, 1999.
993. Cossy, J.; Bouzide, A. Tetrahedron 1997, 53, 5775; Oppolzer, W.; Fürstner, A. Helv. Chim. Acta 1993, 76, 2329; Oppolzer, W.; Schröder, F. Tetrahedron Lett. 1994, 35, 7939.
994. Singleton, D.A.; Hang, C.; Szymanski, M.J.; Meyer, M.P.; Leach, A.G.; Kuwata, K.T.; Chen, J.S.; Greer, A.; Foote, C.S.; Houk, K.N. J. Am. Chem. Soc. 2003, 125, 1319.
995. Eto, M.; Nishimoto, M.; Kubota, S.; Matsuoka, T.; Harano, K. Tetrahedron Lett. 1996, 37, 2445.
996. Fisk, J.S.; Tepe, J.J. J. Am. Chem. Soc. 2007, 129, 3058.
997. Nahm, S.H.; Cheng, H.N. J. Org. Chem. 1986, 51, 5093.
998. See Jenner, G.; Salem, R.B.; El'yanov, B.; Gonikberg, E.M. J. Chem. Soc. Perkin Trans. 2,1989, 1671; Thomas IV, B.E.; Loncharich, R.J.; Houk, K.N. J. Org. Chem. 1992, 57, 1354.
999. Ooi, T.; Maruoka, K.; Yamamoto, H. Tetrahedron 1994, 50, 6505; Thomas IV, B.E.; Houk, K.N. J. Am. Chem. Soc. 1993, 115, 790; Also see, Masaya, K.; Tanino, K.; Kuwajima, I. Tetrahedron Lett. 1994, 35, 7965.
1000. See Chaloner, P.A. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 456–460; Snider, B.B. Acc. Chem. Res. 1980, 13, 426.
1001. Sturla, S.J.; Kablaoui, N.M.; Buchwald, S.L. J. Am. Chem. Soc. 1999, 121, 1976.
1002. Aggarwal, V.K.; Vennall, G.P.; Davey, P.N.; Newman, C. Tetrahedron Lett. 1998, 39, 1997.
1003. Davies, A.G.; Kinart, W.J. J. Chem. Soc. Perkin Trans. 2,1993, 2281.
1004. Molander, G.A.; Corrette, C.P. J. Org. Chem. 1999, 64, 9697.
1005. Hatakeyama, S. Pure Appl. Chem. 2009, 81, 217.
1006. Corkey, B.K.; Toste, F.D. J. Am. Chem. Soc. 2005, 127, 17168.
1007. Hilt, G.; Treutwein, J. Angew. Chem. Int. Ed. 2007, 46, 8500.
1008. Michelet, V.; Galland, J.-C.; Charruault, L.; Savignac, M.; Genêt, J.-P. Org. Lett. 2001, 3, 2065.
1009. Kennedy-Smith, J.J.; Staben, S.T.; Toste, F.D. J. Am. Chem. Soc. 2004, 126, 4526.
1010. Cheng, D.; Zhu, S.; Yu, Z.; Cohen, T. J. Am. Chem. Soc. 2001, 123, 30.
1011. See Snider, B.B.; Ron E. J. Am. Chem. Soc. 1985, 107, 8160.
1012. Shibata, T.; Yamasaki, M.; Kadowaki, S.; Takagi, K. Synlett 2004, 2812.
1013. Jayanth, T.T.; Jeganmohan, M.; Cheng, M.-J.; Chu, S.-Y.; Cheng, C.-H. J. Am. Chem. Soc. 2006, 128, 2232.
1014. See Terada, M.; Machioka, K.; Sorimachi, K. Angew. Chem. Int. Ed. 2006, 45, 2254.
1015. Terada, M.; Machioka, K.; Sorimachi, K. J. Am. Chem. Soc. 2007, 129, 10336.
1016. See Pandey, M.K.; Bisai, A.; Pandey, A.; Singh, V.K. Tetrahedron Lett. 2005, 46, 5039.
1017. For a review, see Clarke, M.L.; France, M.B. Tetrahedron 2008, 64, 9003.
1018. See Achmatowicz, O.; Bialeck-Florjanczyk, E. Tetrahedron 1996, 52, 8827; Marshall, J.A.; Andersen, M.W. J. Org. Chem. 1992, 57, 5851 for mechanistic discussions of this reaction.
1019. See Aggarwal, V.K.; Vennall, G.P.; Davey, P.N.; Newman, C. Tetrahedron Lett. 1998, 39, 1997.
1020. Ruck, R.T.; Jacobsen, E.N. J. Am. Chem. Soc. 2002, 124, 2882.
1021. Grachan,, M.L.; Tudge, M.T.; Jacobsen, E.N. Angew. Chem. Int. Ed. 2008, 47, 1469.
1022. Evans, D.A.; Wu, J. J. Am. Chem. Soc. 2005, 127, 8006.
1023. Zhao, J.-F.; Tsui, H.-Y.; Wu, P.-J.; Lu, J.; Loh, T.-P. J. Am. Chem. Soc. 2008, 130, 16492.
1024. Zheng, K.; Shi, J.; Liu, X.; Feng, X. J. Am. Chem. Soc. 2008, 130, 15770.
1025. Mikami, K.; Kawakami, Y.; Akiyama, K.; Aikawa, K. J. Am. Chem. Soc. 2007, 129, 12950.
1026. Dauben, W.G.; Hendricks, R.T. Tetrahedron Lett. 1992, 33, 603.
1027. Yamanaka, M.; Nishida, A.; Nakagawa, M.; Org. Lett. 2000, 2, 159.
1028. See Yu, Z.-X.; Houk, K.N. J. Am. Chem. Soc. 2003, 125, 13825.
1029. Lu, X. Org. Lett. 2004, 6, 2813. See also, Leach, A.G.; Houk, K.N. J. Am. Chem. Soc. 2002, 124, 14820; Adam, W.; Krebs, O. Chem. Rev. 2003, 103, 4131.
1030. Hansen, E.C.; Lee, D. J. Am. Chem. Soc. 2005, 127, 3252.
1031. See Myers, M.C.; Bharadwaj, A.R.; Milgram, B.C.; Scheidt, K.A. J. Am. Chem. Soc. 2005, 127, 14675.
1032. See Yanovskaya, L.A.; Kryshtal, G.V.; Kulganek, V.V. Russ. Chem. Rev. 1984, 53, 744; Bergmann, E.D.; Ginsburg, D.; Pappo, R. Org. React. 1959, 10, 179;. For a review of α-substitution versus conjugate addition, see Lewandowska, E. Tetrahedron 2007, 63, 2107.
1033. Willis, M.C.; McNally, S.J.; Beswick, P.J. Angew. Chem. Int. Ed. 2004, 43, 340; Chi, Y.; Gellman, S.H. Org. Lett. 2005, 7, 4253.
1034. Andrey, O.; Alexakis, A.; Bernardinelli, G. Org. Lett. 2003, 5, 2559; Harada, S.; Kumagai, N.; Kinoshita, T.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 2582.
1035. Kim, S.-G.; Ahn, K.H. Tetrahedron Lett. 2001, 42, 4175.
1036. Halland, N.; Aburel, P.S.; J![]() rgensen, K.A. Angew. Chem. Int. Ed. 2003, 42, 661.
rgensen, K.A. Angew. Chem. Int. Ed. 2003, 42, 661.
1037. da silva, F.M.; Gomes, A.K.; Jones Jr., J. Can. J. Chem. 1999, 77, 624.
1038. Suzuki, T.; Torii, T. Tetrahedron Asymmetry 2001, 12, 1077; García-Gómez, G.; Moretó, J.M. Eur. J. Org. Chem. 2001, 1359; Kobayashi, S.; Kakumoto, K.; Mori, Y.; Manabe, K. Isr. J. Chem. 2001, 41, 247.
1039. Méou, A.; Lamarque, L.; Brun, P. Tetrahedron Lett. 2002, 43, 5301.
1040. Wolckenhauer, S.A.; Rychnovsky, S.D. Org. Lett. 2004, 6, 2745; Fleming, F.F.; Wang, Q. Chem. Rev. 2003, 103, 2035.
1041. Zhu, Q.; Lu, Y. Org. Lett. 2008, 10, 4803.
1042. Ooi, T.; Fujioka, S.; Maruoka, K. J. Am. Chem. Soc. 2004, 126, 11790. See Strzalko, T.; Seyden-Penne, J.; Wartski, L.; Froment, F.; Corset, J. Tetrahedron Lett. 1994, 35, 3935.
1043. Taylor, M.S.; Jacobsen, E.N. J. Am. Chem. Soc. 2003, 125, 11204.
1044. Gimbert, C.; Lumbierres, M.; Marchi, C.; Moreno-Mañas, M.; Sebastián, R.M.; Vallribera, A. Tetrahedron 2005, 61, 8598.
1045. Alma![]() i, D.; Alonso, D.A.; Nájera, C. Tetrahedron Asymmetry 2007, 18, 299; Vicario, J.L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065.
i, D.; Alonso, D.A.; Nájera, C. Tetrahedron Asymmetry 2007, 18, 299; Vicario, J.L.; Badía, D.; Carrillo, L. Synthesis 2007, 2065.
1046. Chen, X.; She, J.; Shang, Z.; Wu, J.; Zhang, P. Synthesis 2008, 3931.
1047. Holeman, D.S.; Rasne, R.M.; Grossman, R.B. J. Org. Chem. 2002, 67, 3149.
1048. Ballini, R.; Bosica, G.; Fiorini, D. Tetrahedron Lett. 2001, 42, 8471.
1049. Bernardi, L.; López-Cantarero, J.; Niess, B.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2007, 129, 5772.
rgensen, K.A. J. Am. Chem. Soc. 2007, 129, 5772.
1050. Oi, S.; Taira, A.; Honma, Y.; Sato, T.; Inoue, Y. Tetrahedron Asymmetry 2006, 17, 598.
1051. Ballini, R.; Bosica, G.; Fiorini, D.; Palmieri, A.; Petrini, M. Chem. Rev. 2005, 105, 933; Vakulya, B.; Varga, S.; Soós, T. J. Org. Chem. 2008, 73, 3475; Prieto, A.; Halland, N.; J![]() rgensen, K.A. Org. Lett. 2005, 7, 3897.
rgensen, K.A. Org. Lett. 2005, 7, 3897.
1052. See Yoshikoshi, A.; Miyashita, M. Acc. Chem. Res. 1985, 18, 284; Baer, H.H.; UrBas L. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 2, Wiley, NY, 1970, pp. 130–148. See Thayumanavan, R.; Tanaka, F.; Barbas, III, C.F. Org. Lett. 2004, 6, 2527; Ishii, T.; Fujioka, S.; Sekiguchi, Y.; Kotsuki, H. J. Am. Chem. Soc. 2004, 126, 9558; Li, H.; Wang, Y.; Tang, L.; Deng, L. J. Am. Chem. Soc. 2004, 126, 9906; Watanabe, M.; Ikagawa, A.; Wang, H.; Murata, K.; Ikariya, T. J. Am. Chem. Soc. 2004, 126, 11148.
1053. Okino, T.; Hoashi, Y.; Furukawa, T.; Xu, X.; Takemoto, Y. J. Am. Chem. Soc. 2005, 127, 119.
1054. Evans, D.A.; Seidel, D. J. Am. Chem. Soc. 2005, 127, 9958; Evans, D.A.; Mito, S.; Seidel, D. J. Am. Chem. Soc. 2007, 129, 11583.
1055. Terada, M.; Ube, H.; Yaguchi, Y. J. Am. Chem. Soc. 2006, 128, 1454; Xu, D.-Q.; Wang, B.T.; Luo, S.-P.; Yue, H.-D.; Wang, L.-P.; Xu, Z.-Y. Tetrahedron Asymmetry 2007, 18, 1788; Wu, L.-Y.; Yan, Z.-Y.; Xie, Y.-X.; Niu, Y.-N.; Liang, Y.-M. Tetrahedron Asymmetry 2007, 18, 2086; Luo, S.; Mi, X.; Zhang, L.; Liu, S.; Xu, H.; Cheng, J.-P. Angew. Chem. Int. Ed. 2006, 45, 3093.
1056. See Mattson, A.E.; Zuhl, A.M.; Reynolds, T.E.; Scheidt, K.A. J. Am. Chem. Soc. 2006, 128, 4932; Mase, N.; Watanabe, K.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas III, C.F. J. Am. Chem. Soc. 2006, 128, 4966; Huang, H.; Jacobsen, E.N. J. Am. Chem. Soc. 2006, 128, 7170; Pansare, S.V.; Pandya, K. J. Am. Chem. Soc. 2006, 128, 9624; Chi, Y.; Guo, L.; Kopf, N.A.; Gellman, S.H. J. Am. Chem. Soc. 2008, 130, 5608; Wiesner, M.; Revell, J.D.; Tonazzi, S.; Wennemers, H. J. Am. Chem. Soc. 2008, 130, 5610; Malerich, J.P.; Hagihara, K.; Rawal, V.H. J. Am. Chem. Soc. 2008, 130, 14416.
1057. Zhang, Z.; Dong, Y.-W.; Wang, G.-W.; Komatsu, K. Synlett 2004, 61.
1058. Yadav, J.S.; Geetha, V.; Reddy, B.V.S. Synth. Commun. 2002, 32, 3519.
1059. Li, H.; Song, J.; Liu, X.; Deng, L. J. Am. Chem. Soc. 2005, 127, 8948.
1060. See Oare, D.A.; Heathcock, C.H. Top. Stereochem. 1989, 19, 227, pp. 232–236.
1061. See, however, Yamaguchi, M.; Yokota, N.; Minami, T. J. Chem. Soc., Chem. Commun. 1991, 1088.
1062. See Oare, D.A.; Heathcock, C.H. Top. Stereochem. 1991, 20, 87; 1989, 19, 227.
1063. Kim, D.Y.; Huh, S.C. Tetrahedron 2001, 57, 8933.
1064. Kotrusz, P.; Toma, S.; Schamlz, H.-G.; Adler, A. Eur. J. Org. Chem. 2004, 1577.
1065. Bartoli, G.; Bosco, M.; Bellucci, M.C.; Marcantoni, E.; Sambri, L.; Torregiani, E. Eur. J. Org. Chem. 1999, 617.
1066. Ding, R.; Katebzadeh, K.; Roman, L.; Bergquist, K.-E.; Lindström, U.M. J. Org. Chem. 2006, 71, 352.
1067. Varala, R.; Alam, M.M.; Adapa, S.R. Synlett 2003, 720.
1068. For a review, see Christoffers, J. Synlett 2001, 723.
1069. For a discussion of Heck coupling versus Michael addition, see Lin, P.-S.; Jeganmohan, M.; Cheng, C.-H. Chemistry: Asian J. 2007, 2, 1409.
1070. Iguchi, Y.; Itooka, R.; Miyaura, N. Synlett 2003, 1040; Meyer, O.; Becht, J.-M.; Helmchen, G. Synlett 2003, 1539. For a discussion of the mechanism, see Comelles, J.; Moreno-Mañas, M.; Pérez, E.; Roglans, A.; Sebastián, R.M.; Vallribera, A. J. Org. Chem. 2004, 69, 6834.
1071. Kim, Y.S.; Matsunaga, S.; Das, J.; Sekine, A.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2000, 122, 6506.
1072. Watanabe, M.; Murata, K.; Ikariya, T. J. Am. Chem. Soc. 2003, 125, 7508; Wadsworth, K.J.; Wood, F.K.; Chapman, C.J.; Frost, C.G. Synlett 2004, 2022; Hayashi, T.; Yamasaki, K. Chem. Rev. 2003, 103, 2829.
1073. Mori, Y.; Kakumoto, K.; Manabe, K.; Kobayashi, S. Tetahedron Lett. 2000, 41, 3107.
1074. Sreekumar, R.; Rugmini, P.; Padmakumar, R. Tetrahedron Lett. 1997, 38, 6557.
1075. Lubineau, A.; Augé, J. Tetrahedron Lett. 1992, 33, 8073.
1076. Nahm, M.R.; Potnick, J.R.; White, P.S.; Johnson, J.S. J. Am. Chem. Soc. 2006, 128, 2751.
1077. Vo, N.T.; Pace, R.D.M.; O'Hara, F.; Gaunt, M.J. J. Am. Chem. Soc. 2008, 130, 404.
1078. Masson, G.; Cividino, P.; Py, S.; Vallée, Y. Angew. Chem. Int. Ed. 2003, 42, 2265.
1079. Yang, J.; Wang, Y.; Wu, S.; Chen, F.-X. Synlett 2009, 3365; Yang, J.; Shen, Y.; Chen, F.-X. Synthesis 2010, 1325.
1080. Fonseca, M.T.H.; List, B. Angew. Chem. Int. Ed. 2004, 43, 3958.
1081. Bremeyer, N.; Smith, S.C.; Ley, S.V.; Gaunt, M.J. Angew. Chem. Int. Ed. 2004, 43, 2681.
1082. Nazarov, I.N.; Torgov, I.B.; Terekhova, L.N. Izv. Akad. Nauk. SSSR otd. Khim. Nauk,1942, 200; Pellissier, H. Tetrahedron 2005, 61, 6479; Frontier, A.J.; Collison, C. Tetrahedron 2005, 61, 7577; Tius, M.A. Eur. J. Org. Chem. 2005, 2193. See Smith, D.A.; Ulmer II, C.W. J. Org. Chem. 1993, 58, 4118 for a discussion of torquoselectivity and hyperconjugation in this reaction.
1083. Marcus, A.P.; Amy, S.; Lee, A.S.; Davis, R.L.; Tantillo, D.J.; Sarpong, R. Angew. Chem. Int. Ed. 2008, 47, 6379.
1084. Cavalli, A.; Pacetti, A.; Recanatini, M.; Prandi, C.; Scarpi, D.; Occhiato, E.G. Chemistry: European J. 2008, 14, 9292.
1085. Zhang, L.; Wang, S. J. Am. Chem. Soc. 2006, 128, 1442.
1086. Walz, I.; Bertogg, A.; Togni, A. Eur. J. Org. Chem. 2007, 2650.
1087. Kokubo, M.; Kobayashi, S. Chemistry: Asian J. 2009, 4, 526.
1088. Douelle, F.; Tal, L.; Greaney, M.F. Chem. Commun. 2005, 660.
1089. Banaag, A.R.; Tius, M.A. J. Am. Chem. Soc. 2007, 129, 5328; Banaag, A.R.; Tius, M.A. J. Org. Chem. 2008, 73,8133.
1090. Rieder, C.J.; Winberg, K.J.; West, F.G. J. Am. Chem. Soc. 2009, 131, 7504.
1091. Aggarwal, V.K.; Belfield, A.J. Org. Lett. 2003, 5, 5075.
1092. Giese, S.; West, F.G. Tetrahedron Lett. 1998, 39, 8393; Giese, S.; West, F.G. Tetrahedron 2000, 56, 10221.
1093. Harmata, M.; Lee, D.R. J. Am. Chem. Soc. 2002, 124, 14328. For a discussion of the scope and mechanism of the retro-Nazarov reaction, see Harmata, M.; Schreiner, P.R.; Lee, D.R.; Kirchhoefer, P.L. J. Am. Chem. Soc.2004, 126, 10954. For a torquoselective retro-Nazarov, see Harmata, M.; Lee, D.R.; Barnes, C.L. Org. Lett. 2005, 7, 1881.
1094. Magomedev, N.A.; Ruggiero, P.L.; Tang, Y. Org. Lett. 2004, 6, 3373.
1095. Dhoro, F.; Tius, M.A. J. Am. Chem. Soc. 2005, 127, 12472; Dhoro, F.; Kristensen, T.E.; Stockmann, V.; Yap, G.P.A.; Tius, M.A. J. Am. Chem. Soc. 2007, 129, 7256. See also, Grant, T.N.; Rieder, C.J.; West, F.G. Chem. Commun. 2009, 5676.
1096. Huang, J.; Frontier, A.J. J. Am. Chem. Soc. 2007, 129, 8060.
1097. See Oare, D.A.; Heathcock, C.H. Top. Stereochem. 1989, 19, pp. 237.
1098. See Töke, L.; Fenichel, L.; Albert, M. Tetrahedron Lett. 1995, 36, 5951; Enders, D.; Demir, A.S.; Rendenbach, B.E.M. Chem. Ber. 1987, 120, 1731; Hawkins, J.M.; Lewis, T.A. J. Org. Chem. 1992, 57, 2114.
1099. See Krause, N.; Hoffmann-Röder, A. Synthesis 2001, 171.
1100. Desimoni, G.; Quadrelli, P.; Righetti, P.P. Tetrahedron 1990, 46, 2927.
1101. See d'Angelo, J.; Revial, G.; Volpe, T.; Pfau, M. Tetrahedron Lett. 1988, 29, 4427.
1102. Guo, R.; Morris, R.H.; Song, D. J. Am. Chem. Soc. 2005, 127, 516.
1103. Evans, D.A.; Thomson, R.J.; Franco, F. J. Am. Chem. Soc. 2005, 127, 10816.
1104. Agostinho, M.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 2430.
1105. Taylor, M.S.; Zalatan, D.N.; Lerchner, A.M.; Jacobsen, E.N. J. Am. Chem. Soc. 2005, 127, 1313.
1106. Hayashi, Y.; Gotoh, H.; Tamura, T.; Yamaguchi, H.; Masui, R.; Shoji, M. J. Am. Chem. Soc. 2005, 127, 16028; Wang, J.; Li, H.; Zu, L.; Jiang, W.; Xie, H.; Duan, W.; Wang, W. J. Am. Chem. Soc. 2006, 128, 12652; Bartoli, G.; Bosco, M.; Carlone, A.; Cavalli, A.; Locatelli, M.; Mazzanti, A.; Ricci, P.; Sambri, L.; Melchiorre, P. Angew. Chem. Int. Ed. 2006, 45, 4966; Palomo, C.; Landa, A.; Mielgo, A.; Oiarbide, M.; Puente, Á.; Vera, S. Angew. Chem. Int. Ed. 2007, 46, 8431.
1107. Peelen, T.J.; Chi, Y.; Gellman, S.H. J. Am. Chem. Soc. 2005, 127, 11598.
1108. d'Angelo, J.; Desmaële, D.; Dumas, F.; Guingant, A. Tetrahedron Asymmetry 1992, 3, 459.
1109. Weinstain, R.; Lerner, R.A.; Barbas, III, C.F.; Shabat, D. J. Am. Chem. Soc. 2005, 127, 13104.
1110. Svedendahl, M.; Hult, K.; Berglund, P. J. Am. Chem. Soc. 2005, 127, 17988.
1111. Jha, S.C.; Joshi, N.N. Tetrahedron Asymmetry 2001, 12, 2463.
1112. Yamaguchi, M.; Shiraishi, T.; Hirama, M. J. Org. Chem. 1996, 61, 3520.
1113. Xu, F.; Tillyer, R.D.; Tschaen, D.M.; Grabowski, E.J.J.; Reider, P.J. Tetrahedron Assymetry 1998, 9, 1651.
1114. Mirza-Aghayan, M.; Etemad-Moghadam, G.; Zaparucha, A.; Berlan, J.; Loupy, A.; Koenig, M. Tetrahedron Asymmetry 1995, 6, 2643.
1115. See Oare, D.A.; Heathcock, C.H. J. Org. Chem. 1990, 55, 157.
1116. Holton, R.A.; Williams, A.D.; Kennedy, R.M. J. Org. Chem. 1986, 51, 5480.
1117. See Hajos, Z.G.; Parrish, D.R. J. Org. Chem. 1974, 39, 1612; Org. Synth. VII, 363.
1118. Bertrand, S.; Glapski, C.; Hoffmann, N.; Pete, J.-P. Tetrahedron Lett. 1999, 40, 3169.
1119. Sibi, M.P.; Jasperse, C.P.; Ji, J. J. Am. Chem. Soc. 1995, 117, 10779. See Wu, J.H.; Radinov, R.; Porter, N.A. J. Am. Chem. Soc. 1995, 117, 11029 for a related reaction.
1120. Enholm, E.J.; Kinter, K.S. J. Org. Chem. 1995, 60, 4850.
1121. Rudorf, W.-D.; Schwarz, R. Synlett 1993, 369.
1122. Yazaki, R.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2010, 132, 10275.
1123. See Makosza, M. Tetrahedron Lett. 1966, 5489.
1124. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1576–1582; Mukaiyama, T.; Kobayashi, S. J. Organomet. Chem. 1990, 382, 39.
1125. Matsuda, I. J. Organomet. Chem. 1987, 321, 307; Narasaka, K. Org. Synth,. 65, 12. See also, Yoshikoshi, A.; Miyashita, M. Acc. Chem. Res. 1985, 18, 284.
1126. Loh, T.-P.; Wei, L.-L. Tetrahedron 1998, 54, 7615.
1127. Tucker, J.A.; Clayton, T.L.; Mordas, D.M. J. Org. Chem. 1997, 62, 4370.
1128. Kabbara, J.; Flemming, S.; Nickisch, K.; Neh, H.; Westermann, J. Tetrahedron 1995, 51, 743.
1129. Ranu, B.C.; Saha, M.; Bhar, S. Tetrahedron Lett. 1993, 34, 1989.
1130. Yasuda, M.; Chiba, K.; Ohigashi, N.; Katoh, Y.; Baba, A. J. Am. Chem. Soc. 2003, 125, 7291.
1131. See Heathcock, C.H.; Uehling, D.E. J. Org. Chem. 1986, 51, 279; Mukaiyama, T.; Tamura, M.; Kobayashi, S. Chem. Lett. 1986, 1017, 1817, 1821; 1987, 743.
1132. Harada, T.; Adachi, S.; Wang, X. Org. Lett. 2004, 6, 4877.
1133. Anderson, R.J.; Corbin, V.L.; Cotterrell, G.; Cox, G.R.; Henrick, C.A.; Schaub, F.; Siddall, J.B. J. Am. Chem. Soc. 1975, 97, 1197.
1134. Alexakis, A.; Chuit, C.; Commerçon-Bourgain, M.; Foulon, J.P.; Jabri, N.; Mangeney, P.; Normant, J.F. Pure Appl. Chem. 1984, 56, 91.
1135. For reactions with conjugated enynes, see Miginiac, L. J. Organomet. Chem. 1982, 238, 235.
1136. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1599–1613, 1814–1824; Posner, G.H. Org. React. 1972, 19, 1; House, H.O. Acc. Chem. Res. 1976, 9, 59; Yamanaka, M.; Kato, S.; Nakamura, E. J. Am. Chem. Soc. 2004, 126, 6287; Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, NY, 1980, pp. 10–67.
1137. See Nagashima, H.; Ozaki, N.; Washiyama, M.; Itoh, K. Tetrahedron Lett. 1985, 26, 657.
1138. Bennabi, S.; Narkunan, K.; Rousset, L.; Bouchu, D.; Ciufolini, M.A. Tetrahedron Lett. 2000, 41, 8873.
1139. Matsuza, S.; Horiguchi, Y.; Nakamura, E.; Kuwajima, I. Tetrahedron 1989, 45, 349; Horiguchi, Y.; Komatsu, M.; Kuwajima, I. Tetrahedron Lett. 1989, 30, 7087; Linderman, R.J.; McKenzie, J.R. J. Organomet. Chem. 1989, 361, 31; Bertz, S.H.; Smith, R.A.J. Tetrahedron 1990, 46, 4091. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1491–1492.
1140. Lipshutz, B.H.; Ellsworth, E.L.; Dimock, S.H.; Smith, R.A.J. J. Am. Chem. Soc. 1990, 112, 4404; Lipshutz, B.H.; James, B. Tetrahedron Lett. 1993, 34, 6689.
1141. Degl'Innocenti, A.; Stucchi, E.; Capperucci, A.; Mordini, A.; Reginato, G.; Ricci, A. Synlett 1992, 329, 332.
1142. See also Yangand, J.; Dudley, G.B. Tetrahedron Lett. 2007, 48, 7887.
1143. Henze, W.; Vyater, A.; Krause, N.; Gschwind, R.M. J. Am. Chem. Soc. 2005, 127, 17335.
1144. Murphy, M.D.; Ogle, C.A.; Bertz, S.H. Chem. Commun. 2005, 854.
1145. Corey, E.J.; Floyd, D.; Lipshutz, B.H. J. Org. Chem. 1978, 43, 3419.
1146. Posner, G.H.; Whitten, C.E. Tetrahedron Lett. 1973, 1815.
1147. Posner, G.H.; Whitten, C.E.; Sterling, J.J. J. Am. Chem. Soc. 1973, 95, 7788.
1148. See Ledlie, D.B.; Miller, G. J. Org. Chem. 1979, 44, 1006.
1149. See Lipshutz, B.H. Tetrahedron Lett. 1983, 24, 127.
1150. See Lipshutz, B.H.; Wilhelm, R.S.; Kozlowski, J.A. J. Org. Chem. 1984, 49, 3938.
1151. Zinn, F.K.; Ramos, E.C.; Comasseto, J.V. Tetahedron Lett. 2001, 42, 2415.
1152. Yamanaka, M.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 4697.
1153. See also, Kireev, A.S.; Manpadi, M.; Kornienko, A. J. Org. Chem. 2006, 71, 2630.
1154. Bertz, S.H.; Cope, S.; Murphy, M.; Ogle, C.A.; Taylor, B.J. J. Am. Chem. Soc. 2007, 129, 7208. See Hu, H.; Snyder, J.P. J. Am. Chem. Soc. 2007, 129, 7210.
1155. Charonnat, J.A.; Mitchell, A.L.; Keogh, B.P. Tetrahedron Lett. 1990, 31, 315.
1156. House, H.O.; Fischer Jr., W.F. J. Org. Chem. 1969, 34, 3615. See also, Daviaud, G.; Miginiac, P. Tetrahedron Lett. 1973, 3345.
1157. Domínguez, E.; Carretero, J.C. Tetrahedron Lett. 1993, 34, 5803
1158. House, H.O.; Umen, M.J. J. Org. Chem. 1973, 38, 3893.
1159. Truce, W.E.; Lusch, M.J. J. Org. Chem. 1974, 39, 3174; 1978, 43, 2252.
1160. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 456–457.
1161. Lee, P.H.; Park, J.; Lee, K.; Kim, H.-C. Tetrahedron Lett. 1999, 40, 7109.
1162. Lipshutz, B.H.; Ellsworth, E.L.; Siahaan, T.J. J. Am. Chem. Soc. 1988, 110, 4834; 1989, 111, 1351.
1163. Alexander, C.W.; Nice, L.E. Tetrahedron 2000, 56, 2767; Dieter, R.K.; Lu, K.; Velu, S.E. J. Org. Chem. 2000, 65, 8715. See Dieter, R.K.; Topping, C.M.; Nice, L.E. J. Org. Chem. 2001, 66, 2302.
1164. Yamamoto, Y.; Asao, N.; Uyehara, T. J. Am. Chem. Soc. 1992, 114, 5427.
1165. Chapdelaine, M.J.; Hulce, M. Org. React. 1990, 38, 225; Taylor, R.J.K. Synthesis 1985, 364; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1609–1612, 1826.
1166. Coates, R.M.; Sandefur, L.O. J. Org. Chem. 1974, 39, 275; Posner, G.H.; Lentz, C.M. Tetrahedron Lett. 1977, 3215.
1167. Page, P.C.B.; Prodger, J.C.; Hursthouse, M.B.; Mazid, M. J. Chem. Soc. Perkin Trans. 1,1990, 167; Corey, E.J.; Hannon, F.J. Tetrahedron Lett. 1990, 31, 1393.
1168. See Posner, G.H. Acc. Chem. Res. 1987, 20, 72; The articles by Tomioka, K.; Koga, K. in Morrison, J.D. Assymmetric Synthesis Vol. 2, Academic Press, NY, 1983, pp. 201–224; Posner, G. pp. 225–241.
1169. Christenson, B.; Ullenius, C.; Håkansson, M.; Jagner, S. Tetrahedron 1992, 48, 3623.
1170. Wu, J.; Mampreian, D.M.; Hoveyda, A.H. J. Am. Chem. Soc. 2005, 127, 4584; Knöpfel, T.F.; Zarotti, P.; Ichikawa, T.; Carreira, E.M. J. Am. Chem. Soc. 2005, 127, 9682; Fillion, E.; Wilsily, A. J. Am. Chem. Soc. 2006, 128, 2774; De Roma, A.; Ruffo, F.; Woodward, S. Chem. Commun. 2008, 5384.
1171. Kanai, M.; Koga, K.; Tomioka, K. Tetrahedron Lett. 1992, 33, 7193.
1172. Jensen, K.B.; Thorhauge, J.; Hazell, R.G.; J![]() rgensen, K.A. Angew. Chem. Int. Ed. 2001, 40, 160.
rgensen, K.A. Angew. Chem. Int. Ed. 2001, 40, 160.
1173. d'Augustin, M.; Palais, L.; Alexakis, A. Angew. Chem. Int. Ed. 2005, 44, 1376.
1174. Li, K.; Alexakis, A. Angew. Chem. Int. Ed. 2006, 45, 7600.
1175. See Okamoto, M.; Yamamoto, Y.; Sakaguchi, S. Chem. Commun. 2009, 7363.
1176. Han, Y.; Hruby, V.J. Tetrahedron Lett. 1997, 38, 7317.
1177. Bongini, A.; Cardillo, G.; Mingardi, A.; Tomasini, C. Tetrahedron Asymmetry 1996, 7, 1457.
1178. Kung, L.-R.; Tu, C.-H.; Shia, K.-S.; Liu, H.-J. Chem. Commun. 2003, 2490.
1179. Fleming, F.F.; Wang, Q.; Zhang, Z.; Steward, O.W. J. Org. Chem. 2002, 67, 5953.
1180. See Negishi, E. Organometallics in Organic Synthesis Vol. 1, Wiley, NY, 1980, pp. 127–133.
1181. Posner, G.H. Org. React. 1972, 19, 1; López, F.; Harutyanyan, S.R.; Minnaard, A.J.; Feringa, B.L. J. Am. Chem. Soc. 2004, 126, 12784; Martin D.; Kehrli, S.; d'Augustin, M.; Clavier, H.; Mauduit, M.; Alexakis, A. J. Am. Chem. Soc. 2006, 128, 8416.
1182. Bartoli, G.; Bosco, M.; Sambri, L.; Marcantoni, E. Tetrahedron Lett. 1994, 35, 8651.
1183. See Kim, Y.M.; Kwon, T.W.; Chung, S.K.; Smith, M.B. Synth. Commun. 1999, 29, 343.
1184. Schneider, C.; Reese, O. Synthesis 2000, 1689.
1185. López, F.; Minnaard, A.J.; Feringa, B.L. Acc. Chem. Res. 2007, 40, 179; Harutyunyan, S.R.; den Hartog, T.; Geurts, K.; Minnaard, A.J.; Feringa, B.L. Chem. Rev. 2008, 108, 2824; Wang, S.-Y.; Ji, S.-J.; Loh, T.-P. J. Am. Chem. Soc. 2007, 129, 276; Matsumoto, Y.; Yamada, K.-i.; Tomioka, K. J. Org. Chem. 2008, 73, 4578.
1186. See Hunt, D.A. Org. Prep. Proced. Int. 1989, 21, 705–749.
1187. Cohen, T.; Abraham, W.D.; Myers, M. J. Am. Chem. Soc. 1987, 109, 7923.
1188. Cooke Jr., M.P. J. Org. Chem. 1986, 51, 1637.
1189. Roux, M.C.; Wartski, L.; Seyden-Penne, J. Tetrahedron 1981, 37, 1927; Synth. Commun. 1981, 11, 85.
1190. El-Bouz, M.; Wartski, L. Tetrahedron Lett. 1980, 21, 2897.
1191. Sikorski, W.H.; Reich, H.J. J. Am. Chem. Soc. 2001, 123, 6527.
1192. Lucchetti, J.; Dumont, W.; Krief, A. Tetrahedron Lett. 1979, 2695; El-Bouz, M.; Wartski, L. Tetrahedron Lett. 1980, 21, 2897. See also, Bürstinghaus, R.; Seebach, D. Chem. Ber. 1977, 110, 841.
1193. See Maezaki, N.; Sawamoto, H.; Yuyama, S.; Yoshigami, R.; Suzuki, T.; Izumi, M.; Ohishi, H.; Tanaka, T. J. Org. Chem. 2004, 69, 6335.
1194. Sparteine is used as a chiral additive, see Curtis, M.D.; Beak, P. J. Org. Chem. 1999, 64, 2996.
1195. Piers, E.; Harrison, C.L.; Zetina-Rocha, C. Org. Lett. 2001, 3, 3245.
1196. Kikuchi, M.; Niikura, S.; Chiba, N.; Terauchi, N.; Asaoka, M. Chem. Lett. 2007, 36, 736.
1197. Corey, E.J.; Boger, D.L. Tetrahedron Lett. 1978, 9. Also see Sato, T.; Okazaki, H.; Otera, J.; Nozaki, H. Tetrahedron Lett. 1988, 29, 2979.
1198. Santos, R.P.; Lopes, R.S.C.; Lopes, C.C. Synthesis 2001, 845.
1199. Takaya, Y.; Ogasawara, M.; Hayashi, T. Tetrahedron Lett. 1999, 40, 6957.
1200. Liao, W.-W.; Li, K.; Tang, Y. J. Am. Chem. Soc. 2003, 125, 13030.
1201. See Ikeda, S.-i.; Cui, D.-M.; Sato, Y. J. Am. Chem. Soc. 1999, 121, 4712.
1202. Liang, L.; Au-Yeung, T.T.-L.; Chan, A.S.C. Org. Lett. 2002, 4, 3799.
1203. Reetz, M.T.; Gosberg, A.; Moulin, D. Tetrahedron Lett. 2002, 43, 1189.
1204. Liang, L.; Yan, M.; Li, Y.-M.; Chan, A.S.C. Tetrahedron Asymmetry 2004, 15, 2575, and references cited therein; Alexakis, A.; Polet, D.; Rosset, S.; March, S. J. Org. Chem. 2004, 69, 5660, and references cited therein; Duncan, A.P.; Leighton, J.L. Org. Lett. 2004, 6, 4117, and references cited therein; Hajra, A.; Yoshikai, N.; Nakamura, E. Org. Lett. 2006, 8, 4153; Valleix, F.; Nagai, K.; Soeta, T.; Kuriyama, M.; Yamada, K.-i.; Tomioka, K. Tetrahedron 2005, 61, 7420.
1205. Hird, A.W.; Hoveyda, A.H. J. Am. Chem. Soc. 2005, 127, 14988.
1206. Takahashi, Y.; Yamamoto, Y.; Katagiri, K.; Danjo, H.; Yamaguchi, K.; Imamoto, T. J. Org. Chem. 2005, 70, 9009; Ito, K.; Eno, S.; Saito, B.; Katsuki, T. Tetrahedron Lett. 2005, 46, 3981. For the use of Pd or Cu catalysts, see Marshall, J.A.; Herold, M.; Eidam, H.S.; Eidam, P. Org. Lett. 2006, 8, 5505.
1207. Malhotra, S.V.; Wang, Y. Tetrahedron Asymmetry 2006, 17, 1032.
1208. Choi, H.; Hua, Z.; Ojima, I. Org. Lett. 2004, 6, 2689; Mampreian, D.M.; Hoveyda, A.H. Org. Lett. 2004, 6, 2829; Duursma, A.; Minnaard, A.J.; Feringa, B.L. J. Am. Chem. Soc. 2003, 125, 3700.
1209. Hayashi, T.; Yamamoto, S.; Tokunaga, N. Angew. Chem. Int. Ed. 2005, 44, 4224.
1210. Hirao, T.; Takada, T.; Sakurai, H. Org. Lett. 2000, 2, 3659; Yin, Y.; Li, X.; Lee, D.-S.; Yang, T.-K. Tetrahedron Asymmetry 2000, 11, 3329; Shadakshari, U.; Nayak, S.K. Tetrahedron 2001, 57, 8185.
1211. Shitani, R.; Tokunaga, N.; Doi, H.; Hayashi, T. J. Am. Chem. Soc. 2004, 126, 6240.
1212. Sarandeses, L.A.; Mouriño, A.; Luche, J.-L. J. Chem. Soc., Chem. Commun. 1992, 798. See also, Das, B.; Banerjee, J.; Mahender, G.; Majhi, A. Org. Lett. 2004, 6, 3349.
1213. Pétrier, C.; de Souza Barboza, J.C.; Dupuy, C.; Luche, J. J. Org. Chem. 1985, 50, 5761.
1214. Berger, S.; Langer, F.; Lutz, C.; Knochel, P.; Mobley, T.A.; Reddy, C.K. Angew. Chem. Int. Ed. 1997, 36, 1496.
1215. Knochel, P.; Normant, J.F. J. Organomet. Chem. 1986, 309, 1.
1216. Wang, C.-C.; Lin, P.-S.; Cheng, C.-H. J. Am. Chem. Soc. 2002, 124, 9696.
1217. Bagnell, L.; Meisters, A.; Mole, T. Aust. J. Chem. 1975, 28, 817; Ashby, E.C.; Heinsohn, G. J. Org. Chem. 1974, 39, 3297. See also, Kunz, H.; Pees, K.J. J. Chem. Soc. Perkin Trans. 1,1989, 1168.
1218. Su, L.; Li, X.; Chan, W.L.; Jia, X.; Chan, A.S.C. Tetrahedron Asymmetry 2003, 24, 1865.
1219. Koltunov, K.Yu.; Walspurger, S.; Sommer, J. Tetrahedron Lett. 2004, 45, 3547.
1220. Liu, J.-Y.,; Jang, Y.-J.; Lin, W.-W.; Liu, J.-T.; Yao, C.-F. J. Org. Chem. 2003, 68, 4030.
1221. In the presence of Mn, see Amatore, M.; Gosmini, C.; Périchon, J. J. Org. Chem. 2006, 71, 6130.
1222. Shen, Z.-L.; Cheong, H.-L.; Loh, T.-P. Tetrahedron Lett. 2009, 50, 1051.
1223. Nishimura, T.; Guo, X.-X.; Uchiyama, N.; Katoh, T.; Hayashi, T.J. Am. Chem. Soc. 2008, 130, 1576.
1224. Chen, L.; Li, C.-J. Chem. Commun. 2004, 2362.
1225. Herath, A.; Thompson, B.B.; Montgomery, J. J. Am. Chem. Soc. 2007, 129, 8712; Herath, A.; Montgomery, J. J. Am. Chem. Soc. 2008, 130, 8132.
1226. Hayashi, T.; Tokunaga, N.; Yoshida, K.; Han, J.W. J. Am. Chem. Soc. 2002, 124, 12102; Lerum, R.V.; Chisholm, J.D. Tetrahedron Lett. 2004, 45, 6591.
1227. Chen, Y.; Lee, C. J. Am. Chem. Soc. 2006, 128, 15598.
1228. Han, Y.; Huang, Y.-Z.; Fang, L.; Tao, W.-T. Synth. Commun. 1999, 29, 867.
1229. Oi, S.; Moro, M.; Ito, H.; Honma, Y.; Miyano, S.; Inoue, Y. Tetrahedron 2002, 58, 91.
1230. Ohe, T.; Uemura, S. Tetrahedron Lett. 2002, 43, 1269.
1231. Venkatraman, S.; Li, C.-J. Tetrahedron Lett. 2001, 42, 781.
1232. Nishikata, T.; Yamamoto, Y.; Miyaura, N. Chem. Commun. 2004, 1822.
1233. Williams, D.R.; Mullins, R.J.; Miller, N.A. Chem. Commun. 2003, 2220.
1234. Augé, J.; Gil, R.; Kalsey, S. Tetrahedron Lett. 1999, 40, 67.
1235. Condon, S.; Dupré, D.; Falgayrac, G.; Nédélec, J.-Y. Eur. J. Org. Chem. 2002, 105.
1236. Kakuuchi, A.; Taguchi, T.; Hanzawa, Y. Tetrahedron 2004, 60, 1293.
1237. This example is from Schmidlin, J.; Wohl, J. Ber. 1910, 43, 1145; Mosher, W.A.; Huber, M.B. J. Am. Chem. Soc. 1953, 75, 4604. See Fuson, R.C. Adv. Organomet. Chem. 1964, 1, 221.
1238. See Bartoli, G. Acc. Chem. Res. 1984, 17, 109; Bartoli, G.; Dalpozzo, R.; Grossi, L. J. Chem. Soc. Perkin Trans. 2,1989, 573. For a study of the mechanism, see Bartoli, G.; Bosco, M.; Cantagalli, G.; Dalpozzo, R.; Ciminale, F. J. Chem. Soc. Perkin Trans. 2,1985, 773.
1239. House, H.O.; Thompson, H.W. J. Org. Chem. 1963, 28, 360; Klein, J. Tetrahedron 1964, 20, 465. See, however, Marets, J.; Rivière, H. Bull. Soc. Chim. Fr. 1970, 4320.
1240. Kingsbury, C.L.; Smith, R.A.J. J. Org. Chem. 1997, 62, 4629. See, Bertz, S.H.; Miao, G.; Rossiter, B.E.; Snyder, J.P. J. Am. Chem. Soc. 1995, 117, 11023; Snyder, J.P. J. Am. Chem. Soc. 1995, 117, 11025.
1241. See Yamamoto, Y.; Yamada, J.; Uyehara, T. J. Am. Chem. Soc. 1987, 109, 5820; Ullenius, C.; Christenson, B. Pure Appl. Chem. 1988, 60, 57; Christenson, B.; Olsson, T.; Ullenius, C. Tetrahedron 1989, 45, 523; Krause, N. Tetrahedron Lett. 1989, 30, 5219.
1242. See Wigal, C.T.; Grunwell, J.R.; Hershberger, J. J. Org. Chem. 1991, 56, 3759.
1243. Whitesides, G.M.; Kendall, P.E. J. Org. Chem. 1972, 37, 3718.
1244. Corey, E.J.; Hannon, F.J.; Boaz, N.W. Tetrahedron 1989, 45, 545.
1245. Bertz, S.H.; Smith, R.A.J. J. Am. Chem. Soc. 1989, 111, 8276.
1246. Sakurai, H.; Hosomi, A.; Hayashi, J. Org. Synth. VII, 443; Kuhnert, N.; Peverley, J.; Robertson, J. Tetrahedron Lett. 1998, 39, 3215; Fleming, I.; Dunoguès, J.; Smithers, R. Org. React. 1989, 37, 57, see p. 127, 335–370; Schinzer, D. Synthesis 1988, 263.
1247. Majetich, G.; Casares, A.; Chapman, D.; Behnke, M. J. Org. Chem. 1986, 51, 1745.
1248. See Lee, P.H.; Lee, K.; Sung, S.-y.; Chang, S. J. Org. Chem. 2001, 66, 8646.
1249. Denmark, S.E.; Amishiro, N. J. Org. Chem. 2003, 68, 6997.
1250. Oi, S.; Taira, A.; Honma, Y.; Inoue, Y. Org. Lett. 2003, 5, 97.
1251. Wadamoto, M.; Yamamoto, H. J. Am. Chem. Soc. 2005, 127, 14556.
1252. Huang, T.-S.; Li, C.-J. Chem. Commun. 2001, 2348.
1253. Koike, T.; Du, X.; Mori, A.; Osakada, K. Synlett 2002, 301.
1254. Pospíšil, J.; Kumamoto, T.; Markó, I.E. Angew. Chem. Int. Ed. 2006, 45, 3357.
1255. Köster, R.; Zimmermann, H.; Fenzl, W. Liebigs Ann. Chem. 1976, 1116. See Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 301–305, 318–323; Brown, H.C. Boranes in Organic Chemistry Cornell University Press, Ithica, NY, 1972, pp. 413–433.
1256. Brown, H.C.; Kabalka, G.W. J. Am. Chem. Soc. 1970, 92, 712, 714. See also, Miyaura, N.; Kashiwagi, M.; Itoh, M.; Suzuki, A. Chem. Lett. 1974, 395.
1257. Brown, H.C.; Negishi, E. J. Am. Chem. Soc. 1971, 93, 3777.
1258. Brown, H.C.; Bhat, N.G.; Rajagopalan, S. Organometallics 1986, 5, 816.
1259. Satoh, Y.; Serizawa, H.; Hara, S.; Suzuki, A. J. Am. Chem. Soc. 1985, 107, 5225. See also, Hara, S.; Hyuga, S.; Aoyama, M.; Sato, M.; Suzuki, A. Tetrahedron Lett. 1990, 31, 247.
1260. Sieber, J.D.; Liu, S.; Morken, J.P. J. Am. Chem. Soc. 2007, 129, 2214.
1261. Hirano, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2007, 9, 1541.
1262. Sinclair, J.A.; Molander, G.A.; Brown, H.C. J. Am. Chem. Soc. 1977, 99, 954. See also, Molander, G.A.; Brown, H.C. J. Org. Chem. 1977, 42, 3106.
1263. Takaya, Y.; Ogasawara, M.; Hayashi, T. Tetrahedron Lett. 1998, 39, 8479.
1264. Fujishima, H.; Takada, E.; Hara, S.; Suzuki, A. Chem. Lett. 1992, 695.
1265. Kabalka, G.W.; Das, B.C.; Das, S. Tetrahedron Lett. 2002, 43, 2323.
1266. Zeng, H.; Hua, R. J. Org. Chem. 2008, 73, 558.
1267. Wu, T.R.; Chong, J.M. J. Am. Chem. Soc. 2005, 127, 3244; Pellegrinet, S.C.; Goodman, J.M. J. Am. Chem. Soc. 2006, 128, 3116.
1268. Chong, J.M.; Shen, L.; Taylor, N.J. J. Am. Chem. Soc. 2000, 122, 1822.
1269. Paquin, J.-F.; Defieber, C.; Stephenson, C.R.J.; Carreira, E.M. J. Am. Chem. Soc. 2005, 127, 10850; Shintani, R.; Duan, W.-L.; Hayashi, T. J. Am. Chem. Soc. 2006, 128, 5628; Duan, W.-L.; Iwamura, H.; Shintani, R.; Hayashi, T. J. Am. Chem. Soc. 2007, 129, 2130; Mariz, R.; Luan, X.; Gatti, M.; Linden, A.; Dorta, R. J. Am. Chem. Soc. 2008, 130 2172; Otomaru, Y.; Okamoto, K.; Shintani, R.; Hayashi, T. J. Org. Chem. 2005, 70, 2503; Stemmler, R.T.; Bolm, C. J. Org. Chem. 2005, 70, 9925; Paquin, J.-F.; Stephenson, C.R.J.; Defieber, C.; Carreira, E.M. Org. Lett. 2005, 7, 3821; Martina, S.L.X.; Minnaard, A.J.; Hessen, B.; Feringa, B.L. Tetrahedron Lett. 2005, 46, 7159.
1270. Lu, X.; Lin, S. J. Org. Chem. 2005, 70, 9651.
1271. Sakuma, S.; Miyaura, N. J. Org. Chem. 2001, 66, 8944.
1272. Dong, L.; Xu, Y.-J.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z. Synthesis 2004, 1057.
1273. With a chiral ligand, see Mauleón, P.; Carretero, J.C. Org. Lett. 2004, 6, 3195.
1274. Wu, T.R.; Chong, J.M. J. Am. Chem. Soc. 2007, 129, 4908.
1275. Nishimura, T.; Yasuhara, Y.; Hayashi, T. Angew. Chem. Int. Ed. 2006, 45, 5164.
1276. Yamamoto, Y.; Kirai, N.; Harada, Y. Chem. Commun. 2008, 2010.
1277. Sugiura, M.; Tokudomi, M.; Nakajima, M. Chem. Commun. 2010, 7799.
1278. Duursma, A.; Boiteau, J.-G.; Lefort, L.; Boogers, J.A.F.; de Vries, A.H.M.; de Vires, J.G.; Minnaard, A.J.; Feringa, B.L. J. Org. Chem. 2004, 69, 8045.
1279. Navarre, L.; Martinez, R.; Genet, J.-P.; Darses, S. J. Am. Chem. Soc. 2008, 130, 6159; Navarre, L.; Pucheault, M.; Darses, S.; Genêt, J.-P. Tetrahedron Lett 2005, 46, 4247.
1280. Lalic, G.; Corey, E.J. Tetrahedron Lett. 2008, 49, 4894; Gendrineau, T.; Genet, J.-P.; Darses, S. Org. Lett. 2009, 11, 3486.
1281. Takaya, Y.; Senda, T.; Kurushima, H.; Ogasawara, M.; Hayashi, T. Tetrahedron Asymmetry 1999, 10, 4047.
1282. Kabalka, G.W.; Brown, H.C.; Suzuki, A.; Honma, S.; Arase, A.; Itoh, M. J. Am. Chem. Soc. 1970, 92, 710. See also, Arase, A.; Masuda, Y.; Suzuki, A. Bull. Chem. Soc. Jpn. 1976, 49, 2275.
1283. Giese, B. Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Elmsford, NY, 1986, pp. 36–68; Giese, B. Angew. Chem. Int. Ed. 1985, 24, 553; Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 263–273. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1809–1813.
1284. Srikanth, G.S.C.; Castle, S.L. Tetrahedron 2005, 61, 10377.
1285. Hayen, A.; Koch, R.; Metzger, J.O. Angew. Chem. Int. Ed. 2000, 39, 2758.
1286. Ollivier, C.; Renaud, P. Chem. Eur. J. 1999, 5, 1468.
1287. See Jasperse, C.P.; Curran, D.P.; Fevig, T.L. Chem. Rev. 1991, 91, 1237; Curran, D.P. Adv. Free Radical Chem. (Greenwich, Conn.) 1990, 1, 121; Giese, B. Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Elmsford, NY, 1986, pp. 151–169. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 413–418.
1288. See Porter, N.A.; Chang, V.H. J. Am. Chem. Soc. 1987, 109, 4976.
1289. Sibi, M.P.; Petrovic, G.; Zimmerman, J. J. Am. Chem. Soc. 2005, 127, 2390; Sibi, M.P.; Patil, K. Org. Lett. 2005, 7, 1453; He, L.; Srikanth, G.S.C.; Castle, S.L. J. Org. Chem. 2005, 70, 8140. Also see Sibi, M.P.; Zimmerman, J. J. Am. Chem. Soc. 2006, 128, 13346.
1290. Lee, S.; Lim, C.J.; Kim, S.; Subramaniam, R.; Zimmerman, J.; Sibi, M.P. Org. Lett. 2006, 8, 4311.
1291. Moran, J.; Suen, T.; Beauchemin, A.M. J. Org. Chem. 2006, 71, 676.
1292. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1278–1282.
1293. See Curran, D.P. Synthesis 1988, 489 (see pp. 497–498).
1294. Deng, L.X.; Kutateladze, A.G. Tetrahedron Lett. 1997, 38, 7829.
1295. Jang, D.O.; Cho, D.H.; Chung, C.-M. Synlett 2001, 1923.
1296. Bałczewski, P.; Mikołajczyk, M. Synthesis 1995, 392.
1297. Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K.; Omoto, K.; Fujimoto, H. J. Org. Chem. 2001, 66, 7776.
1298. Mero, C.L.; Porter, N.A. J. Am. Chem. Soc. 1999, 121, 5155.
1299. Yorimitsu, H.; Wakabayashi, K.; Shinokubo, H.; Oshima, K. Bull. Chem. Soc. Jpn. 2001, 74, 1963.
1300. Ouvry, G.; Zard, S.Z. Chem. Commun. 2003, 778.
1301. Charrier, N.; Quiclet-Sire, B.; Zard, S.Z. J. Am. Chem. Soc. 2008, 130, 8898.
1302. Hirase, K.; Iwahama, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2002, 67, 970.
1303. Zytowski, T.; Fischer, H. J. Am. Chem. Soc. 1996118, 437.
1304. Avila, D.V.; Ingold, K.U.; Lusztyk, J.; Dolbier, Jr., W.R.; Pan, H.-Q. J. Org. Chem. 1996, 61, 2027.
1305. Leach, A.G.; Wang, R.; Wohlhieter, G.E.; Khan, S.I.; Jung, M.E.; Houk, K.N. J. Am. Chem. Soc. 2003, 125, 4271.
1306. Giese, B.; Lachhein, S. Angew. Chem. Int. Ed. 1982, 21, 768.
1307. Dickstein, J.I.; Miller, G.I. in The Chemistry of Carbon Carbon Triple Bonds, Vol. 2, Patai, S. (Ed.), Wiley, NY 1978.
1308. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1283–1295; Rheault, T.R.; Sibi, M.P. Synthesis 2003, 803.
1309. See Pandey, G.; Reddy, G.D.; Chakrabarti, D. J. Chem. Soc., Perkin Trans. 1 1996, 219.
1310. Chang, S.-Y.; Jiang, W.-T.; Cherng, C.-D.; Tang, K.-H.; Huang, C.-H.; Tsai, Y.-M. J. Org. Chem. 1997, 62, 9089. See McCarroll, A.J.; Walton, J.C. J. Chem. Soc., Perkin Trans. 1 2001, 3215.
1311. Chatgilialoglu, C.; Ferreri, C.; Guerra, M.; Timokhin, V.; Froudakis, G.; Gimisis, Z.T. J. Am. Chem. Soc. 2002, 124, 10765. See Guan, X.; Phillips, D.L.; Yang, D. J. Org. Chem. 2006, 71, 1984.
1312. See Ishibashi, H.; Sato, T.; Ikeda, M. Synthesis 2002, 695.
1313. See Ha, C.; Horner, J.H.; Newcomb, M.; Varick, T.R.; Arnold, B.R.; Lusztyk, J. J. Org. Chem. 1993, 58 1194.
1314. See Furxhi, E.; Horner, J.H.; Newcomb, M. J. Org. Chem. 1999, 64, 4064; Tauh, P.; Fallis, A.G. J. Org. Chem. 1999, 64, 6960.
1315. Cook, G.R.; Hayashi, R. Org. Lett. 2006, 8, 1045.
1316. Clark, A.J.; Wilson, P. Tetrahedron Lett. 2008, 49, 4848.
1317. Smith, D.M.; Pulling, M.E.; Norton, J.R. J. Am. Chem. Soc. 2007, 129, 770.
1318. Mueller, A.M.; Chen, P. J. Org. Chem. 1998, 63, 4581.
1319. Bogen, S; Malacria, M. J. Am. Chem. Soc. 1996118, 3992.; Beckwith, A.L.J.; Ingold, K.U. in Vol. 1 of Rearrangements in Ground States and Excited States, de Mayo, P., Ed., Academic Press, NY 1980, pp. 162–283. Gómez, A.M.; Company, M.D.; Uriel, C.; Valverde, S.; López, J.C. Tetrahedron Lett. 2002, 43, 4997.
1320. Mayon, P.; Chapleur, Y. Tetrahedron Lett. 1994, 35, 3703; Marco-Contelles, J.; Sánchez, B. J. Org. Chem. 1993, 58, 4293.
1321. Jung, M.E.; Marquez, R.; Houk, K.N. Tetrahedron Lett. 1999, 40, 2661.
1322. Kamimura, A.; Taguchi, Y. Tetrahedron Lett. 2004, 45, 2335.
1323. Wang, Li.C. J. Org. Chem. 2002, 67, 1271.
1324. Kraus, G.A.; Wu, Y. J. Am. Chem. Soc. 1992114, 8705.
1325. Zhang, W.; Dowd, P. Tetrahedron Lett. 1995, 36, 8539.
1326. Bailey, W.F.; Carson, M.W. Tetrahedron Lett. 1999, 40, 5433.
1327. Alabugin, I.V.; Manoharan, M. J. Am. Chem. Soc. 2005, 127, 9534
1328. Afor a polymer-bound Sn catalyst see Hernán, A.G.; Kilburn, J.D. Tetrahedron Lett. 2004, 45, 831.
1329. Clark, A.J.; Davies, D.I.; Jones, K.; Millbanks, C. J. Chem. Soc., Chem. Commun. 1994, 41.
1330. Barrero, A.F.; Oltra, J.E.; Cuerva, J.M.; Rosales, A. J. Org. Chem. 2002, 67, 2566.
1331. Molander, G.A.; St. Jean, Jr., D.J. J. Org. Chem. 2002, 67, 3861.
1332. Becattini, B.; Ollivier, C.; Renaud, P. Synlett 2003, 1485.
1333. Tamura, O.; Matsukida, H.; Toyao, A.; Takeda, Y.; Ishibashi, H. J. Org. Chem. 2002, 67, 5537.
1334. See Ikeda, M.; Shikaura, J.; Maekawa, N.; Daibuzono, K.; Teranishi, H.; Teraoka, Y.; Oda, N.; Ishibashi, H. Heterocycles 1999, 50, 31.
1335. See Ericsson, C.; Engman, L. Org. Lett. 2001, 3, 3459.
1336. Quirante, J.; Vila, X.; Escolano, C.; Bonjoch, J. J. Org. Chem. 2002, 67, 2323.
1337. Crich, D.; Ranganathan, K.; Huang, X. Org. Lett. 2001, 3, 1917.
1338. Andrukiewicz, R.; Loska, R.; Prisyahnyuk, V.; Stalinski, K. J. Org. Chem. 2003, 68, 1552.
1339. See Bouvier, J.-P.; Jung, G.; Liu, Z.; Guérin, B.; Guindon, Y. Org. Lett. 2001, 3, 1391; Bailey, W.F.; Longstaff, S.C. Org. Lett. 2001, 3, 2217; Stalinski, K.; Curran, D.P. J. Org. Chem. 2002, 67, 2982.
1340. See Sha, C.-K.; Shen, C.-Y.; Jean, T.-S.; Chiu, R.-T.; Tseng, W.-H. Tetrahedron Lett. 1993, 34, 764; Miyabe, H.; Takemoto, Y. Chemistry: European J. 2007, 13, 7280.
1341. Kano, S.; Yuasa, Y.; Asami, K.; Shibuya, S. Chem. Lett. 1986, 735; Robertson, J.; Lam, H.W.; Abazi, S.; Roseblade, S.; Lush, R.K. Tetrahedron 2000, 56, 8959.
1342. Petit, M.; Lapierre, A.J.B.; Curran, D.P. J. Am. Chem. Soc. 2005, 127, 14994.
1343. Majumdar, K.C.; Basu, P.K.; Mukhopadhyay, P.P. Tetrahedron 2005, 61, 10603, Tetrahedron 2007, 63, 793.
1344. Yorimitsu, H.; Shinokubo, H.; Oshima, K. Chem. Lett. 2000, 104.
1345. Sha, C.-K.; Zhan, Z.-P.; Wang, F.-S. Org. Lett. 2000, 2, 2011.
1346. Wartenberg, F.-H.; Junga, H.; Blechert, S. Tetrahedron Lett. 1993, 34, 5251. See Shi, J.; Zhang, M.; Fu, Y.; Liu, L.; Guo, Q.-X. Tetrahedron 2007, 63, 12681.
1347. Gilbert, B.C.; Kalz, W.; Lindsay, C.I.; McGrail, P.T.; Parsons, A.F.; Whittaker, D.T.E. J. Chem. Soc., Perkin Trans. 1 2000, 1187. See El Bialy, S.A.A.; Ohtani, S.; Sato, T.; Ikeda, M. Heterocycles 2001, 54, 1021; Liu, L.; Wang, X.; Li, C. Org. Lett. 2003, 5, 361.
1348. Keusenkothen, P.F.; Smith, M.B. J. Chem. Soc., Perkin Trans. 1 1994, 2485.
1349. Padwa, A.; Rashatasakhon, P.; Ozdemir, A.D.; Willis, J. J. Org. Chem 2005, 70, 519.
1350. Beckwith, A.L.J.; Joseph, S.P.; Mayadunne, R.T.A. J. Org. Chem. 1993, 58, 4198.
1351. D'Annibale, A.; Nanni, D.; Trogolo, C.; Umani, F. Org. Lett. 2000, 2, 401. See Lee, E.; Kim, S.K.; Kim, J.Y.; Lim, J. Tetrahedron Lett. 2000, 41, 5915. For a related reaction with tin hydride, see Curran, D.P.; Guthrie, D.B.; Geib, S.J. J. Am. Chem. Soc. 2008, 130, 8437. For a discussion of mechanism, see Snider, B.B. Tetrahedron 2009, 65, 10738.
1352. Glover, S.A.; Warkentin, J. J. Org. Chem. 1993, 58, 2115.
1353. Kitamura, M.; Narasaka, K. Bull. Chem. Soc. Jpn. 2008, 81, 539.
1354. Gupta, V.; Besev, M.; Engman, L. Tetrahedron Lett. 1998, 39, 2429.
1355. Ryu, I.; Nagahara, K.; Yamazaki, H.; Tsunoi, S.; Sonoda, N. Synlett 1994, 643.
1356. Ollivier, C.; Renaud, P. J. Am. Chem. Soc. 2000, 122, 6496.
1357. Ollivier, C.; Bark, T.; Renaud, P. Synthesis 2000, 1598.
1358. Mikami, S.; Fujita, K.; Nakamura, T.; Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K. Org. Lett. 2001, 3, 1853.
1359. Villar, F.; Equey, O.; Renaud, P. Org. Lett. 2000, 2, 1061.
1360. Fujioka, T.; Nakamura, T.; Yorimitsu, H.; Oshima, K. Org. Lett. 2002, 4, 2257.
1361. Rainer, J.D.; Kennedy, A.R.; Chase, E. Tetrahedron Lett. 1999, 40, 6325.
1362. Kizil, M.; Patro, B.; Callaghan, O.; Murphy, J.A.; Hursthouse, M.B.; Hobbs, D. J. Org. Chem. 1999, 64, 7856.
1363. Sadasivam, D.V.; Antharjanam, P.K.S.; Prasad, E.; Flowers II, R.A. J. Am. Chem. Soc. 2008, 130, 7228.
1364. See Jiaang, W.-T.; Lin, H.-C.; Tang, K.-H.; Chang, L.-B.; Tsai, Y.-M. J. Org. Chem. 1999, 64, 618.
1365. Schiesser, C.H.; Matsubara, H.; Ritsner, I.; Wille, U. Chem. Commun. 2006, 1067.
1366. Pattenden, G.; Roberts, L.; Blake, A.J. J. Chem. Soc., Perkin Trans. 1 1998, 863;. Also see, Pattenden, G.; Smithies, A.J.; Tapolczay, D.; Walter, D.S. J. Chem. Soc., Perkin Trans. 1 1996, 7.
1367. Rigby, J.H.; Danca, D.M.; Horner, J.H. Tetrahedron Lett. 1998, 39, 8413.
1368. Devin, P.; Fensterbank, L.; Malacria, M. Tetrahedron Lett. 1999, 40, 5511.
1369. Hays, D.S.; Fu, G.C. Tetrahedron. 1999, 55, 8815.
1370. Naito, T.; Nakagawa, K.; Nakamura, T.; Kasei, A.; Ninomiya, I.; Kiguchi, T. J. Org. Chem. 1999, 64, 2003.
1371. Inokuchi, T.; I.; Kawafuchi, H. Synlett 2001, 421.
1372. Tojino, M.; Otsuka, N.; Fukuyama, T.; Matsubara, H.; Ryu, I. J. Am. Chem. Soc. 2006, 128, 7712.
1373. See Majumdar, K.C.; Basu, P.K.; Mukhopadhyay, P.P. Tetrahedron 2004, 60, 6239; Bowman, W.R.; Cloonan, M.O.; Krintel, S.L. J. Chem. Soc., Perkin Trans. 1 2001, 2885.
1374. Clark, A.J.; Peacock, J.L. Tetrahedron Lett. 1998, 39, 6029. See Prabhakaran, E.N.; Nugent, B.M.; Williams, A.L.; Nailor, K.E.; Johnston, J.N. Org. Lett. 2002, 4, 4197.
1375. Martinez, II, E.; Newcomb, M. J. Org. Chem. 2006, 71, 557.
1376. Hemmerling, M.; Sjöholm, Å,; Somfai, P. Tetrahedron Asymmetry 1999, 10, 4091.
1377. Guindon, Y.; Guérin, B.; Landry, S.R. Org. Lett. 2001, 3, 2293.
1378. Lin, X.; Stien, D.; Weinreb, S.M. Org. Lett. 1999, 1, 637.
1379. For a review, see Hartung, J. Eur. J. Org. Chem. 2001, 619.
1380. Crich, D.; Huang, X.; Newcomb, M. Org. Lett. 1999, 1, 225.
1381. See Cossu, S.; DeLucchi, O.; Durr, R. Synth. Commun. 1996, 26, 4597.
1382. Yamagiwa, N.; Qin, H.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 13419. See Munro-Leighton, C.; Blue, E.D.; Gunnoe, T.B. J. Am. Chem. Soc. 2006, 128, 1446.
1383. Loh, T.-P.; Wei, L.-L. Synlett 1998, 975.
1384. Takasu, K.; Nishida, N.; Ihara, M. Synlett 2004, 1844.
1385. Yadav, J.S.; Reddy, A.R.; Rao, Y.G.; Narsaiah, A.V.; Reddy, B.V.S. Synthesis 2007, 3447.
1386. Srivastava, N.; Banik, B.K. J. Org. Chem. 2003, 68, 2109.
1387. Xu, L.-W.; Wei, J.-W.; Xia, C.-G.; Zhou, S.-L.; Hu, X.-X. Synlett 2003, 2425.
1388. Bartoli, G.; Bosco, M.; Marcantoni, E.; Petrini, M.; Sambri, L.; Torregiani, E. J. Org. Chem. 2001, 66, 9052.
1389. Matsubara, S.; Yoshioka, M.; Utimoto, K. Chem. Lett. 1994, 827.
1390. Jenner, G. Tetrahedron Lett. 1995, 36, 233.
1391. Das, S.; Kumar, J.S.D.; Shivaramayya, K.; George, M.V. J. Chem. Soc. Perkin Trans. 1,1995, 1797.
1392. Moghaddam, F.M.; Mohammadi, M.; Hosseinnia, A. Synth. Commn. 2000, 30, 643.
1393. Markó, I.E.; Chesney, A. Synlett 1992, 275.
1394. Yeom, C.-E.; Kim, M.J.; Kim, B.M. Tetrahedron 2007, 63, 904.
1395. Doi, H.; Sakai, T.; Iguchi, M.; Yamada, K.-i.; Tomioka, K. J. Am. Chem. Soc. 2003, 125, 2886.
1396. Bertz, S.H.; Ogle, C.A.; Rastogi, A. J. Am. Chem. Soc. 2005, 127, 1372.
1397. Bartoli, G.; Bartolacci, M.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Torregiani, E. J. Org. Chem. 2005, 70, 169.
1398. Chaudhuri, M.K.; Hussain, S.; Kantam, M.L.; Neelima, B. Tetrahedron Lett. 2005, 46, 8329.
1399. Zhang, X.; Jung, Y.S.; Mariano, P.S.; Fox, M.A.; Martin, P.S.; Merkert, J. Tetrahedron Lett. 1993, 34, 5239.
1400. Sosnicki, J.G.; Jagodzinski, T.S.; Liebscher, J. J. Heterocyclic Chem. 1997, 34, 643.
1401. Vicario, J.L.; Badía, D.; Carrillo, L.; Etxebarria, J.; Reyes, E.; Ruiz, N. Org. Prep. Proceed. Int. 2005, 37, 513; Krishna, P.R.; Sreeshailam, A.; Srinivas, R. Tetrahedron 2009, 65, 9657.
1402. Chen, Y.K.; Yoshida, M.; MacMillan, D.W.C. J. Am. Chem. Soc. 2006, 128, 9328; Sibi, M.P.; Itoh, K. J. Am. Chem. Soc. 2007, 129, 8064; Vesely, J.; Ibrahem, I.; Rios, R.; Zhao, G.-L.; Xu, Y.; Córdova, A. Tetrahedron Lett.2007, 48, 2193; Lu, X.; Deng, L. Angew. Chem. Int. Ed. 2008, 47, 7710; Fadini, L.; Togni, A. Helv. Chim. Acta 2007, 90, 411.
1403. Sugihara, H.; Daikai, K.; Jin, X.L.; Furuno, H.; Inanaga, J. Tetrahedron Lett. 2002, 43, 2735.
1404. Jew, S.-s.; Jeong, B.S.; Yoo, M.-S.; Huh, H.; Park, H.-g. Chem. Commun. 2001, 1244.
1405. Yamagiwa, N.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2003, 125, 16178.
1406. Ambroise, L.; Desmaële, D.; Mahuteau, J.; d'Angelo, J. Tetrahedron Lett. 1994, 35, 9705.
1407. Palomo, C.; Oiarbide, M.; Halder, R.; Kelso, M.; Gómez-Bengoa, E.; García, J.M. J. Am. Chem. Soc. 2004, 126, 9188.
1408. Ganesh, M.; Seidel, D. J. Am. Chem. Soc. 2008, 130, 16464.
1409. Dinér, P.; Nielsen, M.; Marigo, M.; J![]() rgensen, K.A. Angew. Chem. Int. Ed. 2007, 46, 1983.
rgensen, K.A. Angew. Chem. Int. Ed. 2007, 46, 1983.
1410. Ahn, K.H.; Lee, S.J. Tetrahedron Lett. 1994, 35, 1875.
1411. Aoyagi, K.; Nakamura, H.; Yamamoto, Y. J. Org. Chem. 2002, 67, 5977.
1412. Naidu, B.N.; Sorenson, M.E.; Connolly, J.P.; Ueda, Y. J. Org. Chem. 2003, 68, 10098.
1413. Panarina, A.E.; Dogadina, A.V.; Zakharov, V.I.; Ionin, B.I. Tetrahedron Lett. 2001, 42, 4365.
1414. O'Neil, I.A.; Cleator, E.; Southern, J.M.; Bickley, J.F.; Tapolczay, D.J. Tetrahedron Lett. 2001, 42, 8251.
1415. Cardillo, G.; Gentilucci, L.; Gianotti, M.; Kim, H.; Perciaccante, R.; Tolomelli, A. Tetrahedron Asymmetry 2001, 12, 2395.
1416. Guerin, D.J.; Horstmann, T.E.; Miller, S.J. Org. Lett. 1999, 1, 1107.
1417. Xu, L.-W.; Xia, C.-G.; Li, J.-W.; Zhou, S.-L. Synlett 2003, 2246.
1418. Sriramurthy, V.; Barcan, G.A.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 12928.
1419. Kakumoto, K.; Kobayashi, S.; Sugiura, M. Org. Lett. 2002, 4, 1319.
1420. Gaunt, M.J.; Spencer, J.B. Org. Lett. 2001, 3, 25.
1421. Wabnitz, T.C.; Spencer, J.B. Tetrahedron Lett. 2002, 43, 3891.
1422. Wabnitz, T.C.; Spencer, J.B. Org. Lett. 2003, 5, 2141.
1423. Wabnitz, T.C.; Yu, J.-Q.; Spencer, J.B. Synlett 2003, 1070.
1424. Xu, L.-W.; Li, L.; Xia, C.-G.; Zhou, S.-L.; Li, J.-W.; Hu, X.-X. Synlett 2003, 2337.
1425. Rao, H.S.P.; Jothilingam, S. Tetrahedron Lett. 2004, 42, 6595.
1426. Sadow, A.D.; Haller, I.; Fadini, L.; Togni, A. J. Am. Chem. Soc. 2004, 126, 14704.
1427. Feng, J.-J.; Chen, X.-F.; Shi, M.; Duan, W.-L. J. Am. Chem. Soc. 2010, 132, 5562.
1428. Terada, M.; Ikehara, T.; Ube, H. J. Am. Chem. Soc. 2007, 129, 14112.
1429. Stewart, I.C.; Bergman, R.G.; Toste, F.D. J. Am. Chem. Soc. 2003, 125, 8696.
1430. Also called hydroalkoxylation. See Ramachary, D.B.; Mondal, R. Tetrahedron Lett. 2006, 47, 7689.
1431. Phillips, E.M.; Riedrich, M.; Scheidt, K.A. J. Am. Chem. Soc. 2010, 132, 13179.
1432. Kano, T.; Tanaka, Y.; Maruoka, K. Tetrahedron Lett. 2006, 47, 3039.
1433. Kano, T.; Tanaka, Y.; Maruoka, K. Tetrahedron 2007, 63, 8658.
1434. Baker-Glenn, C.; Hodnett, N.; Reiter, M.; Ropp, S.; Ancliff, R.; Gouverneur, V. J. Am. Chem. Soc. 2005, 127, 1481.
1435. Kamimura, A.; Kawahara, F.; Omata, Y.; Murakami, N.; Morita, R.; Otake, H.; Mitsudera, H.; Shirai, M.; Kakehi, A. Tetrahedron Lett. 2001, 42, 8497.
1436. Zeni, G.; Stracke, M.P.; Nogueira, C.W.; Braga, A.L.; Menezes, P.H.; Stefani, H.A. Org. Lett. 2004, 6, 1135.
1437. Kobayashi, S.; Ogawa, C.; Kawamura, M.; Sugiura, M. Synlett 2001, 983.
1438. Yadav, J.S.; Reddy, B.V.S.; Baishya, G. J. Org. Chem. 2003, 68, 7098.
1439. Marigo, M.; Schulte, T.; Franzén, J.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 15710; Kumar, A.; Akanksha Tetrahedron 2007, 63, 11086.
rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 15710; Kumar, A.; Akanksha Tetrahedron 2007, 63, 11086.
1440. Chu, C.-M.; Huang, W.-J.; Lu, C.; Wu, P.; Liu, J.-T.; Yao, C.-F. Tetrahedron Lett. 2006, 47, 7375.
1441. Khatik, G.L.; Kumar, R.; Chakraborti, A.K. Org. Lett. 2006, 8, 2433.
1442. Kamal, A.; Reddy, D.R.; Rajendar Tetrahedron Lett. 2005, 46, 7951.
1443. Me![]() iarová, M.; Toma, Š.; Kotrusz, P. Org. Biomol. Chem. 2006, 4, 1420.
iarová, M.; Toma, Š.; Kotrusz, P. Org. Biomol. Chem. 2006, 4, 1420.
1444. Chu, C.-M.; Gao, S.; Sastry, M.N.V.; Yao, C.-F. Tetrahedron Lett. 2005, 46, 4971; Gao, S.; Tzeng, T.; Sastry, M.N.V.; Chu, C.-M.; Liu, J.-T.; Lin, C.; Yao, C.-F. Tetrahedron Lett. 2006, 47, 1889.
1445. Chu, C.-M.; Gao, S.; Sastry, M.N.V.; Kuo, C.-W.; Lu, C.; Liu, J.-T.; Yao, C.-F. Tetrahedron 2007, 63, 1863.
1446. Taniguchi, Y.; Maruo, M.; Takaki, K.; Fujiwara, Y. Tetrahedron Lett. 1994, 35, 7789.
1447. McDaid, P.; Chen, Y.; Deng, L. Angew. Chem. Int. Ed. 2002, 41, 338.
1448. Ranu, B.C.; Mandal, T. Synlett 2004, 1239.
1449. Nishimura, K.; Tomioka, K. J. Org. Chem. 2002, 67, 431.
1450. Azizi, N.; Aryanasab, F.; Torkiyan, L.; Ziyaei, A.; Saidi, M.R. J. Org. Chem. 2006, 71, 3634.
1451. Ruano, J.L.G.; García, M.C.; Laso, N.M.; Castro, A.M.M.; Ramos, J.H.R. Angew. Chem. Int. Ed. 2001, 40, 2507.
1452. Ohno, T.; Sakai, M.; Ishino, Y.; Shibata, T.; Maekawa, H.; Nishiguchi, I. Org. Lett. 2001, 3, 3439.
1453. Kyoda, M.; Yokoyama, T.; Maekawa, H.; Ohno, T.; Nishiguchi, I. Synlett 2001, 1535.
1454. Blakskjær, P.; H![]() j, B.; Riber, D.; Skrydstrup, T. J. Am. Chem. Soc. 2003, 125, 4030.
j, B.; Riber, D.; Skrydstrup, T. J. Am. Chem. Soc. 2003, 125, 4030.
1455. Mattson, A.E.; Bharadwaj, A.R.; Scheidt, K.A. J. Am. Chem. Soc. 2004, 126, 2314.
1456. Yadav, J.S.; Anuradha, K.; Reddy, B.V.S.; Eeshwaraiah, B. Tetrahedron Lett. 2003, 44, 8959.
1457. Hanzawa, Y.; Tabuchi, N.; Narita, K.; Kakuuchi, A.; Yabe, M.; Taguchi, T. Tetrahedron 2002, 58, 7559.
1458. Corey, E.J.; Hegedus, L.S. J. Am. Chem. Soc. 1969, 91, 4926.
1459. Sawa, Y.; Hashimoto, I.; Ryang, M.; Tsutsumi, S. J. Org. Chem. 1968, 33, 2159.
1460. Seyferth, D.; Hui, R.C. J. Am. Chem. Soc. 1985, 107, 4551. See also, Lipshutz, B.H.; Elworthy, T.R. Tetrahedron Lett. 1990, 31, 477.
1461. Seyferth, D.; Hui, R.C. Tetrahedron Lett. 1986, 27, 1473.
1462. See Stetter, H.; Kuhlmann, H. Org. React. 1991, 40, 407–496; Stetter, H.; Kuhlmann, H.; Haese, W. Org. Synth., 65, 26.
1463. Enders, D.; Gerdes, P.; Kipphardt, H. Angew. Chem. Int. Ed. 1990, 29, 179.
1464. Herrmann, J.L.; Richman, J.E.; Schlessinger, R.H. Tetrahedron Lett. 1973, 3271, 3275.
1465. Beockman Jr., R.K.; Bruza, K.J.; Baldwin, J.E.; Lever, Jr., O.W. J. Chem. Soc., Chem. Commun. 1975, 519.
1466. Boeckman Jr., R.K; Bruza, K.J. J. Org. Chem. 1979, 44, 4781.
1467. Stork, G.; Maldonado, L. J. Am. Chem. Soc. 1974, 96, 5272.
1468. Sato, K.; Yamazoe, S.; Yamamoto, R.; Ohata, S.; Tarui, A.; Omote, M.; Kumadaki, I.; Ando, A. Org. Lett. 2008, 10, 2405.
1469. Boger, D.L.; Mathvink, R.J. J. Org. Chem. 1989, 54, 1777.
1470. See Giese, B. Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon, Elmsford, NY, 1986, pp. 69–77; Vogel, H. Synthesis 1970, 99; Dang, H.-S.; Roberts, B.P. Chem. Commun. 1996, 2201.
1471. Elad, D. Fortschr. Chem. Forsch. 1967, 7, 528, see pp. 530–543.
1472. See Hájek, M.; Málek, J. Coll. Czech. Chem. Commun. 1979, 44, 3695.
1473. de Klein, W.J. Recl. Trav. Chim. Pays-Bas 1975, 94, 48.
1474. Cadogan, J.I.G. Pure Appl. Chem. 1967, 15, 153, pp. 153–158. See also, Giese, B.; Zwick, W. Chem. Ber. 1982, 115, 2526; Giese, B.; Erfort, U. Chem. Ber. 1983, 116, 1240.
1475. See DiPietro, J.; Roberts, W.J. Angew. Chem. Int. Ed. 1966, 5, 415.
1476. Hua, R.; Takeda, H.; Onozawa, S.-y.; Abe, Y.; Tanaka, M. J. Am. Chem. Soc. 2001, 123, 2899.
1477. Kokubo, K.; Matsumasa, K.; Miura, M.; Nomura, M. J. Org. Chem. 1997, 62, 4564.
1478. Fairlie, D.P.; Bosnich, B. Organometallics 1988, 7, 936, 946. Also see, Barnhart, R.W.; Wang, X.; Noheda, P.; Bergens, S.H.; Whelan, J.; Bosnich, B. J. Am. Chem. Soc. 1994, 116, 1821.
1479. Yamane, M.; Amemiya, T.; Narasaka, K. Chem. Lett. 2001, 1210.
1480. Fujihara, T.; Katafuchi, Y.; Iwai, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2010, 132, 2094.
1481. Willis, M.C.; Randell-Sly, H.E.; Woodward, R.L.; McNally, S.J.; Currie, G.S. J. Org. Chem 2006, 71, 5291; Imai, M.; Tanaka, M.; Nagumo, S.; Kawahara, N.; Suemune, H. J. Org. Chem. 2007, 72, 2543. For a discussion of the mechanism, see Roy, A.H.; Lenges, C.P.; Brookhart, M. J. Am. Chem. Soc. 2007, 129, 2082.
1482. Curini, M.; Epifano, F.; Maltese, F.; Rosati, O. Synlett 2003, 552.
1483. See Jo, E.-A.; Jun, C.-H. Tetrahedron Lett. 2009, 50, 3338.
1484. Barnhart, R.W.; McMorran, D.A.; Bosnich, B. Chem. Commun. 1997, 589.
1485. de Alaniz, J.R.; Rovis, T. J. Am. Chem. Soc. 2005, 127, 6284; Liu, Q.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 2552; Kundu, K.; McCullagh, J.V.; Morehead, Jr., A.T. J. Am. Chem. Soc. 2005, 127, 16042; Enders, D.; Han, J.; Henseler, A. Chem. Commun. 2008, 3989.
1486. Omura, S.; Fukuyama, T.; Horiguchi, J.; Murakami, Y.; Ryu, I. J. Am. Chem. Soc. 2008, 130, 14094. For a related reaction, see Shibahara, F.; Bower, J.F.; Krische, M.J. J. Am. Chem. Soc. 2008, 130, 14120.
1487. Stetter, H.; Kuhlmann, H. Org. React. 1991, 40, 407; Kerr, M.S.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 8876; Pesch, J.; Harms, K.; Bach, T. Eur. J. Org. Chem. 2004, 2025; Mennen, S.; Blank, J.; Tran-Dube, M.B.; Imbriglio, J.E.; Miller, S.J. Chem. Commun. 2005, 195. See also, Mattson, A.E.; Bharadwaj, A.R.; Scheidt, K.A. J. Am. Chem. Soc. 2004, 126, 2314.
1488. Tanaka, K.; Fu, G.C. J. Am. Chem. Soc. 2002, 124, 10296.
1489. See Lee, E.; Tae, J.S.; Chong, Y.H.; Park, Y.C.; Yun, M.; Kim, S. Tetrahedron Lett. 1994, 35, 129 for an example.
1490. Rhee, J.U.; Krische, M.J. Org. Lett. 2005, 7, 2493.
1491. Kraus, G.A.; Liu, P. Tetrahedron Lett. 1994, 35, 7723.
1492. Yang, X.-F.; Wang, M.; Varma, R.S.; Li, C.-J. Org. Lett. 2003, 5, 657.
1493. Na, Y.; Ko, S.; Hwang, L.K.; Chang, S. Tetrahedron Lett. 2003, 44, 4475.
1494. See Lapidus, A.L.; Pirozhkov, S.D. Russ. Chem. Rev. 1989, 58, 117; Anderson, G.K.; Davies, J.A. in Hartley, F.R.; Patai, S. The Chemistry of the Metal-Carbon Bond, Vol. 3, Wiley, NY, 1985, pp. 335–359, 335–348; in Falbe, J. New Syntheses with Carbon Monoxide, Springer, NY, 1980, the articles by Mullen, A. pp. 243–308; and Bahrmann, H. pp. 372–413; Falbe, J. Carbon Monoxide in Organic Synthesis, Springer: Berlin, 1970, pp. 78–174.
1495. Haaf, W. Chem. Ber. 1966, 99, 1149; Christol, H.; Solladié, G. Bull. Soc. Chim. Fr. 1966, 1307.
1496. Sternberg, H.W.; Markby, R.; Wender, P. J. Am. Chem. Soc. 1960, 82, 3638.
1497. Tsuji, J. Palladium Reagents and Catalysts Wiley, NY, 1999; Hohn, A. in Applied Homogeneous Catalysis with Organometallic Compounds, Vol. 1 VCH, NY, 1996, p. 137; Beller, M.; Tafesh, A.M. in Applied Homogeneous Catalysis with Organometallic Compounds, Vol. 1 VCH, NY, 1996, p. 187; Drent, E.; Jager, W.W.; Keijsper, J.J.; Niele, F.G.M. in Applied Homogeneous Catalysis with Organometallic Compounds, Vol. 1 VCH, NY, 1996, p. 1119.; Bertoux, F.; Monflier, E.; Castanet, Y.; Mortreux, A. J. Mol. Catal. A: Chem. 1999, 143, 11; Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpaintner, C.W. J. Mol. Catal. A: Chem. 1995, 104, 17; Milstein, D. Acc. Chem. Res. 1988, 21, 428; Tsuji, J. Acc. Chem. Res. 1969, 2, 144; Bird, C.W. Chem. Rev. 1962, 62, 283.
1498. For a review, see Kiss, G. Chem. Rev. 2001, 101, 3435.
1499. See Heck, R.F. Palladium Reagents in Organic Synthesis Academic Press, NY, 1985, pp. 381–395; Mukhopadhyay, K.; Sarkar, B.R.; Chaudhari, R.V. J. Am. Chem. Soc. 2002, 124, 9692; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2008, 130, 15254.
1500. Xu, Q.; Fujiwara, M.; Tanaka, M.; Souma, Y. J. Org. Chem. 2000, 65, 8105.
1501. Xu, Q.; Nakatani, H.; Souma, Y. J. Org. Chem. 2000, 65, 1540.
1502. Alper, H.; Hamel, N. J. Am. Chem. Soc. 1990, 112, 2803.
1503. Brunet, J.-J.; Neibecker, D.; Srivastava, R.S. Tetrahedron Lett. 1993, 34, 2759.
1504. Senboku, H.; Komatsu, H.; Fujimura, Y.; Tokuda, M. Synlett 2001, 418.
1505. Williams, C.M.; Johnson, J.B.; Rovis, T. J. Am. Chem. Soc. 2008, 130, 14936.
1506. Duñach, E.; Dérien, S.; Périchon, J. J. Organomet. Chem. 1989, 364, C33.
1507. Periasamy, M.; Radhakrishnan, U.; Rameshkumar, C.; Brunet, J.-J. Tetrahedron Lett. 1997, 38, 1623.
1508. Takeuchi, R.; Sugiura, M. J. Chem. Soc. Perkin Trans. 1,1993, 1031.
1509. Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. J. Org. Chem. 1999, 64, 3975. See also, Takimoto, M.; Shimizu, K.; Mori, M. Org. Lett. 2001, 3, 3345.
1510. See Hogeveen, H. Adv. Phys. Org. Chem. 1973, 10, 29.
1511. Bird, C.W.; Cookson, R.C.; Hudec, J.; Williams, R.O. J. Chem. Soc. 1963, 410.
1512. Chen, A.; Ren, L.; Crudden, C.M.; J. Org. Chem. 1999, 64, 9704.
1513. See Fallis, A.G.; Forgione, P. Tetrahedron 2001, 57, 5899.
1514. Kishimoto, Y.; Ikariya, T. J. Org. Chem. 2000, 65, 7656.
1515. Li, J.; Jiang, H.; Feng, A.; Jia, L. J. Org. Chem. 1999, 64, 5984. See also, Clarke, M.L. Tetrahedron Lett. 2004, 45, 4043.
1516. Xiao, W.-J.; Alper, H. J. Org. Chem. 2001, 66, 6229.
1517. Zhou, D.-Y.; Yoneda, E.; Onitsuka, K.; Takahashi, S. Chem. Commun. 2002, 2868.
1518. Xiao, W.-J.; Vasapollo, G.; Alper, H. J. Org. Chem. 1999, 64, 2080.
1519. Kawakami, J.-i.; Mihara, M.; Kamiya, I.; Takeba, M.; Ogawa, A.; Sonoda, N. Tetrahedron 2003, 59, 3521.
1520. Li, J.; Jiang, H.; Chen, M. Synth. Commun. 2001, 31, 3131; El Ali, B.; Tijani, J.; El-Ghanam, A.; Fettouhi, M. Tetrahedron Lett. 2001, 42, 1567.
1521. Yokota, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2002, 67, 5005.
1522. Dai, M.; Wang, C.; Dong, G.; Xiang, J.; Luo, J.; Liang, B.; Chen, J.; Yang, Z. Eur. J. Org. Chem. 2003, 4346.
1523. Ko, S.; Na, Y.; Chang, S. J. Am. Chem. Soc. 2002, 124, 750.
1524. Cho, S.H.; Yoo, E.J.; Bae, I.; Chang, S. J. Am. Chem. Soc. 2005, 127, 16046.
1525. Yoneda, E.; Kaneko, T.; Zhang, S.-W.; Onitsuka, K.; Takahashi, S. Tetrahedron Lett. 1999, 40, 7811.
1526. Gagnier, S.V.; Larock, R.C. J. Am. Chem. Soc. 2003, 125, 4804.
1527. Murakami, M.; Itami, K.; Ito, Y. J. Am. Chem. Soc. 1999, 121, 4130.
1528. Jeong, N.; Hwang, S.H. Angew. Chem. Int. Ed. 2000, 39, 636.
1529. Lee, S.I.; Park, J.H.; Chung, Y.K.; Lee, S.-G. J. Am. Chem. Soc. 2004, 126, 2714.
1530. Mazzola, Jr., R.D.; Giese, S.; Benson, C.L.; West, F.G. J. Org. Chem. 2004, 69, 220.
1531. Rameshkumar, C.; Periasamy, M. Tetrahedron Lett. 2000, 41, 2719.
1532. Plietker, B. J. Org. Chem. 2004, 69, 8287.
1533. See Krafft, M.E.; Hirosawa, C.; Bonaga, L.V.R. Tetrahedron Lett. 1999, 40, 9177.
1534. See Krafft, M.E.; Boñaga, L.V.R.; Hirosawa, C. J. Org. Chem. 2001, 66, 3004.
1535. Koga, Y.; Kobayashi, T.; Narasaka, K. Chem. Lett. 1998, 249. An entrapped-Rh catalyst has been used: Park, K.H.; Son, S.U.; Chung, Y.K. Tetrahedron Lett. 2003, 44, 2827.
1536. Hicks, F.A.; Kablaoui, N.M.; Buchwald, S.L. J. Am. Chem. Soc. 1997, 118, 9450; Hicks, F.A.; Kablaoui, N.M.; Buchwald, S.L. J. Am. Chem. Soc. 1999, 121, 5881.
1537. Adrio, J.; Carretero, J.C. J. Am. Chem. Soc. 2007, 129, 778; Adrio, J.; Rivero, M.R.; Carretero, J.C. Org. Lett. 2005, 7, 431.
1538. Hoye, T.R.; Suriano, J.A. J. Am. Chem. Soc. 1993, 115, 1154.
1539. Khand, I.U.; Pauson, P.L.; Habib, M.J. J. Chem. Res. (S) 1978, 348; Khand, I.U; Pauson, P.L. J. Chem. Soc. Perkin Trans. 1,1976, 30. Gibson, S.E.; Stevenazzi, A. Angew. Chem. Int. Ed. 2003, 42, 1800; Gibson, S.E.; Mainolfi, N. Angew. Chem. Int. Ed. 2005, 44, 3022; Lee, H.-W.; Kwong, F.-Y. Eur. J. Org. Chem. 2010, 789.
1540. See de Bruin, T.J.M.; Milet, A.; Greene, A.E.; Gimbert, Y. J. Org. Chem. 2004, 69, 1075. See also, Rivero, M.R.; Adrio, J.; Carretero, J.C. Eur. J. Org. Chem. 2002, 2881.
1541. Magnus, P.; Principe, L.M. Tetrahedron Lett. 1985, 26, 4851.
1542. Itami, K.; Mitsudo, K.; Fujita, K.; Ohashi, Y.; Yoshida, J.-i. J. Am. Chem. Soc. 2004, 126, 11058.
1543. Wender, P.A.; Croatt, M.P.; Deschamps, N.M. J. Am. Chem. Soc. 2004, 126, 5948.
1544. Wender, P.A; Deschamps, N.M.; Gamber, G.G. Angew. Chem. Int. Ed. 2003, 42, 1853.
1545. Pagenkopf, B.L.; Livinghouse, T. J. Am. Chem. Soc. 1996, 118, 2285.
1546. Sugihara, T.; Yamada, M.; Ban, H.; Yamaguchi, M.; Kaneko, C. Angew. Chem. Int. Ed. 1997, 36, 2801.
1547. Krafft, M.E.; Scott, I.L.; Romero, R.H. Tetrahedron Lett. 1992, 33, 3829.
1548. Kerr, W.J.; Lindsay, D.M.; McLaughlin, M.; Pauson, P.L. Chem. Commun. 2000, 1467; Brown, D.S.; Campbell, E.; Kerr, W.J.; Lindsay, D.M.; Morrison, A.J.; Pike, K.G.; Watson, S.P. Synlett 2000, 1573.
1549. Krafft, M.E.; Boñaga, L.V.R.; Wright, J.A.; Hirosawa, C. J. Org. Chem. 2002, 67, 1233; Blanco- Urgoiti, J.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. Tetrahedron Lett. 2002, 43, 5763. The reaction has been done in aqueous media: Krafft, M.E.; Wright, J.A.; Boñaga, L.V.R. Tetrahedron Lett. 2003, 44, 3417.
1550. Kim, S.-W.; Son, S.U.; Lee, S.I.; Hyeon, T.; Chung, Y.K. J. Am. Chem. Soc. 2000, 122, 1550.
1551. Kim, S.-W.; Son, S.U.; Lee, S.S.; Hyeon, T.; Chung, Y.K. Chem. Commun. 2001, 2212; Son, S.U.; Lee, S.I.; Chung, Y.K.; Kim, S.-W.; Hyeon, T. Org. Lett. 2002, 4, 277.
1552. Krafft, M.E.; Wright, J.A.; Llorente, V.R.; Boñaga, L.V.R. Can. J. Chem. 2005, 83, 1006.
1553. Dahan, A.; Portnoy, M. Chem. Commun. 2002, 2700.
1554. Ford, J.G.; Kerr, W.J.; Kirk, G.G.; Lindsay, D.M.; Middlemiss, D. Synlett 2000, 1415.
1555. Iqbal, M.; Vyse, N.; Dauvergne, J.; Evans, P. Tetrahedron Lett. 2002, 43, 7859.
1556. Ishizaki, M.; Iwahara, K.; Niimi, Y.; Satoh, H.; Hoshino, O. Tetrahedron 2001, 57, 2729; Son, S.U.; Yoon, Y.A.; Choi, D.S.; Park, J.K.; Kim, B.M.; Chung, Y.K. Org. Lett. 2001, 3, 1065.
1557. Verdaguer, X.; Moyano, A.; Pericás, M.A.; Riera, A.; Maestro, M.A.; Mahía, J. J. Am. Chem. Soc. 2000, 122, 10242l; Konya, D.; Robert, F.; Gimbert, Y.; Greene, A.E. Tetrahedron Lett. 2004, 45, 6975.
1558. Pérez-Serrano, L.; Banco-Urgoiti, J.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. J. Org. Chem. 2000, 65, 3513. For a review, see Suh. W.H.; Choi, M.; Lee, S.I.; Chung, Y.K. Synthesis 2003, 2169.
1559. Krafft, M.E.; Boñaga, L.V.R. Angew. Chem. Int. Ed. 2000, 39, 3676, and references cited therein; Jeong, N.; Sung, B.S.; Choi, Y.K. J. Am. Chem. Soc. 2000, 122, 6771; Sturla, S.J.; Buchwald, S.L. J. Org. Chem. 2002, 67, 3398.
1560. Comely, A.C.; Gibson, S.E.; Stevenazzi, A.; Hales, N.J. Tetrahedron Lett. 2001, 42, 1183.
1561. Blanco-Urgoiti, J.; Casarrubios, L.; Domínguez, G.; Pérez-Castells, J. Tetrahedron Lett. 2001, 42, 3315.
1562. Mukai, C.; Kim, J.S.; Sonobe, H.; Hanaoka, M. J. Org. Chem. 1999, 64, 6822.
1563. Pérez-Serrano, L.; Domínguez, G.; Pérez-Castells, J. J. Org. Chem. 2004, 69, 5413.
1564. Brummond, K.M.; Sill, P.C.; Rickards, B.; Geib, S.J. Tetrahedron Lett. 2002, 43, 3735; Reichwein, J.F.; Iacono, S.T.; Patel, U.C.; Pagenkopf, B.L. Tetrahedron Lett. 2002, 43, 3739.
1565. Brummond, K.M.; Chen, H.; Fisher, K.D.; Kerekes, A.D.; Rickards, B.; Sill, P.C.; Geib, A.D. Org. Lett. 2002, 4, 1931. See Shibata, T.; Kadowaki, S.; Hirase, M.; Takagi, K. Synlett 2003, 573.
1566. Trost, B.M.; Brown, R.E.; Toste, F.D. J. Am. Chem. Soc. 2000, 122, 5877.
1567. Rausch, B.J.; Gleiter, R. Tetrahedron Lett. 2001, 42, 1651.
1568. See Shibata, T.; Toshida, N.; Takagi, K. J. Org. Chem. 2002, 67, 7446; Morimoto, T.; Tsutsumi, K.; Kakiuchi, K. Tetrahedron Lett. 2004, 45, 9163.
1569. Magnus, P.; Principe, L.M. Tetrahedron Lett. 1985, 26, 4851.
1570. For a review, see Brummond, K.M.; Kent, J.L. Tetrahedron 2000, 56, 3263.
1571. Krafft, M.E. Tetrahedron Lett. 1988, 29, 999.
1572. Gimbert, Y.; Lesage, D.; Milet, A.; Fournier, F.; Greene, A.E.; Tabet, J.-C. Org. Lett. 2003, 5, 4073. See Robert, F.; Milet, A.; Gimbert, Y.; Konya, D.; Greene, A.E. J. Am. Chem. Soc. 2001, 123, 5396.
1573. de Bruin, T.J.M.; Milet, A.; Greene, A.E.; Gimbert, Y. J. Org. Chem.,200469, 1075.
1574. Sauthier, M.; Castanet, Y.; Mortreux, A. Chem. Commun. 2004 1520.
1575. Xi, Z.; Song, Q. J. Org. Chem. 2000, 65, 9157.
1576. Song, Q.; Chen, J.; Jin, X.; Xi, Z. J. Am. Chem. Soc. 2001, 123, 10419; Song, Q.; Li, Z.; Chen, J.; Wang, C.; Xi, Z. Org. Lett. 2002, 4, 4627.
1577. Shibata, T.; Yamashita, K.; Katayama, E.; Takagi, K. Tetrahedron 2002, 58, 8661.
1578. Sugihara, T.; Wakabayashi, A.; Takao, H.; Imagawa, H.; Nishizawa, M. Chem. Commun. 2001, 2456.
1579. Dong, C.; Alper, H. J. Org. Chem. 2004, 69, 5011.
1580. See Ohshiro, Y.; Hirao, T. Heterocycles 1984, 22, 859; Falbe, J. New Syntheses with Carbon Monoxide, Springer, NY, 1980, pp. 147–174. See Krafft, M.E.; Wilson, L.J.; Onan, K.D. Tetrahedron Lett. 1989, 30, 539.
1581. Kablaoui, N.M.; Hicks, F.A.; Buchwald, S.L. J. Am. Chem. Soc. 1997, 119, 4424.
1582. El Ali, B.; Okuro, K.; Vasapollo, G.; Alper, H. J. Am. Chem. Soc. 1996, 118, 4264. Also see, Brunner, M.; Alper, H. J. Org. Chem. 1997, 62, 7565.
1583. Kondo, T.; Kodoi, K.; Mitsudo, T.-a.; Watanabe, Y. J. Chem. Soc., Chem. Commun. 1994, 755.
1584. Yoneda, E.; Kaneko, T.; Zhang, S.-W.; Takahashi, S. Tetrahedron Lett. 1998, 39, 5061.
1585. Yoneda, E.; Kaneko, T.; Zhang, S.-W.; Onitsuka, K.; Takahashi, S. Org. Lett. 2000, 2, 441.
1586. Yoneda, E.; Zhang, S.-W.; Onitsuka, K.; Takahashi, S. Tetrahedron Lett. 2001, 42, 5459.
1587. Ma, S.; Wu, B.; Zhao, S. Org. Lett. 2003, 5, 4429.
1588. Fukuta, Y.; Matsuda, I.; Itoh, K. Tetrahedron Lett. 2001, 42, 1301.
1589. Kang, S.-K.; Kim, K.-J.; Yu, C.-M.; Hwang, J.-W.; Do, Y.-K. Org. Lett. 2001, 3, 2851.
1590. Chatani, N.; Kamitani, A.; Murai, S. J. Org. Chem. 2002, 67, 7014.
1591. Good, G.M.; Kemp, M.I.; Kerr, W.J. Tetahedron Lett. 2000, 41, 9323.
1592. Grigg, R.; Monteith, M.; Sridharan, V.; Terrier, C. Tetrahedron 1998, 54, 3885.
1593. Matsuda, I.; Takeuchi, K.; Itoh, K. Tetrahedron Lett. 1999, 40, 2553.
1594. Okuro, K.; Kai, H.; Alper, H. Tetrahedron Asymmetry 1997, 8, 2307.
1595. Shiba, T.; Zhou, D.-Y.; Onitsuka, K.; Takahashi, S. Tetrahedron Lett. 2004, 45, 3211.
1596. Berger, D.; Imhof, W. Tetrahedron 2000, 56, 2015.
1597. See Kalck, P.; Peres, Y.; Jenck, J. Adv. Organomet. Chem. 1991, 32, 121; Davies, J.A. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 3, Wiley, NY, 1985, pp. 361–389; Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA 1987, pp. 621–632; Pino, P. J. Organomet. Chem. 1980, 200, 223; Falbe, J. Carbon Monoxide in Organic Synthesis Springer, NY, 1980, pp. 3–77. See Ohshiro, Y.; Hirao, T. Heterocycles 1984, 22, 859.
1598. See Amer, I.; Alper, H. J. Am. Chem. Soc. 1990, 112, 3674; Jardine, F.H. in Hartley, F.R. The Chemistry of the Metal-Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 733–818, pp. 778–784.
1599. Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, CA 1987, p. 630.
1600. Chan, A.S.C.; Pai, C.-C.; Yang, T.-K.; Chen, S.M. J. Chem. Soc., Chem. Commun. 1995, 2031; Doyle, M.P.; Shanklin, M.S.; Zlokazov, M.V. Synlett 1994, 615.
1601. Breit, B.; Seiche, W. J. Am. Chem. Soc. 2003, 125, 6608.
1602. Simaan, S.; Marek, I. J. Am. Chem. Soc. 2010, 132, 4066.
1603. Johnson, J.R.; Cuny, G.D.; Buchwald, S.L. Angew. Chem. Int. Ed. 1995, 34, 1760.
1604. van der Veen, L.A.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Angew. Chem. Int. Ed. 1999, 38, 336.
1605. Fell, B.; Rupilius, W. Tetrahedron Lett. 1969, 2721.
1606. See Mullen, A. in Falbe, J. New Syntheses with Carbon Monoxide, Springer, NY, 1980, pp. 414–439. See also, Eilbracht, P.; Hüttmann, G.; Deussen, R. Chem. Ber. 1990, 123, 1063, and other papers in this series.
1607. See Botteghi, C.; Salomon, C. Tetrahedron Lett. 1974, 4285. For an indirect method, see Campi, E.; Fitzmaurice, N.J.; Jackson, W.R.; Perlmutter, P.; Smallridge, A.J. Synthesis 1987, 1032.
1608. van den Hoven, B.G.; Alper, H. J. Org. Chem. 1999, 64, 3964.
1609. Kuil, M.; Soltner, T.; van Leeuwen, P.W.N.M.; Reek, J.N.H. J. Am. Chem. Soc. 2006, 128, 11344.
1610. See Hu, Y.; Chen, W.; Osuna, A.M.B.; Stuart, A.M.; Hope, E.G.; Xiao, J. Chem. Commun. 2001, 725.
1611. See Haelg, P.; Consiglio, G.; Pino, P. Helv. Chim. Acta 1981, 64, 1865.
1612. Krauss, I.J.; Wang, C.C-Y.; Leighton, J.L. J. Am. Chem. Soc. 2001, 123, 11514.
1613. Ojima, I.; Hirai, K. in Morrison, J.D. Organic Synthesis Vol. 5, Wiley, NY, 1985, pp. 103–145, pp. 125–139; Breit, B.; Seiche, W. Synthesis 2001, 1; Clark, T.P.; Landis, C.R.; Freed, S.L.; Klosin, J.; Abboud, K.A. J. Am. Chem. Soc. 2005, 127, 5040; Yan, Y.; Zhang, X. J. Am. Chem. Soc. 2006, 128, 7198; Watkins, A.L.; Hashiguchi, B.G.; Landis, C.R. Org. Lett. 2008, 10, 4553.
1614. Sakai, N.; Nozaki, K.; Takaya, H. J. Chem. Soc., Chem. Commun. 1994, 395. See Gladiali, S.; Bayón, J.C.; Claver, C. Tetrahedron Asymmetry 1995, 6, 1453.
1615. Klosin, J.; Landis, C.R. Acc. Chem. Res. 2007, 40, 1251.
1616. Anastasiou, D.; Campi, E.M.; Chaouk, H.; Jackson, W.R.; McCubbin, Q.J. Tetrahedron Lett. 1992, 33, 2211
1617. Mirbach, M.F. J. Organomet. Chem. 1984, 265, 205. For the mechanism see Orchin, M. Acc. Chem. Res. 1981, 14, 259; Versluis, L.; Ziegler, T.; Baerends, E.J.; Ravenek, W. J. Am. Chem. Soc. 1989, 111, 2018.
1618. Ungváry, F.; Markó, L. Organometallics 1982, 1, 1120.
1619. See Kovács, I.; Ungváry, F.; Markó, L. Organometallics 1986, 5, 209.
1620. Bockman, T.M.; Garst, J.F.; King, R.B.; Markó, L.; Ungváry, F. J. Organomet. Chem. 1985, 279, 165.
1621. Godard, C.; Duckett, S.B.; Polas, S.; Tooze, R.; Whitwood, A.C. J. Am. Chem. Soc. 2005, 127, 4994.
1622. Aldridge, C.L.; Jonassen, H.B. J. Am. Chem. Soc. 1963, 85, 886.
1623. See Friedrich, K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 2, Wiley, NY, 1983, pp. 1345–1390; Nagata, W.; Yoshioka, M. Org. React. 1977, 25, 255; Brown, E.S. in Wender, I.; Pino, P. Organic Syntheses via Metal Carbonyls, Vol. 2, Wiley, NY, 1977, pp. 655–672.
1624. Arthur, Jr., P.; England, D.C.; Pratt, B.C.; Whitman, G.M. J. Am. Chem. Soc. 1954, 76, 5364.
1625. See Brown, E.S. in Wender, P.; Pino, P. Organic Syntheses via Metal Carbonyls, Vol. 2, Wiley, NY, 1977, pp. 658–667; Tolman, C.A.; McKinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Adv. Catal. 1985, 33, 1. For studies of the mechanism see McKinney, R.J.; Roe, D.C. J. Am. Chem. Soc. 1986, 108, 5167; Funabiki, T.; Tatsami, K.; Yoshida, S. J. Organomet. Chem. 1990, 384, 199. See also, Bini, L.; Müller, C.; Vogt, D. Chem. Commun.2010, 8325.
1626. Yanagisawa, A.; Nezu, T.; Mohri, S.-i. Org. Lett. 2009, 11, 5286.
1627. Jackson, W.R.; Lovel, C.G. Aust. J. Chem. 1983, 36, 1975.
1628. Jackson, W.R.; Perlmutter, P. Chem. Br. 1986, 338.
1629. Yan, M.; Xu, Q.-Y.; Chan, A.S.C. Tetrahedron Asymmetry 2000, 11, 845.
1630. See Nagata, W.; Yoshioka, M. Org. React. 1977, 25, 255.
1631. Buchwald, S.L.; LeMaire, S.J. Tetrahedron Lett. 1987, 28, 295.
1632. Ito, Y.; Kato, H.; Imai, H.; Saegusa, T. J. Am. Chem. Soc. 1982, 104, 6449.
1633. Luo, F.-T.; Ko, S.-L.; Chao, D.-Y. Tetrahedron Lett. 1997, 38, 8061.
1634. Kitano, Y.; Chiba, K.; Tada, M. Synlett 1999, 288.
1635. Mita, T.; Kazuki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 514.
1636. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 629–632.
1637. de la Mare, P.B.D. Electrophilic Halogenation Cambridge University Press, Cambridge, 1976; House, H.O. Modern Synthetic Reaction, 2nd ed., W.A. Benjamin, NY, 1972, pp. 422–431.
1638. McMillen, D.W.; Grutzner, J.B. J. Org. Chem. 1994, 59, 4516.
1639. Zanger, M.; Rabinowitz, J.L. J. Org. Chem. 1975, 40, 248.
1640. Ayres, R.L.; Michejda, C.J.; Rack, E.P. J. Am. Chem. Soc. 1971, 93, 1389.
1641. See Lenoir, D.; Chiappe, C. Chem. Eur. J. 2003, 9, 1037. For a theoretical study of these intermediates, see Okazaki, T.; Laali, K.K. J. Org. Chem. 2005, 70, 9139. Also see Zabalov, M.V.; Karlov, S.S.; Lemenovskii, D.A.; Zaitseva, G.S. J. Org. Chem. 2005, 70, 9175.
1642. See Dessau, R.M. J. Am. Chem. Soc. 1979, 101, 1344.
1643. See Cais, M. in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, pp. 993.
1644. See Fuller, G.; Stacey, F.W.; Tatlow, J.C.; Thomas, C.R. Tetrahedron 1962, 18, 123.
1645. Rozen, S.; Brand, M. J. Org. Chem. 1986, 51, 3607.
1646. Hara, S.; Nakahigashi, J.; Ishi-i, K.; Sawaguchi, M.; Sakai, H.; Fukuhara, T.; Yoneda, N. Synlett 1998, 495.
1647. Bissell, E.R.; Fields, D.B. J. Org. Chem. 1964, 29, 1591.
1648. Akana, J.A.; Bhattacharyya, K.X.; Müller, P.; Sadighi, J.P. J. Am. Chem. Soc. 2007, 129, 7736.
1649. See Bellucci, G.; Chiappe, C. J. Org. Chem. 1993, 58, 7120.
1650. Zheng, C.Y.; Slebocka-Tilk, H.; Nagorski, R.W.; Alvarado, L.; Brown, R.S. J. Org. Chem. 1993, 58, 2122.
1651. Bellucci, G.; Chiappe, C.; Bianchini, R.; Lenoir, D.; Herges, R. J. Am. Chem. Soc. 1995, 117, 12001.
1652. See Kuchar, E.J. in Patai, S. The Chemistry of Alkenes, Vol. 1, Wiley, NY, 1964, pp. 273–280.
1653. Chiappe, C.; Capraro, D.; Conte, V.; Picraccini, D. Org. Lett. 2001, 3, 1061.
1654. Bellesia, F.; Ghelfi, F.; Pagnoni, U.M.; Pinetti, A. J. Chem. Res. (S) 1989, 108, 360.
1655. Markó, I.E.; Richardson, P.R.; Bailey, M.; Maguire, A.R.; Coughlan, N. Tetrahedron Lett. 1997, 38, 2339.
1656. Markó, I.E.; Richardson, P.F. Tetrahedron Lett. 1991, 32, 1831.
1657. Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis Vol. 1, Wiley, NY, 1967, pp. 967–970. For a discussion of the mechanism, see Bellucci, G.; Bianchini, R.; Vecchiani, S. J. Org. Chem. 1986, 51, 4224.
1658. Nair, V.; Panicker, S.B.; Augstine, A.; George, T.G.; Thomas, S.; Vairamani, M. Tetrahedron 2001, 57, 7417.
1659. Ye, C.; Shreeve, J.M. J. Org. Chem. 2004, 69, 8561.
1660. El-Quisairi, A.K.; Qaseer, H.A.; Katsigras, G.; Lorenzi, P.; Tribedi, U.; Tracz, S.; Hartman, A.; Miller, J.A.; Henry, P.M. Org. Lett. 2003, 5, 439.
1661. Uemura, S.; Okazaki, H.; Onoe, A.; Okano, M. J. Chem. Soc. Perkin Trans. 1,1977, 676.
1662. Das, B.; Srinivas, Y.; Sudhakar, C.; Damodar, K.; Narender, R. Synth. Commun. 2009, 39, 220.
1663. See McCleland, C.W. in Pizey, J.S. Synthetic Reagents, Vol. 5, Wiley, NY, 1983, pp. 85–164.
1664. White, E.P.; Robertson, P.W. J. Chem. Soc. 1939, 1509.
1665. Buckles, R.E.; Forrester, J.L.; Burham, R.L.; McGee, T.W. J. Org. Chem. 1960, 25, 24.
1666. Negoro, T.; Ikeda, Y. Bull. Chem. Soc. Jpn. 1986, 59, 3519.
1667. Zefirov, N.S.; Sereda, G.A.; Sosounk, S.E.; Zyk, N.V.; Likhomanova, T.I. Synthesis 1995, 1359.
1668. See Sharts, C.M.; Sheppard, W.A. Org. React. 1974, 21, 125, see pp. 137–157; Boguslavskaya. L.S. Russ. Chem. Rev. 1984, 53, 1178.
1669. Evans, R.D.; Schauble, J.H. Synthesis 1987, 551; Kuroboshi, M.; Hiyama, T. Synlett 1991, 185.
1670. Pattison, F.L.M.; Peters, D.A.V.; Dean, F.H. Can. J. Chem. 1965, 43, 1689. For other methods, see Shimizu, M.; Nakahara, Y.; Yoshioka, H. J. Chem. Soc., Chem. Commun. 1989, 1881.
1671. Nojima, M.; Kerekes, I.; Olah, J.A. J. Org. Chem. 1979, 44, 3872. See Camps, F.; Chamorro, E.; Gasol, V.; Guerrero, A. J. Org. Chem. 1989, 54, 4294; Ichihara, J.; Funabiki, K.; Hanafusa, T. Tetrahedron Lett. 1990, 31, 3167.
1672. Barluenga, J.; González, J.M.; Campos, P.J.; Asensio, G. Angew. Chem. Int. Ed. 1985, 24, 319.
1673. Barluenga, J.; Rodríguez, M.A.; González, J.M.; Campos, P.J.; Asensio, G. Tetrahedron Lett. 1986, 27, 3303.
1674. Sinn, H.; Hopperdietzel, S.; Sauermann, D. Monatsh. Chem. 1965, 96, 1036.
1675. Hondrogiannis, G.; Lee, L.C.; Kabalka, G.W.; Pagni, R.M. Tetrahedron Lett. 1989, 30, 2069.
1676. Dabdoub, M.J.; Dabdoub, V.B.; Baroni, A.C.M. J. Am. Chem. Soc. 2001, 123, 9694.
1677. Kabalka, G.W.; Yang, K. Synth. Commun. 1998, 28, 3807; Kabalka, G.W.; Yang, K.; Reddy, N.K.; Narayana, A. Synth. Commun. 1998, 28, 925.
1678. See Jacobs, T.L. in Landor, S.R. The Chemistry of Allenes, Vol. 2, Acaademic Press, NY, 1982, pp. 466–483.
1679. Addends are listed in order of priority in the Cahn–Ingold–Prelog system (Sec. 4.E.i).
1680. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 638–642.
1681. See Boguslavskaya, L.S. Russ. Chem. Rev. 1972, 41, 740.
1682. Cambie, R.C.; Noall, W.I.; Potter, G.J.; Rutledge, P.S.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 1,1977, 266.
1683. See Antonioletti, R.; D'Auria, M.; De Mico, A.; Piancatelli, G.; Scettri, A. Tetrahedron 1983, 39, 1765.
1684. Horiuchi, C.A.; Ikeda, A.; Kanamori, M.; Hosokawa, H.; Sugiyama, T.; Takahashi, T.T. J. Chem. Res. (S) 1997, 60.
1685. Myint, Y.Y.; Pasha, M.A. Synth. Commun. 2004, 34, 4477.
1686. Masuda, H.; Takase, K.; Nishio, M.; Hasegawa, A.; Nishiyama, Y.; Ishii, Y. J. Org. Chem. 1994, 59, 5550.
1687. SeeDalton, D.R.; Dutta, V.P. J. Chem. Soc. B 1971, 85; Sisti, A.J. J. Org. Chem. 1970, 35, 2670.
1688. Smietana, M.; Gouverneur, V.; Mioskowski, C. Tetahedron Lett. 2000, 41, 193.
1689. Klunder, J.M.; Caron M.; Uchiyama, M.; Sharpless, K.B. J. Org. Chem. 1985, 50, 912.
1690. See Bremner, D.H. in Pizey, J.S. Synthetic Reagents, Vol. 6, Wiley, NY, 1985, pp. 9–59; Campbell, M.M.; Johnson, G. Chem. Rev. 1978, 78, 65.
1691. Damin, B.; Garapon, J.; Sillion, B. Synthesis 1981, 362.
1692. Ohta, M.; Sakata, Y.; Takeuchi, T.; Ishii, Y. Chem. Lett. 1990, 733.
1693. Carrera, I.; Brovetto, M.C.; Seoane, G.A. Tetrahedron Lett. 2006, 47, 7849.
1694. Sakurada, I.; Yamasaki, S.; Göttlich, R.; Iida, T.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2000, 122, 1245.
1695. DeCorso, A.R.; Panunzi, B.; Tingoli, M. Tetrahedron Lett. 2001, 42, 7245.
1696. Yadav, J.S.; Reddy, B.V.S.; Baishya, G.; Harshavardhan, S.J.; Chary, Ch.J.; Gupta, M.K. Tetrahedron Lett. 2005, 46, 3569.
1697. Urankar, D.; Rutar, I.; Modec, B.; Dolenc, D. Eur. J. Org. Chem. 2005, 2349.
1698. Migliorese, K.G.; Appelman, E.H.; Tsangaris, M.N. J. Org. Chem. 1979, 44, 1711.
1699. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 642–643.
1700. Zefirov, N.S.; Koz'min, A.S. Acc. Chem. Res. 1985, 18, 154; Sov. Sci. Rev. Sect. B 1985, 7, 297.
1701. Buss, E.; Rockstuhl, A.; Schnurpfeil, D. J. Prakt. Chem. 1982, 324, 197.
1702. Chirskaya, M.V.; Vasil'ev, A.A.; Sergovskaya, N.L.; Shovshinev, S.V.; Sviridov, S.I. Tetrahedron Lett. 2004, 45, 8811.
1703. Glover, S.A.; Goosen, A. Tetrahedron Lett. 1980, 21, 2005.
1704. Sanseverino, A.M.; de Mattos, M.C.S. Synthesis 1998, 1584. See Horiuchi, C.A.; Hosokawa, H.; Kanamori, M.; Muramatsu, Y.; Ochiai, K.; Takahashi, E. Chem. Lett. 1995, 13.
1705. Iranpoor, N.; Shekarriz, M. Tetahedron 2000, 56, 5209.
1706. Bresson, A.; Dauphin, G.; Geneste, J.; Kergomard, A.; Lacourt, A. Bull. Soc. Chim. Fr. 1970, 2432; 1971, 1080.
1707. Weissermel, K.; Lederer, M. Chem. Ber. 1963, 96, 77.
1708. Wilson, M.A.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 2,1976, 141. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 643–644.
1709. Rozen, S.; Lerman, O.; Kol, M.; Hebel, D. J. Org. Chem. 1985, 50, 4753.
1710. See Cardillo, G.; Orena, M. Tetrahedron 1990, 46, 3321; Dowle, M.D.; Davies, D.I. Chem. Soc. Rev. 1979, 8, 171. For a list of reagents that accomplish this, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1870–1876. Also see Bartlett, P.A. in Morrison, J.D. Organic Synthesis Vol. 3, Wiley, NY, 1984, pp. 411–454, 416–425.
1711. Corey, E.J.; Albonico, S.M.; Koelliker, V.; Schaaf, T.K.; Varma, R.K. J. Am. Chem. Soc. 1971, 93, 1491.
1712. Yaguchi, Y.; Akiba, M.; Harada, M.; Kato, T. Heterocycles 1996, 43, 601.
1713. Homsi, F.; Rousseau, G. J. Org. Chem. 1998, 63, 5255.
1714. Royer, A.C.; Mebane, R.C.; Swafford, A.M. Synlett 1993, 899.
1715. See Cambie, R.C.; Rutledge, P.S.; Somerville, R.F.; Woodgate, P.D. Synthesis 1988, 1009, and references cited therein.
1716. Genovese, S.; Epifano, F.; Pelucchini, C.; Procopio, A.; Curini, M. Tetrahedron Lett. 2010, 51, 5992.
1717. Garnier, J.M.; Robin, S.; Rousseau, G. Eur. J. Org. Chem. 2007, 3281.
1718. Inoue, T.; Kitagawa, O.; Kurumizawa, S.; Ochiai, O.; Taguchi, T. Tetrahedron Lett. 1995, 36, 1479.
1719. Haas, J.; Piguel, S.; Wirth, T. Org. Lett. 2002, 4, 297.
1720. Whitehead, D.C.; Yousefi, R.; Jaganathan, A.; Borhan, B. J. Am. Chem. Soc. 2010, 132, 3298; Zhou, L.; Tan, C.K.; Jiang, X.; Chen, F.; Yeung, Y.-Y. J. Am. Chem. Soc. 2010, 132, 15474; Murai, K.; Matsushita, T.; Nakamura, A.; Fukushima, S.; Shimura, M.; Fujioka, H. Angew. Chem. Int. Ed. 2010, 49, 9174.
1721. Ning, Z.; Jin, R.; Ding, J.; Gao, L. Synlett 2009, 2291.
1722. Simonot, B.; Rousseau, G. Tetrahedron Lett. 1993, 34, 4527.
1723. Knapp, S.; Rodriques, K.E. Tetrahedron Lett. 1985, 26, 1803.
1724. Tamaru, Y.; Kawamura, S.; Tanaka, K.; Yoshida, Z. Tetrahedron Lett. 1984,25, 1063.
1725. Tseng, C.K.; Teach, E.G.; Simons, R.W. Synth. Commun. 1984, 14, 1027.
1726. When a general group (e.g., halo) is used, its priority is that of the lowest member of its group (see Ref. 1680). Thus the general name for this transformation is halo-alkylsulfonyl addition because “halo” has the same priority as “fluoro”, its lowest member.
1727. Sinnreich, J.; Asscher, M. J. Chem. Soc. Perkin Trans. 1,1972, 1543.
1728. Quebatte, L.; Thommes, K.; Severin, K. J. Am. Chem. Soc. 2006, 128, 7440.
1729. Nair, V.; Augustine, A.; George, T.G.; Nair, L.G. Tetrahedron Lett. 2001, 42, 6763.
1730. Amiel, Y. J. Org. Chem. 1974, 39, 3867; Okuyama, T.; Izawa, K.; Fueno, T. J. Org. Chem. 1974, 39, 351.
1731. See Rasteikiene, L.; Greiciute, D.; Lin'kova, M.G.; Knunyants, I.L. Russ. Chem. Rev. 1977, 46, 548; Kühle, E. Synthesis 1971, 563.
1732. Bellesia, F.; Ghelfi, F.; Pagnoni, U.M.; Pinetti, A. J. Chem. Res. (S) 1987, 238. See also, Liu, H.; Nyangulu, J.M. Tetrahedron Lett. 1988, 29, 5467.
1733. Woodgate, P.D.; Janssen, S.J.; Rutledge, P.S.; Woodgate, S.D.; Cambie, R.C. Synthesis 1984, 1017, and references cited therein. See also, Watanabe, N.; Uemura, S.; Okano, M. Bull. Chem. Soc. Jpn. 1983, 56, 2458.
1734. Cambie, R.C.; Larsen, D.S.; Rutledge, P.S.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 1,1981, 58.
1735. Prakash, O.; Sharma, V.; Batra, H.; Moriarty, R.M. Tetrahedron Lett. 2001, 42, 553.
1736. Yadav, J.S.; Reddy, B.V.S.; Gupta, M.K. Synthesis 2004, 1983.
1737. For a discsssion of substituent effects, see Brammer, C.N.; Nelson, D.J.; Li, R. Tetrahedron Lett. 2007, 48, 3237.
1738. Fujisawa, T.; Kobori, T. Chem. Lett. 1972, 935; Capozzi, F.; Capozzi, G.; Menichetti, S. Tetrahedron Lett. 1988, 29, 4177.
1739. See Mirskova, A.N.; Drozdova, T.I.; Levkovskaya, G.G.; Voronkov, M.G. Russ. Chem. Rev. 1989, 58, 250; Neale, R.S. Synthesis 1971, 1.
1740. Tuaillon, J.; Couture, Y.; Lessard, J. Can. J. Chem. 1987, 65, 2194, and other papers in this series. For a review, see Labeish, N.N.; Petrov, A.A. Russ. Chem. Rev. 1989, 58, 1048.
1741. Yeung, Y.-Y.; Gao, X.; Corey, E.J. J. Am. Chem. Soc. 2006, 128, 9644.
1742. Wei, H.-X.; Ki, S.H.; Li, G. Tetrahedron 2001, 57, 3869.
1743. Minakata, S.; Yoneda, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2006, 8, 967.
1744. See Chow, Y.L.; Danen, W.C.; Nelson, S.F.; Rosenblatt, D.H. Chem. Rev. 1978, 78, 243.
1745. Besson, L.; Goré, J.; Cazes, B. Tetrahedron Lett. 1995, 36, 3857.
1746. Li, G.; Wei, H.-X.; Kim, S.H. Tetrahedron 2001, 57, 8407.
1747. Xu, X.; Kotti, S.R.S.S.; Liu, J.; Cannon, J.F.; Headley, A.D.; Li, G. Org. Lett. 2004, 6, 4881.
1748. See Kadzyauskas, P.P.; Zefirov, N.S. Russ. Chem. Rev. 1968, 37, 543.
1749. See Gowenlock, B.G.; Richter-Addo, G.B. Chem. Rev. 2004, 104, 3315.
1750. Shvekhgeimer, G.A.; Smirnyagin, V.A.; Sadykov, R.A.; Novikov, S.S. Russ. Chem. Rev. 1968, 37, 351.
1751. See Meinwald, J.; Meinwald, Y.C.; Baker, III, T.N. J. Am. Chem. Soc. 1964, 86, 4074.
1752. Shechter, H. Rec. Chem. Prog.,1964, 25, 55–76.
1753. Schlubach, H.H.; Braun, A. Liebigs Ann. Chem. 1959, 627, 28.
1754. Sharts, C.M.; Sheppard, W.A. Org. React. 1974, 21, 125–406, see pp. 236–243.
1755. Knunyants, I.L.; German, L.S.; Rozhkov, I.N. Bull Acad. Sci. USSR Div. Chem. Sci. 1963, 1794.
1756. Olah, G.A.; Nojima, M. Synthesis 1973, 785.
1757. Dehnicke, K. Angew. Chem. Int. Ed. 1979, 18, 507; Hassner, A. Acc. Chem. Res. 1971, 4, 9; Biffin, M.E.C.; Miller, J.; Paul, D.B. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 136–147. See Nair, V.; George, T.G.; Sheeba, V.; Augustine, A.; Balagopal, L.; Nair, L.G. Synlett 2000, 1597.
1758. Curini, M.; Epifano, F.; Marcotullio, M.C.; Rosati, O. Tetrahedron Lett. 2002, 43, 1201.
1759. See, however, Cambie, R.C.; Hayward, R.C.; Rutledge, P.S.; Smith-Palmer, T.; Swedlund, B.E.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 1,1979, 180.
1760. Hassner, A.; Keogh, J. J. Org. Chem. 1986, 51, 2767.
1761. Olah, G.A.; Wang, Q.; Li, X.; Prakash, G.K.S. Synlett 1990, 487.
1762. Hassner, A.; Keogh, J. Tetrahedron Lett. 1975, 1575.
1763. Hassner, A.; Teeter, J.S. J. Org. Chem. 1971, 36, 2176.
1764. See Cambie, R.C.; Jurlina, J.L.; Rutledge, P.S.; Swedlund, B.E.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 1,1982, 327. Also see Hassner, A. Intra-Sci. Chem. Rep.,1970, 4, 109.
1765. Hassner, A.; Isbister, R.J.; Friederang, A. Tetrahedron Lett. 1969, 2939.
1766. Hassner, A.; Matthews, G.J.; Fowler, F.W. J. Am. Chem. Soc. 1969, 91, 5046.
1767. Levy, A.B.; Brown, H.C. J. Am. Chem. Soc. 1973, 95, 4067.
1768. Ali, S.I.; Nikalje, M.D.; Sudalai, A. Org. Lett. 1999, 1, 705.
1769. Schmerling, L. in Olah, G.A. Friedel–Crafts and Related Reactions, Vol. 2, Wiley, NY, 1964, pp. 1133–1174; Mayr, H.; Schade, C.; Rubow, M.; Schneider, R. Angew. Chem. Int. Ed. 1987, 26, 1029.
1770. See Pock, R.; Mayr, H.; Rubow, M.; Wilhelm, E. J. Am. Chem. Soc. 1986, 108, 7767.
1771. Kolyaskina, Z.N.; Petrov, A.A. J. Gen. Chem. USSR 1962, 32, 1067.
1772. See Maroni, R.; Melloni, G.; Modena, G. J. Chem. Soc. Perkin Trans. 1,1973, 2491; 1974, 353.
1773. See Freidlina, R.Kh.; Velichko, F.K. Synthesis 1977, 145; Freidlina, R.Kh.; Chukovskaya, E.C. Synthesis 1974, 477.
1774. For other initiators, see Tsuji, J.; Sato, K.; Nagashima, H. Tetrahedron 1985, 41, 393; Phelps, J.C.; Bergbreiter, D.E.; Lee, G.M.; Villani, R.; Weinreb, S.M. Tetrahedron Lett. 1989, 30, 3915.
1775. See Martin, P.; Steiner, E.; Streith, J.; Winkler, T.; Bellus, D. Tetrahedron 1985, 41, 4057. Also see Mitani, M.; Nakayama, M.; Koyama, K. Tetrahedron Lett. 1980, 21, 4457.
1776. Simal, F.; Wlodarczak, L.; Demonceau, A.; Noels, A.F. Eur. J. Org. Chem. 2001, 2689.
1777. For a review with respect to fluoroalkenes, see Paleta, O. Fluorine Chem. Rev. 1977, 8, 39.
1778. Nakamura, T.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Synlett 1998, 1351.
1779. Yorimitsu, H.; Nakamura, T.; Shinokubo, H.; Oshima, K. J. Org. Chem. 1998, 63, 8604.
1780. Baciocchi, E.; Muraglia, E. Tetrahedron Lett. 1994, 35, 2763.
1781. See Groves, J.K. Chem. Soc. Rev. 1972, 1, 73; Nenitzescu, C.D.; Balaban, A.T. in Olah, G.A. Friedel–Crafts and Related Reactions, Vol. 3, Wiley, NY, 1964, pp. 1033–1152.
1782. Jones, N.; Taylor, H.T.; Rudd, E. J. Chem. Soc. 1961, 1342.
1783. See Melikyan, G.G.; Babayan, E.V.; Atanesyan, K.A.; Badanyan, Sh.O. J. Org. Chem. USSR 1984, 20, 1884.
1784. Bedekar, A.V.; Nair, K.B.; Soman, R. Synth. Commun. 1994, 24, 2299.
1785. Hua, R.; Onozawa, S.-y.; Tanaka, M. Chemistry: European J. 2005, 11, 3621.
1786. See Brownstein, S.; Morrison, A.; Tan, L.K. J. Org. Chem. 1985, 50, 2796.
1787. Yen, V.Q. Ann. Chim. (Paris) 1962, [13] 7, 785.
1788. Hua, R.; Tanaka, M. Tetrahedron Lett. 2004, 45, 2367.
1789. See Hudlicky, M. Oxidations in Organic Chemistry American Chemical Society, Washington, 1990, pp. 67–73; Haines, A.H. Methods for the Oxidation of Organic Compounds Academic Press, NY, 1985, pp. 73–98, 278–294; Sheldon, R.A.; Kochi, J.K. Metal-Catalyzed Oxidations of Organic Compounds Academic Press, NY, 1981, pp. 162–171, 294–296. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 996–1003.
1790. See Schröder, M. Chem. Rev. 1980, 80, 187. Also see, Norrby, P.-O.; Gable, K.P. J. Chem. Soc. Perkin Trans. 2,1996, 171; Lohray, B.B.; Bhushan, V. Tetrahedron Lett. 1992, 33, 5113.
1791. Criegee, R. Liebigs Ann. Chem. 1936, 522, 75.
1792. See Fatiadi, A.J. Synthesis 1987, 85; Nelson, D.J.; Henley, R.L. Tetrahedron Lett. 1995, 36, 6375.
1793. Crispino, G.A.; Jeong, K.-S.; Kolb, H.C.; Wang, Z.-M.; Xu, D.; Sharpless, K.B. J. Org. Chem. 1993, 58, 3785.
1794. Wiberg, K.B.; Wang, Y.-g.; Sklenak, S.; Deutsch, C.; Trucks, G. J. Am. Chem. Soc. 2006, 128, 11537.
1795. de Boer, J.W.; Brinksma, J.; Browne, W.R.; Meetsma, A.; Alsters, P.L.; Hage, R.; Feringa, B.L. J. Am. Chem. Soc. 2005, 127, 7990.
1796. See J![]() rgensen, K.A.; Hoffmann, R. J. Am. Chem. Soc. 1986, 108, 1867.
rgensen, K.A.; Hoffmann, R. J. Am. Chem. Soc. 1986, 108, 1867.
1797. See Ogino, T.; Hasegawa, K.; Hoshino, E. J. Org. Chem. 1990, 55, 2653. See, however, Freeman, F.; Kappos, J.C. J. Org. Chem. 1989, 54, 2730, and other papers in this series.
1798. Iwasawa, N.; Kato, T.; Narasaka, K. Chem. Lett. 1988, 1721. See also, Ray, R.; Matteson, D.S. Tetrahedron Lett. 1980, 449.
1799. Akashi, K.; Palermo, R.E.; Sharpless, K.B. J. Org. Chem. 1978, 43, 2063.
1800. See Usui, Y.; Sato, K.; Tanaka, M. Angew. Chem. Int. Ed. 2003, 42, 5623.
1801. Bergstad, K.; Piet, J.J.N.; Bäckvall, J.-E. J. Org. Chem. 1999, 64, 2545.
1802. Torii, S.; Liu, P.; Tanaka, H. Chem. Lett. 1995, 319; Soderquist, J.A.; Rane, A.M.; López, C.J. Tetrahedron Lett. 1993, 34, 1893. See Corey, E.J.; Noe, M.C.; Grogan, M.J. Tetrahedron Lett. 1994, 35, 6427; Imada, Y.; Saito, T.; Kawakami, T.; Murahashi, S.-I. Tetrahedron Lett. 1992, 33, 5081 for oxidation using an asymmetric ligand.
1803. Chen, K.; Costas, M.; Kim, J.; Tipton, A.K.; Que, Jr., L. J. Am. Chem. Soc. 2002, 124, 3026.
1804. Ley, S.V.; Ramarao, C.; Lee, A.-L.; Ostergaard, N.; Smith, S.C.; Shirley, I.M. Org. Lett. 2003, 5, 185.
1805. Nagayama, S.; Endo, M.; Kobayashi, S. J. Org. Chem. 1998, 63, 6094.
1806. Choudary, B.M.; Chowdari, N.S.; Jyothi, K.; Kantam, M.L. J. Am. Chem. Soc. 2002, 124, 5341.
1807. Closson, A.; Johansson, M.; Bäckvall, J.-E. Chem. Commun. 2004, 1494; Branco, L.C.; Serbanovic, A.; da Ponte, M.N.; Afonso, C.A.M. Chem. Commun. 2005, 107.
1808. Oldenburg, P.D.; Shteinman, A.A.; Que, Jr., L. J. Am. Chem. Soc. 2005, 127, 15672.
1809. Yip, W.-P.; Ho, C.-M.; Zhu, N.; Lau, T.-C.; Che, C.-M. Chemistry: Asian J. 2008, 3, 70.
1810. Motorina, I.; Crudden, C.M. Org. Lett. 2001, 3, 2325.
1811. See Wolfe, S.; Ingold, C.F.; Lemieux, R.U. J. Am. Chem. Soc. 1981, 103, 938; Wolfe, S.; Ingold, C.F. J. Am. Chem. Soc. 1981, 103, 940. Also see, Lohray, B.B.; Bhushan, V.; Kumar, R.K. J. Org. Chem. 1994, 59, 1375.
1812. See Taylor, J.E.; Green, R. Can. J. Chem. 1985, 63, 2777.
1813. See Ogino, T.; Mochizuki, K. Chem. Lett. 1979, 443.
1814. Taylor, J.E.; Williams, D.; Edwards, K.; Otonnaa, D.; Samanich, D. Can. J. Chem. 1984, 62, 11; Taylor, J.E. Can. J. Chem. 1984, 62, 2641.
1815. Varma, R.S.; Naicker, K.P. Tetrahedron Lett. 1998, 39, 7463.
1816. Donohoe, T.J.; Blades, K.; Moore, P.R.; Waring, M.J.; Winter, J.J.G.; Helliwell, M.; Newcombe, N.J.; Stemp, G. J. Org. Chem. 2002, 67, 7946.
1817. Fringuelli, F.; Germani, R.; Pizzo, F.; Savelli, G. Synth. Commun. 1989, 19, 1939.
1818. See Brimble, M.A.; Nairn, M.R. J. Org. Chem. 1996, 61, 4801.
1819. For another possible mechanism: Woodward, R.B.; Brutcher, Jr., F.V. J. Am. Chem. Soc. 1958, 80, 209.
1820. Also see Corey, E.J.; Das, J. Tetrahedron Lett. 1982, 23, 4217.
1821. See Horiuchi, C.A.; Satoh, J.Y. Chem. Lett. 1988, 1209; Campi, E.M.; Deacon, G.B.; Edwards, G.L.; Fitzroy, M.D.; Giunta, N.; Jackson, W.R.; Trainor, R. J. Chem. Soc., Chem. Commun. 1989, 407.
1822. Cambie, R.C.; Hayward, R.C.; Roberts, J.L.; Rutledge, P.S. J. Chem. Soc. Perkin Trans. 1,1974, 1858, 1864; Cambie, R.C.; Rutledge, P.S. Org. Synth. VI, 348.
1823. Robinson, R.I.; Woodward, S. Tetrahedron Lett. 2003, 44, 1655.
1824. Seayad, J.; Seayad, A.M.; Chai, C.L.L. Org. Lett. 2010, 12, 1412.
1825. Schultz, M.J.; Sigman, M.S. J. Am. Chem. Soc. 2006, 128, 1460.
1826. Zhang, Y.; Sigman, M.S. J. Am. Chem. Soc. 2007, 129, 3076.
1827. For diastereoselective, but not enantioselective, addition of OsO4, see Vedejs, E.; McClure, C.K. J. Am. Chem. Soc. 1986, 108, 1094; Evans, D.A.; Kaldor, S.W. J. Org. Chem. 1990, 55, 1698.
1828. Lohray, B.B. Tetrahedron Asymmetry 1992, 3, 1317; Zaitsev, A.B.; Adolfsson, H. Synthesis 2006, 1725.
1829. McNamara, C.A.; King, F.; Bradley, M. Tetrahedron Lett. 2004, 45, 8527; Jiang, R.; Kuang, Y.; Sun, X.; Zhang, S. Tetrahedron Asymmetry 2004, 15, 743.
1830. Wai, J.S.M.; Marko, I.; Svendsen, J.S.; Finn, M.G.; Jacobsen, E.N.; Sharpless, K.B. J. Am. Chem. Soc. 1989, 111, 1123; Sharpless, K.B.; Amberg, W.; Beller, M.; Chens, H.; Hartung, J.; Kawanami, Y.; Lübben, D.; Manoury, E.; Ogino, Y.; Shibata, T.; Ukita, T. J. Org. Chem. 1991, 56, 4585.
1831. Kolb, H.C.; Van Nieuwenhze, M.S.; Sharpless, K.B. Chem. Rev. 1994, 94, 2483. Also see, Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 294–301.
1832. Wang, L.; Sharpless, K.B. J. Am. Chem. Soc. 1992, 114, 7568; Xu, D.; Crispino, G.A.; Sharpless, K.B. J. Am. Chem. Soc. 1992, 114, 7570; Rosini, C.; Tanturli, R.; Pertici, P.; Salvadori, P. Tetrahedron Asymmetry 1996, 7, 2971; Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.-S.; Kwong, H.-L.; Morikawa, K.; Wang, Z.-M.; Xu, D.; Zhang, X.-L. J. Org. Chem. 1992, 57, 2768.
1833. Bolm, C.; Gerlach, A. Eur. J. Org. Chem. 1998, 21. For a review, see Karjalainen, J.K.; Hormi, O.E.O.; Sherrington, D.C. Tetrahedron Asymmetry 1998, 9, 1563.
1834. Song, C.E.; Yang, J.W.; Ha, H.-J. Tetrahedron Asymmetry 1997, 8, 841.
1835. See Corey, E.J.; Noe, M.C. J. Am. Chem. Soc. 1996, 118, 319; Norrby, P.-O.; Kolb, H.C.; Sharpless, K.B. J. Am. Chem. Soc. 1994, 116, 8470; Wu, Y.-D.; Wang, Y.; Houk, K.N. J. Org. Chem. 1992, 57, 1362. Also see Nelson, D.W.; Gypser, A.; Ho, P.T.; Kolb, H.C.; Kondo, T.; Kwong, H.-L.; McGrath, D.V.; Rubin, A.E.; Norrby, P.-O.; Gable, K.P.; Sharpless, K.B. J. Am. Chem. Soc. 1997, 119, 1840.
1836. See Annunziata, R.; Cinquini, M.; Cozzi, F.; Raimondi, L.; Stefanelli, S. Tetrahedron Lett. 1987, 28, 3139; Hirama, M.; Oishi, T.; Itô, S. J. Chem. Soc., Chem. Commun. 1989, 665.
1837. Walsh, P.J.; Sharpless, K.B. Synlett 1993, 605.
1838. Park, C.Y.; Kim, B.M.; Sharpless, K.B. Tetrahedron Lett. 1991, 32, 1003.
1839. Oishi, T.; Iida, K.; Hirama, M. Tetrahedron Lett. 1993, 34, 3573.
1840. See Jacobsen, E.N.; Marko, I.; France, M.B.; Svendsen, J.S.; Sharpless, K.B. J. Am. Chem. Soc. 1989, 111, 737.
1841. Jacobsen, E.N.; Marko, I.; Mungall, W.S.; Schröder, G.; Sharpless, K.B. J. Am. Chem. Soc. 1988, 110, 1968.
1842. See Branco, L.C.; Afonso, C.A.M. J. Org. Chem. 2004, 69, 4381.
1843. Sharpless, K.B.; Amberg, W.; Bennani, Y.L.; Crispino, G.A.; Hartung, J.; Jeong, K.-S.; Kwong, H.-L.; Morikawa, K.; Wang, Z.-M.; Xu, D.; Zhang, X.-L. J. Org. Chem. 1992, 57, 2768.
1844. For a review, see Français, A.; Bedel, O.; Haudrechy, A. Tetrahedron 2008, 64, 2495.
1845. Kobayashi, S.; Ishida, T.; Akiyama, R. Org. Lett. 2001, 3, 2649.
1846. Kuang, Y.-Q.; Zhang, S.-Y.; Wei, L.-L. Tetrahedron Lett. 2001, 42, 5925.
1847. Lee, B.S.; Mahajan, S.; Janda, K.D. Tetrahedron Lett. 2005, 46, 4491.
1848. Jonsson, S.Y.; Adolfsson, H.; Bäckvall, J.-E. Org. Lett. 2001, 3, 3463.
1849. Krief, A.; Colaux-Castillo, C. Tetrahedron Lett. 1999, 40, 4189.
1850. Junttila, M.H.; Hormi, O.E.O. J. Org. Chem. 2004, 69, 4816.
1851. Aladro, F.J.; Guerra, I.M.; Moreno-Dorado, F.J.; Bustamante, J.M.; Jorge, Z.D.; Massanet, G.M. Tetrahedron Lett. 2000, 41, 3209.
1852. Kokubo, T.; Sugimoto, T.; Uchida, T.; Tanimoto, S.; Okano, M. J. Chem. Soc., Chem. Commun. 1983, 769.
1853. Hauser, F.M.; Ellenberger, S.R.; Clardy, J.C.; Bass, L.S. J. Am. Chem. Soc. 1984, 106, 2458; Johnson, C.R.; Barbachyn, M.R. J. Am. Chem. Soc. 1984, 106, 2459.
1854. Trudeau, S.; Morgan, J.B.; Shrestha, M.; Morken, J.P. J. Org. Chem. 2005, 70, 9538.
1855. For a review, see Moriarty, R.M. Sel Org. Transform. 1972, 2, 183–237.
1856. See Uemura, S.; Miyoshi, H.; Tabata, A.; Okano, M. Tetrahedron 1981, 37, 291; Uemura, S. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wley, NY, 1987, pp. 473–538, 497–513; Uemura, S. in Pizey, J.S. Synthetic Reagents, Vol. 5, Wiley, NY, 1983, pp. 165–187.
1857. For another method see Fristad, W.E.; Peterson, J.R. Tetrahedron 1984, 40, 1469.
1858. See Bäckvall, J.E.; Awasthi, A.K.; Renko, Z.D. J. Am. Chem. Soc. 1987, 109, 4750 and references cited therein; Bäckvall, J.E. Bull. Soc. Chim. Fr. 1987, 665; New. J. Chem. 1990, 14, 447. For another method, see Uemura, S.; Fukuzawa, S.; Patil, S.R.; Okano, M. J. Chem. Soc. Perkin Trans. 1,1985, 499.
1859. Shimada, T.; Mukaide, K.; Shinohara, A.; Han, J.W.; Hayashi, T. J. Am. Chem. Soc. 2004, 124, 1584.
1860. DeBergh, J.R.; Spivey, K.M.; Ready, J.M. J. Am. Chem. Soc. 2008, 130, 7828.
1861. Sarma, K.; Borthakur, N.; Goswami, A. Tetrahedron Lett. 2007, 48, 6776.
1862. Wang, A.; Jiang, H.; Chen, H. J. Am. Chem. Soc. 2009, 131, 3846.
1863. Elgemeie, G.H.; Sayed, S.H. Synthesis 2001, 1747.
1864. Gibson, D.T.; Koch, J.R.; Kallio, R.E. Biochemistry 1968, 7, 2653; Brown, S.M. in Hudlicky, T. Organic Synthesis: Theory and Practice JAI Press, Greenwich, CT., 1993, Vol. 2, p. 113; Carless, H.A.J. Tetrahedron Asymmetry 1992, 3, 795.
1865. Gibson, D.T.; Koch, J.R.; Schuld, C.L.; Kallio, R.E. Biochemistry 1968, 7, 3795; Hudlicky, T.; Price, J.D. Synlett. 1990, 159.
1866. Gibson, D.T.; Hensley, M.; Yoshioka, H.; Mabry, T.J. Biochemsitry,1970, 9, 1626.
1867. Hudlicky, T.; Gonzalez, D.; Gibson, D.T. Aldrichimica Acta 1999, 32, 35; Ley, S.V.; Redgrave, A.J. Synlett 1990, 393. Also see, Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 303–306.
1868. For a list of reagents, including peroxyacids and others, used for epoxidation, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 915–927.
1869. See Hudlicky, M. Oxidations in Organic Chemistry American Chemical Society, Washington, 1990, pp. 60–64; Haines, A.H. Methods for the Oxidation of Organic Compunds, Academic Press, NY, 1985, pp. 98–117, 295–303; Dryuk, V.G. Russ. Chem. Rev. 1985, 54, 986; Plesnicar, B. in Trahanovsky, W.S. Oxidation in Organic Chemistry pt. C, Academic Press, NY, 1978, pp. 211–252; Hiatt, R. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 2; Marcel Dekker, NY, 1971; pp. 113–140.
1870. Emmons, W.D.; Pagano, A.S. J. Am. Chem. Soc. 1955, 77, 89.
1871. Rastetter, W.H.; Richard, T.J.; Lewis, M.D. J. Org. Chem. 1978, 43, 3163.
1872. Foti, C.J.; Fields, J.D.; Kropp, P.J. Org. Lett. 1999, 1, 903.
1873. Stack, T.D.P. Org. Lett. 2003, 5, 2469; Murphy, A.; Pace, A.; Stack, T.D.P. Org. Lett. 2004, 6, 3119.
1874. See Finn, M.G.; Sharpless, K.B. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Wiley, NY, 1985, pp. 247–308; Bach, R.D.; Canepa, C.; Winter, J.E.; Blanchette, P.E. J. Org. Chem. 1997, 62, 5191; Freccero, M.; Gandolfi, R.; Sarzi-Amadè, M.; Rastelli, A. J. Org. Chem. 2002, 67, 8519.
1875. Bartlett, P.D. Rec. Chem. Prog.,1957, 18, 111. For other proposed mechanisms see Kwart, H.; Hoffman, D.M. J. Org. Chem. 1966, 31, 419; Hanzlik, R.P.; Shearer, G.O. J. Am. Chem. Soc. 1975, 97, 5231.
1876. Freccero, M.; Gandolfi, R.; Sarzi-Amadè, M.; Rastelli, A. J. Org. Chem. 2004, 69, 7479. See also, Vedejs, E.; Dent III, W.H.; Kendall, J.T.; Oliver, P.A. J. Am. Chem. Soc. 1996, 118, 3556.
1877. See Gisdakis, P.; Rösch, N. Eur. J. Org. Chem. 2001, 719.
1878. Schneider, H.; Becker, N.; Philippi, K. Chem. Ber. 1981, 114, 1562; Batog, A.E.; Savenko, T.V.; Batrak, T.A.; Kucher, R.V. J. Org. Chem. USSR 1981, 17, 1860.
1879. See Freccero, M.; Gandolfi, R.; Sarzi-Amade, M.; Rastelli, A. J. Org. Chem. 2000, 65, 8948.
1880. See Houk, K.N.; Liu, J.; DeMello, N.C.; Condroski, K.R. J. Am. Chem. Soc. 1997, 119, 10147.
1881. Arends, I.W.C.E. Angew. Chem. Int. Ed. 2006, 45, 6250.
1882. See Deubel, D.V.; Frenking, G.; Gisdakis, P.; Herrmann, W.A.; Rösch, N.; Sundermeyer, J. Acc. Chem. Res. 2004, 37, 645.
1883. La: Nemoto, T.; Kakei, H.; Gnanadesikan, V.; Tosaki, S.-y.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2002, 124, 14544. Mn: Lane, B.S.; Vogt, M.; De Rose, V.T.; Burgess, K. J. Am. Chem. Soc. 2002, 124, 11946. Ti: Lattanzi, A.; Iannece, P.; Screttri, A. Tetrahedron Lett. 2002, 43, 5629. Pd: Yu, J.-Q.; Corey, E.J. Org. Lett. 2002, 4, 2727. Ru: Adam, W.; Alsters, P.L.; Neumann, R.; Saha-Möller, C.; Sloboda-Rozner, D.; Zhang, R. Synlett 2002, 2011. V: Sharpless, K.B.; Verhoeven, T.R. Aldrichimica Acta 1979, 12, 63; Torres, G.; Torres, W.; Prieto, J.A. Tetrahedron 2004, 60, 10245.
1884. See Hiatt, R. in Augustine, R.L.; Trecker, D.J. Oxidation, Vol. 2, Marcel Dekker, NY, 1971, p 124.
1885. Anilkumar, G.; Bitterlich, B.; Gelalcha, F.G.; Tse, M.K.; Beller, M. Chem. Commun. 2007, 289; Bitterlich, B.; Schröder, K.; Tse, M.K.; Beller, M. Eur. J. Org. Chem. 2008, 4867.
1886. Sawada, Y.; Matsumoto, K.; Katsuki, T. Angew. Chem. Int. Ed. 2007, 46, 4559; Malkov, A.V.; Bourhani, Z.; Ko![]() ovský, P. Org. Biomol. Chem. 2005, 3, 3194; Matsumoto, K.; Sawada, Y.; Katsuki, T. Pure Appl. Chem. 2008, 80, 1071.
ovský, P. Org. Biomol. Chem. 2005, 3, 3194; Matsumoto, K.; Sawada, Y.; Katsuki, T. Pure Appl. Chem. 2008, 80, 1071.
1887. Zeng, W.; Ballard, T.E.; Melander, C. Tetrahedron Lett. 2006, 47, 5923. See Malkov, A.V.; Czemerys, L.; Malyshev, D.A. J. Org. Chem. 2009, 74, 3350.
1888. See Adam, W.; Curci, R.; Edwards, J.O. Acc. Chem. Res. 1989, 22, 205.
1889. Majetich, G.; Hicks, R.; Sun, G.-r.; McGill, P. J. Org. Chem. 1998, 63, 2564; Murray, R.W.; Iyanar, K. J. Org. Chem. 1998, 63, 1730.
1890. Yamaguchi, K.; Ebitani, K.; Kaneda, K. J. Org. Chem. 1999, 64, 2966.
1891. Chan, W.-K.; Liu, P.; Yu, W.-Y.; Wong, M.-K.; Che, C.-M. Org. Lett. 2004, 6, 1597.
1892. For an example without microwave irradiation, see Pillai, U.R.; Sahle-Demessie, E.; Varma, R.S. Synth. Commun. 2003, 33, 2017.
1893. Pillai, U.R.; Sahle-Demessie, E.; Varma, R.S. Tetrahedron Lett. 2002, 43, 2909.
1894. Van Vliet, M.C.A.; Arends, I.W.C.E.; Sheldon, R.A. Tetrahedron Lett. 1999, 40, 5239.
1895. Yamazaki, S. Tetrahedron 2008, 64, 9253; Yamazaki, S. Org. Biomol. Chem. 2007, 5, 2109–2113; Saladino, R.; Neri, V.; Pelliccia, A.R.; Caminiti, R.; Sadun, C. J. Org. Chem. 2002, 67, 1323.
1896. Vaino, A.R. J. Org. Chem. 2000, 65, 4210.
1897. See Iskra, J.; Bonnet-Delpon, D.; Bégué, J.-P. Tetrahedron Lett. 2002, 43, 1001.
1898. Owens, G.S.; Abu-Omar, M.M. Chem. Commun. 2000, 1165.
1899. Tong, K.-H.; Wong, K.-Y.; Chan, T.H. Org. Lett. 2003, 5, 3423.
1900. Srinivas, K.A.; Kumar, A.; Chauhan, S.M.S. Chem. Commun. 2002, 2456.
1901. Tse, M.K.; Bhor, S.; Klawonn, M.; Döbler, C.; Beller, M. Tetrahedron Lett. 2003, 44, 7479.
1902. Li, Z.; Xia, C.-G. Tetrahedron Lett. 2003, 44, 2069.
1903. Geng, X.-L.; Wang, Z.; Li, X.-Q.; Zhang, C. J. Org. Chem. 2005, 70, 9610.
1904. Bogdal, D.; Lukasiewicz, M.; Pielichowski, J.; Bednarz, S. Synth. Commun. 2005, 35, 2973.
1905. Orendt, A.M.; Roberts, S.W.; Rainier, J.D. J. Org. Chem. 2006, 71, 5565.
1906. Xia, Q.-H.; Ge, H.-Q.; Ye, C.-P.; Liu, Z.-M.; Su, K.-X. Chem. Rev. 2005, 105, 1603.
1907. Kubo, T.; Peters, M.W.; Meinhold, P.; Arnold, F.H. Chemistry: European J. 2006, 12, 1216. For a method of electrochemical regenration of monooxygenase, see Hollmann, F.; Hofstetter, K.; Habicher, T.; Hauer, B.; Schmid, A. J. Am. Chem. Soc. 2005, 127, 6540.
1908. Chen, Y.; Reymond, J.-L. Synthesis 2001, 934.
1909. Bulman Page, P.C.; Buckley, B.R.; Rassias, G.A.; Blacker, A.J. Eur. J. Org. Chem. 2006, 803.
1910. Weitz, E.; Scheffer, A. Chem. Ber. 1921, 54, 2327. See Enders, D.; Zhu, J.; Raabe, G. Angew. Chem. Int. Ed. 1996, 35, 1725.
1911. Helder, R.; Hummelen, J.C.; Laane, R.W.P.M.; Wiering, J.S.; Wynberg, H. Tetrahedron Lett. 1976, 17, 1831; Wynberg, H.; Marsman, B. J. Org. Chem. 1980, 45, 158; Pluim, H.; Wynberg, H. J. Org. Chem. 1980, 45, 2498.
1912. Arai, S.; Shirai, Y.; Ishida, T.; Shioiri, T. Tetrahedron 1999, 55, 6375; Corey, E.J.; Zhang, F.-Y. Org. Lett. 1999, 1, 1287; Lygo, B.; Wainwright, P.G. Tetrahedron 1999, 55, 6289. See Adam, W.; Rao, P.B.; Degen, H.-G.; Levai, A.; Patonay, T.; Saha-Moller, C.R. J. Org. Chem. 2002, 67, 259.
1913. Watanabe, S.; Arai, T.; Sasai, H.; Bougauchi, M.; Shibasaki, M. J. Org. Chem. 1998, 63, 8090.
1914. Lygo, B.; Wainwright, P.G. Tetrahedron Lett. 1998, 39, 1599.
1915. Lygo, B.; To, D.C.M. Tetrahedron Lett. 2001, 42, 1343.
1916. Adam, W.; Rao, P.B.; Degen, H.-G.; Saha-Möller, C.R. Tetrahedron Asymmetry 2001, 12, 121.
1917. For example, see Ledon, H.J.; Durbut, P.; Varescon, F. J. Am. Chem. Soc. 1981, 103, 3601; Mimoun, H.; Mignard, M.; Brechot, P.; Saussine, L. J. Am. Chem. Soc. 1986, 108, 3711; Laszlo, P.; Levart, M.; Singh, G.P. Tetrahedron Lett. 1991, 32, 3167.
1918. Nemoto, T.; Ohshima, T.; Shibasaki, M. J. Am. Chem.Soc. 2001, 123, 9474.
1919. Kakei, H.; Tsuji, R.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 8962.
1920. See J![]() rgensen, K.A. Chem. Rev. 1989, 89, 431.
rgensen, K.A. Chem. Rev. 1989, 89, 431.
1921. Wang, X.; Reisinger, C.M.; List, B. J. Am. Chem. Soc. 2008, 130, 6070; Lu, X.; Liu, Y.; Sun, B.; Cindric, B.; Deng, L. J. Am. Chem. Soc. 2008, 130, 8134.
1922. Lu, J.; Xu, Y.-H.; Liu, F.; Loh, T.-P. Tetrahedron Lett. 2008, 49, 6007.
1923. Ko![]() nik, W.; Bocian, W.; Kozerski, L.; Tvaroška, I.; Chmielewski, M. Chemistry: European J. 2008, 14, 6087.
nik, W.; Bocian, W.; Kozerski, L.; Tvaroška, I.; Chmielewski, M. Chemistry: European J. 2008, 14, 6087.
1924. Murray, R.W. Chem. Rev. 1989, 89, 1187; Adam, W.; Curci, R.; Edwards, J.O. Acc. Chem. Res. 1989, 22, 205; Curci, R.; Dinoi, A.; Rubino, M.E Pure Appl. Chem. 1995, 67, 811; Clennan, E.L. Trends in Organic Chemistry1995, 5, 231; Denmark, S.E.; Wu, Z. Synlett 1999, 847; Annese, C.; D'Accolti, L.; Dinoi, A.; Fusco, C.; Gandolfi, R.; Curci, R. J. Am. Chem. Soc. 2008, 130, 1197.
1925. Frohn, M.; Wang, Z.-X.; Shi, Y. J. Org. Chem. 1998, 63, 6425. See Angelis, Y.; Zhang, X.; Organopoulos, M. Tetrahedron Lett. 1996, 37, 5991 for a discussion of the mechanism of this oxidation.
1926. See Denmark, S.E.; Wu, Z. J. Org. Chem. 1998, 63, 2810 and references cited therein; Yang, D.; Yip, Y.-C.; Tang, M.-W.; Wong, M.-K.; Cheung, K.-K. J. Org. Chem. 1998, 63, 9888 and references cited therein; Masuyama, A.; Yamaguchi, T.; Abe, M.; Nojima, M. Tetrahedron Lett. 2005, 46, 213.
1927. Adam, W.; Prechtl, F.; Richter, M.J.; Smerz, A.K. Tetrahedron Lett. 1993, 34, 8427.
1928. Düfert,, A.; Werz, D.B. J. Org. Chem. 2008, 73, 5514.
1929. Nieto, N.; Munslow, I.J.; Fernández-Pérez, H.; Vidal-Ferran, A. Synlett 2008, 2856.
1930. Sartori, G.; Armstrong, A.; Maggi, R.; Mazzacani, A.; Sartorio, R.; Bigi, F.; Dominguez-Fernandez, B. J. Org. Chem. 2003, 68, 3232.
1931. Carnell, A.J.; Johnstone, R.A.W.; Parsy, C.C.; Sanderson, W.R. Tetrahedron Lett. 1999, 40, 8029.
1932. Shi, Y. Acc. Chem. Res. 2004, 37, 488; Yang, D. Acc. Chem. Res. 2004, 37, 497; Goeddel, D.; Shu, L.; Yuan, Y.; Wong, O.A.; Wang, B.; Shi, Y. J. Org. Chem. 2006, 71, 1715; Crane, Z.; Goeddel, D.; Gan, Y.; Shi, Y. Tetrahedron 2005, 61, 6409; Armstrong, A.; Tsuchiya, T. Tetrahedron 2006, 62, 257; Boutureira, O.; McGouran, J.F.; Stafford, R.L.; Emmerson, D.P.G.; Davis, B. Org. Biomol. Chem. 2009, 7, 4285; Armstrong, A.; Bettati, M.; White, A.J.P. Tetrahedron 2010, 66, 6309.
1933. See Tian, H.; She, X.; Yu, H.; Shu, L.; Shi, Y. J. Org. Chem. 2002, 67, 2435; Denmark, S.E.; Matsuhashi, H. J. Org. Chem. 2002, 67, 3479; Arsmtrong, A.; Ahmed, G.; Dominguez-Fernandez, B.; Hayter, B.R.; Wailes, J.S. J. Org. Chem. 2002, 67, 8610; Wu, X.-Y.; She, X.; Shi, Y. J. Am. Chem. Soc. 2002, 124, 8792; Bez, G.; Zhao, C.-G. Tetrahedron Lett. 2003, 44, 7403; Chan, W.-K.; Yu, W.-y.; Che, C.-M.; Wong, M.-K. J. Org. Chem. 2003, 68, 6576.
1934. Shu, L.; Shi, Y. Tetrahedron Lett. 1999, 40, 8721. DFT modeling of the ee is discussed in Schneebeli, S.T.; Hall, M.L.; Breslow, R.; Friesner, R. J. Am. Chem. Soc. 2009, 131, 3965.
1935. Burke, C.P.; Shi, Y. Angew. Chem. Int. Ed. 2006, 45, 4475.
1936. Wang, Z.-X.; Tu, Y.; Frohn, M.; Zhang, J.-R.; Shi, Y. J. Am. Chem. Soc. 1997, 119, 11224; Frohn, M.; Shi, Y. Synthesis 2000, 1979; Shi, Y. Acc. Chem. Res. 2004, 37, 488; Hickey, M.; Goeddel, D.; Crane, Z.; Shi, Y. Proc. Natl. Acad. Sci. USA. 2004, 101, 5794.
1937. Arias, L.A.; Adkins, S.; Nagel, C.J.; Bach, R.D. J. Org. Chem. 1983, 48, 888.
1938. See Rudolph, J.; Sennhenn, P.C.; Vlaar, C.P.; Sharpless, K.B. Angew. Chem. Int. Ed. 1996, 35, 2810.
1939. Yang, D.; Zhang, C.; wang, X.-C. J. Am. Chem. Soc. 2000, 122, 4039.
1940. See Bohé, L.; Kammoun, M. Tetrahedron Lett. 2002, 43, 803; Bohé, L.; Kammoun, M. Tetrahedron Lett. 2004, 45, 747.
1941. See Washington, I.; Houk, K. N. J. Am. Chem. Soc. 2000, 122, 2948.
1942. See Jacobson, E. N. in Ojima, I. Catalytic Asymmetric Synthesis VCH, NY, 1993, pp. 159–203; Page, P.C.B.; Barros, D.; Buckley, B.R.; Ardakani, A.; Marples, B.A. J. Org. Chem. 2004, 69, 3595; Page, P.C.B.; Buckley, B.R.; Blacker, A.J. Org. Lett. 2004, 6, 1543.
1943. Wong, O.A.; Shi, Y. Chem. Rev. 2008, 108, 3958; Page, P.C.B.; Buckley, B.R.; Farah, M.M.; Blacker, A.J. Eur. J. Org. Chem. 2009, 3413.
1944. Biscoe, M.R.; Breslow, R. J. Am. Chem. Soc. 2005, 127, 10812.
1945. Lo, C.-Y.; Pal, S.; Odedra, A.; Liu, R.-S. Tetrahedron Lett. 2003, 44, 3143.
1946. See Lewars, E.G. Chem. Rev. 1983, 83, 519.
1947. Torres, M.; Bourdelande, J.L.; Clement, A.; Strausz, O.P. J. Am. Chem. Soc. 1983, 105, 1698. See also, Laganis, E.D.; Janik, D.S.; Curphey, T.J.; Lemal, D.M. J. Am. Chem. Soc. 1983, 105, 7457.
1948. Ibne-Rasa, K.M.; Pater, R.H.; Ciabattoni, J.; Edwards, J.O. J. Am. Chem. Soc. 1973, 95, 7894; Ogata, Y.; Sawaki, Y.; Inoue, H. J. Org. Chem. 1973, 38, 1044.
1949. Exceptions are known. See Hart, H.; Verma, M.; Wang, I. J. Org. Chem. 1973, 38, 3418. For diiron-catalysis, see Marchi-Delapierre, C.; Jorge-Robin, A.; Thibon, A.; Ménage, S. Chem. Commun. 2007, 1166.
1950. Weitz, E.; Scheffer, A. Ber. Dtsch. Chem. Ges. 1921, 54, 2327.
1951. See Zwanenburg, B.; ter Wiel, J. Tetrahedron Lett. 1970, 935.
1952. Apeloig, Y.; Karni, M.; Rappoport, Z. J. Am. Chem. Soc. 1983, 105, 2784. See Patai, S.; Rappoport, Z. in Patai, S. The Chemistry of Alkenes, pt. 1, Wiley, NY, 1964, pp. 512–517.
1953. Arai, S.; Tsuge, H.; Oku, M.; Miura, M.; Shioiri, T. Tetrahedron 2002, 58, 1623; Bortolini, O.; Fogagnolo, M.; Fantin, G.; Maietti, S.; Medici, A. Tetrahedron Asymmetry 2001, 12, 1113; Honma, T.; Nakajo, M.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Tetrahedron Lett. 2002, 43, 6229.
1954. See Marigo, M.; Franzén, J.; Poulsen, T.B.; Zhuang, W.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 6964.
rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 6964.
1955. Oguchi, T.; Sakata, Y.; Takeuchi, N.; Kaneda, K.; Ishii, Y.; Ogawa, M. Chem. Lett. 1989, 2053.
1956. Anand, R.V.; Singh, V.K. Synlett 2000, 807.
1957. For a mechanistic discussion of polypeptide catalyzed epoxidation, see Mathew, S.P.; Gunathilagan, S.; Roberts, S.M.; Blackmond, D.G. Org. Lett. 2005, 7, 4847.
1958. Allen, J.V.; Drauz, K.-H.; Flood, R.W.; Roberts, S.M.; Skidmore, J. Tetrahedron Lett. 1999, 40, 5417; Geller, T.; Roberts, S.M. J. Chem. Soc., Perkin Trans. 1,1999, 1397; Bentley, P.A.; Bickley, J.F.; Roberts, S.M.; Steiner, A. Tetrahedron Lett. 2001, 42, 3741.
1959. Banfi, S.; Colonna, S.; Molinari, H.; Juliá, S.; Guixer, J. Tetrahedron,1984, 40, 5207. For reviews, see Lin, P. Tetrahedron: Asymmetry 1998, 9, 1457; Ebrahim, S.; Wills, M. Tetrahedron; Asymmetry 1997, 8, 3163.
1960. Lygo, B.; Wainwright, P.G. Tetrahedron 1999, 55, 6289.
1961. Geller, T.; Krüger, C.M.; Militzer, H.-C. Tetrahedron Lett. 2004, 45, 5069.
1962. Coffey, P.E.; Drauz, K.-H.; Roberts, S.M.; Skidmore, J.; Smith, J.A. Chem. Commun. 2001, 2330.
1963. See Jacobs, T.L. in Landor, S.R. The Chemistry of Allenes Vol. 2, Academic Press, NY, 1982, pp. 417–510, 483–491.
1964. For a review of allene oxides, see Chan, T.H.; Ong, B.S. Tetrahedron 1980, 36, 2269.
1965. Crandall, J.K.; Batal, D.J. J. Org. Chem. 1988, 53, 1338.
1966. See Crandall, J.K.; Rambo, E. J. Org. Chem. 1990, 55, 5929.
1967. Antonioletti, R.; Bonadies, F.; Locati, L.; Scettri, A. Tetrahedron Lett. 1992, 33, 3205.
1968. Fringuelli, F.; Germani, R.; Pizzo, F.; Santinelli, F.; Savelli, G. J. Org. Chem. 1992, 57, 1198.
1969. See Pfenninger, A. Synthesis 1986, 89; Rossiter, B.E. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, pp. 193–246. For histories of its discovery, see Sharpless, K.B. Chem. Br. 1986, 38. Also see, Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 284–290.
1970. Sharpless, K.B.; Woodard, S.S.; Finn, M.G. Pure Appl. Chem. 1983, 55, 1823 and references cited therein.
1971. Gao, Y.; Hanson, R.M.; Klunder, J.M.; Ko, S.Y.; Masamune, H.; Sharpless, K.B. J. Am. Chem. Soc. 1987, 109, 5765. See Massa, A.; D'Ambrosi, A.; Proto, A.; Screttri, A. Tetrahedron Lett. 2001, 42, 1995. For another improvement, see Wang, Z.; Zhou, W. Tetrahedron 1987, 43, 2935.
1972. Canali, L.; Karjalainen, J.K.; Sherrington, D.C.; Hormi, O. Chem. Commun. 1997, 123.
1973. Reed, N.N.; Dickerson, T.J.; Boldt, G.E.; Janda, K.D. J. Org. Chem. 2005, 70, 1728.
1974. See the table in Finn, M.G.; Sharpless, K.B. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, pp. 249–250. See also, Schweiter, M.J.; Sharpless, K.B. Tetrahedron Lett. 1985, 26, 2543.
1975. See Williams, I.D.; Pedersen, S.F.; Sharpless, K.B.; Lippard, S.J. J. Am. Chem. Soc. 1984, 106, 6430.
1976. See Finn, M.G.; Sharpless, K.B. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, p. 247. Also see Corey, E.J. J. Org. Chem. 1990, 55, 1693; Woodard, S.S.; Finn, M.G.; Sharpless, K.B. J. Am. Chem. Soc. 1991, 113, 106; Finn, M.G.; Sharpless, K.B. J. Am. Chem. Soc. 1991, 113, 113; Takano, S.; Iwebuchi, Y.; Ogasawara, K. J. Am. Chem. Soc. 1991, 113, 2786; Cui, M.; Adam, W.; Shen, J.H.; Luo, X.M.; Tan, X.J.; Chen, K.X.; Ji, R.Y.; Jiang, H.L. J. Org. Chem. 2002, 67, 1427.
1977. Zhang, W.; Yamamoto, H. J. Am. Chem. Soc. 2007, 129, 286.
1978. Jacobsen, E.N.; Zhang, W.; Muci, A.R.; Ecker, J.R.; Deng, L. J. Am. Chem. Soc. 1991, 113, 7063. See also, Irie, R.; Noda, K.; Ito, Y.; Katsuki, T. Tetrahedron Lett. 1991, 32, 1055; Halterman, R.L.; Jan, S. J. Org. Chem.1991, 56, 5253.
1979. Kureshy, R.I.; Khan, N.H.; Abdi, S.H.R.; Patel, S.T.; Jasra, R.V. Tetrahedron Asymmetry 2001, 12, 433.
1980. Wu, M.; Wang, B.; Wang, S.; Xia, C.; Sun, W. Org. Lett. 2009, 11, 3622. See Adam, W.; Mock-Knoblauch, C.; Saha-Moller, C.R.; Herderich, M. J. Am. Chem. Soc. 2000, 122, 9685.
1981. Brandes, B.D.; Jacobsen, E.N. Tetrahedron Lett. 1995, 36, 5123; Nishikori, H.; Ohta, C.; Katsuki, T. Synlett 2000, 1557; Tangestaninejad, S.; Habibi, M.H.; Mirkhani, V.; Moghadam, M. Synth. Commun. 2002, 32, 3331. See Fristrup, P.; Dideriksen, B.B.; Tanner, D.; Norrby, P.-O. J. Am. Chem. Soc. 2005, 127, 13672. For a discussion on the origin of enantioselectivity, see Kürti, L.; Blewett, M.M.; Corey, E.J. Org. Lett. 2009, 11, 4592.
1982. See Nishida, T.; Miyafuji, A.; Ito, Y.N.; Katsuki, T. Tetrahedron Lett. 2000, 41, 7053.
1983. Daly, A.M.; Renehan, M.F.; Gilheany, D.G. Org. Lett. 2001, 3, 663; O'Mahony, C.P.; McGarrigle, E.M.; Renehan, M.F.; Ryan, K.M.; Kerrigan, N.J.; Bousquet, C.; Gilheany, D.G. Org. Lett. 2001, 3, 3435. See the references cited therein.
1984. Matsumoto, K.; Oguma, T.; Katsuki, T. Angew. Chem. Int. Ed. 2009, 48, 7432.
1985. Nakata, K.; Takeda, T.; Mihara, J.; Hamada, T.; Irie, R.; Katsuki, T. Chem. Eur. J. 2001, 7, 3776.
1986. McGarrigle, E.M.; Gilheany, D.G. Chem. Rev. 2005, 105, 1563.
1987. See Linker, T. Angew. Chem,. Int. Ed. 1997, 36, 2060; Adam, W.; Roschmann, K.J.; Saha-Möller, C.R. Eur. J. Org. Chem. 2000, 3519. Also see Cavallo, L.; Jacobsen, H. J. Org. Chem. 2003, 68, 6202.
1988. Cavallo, L.; Jacobsen, H. Angew. Chem. Int. Ed. 2000, 39, 589.
1989. Song, C.E.; Roh, E.J.; Yu, B.M.; Chi, D.Y.; Kim, S.C.; Lee, K.J. Chem. Commun. 2000, 615; Ahn, K.-H.; Park, S.W.; Choi, S.; Kim, H.-J.; Moon, C.J. Tetrahedron Lett. 2001, 42, 2485.
1990. Konishi, K.; Oda, K.; Nishida, K.; Aida, T.; Inoue, S. J. Am. Chem. Soc. 1992, 114, 1313.
1991. Takai, T.; Hata, E.; Yorozu, K.; Mukaiyama, T. Chem. Lett. 1992, 2077.
1992. Saalfrank, R.W.; Reihs, S.; Hug, M. Tetrahedron Lett,1993, 34, 6033.
1993. Savle, P.S.; Lamoreaux, M.J.; Berry, J.F.; Gandour, R.D. Tetrahedron Asymmetry 1998, 9, 1843.
1994. Adam, W.; Braun, M.; Griesbeck, A.; Lucchini, V.; Staab, E.; Will, B. J. Am. Chem. Soc. 1989, 111, 203.
1995. See Iwahama, T.; Hatta, G.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2000, 163.
1996. Ragagnin, G.; Knochel, P. Synlett 2004, 951.
1997. Srikanth, A.; Nagendrappa, G.; Chandrasekaran, S. Tetrahedron 2003, 59, 7761; Qi, J.Y.; Qiu, L.Q.; Lam, K.H.; Yip, C.W.; Zhou, Z.Y.; Chan, A.S.C. Chem. Commun. 2003, 1058.
1998. Adam, W.; Bargon, R.M. Eur. J. Org. Chem. 2001, 1959; See Sugihara, Y.; Onda, K.; Sato, M.; Suzu, T. Tetrahedron Lett. 2010, 51, 4110.
1999. Tangestaninejad, S.; Mirkhani, V. Synth. Commun. 1999, 29, 2079.
2000. Nakayama, J.; Takahashi, K.; Watanabe, T.; Sugihara, Y.; Ishii, A. Tetrahedron Lett. 2000, 41, 8349.
2001. Iranpoor, N.; Tamami, B.; Shekarriz, M. Synth. Commun. 1999, 29, 3313.
2002. Adam, W.; Bargon, R.M.; Schenk, W.A. J. Am. Chem. Soc. 2003, 125, 3871.
2003. Trost, B.M.; Ochiai, M.; McDougal, P.G. J. Am. Chem. Soc. 1978, 100, 7103; See Zefirov, N.S.; Zyk, N.V.; Kutateladze, A.G.; Kolbasenko, S.I.; Lapin, Yu.A. J. Org. Chem. USSR 1986, 22, 190.
2004. Bewick, A.; Mellor, J.M.; Owton, W.M. J. Chem. Soc. Perkin Trans. 1,1985, 1039; Bewick, A.; Mellor, J.M.; Milano, D.; Owton, W.M. J. Chem. Soc. Perkin Trans. 1,1985, 1045; Samii, Z.K.M.A.E.; Ashmawy, M.I.A.; Mellor, J.M. Tetrahedron Lett. 1986, 27, 5289.
2005. Chung, M.; D'Souza, V.T.; Szmant, H.H. J. Org. Chem. 1987, 52, 1741, and other papers in this series.
2006. Inoue, H.; Murata, S. Heterocycles 1997, 45, 847.
2007. Tuladhar, S.M.; Fallis, A.G. Tetrahedron Lett. 1987, 28, 523. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 905–908.
2008. Bosman, C.; D'Annibale, A.; Resta, S.; Trogolo, C. Tetrahedron Lett. 1994, 35, 6525. See Ogawa, A.; Tanaka, H.; Yokoyama, H.; Obayashi, R.; Yokoyama, K.; Sonoda, N. J. Org. Chem. 1992, 57, 111.
2009. Hermans, B.; Colard, N.; Hevesi, L. Tetrahedron Lett. 1992, 33, 4629.
2010. Brunel, Y.; Rousseau, G. Synlett 1995, 323.
2011. See Bäckvall, J.E.; Oshima, K.; Palermo, R.E.; Sharpless, K.B. J. Org. Chem. 1979, 44, 1953.
2012. Sharpless, K.B.; Chong, A.O.; Oshima, K. J. Org. Chem. 1976, 41, 177. See Rudolph, J.; Sennhenn, P.C.; Vlaar, C.P.; Sharpless, K.B. Angew. Chem. Int. Ed. 1996, 35, 2810.
2013. See Fokin, V.V.; Sharpless, K.B. Angew. Chem. Int. Ed. 2001, 40, 3455.
2014. Herranz, E.; Sharpless, K.B. J. Org. Chem. 1978, 43, 2544.
2015. Hassine, B.B.; Gorsane, M.; Pecher, J.; Martin, R.H. Bull. Soc. Chim. Belg. 1985, 94, 759.
2016. For a review, see Bodkin, J.A.; McLeod, M.D. J. Chem. Soc., Perkin Trans. 1 2002, 2733.
2017. Michaelis, D.J.; Shaffer, C.J.; Yoon, T.P. J. Am. Chem. Soc. 2007, 129, 1866.
2018. Kim, J.D.; Kim, I.S.; Hua, J.C.; Zee, O.P.; Jung, Y.H. Tetrahedron Lett. 2005, 46, 1079.
2019. Kalita, B.; Nicholas, K.M. Tetrahedron Lett. 2005, 46, 1451.
2020. Li, G.; Chang, H.-T.; Sharpless, K.B. Angew. Chem., Int. Ed. 1996, 35, 451.
2021. Streuff, J.; Osterath, B.; Nieger, M.; Muñiz, K. Tetrahedron Asymmetry 2005, 16, 3492.
2022. Demko, Z.P.; Bartsch, M.; Sharpless, K.B. Org. Lett. 2000, 2, 2221.
2023. Bäckvall, J.E.; Björkman, E.E. Acta Chem. Scand. Ser. B 1984, 38, 91; Bäckvall, J.E.; Bystrom, S.E. J. Org. Chem. 1982, 47, 1126.
2024. Ryu, J.-S.; Li, G.Y.; Marks, T.J. J. Am. Chem. Soc. 2003, 125, 12584. For a review, see Hong, S.; Marks, T.J. Acc. Chem. Res. 2004, 37, 673.
2025. Gagné, M.R.; Stern, C.L.; Marks, T.J. J. Am Chem. Soc. 1992, 114, 275.
2026. Li, Y.; Marks, T.J. J. Am. Chem. Soc. 1998, 120, 1757.
2027. Douglass, M.R.; Ogasawara, M.; Hong, S.; Metz, M.V.; Marks, T.J. Organometallics 2002, 21, 283; Giardello, M.A.; Conticello, V.P.; Brard, L.; Gagné, M.R.; Marks, T.J. J. Am. Chem. Soc. 1994, 116, 10241; Giardello, M.A.; Conticello, V.P.; Brard, L.; Sabat, M.; Rheingold, A.L.; Stern, C.L.; Marks, T.J. J. Am. Chem. Soc. 1994, 116, 10212.
2028. Hong, S.; Tian, S.; Metz, M.V.; Marks, T.J. J. Am. Chem. Soc. 2003, 125, 14768.
2029. Barluenga, J.; Alonso-Cires, L.; Asensio, G. Synthesis 1981, 376.
2030. Sakurada, I.; Yamasaki, S.; Kanai, M.; Shibasaki, M. Tetrahedron Lett. 2000, 41, 2415.
2031. Gómez Aranda, V.; Barluenga, J.; Aznar, F. Synthesis 1974, 504.
2032. Chong, A.O.; Oshima, K.; Sharpless, K.B. J. Am. Chem. Soc. 1977, 99, 3420. See also, Sharpless, K.B.; Singer, S.P. J. Org. Chem. 1976, 41, 2504.
2033. For a X-ray structure of the osmium intermediate, see Muñiz, K.; Iesato, A.; Nieger, M. Chem. Eur.J. 2003, 9, 5581.
2034. Bäckvall, J. Tetrahedron Lett. 1978, 163.
2035. See Osowska-Pacewicka, K.; Zwierzak, A. Synthesis 1990, 505.
2036. Barluenga, J.; Alonso-Cires, L.; Asensio, G. Synthesis 1979, 962.
2037. Li, G.; Kim, S.H.; Wei, H.-X. Tetrahedron Lett. 2000, 41, 8699.
2038. Iglesias, Á.; Pérez, E.G.; Muñiz, K. Angew. Chem. Int. Ed. 2010, 49, 8109.
2039. Moriarty, R.M.; Khosrowshahi, J.S. Tetrahedron Lett. 1986, 27, 2809. See Fristad, W.E.; Brandvold, T.A.; Peterson, J.R.; Thompson, S.R. J. Org. Chem. 1985, 50, 3647.
2040. Bar, G.L.J.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. J. Am. Chem. Soc. 2005, 127, 7308.
2041. Du, H.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 762; Du, H.; Yuan, W.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 7496; Du, H.; Yuan, W.; Zhao, B.; Shi, Y. J. Am. Chem. Soc. 2007, 129, 11688.
2042. Fukudome, Y.; Naito, H.; Hata, T.; Urabe, H. J. Am. Chem. Soc. 2008, 130, 1820.
2043. See Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines Academic Press, NY, 1969, pp. 68–79; Muller, L.L.; Hamer, J. 1,2-Cycloaddition Reactions, Wiley, NY, 1967. See Singh, G.S.; D'hooghe, M.; De Kimpe, N. Chem. Rev. 2007, 107, 2080.
2044. Fioravanti, S.; Morreale, A.; Pellacani, L.; Tardella, P.A. Synthesis 2001, 1975. For an enantioselective version, see Fioravanti, S.; Morreale, A.; Pellacani, L.; Tardella, P.A. J. Org. Chem. 2002, 67, 4972.
2045. Sanders, C.J.; Gillespie, K.M.; Scott, P. Tetrahedron Asymmetry 2001, 12, 1055; Ma, J.-A.; Wang, L.-X.; Zhang, W.; Zhou, W.; Zhou, Q.-L. Tetrahedron Asymmetry 2001, 12, 2801.
2046. Ikeno, T.; Nishizuka, A.; Sato, M.; Yamada, T. Synlett 2001, 406.
2047. Mohan, J.M.; Uphade, T.S.S.; Choudhary, V.R.; Ravindranathan, T.; Sudalai, A. Chem. Commun. 1997, 1429; Moran, M.; Bernardinelli, G.; Müller, P. Helv. Chim. Acta 1995, 78, 2048.
2048. Ishihara, H.; Ito, Y.N.; Katsuki, T. Chem. Lett. 2001, 984.
2049. Carducci, M.; Fioravanti, S.; Loreta, M.A.; Pellacani, L.; Tardella, P.A. Tetrahedron Lett. 1996, 37, 3777.
2050. Fioravanti, S.; Morreale, A.; Pellacani, L.; Tardella, P.A. Tetrahedron Lett. 2003, 44, 3031.
2051. Aires-de-Sousa, J.; Labo, A.M.; Prabhakar, S. Tetrahedron Lett. 1996, 37, 3183.
2052. Siu, T.; Yudin, A.K. J. Am. Chem. Soc. 2002, 124, 530.
2053. Anderson, D.J.; Gilchrist, T.L.; Rees, C.W. Chem. Commun. 1969, 147.
2054. Siu, T.; Picard, C.J.; Yudin, A.K. J. Org. Chem. 2005, 70, 932.
2055. Mahoney, J.M.; Smith, C.R.; Johnston, J.N. J. Am. Chem. Soc. 2005, 127, 1354.
2056. Lebel, H.; Huard, K.; Lectard, S. J. Am. Chem. Soc. 2005, 127, 14198.
2057. Catino, A.J.; Nichols, J.M.; Forslund, R.E.; Doyle, M.P. Org. Lett. 2005, 7, 2787.
2058. Fan, R.; Pu, D.; Gan, J.; Wang, B. Tetrahedron Lett. 2008, 49, 4925.
2059. Espino, C.G.; Wehn, P.M.; Chow, J.; Du Bois, J. J. Am. Chem. Soc. 2001, 123, 6935; Wehn, P.M.; Lee, J.; Du Bois, J. Org. Lett. 2003, 5, 4823; Espino, C.G.; Fiori, K.W.; Kim, M.; Du Bois, J. J. Am. Chem. Soc. 2004, 126, 15378; Guthikonda, K.; Du Bois, J. J. Am. Chem. Soc. 2002, 124, 13672; Keaney, G.F.; Wood, J.L. Tetrahedron Lett. 2005, 46, 4031; Guthikonda, K.; Wehn, P.M.; Caliando, B.J.; Du Bois, J. Tetrahedron 2006, 62, 11331.
2060. Roth, P.; Andersson, P.G.; Somfai, P. Chem. Commun. 2002, 1752.
2061. Wu, H.; Xu, L.-W.; Xia, C.-G.; Ge, J.; Yang, L. Synth. Commun. 2005, 35, 1413; Karabal, P.U.; Chouthaiwale, P.V.; Shaikh, T.M.; Suryavanshi, G.; Sudalai, A. Tetrahedron Lett. 2010, 51, 6460. Also see Chen, D.; Timmons, C.; Guo, L.; Xu, X.; Li, G. Synthesis 2004, 2479.
2062. Thakur, V.V.; Sudalai, A. Tetrahedron Lett. 2003, 44, 989.
2063. Vyas, R.; Chanda, B.M.; Bedekar, A.V. Tetrahedron Lett. 1998, 39, 4715; Hayer, M.F.; Hossain, M.M. J. Org. Chem. 1998, 63, 6839. See Antunes, A.M.M.; Bonifácio, V.D.B.; Nascimento, S.C.C.; Lobo, A.M.; Branco, P.S.; Prabhakar, S. Tetrahedron 2007, 63, 7009.
2064. Jain, S.L.; Sain, B. Tetrahedron Lett. 2003, 44, 575.
2065. Casarrubios, L.; Pérez, J.A.; Brookhart, M.; Templeton, J.L. J. Org. Chem. 1996, 61, 8358.
2066. Müller, P.; Baud, C.; Jacquier, Y. Tetrahedron 1996, 52, 1543. Also see, Södergren, M.J.; Alonso, D.A.; Bedekar, A.V.; Andersson, P.G. Tetrahedron Lett. 1997, 38, 6897.
2067. Knight, J.G.; Muldowney, M.P. Synlett 1995, 949. See also, Mohr, F.; Binfield, S.A.; Fettinger, J.C.; Vedernikov, A.N. J. Org. Chem. 2005, 70, 4833.
2068. See GuthiKonda, K.; Du Bois, J. J. Am. Chem. Soc. 2002, 124, 13672. See also, Dichenna, P.H.; Robert-Peillard, F.; Dauban, P.; Dodd, R.H. Org. Lett. 2004, 6, 4503; Kwong, H.-L.; Liu, D.; Chan, K.-Y.; Lee, C.-S.; Huang, K.-H.; Che, C.-M. Tetrahedron Lett. 2004, 45, 3965.
2069. Vedernikov, A.N.; Caulton, K.G. Org. Lett. 2003, 5, 2591; Cui, Y.; He, C. J. Am. Chem. Soc. 2003, 125, 16202. See Nishimura, M.; Minakata, S.; Takahashi, T.; Oderaotoshi, Y.; Komatsu, M. J. Org. Chem. 2002, 67, 2101.
2070. See Gillespie, K.M.; Sanders, C.J.; O'Shaughnessy, P.; Westmoreland, I.; Thickitt, C.P.; Cott, P. J. Org. Chem. 2002, 67, 3450.
2071. Kantam, M.L.; Neeraja, V.; Kavita, B.; Haritha, Y. Synlett 2004, 525.
2072. Antunes, A.M.M.; Marto, S.J.L.; Branco, P.S.; Prabhakar, S.; Lobo, A.M. Chem. Commun. 2001, 405.
2073. Jean, H.-J.; Nguyen, S.B.T. Chem. Commun. 2001, 235.
2074. Nishikori, H.; Katsuki, T. Tetrahedron Lett. 1996, 37, 9245.
2075. Nishimura, M.; Minakata, S.; Thonchant, S.; Ryu, I.; Komatsu, M. Tetrahedron Lett. 2000, 41, 7089.
2076. Li, Z.; Ding, X.; He, C. J. Org. Chem. 2006, 71, 5876.
2077. Jain, S.L.; Sharma, V.B.; Sain, B. Synth. Commun. 2005, 35, 9.
2078. Vesely, J.; Ibrahem, I.; Zhao, G.-L.; Rios, R.; Córdova, A. Angew. Chem. Int. Ed. 2007, 46, 778.
2079. See Lwowski, W.; Johnson, R.L. Tetrahedron Lett. 1967, 891.
2080. Benati, L.; Montavecchi, P.C.; Spagnolo, P. Tetrahedron Lett. 1984, 25, 2039. See also, Brownbridge, P. Tetrahedron Lett. 1984, 25, 3759.
2081. See Ref. 21.
2082. Trost, B.M.; Shibata, T. J. Am. Chem. Soc. 1982, 104, 3225; Caserio, M.C.; Kim., J.K. J. Am. Chem. Soc. 1982, 104, 3231.
2083. Trost, B.M.; Shibata, T.; Martin, S.J. J. Am. Chem. Soc. 1982, 104, 3228; Trost, B.M.; Shibata, T. J. Am. Chem. Soc. 1982, 104, 3225. For an extension that allows A to be C![]() CR, see Trost, B.M.; Martin, S.J. J. Am. Chem. Soc. 1984, 106, 4263.
CR, see Trost, B.M.; Martin, S.J. J. Am. Chem. Soc. 1984, 106, 4263.
2084. Sreekumar, R.; Padmakumar, R.; Rugmini, P. Chem. Commun. 1997, 1133.
2085. Bewick, A.; Coe, D.E.; Mellor, J.M.; Owton, M.W. J. Chem. Soc. Perkin Trans. 1,1985, 1033.
2086. Shastin, A.V.; Balenkova, E.S. J. Org. Chem. USSR 1984, 20, 870.
2087. Hashem, Md.A.; Jung, A.; Ries, M.; Kirschning, A. Synlett 1998, 195.
2088. Gridnev, I.D.; Balenkova, E.S. J. Org. Chem. USSR 1988, 24, 1447.
2089. Shundo, R.; Nishiguchi, I.; Matsubara, Y.; Hirashima, T. Tetrahedron 1991, 47, 831. See also, Corey, E.J.; Gross, A.W. Tetrahedron Lett. 1985, 26, 4291.
2090. D'Annibale, A.; Trogolo, C. Tetrahdron Lett. 1994, 35, 2083.
2091. Lamarque, L.; Méou, A.; Brun, P. Tetrahedron 1998, 54, 6497.
2092. Allegretti, M.; D'Annibale, A.; Trogolo, C. Tetrahedron 1993, 49, 10705.
2093. Nakano, T.; Kayama, M.; Nagai, Y. Bull. Chem. Soc. Jpn. 1987, 60, 1049. See also, Kraus, G.A.; Landgrebe, K. Tetrahedron Lett. 1984, 25, 3939.
2094. Wang, S.L.B.; Su, J.; Wulff, W.D. J. Am. Chem. Soc. 1992, 114, 10665.
2095. See Bäuml, E.; Tscheschlok, K.; Pock, R.; Mayr, H. Tetrahedron Lett. 1988, 29, 6925.
2096. Caldwell, J.J.; Harrity, J.P.A.; Heron, N.M.; Kerr, W.J.; McKendry, S.; Middlemiss, D. Tetrahedron Lett. 1999, 40, 3481; Caldwell, J.J.; Kerr, W.J.; McKendry, S. Tetrahedron Lett. 1999, 40, 3485.
2097. Nguyen, R.V.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 17184.
2098. López-López, J.A.; Guerra, F.M.; Moreno-Dorado, F.J.; Jorge, Z.D.; Massanet, G.M. Tetrahedron Lett. 2007, 48, 1749.
2099. Tellitu, I.; Serna, S.; Herrero, M.T.; Moreno, I.; Domínguez, E.; SanMartin, R. J. Org. Chem. 2007, 72, 1526;
2100. Browne, D.M.; Niyomura, O.; Wirth, T. Org. Lett. 2007, 9, 3169.
2101. Harkat, H.; Weibel, J.-M.; Pale, P. Tetrahedron Lett. 2006, 47, 6273.
2102. Davies, D.T.; Kapur, N.; Parsons, A.F. Tetrahedron Lett. 1998, 39, 4397.
2103. See Broggini, G.; Zecchi, G. Synthesis 1999, 905.
2104. Karlsson, S.; Högberg, H.-E. Org. Prep. Proceed. Int. 2001, 33, 103.
2105. See Carruthers, W. Cycloaddition Reactions in Organic Synthesis Pergamon, Elmsford, NY, 1990; Huisgen, R. Helv. Chim. Acta 1967, 50, 2421; Angew. Chem. Int. Ed. 1963, 2, 565, 633; Torssell, K.B.G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis VCH, NY, 1988; Scriven, E.F.V. Azides and Nitrenes Academic Press, NY, 1984; Stanovnik, B. Tetrahedron 1991, 47, 2925 (diazoalkanes); Kanemasa, S.; Tsuge, O. Heterocycles 1990, 30, 719 (nitrile oxides); Paton, R.M. Chem. Soc. Rev. 1989, 18, 33 (nitrile sulfides); Terao, Y.; Aono, M.; Achiwa, K. Heterocycles 1988, 27, 981; Coldham, I.; Hufton, R. Chem. Rev. 2005, 105, 2765 (azomethine ylids); Vedejs, E. Adv. Cycloaddit. 1988, 1, 33 (azomethine ylids); DeShong, P.; Lander, Jr., S.W.; Leginus, J.M.; Dicken, C.M. Adv. Cycloaddit. 1988, 1, 87 (nitrones); Balasubramanian, N. Org. Prep. Proced. Int. 1985, 17, 23 (nitrones); Confalone, P.N.; Huie, E.M. Org. React. 1988, 36, 1 (nitrones); Padwa, A. in Horspool, W.M. Synthetic Organic Photochemistry, Plenum, NY, 1984, pp. 313–374 (nitrile ylids); Bianchi, G.; Gandolfi, R.; Grünanger, P. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 1, Wiley, NY, 1983, pp. 752–784 (nitrile oxides); Stuckwisch, C.G. Synthesis 1973, 469 (azomethine ylids, azomethine imines). For reviews of intramolecular 1,3-dipolar additions see Padwa, A. in Padwa, A. treatise cited above, Vol. 2, pp. 277–406; Padwa, A.; Schoffstall, A.M. Adv. Cycloaddit. 1990, 2, 1; Tsuge, O.; Hatta, T.; Hisano, T. in Patai, S. Supplement A: The Chemistry of Double-bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 345–475; Padwa, A. Angew. Chem. Int. Ed. 1976, 15, 123. For a review of azomethine ylids, see Tsuge, O.; Kanemasa, S. Adv. Heterocycl. Chem. 1989, 45, 231. For reviews of 1,3-dipolar cycloreversions, see Bianchi, G.; De Micheli, C.; Gandolfi, R. Angew. Chem. Int. Ed. 1979, 18, 721. For the use of this reaction to synthesize natural products, see papers in Tetrahedron 1985, 41, 3447.
2106. Houk, K.N.; Strassner, T. J. Org. Chem. 1999, 64, 800.
2107. See Gisdakis, P.; Rösch, N. J. Am. Chem. Soc. 2001, 123, 697.
2108. Kobayashi, S.; Hirabayashi, R.; Shimizu, H.; Ishitani, H.; Yamashita, Y. Tetrahedron Lett. 2003, 44, 3351.
2109. See Baskaran, S.; Vasu, J.; Prasad, R.; Kodukulla, K.; Trivedi, G.K. Tetrahedron 1996, 52, 4515.
2110. Sibi, M.P.; Stanley, L.M.; Jasperse, C.P. J. Am. Chem. Soc. 2005, 127, 8276.
2111. Raposo, C.; Wilcox, C.S. Tetrahedron Lett. 1999, 40, 1285.
2112. See Nishiwaki, N.; Uehara, T.; Asaka, N.; Tohda, Y.; Ariga, M.; Kanemasa, S. Tetrahedron Lett. 1998, 39, 4851; Jung, M.E.; Vu, B.T. Tetrahedron Lett. 1996, 37, 451. See Muri, D.; Bode, J.W.; Carreira, E.M. Org. Lett.2000, 2, 539; Gissot, A.; Wagner, A.; Mioskowski, C. Tetrahedron Lett. 2000, 41, 1191. For a discussion of transition structures, see Luft, J.A.R.; Meleson, K.; Houk, K.N. Org. Lett. 2007, 9, 555.
2113. For a reaction using a chiral Si Lewis acid, see Shirakawa, S.; Lombardi, P.J.; Leighton, J.L. J. Am. Chem. Soc. 2005, 127, 9974.
2114. See Cabrera, S.; Arrayás, R.G.; Carretero, J.C. J. Am. Chem. Soc. 2005, 127, 16394; Wang, C.-J.; Liang, G.; Xue, Z.-Y.; Gao, F. J. Am. Chem. Soc. 2008, 130, 17250; Nájera, C.; Sansano, J.M. Angew. Chem. Int. Ed. 2005, 44, 6272.
2115. See Iesce, M.R.; Cermola, F.; Giordano, F.; Scarpati, R.; Graziano, M.L. J. Chem. Soc. Perkin Trans. 1,1994, 3295; MuCullough, K.J.; Sugimoto, T.; Tanaka, S.; Kusabayashi, S.; Nojima, M. J. Chem. Soc. Perkin Trans. 1,1994, 643.
2116. Kusama, H.; Funami, H.; Shido, M.; Hara, Y.; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2005, 127, 2709; Diev, V.V.; Kostikov, R.R.; Gleiter, R.; Molchanov, A.P. J. Org. Chem. 2006, 71, 4066.
2117. For a review of some aspects of this, see Grigg, R. Chem. Soc. Rev. 1987, 16, 89.
2118. See Bianchi, G.; De Micheli, C.; Gandolfi, R. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, pt. 1, Wiley, NY, 1977, pp. 369–532. Also see Dunn, A.D.; Rudorf, W. Carbon Disulfide in Organic Chemistry, Wiley, NY, 1989, pp. 97–119.
2119. Di Valentin, C.; Freccero, M.; Gandolfi, R.; Rastelli, A. J. Org. Chem. 2000, 65, 6112. For a theoretical study of transition states, see Lu, X.; Xu, X.; Wang, N.; Zhang, Q. J. Org. Chem. 2002, 67, 515; DiValentin, C.; Freccero, M.; Gandolfi, R.; Rastelli, A. J. Org. Chem. 2000, 65, 6112. For a discussion of loss of concertedness in reactions of azomethine ylids, see Vivanco, S.; Lecea, B.; Arrieta, A.; Prieto, P.; Morao, I.; Linden, A.; Cossío, F.P. J. Am. Chem. Soc. 2000, 122, 6078. See Ess, D.H.; Houk, K.N. J. Am. Chem. Soc. 2007, 129, 10646.
2120. For Huisgen, R. Adv. Cycloaddit. 1988, 1, 1; Al-Sader, B.H.; Kadri, M. Tetrahedron Lett. 1985, 26, 4661; Houk, K.N.; Firestone, R.A.; Munchausen, L.L.; Mueller, P.H.; Arison, B.H.; Garcia, L.A. J. Am. Chem. Soc. 1985, 107, 7227; Majchrzak, M.W.; Warkentin, J. J. Phys. Org. Chem. 1990, 3, 339.
2121. Caramella, P.; Gandour, R.W.; Hall, J.A.; Deville, C.G.; Houk, K.N. J. Am. Chem. Soc. 1977, 99, 385 and references cited therein.
2122. Engels, B.; Christl, M. Angew. Chem. Int. Ed. 2009, 48, 7968.
2123. Cossío, F.P.; Marao, I.; Jiao, H.; Schleyer, P.v.R. J. Am. Chem. Soc. 1999, 121, 6737.
2124. For a review of the role of solvents in this reaction, see Kadaba, P.K. Synthesis 1973, 71.
2125. Dubreuil, J.F.; Bazureau, J.P. Tetrahedron Lett. 2000, 41, 7351.
2126. Lee, C.K.Y; Holmes, A.B.; Al-Duri, B.; Leeke, G.A.; Santos, R.C.D.; Seville, J.P.K. Chem. Commun. 2004, 2622.
2127. See Houk, K.N.; Yamaguchi, K. in Padwa, A. 1,3-Dipolar Cycloaddition Chemistry Vol. 2, Wiley, NY, 1984, pp. 407–450. See also, Burdisso, M.; Gandolfi, R.; Quartieri, S.; Rastelli, A. Tetrahedron 1987, 43, 159.
2128. Mloston, G.; Langhals, E.; Huisgen, R. Tetrahedron Lett. 1989, 30, 5373; Huisgen, R.; Mloston, G. Tetrahedron Lett. 1989, 30, 7041.
2129. Karadakov, P.B.; Cooper, D.L.; Gerratt, J. Theor. Chem. Acc. 1998, 100, 222.
2130. Blavins, J.J.; Karadakov, P.B.; Cooper, D.L. J. Org. Chem. 2001, 66, 4285.
2131. Toker, J.D.; Wentworth, Jr., P.; Hu, Y.; Houk, K.N.; Janda, K.D. J. Am. Chem. Soc. 2000, 122, 3244.
2132. Kanemasa, S. Synlett 2002, 1371.
2133. Barluenga, J.; Barrio, P.; Riesgo, L.; López, L.A.; Tomás, M. J. Am. Chem. Soc. 2007, 129, 14422.
2134. See Fischer, H. Chem. Ber. 1980, 113, 193.
2135. For a discussion of reactivity and regioselectivity with strained alkenes and alkynes, see Schoenebeck, F.; Ess, D.H.; Jones, G.O.; Houk, K.N. J. Am. Chem. Soc. 2009, 131, 8121.
2136. Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. J. Am. Chem. Soc. 2005, 127, 15998; Kamata, K.; Nakagawa, Y.; Yamaguchi, K.; Mizuno, N. J. Am. Chem. Soc. 2008, 130, 15304.
2137. da Silva, G.; Bozzelli, J.W. J. Org. Chem. 2008, 73, 1343.
2138. Feldman, K.S.; Iyer, M.R. J. Am. Chem. Soc. 2005, 127, 4590.
2139. See Padwa, A. Angew. Chem. Int. Ed. 1976,15, 123; Oppolzer, W. Angew. Chem. Int. Ed. 1977, 16, 10 (see pp. 18–22).
2140. Dolle, R.E.; Barden, M.C.; Brennan, P.E.; Ahmed, G.; Tran, V.; Ho, D.M. Tetrahedron Lett. 1999, 40, 2907.
2141. For an enantioselective reaction, see Kano, T.; Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 2174.
2142. Jan, D.; Simal, F.; Demonceau, A.; Noels, A.F.; Rufanov, K.A.; Ustynyuk, N.A.; Gourevitch, D.N. Tetrahedron Lett. 1999, 40, 5695.
2143. For a discussion of the mechansim and origin of enantioselectivity, see Nowlan, III, D.T.; Singleton, D.A. J. Am. Chem. Soc. 2005, 127, 6190. Also see Jiao, L.; Ye, S.; Yu, Z.-X. J. Am. Chem. Soc. 2008, 130, 7178.
2144. Stanley, L.M.; Sibi, M.P. Chem. Rev. 2008, 108, 2887. See B![]() doiu, A.; Brinkmann,Y.; Viton, F.; Kündig, E.P. Pure Appl. Chem. 2008, 80, 1013.
doiu, A.; Brinkmann,Y.; Viton, F.; Kündig, E.P. Pure Appl. Chem. 2008, 80, 1013.
2145. Niimi, T.; Uchida, T.; Irie, R.; Katsuki, T. Tetrahedron Lett. 2000, 41, 3647.
2146. Sibi, M.P.; Ma, Z.; Jasperse, C.P. J. Am. Chem. Soc. 2004, 126, 718.
2147. Sibi, M.P.; Ma, Z.; Jasperse, C.P. J. Am. Chem. Soc. 2005, 127, 5764.
2148. Kano, T.; Hashimoto, T.; Maruoka, K. J. Am. Chem. Soc. 2005, 127, 11926.
2149. Baran, J.; Mayr, H. J. Am. Chem. Soc. 1987, 109, 6519.
2150. See Bastide, J.; Hamelin, J.; Texier, F.; Quang, Y.V. Bull. Soc. Chim. Fr. 1973, 2555; 2871; Fuks, R.; Viehe, H.G. in Viehe, H.G. Acetylenes Marcel Dekker, NY, 1969, p. 460–477.
2151. Lown, J.W. in Padwa, A. 1,3-Dipolar Cycloaddition Chemistry, vol 1. Wley, NY, 1984, pp. 683–732.
2152. See Lown, J.W. Rec. Chem. Prog. 1971, 32, 51; Gladysheva, F.N.; Sineokov, A.P.; Etlis, V.S. Russ. Chem. Rev. 1970, 39, 118.
2153. For a system of classification of cycloaddition reactions, see Huisgen, R. Angew. Chem. Int. Ed. 1968, 7, 321. See Posner, G.H. Chem. Rev. 1986, 86, 831. See also, the series Advances in Cycloaddition.
2154. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1101–1116.
2155. See Trost, B.M.; Seoane, P.; Mignani, S.; Acemoglu, M. J. Am. Chem. Soc. 1989, 111, 7487.
2156. Wang, J.-C.; Ng, S.-S.; Krische, M.J. J. Am. Chem. Soc. 2003, 125, 3682.
2157. See Trost, B.M. Pure Appl. Chem. 1988, 60, 1615; Angew. Chem. Int. Ed. 1986, 25, 1.
2158. See Trost, B.M.; Lynch, J.; Renaut, P.; Steinman, D.H. J. Am. Chem. Soc. 1986, 108, 284.
2159. Trost, B.M.; MacPherson, D.T. J. Am. Chem. Soc. 1987, 109, 3483; Trost, B.M. Angew. Chem. Int. Ed. 1986, 25, 1; Trost, B.M.; Stambuli, J.P.; Silverman, S.M.; Schwörer, U. J. Am. Chem. Soc. 2006, 128, 13328; Trost, B.M.; Cramer, N.; Silverman, S.M. J. Am. Chem. Soc. 2007, 129, 12396.
2160. Trost, B.M.; Silverman, S.M.; Stambuli, J.P. J. Am. Chem. Soc. 2007, 129, 12398.
2161. Greco, G.E.; Gleason, B.L.; Lowery, T.A.; Kier, M.J.; Hollander, L.B.; Gibbs, S.A.; Worthy, A.D. Org. Lett. 2007, 9, 3817.
2162. Hedley, S. J.; Moran, W. J.; Price, D.A.; Harrity, J.P.A. J. Org. Chem. 2003, 68, 4286.
2163. See Yamago, S.; Nakamura, E. J. Am. Chem. Soc. 1989, 111, 7285.
2164. See Chaigne, F.; Gotteland, J.; Malacria, M. Tetrahedron Lett. 1989, 30, 1803.
2165. Kauffmann, T. Top. Curr. Chem. 1980, 92, 109, pp. 111–116; Angew. Chem. Int. Ed. 1974, 13, 627.
2166. Eidenschink, R.; Kauffmann, T. Angew. Chem. Int. Ed. 1972, 11, 292.
2167. See Beak, P.; Burg, D.A. J. Org. Chem. 1989, 54, 1647.
2168. See Noyori, R.; Hayakawa, Y. Tetrahedron 1985, 41, 5879.
2169. Wasserman, A. Diels–Alder Reactions Elsevier, NY, 1965; Roush, W.R. Adv. Cycloaddit. 1990, 2, 91; Carruthers, W. Cycloaddition Reactions in Organic Synthesis, Pergamon, Elmsford, NY, 1990; Brieger, G.; Bennett, J.N. Chem. Rev. 1980, 80, 63; Oppolzer, W. Angew. Chem. Int. Ed. 1977, 16, 10; Sauer, J. Angew. Chem. Int. Ed. 1966, 5, 211; 1967, 6, 16; Taber, D.F. Intramolecular Diels–Alder and Alder Ene Reactions, Springer, NY, 1984; Deslongchamps, P. Aldrichimica Acta 1991, 24, 43; Craig, D. Chem. Soc. Rev. 1987, 16, 187. For a long list of references to various aspects of the Diels–Alder reaction, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 523–544.
2170. See Konovalov, A.I. Russ. Chem. Rev. 1983, 52, 1064.
2171. See Nelson, D.J.; Li, R.; Brammer, C. J. Org. Chem. 2001, 66, 2422.
2172. Spino, C.; Rezaei, H.; Dory, Y.L. J. Org. Chem. 2004, 69, 757. For tables of HOMOs and LUMOs for dienes and dienophiles, see Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 999–1032, especially pp. 1003–1004 and 1008–1009.
2173. See Domingo, L.R. Eur. J. Org. Chem. 2004, 4788.
2174. Fringuelli, F.; Taticchi, A.; Wenkert, E. Org. Prep. Proced. Int. 1990, 22, 131.
2175. See Butskus, P.F. Russ. Chem. Rev. 1962, 31, 283; Also see Ciganek, E.; Linn, W.J.; Webster, O.W. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 449–453.
2176. See Novikov, S.S.; Shuekhgeimer, G.A.; Dudinskaya, A.A. Russ. Chem. Rev. 1960, 29, 79.
2177. McClure, C.K.; Herzog, K.J.; Bruch, M.D. Tetrahedron Lett. 1996, 37, 2153.
2178. Johnstone, R.A.W.; Quan, P.M. J. Chem. Soc. 1963, 935.
2179. Carr, R.V.C.; Williams, R.V.; Paquette, L.A. J. Org. Chem. 1983, 48, 4976; Kinney, W.A.; Crouse, G.D.; Paquette, L.A. J. Org. Chem. 1983, 48, 4986.
2180. De Lucchi, O.; Lucchini, V.; Pasquato, L.; Modena, G. J. Org. Chem. 1984, 49, 596; Hermeling, D.; Schäfer, H.J. Angew. Chem. Int. Ed. 1984, 23, 233; De Lucchi, O.; Pasquato, L. Tetrahedron 1988, 44, 6755.
2181. See Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. Tetrahedron 2002, 58, 4417.
2182. For ground state conformations, see Bur, S.K.; Lynch, S.M.; Padwa, A. Org. Lett. 2002, 4, 473.
2183. For a discussion of conformational thermodynamic and kinetic parameters of methyl 1,3-butadienes, see Squillacote, M.E.; Liang, F. J. Org. Chem. 2005, 70, 6564.
2184. Sauer, J.; Lang, D.; Mielert, A. Angew. Chem. Int. Ed. 1962, 1, 268; Sauer, J.; Wiest, H. Angew. Chem. Int. Ed. 1962, 1, 269. See, however, Scharf, H.; Plum, H.; Fleischhauer, J.; Schleker, W. Chem. Ber. 1979, 112, 862.
2185. See Charlton, J.L.; Alauddin, M.M. Tetrahedron 1987, 43, 2873; Oppolzer, W. Synthesis 1978, 793.
2186. For a monograph on dienes, with tables showing > 800 types, see Fringuelli, F.; Taticchi, A. Dienes in the Diels–Alder Reaction, Wiley, NY, 1990. See Danishefsky, S. Chemtracts: Org. Chem. 1989, 2, 273; Petrzilka, M.; Grayson, J.I. Synthesis 1981, 753; Smith, M.B. Org. Prep. Proced. Int. 1990, 22, 315; Robiette, R.; Cheboub-Benchaba, K.; Peeters, D.; Marchand-Brynaert, J. J. Org. Chem. 2003, 68, 9809; Huang, Y.; Iwama, T.; Rawal, V.H. J. Am. Chem. Soc. 2002, 122, 5950.
2187. Avalos, M.; Babiano, R.; Bravo, J.L.; Cintas, P.; Jiménez, J.L.; Palacios, J.C.; Silva, M.A. J. Org. Chem. 2000, 65, 6613; Kiselev, V.D.; Konovalov, A.I. Russ. Chem. Rev. 1989, 58, 230; Zheng, M.; Zhang, M.-H.; Shao, J.-G.; Zhong, Q. Org. Prep. Proceed. Int. 1996, 28, 117; Yamabe, S.; Minato, T. J. Org. Chem. 2000, 65, 1830; Mathieu, B.; de Fays, L.; Ghosez, L. Tetrahedron Lett. 2000, 41, 9561.
2188. For a discussion of the effect of Lewis acids, see Celebi-Olcum, N.; Ess, D.H.; Aviyente, V.; Houk, K.N. J. Org. Chem. 2008, 73, 7472.
2189. Nakashima, D.; Yamamoto, H. Org. Lett. 2005, 7, 1251; Shen, J.; Tan. C.-H. Org. Biomol. Chem. 2008, 6, 3229.
2190. See Alston, P.V.; Ottenbrite, R.M. J. Org. Chem. 1975, 40, 1111.
2191. Reymond, S.; Cossy, J. Chem. Rev. 2008, 108, 5359.
2192. Ishihara, K.; Kurihara, H.; Yamamoto, H. J. Am. Chem. Soc,1996, 118, 3049.
2193. For a study of the influence of Lewis acids in ionic liquids, see Silvero, G.; Arévalo, M.J.; Bravo, J.L.; Ávalos, M.; Jiménez, J.L.; López, I. Tetrahedron 2005, 61, 7105. See López, I.; Silvero, G.; Arévalo, M.J.; Babiano, R.; Palacios, J.C.; Bravo, J.L. Tetrahedron 2007, 63, 2901.
2194. Kobayashi, S.; Hachiya, I.; Takahori, T.; Araki, M.; Ishitani, H. Tetrahedron Lett. 1992, 33, 6815.
2195. Mathieu, B.; Ghosez, L. Tetrahedron 2002, 58, 8219.
2196. See Sprott, K.T.; Corey, E.J. Org. Lett. 2003, 5, 2465; Corey, E.J.; Shibata, T.; Lee, T.W. J. Am. Chem. Soc. 2002, 124, 3808; Ryu, D.H.; Lee, T.W.; Corey, E.J. J. Am. Chem. Soc. 2002, 124, 9992.
2197. Gao, D.; Bauld, N.L. J. Org. Chem. 2000, 65, 6276. See Saettel, N.J.; Oxgaard, J.; Wiest, O. Eur. J. Org. Chem. 2001, 1429.
2198. For a review, see Bauld, N.L. Tetrahedron 1989, 45, 5307.
2199. Wipf, P.; Xu, W. Tetrahedron 1995, 51, 4551.
2200. Zhang, X.; Deng, Q.; Yoo, S.H.; Houk, K.N. J. Org. Chem. 2002, 67, 9043.
2201. Pandey, B.; Dalvi, P.V. Angew. Chem. Int. Ed. 1993, 32, 1612.
2202. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1037–1049.
2203. Jankowski, C.K.; LeClair, G.; Bélanger, J.M.R.; Paré, J.R.J.; Van Calsteren, M.-R. Can. J. Chem. 2001, 79, 1906. For a review, see de la Hoz, A.; Díaz-Ortis, A.; Moreno, A.; Langa, F. Eur. J. Org. Chem. 2000, 3659; Kaval, N.; Dehaen, W.; Kappe, C.O.; van der Eycken, E. Org. Biomol. Chem. 2004, 2, 154.
2204. Raj. C.P.; Dhas, N.A.; Cherkinski, M.; Gedanken, A.; Braverman, S. Tetrahedron Lett. 1998, 39, 5413.
2205. Veselovsky, V.V.; Gybin, A.S.; Lozanova, A.V.; Moiseenkov, A.M.; Smit, W.A.; Caple, R. Tetrahedron Lett. 1988, 29, 175.
2206. Kang, J.; Hilmersson, G.; Sartamaría, J.; Rebek Jr., J. J. Am. Chem. Soc. 1998, 120, 3650; Diego-Castro, M.J.; Hailes, H.C. Tetrahedron Lett. 1998, 39, 2211.
2207. Dolata, D.P.; Bergman, R. Tetrahedron Lett. 1987, 28, 707.
2208. For reviews, see Isaacs, N.S.; George, A.V. Chem. Br. 1987, 47–54; Asano, T.; le Noble, W.J. Chem. Rev. 1978, 78, 407. See also, Firestone, R.A.; Smith, G.M. Chem. Ber. 1989, 122, 1089.
2209. Kim, J.H.; Hubig, S.M.; Lindeman, S.V.; Kochi, J.K. J. Am. Chem. Soc. 2001, 123, 87.
2210. Breslow, R. Acc. Chem. Res. 1991, 24, 159; Breslow, R.; Rizzo, C.J. J. Am. Chem. Soc. 1991, 113, 4340; Engberts, J.B.F.N. Pure Appl. Chem. 1995, 67, 823; Pindur, U.; Lutz, G.; Otto, C. Chem. Rev. 1993, 93, 741; Otto, S.; Blokzijl, W.; Otto, S.; Egberts, J.B.F.N. Pure. Appl. Chem. 2000, 72, 1365; Deshpande, S.S.; Kumar, A. Tetrahedron 2005, 61, 8025; Hayashi, Y.; Takeda, M.; Shoji, M.; Morita, M. Chem. Lett. 2007, 36, 68.
2211. See Fringuelli, F.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Eur. J. Org. Chem. 2001, 439; Otto, S.; Engberts, J.B.F.N. Tetrahedron Lett. 1995, 36, 2645
2212. Chavan, S.P.; Sharma, P.; Krishna, G.R.; Thakkar, M. Tetrahedron Lett. 2003, 44, 3001.
2213. Pearson, R.J.; Kassianidis, E.; Philip, D. Tetrahedron Lett. 2004, 45, 4777.
2214. Meijer, A.; Otto, S.; Engberts, J.B.F.N. J. Org. Chem. 1998, 63, 8989.
2215. Jaeger, D.A.; Wang, J. Tetrahedron Lett. 1992, 33, 6415.
2216. Grieco, P.A.; Handy, S.T.; Beck, J.P. Tetrahedron Lett. 1994, 35, 2663. See Handy, S.T.; Grieco, P.A.; Mineur, C.; Ghosez, L. Synlett 1995, 565.
2217. Augé, J.; Gil, R.; Kalsey, S.; Lubin-Germain, N. Synlett 2000, 877.
2218. Pai, C.K.; Smith, M.B. J. Org. Chem. 1995, 60, 3731.
2219. Wijnen, J.W.; Engberts, J.B.F.N. J. Org. Chem. 1997, 62, 2039.
2220. Oakes, R.S.; Heppenstall, T.J.; Shezad, N.; Clifford, A.A.; Rayner, C.M. Chem. Commun. 1999, 1459. For an asymmetric cycloaddition, see Fukuzawa, S.-i.; Metoki, K.; Esumi, S.-i. Tetrahedron 2003, 59, 10445.
2221. Harano, Y.; Sato, H.; Hirata, F. J. Am. Chem. Soc. 2000, 122, 2289.
2222. Yli-Kauhaluoma, J. Tetrahedron 2001, 57, 7053.
2223. Eklund, L.; Axelsson, A.-K.; Nordahl, Å.; Carlson, R. Acta Chem. Scand. 1993, 47, 581.
2224. Pagni, R.M.; Kabalka, G.W.; Hondrogiannis, G.; Bains, S.; Anosike, P.; Kurt, R. Tetrahedron 1993, 49, 6743.
2225. Bini, R.; Chiappe, C.; Mestre, V.L.; Pomelli, C.S.; Welton, T. Org. Biomol. Chem. 2008, 6, 2522. For reactions in low-melting sugar–urea-salt mixtures, see Imperato, G.; Eibler, E.; Niedermaier, J.; König, B. Chem. Commun. 2005, 1170.
2226. Meracz, I.; Oh, T. Tetrahedron Lett. 2003, 44, 6465.
2227. Tiwari, S.; Kumar, A. Angew. Chem. Int. Ed. 2006, 45, 4824.
2228. Murali, R.; Scheeren, H.W. Tetrahedron Lett. 1999, 40, 3029.
2229. Alston, P.V.; Gordon, M.D.; Ottenbrite, R.M.; Cohen, T. J. Org. Chem. 1983, 48, 5051; Kahn, S.D.; Pau, C.F.; Overman, L.E.; Hehre, W.J. J. Am. Chem. Soc. 1986, 108, 7381.
2230. Ono, N.; Miyake, H.; Kamimura, A.; Kaji, A. J. Chem. Soc. Perkin Trans. 1,1987, 1929. For another method of controlling regioselectivity, see Kraus, G.A.; Liras, S. Tetrahedron Lett. 1989, 30, 1907.
2231. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1023–1032, 1066–1075; Bakalova, S.M.; Santos, A.G. J. Org. Chem. 2004, 69, 8475.
2232. For an exception, see Meier, H.; Eckes, H.; Niedermann, H.; Kolshorn, H. Angew. Chem. Int. Ed. 1987, 26, 1046.
2233. Robiette, R.; Marchand-Brynaert, J.; Peeters, D. J. Org. Chem. 2002, 67, 6823.
2234. See Baldwin, J.E.; Reddy, V.P. J. Org. Chem. 1989, 54, 5264. For a theoretical study for endo selectivity, see Imade, M.; Hirao, H.; Omoto, K.; Fujimoto, H. J. Org. Chem. 1999, 64, 6697.
2235. See Mülle, P.; Bernardinelli, G.; Rodriguez, D.; Pfyffer, J.; Schaller, J. Chimia 1987, 41, 244.
2236. Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. J. Am. Chem. Soc. 2000, 122, 4243.
2237. Hoffmann, R.; Woodward, R.B. J. Am. Chem. Soc. 1965, 87, 4388, 4389.
2238. García, J.I.; Mayoral, J.A.; Salvatella, L. Acc. Chem. Res. 2000, 33, 658; García, J.-I.; Mayoral, J.A.; Salvatella, L. Eur. J. Org. Chem. 2005, 85.
2239. Arrieta, A.; Cossío, F.P.; Lecea, B. J. Org. Chem. 2001, 66, 6178.
2240. Hickey, E.R.; Paquette, L.A. Tetrahedron Lett. 1994, 35, 2309, 2313.
2241. Cainelli, G.; Galletti, P.; Giacomini, D.; Quintavalla, A. Tetrahedron Lett. 2003, 44, 93.
2242. Domingo, L.R.; Picher, M.T.; Andrés, J.; Safont, V.S. J. Org. Chem. 1997, 62, 1775. Also see, Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 1023–1032, 1066–1075; Mörschel, P.; Janikowski, J.; Hilt, G.; Frenking, G. J. Am. Chem. Soc. 2008, 130, 8952.
2243. See Corey, E.J.; Sarshar, S.; Lee, D.-H. J. Am. Chem. Soc. 1994, 116, 12089; Taschner, M.J. Org. Synth: Theory Appl. 1989, 1, 1; Helmchen, G.; Karge, R.; Weetman, J. Mod. Synth. Methods 1986, 4, 261; Oppolzer, W. Angew. Chem. Int. Ed. 1984, 23, 876. See also Macaulay, J.B.; Fallis, A.G. J. Am. Chem. Soc. 1990, 112, 1136.
2244. Ruiz-López, M.F.; Assfeld, X.; García, J.I.; Mayoral, J.A.; Salvatella, L. J. Am. Chem. Soc. 1993, 115, 8780.
2245. Sustmann, R.; Sicking, W. J. Am. Chem. Soc. 1996, 118, 12562.
2246. For the use of chiral dienes, see Tripathy, R.; Carroll, P.J.; Thornton, E.R. J. Am. Chem. Soc. 1990, 112, 6743; 1991, 113, 7630; Rieger, R.; Breitmaier, E. Synthesis 1990, 697.
2247. See Xidos, J.D.; Poirier, R.A.; Pye, C.C.; Burnell, D.J. J. Org. Chem. 1998, 63, 105.
2248. Tomioka, K.; Hamada, N.; Suenaga, T.; Koga, K. J. Chem. Soc. Perkin Trans. 1,1990, 426; Cativiela, C.; López, P.; Mayoral, J.A. Tetrahedron: Asymmetry 1990, 1, 61.
2249. Narasaka, K. Synthesis 1991, 1; Corey, E.J.; Imai, N.; Zhang, H. J. Am. Chem. Soc. 1991, 113, 728; Narasaka, K.; Tanaka, H.; Kanai, F. Bull. Chem. Soc. Jpn. 1991, 64, 387; Hawkins, J.M.; Loren, S. J. Am. Chem. Soc. 1991, 113, 7794; Evans, D.A.; Barnes, D.M.; Johnson, J.S.; Lectka, T.; von Matt, P.; Miller, S.J.; Murry, J.A.; Norcross, R.D.; Shaughnessy, E.A.; Campos, K.R. J. Am. Chem. Soc. 1999, 121, 7582.
2250. See Corey, E.J. Angew. Chem. Int. Ed. 2002, 41, 1651; Doyle, M.P.; Phillips, I.M.; Hu, W. J. Am. Chem. Soc. 2001, 123, 5366; Owens, T.D.; Hollander, F.J.; Oliver A.G.; Ellman, J.A. J. Am. Chem. Soc. 2001, 123, 1539; Faller, J.W.; Grimmond, B.J.; D'Alliessi, D.G. J. Am. Chem. Soc. 2001, 123, 2525; Bolm, C.; Simic, O. J. Am. Chem. Soc. 2001, 123, 3830; Fukuzawa, S.; Komuro, Y.; Nakano, N.; Obara, S. Tetrahedron Lett. 2003, 44, 3671.
2251. Hawkins, J.M.; Loren, S.; Nambu, M. J. Am. Chem Soc. 1994, 116, 1657. See Sibi, M.P.; Venkatraman, L.; Liu, M.; Jaspersé, C.P. J. Am. Chem. Soc. 2001, 123, 8444.
2252. Ishihara, K.; Nakano, K. J. Am. Chem. Soc. 2005, 127, 10504; Liu, D.; Canales, E.; Corey, E.J. J. Am. Chem. Soc. 2007, 129, 1498; Wang, Y.; Li, H.; Wang, Y.-Q.; Liu, Y.; Foxman, B.M.; Deng, L. J. Am. Chem. Soc. 2007, 129, 6364; Payette, J.N.; Yamamoto, H. J. Am. Chem. Soc. 2007, 129, 9536; Singh, R.P.; Bartelson, K.; Wang, Y.; Su, H.; Lu, X.; Deng, L. J. Am. Chem. Soc. 2008, 130, 2422; Futatsugi, K.; Yamamoto, H. Angew. Chem. Int. Ed.2005, 44, 1484; Paddon-Row, M.N.; Kwan, L.C.H.; Willis, A.C.; Sherburn, M.S. Angew. Chem. Int. Ed. 2008, 47, 7013.
2253. See Bastide, J.; Henri-Rousseau, O. in Patai, S. The Chemistry of the Carbon-Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 447–522, Fuks, R.; Viehe, H.G. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 477–508.
2254. See Paik, S.-J.; Son, S.U.; Chung, Y.K. Org. Lett. 1999, 1, 2045.
2255. This type of Diels–Alder reaction has been called the Dehydro-Diels–Alder. See Wessig, P.; Gunnar Müller, G. Chem. Rev. 2008, 108, 2051; Dunetz, J.R.; Danheiser, R.L. J. Am. Chem. Soc. 2005, 127, 5776; Dai, M.; Sarlah, D.; Yu, M.; Danishefsky, S.J.; Jones, G.O.; Houk, K.N. J. Am. Chem. Soc. 2007, 129, 645.
2256. See Hopf, H. in Landor, S.R. The Chemistry of Allenes, Vol. 2, Academic Press, NY, 1982, pp. 563–577. See Nendel, M.; Tolbert, L.M.; Herring, L.E.; Islam, Md.N.; Houk, K.N. J. Org. Chem. 1999, 64, 976.
2257. Ketenes react with conjugated dienes to give 1,2-addition (see Reaction 15-49).
2258. See Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. Angew. Chem. Int. Ed. 2002, 41, 1669.
2259. For a review, see Wagner-Jauregg, T. Synthesis 1980, 165, 769. See also, Balaban, A.T.; Biermann, D.; Schmidt, W. Nouv. J. Chim. 1985, 9, 443.
2260. However, see Chordia, M.D.; Smith, P.L.; Meiere, S.H.; Sabat, M.; Harman, W.D. J. Am. Chem. Soc. 2001, 123, 10756.
2261. Friedman, L. J. Am. Chem. Soc. 1967, 89, 3071; Liu, R.S.H.; Krespan, C.G. J. Org. Chem. 1969, 34, 1271.
2262. See Hoffmann, R.W. Dehydrobenzene and Cycloalkynes; Academic Press, NY, 1967, pp. 200–239; Bryce, M.R.; Vernon, J.M. Adv. Heterocycl. Chem. 1981, 28, 183–229. Also see Liu, W.; You, F.; Mocella, C.J.; Harman, W.D. J. Am. Chem. Soc. 2006, 128, 1426.
2263. Wittig, G.; Niethammer, K. Chem. Ber. 1960, 93, 944; Wittig, G.; Härle, H.; Knauss, E.; Niethammer, K. Chem. Ber. 1960, 93, 951.
2264. Plieninger, H.; Wild, D.; Westphal, J. Tetrahedron 1969, 25, 5561.
2265. See Paquette, L.A.; Kesselmayer, M.A.; Künzer, H. J. Org. Chem. 1988, 53, 5183.
2266. Hilt, G.; du Mesnil, F.-X. Tetrahedron Lett. 2000, 41, 6757.
2267. For a review of natural product syntheses using Diels–Alder reactions, see Takao, K.-i.; Munakata, R.; Tadano, K.-i. Chem. Rev. 2005, 105, 4779.
2268. Oppolzer, W. Angew. Chem. Int. Ed. 1977, 16, 1; Brieger, G.; Bennett, J.N. Chem. Rev. 1980, 80, 63 (see p. 67); Fallis, A.G. Can. J. Chem. 1984, 62, 183; Smith, M.B. Org. Prep. Proceed. Int. 1990, 22, 315.
2269. Au catalyst: Nieto-Oberhuber, C.; López, S.; Echavarren, A.M. J. Am. Chem. Soc. 2005, 127, 6178.
2270. Paddon-Row, M.N.; Moran, D.; Jones, G.A.; Sherburn, M.S. J. Org. Chem. 2005, 70, 10841.
2271. Khuong, K.S.; Beaudry, C.M.; Trauner, D.; Houk, K.N. J. Am. Chem. Soc. 2005, 127, 3688.
2272. See Tantillo, D.J.; Houk, K.N.; Jung, M.E. J. Org. Chem. 2001, 66, 1938.
2273. Stork, G.; Chan, T.Y.; Breault, G.A. J. Am. Chem. Soc. 1992, 114, 7578.
2274. Craig, D.; Reader, J.C. Tetrahedron Lett. 1992, 33, 6165.
2275. Ishikawa, T.; Senzaki, M.; Kadoya, R.; Morimoto, T.; Miyake, N.; Izawa, M.; Saito, S.; Kobayashi, H. J. Am. Chem. Soc. 2001, 123, 4607.
2276. Paddon-Row, M.N.; Longshaw, A.I.; Willis, A.C.; Sherburn, M.S. Chemistry: Asian J. 2009, 4, 126.
2277. Bertozzi, F.; Olsson, R.; Frejd, T. Org. Lett. 2000, 2, 1283.
2278. See Ichihara, A. Synthesis 1987, 207; Lasne, M.; Ripoll, J.L. Synthesis 1985, 121; Kwart, H.; King, K. Chem. Rev. 1968, 68, 415.
2279. Sample, Jr., T.E.; Hatch, L.F. Org. Synth. VI, 454. For a review, see Chou, T.; Tso, H. Org. Prep. Proced. Int. 1989, 21, 257.
2280. See Sauer, J.; Sustmann, R. Angew. Chem. Int. Ed. 1980, 19, 779; Houk, K.N. Top. Curr. Chem. 1979, 79, 1; Babichev, S.S.; Kovtunenko, V.A.; Voitenko, Z.V.; Tyltin, A.K. Russ. Chem. Rev. 1988, 57, 397. For a discussion of synchronous versus nonsynchronous mechanisms see Beno, B.R.; Houk, K.N.; Singleton, D.A. J. Am. Chem. Soc. 1996, 118, 9984; Singleton, D.A.; Schulmeier, B.E.; Hang, C.; Thomas, A.A.; Leung, S.-W.; Merrigan, S.R. Tetrahedron 2001, 57, 5149. Also see, Li, Y.; Houk, K.N. J. Am. Chem. Soc. 1993, 115, 7478.
2281. See Orlova, G.; Goddard, J.D. J. Org. Chem. 2001, 66, 4026.
2282. Domingo, L.R.; Sáez, J.A. Org. Biomol. Chem. 2009, 7, 3576. See also, Soto-Delgado, J.; Domingo, L.R.; Contreras, R. Org. Biomol. Chem. 2010, 8, 3678.
2283. For a contrary view, see Dewar, M.J.S.; Olivella, S.; Stewart, J.J.P. J. Am. Chem. Soc. 1986, 108, 5771. For arguments against this view, see Houk, K.N.; Lin, Y.; Brown, F.K. J. Am. Chem. Soc. 1986, 108, 554; Gajewski, J.J.; Peterson, K.B.; Kagel, J.R.; Huang, Y.C.J. J. Am. Chem. Soc. 1989, 111, 9078.
2284. See Van Mele, B.; Huybrechts, G. Int. J. Chem. Kinet. 1987, 19, 363; 1989, 21, 967.
2285. See Gassman, P.G.; Gorman, D.B. J. Am. Chem. Soc. 1990, 112, 8624.
2286. Haberl, U.; Wiest, O.; Steckhan, E. J. Am. Chem. Soc. 1999, 121, 6730.
2287. Seltzer, S. J. Am. Chem. Soc. 1965, 87, 1534; Gajewski, J.J. Isot. Org. Chem. 1987, 7, 115–176.
2288. Van Sickle, D.E.; Rodin, J.O. J. Am. Chem. Soc. 1964, 86, 3091.
2289. See Rücker, C.; Lang, D.; Sauer, J.; Friege, H.; Sustmann, R. Chem. Ber. 1980, 113, 1663; Tolbert, L.M.; Ali, M.B. J. Am. Chem. Soc. 1981, 103, 2104.
2290. For an example of a study of a reaction that is concerted but asynchronous, see Avalos, M.; Babiano, R.; Clemente, F.R.; Cintas, P.; Gordillo, R.; Jiménez, J.L.; Palacios, J.C. J. Org. Chem. 2000, 65, 8251
2291. Houk, K.N.; Loncharich, R.J.; Blake, J.F.; Jorgensen, W.L. J. Am. Chem. Soc. 1989, 111, 9172; Lehd, M.; Jensen, F. J. Org. Chem. 1990, 55, 1034.
2292. See Domingo, L.R.; Aurell, M.J.; Pérez, P.; Contreras, R. J. Org. Chem. 2003, 68, 3884.
2293. de Echagüen, C.O.; Ortuño, R.M. Tetrahedron Lett. 1995, 36, 749. See Li, Y.; Padias, A.B.; Hall, Jr., H.K. J. Org. Chem. 1993, 58, 7049 for a discussion of diradicals in concerted Diels–Alder reactions.
2294. Sauer, J.; Wiest, H. Angew. Chem. Int. Ed. 1962, 1, 269.
2295. Boger, D.L.; Patel, M. Prog. Heterocycl. Chem. 1989, 1, 30. Also see, Pugnaud, S.; Masure, D.; Hallé, J.-C.; Chaquin, P. J. Org. Chem,. 1997, 62, 8687; Wan, Z.-K.; Snyder, J.K. Tetrahedron Lett. 1998, 39, 2487.
2296. For a mechanistic study, see Gomez-Bengoa, E.; Helm, M.D.; Plant, A.; Harrity, J.P.A. J. Am. Chem. Soc. 2007, 129, 2691.
2297. Fleming, I. Pericyclic Reactions Oxford University Press, Oxford, 1999, pp. 31–56; Gilchrist, T.L.; Storr, R.C. Organic Reactions and Orbital Symmetry 2nd ed., Cambridge University Press, Cambridge, 1979; Fleming, I. Frontier Orbitals and Organic Chemical Reactions, Wiley, NY, 1976; Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970 [the text of this book also appears in Angew. Chem. Int. Ed.1969, 8, 781]; Simonetta, M. Top. Curr. Chem. 1973, 42, 1; Houk, K.N. Surv. Prog. Chem. 1973, 6, 113; Gill, G.B. Q. Rev. Chem. Soc. 1968, 22, 338; Miller, S.I. Adv. Phys. Org. Chem. 1968, 6, 185; Miller, S.I. Bull. Soc. Chim. Fr.1966, 4031.
2298. Chattaraj, P.K.; Fuentealba, P.; Gómez, B.; Contreras, R. J. Am. Chem. Soc. 2000, 122, 348.
2299. See Epiotis, N.D. Theory of Organic Reactions, Springer, NY, 1978; Ponec, R. Collect. Czech. Chem. Commun. 1984, 49, 455; 1985, 50, 1121; Hua-ming, Z.; De-xiang, W. Tetrahedron 1986, 42, 515; Bernardi, F.; Olivucci, M.; Robb, M.A. Res. Chem. Intermed. 1989, 12, 217; Acc. Chem. Res. 1990, 23, 405.
2300. See Woodward, R.B.; Hoffmann, R. The Conservation of Orbital Symmetry, Academic Press, NY, 1970; Angew. Chem. Int. Ed. 1969, 8, 781; Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge University Press, Cambridge, 1984, pp. 352–366; Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982, pp. 378–389; Yates, K. Hückel Molecular Orbital Theory, Academic Press, NY, 1978, pp. 263–276.
2301. Fukui, K.; Fujimoto, H. Bull. Chem. Soc. Jpn. 1967, 40, 2018; 1969, 42, 3399; Fukui, K. Acc. Chem. Res. 1971, 4, 57; Houk, K.N. Acc. Chem. Res. 1975, 8, 361. See Chu, S. Tetrahedron 1978, 34, 645; Fleming, I. Pericyclic Reactions, Oxford University Press, Oxford, 1999; Fukui, K. Angew. Chem. Int. Ed. 1982, 21, 801.
2302. For a discussion of molecules with small HOMO–LUMO gaps, see Perepichka, D.F.; Bryce, M.R. Angew. Chem. Int. Ed.2005, 44, 5370.
2303. Zimmerman, H.E. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 53–107; Acc. Chem. Res. 1971, 4, 272; Dewar, M.J.S. Angew. Chem. Int. Ed. 1971, 10, 761; Jefford, C.W.; Burger, U. Chimia,1971, 25, 297; Herndon, W.C. J. Chem. Educ. 1981, 58, 371.
2304. See Morao, I.; Cossío, F.P. J. Org. Chem. 1999, 64, 1868.
2305. See Hennigar, K.H.R.; Langler, R.F. Austr. J. Chem. 2010, 63, 490.
2306. Rzepa, H.S. Chem. Commun. 2005, 5220.
2307. See Lehr, R.E.; Marchand, A.P. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 1, Academic Press, NY, 1977, pp. 1–51.
2308. See Baldwin, J.E.; Andrist, A.H.; Pinschmidt, Jr., R.K. Acc. Chem. Res. 1972, 5, 402; Berson, J.A. Acc. Chem. Res. 1972, 5, 406; Baldwin, J.E. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions, Vol. 2, Academic Press, NY, 1977, pp. 273–302.
2309. See Sieber, W.; Heimgartner, H.; Hansen, H.; Schmid, H. Helv. Chim. Acta 1972, 55, 3005; Bartlett, P.D.; Helgeson, R.; Wersel, O.A. Pure Appl. Chem. 1968, 16, 187; Seeley, D.A. J. Am. Chem. Soc. 1972, 94, 4378; Kaupp, G. Angew. Chem. Int. Ed. 1972, 11, 313, 718.
2310. A possible photochemical [2 + 4] cycloaddition has been reported: Hart, H.; Miyashi, T.; Buchanan, D.N.; Sasson, S. J. Am. Chem. Soc. 1974, 96, 4857.
2311. Hoffmann, R.; Woodward, R.B. J. Am. Chem. Soc. 1965, 87, 4388.
2312. See Ginsburg, D. Tetrahedron 1983, 39, 2095; Gleiter, R.; Paquette, L.A. Acc. Chem. Res. 1983, 16, 328. See Singleton, D.A. J. Am. Chem. Soc. 1992, 114, 6563.
2313. See Apeloig, Y.; Matzner, E. J. Am. Chem. Soc. 1995, 117, 5375.
2314. See Fox, M.A.; Cardona, R.; Kiwiet, N.J. J. Org. Chem. 1987, 52, 1469.
2315. See McCarrick, M.A.; Wu, Y.-D.; Houk, K.N. J. Org. Chem. 1993, 58, 3330.
2316. Anniyappan, M.; Muralidharan, D.; Perumal, P.T. Tetrahedron Lett. 2003, 44, 3653. For a review see Buonora, P.; Olsen, J.-C.; Oh, T. Tetrahedron 2001, 57, 6099.
2317. Domingo, L.R. J. Org. Chem. 2001, 66, 3211; Chou, S.-S.P.; Hung, C.-C. Synth. Commun. 2001, 31, 1097.
2318. Martin, S.F.; Hartmann, M.; Josey, J.A. Tetrahedron Lett. 1992, 33, 3583.
2319. Boger, D.L.; Weinreb, S.M. Hetero Diels–Alder Methodology in Organic Synthesis; Academic Press, NY, 1987; Weinreb, S.M.; Scola, P.M. Chem. Rev. 1989, 89, 1525; Kametani, T.; Hibino, S. Adv. Heterocycl. Chem. 1987, 42, 245; Boger, D.L. Tetrahedron 1983, 39, 2869; Weinreb, S.M.; Staib, R.R. Tetrahedron 1982, 38, 3087; Desimoni, G.; Tacconi, G. Chem. Rev. 1975, 75, 651; Katritzky, A.R.; Dennis, N. Chem. Rev. 1989, 89, 827; Schmidt, R.R. Acc. Chem. Res. 1986, 19, 250; Boger, D.L. Chem. Rev. 1986, 86, 781.
2320. See Molander, G.A.; Rzasa, R.M. J. Org. Chem. 2000, 65, 1215.
2321. Amos, D.T.; Renslo, A.R.; Danhesier, R.L. J. Am. Chem. Soc. 2003, 125, 4970.
2322. See Danishefsky, S.; Schuda, P.F.; Kitahara, T. Etheredge, S.J. J. Am. Chem. Soc. 1977, 99, 6066. Also see Sudo, Y.; Shirasaki, D.; Harada, S.; Nishida, A. J. Am. Chem. Soc. 2008, 130, 12588.
2323. See Zhang, X.; Du, H.; Wang, Z.; Wu, Y.-D.; Ding, K. J. Org. Chem. 2006, 71, 2862.
2324. Kezuka, S.; Mita, T.; Ohtsuki, N.; Ikeno, T.; Yamada, T. Bull. Chem. Soc. Jpn. 2001, 74, 1333.
2325. Ali, T.; Chauhan, K.K.; Frost, C.G. Tetrahedron Lett. 1999, 40, 5621.
2326. Wang, B.; Feng, X.; Huang, Y.; Liu, H.; Cui, X.; Jiang, Y. J. Org. Chem. 2002, 67, 2175.
2327. Yamashita, Y.; Saito, S.; Ishitani, H.; Kobayashi, S. J. Am. Chem. Soc. 2003, 125, 3793.
2328. Chen, I.-H.; Oisaki, K.; Kanai, M.; Shibasaki, M. Org. Lett. 2008, 10, 5151.
2329. In an ionic liquid, see Pégot, B.; Vo-Thanh, G. Synlett 2005, 1409/
2330. Yuan, Y.; Li, X.; Ding, K. Org. Lett. 2002, 4, 3309.
2331. See J![]() rgensen, K.A. Eur. J. Org. Chem. 2004, 2093.
rgensen, K.A. Eur. J. Org. Chem. 2004, 2093.
2332. Babu, G.; Perumal, P.T. Tetrahedron 1998, 54, 1627.
2333. Lubineau, A.; Augé, J.; Grand, E.; Lubin, N. Tetrahedron 1994, 50, 10265.
2334. Villacampa, M.; Pérez, J.M.; Avendaño, C.; Menéndez, J.C. Tetrahedron 1994, 50, 10047.
2335. Pierres, C.; George, P.; van Hijfte, L.; Ducep, J.-B.; Hibert, M.; Mann, A. Tetrahedron Lett. 2003, 44, 3645.
2336. Yao, S.; Johannsen, M.; Audrain, H.; Hazell, R.G.; J![]() rgensen, K.A. J. Am. Chem. Soc. 1998, 120, 8599.
rgensen, K.A. J. Am. Chem. Soc. 1998, 120, 8599.
2337. Pellissier, H. Tetrahedron 2009, 65, 2839.
2338. Beaudegnies, R.; Ghosez, L. Tetrahedron Asymmetry 1994, 5, 557.
2339. Wurz, R.P.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 12234.
2340. See He, M.; Struble, J.R.; Bode, J.W. J. Am. Chem. Soc. 2006, 128, 8418.
2341. Jensen, K.H.; Sigman, M.S. Angew. Chem. Int. Ed. 2008, 47, 4748.
2342. Wurz, R.P.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 12234.
2343. Kawasaki, M.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 16482.
2344. See Sarkar, N.; Banerjee, A.; Nelson, S.G. J. Am. Chem. Soc. 2008, 130, 9222.
2345. Jayakumar, S.; Ishar, M.P.S.; Mahajan, M.P. Tetrahedron 2002, 58, 379.
2346. Yadav, J.S.; Reddy, B.V.S.; Reddy, J.S.S.; Rao, R.S. Tetrahedron 2003, 59, 1599.
2347. Suga, S.; Nagaki, A.; Tsutsui, Y.; Yoshida, J.-i. Org. Lett. 2003, 5, 945.
2348. Sheu, J.; Smith, M.B.; Matsumoto, K. Synth. Commun,1993, 23, 253.
2349. Akiyama, T.; Morita, H.; Fuchibe, K. J. Am. Chem. Soc. 2006, 128, 13070; Esquivias, J.; Arrayás, R.G.; Carretero, J.C. J. Am. Chem. Soc. 2007, 129, 1480.
2350. Schatz, J.; Sauer, J. Tetrahedron Lett. 1994, 35, 4767.
2351. Degnan, A.P.; Kim, C.S.; Stout, C.W.; Kalivretenos, A.G. J. Org. Chem. 1995, 60, 7724.
2352. Katritzky, A.R.; Dennis, N. Chem. Rev. 1989, 89, 827; Schmidt, R.R. Acc. Chem. Res. 1986, 19, 250; Boger, D.L. Chem. Rev. 1986, 86, 781. See Hayashi, Y.; Nakamura, M.; Nakao, S.; Inoue, T.; Shoji, M. Angew. Chem. Int. Ed. 2002, 41, 4079.
2353. Bach, P.; Bols, M. Tetrahedron Lett. 1999, 40, 3461.
2354. Gademann, K.; Chavez, D.E.; Jacobsen, E.N. Angew. Chem. Int. Ed. 2002, 41, 3059.
2355. Ruano, J.L.G.; Clemente, F.R.; Gutiérrez, L.G.; Gordillo, R.; Castro, A.M.M.; Ramos, J.H.R. J. Org. Chem. 2002, 67, 2926.
2356. See Clennan, E.L. Tetrahedron 1991, 47, 1343; Adv. Oxygenated Processes 1988, 1, 85; Wasserman, H.H.; Ives, J.L. Tetrahedron 1981, 37, 1825; Denny, R.W.; Nickon, A. Org. React. 1973, 20, 133; Schönberg, A. Preparative Organic Photochemistry, Springer, NY, 1968, pp. 382–397; Gollnick, K.; Schenck, G.O. in Hamer, J. 1,4-Cycloaddition Reactions, Academic Press, NY, 1967, pp. 255–344.
2357. Matsumoto, M.; Dobashi, S.; Kuroda, K.; Kondo, K. Tetrahedron 1985, 41, 2147.
2358. See Saito, I.; Nittala, S.S. in Patai, S. The Chemistry of Peroxides, Wiley, NY, 1983, pp. 311–374; Balci, M. Chem. Rev. 1981, 81, 91; Adam, W.; Bloodworth, A.J. Top. Curr. Chem. 1981, 97, 121.
2359. Onitsuka, S.; Nishino, H.; Kurosawa, K. Tetrahedron 2001, 57, 6003.
2360. See in Wasserman, H.H.; Murray, R.W. Singlet Oxygen, Academic Press, NY, 1979, the articles by Wasserman, H.H.; Lipshutz, B.H. pp. 429–509; Saito, I.; Matsuura, T. pp. 511–574; Rigaudy, J. Pure Appl. Chem. 1968, 16, 169.
2361. Frimer, A.A. Singlet O2, 4 Vols., CRC Press, Boca Raton, FL, 1985; Wasserman, H.H.; Murray, R.W. Singlet Oxygen, Academic Press, NY, 1979. See Frimer, A.A. in Patai, S. The Chemistry of Peroxides, Wiley, NY, 1983, pp. 201–234; Gorman, A.A.; Rodgers, M.A.J. Chem. Soc. Rev. 1981, 10, 205; Ohloff, G. Pure Appl. Chem. 1975, 43, 481; Kearns, D.R. Chem. Rev. 1971, 71, 395; Wayne, R.P. Adv. Photochem. 1969, 7, 311.
2362. Turro, N.J.; Ramamurthy, V. in de Mayo, P. Rearrangements in Ground and Excited States, Vol. 3, Academic Press, NY, 1980, pp. 1–23; Murray, R.W. in Wasserman, H.H.; Murray, R.W. Singlet Oxygen, Academic Press, NY, 1979, pp. 59–114; Adam, W.; Cilento, G. Chemical and Biological Generation of Excited States, Academic Press, NY, 1982.
2363. Monroe, B.M. J. Am. Chem. Soc. 1981, 103, 7253. See also, Hathaway, S.J.; Paquette, L.A. Tetrahedron Lett. 1985, 41, 2037; O'Shea, K.E.; Foote, C.S. J. Am. Chem. Soc. 1988, 110, 7167.
2364. Adam, W.; Cilento, G. Angew. Chem. Int. Ed. 1983, 22, 529; Schaap, A.; Zaklika, K.A. in Wasserman, H.H.; Murray, R.W. Singlet Oxygen Academic Press, NY, 1979, pp. 173–242; Bartlett, P.D. Chem. Soc. Rev. 1976, 5, 149. For discussions of the mechanisms see Frimer, A.A. Chem. Rev. 1979, 79, 359; Clennan, E.L.; Nagraba, K. J. Am. Chem. Soc. 1988, 110, 4312.
2365. Kearns, D.R. Chem. Rev. 1971, 71, 395, pp. 422–424; Foote, C.S. Pure Appl. Chem. 1971, 27, 635.
2366. Adam, W. in Patai, S. The Chemistry of Peroxides, Wiley, NY, 1983, pp. 829–920; Bartlett, P.D.; Landis, M.E. in Wasserman, H.H.; Murray, R.W. Singlet Oxygen, Academic Press, NY, 1979, pp. 243–286; Adam, W. Adv. Heterocycl. Chem. 1977, 21, 437. See also, Adam, W.; Encarnación, L.A.A. Chem. Ber. 1982, 115, 2592; Adam, W.; Baader, W.J. Angew. Chem. Int. Ed. 1984, 23, 166.
2367. See Nelson, S.F.; Teasley, M.F.; Kapp, D.L. J. Am. Chem. Soc. 1986, 108, 5503.
2368. See Nelson, S.F. Acc. Chem. Res. 1987, 20, 269.
2369. See Curci, R.; Lopez, L.; Troisi, L.; Rashid, S.M.K.; Schaap, A.P. Tetrahedron Lett. 1987, 28, 5319.
2370. Posner, G.H.; Weitzberg, M.; Nelson, W.M.; Murr, B.L.; Seliger, H.H. J. Am. Chem. Soc. 1987, 109, 278.
2371. Carruthers, W. Cycloaddition Reactions in Organic Synthesis Pergamon, Elmsford, NY, 1990; Reinhoudt, D.N. Adv. Heterocycl. Chem. 1977, 21, 253; Roberts, J.D.; Sharts, C.M. Org. React. 1962, 12, 1; Gilchrist, T.L.; Storr, R.C. Organic Reactions and Orbital Symmetry, 2nd ed., Cambridge University Press, Cambridge, 1979, pp. 173–212; Dilling, W.L. Chem. Rev. 1983, 83, 1. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 546–647, 1341–1344.
2372. See Takasu, K.; Ueno, M.; Inanaga, K.; Ihara, M. J. Org. Chem. 2004, 69, 517.
2373. See Mariet, N.; Ibrahim-Ouali, M.; Santelli, M. Tetrahedron Lett. 2002, 43, 5789.
2374. Dolbier, Jr., W.R.; Lomas, D.; Garza, T.; Harmon, C.; Tarrant, P. Tetrahedron 1972, 28, 3185.
2375. López-Carrillo, V.; Echavarren, A.M. J. Am. Chem. Soc. 2010, 132, 9292.
2376. Jordan, R.W.; Tam, W. Org. Lett. 2000, 2, 3031.
2377. Saito, S.; Hirayama, K.; Kabuto, C.; Yamamoto, Y. J. Am. Chem. Soc. 2000, 122, 10776.
2378. Dehmlow, E.V.; Pickardt, J.; Slopianka, M.; Fastabend, U.; Drechsler, K.; Soufi, J. Liebigs Ann. Chem. 1987, 377.
2379. Bouwkamp, M.W.; Bowman, A.C.; Lobkovsky, E.; Chirik, P.J. J. Am. Chem. Soc. 2006, 128, 13340.
2380. Bandin, E.; Favi, G.; Martelli, G.; Panunzio, M.; Piersanti, G. Org. Lett. 2000, 2, 1077.
2381. Baik, T.-G.; Luis, A.L.; Wang, L.-C.; Krische, M.J. J. Am. Chem. Soc. 2001, 123, 6716.
2382. Delas, C.; Urabe, H.; Sato, F. Tetrahedron Lett. 2001, 42, 4147.
2383. See de Faria, A.R.; Matos, C.R.; Correia, C.R.D. Tetrahedron Lett. 1993, 34, 27.
2384. Martin, P.; Greuter, H.; Bellus, D. Helv. Chim. Acta.,1984, 64, 64.
2385. Sustmann, R.; Ansmann, A.; Vahrenholt, F. J. Am. Chem. Soc. 1972, 94, 8099; Desimoni, G.; Tacconi, G.; Barco, A.; Pollini, G.P. Natural Product Synthesis Through Pericyclic Reactions, American Chemical Society Washington, DC, 1983, pp. 119–254, p. 39.
2386. Brown, M.J. Heterocycles 1989, 29, 2225; Isaacs, N.S. Chem. Soc. Rev. 1976, 5, 181; Mukerjee, A.K.; Srivastava, R.C. Synthesis 1973, 327; Sandhu, J.S.; Sain, B. Heterocycles 1987, 26, 777.; Wack, H.; France, S.; Hafez, A.M.; Drury, III, W.J.; Weatherwax, A.; Lectka, T. J. Org. Chem. 2004, 69, 4531.
2387. Townes, J.A.; Evans, M.A.; Queffelec, J.; Taylor, S.J.; Morken, J.P. Org. Lett. 2002, 4, 2537.
2388. De Cock, C.; Piettre. S.; Lahousse, F.; Janousek, Z.; Merényi, R.; Viehe, H.G. Tetrahedron 1985, 41, 4183.
2389. Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis Wley, NY, 1984, pp. 286–317; Hopf, H. in Landor, S.R. The Chemistry of Allenes, Vol. 2, Academic Press, NY, 1982, pp. 525–562; Ghosez, L.; O'Donnell, M.J. in Marchand, A.P.; Lehr, R.E. Pericyclic Reactions Vol. 2, Academic Press, NY, 1977, pp. 79–140; Luzung, M.R.; Mauleón, P.; Toste, F.D. J. Am. Chem. Soc. 2007, 129, 12402.
2390. Ghosez, L.; O'Donnell, M.J. in Marchand, A.P.; Lehr, R.E. Pericyclic Reaction, Vol. 2, Academic Press, NY, 1977; Brady, W.T. Synthesis 1971, 415; Snider, B.B. Chem. Rev. 1988, 88, 793. See Ussing, B.R.; Hang, C.; Singleton, D.A. J. Am. Chem. Soc. 2006, 128, 7594.
2391. See Huisgen, R.; Feiler, L.A.; Otto, P. Tetrahedron Lett. 1968, 4491; Chem. Ber. 1969, 102, 3475; Corey, E.J.; Ravindranathan, T.; Terashima, S. J. Am. Chem. Soc. 1971, 93, 4326. For a review of ketene equivalents, see Ranganathan, S.; Ranganathan, D.; Mehrotra, A.K. Synthesis 1977, 289.
2392. Bak, D.A.; Brady, W.T. J. Org. Chem. 1979, 44, 107.
2393. Gras, J.; Bertrand, M. Nouv. J. Chim. 1981, 5, 521.
2394. Cook, A.G. in Cook, A.G. Enamines, 2nd ed.; Marcel Dekker, NY, 1988, pp. 347–440.
2395. Brannock, K.C.; Bell, A.; Goodlett, V.W.; Thweatt, J.G. J. Org. Chem. 1964, 29, 813.
2396. Hasek, R.H.; Gott, P.G.; Martin, J.C. J. Org. Chem. 1966, 31, 1931.
2397. See Inanaga, K.; Takasu, K.; Ihara, M. J. Am. Chem. Soc. 2005, 127, 3668.
2398. Scheeren, J.W. Recl. Trav. Chim. Pays-Bas 1986, 105, 71-84.
2399. Williams, J.K., Wiley, D.W.; McKusick, B.C. J. Am. Chem. Soc. 1962, 84, 2210.
2400. Nishida, S.; Moritani, I.; Teraji, T. J. Org. Chem. 1973, 38, 1878.
2401. Nagata, J.; Shirota, Y.; Nogami, T.; Mikawa, H. Chem. Lett. 1973, 1087; Shirota, Y.; Yoshida, K.; Nogami, T.; Mikawa, H. Chem. Lett. 1973, 1271.
2402. Canales, E.; Corey, E.J. J. Am. Chem. Soc. 2007, 129, 12686; Butenschön, H. Angew. Chem. Int. Ed. 2008, 47, 3492; Ishihara, K.; Fushimi, M. J. Am. Chem. Soc. 2008, 130, 7532.
2403. Ng, S.M.; Bader, S.J.; Snapper, M.L. J. Am. Chem. Soc. 2006, 128, 7315.
2404. Treutwein, J.; Hilt, G. Angew. Chem. Int. Ed. 2008, 47, 6811.
2405. Yamazaki, S.; Fujitsuka, H.; Yamabe, S.; Tamura, H. J. Org. Chem. 1992, 57, 5610.
2406. Yoshikawa, S.; Aoki, K.; Kiji, J.; Furukawa, J. Tetrahedron 1974, 30, 405.
2407. See Mango, F.D. Top. Curr. Chem. 1974, 45, 39; Tetrahedron Lett. 1973, 1509.
2408. See Bachrach, S.M.; Gilbert, J.C. J. Org. Chem. 2004, 69, 6357; Ozkan, I.; Kinal, A. J. Org. Chem. 2004, 69, 5390.
2409. See Grubbs, R.H.; Miyashita, A.; Liu, M.M.; Burk, P.L. J. Am. Chem. Soc. 1977, 99, 3863.
2410. Fraser, A.R.; Bird, P.H.; Bezman, S.A.; Shapley, J.R.; White, R.; Osborn, J.A. J. Am. Chem. Soc. 1973, 95, 597.
2411. See Prinzbach, H.; Sedelmeier, G.; Martin, H. Angew. Chem. Int. Ed. 1977, 16, 103.
2412. Ischay, M.A.; Anzovino, M.E.; Du, J.; Yoon, T.P. J. Am. Chem. Soc. 2008, 130, 12886.
2413. Gassman, P.G.; Mansfield, K.T. J. Am. Chem. Soc. 1968, 90, 1517, 1524.
2414. For a review, see Gassman, P.G. Acc. Chem. Res. 1971, 4, 128.
2415. Cairncross, A.; Blanchard, E.P. J. Am. Chem. Soc. 1966, 88, 496.
2416. Gassman, P.G.; Mansfield, K.T.; Murphy, T.J. J. Am. Chem. Soc. 1969, 91, 1684.
2417. For a review, see Bartlett, P.D. Q. Rev. Chem. Soc. 1970, 24, 473. See Check, C.E.; Gilbert, T.M. J. Org. Chem. 2005, 70, 9828.
2418. See Gilbert, J.C.; Baze, M.E. J. Am. Chem. Soc. 1984, 106, 1885.
2419. See Bartlett, P.D.; Cohen, G.M.; Elliott, S.P.; Hummel, K.; Minns, R.A.; Sharts, C.M.; Fukunaga, J.Y. J. Am. Chem. Soc. 1972, 94, 2899.
2420. Also see Gheorghiu, M.D.; Pârvulescu, L.; Drâghici, C.; Elian, M. Tetrahedron 1981, 37 Suppl., 143. See, however, Holder, R.W.; Graf, N.A.; Duesler, E.; Moss, J.C. J. Am. Chem. Soc. 1983, 105, 2929.
2421. See, however, Wang, X.; Houk, K.N. J. Am. Chem. Soc. 1990, 112, 1754; Bernardi, F.; Bottoni, A.; Robb, M.A.; Venturini, A. J. Am. Chem. Soc. 1990, 112, 2106; Valentí, E.; Pericàs, M.A.; Moyano, A. J. Org. Chem. 1990, 55, 3582.
2422. Bertrand, M.; Gras, J.L.; Goré, J. Tetrahedron 1975, 31, 857; Marchand-Brynaert, J.; Ghosez, L. J. Am. Chem. Soc. 1972, 94, 2870; Huisgen, R.; Mayr, H. Tetrahedron Lett. 1975, 2965, 2969.
2423. Rey, M.; Roberts, S.; Dieffenbacher, A.; Dreiding, A.S. Helv. Chim. Acta 1970, 53, 417. See Rey, M.; Roberts, S.M.; Dreiding, A.S.; Roussel, A.; Vanlierde, H.; Toppet, S.; Ghosez, L. Helv. Chim. Acta 1982, 65, 703.
2424. Brady, W.T.; Roe Jr., R. J. Am. Chem. Soc. 1970, 92, 4618.
2425. Brook, P.R.; Harrison, J.M.; Duke, A.J. Chem. Commun. 1970, 589
2426. Brady, W.T.; O'Neal, H.R. J. Org. Chem. 1967, 32, 612; Huisgen, R.; Feiler, L.A.; Otto, P. Tetrahedron Lett. 1968, 4485, Chem. Ber. 1969, 102, 3444; Sterk, H. Z. NaturForsch. Teil B 1972, 27, 143.
2427. Isaacs, N.S.; Stanbury, P. J. Chem. Soc. Perkin Trans. 2,1973, 166.
2428. See Baldwin, J.E.; Roy, U.V. Chem. Commun. 1969, 1225; Moore, W.R.; Bach, R.D.; Ozretich, T.M. J. Am. Chem. Soc. 1969, 91, 5918.
2429. Pasto, D.J.; Yang, S.H. J. Org. Chem. 1986, 51, 1676; Dolbier, D.W.; Seabury, M. J. Am. Chem. Soc. 1987, 109, 4393; Dolbier, Jr., W.R.; Weaver, S.L. J. Org. Chem. 1990, 55, 711; Becker, D.; Denekamp, C.; Haddad, N. Tetrahedron Lett. 1992, 33, 827.
2430. See, however, Roberts, D.W. Tetrahedron 1985, 41, 5529.
2431. Bartlett, P.D.; Hummel, K.; Elliott, S.P.; Minns, R.A. J. Am. Chem. Soc. 1972, 94, 2898.
2432. See De Cock, C.; Piettre. S.; Lahousse, F.; Janousek, Z.; Merényi, R.; Viehe, H.G. Tetrahedron 1985, 41, 4183; Doering, W. von E.; Guyton, C.A. J. Am. Chem. Soc. 1978, 100, 3229.
2433. See Huisgen, R. Acc. Chem. Res. 1977, 10, 117, 199; Huisgen, R.; Schug, R.; Steiner, G. Bull. Soc. Chim. Fr. 1976, 1813.
2434. See Gompper, R. Angew. Chem. Int. Ed. 1969, 8, 312.
2435. The reactions of ketenes with enamines are apparently not concerted but take place by the diionic mechanism: Otto, P.; Feiler, L.A.; Huisgen, R. Angew. Chem. Int. Ed. 1968, 7, 737.
2436. See Moore, H.W.; Wilbur, D.S. J. Am. Chem. Soc. 1978, 100, 6523.
2437. Proskow, S.; Simmons, H.E.; Cairns, T.L. J. Am. Chem. Soc. 1966, 88, 5254. See also, Huisgen, R. Pure Appl. Chem. 1980, 52, 2283.
2438. Huisgen, R.; Steiner, G. J. Am. Chem. Soc. 1973, 95, 5054, 5055.
2439. Hall Jr., H.K. Angew. Chem. Int. Ed. 1983, 22, 440.
2440. See Frey, H.M. Adv. Phys. Org. Chem. 1966, 4, 147, see pp. 170–175, 180–183.
2441. See Schaumann, E.; Ketcham, R. Angew. Chem. Int. Ed. 1982, 21, 225. See also, Reddy, G.D.; Wiest, O.; Hudlicky, T.; Schapiro, V.; Gonzalez, D. J. Org. Chem. 1999, 64, 2860.
2442. See Paquette, L.A.; Carmody, M.J. J. Am. Chem. Soc. 1976, 98, 8175. See however Doering, W. von E.; Roth, W.R.; Breuckmann, R.; Figge, L.; Lennartz, H.; Fessner, W.; Prinzbach, H. Chem. Ber. 1988, 121, 1.
2443. See Fuks, R.; Viehe, H.G. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 435–442.
2444. D'Angelo, J.; Ficini, J.; Martinon, S.; Riche, C.; Sevin, A. J. Organomet. Chem. 1979, 177, 265. See Hogeveen, H.; Kok, D.M. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C pt. 2, Wiley, NY, 1983, pp. 981–1013.
2445. Demuth, M.; Mikhail, G. Synthesis 1989, 145; Ninomiya, I.; Naito, T. Photochemical Synthesis, Academic Press, NY, 1989, pp. 58–109; Ramamurthy, V.; Venkatesan, K. Chem. Rev. 1987, 87, 433; Wender, P.A. in Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986, pp. 163–188; Schreiber, S.L. Science 1985, 227, 857; Baldwin, S.W. Org. Photochem. 1981, 5, 123; Kricka, L.J.; Ledwith, A. Synthesis 1974, 539; Herndon, W.C. Top. Curr. Chem. 1974, 46, 141; Turro, N.J.; Dalton, J.C.; Weiss, D.S. Org. Photochem. 1969, 2, 1; Trecker, D.J. Org. Photochem. 1969, 2, 63.
2446. See Arnold, D.R.; Abraitys, V.Y. Chem. Commun. 1967, 1053; Yamazaki, H.; Cvetanovic, R.J. J. Am. Chem. Soc. 1969, 91, 520.
2447. See Dilling, W.L. Chem. Rev. 1969, 69, 845.
2448. Schuster, D.I.; Lem, G.; Kaprinidis, N.A. Chem. Rev. 1993, 93, 3; Cossy, J.; Carrupt, P.; Vogel, P. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, Vol. 2, pt. 2, Wiley, NY, 1989, pp. 1369–1565; Weedon, A.C. in Horspool, W.M. Synthetic Organic Photochemistry Plenum, NY, 1984, pp. 61–143; Bauslaugh, P.G. Synthesis 1970, 287; Eaton, P.E. Acc. Chem. Res. 1968, 1, 50; Erickson, J.A.; Kahn, S.D. Tetrahedron1993, 49, 9699.
2449. Liu, R.S.H.; Turro, N.J.; Hammond, G.S. J. Am. Chem. Soc. 1965, 87, 3406; Cundall, R.B.; Griffiths, P.A. Trans. Faraday Soc. 1965, 61, 1968; DeBoer, C.D.; Turro, N.J.; Hammond, G.S. Org. Synth. V, 528.
2450. Pappas, S.P.; Pappas, B.C. Tetrahedron Lett. 1967, 1597.
2451. See Becker, D.; Haddad, N. Org. Photochem. 1989, 10, 1-162; Crimmins, M.T. Chem. Rev. 1988, 88, 1453; Oppolzer, W. Acc. Chem. Res. 1982, 15, 135; Prinzbach, H. Pure Appl. Chem. 1968, 16, 17.
2452. Hammond, G.S.; Turro, N.J.; Fischer, A. J. Am. Chem. Soc. 1961, 83, 4674; Dauben, W.G.; Cargill, R.L. Tetrahedron 1961, 15, 197. See also, Cristol, S.J.; Snell, R.L. J. Am. Chem. Soc. 1958, 80, 1950.
2453. Ciamician, G.; Silber, P. Ber. 1908, 41, 1928; Büchi, G.; Goldman, I.M. J. Am. Chem. Soc. 1957, 79, 4741.
2454. Paternò, E.; Chieffi, C. Gazz. Chim. Ital. 1909, 39, 341; Büchi, G.; Inman, C.G.; Lipinsky, E.S. J. Am. Chem. Soc. 1954, 76, 4327. See García-Expósito, E.; Bearpark, M.J.; Ortuño, R.M.; Robb, M.A.; Branchadell, V. J. Org. Chem. 2002, 67, 6070.
2455. Wang, Y.; Zhao, C.; Romo, D. Org. Lett. 1999, 1, 1197.
2456. Reactions between two excited molecules are extremely rare.
2457. Yamazaki, H.; Cvetanovic, R.J.; Irwin, R.S. J. Am. Chem. Soc. 1976, 98, 2198; Lewis, F.D.; Kojima, M. J. Am. Chem. Soc. 1988, 110, 8660.
2458. Maradyn, D.J.; Weedon, A.C. Tetrahedron Lett. 1994, 35, 8107.
2459. Becker, D.; Haddad, N.; Sahali, Y. Tetrahedron Lett. 1989, 30, 2661.
2460. This is an example of the Wigner spin conservation rule (Sec. 7.A.vi, category 5). Note that spin conservation is something entirely different from symmetry conservation.
2461. See, for example, Kramer, B.D.; Bartlett, P.D. J. Am. Chem. Soc. 1972, 94, 3934.
2462. See Caldwell, R.A.; Creed, D. Acc. Chem. Res. 1980, 13, 45; Mattes, S.L.; Farid, S. Acc. Chem. Res. 1982, 15, 80; Swapna, G.V.T.; Lakshmi, A.B.; Rao, J.M.; Kunwar, A.C. Tetrahedron 1989, 45, 1777.
2463. For a review of exciplexes, see Davidson, R.S. Adv. Phys. Org. Chem. 1983, 19, 1–130.
2464. Schuster, D.I.; Heibel, G.E.; Brown, P.B.; Turro, N.J.; Kumar, C.V. J. Am. Chem. Soc. 1988, 110, 8261.
2465. See Schäfer, W.; Criegee, R.; Askani, R.; Grüner, H. Angew. Chem. Int. Ed. 1967, 6, 78.
2466. See Schäfer, W.; Hellmann, H. Angew. Chem. Int. Ed. 1967, 6, 518.
2467. With transition metal catalysts: Landis, M.E.; Gremaud, D.; Patrick, T.B. Tetrahedron Lett. 1982, 23, 375; Maruyama, K.; Tamiaki, H. Chem. Lett. 1987, 683.
2468. See Oth, J.F.M. Recl. Trav. Chim. Pays-Bas 1968, 87, 1185.
2469. Kobayashi, Y.; Kumadaki, I. Adv. Heterocycl. Chem. 1982, 31, 169; Acc. Chem. Res. 1981, 14, 76; van Tamelen, E.E. Acc. Chem. Res. 1972, 5, 186; Angew. Chem. Int. Ed. 1965, 4, 738; Schäfer, W.; Hellmann, H. Angew. Chem. Int. Ed. 1967, 6, 518.
2470. See Rappoport, Z. The Chemistry of the Cyclopropyl Group, Wiley, NY, 1987, the reviews by Tsuji, T.; Nishida, S. pt. 1, pp. 307–373; Verhé, R.; De Kimpe, N. pt. 1, pp. 445–564; Marchand, A.P. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, pt. 1, Wiley, NY, 1977, pp. 534–607, 625–635;; Kirmse, W. Carbene Chemistry 2nd ed.; Academic Press, NY, 1971, pp. 85–122, 267–406. For a review of certain intramolecular additions, see Burke, S.D.; Grieco, P.A. Org. React. 1979, 26, 361. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 135–153.
2471. See Schöllkopf, U. Angew. Chem. Int. Ed. 1968, 7, 588.
2472. See Parham, W.E.; Schweizer, E.E. Org. React. 1963, 13, 55.
2473. See Dave, V.; Warnhoff, E.W. Org. React. 1970, 18, 217.
2474. See Frey, H.M. J. Chem. Soc. 1962, 2293.
2475. Doyle, M.P.; Phillips, I.M. Tetrahedron Lett. 2001, 42, 3155. For a review, see Merlic, C.A.; Zechman, A.L. Synthesis 2003, 1137.
2476. Moss, R.A. Acc. Chem. Res. 1989, 22, 15; Kostikov, R.R.; Molchanov, A.P.; Khlebnikov, A.F. Russ. Chem. Rev. 1989, 58, 654.
2477. Bykowski, D.; Wu, K.-H.; Doyle, M.P. J. Am. Chem. Soc. 2006, 128, 16038.
2478. Moss, R.A.; Ge, C.-S.; Wlostowska, J.; Jang, E.G.; Jefferson, E.A.; Fan, H. Tetrahedron Lett. 1995, 36, 3083.
2479. Moss, R.A.; Wang, L.; Zhang, M.; Skalit, C.; Krogh-Jespersen, K. J. Am. Chem. Soc. 2008, 130, 5634.
2480. Seyferth, D.; Haas, C.K.; Dagani, D. J. Organomet. Chem. 1976, 104, 9.
2481. See also Kirmse, W. Carbene Chemistry Carbene Chemistry 2nd ed., Academic Press, NY, 1971, pp. 313–319; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 135–143.
2482. See Seyferth, D. Acc. Chem. Res. 1972, 5, 65; Larock, R.C. Organomercurcury Compounds in Organic Synthesis Springer, NY, 1985, pp. 341–380.
2483. Seyferth, D. in Moss, R.A.; Jones, Jr., M. Carbenes, Vol. 2, Wiley, NY, 1975, pp. 101–158; Sheppard, W.A.; Sharts, C.M. Organic Fluorine Chemistry, W.A. Benjamin, NY, 1969, pp. 237–270.
2484. Léonel, E.; Paugam, J.P.; Condon-Gueugnot, S.; Nédélec, Y.-Y. Tetrahedron 1998, 54, 3207.
2485. See Starks, C.M.; Liotta, C. Phase Transfer Catalysis Academic Press, NY, 1978, pp. 224–268; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 18–43, 58–62. For a discussion of the mechanism, see Gol'dberg, Yu.Sh.; Shimanskaya, M.V. J. Org. Chem. USSR 1984, 20, 1212.
2486. Moss, R.A.; Lawrynowicz, W. J. Org. Chem. 1984, 49, 3828.
2487. Takai, K.; Toshikawa, S.; Inoue, A.; Kokumai, R. J. Am. Chem. Soc. 2003, 125, 12990.
2488. Banwell, M.G.; Reum, M.E. Adv. Strain Org. Chem. 1991, 1, 19–64; Kostikov, R.R.; Molchanov, A.P.; Hopf, H. Top. Curr. Chem. 1990, 155, 41–80.
2489. Dehmlow, E.V.; Eulenberger, A. Liebigs Ann. Chem. 1979, 1112.
2490. Cairns, T.L.; McKusick, B.C. Angew. Chem. 1961, 73, 520.
2491. Woodworth, R.C.; Skell, P.S. J. Am. Chem. Soc. 1957, 79, 2542.
2492. See Skatteb![]() l, L. J. Org. Chem. 1964, 29, 2951.
l, L. J. Org. Chem. 1964, 29, 2951.
2493. See Hudlicky, T.; Seoane, G.; Price, J.D.; Gadamasetti, K.G. Synlett 1990, 433; Lambert, J.B.; Ziemnicka-Merchant, B.T. J. Org. Chem. 1990, 55, 3460.
2494. Rothgery, E.F.; Holt, R.J.; McGee Jr., H.A. J. Am. Chem. Soc. 1975, 97, 4971; Wasserman, H.H.; Berdahl, D.R.; Lu, T. in Rappoport, Z. The Chemistry of the Cyclopropyl Group, Wiley, NY, 1987, pt. 2, pp. 1455–1532.
2495. Landor, S.R. in Landor, S.R. The Chemistry of Allenes, Vol. 2, Acaademic Press, NY, 1982, pp. 351–360; Binger, P.; Büch, H.M. Top. Curr. Chem. 1987, 135, 77.
2496. See Krapcho, A.P. Synthesis 1978, 77–126.
2497. Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, NY, 1998.
2498. See Helquist, P. Adv. Met.-Org. Chem. 1991, 2, 143; Brookhart, M.; Studabaker, W.B. Chem. Rev. 1987, 87, 411; Syatkovskii, A.I.; Babitskii, B.D. Russ. Chem. Rev. 1984, 53, 672.
2499. Brookhart, M.; Tucker, J.R.; Husk, G.R. J. Am. Chem. Soc. 1983, 105, 258.
2500. Barluenga, J.; Aznar, F.; Gutiérrez, I.; García-Granda, S.; Llorca-Baragaño, M.A. Org. Lett. 2002, 4, 4233.
2501. Beaufort, L.; Demonceau, A.; Noels, A.F. Tetrahedron 2005, 61, 9025. Also see Rodríguez-García, C.; Oliva, A.; Ortuño, R.M.; Branchadell, V. J. Am. Chem. Soc. 2001, 123, 6157.
2502. Thompson, J.L.; Davies, H.M.L. J. Am. Chem. Soc. 2007, 129, 6090.
2503. Nishiyama, Y.; Tanimizu, H.; Tomita, T. Tetrahedron Lett. 2007, 48, 6405,
2504. See Panne, P.; DeAngelis, A.; Fox, J.M. Org. Lett. 2008, 10, 2987.
2505. See Adams, J.; Spero, D.M. Tetrahedron 1991, 47, 1765; Maas, G. Top. Curr. Chem. 1987, 137, 75; Doyle, M.P. Chem. Rev. 1986, 86, 919; Acc. Chem. Res. 1986, 19, 348; Heck, R.F. Palladium Reagents in Organic Synthesis Academic Press, NY, 1985, pp. 401–407; Wulfman, D.S.; Poling, B. React. Intermed. (Plenum) 1980, 1, 321; Müller, E.; Kessler, H.; Zeeh, B. Fortschr. Chem. Forsch. 1966, 7, 128.
2506. Green, G.M.; Peet, N.P.; Metz, W.A. J. Org. Chem. 2001, 66, 2509.
2507. Díaz-Requejo, M.M.; Belderraín, T.R.; Trofimenko, S.; Pérez, P.J. J. Am. Chem. Soc. 2001, 123, 3167. See Bühl, M.; Terstegen, F.; Löffler, F.; Meynhardt, B.; Kierse, S.; Müller, M.; Näther, C.; Lüning, U. Eur. J. Org. Chem.2001, 2151.
2508. See Panne, P.; Fox, J.M. J. Am. Chem. Soc. 2007, 129, 22.
2509. Li, Y.; Huang, J.-S.; Zhou, Z.-Y.; Che, C.-M. J. Am. Chem. Soc. 2001, 123, 4843.
2510. See Davies, H.M.L.; Xiang, B.; Kong, N.; Stafford, D.G. J. Am. Chem. Soc. 2001, 123, 7461.
2511. Pellissier, H. Tetrahedron 2008, 64, 7041.
2512. Singh, V.K.; DattaGupta, A.; Sekar, G. Synthesis 1997, 137; Davies, H.M.L.; Bruzinski, P.R.; Fall, M.J. Tetrahedron Lett. 1996, 37, 4133.
2513. Davies, H.M.L.; Rusiniak, L. Tetrahedron Lett. 1998, 39, 8811; Haddad, N.; Galili, N. Tetrahedron Asymmetry 1997, 8, 3367; Ichiyanagi, T.; Shimizu, M.; Fujisawa, T. Tetrahedron 1997, 53, 9599; Fukuda, T.; Katsuki, T. Tetrahedron 1997, 53, 7201.
2514. Bayardon, J.; Holczknecht, O.; Pozzi, G.; Sinou, D. Tetrahedron Asymmetry 2006, 17, 1568.
2515. Suematsu, H.; Kanchiku, S.; Uchida, T.; Katsuki, T. J. Am. Chem. Soc. 2008, 130, 10327.
2516. Chen, Y.; Ruppel, J.V.; Zhang, X.P. J. Am. Chem. Soc. 2007, 129, 12074; Doyle, M.P. Angew. Chem. Int. Ed. 2009, 48, 850.
2517. Prieto, A.; Fructos, M.R.; Díaz-Requejo, M.M.; Pérez, P.J.; Pérez-Galán, P.; Delpont, N.; Echavarren, A.M. Tetrahedron 2009, 65, 1790.
2518. Iwasa, S.; Takezawa, F.; Tuchiya, Y.; Nishiyama, H. Chem. Commun. 2001, 59. For a discussion of the mechanism, see Oxgaard, J.; Goddard III, W.A. J. Am. Chem. Soc. 2004, 126, 442.
2519. Ye, T.; Zhou, C. New J. Chem. 2005, 29, 1159.
2520. Ferreira, V.F.; Leão, R.A.C.; da Silva, F. de C.; Pinheiro, S.; Lhoste, P.; Sinou, D. Tetrahedron Asymmetry 2007, 18, 1217; Miller, J.A.; Gross, B.A.; Zhuravel, M.A.; Jin, W.; Nguyen, S.B.T. Angew. Chem. Int. Ed. 2005, 44, 3885.
2521. Doyle, M.P. Pure Appl. Chem. 1998, 70 1123; Davies, H.M.L.; Hansen, T.; Churchill, M.R. J. Am. Chem. Soc. 2000, 122, 3063. See also, Davies, H.M.L.; Lee, G.H. Org. Lett. 2004, 6, 2117.
2522. Davies, H.M.L.; Townsend, R.J. J. Org. Chem. 2001, 66, 6595.
2523. Johansson, C.C.C.; Bremeyer, N.; Ley, S.V.; Owen, D.R.; Smith, S.C.; Gaunt, M.J. Angew. Chem. Int. Ed. 2006, 45, 6024.
2524. Fraile, J.M.; García, J.I.; Herrerías, C.I.; Mayoral, J.A.; Carrié, D.; Vaultier, M. Tetrahedron Asymmetry 2001, 12, 1891.
2525. Ferrand, Y.; Le Maux, P.; Simonneaux, G. Org. Lett. 2004, 6, 3211.
2526. Barluenga, J.; Suero, M.G.; Pérez-Sánchez, I.; Flórez, J. J. Am. Chem. Soc. 2008, 130, 2708.
2527. Concellón, J.M.; Rodríguez-Solla, H.; Méjica, C.; Blanco, E.G.; García-Granda, S.; Diaz, M.R. Org. Lett. 2008, 10, 349.
2528. See Honma, M.; Sawada, T.; Fujisawa, Y.; Utsugi, M.; Watanabe, H.; Umino, A.; Matsumura, T.; Hagihara, T.; Takano, M.; Nakada, M. J. Am. Chem. Soc. 2003, 125, 2860.
2529. Doyle, M.P.; May, E.J. Synlett 2001, 967.
2530. See Fuks, R.; Viehe, H.G. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 427–434; Closs, G.L. Adv. Alicyclic Chem. 1966, 1, 53–127, see pp. 58–65.
2531. Frey, H.M. Chem. Ind. (London) 1960, 1266.
2532. Vol'pin, M.E.; Koreshkov, Yu.D.; Kursanov, D.N. Bull. Acad. Sci. USSR Div. Chem. Sci. 1959, 535.
2533. Mitsch, R.A.; Rodgers, A.S. Int. J. Chem. Kinet. 1969, 1, 439.
2534. Moss, R.A. in Jones Jr., M.; Moss, R.A. Carbenes, Vol. 1, Wiley, NY, 1973, pp. 153–304. See also, Cox, D.P.; Gould, I.R.; Hacker, N.P.; Moss, R.A.; Turro, N.J. Tetrahedron Lett. 1983, 24, 5313.
2535. Ando, W.; Hendrick, M.E.; Kulczycki, Jr., A.; Howley, P.M.; Hummel, K.F.; Malament, D.S. J. Am. Chem. Soc. 1972, 94, 7469.
2536. See Giese, B.; Lee, W.; Neumann, C. Angew. Chem. Int. Ed. 1982, 21, 310.
2537. Nefedov, O.M.; Zuev, P.S.; Maltsev, A.K.; Tomilov, Y.V. Tetrahedron Lett. 1989, 30, 763.
2538. Skell, P.S.; Klebe, J. J. Am. Chem. Soc. 1960, 82, 247. See also, Jones, Jr., M.; Tortorelli, V.J.; Gaspar, P.P.; Lambert, J.B. Tetrahedron Lett. 1978, 4257.
2539. Moss, R.A. Sel. Org. Transform.,1970, 1, 35-88; Closs, G.L. Top Stereochem. 1968, 3, 193–235. For a discussion of enantioselectivity in this reaction, see Nakamura, A. Pure Appl. Chem. 1978, 50, 37.
2540. See Giese, C.M.; Hadad, C.M. J. Org. Chem. 2002, 67, 2532.
2541. Doering, W. von E.; Knox, L.H. J. Am. Chem. Soc. 1951, 75, 297.
2542. It has been detected by UV spectroscopy: Rubin, M.B. J. Am. Chem. Soc. 1981, 103, 7791.
2543. Ciganek, E. J. Am. Chem. Soc. 1967, 89, 1454.
2544. See Kawase, T.; Iyoda, M.; Oda, M. Angew. Chem. Int. Ed. 1987, 26, 559.
2545. Wittig, G.; Schwarzenbach, K. Liebigs Ann. Chem. 1961, 650, 1; Müller, E.; Fricke, H. Liebigs Ann. Chem. 1963, 661, 38; Müller, E.; Kessler, H.; Fricke, H.; Kiedaisch, W. Liebigs Ann. Chem. 1961, 675, 63.
2546. Khan, M.I.; Goodman, J.L. J. Am. Chem. Soc. 1995, 117, 6635.
2547. See Rees, C.W.; Smithen, C.E. Adv. Heterocycl. Chem. 1964, 3, 57–78.
2548. Jefford, C.W.; Gunsher, J.; Hill, D.T.; Brun, P.; Le Gras, J.; Waegell, B. Org. Synth. VI, 142. For a review of the addition of halocarbenes to bridged bicyclic alkenes see Jefford, C.W. Chimia,1970, 24, 357–363.
2549. See Simmons, H.E.; Cairns, T.L.; Vladuchick, S.A.; Hoiness, C.M. Org. React. 1973, 20, 1–131; Furukawa, J.; Kawabata, N. Adv. Organomet. Chem. 1974, 12, 83–134, see pp. 84–103.
2550. Bull, J.A.; Charette, A.B. J. Am. Chem. Soc. 2010, 132, 1895.
2551. Simmons, H.E.; Smith, R.D. J. Am. Chem. Soc. 1959, 81, 4256.
2552. LeGoff, E. J. Org. Chem. 1964, 29, 2048; Denis, J.M.; Girard, C.; Conia, J.M. Synthesis 1972, 549.
2553. Rawson, R.J.; Harrison, I.T. J. Org. Chem. 1970, 35, 2057.
2554. Repic, O.; Lee, P.G.; Giger, U. Org. Prep. Proced. Int. 1984, 16, 25.
2555. Friedrich, E.C.; Lunetta, S.E.; Lewis, E.J. J. Org. Chem. 1989, 54, 2388.
2556. Blanchard, E.P.; Simmons, H.E. J. Am. Chem. Soc. 1964, 86, 1337. For an analysis of the reaction by density functional theory, see Fang, W.-H.; Phillips, D.L.; Wang, D.-q.; Li, Y.-L. J. Org. Chem. 2002, 67, 154.
2557. Denmark, S.E.; Edwards, J.P.; Wilson, S.R. J. Am. Chem. Soc. 1991, 113, 723.
2558. Dargel, T.K.; Koch, W. J. Chem. Soc. Perkin Trans. 2,1996, 877.
2559. Simmons, H.E.; Blanchard, E.P.; Smith, R.D. J. Am. Chem. Soc. 1964, 86, 1347. For the transition state and intermediate see Bernardi, F.; Bottoni, A.; Miscione, G.P. J. Am. Chem. Soc. 1997, 119, 12300.
2560. Lacasse, M.-C.; Poulard, C.; Charette, A.B. J. Am. Chem. Soc. 2005, 127, 12440.
2561. Virender; Jain, S.L.; Sain, B. Tetrahedron Lett. 2005, 46, 37.
2562. Balsells, J.; Walsh, P.J. J. Org. Chem. 2000, 65, 5005; Du, H.; Long, J.; Shi, Y. Org. Lett. 2006, 8, 2827; Long, J.; Du, H.; Li, K.; Shi, Y. Tetrahedron Lett. 2005, 46, 2737.
2563. Song, Z.; Lu, T.; Hsung, R.P.; Al-Rashid, Z.F.; Ko, C.; Tang, Y. Angew. Chem. Int. Ed. 2007, 46, 4069; Shitama, H.; Katsuki, T. Angew. Chem. Int. Ed. 2008, 47, 2450; Zimmer, L.E.; Charette, A.B. J. Am. Chem. Soc. 2009, 131, 15624.
2564. Long, J.; Xu, L.; Du, H.; Li, K.; Shi, Y. Org. Lett. 2009, 11, 5226.
2565. Overberger, C.G.; Halek, G.W. J. Org. Chem. 1963, 28, 867.
2566. Charette, A.B.; Jolicoeur, E.; Bydlinski, G.A.S. Org. Lett. 2001, 3, 3293.
2567. See Zhao, C.; Wang, D.; Phillips, D.L. J. Am. Chem. Soc. 2002, 124, 12903.
2568. Friedrich, E.C.; Biresaw, G. J. Org. Chem. 1982, 47, 1615.
2569. Charette, A.B.; Beauchemin, A.; Fraancoeur, S. J. Am. Chem. Soc. 2001, 123, 8139.
2570. Maruoka, K.; Fukutani, Y.; Yamamoto, H. J. Org. Chem. 1985, 50, 4412; Org. Synth. 67, 176.
2571. Charette, A.B.; Molinaro, C.; Brochu, C. J. Am. Chem. Soc. 2001, 123, 12168.
2572. Concellón, J.M.; Rodríguez-Solla, H.; Gómez, C. Angew. Chem. Int. Ed. 2002, 41, 1917.
2573. Imamoto, T.; Takiyama, N. Tetrahedron Lett. 1987, 28, 1307. See also, Molander, G.A.; Harring, L.S. J. Org. Chem. 1989, 54, 3525.
2574. Bolm, C.; Pupowicz, D. Tetrahedron Lett. 197, 38, 7349.
2575. See Wenkert, E.; Mueller, R.A.; Reardon, Jr., E.J.; Sathe, S.S.; Scharf, D.J.; Tosi, G. J. Am. Chem. Soc. 1970, 92, 7428 for the enol ether procedure; Kuehne, M.E.; King, J.C. J. Org. Chem. 1973, 38, 304 for the enamine procedure; Conia, J.M. Pure Appl. Chem. 1975, 43, 317–326 for the silyl ether procedure.
2576. See Ito, Y.; Fujii, S.; Saegusa, T. J. Org. Chem. 1976, 41, 2073; Org. Synth. VI, 327.
2577. Brogan, J.B.; Zercher, C.K. J. Org. Chem. 1997, 62, 6444.
2578. Lehnert, E.K.; Sawyer, J.S.; Macdonald, T.L. Tetrahedron Lett. 1989, 30, 5215.
2579. Yang, M.; Wang, X.; Li, H.; Livant, P. J. Org. Chem. 2001, 66, 6729.
2580. Doyle, M.P.; Hu, W.; Timmons, D.J. Org. Lett. 2001, 3, 933.
2581. Dias, E.L.; Brookhart, M.; White, P.S. J. Am. Chem. Soc. 2001, 123, 2442.
2582. For a review, see Rubin, M.; Sromek, A.W.; Gevorgyan, V. Synlett 2003, 2265.
2583. Kociolek, M.G.; Johnson, R.P. Tetrahedron Lett. 1999, 40, 4141.
2584. Viehe, H.G.; Merényi, R.; Oth, J.F.M.; Valange, P. Angew. Chem. Int. Ed. 1964, 3, 746; Viehe, H.G.; Merényi, R.; Oth, J.F.M.; Senders, J.R.; Valange, P. Angew. Chem. Int. Ed. 1964, 3, 755.
2585. See also, Wingert, H.; Regitz, M. Chem. Ber. 1986, 119, 244.
2586. See Winter, M.J. in Hartley, F.R.; Patai, S. The Chemistry of the Metal-Carbon Bond, Vol. 3, Wiley, NY, 1985, pp. 259–294; Vollhardt, K.P.C. Angew. Chem. Int. Ed. 1984, 23, 539; Acc. Chem. Res. 1977, 10, 1; Maitlis, P.M. J. Organomet. Chem. 1980, 200, 161; Reppe, W.; Kutepow, N.V.; Magin, A. Angew. Chem. Int. Ed. 1969, 8, 727; Schore, N.E. Chem. Rev. 1988, 88, 1081. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 198–201.
2587. Sigman, M.S.; Fatland, A.W.; Eaton, B.E. J. Am. Chem. Soc. 1998, 120, 5130; Larock, R.C.; Tian, Q. J. Org. Chem. 1998, 63, 2002.
2588. Yong, L.; Butenschön, H. Chem. Commun. 2002, 2852; Marchueta, I.; Olivella, S.; Solà, L.; Moyano, A.; Pericàs, M.A.; Riera, A. Org. Lett. 2001, 3, 3197. See also, Agenet, N.; Gandon, V.; Vollhardt, K.P.C.; Malacria, M.; Aubert, C. J. Am. Chem. Soc. 2007, 129, 8860.
2589. See Hopff, H.; Gati, A. Helv. Chim. Acta 1965, 48, 509.
2590. See Yoshida, K.; Morimoto, I.; Mitsudo, K.; Tanaka, H. Chem. Lett. 2007, 36, 998.
2591. Mori, N.; Ikeda, S.-i.; Odashima, K. Chem. Commun. 2001, 181.
2592. Tanaka, R.; Nakano, Y.; Suzuki, D.; Urabe, H.; Sato, F. J. Am. Chem. Soc. 2002, 124, 9682.
2593. Nishida, M.; Shiga, H.; Mori, M. J. Org. Chem. 1998, 63, 8606.
2594. Yamamoto, Y.; Ishii, J.-i.; Nishiyama, H.; Itoh, K. J. Am. Chem. Soc. 2004, 126, 3712.
2595. Sugihara, T.; Wakabayashi, A.; Nagai, Y.; Takao, H.; Imagawa, H.; Nishizawa, M. Chem. Commun. 2002, 576.
2596. Gevorgyan, V.; Quan, L.G.; Yamamoto, Y. J. Org. Chem. 2000, 65, 568. Also see Kawasaki, S.; Satoh, T.; Miura, M.; Nomura, M. J. Org. Chem. 2003, 68, 6836; Li, J.-H.; Xie, Y.-X. Synth. Commun. 2004, 34, 1737.
2597. Shanmugasundaram, M.; Wu, M.-S.; Cheng, C.-H. Org. Lett. 2001, 3, 4233.
2598. Ikeda, S.; Kondo, H.; Arii, T.; Odashima, K. Chem. Commun. 2002, 2422.
2599. Mori, N.; Ikeda, S.-i.; Sato, Y. J. Am. Chem. Soc. 1999, 121, 2722.
2600. Amii, H.; Kishikawa, Y.; Uneyama, K. Org. Lett. 2001, 3, 1109.
2601. Sangu, K.; Fuchibe, K.; Akiyama, T. Org. Lett. 2004, 6, 353.
2602. See Kawathar, S.P.; Schreiner, P.R. Org. Lett. 2002, 4, 3643.
2603. Gevorgyan, V.; Radhakrishnan, U.; Takeda, A.; Rubina, M.; Rubin, M.; Yamamoto, Y. J. Org. Chem. 2001, 66, 2835. See also, Tsukada, N.; Sugawara, S.; Nakaoka, K.; Inoue, Y. J. Org. Chem. 2003, 68, 5961.
2604. Hara, R.; Guo, Q.; Takahashi, T. Chem. Lett. 2000, 140.
2605. Jeevanandam, A.; Korivi, R.P.; Huang, I.-w.; Cheng, C.-H. Org. Lett. 2002, 4, 807.
2606. Witulski, B.; Zimmermann, A. Synlett 2002, 1855.
2607. Shibata, T.; Fujimoto, T.; Yokota, K.; Takagi, K. J. Am. Chem. Soc. 2004, 126, 8382.
2608. Zhao, J.; Hughes, C.O.; Toste, F.D. J. Am. Chem. Soc. 2006, 128, 7436.
2609. Hilt, G.; Vogler, T.; Hess, W.; Galbiati, F. Chem. Commun. 2005, 1474.
2610. Yamamoto, Y.; Hattori, K.; Nishiyama, H. J. Am. Chem. Soc. 2006, 128, 8336.
2611. Kinoshita, H.; Shinokubo, H.; Oshima, K. J. Am. Chem. Soc. 2003, 125, 7784.
2612. Chouraqui, G.; Petit, M.; Aubert, C.; Malacria, M. Org. Lett. 2004, 6, 1519.
2613. Shanmugasundaram, M.; Wu, M.-S.; Jeganmohan, M.; Huang, C.-W.; Cheng, C.-H. J. Org. Chem. 2002, 67, 7724.
2614. Young, D.D.; Senaiar, R.S.; Deiters, A. Chemistry: European J. 2006, 12, 5563.
2615. Kawasaki, T.; Saito, S.; Yamamoto, Y. J. Org. Chem. 2002, 67, 2653.
2616. Odedra, A.; Wu, C.-J.; Pratap, T.B.; Huang, C.-W.; Ran, Y.-F.; Liu, R.-S. J. Am. Chem. Soc. 2005, 127, 3406.
2617. Lian, J.-J.; Odedra, A.; Wu, C.-J.; Liu, R.-S. J. Am. Chem. Soc. 2005, 127, 4186.
2618. Yao, T.; Campo, M.A.; Larock, R.C. J. Org. Chem. 2005, 70, 3511.
2619. Ojima, I.; Vu, A.T.; McCullagh, J.V.; Kinoshita, A. J. Am. Chem. Soc. 1999, 121, 3230.
2620. Atienza, C.; Mateo, C.; de Frutos, Ó.; Echavarren, A.M. Org. Lett. 2001, 3, 153.
2621. Landis, C.A.; Payne, M.M.; Eaton, D.L.; Anthony, J.E. J. Am. Chem. Soc. 2004, 126, 1338.
2622. Klumpp, D.A.; Beauchamp, P.S.; Sanchez, Jr., G.V.; Aguirre, S.; de Leon, S. Tetrahedron Lett. 2001, 42, 5821.
2623. Gandon, V.; Leca, D.; Aechtner, T.; Vollhardt, K.P.C.; Malacria, M.; Aubert, C. Org. Lett. 2004, 6, 3405.
2624. Roesch, K.R.; Larock, R.C. J. Org. Chem. 2002, 67, 86.
2625. Huang, Q.; Hunter, J.A.; Larock, R.C. Org. Lett. 2001, 3, 2973.
2626. Dai, G.; Larock, R.C. J. Org. Chem. 2003, 68, 920; Dai, G.; Larock, R.C. Org. Lett. 2002, 4, 193.
2627. Varela, J.A.; Castedo, L.; Saá, C. J. Org. Chem. 2003, 68, 8595.
2628. Boñaga, L.V.R.; Zhang, H.-C.; Maryanoff, B.E. Chem. Commun. 2004, 2394.
2629. Yamamoto, Y.; Takagishi, H.; Itoh, K. Org. Lett. 2001, 3, 2117.
2630. Kamijo, S.; Kanazawa, C.; Yamamoto, Y. Tetrahedron Lett. 2005, 46, 2563.
2631. Madhusaw, R.J.; Lin, M.-Y.; Shoel, S.Md.A.; Liu, R.-S. J. Am. Chem. Soc. 2004, 126, 6895.
2632. Heller, B.; Oehme, G. J. Chem. Soc., Chem. Commun. 1995, 179.
2633. Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. J. Org. Chem. 2003, 68, 6959.
2634. Takahashi, T.; Tsai, F.Y.; Kotora, M. J. Am. Chem. Soc. 2000, 122, 4994.
2635. Moretto, A.F.; Zhang, H.-C.; Maryanoff, B.E. J. Am. Chem. Soc. 2001, 123, 3157.
2636. McCormick, M.M.; Duong, H.A.; Zuo, G.; Louie, J. J. Am. Chem. Soc. 2005, 127, 5030.
2637. Bagley, M.C.; Dale, J.W.; Hughes, D.D.; Ohnesorge, M.; Philips, N.G.; Bower, J. Synlett 2001, 1523.
2638. Kikiya, H.; Yagi, K.; Shinokubo, H.; Oshima, K. J. Am. Chem. Soc. 2002, 124, 9032.
2639. Mori, M.; Hori, M.; Sato, Y. J. Org. Chem. 1998, 63, 4832; Mori, M.; Hori, K.; Akashi, M.; Hori, M.; Sato, Y.; Nishida, M. Angew. Chem. Int. Ed. 1998, 37, 636.
2640. See Saito, S.; Kawasaki, T.; Tsuboya, N.; Yamamoto, Y. J. Org. Chem. 2001, 66, 796.
2641. See Colborn, R.E.; Vollhardt, K.P.C. J. Am. Chem. Soc. 1986, 108, 5470; Lawrie, C.J.; Gable, K.P.; Carpenter, B.K. Organometallics 1989, 8, 2274.
2642. Colborn, R.E.; Vollhardt, K.P.C. J. Am. Chem. Soc. 1981, 103, 6259; Kochi, J.K. Organometallic Mechanisms and Catalysis Academic Press, NY, 1978, pp. 428–432; Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry University Science Books, Mill Valley, CA 1987, pp. 870–877; Eisch, J.J.; Sexsmith, S.R. Res. Chem. Intermed. 1990, 13, 149–192.
2643. Takahahsi, T.; Ishikawa, M.; Huo, S. J. Am. Chem. Soc. 2002, 124, 388.
2644. See Eisch, J.J.; Galle, J.E. J. Organomet. Chem. 1975, 96, C23; McAlister, D.R.; Bercaw, J.E.; Bergman, R.G. J. Am. Chem. Soc. 1977, 99, 1666.
2645. See, however, Bianchini, C.; Caulton, K.G.; Chardon, C.; Eisenstein, O.; Folting, K.; Johnson, T.J.; Meli, A.; Peruzzini, M.; Raucher, D.J.; Streib, W.E.; Vizza, F. J. Am. Chem. Soc. 1991, 113, 5127.
2646. Mauret, P.; Alphonse, P. J. Organomet. Chem. 1984, 276, 249. See also, Pepermans, H.; Willem, R.; Gielen, M.; Hoogzand, C. Bull. Soc. Chim. Belg. 1988, 97, 115.
2647. Suzuki, D.; Urabe, H.; Sato, F. J. Am. Chem. Soc. 2001, 123, 7925.
2648. Li, J.; Jiang, H.; Chen, M. J. Org. Chem. 2001, 66, 3627.
2649. Jackson, T.J.; Herndon, J.W. Tetrahedron 2001, 57, 3859.
2650. Davies, M.W.; Johnson, C.N.; Harrity, J.P.A. J. Org. Chem. 2001, 66, 3525.
2651. Jiang, M.X.-W.; Rawat, M.; Wulff, W.D. J. Am. Chem. Soc. 2004, 126, 5970.
2652. Schirmer, H.; Flynn, B.L.; de Meijere, A. Tetrahedron 2000, 56, 4977.
2653. Herndon, J.W.; Zhang, Y.; Wang, H.; Wang, K. Tetrahedron Lett. 2000, 41, 8687.
2654. Barluenga, J.; López, L.A.; Martínez, S.; Tomás, M. Tetrahedron 2000, 56, 4967.
2655. Campos, P.J.; Sampedro, D.; Rodríquez, M.A. J. Org. Chem. 2003, 68, 4674.
2656. Dötz, K.H. Angew. Chem. Int. Ed. 1975, 14, 644.
2657. Barluenga, J.; Aznar, F.; Palomero, M.A. Angew. Chem. Int. Ed. 2000, 39, 4346.
2658. Parker, W.L.; Hexter, R.M.; Siedle, A.R. J. Am. Chem. Soc. 1985, 107, 4584.
2659. Iranpoor, N.; Zeynizaded, B. Synlett 1998, 1079.
2660. Li, Z.; Sun, W.-H.; Jin, X.; Shao, C. Synlett 2001, 1947.
2661. Wilke, G. Angew. Chem. Int. Ed. 1988, 27, 186; Heimbach, P.; Schenkluhn, H. Top Curr. Chem. 1980, 92, 45; Baker, R. Chem. Rev. 1973, 73, 487, see pp. 489–512; Semmelhack, M.F. Org. React. 1972, 19, 115, pp. 128–143; Khan, M.M.T.; Martell, A.E. Homogeneous Catalysis by Metal Complexes, Vol. 2 Academic Press, NY, 1974, pp. 159–163; Heck, R.F. Organotransition Metal Chemistry Academic Press, NY, 1974, pp. 157–164.
2662. See Rona, P. Intra-Sci. Chem. Rep. 1971, 5, 105.
2663. See Graham, G.R.; Stephenson, L.M. J. Am. Chem. Soc. 1977, 99, 7098.
2664. Davies, H.M.L.; Calvo, R.L.; Townsend, R.-J.; Ren, P.; Churchill, R.M. J. Org. Chem. 2000, 65, 4261. For reviews of [3 + 4] cycloadditions see Mann, J. Tetrahedron 1986, 42, 4611; Hoffmann, H.M.R. Angew. Chem. Int. Ed. 1984, 23, 1; 1973, 12, 819; Noyori, R. Acc. Chem. Res. 1979, 12, 61.
2665. Garst, M.E.; Roberts, V.A.; Houk, K.N.; Rondan, N.G. J. Am. Chem. Soc. 1984, 106, 3882.
2666. Reddy, R.P.; Davies, H.M.L. J. Am. Chem. Soc. 2007, 129, 10312.
2667. Gulías, M.; Durán, J.; López, F.; Castedo, L.; Mascareñas, J.L. J. Am. Chem. Soc. 2007, 129, 11026.
2668. Evans, P.A.; Inglesby, P.A. J. Am. Chem. Soc. 2008, 130, 12838.
2669. Deng, L.; Giessert, A.J.; Gerlitz, O.O.; Dai, X.; Diver, S.T.; Davies, H.M.L. J. Am. Chem. Soc. 2005, 127,1342.
2670. Huang, J.; Hsung, R.P. J. Am. Chem. Soc. 2005, 127, 50. See Harmata, M. Chem. Commun. 2010, 8886, 8904.
2671. Wender, P.A.; Haustedt, L.O.; Lim, J.; Love, J.A.; Williams, T.J.; Yoon, J.-Y. J. Am. Chem. Soc. 2006, 128, 6302.
2672. Son, S.; Fu, G.C. J. Am. Chem. Soc. 2007, 129, 1046.
2673. Laroche, C.; Bertus, P.; Szymoniak, J. Chem. Commun. 2005, 3030.
2674. Achard, M.; Tenaglia, A.; Buono, G. Org. Lett. 2005, 7, 2353.
2675. Shönberg, A. Preparative Organic Photochemistry Springer, NY, 1968, pp. 97–99. Also see Zhu, M.; Qiu, Z.; Hiel, G.P.; Sieburth, S.Mc.N. J. Org. Chem. 2002, 67, 3487.
2676. Wender, P.A.; Gamber, G.G.; Scanio, M.J.C. Angew. Chem. Int. Ed. 2001, 40, 3895; Wender, P.A.; Pedersen, T.M.; Scanio, M.J.C. J. Am. Chem. Soc. 2002, 124, 15154; Wender, P.A.; Love, J.A.; Williams, T.J. Synlett 2003, 1295.
2677. Rigby, J.H.; Mann, L.W.; Myers, B.J. Tetrahedron Lett. 2001, 42, 8773. See Rigby, J.H.; Ateeq, H.S.; Charles, N.R.; Henshilwood, J.A.; Short, K.M.; Sugathapala, P.M. Tetrahedron 1993, 49, 5495.
2678. Farrant, G.C.; Feldmann, R. Tetrahedron Lett. 1970, 4979.
2679. See Wender, P.A.; Ternansky, R.; deLong, M.; Singh, S.; Olivero, A.; Rice, K. Pure Appl. Chem. 1990, 62, 1597; Gilbert, A. in Horspool, W.M. Synthetic Organic Photochemistry, Plenum, NY, 1984, pp. 1–60. For a review of this and related reactions, see McCullough, J.J. Chem. Rev. 1987, 87, 811.
2680. See the table in Wender, P.A.; Siggel, L.; Nuss, J.M. Org. Photochem. 1989, 10, 357, pp. 384–415.
2681. For a review, see Kotha, S.; Brahmachary, E.; Lahiri, K. Eur. J. Org. Chem. 2005, 4741.
2682. Louie, J.; Gibby, J.E.; Farnsworth, M.V.; Tekavec, T.N. J. Am. Chem. Soc. 2002, 124, 15188.
2683. Cadierno, V.; García-Garrido, S.E.; Gimeno, J. J. Am. Chem. Soc. 2006, 128, 15094; Tanaka, D.; Sato, Y.; Mori, M. J. Am. Chem. Soc. 2007, 129, 7730; Mallagaray, Á.; Medina, S.; Domínguez, G.; Pérez-Castells, J. Synlett2010, 2114.
2684. Hilt, G.; Paul, A.; Harms, K. J. Org. Chem. 2008, 73, 5187.
2685. Varela, J.A.; Rubín, S.G.; Castedo, L.; Saá, C. J. Org. Chem. 2008, 73, 1320.
2686. Boñaga, L.V.R.; Zhang, H.-C.; Moretto, A.F.; Ye, H.; Gauthier, D.A.; Li, J.; Leo, G.C.; Maryanoff, B.E. J. Am. Chem. Soc. 2005, 127, 3473.
2687. Yu, R.T.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 2782; Yu, R.T.; Rovis, T. J. Am. Chem. Soc. 2006, 128, 12370.
2688. Varela, J.A.; Saá, C. Synlett 2008, 2571.
2689. Kondo, T.; Nomura, M.; Ura, Y.; Wada, K.; Mitsudo, T.-a. J. Am. Chem. Soc. 2006, 128, 14816.
2690. Knölker, H.-J.; Braier, A.; Bröcher, D.J.; Jones, P.G.; Piotrowski, H. Tetrahedron Lett. 1999, 40, 8075; Chatani, N.; Tobisu, M.; Asaumi, T.; Fukumoto, Y.; Murai, S. J. Am. Chem. Soc. 1999, 121, 7160.
2691. Kiattansakul, R.; Snyder, J.K. Tetrahedron Lett. 1999, 40, 1079.
2692. Gilbertson, S.R.; DeBoef, B. J. Am. Chem. Soc. 2002, 124, 8784; Wender, P.A.; Christy, J.P. J. Am. Chem. Soc. 2006, 128, 5354. For a computational study, see Baik, M.-H.; Baum, E.W.; Burland, M.C.; Evans, P.A. J. Am. Chem. Soc. 2005, 127, 1602.
2693. Ni, Y.; Montgomery, J. J. Am. Chem. Soc. 2006, 128, 2609.
2694. Kündig, E.P.; Robvieux, F.; Kondratenko, M. Synthesis 2002, 2053.
2695. Bennacer, B.; Fujiwara, M.; Lee, S.-Y.; Ojima, I. J. Am. Chem. Soc. 2005, 127, 17756.
2696. Wender, P.A.; Christy, J.P. J. Am. Chem. Soc. 2007, 129, 13402.
2697. Wegner, H.A.; de Meijere, A.; Wender, P.A. J. Am. Chem. Soc. 2005, 127, 6530; Wang, Y.; Wang, J.; Su, J.; Huang, F.; Jiao, L.; Liang, Y.; Yang, D.; Zhang, S.; Wender, P.A.; Yu, Z.-X. J. Am. Chem. Soc. 2007, 129, 10060.
2698. Yu, Z.X.; Cheong, P.h.-Y.; Liu, P.; Legault, C.Y.; Wender, P.A.; Houk, K.N. J. Am. Chem. Soc. 2008, 130, 2378.