March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 16. Addition to Carbon–Hetero Multiple Bonds
16.B. Reactions
Many of the reactions in this chapter are simple additions to carbon–hetero multiple bonds, with the reaction ending when the two groups have been added. But in many other cases subsequent reactions take place. There are generally two types:
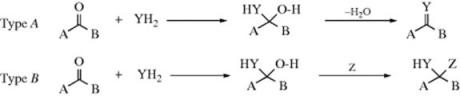
In type A, the initially formed adduct loses water (or, in the case of addition to C=NH, ammonia, etc.), and the net result of the reaction is the substitution of C=Y for C=O (or C=NH, etc.). In type B there is a rapid substitution, and the OH (or NH2, etc.) is replaced by another group Z, which is often another YH moiety. This substitution is nucleophilic is most cases: Y usually has an unshared pair and SN1 reactions occur very well on this type of compound (Sec. 10.G.i, category 2), even when the leaving group is as poor as OH or NH2. In this chapter, reactions will be classified according to what is initially adding to the carbon–heteroatom multiple bond, even if subsequent reactions take place so rapidly that it is impossible to isolate the initial adduct.
Most of the reactions considered in this chapter can be reversed. In many cases, we will consider the reverse reactions with the forward ones, in the same section. The reverse of some of the other reactions are considered in other chapters. In still other cases, one of the reactions in this chapter is the reverse of another (e.g., 16-2 and 16-13). For reactions that are reversible, the principle of microscopic reversibility (Sec. 6.H) applies.
First, reactions in which hydrogen or a metallic ion (or in one case phosphorus or sulfur) adds to the heteroatom will be discussed. Second, reactions in which carbon adds to the heteroatom will be discussed. Within each group, the reactions are classified by the nature of the nucleophile. Additions to isocyanides, which are different in character, follow. Acyl substitution reactions that proceed by the tetrahedral mechanism, which mostly involve derivatives of carboxylic acids, are treated at the end.
16.B.i. Reactions in which Hydrogen or a Metallic Ion Adds to the Heteroatom
A. Attack by OH (Addition of H2O)
16-1 The Addition of Water to Aldehydes and Ketones: Formation of Hydrates
O-Hydro-C-hydroxy-addition
![]()
The adduct formed upon addition of water to an aldehyde or ketone is called a hydrate or gem-diol.38 These compounds are usually stable only in water solution and decompose on distillation; that is, the equilibrium shifts back toward the carbonyl compound, usually via formation of an enol and tautomerization to the carbonyl. The position of the equilibrium is greatly dependent on the structure of the hydrate. Thus, formaldehyde in water at 20 °C exists 99.99% in the hydrated form, while for acetaldehyde this figure is 58%, and for acetone the hydrate concentration is negligible.39 It has been found, by exchange with 18O, that the reaction with acetone is quite rapid when catalyzed by acid or base, but the equilibrium lies on the side of acetone and water.40 Since methyl, a +I group, inhibits hydrate formation, it may be expected that electron-attracting groups would have the opposite effect, and this is indeed the case. The hydrate of chloral (trichloroacetaldehyde)41 is a stable crystalline substance. In order for it to revert to chloral, −OH or H2O must leave. This is made difficult by the electron-withdrawing character of the Cl3C group and by the absence of a proton on the α carbon, which is required for loss of water to form an enol. Some other42 polychlorinated and polyfluorinated aldehydes and ketones43 and α-keto aldehydes also form stable hydrates, as do cyclopropanones.44 In the last case,45 formation of the hydrate relieves some of the I strain (Sec. 9.B) of the parent ketone.
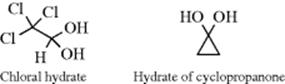
The reaction is subject to both general-acid and general-base catalysis; the following mechanisms can be written for basic (B) and acidic (BH) catalysis, respectively:46
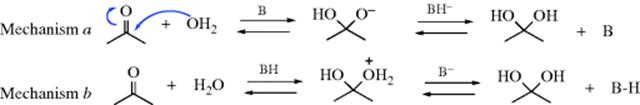
In mechanism a, as the H2O attacks, the base pulls off a proton, and the net result is addition of −OH. This can happen because the base is already hydrogen bonded to the H2O molecule before the attack. In mechanism b, because HB is already hydrogen bonded to the oxygen of the carbonyl group, it gives up a proton to the oxygen as the water attacks. In this way, B and HB accelerate the reaction even beyond the extent that they form –OH or H3O+ by reaction with water. Reactions in which the catalyst donates a proton to the electrophilic reagent (in this case the aldehyde or ketone) in one direction and removes it in the other are called class e reactions. Reactions in which the catalyst does the same to the nucleophilic reagent are called class n reactions.47 Thus the acid-catalyzed process here is a class e reaction, while the base-catalyzed process is a class n reaction.
For the reaction between ketones and H2O2, see 17-37.
There are no OS references, but see OS VIII, 597, for the reverse reaction.
16-2 Hydrolysis of the Carbon–Nitrogen Double Bond48
Oxo-de-alkylimino-bisubstitution, and so on
![]()
Compounds containing carbon–nitrogen double bonds can be hydrolyzed to the corresponding aldehydes or ketones.49 For imines (W = R or H) the hydrolysis is easy and can be carried out with water. When W = H, the imine is seldom stable enough for isolation, and in aqueous media hydrolysis usually occurs in situ, without isolation. The hydrolysis of Schiff bases (W = Ar) is more difficult and requires acid or base catalysis. Oximes (W = OH), arylhydrazones (W = NHAr), and, most easily, semicarbazones (W = NHCONH2) can also be hydrolyzed. Often a reactive aldehyde (e.g., formaldehyde), is added to combine with the liberated amine.
A number of reagents50 have been used to cleave C=N bonds, especially those not easily hydrolyzable with acidic or basic catalysts or those that contain other functional groups that are attacked under these conditions.
Oximes have been converted to the corresponding aldehyde or ketone51 by treatment with aq phosphoric acid without an organic cosolvent,52 periodic acid,53 DABCO–Br2,54 NBS in water,55 Chloramine-T56 HCO2H on SiO2 with microwave irradiation,57 and 20% I2 in water containing SDS (sodium dodecyl sulfate)58 or in an ionic liquid on SiO2.59 Transition metal compounds have been used, including those of Sb,60 Co61 Hg,62 Bi,63 Cu,64 or Zn.65 Oxidizing agents can be quite effective, including KMnO4 on Al2O3,66 tetraalkylammonium permanganates,67 and quinolinium dichromate.68 Alkaline H2O269 has also been used.
Phenylhydrazones can be converted to a ketone using Oxone and KHCO3,70 polymer-bound iodonium salts,71 or KMnO4 on wet SiO2.72 Dimethylhydrazones have been converted to ketones with FeSO4·7 H2O in chloroform,73and Me3SiCl/NaI in acetonitrile with 1% water.74 Hydrazones (e.g., RAMP or SAMP, Reaction 10-68, category 4) can be hydrolyzed with aq CuCl2.75 Tosylhydrazones can be hydrolyzed to the corresponding ketones with aq acetone and BF3–etherate,76 as well as with other reagents.77
Semicarbazones have been cleaved with ammonium chlorochromates on alumina78 (Bu4N)2S2O8,79 Mg(HSO4)2 on wet silica,80 or by SbCl3 with microwave irradiation.81
The hydrolysis of carbon–nitrogen double bonds involves initial addition of water and elimination of a nitrogen moiety:
![]()
It is thus an example of reaction type A (see above). The sequence shown is generalized.82 In specific cases, there are variations in the sequence of the steps, depending on acid or basic catalysis or other conditions.83 Which step is rate determining also depends on acidity and on the nature of W and of the groups connected to the carbonyl.84

Iminium ions (10)85 would be expected to undergo hydrolysis quite readily, since there is a resonance contributor with a positive charge on the carbon. Indeed, they react with water at room temperature.86 Acid-catalyzed hydrolysis of enamines (the last step of the Stork enamine reaction, 10-69, involves conversion to iminium ions).87
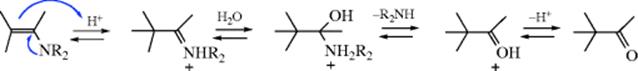
The mechanism of enamine hydrolysis is thus similar to that of vinyl ether hydrolysis (Reaction 10-6).
OS I, 217, 298, 318, 381; II, 49, 223, 234, 284, 310, 333, 395, 519, 522; III, 20, 172, 626, 818; IV, 120; V, 139, 277, 736, 758; VI, 1, 358, 640, 751, 901, 932; VII, 8; 65, 108, 183; 67, 33; 76, 23.
OS II, 24; IV, 819; V, 273; VI, 910.
16-3 Hydrolysis of Aliphatic Nitro Compounds
Oxo-de-hydro,nitro-bisubstitution
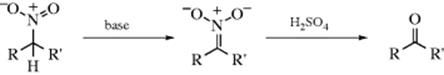
Primary or secondary aliphatic nitro compounds can be hydrolyzed, respectively, to aldehydes or ketones, by treatment of their conjugate bases with sulfuric acid. This is called the Nef reaction.88 Tertiary aliphatic nitro compounds do not give the reaction because they cannot be converted to their conjugate bases. Like 16-2, this reaction involves hydrolysis of a C=N double bond. A possible mechanism involves initial formation of the aci form of the nitro compound (11):89
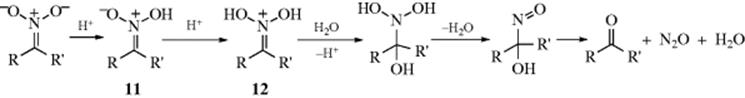
Intermediates of type 12 have been isolated in some cases.90
The conversion of nitro compounds to aldehydes or ketones has been carried out with better yields and fewer side reactions by several alternative methods.91 Among these are treatment of the nitro compound with basic H2O2 in an ionic liquid,92 or 30% H2O2–K2CO3,93 DBU in acetonitrile,94 or CAN.95
When primary nitro compounds are treated with sulfuric acid without previous conversion to the conjugate bases, they give carboxylic acids. Hydroxamic acids are intermediates and can be isolated, so that this is also a method for their preparation.96 Both the Nef reaction and the hydroxamic acid process involve the aci form; the difference in products arises from higher acidity. For example, a difference in sulfuric acid concentration from 2 to 15.5 M changes the product from the aldehyde to the hydroxamic acid.97 The mechanism of the hydroxamic acid reaction is not known with certainty, but if higher acidity is required, it may be that the protonated aci form of the nitro compound is further protonated.
OS VI, 648; VII, 414. See also, OS IV, 573.
16-4 Hydrolysis of Nitriles
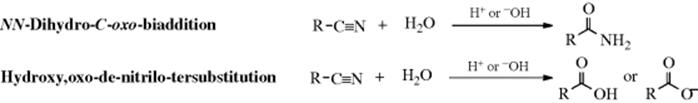
Nitriles can be hydrolyzed to give either amides or carboxylic acids.98 The amide is formed initially, but since amides are also hydrolyzed with acid or basic treatment, the carboxylic acid is readily formed. When the acid is desired,99 the reagent of choice is aq NaOH containing ~ 6–12% H2O2, but acid-catalyzed hydrolysis is also carried out. The reaction of nitriles with TFA–acetic acid–sulfuric acid, followed by treatment with water gives the corresponding amide.100 A Rh catalyzed hydrolysis with aq isopropyl alcohol leads to the amide.101 A "dry" hydrolysis of nitriles has been reported.102 Enzymatic hydrolysis to give the amide was reported with nitrilase (ZmNIT2).103 Nitriles can be hydrolyzed to the carboxylic acids without disturbing carboxylic ester functions also present, by the use of tetrachloro- or tetrafluorophthalic acid.104
Hydrolysis of cyanohydrins [RCH(OH)CN] is usually carried out under acidic conditions, because basic solutions cause competing reversion of the cyanohydrin to the aldehyde and CN−. However, cyanohydrins have been hydrolyzed under basic conditions with borax or alkaline borates.105
There are a number of procedures for stopping at the amide stage,106 among them the use of concentrated H2SO4; 2 molar equivalents of chlorotrimethylsilane followed by H2O,107 aq NaOH with PEG-400 and microwave irradiation,108 heating on neutral alumina,109 or dry HCl followed by H2O. The same result can also be obtained by use of water and certain metal ions or complexes110 including an In,111 Au,112 or a Ru catalyst.113 Other reagents include MnO2/SiO2 with microwave irradiation,114 or Hg(OAc)2 in HOAc.115 The reaction of ferric nitrate and an amine generates the amide.116
Nitriles are converted to thioamides [ArC(=S)NH2] with ammonium sulfide [(NH4)2S] in methanol, with microwave irradiation.117 Thioamides are also prepared using phosphorus pentasulfide.118
Thiocyanates are converted to thiocarbamates in a similar reaction:119 R–S–C![]() N + H2O → R–S–CO– NH2. Hydrolysis of cyanamides gives amines, produced by the breakdown of the unstable carbamic acid intermediates: R2NCN → [R2NCOOH] → R2NH.
N + H2O → R–S–CO– NH2. Hydrolysis of cyanamides gives amines, produced by the breakdown of the unstable carbamic acid intermediates: R2NCN → [R2NCOOH] → R2NH.
OS I, 21, 131, 201, 289, 298, 321, 336, 406, 436, 451; II, 29, 44, 292, 376, 512, 586 (see, however, V, 1054), 588; III; 34, 66, 84, 88, 114, 221, 557, 560, 615, 851; IV, 58, 93, 496, 506, 664, 760, 790; V, 239; VI, 932; 76, 169. Also see, OS III, 609; IV, 359, 502; 66, 142.
B. Attack by OR (Addition of ROH)
16-5 The Addition of Alcohols to Aldehydes and Ketones
Dialkoxy-de-oxo-bisubstitution
Dithioalkyl-de-oxo-bisubstitution
![]()
Acetals and ketals are formed by treatment of aldehydes and ketones, respectively, with alcohols in the presence of acid catalysts.120 Lewis acid derivatives of Ti,121 Cu,122 In,123 Ru,124 or Co125 can be used in conjunction with alcohols. Organocatalysts have been used for this conversion under acid-free conditions.126 This reaction is reversible, and acetals and ketals can be hydrolyzed by treatment with acid.127 With small unbranched aldehydes the equilibrium lies to the right. If ketals or acetals of larger molecules must be prepared the equilibrium must be shifted, usually by removal of water. This can be done by azeotropic distillation, ordinary distillation, or the use of a drying agent (e.g., Al2O3 or a molecular sieve).128 The reaction is not catalyzed in either direction by bases, so most acetals and ketals are quite stable to bases, though they are easily hydrolyzed by acids. This reaction is therefore a useful method of protection of aldehyde or ketone functions from attack by bases. The reaction is of wide scope.
Most aldehydes are easily converted to acetals,129 but the reaction with ketones is more difficult, presumably for steric reasons. While the reaction often fails, many ketals, especially from cyclic ketones, have been made in this manner.130 Many functional groups may be present without being affected. 1,2- and 1,3-Diols form cyclic acetals and ketals (1,3-dioxolanes131 and 1,3-dioxanes,132 respectively), and these are often used to protect aldehydes and ketones. Chiral dioxolanes have been prepared from chiral diols.133 Dioxolanes have been prepared from ketones in ionic liquids.134 Ketones are converted with dimethyl ketals by electrolysis with NaBr in methanol.135Intramolecular reactions are possible in which a keto diol or an aldehyde diol generates a bicyclic ketal or acetal.
The conversion of acetals back to aldehydes or ketones is accomplished by many reagents, including aq acid. Heating with water under microwave irradiation converts acetals to the corresponding carbonyl compound.136Transition metal compounds of Bi137 catalyze this conversion as well.
The mechanism for acetal/ketal formation involves initial formation of a hemiacetal,138 and it is the reverse of that given for acetal hydrolysis:
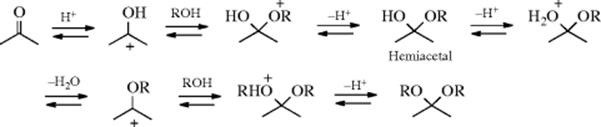
In a study of the acid-catalyzed formation of the hemiacetal, Grunwald139 showed that the data best fit a mechanism in which the three steps shown here are actually all concerted; that is, the reaction is simultaneously catalyzed by acid and base, with water acting as the base:140
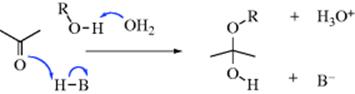
Hemiacetals themselves are no more stable than the corresponding hydrates (Reaction 16-1). If the original aldehyde or ketone has an α hydrogen, it is possible to lose water from the hemiacetal, and enol ethers can be prepared in this manner:

Similarly, treatment with an anhydride and a catalyst can give an enol ester (see Reaction 16-6).141 As with hydrates, it is noted that hemiacetals of cyclopropanones142 and of polychloro and polyfluoro aldehydes and ketones may be quite stable.
When acetals or ketals are treated with an alcohol of higher molecular weight than the one already there, transacetalation is possible (see Reaction 10-13). In another type of transacetalation, aldehydes or ketones can be converted to acetals or ketals by treatment with another acetal or ketal or with an ortho ester,143 in the presence of an acid catalyst (shown for an ortho ester):
![]()
This method is useful for the conversion of ketones to ketals, since the direct reaction of a ketone with an alcohol often gives poor results. Alternatively, the substrate is treated with an alkoxysilane (ROSiMe3) in the presence of trimethylsilyl trifluoromethanesulfonate.144 Formic acid reacts with alcohols to give orthoformates.
1,4-Diketones give furans when treated with acids. This is actually an example of an intramolecular addition of an alcohol to a ketone, since it is the enol form that adds:
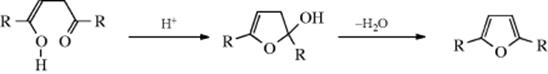
Similarly, 1,5-diketones give pyrans. Conjugated 1,4-diketones (e.g., 1,4-diphenylbut-2-en-1,4-dione) are converted to 2,5-diphenylfuran with formic acid, 5% Pd/C, PEG-200, and a sulfuric acid catalyst with microwave irradiation.145 Note that alkynyl ketones are converted to furans with palladium(II) acetate.146
OS I, 1, 298, 364, 381; II, 137; III, 123, 387, 502, 536, 644, 731, 800; IV, 21, 479, 679; V, 5, 292, 303, 450, 539; VI, 567, 666, 954; VII, 59, 149, 168, 177, 241, 271, 297; VIII, 357. Also see, OS IV, 558, 588; V, 25; VIII, 415.
16-6 Acylation of Aldehydes and Ketones
O-Acyl-C-acyloxy-addition
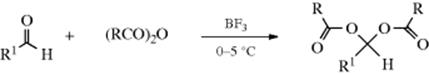
Aldehydes can be converted to acylals by treatment with an anhydride in the presence of proton acids,147 NBS,148 ceric ammonium nitrate,149 BF3, LiBF4,150 and Lewis acid compounds of Fe,151 In,152 Cu,153 Bi,154 W,155 or Zr.156N-Chlorosuccinimide with thiourea is a highly efficient catalyst.157 Silica supported perchloric acid is useful for the preparation of acylals.158 Conjugated aldehydes are converted to the corresponding acylal by reaction with acetic anhydride and a FeCl3 catalyst.159 The reaction cannot normally be applied to ketones, though an exception has been reported when the reagent is trichloroacetic anhydride, which gives acylals with ketones without a catalyst.160
OS IV, 489.
16-7 Reductive Alkylation of Alcohols
C-Hydro-O-alkyl-addition
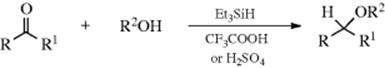
Aldehydes and ketones can be converted to ethers by treatment with an alcohol and triethylsilane in the presence of a strong acid161 or by hydrogenation in alcoholic acid in the presence of a Pt catalyst.162 The process can formally be regarded as addition of ROH to give a hemiacetal [RR′C(OH)OR2], followed by reduction of the OH. In this respect, it is similar to Reaction 16-17. Homoallylic ethers are formed by the Fe catalyzed reaction of acetals and aldehydes,163 and by reactions in ionic liquids.164 The In catalyzed reaction of aldehydes with allyltriethoxysilane leads to the corresponding ether.165
Ethers have also been prepared by the reductive dimerization of two molecules of an aldehyde or ketone (e.g., cyclohexanone → dicyclohexyl ether). This was accomplished by treatment of the substrate with a trialkylsilane and a catalyst.166
16-8 The Addition of Alcohols to Isocyanates
N-Hydro-C-alkoxy-addition

The reaction of an isocyanate with alcohols gives a carbamate (a substituted urethane). This is an excellent reaction of wide scope that gives good yields. Isocyanic acid (HNCO) gives unsubstituted carbamates. Addition of a second equivalent of HNCO gives allophanates.
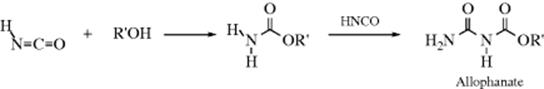
The isocyanate can be generated in situ by the reaction of an amine and oxalyl chloride, and subsequent reaction with HCl and then an alcohol gives the carbamate.167 Combining compounds with two NCO groups with compounds containing two OH groups makes polyurethanes. Cyclic carbamates (e.g., 1,3-oxazine-2-ones), are generated by the reaction of an isocyanate with an oxetane, in the presence of a Pd catalyst.168 Isothiocyanates similarly give thiocarbamates (RNHCSOR′),169 but they react slower than the corresponding isocyanates. Isocyanates react with LiAlHSeH and then iodomethane to give the corresponding selenocarbonate (RNHCOSeMe).170
The details of the mechanism are poorly understood,171 but the oxygen of the alcohol certainly attacks the carbon of the isocyanate. Hydrogen bonding complicates the kinetic picture.172 Metallic compounds, can also catalyze the addition of ROH to isocyanates173 by light,174 or, for tertiary ROH, by lithium alkoxides175 or n-butyllithium.176
OS I, 140; V, 162; VI, 95, 226, 788, 795.
16-9 Alcoholysis of Nitriles
Alkoxy,oxo-de-nitrilo-tersubstitution
![]()
The addition of dry HCl to a mixture of a nitrile and an alcohol in the absence of water leads to the hydrochloride salt of an imino ester (imino esters are also called imidates and imino ethers). This reaction is called the Pinner synthesis.177 The salt can be converted to the free imino ester by treatment with a weak base (e.g., sodium bicarbonate) or it can be hydrolyzed with water and an acid catalyst to the corresponding carboxylic ester. If the latter is desired, water may be present from the beginning, in which case aq HCl can be used and the need for gaseous HCl is eliminated. Imino esters can also be prepared from nitriles with basic catalysts.178
This reaction is of broad scope and is good for aliphatic, aromatic, and heterocyclic R and for nitriles with oxygen-containing functional groups. The application of the reaction to nitriles containing a carboxyl group constitutes a good method for the synthesis of mono esters of dicarboxylic acids with the desired group esterified and with no diester or diacid present.
Cyanogen chloride reacts with alcohols in the presence of an acid catalyst (e.g., dry HCl or AlCl3), to give carbamates:179
![]()
ROH can also be added to nitriles in another manner (Reaction 16-91).
OS I, 5, 270; II, 284, 310; IV, 645; VI, 507; VIII, 415.
16.2.1.2.6 16-10 The Formation of Carbonates and Xanthates
Di-C-alkoxy-addition; S-Metallo-C-alkoxy-addition
![]()
The reaction of phosgene with an alcohol generates haloformic esters, and reaction with a second equivalent of alcohol gives a carbonate. This reaction is related to the acyl addition reactions of acyl chlorides in Reaction 16-98. An important example is the preparation of carbobenzoxy chloride (PhCH2OCOCl; CbzCl) from phosgene and benzyl alcohol. When CbzCl reacts with an amine, the product is the benzyl carbamate, N-Cbz, which is widely used for protection of amino groups during peptide synthesis. When an alcohol reacts with certain alkyl halides (e.g., benzyl chloride) and CO2, in the presence of Cs2CO3 and tetrabutylammonium iodide, a mixed carbonate is formed.180
The addition of alcohols to carbon disulfide (S=C=S) in the presence of a base produces xanthates.181 The base is often HO−, but in some cases better results can be obtained by using methylsulfinyl carbanion (MeSOCH2−).182 If an alkyl halide (RX) is present, the xanthate ester (ROCSSR′) can be produced directly. In a similar manner, alkoxide ions add to CO2 to give carbonate ester salts (ROCO2−).
OS V, 439; VI, 207, 418; VII, 139.
C. Sulfur Nucleophiles
16-11 The Addition of H2S and Thiols to Carbonyl Compounds
O-Hydro-C-mercapto-addition, thioxo-de-oxo-bisubstitution,dimercapto-de-oxo-bisubstitution, and carbonyl-trithiane transformation
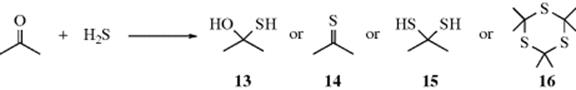
The addition of H2S to an aldehyde or ketone can result in a variety of products. The most usual product is the trithiane 16.183gem-Dithiols (15) are much more stable than the corresponding hydrates or α-hydroxy thiols.184 They have been prepared by the treatment of ketones with H2S under pressure185 and under mild conditions with HCl as a catalyst.186 A much more useful application is the addition of thiols to aldehydes and ketones to give hemi-mercaptals [CH(OH)SR] and dithioacetals [CH(SR)2] (Reaction 16-5). This reaction is generally not a good route to thioketones (14). α-Hydroxythiols (13) can be prepared from polychloro and polyfluoro aldehydes and ketones.187Apparently gem-hydroxy thiols (e.g., 13) are rather unstable and quite difficult to prepare.
Thiols add to aldehydes and ketones to initially give hemi-mercaptals and dithioacetals. Hemi-mercaptals are ordinarily unstable,188 but they are usually more stable than the corresponding hemiacetals and can be isolated in certain cases.189 The isolated product of this reaction is most often the dithioacetal, which like the acetals obtained by reaction with an alcohol, are stable in the presence of base. However, a sufficiently strong base can remove a proton from the carbon between the sulfur atoms (–S–CHR–S–)190 to form the corresponding carbanion (see Reaction 10-71). The pKa of such protons is typically 31–37,191 requiring a strong base, and deprotonation is often quite slow. The reaction of aldehydes or ketones with thiols most commonly uses a Lewis acid catalyst (e.g., boron trifluoride etherate, BF3·OEt2)192 to give the dithioacetal193 or dithioketal. Dithioacetals can also be prepared from aldehydes or ketones by treatment with thiols in the presence of TiCl4,194 SiCl4,195 LiBF4,196 Al(OTf)3,197 tosic acid on silica gel in dichloromethane,198 and oxalic acid promotes the reaction.199 Similarly reactions that use 1,2-ethanedithiol or 1,3-propanedithiol lead to 1,3-dithiolanes (17)200 or 1,3-dithianes.201 3-(1,3-Dithian-2-ylidene)pentane- 2,4-dione has been used as a thioacetalization reagent for reactions in water.202
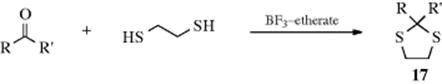
Dithioacetals and dithioketals are used as protecting groups for aldehydes and ketones, and after subsequent reactions involving the R or R′ group, deprotection regenerates the carbonyl.203 Simple hydrolysis is the most common method for converting thiocarbonyls to carbonyls, but there are a variety of reagents for this conversion.204 Lewis acids (e.g., aluminum chloride, AlCl3) and mercuric salts are common reagents (the Corey–Seebach procedure).205Other reagents include BF3·OEt2 in aq THF containing mercuric oxide (HgO),206 ceric ammonium nitrate [Ce(NH4)2(NO3)6],207 chlorotrimethylsilane and H2O2,208 PhI(OAc)2 in aq acetone,209 and Cu salts.210 When aldehydes and ketones react with mercapto–alcohols, mixed acetals or ketals are formed.
Thioamides are converted to amides with Caro's acid (H2SO5) on SiO2.211 The use of 2-mercaptoethanol (HSCH2CH2OH), for example, leads to an oxathiolane212 and 3-mercaptopropanol (HSCH2CH2CH2OH) leads to an oxathiane. Alternatively, the dithioketal can be desulfurized with Raney nickel (Reaction 14-27), giving the overall conversion C=O → CH2 (Reaction 19-70).
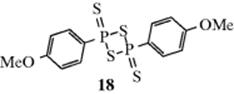
Thioketones (14) can be prepared from certain ketones (e.g., diaryl ketones) by treatment with H2S and an acid catalyst, usually HCl. They are often unstable and usually trimerize (to 16) or react with air. Thioaldehydes213 are even less stable and simple ones214 apparently have never been isolated, although tert-BuCHS has been prepared in solution, where it exists for several hours at 20 °C.215 A high-yield synthesis of thioketones involves treatment of acyclic216 ketones with 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide217 (18, known as Lawesson's reagent).218 Thioketones can also be prepared by treatment of ketones with P4S10,219 P4S10 and hexamethyldisiloxane,220 P4S10 on alumina,221 and from oximes or various types of hydrazone (overall conversion C=N– → C=S).222 Reagent 18 converts the C=O groups of amides and carboxylic esters223 to C=S groups.224Lactones react with 18 in the presence of hexamethyldisiloxane an microwave irradiation to give the thiolactone.225
Other reagents may be used for this transformation including POCl3 followed by S(TMS)2, which converts lactams to thiolactams.226 Treatment with triflic anhydride, and then H2S227 or aq S(NH4)2228 converts amides to thioamides, as does the microwave assisted reaction with PSCl2/H2O/Et3N, without solvent.229 Carboxylic acids (RCOOH) can be converted directly to dithiocarboxylic esters (RCSSR′),230 in moderate yield, with P4S10 and a primary alcohol (R′OH).231
If an aldehyde or ketone possesses an α hydrogen, it can be converted to the corresponding enol thioether (19) by treatment with a thiol in the presence of TiCl4.232 Aldehydes and ketones have been converted to sulfides by treatment with thiols and pyridine–borane, RCOR′ + R2SH → RR′CHSR2,233 in a reductive alkylation reaction, analogous to Reaction 16-7.
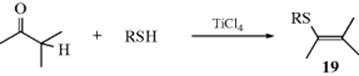
OS II, 610; IV, 927; VI, 109; VII, 124, 372. Also see, OS III, 332; IV, 967; V, 780; VI, 556; VIII, 302.
16-12 Formation of Bisulfite Addition Products
O-Hydro-C-sulfonato-addition
![]()
Bisulfite addition products are formed from aldehydes, methyl ketones, cyclic ketones (generally seven-membered and smaller rings), α-keto esters, and isocyanates, upon treatment with sodium bisulfite (NaHSO3). Most other ketones do not undergo the reaction, probably for steric reasons. The reaction is reversible (by treatment of the addition product with either acid or base)234 and is useful for the purification of the starting compounds, since the addition products are soluble in water and many of the impurities are not.235
OS I, 241, 336; III, 438; IV, 903; V, 437.
D. Attack by NH2, NHR, or NR2 (Addition of NH3, RNH2, R2NH)
16-13 The Addition of Amines to Aldehydes and Ketones
Alkylimino-de-oxo-bisubstitution
The addition of ammonia236 to aldehydes or ketones does not generally give useful products. According to the pattern followed by analogous nucleophiles, the initial products would be expected to be hemiaminals,237 but these compounds are generally unstable. In addition, many imines with a hydrogen on the nitrogen spontaneously polymerize.238
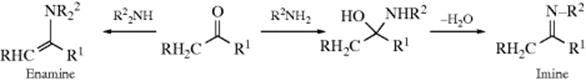
In contrast to ammonia, primary, secondary, and tertiary amines can add to aldehydes239 and ketones to give different kinds of products. Primary amines give imines240 and secondary amines give enamines (for reactions, see 10-69). Such imines are stable enough for isolation, but in some cases, especially with simple R groups, they rapidly decompose or polymerize unless there is at least one aryl group on the nitrogen or the carbon. When there is an aryl group, the compounds are quite stable. They are usually called Schiff bases, and this reaction is the best way to prepare them.241 Even sterically hindered imines can be prepared.242 The initial N-substituted hemiaminals243 lose water to give the stable Schiff bases.
In general, ketones react more slowly than aldehydes, and higher temperatures and longer reaction times are often required.244 In addition, the equilibrium must often be shifted, usually by removal of the water, either azeotropically by distillation, or with a drying agent (e.g., TiCl4),245 or by addition of a molecular sieve.246 Imines have been formed from aldehydes and amines in an ionic liquid.247

The reaction is often used to effect ring closure.248 The Friedländer quinoline synthesis249 is an example where ortho alkenyl aniline derivatives give the quinoline (20).250 The alkene derivative can be prepared in situ from an aldehyde and a suitably functionalized ylid.251 Pyrylium ions react with ammonia or primary amines to give pyridinium ions252 (see Reaction 10-57). Primary amines react with 1,4-diketones, with microwave irradiation, to give N-substituted pyrroles.253 Similar reactions in the presence of Montmorillonite KSF254 or by simply heating the components with tosic acid255 have been reported.
The reaction of secondary amines with ketones leads to enamines (see 10-69).256 When secondary amines are added to aldehydes or ketones, the initially formed N,N-disubstituted hemiaminals (21) cannot lose water in the same way since the iminium ion intermediate does not have a proton on nitrogen, and in some cases it is possible to isolate them.257 However, they are generally unstable, and under the reaction conditions usually react further. If no α hydrogen is present, 21 is converted to the more stable aminal (22).258 However, if an α hydrogen is present, water (from 21) or RNH2 (from 22) can be lost in that direction to give an enamine (24).259 This is the most common method260 for the preparation of enamines and usually takes place when an aldehyde
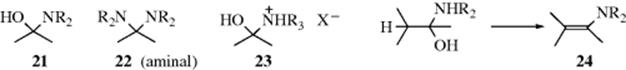
or ketone containing an α hydrogen is treated with a secondary amine.261 The water is usually removed azeotropically or with a drying agent,262 and molecular sieves can also be used.263 Silyl carbamates (e.g., Me2NCO2SiMe3) have been used to convert ketones to enamines.264 Stable primary enamines have also been prepared.265 Enamino–ketones have been prepared from diketones and secondary amines using low-molecular-weight amines in water,266 or using microwave irradiation on silica gel.267 Enamines have been prepared by the reaction of an aldehyde, a secondary amine, and a terminal alkyne in the presence of AgI at 100 °C,268 AgI in an ionic liquid,269 CuI with microwave irradiation,270 or a Au catalyst.271
Tertiary amines can only give salts (23).
OS I, 80, 355, 381; II, 31, 49, 65, 202, 231, 422; III, 95, 328, 329, 332, 358, 374, 513, 753, 827; IV, 210, 605, 638, 824; V, 191, 277, 533, 567, 627, 703, 716, 736, 758, 808, 941, 1070; VI, 5, 448, 474, 496, 520, 526, 592, 601, 818, 901, 1014; VII, 8, 135, 144, 473; VIII, 31, 132, 403, 451, 456, 493, 586, 597. Also see, OS IV, 283, 464; VII, 197; VIII, 104, 112, 241.
16-14 The Addition of Hydrazine Derivatives to Carbonyl Compounds
Hydrazono-de-oxo-bisubstitution
![]()
The product of condensation of a hydrazine and an aldehyde or ketone is called a hydrazone. Hydrazine itself gives hydrazones only with aryl ketones. With other aldehydes and ketones, either no useful product can be isolated, or the remaining NH2 group condenses with a second equivalent of carbonyl compound to give an azine. This type of product is especially important for aromatic aldehydes:
![]()
However, in some cases azines can be converted to hydrazones by treatment with excess hydrazine and NaOH.272 Arylhydrazines, especially phenyl, p-nitrophenyl, and 2,4-dinitrophenyl,273 are used much more often and give the corresponding hydrazones with most aldehydes and ketones.274 Since these are usually solids, they make excellent derivatives and were commonly employed for this purpose in the past, before the advent of modern spectroscopy methods. Cyclic hydrazones are known,275 as are conjugated hydrazones.276 Azides react with N,N-dimethylhydrazine and ferric chloride to give the N,N-dimethylhydrazone.277 Alkenes react with CO/H2, phenylhydrazine and a diphosphine catalyst to give a regioisomeric mixture of phenylhydrazones that favored “anti-Markovnikov” addition.278 Oximes are converted to hydrazones with water and hydrazine in refluxing ethanol.279
α-Hydroxy aldehydes and ketones and α-dicarbonyl compounds give osazones, in which two adjacent carbons have carbon–nitrogen double bonds:
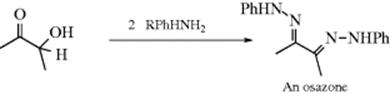
Osazones are particularly important in carbohydrate chemistry, and the osazone test280 with phenylhydrazine is used to test for the presence of sugars with an adjacent sterogenic carbon. In contrast to this behavior, β-diketones and β-keto esters give pyrazoles and pyrazolones, respectively (the latter is illustrated for β-keto esters). No azines are formed under these conditions.
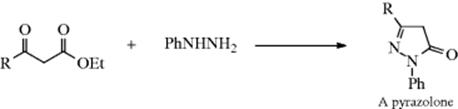
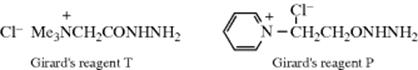
Other hydrazine derivatives frequently used to prepare the corresponding hydrazone are semicarbazide (NH2NHCONH2), in which case the hydrazone is called a semicarbazone: Girard's reagents T and P are hydrazones that are water soluble because of the ionic group. Girard's reagents are often used for purification of carbonyl compounds.281
Simple N-unsubstituted hydrazones can be obtained by an exchange reaction. The N,N-dimethylhydrazone is prepared first and then treated with hydrazine:282
![]()
OS II, 395; III, 96, 351; IV, 351, 377, 536, 884; V, 27, 258, 747, 929; VI, 10, 12, 62, 242, 293, 679, 791; VII, 77, 438. Also see, OS III, 708; VI, 161; VIII, 597.
16-15 The Formation of Oximes
Hydroxyimino-de-oxo-bisubstitution
![]()
In a reaction very much like 16-14, oximes can be prepared by the addition of hydroxylamine (NH2OH) to aldehydes or ketones. Derivatives of hydroxylamine [e.g., H2NOSO3H and HON(SO3Na)2] have also been used. For hindered ketones (e.g., 2,2,4,4-tetramethyl-3-pentanone), high pressures (as high as 10,000 atm) may be necessary.283 The reaction of hydroxylamine with unsymmetrical ketones or with aldehydes leads to a mixture of (E)- and (Z)-isomers. For aromatic aldehydes, heating with K2CO3 led to the (E)- isomer, whereas heating with CuSO4 gave the (Z)-hydroxylamine.284 Hydroxylamines react with ketones in ionic liquids285 and on silica gel.286
It has been shown287 that the rate of formation of oximes is at a maximum at a pH that depends on the substrate but is usually ~ 4. The rate also decreases as the pH is either raised or lowered from this point (a bell-shaped curve). In Section 16.A.i, bell-shaped curves like this were shown to be caused by changes in the rate-determining step in many cases. In this case, at low pH values step 2 is rapid (because it is acid catalyzed), and
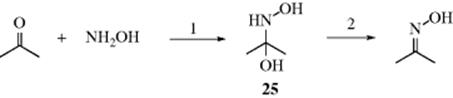
step 1 is slow (and rate determining), because under these acidic conditions most of the NH2OH molecules have been converted to the conjugate +NH3OH ions, which cannot attack the substrate. As the pH is slowly increased, the fraction of free NH2OH molecules increases and consequently so does the reaction rate, until the maximum rate is reached at ~ pH 4. As the rising pH causes an increase in the rate of step 1, it also causes a decrease in the rate of the acid-catalyzed step 2, although this latter process has not affected the overall rate since step 2 was still faster than step 1. However, when the pH goes above ~ 4, step 2 becomes rate determining, and although the rate of step 1 is still increasing (as it will until essentially all the NH2OH is unprotonated), it is now step 2 that determines the rate. This step is slowed by the decrease in acid concentration. Thus the overall rate decreases as the pH rises beyond ~ 4. It is likely that similar considerations apply to the reaction of aldehydes and ketones with amines, hydrazines, and other nitrogen nucleophiles.288 There is evidence that when the nucleophile is 2-methylthiosemicarbazide, there is a second change in the rate-determining step: > pH ~ 10 basic catalysis of step 2 has increased the rate of this step to the point where step 1 is again rate determining.289 Still a third change in the rate-determining step has been found at ~ pH 1, showing that at least in some cases step 1 actually consists of two steps: formation of a zwitterion (e.g., HOH2N+–C–O− in the case shown above) and conversion of this to 25.290 The intermediate 25 has been detected by NMR in the reaction between NH2OH and acetaldehyde.291
OS I, 318, 327; II, 70, 204, 313, 622; III, 690, IV, 229; V, 139, 1031; VII, 149. See also, OS VI, 670.
16-16 The Conversion of Aldehydes to Nitriles
Nitrilo-de-hydro,oxo-tersubstitution
![]()
Aldehydes can be converted to nitriles in one step by treatment with hydroxylamine hydrochloride and either formic acid,292 KF–Al2O3,293 or NaHSO4·SiO2 with microwave irradiation.294 Heating in N-methylpyrrolidinone (NMP) is also effective with aryl aldehydes295 and heating on dry alumina with aliphatic aldehyde.296 The reaction is a combination of 16-15 and 17-29. Direct nitrile formation has also been accomplished with certain derivatives of NH2OH, notably, NH2OSO2OH.297 Treatment with hydroxylamine and NaI298 or certain carbonates299 also converts aldehydes to the nitrile. Another method involves treatment with hydrazoic acid, although the Schmidt reaction(18-16) may compete.300 Microwave irradiation has been used with NH2OH·HCl and another reagent, which includes phthalic anhydride301 or H–Y zeolite.302 The reaction of an aldehyde with hydroxylamine followed by diethyl cholorophosphate (EtOPOCl2) gives the nitrile.303 Heating with hydroxylamine in DMSO304 and using hydroxylamine and oxalyl chloride305 have been used. tert-Butanesulfinyl imines are also used for the conversion of aldehydes to nitriles.306 Other reagents include trimethylsilyl azide,307 hydroxylamine hydrochloride/MgSO4/TsOH,308 or I2 with aq ammonia,309 The reaction of a conjugated aldehyde with ammonia, CuCl, and 50% H2O2 gave the conjugated nitrile.310 Aldehydes with IBX (o-iodoxybenzoic acid) and liquid ammonia gives the nitrile.311 Tetrabutylammonium tribromide in aq ammonia has also been used.312 Trichloroisocyanuric acid with a catalytic amount of TEMPO (Sec. 5.C.i) converts aldehydes to nitriles at 0 °C in dichloromethane.313 Aromatic aldehydes are converted to the nitrile by heating 2.2 molar equivalents of NaN(SiMe3)2 in 1,3-dimethylimidazolidin-2-one, in a sealed tube.314 The aldehydes employed had a hydroxy substituent.
In a related reaction, the reaction of primary alcohols with iodine in ammonia water gives the corresponding nitrile.315 Upon treatment with 2 molar equivalents of dimethylaluminum amide (Me2AlNH2), carboxylic esters give nitriles: RCO2R′ → RCN.316 This is likely a combination of Reaction 16-75 and 17-30 (see Reaction 19-5).
OS V, 656.
16-17 Reductive Alkylation of Ammonia or Amines
Hydro,dialkylamino-de-oxo-bisubstitution
![]()
When an aldehyde or a ketone is treated with ammonia or a primary or secondary amine in the presence of hydrogen gas and an appropriate catalyst (e.g., Rh or Ir; heterogeneous or homogeneous),317reductive alkylation of ammonia or the amine (or reductive amination of the carbonyl compound) takes place.318 The reaction can formally be regarded as occurring in the following manner (shown for a primary amine), which probably does correspond to the actual sequence of steps:319 In this regard, the reaction of an aldehyde with an amine to give an iminium salt (16-31) can be followed in a second chemical step of reduction of the C=N unit (19-42) using NaBH4 or a variety of other reagents.320
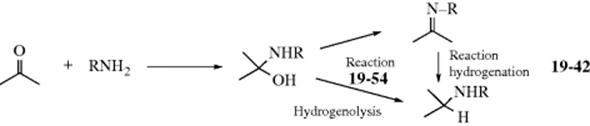
Primary amines have been prepared from many aldehydes with at least five carbons and from many ketones by treatment with ammonia and a reducing agent. Smaller aldehydes are usually too reactive to permit isolation of the primary amine. Secondary amines have been prepared by both possible procedures: 2 molar equivalents of ammonia and 1 molar equivalent of aldehyde or ketone, and 1 molar equivalent of primary amine and 1 molar equivalent of carbonyl compound, the latter method being better for all but aromatic aldehydes. Tertiary amines can be prepared in three ways. In general, they are prepared from primary or secondary amines.321 The method is seldom carried out with 3 molar equivalents of ammonia and 1 molar equivalent of carbonyl compound. When the reagent is ammonia, it is possible for the initial product to react again and for this product to react again, so that secondary and tertiary amines are usually obtained as side products. Similarly, primary amines give tertiary as well as secondary amines. In order to minimize this, the aldehyde or ketone is treated with an excess of ammonia or primary amine (unless of course the higher amine is desired).
For ammonia and primary amines there are two possible pathways, but when secondary amines are involved, only the hydrogenolysis pathway is possible. The reaction is compatible with amino acids, giving the N-alkylated amino acid.322 Other reducing agents323 can be used instead of hydrogen and a catalyst, among them boranes,324 PhSiH3 with 2% Bu2SnCl2,325 triethylsilane with an Ir326 or an In catalyst,327 zinc and HCl, or Zn (with formaldehyde for reductive methylation),328 and polymethylhydrosiloxane.329 Several hydride reducing agents can be used, including NaBH4,330 NaBH4 with Ti(OiPr)4331 or NiCl2,332 NaBH4/H3BO4,333 BER,334 polymer-bound triethylammonium acetoxyborohydride,335 LiBH4,336 ZnBH4-N-methylpiperidine,337 ZrBH4,338 NaBH3CN,339 or sodium triacetoxyborohydride,340 Aldehydes and primary amines react with allylic halides, in the presence of Zn dust, to give a homoallylic secondary amine.341 A Hantzsch dihydropyridine in conjunction with a Sc catalyst has been used,342 and the use of a Hantzsch ester in a reductive amination is sometimes called a hydrogen-bond catalyzed reaction.343The reaction of an aldehyde and an amine in isopropyl alcohol, in the presence of Ni nanoparticles, undergoes reductive amination via hydrogen transfer.344
Formic acid is an effective reagent for reductive amination345 in what is called the Wallach reaction. Secondary amines react with formaldehyde and NaH2PO3 to give the N-methylated tertiary amine346 and microwave irradiation has also been used.347 Conjugated aldehydes are converted to alkenyl-amines with the amine/silica gel followed by reduction with zinc borohydride.348 In the particular case where primary or secondary amines are reductively methylated with formaldehyde and formic acid, the method is called the Eschweiler–Clarke procedure. Heating with paraformaldehyde and oxalyl chloride has been used to give the same result.349 It is possible to use ammonium (or amine) salts of formic acid,350 or formamides, as a substitute for the Wallach conditions. This method is called the Leuckart reaction,351 and in this case the products obtained are often the N-formyl derivatives of the amines instead of the free amines. A Rh catalyzed variation has been reported.352
Allylic silanes react with aldehydes and carbamates, in the presence of Bi catalysts,353 or BF3·OEt2354 to give the corresponding allylic N-carbamoyl derivative. The reaction can be done with aromatic amines in the presence of vinyl ethers and a Cu complex to give β-amino ketones.355 Reductive amination of an aryl amine and an aryl aldehyde that contains an ortho conjugated ketone substituent gives the amine, which adds 1,4- (Reaction 15-AA) to the α,β-unsaturated ketone unit to give a bicyclic amine.356 Alternative methods of reductive alkylation have been developed. Alkylation of an imine formed in situ is also possible.357
Enantioselective reductive amination reactions are known, generating chiral amines. Ketones and anilines react in the presence of an organocatalyst and a catalytic amount of a chiral phosphoric acid to give the chiral amine.358The reaction of an aldehyde with a chiral amine initiated a reaction that gave a chiral primary amine.359 A Yb catalyzed reaction with a ketone gave a chiral secondary amine.360 Aldehydes react with N-diphenylphosphinoylimines and Et2Zn, in the presence of a chiral Cu precatalyst, to give a chiral amine.361 Asymmetric biocatalytic reductive amination reactions are known.362 Asymmetric reductive amination has been attempted using a Hantzsch estermediated reaction.363
Reductive alkylation has also been carried out on nitro, nitroso, azo, and other compounds that are reduced in situ to primary or secondary amines. Azo compounds react with aldehydes, in the presence of proline, and subsequent reduction with NaBH4 gives the chiral hydrazine derivative.364
OS I, 347, 528, 531; II, 503; III, 328, 501, 717, 723; IV, 603; V, 552; VI, 499; VII, 27.
16-18 Addition of Amides to Aldehydes
Alkylamido-de-oxo-bisubstitution
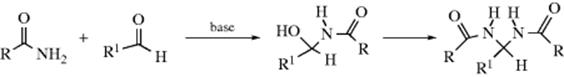
Amides can add to aldehydes in the presence of bases (so the nucleophile is actually RCONH−) or acids to give acylated amino alcohols, which often react further to give alkylidene or arylidene bisamides.365 If the R′ group contains an α hydrogen, water may split out.
Sulfonamides add to aldehydes to give the N-sulfonyl imine. Benzaldehyde reacts with TsNH2 (e.g., with trifluoroacetic anhydride, TFAA) in CH2Cl2 at reflux,366 or with TiCl4 in refluxing dichloroethane,367 to give the N-tosylimine (Ts–N=CHPh). In a similar manner, the reaction of TolSO2Na + PhSO2Na with an aldehyde in aq formic acid gives the N-phenylsulfonyl imine.368 The reaction of an aldehyde with Ph3P=NTs and a ruthenium catalyst gives the N-tosyl imine.369
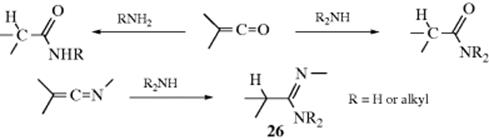
Primary and secondary amines add to ketenes to give, respectively, N-substituted and N,N-disubstituted amides:370 and to ketenimines to give amidines, (26).371
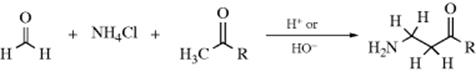
16-19 The Mannich Reaction
Acyl,amino-de-oxo-bisubstitution, and so on
In the Mannich reaction, formaldehyde (or sometimes another aldehyde) is condensed with ammonia, in the form of its salt, and a compound containing an active hydrogen.372 This can formally be considered as an addition of ammonia to give H2NCH2OH, followed by a nucleophilic substitution. The reaction can be carried out with salts of primary or secondary amines,373 or with amides374 rather than ammonia, in which cases the product is substituted on the nitrogen with R, R2, and RCO, respectively. The product is referred to as a Mannich base. The imine can be generated in situ, and the reaction of a ketone, formaldehyde, and diethylamine with microwave irradiation gave the Mannich product, a β-amino ketone.375 Many active hydrogen compounds give the reaction, including ketones and aldehydes, esters, nitroalkanes,376 and nitriles as well as ortho-carbon atoms of phenols, the carbon of terminal alkynes, the oxygen of alcohols and the sulfur of thiols.377 Arylamines do not normally give the reaction, but hydrazines can be used.378 Vinylogous Mannich reactions are known379 (see Sec. 6.B for vinylogy).
The Mannich base product can react further in three ways. If it is a primary or secondary amine, it may condense with one or two additional molecules of aldehyde and an active compound, for example,
![]()
If the active hydrogen compound has two or three active hydrogen atoms, the Mannich base may condense with one or two additional molecules of aldehyde and ammonia or amine, for example,
![]()
Another reaction consists of condensation of the Mannich base with excess formaldehyde:
![]()
Sometimes it is possible to obtain these products of further condensation as the main products of the reaction. At other times, they are side products.
When the Mannich base contains an amino group β to a carbonyl (and it usually does), ammonia is easily eliminated. This is a route to α,β-unsaturated aldehydes, ketones, esters, and so on.
Studies of the reaction kinetics have led to the following proposals for the mechanism of the Mannich reaction.380 The base-catalyzed reaction:
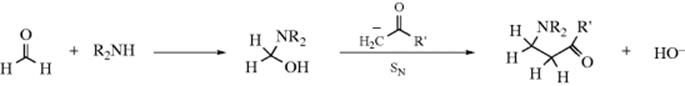
The acid-catalyzed reaction:
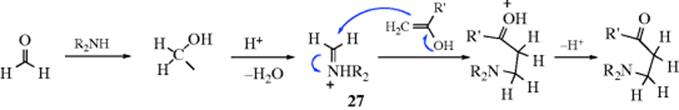
According to this mechanism, it is the free amine, not the salt that reacts, even in acid solution; and the active-hydrogen compound (in the acid-catalyzed process) reacts as the enol when that is possible. This latter step is similar to what happens in Reaction 12-4. There is kinetic evidence for the intermediacy of the iminium ion (27).381
When an unsymmetrical ketone is used as the active-hydrogen component, two products are possible. Regioselectivity has been obtained by treatment of the ketone with preformed iminium ions:382 the use of Me2N+=CH2CF3COO− in CF3COOH gives substitution at the more highly substituted position, while with (iPr)2N+=CH2 ClO4− the reaction takes place at the less highly substituted position.383 The preformed iminium compound dimethyl(methylene)ammonium iodide (CH2=N+Me2 I−), called an Eschenmoser's salt,384 has also been used in Mannich reactions.385 The analogous chloride salt has been condensed with an imine to give a β,β′-dimethylamino ketone after acid hydrolysis.386
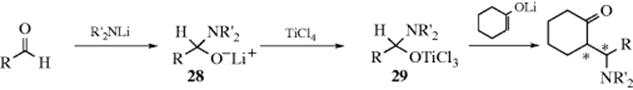
Another type of preformed reagent (29) has been used to carry out diastereoselective Mannich reactions. The lithium salts (28) are treated with TiCl4 to give 29, which is then treated with the enolate anion of a ketone.387 The Pd catalyzed Mannich reaction of enol ethers to imines is also known.388 The reaction of silyl enol ethers and imines389 is catalyzed by HBF4 in aq methanol.390 Similarly, silyl enol ethers react with aldehydes and aniline in the presence of InCl3 to give the β-amino ketone.391
Enantioselective Mannich reactions are known.392 Chiral catalysts are commonly used,393 including proline,394 proline derivatives, or proline analogues,395 a Pybox-La catalyst,396 chiral aminosulfonamides,397 or Cinchona alkaloids,398 and other chiral amines.399 Chiral Br![]() nsted acids are also used as catalysts,400 as well as chiral ammonium salts.401 Chiral diamine402 or phosphine-imine403 ligands have been used, and chiral dinuclear zinc compounds.404 Chiral auxiliaries on the carbonyl fragment can be used.405 Chiral imines, in the form of chiral hydrazones, have been used with silyl enol ethers and a Sc catalyst.406 Chiral amines react with aldehydes, with silyl enol ethers and an InCl3 catalyst in ionic liquids, to give the Mannich product with good enantioselectivity.407 A chiral thiourea catalyst has been used with a vinylogous Mannich reaction408 (see Sec. 6.B for vinylogy).
nsted acids are also used as catalysts,400 as well as chiral ammonium salts.401 Chiral diamine402 or phosphine-imine403 ligands have been used, and chiral dinuclear zinc compounds.404 Chiral auxiliaries on the carbonyl fragment can be used.405 Chiral imines, in the form of chiral hydrazones, have been used with silyl enol ethers and a Sc catalyst.406 Chiral amines react with aldehydes, with silyl enol ethers and an InCl3 catalyst in ionic liquids, to give the Mannich product with good enantioselectivity.407 A chiral thiourea catalyst has been used with a vinylogous Mannich reaction408 (see Sec. 6.B for vinylogy).
The reaction of nitroalkanes and amines, usually in the presence of a metal catalyst (e.g., CuBr), 409 has been called the nitro-Mannich reaction.410 An asymmetric nitro-Mannich reaction used a Cu–Sm catalyst,411 a Cu catalyst,412or a chiral thiourea catalyst.413
Also See, 11-22.
OS III, 305; IV, 281, 515, 816; VI, 474, 981, 987; VII, 34. See also, OS VIII, 358.
16-20 The Addition of Amines to Isocyanates
N-Hydro-C-alkylamino-addition
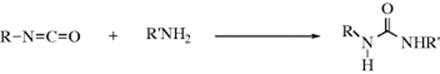
Ammonia and primary and secondary amines can be added to isocyanates414 to give substituted ureas.415 Isothiocyanates give thioureas.416 This is an excellent method for the preparation of ureas and thioureas. These compounds are often used as derivatives for primary and secondary amines. Isocyanic acid (HNCO) also gives the reaction; usually its salts (e.g., NaNCO) are used. Wöhler's famous synthesis of urea involved the addition of ammonia to a salt of this acid.417
OS II, 79; III, 76, 617, 735; IV, 49, 180, 213, 515, 700; V, 555, 801, 802, 967; VI, 936, 951; VIII, 26.
16-21 The Addition of Ammonia or Amines to Nitriles
N-Hydro-C-amino-addition
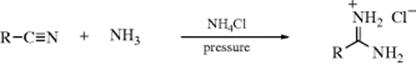
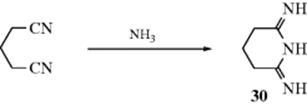
Unsubstituted amidines (in the form of their salts) can be prepared by addition of ammonia to nitriles.418 Many amidines have been made in this way. Dinitriles of suitable chain length can give imidines (30).419
Primary and secondary amines can be used instead of ammonia, to give substituted amidines, but only if the nitrile contains electron-withdrawing groups; for example, Cl3CCN gives the reaction. Ordinary nitriles do not react, and, in fact, acetonitrile is often used as a solvent in this reaction.420 Ordinary nitriles can be converted to amidines by treatment with an alkylchloroaluminum amide [MeAl(Cl)NR2; R = H or Me).421 The addition of ammonia to cyanamide (NH2CN) gives guanidine [(NH2)2C=NH]. Guanidines can also be formed from amines.422
If water is present, in the presence of a Ru423 or a Pt catalyst,424 the addition of a primary or secondary amine to a nitrile gives an amide: RCN + R1NHR2 + H2O → RCONR1R2 + NH3 (R2 may be H). When benzonitrile reacts with H2PO3Se− in aq methanol, a selenoamide [Ph(C=Se)NH2], is formed after treatment with aq potassium carbonate.425
OS I, 302 [but also see, OS V, 589]; IV, 245, 247, 515, 566, 769. See also, OS V, 39.
16-22 The Addition of Amines to Carbon Disulfide and Carbon Dioxide
S-Metallo-C-alkylamino-addition
![]()
Salts of dithiocarbamic acid can be prepared by the addition of primary or secondary amines to carbon disulfide.426 This reaction is similar to 16-10. Hydrogen sulfide can be eliminated from the product, directly or indirectly, to give isothiocyanates (RNCS). Isothiocyanates can be obtained directly by the reaction of primary amines and CS2 in pyridine in the presence of dicyclohexylcarbodiimide.427 A tosyl chloride mediated preparation of isothiocyanates is also known.428 Aniline derivatives react with CS2 and NaOH, and then ethyl chloroformate to give the aryl isothiocyanate.429 In the presence of diphenyl phosphite and pyridine, primary amines add to CO2 and to CS2 to give, respectively, symmetrically substituted ureas and thioureas:430 Isoselenoureas [R2NC(=NR1)SeR2] can also be formed.431
![]()
OS I, 447; III, 360, 394, 599, 763; V, 223.
E. Halogen Nucleophiles
16-23 The Formation of gem-Dihalides from Aldehydes and Ketones
Dihalo-de-oxo-bisubstitution
![]()
Aliphatic aldehydes and ketones can be converted to gem-dichlorides432 by treatment with PCl5. The reaction fails for perhalo ketones.433 If the aldehyde or ketone has an α hydrogen, elimination of HCl may follow and a vinylic chloride is a frequent side product, as shown,434 or even the main product.435 Phosphorus pentabromide (PBr5) does not give good yields of gem-dibromides,436 but these can be obtained from aldehydes, by the use of Br2, and triphenyl phosphite.437gem-Dichlorides can be prepared by reacting an aldehyde with BiCl3.438

The mechanism of gem-dichloride formation involves initial attack on PCl4+, which is present in solid PCl5, by the carbonyl oxygen, followed by addition of Cl− to the carbon:439
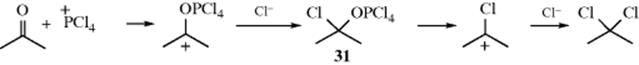
This chloride ion may come from PCl6−, which is also present in solid PCl5. There follows a two-step SN1 process. Alternatively, 31 can be converted to the product without going through the chlorocarbenium ion, by an SNi process.
This reaction has sometimes been performed on carboxylic esters, though these compounds very seldom undergo any addition to the C=O bond. An example is the conversion of F3CCOOPh to F3CCCl2OPh.440 However, formates commonly give the reaction.
Many aldehydes and ketones have been converted to gem-difluoro compounds with sulfur tetrafluoride (SF4),441 including quinones, which give 1,1,4,4-tetrafluorocyclohexadiene derivatives. With ketones, yields can be raised and the reaction temperature lowered, by the addition of anhydrous HF.442 Carboxylic acids, acyl chlorides, and amides react with SF2 to give 1,1,1-trifluorides. In these cases, the first product is the acyl fluoride, which then undergoes the gem-difluorination reaction:
![]()
The acyl fluoride can be isolated. Carboxylic esters also give trifluorides, but more vigorous conditions are required. In this case, the carbonyl group of the ester is attacked first, and RCF2OR′ can be isolated from RCO2R′443 and then converted to the trifluoride. Anhydrides can react in either manner. Both types of intermediate are isolable under the right conditions, and SF4 even converts CO2 to CF4. A disadvantage of reactions with SF4 is that they require a pressure vessel lined with stainless steel. Selenium tetrafluoride (SeF4) gives similar reactions, but atmospheric pressure and ordinary glassware can be used.444 Another reagent that is often used to convert aldehydes and ketones to gem-difluorides is the commercially available diethylaminosulfur trifluoride (DAST, Et2NSF3), and CF2Br2 in the presence of zinc.445 The mechanism with SF4 is probably similar in general nature, if not in specific detail, to that with PCl5. Some dithianes can be converted to gem-difluorides with a mixture of fluorine and iodine in acetonitrile.446 Oximes give gem-difluorides with NO+BF4− and pyridinium polyhydrogen fluoride.447
Treatment with hydrazine to give the hydrazone, and then CuBr2/t-BuOLi, generated the gem-dibromide.448 Oximes give gem-dichlorides upon treatment with chlorine and BF3·OEt2, and then HCl.449
In a related process, α-halo ethers can be prepared by treatment of aldehydes and ketones with an alcohol and HX. The reaction is applicable to aliphatic aldehydes and ketones and to primary and secondary alcohols. The addition of HX to an aldehyde or ketone gives α-halo alcohols, which are usually unstable, although exceptions are known, especially with perfluoro and perchloro species.450
OS II, 549; V, 365, 396, 1082; VI, 505, 845; VIII, 247. Also see, OS I, 506. For α-halo-ethers, see OS I, 377; IV, 101 (see, however, OS V, 218), 748; VI, 101.
F. Attack at Carbon by Organometallic Compounds451
16-24 The Addition of Grignard Reagents and Organolithium Reagents to Aldehydes and Ketones
O-Hydro-C-alkyl-addition
![]()
Organomagnesium compounds, commonly known as Grignard reagents (RMgX), are formed by the reaction of alkyl, vinyl, or aryl halides with magnesium metal, usually in ether solvents (e.g., diethyl ether or THF; Reaction 12-38). Halogen–Mg exchange can generate a Grignard reagent by reaction of aryl halides with reactive aliphatic Grignard reagents.452 Microwave irradiation has been used to facilitate the formation of Grignard reagents from aryl chlorides that are slow to react otherwise.453
The addition of Grignard reagents to aldehydes and ketones454 is known as the Grignard reaction.455 The initial product of reaction with a carbonyl is a magnesium alkoxide, requiring a hydrolysis step to generate the final alcohol product. Formaldehyde gives primary alcohols; other aldehydes give secondary alcohols; and ketones give tertiary alcohols. The reaction is of very broad scope. In many cases, the hydrolysis step is carried out with dilute HCl or H2SO4, but this cannot be done for tertiary alcohols in which at least one R group is alkyl because such alcohols are easily dehydrated under acidic conditions (Reaction 17-1). In such cases (and often for other alcohols as well), an aqueous solution of ammonium chloride is used instead of a strong acid. Grignard reagents have been used in solid-phase synthesis.456 Ionic liquids have been used for the Grignard reaction.457
Transition metal catalysts can promote 1,2-addition of Grignard reagents to ketones. A catalytic amount of Zn(II) compounds promote the reaction, for example.458 In the presence of a catalytic amount of InCl3, Grignard reagentsreact to give a mixture of 1,2- and 1,4-addition products with the 1,4-product predominating, but there was an increased 1,2-addition relative to the uncatalyzed reaction.459
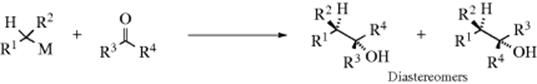
Diastereoselective addition460 has been carried out with achiral reagents and chiral substrates,461 similar to the reduction shown in Reaction 19-36.462 Because the attacking atom in this case is carbon, diastereoselective addition is possible with an achiral substrate and an optically active reagent.463 The use of suitable reactants creates, in the most general case, two new stereogenic centers, so the product can exist as two pairs of enantiomers, as shown. Even if the organometallic compound is racemic, it still may be possible to get a diastereoselective reaction; that is, one pair of enantiomers is formed in greater amount than the other.464
Asymmetric Grignard reactions are possible under certain circumstances.465 Chiral ligands with a chiral Cu catalyst466 or a chiral Ti complex467 give alcohols with good enantioselectivity. An interesting method formed using an alkylmagnesium halide, dibutylmagnesium (Bu2Mg) and a chiral diamine, and subsequent reaction with an aldehyde led to the alcohol derived from acyl addition of a butyl group with good enantioselectivity.468 N-Heterocyclic carbenes have been used as organocatalysts for asymmetric Grignard reactions.469 Aryl iodides undergo halogen–magnesium exchange when pretreated with PhMgCl, and subsequent reaction with an aldehyde gives the alcohol.470
The reaction of aldehydes or ketones with alkyl and aryl Grignard reagents was done in the earliest work without preliminary formation of RMgX, by mixing RX, the carbonyl compound, and magnesium metal in an ether solvent. This approach preceded Grignard's work, and is now known as the Barbier reaction.471 The organolithium analogue of this process is also known.472 Yields were generally satisfactory. Carboxylic ester, nitrile, and imide groups in the R are not affected by the reaction conditions.473 Modern versions of the Barbier reaction employ other metals and/or reaction conditions, and will be discussed in Reaction 16-25. However, Mg–Barbier reactions are catalyzed by other metal complexes (e.g., Cu compounds).474 Some transition metal compounds are stable in water, so some Grignard–Barbier reactions can be done in water.475 A retro-Barbier reaction has been reported in which a cyclic tertiary alcohol was treated to an excess of bromine and potassium carbonate to give 6-bromo-2-hexanone from 1-methylcyclopentanol.476
The reaction of RMgX or RLi with α,β-unsaturated aldehydes or ketones can proceed via 1,4-addition as well as normal 1,2-addition (see Michael addition in Reaction 15-25).477 In general, alkyllithium reagents give less 1,4 addition than the corresponding Grignard reagents.478 In a compound containing both an aldehyde and a ketone, it is possible to add RMgX chemoselectively to the aldehyde without significantly disturbing the carbonyl of the ketone group479 (see also, Reaction 16-24). Grignard reagents have been shown to add to some conjugated cyclic ketones with an α,β-OTf group via 1,2-addition, followed by cleavage to give an alkynyl ketone.480
In some cases, a Grignard reaction can be performed intramolecularly.481 For example, treatment of 5-bromo-2-pentanone with magnesium and a small amount of mercuric chloride in THF produced 1-methyl-1-cyclobutanol in 60% yield.482 Other four- and five-membered ring compounds were also prepared by this procedure. Similar closing of five- and six-membered rings was achieved by treatment of a δ- or ε-halocarbonyl compound, not with a metal, but with a dianion derived from nickel tetraphenyporphine483 (see Reaction 16-25).
![]()
The gem-disubstituted Mg compounds formed from CH2Br2 or CH2I2 (Reaction 12-38) react with aldehydes or ketones to give alkenes in moderate-to-good yields.484Wittig-type reactions also produce alkenes and are discussed in Reaction 16-44. The reaction could not be extended to other gem-dihalides. Similar reactions with gem-dimetallic compounds prepared with metals other than magnesium also have produced alkenes.485
Organolithium reagents (RLi), prepared from alkyl halides and Li metal or by exchange of an alkyl halide with a reactive organolithium (Reaction 12-38) react with aldehydes and ketones by acyl addition to give the alcohol,486after hydrolysis. Organolithium reagents are more basic than the corresponding Grignard reagent, which leads to problems of deprotonation in some cases. Organolithium reagents are generally more nucleophilic, however, and can add to hindered ketones with relative ease when compared to the analogous Grignard reagent.487 These reagents tend to form aggregates, which influences the reactivity and selectivity of the addition reaction.488 The addition of lithium amide–butyllithium mixed aggregates has been studied.489
Alkyl, vinyl,490 and aryl organolithium reagents can be prepared and undergo acyl addition. Structural variations are also possible, including enantioselective 1,2-addition.491 1-Bromo-1-lithioethene was prepared, and reacts with an aldehyde to give an allylic alcohol bearing a vinyl bromide unit.492 An interesting variation of the fundamental acyl addition reaction of organolithium reagents treated an aldehyde with an acyl-lithio amide [LiC(=O)N(Me)CH2Me] to give an α-hydroxy amide derivative.493
As with the reduction of aldehydes and ketones (Reaction 19-36), the addition of organometallic compounds to these substrates can be carried out enantioselectively and diastereoselectively.494 Chiral secondary alcohols have been obtained with high enantioselectivity by addition of Grignard and organolithium compounds to aromatic aldehydes, in the presence of optically active amino alcohols as ligands.495
An interesting variation is the reaction of methyllithium and CH2I2 with an aliphatic aldehyde to give an epoxide.496 A lithio-epoxide was formed by treating an epoxide with sec-butyllithium in the presence of sparteine,497 or with n-butyllithium/TMEDA,498 and subsequent reaction with an aldehyde led to an epoxy alcohol. Alkylidene oxetanes react with lithium, and then with an aldehyde to give a conjugated ketone.499 The reaction of gem-dihalides with a carbonyl compound and Li or BuLi give epoxides500 (see also, Reaction 16-46).
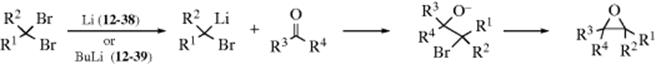
In other uses of gem-dihalo compounds, aldehydes and ketones add the CH2I group [R2CO → R2C(OH)CH2I] when treated with CH2I2 in the presence of SmI2,501 and the CHX2 group when treated with methylene halides and lithium dicyclohexylamide at low temperatures.502
![]()
It is possible to add an acyl group to a ketone to give (after hydrolysis) an α-hydroxy ketone.503 This can be done by adding RLi and CO to the ketone at −110 °C:504
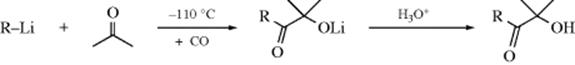
When the same reaction is carried out with carboxylic esters (R′COOR2), α-diketones (RCOCOR′) are obtained.503
Most aldehydes and ketones react with most Grignard reagents, but there are several potential side reactions505 that occur mostly with hindered ketones and with bulky Grignard reagents. The two most important of these are enolization and reduction. The former requires that the aldehyde or ketone have an α hydrogen, and the latter requires that the Grignard reagent have a β hydrogen:
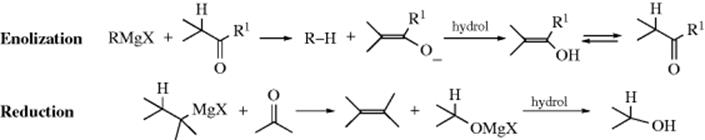
Enolization is an acid–base reaction (12-24) in which a proton is removed from the α carbon by the Grignard reagent, which is a strong base. The carbonyl compound is converted to its enolate anion, which, on hydrolysis, gives the original ketone or aldehyde. Enolization is important not only for hindered ketones but also for those that have a relatively high percentage of enol (e.g., β-keto esters).
The carbonyl compound can be reduced to an alcohol (Reaction 16-24) by the Grignard reagent, which itself undergoes elimination to give an alkene. The Grignard reagent must have a β-carbon that bears a hydrogen atom.
Two other side reactions are condensation (between enolate ion and excess ketone) and Wurtz-type coupling (10-64). Addition of Grignard reagents to ketones cannot be used to prepare highly hindered tertiary alcohols (e.g., triisopropylcarbinol, tri-tert-butylcarbinol, and diisopropylneopentylcarbinol) or they can be prepared only in extremely low yields, because reduction and/or enolization become prominent.506 However, these alcohols can be prepared by the use of alkyllithium reagents at –80 °C507 because enolization and reduction are much less important.508 Other methods of increasing the degree of addition at the expense of reduction include complexing the Grignard reagent with LiClO4 or Bu4N+ Br−,509 or using benzene or toluene instead of ether as solvent.510 Both reduction and enolization can be avoided by adding CeCl3 to the Grignard reagent.511
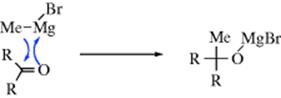
There has been controversy regarding the mechanism of addition of Grignard reagents to aldehydes and ketones.512 The reaction is difficult to study because of the variable nature of the species present in the Grignard solution (Sec. 5.B.ii) and because the presence of small amounts of impurities in the Mg seems to have a great effect on the kinetics of the reaction, making reproducible experiments difficult.513 There seem to be two basic mechanisms, depending on the reactants and the reaction conditions. In one of these, the R group is transferred to the carbonyl carbon with its electron pair. A detailed mechanism of this type has been proposed by Ashby et al.,514 based on the discovery that this reaction proceeds by two paths: one first order in MeMgBr and the other first order in Me2Mg.515 According to this proposal, both MeMgBr and Me2Mg add to the carbonyl carbon, though the exact nature of the step by which MeMgBr or Me2Mg reacts with the substrate is not certain. One possibility is a four-centered cyclic transition state:516
The other type of mechanism is a SET process517 with a ketyl intermediate:518
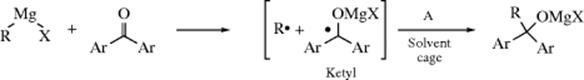
This mechanism, which has been mostly studied with diaryl ketones, is more likely for aromatic and other conjugated aldehydes and ketones than it is for strictly aliphatic ones. Among the evidence519 for the SET mechanism are ESR spectra520 and the fact that Ar2C(OH)C(OH)Ar2 side products are obtained (from dimerization of the ketyl; see pinacol coupling in Reaction 19-76).521 In the case of addition of RMgX to benzil (PhCOCOPh), ESR spectra of two different ketyl radicals were observed, both reported to be quite stable at room temperature.522 Note that a separate study failed to observe freely defusing radicals in the formation of Grignard reagents.523 Carbon isotope effect studies with Ph14COPh showed that the rate-determining step with most Grignard reagents is the carbon–carbon bond-forming step (marked A), although it is the initial electron-transfer step with allylmagnesium bromide.524 In the formation of Grignard reagents from bromocyclopropane, diffusing cyclopropyl radical intermediates were found.525 The concerted versus stepwise mechanism has been probed with chiral Grignard reagents.526
Note that there are similarities in reactivity for the SRN1 (see Sec. 13.A.iv) and Grignard mechanisms.527 Experimental evidence from this work suggests a linear rather than a chain mechanism.
Mechanisms for the addition of organolithium reagents have been investigated much less.528 Addition of a cryptand that binds Li+ inhibited the normal addition reaction, showing that the lithium is necessary for the reaction to take place.529
There is general agreement that the mechanism leading to reduction530 is usually as follows:
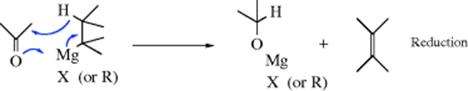
There is evidence that the mechanism leading to enolization is also cyclic, but involves prior coordination with magnesium:531
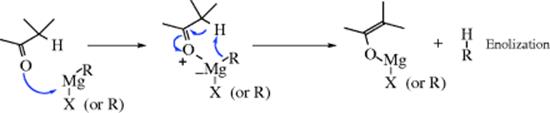
Aromatic aldehydes and ketones can be alkylated and reduced in one reaction vessel by treatment with an alkyl- or aryllithium, followed by lithium and ammonia and then by ammonium chloride.532
![]()
A similar reaction has been carried out with N,N-disubstituted amides: RCONR2' → RR2CHNR2'.533
OS I, 188; II, 406, 606; III, 200, 696, 729, 757; IV, 771, 792; V, 46, 452, 608, 1058; VI, 478, 537, 542, 606, 737, 991, 1033; VII, 177, 271, 447; VIII, 179, 226, 315, 343, 386, 495, 507, 556; IX, 9, 103, 139, 234, 306, 391, 472; 75, 12; 76, 214; X, 200.
16-25 Addition of Other Organometallics to Aldehydes and Ketones
O -Hydro-C-alkyl-addition
![]()
A variety of organometallic reagents other than RMgX and RLi add to aldehydes and ketones. A simple example is formation of Na, or K alkyne anions (e.g., RC![]() C-M, Reaction 16-38), which undergo acyl addition to ketones or aldehydes to give the propargylic alcohol.534 In the reaction with terminal acetylenes,535 sodium acetylides are the most common reagents (when they are used, While Na is the metal of choice for the addition of acetylenic groups, vinylic alanes (prepared as in Reaction 15-17) are the reagents of choice for the addition of vinylic groups.536 The reagent Me3Al/−C
C-M, Reaction 16-38), which undergo acyl addition to ketones or aldehydes to give the propargylic alcohol.534 In the reaction with terminal acetylenes,535 sodium acetylides are the most common reagents (when they are used, While Na is the metal of choice for the addition of acetylenic groups, vinylic alanes (prepared as in Reaction 15-17) are the reagents of choice for the addition of vinylic groups.536 The reagent Me3Al/−C![]() CH Na+ also adds to aldehydes to give the ethynyl alcohol.537 A solvent-free reaction was reported that mixed a ketone, a terminal alkyne and potassium tert-butoxide.538 The reaction is often called the Nef reaction, but Li,539 Mg, and other metallic acetylides have also been used. A particularly convenient reagent is the lithium acetylide–ethylenediamine complex,540 a stable, free-flowing powder that is commercially available. Alternatively, the substrate may be treated with the alkyne itself in the presence of a base, so that the acetylide is generated in situ. This procedure is called the Favorskii reaction, not to be confused with the Favorskii rearrangement (18-7).541 Zinc(II) chloride facilitates the addition of a terminal alkyne to an aldehyde to give a propargylic alcohol.542Zinc(II) triflate can also be used for alkyne addition to aldehydes,543 and in the presence of a chiral ligand leads to good enantioselectivity in the propargyl alcohol product.544
CH Na+ also adds to aldehydes to give the ethynyl alcohol.537 A solvent-free reaction was reported that mixed a ketone, a terminal alkyne and potassium tert-butoxide.538 The reaction is often called the Nef reaction, but Li,539 Mg, and other metallic acetylides have also been used. A particularly convenient reagent is the lithium acetylide–ethylenediamine complex,540 a stable, free-flowing powder that is commercially available. Alternatively, the substrate may be treated with the alkyne itself in the presence of a base, so that the acetylide is generated in situ. This procedure is called the Favorskii reaction, not to be confused with the Favorskii rearrangement (18-7).541 Zinc(II) chloride facilitates the addition of a terminal alkyne to an aldehyde to give a propargylic alcohol.542Zinc(II) triflate can also be used for alkyne addition to aldehydes,543 and in the presence of a chiral ligand leads to good enantioselectivity in the propargyl alcohol product.544
The reagents Et3Al, Et2Zn, and a terminal alkyne react with ketones, and in the presence of a cinchona alkaloid gives the alkynyl alcohol in moderate ee.545 Other enantioselective alkynylation reactions are known using various catalysts.546 Terminal alkynes add to aryl aldehydes in the presence of InBr3 and NEt3,547 SmI2,548 or Me2Zn.549 A Zn mediated reaction using iodoalkynes is known.550 Catalytically generated zinc acetylides add to aldehydes.551 An In catalyzed addition of alkynes to aldehydes used a catalytic amount of BINOL and gave the alkynyl alcohol with high enantioselectivity.552 Other enantioselective addition reactions of terminal alkynes are known.553 The reaction with In is compatible with the presence of a variety of other functional groups in the molecule, including phosphonate,554 propargylic sulfides.555
Many organometallic reagents have been reported for the addition of allylic groups,556 and there are enantioselective reactions.557 One of the most common methods is the Barbier reaction noted in Reaction 16-24, which includes metals and metal compounds other than Mg or Li, although the method is not limited to allylic compounds. Common reagents are allylic In compounds,558 which add to aldehydes or ketones in various solvents,559 Enantioselective In mediated Barbier reactions are known.560 Indium reacts with allylic bromides and ketones in water561 and in aqueous media. The reaction of a propargyl halide, In, and an aldehyde in aq THF leads to an allenic alcohol.562 When allyl iodide is mixed with In and TMSCl, reaction with a conjugated ketone proceed by 1,4-addition, but in the presence of 10% CuI, the major product is that of 1,2-addition.563 Allyl bromide reacts with Mn/TMSCl and an In catalyst in water to give the homoallylic alcohols from aldehydes.564 Indium metal is used for the acyl addition of allylic halides with a variety of aldehydes and ketones, including aliphatic aldehydes,565 aryl aldehydes,566 and α-keto esters.567 Elimination of the homoallylic alcohol to a conjugated diene can accompany the addition in some cases.568
Allyl bromide reacts with a ketone and Sm569 or SmI2,570 to give the homoallylic alcohol. Other metals can be used with allylic halides to give homoallylic alcohols from aldehydes or ketones,571, including Zn,572 La,573 or Mg-Cd574 and compounds of Ti,575 Mn,576 Fe,577 Ga,578 Ge,579 Zr,580 Nb,581 Cd,582 Sb,583 Te,584 Ba,585 Ce,586 Nd,587 Hg,588 Bi,589 In,590 and Pb.591 In addition, BiCl3/NaBH4,592 Mg–BiCl3,593 and CrCl2/NiCl2,594 have been used. Allylic alcohols have been converted to organometallic reagents with diethylzinc and a Pd595 or a Ru catalyst,596 leading to the homoallylic alcohol upon reaction with an aldehyde. Allylic acetates add to aldehydes in the presence of a Ru catalyst.597
Enantioselective reactions are known.598 A Cu catalyzed reaction is known that proceeds with good enantioselectivity.599 Allylzinc bromide adds to aldehydes under solvent-free conditions.600 A chiral Cr–Mn complex has been used with allylic bromides in conjunction with trimethylsilyl chloride.601 Reagents of the type R–Yb have been prepared from RMgX.602 The alkyl group of trialkyl aluminum compounds (e.g., AlEt3) add to aldehydes, enantioselectively in the presence of chiral transition metal complexes.603 Furthermore, organotitanium reagents can be made to add chemoselectively to aldehydes in the presence of ketones.604 Organomanganese compounds are also chemoselective in this way.605 In a related reaction, organocerium reagents, generated from cerium chloride (CeCl3 and a Grignard reagent or an organolithium reagent) gives an organometallic reagent that adds chemoselectively.606 Aryl halides that have a pendant ketone unit react with a Pd catalyst to give cyclization via acyl addition.607
As noted in Reaction 16-24, enolate formation and reduction complicate some Grignard reactions. One way to avoid complications is to add (RO)3TiCl, TiCl4,608 (RO)3ZrCl, or (R2N)3TiX to the Grignard or organolithium reagent. This produces organotitanium or organozirconium compounds that are much more selective than Grignard or organolithium reagents.609 An important advantage of these reagents is that they do not react with NO2 or CN functions that may be present in the substrate, as Grignard and organolithium reagents do. The reaction of a β-keto amide with TiCl4, for example, gives a complex that allows selective reaction of the ketone unit with MeMgCl–CeCl3 to give the corresponding alcohol.610 Premixing an allylic Grignard reagent with ScCl3 prior to reaction with the aldehyde gives direct acyl addition without allylic rearrangement as the major product, favoring the trans-alkene unit.611
Allyltin compounds readily add to aldehydes and ketones.612 Maleic acid promotes the reaction in aqueous media.613 Allylic bromides react with Sn to generate the organometallic in situ, which then adds to aldehydes.614 Allylic chlorides react with aldehydes in the presence of ditin compounds (e.g., Me3Sn–SnMe3 and a Pd catalyst).615 Allyltrialkyltin compounds616 and tetraallyltin react with aldehydes or ketones in the presence of BF3–etherate,617 or compounds of Cu,618 Ce,619 Bi,620 Pb,621 Ag622 Cd,623 Cr,624 Pd,625 Re,626 Gd,627 Ti,628 Rh,629 Zr,630 Co,631 or La.632 Propargylic tin compounds react with aldehydes to give the alcohol, with good antiselectivity.633 Tetraallyltin reacts via 1,2-addition to conjugated ketones in refluxing methanol.634 Tetraallyltin reacts with aldehydes in ionic liquids635 and on wet silica.636 In addition, allyltributyltin adds to aldehydes in ionic liquids with InCl3.637 Tetraallyltin adds to ketones or aldehydes to give homoallylic alcohols with good enantioselectivity in the presence of a chiral Ti complex638 or a chiral In complex.639 Allylic alcohols and homoallylic alcohols add to aldehydes in the presence of Sn(OTf)2640 In/InCl3,641 or with a Rh catalyst.642
The tin compound can be prepared in situ using an α-iodo ketone with an aldehyde and Bu2SnI2–LiI.643 A similar addition occurs with (allyl)2SnBr2 in water.644 Asymmetric induction has been reported.645 The use of a chiral Rh646 or Ti647 catalyst leads to enantioselective addition of allyltributyltin to aldehydes. Allyltributyltin reacts with aldehydes in the presence of SiCl4 and a chiral phosphoramide to give the homoallylic alcohol with moderate enantioselectivity.648 Tetravinyltin adds to ketones in the presence of an In catalyst.649 Allenyl tin compounds (CH2=C=CHSnBu3) also react with aldehydes in the presence of BF3·OEt2 to give a 2-dienyl alcohol.650 Selectfluor has been used to induce 1,2-addition of the allyl group of allyltributyltin to a conjugated aldehyde.651
Both aluminum and titanium reagents have been used. Aluminum catalysts, (e.g., [methylaluminum bis(4-bromo-2,6-di-tert-butylphenoxide)], MABR), facilitate addition of allyltributyltin to aldehydes.652 Triphenylaluminum reacts with aryl aldehydes in the presence of a Ti catalyst.653 Trimethylaluminum654 and dimethyltitanium dichloride655 exhaustively methylate ketones to give gem-dimethyl compounds656 (see also, Reaction 10-63):
![]()
The Ti reagent also dimethylates aromatic aldehydes.657 Triethylaluminum reacts with aldehydes, however, to give the mono-ethyl alcohol, and in the presence of a chiral additive the reaction proceeds with good asymmetric induction.658 A complex of Me3Ti·MeLi has been shown to be selective for 1,2-addition with conjugated ketones, in the presence of nonconjugated ketones.659
Chiral amides react with aldehydes in the presence of TiCl4 to give syn-selective addition products.660 Titanium-catalyzed enantioselective additions are also known.661 High %ee values have been obtained with organometallics,662including organotitanium compounds (methyl, aryl, allylic) in which an optically active ligand is coordinated to the Ti,663 allylic boron compounds, and organozinc compounds. Chiral dendritic Ti catalysts have been used to give moderate enantioselectivity.664
Organozinc compounds add to aldehydes and ketones. An example is the reaction of an alkylzinc chloride (RZnCl) to give the corresponding alcohol.665 Enantioselective reaction of a carbonyl with a dialkylzinc is possible when chiral catalysts are employed,666 or when chiral ligands667 are employed.668 A comparison of the stereoselectivity for reactions of diphenylzinc and diethylzinc has been reported.669 Dialkylzinc reagents, in the presence of a chiral Ti complex670 a Zn complex,671 an Al complex,672 a chiral Cr complex,673 a chiral Schiff base,674 or a chiral bis(sulfonamide675 and other chiral complexes676 react with aldehydes or ketones to give the corresponding alcohol with good enantioselectivity.677 High enantioselectivity was obtained from R2Zn reagents (R = alkyl)678 and aromatic679 aldehydes by the use of a small amount of various catalysts.680 The enantioselectivity is influenced by additives (e.g., LiCl).681 Silica-immobilized chiral ligands682 can be used in conjunction with dialkylzinc reagents, and polymer-supported ligands have been used.683
Propargylic acetate adds to aldehydes with good antiselectivity in the presence of Et2Zn and a Pd catalyst.684 With other organometallic compounds, active metals (e.g., alkylzinc reagents)685 are useful and compounds, such as alkylmercurys, do not react. Dimethylzinc and diethylzinc are probably the most common reagents. An intramolecular version is possible by reaction of an allene–aldehyde with dimethylzinc. Aryl halides react with Zn–Ni complexes to give acyl addition of the aryl group to an aldehyde.686 The reaction of an allylic halide and Zn687 or Zn/TMSCl688 leads to acyl addition of aldehydes.
Organochromium compounds add to aldehydes or ketones.689 The reaction of an organochromium reagent with an aldehyde or ketone is known as the Nozaki–Hiyama reaction. In the original version, a Cr(II) solution was prepared by reduction of chromic chloride by LiAlH4. This product was subsequently treated with an aldehyde and an allylic halide.690 The coupling of allylic halides and aldehydes or ketones in the presence of a Cr catalyst and a chiral ligand gives products with good enantioselectivity.691 Enantioselective coupling reactions catalyzed by Cr compounds are of increasing interest.692 The organochromium reagent may be derived from vinyl halides, triflates, or aryl derivatives.693
Other metals facilitate addition of groups to an aldehyde, including the coupling of an alkene to an aldehyde using a Ni catalyst.694 Vinyl bromides react with NiBr2/CrCl3/TMSCl to give a reagent that adds to aldehydes to give the allylic alcohol.695 Vinyl complexes generated from alkynes and SmI2 add intramolecularly, and eight-membered rings have been formed in this way.696 Vinyl reagents are formed in situ via organozirconium compounds with Me2Zn, Ti compounds, and terminal alkynes.697
Lithium dimethylcopper (Me2CuLi) reacts with aldehydes698 and with certain ketones699 to give the expected alcohols. The RCu(CN)ZnI reagents also react with aldehydes, in the presence of BF3–etherate, to give secondary alcohols. Vinyltellurium compounds react with BF3·OEt2 and cyanocuprates [R(2-thienyl)CuCNLi2] to give a reagent that adds 1,2- to the carbonyl of a conjugated ketone.700 Vinyl tellurium compounds also react with n-butyllithium to give a reagent that adds to nonconjugated ketones.701 In conjunction with BeCl2, organolithium reagents add to conjugated ketones. In THF, 1,4- addition is observed, but in diethyl ether the 1,2-addition product is formed.702
Alkenes and alkynes add to aldehydes or ketones via the π bond by conversion to a reactive organometallic. A radical-type addition is possible using alkenes with BEt3. Alkynes add to aldehydes elsewhere in the same molecule in the presence of BEt3 and a Ni catalyst to give a cyclic allylic alcohol.703 Alkene aldehydes react similarly using Me3SiOTf.704 In a similar manner, dienes705 or alkynes706 add to aldehydes in the presence of a Ni catalyst. Allenes add to aldehydes in the presence of a Ni catalyst, using a chiral imidazolinyl carbene ligand, and the product is trapped as the triethylsilyl ether by addition of Et3SiI.707 Ketenimines add to aldehydes, in the presence of SiCl4 and an achiral ligand, to give the β-cyanohydrin with good enantioselectivity.708 Allylic acetates react with ketones to give the homoallylic alcohol under electrochemical conditions that include bipyridyl, tetrabutylammonium tetrafluoroborate, and FeBr2.709 Terminal alkynes react with Zr complexes and Me2Zn to give an allylic tertiary alcohol.710 Internal alkynes also give allylic alcohols in the presence of BEt3 and a Ni catalyst.711 Reaction of an aldehyde containing a conjugated diene unit with diethylzinc and a Ni catalyst leads to cyclic alcohols having a pendant allylic unit.712 A similar reaction was reported using a Cu catalyst.713 The intramolecular addition of an alkene to an aldehyde leads to a saturated cyclic alcohol using PhSiH3 and a Co catalyst.714
Aryl halides react with a Ni complex under electrolytic conditions to add the aryl group to aldehydes.715 The C position of an indole adds to aldehydes in the presence of a Pd catalyst.716 The addition of trifluoromethyl to an aldehyde was accomplished photochemically using CF3I and (Me2N)2C=C(NMe2)2.717 α-Iodo phosphonate esters react with aldehydes and SmI2 to give a β-hydroxy phosphonate ester.718 Addition to the allene in the presence of a Ni catalyst719 or a CeCl3 catalyst720 is followed by addition of the intermediate organometallic to the aldehyde to give the cyclic product.
Intramolecular addition of a conjugated ester (via the β-carbon) to an aldehyde generates a cyclic ketone.721 This type of coupling has been called the Stetter reaction,722 which actually involves the addition of aldehydes to activated double bonds (Reaction 15-34), mediated by a catalytic amount of thiazolium salt in the presence of a weak base. The intramolecular addition of the allene moiety to an aldehyde is catalyzed by a Pd complex in the presence of Me3SiSnBu3.723 A highly enantio- and diastereoselective intramolecular Stetter reaction has been developed.724 Alkynyl aldehydes react with silanes (e.g., Et3SiH) and a nickel catalyst to give a cyclic compound having a silyl ether and an exocyclic vinylidene unit.725 Alkene-aldehydes give cyclic alcohols via intramolecular addition of the C=C unit to the carbonyl under electrolytic conditions using a phase-transfer catalyst.726 A similar cyclization was reported using SnCl4.727 Vinylidene cycloalkanes react with aldehydes in the presence of a Pd catalyst to give a homoallylic alcohol where addition occurs at the carbon exocyclic to the ring.728 Allenes react with benzaldehyde using HCl–SnCl2 with a Pd catalyst.729 Silyl allenes react with aldehydes in the presence of a chiral Sc catalyst to give homopropargylic alcohols with good enantioselectivity.730 Intramolecular addition of an allene to aldehyde via addition of phenyl when treated with PhI and a Pd catalyst.731 Allenes add to ketones to give homoallylic alcohols in the presence of SmI2 and HMPA.732 Alkenes having an allylic methyl group in the presence of BF3·OEt2 add to formaldehyde to give a homoallylic alcohol.733 Conjugated dienes react with aldehyde via acyl addition of a terminal carbon of the diene, in the presence of Ni(acac)2 and Et2Zn.734
Although organoboranes do not generally add to aldehydes and ketones,735 allylic boranes are exceptions.736 When they add, an allylic rearrangement always takes place. Allylic rearrangements may take place with the other reagents as well. The use of a chiral catalyst leads to asymmetric induction737 and chiral allylic boranes have been prepared.738 Addition of trialkylboranes to aldehydes is catalyzed by a Ni complex.739 Note that chloroboranes (R2BCl) react with aldehydes via acyl addition of the alkyl group, giving the corresponding alcohol after treatment with water.740 Treatment with catecholborane gives addition to the conjugated ketone, and subsequent cyclization of the resulting organometallic at the nonconjugated ketone gives a cyclic alcohol with a pendant ketone unit, after treatment with methanol.741 In the presence of Ru742 Cu,743 Ni,744 or Pd745 compounds, RB(OH)2 and arylboronic acids [ArB(OH)2, see Reaction 12-28] add to aldehydes to give the corresponding alcohol. Arylboronic acids add to aldehydes in the presence of a chiral ligand to give an alcohol with good enantioselectivity.746 An enantioselective intramolecular reaction of an arylboronic acid to a pendant ketone moiety used a Pd catalyst.747 Polymer-bound aryl borates add an aryl group to aldehydes in the presence of a Rh catalyst.748 An intramolecular version of the phenylboronic acid induced reaction is known, where a molecule with ketone and conjugated ketone units is converted to a cyclic alcohol using a chiral Rh catalyst.749 Allylic boronates add to aldehydes,750 and there are enantioselective reactions.751
A number of optically active allylic boron compounds have been used, including752 B-allylbis(2-isocaranyl)borane (32),753 (E)- and (Z)-crotyl-(R,R)-2,5-dimethylborolanes (33),754 and the borneol derivative 34,755 all of which add an allyl group to aldehydes, with good enantioselectivity. A recyclable 10-TMS-9-borabicyclo[3.3.2]decane has been used for asymmetric allyl and crotyl addition to aldehydes.756 Where the substrate possesses an aryl group or a triple bond, using a metal carbonyl complex of the substrate enhances enantioselectivity.757
Potassium alkynyltrifluoroborates (Reaction 12-28) react with aldehydes and a secondary amine, in an ionic liquid, to give a propargylic amine.758 Allylic trifluoroborates (Reaction 12-28) react with aldehydes to give the homoallylic alcohol. The Pd catalyzed reaction of aryl aldehydes with PhBF3K gave a diaryl alcohol.759 A Ru catalyzed reaction of aryltrifluoroborates led to sterically hindered diaryl ketones.760 Aliphatic aldehydes react with this reagent, in the presence of BF3·OEt2, to give the homoallylic alcohol with allylic rearrangement and a preference for the syn diastereomer.761 Aryl aldehydes react as well.762
16-26 Addition of Trialkylallylsilanes to Aldehydes and Ketones
O-Hydro-C-alkyl-addition
Allylic trialkyl, trialkoxy, and trihalosilanes add to aldehydes to give the homoallylic alcohols in the presence of a Lewis acid763 (including TaCl5764 and YbCl3765), fluoride ion,766 proazaphosphatranes,767 or a catalytic amount of iodine.768 A Ru catalyst has been used in conjunction with an arylsilane and an aldehyde.769 Allyl(trimethoxy)silane adds an allyl group to aldehydes using a CdF2770 catalyst or a chiral AgF complex.771 Allyltrichlorosilanes have also been used in addition reactions with aldehydes.772 Hünig's base (iPr2NEt) and a sulfoxide have also been used to facilitate the addition of an allyl group to an aldehyde from allyltrichlorosilane.773 The mechanism of this reaction has been examined.774
Allyltrichlorosilane reacts with aldehydes in the presence of certain additives to give the corresponding alcohol,775 and the reaction proceeds with good enantioselectivity by addition of a chiral additive.776 Other chiral additives have been used,777 as well as chiral catalysts,778 and chiral complexes of allyl silanes.779 Trimethoxyallylsilanes react with aldehydes in the presence of a Cu catalyst and a chiral ligand to give the chiral alcohol.780 Chiral allylic silyl derivatives add to aldehydes to give the chiral homoallylic alcohol.781
Allylic silanes react with gem-diacetates in the presence of InCl3 to give a homoallylic acetate782 or with dimethyl acetals and TMSOTf in an ionic liquid to give the homoallylic methyl ether.783 Allylic alcohols can be treated with TMS–Cl and NaI, and then Bi to give an organometallic reagent that adds to aldehydes.784
16-27 Addition of Conjugated Alkenes to Aldehydes (the Baylis–Hillman Reaction)785
O-Hydro-C-alkenyl-addition
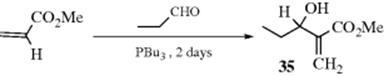
In the presence of a base786 (1,4-diazabicyclo[2.2.2]octane, DABCO) or trialkylphosphines, conjugated carbonyl compounds (ketones, esters, including lactones)787 thioesters,788 and amides789) add to aldehydes via the α carbon to give α-alkenyl-β-hydroxy esters or amides. This sequence is called the Baylis–Hillman reaction (or Morita–Baylis–Hillman),790 and a simple example is the formation of 35.791 Mechanistic investigations of the reaction have been reported.792 Methyl vinyl ketone gave other products in the Baylis–Hillman reaction, whereas conjugated esters do not.793 Methods that are catalytic in base have been developed for the Baylis–Hillman reaction.794 Both microwave irradiation795 and ultrasound796 have been used to induce the reaction.797 Baylis–Hillman reactions have been done in aqueous acidic media.798 There is a protein-catalyzed reaction.799 Under certain conditions, rate enhancements have been observed.800 The reaction has been done in ionic liquids801 and PEG802 or sulfolane,803 and without an organic solvent.804 Rate acceleration occurs with bis(arylthio)ureas in a DABCO promoted reaction.805 Transition metal compounds can facilitate the Baylis–Hillman reaction,806 and BF3·OEt2 has been used.807 A sila variation is known, involving the reaction of vinylsilanes and aldehydes.808 An aza-Balyis–Hillman reaction is discussed in Reaction 16-31.
An intramolecular version of the Baylis–Hillman reaction generated cyclopentenone derivatives from alkyne-aldehydes and a Rh catalyst.809 Other intramolecular cyclization reactions are known.810 Cyclization of a conjugated ester using DABCO, where the “alcohol” group contained an aldehyde unit (an α-hydroxy aldehyde derivative) gave a lactone with a hydroxy unit at C-3 relative to the carbonyl and an α-vinylidene.811
Enantioselective Baylis–Hillman reactions812 are possible using a chiral auxiliary via an amide813 or ester.814 Organocatalysts can be used to give products with good enantioselectivity.815 Sugars have been used as ester auxiliaries, and in reaction with aryl aldehydes and 20% DABCO gave the allylic alcohol with modest enantioselectivity.816
Another variation is the Rauhut–Currier cyclization reaction,817 which involves the reaction of a conjugated carbonyl with the allylic site of a second conjugated system. Intramolecular variations of this latter method are known.818 With the boron trifluoride induced reaction between an aldehyde and a conjugated ketone, a saturated β-hydroxy ketone was formed with good antiselectivity.819 The coupling of aldehydes with conjugated ketones used TiCl4,820 dialkylaluminum halides,821 and with (polymethyl)hydrosiloxane and a Cu catalyst.822 Conjugated esters were coupled to aldehydes with DABCO and a La catalyst.823 Aldehydes are coupled to conjugated esters with a chiral quinuclidine catalyst and a Ti catalyst, as well as in the presence of tosylamine, the final product was the allylic N-tosylamine formed with modest enantioselectivity.824
Alkyl halides are coupled with conjugated carbonyls to give the alkylated derivative in what is known as Morita–Baylis–Hillman alkylation.825 α-Bromomethyl esters react with conjugated ketones and DABCO to give a coupling product, (34).826 A similar DBU induced reaction was reported using α-bromomethyl esters and conjugated nitro compounds.827

The reaction can be modified to give additional products, as with the reaction of o-hydroxybenzaldehyde and methyl vinyl ketone with DABCO, where the initial Baylis–Hillman product cyclized via conjugate addition of the phenolic oxygen to the conjugated ketone (Reaction 15-31).828 Aldehydes and conjugated esters can be coupled with a sulfonamide to give an allylic amine.829
See OS 2010, 87, 201
16-28 The Reformatsky Reaction
O-Hydro-C-α-ethoxycarbonylalkyl-addition
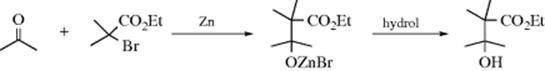
The Reformatsky reaction830 is very similar to Reaction 16-24. An aldehyde or ketone is treated with Zn and a halide; the halide is usually an α-halo ester or a vinylog (see Sec. 6.B) of an α-halo ester (e.g., RCHBrCH=CHCOOEt), although α-halo nitriles,831 α-halo ketones,832 and α-halo N,N-disubstituted amides have also been used. Especially high reactivity can be achieved with activated Zn,833 and with Zn and ultrasound.834 The reaction is catalytic in Zn in the presence of iodine and ultrasound.835 Metals other than Zn can be used, including In,836 Mn,837 low valent Ti,838 metal compounds of Ti,839 Sn,840 Sm841 or Sc.842 The use of additives (e.g., Ge843 or Me2Zn)844 can lead to highly selective reactions.845 A combination of Zn and an α-bromo ester can be used in conjunction with BF3·OEt2, followed by reaction with dibenzoyl peroxide.846 The aldehyde or ketone can be aliphatic, aromatic, or heterocyclic, or contain various functional groups. Solvents used are generally ethers, including Et2O, THF, and 1,4-dioxane, although the reaction can be done in water847 using dibenzoyl peroxide and MgClO4.
With the use of chiral auxiliaries848 or chiral catalysts,849 good enantioselectivity850 can be achieved.
Dialkylzinc compounds are an alternative source of Zn in the Reformatsky reaction. The reaction of an α-bromo ester, an aldehyde, and diethylzinc in THF with a Rh catalyst, gave a β-hydroxy ester.851
Formally, the reaction is somewhat analogous to the Grignard reaction (16-24), with EtO2C–C–ZnBr (37) as an intermediate analogous to RMgX.852 There is an intermediate derived from Zn and the ester, the structure of which has been shown to be 38, by X-ray crystallography of the solid intermediate prepared from t-BuOCOCH2Br and Zn.853 As can be seen, it has some of the characteristics of 37.
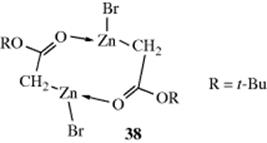
After hydrolysis, the alcohol is the usual product, but sometimes (especially with aryl aldehydes) elimination follows directly and the product is an alkene. By the use of Bu3P along with Zn, the alkene can be made the main product,854 making this an alternative to the Wittig reaction (16-44). The alkene is also the product when K2CO3/NaHCO3 is used with 2% PEG–telluride.855 Since Grignard reagents cannot be formed from α-halo esters, the method is quite useful, but competing reactions sometimes lead to low yields. A similar reaction (called the Blaise reaction) has been carried out on nitriles856:
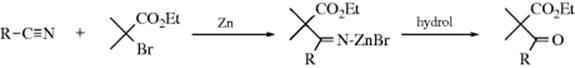
Carboxylic esters have also been used as substrates, but then, as might be expected (Sec. 16.A), the result is substitution and not addition:
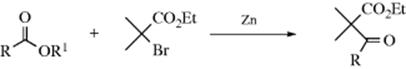
The product in this case is the same as with the corresponding nitrile, though the pathways are different. The reaction is compatible with amine substituents, and α-(N,N-dibenzyl)amino aldehydes have been used to prepare β-hydroxy-γ-(N,N-dibenzylamino) esters with good antiselectivity.857
For an alternative approach involving ester enolates, see Reaction 16-36.
OS 3, 408; 4, 120, 444; 6, 598; IX, 275.
16-29 The Conversion of Carboxylic Acid Salts to Ketones with Organometallic Compounds
Alkyl-de-oxido-substitution
![]()
Good yields of ketones can often be obtained by treatment of the lithium salt of a carboxylic acid with an alkyllithium reagent, followed by hydrolysis.858 The carboxylate salt is formed by reaction of the carboxylic acid with 1 molar equivalent of R1Li. The R′ group may be aryl or primary, secondary, or tertiary alkyl and R may be alkyl or aryl. The compounds MeLi and PhLi have been employed most often. Tertiary alcohols are side products. Lithium acetate can be used, but generally gives low yields. Using R(PrNH)Mg, carboxylic acid salts react to form ketones.859
A variation of this transformation reacts the acid with lithium naphthalenide in the presence of 1-chlorobutane. The product is the ketone.860 A related reaction treats the lithium carboxylate with lithium metal and the alkyl halide, with sonication, to give the ketone.861 Phenylboronic acid (Reaction 12-28) reacts with aryl carboxylic acids in the presence of a Pd catalyst and disuccinoyl carbonate to give a diaryl ketone.862
OS V, 775.
16-30 The Addition of Organometallic Compounds to CO2 and CS2
C-Alkyl-O-halomagnesio-addition
![]()
Grignard reagents add to one C=O bond of CO2 exactly as they do to an aldehyde or a ketone,863 but the product is the salt of a carboxylic acid. The reaction is usually performed by adding the Grignard reagent to dry ice. Many carboxylic acids have been prepared in this manner, and this constitutes an important way of increasing a carbon chain by one unit. Since labeled CO2 is commercially available, this is a good method for the preparation of carboxylic acids labeled in the carboxyl group. Other organometallic compounds have also been used (RLi,864 RNa, RCaX, RBa,865 etc.), but much less often. The formation of the salt of a carboxylic acid after the addition of CO2 to a reaction mixture is regarded as a positive test for the presence of a carbanion or of a reactive organometallic intermediate in that reaction mixture (see also, Reaction 16-42).
Aryl organoboronates react with CO2, in the presence of a Ru catalyst, to give the corresponding aryl carboxylic acid.866 Aryl and alkenyl boronic esters are carboxylated in the presence of a Cu catalyst.867 Vinyl halides react with CO and water, with a Pd catalyst in an ionic liquid, to give the conjugated carboxylic acid.868 Organozinc compounds are carboxylated with CO2 and a Ni catalyst.869 Metal-free carboxylation of organozinc reagents is also known.870In the presence of CO2 and an organocatalyst, aromatic aldehydes are converted to the corresponding carboxylic acid.871 Direct carboxylation of aryl bromides is possible using CO2 and a Pd catalyst.872 Allenes are converted to β,γ-unsaturated acids with CO2 in the presence of Et2Zn.873
When chiral additives (e.g., (−)-sparteine) have added to the initial reaction with the organolithium reagent, quenching with CO2 produces carboxylic acids with good asymmetric induction.874
In a closely related reaction, Grignard reagents add to CS2 to give salts of dithiocarboxylic acids.875 These salts can be trapped with amines to form thioamides.876 Two other reactions are worthy of note. (1) Lithium dialkylcopper reagents react with dithiocarboxylic esters to give tertiary thiols.877 (2) Thiono lactones can be converted to cyclic ethers,878 for example:
This is a valuable procedure because medium and large ring ethers are not easily made, while the corresponding thiono lactones can be prepared from the readily available lactones (see, e.g., Reaction 16-63) by reaction 16-11.
A terminal alkyne can be converted to the anion under electrolytic conditions, in the presence of CO2, to give propargylic acids (R–C![]() C–CO2H).879
C–CO2H).879
OS I, 361, 524; II, 425; III, 413, 553, 555; V, 890, 1043; VI, 845; IX, 317.
16-31 The Addition of Organometallic Compounds to C=N Compounds
N-Hydro-C-alkyl-addition
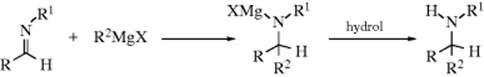
Aldimines can be converted to secondary amines by treatment with Grignard reagents.880 Ketimines generally react with Grignard reagents to give reduction instead of addition. However, organolithium compounds give the normal addition product with both aldimines and ketimines.881 The solvent and the aggregation state of the organolithium play a role in the addition, however.882 For the addition of an organometallic compound to an imine to give a primary amine, R′ in RCH=NR′ would have to be H, and such compounds are seldom stable. However, the conversion has been done for R = aryl, by the use of the masked reagents (ArCH=N)2SO2 [prepared from an aldehyde, RCHO, and sulfamide, (NH2)2SO2]. Addition of R2MgX or R2Li to these compounds gives ArCHR2NH2 after hydrolysis.883 An intramolecular version of the addition of organolithium reagents is known that gave 2-phenylpyrrolidine.884 Grignard reagents add to imines in the presence of various transition metal catalysts, including Sc(OTf)3885 or Cp2ZrCl2.886 Alkynes add to imines yielding propargylic amines.887
When chiral additives are used in conjunction with the organolithium reagent, chiral amines are produced888 with good asymmetric induction.889 Chiral auxiliaries have been used in addition reactions of organometallic compounds to imines,890 and to oxime derivatives.891 Transition metal asymmetric alkylation of imines uses metal compounds of Cu.892 Chiral catalysts lead to enantioselective addition of alkynes to imines to give the amine.893 Chiral N-sulfinylimines reaction with lithium silanes give the α-silylsulfinylamine.894
Zinc metal reacts with allylic bromides to form an allylic zinc complex, which reacts with imines to give the homoallylic amine.895 This reaction is catalyzed by TMSCl.896 Dialkylzinc reagents add to functionalized imines to give the functionalized amine, often with transition metal catalysts (e.g., Ni compounds).897 Dialkylzinc reagents add to N-tosyl imines to give the alkylated tosylamine.898 In the presence of a chiral ligand, the metal-catalyzed reaction proceeds with good enantioselectivity.899 With a Cu catalyst and a chiral ligand the product is formed with good enantioselectivity.900 The reaction of imines (e.g., ArN=CHCO2Et), where R = a chiral benzylic substituent and ZnBr2, followed by R′ZnBr leads to a chiral α-amino ester.901 Terminal alkynes add to imines using ZnCl2 and TMSCl, and with a chiral ligand attached to nitrogen the reaction proceeds with some enantioselectivity.902 Dimethylzinc has been used to mediate the addition of terminal alkynes to N-tosylimines.903
Other organometallic compounds add to aldimines,904 including Sn,905 Sm,906 Ge,907 Zr,908 Ga metal with ultrasound,909 Yb with Me3SiCl,910and In.911 Catalytic amounts of the metal compound can be used with an allylic stannane.912 Catalytic enantioselective addition reactions with these organometallics are well known,913 including reactions in an ionic liquid.914 Aryltrialkylstannanes add the aryl group to N-tosyl imines using a Rh catalyst and sonication.915 Allylic halides react with imines in the presence of In metal916 or InCl3917 to give the homoallylic amine, and with N-sulfonyl imines to give the homoallylic sulfonamide.918 In this latter reaction, antiselectivity was observed when the reaction was done in water, and syn selectivity when done in aq THF.919 Aryl iodides add to N-aryl imines in the presence of a Rh catalyst.920 Arylation of N-tosyl ketimines proceeds with good enantioselectivity using a Rh catalyst.921 Propargylic halides add to imines in the presence of In metal, in aq THF.922
Terminal alkynes react with aryl aldehydes and aryl amines to give propargylic amine without a catalyst.923 Alternatively, with an Ir924 or a Cu catalyst925 they also lead to a propargylic amine.926 Terminal alkynes add to N-substituted imines to give a propargylic amine with good enantioselectivity using a chiral Cu complex.927 Lithium alkyne anions add to chiral N-sulfinylimines, in the presence of Me3Al, to give the chiral propargyl compound.928Diynes add to N-sulfinyl imines in the presence of a chiral Rh catalyst to give the corresponding allylic sulfinylamine.929 Alkenes add to N-sulfonyl imines in the presence of a chiral Rh catalyst to give the alkylated sulfonamide.930
Triethylaluminum adds an ethyl group to an imine in the presence of a Eu catalyst. Reaction with PhSnMe3 and N-tosylimines with a Rh catalyst leads to addition of a phenyl group to the carbon of the C=N bond.931 Other N-sulfonyl imines react similarly to give the corresponding sulfonamide, and in the presence of a chiral ligand the reaction proceeds to good enantioselectivity.932N-Tosyl imines also react with dialkylzinc reagents, giving the sulfonamide with modest enantioselectivity.933N-Sulfinyl imines [R2CH=NS(=O)R′]934 react with Grignard reagents at carbon to give the corresponding N-sulfinylamine.935 Furan derivatives add via C-2 with good enantioselectivity using a chiral phosphoric acid catalyst.936 Alkenes add to N-tosyl imines with a Yb catalyst937 and allenes add to N-carbamoyl imines in the presence of a V catalyst.938N-Carbamoyl imines, formed in situ, react with allylic silanes in the presence of an iodine catalyst.939N-Carbamoyl imines add acetonitrile (via carbon) using DBU and a Ru catalyst.940
Arylboronates (Reaction 12-28) add to N-sulfonyl imines in the presence of a Rh catalyst to give the corresponding sulfonamide.941 Chiral Cu complexes have also been used for effective allylation of ketimines.942 Aryl boronic acids (Reaction 12-28) add the aryl group to N-tosyl imines using a Rh943 or Pd944 catalyst. Arylboronic acids react similarly, and in the presence of a chiral Rh945 or Ir946 catalyst give a chiral sulfinamide or sulfonamide. Allylic boronates also add to aldehydes, and subsequent treatment with ammonia gives the homoallylic amine.947 Vinyl boronates add to nitrones in the presence of Me2Zn, transferring the vinyl group to the C=N unit.948 Potassium allyltrifluoroborates react with N-tosyllimines in the presence of a Pd catalyst.949
Allylic silanes (e.g., allyltrimethylsilane) add to N-substituted imines in the presence of a Pd catalyst to give the homoallylic amine.950 Similar results are obtained when the allylic silane and imine are treated with a catalytic amount of tetrabutylammonium fluoride.951 Allylic trichlorosilanes add to hydrazones to give homoallylic hydrazine derivatives with excellent antiselectivity952 and with good enantioselectivity using a chiral ligand.953 Chiral allylic silane derivatives have been developed, and add to hydrazones with good enantioselectivity.954
There is an aza-Balyis–Hillman reaction that converts imines and conjugated carbonyl derivatives to the α-amino conjugated derivative.955N-Tosyl imines can be used in place of aldehydes, and the reaction of the imine, a conjugated ester and DABCO gave the allylic N-tosylimine.956 A “double Baylis–Hillman” reaction has also been reported using N-tosylimines and conjugated ketones.957 The use of chiral catalysts leads enantioselective product formation.958 An enantioselective aza-Baylis–Hillman reaction959 was reported using as chiral reaction medium.960 Aldehydes add via the α carbon using proline, to give β-amino aldehydes with good selectivity to give chiral β-amino aldehydes.961
Silyl enol ethers add to hydrazones in the presence of ZnF2 and a chiral ligand to give chiral β-hydrazino ketones.962 Similar addition to imine derivatives was accomplished using ketene silyl acetals and Amberlyst-15.963Alternatively, an imine is reacted first with Zn(OTf)2 and then with a ketene silyl acetal.964
Nitro compounds add to N-carbamoyl imines with a chiral diamine catalyst with some enantioselectivity.965 Nitro compounds add via carbon using a Cu catalyst, and with good enantioselectivity when a chiral ligand is used.966The conjugate bases of nitro compounds (formed by treatment of the nitro compound with BuLi) react with Grignard reagents in the presence of ClCH=NMe2+ Cl− to give oximes: RCH=N(O)OLi + R′MgX → RR′C=NOH.967
![]()
Iminium salts968 give tertiary amines directly, via 1,2-addition to the C=N unit. Chloroiminium salts [ClCH=NR2'Cl−, generated in situ from an amide (HCONR2') and phosgene (COCl2)] react with 2 molar equivalents of a Grignard reagent RMgX, one adding to the C=N and the other replacing the Cl, to give tertiary amines R2CHNR2'.969
Many other C=N systems (phenylhydrazones, oxime ethers, etc.) give 1,2-addition when treated with Grignard reagents, while some gave reductions and others gave miscellaneous reactions. Organocerium reagents add to hydrazones.970 Indium metal promotes the addition of alkyl iodides to hydrazones.971 A hydrazone can be formed in situ by reacting an aldehyde with a hydrazine derivative. In the presence of tetrallyltin and a Sc catalysts, homoallylic hydrazine derivatives are formed.972 Hydrazone derivatives react with iodoalkenes in the presence of InCl3 and Mn2(CO)10 under photochemical conditions to give the hydrazine derivative.973 Ketene dithioacetals add to hydrazones using a chiral Zr catalyst to give a pyrazolidine.974 Oximes can be converted to hydroxylamines (39) by treatment with 2 molar equivalents of an alkyllithium reagent, followed by methanol.975 Oxime ethers add an allyl group upon reaction with allyl bromide and In metal in water.976 Nitrones [R2C=N+(R′)–O−] react with allylic bromides and Sm to give homoallylic oximes,977 and with terminal alkynes and a Zn catalyst to give propargylic oximes.978 Grignard reagents also add to nitrones.979 Nitrones react with CH2=CHCH2InBr in aq DMF to give the homoallylic oxime980 and silyl ketene acetals add in the presence of a chiral Ti catalyst to good enantioselectivity.981
Allylic alcohols add to imines in the presence of a Pd catalyst to give the homoallylic amine.982
OS IV, 605; VI, 64. Also see, OS III, 329.
16-32 Addition of Carbenes and Diazoalkanes to C=N Compounds
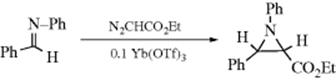
In the presence of metal catalysts [e.g., Yb(OTf)3], diazoalkanes add to imines to generate aziridines.983 The reaction is somewhat selective for the cis-diastereomer. The use of chiral additives in this reaction leads to aziridines enantioselectively.984 Imines can be formed by the reaction of an aldehyde and an amine, and subsequent treatment with Me3SiI and butyllithium gives an aziridine.985N-Tosyl imines react with diazoalkenes to form N-tosyl aziridines, with good cis selectivity986 and modest enantioselectivity in the presence of a chiral Cu catalyst,987 but give excellent enantioselectivity with a chiral Rh catalyst988 or with the use of an organocatalyst.989 Note that N-tosyl aziridines are formed by the reaction of an alkene with PhI=NTs and a Cu catalyst.990N-Acylimines react with diazoesters via C–H insertion using a Pt catalyst.991 The reaction of alkenes with diazo compounds is discussed in Reaction 15-53.
16-33 The Addition of Grignard Reagents to Nitriles and Isocyanates
Alkyl,oxo-de-nitrilo-tersubstitution (Overall transformation)
![]()
N-Hydro-C-alkyl-addition
![]()
Ketones can be prepared by addition of Grignard reagents to nitriles, followed by hydrolysis of the initially formed imine anion. Many ketones have been made in this manner, though when both R groups are alkyl, yields are not high.992 Yields can be improved by the use of Cu(I) salts993 or by using benzene containing 1 equiv of ether as the solvent, rather than ether alone.994 In general, the ketimine salt does not react with Grignard reagents: Hence, tertiary alcohols or tertiary alkyl amines are not often side products.995 By careful hydrolysis of the salt, it is sometimes possible to isolate ketimines (RR′C=NH),996 especially when R and R′ = aryl. The addition of Grignard reagents to the C![]() N group is normally slower than to the C=O group, and cyano group containing aldehydes add the Grignard reagent without disturbing the CN group.997 Organolithium reagents add to nitriles, mediated by LiBr, to form N-acetyl enamines.998 Other metal compounds have been used, including Sm with allylic halides999 and organocerium compounds (e.g., MeCeCl2).1000 Allylic halides react with an excess of Zn metal in the presence of 40% AlCl3 and in the presence of a nitrile, to give homoallylic ketones after hydrolysis.1001
N group is normally slower than to the C=O group, and cyano group containing aldehydes add the Grignard reagent without disturbing the CN group.997 Organolithium reagents add to nitriles, mediated by LiBr, to form N-acetyl enamines.998 Other metal compounds have been used, including Sm with allylic halides999 and organocerium compounds (e.g., MeCeCl2).1000 Allylic halides react with an excess of Zn metal in the presence of 40% AlCl3 and in the presence of a nitrile, to give homoallylic ketones after hydrolysis.1001
Addition of Grignard reagents1002 or organolithium reagents1003 to ω-halo nitriles leads to 2-substituted cyclic imines.
The Blaise reaction is the reaction of the organozinc reagent derived from a α-bromoester with Zn metal and a nitrile to give the corresponding β-ketoester.1004
The following mechanism has been proposed for the reaction of the methyl Grignard reagent with benzonitrile1005:
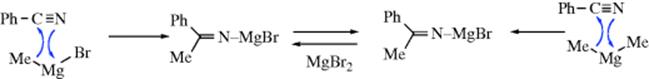
Arylboronic acids add to nitriles in the presence of a Pd catalyst.1006 Aryl and alkenylboronic acids add to isocyanates in the presence of a Pd1007 or Rh1008 catalyst.
Arenes add to nitriles in the presence of a Pd catalyst in DMSO/trifluoroacetic acid to give a diaryl ketone.1009
The addition of Grignard reagents to isocyanates gives, after hydrolysis, N-substituted amides.1010 This is a very good reaction and can be used to prepare derivatives of alkyl and aryl halides. The reaction has also been performed with alkyllithium compounds.1011 Isothiocyanates give N-substituted thioamides. Other organometallic compounds add to isocyanates. Vinyltin reagents lead to conjugate amides.1012
Note that terminal alkynes add to the carbon of an isonitrile in the presence of a uranium complex, giving a propargylic imine.1013
OSCV III, 26, 562; V, 120, 520.
G. Carbon Attack by Active Hydrogen Compounds
Reactions 16-34–16-50 are base-catalyzed condensations (although some of them are also catalyzed to acids).1014 In Reactions 16-34–16-44, a base removes a C–H proton to give a carbanion, which then adds to a C=O. The oxygen acquires a proton, and the resulting alcohol may or may not be dehydrated, depending on whether an α hydrogen is present and on whether the new double bond would be in conjugation with double bonds already present:
The reactions differ in the nature of the active hydrogen component and the carbonyl component. Table 16.2 illustrates the differences. Reaction 16-50 is an analogous reaction involving addition to C![]() N.
N.
Table 16.2 Base-Catalyzed Condensations Showing the Active-Hydrogen Components and the Carbonyl Compounds.
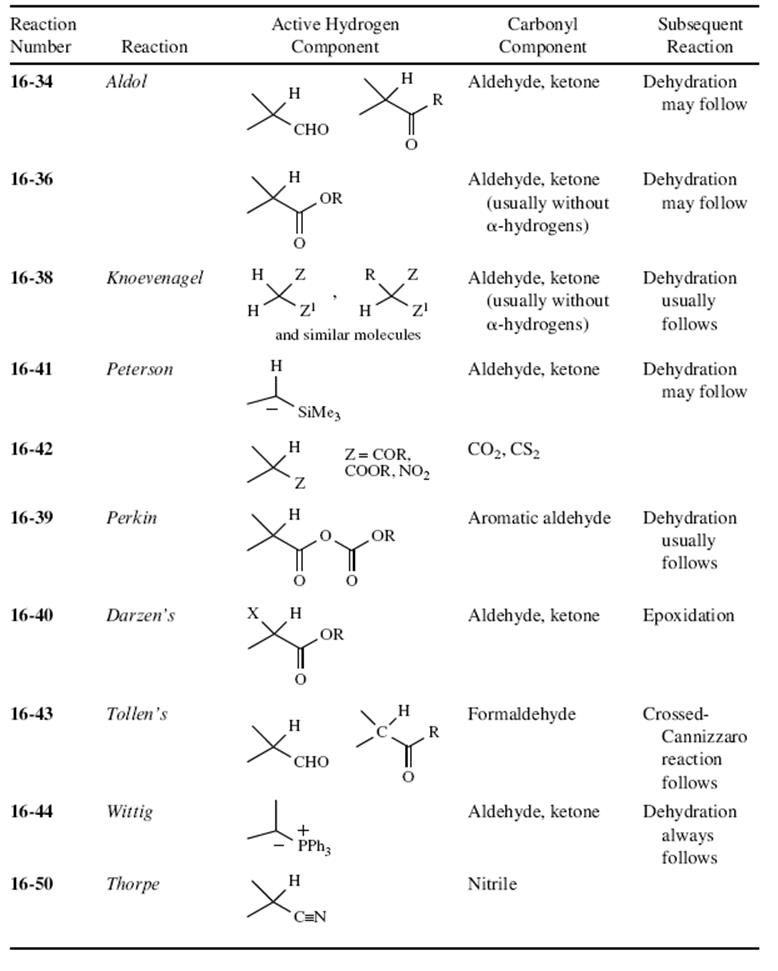
16-34 The Aldol Condensation1015
O-Hydro-C-(α-acylalkyl)-addition; α-Acylalkylidine-de-oxo-bisubstitution
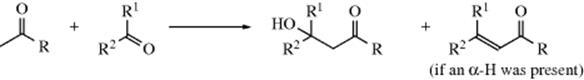
In the aldol reaction or aldol condensation1016 the α carbon of one aldehyde or ketone molecule adds to the carbonyl carbon of another.1017 Although acid-catalyzed aldol reactions are known,1018 the most common form of the reaction uses a base. There is evidence that an SET mechanism can intervene when the substrate is an aromatic ketone.1019 Although hydroxide was commonly used in early versions of this reaction, stronger bases [e.g., alkoxides (RO−) or amides (R2N−)] are also common. Amine bases have been used to catalyze the aldol condensation.1020 Hydroxide ion is not a strong enough base to convert substantially all of an aldehyde or ketone molecule to the corresponding enolate ion; that is, the equilibrium lies well to the left, for both aldehydes
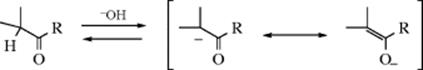
and ketones. Nevertheless, enough enolate ion is present for the reaction to proceed:
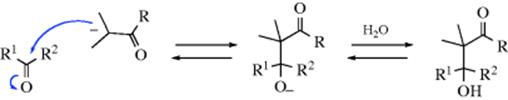
This equilibrium lies further to the right with alkoxide relative to hydroxide, but the equilibrium still lies predominantly to the left. With amide bases, and particularly with aprotic solvents, the equilibrium usually lies much more to the right when compared with alkoxides or hydroxide. Protic solvents, (e.g., water or alcohol) are acidic enough to react with the enolate anion and shift the equilibrium to the left. As noted, in an aprotic solvent (e.g., ether or THF), with a strong amide base (e.g., LDA, sec. Sec. 8.F, category 7), the equilibrium lies more to the right.1021 A variety of amide bases can be used to deprotonate the ketone or aldehyde, and in the case of an unsymmetrical ketone removal of the more acidic proton leads to the kinetic enolate anion.1022 Note that a polymer-bound amide base has been used1023 and solid-phase chiral lithium amides are known.1024 A polymer-supported phosphoramide has been used as a catalyst for the aldol condensation.1025
The product of an aldol condensation is a β-hydroxy aldehyde (called an aldol) or a β-hydroxy ketone, which in some cases is dehydrated during the course of the reaction. An aldol is readily isolated unless the substrate is an aromatic aldehyde or ketone when the reaction is done in aprotic solvents with a mild workup procedure. The aldol reaction has been done in ionic liquids.1026 Even if the dehydration is not spontaneous, it can usually be done easily, since the new double bond is in conjugation with the C=O bond; so that this is a method of preparing α,β-unsaturated aldehydes and ketones, as well as β-hydroxy aldehydes and ketones. One-pot procedures have been reported to give the conjugated product.1027 The entire reaction is an equilibrium (including the dehydration step), and α,β-unsaturated and β-hydroxy aldehydes and ketones can be cleaved by treatment with −OH (the retrograde aldol reaction). The retro-aldol condensation has been exploited for crossed-aldol reactions.1028 A vinylogous (Sec. 6.B) aldol reaction is known1029 as is a ‘double’ aldol.1030 Enzyme-mediated aldol reactions have been reported using two aldehydes, including formaldehyde.1031
Under the principle of vinylogy (Sec. 6.B), the active hydrogen can be one in the γ position of an α,β-unsaturated carbonyl compound:
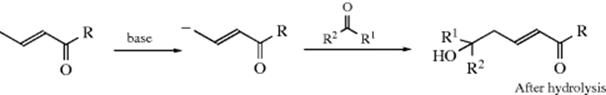
The scope of the aldol reaction may be discussed under five headings:
1. Reaction between Two Molecules of the Same Aldehyde. Homo-coupling. Hydroxide or alkoxide bases are used in protic solvents,1032 and the reaction is quite feasible. Nowadays, the use of dialkylamide bases in aprotic solvents (e.g., ether or THF) is more common. Many aldehydes have been converted to aldols and/or their dehydration products in this manner. The most effective catalysts are basic ion-exchange resins. Of course, the aldehyde must possess an α hydrogen.
2. Reaction between Two Molecules of the Same Ketone. Homo-coupling. With hydroxide or alkoxide bases in protic solvents the equilibrium lies well to the left,1033 and the reaction is feasible only if the equilibrium can be shifted. This can often be done by allowing the reaction to proceed in a Soxhlet extractor (e.g., See OS I, 199). As with aldehydes, the use of dialkylamide bases (e.g., LDA or lithium hexamethyldisilazide, Sec. 8.F., category 7) in aprotic solvents (e.g., ether or THF) are more common. Unsymmetrical ketones condense on the side that has the most hydrogens with dialkylamide bases in aprotic solvents, but on the side with the fewest hydrogens with alkoxide bases in alcohol solvents.
3. Reaction between Two Different Aldehydes. Cross-coupling. In protic solvents with an alkoxide base, this will produce a mixture of four products (eight, if the alkenes are counted). However, if one aldehyde does not have an α hydrogen, only two aldols are possible, and in many cases the crossed product is the main one. The crossed-aldol reaction is often called the Claisen–Schmidt reaction.1034 The crossed-aldol reaction is readily accomplished using amide bases in aprotic solvent. The first aldehyde is treated with LDA in THF at –78 °C, for example, to form the enolate anion. Subsequent treatment with a second aldehyde leads to the mixed-aldol product. The crossed aldol of two aldehydes has been done using potassium tert-butoxide and Ti(OBu)4.1035
4. Reaction between Two Different Ketones. Cross-coupling. This is seldom attempted with hydroxide or alkoxide bases in protic solvents since similar considerations apply to those discussed for aldehydes. This reaction is commonly done with amide bases in aprotic solvents, but with somewhat more difficulty than with aldehydes.
5. Reaction between an Aldehyde and a Ketone. This is usually feasible with hydroxide or alkoxides bases in protic solvents when the aldehyde has no α hydrogen, since there is no competition from ketone condensing with itself.1036 This is also called the Claisen–Schmidt reaction. Even when the aldehyde has an α hydrogen, it is generally the α carbon of the ketone that adds to the carbonyl of the aldehyde, not the other way around. Mixtures are usually produced, however. If the ketone or the aldehyde is treated with an amide base in aprotic solvents, a second aldehyde or ketone can be added to give the aldolate with high regioselectivity. The reaction can be also made regioselective by preparing an enol derivative of the ketone separately1037 and then adding this to the aldehyde (or ketone). Other types of preformed derivatives that react with aldehydes and ketones are enamines (with a Lewis acid catalyst),1038 and enol borinates (R′CH=CR2–OBR2),1039 which can be synthesized by Reaction 15-27 or directly from an aldehyde or ketone1040. Preformed metallic enolates are also used. For example, lithium enolates1041(prepared by Reaction 12-23) react with the substrate in the presence of ZnCl2.1042 In this case, the aldol product is stabilized by chelation of its two oxygen atoms with the zinc ion.1043 Other metallic enolates can be used for aldol reactions, either preformed or generated in situ with a catalytic amount of a metal compound. Compounds used for this purpose include metal complexes of Mg,1044 Ti,1045 Zr,1046 Pd,1047 In,1048 Sn,1049 La,1050 and Sm,1051all of which give products with moderate-to-excellent diastereoselectivity1052 and regioselectivity. α-Alkoxy ketones react with lithium enolates particularly fast.1053 A bis(aldol) condensation has been reported with epoxy ketones and aldehydes using SmI2.1054
There are discussions that relate to the transition state of the aldol condensation. There is experimental evidence for chair-like transition states in the aldol reactions of methyl ketone lithium enolate anions.1055A computational study gave the gas-phase activation energies for lithium enolate anions in an aldol-type reaction.1056 There is a computational study of the mechanism for an aldol reaction in pure water.1057
The reactions with pre-formed enol derivatives provide a way to control the stereoselectivity of the aldol reaction.1058 As with the Michael reaction (15-24), the aldol reaction creates two new stereogenic centers. In the most general case, there are four stereoisomers of the aldol product (two racemic diastereomers), which can be represented as the syn and anti diastereomers shown. The reaction may be diastereoselective, however, if one is preferred over the other.
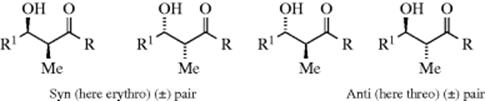
Among the preformed enol, derivatives used for diastereoselective aldol condensations have been enolates of Li,1059 Mg, Ti,1060 and Sn,1061 silyl enol ethers,1062 enol borinates,1063 and enol borates [R′CH=CR2OB(OR)2].1064 The nucleophilicity of silyl enol ethers has been examined1065 and reactions of these compounds are discussed in Reaction 16-35.
Base-induced formation of the enolate anion generally leads to a mixture of (E)- and (Z)-isomers, and dialkyl amide bases are used in most cases. The (E/Z) stereoselectivity depends on the structure of the lithium dialkylamide base, with the highest (E/Z) ratios obtained with LiTMP–butyllithium mixed aggregates in THF.1066 The use of LiHMDS resulted in a reversal of the (E/Z) selectivity. In general, metallic (Z)-enolates give the syn (or erythro) pair, and this reaction is highly useful for the diastereoselective synthesis of these products.1067 The (E) isomers generally react nonstereoselectively. However, anti-stereoselectivity has been achieved in a number of cases, with Ti enolates,1068 with Mg enolates,1069 with certain enol borinates,1070 and with lithium enolates at –78 °C.1071 Enolization accounts for syn–anti isomerization of aldols.1072 In another variation, a β-keto Weinreb amide (see Reaction 16-82) reacted with TiCl4 and Hünig's base (iPr2NEt) and then an aldehyde to give the β-hydroxy ketone.1073
These reactions are enantioselective1074 (in which case only one of the four isomers predominates)1075 by using1076 chiral enol derivatives,1077 chiral aldehydes or ketones,1078 or both.1079 Chiral bases1080 can be used (e.g., proline),1081 proline derivatives,1082 or chiral additives used in conjunction with an organobase.1083 Indeed, chiral organocatalysts are increasingly important,1084 including those that can be used in aqueous media.1085 Chiral auxiliaries1086 have been developed that can be used in conjunction with the aldol condensation, as well as chiral transition metal complexes1087 and chiral ligands1088 in catalytic reactions. The enantioselective condensation of methyl vinyl ketone and an aldehyde used a chiral Zn catalyst.1089 Structural variations in the aldehyde or ketone are compatible with many enantioselective condensation reactions. An α-hydroxy ketone was condensed with an aldehyde using a chiral Zn catalyst to give the aldol (an α,β-dihydroxy ketone) with good syn selectivity and good enantioselectivity.1090 Chiral vinylogous aldol reactions (Sec. 6.B) have been reported.1091 Formation of the magnesium enolate anion of a chiral amide, adds to aldehydes to give the alcohol enantioselectively.1092 Diamine protonic acids have been used for catalytic asymmetric aldol reactions.1093
Silyl enol ethers react with aldehydes in the presence of chiral boranes1094 or other additives1095 to give aldols with good asymmetric induction (see the Mukaiyama aldol Reaction in 16-35). Chiral boron enolates have been used.1096 Since both new stereogenic centers are formed enantioselectively, this kind of process is called double asymmetric synthesis.1097 Where both the enolate derivative and substrate were achiral, carrying out the reaction in the presence of an optically active boron compound1098 or a diamine coordinated with a Sn compound1099 gave the aldol product with excellent enantioselectivity for one stereoisomer. Boron triflate (R2BOTf) derivatives have been used for the condensation of ketals and ketone to give β-alkoxy ketones.1100
It is possible to make the α carbon of the aldehyde add to the carbonyl carbon of the ketone, by using an imine instead of an aldehyde, and LiN(iPr)2 as the base to form the α-lithio imine (40).1101 This is known as a directed aldol reaction. Similar reactions have been performed with α-lithiated dimethylhydrazones of aldehydes or ketones1102 and with α-lithiated aldoximes.1103
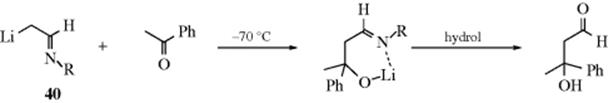
The aldol reaction can also be performed with acid catalysts, as mentioned above, in which case dehydration usually follows. Here there is initial protonation of the carbonyl group, which attacks the α carbon of the enol form of the other molecule1104:
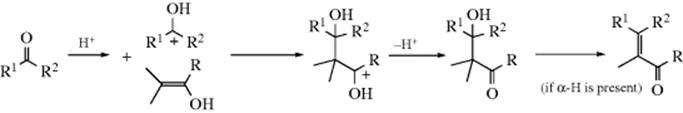
With respect to the enol, this mechanism is similar to that of halogenation (Reaction 12-4). A side reaction that is sometimes troublesome is further condensation, since the product of an aldol reaction is still an aldehyde or ketone. The aldol condensation of aldehydes has also been done using a mixture of pyrrolidine and benzoic acid.1105
The intramolecular aldol condensation is well known, and aldol reactions are often used to close five- and six-membered rings. Because of the favorable entropy (Sec. 6.D), such ring closures generally take place with ease1106when using hydroxide or alkoxide bases in protic solvents. In aprotic solvents with amide bases, formation of the enolate anion occurs by deprotonation of the more acidic site, followed by cyclization to the second carbonyl. The acid-catalyzed intramolecular aldol condensation is known, and the mechanism has been studied.1107 Stereoselective proline-catalyzed intramolecular aldol reactions give the cyclized product with good enantioselectivity.1108 Chiral ligands, in conjugation with transition metal compounds of Cu1109, lead to asymmetric intramolecular aldol condensation reactions. An asymmetric intramolecular aldol reaction was catalyzed by a chiral amine.1110 The regioselectivity of an intramolecular aldol condensation of unsaturated 1,5-diketones is strongly influenced by the presence or absence of a trialkylphosphine.1111
An important extension of the intramolecular aldol condensation is the Robinson annulation reaction,1112 which has often been used in the synthesis of steroids and terpenes. In original versions of this reaction, a cyclic ketone is converted to another cyclic ketone under equilibrium conditions using hydroxide or alkoxide bases in a protic solvent, forming one additional six-membered ring containing a double bond. The reaction can be done in a stepwise manner using amide bases in aprotic solvents. In the reaction with hydroxide or alkoxide bases in alcohol or water solvents, the substrate is treated with methyl vinyl ketone (or a simple derivative of methyl vinyl ketone) and a base.1113 The enolate ion of the substrate adds to the methyl vinyl ketone in a Michael reaction (15-24) to give a diketone that undergoes or is made to undergo an internal aldol reaction and subsequent dehydration to give the product.1114 The Robinson annulation can be combined with alkylation.1115 Enantioselective Robinson annulation techniques have been developed, including a proline-catalyzed reaction.1116 The Robinson annulation has been done in ionic liquids1117 and a solvent-free version of the reaction is known.1118
![]()
Because methyl vinyl ketone has a tendency to polymerize, surrogates are often used instead (i.e., compounds that will give methyl vinyl ketone when treated with a base). One common example, MeCOCH2CH2NEt2Me+ I− (see Reaction 17-9), is easily prepared by quaternization of MeCOCH2CH2NEt2, which itself is prepared by a Mannich Reaction (16-19) involving acetone, formaldehyde, and diethylamine. α-Silylated vinyl ketones [RCOC(SiMe3)=CH2] have also been used successfully in annulation reactions1119 because the SiMe3 group is easily removed. When the ring closure of a 1,5-diketone is catalyzed by the amino acid (S)-proline, the product is optically active with high ee.1120Stryker's reagent,1121[(Ph3P)CuH]6, has been used for an intramolecular addition where a ketone enolate anion adds to a conjugated ketone, giving cyclic alcohol with a pendant ketone unit.1122
OS I, 77, 78, 81, 199, 283, 341; II, 167, 214; III, 317, 353, 367, 747, 806, 829; V, 486, 869; VI, 496, 666, 692, 781, 901; VII, 185, 190, 332, 363, 368, 473; VIII, 87, 208, 241, 323, 339, 620; IX, 432, 610; X, 339.
16-35 Mukaiyama Aldol and Related Reactions1123
O-Hydro-C-(α-acylalkyl)-addition
An important variation of the aldol condensation involves treatment of an aldehyde or ketone with a silyl ketene acetal [R2C=C(OSiMe3)OR′]1124 or a silyl enol ether, in the presence of TiCl41125, to give 41. This variation is known as the Mukaiyama aldol reaction, or simply the Mukaiyama reaction. The silyl ketene acetal can be considered a pre-formed enolate that gives aldol product with TiCl4 in aqueous solution, or with no
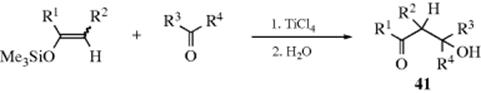
catalyst at all.1126 A combination of TiCl4 and N-tosyl imine has also been used to facilitate the Mukaiyama aldol reaction.1127 Reaction at the carbonyl of saturated carbonyl compounds is significantly faster than 1,2-addition to unsaturated carbonyl compounds.1128 The mechanism of this reaction has been explored.1129 Other catalysts have been used for this reaction, including InCl3,1130 SmI2,1131 HgI2,1132 Yb(OTf)3,1133 Cu(OTf)2,1134 LiClO4,1135 VOCl3,1136an Fe catalyst,1137 and Bi(OTf)3.1138 Lithium perchlorate in acetonitrile (5 M) can be used for the reaction of an aldehyde and a silyl enol ether.1139 The reaction can be done in water using a Sc catalyst1140 or on a Montmorillonite K-10 clay.1141 Silyl enol ethers react with aq formaldehyde in the presence of TBAF to give the aldol product.1142 A catalytic amount of Me3SiCl facilitates the Ti mediated reaction.1143 Sulfonamides (e.g., HNTf2) have been used as a catalyst1144 as has pyridine N-oxide1145 and N-methylimidazole.1146 An ab initio study of the uncatalyzed Mukaiyama aldol reaction showed that the nucleophilicity of silyl enol ether and the electrophilicity of the aldehyde are important in promoting the reactivity.1147 This reaction can also be run with the aldehyde or ketone in the form of its acetal [R3R4C(OR′)2], in which case the product is the ether R1COCHR2CR3R4OR′ instead of 41.1148 Vinylogous (Sec. 6.B) silyl ketene acetals with Ti,1149 Cu,1150 In,1151 Fe,1152 or Zn1153 catalysts or an organocatalyst,1154 give the product with good enantioselectivity.
Silyl enol ethers1155 derived from esters (silyl ketene acetals) react with aldehydes in the presence of various catalysts to give β-hydroxy esters. Water accelerates the reaction of an aldehyde and a ketene silyl acetal with no other additives.1156 The reaction is catalyzed by triphenylphosphine1157 and also by SiCl4 with a chiral bis(phosphoramide) catalyst.1158 The reaction was done without a catalyst in an ionic liquid.1159 A vinylogous reaction (Sec. 6.B) is known that gives δ-hydroxy-α,β-unsaturated esters.1160 An interesting variation in this reaction combined an intermolecular Mukaiyama aldol followed by an intramolecular reaction (a “domino” Mukaiyama aldol) that gave cyclic conjugated ketone products.1161 Under different conditions, silyl ketene acetals of conjugated esters react with aldehydes to give conjugated lactones.1162 Imines react with silyl ketene acetals in the presence of SmI3 to give β-amino esters.1163 Imines react with silyl enol ethers in the presence of BF3·OEt2 to give β-amino ketones.1164 Silyl ketene acetals also undergo conjugate addition in reactions with conjugated ketones.1165 Propargylic acetals react with silyl enol ethers and a Sc catalyst to give β-alkoxy ketones.1166 α-Silyl silyl enol ethers [RCH=CH(OTMS)SiMe3] react with acetals in the presence of SnCl4 to give β-alkoxy silyl ketones.1167 Borane derivatives [e.g., C=C–OB(NMe2)2] react with aldehydes to give β-amino ketones.1168
Asymmetric Mukaiyama aldol reactions and reactions of silyl ketene acetals have been reported,1169 usually using chiral additives1170 although chiral auxiliaries have also been used.1171 Chiral catalysts, usually transition metal complexes using chiral ligands, are quite effective1172 but chiral organocatalysts1173 are increasingly important.
Enol acetates and enol ethers also give this product when treated with acetals and TiCl4 or a similar catalyst.1174 A variation of this condensation uses an enol acetate with an aldehyde in the presence of Et2AlOEt to give the aldol product.1175
16-36 Aldol-Type Reactions between Carboxylic Acid Derivatives and Aldehydes or Ketones
O-Hydro-C-(α-alkoxycarbonylalkyl)-addition; α-Alkoxycarbonylalkylidene-de-oxo-bisubstitution
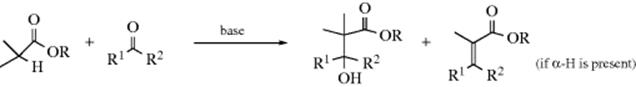
In the presence of a strong base, removal of a proton from the α carbon of a carboxylic ester or other acid derivative generates an enolate anion that can condense with the carbonyl carbon of an aldehyde or ketone to give a β-hydroxy ester,1176 amide, and so on. These products may or may not be dehydrated to the α,β-unsaturated derivative. This reaction is sometimes called the Claisen reaction,1177 an unfortunate usage since that name is more firmly connected to Reaction 16-85. Claisen condensation is a better descriptor. Early reactions used hydroxide or an alkoxide base in water or alcohol solvents, where self-condensation was the major process. Under such conditions, the aldehyde or ketone was usually chosen for its lack of an α-proton. Much better control of the reaction was achieved when dialkylamide bases in aprotic solvents (e.g., ether or THF) were used. The reaction of tert-butyl acetate and LDA1178 in hexane or more commonly THF at –78 °C gives the enolate anion of tert-butyl acetate,1179 (Reaction 12-23, e.g., although self-condensation is occasionally a problem even here). Additives play an important role in the LDA mediated enolization of esters1180. Subsequent reaction of a ketone provides a simple rapid alternative to the Reformatsky Reaction (16-28) as a means of preparing β-hydroxy tert-butyl esters. It is also possible for the α carbon of an aldehyde or ketone to add to the carbonyl carbon of a carboxylic ester, but this is a different reaction (16-86) involving nucleophilic substitution and not addition to a C=O bond. It can, however, be a side reaction if the aldehyde or ketone has an α hydrogen.
Transition metal mediated condensation of esters with aldehydes is known. The reaction of a thioester and an aryl aldehyde with TiCl4–NBu3, for example, gave a β-hydroxy thioester with good syn selectivity.1181 Selenoamides [RCH2C(=Se)NR′2] react with LDA and then an aldehyde to give β-hydroxy selenoamides.1182 The reaction of an α,β-unsaturated ester and benzaldehyde with a chiral Rh catalyst gave a β-hydroxy ester with good diastereoselectivity and good enantioselectivity.1183
Besides ordinary esters (containing an α hydrogen), the reaction can also be carried out with lactones and, as in Reaction 16-34, with the γ position of α,β-unsaturated esters (vinylogy; Sec. 6.B). The enolate anion of an amide can be condensed with an aldehyde.1184 Thioesters undergo aldol-type condensations.1185
For most esters, a much stronger base is needed than for aldol reactions; (iPr)2NLi (LDA, Sec. 8.F, category 7), Ph3CNa and LiNH2 are among those employed. However, esters of malonic and succinic acid react more easily, and such strong bases are not needed. For example, diethyl succinate and its derivatives condense with aldehydes and ketones in the presence of bases (e.g., NaOEt, NaH, or KOCMe3). This reaction is called the Stobbe condensation.1186One of the ester groups (sometimes both) is hydrolyzed in the course of the reaction. The following mechanism accounts for (1) the fact the succinic esters react so much better than others; (2) one ester group is always cleaved; and (3) the alcohol is not the product but the alkene. In addition, intermediate lactones (42) have been isolated from the mixture.1187 The Stobbe condensation has been extended to di-tert-butyl esters of glutaric acid.1188 The boron-mediated reaction is known.1189
The intramolecular reaction of an aldehyde–carboxylic acid in the presence of triethylamine and a pyridinium salt led to the ring-forming condensation reaction, followed by formation of a β-lactone.1190
Amides participate in this condensation reaction, reacting with aldehydes in the presence of a Ba catalyst to give a β-hydroxy amide derivative.1191 The reaction of an amide with LDA in the presence of an acyl silane, followed by reaction with an alkyl halide, leads to the β-hydroxy amide with the additional alkyl group at the β-carbon.1192
Chiral additives (e.g., diazaborolidines) can be added to an ester, and subsequent treatment with a base, and then an aldehyde leads to a chiral β-hydroxy ester.1193 A variety of chiral amide or oxazolidinone derivatives have been used to form amide linkages to carboxylic acid derivatives. These chiral auxiliaries lead to chirality transfer from the enolate anion of such derivatives, in both alkylation reactions and acyl substitution reactions with aldehydes and ketones. The so-called Evans auxiliaries (43–45) are commonly used and give good enantioselectivity.1194 A variation is the magnesium halide-catalyzed anti-aldol reaction of chiral N-acylthiazolidinethiones (see 46).1195 The use of chiral N-acyloxazolidinthiones with TiCl4 and sparteine also gave good selectivity in the acyl addition.1196 Chiral diazaboron derivatives have also been used to facilitate the condensation of a α-phenylthio ester with an aldehyde.1197
The condensation of an ester enolate and a ketone1198 can be used as part of a Robinson annulation-like sequence (see Reaction 16-34).
OS I, 252; III, 132; V, 80, 564; 70, 256; X, 437; 81, 157. Also see, OS IV, 278, 478; V, 251.
16-37 The Henry Reaction1199
![]()
The classical condensation of an aliphatic nitro compound with an aldehyde or ketone is usually called the Henry reaction1200 or the Kamlet reaction, and is essentially a nitro aldol reaction. A variety of conditions have been reported, including the use of a recoverable polymer catalyst,1201 a silica catalyst,1202 a tetraalkylammonium hydroxide,1203 proazaphosphatranes,1204 and it has been done in an aqueous media1205 or an ionic liquid.1206 A solvent-free Henry reaction was reported in which a nitroalkane and an aldehyde were reacted on KOH powder.1207 A solvent-free microwave assisted reaction was reported.1208 Potassium phosphate has been used with nitromethane and aryl aldehydes.1209 The Henry reaction has been done using ZnEt2 and 20% ethanolamine.1210 A gel-entrapped base has been used to catalyze this reaction.1211 Biocatalysts have been used.1212
Catalytic enantioselective Henry reactions are known,1213 including the use of a chiral Cu,1214 Zn,1215 Nd,1216 or Ti catalyst.1217 The Henry reaction of nitromethane and a chiral aldehyde under high pressure gives the β-nitro alcohol with excellent enantioselectivity.1218 Enantioselective nitro–aldol reactions are catalyzed by organocatalysts (e.g., Cinchona alkaloids1219 or other organocatalysts).1220
A variation of this reaction converts nitro compounds to nitronates [RCH=N+(OTMS)–O−], which subsequently react with aldehydes in the presence of a Cu catalyst to give the β-nitro alcohol.1221 aza-Henry reactions condense nitroalkanes with imine derivatives, and the resulting amino nitro compounds are formed with good enantioselectivity in the presence of organocatalysts1222 or Br![]() nsted acid catalysts.1223 Aza-Henry products are also formed by the reaction of amines with activated unsaturated compounds.1224
nsted acid catalysts.1223 Aza-Henry products are also formed by the reaction of amines with activated unsaturated compounds.1224
16-38 The Knoevenagel Reaction
Bis(ethoxycarbonyl)methylene-de-oxo-bisubstitution, and so on
The condensation of aldehydes or ketones, usually not containing an α hydrogen, with compounds of the form Z–CH2–Z' or Z–CHR–Z' is called the Knoevenagel reaction.1225 Both Z and Z' may be CHO, COR, CO2H, CO2R, CN, NO2, SOR, SO2R, SO2OR, or similar groups. The presence of two electron-withdrawing groups makes the α-proton much more acidic (Table 8.1 in Sec. 8.A.i), and such compounds have a significantly higher enol content.1226 When Z = CO2H, decarboxylation of the product often takes place in situ.1227 As shown in the example, the reaction of β-keto esters and aldehydes to give 47 is promoted by diethylamine at 0 °C. Nitroalkanes,1199 as well as β-keto sulfoxides,1228 undergo the reaction.
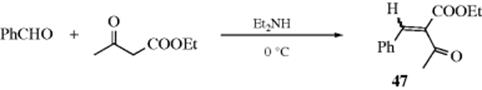
As with Reaction 16-34, these reactions have sometimes been mediated by an acid catalyst.1229 Ionic liquid solvents have been used,1230 and heating on quaternary ammonium salts without solvent leads to a Knoevenagel reaction.1231 Other solvent-free reactions are known.1232 Ultrasound has been used to promote the reaction,1233 and it has also been done using microwave irradiation1234 or on silica,1235 with microwave irradiation. Another solid-state variation is done on moist LiBr,1236 heating with sodium carbonate and molecular sieves 4 Å promotes the reaction,1237 as do zeolites.1238 High-pressure conditions have been used.1239 Transition metal compounds of Pd,1240Sm1241 Ce,1242 Ti,1243 or Bi1244 have been used to promote the Knoevenagel reaction.
With most of these reagents the alcohol is not isolated (only the alkene) if the alcohol has a hydrogen in the proper position,1245 but with a careful workup the alcohol may be the major product. With suitable reactants, the Knoevenagel reaction, like the aldol condensation (16-34), has been carried out diastereoselectively1246 and enantioselectively.1247 When the reactant is of the form ZCH2Z', aldehydes react much better than ketones and few successful reactions with ketones have been reported. However, it is possible to get good yields of the alkene from the condensation of diethyl malonate [CH2(CO2Et)2] with ketones, as well as with aldehydes, if the reaction is run with TiCl4 and pyridine in THF.1248 In reactions with ZCH2Z', the catalyst is most often a secondary amine (piperidine is the most common, but see formation of 47), but many other catalysts have been used. Alkoxides are also common catalysts. When the catalyst is pyridine (to which piperidine may or may not be added) the reaction is known as the Doebner modification of the Knoevenagel reaction and the product is usually the conjugated acid (48). Microwave-induced Doebner condensation reactions are known.1249
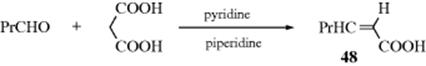
A number of special applications of the Knoevenagel reaction follow:
1. The dilithio derivative of N-methanesulfinyl-p-toluidine1250 (49) adds to aldehydes and ketones to give, after hydrolysis, the hydroxysulfinamides (50). Subsequent heating leads to a stereospecific syn eliminations to give an alkene.1251 The reaction is thus a method for achieving the conversion RR′CO → RR′C=CH2 and represents an alternative to the Wittig reaction.1252 Note that sulfones with an amide group at the α-position, [ArSO2CH(R)N(R)C=O] react with ketones via acyl addition in the presence of SmI2.1253
2. The reaction of ketones with tosylmethylisocyanide (51) gives different products,1254 depending on the reaction conditions. When the reaction is run with potassium tert-butoxide in THF at –5 °C, one obtains (after hydrolysis) the normal Knoevenagel product (52), except that the isocyano group has been hydrated (Reaction 16-97).1255 With the same base but with 1,2-dimethoxyethane (DME) as solvent the product is the nitrile (53).1256When the ketone is treated with 51 and thallium(I) ethoxide in a 4: 1 mixture of absolute ethanol and DME at room temperature, the product is a 4-ethoxy-2-oxazoline (54).1257 Since 53 can be hydrolyzed to a carboxylic acid1198 and 54 to an α-hydroxy aldehyde,1257 this versatile reaction provides a means for achieving the conversion of RCOR′ to RCHR′CO2H, RCHR′CN, or RCR′(OH)CHO. The conversions to RCHR′COOH and to RCHR′CN1258 have also been carried out with certain aldehydes (R′ = H).
3. Aldehydes and ketones (RCOR′) react with α-methoxyvinyllithium [CH2=C(Li)OMe] to give hydroxy enol ethers [RR′C(OH)C(OMe)=CH2], which are easily hydrolyzed to acyloins [RR′C(OH)COMe].1259 In this reaction, the CH2=C(Li)OMe is a synthon for the unavailable H3C–C=O,1260 and is termed an acyl anion equivalent. The reagent also reacts with esters (RCOOR′) to give RC(OH)(COMe=CH2)2. A synthon for the Ph–C=O ion is PhC(CN)OSiMe3, which adds to aldehydes and ketones (RCOR′) to give, after hydrolysis, the α-hydroxy ketones [RR′C(OH)COPh].1261
4. Lithiated allylic carbamates (55) (prepared as shown) react with aldehydes or ketones (R6COR7), in a reaction accompanied by an allylic rearrangement, to give (after hydrolysis) γ-hydroxy aldehydes or ketones.1262 The reaction is called the homoaldol reaction, since the product is a homologue of the product of Reaction 16-34. The reaction has been performed enantioselectively.1263
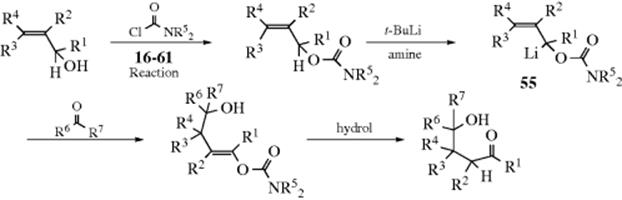
5. The lithium salt of an active hydrogen compound adds to the lithium salt of the tosylhydrazone of an aldehyde to give product 56. If X = CN, SPh, or SO2R, 56 spontaneously loses N2 and LiX to give the alkene 57. The entire process is done in one reaction vessel: The active hydrogen compound is mixed with the tosylhydrazone and the mixture is treated with (iPr)2NLi to form both salts at once.1264 This process is another alternative to the Wittig reaction for forming double bonds.
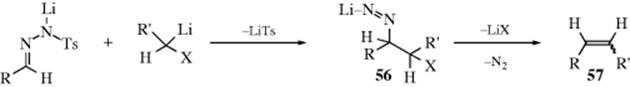
OS I, 181, 290, 413; II, 202; III, 39, 165, 317, 320, 377, 385, 399, 416, 425, 456, 479, 513, 586, 591, 597, 715, 783; IV, 93, 210, 221, 234, 293, 327, 387, 392, 408, 441, 463, 471, 549, 573, 730, 731, 777; V, 130, 381, 572, 585, 627, 833, 1088, 1128; VI, 41, 95, 442, 598, 683; VII, 50, 108, 142, 276, 381, 386, 456; VIII, 258, 265, 309, 353, 391, 420; X, 271. Also see, OS III, 395; V, 450.
16-39 The Perkin Reaction
α-Carboxyalkylidene-de-oxo-bisubstitution
![]()
The condensation of aromatic aldehydes with anhydrides is called the Perkin reaction.1265 When the anhydride has two α hydrogen atoms (as shown), dehydration almost always occurs; the β-hydroxy acid salt is rarely isolated. In some cases, anhydrides of the form (R2CHCO)2O have been used, and then the hydroxy compound is the product since dehydration cannot take place. The base in the Perkin reaction is nearly always the salt of the acid corresponding to the anhydride. Although the Na and K salts have been most frequently used, higher yields and shorter reaction times have been reported for the Cs salt.1266 Besides aromatic aldehydes, their vinylogs (ArCH=CHCHO) also give the reaction (see Sec. 6.B). Otherwise, the reaction is not suitable for aliphatic aldehydes.1267
OS I, 398; II, 61, 229; III, 426.
16-40 Darzens Glycidic Ester Condensation
(2+1) OC,CC-cyclo-α-Alkoxycarbonylmethylene-addition
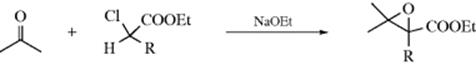
Aldehydes and ketones condense with α-halo esters in the presence of bases to give α,β-epoxy esters, called glycidic esters. This is called the Darzens condensation.1268 The reaction consists of an initial Knoevenagel-typeReaction (16-38), followed by an internal SN2 Reaction (10-9)1269:
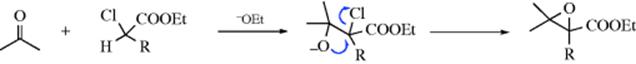
Although the intermediate halo alkoxide is generally not isolated,1270 it has been done, not only with α-fluoro esters (since fluorine is such a poor leaving group in nucleophilic substitutions), but also with α-chloro esters.1271 This is only one of several types of evidence that rule out a carbene intermediate.1272 Sodium ethoxide is often used as the base, but other bases, including sodium amide, are sometimes used. Aromatic aldehydes and ketones give good yields. The reaction can be made to give good yields (~ 80%) with simple aliphatic aldehydes, as well as with aromatic aldehydes and ketones, by treatment of the α-halo ester with the base lithium bis(trimethylsilyl)amide [LiN(SiMe3)2] in THF at –78 °C (to form the conjugate base of the ester) and addition of the aldehyde or ketone to this solution.1273 If a preformed dianion of an α-halo carboxylic acid (Cl−CRCO2−) is used instead, α,β-epoxy acids are produced directly.1274 The Darzens reaction has also been carried out on α-halo ketones, α-halo nitriles,1275 α-halo sulfoxides1276 and sulfones,1277 α-halo N,N-disubstituted amides,1278 α-halo ketimines,1279 and even on allylic1280and benzylic halides. Phase-transfer catalysis has been used.1281 Note that the reaction of a β-bromo-α-oxo ester and a Grignard reagent leads to the glycidic ester.1282 Acid-catalyzed Darzens reactions have also been reported.1283(see also, Reaction 16-46).
Diastereoselective Darzens condensations are possible.1284 The Darzens reaction has been performed with good enantioselectivity,1285 and chiral additives have proven to be effective.1286 Chiral phase-transfer agents have been used to give epoxy ketones with modest enantioselectivity.1287
Glycidic esters can easily be converted to aldehydes (Reaction 12-40). The reaction has been extended to the formation of analogous aziridines by treatment of an imine with an α-halo ester or an α-halo N,N-disubstituted amide and t-BuOK in the solvent 1,2-dimethoxyethane.1288 However, yields were not high.
OS III, 727; IV, 459, 649.
16-41 The Peterson Alkenylation Reaction
Alkylidene-de-oxo-bisubstitution
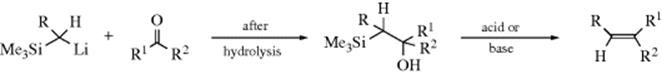
In the Peterson alkenylation reaction1289, the lithio (or sometimes magnesio) derivative of a trialkylsilane adds to an aldehyde or ketone to give a β-hydroxysilane, which spontaneously eliminates water, or can be made to do so by treatment with acid or base, to produce an alkene. This reaction is still another alternative to the Wittig reaction (16-44), and is sometimes called the silyl–Wittig reaction.1290 The R group can also be a COOR group, in which case the product is an α,β-unsaturated ester,1291 or an SO2Ph group, in which case the product is a vinylic sulfone.1292 The stereochemistry of the product can often be controlled by whether an acid or a base is used to achieve elimination. The role of Si–O interactions has also been examined.1293 Use of a base generally gives syn elimination (Ei mechanism, see Sec. 17.C.i), while an acid usually results in anti elimination (E2 mechanism, see Sec. 17.A.i).1294 Samarium(II) iodide in HMPA has also been used for elimination of the hydroxy sulfone.1295
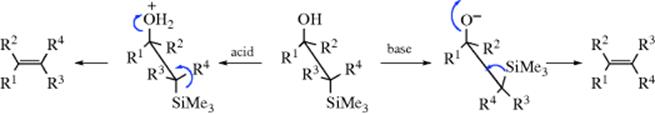
When aldehydes or ketones are treated with reagents of the form 58, the product is an epoxy silane (Reaction 16-46), which can be hydrolyzed to a methyl ketone.1296 For aldehydes, this is a method for converting RCHO to a methyl ketone (RCH2COMe).
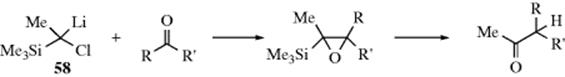
The reagents Me3SiCHRM (M = Li or Mg) are often prepared from Me3SiCHRCl1297 (by Reaction 12-38 or 12-39), but they have also been made by Reaction 12-22 and other procedures.1298 Lithio alkenylsilanes have been used for this reaction.1299
A new version of the reaction has been developed, reacting Me3SiCH2CO2Et with an aldehyde and a catalytic amount of CsF in DMSO.1300 A seleno-amide derivative has been used in a similar manner.1301
There are no references in Organic Syntheses, but see OS VIII, 602, for a related reaction.
16-42 The Addition of Active Hydrogen Compounds to CO2 and CS2
α-Acylalkyl-de-methoxy-substitution (Overall reaction)
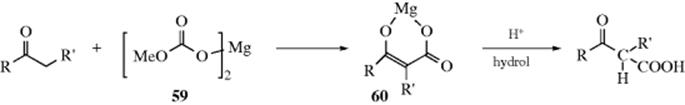
Ketones of the form RCOCH3 and RCOCH2R′ can be carboxylated indirectly by treatment with magnesium methyl carbonate (59).1302 Because formation of the chelate (60) provides the driving force of the reaction, carboxylation cannot be achieved at a disubstituted α position. The reaction has also been performed on CH3NO2, on compounds of the form RCH2NO21303 and on certain lactones.1304 Direct carboxylation has been reported in a number of instances. Ketones have been carboxylated in the α position to give β-keto acids.1305 The base here was lithium 4-methyl-2,6-di-tert-butylphenoxide.
Ketones (RCOCH2R′), as well as other active hydrogen compounds, undergo base-catalyzed addition to CS21306 to give a dianion intermediate (RCOC−R′CSS2-), which can be dialkylated with a halide (R2X) to produce α-dithiomethylene ketones [RCOCR′=C(SR2)2].1307 Compounds of the form ZCH2Z' also react with bases and CS2 to give analogous dianions.1308
Although reactions with N=O derivatives do not formally fall into this category of reactions, it is somewhat related. Nitroso compounds react with activated nitriles in the presence of LiBr and microwave irradiation to give a cyano imine [ArN=C(CN)Ar].1309 This transformation has been called the Ehrlich–Sachs reaction.1310
OS VII, 476. See also, OS VIII, 578.
16-43 Tollens' Reaction
O-Hydro-C (β-hydroxyalkyl)-addition
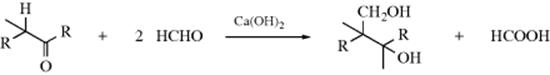
In the Tollens' reaction, an aldehyde or ketone containing an α hydrogen is treated with formaldehyde in the presence of Ca(OH)2 or a similar base. The first step is a mixed-aldol reaction (16-34).
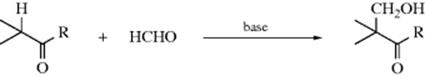
The reaction can be stopped at this point, but more often a second equivalent of formaldehyde is permitted to reduce the newly formed aldol to a 1,3-diol, in a crossed-Cannizzaro Reaction (19-81). If the aldehyde or ketone has several α hydrogen atoms, they can all be replaced. An important use of the reaction is to prepare pentaerythritol from acetaldehyde:
![]()
OS I, 425; IV, 907; V, 833.
16-44 The Wittig Reaction
Alkylidene-de-oxo-bisubstitution
![]()
In the Wittig reaction, an aldehyde or ketone is treated with a phosphorus ylid (a phosphorane; also spelled ylide) to give an alkene.1311 The conversion of a carbonyl compound to an alkene with a phosphorus ylid is called the Wittig reaction. Phosphorus ylids are usually prepared by treatment of a phosphonium salt with a base,1312 and phosphonium salts (61) are usually prepared from a triaryl phosphine and an alkyl halide (Reaction 10-31):
The reaction of triphenylphosphine and an alkyl halides is facilitated by the use of microwave irradiation.1313 Indeed, the Wittig reaction itself is assisted by microwave irradiation.1314 Phosphonium salts are also prepared by addition of phosphines to Michael alkenes (like Reaction 15-8) and in other ways.
The phosphonium salts are most often converted to the ylids by treatment with a strong base (e.g., butyllithium, sodium amide,1315 sodium hydride, or a sodium alkoxide, though weaker bases can be used if the salt is acidic enough. In some cases, and excess of fluoride ion is sufficient.1316 For (Ph3P+)2CH2, sodium carbonate is a strong enough base.1317 When the base used does not contain lithium, the ylid is said to be prepared under “salt-free” conditions1318 because the lithium halide (where the halide counterion comes from the phosphonium salt) is absent. Wittig reactions can be done in aqueous media in the presence of surfactants.1319
When the phosphorus ylid reacts with the aldehyde or ketone to form an alkene, a phosphine oxide is also formed. When triphenylphosphine is used to give Ph3P=CRR′, for example, the byproduct is triphenylphosphine oxide (Ph3PO), which is sometimes difficult to separate from the other reaction products. Other triarylphosphines1320 and trialkylphosphines1321 have been used. Phosphines that have an α-hydrogen should be avoided, so that reaction with the chosen alkyl halide will lead to a phosphonium salt (61) with the α-proton at the desired position. This limitation is essential if a specific ylid is to be formed from the alkyl halide precursor. The Wittig reaction has been carried out with polymer-supported ylids.1322 It has also been done on silica gel.1323 Polymer-bound aryldiphenylphosphino compounds1324 have been used in reactions with alkyl halides to complete a Wittig reaction.
If an alkyl halide is viewed as the starting material (alkyl halide phosphonium salt → phosphorus ylid → alkene), the halogen-bearing carbon of an alkyl halide must contain at least one hydrogen, as in 62 (for deprotonation at the phosphonium salt stage).
The reaction is very general.1325 The aldehyde or ketone may be aliphatic, alicyclic, or aromatic (including diaryl ketones). Wittig reactions are known in which the ylid and/or the carbonyl substrate contain double or triple bonds; various functional groups may be present (e.g., OH, OR, NR2, aromatic nitro or halo, acetal, amide,1326 or even ester groups).1327 Note, however, that a Wittig reaction has been reported in which the carbonyl group of an ester was converted to a vinyl ether.1328 An important advantage of the Wittig reaction is that the position of the new double bond is always certain, in contrast to the result in most of the base-catalyzed condensations (Reactions 16-34–16-43). Ylids have been shown to react with lactones, however, to form ω-alkenyl alcohols.1329 β-Lactams have also been converted to alkenyl-azetidine derivatives using phosphorus ylids.1330 Double or triple bonds conjugated with the carbonyl also do not interfere, the attack being at the C=O carbon. The carbonyl partner can be generated in situ, in the presence of an ylid; the reaction of an alcohol with a mixture of an oxidizing agent and an ylid generates an alkene. Oxidizing agents used in this manner include BaMnO4,1331 MnO2,1332 and PhI(OAc)2.1333 Weinreb amides (Reaction 16-82) can be converted to the corresponding aldehyde via the Wittig reaction.1334
As noted above, the phosphorus ylid may also contain double or triple bonds and certain functional groups. Simple ylids (R, R′ = hydrogen or alkyl) are highly reactive, reacting with oxygen, water, hydrohalic acids, and alcohols, as well as carbonyl compounds and carboxylic esters, so the reaction must be run under conditions where these materials are absent. When an electron-withdrawing group (e.g., COR, CN, CO2R, CHO) is present in the α position, the ylids are much more stable, because the charge on the carbon is delocalized by
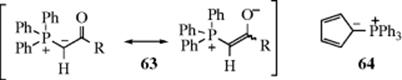
resonance as in 63. Such ylids react readily with aldehydes, but slowly or not at all with ketones.1335 In extreme cases (e.g., 64 where the carbanion unit is part of the aromatic cyclopentadienyl anion), the ylid does not react with ketones or aldehydes. Besides these groups, the ylid may contain one or two α halogens1336 or
![]()
an α OR or OAr group. In the latter case, the product is an enol ether, which can be hydrolyzed (Reaction 10-6) to an aldehyde,1337 so that this reaction is a means of achieving the conversion RCOR′ → RR′CHCHO.1338 However, the ylid may not contain an α nitro group. If the phosphonium salt contains a potential leaving group (e.g., Br or OMe) in the β position, treatment with a base gives elimination, instead of the ylid:
![]()
However, a β COO− group may be present, and the product is a β,γ-unsaturated acid:1339 This is the only convenient way to make these compounds, since elimination by any other route gives the thermodynamically more stable α,β-unsaturated isomers. This is an illustration of the utility of the Wittig method for the specific location of a double bond. Another illustration is the conversion of cyclohexanones to alkenes containing exocyclic double bonds, for example:1340
![]()
Still another example is the formation of anti-Bredt bicycloalkenones1341 (see Sec. 4.Q.iii). As indicated above, α,α'-dihalophosphoranes can be used to prepare 1,1-dihaloalkenes. Another way to prepare haloalkenes1342 is to treat the carbonyl compound with a mixture of CX4 (X = Cl, Br, or I) and triphenylphosphine, either with or without the addition of zinc dust, which allows less Ph3P to be used.1343 Aryl aldehydes react with these dihalophosphoranes to give aryl alkynes after treatment of the initially formed vinyl halide with potassium tert-butoxide.1344
The mechanism1345 of the key step of the Wittig reaction is as follows:1346
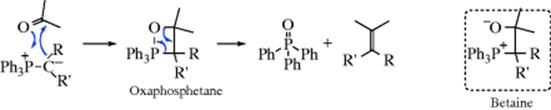
The energetics of ylid formation and their reaction in solution has been studied.1347 For many years it was assumed that a diionic compound, called a betaine, is an intermediate on the pathway from the starting compounds to the oxaphosphetane, but it has been argued that there is little evidence for it.1348 However, “betaine” precipitates have been isolated in certain Wittig reactions,1349 although these are betaine–lithium halide adducts, and might just as well have been formed from the oxaphosphetane as from a true betaine.1350 There is one report of an observed betaine lithium salt during the course of a Wittig reaction.1351 An X-ray structure was determined for a gauche betaine from a thio-Wittig reaction.1352 In contrast, there is much evidence for the presence of the oxaphosphetane intermediates, at least with unstable ylids. For example, 31P NMR spectra taken of the reaction mixtures at low temperatures1353 are compatible with an oxaphosphetane structure that persists for some time but not with a tetra-coordinated phosphorus species. Since a betaine, an ylid, and a phosphine oxide all have tetracoordinated phosphorus, these species could not be causing the spectra, leading to the conclusion that an oxaphosphetane intermediate is present in the solution. In certain cases, oxaphosphetanes have been isolated.1354 It has even been possible to detect cis and trans isomers of the intermediate oxaphosphetanes by NMR spectroscopy.1355 According to this mechanism, an optically active phosphonium salt (RR1R2P+CHR2) should retain its configuration all the way through the reaction, and it should be preserved in the phosphine oxide (RR1R2PO). This has been shown to be the case.1356
Note that the proposed betaine intermediates can be formed, in a completely different manner, by nucleophilic substitution by a phosphine on an epoxide (10-35):

Betaines formed in this way can then be converted to the alkene. This is one reason why betaine intermediates were long accepted in the Wittig reaction. It is also noteworthy that stable phosphonium enolate zwitterions have been formed by the reaction of an aryl aldehyde, a phosphine, and a propargylic ester.1357
Phosphorus is not the only key element used to produce useful ylids. Triphenylarsine1358 has been used. Tellurium ylids have been prepared in situ from α-halo esters and BrTeBu2OTeBu2Br and react with aldehydes to give conjugated esters.1359
The Wittig reaction has been carried out with phosphorus ylids other than phosphoranes, the most important being prepared from phosphonate esters (e.g., 65).1360
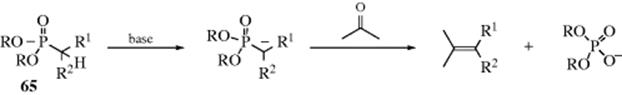
This method, sometimes called the Horner–Emmons, Wadsworth–Emmons, Wittig–Horner, or Horner–Wadsworth–Emmons reaction,1361 has several advantages over the use of phosphoranes, including selectivity.1362 These ylids are more reactive than the corresponding phosphoranes, and when R1 or R2 is an electron-withdrawing group, these compounds often react with ketones that are inert to phosphoranes. High pressure has been used to facilitate this reaction.1363 In addition, the phosphorus product is a phosphate ester, and hence soluble in water, unlike Ph3PO, which makes it easy to separate it from the alkene product. Phosphonates are also cheaper than phosphonium salts and can easily be prepared by the Arbuzov reaction:1364

Phosphonates have also been prepared from alcohols and (ArO)2P(=O)Cl, NEt3, and a TiCl4 catalyst.1365 The reaction of (RO)2P(=O)H and aryl iodides with a CuI catalyst leads to aryl phosphonates.1366 Polymer-bound phosphonate esters have been used for olefination.1367 Dienes are produced when allylic phosphonate esters react with aldehydes.1368 Nucleophilicity parameters have been determined for phosphoryl-stabilized carbanions.1369 A Zn promoted reaction is known using diprotic phosphonates.1370 Wittig reactions of stabilized phosphorus ylids have also been done in water.1371
Stereoselective alkenylation reactions have been achieved using chiral additives1372 or auxiliaries.1373 Ylids formed from phosphine oxides [Ar2P(=O)CHRR′], phosphonic acid bis(amides), (R22N)2POCHRR1],1374 and alkyl phosphonothionates [(MeO)2PSCHRR1],1375 share some of these advantages. Reagents (e.g., Ph2POCH2NR2') react with aldehydes or ketones (R2COR3) to give good yields of enamines (R2R3C=CHNR).1376 (Z)-Selective reagents are also known,1377 including the use of a di(2,2,2-trifluoroethoxy)phosphonate with KHMDS and 18-crown-6.1378 An interesting intramolecular version of the Horner–Emmons reaction leads to alkynes.1379 The reaction of a functionalized aldehyde (R–CHO) with (MeO)2POCHN2, leads to an alkyne (R–C![]() CH).1380
CH).1380
Some Wittig reactions give the (Z)-alkene; some the (E), and others give mixtures. The question of which factors determine the stereoselectivity has been much studied.1381 It is generally found that ylids containing stabilizing groups or formed from trialkylphosphines give (E)-alkenes. The origin of the (E) selectivity in salt-free stabilized ylids may be related to dipole–dipole interactions.1382 The energy of the elimination transition state must also be taken into account.1383 It has been shown that ylids formed from triarylphosphines and not containing stabilizing groups often give (Z) or a mixture of (Z)- and (E)-alkenes.1384 One explanation for this1356 is that the reaction of the ylid with the carbonyl compound is a [2+2]-cycloaddition, which in order to be concerted must adopt the [π2s + π2a]-pathway. As discussed in Reaction 15-63, this pathway leads to the formation of the more sterically crowded product, in this case the (Z)-alkene. If this explanation is correct, it is not easy to explain the predominant formation of (E) products from stable ylids, but (E) compounds are of course generally thermodynamically more stable than the (Z) isomers, and the stereochemistry seems to depend on many factors.
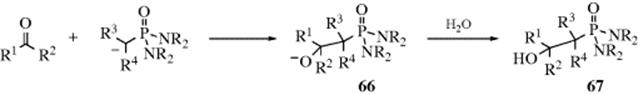
The (E/Z) ratio of the product can often be changed by a change in solvent or by the addition of salts.1385 Another way of controlling the stereochemistry of the product is by use of the aforementioned phosphonic acid bis(amides). In this case, the betaine (66) does form and when treated with water gives the β-hydroxyphosphonic acid bis(amides) (67), which can be crystallized and then cleaved to R1R2C=CR3R4 by refluxing in benzene or toluene in the presence of silica gel.1374 β-Hydroxy products (67) are generally formed as mixtures of diastereomers, and these mixtures can be separated by recrystallization. Each diastereomer will give one of the two isomeric alkenes. Optically active phosphonic acid bis(amides) have been used to give optically active alkenes.1386 Another method of controlling the stereochemistry of the alkene [to obtain either the (Z)- or (E)-isomer] starting with a phosphine oxide (Ph2POCH2R) has been reported.1387
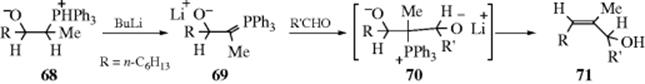
In reactions where the betaine–lithium halide intermediate is present, it is possible to extend the chain further if a hydrogen is present α to the phosphorus. For example, reaction of ethylidenetriphenylphosphorane with heptanal at −78 °C gave 68, and subsequent treatment with butyllithium gave the ylid, (69). Treatment of 69 with an aldehyde (R′CHO) gave the intermediate (70) that gave, after workup, 711388 stereoselectively. Alkoxide 69 also reacts with other electrophiles. For example, treatment of 69 with NCS or PhICl2 gave the vinylic chloride (RCH=CMeCl) stereoselectively: NCS gave the cis and PhICl2 the trans-isomer.1389 The use of Br2 and FClO3 (several explosions1390have been observed with this reagent) gives the corresponding bromide or fluoride, respectively.1391 Reactions of 69 with electrophiles have been called scoopy reactions (α substitution plus carbonyl alkenylation via β-oxido phosphorus ylids).1392
The reaction of a phosphonate ester, DBU, NaI, and HMPA with an aldehyde leads to a conjugated ester with excellent (Z)-selectivity.1393 A (Z)-selective reaction was reported using a trifluoroethyl phosphonate in a reaction with an aldehyde and potassium tert-butoxide.1394
The Wittig reaction has been carried out intramolecularly, to prepare rings containing from 5 to 16 carbons,1395 both by single-ring closure to give alkenes (e.g., 70), or by double-ring closure, as in the conversion of 71 to 72).1396
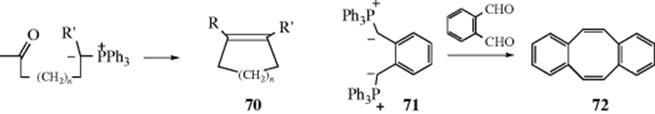
The Wittig reaction has proved very useful in the synthesis of natural products, some of which are quite difficult to prepare in other ways.1397
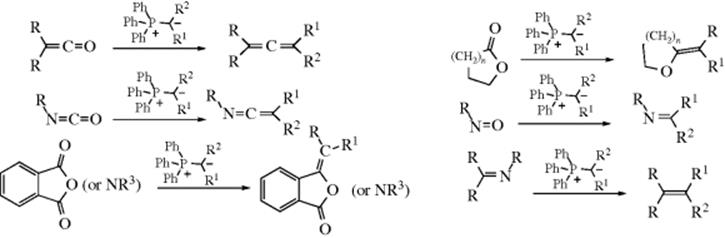
Phosphorus ylids also react in a similar manner with the C=O bonds of ketenes,1398 isocyanates,1399 certain anhydrides1400 lactones,1401 and imides,1402 the N=O of nitroso groups, and the C=N of imines,1403 as shown in the composite reaction. Phosphorus ylids react with carbon dioxide to give the isolable salts (74),1404 which can be hydrolyzed to the carboxylic acids (75) (thus achieving the conversion RR′CHX → RR′CHCOOH) or (if neither R nor R′ is hydrogen) dimerized to allenes (76).
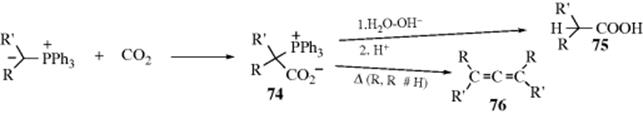
Although phosphorus ylids are most commonly used for alkenylation reactions, nitrogen ylids can occasionally be used. However, nitrogen ylids are often difficult to form, are unstable and highly reactive. The reaction of N-benzyl-N-phenylpiperidinium bromide with base to give an N-ylid is one example, and it reacted with benzaldehyde to form styrene.1405 The structure has been determined for an intermediate in an aza-Wittig reaction.1406 An aza-Wittig reaction1407 has been used to prepare pyrrolines and tetrahydropyridines.1408
OS V, 361, 390, 499, 509, 547, 751, 949, 985; VI, 358; VII, 164, 232; VIII, 265, 451; 75, 139, OS IX, 39, 230.
16-45 Tebbe, Petasis, and Alternative Alkenylations
Methylene-de-oxo-bisubstitution

A useful alternative to phosphorus ylids are Ti reagents (e.g., 77) prepared from dicyclopentadienyltitanium dichloride and trimethylaluminum.1409 Treatment of a carbonyl compound with 77 (Tebbe reagent) in toluene–THF containing a small amount of pyridine1410 leads to the alkene. Dimethyltitanocene (Me2TiCp2), called the Petasis reagent, is a convenient and highly useful alternative to 77.1411 The mechanism of Petasis olefination has been examined.1412 Both the Tebbe and the Petasis reagent give good results with ketones.1413 An important feature of these new reagents is that carboxylic esters and lactones1414 can be converted to the corresponding enol ethers in good yields. The enol ether can be hydrolyzed to a ketone (Reaction 10-6), so this is also an indirect method for making the conversion RCO2R′ → RCOCH3 (see also, Reaction 16-82). Conjugated esters are converted to alkoxy-dienes with this reagent.1415 Lactams, including β-lactams, are converted with alkylidene cycloamines (alkylidene azetidines from β-lactams, which are easily hydrolyzed to β-amino ketones).1416
Besides stability and ease of preparation, another advantage of the Petasis reagent is that structural analogues can be prepared, including Cp2Ti(C3H5)21417 (C3H5 = cyclopropyl), CpTi(CH2SiMe3)3,1418 and Cp2TiMe(CH=CH2).1419In another variation, 2 molar equivalents of Cp2Ti[P(OEt)3]2 reacted with a ketone in the presence of 1,1-diphenylthiocyclobutane to give the alkenylcyclobutane derivative.1420 An alternative Ti reagent was prepared using TiCl4, Mg metal and dichloromethane, reacting with both ketones1421 and esters1422 to give alkenes or vinyl ethers, respectively. Alkenes are generated form ketones and alkyl iodides in the presence of a catalytic amount of Cp2Ti[POEt)3]2.1423
Carboxylic esters undergo the conversion C=O → C=CHR (R = primary or secondary alkyl) when treated with RCHBr2, Zn,1424 and TiCl4 in the presence of N,N,N',N'-tetramethylethylenediamine.1425 Metal carbene complexes1426R2C=MLn (L = ligand), where M is a transition metal (e.g., Zr, W, or Ta) have also been used to convert the C=O of carboxylic esters and lactones to CR2.1427 It is likely that the complex Cp2Ti=CH2 is an intermediate in the reaction with the Tebbe reagent. Indeed, Ti carbenoids have been used to convert carbonyl groups to the corresponding alkene.1428
There are a few other methods for converting ketones or aldehydes to alkenes.1429 Carbonyl compounds react with bis(iodozincio)methane to give alkenes.1430 When a ketone is treated with CH3CHBr2/Sm/SmI2, with a catalytic amount of CrCl3, the alkene is formed.1431 α-Halo esters also react with CrCl2 in the presence of a ketone to give vinyl halides.1432 Organozinc reagents have been used to convert carbonyl compounds to alkenes in the presence of Lewis acids.1433 α-Diazo esters react with ketones in the presence of an iron catalyst to give the corresponding alkene.1434 α-Diazo silylalkanes react similarly in the presence of a Rh catalyst.1435 Ketone olefination has been accomplished using methyltrioxorhenium.1436 α-Halosulfones react with aldehydes in the presence of LiHMDS and MgBr2·OEt2 to give a vinyl chloride.1437
OS VIII, 512, IX, 404; X, 355.
16-46 The Formation of Epoxides from Aldehydes and Ketones
(1+2)OC,CC-cyclo-Methylene-addition
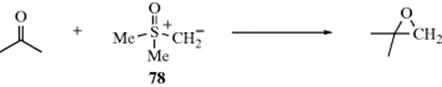
Aldehydes and ketones can be converted to epoxides1438 in good yields with the sulfur ylids1439 dimethyloxosulfonium methylid (78)1440 and dimethylsulfonium methylid (79).1441 Ylid 79 is much less stable
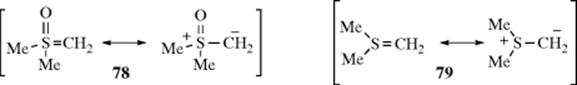
and ordinarily must be used as soon as it is formed, while 78 can be stored several days at room temperature. When diastereomeric epoxides can be formed, 79 usually attacks from the more hindered and 78 from the less-hindered side. Thus, 4-tert-butylcyclohexanone, treated with 78 gave exclusively 80 while 79 gave mostly 81.1442 Another difference in behavior between the two reagents is that with α,β-unsaturated ketones, 78 gives
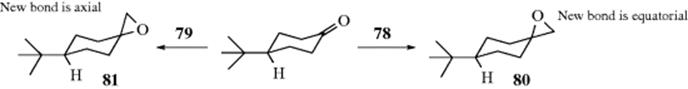
only cyclopropanes (Reaction 15-64), while 79 gives oxirane formation. Other sulfur ylids have been used in an analogous manner, to transfer CHR or CR2.1443 Other sulfur ylids convert aldehydes to epoxides.1444 High yields have been achieved by the use of sulfonium ylids anchored to insoluble polymers under phase-transfer conditions.1445 A solvent-free version of this reaction has been developed using powdered K tert-butoxide and Me3S+I–.1446 Note that treatment of epoxides with 2 equiv of Me2S=CH2 leads to allylic alcohols.1447
Chiral sulfur ylids1448 have been prepared, giving the epoxide with good asymmetric induction,1449 and chiral additives have also been used.1450 Chiral Se ylids have been used in a similar manner.1451
The generally accepted mechanism for the reaction between sulfur ylids and aldehydes or ketone is formation of 82, with displacement of the Me2S leaving group by the alkoxide.1452 This mechanism is similar to that of the reaction of sulfur ylids with C=C double bonds (Reaction 15-64).1453 The stereochemical difference in the behavior of 78 and 79 has been attributed to formation of the betaine (82) being reversible for 78, but not for the less stable 79, so that the more-hindered product is the result of kinetic control and the less hindered of thermodynamic control.1454
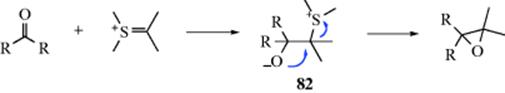
Phosphorus ylids do not give this reaction, but give 16-44 instead.
Aldehydes and ketones can also be converted to epoxides by treatment with a diazoalkane.1455 Most commonly it is diazomethane, but an important side reaction is the formation of an aldehyde or ketone with one more carbon than the starting compound (Reaction 18-9). The reaction can be carried out with many aldehydes, ketones, and quinones, usually with a Rh catalyst.1456 A mechanism that accounts for both products follows:
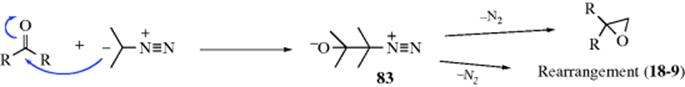
Compound 83 or nitrogen-containing derivatives of it have sometimes been isolated.
An alternative route to epoxides from ketones uses α-chloro sulfones and potassium tert-butoxide to give α,β-epoxy sulfones.1457 A similar reaction was reported using KOH and 10% of a chiral phase-transfer agent, giving moderate enantioselectivity in the epoxy sulfone product.1458
Dihalocarbenes and carbenoids, which readily add to C=C bonds (Reaction 15-64), do not generally add to the C=O bonds of ordinary aldehydes and ketones.1459 See also, Reaction 16-91.
There is a report of a strained azetidinium ylid that has been used for epoxidation.1460
OS V, 358, 755.
16-47 The Formation of Aziridines from Imines
(1+2)NC,CC-cyclo-Methylene-addition

Just as sulfur ylids (e.g., 78) react with the carbonyl of an aldehyde or ketone to give an epoxide, Te ylids react with imines to give an aziridine. The reaction of an allylic Te salt (RCH=CHCH2Te+Bu2 Br−), with lithium hexamethyldisilazide in HMPA/toluene leads to the tellurium ylid via deprotonation. In the presence of an imine, the ylid add to the imine and subsequent displacement of Bu2Te generates an aziridine with a pendant vinyl group.1461Catalytic aziridination of tosylimines was reported and mediated by arsonium ylids.1462
16-48 The Formation of Episulfides and Episulfones1463

Epoxides can be converted directly to episulfides by treatment with NH4SCN and ceric ammonium nitrate.1464 Diazoalkanes, treated with sulfur, give episulfides.1465 It is likely that R2C=S is an intermediate, which is attacked by another molecule of diazoalkane, in a process similar to that shown in Reaction 16-46. Thioketones do react with diazoalkanes to give episulfides,1466 and have also been converted to episulfides with sulfur ylids.1442 Carbenes (e.g., the dichlorocarbene from CHCl3) and base react with thioketones to give an α,α-dichloro episufide.1467
![]()
Alkanesulfonyl chlorides, when treated with diazomethane in the presence of a base (usually a tertiary amine), give episulfones (85).1468 The base removes HCl from the sulfonyl halide to produce the highly reactive sulfene (84) (see Reaction 17-14), which then adds CH2. The episulfone can then be heated to give off SO2 (see Reaction 17-20), making the entire process a method for achieving the conversion RCH2SO2Cl → RCH=CH2.1469
OS V, 231, 877.
16-49 Cyclopropanation of Conjugated Carbonyl Compounds
Double-bond compounds that undergo the Michael reaction (15-24) can be converted to cyclopropane derivatives with sulfur ylids.1470 Among the most common of these is dimethyloxosulfonium methylid (78),1471
which is widely used to transfer CH2 to activated double bonds, but other sulfur ylids have also been used. A combination of DMSO and KOH in an ionic liquid converts conjugated ketones to α,β-cyclopropyl ketones.1472 Both CHR and CR2 can be added in a similar manner with certain nitrogen-containing compounds. For example, ylids1473 (e.g., 86), add various groups to activated double bonds.1474 Sulfur ylids react with allylic alcohols in the presence of MnO2 and molecular sieve 4 Å to give the cyclopropyl aldehyde.1475 Similar reactions have been performed with phosphorus ylids,1476 with pyridinium ylids,1477 and with the compounds (PhS)3CLi and Me3Si(PhS)2CLi.1478 The reactions with ylids such as these of course involve nucleophilic acyl addition. Enantioselective cyclopropanation occurs in the presence of certain organocatalysts1479 or chiral metal catalysts.1480
Other reagents can be used to convert an aldehyde or ketone to a cyclopropane derivative. Tellurium ylids react with conjugated imines to the cyclopropyl aldehyde after hydrolysis of the imine.1481 Conjugated ketones react with Cp2Zr(CH2=CH2) and PMe3 to give a vinyl cyclopropane derivative after treatment with aq H2SO4.1482
16-50 The Thorpe Reaction
N-Hydro-C-(α-cyanoalkyl)-addition
In the Thorpe reaction, the α carbon of one nitrile molecule is added to the CN carbon of another, so this reaction has analogies with the aldol reaction (16-34). The C=NH bond is, of course, hydrolyzable (Reaction 16-2), so β-keto nitriles can be prepared in this manner. The Thorpe reaction can be done intramolecularly, in which case it
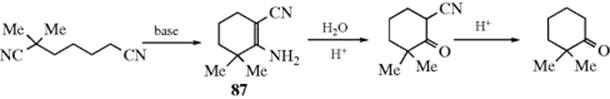
is called the Thorpe–Ziegler reaction.1483 As with other cyclization methods, yields are high for 5–8-membered rings, fall off to about zero for rings of 9–13 members, but are high again for 14-membered and larger rings, if high-dilution techniques are employed. The product in the Thorpe–Ziegler reaction is not the imine, but the tautomeric enamine (e.g., 87); if desired this can be hydrolyzed to an α-cyano ketone (Reaction 16-2), which can in turn be hydrolyzed and decarboxylated (Reactions 16-4 and 12-40). Other active-hydrogen compounds can also be added to nitriles.1484
OS VI, 932.
H Other Carbon or Silicon Nucleophiles
16-51 Addition of Silanes
O-Hydro-C-alkyl-addition
![]()
Allylic silanes react with aldehydes, in the presence of Lewis acids, to give a homoallylic alcohol.1485 In the case of benzylic silanes, this addition reaction has been induced with Mg(ClO4)2 under photochemical conditions.1486Cyclopropylcarbinyl silanes add to acetals in the presence of TMSOTf to give a homoallylic alcohol.1487 Allyltrichlorosilane adds an allyl group to an aldehyde in the presence of a cyclic urea and AgOTf.1488 In a related reaction, allylic silanes react with acyl halides to produce the corresponding carbonyl derivative. The reaction of phenyl chloroformate, allyltrimethylsilane, and AlCl3, for example, gave phenyl but-3-enoate.1489 The use of chiral additives leads to the alcohol with good asymmetric induction.1490
Allylic silanes also add to imines, in the presence of TiCl4, to give amines.1491 Silanes also add to iminium salts, mediated by alkyltin compounds.1492
16-52 The Formation of Cyanohydrins
O-Hydro-C-cyano-addition
![]()
The addition of HCN to aldehydes or ketones produces cyanohydrins.1493 This is an equilibrium reaction, and for aldehydes and aliphatic ketones the equilibrium lies to the right; therefore the reaction is quite feasible, except with sterically hindered ketones (e.g., diisopropyl ketone). However, ketones (ArCOR) give poor yields, and the reaction cannot be carried out with ArCOAr since the equilibrium lies too far to the left. With aromatic aldehydes the benzoin condensation (Reaction 16-55) competes. With α,β-unsaturated aldehydes and ketones, 1,4-addition competes (Reaction 15-38).
Ketones of low reactivity (e.g., ArCOR) can be converted to cyanohydrins by treatment with diethylaluminum cyanide (Et2AlCN) (see OS VI, 307) or, indirectly, with cyanotrimethylsilane (Me3SiCN)1494 in the presence of a Lewis acid or base,1495 followed by hydrolysis of the resulting O-trimethylsilyl cyanohydrin (88). Both direct formation of the cyanohydrin (hydrocyanation) and formation of the cyano-O-silyl ether have been carried out enantioselectively using chiral catalysts,1496 including chiral organocatalysts,1497 or chiral additives.1498 Biocatalysts have been used.1499 Hydrogen cyanide adds to aldehydes in the presence of a lyase to give the cyanohydrin with good enantioselectivity.1500 Cyanohydrins have been formed using a lyase in an ionic liquid.1501
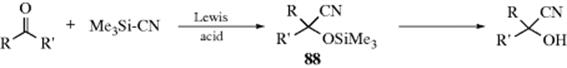
Solvent-free conditions have been reported using TMSCN, an aldehyde, and potassium carbonate.1502 Amine N-oxides catalyze the reaction,1503 as does tetrabutylammonium cyanide.1504 Lithium perchlorate in ether facilitates this reaction,1505 and LiCl catalyzes the reaction with Me3SiCN.1506N-Heterocyclic carbenes catalyze the reaction,1507 as do certain ionic liquids.1508 With MgBr2 as a catalyst, the reaction proceeds with good syn selectivity.1509 Other useful catalysts include Pt1510 Au,1511 or Ti1512 compounds, and InBr3.1513 The use of chiral additives leads to cyanohydrins with good asymmetric induction.1514 Chiral transition metal catalysts have been used to give O-trialkylsilyl cyanohydrins with good enantioselectivity.1515 A venedium catalyst has been used in an ionic liquid.1516 Note that the reaction of an aldehyde and TMSCN in the presence of aniline and a BiCl3 catalyst leads to an α-cyano amine.1517Potassium cyanide and acetic anhydride react with an aldehyde in the presence of a chiral Ti catalyst to give an α-acetoxy nitrile.1518
Rather than direct reaction with an aldehyde or ketone, the bisulfite addition product is often treated with cyanide. The addition is nucleophilic and the actual nucleophile is −CN, so the reaction rate is increased by the addition of base.1519 This was demonstrated by Lapworth in 1903, and consequently this was one of the first organic mechanisms to be known.1520 This method is especially useful for aromatic aldehydes, since it avoids competition from the benzoin condensation. If desired, it is possible to hydrolyze the cyanohydrin in situ to the corresponding α-hydroxy acid. This reaction is important in the Kiliani–Fischer method of extending the carbon chain of a sugar.
A particularly useful variation of this reaction uses cyanide rather than HCN. α-Amino nitriles1521 can be prepared in one step by the treatment of an aldehyde or ketone with NaCN and NH4Cl. This is called the Strecker synthesis;1522 and it is a special case of the Mannich reaction (16-19). Since the CN group is easily hydrolyzed to the acid, this is a convenient method for the preparation of α-amino acids. The reaction has also been carried out with NH3 + HCN and with NH4CN. Salts of primary and secondary amines can be used instead of NH4+ to obtain N-substituted and N,N-disubstituted α-amino nitriles. Br![]() nsted acids can also be used.1523 Unlike Reaction 16-52, the Strecker synthesis is useful for aromatic as well as aliphatic ketones. As in Reaction 16-52, the Me3SiCN method has been used; 76 is converted to the product with ammonia or an amine.1524 The effect of pressure on the Strecker synthesis has been studied.1525 A catalyst-free multi-component Strecker reaction is known.1526 There is also an In mediated Strecker reaction in aq media.1527 Enantioselective Strecker syntheses are possible using chiral ammonium salts1528 and other organocatalysts,1529 chiral acids,1530 or chiral metal complexes.1531 There is a radical version of the Strecker synthesis.1532
nsted acids can also be used.1523 Unlike Reaction 16-52, the Strecker synthesis is useful for aromatic as well as aliphatic ketones. As in Reaction 16-52, the Me3SiCN method has been used; 76 is converted to the product with ammonia or an amine.1524 The effect of pressure on the Strecker synthesis has been studied.1525 A catalyst-free multi-component Strecker reaction is known.1526 There is also an In mediated Strecker reaction in aq media.1527 Enantioselective Strecker syntheses are possible using chiral ammonium salts1528 and other organocatalysts,1529 chiral acids,1530 or chiral metal complexes.1531 There is a radical version of the Strecker synthesis.1532
OS I, 336; II, 7, 29, 387; III, 436; IV, 58, 506; VI, 307; VII, 20, 381, 517, 521. For the reverse reaction, see OS III, 101. For the Strecker synthesis, see OS I, 21, 355; III, 66, 84, 88, 275; IV, 274; V, 437; VI, 334.
16-53 The Addition of HCN to C=N and C![]() N Bonds
N Bonds
N-Hydro-C-cyano-addition
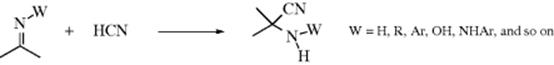
Hydrogen cyanide adds to imines, Schiff bases, hydrazones, oximes, and similar compounds. Cyanide can be added to iminium ions to give α-cyano amines (89). As in Reaction 16-50, the addition to imines has been carried out enantioselectively.1533 Chiral ammonium salts have been used with HCN.1534 Trimethylsilyl cyanide (TMSCN) reacts with N-tosyl

imines in the presence of BF3·OEt2 to give the α-cyano N-tosyl amine.1535 In the presence of a chiral Zr1536 or Al1537 catalyst, Bu3SnCN reacts with imines to give α-cyanoamines enantioselectively. The imine can be formed in situ by reaction of an aldehyde or ketone with an amine, in the present of TMSCN and a suitable promoter.1538 The reaction of an imine and TMSCN gives the cyano amine with good enantioselectivity using a chiral Sc catalyst.1539Titanium catalysts have been used in the presence of a chiral Schiff base.1540
The addition of KCN to triisopropylbenzenesulfonyl hydrazones (90) provides an indirect method for achieving the conversion RR′CO → RR′CHCN.1541 The reaction is successful for hydrazones of aliphatic aldehydes and ketones.
![]()
Hydrogen cyanide can also be added to the C![]() N bond to give iminonitriles or α-aminomalononitriles.1542
N bond to give iminonitriles or α-aminomalononitriles.1542
![]()
The acylcyanation of imines is known, and enantioselectivity is achieved with a suitable catalyst.1543
OS V, 344. See also, OS V, 269.
16-54 The Prins Reaction
The addition of an alkene to formaldehyde in the presence of an acid1544 catalyst is called the Prins reaction.1545 Three main products are possible; which one predominates depends on the alkene and the conditions. When the product is the 1,3-diol or the dioxane,1546 the reaction involves addition to the C=C as well as to the C=O. The mechanism is one of electrophilic attack on both double bonds. The acid first protonates the C=O, and the resulting carbocation is attacked by the C=C to give 91. The cation product 91 can
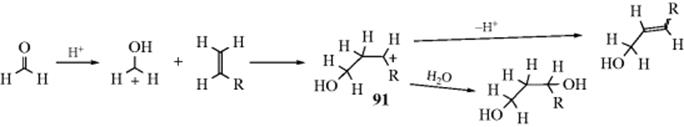
undergo loss of H+ to give the alkene or add water to give the diol.1547 It has been proposed that 91 is stabilized by neighboring-group attraction, with either the oxygen1548 or a carbon1549 stabilizing the charge (92 and 94, respectively). This stabilization is postulated to explain the fact that with 2-butenes1550 and with cyclohexenes the addition is anti. A backside attack of H2O on the three- or four-membered ring would account for it. Other products are obtained too, which can be explained on the basis of 92 or 93.1548,1549 Additional evidence for the intermediacy of 92 is the finding that oxetanes (94) subjected to the reaction conditions, which would protonate 94 to give 92, give essentially the same product ratios as the corresponding alkenes.1551 An argument against the intermediacy of 92 and 93 is that not all alkenes show the anti-stereoselectivity mentioned above. Indeed, the stereochemical results are often quite complex, with syn, anti, and nonstereoselective addition reported, depending on the nature of the reactants and the reaction conditions.1552 Since addition to the C=C bond is electrophilic, the reactivity of the alkene increases with alkyl substitution and Markovnikov's rule is followed. The dioxane product may arise from a reaction between the 1,3-diol and formaldehyde1553 (16-5) or between 92 and formaldehyde. Racemization may occur in the Prins cyclization reaction by 2-oxonia-Cope rearrangements (see 18-32) by way of a (Z)-oxocarbenium ion intermediate.1554
Iodine can promote the Prins reaction.1555 Lewis acids (e.g., SnCl4) also catalyze the reaction, in which case the species that adds to the alkenes is H2C+–O–SnCl4.1556 The reaction can also be catalyzed by peroxides, in which case the mechanism is probably a free radical one. Other transition metal complexes can be used to form homoallylic alcohols. A typical example is the reaction of methylenecyclohexane with an aryl aldehyde to give 95.1557
Samarium iodide promotes this addition reaction.1558 In a related reaction, simple alkene units add to esters in the presence of sodium and liquid ammonia to give an alcohol.1559 Dienes react with alcohols in the presence of a transition metal compound, to give alkenyl alcohols.1560 Allenes also add to aldehydes.1561 Enynes undergo Prins cyclization with Au catalysts.1562
Structural variations in the alkene lead to different products. Homoallylic alcohols react with aldehydes in the presence of Montmorillonite-KSF clay to give 4-hydroxytetrahydropyrans.1563 A variation of this reaction converts an aryl aldehyde and a homoallylic alcohol to a 4-chlorotetrahydropyran in the presence of InCl3.1564 Homoallylic alcohols, protected as –O(CHMeOAc) react with BF3·OEt2 and acetic acid to give 4-acetoxytetrahydropyrans or with SnBr4 to give 4-bromotetrahydropyrans.1565 Homoallylic alcohols with a vinyl silane moiety react with InCl3 and an aldehyde to give a dihydropyran.1566
A closely related reaction has been performed with activated aldehydes or ketones; without a catalyst (e.g., chloral and acetoacetic ester), but with heat.1567 The product in these cases is a β-hydroxy alkene, and the mechanism is pericyclic:1568
This reaction is reversible and suitable β-hydroxy alkenes can be cleaved by heat (17-32). There is evidence that the cleavage reaction occurs by a cyclic mechanism (see 17-32), and, by the principle of microscopic reversibility, the addition mechanism should be cyclic too.1569 Note that this reaction is an oxygen analogue of the ene synthesis (15-23). This reaction can also be done with unactivated aldehydes1570 and ketones1571 if Lewis acid catalysts [e.g., dimethylaluminum chloride (Me2AlCl) or ethylaluminum dichloride (EtAlCl2)] are used.1572 Lewis acid catalysts also increase rates with activated aldehydes.1573 The use of optically active catalysts has given optically active products with a high % ee.1574
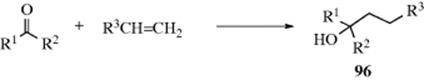
In a related reaction, alkenes can be added to aldehydes and ketones to give reduced alcohols (96). This has been accomplished by several methods,1575 including treatment with SmI21576 or Zn and Me3SiCl,1577 and by electrochemical1578 and photochemical1579 methods. Most of these methods have been used for intramolecular addition and most or all involve free radical intermediates.
There is an aza-Prins reaction, promoted by TiI4 and I2.1580
OS IV, 786. See also, OS VII, 102.
16-55 The Benzoin Condensation
Benzoin aldehyde condensation
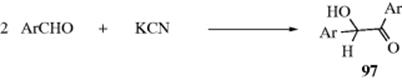
When certain aldehydes are treated with cyanide ion, benzoins (97) are produced in a reaction called the benzoin condensation. The condensation can be regarded as involving the addition of one molecule of aldehyde to the C=O group of another. The reaction only occurs with aromatic aldehydes, but not all of them,1581 and for glyoxals (RCOCHO). The two molecules of aldehyde obviously perform different functions. The one that no longer has a C–H bond in the product is called the donor, because it has “donated” its hydrogen to the oxygen of the other molecule, the acceptor. Some aldehydes can perform only one of these functions, and hence cannot be self-condensed, though they can often be condensed with a different aldehyde. For example, p-dimethylaminobenzaldehyde is not an acceptor, but only a donor. Thus it cannot condense with itself. It can condense with benzaldehyde, which can perform both functions, but is a better acceptor than it is a donor. N-alkyl-3-methylimidazolium salts catalyze the reaction,1582 as does an imidazole-based solid supported catalyst.1583
The following is the accepted mechanism1584 for this reversible reaction, which was originally proposed by Lapworth in 1903:1585
The key step, the loss of the aldehyde proton, can take place because the acidity of this C–H bond is increased by the electron-withdrawing power of the CN group. Thus, cyanide is a highly specific catalyst for this reaction, because, almost uniquely, it can perform three functions: (1) It acts as a nucleophile; (2) its electron-withdrawing ability permits loss of the aldehyde proton; and (3) having done this, it then acts as a leaving group. Certain thiazolium salts can also catalyze the reaction.1586 In this case, aliphatic aldehydes can also be used1587 (the products are called acyloins), and mixtures of aliphatic and aromatic aldehydes give mixed α-hydroxy ketones.1588 The reaction has also been carried out without cyanide, by using the benzoylated cyanohydrin as one of the components in a phase-transfer catalyzed process. By this means, products can be obtained from aldehydes that normally fail to self-condense.1589 The condensation has also been done with excellent enantioselectivity using benzoyl formate decarboxylase.1590 Enantiopure triazolium salts have been evaluated as catalysts in the enantioselective benzoin condensation.1591N-Heterocyclic carbene catalysts have been used for asymmetric induction.1592
A “mixed”-benzoin condensation has been accomplished by using aryl silyl ketones [ArC(=O)SiMe2Ph] and aldehydes with a La catalyst.1593 The reaction of acylsilanes and aldehydes, catalyzed by metal cyanides, is known as the silyl-benzoin reaction.1594
OS I, 94; VII, 95.
16-56 Addition of Radicals to C=O, C=S, C=N Compounds
![]()
Radical cyclization is not limited to reaction with a C=C unit (see 15-29 and 15-30), and reactions with both C=N and C=O moieties are known. Reaction of MeON=CH(CH2)3CHO with Bu3SnH and AIBN, for example, led to trans-2-(methoxyamino)cyclopentanol in good yield.1595 Conjugated ketones add to aldehyde via the β carbon under radical conditions (2 molar equivalents of Bu3SnH and 0.1 equiv of CuCl) to give a β-hydroxy ketone.1596
In a related reaction, diazo compounds add to ketenes to give allenes.1597
Addition of radical to the C=N unit of R–C=N–SPh1598 or R–C=N–OBz1599 led to cyclic imines. Radical addition to simple imines leads to aminocycloalkenes.1600 Radicals also add to the carbonyl unit of phenylthio esters to give cyclic ketones.1601 Carbon-centered radicals add to imines.1602 The reaction of an alkyl halide with BEt3 in aq methanol, for example, gives the imine addition product, an alkylated amine.1603
Secondary alkyl iodides add to O-alkyl oximes in the presence of BEt3 and AIBN. This methodology was used to convert MeO2C-CH=NOBn to MeO2C–CH(R)NOBn.1604 Benzylic halides add to imines under photochemical conditions, and in the presence of 1-benzyl-1,4-dihydronicotinamide1605 or with BEt3 in aq methanol.1606 Tertiary alkyl iodides add to oxime ethers using BF3·OEt2 in the presence of BEt3/O2.1607 O-Trityl oximes of 5- and 6-iodoaldehydes undergo radical cyclization to give oximes1608 (also see Reaction 15-30). Enantioselective radical addition reactions to N-benzoyl hydrazones used chiral ammonium salts.1609 The Mn mediated reaction of allyl iodides with chiral N-acylhydrazones leads to chiral hydrazine derivatives.1610
N,N-Dimethylaniline reacts with aldehydes under photochemical conditions to give acyl addition via the carbon atom of one of the methyl groups.1611 The reaction of PhNMe2 and benzaldehyde, for example, gave PhN(Me)CH2CH(OH)Ph upon photolysis.
16.B.ii. Acyl Substitution Reactions
A. O,N, and S Nucleophiles
16-57 Hydrolysis of Acyl Halides
Hydroxy-de-halogenation
![]()
Acyl halides are so reactive that hydrolysis is easily carried out.1612 In fact, most simple acyl halides must be stored under anhydrous conditions or they may react with water in the air. Consequently, water is usually a strong enough nucleophile for the reaction, but in unreactive systems hydroxide ion may be required. The reaction is seldom synthetically useful, because acyl halides are normally prepared from acids. The reactivity order is F < Cl < Br < I.1613 If a carboxylic acid is used as the nucleophile, an exchange may take place (see Reaction 16-79). The mechanism1613 of hydrolysis can be either SN1 or tetrahedral, the former occurring in highly polar solvents and in the absence of strong nucleophiles.1614 There is also evidence for the SN2 mechanism in some cases.1615
Hydrolysis of acyl halides is not usually catalyzed by acids, except for acyl fluorides, where hydrogen bonding can assist in the removal of F.1616 There are several methods available for the hydrolysis of acyl fluorides.1617
OS II, 74.
16-58 Hydrolysis of Anhydrides
Hydroxy-de-acyloxy-substitution
![]()
Anhydrides are somewhat more difficult to hydrolyze than acyl halides, but here too water is usually a strong enough nucleophile. The mechanism is usually tetrahedral.1618 The SN1 mechanism only occurs with acid catalysis and seldom even then.1619 Anhydride hydrolysis can also be catalyzed by bases. Of course, hydroxide ion attacks more readily than water, but other bases can also catalyze the reaction. This phenomenon, called nucleophilic catalysis(Sec. 16.A.i, category 4), is actually the result of two successive tetrahedral mechanisms. For example, pyridine catalyzes the hydrolysis of acetic anhydride in this manner.1620
Many other nucleophiles similarly catalyze the reaction.
OS I, 408; II, 140, 368, 382; IV, 766; V, 8, 813.
16-59 Hydrolysis of Carboxylic Esters
Hydroxy-de-alkoxylation
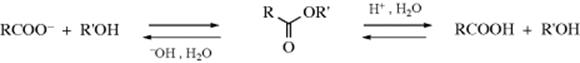
Ester hydrolysis is usually catalyzed by acids or bases. Since OR is a much poorer leaving group than halide or OCOR, water alone does not hydrolyze most esters. When bases catalyze the reaction, the attacking species is the more powerful nucleophile −OH. This reaction is called saponification and gives the salt of the acid. Acids catalyze the reaction by making the carbonyl carbon more positive, and therefore more susceptible to attack by the nucleophile. Both reactions are equilibrium reactions, so there must be a way to shift the equilibrium to the right for this to be useful. Since formation of the salt does just this, ester hydrolysis is almost always done for preparative purposes in basic solution, unless the compound is base sensitive. Even in the case of 98, however, selective base hydrolysis of the ethyl ester gave an 80% yield of the acid–dimethyl ester (99).1621

Ester hydrolysis can also be catalyzed1622 by metal ions, by cyclodextrins,1623 by enzymes,1624 and by nucleophiles.1621 Other reagents used to cleave carboxylic esters include Dowex-50,1625 Me3SiI,1626 and InCl3 on moist silica gel using microwave irradiation.1627 Cleavage of phenolic esters is usually faster than carboxylic esters derived from aliphatic acids. The reagent Sm/I2 at -78 °C has been used,1628 ammonium acetate in aq methanol,1629 Amberlyst 15 in methanol,1630 and phenolic esters have been selectively hydrolyzed in the presence of alkyl esters on alumina with microwave irradiation.1631 Allylic esters were cleaved with DMSO–I2.1632 Thiophenol with K2CO3 in NMP quantitatively converted methyl benzoate to benzoic acid.1633 Allylic esters were cleaved with 2% Me3SiOTf in dichloromethane,1634 with CeCl3·7 H2O–NaI,1635 and with NaHSO4·silica gel.1636 Lactones also undergo the reaction1637 (but if the lactone is five- or six-membered, the hydroxy acid often spontaneously re-forms the lactone) and thiol esters (RCOSR′) give thiols R′SH. Typical reagents for this latter transformation include NaSMe in methanol,1638 borohydride exchange resin–Pd(OAc)2 for reductive cleavage of thiol esters to thiols,1639 and TiCl4/Zn for the conversion of phenylthioacetates to thiophenols.1640 Sterically hindered esters are hydrolyzed with difficulty (Sec. 10.G.i), but reaction of 2 equiv of t-BuOK with 1 equiv of water is effective.1641 Hindered esters can also be cleaved by sequential treatment with zinc bromide and then water,1642 with silica gel in refluxing toluene,1643 and on alumina when irradiated with microwaves.1644 For esters insoluble in water the rate of two-phase ester saponification can be greatly increased by the application of ultrasound,1645 and phase-transfer techniques have been applied.1646
Enzymatic hydrolysis of diesters with esterase has been shown to give the hydroxy-ester,1647 and selective hydrolysis of dimethyl succinate to monomethyl succinic acid was accomplished with aq NaOH in THF.1648 Hydrolysis of vinyl esters leads to ketones, and the reaction of C-substituted vinyl acetates with an esterase derived from Marchantia polymorpha gave substituted ketones with high enantioselectivity.1649 Scandium triflate was shown to hydrolyze α-acetoxy ketones to α-hydroxy ketones.1650
Ingold1651 has classified the acid- and base-catalyzed hydrolyses of esters (and the formation of esters, since these are reversible reactions and thus have the same mechanisms) into eight possible mechanisms (Table 16.3),1651,1652depending on the following criteria: (1) acid- or base catalyzed, (2) unimolecular or bimolecular, and (3) acyl cleavage or alkyl cleavage.1653 All eight of these are SN1, SN2, or tetrahedral mechanisms. The acid-catalyzed mechanisms are shown with reversible arrows. They are not only reversible, but also symmetrical; that is, the mechanisms for ester formation are exactly the same as for hydrolysis, except that H replaces R. Internal proton transfers, such as shown for B and C, may not actually be direct but may take place through the solvent. There is much physical evidence to show that esters are initially protonated on the carbonyl and not on the alkyl oxygen (Chap 8, Ref. 17). Nevertheless the AAC1 mechanism is shown as proceeding through the ether-protonated intermediate A, since it is difficult to envision OR′ as a leaving group here. It is of course possible for a reaction to proceed through an intermediate even if only a tiny concentration is present. The designations AAC1, and so on, are those of Ingold. The AAC2 and AAC1 mechanisms are also called A2 and A1, respectively. Note that the AAC1 mechanism is actually the same as the SN1cA mechanism for this type of substrate and that AAL2 is analogous to SN2cA. Some authors use A1 and A2 to refer to all types of nucleophilic substitution in which the leaving group first acquires a proton. The base-catalyzed reactions are not shown with reversible arrows, since they are reversible only in theory and not in practice. Hydrolyses taking place under neutral conditions are classified as B mechanisms. Molecular dynamics has shown that “the rate of hydrolysis of methyl formate in pure water is consistent with mechanisms involving cooperative catalysis by autoionization-generated hydroxide and hydronium, a process known to have an activation free energy of 23.8 kcal mol–1 (99.6 kJ mol–1).”1654
Table 16.3 Classification of the Eight Mechanisms for Ester Hydrolysis and Formation.
Name
Ingold
IUPACa
Type
AAC1
Ah+DN+AN+Dh
SN1
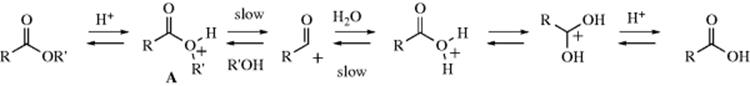
AAC2
Ah+AN+AhDh+Dh
Tetrahedral
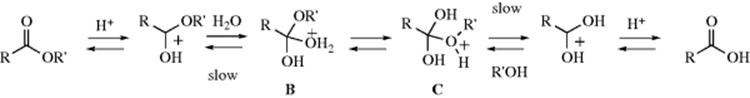
AAL1
Ah+DN+AN+Dh
SN1
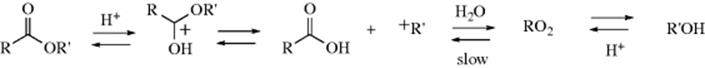
AAL2
Ah+ANDN+Dh
SN2
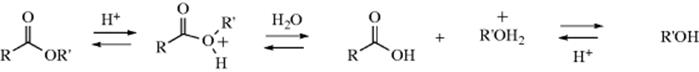
BAC1
DN+AN+AxhDh
SN1
![]()
BAC2
AN+DN+AxhDh
Tetrahedral

BAL1
DN+AN+AxhDh
SN1
![]()
BAL2
ANDN
SN2
![]()
Adapted material from Structure and Mechanism in Organic Chemistry, 2d ed., Cornell University Press, Ithaca, NY, 1969, pp. 1129–1131, edited by Ingold, C.K. Copyright ©1969 by Cornell University. Used by permission of the publisher, Cornell University Press.
a. See Ref. 1652
Of the eight mechanisms, seven have actually been observed in the hydrolysis of carboxylic esters. The one that has not been observed is the BAC1 mechanism.1655 The most common mechanisms are the BAC2 for basic catalysis and the AAC21656 for acid catalysis, that is, the two tetrahedral mechanisms. Both involve acyl–oxygen cleavage. The evidence is (1) hydrolysis with H218O results in the 18O appearing in the acid and not in the alcohol;1657 (2) esters with chiral R′ groups give alcohols with retention of configuration;1658 (3) allylic R′ gives no allylic rearrangement;1659 (4) neopentyl R′ gives no rearrangement1660; all these facts indicate that the O–R′ bond is not broken. It has been concluded that two molecules of water are required in the AAC2 mechanism, as shown:
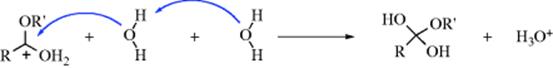
If this were so, the protonated derivatives B and C would not appear at all. This conclusion stems from a value of w (see Sec. 8.C) of ~ 5, indicating that water acts as a proton donor here as well as a nucleophile.1661 Termolecular processes are rare, but in this case the two water molecules are already connected by a hydrogen bond. (A similar mechanism, called BAC3, also involving two molecules of water, has been found for esters that hydrolyze without a catalyst.1662 Such esters are mostly those containing halogen atoms in the R group.)
The other mechanism involving acyl cleavage is the AAC1 mechanism. This is rare, being found only where R is very bulky, so that bimolecular attack is sterically hindered, and only in ionizing solvents. The mechanism has been demonstrated for esters of 2,4,6-trimethylbenzoic acid (mesitoic acid). This acid depresses the freezing point of sulfuric acid four times as much as would be predicted from its molecular weight, which is evidence for the equilibrium
![]()
In a comparable solution of benzoic acid, the freezing point is depressed only twice the predicted amount, indicating only a normal acid–base reaction. Further, a sulfuric acid solution of methyl mesitoate when poured into water gave mesitoic acid, while a similar solution of methyl benzoate similarly treated did not.1663 The AAC1 mechanism is also found when acetates of phenols or of primary alcohols are hydrolyzed in concentrated (>90%) H2SO4 (the mechanism under the more usual dilute acid conditions is the normal AAC2).1664
The mechanisms involving alkyl–oxygen cleavage are ordinary SN1 and SN2 mechanisms in which OCOR (an acyloxy group) or its conjugate acid is the leaving group. Two of the three mechanisms, the BAL1 and AAL1 mechanisms, occur most readily when R′ comes off as a stable carbocation (i.e., when R′ is tertiary alkyl, allylic, benzylic, etc.). For acid catalysis, most esters with this type of alkyl group (especially tertiary alkyl) cleave by this mechanism, but even for these substrates, the BAL1 mechanism occurs only in neutral or weakly basic solution, where the rate of attack by hydroxide is so slowed that the normally slow (by comparison) unimolecular cleavage takes over. These two mechanisms have been established by kinetic studies, 18O labeling, and isomerization of R′.1665 Secondary and benzylic acetates hydrolyze by the AAC2 mechanism in dilute H2SO4, but in concentrated acid the mechanism changes to AAL1.1665 Despite its designation, the BAL1 mechanism is actually uncatalyzed (as is the unknown BAC1 mechanism).
The two remaining mechanisms, BAL2 and AAL2, are very rare. The BAL2 mechanism because it requires hydroxide ion to attack an alkyl carbon when an acyl carbon is also available,1666 and the AAL2 because it requires water to be a nucleophile in an SN2 process. Both have been observed, however. The BAL2 has been seen in the hydrolysis of β-lactones under neutral conditions1667 (because cleavage of the C–O bond in the transition state opens the four-membered ring and relieves strain), the alkaline hydrolysis of methyl 2,4,6-tri-tert-butyl benzoate,1668 and in the unusual reaction:1669
![]()
When it does occur, the BAL2 mechanism is easy to detect, since it is the only one of the base-catalyzed mechanisms that requires inversion at R′. However, in the last example given, the mechanism is evident from the nature of the product, since the ether could have been formed in no other way. The AAL2 mechanism has been reported in the acid cleavage of γ-lactones.1670
To sum up the acid-catalysis mechanisms, AAC2 and AAL1 are the common mechanisms, the latter for R′ that give stable carbocations, the former for practically all the rest. The AAC1 mechanism is rare, being found mostly with strong acids and sterically hindered R. The AAL2 mechanism is even rarer. For basic catalysis, BAC2 is almost universal; BAL1 occurs only with R′ that give stable carbocations and then only in weakly basic or neutral solutions; BAL2 is very rare; and BAC1 has never been observed.
The above results pertain to reactions in solution. In the gas phase,1671 reactions can take a different course, as illustrated by the reaction of carboxylic esters with MeO−, which in the gas phase was shown to take place only by the BAL2 mechanism,1672 even with aryl esters,1673 where this means that an SN2 mechanism takes place at an aryl substrate. However, when the gas-phase reaction of aryl esters was carried out with MeO− ions, each of which was solvated with a single molecule of MeOH or H2O, the BAC2 mechanism was observed.1672
In the special case of alkaline hydrolysis of N-substituted aryl carbamates, there is another mechanism1674 involving elimination–addition:1675
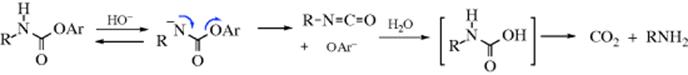
This mechanism does not apply to unsubstituted or N,N-disubstituted aryl carbamates, which hydrolyze by the normal mechanisms. Carboxylic esters substituted in the α position by an electron-withdrawing group (e.g., CN or CO2Et) can also hydrolyze by a similar mechanism involving a ketene intermediate.1676 These elimination–addition mechanisms usually are referred to as E1cB mechanisms, because that is the name given to the elimination portion of the mechanism (Sec. 17.A.iii).
The acid-catalyzed hydrolysis of enol esters (RCOOCR′=CR) can take place either by the normal AAC2 mechanism or by a mechanism involving initial protonation on the double-bond carbon, similar to the mechanism for the hydrolysis of enol ethers given in Reaction 10-6,1677 depending on reaction conditions.1678 In either case, the products are the carboxylic acid (RCO2H) and the aldehyde or ketone (R–CHCOR′).
OS I, 351, 360, 366, 379, 391, 418, 523; II, 1, 5, 53, 93, 194, 214, 258, 299, 416, 422, 474, 531, 549; III, 3, 33, 101, 209, 213, 234, 267, 272, 281, 300, 495, 510, 526, 531, 615, 637, 652, 705, 737, 774, 785, 809 (but see OS V, 1050), 833, 835; IV, 15, 55, 169, 317, 417, 444, 532, 549, 555, 582, 590, 608, 616, 628, 630, 633, 635, 804; V, 8, 445, 509, 687, 762, 887, 985, 1031; VI, 75, 121, 560, 690, 824, 913, 1024; VII, 4, 190, 210, 297, 319, 323, 356, 411; VIII, 43, 141, 219, 247, 258, 263, 298, 486, 516, 527. Ester hydrolyses with concomitant decarboxylation are listed at Reaction 12-40.
16-60 Hydrolysis of Amides
Hydroxy-de-amination
![]()
Unsubstituted amides (RCONH2) can be hydrolyzed with either acidic or basic catalysis, and the products are, respectively, the free acid and the ammonium ion or the salt of the acid and ammonia. N-Substituted (RCONHR′) and N,N-disubstituted (RCONR2′) amides can be hydrolyzed analogously, and the product is the primary or secondary amine, respectively (or their salts), rather than ammonia. Twisting of the amide bond leads to an acceleration of water-promoted hydrolysis reactions.1679 Lactams, imides, cyclic imides, hydrazides, and so on, also undergo the reaction.
Water alone is not sufficient to hydrolyze most amides,1680 since NH2 is even a poorer leaving group than OR.1681 Prolonged heating is often required, even with acidic or basic catalysts.1682 Treatment of primary amides with phthalic anhydride at 250 °C and 4 atm gives the carboxylic acid and phthalimide.1683 Hydrolysis of carbamates (RNHCO2R) to the corresponding amine can be categorized in this section. Although the product is an amine and the carboxyl unit fragments, this reaction is simply a variation of amide hydrolysis. Strong acids [e.g., trifluoroacetic acid (in dichloromethane)] are usually employed.1684 Treatment of N-Boc derivatives (RNHCO2t-Bu) with AlCl31685or with aq sodium tert-butoxide1686 gave the amine. The byproducts of this reaction are typically carbon dioxide and isobutylene.
![]()
In difficult cases, nitrous acid, NOCl, N2O4,1687 or a similar compound can be used (unsubstituted amides only1688). These reactions involve a diazonium ion (see Reaction 13-19) and are much faster than ordinary hydrolysis. The benzamide–nitrous acid reaction took place 2.5 × 107 times faster than ordinary hydrolysis, for example.1689 Another procedure for difficult cases involves treatment with aq sodium peroxide.1690 In still another method, the amide is treated with water and t-BuOK at room temperature.1691 A kinetic study has been done on the alkaline hydrolyses of N-trifluoroacetyl aniline derivatives.1692 Amide hydrolysis can also be catalyzed by nucleophiles (see Sec. 16.A.i, category 4).
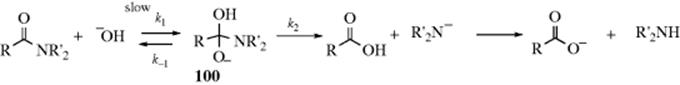
The same framework of eight possible mechanisms discussed for ester hydrolysis in Reaction 16-59 can also be applied to amide hydrolysis.1693 Both the acid- and base-catalyzed hydrolyses are essentially irreversible, since salts are formed in both cases. For basic catalysis1694 the mechanism is BAC2. There is much evidence for this mechanism, similar to that discussed for ester hydrolysis. Molecular orbital studies on the mechanism of amide hydrolysis suggest a highly tetrahedral transition state.1695 In certain cases, kinetic studies have shown that the reaction is second order in −OH indicating that 100 can lose a proton to give 101.1696 Depending on the nature of R′, 101 can cleave directly to give the two negative ions (path a) or become N-protonated prior to or during the act of cleavage (path b), in which case the products are obtained directly and a final proton transfer is not necessary.1697 Studies of the effect, on the rate of hydrolysis and on the ratio k-1/k2, of substituents on the aromatic rings in a series of amides (CH3CONHAr) led to the conclusion that path a is taken when Ar contains
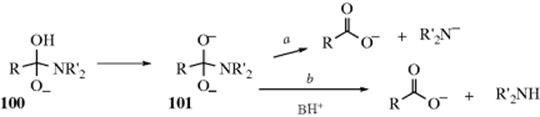
electron-withdrawing substituents and path b when electron-donating groups are present.1698 The presence of electron-withdrawing groups helps stabilize the negative charge on the nitrogen, so that NR2′− can be a leaving group (path a). Otherwise, the C–N bond does not cleave until the nitrogen is protonated (either prior to or in the act of cleavage), so that the leaving group, even in the base-catalyzed reaction, is not NR2′− but the conjugate NHR2′ (path b). It is known that formation of 100 is the rate-determining step in the BAC2 mechanism, but only at high-base concentrations. At lower concentrations of base, the cleavage of 100 or 101 becomes rate determining.1699
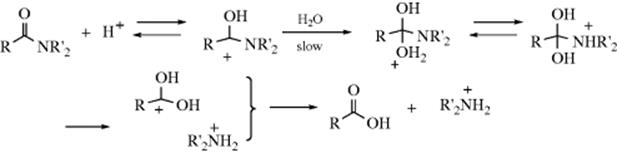
For acid catalysis, matters are less clear. The reaction is generally second order, and it is known that amides are primarily protonated on the oxygen. Because of these facts, it has been generally agreed that most acid-catalyzed amide hydrolysis takes place by the AAC2 mechanism. Further evidence for this mechanism is that a small but detectable amount of 18O exchange (see Sec. 16.A.i, category 3) has been found in the acid-catalyzed hydrolysis of benzamide1700 (18O exchange has also been detected for the base-catalyzed process,1701 in accord with the BAC2 mechanism). Kinetic data have shown that three molecules of water are involved in the rate-determining step,1702suggesting that, as in the AAL2 mechanism for ester hydrolysis (Reaction 16-59), additional water molecules take part in a process, such as:
![]()
The four mechanisms involving alkyl–N cleavage (the AL mechanisms) do not apply to this reaction. They are not possible for unsubstituted amides, since the only N–C bond is the acyl bond. They are possible for N-substituted and N,N-disubstituted amides, but in these cases they give entirely different products and are not amide hydrolyses at all.
![]()
The reaction shown involves attack by the base on an N-alkyl group to give an alcohol. While rare, it has been observed for various N-tert-butylamides in 98% sulfuric acid, where the mechanism was AAL1,1703 and for certain amides containing an azo group, where a BAL1 mechanism was postulated.1704 Of the two first-order acyl cleavage mechanisms, only the AAC1 has been observed, in concentrated sulfuric acid solutions.1705 Of course, the diazotization of unsubstituted amides might be expected to follow this mechanism, and there is evidence that this is true.1689
OS I, 14, 111, 194, 201, 286; II, 19, 25, 28, 49, 76, 208, 330, 374, 384, 457, 462, 491, 503, 519, 612; III, 66, 88, 154, 256, 410, 456, 586, 591, 661, 735, 768, 813; IV, 39, 42, 55, 58, 420, 441, 496, 664; V, 27, 96, 341, 471, 612, 627; VI, 56, 252, 507, 951, 967; VII, 4, 287; VIII, 26, 204, 241, 339, 451.
The oxidation of aldehydes to carboxylic acids can proceed by a nucleophilic mechanism, but more often it does not. The reaction is considered in Reaction 19-23. Basic cleavage of β-keto esters and the haloform reaction could be considered at this point, but they are also electrophilic substitutions and are treated in Reactions 12-43 and 12-44.
B. Attack by OR at an Acyl Carbon
16-61 Alcoholysis of Acyl Halides
Alkoxy-de-halogenation
![]()
The reaction between acyl halides and alcohols or phenols is the best general method for the preparation of carboxylic esters. It is believed to proceed by a SN2 mechanism.1706 As with Reaction 16-57, however, the mechanism can be SN1 or tetrahedral.1613 Lewis acids (e.g., lithium perchlorate) can be used.1707 The reaction is of wide scope, and many functional groups do not interfere. A base is frequently added to combine with the HX formed. When aq alkali is used, this is called the Schotten–Baumann procedure, but pyridine is also frequently used. Indeed, pyridine catalyzes the reaction by the nucleophilic catalysis route (see Reaction 16-58). Both R and R′ may be primary, secondary, or tertiary alkyl or aryl. Enol esters can also be prepared by this method, though C-acylation competes in these cases. In difficult cases, especially with hindered acids or tertiary R′, the alkoxide can be used instead of the alcohol.1708 Activated alumina has also been used as a catalyst, for tertiary R′.1709 Thallium salts of phenols give very high yields of phenolic esters,1710 and BiOCl is very effective for the preparation of phenolic acetates.1711 Phase-transfer catalysis has been used for hindered phenols.1712 Zinc has been used to couple alcohols and acyl chlorides,1713 Zr compounds were used,1714 and catalytic Cu(acac)2 and benzoyl chloride was used to prepare the monobenzoate of ethylene glycol.1715 Selective acylation is possible in some cases.1716
Acyl halides react with thiols, in the presence of Zn, to give the corresponding thio-ester.1717 The reaction of acid chlorides or anhydrides (see 16-62) with diphenyldiselenide, in the presence of Sm/CoCl21718 or Sm/CrCl31719 gave the corresponding seleno ester (PhSeCOMe).
Acyl halides can also be converted to carboxylic acids by using ethers instead of alcohols, as shown, in MeCN in the presence of certain catalysts [e.g., cobalt(II) chloride].1720 A variation of this reaction has been reported that uses acetic anhydride (also see 16-62).1721
![]()
This is a method for the cleavage of ethers (see also, Reaction 10-49).
OS I, 12; III, 142, 144, 167, 187, 623, 714; IV, 84, 263, 478, 479, 608, 616, 788; V, 1, 166, 168, 171; VI, 199, 259, 312, 824; VII, 190; VIII, 257, 516.
16-62 Alcoholysis of Anhydrides
Alkoxy-de-acyloxy-substitution
![]()
The scope of this reaction is similar to that of 16-61. Anhydrides are somewhat less reactive than acyl halides, but they are often used to prepare carboxylic esters. Acids,1722 Lewis acids,1723 and bases (e.g., pyridine) are often used as catalysts.1724 Acetic anhydride and NiCl2 with microwave irradiation converts benzylic alcohols to the corresponding acetate.1725 The monoacetates of 1,2-diols have been prepared using CeCl3 as a catalyst.1726 Pyridine is a nucleophilic-type catalyst (see Reaction 16-58), but DMAP is superior and can be used in cases where pyridine fails.1727 Nonaromatic amidine derivatives have been used to catalyze the reaction with acetic anhydride.1728 Formic anhydride is not a stable compound but esters of formic acid can be prepared by treating alcohols1729 or phenols1730 with acetic-formic anhydride. Cyclic anhydrides give monoesterified dicarboxylic acids (e.g., 102).1731 The asymmetric alcoholysis of cyclic anhydrides has been reviewed.1732
![]()
Alcohols can also be acylated by mixed organic–inorganic anhydrides [e.g., acetic-phosphoric anhydride, MeCOOPO(OH)2,1733 see Reaction 16-68]. Thioesters of the type ArS(C=O)Me have been prepared by simple reaction of thiols and anhydrides in the presence of potassium carbonate,1734 and from diphenyl disulfide and PBu3, followed by treatment with acetic anhydride.1735
OS I, 285, 418; II, 69, 124; III, 11, 127, 141, 169, 237, 281, 428, 432, 690, 833; IV, 15, 242, 304; V, 8, 459, 591, 887; VI, 121, 245, 560, 692; 486; VIII, 141, 258.
16-63 Esterification of Carboxylic Acids
Alkoxy-de-hydroxylation
![]()
The acid-catalyzed esterification of carboxylic acids with alcohols1736 is the reverse of Reaction 16-60 and can be accomplished only if a means is available to drive the equilibrium to the right.1737 There are many ways of doing this, among which are (1) addition of an excess of one of the reactants, usually the alcohol; (2) removal of the ester or the water by distillation; (3) removal of water by azeotropic distillation; and (4) removal of water by use of a dehydrating agent, silica gel,1738 or a molecular sieve. When R′ is methyl, the most common way of driving the equilibrium is by adding excess MeOH; when R′ is ethyl or larger, it is preferable to remove water by azeotropic distillation.1739 The most common catalysts are H2SO4 and TsOH, but some reactive carboxylic acids (e.g., formic,1740 trifluoroacetic1741) do not require a catalyst. Ammonium salts have been used to initiate esterification,1742 and boric acid has been used to esterify α-hydroxy acids.1743 The R′ group may be primary or secondary alkyl groups other than methyl or ethyl, but tertiary alcohols usually give carbocations and elimination. Phenols can sometimes be used to prepare phenolic esters, but yields are generally very low. Selective esterification of an aliphatic carboxylic acid in the presence of an aromatic acid was accomplished with NaHSO4·SiO2 and methanol.1744Diphenylammonium triflate was useful for direct esterification of carboxylic acids with longer chain aliphatic alcohols.1745 Photoirradiation of carboxylic acid with CBr41746 or CCl41747 in methanol was shown to give the methyl ester, with high selectivity for nonconjugated acids in the case of CBr4. Esterification has been accomplished in ionic liquids.1748 A solid-state esterification was reported on P2O5/SiO2.1749 Diols are converted to the monoacetate by heating with acetic acid on a zeolite.1750 Vinyl acetate and iodine has been used for the acetylation of alcohols.1751
O-Alkylisoureas react with conjugated carboxylic acids to give the corresponding ester with microwave irradiation,1752 and a polymer-bound O-alkylurea has been used as well.1753 Transition metal compounds of Ti,1754 or Co1755catalyze esterification. Allylic sulfonium salts react with carboxylic acids to give allylic esters in the presence of CuBr.1756 Triphenylphosphine dibromide is a useful esterification reagent.1757 Phenols can be esterified using amide acetals.1758
The reaction of a carboxylic acid with an alcohol, in the presence of triphenylphosphine and DEAD gives the corresponding ester. This reaction is known as the Mitsunobu reaction (see 10-17). A variation of this esterification reaction used azopyridines to mediate formation of the ester.1759

Both γ- and δ-hydroxy acids (e.g., 103) are easily converted to a lactone by treatment with acids, or often simply on standing, but larger and smaller lactone rings cannot be made in this manner, because polyester formation occurs more readily.1760 Often the conversion of one group to a hydroxyl group, gives the lactone directly, since the hydroxy acid cyclizes too rapidly for isolation. Such groups include keto or halogen that are γ or δ to a carbotyl group. β-Substituted β-hydroxy acids can be converted to β-lactones by treatment with benzenesulfonyl chloride in pyridine at 0–5 °C.1761 ε-Lactones (seven-membered rings) have been made by cyclization of ε-hydroxy acids at high dilution.1762 Macrocyclic lactones1763 can be prepared indirectly in very good yields by conversion of the hydroxy acids to 2-pyridinethiol esters and adding these to refluxing xylene.1764
A closely related method, which often gives higher yields of a macrocyclic lactone, involves treatment of the hydroxy acids with 1-methyl- or 1-phenyl-2-halopyridinium salts, especially 1-methyl-2-chloropyridinium iodide (Mukaiyama's reagent).1765 A macrocyclization technique has been developed based on formation of a mixed anhydride. The Yamaguchi protocol1766 reacts a seco acid (the hydroxy acid precursor of a macrocyclic lactone) with 2,4,6-trichlorobenzoyl chloride. The resulting mixed anhydride is heated with DMAP in toluene.
Esterification is catalyzed by acids (not bases) in ways that were presented in Table 16.3 in Reaction 16-59.1653 The mechanisms are usually AAC2, but AAC1 and AAL1 have also been observed.1767 Certain acids (e.g., 2,6-di-ortho-substituted benzoic acids), cannot be esterified by the AAC2 mechanism because of steric hindrance (Sec. 10.G.i, category 1). In such cases, esterification can be accomplished by dissolving the acid in 100% H2SO4 (forming the ion RCO+) and pouring the solution into the alcohol (AAC1 mechanism). The reluctance of hindered acids to undergo the normal AAC2 mechanism can sometimes be put to advantage when, in a molecule containing two CO2H groups, only the less hindered one is esterified. The AAC1 pathway cannot be applied to unhindered carboxylic acids.
Another way to esterify a carboxylic acid is to treat it with an alcohol in the presence of a dehydrating agent.1738 One of these is dicyclohexylcarbodiimide (DCC), which is converted in the process to dicyclohexylurea (DHU). The mechanism1768 has much in common with the nucleophilic catalysis mechanism; the acid is converted to a compound with a better leaving group, but the conversion is not by a tetrahedral mechanism (as it is in nucleophilic catalysis), since the C–O bond remains intact during this step:
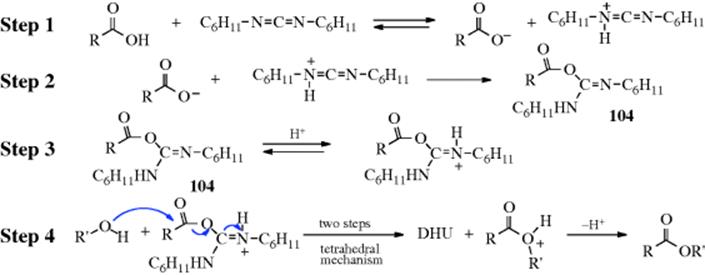
Evidence for this mechanism was the preparation of O-acylureas similar to 104 and the finding that when catalyzed by acids they react with alcohols to give esters.1769 Hindered tertiary alcohols can be coupled via DCC to give the hindered ester.1770 A polymer-bound carbodiimide has been used to prepare macrocyclic lactones.1771 In at least one case, the reaction of HOOCCH2CN with DCC and tert-butanol gave the tert-butyl ester via a ketene intermediate.1772
There are limitations to the use of DCC; yields are variable and N-acylureas are side products. Many other dehydrating agents1773 have been used, including DCC and an aminopyridine,1774 chlorosilanes,1775 and N,N'-carbonyldiimidazole (105).1776 In the latter case, imidazolides (106) are intermediates that react with alcohols.
It is known that the Lewis acid BF3 promotes the esterification by converting the acid to RCO+−BF3–OH, so the reaction proceeds by an AAC1 type of mechanism. The use of BF3–etherate is simple and gives high yields.1777 Other Lewis acids can be used.1778
Carboxylic esters can also be prepared by treating carboxylic acids with tert-butyl ethers and acid catalysts:1779
![]()
Carboxylic esters can be formed from the carboxylate anion and a suitable alkylating agent (Reaction 10-26).
Thioesters of the type RSC(=S)R′ (a dithiocarboxylic ester) and RSC(C=O)R′ (a thiocarboxylic ester) can be generated by reaction of carboxylic acids with thiols. In one example, phosphorous pentasulfide was used in conjunction with a thiol to make dithiocarboxylic esters1780 or thiocarboxylic esters.1781 Thiocarboxylic esters were prepared from thiols and triflic acid.1782
OS I, 42, 138, 237, 241, 246, 254, 261, 451; II, 260, 264, 276, 292, 365, 414, 526; III, 46, 203, 237, 381, 413, 526, 531, 610; IV, 169, 178, 302, 329, 390, 398, 427, 506, 532, 635, 677; V, 80, 762, 946; VI, 471, 797; VII, 93, 99, 210, 319, 356, 386, 470; VIII, 141, 251, 597; IX, 24, 58; 75, 116; 75, 129. Also see, OS III, 536, 742.
16-64 Transesterification
Alkoxy-de-alkoxylation
![]()
Transesterification1783 is catalyzed1784 by acids1785 or bases,1786 or done under neutral conditions.1787 It is an equilibrium reaction that must be shifted in the desired direction.1788 In many cases, low-boiling esters can be converted to higher-boiling ones by the distillation of the lower-boiling alcohol as fast as it is formed. Reagents used to catalyze1789 transesterification include various Lewis acids.1790 Zwitterionic salts have been used as organocatalysts for this reaction.1791 A polymer-bound siloxane has been used to induce transesterification.1792 Vinyl acetate has been used for transesterification, usually with a coreagent or metal mediator.1793 This reaction has been used as a method for the acylation of a primary OH in the presence of a secondary OH.1794 Regioselectivity has also been accomplished by using enzymes (lipases) as catalysts.1795 Lactones, (e.g., 107) are easily opened by treatment with alcohols1796to give open-chain hydroxy esters.
Transesterification has been carried out with phase-transfer catalysts, without an added solvent.1797 Nonionic superbases (see Sec. 8.A.i) of the type P(RNCH2CH2)3N catalyze the transesterification of carboxylic acid esters at 25 °C.1798 Silyl esters (R′CO2SiR3) have been converted to alkyl esters (R′CO2R) via reaction with alkyl halides and tetrabutylammonium fluoride.1799 Thioesters are converted to phenolic esters by treatment with triphosgene–pyridine and then phenol.1800
Transesterification occurs by mechanisms1801 that are identical with those of ester hydrolysis, except that ROH replaces HOH (by the acyl–oxygen fission mechanisms). When alkyl fission takes place, the products are the acid and the ether:
![]()
Therefore, transesterification reactions frequently fail when R′ is tertiary, since this type of substrate most often reacts by alkyl–oxygen cleavage. In such cases, the reaction is of the Williamson type with OCOR as the leaving group (see Reaction 10-10).
With enol esters (e.g., 108), reaction with an alcohol gives an ester and the enol of a ketone, which readily tautomerizes to the ketone as shown. Hence, enol esters are good acylating agents for alcohols.1802 This transformation has been accomplished in ionic liquid media,1803 and there is a PdCl2/CuCl2 mediated version.1804 Isopropenyl acetate can also be used to convert other ketones to the corresponding enol acetates in an exchange reaction:1805
![]()
Enol esters can also be prepared in the opposite type of exchange reaction, catalyzed by mercuric acetate1806 or Pd(II) chloride,1807 for example,
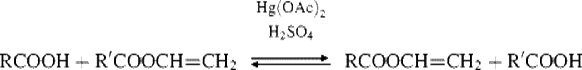
A closely related reaction is equilibration of a dicarboxylic acid and its diester to produce monoesters: The reaction of a carboxylic acid with ethyl acetate, in the presence of NaHSO4·SiO2, was shown to give the corresponding ethyl ester.1808 Iodine catalyzes the transesterification of β-keto esters.1809
OS II, 5, 122, 360; III, 123, 146, 165, 231, 281, 581, 605; IV, 10, 549, 630, 977; V, 155, 545, 863; VI, 278; VII, 4, 164, 411; VIII, 155, 201, 235, 263, 350, 444, 528. See also, OS VII, 87; VIII, 71.
16-65 Alcoholysis of Amides
Alkoxy-de-amidation
![]()
Alcoholysis of amides is possible,1810 although it is usually difficult. It has been most common with the imidazolide type of amides (e.g., 100). For other amides, an activating agent is usually necessary before the alcohol will replace the NR2 unit. N, N-Dimethylformamide, however, reacted with primary alcohols in the presence of 2,4,6-trichloro-1,3,5-pyrazine (cyanuric acid) to give the corresponding formate ester.1811 Treatment of an amide with triflic anhydride (CF3SO2OSO2CF3) in the presence of pyridine, and then with an excess of alcohol, leads to the ester,1812 as does treatment with Me2NCH(OMe)2 followed by the alcohol.1813 Trimethyloxonium tetrafluoroborate converted primary amides to methyl esters.1814 The reaction of acetanilide derivatives with sodium nitrite in the presence of acetic anhydride–acetic acid leads to phenolic acetates.1815 Acyl hydrazides (RCONHNH2) were converted to esters by reaction with alcohols and various reagents,1816 and methoxyamides (RCONHOMe) were converted to esters with TiCl4/ROH.1817 The reaction of an oxazolidinone amide (109) with methanol and 10% MgBr2 gave the corresponding methyl ester.1818
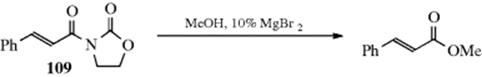
C. Attack by OCOR at an Acyl Carbon
16-66 Acylation of Carboxylic Acids with Acyl Halides
Acyloxy-de-halogenation
![]()
Unsymmetrical, as well as symmetrical, anhydrides are often prepared by the treatment of an acyl halide with a carboxylic acid salt. If a metallic salt is used, Na+, K+, or Ag+ are the most common cations, but more often pyridine or another tertiary amine is added to the free acid. The resulting salt is subsequently treated with the acyl halide. Zinc–DMF has been used to mediate the synthesis of symmetrical anhydrides from acid chlorides.1819 Cobalt(II) chloride (CoCl2) has been used as a catalyst.1820 Mixed formic anhydrides are prepared from sodium formate and an aryl halide, by use of a solid-phase copolymer of pyridine-1-oxide.1821 Symmetrical anhydrides can be prepared by reaction of the acyl halide with aq NaOH or NaHCO3 under phase-transfer conditions,1822 or with sodium bicarbonate with ultrasound.1823
OS III, 28, 422, 488; IV, 285; VI, 8, 910; VIII, 132. See also, OS VI, 418.
16-67 Acylation of Carboxylic Acids with Carboxylic Acids
Acyloxy-de-hydroxylation
![]()
Anhydrides can be formed from two molecules of an ordinary carboxylic acid only if a dehydrating agent is present so that the equilibrium can be driven to the right. Common dehydrating agents1824 are acetic anhydride, trifluoroacetic anhydride, dicyclohexylcarbodiimide,1825 and P2O5. Triphenylphosphine/CCl3CN with triethylamine has also been used with benzoic acid derivatives.1826 The method is very poor for the formation of mixed anhydrides, which in any case generally undergo disproportionation to the two simple anhydrides when they are heated. However, simple heating of dicarboxylic acids does give cyclic anhydrides, provided that the ring formed contains five, six, or seven members, for example:
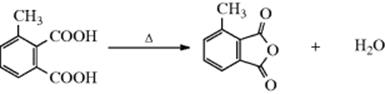
Malonic acid and its derivatives, which would give four-membered cyclic anhydrides, do not give this reaction when heated, but undergo decarboxylation (12-40) instead.
Carboxylic acids exchange with amides and esters; these methods are sometimes used to prepare anhydrides if the equilibrium can be shifted. Enolic esters are especially good for this purpose, because the equilibrium is shifted by formation of the ketone.
![]()
The combination of KF with 2-acetoxypropene under microwave conditions was effective.1827 Carboxylic acids also exchange with anhydrides; indeed, this is how acetic anhydride acts as a dehydrating agent in this reaction.
Anhydrides can be formed from certain carboxylic acid salts (e.g., by treatment of trimethylammonium carboxylates with phosgene):1828
![]()
or of thallium(I) carboxylates with thionyl chloride,1710 or of sodium carboxylates with CCl4 and a catalyst (e.g., CuCl or FeCl2).1829
OS I, 91, 410; II, 194, 368, 560; III, 164, 449; IV, 242, 630, 790; V, 8, 822; IX, 151. Also see, OS VI, 757; VII, 506.
16-68 Preparation of Mixed Organic–Inorganic Anhydrides
Nitrooxy-de-acyloxy-substitution
![]()
Mixed organic–inorganic anhydrides are seldom isolated, although they are often intermediates when acylation is carried out with acid derivatives catalyzed by inorganic acids. Sulfuric, perchloric, phosphoric, and other acids form similar anhydrides, most of which are unstable or not easily obtained because the equilibrium lies in the wrong direction. These intermediates are formed from amides, carboxylic acids, and esters, as well as anhydrides. Organic anhydrides of phosphoric acid are more stable than most others and, for example, RCOOPO(OH)2 can be prepared in the form of its salts.1830 Mixed anhydrides of carboxylic and sulfonic acids (RCOOSO2R′) are obtained in high yields by treatment of sulfonic acids with acyl halides or (less preferred) anhydrides.1831
OS I, 495; VI, 207; VII, 81.
16-69 Attack by SH or SR at an Acyl Carbon1832
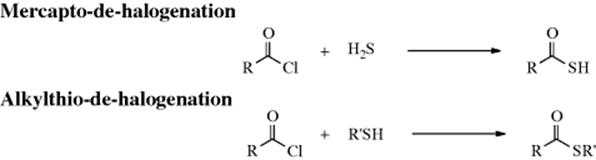
Thiol acids and thiol esters1833 can be prepared in this manner, which is analogous to Reaction 16-57 and 16-64. Anhydrides1834 and aryl esters (RCOOAr)1835 are also used as substrates, but the reagents in these cases are usually HS− and RS−. Thiol esters can also be prepared by treatment of carboxylic acids with P4S10–Ph3SbO,1836 or with a thiol (RSH) and either polyphosphate ester or phenyl dichlorophosphate (PhOPOCl2).1837 Carboxylic acids are converted to thioacids with Lawesson's reagent (structure 18 in Reaction 16-11).1838 Esters RCOOR′ can be converted to thiol esters (RCOSR2) by treatment with trimethylsilyl sulfides (Me3SiSR2) and AlCl3.1839
Alcohols, when treated with a thiol acid and zinc iodide, give thiol esters (R′COSR)1840
OS III, 116, 599; IV, 924, 928; VII, 81; VIII, 71.
16-70 Transamidation
Alkylamino-de-amidation
![]()
It is sometimes necessary to replace one amide group with another, particularly when the group attached to nitrogen functions as a protecting group1841N-Benzyl amides can be converted to the corresponding N-allyl amide with allylamine and Ti catalysts.1842 Reaction of N-Boc 2-phenylethylamine with Ti(OiPr)4 and benzyl alcohol, for example, gives the N-Cbz derivative.1843N-Carbamoyl amines were converted to N-acetyl amines with acetic anhydride, Bu3SnH, and a Pd catalyst1844 Triethylaluminum converts methyl carbamates (ArNHCO2Me) to the corresponding propanamide.1845
A related process reacts acetamide with amines and aluminum chloride to give the N-acetyl amine.1846 Another related process converted imides to O-benzyloxy amides by the Sm catalyzed reaction with O-benzylhydroxylamine.1847
Thioamides can be prepared from amide by reaction with an appropriate sulfur reagent. The reaction of N,N-dimethylacetamide under microwave irradiation, with the polymer-bound reagent 110 (where![]() = polymeric backbone) gave 111.1848 Reaction of the thioamide with Bi(NO3)3·5 H2O regenerates the amide.1849 Other methods are known to convert a thioamide to an amide.1850 Selenoamides [RC(=Se)NR′2] have also been prepared from amides.1851
= polymeric backbone) gave 111.1848 Reaction of the thioamide with Bi(NO3)3·5 H2O regenerates the amide.1849 Other methods are known to convert a thioamide to an amide.1850 Selenoamides [RC(=Se)NR′2] have also been prepared from amides.1851
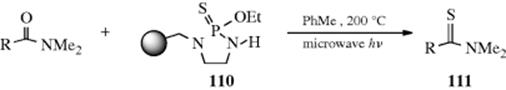
D. Attack by Halogen
16-71 The Conversion of Carboxylic Acids to Halides
Halo-de-oxido,oxo-tersubstitution
![]()
In certain cases, carboxyl groups can be replaced by halide. Acrylic acid derivatives (ArCH=CHCOOH), for example, react with 3 molar equivalents of Oxone in the presence of NaBr to give a vinyl bromide (ArCH=CHBr).1852Diphosphorus tetraiodide/tetraethylammonium bromide (TEAB) readily converts conjugated acids to vinyl bromides.1853 In other cases, conjugated acids, (e.g., 112), have been converted to the bromide by reaction with (NBS, Reaction 14-3) and LiOAc.1854

E. Attack by Nitrogen at an Acyl Carbon1855
16-72 Acylation of Amines by Acyl Halides
Amino-de-halogenation
![]()
The treatment of acyl halides with ammonia or amines is a very general reaction for the preparation of amides.1856 The reaction is exothermic and must be carefully controlled, usually by cooling or dilution. Ammonia gives unsubstituted amides, primary amines give N-substituted amides,1857 and secondary amines give N,N-disubstituted amides. Arylamines can be similarly acylated. Hydroxamic acids have been prepared by this route.1858 In some cases, aq alkali is added to combine with the liberated HCl. This is called the Schotten–Baumann procedure, as in Reaction 16-61. Activated Zn can be used to increase the rate of amide formation when hindered amines and/or acid chlorides are used.1859 A solvent-free reaction was reported using DABCO and methanol.1860 Metal-mediated reactions using In,1861 Sm,1862 or a BiOCl mediated reaction1863 have been reported. A variation of this basic reaction uses DMF with acyl halides to give N,N-dimethylamides.1864 Formic acid and iodine react with amines to give the formamide.1865
Hydrazine and hydroxylamine also react with acyl halides to give, respectively, hydrazides (RCONHNH2)1866 and hydroxamic acids (RCONHOH).1867 When phosgene is the acyl halide, both aliphatic and aromatic primary amines give chloroformamides (ClCONHR) that lose HCl to give isocyanates (RNCO).1868 This is one of the most common methods for the preparation of isocyanates.1869 Similar
![]()
treatment with thiophosgene1870 gives isothiocyanates. A safer substitute for phosgene in this reaction is trichloromethyl chloroformate (CCl3OCOCl).1871 When chloroformates (ROCOCl) are treated with primary amines, carbamates (ROCONHR′) are obtained.1872 An example of this reaction is the use of benzyl chloroformate to protect the amino group of amino acids and peptides.
The PhCH2OCO group in 113 has been called the carbobenzoxy group,1873 and is often abbreviated Cbz or Z, but it is really a benzyl carbamate. Another important group similarly used is Boc, which is a tert-butyl carbamate. In this case, the chloride (Me3COCOCl) is unstable, so the anhydride [(Me3COCO)2O] is used instead, in an example of Reaction 16-73. Amino groups in general are often protected by conversion to amides.1874 The reactions proceed by the tetrahedral mechanism.1875
An interesting variation of this transformation reacts carbamoyl chlorides with organocuprates to give the corresponding amide.1876
OS I, 99, 165; II, 76, 208, 278, 328, 453; III, 167, 375, 415, 488, 490, 613; IV, 339, 411, 521, 620, 780; V, 201, 336; VI, 382, 715; VII, 56, 287, 307; VIII, 16, 339; IX, 559; 81, 254. See also, OS VII, 302.
16-73 Acylation of Amines by Anhydrides
Amino-de-acyloxy-substitution
![]()
This reaction, similar in scope and mechanism1877 to Reaction 16-72, can be carried out with ammonia or primary or secondary amines.1878 Note that there is a report where a tertiary amine (an N-alkylpyrrolidine) reacted with acetic anhydride at 120 °C, in the presence of a BF3·etherate catalyst, to give N-acetylpyrrolidine (an acylative dealkylation).1879 Amino acids can be N-acylated using acetic anhydride and ultrasound.1880 However, ammonia and primary amines can also give imides, in which two acyl groups are attached to the nitrogen. The conversion of cyclic anhydrides to cyclic imides is generally facile,1881 although elevated temperatures are occasionally required to generate the imide.1882 Microwave irradiation of formamide and a cyclic anhydride generates the cyclic imide.1883 Cyclic imides have also been formed in ionic liquids.1884 Cyclic imides were also formed by microwave irradiation of a polymer-bound phthalate after initial reaction with an amine.1885
The second step for imide formation, which is much slower than the first, is the attack of the amide nitrogen on the carboxylic carbon. Unsubstituted and N-substituted amides have been used instead of ammonia. Since the other product of this reaction is RCOOH, this is a way of “hydrolyzing” such amides in the absence of water.1886
Even though formic anhydride is not a stable compound (see Reaction 11-17), amines can be formylated with the mixed anhydride of acetic and formic acids (HCOOCOMe)1887 or with a mixture of formic acid and acetic anhydride. Acetamides are not formed with these reagents. Secondary amines can be acylated in the presence of a primary amine by conversion to their salts and addition of 18-crown-6.1888 The crown ether complexes the primary ammonium salt, preventing its acylation, while the secondary ammonium salts, which do not fit easily into the cavity, are free to be acylated. Dimethyl carbonate can be used to prepare methyl carbamates in a related procedure.1889N-Acetylsulfonamides were prepared from acetic anhydride and a primary sulfonamide, catalyzed by Montmorillonite K10–FeO1890 or sulfuric acid.1891
There are acylating reagents other than anhydrides of course. The reaction with acyl halides is discussed in Reaction 16-72. There are a few specialized reagents. Kinetic resolution of racemic amines was accomplished using (1S,2S)-N-acetyl-1,2- bis(trifluoromethanesulfonamido)cyclohexane.1892
OS I, 457; II, 11; III, 151, 456, 661, 813; IV, 5, 42, 106, 657; V, 27, 373, 650, 944, 973; VI, 1; VII, 4, 70; VIII, 132; 76, 123.
16-74 Acylation of Amines by Carboxylic Acids
Amino-de-hydroxylation
![]()
When carboxylic acids are treated with ammonia or amines, salts are obtained. The salts of ammonia or primary or secondary amines can be pyrolyzed to give amides,1893 but the method is less convenient than Reaction 16-72, 16-73, and 16-75 and is seldom of preparative value.1894 Heating in the presence of a base (e.g., hexamethyldisilazide) makes the amide-forming process more efficient.1895 Boronic acids catalyze the direct conversion of carboxylic acid and amine to amides.1896 Polymer-bound reagents have also been used.1897 Triphenylphosphine/trichloroisocyanuric acid converts acids and amides to the amide.1898 The Burgess reagent (Et3N+–SO2N–CO2Me; see Reaction 17-29) activates carboxylic acids for amide formation.1899 The reaction of a carboxylic acid and imidazole under microwave irradiation gives the amide.1900 Microwave irradiation of a secondary amine, formic acid, 2-chloro-4,6-dimethoxy[1,3,5]triazine, and a catalytic amount of DMAP leads to the formamide.1901 Ammonium bicarbonate and formamide converts acids to amides with microwave irradiation.1902 Formamides are produced from formic acid and anion nitriles in the presence of ZnO.1903
Lactams are readily produced from γ- or δ-amino acids,1904 for example,

This lactonization process can be promoted by enzymes (e.g., pancreatic porcine lipase).1905 Reduction of ω-azide carboxylic acids leads to macrocyclic lactams.1906
Although treatment of carboxylic acids with amines does not directly give amides, the reaction can be made to proceed in good yield at room temperature or slightly above by the use of coupling agents,1907 the most important of which is dicyclohexylcarbodiimide. This reagent is very convenient and is used1908 a great deal in peptide synthesis.1909 A polymer-supported carbodiimide has been used.1910 The mechanism is probably the same as in Reaction 16-63 up to the formation of 114. This intermediate is then attacked by another molecule of RCOO− to give the anhydride (RCO)2O, which is the actual species that reacts with the amine:

The anhydride has been isolated from the reaction mixture and then used to acylate an amine.1911
The synthetically important Weinreb amides [RCON(Me)OMe, see Reaction 16-82] can be prepared from the carboxylic acid and MeO(Me)NH·HCl in the presence of tributylphosphine and 2-pyridine-N-oxide disulfide.1912 Di(2-pyridyl)carbonate has been used in a related reaction that generates amides directly.1913 Other promoting agents1914 are ArB(OH)2 reagents,1915N,N'-carbonyldiimidazole (115, in Reaction 16-63),1916 POCl3,1917 TiCl4,1918 molecular sieves,1919Lawesson's reagent (Reaction 16-11),1920 and (MeO)2POCl.1921 Certain dicarboxylic acids form amides simply on treatment with primary aromatic amines. In these cases, the cyclic anhydride is an intermediate and is the species actually attacked by the amine.1922 Carboxylic acids can also be converted to amides by heating with amides of carboxylic acids (exchange),1923 sulfonic acids, or phosphoric acids, for example,1924
or by treatment with trisalkylaminoboranes [B(NHR′)3], with trisdialkylaminoboranes [B(NR2')3],1925
![]()
or with bis(diorganoamino)magnesium reagents [(R2N)2Mg].1926 The reaction of thiocarboxylic acids and azides, in the presence of triphenylphosphine, gives the corresponding amide.1927
An important technique, discovered by R.B. Merrifield1928 and since used for the synthesis of many peptides,1929 is called solid-phase synthesis or polymer-supported synthesis.1930 The reactions used are the same as in ordinary synthesis, but one of the reactants is anchored onto a solid polymer. For example, if it is desired to couple two amino acids (to form a dipeptide), the polymer selected might be polystyrene with CH2Cl side chains. One of the amino acids, protected by (Boc), would then be coupled to the side chains. It is not necessary that all the side chains be converted, but a random selection will be converted. The Boc group is then removed by hydrolysis with trifluoroacetic acid in CH2Cl2 and the second amino acid is coupled to the first, using DCC or some other coupling agent. The second Boc group is removed, resulting in a dipeptide that is still anchored to the polymer. If this dipeptide is the desired product, it can be cleaved from the polymer by various methods,1931 one of which is treatment with HF. If a longer peptide is wanted, additional amino acids can be added by repeating the requisite steps.
The basic advantage of the polymer support techniques is that the polymer (including all chains attached to it) is easily separated from all other reagents, because it is insoluble in the solvents used. Excess reagents, other reaction products (e.g., dicyclohexylurea), side products, and the solvents themselves are quickly washed away. Purification of the polymeric species is rapid and complete. The process can even be automated,1932 to the extent that six or more amino acids can be added to a peptide chain in one day. Commercial automated peptide synthesizers are now available.1933
Although the solid-phase technique was first developed for the synthesis of peptide chains and has seen considerable use for this purpose, it has also been used to synthesize chains of polysaccharides and polynucleotides; in the latter case, solid-phase synthesis has almost completely replaced synthesis in solution.1934 The technique has been applied less often to reactions in which only two molecules are brought together (nonrepetitive syntheses), but many examples have been reported.1935 Combinatorial chemistry in some ways can be viewed as an extension of the Merrifield synthesis, particularly when applied to peptide synthesis, and continues as an important part of modern organic chemistry.1936
OS I, 3, 82, 111, 172, 327; II, 65, 562; III, 95, 328, 475, 590, 646, 656, 768; IV, 6, 62, 513; V, 670, 1070; VIII, 241; 81, 262. Also see, OS III, 360; VI, 263; VIII, 68.
16-75 Acylation of Amines by Carboxylic Esters
Amino-de-alkoxylation
![]()
The conversion of carboxylic esters to amides is a useful reaction, and unsubstituted, N-substituted, and N,N-disubstituted amides can be prepared this way from the appropriate amine1937 or ammonia.1938 Both R and R′ can be alkyl or aryl, but an especially good leaving group is p-nitrophenyl. Ethyl trifluoroacetate was found to react selectively with primary amines to form the corresponding trifluoroacetyl amide.1939 Many simple esters (R = Me, Et, etc.) are not very reactive, and strongly basic catalysis has been used in such cases,1940 but catalysis by cyanide ion1941 MgBr2,1942 InI3,1943 and acceleration by high pressure1944 have been reported. Methyl esters1945 and ethyl esters1946have been converted to the corresponding amide under microwave irradiation. Lithium amides have been used to convert esters to amides as well.1947 β-Keto esters undergo the reaction especially easily.1948 Aniline was treated with n-butyllithium to form the lithium amide, which reacted with an ester to give the amide.1949 An enzyme-mediated amidation is known using amino cyclase I.1950 The reaction of dimethyl carbonate and an amine is an effective way to prepare methyl carbamates.1951
Lactones give lactams when treated with ammonia or primary amines. Lactams are also produced from γ- and δ-amino esters in an internal example of this reaction. Lactonization has been accomplished in ionic liquids.1952
As in Reaction 16-72, hydrazides and hydroxamic acids can be prepared from carboxylic esters, with hydrazine and hydroxylamine,1953 respectively. Both hydrazine and hydroxylamine react more rapidly than ammonia or primary amines (the alpha effect, Sec. 10.G.ii). Imidates [RC(=NH)OR′] give amidines [RC(=NH)NH2]. Isopropenyl formate is a useful compound for the formylation of primary and secondary amines.1954
![]()
Although more studies have been devoted to the mechanism of the acylation of amines with carboxylic esters than with other reagents, the mechanistic details are not yet entirely clear.1955 In its broad outlines, the mechanism appears to be essentially BAC2.1956 Under normal basic conditions, the reaction is general base catalyzed,1957 indicating that a proton is being transferred in the rate-determining step and that two molecules
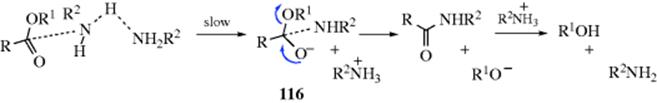
of amine are involved.1958 Alternatively, another base (e.g., H2O or OH−) can substitute for the second molecule of amine. With some substrates and under some conditions, especially at low pH, the breakdown of 116 can become rate determining.1959 The reaction also takes place under acidic conditions and is general acid catalyzed, so that breakdown of 116 is rate determining and proceeds as follows:1960
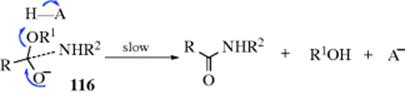
Here HA may be R2NH3+ or another acid. Intermediate 116 may or may not be further protonated on the nitrogen. Even under basic conditions, a proton donor may be necessary to assist leaving-group removal. Evidence for this is that the rate is lower with NR2− in liquid ammonia than with NHR2 in water, apparently owing to the lack of acids to protonate the leaving oxygen.1961
In the special case of β-lactones, where small-angle strain is an important factor, alkyl–oxygen cleavage is observed (BAL2 mechanism, as in the similar case of hydrolysis of β-lactones, Reaction 16-59), and the product is not an amide but a β-amino acid (β-alanine).
![]()
A similar result has been found for certain sterically hindered esters.1962 This reaction is similar to 10-31, with OCOR as the leaving group. Other lactones have been opened to ω-hydroxy amides with Dibal-H:BnNH2.1963
OS I, 153, 179; II, 67, 85; III, 10, 96, 108, 404, 440, 516, 536, 751, 765; IV, 80, 357, 441, 486, 532, 566, 819; V, 168, 301, 645; VI, 203, 492, 620, 936; VII, 4, 30, 41, 411; VIII, 26, 204, 528. Also see, OS I, 5; V, 582; VII, 75.
16-76 Acylation of Amines by Amides
Alkylamino-de-amination
![]()
This is an exchange reaction and is usually carried out with the salt of the amine.1964 The leaving group is usually NH2 rather than NHR or NR2 and primary amines (in the form of their salts) are the most common reagents. Boron trifluoride can be added to complex with the leaving ammonia. Neutral amines also react in some cases to give the new amide.1965 The reaction is catalyzed by Al(III).1966 The reaction is often used to convert urea to substituted ureas: NH2CONH2 + RNH3+ → NH2CONHR + NH4+.1967 An N-aryl group of a urea can be converted to a N,N-dialkyl group by heating the urea with the amine in an autoclave.1968N-alkyl substituted amides (alkyl = R) are converted to N-R′-substituted amides, where alkyl = R′, by treatment with N2O4 to give an N-nitroso compound, followed by treatment of this with a primary amine (R′NH2).1969 Lactams can be converted to ring-expanded lactams (e.g., 117) if a side chain containing an amino group is present on the nitrogen. A strong base is used to convert the NH2 to NH–, which then acts as a nucleophile, expanding the ring by means of a transamidation.1970 The discoverers call it the Zip reaction, by analogy with the action of zippers.1971
Lactams can be opened to ω-amino amides by reaction with amines at 10 kbar.1972
OS I, 302 (but see V, 589), 450, 453; II, 461; III, 151, 404; IV, 52, 361. See also, OS VIII, 573.
16-77 Acylation of Amines by Other Acid Derivatives
Acylamino-de-halogenation or dealkoxylation
![]()
Acid derivatives that can be converted to amides include thiol acids (RCOSH), thiol esters (RCOSR),1973 acyloxyboranes [RCOB(OR′)2],1974 α-keto nitriles, acyl azides, and nonenolizable ketones (see the Haller–Bauer Reaction12-34). N-Acylsulfonamides react with primary amines to the amide (AcNHR).1975 Carbonylation reactions can be used to prepare amides and related compounds. The reaction of a primary amine, an alkyl halide with CO2, in the presence of Cs2CO3/Bu4NI, gave the corresponding carbamate.1976
OS III, 394; IV, 6, 569; V, 160, 166; VI, 1004.
Imides can be prepared by the attack of amides or their salts on acyl halides, anhydrides, and carboxylic acids or esters.1977 A good synthetic method for the preparation of acyclic imides is the reaction between an amide and an anhydride at 100 °C catalyzed by H2SO4.1978 When acyl chlorides are treated with amides in a 2: 1 molar ratio at low temperatures in the presence of pyridine, the products are N,N-diacylamides [(RCO)3N].1979
This reaction is often used to prepare urea derivatives, an important example being the preparation of barbituric acid (118).1980
When the substrate is oxalyl chloride (ClCOCOCl) and the reagent an unsubstituted amide, an acyl isocyanate (RCONCO) is formed. The “normal” product (RCONHCOCOCl) does not form, or if it does, it rapidly loses CO and HCl.1981
OS II, 60, 79, 422; III, 763; IV, 245, 247, 496, 566, 638, 662, 744; V, 204, 944.
16-78 Acylation of Azides
![]()
The reaction of an aldehyde with sodium azide and Et4+ I(OAc)2 or polymer-bound PhI(OAc)2 leads to an acyl azide.1982 Acyl azides are also prepared directly from aldehydes using tert-butyl hypochlorite.1983
F. Attack by Halogen at an Acyl Carbon
16-79 Formation of Acyl Halides from Carboxylic Acids
Halo-de-hydroxylation
![]()
The same inorganic acid halides that convert alcohols to alkyl halides (Reaction 10-48) also convert carboxylic acids to acyl halides.1984 The reaction is the best and the most common method for the preparation of acyl chlorides. Bromides and iodides1985 are also made in this manner, but much less often. Acyl bromides can be prepared with BBr3 on alumina,1986 or with ethyl tribromoacetae/PPh3.1987 Thionyl chloride1988 is a good reagent, since the byproducts are gases and the acyl halide is easily isolated, but PX3 and PX5 (X = Cl or Br) are also commonly used.1989 Hydrogen halides do not give the reaction. A particularly mild procedure, similar to one mentioned in Reaction 10-48, involves reaction of the acid with Ph3P in CCl4, whereupon acyl chlorides are produced without obtaining any acidic compound as a byproduct.1990
Oxalyl chloride (113) and oxalyl bromide are mild and often superior reagents, since the oxalic acid byproduct decomposes to CO and CO2, and the equilibrium is thus driven to the side of the other acyl halide.1991 These reagents are commonly the reagent of choice, particularly when sensitive functionality is present elsewhere in the molecule.
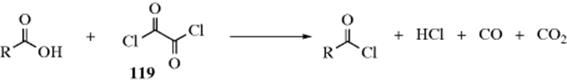
Acyl fluorides can be prepared by treatment of carboxylic acids with cyanuric fluoride.1992C,N-Chelated di-n-butyltin(IV) fluoride has been used to prepare acyl fluorides.1993 Acid salts are also sometimes used as substrates and acyl halides are used as reagents in an exchange reaction:
![]()
which probably involves an anhydride intermediate. This is an equilibrium reaction that must be driven to the desired side.
OS I, 12, 147, 394; II, 74, 156, 169, 569; III, 169, 490, 547, 555, 613, 623, 712, 714; IV, 34, 88, 154, 263, 339, 348, 554, 608, 616, 620, 715, 739, 900; V, 171, 258, 887; VI, 95, 190, 549, 715; VII, 467; VIII, 441, 486, 498.
16-80 Formation of Acyl Halides from Acid Derivatives
Halo-de-acyloxy-substitution
Halo-de-halogenation
![]()
These reactions are most important for the preparation of acyl fluorides.1994 Acyl chlorides and anhydrides can be converted to acyl fluorides by treatment with polyhydrogen fluoride–pyridine solution1995 or with liquid HF at –10 °C.1996 Formyl fluoride, which is a stable compound, was prepared by the latter procedure from the mixed anhydride of formic and acetic acids.1997 Acyl fluorides can also be obtained by reaction of acyl chlorides with KF in acetic acid1998 or with diethylaminosulfur trifluoride (DAST).1999 Carboxylic esters and anhydrides can be converted to acyl halides other than fluorides by the inorganic acid halides mentioned in Reaction 16-79, as well as with Ph3PX2 (X = Cl or Br),2000 but this is seldom done. Halide exchange can be carried out in a similar manner. When halide exchange is done, it is always acyl bromides and iodides that are made from chlorides, since chlorides are by far the most readily available.2001
OS II, 528; III, 422; V, 66, 1103; IX, 13. See also, OS IV, 307.
G. Attack by Carbon at an Acyl Carbon2002
16-81 The Conversion of Acyl Halides to Ketones with Organometallic Compounds2003
Alkyl-de-halogenation
![]()
Acyl halides react cleanly and under mild conditions with lithium dialkylcopper reagents (see Reaction 10-58)2004 to give high yields of ketones.2005 The R′ group may be primary, secondary, or tertiary alkyl or aryl and may contain iodo, keto, ester, nitro, or cyano groups. The R groups that have been used successfully are methyl, primary alkyl, and vinylic. Secondary and tertiary alkyl groups can be introduced by the use of PhS(R)CuLi (Reaction 10-58) instead of R2CuLi,2006 or by the use of either the mixed homocuprate [(R′SO2CH2CuR)− Li+],2007 or a magnesium dialkylcopper reagent ‘(RMeCuMgX).’2008 Secondary alkyl groups can also be introduced with the copper–zinc reagents [RCu(CN)ZnI].2009 The R group may be alkynyl if a cuprous acetylide (R2C≡CCu) is the reagent.2010 Organocopper reagents generated in situ from highly reactive copper, and containing such functional groups as cyano, chloro, and ester, react with acyl halides to give ketones.2011
When the organometallic compound is a Grignard reagent,2012 ketones are generally not obtained because the initially formed ketone reacts with a second molecule of RMgX to give the salt of a tertiary alcohol (Reaction 16-82). Ketones have been prepared in this manner by the use of low temperatures, inverse addition (i.e., addition of the Grignard reagent to the acyl halide rather than the other way), excess acyl halide, and so on, but the yields are usually low, although high yields have been reported in THF at –78 °C.2013 Pretreatment with a trialkylphosphine followed by the Grignard reagent gave the ketone.2014 Using CuBr2015 or a Ni catalyst2016 with the Grignard reagent can lead to the ketone. Some ketones are unreactive toward Grignard reagents for steric or other reasons; these can be prepared in this way.2017 Other methods involve running the reaction in the presence of Me3SiCl2018, which reacts with the initial adduct in the tetrahedral mechanism (Sec. 16.A.i), and the use of a combined Grignard–LiNEt2 reagent.2019 Certain metallic halides, notably ferric and cuprous halides, are catalysts that improve the yields of ketone at the expense of tertiary alcohol.2020 For this catalysis, both free radical and ionic mechanisms have been proposed.2021
Grignard reagents react with ethyl chloroformate to give carboxylic esters [EtOCOCl + RMgX → EtOCOR].
The Pd catalyzed reaction of an acyl chloride and an arylboronic acid2022 or an alkenylboronic acid2023 gives a ketone. Surfactants are known to promote arylboronic acid coupling reactions.2024 Arylboronic esters add to carbamoyl halides, in the presence of a Pd catalyst, to give the corresponding benzamide.2025 Arylboronic acids also react with anhydrides to give a ketone in the presence of a Pd catalyst.2026 Similar reaction of acid chlorides, NaBPh4, KF, and a Pd catalyst gave the aryl ketone.2027
Other organometallic reagents2028 give good yields of ketones when treated with acyl halides because, as with R2CuLi or R2Cd, these compounds do not generally react with the ketone product. A particularly useful class of organometallic reagent are the organocadmium reagents (R2Cd), prepared from Grignard reagents (Reaction 12-22). In this case, R may be an aryl or primary alkyl. In general, secondary and tertiary alkylcadmium reagents are not stable enough to be useful in this reaction.2029 An ester group may be present in either R′COX or R2Cd. Direct treatment of the acid chloride with an alkyl halide and Cd metal leads to the ketone in some cases.2030 Organozinc compounds behave similarly to dialkylcadmium reagents, but are used less often.2031 Organotin reagents (R4Sn) react with acyl halides to give high yields of ketones, if a Pd complex is present.2032 Organolead reagents (R4Pb) behave similarly.2033 Allylic halides and In metal react with acyl chlorides to give the ketone.2034 Various other groups (e.g., nitrile, ester, and aldehyde) can be present in the acyl halide without interference. Other reagents include organomanganese compounds2035 (R can be primary, secondary, or tertiary alkyl, vinylic, alkynyl, or aryl), organozinc,2036 organobismuth,2037 and organothallium compounds (R can be primary alkyl or aryl).2038 The reaction of an α-halo-ketone and an acyl chloride with SmI2 leads to a β-diketone.2039
Antimony alkynes (e.g., Ph2Sb–C![]() C–Ph) react with acid chloride in the presence of a Pd catalyst to give the conjugated alkynyl ketone.2040 Such conjugated ketones can also be prepared from an acyl halide, a terminal alkyne, and a CuI,2041 Pd,2042 or Fe catalyst,2043 or with In metal.2044 Terminal alkynes react with chloroformates and a Pd catalyst to give the corresponding propargyl ester.2045 Similar reaction of an alkyne with an acid chloride and a Pd–Cu2046 or CuI catalyst,2047 both with microwave irradiation, gave alkynyl ketones.
C–Ph) react with acid chloride in the presence of a Pd catalyst to give the conjugated alkynyl ketone.2040 Such conjugated ketones can also be prepared from an acyl halide, a terminal alkyne, and a CuI,2041 Pd,2042 or Fe catalyst,2043 or with In metal.2044 Terminal alkynes react with chloroformates and a Pd catalyst to give the corresponding propargyl ester.2045 Similar reaction of an alkyne with an acid chloride and a Pd–Cu2046 or CuI catalyst,2047 both with microwave irradiation, gave alkynyl ketones.
Acyl halides can also be converted to ketones by treatment with Na2Fe(CO)4 followed by R′X (Reaction 10-76).
OS II, 198; III, 601; IV, 708; VI, 248, 991; VII, 226, 334; VIII, 268, 274, 371, 441, 486.
16-82 The Conversion of Anhydrides, Carboxylic Esters, or Amides to Ketones with Organometallic Compounds2048
Dialkyl,hydroxy-de-alkoxy,oxo-tersubstitution; Alkyl-de-acyloxy- or de-amido substitution
![]()
When carboxylic esters are treated with Grignard reagents, addition to the carbonyl (Reaction 16-24) generates a ketone. Under the reaction conditions, the initially formed ketone usually undergoes acyl substitution of R2 for OR′ (Reaction 16-81), so that tertiary alcohols are formed in which two R groups are the same. Isolation of the ketone as the major product is possible in some cases, particularly when the reaction is done at low temperature2049 and when there is steric hindrance to the carbonyl in the first-formed ketone. Esters (RCO2Me) react with Zn(BH4)2/EtMgBr to give an alcohol [RCH(OH)Et].2050 Formates give secondary alcohols and carbonates give tertiary alcohols in which all three R groups are the same: (EtO)2C=O + RMgX → R3COMgX. Acyl halides and anhydrides behave similarly, though these substrates are employed less often.2051 Many side reactions are possible, especially when the acid derivative or the Grignard reagent is branched: enolizations, reductions (not for esters, but for halides), condensations, and cleavages, but the most important is simple substitution (16-81), which in some cases can be made to predominate. When 1,4-dimagnesium compounds are used, carboxylic esters are converted to cyclopentanols.2052 1,5-Dimagnesium compounds give cyclohexanols, but in lower yields.2053
![]()
As is the case with acyl halides (Reaction 16-81), anhydrides and carboxylic esters give tertiary alcohols (Reaction 16-82) when treated with Grignard reagents. Low temperatures,2054 the solvent HMPA,2055 and inverse addition have been used to increase the yields of ketone.2056 Amides give better yields of ketone at room temperature, but still not very high.2057 Anhydrides can react with arylmagnesium halides at low temperature, and in the presence of (−)-sparteine, to give a keto acid with good enantioselectivity.2058 Organocadmium reagents are less successful with these substrates than with acyl halides (Reaction 16-81). Esters of formic acid, dialkylformamides, and lithium or sodium formate2059 give good yields of aldehydes, when treated with Grignard reagents.
![]()
Organolithium compounds have been used to give ketones from carboxylic esters. The reaction must be carried out in a high-boiling solvent (e.g., toluene), since reaction at lower temperatures gives tertiary alcohols.2060Organolithium reagents also give good yields of carbonyl compounds with N,N-disubstituted amides.2061 Dialkylformamides react to give aldehydes, other disubstituted amides give ketones and other acid derivatives have been used.2062
Ketones can also be obtained by treatment of the lithium salt of a carboxylic acid with an organolithium reagent (Reaction 16-28). For an indirect way to convert carboxylic esters to ketones, see Reaction 16-82. A similar reaction with hindered aryl carboxylic acids has been reported.2063 Carboxylic acids can be treated with 2-chloro-4,6-dimethoxy[1,3,5]triazine and the RMgX/CuI to give ketones.2064
![]()
Disubstituted formamides can give addition of 2 molar equivalents of Grignard reagent. The products of this reaction (called Bouveault reaction) are an aldehyde and a tertiary amine.2065 The use of an amide other than a formamide can give a ketone instead of an aldehyde, but yields are generally low. The addition of 2 molar equivalents of phenyllithium to a carbamate gave good yields of the ketone, however.2066 The reaction of N-(3-bromopropyl) lactams with tert-butyllithium gave cyclization to the bicyclic amino alcohol, and subsequent reduction with LiAlH4 (Reaction 19-64) gave the bicyclic amine.2067 Ketones can also be prepared by treatment of thioamides with organolithium compounds (alkyl or aryl).2068 Cerium reagents (e.g., MeCeCl2) also add two R groups to an amide.2069 More commonly, an organolithium reagent is treated with CeCl3 to generate the organocerium reagent in situ.2070It has proven possible to add two different R groups by sequential addition of two Grignard reagents.2071 Diketones have also been produced by using the bis(imidazole) derivative of oxalic acid.2072 When an amide having a gem-dibromocyclopropyl unit elsewhere in the molecule was treated with methyllithium, Li–Br exchange was accompanied by intramolecular acyl addition to the amide carbonyl, giving a bicyclic amino alcohol.2073
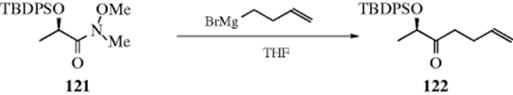
N-Methoxy-N-methyl amides, (e.g., 120) are referred to as a Weinreb amide.2074 When a Weinreb amide reacts with a Grignard reagent or an organolithium reagent,2075 the product is the ketone. The reaction of 121 (TBDPSO = tertiarybutyldiphenylsilyl) with 3-butenylmagnesium bromide to give ketone 122 is a typical example.2076 Aryloxy carbamates with a Weinreb amide unit [ArO2C-NMe(OMe)] react with RMgBr and then R′Li to give an unsymmetrical ketone [RC(=O)R′].2077 Intramolecular displacement of a Weinreb amide by an organolithium reagent generated in situ from an iodide precursor leads to cyclic ketones.2078 Reaction with vinylmagnesium bromide led to a β-N-methoxy-N-methylamino ketone, presumably by initial formation of the conjugated ketone followed by Michael addition (Reaction 15-24) of the liberated amine.2079 By the use of the compound N-methoxy-N,N',N'-trimethylurea, it is possible to add two R groups as RLi, the same or different, to a CO group.2080 Another variant used organocerium reagents with (Z)-α,β-unsaturated Weinreb amides to give (Z)-α,β-unsaturated ketones.2081
N,N-Disubstituted amides can be converted to alkynyl ketones by treatment with alkynylboranes: RCONR22 + (R′C![]() C)3B → RCOC
C)3B → RCOC![]() CR′.2082 Lactams react with triallylborane to give cyclic 2,2-diallyl amines after treatment with methanol and then aq hydroxide.2083 Triallylborane reacts with the carbonyl group of lactams, and after treatment with methanol and then aq NaOH gives the gem-diallyl amine: 2-pyrrolidinone → 2,2-diallylpyrrolidine.2084N,N-Disubstituted carbamates (X = OR2) and carbamoyl chlorides (X = Cl) react with 2 molar equivalents of an alkyl- or aryllithium or Grignard reagent to give symmetrical ketones, in which both R groups are derived from the organometallic compound: R2'NCOX + 2 RMgX → R2CO.2085N,N-Disubstituted amides give ketones in high yields when treated with alkyllanthanum triflates [RLa(OTf)2].2086
CR′.2082 Lactams react with triallylborane to give cyclic 2,2-diallyl amines after treatment with methanol and then aq hydroxide.2083 Triallylborane reacts with the carbonyl group of lactams, and after treatment with methanol and then aq NaOH gives the gem-diallyl amine: 2-pyrrolidinone → 2,2-diallylpyrrolidine.2084N,N-Disubstituted carbamates (X = OR2) and carbamoyl chlorides (X = Cl) react with 2 molar equivalents of an alkyl- or aryllithium or Grignard reagent to give symmetrical ketones, in which both R groups are derived from the organometallic compound: R2'NCOX + 2 RMgX → R2CO.2085N,N-Disubstituted amides give ketones in high yields when treated with alkyllanthanum triflates [RLa(OTf)2].2086
Other organometallic reagents give acyl substitution. Sodium naphthalenide reacts with esters to give naphthyl ketones.2087 Trimethylaluminum, which exhaustively methylates ketones (Reaction 16-24), also exhaustively methylates carboxylic acids to give tert-butyl compounds2088 (see also, Reaction 10-63). Trimethylaluminum reacts with esters to form ketones, in the presence of N,N'-dimethylethylenediamine.2089 Trialkylboranes have been used to convert thioesters to ketones.2090 Thioesters (RCOSR′) react with arylboronic acids, in the presence of a Pd catalyst, to give the corresponding ketone,2091 and esters react similarly with arylboronic acids (a Pd catalyst)2092 or arylboronates (a Ru catalyst).2093 Arylboronic acids also react with dialkyl anhydrides, with a Rh2094 or a Pd catalyst,2095 to give the ketone. Thioesters are converted to ketones with organoindium compounds.2096 Thioesters give good yields of ketones when treated with lithium dialkylcopper reagents (R22CuLi, R″ = primary or secondary alkyl or aryl).2097 Organozinc reagents convert thioesters to ketones.2098 Diaryl- or dialkylzinc reagents react with anhydrides and a Pd2099 or a Ni catalyst2100 to give the ketone. The reaction of alkylzinc halides and thioesters leads to ketones in the presence of 1.5% Pd/C,2101 in what has been called Fukuyama coupling.2102 Note that in the presence of a SmI2 catalyst and 2 molar equivalents of allyl bromide, lactones were converted to the diallyl diol.2103 Aryl iodides react with acetic anhydride, with a Pd catalyst, to give the aryl methyl ketone.2104
Carboxylic esters can be converted to their homologues (RCOOEt → RCH2COOEt) by treatment with Br2CHLi followed by BuLi at -90 °C. The ynolate (RC![]() COLi) is an intermediate.2105 If the ynolate is treated with 1,3-cyclohexadiene, followed by NaBH4, the product is the alcohol RCH2CH2OH.2106
COLi) is an intermediate.2105 If the ynolate is treated with 1,3-cyclohexadiene, followed by NaBH4, the product is the alcohol RCH2CH2OH.2106
Note that acyl benzotriazoles react with β-keto esters to give diketones via acyl substitution.2107 Acyl cyanides [RC(=O)CN] react with allylic bromides and In metal to give the corresponding ketone.2108 Acyl benzotriazoles have been coupled with SmI2 to give the 1,2-diketone.2109 α-Cyanoketones (acyl nitriles) were coupled with YbI2 in a similar manner.2110
Vinyl organometallic reagents can be added to acyl derivatives. Reaction of an alkyne with Cp2ZrEt2 generates the vinyl zirconium reagent, which react with ethyl chloroformate to give an α,β-unsaturated ester.2111
OS I, 226; II, 179, 602; III, 237, 831, 839; IV, 601; VI, 240, 278; VIII, 474, 505.
OS II, 282; 72, 32; III, 353; IV, 285; VI, 611; VII, 323, 451; 81, 14.
16-83 The Coupling of Acyl Halides
De-halogen-coupling
![]()
Acyl halides can be coupled with pyrophoric lead to give symmetrical α-diketones in a Wurtz-type reaction.2112 The reaction has been performed with R = Me and Ph. Samarium iodide2113 gives the same reaction. The photochemical coupling of acyl iodides gives α-diketones.2114 Benzoyl chloride was coupled to give benzil by subjecting it to ultrasound in the presence of Li wire: 2 PhCOCl + Li → PhCOCOPh.2115
Unsymmetrical α-diketones (RCOCOR′) have been prepared by treatment of an acyl halide (RCOCl) with an acyltin reagent R′COSnBu3, with a Pd complex catalyst.2116
16-84 Acylation at a Carbon Bearing an Active Hydrogen
Bis(ethoxycarbonyl)methyl-de-halogenation, and so on

This reaction is similar to 10-67, but there are fewer examples.2117 Either Z or Z′ may be any of the electron-withdrawing groups listed in Reaction 10-67 (CO2R, COR, CN, etc.)2118 Anhydrides react similarly but are used less often. The product contains three Z groups, since RCO is a Z group. One or two of these can be cleaved (Reactions 12-40 and 12-43). In this way, a compound ZCH2Z′ can be converted to ZCH2Z2 or an acyl halide (RCOCl) to a methyl ketone (RCOCH3). O-Acylation is sometimes a side reaction.2119 When thallium(I) salts of ZCH2Z′ are used, it is possible to achieve regioselective acylation at either the C or the O position. For example, treatment of the thallium(I) salt of MeCOCH2COMe with acetyl chloride at -78 °C gave >90% O-acylation, while acetyl fluoride at room temperature gave >95% C-acylation.2120 The use of an alkyl chloroformate gives triesters.2121
The application of this reaction to simple ketones2122 (in parallel with Reaction 10-68) requires a strong base (e.g., NaNH2 or Ph3CNa) and is often complicated by O-acylation, which in many cases becomes the principal pathway because acylation at the oxygen is usually much faster. It is possible to increase the proportion of C-acylated product by employing an excess (2–3 molar equivalents) of enolate anion (and adding the substrate to this, rather than vice versa), by the use of a relatively nonpolar solvent and a metal ion (e.g., Mg2+), which is tightly associated with the enolate oxygen atom, by the use of an acyl halide rather than an anhydride,2123 and by working at low temperatures.2124 In cases where the use of an excess of enolate anion results in C-acylation, it is because O-acylation takes place first, and the O-acylated product (an enol ester) is then C-acylated. Simple ketones can also be acylated by treatment of their silyl enol ethers with an acyl chloride in the presence of ZnCl2 or SbCl3.2125 Ketones can be acylated by anhydrides to give β-diketones, with BF3 as catalyst.2126 Simple esters (RCH2CO2Et) can be acylated at the α carbon (at –78 °C) if a strong base (e.g., lithium N-isopropylcyclohexylamide) is used to remove the proton.2127
Silyl enol esters react with acetic anhydride, in the presence of a chiral Fe complex, to give a chiral β-keto ester.2128
OS II, 266, 268, 594, 596; III, 16, 390, 637; IV, 285, 415, 708; V, 384, 937; VI, 245; VII, 213, 359; VIII, 71, 326, 467. See also, OS VI, 620.
16-85 Acylation of Carboxylic Esters by Carboxylic Esters: The Claisen and Dieckmann Condensations
Alkoxycarbonylalkyl-de-alkoxy-substitution
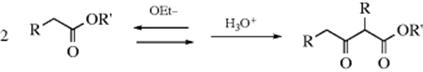
When carboxylic esters containing an α hydrogen are treated with a strong base (e.g., sodium ethoxide), a condensation occurs to give a β-keto ester via an ester enolate anion.2129 This reaction is called the Claisen condensation. When it is carried out with a mixture of two different esters, each of which possesses an α hydrogen (this reaction is called a mixed-Claisen or a crossed-Claisen condensation), a mixture of all four products is generally obtained and the reaction is seldom useful synthetically.2130 However, if only one of the esters has an α hydrogen, the mixed reaction is frequently satisfactory. Among esters lacking α hydrogen atoms (hence acting as the substrate ester) that are commonly used in this way are esters of aromatic acids, and ethyl carbonate and ethyl oxalate. When the ester enolate reacts with ethyl carbonate, the product is a malonic ester, and reaction with ethyl formate introduces a formyl group. Claisen condensation of phenyl esters with ZrCl4 and diisopropylethylamine (Hünigs base) give the corresponding keto ester.2131 Titanium compounds catalyze a crossed-Claisen condensation.2132 Boron(III) compounds also catalyzed ester condensation reactions.2133
As with ketone enolate anions (see Reaction 16-34), the use of amide bases under kinetic control conditions (strong base with a weak conjugate acid, aprotic solvents, low temperatures), allows the mixed-Claisen condensation to proceed. Self-condensation of the lithium enolate with the parent ester is a problem when LDA is used as a base,2134 but this is minimized with LICA.2135 Note that solvent-free Claisen condensation reactions have been reported.2136There is a retro-Claisen condensation, catalyzed by indium.2137
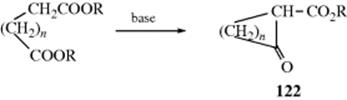
When the two ester groups involved in the condensation are in the same molecule, the product is a cyclic β-keto ester (122) and the reaction is called the Dieckmann condensation:2138 The Dieckmann condensation is most successful for the formation of 5-, 6-, and 7-membered rings, but yields for rings of 9–12 members are very low or nonexistent. Reactions that form large rings are generally assisted by high dilution. A solvent-free Dieckmann condensation has been reported on solid potassium tert-butoxide.2139 Dieckmann condensation of unsymmetrical substrates can be made regioselective by the use of solid-phase supports.2140 The Dieckmann condensation has also been done using TiCl3/NBu3 with a TMSOTf catalyst.2141 A Dieckmann-like condensation was reported where an α,ω-dicarboxylic acid was heated to 450 °C on graphite, with microwave irradiation, to give the cyclic ketone.2142
The mechanism of the Claisen and Dieckmann condensations (steps 1–3) is the ordinary tetrahedral mechanism,2143 with one molecule of ester being converted to a nucleophile by the base and the other serving as the substrate. This reaction illustrates the striking difference in behavior between carboxylic esters on the one hand and aldehydes and ketones on the other. When a carbanion (e.g., an enolate anion) is added to the carbonyl group of an aldehyde or ketone (Reaction 16-38), the H or R is not lost, since these groups are much poorer leaving groups than OR. Instead, the intermediate similar to 123 adds a proton at the oxygen to give a hydroxy compound.
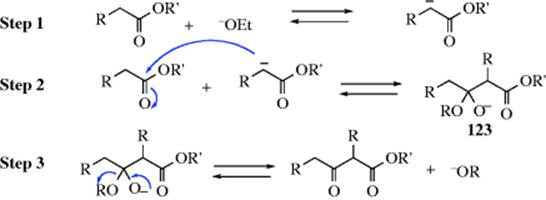
In contrast to Reaction 10-67, ordinary esters react quite well; that is, two Z groups are not needed. A lower degree of acidity is satisfactory because it is not necessary to convert the attacking ester entirely to its ion. Step 1 is an equilibrium that lies well to the left, but the small amount of enolate anion formed is sufficient to attack the readily approachable ester substrate. All the steps are in equilibria. The reaction proceeds because the product is converted to its conjugate base by the base present (i.e., a β-keto ester is a stronger acid than an alcohol):

The use of a stronger base (e.g., NaNH2, NaH, or KH),2144 often increases the yield. For some esters, stronger bases must be used, since sodium ethoxide is ineffective. Among these are esters of the type R2CHCOOEt, the products of which (R2CHCOCR2CO2Et) lack an acidic hydrogen, so that they cannot be converted to enolate anions by sodium ethoxide.2145
OS I, 235; II, 116, 194, 272, 288; III, 231, 300, 379, 510; IV, 141; V, 288, 687, 989; VIII, 112.
16-86 Acylation of Ketones and Nitriles by Carboxylic Esters
α-Acylalkyl-de-alkoxy-substitution
Carboxylic esters can be treated with ketones to give β-diketones. The reaction is so similar that it is sometimes also called the Claisen reaction, but this usage may be confusing. A strong base (e.g., sodium amide or sodium hydride) is required. Yields can be increased by the catalytic addition of crown ethers.2146 Esters of formic acid (R = H) give β-keto aldehydes and ethyl carbonate gives β-keto esters. β-Keto esters can also be obtained by treating the lithium enolates of ketones with methyl cyanoformate (MeOCOCN,2147 in this case CN is the leaving group) and by treating ketones with KH and diethyl dicarbonate [(EtOCO)2O].2148 This reaction has been used to effect cyclization, especially to prepare five- and six-membered rings. Nitriles are frequently used instead of ketones, the products being β-keto nitriles, as shown.
Other nucleophilic carbon reagents (e.g., acetylide ions) and ions derived from α-methylpyridines have also been used. A particularly useful nucleophile is the methylsulfinyl carbanion (CH3SOCH2−),2149 the conjugate base of DMSO, since the β-keto sulfoxide produced can easily be reduced to a methyl ketone (see Reaction 10-67). The methylsulfonyl carbanion (CH2SO2CH2−), the conjugate base of dimethyl sulfone, behaves similarly,2150 and the product can be similarly reduced. Certain carboxylic esters, acyl halides, and DMF will acylate 1,3-dithianes2151 (see Reaction 10-71) to give, after oxidative hydrolysis with NBS or NCS, α-keto aldehydes or α-diketones.2152
As in Reaction 10-67, a ketone is deprotonated at the most acidic proton first and the second most acidic position after that if 2 equivs of base are used, to give dianion 124. Thus, β-diketones have been converted to 1,3,5-triketones.2153
Side reactions are condensation of the ketone with itself (16-34), of the ester with itself, and of the ketone with the ester but with the ester supplying the α-position (16-36). The mechanism is the same as in Reaction 16-85.2154
OS I, 238; II, 126, 200, 287, 487, 531; III, 17, 251, 291, 387, 829; IV, 174, 210, 461, 536; V, 187, 198, 439, 567, 718, 747; VI, 774; VII, 351.
16-87 Acylation of Carboxylic Acid Salts
α-Carboxyalkyl-de-alkoxy-substitution
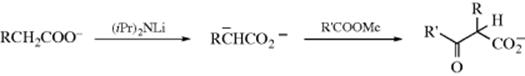
As seen previously (Reaction 10-70), dianions of carboxylic acids can be alkylated in the α position. These ions can also be acylated on treatment with a carboxylic ester2155 to give salts of β-keto acids. As in Reaction 10-70, the carboxylic acid can be of the form RCH2CO2H or RR2CHCO2H. Since β-keto acids are so easily converted to ketones (Reaction 12-40), this is also a method for the preparation of ketones (R′COCH2R and R′COCHRR2), where R′ can be primary, secondary, or tertiary alkyl, or aryl. If the ester is ethyl formate, an α-formyl carboxylate salt (R′ = H) is formed, which on acidification spontaneously decarboxylates into an aldehyde.2156 This method accomplishes the conversion RCH2CO2H → RCH2CHO, and is an alternative to the reduction methods discussed in Reaction 19-39. When the carboxylic acid is of the form RR2CHCO2H, better yields are obtained by acylating with acyl halides rather than esters.2157
16-88 Preparation of Acyl Cyanides
Cyano-de-halogenation
![]()
Acyl cyanides2158 can be prepared by treatment of acyl halides with copper cyanide. The mechanism could be free radical or nucleophilic substitution. The reaction has also been accomplished with thallium(I) cyanide,2159 with Me3SiCN and an SnCl4 catalyst,2160 and with Bu3SnCN,2161 but these reagents are successful only when R = aryl or tertiary alkyl. Potassium cyanide has also been used, along with ultrasound,2162 as has NaCN with phase-transfer catalysts.2163
OS III, 119.
16-89 Preparation of Diazo Ketones
Diazomethyl-de-halogenation
![]()
The reaction between acyl halides and diazomethane is of wide scope and is the best way to prepare diazo ketones.2164 Diazomethane must be present in excess or the HX produced will react with the diazo ketone (Reaction 10-52). This reaction is the first step of the Arndt–Eistert synthesis (18-8). Diazo ketones can also be prepared directly from a carboxylic acid and diazomethane or diazoethane in the presence of DCC.2165
OS III, 119; VI, 386, 613; VIII, 196.
16-90 Ketonic Decarboxylation2166
Alkyl-de-hydroxylation
![]()
Carboxylic acids can be converted to symmetrical ketones by pyrolysis in the presence of thorium oxide. In a mixed reaction, formic acid and another acid heated over thorium oxide give aldehydes. Mixed alkyl aryl ketones have been prepared by heating mixtures of ferrous salts.2167 When the R group is large, the methyl ester rather than the acid can be decarbmethoxylated over thorium oxide to give the symmetrical ketone.
The reaction has been performed on dicarboxylic acids, whereupon cyclic ketones are obtained:
This process, called Ruzicka cyclization, is good for the preparation of rings of six and seven members and, with lower yields, of C8–C10 to C30 cyclic ketones.2168
Not much work has been done on the mechanism of this reaction. However, a free radical mechanism has been suggested on the basis of a thorough study of all the side products.2169
OS I, 192; II, 389; IV, 854; V, 589. Also see, OS IV, 55, 560.
16.B.iii. Reactions in which Carbon Adds to the Heteroatom
A. Oxygen Adding to the Carbon
16-91 The Ritter Reaction
N-Hydro,N-alkyl-C-oxo-biaddition
Alcohols can be added to nitriles in an entirely different manner from that seen in Reaction 16-9. In this reaction, the alcohol is converted by a strong acid to a carbocation, which is attacked by the nucleophilic nitrogen atom to give 125. Subsequent addition of water to the electrophilic carbon atom leads to the enol form of the amide (see Reaction 126), which tautomerizes (Sec. 2.N.i) to the N-alkyl amide.

Only alcohols that give rise to fairly stable carbocations react (secondary, tertiary, benzylic, etc.); non-benzylic primary alcohols do not give the reaction. The carbocation need not be generated from an alcohol, but may come from protonation of an alkene or from other sources. In any case, the reaction is called the Ritter reaction.2170 Lewis acids [e.g., Mg(HSO4)2] have been used to promote the reaction.2171 Highly sterically hindered nitriles have been converted to N-methyl amides by heating with methanol and sulfuric acid.2172 Hydrogen cyanide also gives the reaction, the product being a formamide. Trimethylsilyl cyanide has also been used.2173
Since the amides (especially the formamides) are easily cleaved to amines under hydrolysis conditions, the Ritter reaction provides a method for achieving the conversions R′OH → R′NH2 (see 10-32) and alkene → R′NH2 (see 15-8) in those cases, where R′ can form a relatively stable carbocation. The reaction is especially useful for the preparation of tertiary alkyl amines because there are few alternate ways of preparing these compounds. The reaction can be extended to primary alcohols by treatment with triflic anhydride2174 or Ph2CCl+ SbCl6− or a similar salt2175 in the presence of the nitrile. A mixture of P2O5 and silica gel has been used to mediate the Ritter reaction.2176 There is a Nafion-catalyzed, microwave assisted variation,2177 as well as FeCl3 catalyzed2178 and iodine-catalyzed2179 reactions.
Alkenes of the form RCH=CHR′ and RR′C=CH2 add to nitriles in the presence of mercuric nitrate to give, after treatment with NaBH4, the same amides that would be obtained by the Ritter reaction.2180 This method has the advantage of avoiding strong acids.
Benzylic compounds (e.g., ethylbenzene) react with alkyl nitriles, ceric ammonium nitrate, and a catalytic amount of N-hydroxysuccinimide to give the Ritter product, the amide.2181
The Ritter reaction can be applied to cyanamides RNHCN to give ureas (RNHCONHR′).2182
OS V, 73, 471.
16-92 The Addition of Aldehydes to Aldehydes
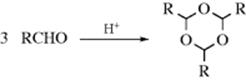
When catalyzed by acids, low-molecular-weight aldehydes add to each other to give cyclic acetals, the most common product being the trimer.2183 The cyclic trimer of formaldehyde is called trioxane,2184 and that of acetaldehyde is known as paraldehyde. Under certain conditions, it is possible to get tetramers2185 or dimers. Aldehydes can also polymerize to linear polymers, but a small amount of water is required to form hemiacetal groups at the ends of the chains. The linear polymer formed from formaldehyde is called paraformaldehyde. Since trimers and polymers of aldehydes are acetals, they are stable to bases, but can be hydrolyzed by acids. Because formaldehyde and acetaldehyde have low boiling points, it is often convenient to use them in the form of their trimers or polymers.
A slightly related reaction involves nitriles, which can be trimerized with various acids, bases, or other catalysts to give triazines (see OS III, 71).2186 Here HCl is most often used. Most nitriles with an α hydrogen do not give the reaction.
B. Nitrogen Adding to the Carbon
16-93 The Addition of Isocyanates to Isocyanates (Formation of Carbodiimides)
Alkylimino-de-oxo-bisubstitution
The treatment of isocyanates with 3-methyl-1-ethyl-3-phospholene-1-oxide (127) is a useful method for the synthesis of carbodiimides2187 in good yields.2188 The mechanism does not simply involve the addition of one molecule of isocyanate to another, since the kinetics are first order in isocyanate and first order in catalyst. The following mechanism has been proposed (the catalyst is here represented as R3P+–O−:2189
According to this mechanism, one molecule of isocyanate undergoes addition to C=O, and the other addition to C=N. Evidence is that 18O labeling experiments have shown that each molecule of CO2 produced contains one oxygen atom derived from the isocyanate and one from 127,2190 precisely what is predicted by this mechanism. Certain other catalysts are also effective.2191 High load, soluble oligomeric carbodiimides have been prepared.2192
OS V, 501.
16-94 The Conversion of Carboxylic Acid Salts to Nitriles
Nitrilo-de-oxido,oxo-tersubstitution
![]()
Salts of aliphatic or aromatic carboxylic acids can be converted to the corresponding nitriles by heating with BrCN or ClCN. Heating with acetonitrile in sulfuric acid also gave the nitrile.2193 Despite appearances, this is not a substitution reaction. When R14COO− was used, the label appeared in the nitrile, not in the CO2,2194 and optical activity in R was retained.2195 The acyl isocyanate (RCON=C=O) could be isolated from the reaction mixture; hence the following mechanism was proposed:2194
C. Carbon Adding to the Carbon
The reactions in this group are cycloadditions.
16-95 The Formation of β-Lactones and Oxetanes
(2+2)OC,CC-cyclo-[oxoethylene]-1/2/addition
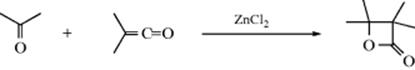
Aldehydes, ketones, and quinones react with ketenes to give β-lactones,2196 diphenylketene being used most often.2197 The reaction is catalyzed by Lewis acids, and without them most ketenes do not give adducts because the adducts decompose at the high temperatures necessary when no catalyst is used. When ketene was added to chloral (Cl3CCHO) in the presence of the chiral catalyst (+)-quinidine, one enantiomer of the β-lactone was produced with excellent enantioselectivity.2198 Enantioselective β-lactone formation was accomplished using chiral oxazaborolidines.2199 The use of a chiral Al catalyst also led to β-lactones with good syn selectivity and good enantioselectivity.2200Other di- and trihalo aldehydes and ketones also give the reaction enantioselectively, with somewhat lower enantioselectivity.2201 Ketene adds to another molecule of itself:
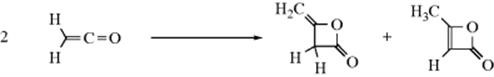
This dimerization is so rapid that ketene does not form β-lactones with aldehydes or ketones, except at low temperatures. Other ketenes dimerize more slowly. In these cases, the major dimerization product is not the β-lactone, but a cyclobutanedione (see Reaction 15-63). However, the proportion of ketene that dimerizes to β-lactone can be increased by the addition of catalysts (e.g., triethylamine or triethyl phosphite).2202 Ketene acetals [R2C=C(OR′)2] add to aldehydes and ketones in the presence of ZnCl2 to give the corresponding oxetanes.2203
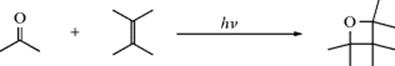
Ordinary aldehydes and ketones can add to alkenes, under the influence of UV light, to give oxetanes. Quinones also react to give spirocyclic oxetanes.2204 This reaction, called the Paterno–Büchi reaction,2205 is similar to the photochemical dimerization of alkenes discussed at Reaction 15-63. In general, the mechanism consists of the addition of an excited state of the carbonyl compound to the ground state of the alkene. Both singlet (S1)2206 and n,π∗ triplet2207 states have been shown to add to alkenes to give oxetanes. A diradical intermediate ·O–C–C–C·2208 has been detected by spectroscopic methods.2209 Yields in the Paterno–Büchi reaction are variable, ranging from very low to fairly high (90%). The reaction can be highly diastereoselective,2210 and allylic alcohols were shown to react with aldehydes to give an oxetane with syn selectivity.2211 There are several side reactions. When the reaction proceeds through a triplet state, it can in general be successful only when the alkene possesses a triplet energy comparable to, or higher than, the carbonyl compound; otherwise energy transfer from the excited carbonyl group to the ground-state alkene can take place (triplet–triplet photosensitization, see Sec. 7.A.vi).2212 In most cases, quinones react normally with alkenes, giving oxetane products, but other α,β-unsaturated ketones usually give preferential cyclobutane formation (Reaction 15-63). Aldehydes and ketones also add photochemically to allenes to give the corresponding alkylideneoxetanes and dioxaspiro compounds:2213 Aldehydes add to silyl enol ethers.2214 An intramolecular reaction of ketones was reported to give a bicyclic oxetane via photolysis on the solid state.2215
![]()
OS III, 508; V, 456. For the reverse reaction, see OS V, 679.
16-96 The Formation of β-Lactams
(2+2)NC,CC-cyclo-[oxoethylene]-1/2/addition
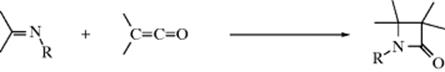
Ketenes add to imines to give β-lactams.2216 The reaction is generally carried out with ketenes of the form R2C=C=O. It has not been successfully applied to RCH=C=O, except when these are generated in situ by decomposition of a diazo ketone (the Wolff rearrangement, Reaction 18-8) in the presence of the imine. It has been done with ketene, but the more usual course with this reagent is an addition to the enamine tautomer of the substrate. Thioketenes2217 (R2C=C=S) give β-thiolactams.2218 Imines also form β-lactams when treated with (1) zinc (or another metal2219) and an α-bromo ester (Reformatsky conditions, 16-28),2220 or (2) the chromium carbene complexes [(CO)5Cr=C(Me)OMe].2221 The latter method has been used to prepare optically active β-lactams.2222 Ketenes have also been added to certain hydrazones (e.g., PhCH=NNMe2) to give N-amino β-lactams.2223 A polymer-bound pyridinium salt facilitates β-lactam formation from carboxylic acids and imines.2224 α-Chloroimines have been used as chiral inductors in this reaction.2225
N-Tosyl imines react with ketenes, Proton Sponge (Sec. 8.A.i) and a chiral amine to give the N-tosyl β-lactam with good enantioselectivity.2226 A chiral ferrocenyl catalyst also gives good enantioselectivity,2227 and chiral ammonium salts2228 or chiral cinchona alkaloids2229 have been used as catalysts. A catalytic amount of benzoyl quinine gives β-lactams with good enantioselectivity.2230
An intramolecular version of this ketene–imine reaction is known.2231
Like the similar cycloaddition of ketenes to alkenes (Reaction 15-63), most of these reactions probably take place by the diionic mechanism c (See 15-63).2232 β-Lactams have also been prepared in the opposite manner by the addition of enamines to isocyanates:2233
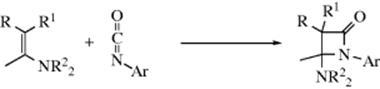
The reactive compound chlorosulfonyl isocyanate2234 (ClSO2NCO) forms β-lactams even with unactivated alkenes,2235 as well as with imines,2236 allenes,2237 conjugated dienes,2238 and cyclopropenes.2239 With microwave irradiation, alkyl isocyanates also react.2240
α-Diazo ketones react with imines and microwave irradiation to give β-lactams.2241 Allylic phosphonate esters react with imines, in the presence of a Pd catalyst, to give β-lactams.2242 Alkynyl reagents (e.g., BuC![]() CO−Li+) react with imines to form β-lactams.2243 Imines and benzylic halides react to give β-lactams in the presence of CO and a Pd catalyst.2244 Conjugated amides react with NBS and 20% sodium acetate to give an α-bromo β-lactam.2245 A different approach to β-lactams heated aziridines with CO and a Co catalyst.2246 Aziridines also react with CO and a dendrimer catalyst to such a β-lactam.2247
CO−Li+) react with imines to form β-lactams.2243 Imines and benzylic halides react to give β-lactams in the presence of CO and a Pd catalyst.2244 Conjugated amides react with NBS and 20% sodium acetate to give an α-bromo β-lactam.2245 A different approach to β-lactams heated aziridines with CO and a Co catalyst.2246 Aziridines also react with CO and a dendrimer catalyst to such a β-lactam.2247
β-Thiolactams are prepared from aryl isothiocyanates.2248
OS V, 673; VIII, 3, 216.
16.B.iv. Addition to Isocyanides2249
Addition to R–+N![]() C− is not a matter of a species with an electron pair adding to one atom and a species without a pair adding to the other, as is addition to the other types of double and triple bonds in this chapter and Chapter 15. In these additions, the electrophile and the nucleophile both add to the carbon. No species add to the nitrogen, which, however, loses its positive charge by obtaining as an unshared pair one of the triple-bond pairs of electrons to give 128. In most of the reactions considered below, 128 undergoes a further reaction, so the product is of the form R–NH−–CR3.
C− is not a matter of a species with an electron pair adding to one atom and a species without a pair adding to the other, as is addition to the other types of double and triple bonds in this chapter and Chapter 15. In these additions, the electrophile and the nucleophile both add to the carbon. No species add to the nitrogen, which, however, loses its positive charge by obtaining as an unshared pair one of the triple-bond pairs of electrons to give 128. In most of the reactions considered below, 128 undergoes a further reaction, so the product is of the form R–NH−–CR3.

16-97 The Addition of Water to Isocyanides
1/ N,2/C-Dihydro-2/C-oxo-biaddition
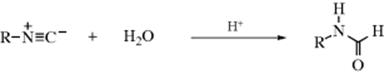
Formamides can be prepared by the acid-catalyzed addition of water to isocyanides. The mechanism is probably2250
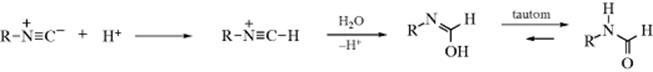
The reaction has also been carried out under alkaline conditions, with hydroxide in aq dioxane.2251 The mechanism here involves nucleophilic attack by hydroxide at the carbon atom. An intramolecular addition of an alkyne (in an ortho alkynyl phenyl isonitrile) to the carbon of an isonitrile occurred with heating in methanol to give quinoline derivatives.2252
16-98 The Passerini and Ugi Reactions2253
1/N-Hydro-2/C-(α-acyloxyalkyl),2/C-oxo-biaddition
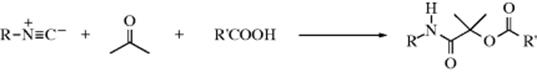
When an isocyanide is treated with a carboxylic acid and an aldehyde or ketone, an α-acyloxy amide is the product in what is called the Passerini reaction. A SiCl4 mediated reaction in the presence of a chiral bis(phosphoramide) gives an α-hydroxy amide with good enantioselectivity.2254 There is a solvent-free Passerini reaction2255 and ionic liquids2256 can be used. The following mechanism has been postulated for the basic reaction:2257
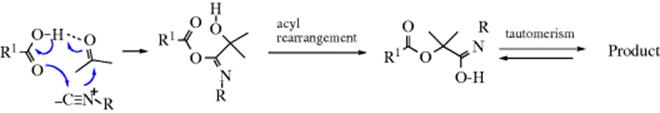
If ammonia or an amine is also added to the mixture (in which case the reaction is known as the Ugi reaction, or the Ugi four-component condensation), the product is R′(C=C)NH–C–(C=O)NHR [the corresponding bis(amide) from NH3] or R′(C=C)NR′–C–(C=O)NHR (from a primary amine R2NH2). There is a catalytic three-component Ugi reaction.2258 Repetitive Ugi reactions are known.2259 This product probably arises from a reaction between the carboxylic acid, the isocyanide, and the imine formed from the aldehyde or ketone and ammonia or the primary amine. “Isocyanide-free” Ugi reactions use alkyl halides/silver cyanide and KCN to generate the isocyanide in situ.2260The use of an N-protected amino acid2261 or peptide as the carboxylic acid component and/or the use of an isocyanide containing a C-protected carboxyl group allows the reaction to be used for peptide synthesis.2262 Rare earth metal triflates catalyze this reaction.2263
16-99 The Formation of Metalated Aldimines
1/1/Lithio-alkyl-addition
![]()
Isocyanides that do not contain an α hydrogen react with alkyllithium compounds,2264 as well as with Grignard reagents, to give lithium (or magnesium) aldimines.2265 These metalated aldimines are versatile nucleophiles and react with various substrates as follows:
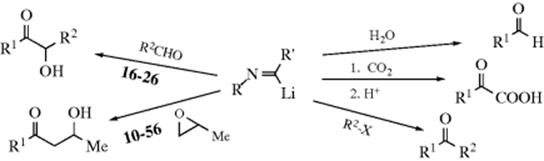
The reaction therefore constitutes a method for converting an organometallic compound (R′M) to an aldehyde (R′CHO, see also, Reaction 12-33), an α-keto acid,2266 a ketone R′COR (see also, Reaction 12-33), an α-hydroxy ketone, or a β-hydroxy ketone. In each case, the C=N bond is hydrolyzed to a C=O bond (Reaction 16-2).
In a related reaction, isocyanides can be converted to aromatic aldimines by treatment with an iron complex followed by irradiation in benzene solution: RNC + C6H6 → PhCH=NR.2267
OS VI, 751.
16.B.v. Nucleophilic Substitution at a Sulfonyl Sulfur Atom2268
Nucleophilic substitution at RSO2X is similar to attack at RCOX. Many of the reactions are essentially the same, though sulfonyl halides are less reactive than halides of carboxylic acids.2269 The mechanisms2270 are not identical, because a “tetrahedral” intermediate in this case (129) would have five groups on the central atom. This is possible since sulfur can accommodate up to 12 electrons in its valence shell, but it seems more likely that these mechanisms more closely resemble the SN2 mechanism, with a trigonal-bipyramidal transition state (130). There are two major experimental results leading to this conclusion.
1. The stereospecificity of this reaction is more difficult to determine than that of nucleophilic substitution at a saturated carbon, where chiral compounds are relatively easy to prepare. Recall (Sec. 4.C, category 2) that optical activity is possible in a compound of the form RSO2X if one oxygen is 16O and the other is 18O. When a sulfonate ester possessing this type of chirality was converted to a sulfone by reaction with a Grignard reagent (16-105), inversion of configuration was found.2271 This is not incompatible with an intermediate (e.g., 129), but it is in good accord with an SN2-like mechanism with backside attack.
2. More direct evidence against 129 (though still not conclusive) was found in an experiment involving acidic and basic hydrolysis of aryl arenesulfonates, where it has been shown by the use of 18O that an intermediate like 129is not reversibly formed, since ester recovered when the reaction was stopped before completion contained no 18O when the hydrolysis was carried out in the presence of labeled water.2272
Other evidence favoring the SN2-like mechanism comes from kinetics and substituent effects.2273 However, evidence for the mechanism involving 129 is that the rates did not change much with changes in the leaving group2274and the ρ values were large, indicating that a negative charge builds up in the transition state.2275
In certain cases in which the substrate carries an α hydrogen, there is strong evidence2276 that at least some of the reaction takes place by an elimination–addition mechanism (E1cB, similar to the one shown in Reaction 16-69), going through a sulfene intermediate,2277 as in the reaction between methanesulfonyl chloride and aniline.
![]()
In the special case of nucleophilic substitution at a sulfonic ester (RSO2OR′), where R′ is alkyl, R′–O cleavage is much more likely than S–O cleavage because the OSO2R group is such a good leaving group (Sec. 10.G.iii).2278Many of these reactions have been considered previously (e.g., Reactions 10-4 and 10-10) as nucleophilic substitutions at an alkyl carbon atom and not at a sulfur atom. However, when R′ is aryl, then the S–O bond is much more likely to cleave because of the low tendency of aryl substrates for nucleophilic substitution.2279
The order of nucleophilicity toward a sulfonyl sulfur has been reported as OH− > RNH2 > N3 > F− > AcO− > Cl− > H2O > I−.2280 This order is similar to that at a carbonyl carbon (Sec. 10.G.ii). Both of these substrates can be regarded as relatively hard acids, compared to a saturated carbon that is considerably softer and has a different order of nucleophilicity (Sec. 10.G.ii).
16-100 Attack by OH: Hydrolysis of Sulfonic Acid Derivatives
S -Hydroxy-de-chlorination, and so on
![]()
Sulfonyl chlorides as well as esters and amides of sulfonic acids can be hydrolyzed to the corresponding acids. Sulfonyl chlorides can be hydrolyzed with water or with an alcohol in the absence of acid or base. Basic catalysis is also used, though of course the salt is the product obtained. Esters are readily hydrolyzed, many with water or dilute alkali. This is the same reaction as 10-4, and usually involves R′–O cleavage, except when R′ is aryl. In some cases, retention of configuration has been shown at alkyl R′, indicating S–O cleavage.2281 Sulfonamides are generally not hydrolyzed by alkaline treatment, not even with hot concentrated alkali. Acids, however, do hydrolyze sulfonamides, but less readily than they do sulfonyl halides or sulfonic esters. Of course, ammonia or the amine appears as the salt. However, sulfonamides can be hydrolyzed with base if the solvent is HMPA.2282
Magnesium in methanol has been used to convert sulfonate esters to the parent alcohol.2283 Likewise, CeCl3·7 H2O–NaI in acetonitrile converted aryl tosylates to the parent phenol derivative.2284
OS I, 14; II, 471; III, 262; IV, 34; V, 406; VI, 652, 727. Also see, OS V, 673; VI, 1016.
16-101 Attack by OR: Formation of Sulfonic Esters
S-Alkoxy-de-chlorination, and so on
Sulfonic esters are most frequently prepared by treatment of the corresponding sulfonyl halides with alcohols in the presence of a base.2285 This procedure is the most common method for the conversion of alcohols to tosylates, brosylates, and similar sulfonic esters. Both R and R′ may be alkyl or aryl. The base is often pyridine or another amine, which functions as a nucleophilic catalyst,2286 as in the similar alcoholysis of carboxylic acyl halides (Reaction 16-61). Primary alcohols react most rapidly, and it is often possible to sulfonate a primary OH group selectively in a molecule that also contains secondary or tertiary OH groups. The reaction with sulfonamides has been much less frequently used and is limited to N,N-disubstituted sulfonamides; that is, R may not be hydrogen. However, within these limits it is a useful reaction. The nucleophile in this case is actually RO−. The R′ may be hydrogen (as well as alkyl) if the nucleophile is a phenol, so that the product is RSO2OAr. Acidic catalysts are used in this case.2287 Sulfonic acids have been converted directly to sulfonates by treatment with triethyl or trimethyl orthoformate [HC(OR)3], without catalyst or solvent;2288 and with a trialkyl phosphite [P(OR)3].2289
Mono-tosylation of a 1,2-diol was achieved using tosyl chloride and triethylamine, with a tin oxide catalyst.2290
OS I, 145; III, 366; IV, 753; VI, 56, 482, 587, 652; VII, 117; 66, 1; 68, 188. Also see, OS IV, 529; VI, 324, 757; VII, 495; VIII, 568.
16-102 Attack by Nitrogen; Formation of Sulfonamides
S-Amino-de-chlorination
![]()
Microwave assisted conversion of sulfonic acids to a 2,4,6-trichloro[1,3,5]triazine derivative is followed by formation of the sulfonamide.2291 The treatment of sulfonyl chlorides with ammonia or amines is the usual way of preparing sulfonamides.2292 Primary amines give N-alkyl sulfonamides, and secondary amines give N,N-dialkyl sulfonamides. The reaction is the basis of the Hinsberg test for distinguishing between primary, secondary, and tertiary amines. N-Alkyl sulfonamides, having an acidic hydrogen, are soluble in alkali, while N,N-dialkyl sulfonamides are not. Since tertiary amines are usually recovered unchanged, primary, secondary, and tertiary amines can be told apart. However, the test is limited for at least two reasons.2293 (1) Many N-alkyl sulfonamides in which the alkyl group has six or more carbons are insoluble in alkali, despite their acidic hydrogen,2294 so that a primary amine may appear to be a secondary amine. (2) If the reaction conditions are not carefully controlled, tertiary amines may not be recovered unchanged.2289
A primary or a secondary amine can be protected by reaction with phenacylsulfonyl chloride (PhCOCH2SO2Cl) to give a sulfonamide (RNHSO2CH2COPh or R2NSO2CH2COPh).2295 The protecting group can be removed when desired with zinc and acetic acid. Sulfonyl chlorides react with azide ion to give sulfonyl azides (RSO2N3).2296 Chlorothioformates, [ROC(=S)Cl] react with triethylamine to give the N,N-diethylthioamide.2297
A quite different synthesis of sulfonamides treated allyltributyltin with PhI=NTs, in the presence of copper(II) triflate.2298 Another alternative method treats silyl enol ethers with sulfur dioxide, and subsequent reaction with a secondary amine gave the β-sulfonamido ester.2299
OS IV, 34, 943; V, 39, 179, 1055; VI, 78, 652; VII, 501; VIII, 104. See also, OS VI, 788.
16-103 Attack by Halogen: Formation of Sulfonyl Halides
S-Halo-de-hydroxylation
![]()
This reaction, parallel with 16-79, is the standard method for the preparation of sulfonyl halides. Both PCl3 and SOCl2 have been used, and sulfonic acid salts can also serve as substrates. Cyanuric acid (2,4,6-trichloro[1,3,5]triazene) serves as a chlorinating agent.2300 Sulfonyl bromides and iodides have been prepared from sulfonyl hydrazides (ArSO2NHNH2, themselves prepared by Reaction 16-102) by treatment with bromine or iodine.2301 Sulfonyl fluorides are generally prepared from the chlorides, by halogen exchange.2302
OS I, 84; IV, 571, 693, 846, 937; V, 196. See also, OS VII, 495.
16-104 Attack by Hydrogen: Reduction of Sulfonyl Chlorides
S-Hydro-de-chlorination or S -Dechlorination
![]()
Sulfinic acids can be prepared by reduction of sulfonyl chlorides. Though mostly done on aromatic sulfonyl chlorides, the reaction has also been applied to alkyl compounds. Zinc, sodium sulfite, hydrazine, sodium sulfide, and other reducing agents have been used. For reduction of sulfonyl chlorides to thiols, see Reaction 19-78.
OS I, 7, 492; IV, 674.
16-105 Attack by Carbon: Preparation of Sulfones
S-Aryl-de-chlorination
![]()
Grignard reagents convert aromatic sulfonyl chlorides or aromatic sulfonates to sulfones. Organolithium reagents react with sulfonyl fluorides at –78 °C to give the corresponding sulfone.2303 Aromatic sulfonates have been converted to sulfones with organolithium compounds,2304 with aryltin compounds,2305 with an Fe catalyst,2306 and with alkyl halides and Zn metal.2307 Vinylic and allylic sulfones have been prepared by treatment of sulfonyl chlorides with a vinylic or allylic stannane and a Pd complex catalyst.2308 Alkynyl sulfones can be prepared by treatment of sulfonyl chlorides with trimethylsilylalkynes, with an AlCl3 catalyst.2309 Note that trifluoromethylsulfones were converted to methyl sulfones by reaction with methylmagnesium bromide.2310
Arylboronic acids (Reaction 12-28) react with sulfonyl chlorides in the presence of PdCl2 to give the corresponding sulfone.2311 Arylboronic acids also react with sulfinate anions (RSO2Na) in the presence of Cu(OAc)2 to give the sulfone.2312
OS VIII, 281.
Notes
1. See Jencks, W.P. Prog. Phys. Org. Chem. 1964, 2, 63
2. See Schaumann, E. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, Vol. 2, pt. 2, Wiley, NY, 1989, pp. 1269–1367; Ohno, A. in Oae. S. Organic Chemistry of Sulfur, Plenum, NY, 1977, pp. 189–229; Mayer, R. in Janssen, M.J. Organosulfur Chemistry, Wiley, NY, 1967, pp. 219–240; Campaigne, E. in Patai, S. The Chemistry of the Carbonyl Group, pt. 1, Wiley, NY, 1966, pp. 917–959.
3. See Wardell, J.L.; Paterson, E.S. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 219–338, pp. 261–267.
4. See Metzner, P.; Vialle, J.; Vibet, A. Tetrahedron 1978, 34, 2289.
5. See Eliel, E.L. The Stereochemistry of Carbon Compounds, McGraw-Hill, NY, 1962, pp. 68–74. Also see Bartlett, P.A. Tetrahedron 1980, 36, 2, 22; Ashby, E.C.; Laemmle, J.T. Chem. Rev. 1975, 75, 521.
6. See Laube, T.; Stilz, H.U. J. Am. Chem. Soc. 1987, 109, 5876.
7. Kumar, V.A.; Venkatesan, K.; Ganguly, B.; Chandrasekhar, J.; Khan, F.A.; Mehta, G. Tetrahedron Lett. 1992, 33, 3069.
8. Yadav, V.K.; Jeyaraj, D.A. J. Org. Chem. 1998, 63, 3474. For a discussion of models, see Priyakumar, U.D.; Sastry, G.N.; Mehta, G. Tetrahedron 2004, 60, 3465.
9. See Beckwith, A.L.J.; Hay, B.P. J. Am. Chem. Soc. 1989, 111, 2674; Clerici, A.; Porta, O. J. Org. Chem. 1989, 54, 3872; Cossy, J.; Pete, J.P.; Portella, C. Tetrahedron Lett. 1989, 30, 7361.
10. See Jencks, W.P.; Gilbert, H.F. Pure Appl. Chem. 1977, 49, 1021.
11. Toromanoff, E. Bull. Soc. Chim. Fr. 1962, 1190.
12. See Brada, B.; Bundhoo, D.; Engels, B.; Hiberty, P.C. Org. Lett. 2008, 10, 1951.
13. Chamberland, S.; Ziller, J.W.; Woerpel, K.A. J. Am. Chem. Soc. 2005, 127, 5322.
14. For a review of the reactivity of nitriles, see Schaefer, F.C. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 239–305.
15. This mechanism has also been called the "addition–elimination mechanism".
16. See Talbot, R.J.E. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 10, Elsevier, NY, 1972, pp. 209–223; Jencks, W.P. Catalysis in Chemistry and Enzymology, McGraw-Hill, NY, 1969, pp. 463–554; Satchell, D.P.N.; Satchell, R.S. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 375–452; Johnson, S.L. Adv. Phys. Org. Chem. 1967, 5, 237.
17. Williams, A. Acc. Chem. Res. 1989, 22, 387. See Bentley, T.W.; Koo, I.S. J. Chem. Soc. Perkin Trans. 2 1989, 1385. See, however, Buncel, E.; Um, I.H.; Hoz, S. J. Am. Chem. Soc. 1989, 111, 971.
18. Bacaloglu, R.; Blaskó, A.; Bunton, C.A.; Ortega, F. J. Am. Chem. Soc. 1990, 112, 9336.
19. SeeJencks, W.P. Acc. Chem. Res. 1976, 9, 425; Chem. Rev. 1972, 72, 705.
20. Also see Guthrie, J.P. J. Am. Chem. Soc. 1978, 100, 5892; Kluger, R.; Chin, J. J. Am. Chem. Soc. 1978, 100, 7382; O'Leary, M.H.; Marlier, J.F. J. Am. Chem. Soc. 1979, 101, 3300.
21. Jencks, W.P.; Gilchrist, M. J. Am. Chem. Soc. 1964, 86, 5616.
22. Kevill, D.N.; Johnson, S.L. J. Am. Chem. Soc. 1965, 87, 928; Leinhard, G.E.; Jencks, W.P. J. Am. Chem. Soc. 1965, 87, 3855; Schowen, R.L.; Jayaraman, H.; Kershner, L.D. J. Am. Chem. Soc. 1966, 88, 3373.
23. Bender, M.L.; Thomas, R.J. J. Am. Chem. Soc. 1961, 83, 4183, 4189.
24. Bender, M.L.; Matsui, H.; Thomas, R.J.; Tobey, S.W. J. Am. Chem. Soc. 1961, 83, 4193. See also, Shain, S.A.; Kirsch, J.F. J. Am. Chem. Soc. 1968, 90, 5848.
25. For evidence for this possibility, see McClelland, R.A. J. Am. Chem. Soc. 1984, 106, 7579.
26. Bender, M.L.; Heck, H. d'A. J. Am. Chem. Soc. 1967, 89, 1211.
27. Fedor, L.R.; Bruice, T.C. J. Am. Chem. Soc. 1965, 87, 4138.
28. See Khouri, F.F.; Kaloustian, M.K. J. Am. Chem. Soc. 1986, 108, 6683.
29. See Capon, B.; Dosunmu, M.I.; Sanchez, M. de N de M. Adv. Phys. Org. Chem. 1985, 21, 37; McClelland, R.A.; Santry, L.J. Acc. Chem. Res. 1983, 16, 394; Capon, B.; Ghosh, A.K.; Grieve, D.M.A. Acc. Chem. Res. 1981, 14, 306. See also, van der Wel, H.; Nibbering, N.M.M. Recl. Trav. Chim. Pays-Bas 1988, 107, 479, 491.
30. See Menger, F.M. Tetrahedron 1983, 39, 1013; Liotta, C.L.; Burgess, E.M.; Eberhardt, W.H. J. Am. Chem. Soc. 1984, 106, 4849.
31. It has also been called the “antiperiplanar lone-pair hypothesis (ALPH)”. For a reinterpretation of this factor in terms of the principle of least nuclear motion (see Reaction 15-10), see Hosie, L.; Marshall, P.J.; Sinnott, M.L. J. Chem. Soc. Perkin Trans. 2 1984, 1121; Sinnott, M.L. Adv. Phys. Org. Chem. 1988, 24, 113.
32. Kirby, A.J. The Anomeric Effect and Related Stereoelectronic Effects at Oxygen, Springer, NY, 1983; Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry, Pergamon, NY, 1983. See Sinnott, M.L. Adv. Phys. Org. Chem. 1988, 24, 113; Gorenstein, D.G. Chem. Rev. 1987, 87, 1047; Deslongchamps, P. Heterocycles 1977, 7, 1271. Also see Ndibwami, A.; Deslongchamps, P.Can. J. Chem. 1986, 64, 1788; Hegarty, A.F.; Mullane, M. J. Chem. Soc. Perkin Trans. 2 1986, 995. For evidence against the theory, see Perrin, C.L.; Nuñez, O. J. Am. Chem. Soc. 1986, 108, 5997; 1987, 109, 522.
33. See Bender, M.L. Mechanisms of Homogeneous Catalysis from Protons to Proteins, Wiley, NY, 1971, pp. 147–179; Johnson, S.L. Adv. Phys. Org. Chem. 1967, 5, 271. For a review where Z = a tertiary amine, see Cherkasova, E.M.; Bogatkov, S.V.; Golovina, Z.P. Russ. Chem. Rev. 1977, 46, 246.
34. Kirby, A.J.; Fersht, A.R. Prog. Bioorg. Chem. 1971, 1, 1; Capon, B. Essays Chem. 1972, 3, 127.
35. Bender, M.L.; Chow, Y.; Chloupek, F.J. J. Am. Chem. Soc. 1958, 80, 5380.
36. See Page, M.I.; Render, D.; Bernáth, G. J. Chem. Soc. Perkin Trans. 2 1986, 867.
37. The compound RCOOH would belong in this sequence just after RCOOAr, but it fails to undergo many reactions for a special reason. Many nucleophiles, instead of attacking the C=O group, are basic enough to take a proton from the acid, converting it to the unreactive RCOO–.
38. See Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell University Press, Ithaca, NY, 1973, pp. 183–187; Adv. Phys. Org. Chem. 1966, 4, 1; Le Hénaff, P. Bull. Soc. Chim. Fr. 1968, 4687.
39. Bell, R.P.; Clunie, J.C. Trans. Faraday Soc. 1952, 48, 439. See also, Bell, R.P.; McDougall, A.O. Trans. Faraday Soc. 1960, 56, 1281.
40. Cohn, M.; Urey, H.C. J. Am. Chem. Soc. 1938, 60, 679.
41. For a review of chloral, see Luknitskii, F.I. Chem. Rev. 1975, 75, 259.
42. Schulman, E.M.; Bonner, O.D.; Schulman, D.R.; Laskovics, F.M. J. Am. Chem. Soc. 1976, 98, 3793.
43. For a review of addition to fluorinated ketones, see Gambaryan, N.P.; Rokhlin, E.M.; Zeifman, Yu.V.; Ching-Yun, C.; Knunyants, I.L. Angew. Chem. Int. Ed. 1966, 5, 947.
44. See Krois, D.; Lehner, H. Monatsh. Chem. 1982, 113, 1019.
45. Turro, N.J.; Hammond, W.B. J. Am. Chem. Soc. 1967, 89, 1028. For a review of cyclopropanone chemistry, see Wasserman, H.H.; Clark, G.M.; Turley, P.C. Top. Curr. Chem. 1974, 47, 73.
46. S![]() rensen, P.E.; Jencks, W.P. J. Am. Chem. Soc. 1987, 109, 4675; Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry, 3rd ed., Harper and Row, NY, 1987, pp. 662–680. A theoretical treatment is in Wolfe, S.; Kim, C.-K.; Yang, K.; Weinberg, N.; Shi, Z. J. Am. Chem. Soc. 1995, 117, 4240.
rensen, P.E.; Jencks, W.P. J. Am. Chem. Soc. 1987, 109, 4675; Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry, 3rd ed., Harper and Row, NY, 1987, pp. 662–680. A theoretical treatment is in Wolfe, S.; Kim, C.-K.; Yang, K.; Weinberg, N.; Shi, Z. J. Am. Chem. Soc. 1995, 117, 4240.
47. Jencks, W.P. Acc. Chem. Res. 1976, 9, 425.
48. For a review, see Khoee, S.; Ruoho, A.E. Org. Prep. Proceed. Int. 2003, 35, 527.
49. For proton affinities of imines, see Hammerum, S.; S![]() lling, T.I. J. Am. Chem. Soc. 1999, 121, 6002.
lling, T.I. J. Am. Chem. Soc. 1999, 121, 6002.
50. For a list of reagents, with references, see Ranu, B.C.; Sarkar, D.C. J. Org. Chem. 1988, 53, 878.
51. For a review, see Corsaro, A.; Chiacchio, U.; Pistarà, V. Synthesis 2001, 1903.
52. Bhar, S.; Guha, S. Synth. Commun. 2005, 35, 1183.
53. Li, Z.; Ding, R.-B.; Xing, Y.-L.; Shi, S.-Y. Synth. Commun. 2005, 35, 2515.
54. Heravi, M.M.; Derikvand, F.; Ghassemzadeh, M. Synth. Commun. 2006, 36, 581.
55. Bandgar, B.P.; Makone, S.S. Org. Prep. Proceed. Int. 2000, 32, 391.
56. Padmavathi, V.; Reddy, K.V.; Padmaja, A.; Venugopalan, P. J. Org. Chem. 2003, 68, 1567.
57. A solvent-free reaction. See Zhou, J.-F.; Tu, S.-J.; Feng, J.-C. Synth. Commun. 2002, 32, 959.
58. Gogoi, P.; Hazarika, P.; Konwar, D. J. Org. Chem. 2005, 70, 1934.
59. Li, D.; Shi, F.; Guo, S.; Deng, Y. Tetrahedron Lett. 2004, 45, 265.; Li, D.; Shi, F.; Deng, Y. Tetrahedron Lett. 2004, 45, 6791.
60. Narsaiah, A.V.; Nagaiah, K. Synthesis 2003, 1881.
61. Mukai, C.; Nomura, I.; Kataoka, O.; Hanaoka, M. Synthesis 1999, 1872.
62. De, S.K. Synth. Commun. 2004, 34, 2289.
63. See Arnold, J.N.; Hayes, P.D.; Kohaus, R.L.; Mohan, R.S. Tetrahedron Lett. 2003, 44, 9173.
64. Hashemi, M.M.; Beni, Y.A. Synth. Commun. 2001, 31, 295; Tamami, B.; Kiasat, A.R. Synth. Commun. 2000, 30, 4129.
65. Tamami, B.; Kiasat, A.R. Synth. Commun. 2000, 30, 4129.
66. See Imanzadeh, G.H.; Hajipour, A.R.; Mallakpour, S.E. Synth. Commun. 2003, 33, 735.
67. Hajipour, A.R.; Mallakpour, S.E.; Khoee, E. Synth. Commun. 2002, 32, 9.
68. Sadeghi, M.M.; Mohammadpoor-Baltork, I.; Azarm, M.; Mazidi, M.R. Synth. Commun. 2001, 31, 435. See also, Zhang, G.-S.; Yang, D.-H.; Chen. M.-F. Org. Prep. Proceed. Int. 1998, 30, 713.
69. Ho, T. Synth. Commun. 1980, 10, 465.
70. Hajipour, A.R.; Mahboubghah, N. Org. Prep Proceed. Int. 1999, 31, 112.
71. Chen, D.-J.; Cheng, D.-P.; Chen, Z.-C. Synth. Commun. 2001, 31, 3847.
72. Hajipour, A.R.; Adibi, H.; Ruoho, A.E. J. Org. Chem. 2003, 68, 4553.
73. Nasreen, A.; Adapa, S.R. Org. Prep. Proceed. Int. 1999, 31, 573.
74. Kamal, A.; Ramana, K.V.; Arifuddin, M. Chem. Lett. 1999, 827.
75. Enders, D.; Hundertmark, T.; Lazny, R. Synth. Commun. 1999, 29, 27.
76. Sacks, C.E.; Fuchs, P.L. Synthesis 1976, 456.
77. See Chandrasekhar, S.; Reddy, Ch.R.; Reddy, M.V. Chem. Lett. 2000, 430; Jiricny, J.; Orere, D.M.; Reese, C.B. Synthesis 1970, 919.
78. Zhang, G.-S.; Gong, H.; Yang, D.-H.; Chen, M.-F. Synth. Commun. 1999, 29, 1165; Gong, H.; Zhang, G.-S. Synth. Commun. 1999, 29, 2591.
79. Chen, F.-E.; Liu, J.-P.; Fu, H.; Peng, Z.-Z.; Shao, L.-Y. Synth. Commun. 2000, 30, 2295.
80. Shirini, F.; Zolfigol, M.A.; Mallakpour, B.; Mallakpour, S.E.; Hajipour, A.R.; Baltork, I.M. Tetrahedron Lett. 2002, 43, 1555.
81. Mitra, A.K.; De, A.; Karchaudhuri, N. Synth. Commun. 2000, 30, 1651.
82. For reviews of the mechanism, see Bruylants, A.; Feytmants-de Medicis, E. in Patai, S.The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 465–504; Salomaa, P. in Patai, S. The Chemistry of the Carbonyl Group pt. 1, Wiley, NY, 1966, pp. 199–205.
83. See Sayer, J.M.; Conlon, E.H. J. Am. Chem. Soc. 1980, 102, 3592.
84. Cordes, E.H.; Jencks, W.P. J. Am. Chem. Soc. 1963, 85, 2843.
85. For a review of iminium ions, see Böhme, H.; Haake, M. Adv. Org. Chem. 1976, 9, pt. 1, 107.
86. Hauser, C.R.; Lednicer, D. J. Org. Chem. 1959, 24, 46. For a study of the mechanism, see Gopalakrishnan, G.; Hogg, J.L. J. Org. Chem. 1989, 54, 768.
87. Sollenberger, P.Y.; Martin, R.B. J. Am. Chem. Soc. 1970, 92, 4261. For a review of enamine hydrolysis see Stamhuis, E.J.; Cook, A.G. in Cook, A.G. Enamines, 2nd ed., Marcel Dekker, NY, 1988, pp. 165–180.
88. See Pinnick, H.W. Org. React. 1990, 38, 655; Haines, A.H. Methods for the Oxidation of Organic Compounds, Academic Press, NY, 1988, pp. 220–231, 416–419.
89. Hawthorne, M.F. J. Am. Chem. Soc. 1957, 79, 2510. Also see van Tamelen, E.E.; Thiede, R.J. J. Am. Chem. Soc. 1952, 74, 2615; Sun, S.F.; Folliard, J.T. Tetrahedron 1971, 27, 323.
90. Feuer, H.; Spinicelli, L.F. J. Org. Chem. 1977, 42, 2091.
91. For a review, see Ballini, R.; Petrini, M. Tetrhaedron 2004, 60, 1017.
92. Bortolini, O.; De Nino, A.; Garofalo, A.; Maiuolo, L.; Russo, B. Synth. Commun. 2010, 40, 2483.
93. Olah, G.A.; Arvanaghi, M.; Vankar, Y.D.; Prakash, G.K.S. Synthesis 1980, 662.
94. Ballini, R.; Bosica, G.; Fiorini, D.; Petrini, M. Tetahedron Lett. 2002, 43, 5233.
95. Olah, G.A.; Gupta, B.G.B. Synthesis 1980, 44.
96. See Sosnovsky, G.; Krogh, J.A. Synthesis 1980, 654.
97. Kornblum, N.; Brown, R.A. J. Am. Chem. Soc. 1965, 87, 1742. See also, Edward, J.T.; Tremaine, P.H. Can J. Chem. 1971, 49, 3483, 3489, 3493.
98. See Zil'berman, E.N. Russ. Chem. Rev. 1984, 53, 900; Compagnon, P.L.; Miocque, M. Ann. Chim. (Paris) 1970, [14] 5, 11, 23.
99. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 1986–1987.
100. Moorthy, J.N.; Singhal, N. J. Org. Chem. 2005, 70, 1926.
101. Goto, A.; Endo, K.; Saito, S. Angew. Chem. Int. Ed. 2008, 47, 3607.
102. Chemat, F.; Poux, M.; Berlan, J. J. Chem. Soc. Perkin Trans. 2 1996, 1781; 1994, 2597.
103. Mukherjee, C.; Zhu, D.; Biehl, E.R.; Parmar, R.R.; Hua, L. Tetrahedron 2006, 62, 6150. Also see Black, G.W.; Gregson, T.; McPake, C.B.; Perry, J.J.; Zhang, M. Tetrahedron Lett. 2010, 51, 1639.
104. Rounds, W.D.; Eaton, J.T.; Urbanowicz, J.H.; Gribble, G.W. Tetrahedron Lett. 1988, 29, 6557.
105. Jammot, J.; Pascal, R.; Commeyras, A. Tetrahedron Lett. 1989, 30, 563.
106. See Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 119–125. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1988–1990.
107. Basu, M.K.; Luo, F.-T. Tetrahedron Lett. 1998, 39, 3005.
108. Bendale, P.M.; Khadilkar, B.M. Synth. Commun. 2000, 30, 1713.
109. Wligus, C.P.; Downing, S.; Molitor, E.; Bains, S.; Pagni, R.M.; Kabalka, G.W. Tetrahedron Lett. 1995, 36, 3469.
110. See McKenzie, C.J.; Robson, R. J. Chem. Soc., Chem. Commun. 1988, 112.
111. Kim, E.S.; Lee, H.S.; Kim, S.H.; Kim, J.N. Tetrahedron Lett. 2010, 51, 1589.
112. Ramón, R.S.; Marion, N.; Nolan, S.P. Chemistry: Eur. J. 2009, 15, 8695.
113. See Polshettiwar, V.; Varma, R.S. Chemistry: Eur. J. 2009, 15, 1582.
114. For a solvent-free reaction. See Khadilkar, B.M.; Madyar, V.R. Synth. Commun. 2002, 32, 1731.
115. Plummer, B.F.; Menendez, M.; Songster, M. J. Org. Chem. 1989, 54, 718.
116. Allen, C.L.; Lapkin, A.A.; Williams, J.M.J. Tetrahedron Lett. 2009, 50, 4262.
117. Bagley, M.C.; Chapaneri, K.; Glover, C.; Merritt, E.A. Synlett 2004 2615.
118. Kaboudin, B.; Elhamifar, D. Synthesis 2006, 224.
119. Zil'berman, E.N.; Lazaris, A.Ya. J. Gen. Chem. USSR 1963, 33, 1012.
120. For reviews, see Meskens, F.A.J. Synthesis 1981, 501; Schmitz, E.; Eichhorn, I. in Patai, S. The Chemistry of the Ether Linkage, Wiley, NY, 1967, pp. 309–351.
121. Clerici, A.; Pastori, N.; Porta, O. Tetrahedron 2001, 57, 217.
122. Kumar, R.; Chakraborti, A.K. Tetrahedron Lett. 2005, 46, 8319.
123. Gregg, B.T.; Golden, K.C.; Quin, J.F. Tetrahedron 2008, 64, 3287.
124. De, S.K.; Gibbs, R.A. Tetrahedron Lett. 2004, 45, 8141.
125. Velusamy, S.; Punniyamurthy, T. Tetrahedron Lett. 2004, 45, 4917.
126. Kotke, M.; Schreiner, P.R. Tetrahedron 2006, 62, 434.
127. See Heravi, M.M.; Tajbakhsh, M.; Habibzadeh, S.; Ghassemzadeh, M. Monat. Chem. 2001, 132, 985.
128. For many examples of each of these methods, see Meskens, F.A.J. Synthesis 1981, 501, pp. 502–505.
129. For other methods, see Ott, J.; Tombo, G.M.R.; Schmid, B.; Venanzi, L.M.; Wang, G.; Ward, T.R. Tetrahedron Lett. 1989, 30, 6151, Liao, Y.; Huang, Y.; Zhu, F. J. Chem. Soc., Chem. Commun. 1990, 493.
130. High pressure has been used to improve the results with ketones: Dauben, W.G.; Gerdes, J.M.; Look, G.C. J. Org. Chem. 1986, 51, 4964. For other methods, see Otera, J.; Mizutani, T.; Nozaki, H. Organometallics 1989, 8,2063; Thurkauf, A.; Jacobson, A.E.; Rice, K.C. Synthesis 1988, 233.
131. See Gopinath, R.; Haque, Sk.J.; Patel, B.K. J. Org. Chem. 2002, 67, 5842.
132. Wu, H.-H.; Yang, F.; Cui, P.; Tang, J.; He, M.-Y. Tetrahedron Lett. 2004, 45, 4963; Ishihara, K.; Hasegawa, A.; Yamamoto, H. Synlett 2002, 1296.
133. Kurihara, M.; Hakamata, W. J. Org. Chem. 2003, 68, 3413.
134. See Li, D.; Shi, F.; Peng, J.; Guo, S.; Deng, Y. J. Org. Chem. 2004, 69, 3582.
135. Elinson, M.N.; Feducovich, S.K.; Dmitriev, D.E.; Dorofeev, A.S.; Vereshchagin, A.N.; Nikishin, G.I. Tetrahedron Lett. 2001, 42, 5557.
136. Procopio, A.; Gaspari, M.; Nardi, M.; Oliverio, M.; Tagarelli, A.; Sindona, G. Tetrahedron Lett. 2007, 48, 8623.
137. Bailey, A.D.; Baru, A.R.; Tasche, K.K.; Mohan, R.S. Tetrahedron Lett. 2008, 49, 691.
138. For a review of hemiacetals, see Hurd, C.D. J. Chem. Educ. 1966, 43, 527.
139. Grunwald, E. J. Am. Chem. Soc. 1985, 107, 4715.
140. See Grunwald, E. J. Am. Chem. Soc. 1985, 107, 4710; Leussing, D.L. J. Org. Chem. 1990, 55, 666.
141. For a list of catalysts, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1484–1485.
142. See Salaun, J. Chem. Rev. 1983, 83, 619.
143. See DeWolfe, R.H. Carboxylic Ortho Ester Derivatives, Academic Press, NY, 1970, pp. 154–164. See Karimi, B.; Ebrahimian, G.R.; Seradj, H. Org. Lett. 1999, 1, 1737; Leonard, N.M.; Oswald, M.C.; Freiberg, D.A.; Nattier, B.A.; Smith, R.C.; Mohan, R.S. J. Org. Chem. 2002, 67, 5202.
144. Kato, J.; Iwasawa, N.; Mukaiyama, T. Chem. Lett. 1985, 743. See also, Torii, S.; Takagishi, S.; Inokuchi, T.; Okumoto, H. Bull. Chem. Soc. Jpn. 1987, 60, 775.
145. Rao, H.S.P.; Jothilingam, S. J. Org. Chem. 2003, 68, 5392.
146. Jeevanandam, A.; Narkunan, K.; Ling, Y.-C. J. Org. Chem. 2001, 66, 6014. See Arcadi, A.; Cerichelli, G.; Chiarini, M.; Di Giuseppe, S.; Marinelli, F. Tetrahedron Lett. 2000, 41, 9195.
147. See Olah, G.A.; Mehrotra, A.K. Synthesis 1982, 962.
148. Karimi, B.; Seradj, H.; Ebrahimian, G.R. Synlett 2000, 623
149. Roy, S.C.; Banerjee, B. Synlett 2002, 1677.
150. Yadav, J.S.; Reddy, B.V.S.; Venugapal, C.; Ramalingam, V.T. Synlett 2002, 604.
151. Trost, B.M.; Lee, C.B. J. Am. Chem. Soc. 2001, 123, 3671; Wang, C.; Li, M. Synth. Commun. 2002, 32, 3469.
152. Smith, B.M.; Graham, A.E. Tetrahedron Lett. 2006, 47, 9317.
153. Chandra, K.L.; Saravanan, P.; Singh, V.K. Synlett 2000, 359.
154. Aggen, D.H.; Arnold, J.N.; Hayes, P.D.; Smoter, N.J.; Mohan, R.S. Tetrahedron 2004, 60, 3675.
155. A solvent-free reaction. See Karimi, B.; Ebrahimian, G.-R.; Seradj, H. Synth. Commun. 2002, 32, 669.
156. Smitha, G.; Reddy, Ch.S. Tetrahedron 2003, 59, 9571.
157. Mei, Y.; Bentley, P.A.; Du, J. Tetrahedron Lett. 2009, 50, 4199.
158. Kamble, V.T.; Jamode, V.S.; Joshi, N.S.; Biradar, A.V.; Deshmukh, R.Y. Tetrahedron Lett. 2006, 47, 5573.
159. Trost, B.M.; Lee, C.B. J. Am. Chem. Soc. 2001, 123, 3671.
160. Libman, J.; Sprecher, M.; Mazur, Y. Tetrahedron 1969, 25, 1679.
161. Doyle, M.P.; DeBruyn, D.J.; Kooistra, D.A. J. Am. Chem. Soc. 1972, 94, 3659.
162. Gooßen, L.J.; Linder, C. Synlett 2006, 3489. For another method, see Loim, L.M.; Parnes, Z.N.; Vasil'eva, S.P.; Kursanov, D.N. J. Org. Chem. USSR 1972, 8, 902.
163. Spafford, M.J.; Anderson, E.D.; Lacey, J.R.; Palma, A.C.; Mohan, R.S. Tetrahedron Lett. 2007, 48, 8665.
164. Anzalone, P.W.; Mohan, R.S. Synthesis 2005, 2661.
165. Yang, M.-S.; Xu, L.-W.; Qiu, H.-Y.; Lai, G.-Q.; Jiang, J.-X. Tetrahedron Lett. 2008, 49, 253.
166. Sassaman, M.B.; Kotian, K.D.; Prakash, G.K.S.; Olah, G.A. J. Org. Chem. 1987, 52, 4314. See also, Kikugawa, Y. Chem. Lett. 1979, 415.
167. Oh, L.M.; Spoors, P.G.; Goodman, R.M. Tetrahedron Lett. 2004, 45, 4769.
168. Larksarp, C.; Alper, H. J. Org. Chem. 1999, 64, 4152.
169. See Walter, W.; Bode, K. Angew. Chem. Int. Ed. 1967, 6, 281. See also, Wynne, J.H.; Jensen, S.D.; Snow, A.W. J. Org. Chem. 2003, 68, 3733.
170. Koketsu, M.; Ishida, M.; Takakura, N.; Ishihara, H. J. Org. Chem. 2002, 67, 486.
171. See Satchell, D.P.N.; Satchell, R.S.Chem. Soc. Rev. 1975, 4, 231.
172. See Donohoe, G.; Satchell, D.P.N.; Satchell, R.S. J. Chem. Soc. Perkin Trans. 2 1990, 1671 and references cited therein. See also, Sivakamasundari, S.; Ganesan, R. J. Org. Chem. 1984, 49, 720.
173. See Kim, Y.H.; Park, H.S. Synlett 1998, 261; Duggan, M.E.; Imagire, J.S. Synthesis 1989, 131.
174. McManus, S.P.; Bruner, H.S.; Coble, H.D.; Ortiz, M. J. Org. Chem. 1977, 42, 1428.
175. Bailey, W.J.; Griffith, J.R. J. Org. Chem. 1978, 43, 2690.
176. Nikoforov, A.; Jirovetz, L.; Buchbauer, G. Liebigs Ann. Chem. 1989, 489.
177. See Compagnon, P.L.; Miocque, M. Ann. Chim. (Paris) 1970, [14] 5, 23, see pp. 24–26. Imino esters: see Neilson, D.G. in Patai, S. The Chemistry of Amidines and Imidates, Wiley, NY, 1975, pp. 385–489.
178. Schaefer, F.C.; Peters, G.A. J. Org. Chem. 1961, 26, 412.
179. See Fuks, R.; Hartemink, M.A. Bull. Soc. Chim. Belg. 1973, 82, 23.
180. Kim, S.i.; Chu, F.; Dueno, E.E.; Jung, K.W. J. Org. Chem. 1999, 64, 4578.
181. See Dunn, A.D.; Rudorf, W. Carbon Disulphide in Organic Chemistry, Ellis Horwood, Chichester, 1989, pp. 316–367.
182. Meurling, P.; Sjöberg, B.; Sjöberg, K. Acta Chem. Scand. 1972, 26, 279.
183. Campaigne, E.; Edwards, B.E. J. Org. Chem. 1962, 27, 3760.
184. For 15, see Mayer, R.; Hiller, G.; Nitzschke, M.; Jentzsch, J. Angew. Chem. Int. Ed. 1963, 2, 370.
185. Cairns, T.L.; Evans, G.L.; Larchar, A.W.; McKusick, B.C. J. Am. Chem. Soc. 1952, 74, 3982.
186. Campaigne, E.; Edwards, B.E. J. Org. Chem. 1962, 27, 3760; Demuynck, M.; Vialle, J. Bull. Soc. Chim. Fr. 1967, 1213.
187. Harris Jr., J.F. J. Org. Chem. 1960, 25, 2259.
188. See, for example, Fournier, L.; Lamaty, G.; Nata, A.; Roque, J.P. Tetrahedron 1975, 31, 809.
189. For example, see Field, L.; Sweetman, B.J. J. Org. Chem. 1969, 34, 1799.
190. Truce, W.E.; Roberts, F.E. J. Org. Chem. 1963, 28, 961.
191. Streitwieser Jr., A.; Caldwell, R.A.; Granger, M.R. J. Am. Chem. Soc. 1964, 86, 3578; Streitwieser, Jr., A.; Maskornick, M.J.; Ziegler, G.R. Tetrahedron Lett. 1971, 3927; Ward, H.R.; Lawler, R.G. J. Am. Chem. Soc. 1967, 89,5517.
192. Fujita, E.; Nagao, Y.; Kaneko, K. Chem. Pharm. Bull. 1978, 26, 3743; Corey, E.J.; Bock, M.G. Tetrahedron Lett. 1975, 2643.
193. See Samajdar, S.; Basu, M.K.; Becker, F.F.; Banik, N.K. Tetrahedron Lett. 2001, 42, 4425.
194. Kumar, V.; Dev, S. Tetrahedron Lett. 1983, 24, 1289.
195. Ku, B.; Oh, D.Y. Synth. Commun. 1989, 433.
196. This reaction is done neat, see Kazaraya, K.; Tsuji, S.; Sato, T. Synlett 2004, 1640.
197. A solvent-free reaction. Firouzabadi, H.; Iranpoor, N.; Kohmarch, G. Synth. Commun. 2003, 33, 167.
198. Ali, M.H.; Goretti Gomes, M. Synthesis 2005, 1326.
199. Miyake, H.; Nakao, Y.; Sasaki, M. Chem. Lett. 2007, 36, 104.
200. See Kamal, A.; Chouhan, G. Synlett 2002, 474. For a review, see Olsen, R.K.; Currie, Jr., J.O. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 521–532.
201. See Laskar, D.D.; De, S.K. Tetrahedron Lett. 2004, 45, 1035, 2339.
202. Dong, D.; Ouyang, Y.; Yu, H.; Liu, Q.; Liu, J.; Wang, M.; Zhu, J. J. Org. Chem. 2005, 70, 4535.
203. See Ganguly, N.C.; Datta, M. Synlett 2004, 659.
204. Corsaro, A.; Pistarà, V. Tetrahedron 1998, 54, 15027.
205. Seebach, D.; Corey, E.J. J. Org. Chem. 1975, 40, 231; Seebach, D. Synthesis 1969, 17.
206. Vedejs, E.; Fuchs, P.L. J. Org. Chem. 1971, 36, 366.
207. Ho, T.–L.; Ho, H.C.; Wong, C.M. J. Chem. Soc., Chem. Commun. 1972, 791a.
208. Bahrami, K.; Khodaei, M.M.; Tajik, M. Synthesis 2010, 4282.
209. Shi, X.-X.; Wu, Q.-Q. Synth. Commun. 2000, 30, 4081.
210. Besra, R.C.; Rudrawar, S.; Chakraborti, A.K. Tetrahedron Lett. 2005, 46, 6213; Oksdath-Mansilla, G.; Peñéñory, A.B. Tetrahedron Lett. 2007, 48, 6150.
211. Movassagh, B.; Lakouraj, M.M.; Ghodrati, K. Synth. Commun. 2000, 30, 2353.
212. See Ballini, R.; Bosica, G.; Maggi, R.; Mazzacani, A.; Righi, P.; Sartori, G. Synthesis 2001,1826; Mondal, E.; Sahu, P.R.; Khan, A.T. Synlett 2002, 463.
213. See Usov, V.A.; Timokhina, L.V.; Voronkov, M.G. Russ. Chem. Rev. 1990, 59, 378.
214. See Muraoka, M.; Yamamoto, T.; Enomoto, K.; Takeshima, T. J. Chem. Soc. Perkin Trans. 1 1989, 1241, and references cited in these papers.
215. Vedejs, E.; Perry, D.A. J. Am. Chem. Soc. 1983, 105, 1683. See also, Baldwin, J.E.; Lopez, R.C.G. J. Chem. Soc., Chem. Commun. 1982, 1029.
216. Cyclopentanone and cyclohexanone gave different products: Scheibye, S.; Shabana, R.; Lawesson, S.; R![]() mming, C. Tetrahedron 1982, 38, 993.
mming, C. Tetrahedron 1982, 38, 993.
217. See Thomsen, I.; Clausen, K.; Scheibye, S.; Lawesson, S. Org. Synth.VII, 372.
218. See Jesberger, M.; Davis, T.P.; Barner, L. Synthesis 2003, 1929. For a study of the mechanism, see Rauchfuss, T.B.; Zank, G.A. Tetrahedron Lett. 1986, 27, 3445. See Ozturk, T.; Ertas, E.; Mert, O. Chem. Rev. 2007, 107, 5210. For reactions with fluorous Lawesson's reagent, see Kaleta, Z.; Makowski, B.T.; Soós, T.; Dembinski, R. Org. Lett. 2006, 8, 1625.
219. See Scheeren, J.W.; Ooms, P.H.J.; Nivard, R.J.F. Synthesis 1973, 149.
220. Curphey, T.J. J. Org. Chem. 2002, 67, 6461.
221. Polshettiwar, V.; Kaushik, M.P. Tetrahedron Lett. 2004, 45, 6255.
222. See Okazaki, R.; Inoue, K.; Inamoto, N. Tetrahedron Lett. 1979, 3673.
223. For a review of thiono esters RC(=S)OR', see Jones, B.A.; Bradshaw, J.S. Chem. Rev. 1984, 84, 17.
224. Yde, B.; Yousif, N.M.; Pedersen, U.S.; Thomsen, I.; Lawesson, S.-O. Tetrahedron 1984, 40, 2047; Thomsen, I.; Clausen, K.; Scheibye, S.; Lawesson, S. Org. Synth. VII, 372.
225. Filippi, J.-J.; Fernandez, X.; Lizzani-Cuvelier, L.; Loiseau, A.-M. Tetrahedron Lett. 2003, 44, 6647.
226. Smith, D.C.; Lee, S.W.; Fuchs, P.L. J. Org. Chem. 1994, 59, 348.
227. Charette, A.B.; Chua, P. Tetrahedron Lett. 1998, 39, 245.
228. Charette, A.B.; Grenon, M. J. Org. Chem. 2003, 68, 5792.
229. Pathak, U.; Pandey, L.K.; Tank, R. J. Org. Chem. 2008, 73, 2890.
230. For a review of dithiocarboxylic esters, see Kato, S.; Ishida, M. Sulfur Rep., 1988, 8, 155.
231. Davy, H.; Metzner, P. Chem. Ind. (London) 1985, 824.
232. Mukaiyama, T.; Saigo, K. Chem. Lett. 1973, 479.
233. Kikugawa, Y. Chem. Lett. 1981, 1157.
234. For cleavage with ion-exchange resins, see Khusid, A.Kh.; Chizhova, N.V. J. Org. Chem. USSR 1985, 21, 37. For a discussion of the mechanism, see Young, P.R.; Jencks, W.P. J. Am. Chem. Soc. 1978, 100, 1228.
235. The reaction has also been used to protect an aldehyde group in the presence of a keto group: Chihara, T.; Wakabayashi, T.; Taya, K. Chem. Lett. 1981, 1657.
236. For a review of this reagent in organic synthesis, see Jeyaraman, R. in Pizey, J.S. Synthetic Reagents, Vol. 5, Wiley, NY, 1983, pp. 9–83.
237. These compounds have been detected by 13C NMR: Chudek, J.A.; Foster, R.; Young, D. J. Chem. Soc. Perkin Trans. 2 1985, 1285.
238. Methanimine CH2=NH is stable in solution for several hours at –95° C, but rapidly decomposes at –80° C: Braillon, B.; Lasne, M.C.; Ripoll, J.L.; Denis, J.M. Nouv. J. Chim., 1982, 6, 121. See also, Bock, H.; Dammel, R. Chem. Ber. 1987, 120, 1961.
239. For a review of the reactions between amines and formaldehyde, see Farrar, W.V. Rec. Chem. Prog., 1968, 29, 85. For a synthesis of imines, see Kwon, M.S.; Kim, S.; Park, S.; Bosco, W.; Chidrala, R.K.; Park, J. J. Org. Chem. 2009, 74, 2877; Kim, J.W.; He, J.; Yamaguchi, K.; Mizuno, N. Chem. Lett. 2009, 38, 920.
240. See Dayagi, S.; Degani, Y. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 64–83; Reeves, R.L. in Patai, S. The Chemistry of the Carbonyl Group, pt. 1, Wiley, NY, 1966, pp. 600–614. Also see Ku, Y.-Y.; Grieme, T.; Pu, Y.-M.; Bhatia, A.V.; King, S.A. Tetrahedron Lett. 2005, 46, 1471; Guzen, K.P.; Guarezemini, A.S.; Órfão, A.T.G.; Cella, R.; Pereira, C.M.P.; Stefani, H.A. Tetrahedron Lett. 2007, 48, 1845.
241. See Lai, J.T. Tetrahedron Lett. 2002, 43, 1965.
242. Love, B.E.; Ren, J. J. Org. Chem. 1993, 58, 5556.
243. See Forlani, L.; Marianucci, E.; Todesco, P.E. J. Chem. Res. (S) 1984, 126.
244. See Eisch, J.J.; Sanchez, R. J. Org. Chem. 1986, 51, 1848.
245. Weingarten, H.; Chupp, J.P.; White, W.A. J. Org. Chem. 1967, 32, 3246.
246. See Roelofsen, D.P.; van Bekkum, H. Recl. Trav. Chim. Pays-Bas 1972, 91, 605.
247. Andrade, C.K.Z.; Takada, S.C.S.; Alves, L.M.; Rodrigues, J.P.; Suarez, P.A.Z.; Branda, R.F.; Soares, V.C.D. Synlett 2004, 2135.
248. For a review, see Katritzky, A.R.; Ostercamp, D.L.; Yousaf, T.I. Tetrahedron 1987, 43, 5171.
249. See Cheng, C.; Yan, S. Org. React. 1982, 28, 37.
250. See Yadav, J.S.; Reddy, B.V.S.; Premalatha, K. Synlett 2004, 963.
251. Hsiao, Y.; Rivera, N.R.; Yasuda, N.; Hughes, D.L.; Reider, P.J. Org. Lett. 2001, 3, 1101.
252. See Zvezdina, E.A.; Zhadonva, M.P.; Dorofeenko, G.N. Russ. Chem. Rev. 1982, 51, 469.
253. Danks, T.N. Tetrahedron Lett. 1999, 40, 3957.
254. Banik, B.K.; Samajdar, S.; Banik, I. J. Org. Chem. 2004, 69, 213.
255. Klappa, J.J.; Rich, A.E.; McNeill, K. Org. Lett. 2002, 4, 435.
256. See Hodgson, D.M.; Bray, C.D.; Kindon, N.D.; Reynolds, N.J.; Coote, S.J.; Um, J.M.; Houk, K.N. J. Org. Chem. 2009, 74, 1019.
257. See Duhamel, P.; Cantacuzène, J. Bull. Soc. Chim. Fr. 1962, 1843.
258. Duhamel, P. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 2, Wiley, NY, 1982, pp. 849–907.
259. See Haynes, L.W.; Cook, A.G. in Cook, A.G. Enamines, 2nd. ed., Marcel Dekker, NY, 1988, pp. 103–163; Pitacco, G.; Valentin, E. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 1, Wiley, NY, 1982, pp. 623–714.
260. For another method, see Katritzky, A.R.; Long, Q.; Lue, P.; Jozwiak, A. Tetrahedron 1990, 46, 8153.
261. Bélanger, G.; Doré, M.; Ménard, F.; Darsigny, V. J. Org. Chem. 2006, 71, 7481.
262. See Nilsson, A.; Carlson, R. Acta Chem. Scand. Ser. B 1984, 38, 523.
263. See Carlson, R.; Nilsson, A.; Strömqvist, M. Acta Chem. Scand. Ser. B 1983, 37, 7.
264. Kardon, F.; Mörtl, M.; Knausz, D. Tetrahedron Lett. 2000, 41, 8937.
265. Erker, G.; Riedel, M.; Koch, S.; Jödicke, T.; Würthwein, E.-U. J. Org. Chem. 1995, 60, 5284.
266. Stefani, H.A.; Costa, I.M.; Silva, D. de O. Synthesis 2000, 1526.
267. Rechsteiner, B.; Texier-Boullet, F.; Hamelin, J. Tetrahedron Lett. 1993, 34, 5071.
268. Wei, C.; Li, Z.; Li, C.-J. Org. Lett. 2003, 5, 4473.
269. Li, Z.; Wei, C.; Chen, L.; Varma, R.S.; Li, C.-J. Tetrahedron Lett. 2004, 45, 2443.
270. Shi, L.; Tu, Y.-Q.; Wang, M.; Zhang, F.-M.; Fan, C.-A. Org. Lett. 2004, 6, 1001.
271. Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2003, 125, 9584.
272. See Day, A.C.; Whiting, M.C. Org. Synth. VI, 10.
273. See Behforouz, M.; Bolan, J.L.; Flynt, M.S. J. Org. Chem. 1985, 50, 1186.
274. For a review of arylhydrazones, see Buckingham, J. Q. Rev. Chem. Soc. 1969, 23, 37.
275. Nakamura, E.; Sakata, G.; Kubota, K. Tetrahedron Lett. 1998, 39, 2157.
276. Palacios, F.; Aparicio, D.; de los Santos, J.M. Tetrahedron Lett. 1993, 34, 3481.
277. Barrett, I.C.; Langille, J.D.; Kerr, M.A. J. Org. Chem. 2000, 65, 6268.
278. Ahmed, M.; Jackstell, R.; Seayad, A.M.; Klein, H.; Beller, M. Tetrahedron Lett. 2004, 45, 869.
279. Pasha, M.A.; Nanjundaswamy, H.M. Synth. Commun. 2004, 34, 3827.
280. See Mester, L.; El Khadem, H.; Horton, D. J. Chem. Soc., C 1970, 2567.
281. See Stachissini, A.S.; do Amaral, L. J. Org. Chem. 1991, 56, 1419.
282. Newkome, G.R.; Fishel, D.L. J. Org. Chem. 1966, 31, 677.
283. Jones, W.H.; Tristram, E.W.; Benning, W.F. J. Am. Chem. Soc. 1959, 81, 2151.
284. Sharghi, H.; Sarvari, M.H. Synlett 2001, 99.
285. Ren, R.X.; Ou, W. Tetrahedron Lett. 2001, 42, 8445.
286. Hajipour, A.R.; Mohammadpoor-Baltork, I.; Nikbaghat, K.; Imanzadeh, G. Synth. Commun. 1999, 29, 1697.
287. Jencks, W.P. J. Am. Chem. Soc. 1959, 81, 475; Prog. Phys. Org. Chem. 1964, 2, 63.
288. See Cockerill, A.F.; Harrison, R.G.in Patai, S. The Chemistry of Functional Groups, Supplement A, pt. 1, Wiley, NY, 1977, pp. 288–299; Sollenberger, P.Y.; Martin, R.B.in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 367–392. For isotope effect studies, see Rossi, M.H.; Stachissini, A.S.; do Amaral, L. J. Org. Chem. 1990, 55, 1300.
289. Sayer, J.M.; Jencks, W.P. J. Am. Chem. Soc. 1972, 94, 3262.
290. Sayer, J.M.; Edman, C. J. Am. Chem. Soc. 1979, 101, 3010.
291. Cocivera, M.; Effio, A. J. Am. Chem. Soc. 1976, 98, 7371.
292. Olah, G.A.; Keumi, T. Synthesis 1979, 112.
293. Movassagh, B.; Shokri, S. Tetrahedron Lett. 2005, 46, 6923.
294. Das, B.; Ramesh, C.; Madhusudhan, P. Synlett 2000, 1599.
295. Kumar, H.M.S.; Reddy, B.V.S.; Reddy, P.T.; Yadav, J.S. Synthesis 1999, 586; Chakraborti, A.K.; Kaur, G. Tetrahedron 1999, 55, 13265.
296. Sharghi, H.; Sarvari, M.H. Tetrahedron 2002, 58, 10323.
297. Streith, J.; Fizet, C.; Fritz, H. Helv. Chim. Acta 1976, 59, 2786.
298. Ballini, R.; Fiorini, D.; Palmieri, A. Synlett 2003, 1841.
299. Bose, D.S.; Goud, P.R. Synth. Commun. 2002, 32, 3621.
300. See Neunhoeffer, H.; Diehl, W.; Karafiat, U. Liebigs Ann. Chem. 1989, 105.
301. Veverková, E.; Toma, Š. Synth. Commun. 2000, 30, 3109.
302. Srinivas, K.V.N.S.; Reddy, E.B.; Das, B. Synlett 2002, 625.
303. Zhu, J.-L.; Lee, F.-Y.; Wu, J.-D.; Kuo, C.-W.; Shia, K.-S. Synlett 2007, 1317.
304. Chill, S.T.; Mebane, R.C. Synth. Commun. 2009, 39, 3601.
305. Movassagh, B.; Fazeli, A. Synth. Commun. 2007, 37, 625.
306. Tanuwidjaja, J.; Peltier, H.M.; Lewis, J.C.; Schenkel, L.B.; Ellman, J.A. Synthesis 2007, 3385.
307. Nishiyama, K.; Oba, M.; Watanabe, A. Tetrahedron 1987, 43, 693.
308. Ganboa, I.; Palomo, C. Synth. Commun. 1983, 13, 219.
309. Talukdar, S.; Hsu, J.-L.; Chou, T.-C.; Fang, J.-M. Tetrahedron Lett. 2001, 42, 1103.
310. Erman, M.B.; Snow, J.W.; Williams, M.J. Tetrahedron Lett. 2000, 41, 6749.
311. Arote, N.D.; Bhalerao, D.S.; Akamanchi, K.G. Tetrahedron Lett. 2007, 48, 3651. Also see Zhu, C.; Ji, L.; Wei, Y. Synthesis 2010, 3121.
312. Zhu, Y.-Z.; Cai, C. Monat. Chemie 2010, 141, 637.
313. Chen, F.-E.; Kuang, Y.-Y.; Dai, H.-F.; Lu, L.; Huo, M. Synthesis 2003, 2629.
314. Hwu, J.R.; Wong, F.F. Eur. J. Org. Chem. 2006, 2513.
315. Mori, N.; Togo, H. Synlett 2005, 1456. Also see Reddy, K.R.; Maheswari, C.U.; Venkateshwar, M.; Prashanthi, S.; Kantam, M.L. Tetrahedron Lett. 2009, 50, 2050.
316. Wood, J.L.; Khatri, N.A.; Weinreb, S.M. Tetrahedron Lett. 1979, 4907.
317. See Kadyrov, R.; Riermeier, T.H.; Dingerdissen, U.; Tararov, V.; Börner, A. J. Org. Chem. 2003, 68, 4067; Chi, Y.; Zhou, Y.-G.; Zhang, X. J. Org. Chem. 2003, 68, 4120.
318. See Rylander, P.N. Hydrogenation Methods Academic Press, NY, 1985, pp. 82–93; Klyuev, M.V.; Khidekel, M.L. Russ. Chem. Rev. 1980, 49, 14; Rylander, P.N. Catalytic Hydrogenation over Platinum Metals Academic Press, NY, 1967, pp. 291–303.
319. See Le Bris, A.; Lefebvre, G.; Coussemant, F. Bull. Soc. Chim. Fr. 1964, 1366, 1374, 1584, 1594.
320. See Bhattacharyya, S. Synth. Commun. 2000, 30, 2001.
321. Spialter, L.; Pappalardo, J.A. The Acyclic Aliphatic Tertiary Amines Macmillan, NY, 1965, pp. 44–52.
322. Song, Y.; Sercel, A.D.; Johnson, D.R.; Colbry, N.L.; Sun, K.-L.; Roth, B.D. Tetrahedron Lett. 2000, 41, 8225.
323. For a list of many of these, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 835–840.
324. Nugent, T.C.; El-Shazly, M.; Wakchaure, V.N. J. Org. Chem. 2008, 73, 1297. For a reaction using ammonia-borane, see Ramachandran, P.V.; Gagare, P.D.; Sakavuyi, K.; Clark. P. Tetrahedron Lett. 2010, 51, 3167. 1,2,3-Triazole-boranes have also been used: Liao, W.; Chen, Y.; Liu, Y.; Duan, H.; Petersen, J.L.; Shi, X. Chem. Commun. 2009, 6436.
325. Apodaca, R.; Xiao, W. Org. Lett. 2001, 3, 1745. For a Mo-catalzyed reaction see Smith, C.A.; Cross, L.E.; Hughes, K.; Davis, R.E.; Judd, D.B.; Merritt, A.T. Tetrahedron Lett. 2009, 50, 4906.
326. Mizuta, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2005, 70, 2195; Lee, O.-Y.; Law, K.-L.; Ho, C.-Y; Yang, D. J. Org. Chem. 2008, 73, 8829.
327. Lee, O.-Y.; Law, K.-L.; Yang, D. Org. Lett. 2009, 11, 3302.
328. da Silva, R.A.; Estevam, I.H.S.; Bieber, L.W. Tetrahedron Lett. 2007, 48, 7680.
329. Chandrasekhar, S.; Reddy, Ch.R.; Ahmed, M. Synlett 2000, 1655.
330. Gribble, G.W.; Nutaitis, C.F. Synthesis 1987, 709. For the use of an ionic liquid-water system, see Nagaiah, K.; Kumar, V.N.; Rao, R.S.; Reddy, B.V.S.; Narsaiah, A.V.; Yadav, J.S. Synth. Commun. 2006, 36, 3345.
331. Neidigh, K.A.; Avery, M.A.; Williamson, J.S.; Bhattacharyya, S. J. Chem. Soc. Perkin Trans. 1 1998, 2527; Bhattacharyya, S. J. Org. Chem. 1995, 60, 4928.
332. Saxena, I.; Borah, R.; Sarma, J.C. J. Chem. Soc., Perkin Trans. 1 2000, 503.
333. This is a solvent-free reaction. See Cho, B.T.; Kang, S.K. Synlett 2004, 1484.
334. Yoon, N.M.; Kim, E.G.; Son, H.S.; Choi, J. Synth. Commun. 1993, 23, 1595.
335. Bhattacharyya, S.; Rana, S.; Gooding, O.W.; Labadie, J. Tetrahedron Lett. 2003, 44, 4957.
336. Cabral, S.; Hulin, B.; Kawai, M. Tetrahedron Lett. 2007, 48, 7134.
337. Alinezhad, H.; Tajbakhsh, M.; Zamani, R. Synlett 2006, 431.
338. Heydari, A.; Khaksar, S.; Esfandyari, M.; Tajbakhsh, M. Tetrahedron 2007, 63, 3363.
339. Mattson, R.J.; Pham, K.M.; Leuck, D.J.; Cowen, K.A. J. Org. Chem. 1990, 55, 2552. See also, Barney, C.L.; Huber, E.W.; McCarthy, J.R. Tetrahedron Lett. 1990, 31, 5547. See Hutchins, R.O.; Natale, N.R. Org. Prep. Proced. Int. 1979, 11, 201; Lane, C.F. Synthesis 1975, 135. See also, Grenga, P.N.; Sumbler, B.L.; Beland, F.; Priefer, R. Tetrahedron Lett. 2009, 50, 6658.
340. Abdel-Magid, A.F.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. J. Org. Chem. 1996, 61, 3849.
341. Fan, R.; Pu, D.; Qin, L.; Wen, F.; Yao, G.; Wu, J. J. Org. Chem. 2007, 72, 3149.
342. Itoh, T.; Nagata, K.; Miyazaki, M.; Ishikawa, H.; Kurihara, A.; Ohsawa, A. Tetrahedron 2004, 60, 6649.
343. Menche, D.; Hassfeld, J.; Li, J.; Menche, G.; Ritter, A.; Rudolph, S. Org. Lett. 2006, 8, 741.
344. Alonso, F.; Riente, P.; Yus, M. Synlett 2008, 1289.
345. For a microwave induced reaction see Torchy, S.; Barbry, D. J. Chem. Res. (S) 2001, 292.
346. Davis, B.A.; Durden, D.A. Synth. Commun. 2000, 30, 3353.
347. Barbry, D.; Torchy, S. Synth. Commun. 1996, 26, 3919.
348. Ranu, B.C.; Majee, A.; Sarkar, A. J. Org. Chem. 1998, 63, 370.
349. Rosenau, T.; Potthast, A.; Röhrling, J.; Hofinger, A.; Sixxa, H.; Kosma, P. Synth. Commun. 2002, 32, 457.
350. For a review of ammonium formate in organic synthesis, see Ram, S.; Ehrenkaufer, R.E. Synthesis 1988, 91. See Byun, E.; Hong, B.; De Castro, K.A.; Lim, M.; Rhee, H. J. Org. Chem. 2007, 72, 9815.
351. Moore, M.L. Org. React. 1949, 5, 301; Awachie, P.I.; Agwada, V.C. Tetrahedron 1990, 46, 1899 and references cited therein; Loupy, A.; Monteux, D.; Petit, A.; Aizpurua, J.M.; Domínguez, E.; Palomo, C. Tetrahedron Lett.1996, 37, 8177; Lejon, T.; Helland, I. Acta Chem. Scand. 1999, 53, 76.
352. Kitamura, M.; Lee, D.; Hayashi, S.; Tanaka, S.; Yoshimura, M. J. Org. Chem. 2002, 67, 8685. See Riermeier, T.H.; Dingerdissen, U.; Börner, A. Org. Prep. Proceed. Int. 2004, 36, 99.
353. Ollevier, T.; Ba, T. Tetrahedron Lett. 2003, 44, 9003.
354. Billet, M.; Klotz, P.; Mann, A. Tetrahedron lett. 2001, 42, 631.
355. Kobayashi, S.; Ueno, M.; Suzuki, R.; Ishitani, H.; Kim, H.-S.; Wataya, Y. J. Org. Chem. 1999, 64, 6833.
356. Suwa, T.; Shibata, I.; Nishino, K.; Baba, A. Org. Lett. 1999, 1, 1579.
357. See Choudary, B.M.; Jyothi, K.; Madhi, S.; Kantam, M.L. Synlett 2004, 231; Yadav, J.S.; Reddy, B.V.S.; Raju, A.K. Synthesis 2003, 883.
358. Storer, R.I.; Carrera, D.E.; Ni, Y.; MacMillan, D.W.C. J. Am. Chem. Soc. 2006, 128, 84. See Hoffmann, S.; Nicoletti, M.; List, B. J. Am. Chem. Soc. 2006, 128, 13074.
359. Sugiura, M.; Mori, C.; Kobayashi, S. J. Am. Chem. Soc. 2006, 128, 11038.
360. Nugent, T.C.; El-Shazly, M.; Wakchaure, V.N. J. Org. Chem. 2008, 73, 1297.
361. Côté, A.; Charette, A.B. J. Org. Chem. 2005, 70, 10864.
362. Koszelewski, D.; Lavandera, I.; Clay, D.; Guebitz, G.M.; Rozzell, D.; Kroutil, W. Angew. Chem. Int. Ed. 2008, 47, 9337.
363. Wakchaure, V.N.; Nicoletti, M.; Ratjen, L.; List, B. Synlett 2010, 2708.
364. List, B. J. Am. Chem. Soc. 2002, 124, 5656; Kumaragurubaran, N.; Juhl, K.; Zhuang, W.; B![]() gevig, A.; J
gevig, A.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2002, 124, 6254.
rgensen, K.A. J. Am. Chem. Soc. 2002, 124, 6254.
365. Challis, B.C.; Challis, J.A. in Zabicky, J.The Chemistry of Amides, Wiley, NY, 1970, pp. 754–759; Zaugg, H.E.; Martin, W.B. Org. React. 1965, 14, 52, pp. 91–95, 104–112; Gilbert, E.E. Synthesis 1972, 30.
366. Lee, K.Y.; Lee, C.G.; Kim, J.N. Tetrahedron Lett. 2003, 44, 1231.
367. Ram, R.N.; Khan, A.A. Synth. Commun. 2001, 31, 841.
368. Chemla, F.; Hebbe, V.; Normant, J.-F. Synthesis 2000, 75.
369. Jain, S.L.; Sharma, V.B.; Sain, B. Tetrahedron Lett. 2004, 45, 4341.
370. Tidwell, T.T. Acc. Chem. Res. 1990, 23, 273; Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev. 1975, 4, 231. For an enantioselective reaction, see Hodous, B.L.; Fu, G.C. J. Am. Chem. Soc. 2002, 124, 10006.
371. Stevens, C.L.; Freeman, R.C.; Noll, K. J. Org. Chem. 1965, 30, 3718.
372. Tramontini, M.; Angiolini, L. Tetrahedron 1990, 46, 1791; Gevorgyan, G.A.; Tramontini, M. Synthesis 1973, 703; House, H.O. Modern Synthetic Reactions, 2nd ed.; W.A. Benjamin, NY, 1972, pp. 654–660; Gevorgyan, G.A.; Agababyan, A.G.; Mndzhoyan, O.L. Russ. Chem. Rev. 1985, 54, 495.
373. Agababyan, A.G.; Gevorgyan, G.A.; Mndzhoyan, O.L. Russ. Chem. Rev. 1982, 51, 387.
374. Hellmann, H. Angew. Chem. 1957, 69, 463; Newer Methods Prep. Org. Chem. 1963, 2, 277.
375. Gadhwal, S.; Baruah, M.; Prajapati, D.; Sandhu, J.S. Synlett 2000, 341.
376. Qian, C.; Gao, F.; Chen, R. Tetrahedron Lett. 2001, 42, 4673. See Baer, H.H.; Urbas, L., in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, Wiley, NY, 1970, pp. 117–130.
377. see Massy, D.J.R. Synthesis 1987, 589; Dronov, V.I.; Nikitin, Yu.E. Russ. Chem. Rev. 1985, 54, 554
378. El Kaim, L.; Grimaud, L.; Perroux, Y.; Tirla, C. J. Org. Chem. 2003, 68, 8733.
379. Bur, S.; Martin, S.F. Tetrahedron 2001, 57, 3221; Martin, S.F. Acc. Chem. Res. 2002, 35, 895.
380. Cummings, T.F.; Shelton, J.R. J. Org. Chem. 1960, 25, 419.
381. Benkovic, S.J.; Benkovic, P.A.; Comfort, D.R. J. Am. Chem. Soc. 1969, 91, 1860.
382. See Schreiber, J.; Maag, H.; Hashimoto, N.; Eschenmoser, A. Angew. Chem. Int. Ed. 1971, 10, 330.
383. Jasor, Y.; Luche, M.; Gaudry, M.; Marquet, A. J. Chem. Soc., Chem. Commun. 1974, 253; Gaudry, M.; Jasor, Y.; Khac, T.B. Org. Synth. VI, 474.
384. Schreiber, J.; Maag, H.; Hashimoto, N.; Eschenmoser, A. Angew. Chem. Int. Ed. 1971, 10, 330.
385. See Bryson, T.A.; Bonitz, G.H.; Reichel, C.J.; Dardis, R.E. J. Org. Chem. 1980, 45, 524, and references cited therein.
386. Arend, M.; Risch, N. Tetrahedron Lett. 1999, 40, 6205.
387. Seebach, D.; Schiess, M.; Schweizer, W.B. Chimia 1985, 39, 272. See also, Katritzky, A.R.; Harris, P.A. Tetrahedron 1990, 46, 987.
388. See Fujii, A.; Hagiwara, E.; Sodeoka, M. J. Am. Chem. Soc. 1999, 121, 5450.
389. See Fujisawa, H.; Takahashi, E.; Mukaiyama, T. Chemistry: European J. 2006, 12, 5082.
390. Akiyama, T.; Takaya, J.; Kagoshima, H. Tetrahedron Lett. 2001, 42, 4025.
391. Loh, T.-P.; Wei, L.L. Tetrahedron Lett. 1998, 39, 323.
392. See Córdova, A. Acc. Chem. Res. 2004, 37, 102; Marques, M.M.B. Angew. Chem. Int. Ed. 2006, 45, 348; Ibrahem, I.; Córdova, A. Chem. Commun. 2006, 1760; Amedjkouh, M.; Brandberg, M. Chem. Commun. 2008, 3043.
393. See Rodríguez, B.; Bolm, C. J. Org. Chem. 2006, 71, 2888.
394. List, B.; Pojarliev, P.; Biller, W.T.; Martin, H.J. J. Am. Chem. Soc. 2004, 124, 827; Ibrahem, I.; Casas, J.; Córdova, A. Angew. Chem. Int. Ed. 2004, 43, 6528; Yang, J.W.; Stadler, M.; List, B. Angew. Chem. Int. Ed. 2007, 46,609.
395. Mitsumori, S.; Zhang, H.; Cheong, P.H.-Y.; Houk, K.N.; Tanaka, F.; Barbas, III, C.F. J. Am. Chem. Soc. 2006, 128, 1040; Zhang, H.; Mifsud, M.; Tanaka, F.; Barbas, III, C.F. J. Am. Chem. Soc. 2006, 128, 9630. See also, Hayashi, Y.; Aratake, S.; Imai, Y.; Hibino, K.; Chen, Q.-Y.; Yamaguchi, J.; Uchimaru, T. Chemistry: Asian J. 2008, 3, 225.
396. Morimoto, H.; Lu, G.; Aoyama, N.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 9588; Cutting, G.A.; Stainforth, N.E.; John, M.P.; Kociok-Köhn, G.; Willis, M.C. J. Am. Chem. Soc. 2007, 129, 10632.
397. Kano, T.; Yamaguchi, Y.; Tokuda, O.; Maruoka, K. J. Am. Chem. Soc. 2005, 127, 16408; Kano, T.; Hato, Y.; Yamamoto, A.; Maruoka, K. Tetrahedron 2008, 64, 1197.
398. Lou, S.; Taoka, B.M.; Ting, A.; Schaus, S.E. J. Am. Chem. Soc. 2005, 127, 11256; Song, J.; Wang, Y.; Deng, L. J. Am. Chem. Soc. 2006, 128, 6048.
399. Haurena, C.; LeGall, E.; Sengmany, S.; Martens, T. Tetrahedron 2010, 66, 9902.
400. Guo, Q.-X.; Liu, H.; Guo, C.; Luo, S.-W.; Gu, Y.; Gong, L.-Z. J. Am. Chem. Soc. 2007, 129, 3790; Yamanaka, M.; Itoh, J.; Fuchibe, K.; Akiyama, T. J. Am. Chem. Soc. 2007, 129, 6756; Rueping, M.; Sugiono, E.; Schoepke, F.R. Synlett 2007, 1441.
401. Uraguchi, D.; Koshimoto, K.; Ooi, T. J. Am. Chem. Soc. 2008, 130, 10878.
402. Kobayashi, S.; Hamada, T.; Manabe, K. J. Am. Chem. Soc. 2002, 124, 5640; Trost, B.M.; Terrell, C.R. J. Am. Chem. Soc. 2003, 125, 338.
403. Suto, Y.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 500.
404. Trost, B.M.; Jaratjaroonphong, J.; Reutrakul, V. J. Am. Chem. Soc. 2006, 128, 2778.
405. Hata, S.; Iguchi, M.; Iwasawa, T.; Yamada, K.-i.; Tomioka, K. Org. Lett. 2004, 6, 1721.
406. Jacobsen, M.F.; Ionita, L.; Skrydstrup, T. J. Org. Chem. 2004, 69, 4792.
407. Sun, W.; Xia, C.-G.; Wang, H.-W. Tetrahedron Lett. 2003, 44, 2409.
408. Liu, T.-Y.; Cui, J.-L.; Long, J.; Li, B.-J.; Wu, Y.; Ding, L.-S.; Chen, Y.-C. J. Am. Chem. Soc. 2007, 129, 1878.
409. Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2005, 127, 3672.
410. Anderson, J.C.; Blake, A.J.; Howell, G.P.; Wilson, C. J. Org. Chem. 2005, 70, 549.
411. Handa, S.; Gnanadesikan, V.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 4900.
412. Anderson, J.C.; Howell, G.P.; Lawrence, R.M.; Wilson, C.S. J. Org. Chem. 2005, 70, 5665.
413. Wang, C.-J.; Dong, X.-Q.; Zhang, Z.-H.; Xue, Z.-Y.; Teng, H.-L. J. Am. Chem. Soc. 2008, 130, 8606.
414. For a review of the mechanism, see Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev. 1975, 4, 231.
415. See Vishnyakova, T.P.; Golubeva, I.A.; Glebova, E.V. Russ. Chem. Rev. 1985, 54, 249.
416. Herr, R.J.; Kuhler, J.L.; Meckler, H.; Opalka, C.J. Synthesis 2000, 1569.
417. See Shorter, J. Chem. Soc. Rev. 1978, 7, 1. See also, Williams, A.; Jencks, W.P. J. Chem. Soc. Perkin Trans. 2 1974, 1753, 1760; Hall, K.J.; Watts, D.W. Aust. J. Chem. 1977, 30, 781, 903.
418. For reviews of amidines, see Granik, V.G. Russ. Chem. Rev. 1983, 52, 377; Gautier, J.; Miocque, M.; Farnoux, C.C. in Patai, S. The Chemistry of Amidines and Imidates, Wiley, NY, 1975, pp. 283–348.
419. Elvidge, J.A.; Linstead, R.P.; Salaman, A.M. J. Chem. Soc. 1959, 208.
420. Grivas, J.C.; Taurins, A. Can. J. Chem. 1961, 39, 761.
421. Garigipati, R.S. Tetrahedron Lett. 1990, 31, 1969.
422. Dräger, G.; Solodenko, W.; Messinger, J.; Schön, U.; Kirschning, A. Tetrahedron Lett. 2002, 43, 1401.
423. Murahashi, S.; Naota, T.; Saito, E. J. Am. Chem. Soc. 1986, 108, 7846.
424. Cobley, C.J.; van den Heuvel, M.; Abbadi, A.; de Vries, J.G. Tetrahedron Lett. 2000, 41, 2467.
425. Kamiñski, R.; Glass, R.S.; Skowroñska, A. Synthesis 2001, 1308.
426. Dunn, A.D.; Rudorf, W. Carbon Disuphide in Organic Chemistry, Ellis Horwood, Chichester, 1989, pp. 226–315; Katritzky, A.R.; Faid-Allah, H.; Marson, C.M. Heterocycles 1987, 26, 1657; Yokoyama, M.; Imamoto, T. Synthesis 1984, 797, see pp. 804–812; Katritzky, A.R.; Marson, C.M.; Faid-Allah, H. Heterocycles 1987, 26, 1333.
427. Jochims, J.C. Chem. Ber. 1968, 101, 1746.; Molina, P.; Alajarin, M.; Arques, A. Synthesis 1982, 596.
428. Wong, R.; Dolman, S.J. J. Org. Chem. 2007, 72, 3969.
429. Li, Z.; Qian, X.; Liu, Z.; Li, Z.; Song, G. Org. Prep. Proceed. Int. 2000, 32, 571.
430. Fournier, J.; Bruneau, C.; Dixneuf, P.H.; Lécolier, S. J. Org. Chem. 1991, 56, 4456. See Chiarotto, I.; Feroci, M. J. Org. Chem. 2003, 68, 7137; Lemoucheux, L.; Rouden, J.; Ibazizene, M.; Sobrio, F.; Lasne, M.-C. J. Org. Chem. 2003, 68, 7289.
431. Asanuma, Y.; Fujiwara, S.-i.; Shi-ike, T.; Kambe, N. J. Org. Chem. 2004, 69, 4845.
432. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 719–722.
433. Farah, B.S.; Gilbert, E.E. J. Org. Chem. 1965, 30, 1241.
434. See Nikolenko, L.N.; Popov, S.I. J. Gen. Chem. USSR 1962, 32, 29.
435. See Newman, M.S.; Fraenkel, G.; Kirn, W.N. J. Org. Chem. 1963, 28, 1851.
436. See Napolitano, E.; Fiaschi, R.; Mastrorilli, E. Synthesis 1986, 122.
437. Hoffmann, R.W.; Bovicelli, P. Synthesis 1990, 657. See also, Lansinger, J.M.; Ronald, R.C. Synth. Commun. 1979, 9, 341.
438. Kabalka, G.W.; Wu, Z. Tetrahedron Lett. 2000, 41, 579.
439. Newman, M.S. J. Org. Chem. 1969, 34, 741.
440. Clark, R.F.; Simons, J.H. J. Org. Chem. 1961, 26, 5197.
441. Wang, C.J. Org. React. 1985, 34, 319; Boswell, Jr., G.A.; Ripka, W.C.; Scribner, R.M.; Tullock, C.W. Org. React. 1974, 21, 1.
442. Muratov, N.N.; Mohamed, N.M.; Kunshenko, B.V.; Burmakov, A.I.; Alekseeva, L.A.; Yagupol'skii, L.M. J. Org. Chem. USSR 1985, 21, 1292.
443. See Bunnelle, W.H.; McKinnis, B.R.; Narayanan, B.A. J. Org. Chem. 1990, 55, 768.
444. Olah, G.A.; Nojima, M.; Kerekes, I. J. Am. Chem. Soc. 1974, 96, 925.
445. Hu, C.-M.; Qing, F.-L.; Shen, C.-X. J. Chem. Soc. Perkin Trans. 1 1993, 335.
446. Chambers, R.D.; Sandford, G.; Atherton, M. J. Chem. Soc., Chem. Commun. 1995, 177.
447. York, C.; Prakash, G.K.S.; Wang, Q.; Olah, G.A. Synlett 1994, 425.
448. Takeda, T.; Sasaki, R.; Nakamura, A.; Yamauchi, S.; Fujiwara, T. Synlett 1996, 273.
449. Tordeux, M.; Boumizane, K.; Wakselman, C. J. Org. Chem. 1993, 58, 1939.
450. For example, see Clark, D.R.; Emsley, J.; Hibbert, F. J. Chem. Soc. Perkin Trans. 2 1988, 1107.
451. See Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vols. 2–4, Wiley, NY, 1985–1987.
452. Song, J.J.; Yee, N.K.; Tan, Z.; Xu, J.; Kapadia, S.R.; Senanayake, C.H. Org. Lett. 2004, 6, 4905.
453. Gold, H.; Larhed, M.; Nilsson, P. Synlett 2005, 1596.
454. See Leung, S.S.-W.; Streitwieser, A. J. Org. Chem. 1999, 64, 3390.
455. See Eicher, T. in Patai, S. The Chemistry of the Carbonyl Group, pt. 1, Wiley, NY, 1966, pp. 621–693; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 138–528; Stowell, J.C. Chem. Rev. 1984, 84, 409. For a computational study of this reaction, see Yamazaki, S.; Yamabe, S. J. Org. Chem. 2002, 67, 9346.
456. Franzén, R.G. Tetrahedron 2000, 56, 685.
457. Handy, S.T. J. Org. Chem. 2006, 71, 4659.
458. Hatano, M.; Suzuki, S.; Ishihara, K. J. Am. Chem. Soc. 2006, 128, 9998; Hatano, M.; Ito, O.; Suzuki, S.; Ishihara, K. J. Org. Chem. 2010, 75, 5008; Hatano, M.; Ito, O.; Suzuki, S.; Ishihara, K. Chem. Commun. 2010, 2674.
459. Kelly, B.G.; Gilheany, D.G. Tetrahedron Lett. 2002, 43, 887.
460. Yamamoto, Y.; Maruyama, K. Heterocycles 1982, 18, 357. Also see Tomoda, S.; Senju, T. Tetrahedron 1999, 55, 3871. See Schulze, V.; Nell, P.G.; Burton, A.; Hoffmann, R.W. J. Org. Chem. 2003, 68, 4546.
461. See Reetz, M.T. Angew. Chem. Int. Ed. 1984, 23, 556. See also, Keck, G.E.; Castellino, S. J. Am. Chem. Soc. 1986, 108, 3847.
462. See Soai, K.; Niwa, S.; Hatanaka, T. Bull. Chem. Soc. Jpn. 1990, 63, 2129. Also see Hoffmann, R.W.; Dresely, S.; Hildebrandt, B. Chem. Ber. 1988, 121, 2225; Paquette, L.A.; Learn, K.S.; Romine, J.L.; Lin, H. J. Am. Chem. Soc. 1988, 110, 879; Brown, H.C.; Bhat, K.S.; Randad, R.S. J. Org. Chem. 1989, 54, 1570.
463. See Denmark, S.E.; Weber, E.J. J. Am. Chem. Soc. 1984, 106, 7970. See Greeves, N.; Pease, J.E. Tetrahedron Lett. 1996, 37, 5821; Zweifel, G.; Shoup, T.M. J. Am. Chem. Soc. 1988, 110, 5578.
464. See Masuyama, Y.; Takahara, J.P.; Kurusu, Y. Tetrahedron Lett. 1989, 30, 3437.
465. Luderer, M.R.; Bailey, W.F.; Luderer, M.R.; Fair, J.D.; Dancer, R.J.; Sommer, M.B. Tetrahedron Asymm. 2009, 20, 981.
466. Cotton, H.K.; Norinder, J.; Bäckvall, J.-E. Tetrahedron 2006, 62, 5632; Yorimitsu, H.; Oshima, K. Angew. Chem. Int. Ed. 2005, 44, 4435; López, F.; van Zijl, A.W.; Minnaard, A.J.; Feringa, B.L. Chem. Commun. 2006, 409.
467. Muramatsu, Y.; Harada, T. Angew. Chem. Int. Ed. 2008, 47, 1088.
468. Yong, K.H.; Taylor, N.J.; Chong, J.M. Org. Lett. 2002, 4, 3553.
469. Xiao, K.-J.; Luo, J.-M.; Ye, K.-Y.; Wang, Y.; Huang, P.-Q. Angew. Chem. Int. Ed. 2010, 49, 3037.
470. Sapountzis, I.; Dube, H.; Lewis, R.; Gommermann, N.; Knochel, P. J. Org. Chem. 2005, 70, 2445.
471. Barbier, P. Compt. Rend., 1899, 128, 110. See Blomberg, C.; Hartog, F.A. Synthesis 1977, 18; Molle, G.; Bauer, P. J. Am. Chem. Soc. 1982, 104, 3481. For a list of Barbier-type reactions, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1125–1134.
472. Guijarro, A.; Yus, M. Tetrahedron Lett. 1993, 34, 3487; de Souza-Barboza, J.D.; Pétrier, C.; Luche, J. J. Org. Chem. 1988, 53, 1212.
473. Yeh, M.C.P.; Knochel, P.; Santa, L.E. Tetrahedron Lett. 1988, 29, 3887.
474. Erdik, E.; Koço![]() lu, M. Tetrahedron Lett. 2007, 48, 4211.
lu, M. Tetrahedron Lett. 2007, 48, 4211.
475. Li, C.-J. Tetrahedron 1996, 52, 5643.
476. Zhang, W.-C.; Li, C.-J. J. Org.Chem. 2000, 65, 5831.
477. For a discussion of the mechanism of this reaction, see Holm, T. Acta Chem. Scand. 1992, 46, 985.
478. An example was given in Reaction 15-25.
479. Vaskan, R.N.; Kovalev, B.G. J. Org. Chem. USSR 1973, 9, 501.
480. Kamijo, S.; Dudley, G.B. J. Am. Chem. Soc. 2005, 127, 5028.
481. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1134–1135.
482. Leroux, Y. Bull. Soc. Chim. Fr. 1968, 359.
483. Corey, E.J.; Kuwajima, I. J. Am. Chem. Soc. 1970, 92, 395. For another method, see Molander, G.A.; McKie, J.A. J. Org. Chem. 1991, 56, 4112, and references cited therein.
484. Bertini, F.; Grasselli, P.; Zubiani, G.; Cainelli, G.Tetrahedron 1970, 26, 1281.
485. See Piotrowski, A.M.; Malpass, D.B.; Boleslawski, M.P.; Eisch, J.J. J. Org. Chem. 1988, 53, 2829; Tour, J.M.; Bedworth, P.V.; Wu, R. Tetrahedron Lett. 1989, 30, 3927; Lombardo, L. Org. Synth. 65, 81.
486. For a study of Hammett ρ values, see Maclin, K.M.; Richey, Jr., H.G. J. Org. Chem. 2002, 67, 4370.
487. Lecomte, V.; Stéphan, E.; Le Bideau, F.; Jaouen, G. Tetrahedron 2003, 59, 2169.
488. See Granander, J.; Sott, R.; Hilmersson, G. Tetrahedron 2002, 58, 4717.
489. Liu, J.; Li, D.; Sun, C.; Williard, P.G. J. Org. Chem. 2008, 73, 4045.
490. For a discussion of selectivity, see Spino, C.; Granger, M.-C.; Tremblay, M.-C. Org. Lett. 2002, 4, 4735.
491. Granander, J.; Eriksson, J.; Hilmersson, G. Tetrahedron Asymmetry 2006, 17, 2021.
492. Novikov, Y.Y.; Sampson, P. J. Org. Chem. 2005, 70, 10247.
493. Cunico, R.F. Tetrahedron Lett. 2002, 43, 355.
494. See Solladié, G. in Morrison, J.D. Asymmetric Synthesis, Vol. 2, Academic Press, NY, 1983, pp. 157–199, 158–183; Nógrádi, M. Stereoselective Synthesis VCH, NY, 1986, pp. 160–193; Noyori, R.; Kitamura, M. Angew. Chem. Int. Ed. 1991, 30, 49.
495. Schön, M.; Naef, R. Tetrahedron Asymmetry 1999, 10, 169; Arvidsson, P.I.; Davidsson, Ö.; Hilmersson, G. Tetrahedron Asymmetry 1999, 10, 527.
496. Concellón, J.M.; Cuervo, H.; Fernándex-Fano, R. Tetrahedron 2001, 57, 8983.
497. Hodgson, D.M.; Reynolds, N.J.; Coote, S.J. Org. Lett. 2004, 6, 4187.
498. Florio, S.; Aggarwal, V.; Salomone, A. Org. Lett. 2004, 6, 4191.
499. Hashemsadeh, M.; Howell, A.R. Tetrahedron Lett. 2000, 41, 1855, 1859.
500. Cainelli, G.; Tangari, N.; Umani-Ronchi, A. Tetrahedron 1972, 28, 3009, and references cited therein.
501. Imamoto, T.; Takeyama, T.; Koto, H. Tetrahedron Lett. 1986, 27, 3243.
502. Taguchi, H.; Yamamoto, H.; Nozaki, H. Bull. Chem. Soc. Jpn. 1977, 50, 1588.
503. See Seyferth, D.; Weinstein, R.M.; Wang, W.; Hui, R.C.; Archer, C.M. Isr. J. Chem. 1984, 24, 167.
504. Seyferth, D.; Weinstein, R.M.; Wang, W. J. Org. Chem. 1983, 48, 1144; Seyferth, D.; Weinstein, R.M.; Wang,W.; Hui, R.C. Tetrahedron Lett. 1983, 24, 4907.
505. Lajis, N. Hj.; Khan, M.N.; Hassan, H.A. Tetrahedron 1993, 49, 3405.
506. Whitmore, F.C.; George, R.S. J. Am. Chem. Soc. 1942, 64, 1239.
507. Bartlett P.D.; Tidwell, T.T. J. Am. Chem. Soc. 1968, 90, 4421. See also, Lomas, J.S. Nouv. J. Chim., 1984, 8, 365; Molle, G.; Briand, S.; Bauer, P.; Dubois, J.E. Tetrahedron 1984, 40, 5113.
508. Buhler, J.D. J. Org. Chem. 1973, 38, 904.
509. Chastrette, M.; Amouroux, R. Chem. Commun. 1970, 470; Bull. Soc. Chim. Fr. 1970, 4348. See also, Richey, Jr., H.G.; DeStephano, J.P. J. Org. Chem. 1990, 55, 3281.
510. Canonne, P.; Foscolos, G.; Caron H.; Lemay, G. Tetrahedron 1982, 38, 3563.
511. Imamoto, T.; Takiyama, N.; Nakamura, K.; Hatajima, T.; Kamiya, Y. J. Am. Chem. Soc. 1989, 111, 4392.
512. See Holm, T. Acta Chem. Scand. Ser. B 1983, 37, 567; Ashby, E.C. Pure Appl. Chem. 1980, 52, 545; Ashby, E.C.; Laemmle, J.; Neumann, H.M. Acc. Chem. Res. 1974, 7, 272. Also see Ashby, E.C.; Laemmle, J. Chem. Rev.1975, 75, 521; Solv'yanov, A.A.; Beletskaya, I.P. Russ. Chem. Rev. 1987, 56, 465.
513. See, for example, Ashby, E.C.; Neumann, H.M.; Walker, F.W.; Laemmle, J.; Chao, L. J. Am. Chem. Soc. 1973, 95, 3330.
514. Ashby, E.C.; Laemmle, J.; Neumann, H.M. J. Am. Chem. Soc. 1972, 94, 5421.
515. Ashby, E.C.; Laemmle, J.; Neumann, H.M. J. Am. Chem. Soc. 1971, 93, 4601; Laemmle, J.; Ashby, E.C.; Neumann, H.M. J. Am. Chem. Soc. 1971, 93, 5120.
516. Ashby, E.C.; Yu, S.H.; Roling, P.V. J. Org. Chem. 1972, 37, 1918. See also, Lasperas, M.; Perez-Rubalcaba, A.; Quiroga-Feijoo, M.L. Tetrahedron 1980, 36, 3403.
517. For a review, see Dagonneau, M. Bull. Soc. Chim. Fr. 1982, II-269.
518. See Walling, C. J. Am. Chem. Soc. 1988, 110, 6846.
519. Also see Holm, T. Acta Chem. Scand. Ser. B 1988, 42, 685; Liotta, D.; Saindane, M.; Waykole, L. J. Am. Chem. Soc. 1983, 105, 2922; Yamataka, H.; Miyano, N.; Hanafusa, T. J. Org. Chem. 1991, 56, 2573.
520. Maruyama, K.; Katagiri, T. Chem. Lett. 1987, 731, 735; J. Phys. Org. Chem. 1988, 1, 21.
521. See Holm, T.; Crossland, I. Acta Chem. Scand. 1971, 25, 59.
522. Maruyama, K.; Katagiri, T. J. Am. Chem. Soc. 1986, 108, 6263; J. Phys. Org. Chem. 1989, 2, 205. See also, Maruyama, K.; Katagiri, T. J. Phys. Org. Chem. 1991, 4, 158.
523. Walter, R.I. J. Org. Chem. 2000, 65, 5014.
524. Yamataka, H.; Matsuyama, T.; Hanafusa, T. J. Am. Chem. Soc. 1989, 111, 4912.
525. Garst, J.F.; Ungváry, F. Org. Lett. 2001, 3, 605.
526. Hoffmann, RW.; Hölzer, B. Chem. Commun. 2001, 491.
527. Bodineau, N.; Mattalia, J.-M.; Hazimeh, H.; Handoo, K.L.; Timokhin, V.; Négrel, J.-C.; Chanon, M. Eur. J. Org. Chem. 2010, 2476.
528. See Yamataka, H.; Kawafuji, Y.; Nagareda, K.; Miyano, N.; Hanafusa, T. J. Org. Chem. 1989, 54, 4706.
529. Perraud, R.; Handel, H.; Pierre, J. Bull. Soc. Chim. Fr. 1980, II-283.
530. See Cabaret, D.; Welvart, Z. J. Organomet. Chem. 1974, 80, 199; Holm, T. Acta Chem. Scand. 1973, 27, 1552; Morrison, J.D.; Tomaszewski, J.E.; Mosher, H.S.; Dale, J.; Miller, D.; Elsenbaumer, R.L. J. Am. Chem. Soc.1977, 99, 3167; Okuhara, K. J. Am. Chem. Soc. 1980, 102, 244.
531. Pinkus, A.G.; Sabesan, A. J. Chem. Soc. Perkin Trans. 2 1981, 273.
532. Lipsky, S.D.; Hall, S.S. Org. Synth. VI, 537; McEnroe, F.J.; Sha, C.; Hall, S.S. J. Org. Chem. 1976, 41, 3465.
533. Hwang, Y.C.; Chu, M.; Fowler, F.W. J. Org. Chem. 1985, 50, 3885.
534. See Guillarme, S.; Plé, K.; Banchet, A.; Liard, A.; Haudrechy, A. Chem. Rev. 2006, 106, 2355.
535. Ziegenbein, W. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 207–241; Ried, W. Newer Methods Prep. Org. Chem. 1968, 4, 95.
536. Newman, H. Tetrahedron Lett. 1971, 4571. See Jacob, III, P.; Brown, H.C. J. Org. Chem. 1977, 42, 579.
537. Joung, M.J.; Ahn, J.H.; Yoon, N.M. J. Org. Chem. 1996, 61, 4472.
538. Miyamoto, H.; Yasaka, S.; Tanaka, K. Bull. Chem. Soc. Jpn. 2001, 74, 185.
539. See Midland, M.M. J. Org. Chem. 1975, 40, 2250, for the use of amine-free monolithium acetylide.
540. Beumel Jr., O.F.; Harris, R.F. J. Org. Chem. 1963, 28, 2775.
541. See Kondrat'eva, L.A.; Potapova, I.M.; Grigina, I.N.; Glazunova, E.M.; Nikitin, V.I. J. Org. Chem. USSR 1976, 12, 948.
542. Jiang, B.; Si, Y.-G. Tetrahedron Lett. 2002, 43, 8323
543. Frantz, D.E.; Fässler, R.; Carreira, E.M. J. Am. Chem. Soc. 2000, 122, 1806.
544. Boyall, D.; Frantz, D.E.; Carreira, E.M. Org. Lett. 2002, 4, 2605.; Xu, Z.; Chen, C.; Xu, J.; Miao, M.; Yan, W.; Wang, R. Org. Lett. 2004, 6, 1193; Jiang, B.; Chen, Z.; Xiong, W. Chem. Commun. 2002, 1524. For an example using zinc(II) diflate, see Chen, Z.; Xiong, W.; Jiang, B. Chem. Commun. 2002, 2098.
545. Liu, L.; Wang, R.; Kang, Y.-F.; Chen, C.; Xu, Z.-Q.; Zhou, Y.-F.; Ni, M.; Cai, H.-Q.; Gong, M.-Z. J. Org. Chem. 2005, 70, 1084.
546. See Li, H.; Huang, Y.; Jin, W.; Xue, F.; Wan, B. Tetrahedron Lett. 2008, 49, 1686; Mao, J.; Bao, Z.; Guo, J.; Ji, S. Tetrahedron 2008, 64, 9901; Yang, X.-F.; Hirose, T.; Zhang, G.-Y. Tetrahedron Asymmetry 2007, 18, 2668.
547. Sakai, N.; Hirasawa, M.; Konakahara, T. Tetrahedron Lett. 2003, 44, 4171.
548. Kwon, D.W.; Cho, M.S.; Kim, Y.H. Synlett 2001, 627.
549. Wolf, C.; Liu, S. J. Am. Chem. Soc. 2006, 128, 10996; Cozzi, P.G.; Rudolph, J.; Bolm, C.; Norrby, P.-O.; Tomasini, C. J. Org. Chem. 2005, 70, 5733.
550. Srihari, P.; Singh, V.K.; Bhunia, D.C.; Yadav, J.S. Tetrahedron Lett. 2008, 49, 7132.
551. Downey, C.W.; Mahoney, B.D.; Lipari, V.R. J. Org. Chem. 2009, 74, 2904.
552. Takita, R.; Yakura, K.; Ohshima, T.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 13760.
553. Ekström, J.; Zaitsev, S.B.; Adolfsson, H. Synlett 2006, 885.
554. Ranu, B.C.; Samanta, S.; Hajra, A. J. Org. Chem. 2001, 66, 7519.
555. Mitzel, T.M.; Palomo, C.; Jendza, K. J. Org. Chem. 2002, 67, 136.
556. For a list of reagents and references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1156–1170. Also see Gajewski, J.J.; Bocian, W.; Brichford, N.L.; Henderson, J.L. J. Org. Chem. 2002, 67, 4236.
557. See Denmark, S.E.; Fu, J. Chem. Rev. 2003, 103, 2763.
558. For a review, see Cintas, P. Synlett 1995, 1087.
559. Yi, X.-H.; Haberman, J.X.; Li, C.-J. Synth. Commun. 1998, 28, 2999; Lloyd-Jones, G.C.; Russell, T. Synlett 1998, 903; Li, C.-J.; Lu, Y.-Q. Tetrahedron Lett. 1995, 36, 2721.
560. Hirayama, L.C.; Gamsey, S.; Knueppel, D.; Steiner, D.; DeLaTorre, K.; Singaram, B. Tetrahedron Lett. 2005, 46, 2315; Haddad, T.D.; Hirayama, L.C.; Taynton, P.; Singaram, B. Tetrahedron Lett. 2008, 49, 508; Preite, M.D.; Pérez-Carvajal, A. Synlett 2006, 3337. For an example in ionic liquids, see Teo, Y.-C.; Goh, E.-L.; Loh, T.-P. Tetrahedron Lett. 2005, 46, 4573. For an example with propargylic halides, see Lacie, C. Hirayama, L.C.; Dunham, K.K.; Singaram, B. Tetrahedron Lett. 2006, 47, 5173.
561. Chan, T.H.; Yang, Y. J. Am. Chem. Soc. 1999, 121, 3228; Paquette, L.A.; Bennett, G.D.; Isaac, M.B.; Chhatriwalla, A. J. Org. Chem. 1998, 63, 1836; Li, X.-R.; Loh, T.-P. Tetrahedron Asymmetry 1996, 7, 1535.
562. Lin, M.-J.; Loh, T.-P. J. Am. Chem. Soc. 2003, 125, 13042.
563. Lee, P.H.; Ahn, H.; Lee. K.; Sung, S.-y.; Kim, S. Tetrahedron Lett. 2001, 42, 37.
564. Augé, J.; Lubin-Germain, N.; Thiaw-Woaye, A. Tetrahedron Lett. 1999, 40, 9245.
565. Loh, T.-P.; Tan, K.-T.; Yang, J.-Y.; Xiang, C.-L. Tetrahedron Lett. 2001, 42, 8701.
566. Khan, F.A.; Prabhudas, B. Tetrahedron 2000, 56, 7595.
567. Kumar, S.; Kaur, P.; Chimni, S.S. Synlett 2002, 573.
568. Kumar, V.; Chimni, S.; Kumar, S. Tetrahedron Lett. 2004, 45, 3409.
569. Basu, M.K.; Banik, B.K. Tetrahedron Lett. 2001, 42, 187.
570. Hélion, F.; Namy, J.-L. J. Org. Chem. 1999, 64, 2944.
571. See Knochel, P.; Rao, S.A. J. Am. Chem. Soc. 1990, 112, 6146; Wada, M.; Ohki, H.; Akiba, K. Bull. Chem. Soc. Jpn. 1990, 63, 1738; Marton, D.; Tagliavini, G.; Zordan, M.; Wardell, J.L. J. Organomet. Chem. 1990, 390, 127; Wang, W.; Shi, L.; Xu, R.; Huang, Y. J. Chem. Soc. Perkin Trans. 1 1990, 424.
572. Wang, J.-x.; Jia, X.; Meng, T.; Xin, L. Synthesis 2005, 2838.
573. In water: Bian, Y.-J.; Zhang, J.-Q.; Xia, J.-P.; Li, J.-T. Synth. Commun. 2006, 36, 2475.
574. Narsaiah, A.V.; Reddy, A.R.; Rao, Y.G.; Kumar, E.V.; Prakasham, R.S.; Reddy, B.V.S.; Yadav, J.S. Synthesis 2008, 3461.
575. See Bareille, L.; Le Gendre, P.; Moïse, C. Chem. Commun. 2005, 775; Estévez, R.E.; Justicia, J.; Bazdi, B.; Fuentes, N.; Paradas, M.; Choquesillo-Lazarte, D.; García-Ruiz, J.M.; Robles, R.; Gansäuer, A.; Cuerva, J.M.; Oltra, J.E. Chemistry: Eur. J. 2009, 15, 2774.
576. Kakiya, H.; Nishimae, S.; Shinokubo, H.; Oshima, K. Tetrahedron 2001, 57, 8807; Berkessel, A.; Menche, D.; Sklorz, C.A.; Schröder, M.; Paterson, I. Angew. Chem. Int. Ed. 2003, 42, 1032.
577. Chan, T.C.; Lau, C.P.; Chan, T.H. Tetrahedron Lett. 2004, 45, 4189.
578. Wang, Z.; Yuan, S.; Li, C.-J. Tetrahedron Lett. 2002, 43, 5097.
579. Hashimoto, Y.; Kagoshima, H.; Saigo, K. Tetrahedron Lett. 1994, 35, 4805.
580. Hanzawa, Y.; Tabuchi, N.; Saito, K.; Noguchi, S.; Taguchi, T. Angew. Chem. Int. Ed. 1999, 38, 2395.
581. Andrade, C.K.Z.; Azevedo, N.R.; Oliveira, G.R. Synthesis 2002, 928.
582. Zheng, Y.; Bao, W.; Zhang, Y. Synth. Commun. 2000, 30, 3517.
583. Li, L.-H.; Chan, T.H. Can. J. Chem. 2001, 79, 1536.
584. See Avilov, D.V.; Malasare, M.G.; Arslancan, E.; Dittmer, D.L. Org. Lett. 2004, 6, 2225.
585. Yanagisawa, A.; Habaue, S.; Yasue, K.; Yamamoto, H. J. Am. Chem. Soc. 1994, 116, 6130.
586. Loh, T.-P.; Zhou, J.-R. Tetrahedron Lett. 1999, 40, 9115.
587. Evans, W.J.; Workman, P.S.; Allen, N.T. Org. Lett. 2003, 5, 2041.
588. Chan, T.H.; Yang, Y. Tetrahedron Lett. 1999, 40, 3863.
589. Smith, K.; Lock, S.; El-Hiti, G.A.; Wada, M.; Miyoshi, N. Org. Biomol. Chem. 2004, 2, 935; Xu, X.; Zha, Z.; Miao, Q.; Wang, Z. Synlett 2004, 1171.
590. Lu, J.; Hong, M.-L.; Ji, S.-J.; Loh, T.-P. Chem. Commun. 2005, 1010; Teo, Y.-C.; Tan, K.-T.; Loh, T.-P. Chem. Commun. 2005, 1318; Lu, J.; Hong, M.-L.; Ji, S.-J.; Teo, Y.-C.; Loh, T.-P. Chem. Commun. 2005, 4217; Masuyama, Y.; Chiyo, T.; Kurusu, Y. Synlett 2005, 2251.
591. Zhou, J.-Y.; Jia, Y.; Sun, G.-F.; Wu, S.-H. Synth. Commun. 1997, 27, 1899.
592. Ren, P.-D.; Shao, D.; Dong, T.-W. Synth. Commun. 1997, 27, 2569.
593. Wada, M.; Fukuma, T.; Morioka, M.; Takahashi, T.; Miyoshi, N. Tetrahedron Lett. 1997, 38, 8045.
594. Taylor, R.E.; Ciavarri, J.P. Org. Lett. 1999, 1, 467.
595. Kimura, M.; Shimizu, M.; Shibata, K.; Tazoe, M.; Tamaru, Y. Angew. Chem. Int. Ed. 2003, 42, 3392.
596. Wang, M.; Yang, X.-F.; Li, C.-J. Eur. J. Org. Chem. 2003, 998.
597. Denmark, S.E.; Nguyen, S.T. Org. Lett. 2009, 11, 781.
598. Appelt, H.R.; Limberger, J.B.; Weber, M.; Rodrigues, O.E.D.; Oliveira, J.S.; Lüdtke, D.S.; Braga, A.L. Tetrahedron Lett. 2008, 49, 4956.
599. Fandrick, D.R.; Fandrick, K.R.; Reeves, J.T.; Tan, Z.; Tang, W.; Capacci, A.G.; Rodriguez, S.; Song, J.J.; Lee, H.; Yee, N.K.; Senanayake, C.H. J. Am. Chem. Soc. 2010, 132, 7600.
600. Zhang, Y.; Jia, X.; Wang, J.-X. Eur. J. Org. Chem. 2009, 2983.
601. Inoue, M.; Suzuki, T.; Nakada, M. J. Am. Chem. Soc. 2003, 125, 1140.
602. Matsubara, S.; Ikeda, T.; Oshima, K.; Otimoto, K. Chem. Lett. 2001, 1226.
603. Lu, J.-F.; You, J.-S.; Gau, H.-M. Tetrahedron Asymmetry 2000, 11, 2531.
604. Reetz, M.T. Organotitanium Reagents in Organic Synthesis, Springer, NY, 1986 (monograph), pp. 75–86. See also, Kim, S.-H.; Rieke, R.D. Tetrahedron Lett. 1999, 40, 4931.
605. Cahiez, G.; Figadere, B. Tetrahedron Lett. 1986, 27, 4445. See Soai, K.; Watanabe, M.; Koyano, M. Bull. Chem. Soc. Jpn. 1989, 62, 2124.
606. Bartoli, G.; Marcantoni, E.; Petrini, M. Angew. Chem. Int. Ed. 1993, 32, 1061; Dimitrov, V.; Bratovanov, S.; Simova, S.; Kostova, K. Tetrahedron Lett. 1994, 35, 6713.
607. Quan, L.G.; Lamrani, M.; Yamamoto, Y. J. Am. Chem. Soc. 2000, 122, 4827.
608. See Reetz, M.T.; Kyung, S.H.; Hüllmann, M. Tetrahedron 1986, 42, 2931.
609. Reetz, M.T. Organotitanium Reagents in Organic Synthesis, Springer, NY, 1986. See Weidmann, B.; Seebach, D. Angew. Chem. Int. Ed. 1983, 22, 31; Reetz, M.T. Top. Curr. Chem. 1982, 106, 1.
610. Bartoli, G.; Bosco, M.; Marcantoni, E.; Massaccesi, M.; Rinaldi, S.; Sambri, L. Tetrahedron Lett. 2001, 42, 6093.
611. Matsukawa, S.; Funabashi, Y.; Imamoto, T. Tetrahedron Lett. 2003, 44, 1007.
612. Marshall, J.A.; Palovich, M.R. J. Org. Chem. 1998, 63, 4381; Zha, Z.; Qiao, S.; Jiang, J.; Wang, Y.; Miao, Q.; Wang, Z. Tetrahedron 2005, 61, 2521. See Yasuda, M.; Hirata, K.; Nishino, M.; Yamamoto, A.; Baba, A. J. Am. Chem. Soc. 2002, 124, 13442; Masuyama, Y.; Ito, T.; Tachi, K.; Ito, A.; Kurusu, Y. Chem. Commun. 1999, 1261. Also see Li, G.-l.; Zhao, G. J. Org. Chem. 2005, 70, 4272.
613. Li, G.-l.; Zhao, G. Synlett 2005, 2540.
614. Tan, K.-T.; Chng, S.-S.; Cheng, H.-S.; Loh, T.-P. J. Am. Chem. Soc. 2003, 125, 2958; Andres, P.C.; Peatt, A.C.; Raston, C.L. Tetraheron Lett. 2002, 43, 7541.
615. Wallner, O.A.; Szabó, K.J. J. Org. Chem. 2003, 68, 2934.
616. See Issacs, N.S.; Maksimovic, L.; Rintoul, G.B.; Young, D.J. J. Chem. Soc., Chem. Commun. 1992, 1749; Isaacs, N.S.; Marshall, R.L.; Young, D.J. Tetrahedron Lett. 1992, 33, 3023.
617. Naruta, Y.; Ushida, S.; Maruyama, K. Chem. Lett. 1979, 919. For a review, see Yamamoto, Y. Aldrichimica Acta 1987, 20, 45.
618. Kalita, H.R.; Borah, A.J.; Phukan, P. Tetrahedron Lett. 2007, 48, 5047.
619. Yadav, J.S.; Reddy, B.V.S.; Kondaji, G.; Reddy, J.S.S. Tetrahedron 2005, 61, 879.
620. Choudary, B.M.; Chidara, S.; Sekhar, Ch.V.R. Synlett 2002, 1694.
621. Shibata, I.; Yoshimura, N.; Yabu, M.; Baba, A. Eur. J. Org. Chem. 2001, 3207.
622. Yanagisawa, A.; Nakashima, H.; Nakatsuka, Y.; Ishiba, A.; Yamamoto, H. Bull. Chem. Soc. Jpn. 2001, 74, 1129.
623. Kobayashi, S.; Aoyama, N.; Manabe, K. Synlett 2002, 483.
624. Kwiatkowski, P.; Chaładaj, W.; Jurczak, J. Tetrahedron 2006, 62, 5116.
625. Zhang, T.; Shi, M.; Zhao, M. Tetrahedron 2008, 64, 2412.
626. Nishiyama, Y.; Kakushou, F.; Sonoda, N. Tetrahedron Lett. 2005, 46, 787.
627. Lingaiah, B.V.; Ezikiel, G.; Yakaiah, T.; Reddy, G.V.; Rao, P.S. Tetrahedron Lett. 2006, 47, 4315.
628. Kii, S.; Maruoka, K. Tetrahedron Lett. 2001, 42, 1935.
629. Andrade, C.K.Z.; Azevedo, N.R. Tetrahedron Lett. 2001, 42, 6473.
630. Kurosa, M.; Lorca, M. Tetrahedron Lett. 2002, 43, 1765.
631. Chaudhuri, M.K.; Dehury, S.K.; Hussain, S. Tetrahedron Lett. 2005, 46, 6247. Also see Tang, L.; Ding, L.; Chang, W.-X.; Li, J. Tetrahedron Lett. 2006, 47, 303.
632. Aspinall, H.C.; Bissett, J.S.; Greeves, N.; Levin, D. Tetrahedron Lett. 2002, 43, 319.
633. Savall, B.M.; Powell, N.A.; Roush, W.R. Org. Lett. 2001, 3, 3057.
634. Khan, A.T.; Mondal, E. Synlett 2003, 694.
635. Gordon, C.M.; McCluskey, A. Chem. Commun. 1999, 1431.
636. Jin, Y.Z.; Yasuda, N.; Furuno, H.; Inanaga, J. Tetrahedron Lett. 2003, 44, 8765.
637. Lu, J.; Ji, S.-J.; Qian, R.; Chen, J.-P.; Liu, Y.; Loh, T.-P. Synlett 2004, 534.
638. Kim, J.G.; Waltz, K.M.; Garcia, I.F.; Kwiatkowski, D.; Walsh, P.J. J. Am. Chem. Soc. 2004, 126, 12580.
639. Lu, J.; Ji, S.-J.; Teo, Y.-C.; Loh, T.-P. Org. Lett. 2005, 7, 159; Teo, Y.-C.; Goh, J.-D.; Loh, T.-P. Org. Lett. 2005, 7, 2743.
640. Nokami, J.; Yoshizane, K.; Matsuura, H.; Sumida, S.-i. J. Am. Chem. Soc. 1998, 120, 6609.
641. Jang, T.-S.; Keum, G.; Kang, S.B.; Chung, B.Y.; Kim, Y. Synthesis 2003, 775.
642. Masuyama, Y.; Kaneko, Y.; Kurusu, Y. Tetrahedron Lett. 2004, 45, 8969.
643. Shibata, I.; Suwa, T.; Sakakibara, H.; Baba, A. Org. Lett. 2002, 4, 301.
644. Chan, T.H.; Yang, Y.; Li, C.J. J. Org. Chem. 1999, 64, 4452.
645. Yanagisawa, A.; Nakashima, H.; Ishiba, A.; Yamamoto, H. J. Am. Chem. Soc. 1996,118, 4723; Motoyama, Y.; Nishiyama, H. Synlett 2003, 1883; Xia, G.; Shibatomi, K.; Yamamoto, H. Synlett 2004, 2437; Jennequin, T.; Wencel-Delord, J.; Rix, D.; Daubignard, J.; Crévisy, C.; Mauduit, M. Synlett 2010, 1661.
646. Moloyama, Y.; Narusawa, H.; Nishiyama, H. Chem. Commun. 1999, 131.
647. Doucet, H.; Santelli, M. Tetrahedron Asymmetry 2000, 11, 4163.
648. Denmark, S.E.; Wynn, T. J. Am. Chem. Soc. 2001, 123, 6199.
649. Zhang, X.; Chen, D.; Liu, X.; Feng, X. J. Org. Chem. 2007, 72, 5227.
650. Luo, M.; Iwabuchi, Y.; Hatakeyama, S. Chem. Commun. 1999, 267; Yu, C.-M.; Lee, S.-J.; Jeon, M. J. Chem. Soc., Perkin Trans. 1 1999, 3557.
651. Liu, J.; Wong, C.-H. Tetrahedron Lett. 2002, 43, 3915.
652. Marx, A.; Yamamoto, H. Synlett 1999, 584.
653. Wu, K.-H.; Gau, H.-M. J. Am. Chem. Soc. 2006, 128, 14808.
654. Meisters, A.; Mole, T. Aust. J. Chem. 1974, 27, 1655. See also, Jeffery, E.A.; Meisters, A.; Mole, T. Aust. J. Chem. 1974, 27, 2569. For discussions of the mechanism of this reaction, see Ashby, E.C.; Smith, R.S. J. Organomet. Chem. 1982, 225, 71. See Maruoka, H.; Yamamoto, H. Tetrahedron 1988, 44, 5001.
655. Reetz, M.T.; Westermann, J.; Kyung, S. Chem. Ber. 1985, 118, 1050.
656. See Araki, S.; Katsumura, N.; Ito, H.; Butsugan, Y. Tetrahedron Lett. 1989, 30, 1581.
657. Reetz, M.T.; Kyung, S. Chem. Ber. 1987, 120, 123.
658. Chan, A.S.C.; Zhang, F.-Y.; Yip, C.-W. J. Am. Chem. Soc. 1997, 119, 4080.
659. Markó, I.E.; Leung, C.W. J. Am. Chem. Soc. 1994, 116, 371.
660. Crimmins, M.T.; Chaudhary, K. Org. Lett. 20002, 775.
661. Walsh, P.J. Acc. Chem. Res. 2003, 36, 739.
662. See Takai, Y.; Kataoka, Y.; Utimoto, K. J. Org. Chem. 1990, 55, 1707.
663. Wang, J.; Fan, X.; Feng, X.; Quian, Y. Synthesis 1989, 291; Riediker, M.; Duthaler, R.O. Angew. Chem. Int. Ed. 1989, 28, 494; Riediker, M.; Hafner, A.; Piantini, U.; Rihs, G.; Togni, A. Angew. Chem. Int. Ed. 1989, 30, 499.
664. Fan, O.-H.; Liu, G.-H.; Chen, X.-M.; Deng, G.-J.; Chan, A.S.C. Tetrahedron Asymmetry 2001, 12, 1559.
665. Ren, H.; Dunet, G.; Mayer, P.; Knochel, P. J. Am. Chem. Soc. 2007, 129, 5376.
666. Costa, A.M.; García, C.; Carroll, P.J.; Walsh, P.J. Tetrahedron 2005, 61, 6442; Roudeau, R.; Pardo, D.G.; Cossy, J. Tetrahedron 2006, 62, 2388; Parrott, II, R.W.; Dore, D.D.; Chandrashekar, S.P.; Bentley, J.T.; Morgan, B.S.; Hitchcock, S.R. Tetrahedron Asymmetry 2008, 19, 607; Hatano, M.; Miyamoto, T.; Ishihara, K. Synlett 2006, 1762. See Ianni, J.C.; Annamalai, V.; Phuan, P.-W.; Panda, M.; Kozlowski, M.C. Angew. Chem. Int. Ed. 2006, 45, 5502. For organocatalysts, see Kang, Y.-f.; Liu, L.; Wang, R.; Ni, M.; Han, Z.-j. Synth. Commun. 2005, 35, 1819; Hui, A.; Zhang, J.; Wang, Z. Synth. Commun. 2008, 38, 2374; Godoi, M.; Alberto, E.E.; Paixão, M.W.; Soares, L.A.; Schneider, P.H.; Braga, A.L. Tetrahedron 2010, 66, 1341; Wu, Z.-L.; Wu, H.-L.; Wu, P.-Y.; Uang, B.-J. Tetrahedron Asymm. 2009, 20, 1556; Banerjee, S.; Ferrence, G.M.; Hitchcock, S.R. Tetrahedron Asymm. 2010, 21, 837.
667. Zhu, H.J.; Jiang, J.X.; Saebo, S.; Pittman, Jr., C.U. J. Org. Chem. 2005, 70, 261; Bisai, A.; Singh, P.K.; Singh, V.K. Tetrahedron 2007, 63, 598; Burguete, M.I.; Escorihuela, J.; Luis, S.V.; Lledós, A.; Ujaque, G. Tetrahedron2008, 64, 9717; Bulut, A.; Aslan, A.; Izgü, E.Ç.; Dogan, Ö Tetrahedron Asymmetry 2007, 18, 1013; Qin, Y.-C.; Pu, L. Angew. Chem. Int. Ed. 2005, 45, 273; Park, J.K.; Lee, H.G.; Bolm, C.; Kim, B.M. Chemistry: European J.2005, 11, 945.
668. See Dean, M.A.; Hitchcock, S.R. Tetrahedron Asymmetry 2008, 19, 2563.
669. Rudolph, J.; Bolm, C.; Norrby, P.-O. J. Am. Chem. Soc. 2005, 127, 1548.
670. For a review, see Pu, L. Tetrahedron 2003, 59, 9873. See Li, Z.-B.; Pu, L. Org. Lett. 2004, 6, 1065; Dahmen, S. Org. Lett. 2004, 6, 2113; Kang, Y.-F.; Liu, L.; Wang, R.; Yan, W.-J.; Zhou, Y.-F. Tetrahedron Asymmetry 2004, 15, 3155; Lu, G.; Li, X.; Jia, X.; Chan, W.L.; Chan, A.S.C. Angew. Chem. Int. Ed. 2003, 42, 5057; Xu, Z.; Wang, R.; Xu, J.; Da, C.-s.; Yan, W.-j.; Chen, C. Angew. Chem. Int. Ed. 2003, 42, 5747.
671. Hatano, M.; Miyamoto, T.; Ishihara, K. Org. Lett. 2007, 9, 4535.
672. Wieland, L.C.; Deng, H.; Snapper, M.L.; Hoveyda, A.H. J. Am. Chem. Soc. 2005, 127, 1545. Alse see Friel, D.K.; Snapper, M.L.; Hoveyda, A.H. J. Am. Chem. Soc. 2008, 130, 9942.
673. Cozzi P.G.; Kotrusz, P. J. Am. Chem. Soc. 2006, 128, 4940.
674. Tanaka, T.; Yasuda, Y; Hayashi, M. J. Org. Chem. 2006, 71, 7091.
675. With Ti(OiPr)4, see Jeon, S.-J.; Li, H.; Walsh, P.J. J. Am. Chem. Soc. 2005, 127, 16416; Jeon, S.-J.; Li, H.; García, C.; LaRochelle, L.K.; Walsh, P.J. J. Org. Chem. 2005, 70, 448. See Huang, Z.; Lai, H.; Qin, Y. J. Org. Chem.2007, 72, 1373.
676. Milburn, R.R.; Hussain, S.M.S.; Prien, O.; Ahmed, Z.; Snieckus, V. Org. Lett. 2007, 9, 4403.
677. See Braga, A.L.; Paixão, M.W.; Westermann, B.; Schneider, P.H.; Wessjohann, L.A. J. Org. Chem. 2008, 73, 2879.
678. See Rasmussen, T.; Norrby, P.-O. J. Am. Chem. Soc. 2001, 123, 2464.
679. See Soai, K.; Yokoyam, S.; Hayasaka, T. J. Org. Chem. 1991, 56, 4264.
680. Pu, L.; Yu, H.-B. Chem. Rev. 2001, 101, 757. See also, Danilova, T.I.; Rozenberg, V.I.; Starikova, Z.A.; Bräse, S. Tetrahedron Asymmetry 2004, 15, 223; Scarpi, D.; Lo Galbo, F.; Occhiato, E.G.; Guarna, A. Tetrahedron Asymmetry 2004, 15, 1319; Sibi, M.P.; Stanley, L.M. Tetrahedron Asymmetry 2004, 15, 3353; Tseng, S.-L.; Yang, T.-K. Tetrahedron Asymmetry 2004, 15, 3375; Harada, T.; Kanda, K.; Hiraoka, Y.; Marutani, Y.; Nakatsugawa, M. Tetrahedron Asymmetry 2004, 15, 3879.
681. Sosa-Rivadeneyra, M.; Muñoz-Muñiz, O.; de Parrodi, C.A.; Quintero, L.; Juaristi, E. J. Org. Chem. 2003, 68, 2369; García, C.; La Rochelle, L.K.; Walsh, P.J. J. Am. Chem. Soc. 2002, 124, 10970.
682. Fraile, J.M.; Mayoral, J.A.; Servano, J.; Pericàs, M.A.; Solà, L.; Castellnou, D. Org. Lett. 2003, 5, 4333.
683. See Lipshutz, B.H.; Shin, Y.-J. Tetrahedron Lett. 2000, 41, 9515.
684. Marshall, J.A.; Adams, N.D. J. Org. Chem. 1999, 64, 5201.
685. See Furukawa, J.; Kawabata, N. Adv. Organomet. Chem. 1974, 12, 103. See Sjöholm, R.; Rairama, R.; Ahonen, M. J. Chem. Soc., Chem. Commun. 1994, 1217; Jones, P.R.; Desio, P.J. Chem. Rev. 1978, 78, 491.
686. Majumdar, K.K.; Cheng, C.-H. Org. Lett. 2000, 2, 2295.
687. See Felpin, F.-X.; Bertrand, M.-J.; Lebreton, J. Tetrahedron 2002, 58, 7381.
688. Ito, T.; Ishino, Y.; Mizuno, T.; Ishikawa, A.; Kobyashi, J.-i. Synlett 2002, 2116.
689. For a reaction of aliphatic halides, mediated by Cr(II), see Wessjohann, L.A.; Schmidt, G.; Schrekker, H.S. Tetrahedron 2008, 64, 2134.
690. Okude, Y.; Hirano, S.; Hiyama, T.; Nozaki, H. J. Am. Chem. Soc. 1977, 99, 3179. See Takai, K. Org. React. 2004, 64, 253.
691. Miller, J.J.; Sigman, M.S. J. Am. Chem. Soc. 2007, 129, 2752; Hargaden, G.C.; O'Sullivan, T.P,; Guiry, P.J. Org. Biomol. Chem. 2008, 6, 562; Huang, X.-R.; Chen, C. Tetrahedron Asymm. 2010, 21, 2999.
692. Xia, G.; Yamamoto, H. J. Am. Chem. Soc. 2006, 128, 2554; Shimada, Y.; Katsuki, T. Chem. Lett. 2005, 34, 786.
693. Takai, K.; Kimura, K.; Kuroda, T.; Hiyama, T.; Nozaki, H. Tetrahedron Lett. 1983, 24, 5281.
694. Ng, S.-S.; Ho, C.-Y.; Jamison, T.F. J. Am. Chem. Soc. 2006, 128, 11513.
695. Kuroboshi, M.; Tanaka, M.; Kishimoto, S.; Goto, K.; Mochizuki, M.; Tanaka, H. Tetrahedron Lett. 2000, 41, 81.
696. Hölemann, A.; Reissig, H.-U. Synlett 2004, 2732.
697. Li, H.; Walsh, P.J. J. Am. Chem. Soc. 2005, 127, 8355.
698. See Reetz, M.T.; Rölfing, K.; Griebenow, N. Tetrahedron Lett. 1994, 35, 1969.
699. See Matsuzawa, S.; Isaka, M.; Nakamura, E.; Kuwajima, I. Tetrahedron Lett. 1989, 30, 1975.
700. Araújo, M.A.; Barrientos-Astigarraga, R.E.; Ellensohn, R.M.; Comasseto, J.V. Tetrahedron Lett. 1999, 40, 5115.
701. Dabdoub, M.J.; Jacob, R.G.; Ferreira, J.T.B.; Dabdoub, V.B.; Marques, F.de.A. Tetrahedron Lett. 1999, 40, 7159.
702. Krief, A.; de Vos, M.J.; De Lombart, S.; Bosret, J.; Couty, F. Tetrahedron Lett. 1997, 38, 6295.
703. Miller, K.M.; Luanphaisarnnont, T.; Molinaro, C.; Jamison, T.F. J. Am. Chem. Soc. 2004, 126, 4130.
704. Suginome, M.; Iwanami, T.; Yamamoto, A.; Ito, Y. Synlett 2001, 1042. For a reaction using a Ni catalyst, see Ng, S.-S.; Jamison, T.F. J. Am. Chem. Soc. 2005, 127, 14194.
705. Sawaki, R.; Sato, Y.; Mori, M. Org. Lett. 2004, 6, 1131.
706. Chaulagain, M.R.; Sormunen, G.J.; Montgomery, J. J. Am. Chem. Soc. 2007, 129, 9568; Yang, Y.; Zhu, S.-F.; Zhou, C.-Y.; Zhou, Q.-L. J. Am. Chem. Soc. 2008, 130, 14052.
707. Ng, S.-S.; Jamison, T.F. J. Am. Chem. Soc. 2005, 127, 7320.
708. Denmark, S.E.; Wilson, T.W.; Burk, M.T.; Heemstra, Jr., J.R. J. Am. Chem. Soc. 2007, 129, 14864.
709. Durandetti, M.; Meignein, C.; Périchon, J. J. Org. Chem. 2003, 68, 3121.
710. Li, H.; Walsh, P.J. J. Am. Chem. Soc. 2004, 126, 6538.
711. Miller, K.M.; Jamison, T.F. J. Am. Chem. Soc. 2004, 126, 15342.
712. Shibata, K.; Kimura, M.; Shimizu, M.; Tamaru, Y. Org. Lett. 2001, 3, 2181.
713. Agapiou, K.; Cauble, D.F.; Krische, M.J. J. Am. Chem. Soc. 2004, 126, 4528.
714. Baik, T.-G.; Luis, A.L.; Wang, L.-C.; Krische, M.J. J. Am. Chem. Soc. 2001, 123, 5112.
715. Durandetti, M.; Nédélec, J.-Y.; Périchon, J. Org. Lett. 2001, 3, 2073.
716. Hao, J.; Taktak, S.; Aikawa, K.; Yusa, Y.; Hatano, M.; Mikami, K. Synlett 2001, 1443.
717. Aït-Mohand, S.; Takechi, N.; Médebielle, M.; Dolbier, Jr., W.R. Org. Lett. 2001, 3, 4271.
718. Orsini, F.; Caselli, A. Tetrahedron Lett. 2002, 43, 7255.
719. Montgomery, J.; Song, M. Org. Lett. 2002, 4, 4009.
720. Fischer, S.; Groth, U.; Jeske, M.; Schütz, T. Synlett 2002, 1922.
721. Kerr, M.S.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 8876.
722. Stetter, H.; Schreckenberg, M. Angew. Chem., Int. Ed 1973, 12, 81; Stetter, H.; Kuhlmann, H. Org. React. 1991, 40,407; Kerr, M.S.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 8876; Pesch, J.; Harms, K.; Bach, T. Eur. J. Org. Chem. 2004, 2025; Mennen, S.; Blank, J.; Tran-Dube, M.B.; Imbriglio, J.E.; Miller, S.J. Chem. Commun. 2005, 195. See Mattson, A.E.; Bharadwaj, A.R.; Scheidt, K.A. J. Am. Chem. Soc. 2004, 126, 2314.
723. Kang, S.-K.; Ha, Y.-H.; Ko, B.-S.; Lim, Y.; Jung, J. Angew. Chem. Int. Ed. 2002, 41, 343.
724. Read de Alaniz, J.; Rovis, T. J. Am. Chem. Soc. 2005, 127, 6284.
725. Tang, X.-Q.; Montgomery, J. J. Am. Chem. Soc. 1999, 121, 6098.
726. Locher, C.; Peerzada, N. J. Chem. Soc., Perkin Trans. 1 1999, 179.
727. Alcaide, B.; Pardo, C.; Rodríguez-Ranera, C.; Rodríguez-Vicente, A. Org. Lett. 2001, 3, 4205.
728. Hao, J.; Hatano, M.; Mikami, K. Org. Lett. 2000, 2, 4059.
729. Chang, H.-M.; Cheng, C.-H. Org. Lett. 2000, 2, 3439.
730. Evans, D.A.; Sweeney, Z.K.; Rovis, T.; Tedrow, J.S. J. Am. Chem. Soc. 2001, 123, 12095.
731. Kang, S.-K.; Lee, S.-W.; Jung, J.; Lim, Y. J. Org. Chem. 2002, 67, 4376.
732. Hölemann, A.; Reißig, H.-U. Chem. Eur. J. 2004, 10, 5493.
733. Okachi, T.; Fujimoto, K.; Onaka, M. Org. Lett. 2002, 4, 1667.
734. Loh, T.-P.; Song, H.-Y.; Zhou, Y. Org. Lett. 2002, 4, 2715.
735. See Satoh, Y.; Tayano, T.; Hara, S.; Suzuki, A. Tetrahedron Lett. 1989, 30, 5153.
736. See Hoffmann, R.W.; Niel, G.; Schlapbach, A. Pure Appl. Chem. 1990, 62, 1993; Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents Academic Press, NY, 1988, pp. 310–318; Bubnov, Yu.N. Pure Appl. Chem. 1987, 21, 895; Buynak, J.D.; Geng, B.; Uang, S.; Strickland, J.B. Tetrahedron Lett. 1994, 35, 985.
737. Wada, R.; Oisaki, K.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2004, 126, 8910. For an enantioselective reaction with an alkoxyboranes, see Lou, S.; Moquist, P.N.; Schaus, S.E. J. Am. Chem. Soc. 2006, 128, 12660.
738. See Schneider, U.; Ueno. M.; Kobayashi, S. J. Am. Chem. Soc. 2008, 130, 13824.
739. Hirano, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2005, 7, 4689.
740. Kabalka, G.W.; Wu, Z.; Ju, Y. Tetrahedron 2002, 58, 3243.
741. Huddleston, R.R.; Cauble, D.F.; Krische, M.J. J. Org. Chem. 2003, 68, 11.
742. Ueda, M.; Miyaura, N. J. Org. Chem. 2000, 65, 4450 and references cited therein; Duan, H.-F.; Xie, J.-H.; Shi, W.-J.; Zhang, Q.; Zhou, Q.-L. Org. Lett. 2006, 8, 1479. See Son, S.U.; Kim, S.B.; Reingold, J.A.; Carpenter, G.B.; Sweigart, D.A. J. Am. Chem. Soc. 2005, 127, 12238.
743. Zheng, H.; Zhang, Q.; Chen, J.; Liu, M.; Cheng, S.; Ding, J.; Wu, H.; Su, W. J. Org. Chem. 2009, 74, 943.
744. Zhou, L.; Du, X.; He, R.; Ci, Z.; Bao, M. Tetrahedron Lett. 2009, 50, 406.
745. Yamamoto, T.; Ohta, T.; Ito, Y. Org. Lett. 2005, 7, 4153.
746. Ji, J.-X.; Wu, J.; Au-Yeung, T.T.-L.; Yip, C.-W.; Haynes, R.K.; Chan, A.S.C. J. Org. Chem. 2005, 70, 1093; Wu, P.-Y.; Wu, H.-L.; Uang, B.-J. J. Org. Chem. 2006, 71, 833; Duan, H.-F.; Xie, J.-H.; Qiao, X.-C.; Wang, L.-X.; Zhou, Q.-L. Angew. Chem. Int. Ed. 2008, 47, 4351; Braga, A.L.; Lüdtke, D.S.; Vargas, F.; Paixão, M.W. Chem. Commun. 2005, 2512; Schmidt, F.; Rudolph, J.; Bolm, C. Synthesis 2006, 3625; Jagt, R.B.C.; Toullec, P.Y.; de Vries, J.G.; Feringa, B.L.; Minnaard, A.J. Org. Biomol. Chem. 2006, 4, 773; Jumde, V.R.; Facchetti, S.; Iuliano, A. Tetrahedron Asymm. 2010, 21, 2775.
747. Liu, G.; Lu, X. J. Am. Chem. Soc. 2006, 128, 6504.
748. See Rudolph, J.; Schmidt, F.; Bolm, C. Synthesis 2005, 840.
749. Cauble, D.F.; Gipson, J.D.; Krische, M.J. J. Am. Chem. Soc. 2003, 125, 1110.
750. See Gravel, M.; Lachance, H.; Lu, X.; Hall, D.G. Synthesis 2004, 1290; Bouffard, J.; Itami, K. Org. Lett. 2009, 11, 4410.
751. Chai, Z.; Liu, X.-Y.; Zhang, J.-K.; Zhao, G. Tetrahedron Asymmetry 2007, 18, 724; Schneider, U.; Kobayashi, S. Angew. Chem. Int. Ed. 2007, 46, 5909; Hall, D.G. Synlett 2007, 1644. See Barnett, D.S.; Moquist, P.N.; Schaus, S.E. Angew. Chem. Int. Ed. 2009, 48, 8679.
752. See Roush, W.R.; Ando, K.; Powers, D.B.; Palkowitz, A.D.; Halterman, R.L. J. Am. Chem. Soc. 1990, 112, 6339; Brown, H.C.; Randad, R.S. Tetrahedron Lett. 1990, 31, 455; Stürmer, R.; Hoffmann, R.W. Synlett 1990, 759.
753. Racherla, U.S.; Brown, H.C. J. Org. Chem. 1991, 56, 401, and references cited therein.
754. Garcia, J.; Kim, B.M.; Masamune, S. J. Org. Chem. 1987, 52, 4831.
755. Reetz, M.T.; Zierke, T. Chem. Ind. (London) 1988, 663.
756. Burgos, C.H.; Canales, E.; Matos, K.; Soderquist, J.A. J. Am. Chem. Soc. 2005, 127, 8044.
757. Roush, W.R.; Park, J.C. J. Org. Chem. 1990, 55, 1143.
758. Kabalka, G.W.; Venkataiah, B.; Dong, G. Tetrahedron Lett. 2004, 45, 729.
759. Kuriyama, M.; Shimazawa, R.; Enomoto, T.; Shirai, R. J. Org. Chem. 2008, 73, 6939.
760. Chuzel, O.; Roesch, A.; Genet, J.-P.; Darses, S. J. Org. Chem. 2008, 73, 7800.
761. Batey, R.A.; Thadani, A.N.; Smil, D.V. Tetrahedron Lett. 1999, 40, 4289.
762. Batey, R.A.; Thadani, A.N.; Smil, D.V.; Lough, A.J. Synthesis 2000, 990.
763. Fleming, I.; Dunoguès, J.; Smithers, R. Org. React. 1989, 37, 57, pp. 113–125, 290–328; Parnes, Z.N.; Bolestova, G.I. Synthesis 1984, 991, see pp. 997–1000. See Aggarwal, V.K.; Vennall, G.P. Tetrahedron Lett. 1996, 37, 3745.
764. Chandrasekhar, S.; Mohanty, P.K.; Raza, A. Synth. Commun. 1999, 29, 257.
765. Fang, X.; Watkin, J.G.; Warner, B.P. Tetrahedron Lett. 2000, 41, 447.
766. See Cossy, J.; Lutz, F.; Alauze, V.; Meyer, C. Synlett 2002, 45.
767. Wang, Z.; Kisanga, P.; Verkade, J.G. J. Org. Chem. 1999, 64, 6459.
768. Yadav, J.S.; Chand, P.K.; Anjaneyulu, S. Tetrahedron Lett. 2002, 43, 3783.
769. Fujii, T.; Koike, T.; Mori, A.; Osakada, K. Synlett 2002, 298.
770. Aoyama, N.; Hamada, T.; Manabe, K.; Kobayashi, S. Chem. Commun. 2003, 676.
771. Yanagisawa, A.; Kageyama, H.; Nakatsuka, Y.; Asakawa, K.; Matsumoto, Y.; Yamamoto, H. Angew. Chem. Int. Ed. 1999, 38, 3701.
772. Kobayashi, S.; Nishio, K. J. Org. Chem. 1994, 59, 6620.
773. Massa, A.; Malkov, A.V.; Kocovský, P.; Scettri, A. Tetrahedron Lett. 2003, 44, 7179.
774. Bottoni, A.; Costa, A.L.; Di Tommaso, D.; Rossi, I.; Tagliavini, E. J. Am. Chem. Soc. 1997, 119, 12131; Denmark, S.E.; Weber, E.J.; Wilson, T.; Willson, T.M. Tetrahedron 1989, 45, 1053; Keck, G.E.; Andrus, M.B.; Castellino, S. J. Am. Chem. Soc. 1989, 111, 8136.
775. Malkov, A.V.; Bell, M.; Orsini, M.; Pernazza, D.; Massa, A.; Herrmann, P.; Meghani, P.; Kocovský, P. J. Org. Chem. 2003, 68, 9659.
776. Malkov, A.V.; Ramírez-López, P.; Biedermannová (née Bendová), L.; Rulíšek, L.; Dufková, L.; Kotora, M.; Zhu, F.; Kocovský, P. J. Am. Chem. Soc. 2008, 130, 5341; Pignataro, L.; Benaglia, M.; Annunziata, R.; Cinquini, M.; Cozzi, F. J. Org. Chem. 2006, 71, 1458; Denmark, S.E.; Fu, J.; Coe, D.M.; Su, X.; Pratt, N.E.; Griedel, B.D. J. Org. Chem. 2006, 71, 1513; Denmark, S.E.; Fu, J.; Lawler, M.J. J. Org. Chem. 2006, 71, 1523; Traverse, J.F.; Zhao, Y.; Hoveyda, A.H.; Snapper, M.L. Org. Lett. 2005, 7, 3151; Malkov, A.V.; Bell, M.; Castelluzzo, F.; Kocovský, P. Org. Lett. 2005, 7, 3219; Chai, Q.; Song, C.; Sun, Z.; Ma, Y.; Ma, C.; Dai, Y.; Andrus, M.B. Tetrahedron Lett. 2006, 47, 8611; De Sio, V.; Massa, A.; Scettri, A. Org. Biomol. Chem. 2010, 8, 3055; Oh, Y.S.; Kotani, S.; Sugiura, M.; Nakajima, M. Tetrahedron Asymm. 2010, 21, 1833.
777. Angell, R.M.; Barrett, A.G.M.; Braddock, D.C.; Swallow, S.; Vickery, B.D. Chem. Commun. 1997, 919.
778. Malkov, A.V.; Dufková, L.; Farrugia, L.; Kocovsky, P. Angew. Chem Int. Ed. 2003, 42, 3802.
779. Iseki, K.; Mizuno, S.; Kuroki, Y.; Kobayashi, Y. Tetrahedron 1999, 55, 977
780. Tomita, D.; Wada, R.; Kanai, M.; Shibasaki, M J. Am. Chem. Soc. 2005, 127, 4138; Yamamoto, H.; Wadamoto, M. Chemistry: Asian J. 2007, 2, 692.
781. Kubota, K.; Leighton, J.L. Angew. Chem. Int. Ed. 2003, 42, 946; Hackman, B.M.; Lombardi, P.J.; Leighton, J.L. Org. Lett. 2004, 6, 4375.
782. Yadav, J.S.; Reddy, B.V.S.; Madhuri, Ch.; Sabitha, G. Chem. Lett. 2001, 18.
783. Zerth, H.M.; Leonard, N.M.; Mohan, R.S. Org. Lett. 2003, 5, 55.
784. Miyoshi, N.; Nishio, M.; Murakami, S.; Fukuma, T.; Wada, M. Bull. Chem. Soc. Jpn. 2000, 73, 689.
785. See Basavaiah, D.; Rao, A.J.; Satyanarayana, T. Chem. Rev. 2003, 103, 811.
786. See Luo, S.; Mi, X.; Xu, H.; Wang, P.G.; Cheng, J.-P. J. Org. Chem. 2004, 69, 8413.
787. See Karur, S.; Hardin, J.; Headley, A.; Li, G. Tetrahedron Lett. 2003, 44, 2991.
788. See Tarsis, E.; Gromova, A.; Lim, D.; Zhou, G.; Coltart, D.M. Org. Lett. 2008, 10, 4819.
789. See Faltin, C.; Fleming, E.M.; Connon, S.J. J. Org. Chem. 2004, 69, 6496.
790. Baylis, A.B.; Hillman, M.E.D. Ger. Offen. 2,155,133 Chem. Abstr., 1972, 77, 34174q [U.S. Patent 3,743,668]; Drewes, S.E.; Roos, G.H.P. Tetrahedron 1988, 44, 4653. For a review, see Basavaiah, D.; Rao, P.D.; Hyma, R.S. Tetrahedron 1996, 52, 8001.
791. Rafel, S.; Leahy, J.W. J. Org. Chem. 1997, 62, 1521. Also see, Drewes, S.E.; Rohwer, M.B. Synth. Commun. 1997, 27, 415.
792. Robiette, R.; Aggarwal, V.K.; Jeremy N; Harvey, J.N. J. Am. Chem. Soc. 2007, 129, 15513 (computational); Roy, D.; Sunoj, R.B. Org. Lett. 2007, 9, 4873 (computational). Price, K.E.; Broadwater, S.J.; Walker, B.J.; McQuade, D.T. J. Org. Chem. 2005, 70, 3980.
793. Shi, M.; Li, C.-Q.; Jiang, J.-K. Chem. Commun. 2001, 833.
794. Pereira, S.I.; Adrio, J.; Silva, A.M.S.; Carretero, J.C. J. Org. Chem. 2005, 70, 10175; Lin, Y.-S.; Liu, C.-W.; Tsai, T.Y.-R. Tetrahedron Lett. 2005, 46, 1859; Zhao, S.-H.; Chen, Z.-B. Synth. Commun. 2005, 35, 3045. For an ionic-liquid immobilizied base, see Mi, X.; Luo, S.; Cheng, J.-P. J. Org. Chem. 2005, 70, 2338. See Aggarwal, V.K.; Emme, I.; Fulford, S.Y. J. Org. Chem. 2003, 68, 692.
795. Kundu, M.K.; Mukherjee, S.B.; Balu, N.; Padmakumar, R.; Bhat, S.V. Synlett 1994, 444.
796. Coelho, F.; Almeida, W.P.; Veronese, D.; Mateus, C.R.; Lopes, E.C.S.; Rossi, R.C.; Silveira, G.P.C.; Pavam, C.H. Tetrahedron 2002, 58, 7437.
797. For improved procedures: Zhao, S.-H.; Bie, H.-Y.; Chen, Z.-B. Org. Prep. Proceed. Int. 2005, 37, 231.
798. Caumul, P.; Hailes, H.C. Tetrahedron Lett. 2005, 46, 8125.
799. Reetz, M.T.; Mondière, R.; Carballeira, J.D. Tetrahedron Lett. 2007, 48, 1679.
800. See Rafel, S.; Leahy, J.W. J. Org. Chem. 1997, 62, 1521; Luo, S.; Wang, P.G.; Cheng, J.-P. J. Org. Chem. 2004, 69, 555; Cai, J.; Park, K.S.; Kim, J.; Choo, H.; Chong, Y. Synlett 2007, 395. For a discussion of salt effects, see Kumar, A.; Pawar, S.S. Tetrahedron 2003, 59, 5019.
801. Rosa, J.N.; Afonso, C.A.M.; Santos, A.G. Tetrahedron 2001, 57, 4189. For an example in a chiral ionic liquid, see Pégot, B.; Vo-Thanh, G.; Gori, D.; Loupy, A. Tetrahedron Lett. 2004, 45, 6425.
802. Chandrasekhar, S.; Narsihmulu, Ch.; Saritha, B.; Sultana, S.S. Tetrahedron Lett. 2004, 45, 5865.
803. Krishna, P.R.; Manjuvani, A.; Kannan, V.; Sharma, G.V.M. Tetrahedron Lett. 2004, 45, 1183.
804. Asano, K.; Matsubara, S. Synthesis 2009, 3219.
805. Maher, D.J.; Connon, S.J. Tetrahedron Lett. 2004, 45, 1301.
806. See Nemoto, T.; Fukuyama, T.; Yamamoto, E.; Tamura, S.; Fukuda, T.; Matsumoto, T.; Akimoto, Y.; Hamada, Y. Org. Lett. 2007, 9, 927.
807. Walsh, L.M.; Winn, C.L.; Goodman, J.M. Tetrahedron Lett. 2002, 43, 8219.
808. Chuprakov, S.; Malyshev, D.A.; Trofimov, A.; Gevorgyan, V. J. Am. Chem. Soc. 2007, 129, 14868.
809. Tanaka, K.; Fu, G.C. J. Am. Chem. Soc. 2001, 123, 11492.
810. Keck, G.E.; Welch, D.S. Org. Lett. 2000, 4, 3687.
811. Krishna, P.R.; Kannan, V.; Sharma, G.V.M. J. Org. Chem. 2004, 69, 6467.
812. See Masson, G.; Housseman, C.; Zhu, J. Angew. Chem. Int. Ed. 2007, 46, 4614; Also see, Markó, I.E.; Giles, P.R.; Hindley, N.J. Tetrahedron 1997, 53, 1015.
813. Brzezinski, L.J.; Rafel, S.; Leahy, J.W. J. Am. Chem. Soc. 1997, 119, 4317.
814. See Wei, H.-X.; Chen, D.; Xu, X.; Li, G.; Paré, P.W. Tetrahedron Asymmetry 2003, 14, 971.
815. Imbriglio, J.E.; Vasbinder, M.M.; Miller, S.J. Org. Lett. 2003, 5, 3741. See also, Wang, J.; Li, H.; Yu, X.; Zu, L.; Wang, W. Org. Lett. 2005, 7, 4293; Berkessel, A.; Roland, K.; Neudörfl, J.M. Org. Lett. 2006, 8, 4195; Lattanzi, A. Synlett 2007, 2106.
816. Filho, E.P.S.; Rodrigues, J.A.R.; Moran, P.J.S. Tetrahedron Asymmetry 2001, 12, 847.
817. Rauhut, M. M.; Currier, H. U.S. Patent 307499919630122, American Cyanamid Co., 1963.
818. Aroyan, C.E.; Miller, S.J. J. Am. Chem. Soc. 2007, 129, 256.
819. Chandrasekhar, S.; Narsihmulu, Ch.; Reddy, N.R.; Reddy, M.S. Tetrahedron Lett. 2003, 44, 2583.
820. Li, G.; Wei, H.-X.; Gao, J.J.; Caputo, T.D. Tetrahedron Lett. 2000, 41, 1; Shi, M.; Jiang, J.-K.; Feng, Y.-S. Org. Lett. 2000, 2, 2397.
821. Pei, W.; Wei, H.X.; Li, G. Chem. Commun. 2002, 2412.
822. Arnold, L.A.; Imbos, R.; Mandoli, A.; de Vries, A.H.M.; Naasz, R.; Feringa, B.L. Tetrahedron 2000, 56, 2865.
823. Yang, K.-S.; Lee, W.-D.; Pan, J.-F.; Chen, K. J. Org. Chem. 2003, 68, 915.
824. Balan, D.; Adolfsson, H. Tetrahedron Lett. 2003, 44, 2521.
825. Krafft, M.E.; Haxell, T.F.M.; Seibert, K.A.; Abboud, K.A. J. Am. Chem. Soc. 2006, 128, 4174; Krafft, M.E.; Haxell, T.F.N. J. Am. Chem. Soc. 2005, 127, 10168; Krafft, M.E.; Seibert, K.A.; Haxell, T.F.N.; Hirosawa, C. Chem. Commun. 2005, 5772.
826. Basavaiah, D.; Sharada, D.S.; Kumaragurubaran, N.; Reddy, R.M. J. Org. Chem. 2002, 67, 7135.
827. Ballini, R.; Barboni, L.; Bosica, G.; Fiorini, D.; Mignini, E.; Palmieri, A. Tetrahedron 2004, 60, 4995.
828. Kaye, P.T.; Nocanda, X.W. J. Chem. Soc., Perkin Trans. 1 2000, 1331.
829. Balan, D.; Adolfsson, H. J. Org. Chem. 2002, 67, 2329.
830. See Fürstner, A. Synthesis 1989, 571; Rathke, M.W. Org. React. 1975, 22, 423; Gaudemar, M. Organomet. Chem. Rev. Sect. A 1972, 8, 183; Ocampo, R.; Dolbier Jr., W.R. Tetrahedron 2004, 60, 9325.
831. Palomo, C.; Aizpurua, J.M.; López, M.C.; Aurrekoetxea, N. Tetrahedron Lett. 1990, 31, 2205; Zheng, J.; Yu, Y.; Shen, Y. Synth. Commun. 1990, 20, 3277.
832. See Huang, Y.; Chen, C.; Shen, Y. J. Chem. Soc. Perkin Trans. 1 1988, 2855.
833. Rieke, R.D.; Uhm, S.J. Synthesis 1975, 452; Bouhlel, E.; Rathke, M.W. Synth. Commun. 1991, 21, 133.
834. Han, B.; Boudjouk, P. J. Org. Chem. 1982, 47, 5030.
835. Ross, N.A.; Bartsch, R.A. J. Org. Chem. 2003, 68, 360.
836. Araki, S.; Yamada, M.; Butsugan, Y. Bull. Chem. Soc. Jpn. 1994, 67, 1126.
837. Suh, Y.S.; Rieke, R.D. Tetrahedron Lett. 2004, 45, 1807.
838. Aoyagi, Y.; Tanaka, W.; Ohta, A. J. Chem. Soc., Chem. Commun. 1994, 1225.
839. See Parrish, J.D.; SheHon, D.R.; Little, R.D. Org. Lett. 2003, 5, 3615.
840. Shibata, I.; Kawasaki, M.; Yasuda, M.; Baba, A. Chem. Lett. 1999, 689.
841. Utimoto, K.; Matsui, T.; Takai, T.; Matsubara, S. Chem. Lett. 1995, 197; Arime, T.; Takahashi, H.; Kobayashi, S.; Yamaguchi, S.; Mori, N. Synth. Commun. 1995, 25, 389; Park, H.S.; Lee, I.S.; Kim, Y.H. Tetrahedron Lett.1995, 36, 1673; Molander, G.A.; Etter, J.B. J. Am. Chem. Soc. 1987, 109, 6556.
842. Kagoshima, H.; Hashimoto, Y.; Saigo, K. Tetrahedron Lett. 1998, 39, 8465.
843. Kagoshima, H.; Hashimoto, Y.; Oguro, D.; Saigo, K. J. Org.Chem. 1998, 63, 691.
844. Cozzi, P.G. Angew. Chem. Int. Ed. 2006, 45, 2951.
845. Cozzi, P.G. Angew. Chem. Int. Ed. 2007, 46, 2568.
846. Chattopadhyay, A.; Salaskar, A. Synthesis 2000, 561.
847. Bieber, L.W.; Malvestiti, I.; Storch, E.C. J. Org. Chem. 1997, 62, 9061.
848. See Orsini, F.; Sello, G.; Manzo, A.M.; Lucci, E.M. Tetrahedron Asymmetry 2005, 16, 1913.
849. Kloetzing, R.J.; Thaler, T.; Knochel, P. Org. Lett. 2006, 8, 1125; Shin, E.-k.; Kim, H.J.; Kim, Y.; Kim, Y.; Park, Y.S. Tetrahedron Lett. 2006, 47, 1933; Emmerson, D.P.G.; Hems, W.P.; Davis, B.G. Tetrahedron Asymmetry2005, 16, 213; Fernández-Ibáñez, M.A.; Maciá, B.; Minnaard, A.J.; Feringa, B.L. Angew. Chem. Int. Ed. 2008, 47, 1317; Chem. Commun. 2008, 2571; Cozzi, P.G.; Mignogna, A.; Zoli, L. Pure Appl. Chem. 2008, 80, 891.
850. See Ribeiro, C.M.R.; de S. Santos, E.; de O. Jardim, A.H.; Maia, M.P.; da Silva, F.C.; Moreira, A.P.D.; Ferreira, V.F. Tetrahedron Asymmetry 2002, 13, 1703.
851. Kanai, K.; Wakabyashi, H.; Honda, T. Org. Lett. 2000, 2, 2549.
852. See Maiz, J.; Arrieta, A.; Lopez, X.; Ugalde, J.M.; Cossio, F.P.; Fakultatea, K.; Unibertsitatea, E.H.; Lecea, B. Tetrahedron Lett. 1993, 34, 6111.
853. Dekker, J.; Budzelaar, P.H.M.; Boersma, J.; van der Kerk, G.J.M.; Spek, A.L. Organometallics 1984, 3, 1403.
854. Shen, Y.; Xin, Y.; Zhao, J. Tetrahedron Lett. 1988, 29, 6119. For another method, see Huang, Y.; Shi, L.; Li, S.; Wen, X. J. Chem. Soc. Perkin Trans. 1 1989, 2397.
855. Huang, Z.-Z.; Ye, S.; Xia, W.; Yu, Y.-H.; Tang, Y. J. Org. Chem. 2002, 67, 3096.
856. See Hannick, S.M.; Kishi, Y. J. Org. Chem. 1983, 48, 3833.
857. Andrés, J.M.; Pedrosa, R.; Pérez, A.; Pérez-Encabo, A. Tetrahedron 2001, 57, 8521.
858. See Jorgenson, M.J. Org. React. 1970, 18, 1; Rubottom, G.M.; Kim, C. J. Org. Chem. 1983, 48, 1550.
859. Ohki, M.; Asaoka, M. Chem. Lett. 2009, 38, 856.
860. Alonso, F.; Lorenzo, E.; Yus, M. J. Org. Chem., 1996, 61, 6058.
861. Aurell, M.J.; Danhui, Y.; Einhorn, J.; Einhorn, C.; Luche, J.L. Synlett 1995, 459. Also see, Aurell, M.J.; Einhorn, C.; Einhorn, J.; Luche, J.L. J. Org. Chem. 1995, 60, 8.
862. Gooßen, L.J.; Ghosh, K. Chem. Commun. 2001, 2084.
863. See Volpin, M.E.; Kolomnikov, I.S. Organomet. React. 1975, 5, 313; Sneeden, R.P.A. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 137–173; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 913–948. For a more general review, see Lapidus, A.L.; Ping, Y.Y. Russ. Chem. Rev. 1981, 50, 63.
864. For a kinetic study, see Nudelman, N.S.; Doctorovich, F. J. Chem. Soc. Perkin Trans. 2 1994, 1233.
865. Yanagisawa, A.; Yasue, K.; Yamamoto, H. Synlett 1992, 593.
866. Ukai, K.; Aoki, M.; Takaya, J.; Iwasawa, N. J. Am. Chem. Soc. 2006, 128, 8706.
867. Takaya, J.; Tadami, S.; Ukai, K.; Iwasawa, N. Org. Lett. 2008, 10, 2697.
868. Zhao, X.; Alper, H.; Yu, Z. J. Org. Chem. 2006, 71, 3988.
869. Ochiai, H.; Jang, M.; Hirano, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2008, 10, 2681.
870. Kobayashi, K.; Kondo, Y. Org. Lett. 2009, 11, 2035.
871. Nair, V.; Varghese, V.; Paul, R.P.; Jose, A.; Sinu, C.R.; Menon, R.S. Org. Lett. 2010, 12, 2653.
872. Correa, A.; Martín, R. J. Am. Chem. Soc. 2009, 131, 15974.
873. North, M. Angew. Chem. Int. Ed. 2009, 48, 4104.
874. Park, Y.S.; Beak, P. J. Org. Chem. 1997, 62, 1574.
875. See Ramadas, S.R.; Srinivasan, P.S.; Ramachandran, J.; Sastry, V.V.S.K. Synthesis 1983, 605.
876. Katritzky, A.R.; Moutou, J.-L.; Yang, Z. Synlett 1995, 99.
877. Bertz, S.H.; Dabbagh, G.; Williams, L.M. J. Org. Chem. 1985, 50, 4414.
878. Nicolaou, K.C.; McGarry, D.G.; Somers, P.K.; Veale, C.A.; Furst, G.T. J. Am. Chem. Soc. 1987, 109, 2504.
879. Köster, F.; Dinjus, E.; Duñach, E. Eur. J. Org. Chem. 2001, 2507.
880. See Harada, K. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 266–272; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 1204–1227; Wang, D.-K.; Dai, L.-X.; Hou, X.-L.; Zhang, Y. Tetrahedron Lett. 1996, 37, 4187; Bambridge, K.; Begley, M.J.; Simpkins, N.S. Tetrahedron Lett. 1994, 35, 3391.
881. Huet, J. Bull. Soc. Chim. Fr. 1964, 952, 960, 967, 973.
882. Qu, B.; Collum, D.B. J. Am. Chem. Soc. 2005, 127, 10820; J. Am. Chem. Soc. 2006, 128, 9355.
883. Davis, F.A.; Giangiordano, M.A.; Starner, W.E. Tetrahedron Lett. 1986, 27, 3957.
884. Yus, M.; Soler, T.; Foubelo, F. J. Org. Chem. 2001, 66, 6207.
885. Saito, S.; Hatanaka, K.; Yamamoto, H. Synlett 2001, 1859.
886. Gandon, V.; Bertus, P.; Szymoniak, J. Eur. J. Org. Chem. 2001, 3677.
887. Zani, L.; Bolm, C. Chem. Commun. 2006, 4263.
888. For a review see Enders, D.; Reinhold, U. Tetrahedron Asymmetry, 1997, 8, 1895.
889. Denmark, S.E.; Stiff, C.M. J. Org. Chem. 2000, 65, 5875; Chrzanowska, M.; Sokołowska, J. Tetrahedron Asymmetry 2001, 12, 1435.
890. See Friestad, G.K.; Mathies, A.K. Tetrahedron 2007, 63, 2541; Ferraris, D. Tetrahedron 2007, 63, 9581.
891. Dieter, R.K.; Datar, R. Can. J. Chem. 1993, 71, 814.
892. Yamada, K.-i.; Tomioka, K. Chem. Rev. 2008, 108, 2874.
893. Hatano, M.; Asai, T.; Ishihara, K. Tetrahedron Lett. 2008, 49, 379; Liu, J.; Liu, B.; Jia, X.; Li, X.; Chan, A.S.C. Tetrahedron Asymmetry 2007, 18, 396; Liu, B.; Huang, L.; Liu, J.; Zhong, Y.; Li, X.; Chan, A.S.C. Tetrahedron Asymmetry 2007, 18, 2901; Zani, L.; Eichhorn, T.; Bolm, C. Chem.: Eur. J. 2007, 13, 2587; Ding, H.; Friestad, G.K. Synthesis 2005, 2815; Zhou, C.-Y.; Zhu, S.-F.; Wang, L.-X.; Zhou, Q.-L. J. Am. Chem. Soc. 2010, 132, 10955.
894. Ballweg, D.M.; Miller, R.C.; Gray, D.L.; Scheidt, K.A. Org. Lett. 2005, 7, 1403.
895. Lee, C.-L.K.; Ling, H.-Y.; Loh, T.-P. J. Org. Chem. 2004, 69, 7787. See van der Sluis, M.; Dalmolen, J.; de Lange, B.; Kaptein, B.; Kellogg, R.M.; Broxterman, Q.B. Org. Lett. 2001, 3, 3943.
896. Legros, J.; Meyer, F.; Coliboeuf, M.; Crousse, B.; Bonnet-Delpon, D.; Bégué, J.-P. J. Org. Chem. 2003, 68, 6444.
897. Xiao, X.; Wang, H.; Huang, Z.; Yang, J.; Bian, X.; Qin, Y. Org. Lett. 2006, 8, 139; Almansa, R.; Guijarro, D.; Yus, M. Tetrahedron 2007, 63, 1167.
898. See Dickstein, J.S.; Fennie, M.W.; Norman, A.L.; Paulose, B.J.; Kozlowski, M.C. J. Am. Chem. Soc. 2008, 130, 15794; Gao, F.; Deng, M.; Qian, C. Tetrahedron 2005, 61, 12238.
899. See Fu, P.; Snapper, M.L.; Hoveyda, A.H. J. Am. Chem. Soc. 2008, 130, 5530; Nishimura, T.; Yasuhara, Y.; Hayashi, T. Org. Lett. 2006, 8, 979; Basra, S.; Fennie, M.W.; Kozlowski, M.C. Org. Lett. 2006, 8, 2659; Charette, A.B.; Boezio, A.A.; Côté, A.; Moreau, E.; Pytkowicz, J.; Desrosiers, J.-N.; Legault, C. Pure Appl. Chem. 2005, 77,1259.
900. Fujihara, H.; Nagai, K.; Yomioka, K. J. Am. Chem. Soc. 2000, 122, 12055. See Wang, C.-J.; Shi, M. J. Org. Chem. 2003, 68, 6229.
901. Chiev, K.P.; Roland, S.; Mangeney, P. Tetrahedron Asymmetry 2001, 13, 2205.
902. Jiang, B.; Si, Y.-G. Tetrahedron Lett. 2003, 44, 6767.
903. Zani, L.; Alesi, S.; Cozzi, P.G.; Bolm, C. J. Org. Chem. 2006, 71, 1558.
904. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 847–863.
905. Nakamura, H.; Nakamura, K.; Yamamoto, Y. J. Am. Chem. Soc. 1998, 120, 4242; Kobayashi, S.; Iwamoto, S.; Nagayama, S. Synlett 1997, 1099.
906. See Kim, B.; Han, R.; Park, R.; Bai, K.; Jun, Y.; Baik, W. Synth. Commun. 2001, 31, 2297.
907. Akiyama, T.; Iwai, J.; Onuma, Y.; Kagoshima, H. Chem. Commun. 1999, 2191.
908. With a Cu catalyst, see Sato, A.; Ito, H.; Okada, M.; Nakamura, Y.; Taguchi, T. Tetrahedron Lett. 2005, 46, 8381.
909. Andrews, P.C.; Peatt, A.C.; Raston, C.L. Tetrahedron Lett. 2004, 45, 243.
910. Su, W.; Li, J.; Zhang, Y. Synth. Commun. 2001, 31, 273.
911. Kargbo, R.; Takahashi, Y.; Bhor, S.; Cook, G.R.; Lloyd-Jones, G.C.; Shepperson, I.R. J. Am. Chem. Soc. 2007, 129, 3846. With a Cu catalyst, see Black, D.A.; Arndtsen, B.A. Org. Lett. 2006, 8, 1991.
912. Al: Niwa, Y.; Shimizu, M. J. Am. Chem. Soc. 2003, 125, 3720. La: Aspinall, H.C.; Bissett, J.S.; Greeves, N.; Levin, D. Tetrahedron Lett. 2002, 43, 323. Nb: Andrade, C.K.Z.; Oliveira, G.R. Tetrahedron Lett. 2002, 43, 1935; Akiyama, T.; Onuma, Y. J. Chem. Soc., Perkin Trans. 1 2002, 1157. Pd: Fernandes, R.A.; Yamamoto, Y. J. Org. Chem. 2004, 69, 3562. Ta: Shibata, I.; Nose, K.; Sakamoto, K.; Yasuda, M.; Baba, A. J. Org. Chem. 2004, 69, 2185. Zr: Gastner, T.; Ishitani, H.; Akiyama, R.; Kobayashi, S. Angew. Chem. Int. Ed. 2001, 40, 1896.
913. See Kobayashi, Sh.; Ishitani, H. Chem. Rev. 1999, 99, 1069.
914. Chowdari, N.S.; Ramachary, D.B.; Barbas, III, C.F. Synlett 2003, 1906.
915. Ding, R.; Zhao, C.H.; Chen, Y.J.; Lu, L.; Wang, D.; Li, C.J. Tetrahedron Lett. 2004, 45, 2995.
916. Choucair, B.; Léon, H.; Miré, M.-A.; Lebreton, C.; Mosset, P. Org. Lett. 2000, 2, 1851. See Hirashita, T.; Hayashi, Y.; Mitsui, K.; Araki, S. J. Org. Chem. 2003, 68, 1309.
917. Under electrolysis conditions, see Hilt, G.; Smolko, K.I.; Waloch, C. Tetrahedron Lett. 2002, 43, 1437.
918. Lu, W.; Chan, T.H. J. Org. Chem. 2000, 65, 8589.
919. Lu, W.; Chan, T.H. J. Org. Chem. 2001, 66, 3467.
920. Ishiyama, T.; Hartwig, J. J. Am. Chem. Soc. 2000, 122, 12043.
921. Shintani, R.; Takeda, M.; Tsuji, T.; Hayashi, T. J. Am. Chem. Soc. 2010, 132, 13168.
922. Prajapati, D.; Laskar, D.D.; Gogoi, B.J.; Devi, G. Tetrahedron Lett. 2003, 44, 6755.
923. Li, C.-J.; Wei, C. Chem. Commun. 2002, 268.
924. Fischer, C.; Carreira, E.M. Synthesis 2004, 1497.
925. Koradin, C.; Gommermann, N.; Polborn, K.; Knochel, P. Chem. Eur. J. 2003, 9, 2797.
926. Wei, C.; Li, C.-J. J. Am. Chem. Soc. 2002, 124, 5638.
927. See Colombo, F.; Benaglia, M.; Orlandi, S.; Usuelli, F.; Celentano, G. J. Org. Chem. 2006, 71, 2064.
928. Patterson, A.W.; Ellman, J.A. J. Org. Chem. 2006, 71, 7110.
929. Kong, J.-R.; Cho, C.-W.; Krische, M.J. J. Am. Chem. Soc. 2005, 127, 11269.
930. Komanduri, V.; Grant, C.D.; Krische, M.J. J. Am. Chem. Soc. 2008, 130, 12592.
931. Oi, S.; Moro, M.; Fukuhara, H.; Kawanishi, T.; Inoue, Y. Tetrahedron Lett. 1999, 40, 9259.
932. Hayashi, T.; Ishigedani, M. J. Am. Chem. Soc. 2000, 122, 976.
933. Soeta, T.; Nagai, K.; Fujihara, H.; Kuriyama, M.; Tomioka, K. J. Org. Chem. 2003, 68, 9723.
934. See Ellman, J.A.; Owens, T.D.; Tang, T.P. Acc. Chem. Res. 2002, 35, 984.
935. Tang, T.P.; Volkman, S.K.; Ellman, J.A. J. Org. Chem. 2001, 66, 8772.
936. Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2004, 126, 11804. See also, Spanedda, M.V.; Ourévitch, M.; Crouse, B.; Bégué, J.-P.; Bonnet-Delpon, D. Tetrahedron Lett. 2004, 45, 5023.
937. Yamanaka, M.; Nishida, A.; Nakagawa, M. J. Org. Chem. 2003, 68, 3112.
938. Trost, B.M.; Jonasson, C. Angew. Chem. Int. Ed. 2003, 42, 2063.
939. Phukan, P. J. Org. Chem. 2004, 69, 4005.
940. Kumagai, N.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2004, 126, 13632.
941. Ueda, M.; Saito, A.; Miyaura, N. Synlett 2000, 1637.
942. Wada, R.; Shibuguchi, T.; Makino, S.; Oisaki, M.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 7687.
943. Duan, H.-F.; Jia, Y.-X.; Wang, L.-X.; Zhou, Q.-L. Org. Lett. 2006, 8, 2567; Trincado, M.; Ellman, J.A. Angew. Chem. Int. Ed. 2008, 47, 5623; Marelli, C.; Monti, C.; Gennari, C.; Piarulli, U. Synlett 2007, 2213.
944. Zhang, Q.; Chen, J.; Liu, M.; Wu, H.; Cheng, J.; Qin, C.; Su, W.; Ding, J. Synlett 2008, 935.
945. Weix, D.J.; Shi, Y.; Ellman, J.A. J. Am. Chem. Soc. 2005, 127, 1092; Beenen, M.A.; Weix, D.J.; Ellman, J.A. J. Am. Chem. Soc. 2006, 128, 6304; Wang, Z.-Q.; Feng, C.-G.; Xu, M.-H.; Lin, G.-Q. J. Am. Chem. Soc. 2007, 129,5336.
946. Ngai, M.-Y.; Barchuk, A.; Krische, M.J. J. Am. Chem. Soc. 2007, 129, 12644.
947. Sugiura, M.; Hirano, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 7182.
948. Pandya, A.; Pinet, S.U.; Chavant, P.Y.; Vallée, Y. Eur. J. Org. Chem. 2003, 3621.
949. Solin, N.; Wallner, O.A.; Szabó, K.J. Org. Lett. 2005, 7, 689.
950. Nakamura, K.; Nakamura, H.; Yamamoto, Y. J. Org. Chem. 1999, 64, 2614.
951. See Fernandes, R.A.; Yamamoto, Y. J. Org. Chem. 2004, 69, 735.
952. Hirabayashi, R.; Ogawa, C.; Sugiura, M.; Kobayashi, S. J. Am. Chem. Soc. 2001, 123, 9493.
953. Kobayashi, S.; Ogawa, C.; Konishi, H.; Sugiura, M. J. Am. Chem. Soc. 2003, 125, 6610.
954. Berger, R.; Duff, K.; Leighton, J.L. J. Am. Chem. Soc. 2004, 126, 5686.
955. Declerck, V.; Martinez, J.; Lamaty, F. Chem. Rev. 2009, 109, 1. See Matsui, K.; Takizawa, S.; Sasai, H. J. Am. Chem. Soc. 2005, 127, 3680; Shi, M.; Chen, L.-H.; Li, C.-Q. J. Am. Chem. Soc. 2005, 127, 3790; Gajda, A.; Gajda, T. J. Org. Chem. 2008, 73, 8643.
956. Xu, Y.-M.; Shi, M. J. Org. Chem. 2004, 69, 417.
957. Shi, M.; Xu, Y.-M. J. Org. Chem. 2003, 68, 4784.
958. Qi, M.-J.; Ai, T.; Shi, M.; Li, G. Tetrahedron 2008, 64, 1181; Utsumi, N.; Zhang, H.; Tanaka, F.; Barbas III, C.F. Angew. Chem. Int. Ed. 2007, 46, 1878.
959. See Masson, G.; Housseman, C.; Zhu, J. Angew. Chem. Int. Ed. 2007, 46, 4614.
960. Gausepohl, R.; Buskens, P.; Kleinen, J.; Bruckmann, A.; Lehmann, C.W.; Klankermayer, J.; Leitner, W. Angew. Chem. Int. Ed. 2006, 45, 3689.
961. Notz, W.; Tanaka, F.; Watanabe, S.; Chowdari, N.S.; Turner, J.M.; Thayumanavan, R.; Barbas, III, C.F. J. Org. Chem. 2003, 68, 9624; Chowdari, N.S.; Suri, J.T.; Barbas, III, C.F. Org. Lett. 2004, 6, 2507.
962. Hamada, T.; Manabe, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 7768. For a similar reaction using a Bi catalyst, see Ollevier, T.; Nadeau, E. J. Org. Chem. 2004, 69, 9292.
963. Shimizu, M.; Itohara, S.; Hase, E. Chem. Commun. 2001, 2318.
964. Ishimaru, K.; Kojima, T. J. Org. Chem. 2003, 68, 4959.
965. Nugent, B.M.; Yoder, R.A.; Johnston, J.N. J. Am. Chem. Soc. 2004, 126, 3418.
966. Nishiwaki, N.; Knudson, K.R.; Gothelf, K.V.; J![]() rgensen, K.A. Angew. Chem. Int. Ed. 2001, 40, 2992.
rgensen, K.A. Angew. Chem. Int. Ed. 2001, 40, 2992.
967. Fujisawa, T.; Kurita, Y.; Sato, T. Chem. Lett. 1983, 1537.
968. Paukstelis, J.V.; Cook, A.G. in Cook, A.G. Enamines, 2nd ed., Marcel Dekker, NY, 1988, pp. 275–356.
969. Wieland, G.; Simchen, G. Liebigs Ann. Chem. 1985, 2178.
970. Denmark, S.E.; Edwards, J.P.; Nicaise, O. J. Org. Chem. 1993, 58, 569.
971. Miyabe, H.; Ueda, M.; Nishimura, A.; Naito, T. Tetrahedron 2004, 60, 4227.
972. Kobayashi, S.; Hamada, T.; Manabe, K. Synlett 2001, 1140.
973. Friedstad, G.K.; Qin, J. J. Am. Chem. Soc. 2001, 123, 9922.
974. Yamshita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 11279.
975. Richey Jr., H.G.; McLane, R.C.; Phillips, C.J. Tetrahedron Lett. 1976, 233.
976. Bernardi, L.; Cerè, V.; Femoni, C.; Pollicino, S.; Ricci, A. J. Org. Chem. 2003, 68, 3348.
977. Laskar, D.D.; Prajapati, D.; Sandu, J.S. Tetrahedron Lett. 2001, 42, 7883.
978. Frantz, D.E.; Fässler, R.; Carreira, E.M. J. Am. Chem. Soc. 1999, 121, 11245. See Pinet, S.; Pandya, S.U.; Chavant, P.Y.; Ayling, A.; Vallee, Y. Org. Lett. 2002, 4, 1463.
979. See Merino, P.; Tejero, T. Tetrahedron 2001, 57, 8125.
980. Kumar, H.M.S.; Anjaneyulu, S.; Reddy, E.J.; Yadav, J.S. Tetrahedron Lett. 2000, 41, 9311.
981. Murahashi, S.-I.; Imada, Y.; Kawakami, T.; Harada, K.; Yonemushi, Y.; Tomita, N. J. Am. Chem. Soc. 2002, 124, 2888.
982. Shimizu, M.; Kimura, M.; Watanabe, T.; Tamaru, Y. Org. Lett. 2005, 7, 637.
983. See Nagayama, S.; Kobayashi, S. Chem Lett. 1998, 685. Also see, Rasmussen, K.G.; J![]() rgensen, K.A. J. Chem. Soc., Chem. Commun. 1995, 1401.
rgensen, K.A. J. Chem. Soc., Chem. Commun. 1995, 1401.
984. See Janardanan, D.; Sunoj, R.B. J. Org. Chem. 2008, 73, 8163.
985. Reetz, M.T.; Lee, W.K. Org. Lett. 2001, 3, 3119.
986. Krumper, J.R.; Gerisch, M.; Suh, J.M.; Bergman, R.G.; Tilley, T.D. J. Org. Chem. 2003, 68, 9705; Williams, A.L.; Johnston, J.N. J. Am. Chem. Soc. 2004, 126, 1612.
987. Juhl, K.; Hazell, R.G.; J![]() rgensen, K.A. J. Chem. Soc., Perkin Trans. 1 1999, 2293.
rgensen, K.A. J. Chem. Soc., Perkin Trans. 1 1999, 2293.
988. Aggarwal, V.K.; Alonso, E.; Fang, G.; Ferrara, M.; Hynd, G.; Porcelloni, M. Angew. Chem. Int. Ed. 2001, 40, 1433.
989. Lu, Z.; Zhang, Y.; Wulff, W.D. J. Am. Chem. Soc. 2007, 129, 7185. Also see Branco, P.S.; Raje, V.P.; Dourado, J.; Gordo, J. Org. Biomol. Chem. 2010, 8, 2968.
990. Handy, S.T.; Czopp, M. Org. Lett. 2001, 3, 1423.
991. Uraguchi, D.; Sorimachi, K.; Terada, M. J. Am. Chem. Soc. 2005, 127, 9360.
992. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 767–845.
993. Weiberth, F.J.; Hall, S.S. J. Org. Chem. 1987, 52, 3901.
994. Canonne, P.; Foscolos, G.B.; Lemay, G. Tetrahedron Lett. 1980, 155.
995. See Gauthier, R.; Axiotis, G.P.; Chastrette, M. J. Organomet. Chem. 1977, 140, 245.
996. Pickard, P.L.; Toblert, T.L. J. Org. Chem. 1961, 26, 4886.
997. Cason, J.; Kraus, K.W.; McLeod, Jr., W.D. J. Org. Chem. 1959, 24, 392.
998. Savarin, C.G.; Boice, G.N.; Murry, J.A.; Corley, E.; DiMichele, L.; Hughes, D. Org. Lett. 2006, 8, 3903.
999. Yu, M.; Zhang, Y.; Guo, H. Synth. Commun. 1997, 27, 1495.
1000. Ciganek, E. J. Org. Chem. 1992, 57, 4521.
1001. Lee, A.S.-Y.; Lin, L.-S. Tetrahedron Lett. 2000, 41, 8803.
1002. Fry, D.F.; Fowler, C.B.; Dieter, R.K. Synlett 1994, 836.
1003. Gallulo, V.; Dimas, L.; Zezza, C.A.; Smith, M.B. Org. Prep. Proceed. Int. 1989, 21, 297.
1004. Blaise, E.E. Compt. Rend. 1901, 132, 478; Rao, H.S.P.R.; Rafi, S.; Padmavathy, K. Tetrahedron 2008, 64, 8037.
1005. Ashby, E.C.; Chao, L.; Neumann, H.M. J. Am. Chem. Soc. 1973, 95, 4896, 5186.
1006. Zhao, B.; Lu, X. Tetrahedron Lett. 2006, 47, 6765.
1007. Kianmehr, E.; Rajabi, A.; Ghanbari, M. Tetrahedron Lett. 2009, 50, 1687.
1008. Miura, T.; Takahashi, Y.; Murakami, M. Chem. Commun. 2007, 3577.
1009. Zhou, C.; Larock, R.C. J. Am. Chem. Soc. 2004, 126, 2302.
1010. See Screttas, C.G.; Steele, B.R. Org. Prep. Proced. Int. 1990, 22, 271.
1011. Cooke, Jr., M.P.; Pollock, C.M. J. Org. Chem. 1993, 58, 7474. For another method, see Einhorn, J.; Luche, J.L. Tetrahedron Lett. 1986, 27, 501.
1012. Niestroj, M.; Neumann, W.P.; Thies, O. Chem. Ber. 1994, 127, 1131.
1013. Barnea, E.; Andrea, T.; Kapon, M.; Berthet, J.-C.; Ephritikhine, M.; Eisen, M.S. J. Am. Chem. Soc. 2004, 126, 10860.
1014. See House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972,, pp. 629–682; Reeves, R.L. in Patai, S. The Chemistry of the Carbonyl Group, pt. 1, Wiley, NY, 1966, pp. 567–619. See also, Stowell, J.C. Carbanions in Organic Synthesis, Wiley, NY, 1979.
1015. See Mahrwald, R. Modern Aldol Reactions, 2 Volume Set, Wiley, NJ, 2004; Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 816–823. For a treatise that discloses Aleksandr Borodin as the discoverer of the aldol condensation, see Podlech, J. Angew. Chem. Int. Ed. 2010, 49, 6490.
1016. This reaction is also called the aldol condensation, although, strictly speaking, this term applies to the formation only of the α,β-unsaturated product, and not the aldol.
1017. See Thebtaranonth, C.; Thebtaranonth, Y. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 1, Wiley, NY, 1989, pp. 199–280, 99–212; Hajos, Z.G. in Augustine, R.L. Carbon–Carbon Bond Formation, Vol. 1; Marcel Dekker, NY, 1979; pp. 1–84; Nielsen, A.T.; Houlihan, W.J. Org. React. 1968, 16, 1.
1018. See Mahrwald, R.; Gündogan, B. J. Am. Chem. Soc. 1998, 120, 413.
1019. Ashby, E.C.; Argyropoulos, J.N. J. Org. Chem. 1986, 51, 472.
1020. Markert, M.; Mulzer, M.; Schetter, B.; Mahrwald, R. J. Am. Chem. Soc. 2007, 129, 7258. See Erkkilä, A.; Pihko, P.M. J. Org. Chem. 2006, 71, 2538.
1021. See Cainelli, G.; Galletti, P.; Giacomini, D.; Orioli, P. Tetrahedron Lett. 2001, 42, 7383.
1022. See Zhao, P.; Lucht, B.L.; Kenkre, S.L.; Collum, D.B. J. Org. Chem. 2004, 69, 242; Zhao, P.; Condo, A.; Keresztes, I.; Collum, D.B. J. Am. Chem. Soc. 2004, 126, 3113.
1023. Seki, A.; Ishiwata, F.; Takizawa, Y.; Asami, M. Tetrahedron 2004, 60, 5001.
1024. Johansson, A.; Abrahamsson, P.; Davidsson, Ö. Tetrahedron Asymmetry 2003, 14, 1261.
1025. Flowers, II, R.A.; Xu, X.; Timmons, C.; Li, G. Eur. J. Org. Chem. 2004, 2988.
1026. Zheng, X.; Zhang, Y. Synth. Commun. 2003, 161.
1027. Kourouli, T.; Kefalas, P.; Ragoussis, N.; Ragoussis, V. J. Org. Chem. 2002, 67, 4615.
1028. See Simpura, I.; Nevalainen, V. Angew. Chem. Int. Ed. 2000, 39, 3422.
1029. See Casiraghi, G.; Zanardi, F.; Appendino, G.; Rassu, G. Chem. Rev. 2000, 100, 1929; Casiraghi, G.; Zanardi, E.; Rassu, G. Pure Appl. Chem. 2000, 72, 1645; Denmark, S.E.; Heemstra, Jr., J.R.; Beutner, G.L. Angew. Chem. Int. Ed. 2005, 44, 4782.
1030. See Abiko, A.; Inoue, T.; Masamune, S. J. Am. Chem. Soc. 2002, 124, 10759.
1031. Demir, A.S.; Ayhan, P.; Igdir, A.C.; Duygu, A.N. Tetrahedron 2004, 60, 6509.
1032. For discussions of equilibrium constants in aldol reactions, see Guthrie, J.P.; Wang, X. Can. J. Chem. 1991, 69, 339; Guthrie, J.P. J. Am. Chem. Soc. 1991, 113, 7249, and references cited therein.
1033. The equilibrium concentration of the product from acetone in pure acetone was determined to be 0.01%: Maple, S.R.; Allerhand, A. J. Am. Chem. Soc. 1987, 109, 6609.
1034. For an aqueous version, see Buonora, P.T.; Rosauer, K.G.; Dai, L. Tetrahedron Lett. 1995, 36, 4009.
1035. Han, Z.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Tetraehdron Lett. 2000, 41, 4415.
1036. See Kad, G.L.; Kaur, K.P.; Singh, V.; Singh, J. Synth. Commun. 1999, 29, 2583.
1037. See Mukaiyama, T. Isr. J. Chem. 1984, 24, 162; Caine, D. in Augustine, R.L. Carbon–Carbon Bond Formation, Vol. 1, Marcel Dekker, NY, 1979, pp. 264–276.
1038. Takazawa, O.; Kogami, K.; Hayashi, K. Bull. Chem. Soc. Jpn. 1985, 58, 2427.
1039. See Hooz, J.; Oudenes, J.; Roberts, J.L.; Benderly, A. J. Org. Chem. 1987, 52, 1347; Nozaki, K.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1041. For a review, see Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 324–333. For an ab initio study see Murga, J.; Falomir, E.; Carda, M.; Marco, J.A. Tetrahedron 2001, 57, 6239.
1040. See Brown, H.C.; Ganesan, K. Tetrahedron Lett. 1992, 33, 3421.
1041. See Arnett, E.M.; Fisher, F.J.; Nichols, M.A.; Ribeiro, A.A. J. Am. Chem. Soc. 1990, 112, 801.
1042. House, H.O.; Crumrine, D.S.; Teranishi, A.Y.; Olmstead, H.D. J. Am. Chem. Soc. 1973, 95, 3310.
1043. It has been contended that such stabilization is not required: Mulzer, J.; Brüntrup, G.; Finke, J.; Zippel, M. J. Am. Chem. Soc. 1979, 101, 7723.
1044. Wei, H.-X.; Jasoni, R.L.; Shao, H.; Hu, J.; Paré, P.W. Tetrahedron 2004, 60, 11829.
1045. See Mahrwald, R.; Costisella, B.; Gündogan, B. Synthesis 1998, 262.
1046. Evans, D.A.; McGee, L.R. Tetrahedron Lett. 1980, 21, 3975; J. Am. Chem. Soc. 1981, 103, 2876.
1047. Nokami, J.; Mandai, T.; Watanabe, H.; Ohyama, H.; Tsuji, J. J. Am. Chem. Soc. 1989, 111, 4126.
1048. See Loh, T.-P.; Feng, L.-C.; Wei, L.-L. Tetrahedron 2001, 57, 4231.
1049. Yanagisawa, A.; Kimura, K.; Nakatsuka, Y.; Yamamoto, H. Synlett 1998, 958.
1050. Kobayashi, S.; Hachiya, I.; Takahori, T. Synthesis 1993, 371.
1051. Yokoyama, Y.; Mochida, K. Synlett 1996, 445; Sasai, H.; Arai, S.; Shibasaki, M. J. Org.Chem. 1994, 59, 2661. Also see, Bao, W.; Zhang, Y.; Wang, J. Synth. Commun. 1996, 26, 3025.
1052. For a review, see Mahrwald, R. Chem. Rev. 1999, 99, 1095.
1053. Das, G.; Thornton, E.R. J. Am. Chem. Soc. 1990, 112, 5360.
1054. Mukaiyama, T.; Arai, H.; Shiina, I. Chem. Lett. 2000, 580.
1055. Liu, C.M.; Smith, III, W.J.; Gustin, D.J.; Roush, W.R. J. Am. Chem. Soc. 2005, 127, 5770.
1056. Pratt, L.M.; Nguên, NV.; Ramachandran, B. J. Org. Chem. 2005, 70, 4279.
1057. Zhang, X.; Houk, K.N. J. Org. Chem. 2005, 70, 9712.
1058. Heathcock, C.H. Aldrichimica Acta 1990, 23, 99; Science 1981, 214, 395; Nógrádi, M. Stereoselective Synthesis, VCH, NY, 1986, pp. 193–220; Heathcock, C.H. in Morrison, J.D. Asymmetric Synthesis, Vol. 3, Acaademic Press, NY, 1984, pp. 111–212; Heathcock, C.H. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pt. B, Elsevier, NY, 1984, pp. 177–237; Evans, D.A.; Nelson, J.V.; Taber, T.R. Top. Stereochem. 1982, 13, 1; Evans, D.A. Aldrichimica Acta 1982, 15, 23; Braun, M.; Sacha, H.; Galle, D.; Baskaran, S. Pure Appl. Chem. 1996, 68, 561; Kitamura, M.; Nakano, K.; Miki, T.; Okada, M.; Noyori, R. J. Am. Chem. Soc. 2001, 123, 8939.
1059. Ertas, M.; Seebach, D. Helv. Chim. Acta 1985, 68, 961.
1060. Schetter, B.; Ziemer, B.; Schnakenburg, G.; Mahrwald, R. J. Org. Chem. 2008, 73, 813; Mahrwald, R.; Schetter, B. Org. Lett. 2006, 8, 281.
1061. Labadie, S.S.; Stille, J.K. Tetrahedron 1984, 40, 2329; Yura, T.; Iwasawa, N.; Mukaiyama, T. Chem. Lett. 1986, 187. See also, Nakamura, E.; Kuwajima, I. Tetrahedron Lett. 1983, 24, 3347.
1062. Yamamoto, Y.; Maruyama, K.; Matsumoto, K. J. Am. Chem. Soc. 1983, 105, 6963; Sakurai, H.; Sasaki, K.; Hosomi, A. Bull. Chem. Soc. Jpn. 1983, 56, 3195; Hagiwara, H.; Kimura, K.; Uda, H. J. Chem. Soc., Chem. Commun. 1986, 860.
1063. Walker, M.A.; Heathcock, C.H. J. Org. Chem. 1991, 56, 5747. For reviews, see Paterson, I. Chem. Ind. (London) 1988, 390; Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, p. 324.
1064. Hoffmann, R.W.; Ditrich, K.; Fröch, S. Liebigs Ann. Chem. 1987, 977.
1065. Patz, M.; Mayr, H. Tetrahedron Lett. 1993, 34, 3393.
1066. Pratt, L. M.; Newman, A.; Cyr, J. S.; Johnson, H.; Miles, B.; Lattier, A.; Austin, E.; Henderson, S.; Hershey, B.; Lin, M.; Balamraju, Y.; Sammonds, L.; Cheramie, J.; Karnes, J.; Hymel, E.; Woodford, B.; Carter, C. J. Org. Chem. 2003, 68, 6387.
1067. See Paddon-Row, M.N.; Houk, K.N. J. Org. Chem. 1990, 55, 481; Denmark, S.E.; Henke, B.R. J. Am. Chem. Soc. 1991, 113, 2177.
1068. See Nerz-Stormes, M.; Thornton, E.R. J. Org. Chem. 1991, 56, 2489.
1069. Swiss, K.A.; Choi, W.; Liotta, D.; Abdel-Magid, A.F.; Maryanoff, C.A. J. Org. Chem. 1991, 56, 5978.
1070. Danda, H.; Hansen, M.M.; Heathcock, C.H. J. Org. Chem. 1990, 55, 173. See also, Corey, E.J.; Kim, S.S. Tetrahedron Lett. 1990, 31, 3715.
1071. Hirama, M.; Noda, T.; Takeishi, S.; Itô, S. Bull. Chem. Soc. Jpn. 1988, 61, 2645; Majewski, M.; Gleave, D.M. Tetrahedron Lett. 1989, 30, 5681.
1072. Ward, D.E.; Sales, M.; Sasmal, P.K. J. Org. Chem. 2004, 69, 4808.
1073. Calter, M.A.; Guo, X.; Liao, W. Org. Lett. 2001, 3, 1499.
1074. Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.-Y.; Houk, K.N. Acc. Chem. Res. 2004, 37, 558; Saito, S.; Yamamoto, H. Acc. Chem. res. 2004, 37, 570; Geary, L.M.; Hultin. P.G. Tetrahedron Asymm. 2009, 20, 131. For a discussion of chelation versus nonchelation control, see Yan, T.-H.; Tan, C.-W.; Lee, H.-C.; Lo, H.-C.; Huang, T.-Y. J. Am. Chem. Soc. 1993, 115, 2613. See Majewski, M.; Lazny, R.; Nowak, P. Tetrahedron Lett. 1995, 36, 5465; Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 861–873. For a model for acyclic stereocontrol, see Evans, D.A.; Cee, V.J.; Siska, S.J. J. Am. Chem. Soc. 2006, 128, 9433.
1075. For antiselective aldol reactions see Oppolzer, W.; Lienard, P. Tetrahedron Lett. 1993, 34, 4321. For a “non-Evans” syn-aldol, see Yan, T.-H.; Lee, H.-C.; Tan, C.-W. Tetrahedron Lett. 1993, 34, 3559.
1076. Klein, J. in Patai, S. Supplement A: The Chemistry of Double-Bonded Functional Groups, Vol. 2, pt. 1, Wiley, NY, 1989, pp. 567–677; Braun, M. Angew. Chem. Int. Ed. 1987, 26, 24.
1077. Paterson, I.; Goodman, J.M. Tetrahedron Lett. 1989, 30, 997; Siegel, C.; Thornton, E.R. J. Am. Chem. Soc. 1989, 111, 5722; Faunce, J.A.; Grisso, B.A.; Mackenzie, P.B. J. Am. Chem. Soc. 1991, 113, 3418.
1078. See Reetz, M.T.; Kesseler, K.; Jung, A. Tetrahedron 1984, 40, 4327.
1079. See Short, R.P.; Masamune, S. Tetrahedron Lett. 1987, 28, 2841.
1080. Notz, W.; Tanaka, F.; Barbas III, C.F. Acc. Chem. Res. 2004, 37, 580.
1081. See Northrup, A.B.; MacMillan, D.W.C. J. Am. Chem. Soc. 2002, 124, 6798. See Suri, J.T.; Mitsumori, S.; Albertshofer, K.; Tanaka, F.; Barbas III, C.F. J. Org. Chem. 2006, 71, 3822; Guizzetti, S.; Benaglia, M.; Pignataro, L.; Puglisi, A. Tetrahedron Asymmetry 2006, 17, 2754. See Chimni, S.S.; Mahajan, D. Tetrahedron 2005, 61, 5019.
1082. Tang, Z.; Jiang, F.; Yu, L.-T.; Cui, X.; Gong, L.-Z.; Mi, A.-Q.; Jiang, Y.-Z.; Wu, Y.-D. J. Am. Chem. Soc. 2003, 125, 5262; Zhong, G.; Fan, J.; Barbas, III, C.F. Tetrahedron Lett. 2004, 45, 5681.
1083. See Mahrwald, R. Org. Lett. 2000, 2, 4011; Zhou, Y.; Shan, Z. J. Org. Chem. 2006, 71, 9510.
1084. For a review, see Guillena, G.; Nájera, C.; Ramón, D.J. Tetrahedron Asymmetry 2007, 18, 2249. Tang, Z.; Yang, Z.-H.; Chen, X.-H.; Cun, L.-F.; Mi, A.-Q.; Jiang, Y.-Z.; Gong, L.-Z. J. Am. Chem. Soc. 2005, 127, 9285; Samanta, S.; Zhao, C.-G. J. Am. Chem. Soc. 2006, 128, 7442; Luo, S.; Xu, H.; Li, J.; Zhang, L.; Cheng, J.P. J. Am. Chem. Soc. 2007, 129, 3074; Liu, J.; Yang, Z.; Wang, Z.; Wang, F.; Chen, X.; Liu, X.; Feng, X.; Su, Z.; Hu, C. J. Am. Chem. Soc. 2008, 130, 5654; Denmark, S.E.; Bui, T. J. Org. Chem. 2005, 70, 10393; Guillena, G.; Hita, M.d.C.; Nájera, C.; Viózquez, S.F. J. Org. Chem. 2008, 73, 5933; Wang, W.; Mei, Y.; Li, H.; Wang, J. Org. Lett. 2005, 7,601; Krattiger, P.; Kovasy, R.; Revell, J.D.; Ivan, S.; Wennemers, H. Org. Lett. 2005, 7, 1101; Samanta, S.; Liu, J.; Dodda, R.; Zhao, C.-G. Org. Lett. 2005, 7, 5321; Revell, J.D.; Wennemers, H. Tetrahedron 2007, 63, 8420; Lombardo, M.; Easwar, S.; Pasi, F.; Trombini, C.; Dhavale, D.D. Tetrahedron 2008, 64, 9203; Guillena, G.; Hita, M.d.C.; Nájera, C. Tetrahedron Asymmetry 2006, 17, 1493; Tang, X.; Liégault, B.; Renaud, J.-L.; Bruneau, C. Tetrahedron Asymmetry 2006, 17, 2187; Rodríguez, B.; Bruckmann, A.; Bolm, C. Chemistry: European J. 2007, 13, 4710; Córdova, A.; Zou, W.; Ibrahem, I.; Reyes, E.; Engqvist, M.; Liao, W.-W. Chem. Commun. 2005, 3586; Sun, G.; Fan, J.; Wang, Z.; Li, Y. Synlett 2008, 2491; Rambo, R.S.; Schneider, P.H. Tetrahedron Asymm. 2010, 21, 2254.
1085. See Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas III, C.F. J. Am. Chem. Soc. 2006, 128, 734; Chi, Y.; Scroggins, S.T.; Boz, E.; Fr échet, J.M.J. J. Am. Chem. Soc. 2008, 130, 17287; Guizzetti, S.; Benaglia, M.; Raimondi, L.; Celentano, G. Org. Lett. 2007, 9, 1247; Maya, V.; Raj, M.; Singh, V.K. Org. Lett. 2007, 9, 2593; Chimni, S.S.; Mahajan, D.; Suresh Babu, V.V. Tetrahedron Lett. 2005, 46, 5617; Akagawa, K.; Sakamoto, S.; Kudo, K. Tetrahedron Lett. 2005, 46, 8185; Lei, L.; Shi, L.; Li, G.; Chen, S.; Weihai, W.; Ge, Z.; Cheng, T.; Li, R. Tetrahedron 2007, 63, 7892; Amedjkouh, M. Tetrahedron Asymmetry 2005, 16, 1411; Chimni, S.S.; Mahajan, D. Tetrahedron Asymmetry 2006, 17, 2108; Hayashi, Y.; Sumiya, T.; Takahashi, J.; Gotoh, H.; Urushima, T.; Shoji, M. Angew. Chem. Int. Ed. 2006, 45, 958; Jiang, Z.; Liang, Z.; Wu, X.; Lu, Y. Chem. Commun. 2006, 2801.
1086. Hein, J.E.; Hultin, P.G. Synlett 2003, 635.
1087. See Kantam, M.L.; Ramani, T.; Chakrapani, L.; Kumar, K.V. Tetrahedron Lett. 2008, 49, 1498; Inoue, H.; Kikuchi, M.; Ito, J.-i.; Nishiyama, H. Tetrahedron 2008, 64, 493.
1088. Trost, B.M.; Ito, H. J. Am. Chem. Soc. 2000, 122, 12003.
1089. Trost, B.M.; Shin, S.; Sclafani, J.A. J. Am. Chem. Soc. 2005, 127, 8602.
1090. Yoshikawa, N.; Kumagai, N.; Matsunaga, S.; Moll, G.; Ohshma, T.; Suzuki, T.; Shibasaki, M. J. Am. Chem. Soc. 2001, 123, 2466; Trost, B.M.; Ito, H.; Silcoff, E.R. J. Am. Chem. Soc. 2001, 123, 3367.
1091. Takikawa, H.; Ishihara, K.; Saito, S.; Yamamoto, H. Synlett 2004, 732; Denmark, S.E.; Heemstra Jr., J.R. Synlett 2004, 2411.
1092. Evans, D.A.; Tedrow, J.S.; Shaw, J.T.; Downey, C.W. J. Am. Chem. Soc. 2002, 124, 392.
1093. Trost, B.M.; Fettes, A.; Shireman, B.T. J. Am. Chem. Soc. 2004, 126, 2660.
1094. Ishihara, K.; Maruyama, T.; Mouri, M.; Gao, Q.; Furuta, K.; Yamamoto, H. Bull. Chem. Soc. Jpn. 1993, 66, 3483.
1095. Corey, E.J.; Cywin, C.L.; Roper, T.D. Tetrahedron Lett. 1992, 33, 6907.
1096. See Yoshida, K.; Ogasawara, M.; Hayashi, T. J. Org. Chem. 2003, 68, 1901.
1097. For a review, see Masamune, S.; Choy, W.; Petersen, J.S.; Sita, L.R. Angew. Chem. Int. Ed. 1985, 24, 1.
1098. Furuta, K.; Maruyama, T.; Yamamoto, H. J. Am. Chem. Soc. 1991, 113, 1041; Kiyooka, S.; Kaneko, Y.; Komura, M.; Matsuo, H.; Nakano, M. J. Org. Chem. 1991, 56, 2276. For a review, see Bernardi, A.; Gennari, C.; Goodman, J.M.; Paterson, I. Tetrahedron Asymmetry 1995, 6, 2613.
1099. Mukaiyama, T.; Uchiro, H.; Kobayashi, S. Chem. Lett. 1990, 1147.
1100. Li, L.-S.; Das, S.; Sinha, S.C. Org. Lett. 2004, 6, 127.
1101. Wittig, G.; Frommeld, H.D.; Suchanek, P. Angew. Chem. Int. Ed. 1963, 2, 683. For reviews, see Mukaiyama, T. Org. React. 1982, 28, 203; Wittig, G. Top. Curr. Chem. 1976, 67, 1; Wittig, G.; Reiff, H. Angew. Chem. Int. Ed.1968, 7, 7; Reiff, H. Newer Methods Prep. Org. Chem. 1971, 6, 48.
1102. Corey, E.J.; Enders, D. Tetrahedron Lett. 1976, 11. See also, Sugasawa, T.; Toyoda, T.; Sasakura, K. Synth. Commun. 1979, 9, 515; Depezay, J.; Le Merrer, Y. Bull. Soc. Chim. Fr. 1981, II-306.
1103. Hassner, A.; Näumann, F. Chem. Ber. 1988, 121, 1823.
1104. See Baigrie, L.M.; Cox, R.A.; Slebocka-Tilk, H.; Tencer, M.; Tidwell, T.T. J. Am. Chem. Soc. 1985, 107, 3640.
1105. Ishikawa, T.; Uedo, E.; Okada, S.; Saito, S. Synlett 1999, 450.
1106. See Guthrie, J.P.; Guo, J. J. Am. Chem. Soc. 1996, 118, 11472; Eberle, M.K. J. Org. Chem. 1996, 61, 3844.
1107. Bouillon, J.-P.; Portella, C.; Bouquant, J.; Humbel, S. J. Org. Chem. 2000, 65, 5823.
1108. Pidathala, C.; Hoang, L.; Vignola, N.; List, B. Angew. Chem. Int. Ed. 2003, 42, 2785.
1109. Lipshutz, B.H.; Amorelli, B.; Unger, J.B. J. Am. Chem. Soc. 2008, 130, 14378.
1110. Zhou, J.; Wakchaure, V.; Kraft, P.; List, B. Angew. Chem. Int. Ed. 2008, 47, 7656.
1111. Thalji, R.K.; Roush, W.R. J. Am. Chem. Soc. 2005, 127, 16778.
1112. Gawley, R.E. Synthesis 1976, 777; Jung, M.E. Tetrahedron 1976, 32, 1; Mundy, B.P. J. Chem. Educ. 1973, 50, 110. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1356–1358.
1113. See Heathcock, C.H.; Ellis, J.E.; McMurry, J.E.; Coppolino, A. Tetrahedron Lett. 1971, 4995.
1114. For improved procedures, see Sato, T.; Wakahara, Y.; Otera, J.; Nozaki, H. Tetrahedron Lett. 1990, 31, 1581, and references cited therein.
1115. Tai, C.-L.; Ly, T.W.; Wu, J.-D.; Shia, K.-S.; Liu, H.-J. Synlett 2001, 214.
1116. Rajagopal, D.; Narayanan, R.; Swaminathan, S. Tetrahedron Lett. 2001, 42, 4887.
1117. Morrison, D.W.; Forbes, D.C.; Davis Jr., J.H. Tetrahedron Lett. 2001, 42, 6053.
1118. Miyamoto, H.; Kanetaka, S.; Tanaka, K.; Yoshizawa, K.; Toyota, S.; Toda, F. Chem. Lett. 2000, 888.
1119. Stork, G.; Singh, J. J. Am. Chem. Soc. 1974, 96, 6181; Boeckman, Jr., R.K. J. Am. Chem. Soc. 1974, 96, 6179.
1120. Eder, U.; Sauer, G.; Wiechert, R. Angew. Chem. Int. Ed. 1971, 10, 496; Hajos, Z.G.; Parrish, D.R. J. Org. Chem. 1974, 39, 1615. See Agami, C. Bull. Soc. Chim. Fr. 1988, 499.
1121. Mahoney, W.S.; Brestensky, D.M.; Stryker, J.M. J. Am. Chem. Soc. 1988, 110, 291; Brestensky, D.M.; Stryker, J.M. Tetrahedron Lett. 1989, 30, 5677.
1122. Chiu, P.; Szeto, C.-P.; Geng, Z.; Cheng, K.-F. Org. Lett. 2001, 3, 1901.
1123. See Smith, M.B. Organic Synthesis, 3rd ed., Wavefunction Inc./Elsevier, Irvine, CA/London, England, 2010, pp. 837–841.
1124. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1745–1752. Also see Revis, A.; Hilty, T.K. Tetrahedron Lett. 1987, 28, 4809, and references cited therein.
1125. Mukaiyama, T. Pure Appl. Chem. 1983, 55, 1749; Kohler, B.A.B. Synth. Commun. 1985, 15, 39; Mukaiyama, T.; Narasaka, K. Org. Synth., 65, 6. See Gennari, C.; Colombo, L.; Bertolini, G.; Schimperna, G. J. Org. Chem.1987, 52, 2754; Mukaiyama, T. Angew. Chem. Int. Ed. 1977, 16, 817. See also, Reetz, M.T. Organotitanium Reagents in Organic Synthesis, Spinger, NY, 1986.
1126. Miura, K.; Sato, H.; Tamaki, K.; Ito, H.; Hosomi, A. Tetrahedron Lett. 1998, 39, 2585. For a high pressure, uncatalyzed reaction, see Bellassoued, M.; Reboul, E.; Dumas, F. Tetrahedron Lett. 1997, 38, 5631.
1127. Miura, K.; Nakagawa, T.; Hosomi, A. J. Am. Chem. Soc. 2002, 124, 536.
1128. Shirakawa, S.; Maruoka, K. Tetrahedron Lett. 2002, 43, 1469.
1129. Hollis, T.K.; Bosnich, B. J. Am. Chem. Soc. 1995, 117, 4570. For the transition state geometry, see Denmark, S.E.; Lee, W. J. Org. Chem. 1994, 59, 707.
1130. Muñoz-Muñiz, O.; Quintanar-Audelo, M.; Juaristi, E. J. Org. Chem. 2003, 68, 1622.
1131. Van de Weghe, P.; Collin, J. Tetrahedron Lett. 1993, 34, 3881.
1132. Dicker, I.B. J. Org. Chem. 1993, 58, 2324.
1133. This catalyst is tolerated in water. See Kobayashi, S.; Hachiya, I. J. Org. Chem. 1994, 59, 3590.
1134. Kobayashi, S.; Nagayama, S.; Busujima, T. Chem. Lett. 1997, 959.
1135. Reetz, M.T.; Fox, D.N.A. Tetrahedron Lett. 1993, 34, 1119.
1136. Kurihara, M.; Hayshi, T.; Miyata, N. Chem. Lett. 2001, 1324.
1137. Bach, T.; Fox, D.N.A.; Reetz, M.T. J. Chem. Soc., Chem. Commun. 1992, 1634.
1138. Ollevier, T.; Desyroy, V.; Debailleul, B.; Vaur, S. Eur. J. Org. Chem. 2005, 4971; 2006, 1061; Ollevier, T.; Li, Z. Eur. J. Org. Chem. 2007, 5665.
1139. Sudha, R.; Sankararaman, S. J. Chem. Soc., Perkin Trans. 1 1999, 383.
1140. Manabe, K.; Kobayashi, S. Tetrahedron Lett. 1999, 40, 3773. See Tian, H.-Y.; Chen, Y.-J.; Wang, D.; Bu, Y.-P.; Li, C.-J. Tetrahedron Lett. 2001, 42, 1803; Komoto, I.; Kobayashi, S. J. Org. Chem. 2004, 69, 680
1141. Loh, T.-P.; Li, X.-R. Tetrahedron 1999, 55, 10789.
1142. Ozasa, N.; Wadamoto, M.; Ishihara, K.; Yamamoto, H. Synlett 2003, 2219.
1143. Yoshida, Y.; Matsumoto, N.; Hamasaki, R.; Tanabe, Y. Tetrahedron Lett 1999, 40, 4227.
1144. Ishihara, K.; Hiraiwa, Y.; Yamamoto, H. Synlett 2001, 1851.
1145. Denmark, S.E.; Fan, Y. J. Am. Chem. Soc. 2002, 124, 4233.
1146. Hagiwara, H.; Inoguchi, H.; Fukushima, M.; Hoshi, T.; Suzuki, T. Tetrahedron Lett. 2006, 47, 5371.
1147. Wong, C.T.; Wong, M.W. J. Org. Chem. 2005, 70, 124.
1148. Murata, S.; Suzuki, M.; Noyori, R. Tetrahedron 1988, 44, 4259. For a review of cross-coupling reactions of acetals, see Mukaiyama, T.; Murakami, M. Synthesis 1987, 1043.
1149. Shirokawa, S.-i.; Kamiyama, M.; Nakamura, T.; Okada, M.; Nakazaki, A.; Hosokawa, S.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 13604.
1150. Moreau, X.; Bazán-Tejeda, B.; Campagne, J.-M. J. Am. Chem. Soc. 2005, 127, 7288; Oisaki, K.; Zhao, D.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2006, 128, 7164.
1151. Fu, F.; Teo, Y.-C.; Loh, T.-P. Tetrahedron Lett. 2006, 47, 4267.
1152. Jankowska, J.; Paradowska, J.; Mlynarski, J. Tetrahedron Lett. 2006, 47, 5281.
1153. Jankowska, J.; Mlynarski, J. J. Org. Chem. 2006, 71, 1317.
1154. Denmark, S.E.; Heemstra, Jr., J.R. J. Am. Chem. Soc. 2006, 128, 1038; Jang, H.-Y.; Hong, J.-B.; MacMillan, D.W.C. J. Am. Chem. Soc. 2007, 129, 7004.
1155. See Carswell, E.L.; Hayes, D.; Henderson, K.W.; Kerr, W.J.; Russell, C.J. Synlett 2003, 1017.
1156. Loh, T.-P.; Feng, L.-C.; Wei, L.-L. Tetrahedron 2000, 56, 7309.
1157. Matsukawa, S.; Okano, N.; Imamoto, T. Tetrahedron Lett. 2000, 41, 103.
1158. Denmark, S.E.; Heemstra, Jr., J.R. Org. Lett. 2003, 5, 2303; Denmark, S.E.; Wynn, T.; Beutner, G.L. J. Am. Chem. Soc. 2002, 124, 13405.
1159. Chen, S.-L.; Ji, S.-J.; Loh, T.-P. Tetrahedron Lett. 2004, 45, 375.
1160. Bluet, G.; Campagne, J.-M. J. Org. Chem. 2001, 66, 4293; Christmann, M.; Kalesse, M. Tetrahedron Lett. 2001, 42, 1269.
1161. Langer, P.; Köhler, V. Org. Lett. 2000, 2, 1597.
1162. Bluet, G.; Bazán-Tejeda, B.; Campagne, J.-M. Org. Lett. 2001, 3, 3807.
1163. Hayakawa, R.; Shimizu, M. Chem. Lett. 1999, 591.
1164. Akiyama, T.; Takaya, J.; Kagoshima, H. Chem. Lett. 1999, 947.
1165. Harada, T.; Iwai, H.; Takatsuki, H.; Fujita, K.; Kubu, M.; Oku, A. Org. Lett. 2001, 3, 2101.
1166. Yoshimatsu, M.; Kuribayashi, M.; Koike, T. Synlett 2001, 1799.
1167. Honda, M.; Oguchi, W.; Segi, M.; Nakajima, T. Tetrahedron 2002, 58, 6815.
1168. Suginome, M.; Uehlin, L.; Yamamoto, A.; Murakami, M. Org. Lett. 2004, 6, 1167.
1169. Bach, T. Angew. Chem. Int. Ed. 1994, 33, 417. For a discussion of stereocontrol, see Annunziata, R.; Cinquini, M.; Cozzi, F.; Cozzi, P.G.; Consolandi, E. J. Org. Chem. 1992, 57, 456.
1170. See Mikami, K.; Matsukawa, S. J. Am. Chem. Soc. 1994, 116, 4077; Kaneko, Y.; Matsuo, T.; Kiyooka, S. Tetrahedron Lett. 1994, 35, 4107; Kiyooka, S.; Kido, Y.; Kaneko, Y. Tetrahedron Lett. 1994, 35, 5243.
1171. See Vasconcellos, M.L.; Desmaële, D.; Costa, P.R.R.; d'Angelo, J. Tetrahedron Lett. 1992, 33, 4921.
1172. Ag: Wadamoto, M.; Ozasa, N.; Yanigisawa, A.; Yamamoto, H. J. Org. Chem. 2003, 68, 5593. Ce: Kobayashi, S.; Hamada, T.; Nagayama, S.; Manabe, K. Org. Lett. 2001, 3, 165. Cu: Kobayashi, S.; Nagayama, S.; Busujima, T. Tetrahedron 1999, 55, 8739. Pb: Nagayama, S.; Kobayashi, S. J. Am. Chem. Soc 2000, 122, 11531. Sc: Ishikawa, S.; Hamada, T.; Manabe, K.; Kobayashi, S. J. Am. Chem. Soc. 2004, 126, 12236. Ti: Imashiro, R.; Kuroda, T. J. Org. Chem. 2003, 68, 974. Zr: Kobayashi, S.; Ishitani, H.; Yamashita, Y.; Ueno, M.; Shimizu, H. Tetrahedron 2001, 57, 861.
1173. Denmark, S.E.; Beutner, G.L.; Wynn, T.; Eastgate, M.D. J. Am. Chem. Soc. 2005, 127, 3774; Denmark, S.E.; Bui, T. J. Org. Chem. 2005, 70, 10190; Adachi, S.; Harada, T. Org. Lett. 2008, 10, 4999; Senapati, B.K.; Gao, L.; Lee, S.I.; Hwang, G.-S.; Ryu, D.H. Org. Lett. 2010, 12, 5088.
1174. Kitazawa, E.; Imamura, T.; Saigo, K.; Mukaiyama, T. Chem. Lett. 1975, 569.
1175. Mukaiyama, T.; Shibata, J.; Shimamura, T.; Shiina, I. Chem. Lett. 1999, 951.
1176. See Solladié, G. Chimia 1984, 38, 233.
1177. Because it was discovered by Claisen, L. Ber. 1890, 23, 977.
1178. Huerta, F.F.; Bäckvall, J.-E. Org. Lett. 2001, 3, 1209.
1179. Rathke, M.W.; Sullivan, D.F. J. Am. Chem. Soc. 1973, 95, 3050.
1180. Ramirez, A.; Sun, X.; Collum, D.B. J. Am. Chem. Soc. 2006, 128, 10326.
1181. Tanabe, Y.; Matsumoto, N.; Funakoshi, S.; Manta, N. Synlett 2001, 1959.
1182. Murai, T.; Suzuki, A.; Kato, S. J. Chem. Soc., Perkin Trans. 1 2001, 2711.
1183. Nishiyama, H.; Shiomi, T.; Tsuchiya, Y.; Matsuda, I. J. Am. Chem. Soc. 2005, 127, 6972.
1184. See Shang, X.; Liu, H-.J. Synth. Commun. 1994, 24, 2485.
1185. Yost, J.M.; Zhou, G.; Coltart, D.M. Org. Lett. 2006, 8, 1503.
1186. See Johnson, W.S.; Daub, G.H. Org. React. 1951, 6, 1.
1187. Robinson, R.; Seijo, E. J. Chem. Soc. 1941, 582.
1188. Puterbaugh, W.H. J. Org. Chem. 1962, 27, 4010. See also, El-Newaihy, M.F.; Salem, M.R.; Enayat, E.I.; El-Bassiouny, F.A. J. Prakt. Chem. 1982, 324, 379.
1189. See Abiko, A. Acc. Chem. Res. 2004, 37, 387.
1190. Oh, S.H.; Cortez, G.S.; Romo, D. J. Org. Chem. 2005, 70, 2835.
1191. Saito, S.; Kobayashi, S. J. Am. Chem. Soc. 2006, 128, 8704.
1192. Lettan, II, R.B.; Reynolds, T.E.; Galliford, C.V.; Scheidt, K.A. J. Am. Chem. Soc. 2006, 128, 15566.
1193. Corey, E.J.; Choi, S. Tetrahedron Lett. 2000, 41, 2769.
1194. See Evans, D.A.; Chapman, K.T.; Bisaha, J. Tetrahedron Lett. 1984, 25, 4071.
1195. Evans, D.A.; Downey, C.W.; Shaw, J.T.; Tedrow, J.S. Org. Lett. 2002, 4, 1127.
1196. Crimmins, M.T.; McDougall, P.J. Org. Lett. 2003, 5, 591.
1197. Corey, E.J.; Choi, S. Tetrahedron Lett. 2000, 41, 2769.
1198. Posner, G.H.; Lu, S.; Asirvatham, E.; Silversmith, E.F.; Shulman, E.M. J. Am. Chem. Soc. 1986, 108, 511; Posner, G.H.; Webb, K.S.; Asirvatham, E.; Jew, S.; Degl'Innocenti, A. J. Am. Chem. Soc. 1988, 110, 4754.
1199. Baer, H.H.; Urbas, L. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, Wiley, NY, 1970, pp. 76–117. See also, Rosini, G.; Ballini, R.; Sorrenti, P. Synthesis 1983, 1014; Matsumoto, K. Angew. Chem. Int. Ed.1984, 23, 617; Eyer, M.; Seebach, D. J. Am. Chem. Soc. 1985, 107, 3601. For reviews of the nitroalkenes that are the products of this reaction, see Barrett, A.G.M.; Graboski, G.G. Chem. Rev. 1986, 86, 751; Kabalka, G.W.; Varma, R.S. Org. Prep. Proced. Int. 1987, 19, 283.
1200. Henry, L. Compt. Rend. 1895, 120, 1265; Kamlet, J. U.S. Patent 2,151,171 1939 [Chem. Abstr., 33: 500391939]; Hass, H.B.; Riley, E.F. Chem. Rev. 1943, 32, 373 (see p. 406); Lichtenthaler, F.W. Angew. Chem. Int. Ed. 1964, 3, 211. For a review, see Luzzio, F.A. Tetrahedron 2001, 57, 915.
1201. Yan, S.; Gao, Y.; Xing, R.; Shen, Y.; Liu, Y.; Wu, P.; Wu, H. Tetrahedron 2008, 64, 6294.
1202. Demicheli, G.; Maggi, R.; Mazzacani, A.; Righi, P.; Sartori, G.; Bigi, F. Tetrahedron Lett. 2001, 42, 2401; Hagiwara, H.; Sekifuji, M.; Tsubokawa, N.; Hoshi, T.; Suzuki, T. Chem. Lett. 2009, 38, 790.
1203. Bulbule, V.J.; Jnaneshwara, G.K.; Deshmukh, R.R.; Borate, H.B.; Deshpande, V.H. Synth. Commun. 2001, 31, 3623.
1204. Kisanga, P.B.; Verkade, J.G. J. Org. Chem. 1999, 64, 4298.
1205. Phukan, M.; Borah, K.J.; Borah, R. Synth. Commun. 2008, 38, 3068.
1206. Jiang, T.; Gao, H.; Han, B.; Zhao, G.; Chang, Y.; Wu, W.; Gao, L.; Yang, G. Tetrahedron Lett. 2004, 45, 2699.
1207. Ballini, R.; Bosica, G.; Parrini, M. Chem. Lett. 1999, 1105.
1208. Gan, C.; Chen, X.; Lai, G.; Wang, Z. Synlett 2006, 387.
1209. Desai, U.V.; Pore, D.M.; Mane, R.B.; Solabannavar, S.B.; Wadgaonkar, P.P. Synth. Commun. 2004, 34, 19.
1210. Klein, G.; Pandiaraju, S.; Reiser, O. Tetrahedron Lett. 2002, 43, 7503.
1211. Bandgar, B.P.; Uppalla, L.S. Synth. Commun. 2000, 30, 2071.
1212. Purkarthofer, T.; Gruber, K.; Gruber-Khadjawi, M.; Waich, K.; Skranc, W.; Mink, D.; Griengl, H Angew. Chem. Int. Ed. 2006, 45, 3454.
1213. Christensen, C.; Juhl, K.; Hazell, R.G.; J![]() rgensen, K.A. J. Org. Chem. 2002, 67, 4875. For reviews, see Boruwa, J.; Gogoi, N.; Saikia, P.P.; Barua, N.C. Tetrahedron Asymmetry 2006, 17, 3315; Palomo, C.; Oiarbide, M.; Laso, A. Eur. J. Org. Chem. 2007, 2561.
rgensen, K.A. J. Org. Chem. 2002, 67, 4875. For reviews, see Boruwa, J.; Gogoi, N.; Saikia, P.P.; Barua, N.C. Tetrahedron Asymmetry 2006, 17, 3315; Palomo, C.; Oiarbide, M.; Laso, A. Eur. J. Org. Chem. 2007, 2561.
1214. Jammi, S.; Saha, P.; Sanyashi, S.; Sakthivel, S.; Punniyamurthy, T. Tetrahedron 2008, 64, 11724.
1215. Bulut, A.; Aslan, A.; Dogan, Ö. J. Org. Chem. 2008, 73, 7373.
1216. Nitabaru, T.; Kumagai, N.; Shibasaki, M. Tetrahedron Lett. 2008, 49, 272.
1217. Tur, F.; Saá, J.M. Org. Lett. 2007, 9, 5079.
1218. Misumi, Y.; Matsumoto, K. Angew. Chem. Int. Ed. 2002, 41, 1031.
1219. Li, H.; Wang, B.; Deng, L. J. Am. Chem. Soc. 2006, 128, 732.
1220. Uraguchi, D.; Sakaki, S.; Ooi, T. J. Am. Chem. Soc. 2007, 129, 12392; Mandal, T.; Samanta, S.; Zhao, C.-G. Org. Lett. 2007, 9, 943; Arai, T.; Watanabe, M.; Yanagisawa, A. Org. Lett. 2007, 9, 3595; Liu, S.; Wolf, C. Org. Lett. 2008, 10, 1831; Marcelli, T.; van der Haas, R.N.S.; van Maarseveen, J.H.; Hiemstra, H. Angew. Chem. Int. Ed. 2006, 45, 929; Toussaint, A.; Pfaltz, A. Eur. J. Org. Chem. 2008, 4591.
1221. Risgaard, T.; Gothelf, K.V.; J![]() rgensen, K.A. Org. Biomol. Chem. 2003, 1, 153.
rgensen, K.A. Org. Biomol. Chem. 2003, 1, 153.
1222. Robak, M.T.; Trincado, M.; Ellman, J.A. J. Am. Chem. Soc. 2007, 129, 15110; Singh, A.; Johnston, J.N. J. Am. Chem. Soc. 2008, 130, 5866.
1223. Rueping, M.; Antonchick, A.P. Org. Lett. 2008, 10, 1731.
1224. Ziyaei-Halimehjani, A.; Saidi, M.R. Tetrahedron Lett. 2008, 49, 1244.
1225. For reviews, see Jones, G. Org. React. 1967, 15, 204; Wilk, B.K. Tetrahedron 1997, 53, 7097.
1226. Rochlin, E.; Rappoport, Z. J. Org. Chem. 2003, 68, 1715.
1227. See Tanaka, M.; Oota, O.; Hiramatsu, H.; Fujiwara, K. Bull. Chem. Soc. Jpn. 1988, 61, 2473.
1228. Kuwajima, I.; Iwasawa, H. Tetrahedron Lett. 1974, 107. See also, Huckin, S.N.; Weiler, L. Can. J. Chem. 1974, 52, 2157.
1229. See Bartoli, G.; Beleggia, R.; Giuli, S.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Paoletti, M. Tetrahedron Lett. 2006, 47, 6501.
1230. Harjani, J.R.; Nara, S.J.; Salunkhe, M.M. Tetrahedron Lett. 2002, 43, 1127. See Su, C.; Chen, Z.-C.; Zheng, Q.G. Synthesis 2003, 555.
1231. Bose, D.S.; Narsaiah, A.V. J. Chem. Res. (S) 2001, 36.
1232. See Pillai, M.K.; Singh, S.; Jonnalagadda, S.B. Synth. Commun. 2010, 40, 3710.
1233. Li, J.-T.; Zang, H.-J.; Feng, Y.-Y.; Li, L.-J.; Li, T.-S. Synth. Commun. 2001, 31, 653.
1234. Yadav, J.S.; Reddy, B.V.S.; Basak, A.K.; Visali, B.; Narsaiah, A.V.; Nagaiah, K. Eur. J. Org. Chem. 2004, 546.
1235. Kumar, H.M.S.; Reddy, B.V.S.; Reddy, P.T.; Srinivas, D.; Yadav, J.S. Org. Prep. Proceed. Int. 2000, 32, 81; Peng, Y.; Song, G.; Qian, X. J. Chem. Res. (S) 2001, 188.
1236. Prajapati, D.; Lekhok, K.C.; Sandhu, J.S.; Ghosh, A.C. J. Chem. Soc. Perkin Trans. 1 1996, 959.
1237. Siebenhaar, B.; Casagrande, B.; Studer, M.; Blaser, H.-U. Can. J. Chem. 2001, 79, 566.
1238. Reddy, T.I.; Varma, R.S. Tetrahedron Lett. 1997, 38, 1721.
1239. Jenner, G. Tetrahedron Lett. 2001, 42, 243.
1240. You, J.; Verkade, J.G. J. Org. Chem. 2003, 68, 8003.
1241. Chandrasekhar, S.; Yu, J.; Falck, J.R.; Mioskowski, C. Tetrahedron Lett. 1994, 35, 5441.
1242. Bartoli, G.; Beleggia, R.; Giuli, S.; Giuliani, A.; Marcantoni, E.; Massaccesi, M.; Paoletti, M. Tetrahedron Lett. 2006, 47, 6501.
1243. Yamashita, K.; Tanaka, T.; Haya, M. Tetrahedron 2005, 61, 7981.
1244. A solvent free reaction. See Prajapati, D.; Sandhu, J.S. Chem. Lett. 1992, 1945.
1245. For lists of reagents (with references) see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 317–325, 341–350. For those that give the alcohol product, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1178–1179, 1540–1541, 1717–1724, 1727, 1732–1736, 1778–1780, 1801–1805.
1246. See Barrett, A.G.M.; Robyr, C.; Spilling, C.D. J. Org. Chem. 1989, 54, 1233; Pyne, S.G.; Boche, G. J. Org. Chem. 1989, 54, 2663.
1247. See Togni, A.; Pastor, S.D. J. Org. Chem. 1990, 55, 1649; Sakuraba, H.; Ushiki, S. Tetrahedron Lett. 1990, 31, 5349; Niwa, S.; Soai, K. J. Chem. Soc. Perkin Trans. 1 1990, 937.
1248. Lehnert, W. Tetrahedron 1973, 29, 635; Synthesis 1974, 667 and references cited therein.
1249. Pellón, R.F.; Mamposo, T.; González, E.; Calderón, O. Synth. Commun. 2000, 30, 3769.
1250. For a method of preparing 49, see Bowlus, S.B.; Katzenellenbogen, J.A. Synth. Commun. 1974, 4, 137.
1251. Corey, E.J.; Durst, T. J. Am. Chem. Soc. 1968, 90, 5548, 5553.
1252. See Yamamoto, K.; Tomo, Y.; Suzuki, S. Tetrahedron Lett. 1980, 21, 2861; Martin, S.F.; Phillips, G.W.; Puckette, T.A.; Colapret, J.A. J. Am. Chem. Soc. 1980, 102, 5866; Arenz, T.; Vostell, M.; Frauenrath, H. Synlett 1991, 23.
1253. Yoda, H.; Ujihara, Y.; Takabe, K. Tetrahedron Lett. 2001, 42, 9225.
1254. See Schöllkopf, U. Pure Appl. Chem. 1979, 51, 1347; Angew. Chem. Int. Ed. 1977, 16, 339; Hoppe, D. Angew. Chem. Int. Ed. 1974, 13, 789.
1255. Schöllkopf, U.; Schröder, U.; Blume, E. Liebigs Ann. Chem. 1972, 766, 130; Schöllkopf, U.; Schröder, U. Angew. Chem. Int. Ed. 1972, 11, 311.
1256. Oldenziel, O.H.; van Leusen, D.; van Leusen, A.M. J. Org. Chem. 1977, 42, 3114.
1257. Oldenziel, O.H.; van Leusen, A.M. Tetrahedron Lett. 1974, 163, 167. See, Moskal, J.; van Leusen, A.M. Tetrahedron Lett. 1984, 25, 2585; van Leusen, A.M.; Oosterwijk, R.; van Echten, E.; van Leusen, D. Recl. Trav. Chim. Pays-Bas 1985, 104, 50.
1258. van Leusen, A.M.; Oomkes, P.G. Synth. Commun. 1980, 10, 399.
1259. Baldwin, J.E.; Höfle, G.A.; Lever, Jr., O.W. J. Am. Chem. Soc. 1974, 96, 7125. For a similar reaction, see Tanaka, K.; Nakai, T.; Ishikawa, N. Tetrahedron Lett. 1978, 4809.
1260. Also see Reetz, M.T.; Heimbach, H.; Schwellnus, K. Tetrahedron Lett. 1984, 25, 511.
1261. Hünig, S.; Wehner, G. Synthesis 1975, 391.
1262. For a review, see Hoppe, D. Angew. Chem. Int. Ed. 1984, 23, 932.
1263. Krämer, T.; Hoppe, D. Tetrahedron Lett. 1987, 28, 5149.
1264. Vedejs, E.; Dolphin, J.M.; Stolle, W.T. J. Am. Chem. Soc. 1979, 101, 249.
1265. See Johnson, J.R. Org. React. 1942, 1, 210.
1266. Koepp, E.; Vögtle, F. Synthesis 1987, 177.
1267. Crawford, M.; Little, W.T. J. Chem. Soc. 1959, 722.
1268. See Berti, G. Top. Stereochem. 1973, 7, 93, pp. 210–218. Also see, Bakó, P.; Szöllõsy, Á; Bombicz, P.; Töke, L. Synlett 1997, 291.
1269. See Bansal, R.K.; Sethi, K. Bull. Chem. Soc. Jpn. 1980, 53, 1197.
1270. See Yliniemelä, A.; Brunow, G.; Flügge, J.; Teleman, O. J. Org. Chem. 1996, 61, 6723.
1271. Ballester, M.; Pérez-Blanco, D. J. Org. Chem. 1958, 23, 652; Elkik, E.; Francesch, C. Bull. Soc. Chim. Fr. 1973, 1277, 1281.
1272. See also, Zimmerman, H.E.; Ahramjian, L. J. Am. Chem. Soc. 1960, 82, 5459.
1273. Borch, R.F. Tetrahedron Lett. 1972, 3761.
1274. Johnson, C.R.; Bade, T.R. J. Org. Chem. 1982, 47, 1205.
1275. See White, D.R.; Wu, D.K. J. Chem. Soc., Chem. Commun. 1974, 988.
1276. Satoh, T.; Sugimoto, A.; Itoh, M.; Yamakawa, K. Tetrahedron Lett. 1989, 30, 1083.
1277. Arai, S.; Ishida, T.; Shioiri, T. Tetrahedron Lett. 1998, 39, 8299.
1278. Tung, C.C.; Speziale, A.J.; Frazier, H.W. J. Org. Chem. 1963, 28, 1514.
1279. Mauzé, B. J. Organomet. Chem. 1979, 170, 265.
1280. Sulmon, P.; De Kimpe, N.; Schamp, N.; Declercq, J.; Tinant, B. J. Org. Chem. 1988, 53, 4457.
1281. See Arai, S.; Suzuki, Y.; Tokumaru, K.; Shioiri, T. Tetrahedron Lett. 2002, 43, 833. See Starks, C.M.; Liotta, C. Phase Transfer Catalysis Academic Press, NY, 1978, pp. 197–198.
1282. Jung, M.E.; Mengel, W.; Newton, T.W. Synth. Commun. 1999, 29, 3659.
1283. Sipos, G.; Schöbel, G.; Sirokmán, F. J. Chem. Soc. Perkin Trans. 2 1975, 805.
1284. Achard, T.J.R.; Belokon', Y.N.; Hunt, J.; North, M.; Pizzato, F. Tetrahedron Lett. 2007, 48, 2961.
1285. Achard, T.J.R.; Belokon', Y.N.; Ilyin, M.; Moskalenko, M.; North, M.; Pizzato, F. Tetrahedron Lett. 2007, 48, 2965. For a review, see Ohkata, K.; Kimura, J.; Shinohara, Y.; Takagi, R.; Hiraga, Y. Chem. Commun. 1996, 2411.
1286. Aggarwal, V.K.; Hynd, G.; Picoul, W.; Vasse, J.-L. J. Am. Chem. Soc. 2002, 124, 9964.
1287. Arai, S.; Shirai, Y.; Ishida, T.; Shioiri, T. Tetrahedron 1999, 55, 6375.
1288. Deyrup, J.A. J. Org. Chem. 1969, 34, 2724.
1289. Peterson, D.J. J. Org. Chem. 1968, 33, 780. See Ager, D.J. Org. React. 1990, 38, 1; Synthesis 1984, 384; Colvin, E.W. Silicon Reagents in Organic Synthesis, Academic Press, NY, 1988, pp. 63–75; Weber, W.P. Silicon Reagents for Organic Synthesis, Springer, NY, 1983, pp. 58–78; Magnus, P. Aldrichimica Acta 1980, 13, 43; Chan, T. Acc. Chem. Res. 1977, 10, 442. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 337–341.
1290. See Hudrlik, P.F.; Agwaramgbo, E.L.O.; Hudrlik, A.M. J. Org. Chem. 1989, 54, 5613.
1291. See Strekowski, L.; Visnick, M.; Battiste, M.A. Tetrahedron Lett. 1984, 25, 5603.
1292. Craig, D.; Ley, S.V.; Simpkins, N.S.; Whitham, G.H.; Prior, M.J. J. Chem. Soc. Perkin Trans. 1 1985, 1949.
1293. Bassindale, A.R.; Ellis, R.J.; Taylor, P.G. J. Chem. Res. (S) 1996, 34.
1294. See Colvin, E.W. Silicon Reagents in Organic Synthesis, Academic Press, NY, 1988, pp. 65–69.
1295. Markò, I.E.; Murphy, F.; Kumps, L.; Ates, A.; Touillaux, R.; Craig, D.; Carballares, S.; Dolan, S. Tetrahedron 2001, 57, 2609.
1296. Cooke, F.; Roy, G.; Magnus, P. Organometallics 1982, 1, 893.
1297. For a review of these reagents, see Anderson, R. Synthesis 1985, 717.
1298. See Barrett, A.G.M.; Flygare, J.A. J. Org. Chem. 1991, 56, 638.
1299. Tsubouchi, A.; Kira, T.; Takeda, T. Synlett 2006, 2577.
1300. Bellassoued, M.; Ozanne, N. J. Org. Chem. 1995, 60, 6582.
1301. Murai, T.; Fujishima, A.; Iwamoto, C.; Kato, S. J. Org. Chem. 2003, 68, 7979.
1302. Stiles, M. J. Am. Chem. Soc. 1959, 81, 2598; Ann. N.Y. Acad. Sci. 1960, 88, 332; Crombie, L.; Hemesley, P.; Pattenden, G. Tetrahedron Lett. 1968, 3021.
1303. Finkbeiner, H.L.; Stiles, M. J. Am. Chem. Soc. 1963, 85, 616; Finkbeiner, H.L.; Wagner, G.W. J. Org. Chem. 1963, 28, 215.
1304. Martin, J.; Watts, P.C.; Johnson, F. Chem. Commun. 1970, 27.
1305. Tirpak, R.E.; Olsen, R.S.; Rathke, M.W. J. Org. Chem. 1985, 50, 4877. For an enantioselective version, see Hogeveen, H.; Menge, W.M.P.B. Tetrahedron Lett. 1986, 27, 2767.
1306. See Dunn, A.D.; Rudorf, W. Carbon Disulphide in Organic Chemistry, Ellis Horwood, Chichester, 1989, pp. 120–225; Yokoyama, M.; Imamoto, T. Synthesis 1984, 797, pp. 797–804.
1307. See Corey, E.J.; Chen, R.H.K. Tetrahedron Lett. 1973, 3817.
1308. See Konen, D.A.; Pfeffer, P.E.; Silbert, L.S. Tetrahedron 1976, 32, 2507, and references cited therein.
1309. Laskar, D.D.; Prajapati, D.; Sandhu, J.S. Synth. Commun. 2001, 31, 1427.
1310. Ehrlich, P.; Sachs, F. Chem. Ber. 1899, 32, 2341
1311. See Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979; Johnson, A.W. Ylid Chemistry, Academic Press, NY, 1966. For reviews, see Maryanoff, B.E.; Reitz, A.B. Chem. Rev. 1989, 89, 863; Bestmann, H.J.; Vostrowsky, O. Top. Curr. Chem. 1983, 109, 85; Pommer, H.; Thieme, P.C. Top. Curr. Chem. 1983, 109, 165; Pommer, H. Angew. Chem. Int. Ed. 1977, 16, 423; Maercker, A. Org. React. 1965, 14, 270; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 682–709. For related reviews, see Zbiral, E. Synthesis 1974, 775; Bestmann, H.J. Angew. Chem. Int. Ed. 1965, 4, 583, 645–660, 830–838; Newer Methods Prep. Org. Chem. 1968, 5, 1; Horner, L. Fortschr. Chem. Forsch., 1966, 7, 1. For a historical background, see Wittig, G. Pure Appl. Chem. 1964, 9, 245. For a list of reagents and references for the Wittig and related reactions, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 327–337.
1312. When phosphonium fluorides are used, no base is necessary, as these react directly with the substrate to give the alkene: Schiemenz, G.P.; Becker, J.; Stöckigt, J. Chem. Ber. 1970, 103, 2077.
1313. Kiddle, J.J. Tetrahedron Lett. 2000, 41, 1339.
1314. Wu, J.; Wu, H.; Wei, S.; Dai, W.-M. Tetrahedron Lett. 2004, 45, 4401.
1315. See Schlosser, M.; Schaub, B. Chimia 1982, 36, 396.
1316. Kobayashi, T.; Eda, T.; Tamura, O.; Ishibashi, H. J. Org. Chem. 2002, 67, 3156.
1317. Ramirez, F.; Pilot, J.F.; Desai, N.B.; Smith, C.P.; Hansen, B.; McKelvie, N. J. Am. Chem. Soc. 1967, 89, 6273.
1318. Bestmann, H.J. Angew. Chem. Int. Ed. 1965, 4, 586.
1319. Orsini, F.; Sello, G.; Fumagalli, T. Synlett 2006, 1717.
1320. Schiemenz, G.P.; Thobe, J. Chem. Ber. 1966, 99, 2663.
1321. See Bestmann, H.J.; Kratzer, O. Chem. Ber. 1962, 95, 1894.
1322. Bernard, M.; Ford, W.T.; Nelson, E.C. J. Org. Chem. 1983, 48, 3164.
1323. Patil, V.J.; Mävers, U. Tetrahedron Lett. 1996, 37, 1281.
1324. Betancort, J.M.; Barbas, III, C.F. Org. Lett. 2001, 3, 3737.
1325. See Dunne, E.C.; Coyne, É.J.; Crowley, P.B.; Gilheany, D.G. Tetrahedron Lett. 2002, 43, 2449.
1326. Smith, M.B.; Kwon, T.W. Synth. Commun. 1992, 22, 2865. Also see Matsunaga, S.; Kinoshita, T.; Okada, S.; Harada, S.; Shibasaki, M. J. Am. Chem. Soc. 2004, 126, 7559.
1327. See Harcken, C.; Martin, S.F. Org. Lett. 2001, 3, 3591; Yu, X.; Huang, X. Synlett 2002, 1895. Also see Greenwald, R.; Chaykovsky, M.; Corey, E.J. J. Org. Chem. 1963, 28, 1128.
1328. Tsunoda, T.; Takagi, H.; Takaba, D.; Kaku, H.; Itô, S. Tetrahedron Lett. 2000, 41, 235.
1329. Brunel, Y.; Rousseau, G. Tetrahedron Lett. 1996, 37, 3853.
1330. Baldwin, J.E.; Edwards, A.J.; Farthing, C.N.; Russell, A.T. Synlett 1993, 49.
1331. Shuto, S.; Niizuma, S.; Matsuda, A. J. Org. Chem. 1998, 63, 4489.
1332. Reid, M.; Roman, E.; Taylor, R.J.K. Synlett 2004, 819.
1333. Zhang, P.-F.; Chen, Z.-C. Synth. Commun. 2001, 31, 1619.
1334. Hisler, K.; Tripoli, R.; Murphy, J.A. Tetrahedron Lett. 2006, 47, 6293.
1335. See Isaacs, N.S.; El-Din, G.N. Tetrahedron Lett. 1987, 28, 2191. See also, Dauben, W.G.; Takasugi, J.J. Tetrahedron Lett. 1987, 28, 4377.
1336. Smithers, R.H. J. Org. Chem. 1978, 43, 2833; Miyano, S.; Izumi, Y.; Fujii, K.; Ohno, Y.; Hashimoto, H. Bull. Chem. Soc. Jpn. 1979, 52, 1197; Stork, G.; Zhao, K. Tetrahedron Lett. 1989, 30, 2173.
1337. See Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1441–1444, 1457–1458.
1338. See Ceruti, M.; Degani, I.; Fochi, R. Synthesis 1987, 79; Moskal, J.; van Leusen, A.M. Recl. Trav. Chim. Pays-Bas 1987, 106, 137; Doad, G.J.S. J. Chem. Res. (S) 1987, 370.
1339. Corey, E.J.; McCormick, J.R.D.; Swensen, W.E. J. Am. Chem. Soc. 1964, 86, 1884.
1340. Wittig, G.; Schöllkopf, U. Chem. Ber. 1954, 87, 1318.
1341. Bestmann, H.J.; Schade, G. Tetrahedron Lett. 1982, 23, 3543.
1342. For a list of references to the preparation of haloalkenes by Wittig reactions, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 725–727.
1343. See Li, P.; Alper, H. J. Org. Chem. 1986, 51, 4354.
1344. Michel, P.; Gennet, D.; Rassat, A. Tetrahedron Lett. 1999, 40, 8575. See Michael, P.; Rassat, A. Tetrahedron Lett. 1999, 40, 8579.
1345. See Cockerill, A.F.; Harrison, R.G. in Patai, S. The Chemistry of Functional Groups: Supplement A, pt. 1, Wiley, NY, 1977, pp. 232–240; Vedejs, E.; Marth, C.F. J. Am. Chem. Soc. 1988, 110, 3948.
1346. It has been contended that another mechanism, involving single electron transfer, may be taking place in some cases: Olah, G.A.; Krishnamurthy, V.V. J. Am. Chem. Soc. 1982, 104, 3987; Yamataka, H.; Nagareda, K.; Hanafusa, T.; Nagase, S. Tetrahedron Lett. 1989, 30, 7187. A diradical mechanism has also been proposed for certain cases: Ward, Jr., W.J.; McEwen, W.E. J. Org. Chem. 1990, 55, 493.
1347. Arnett, E.M.; Wernett, P.C. J. Org. Chem. 1993, 58, 301.
1348. See Vedejs, E.; Marth, C.F. J. Am. Chem. Soc. 1990, 112, 3905.
1349. See Schlosser, M.; Christmann, K.F. Liebigs Ann. Chem. 1967, 708, 1.
1350. Maryanoff, B.E.; Reitz, A.B. Chem. Rev. 1989, 89, 863, see p. 865.
1351. Neumann, R.A.; Berger, S. Eur. J. Org. Chem. 1998, 1085.
1352. Puke, C.; Erker, G.; Wibbeling, B.; Fröhlich, R. Eur. J. Org. Chem. 1999, 1831.
1353. Vedejs, E.; Meier, G.P.; Snoble, K.A.J. J. Am. Chem. Soc. 1981, 103, 2823. See also, Nesmayanov, N.A.; Binshtok, E.V.; Reutov, O.A. Doklad. Chem. 1973, 210, 499.
1354. Mazhar-Ul-Haque; Caughlan, C.N.; Ramirez, F.; Pilot, J.F.; Smith, C.P. J. Am. Chem. Soc. 1971, 93, 5229.
1355. Maryanoff, B.E.; Reitz, A.B.; Mutter, M.S.; Inners, R.R.; Almond Jr., H.R.; Whittle, R.R.; Olofson, R.A. J. Am. Chem. Soc. 1986, 108, 7664. See also, Pískala, A.; Rehan, A.H.; Schlosser, M. Coll. Czech. Chem. Commun.1983, 48, 3539.
1356. McEwen, W.E.; Kumli, K.F.; Bladé-Font, A.; Zanger, M.; VanderWerf, C.A. J. Am. Chem. Soc. 1964, 86, 2378.
1357. Zhu, X.-F.; Henry, C.E.; Kwon, O. J. Am. Chem. Soc. 2007, 129, 6722.
1358. For a catalytic version, see Shi, L.; Wang, W.; Wang, Y.; Huang, Y. J. Org. Chem. 1989, 54, 2027; Huang, Z.-Z.; Huang, X.; Huang, Y.-Z. Tetrahedron Lett. 1995, 36, 425.
1359. Huang, Z.-Z.; Tang, Y. J. Org. Chem. 2002, 67, 5320.
1360. Horner, L.; Hoffmann, H.; Wippel, H.G.; Klahre, G. Chem. Ber. 1959, 92, 2499; Wadsworth, Jr., W.S.; Emmons, W.D. J. Am. Chem. Soc. 1961, 83, 1733.
1361. Wadsworth, Jr., W.S. Org. React. 1977, 25, 73; Stec, W.J. Acc. Chem. Res. 1983, 16, 411; Walker, B.J. in Cadogan, J.I.G. Organophosphorous Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 156–205; Boutagy, J.; Thomas, R. Chem. Rev. 1974, 74, 87; Seguineau, P.; Villieras, J. Tetrahedron Lett. 1988, 29, 477, and other papers in this series.
1362. Motoyoshiya, J.; Kasaura, T.; Kokin, K.; Yokoya, S.-i.; Takaguchi, Y.; Narita, S.; Aoyama, H. Tetrahedron 2001, 57, 1715.
1363. Has-Becker, S.; Bodmann, K.; Kreuder, R.; Santoni, G.; Rein, T.; Reiser, O. Synlett 2001, 1395.
1364. Also known as the Michaelis–Arbuzov rearrangement. For reviews, see Petrov, A.A.; Dogadina, A.V.; Ionin, B.I.; Garibina, V.A.; Leonov, A.A. Russ. Chem. Rev. 1983, 52, 1030; Bhattacharya, A.K.; Thyagarajan, G. Chem. Rev. 1981, 81, 415. See also Shokol, V.A.; Kozhushko, B.N. Russ. Chem. Rev. 1985, 53, 98; Brill, T.B.; Landon, S.J. Chem. Rev. 1984, 84, 577; Lherbet, C.; Castonguay, R.; Keillor, J.W. Tetrahedron Lett. 2005, 46, 3565; Huang, C.; Tang, X.; Fu, H.; Jiang, Y.; Zhao, Y. J. Org. Chem. 2006, 71, 5020.
1365. Jones, S.; Selitsianos, D. Org. Lett. 2002, 4, 3671.
1366. Gelman, D.; Jiang, L.; Buchwald, S.L. Org. Lett. 2003, 5, 2315.
1367. Barrett, A.G.M.; Cramp, S.M.; Roberts, R.S.; Zecri, F.J. Org. Lett. 1999, 1, 579.
1368. Wang, Y.; West, F.G. Synthesis 2002, 99.
1369. Appel, R.; Loos, R.; Mayr, H. J. Am. Chem. Soc. 2009, 131, 704.
1370. Schauer, D.J.; Helquist, P. Synthesis 2006, 3654.
1371. Wu, J.; Zhang, D.; Wei. S. Synth. Commun. 2005, 35, 1213; Wu, J.; Li, D.; Zhang, D. Synth. Commun. 2005, 35, 2543. See also McNulty, J.; Das, P. Tetrahedron Lett. 2009, 50, 5737; McNulty, J.; Das, P.; McLeod, D. Chemistry: Eur. J. 2010, 16, 6756.
1372. Mizuno, M.; Fujii, K.; Tomioka, K. Angew. Chem. Int. Ed. 1998, 37, 515. Also see, Arai, S.; Hamaguchi, S.; Shioiri, T. Tetrahedron Lett. 1998, 39, 2997. For a review of asymmetric Wittig-type reactions see Rein, T.; Pedersen, T.M. Synthesis 2002, 579.
1373. Abiko, A.; Masamune, S. Tetrahedron Lett. 1996, 37, 1077.
1374. Corey, E.J.; Cane, D.E. J. Org. Chem. 1969, 34, 3053. For a chiral derivative, see Hanessian, S.; Beaudoin, S. Tetrahedron Lett. 1992, 33, 7655, 7659.
1375. Corey, E.J.; Kwiatkowski, G.T. J. Am. Chem. Soc. 1966, 88, 5654.
1376. Broekhof, N.L.J.M.; van der Gen, A. Recl. Trav. Chim. Pays-Bas 1984, 103, 305; Broekhof, N.L.J.M.; van Elburg, P.; Hoff, D.J.; van der Gen, A. Recl. Trav. Chim. Pays-Bas 1984, 103, 317.
1377. Ando, K. Tetrahedron Lett. 1995, 36, 4105.
1378. Yu, W.; Su, M.; Jin, Z. Tetrahedron Lett. 1999, 40, 6725.
1379. Nangia, A.; Prasuna, G.; Rao, P.B. Tetrahedron Lett. 1994, 35, 3755; Couture, A.; Deniau, E.; Gimbert, Y.; Grandclaudon, P. J. Chem. Soc. Perkin Trans. 1 1993, 2463.
1380. Hauske, J.R.; Dorff, P.; Julin, S.; Martinelli, G.; Bussolari, J. Tetrahedron Lett. 1992, 33, 3715.
1381. See Maryanoff, B.E.; Reitz, A.B. Chem. Rev. 1989, 89, 863; Gosney, I.; Rowley, A.G. in Cadogan, J.I.G. Organophosphorous Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 17–153; Reucroft, J.; Sammes, P.G. Q. Rev. Chem. Soc. 1971, 25, 135, see pp. 137–148, 169; Schlosser, M. Top. Stereochem. 1970, 5, 1. Also see, Takeuchi, K.; Paschal, J.W.; Loncharich, R.J. J. Org. Chem. 1995, 60, 156.
1382. Robiette, R.; Richardson, J.; Aggarwal, V.K.; Harvey, J.N. J. Am. Chem. Soc. 2005, 127, 13468.
1383. Robiette, R.; Richardson, J.; Aggarwal, V.K.; Harvey, J.N. J. Am. Chem. Soc. 2006, 128, 2394.
1384. See Maryanoff, B.E.; Reitz, A.B.; Duhl-Emswiler, B.A. J. Am. Chem. Soc. 1985, 107, 217; Le Bigot, Y.; El Gharbi, R.; Delmas, M.; Gaset, A. Tetrahedron 1986, 42, 3813. Also see Schlosser, M.; Schaub, B.; de Oliveira-Neto, J.; Jeganathan, S. Chimia 1986, 40, 244.
1385. See Reitz, A.B.; Nortey, S.O.; Jordan, Jr., A.D.; Mutter, M.S.; Maryanoff, B.E. J. Org. Chem. 1986, 51, 3302.
1386. See Rein, T.; Reiser, O. Acta Chem. Scand. B, 1996, 50, 369. For a review of asymmetric ylid reactions, see Li, A.-H.; Dai, L.-X.; Aggarwal, V.K. Chem. Rev. 1997, 97, 2341.
1387. Ayrey, P.M.; Warren, S. Tetrahedron Lett. 1989, 30, 4581.
1388. See Schlosser, M.; Tuong, H.B.; Respondek, J.; Schaub, B. Chimia 1983, 37, 10.
1389. See Corey, E.J.; Shulman, J.I.; Yamamoto, H. Tetrahedron Lett. 1970, 447.
1390. Peet, J.H.J.; Rockett, B.W. J. Organomet. Chem. 1974, 82, C57; Adcock, W.; Khor, T. J. Organomet. Chem. 1975, 91, C20.
1391. Schlosser, M.; Christmann, K.-F. Synthesis 1969, 38.
1392. Schlosser, M. Top. Stereochem. 1970, 5, 1, p. 22.
1393. Ando, K.; Oishi, T.; Hirama, M.; Ohno, H.; Ibuka, T. J. Org. Chem. 2000, 65, 4745.
1394. Touchard, F.P. Tetrahedron Lett. 2004, 45, 5519.
1395. For a review, see Becker, K.B. Tetrahedron 1980, 36, 1717.
1396. For a review of these double ring closures, see Vollhardt, K.P.C. Synthesis 1975, 765.
1397. See Bestmann, H.J.; Vostrowsky, O. Top. Curr. Chem. 1983, 109, 85.
1398. See Aksnes, G.; Fr![]() yen, P. Acta Chem. Scand. 1968, 22, 2347.
yen, P. Acta Chem. Scand. 1968, 22, 2347.
1399. See Fr![]() yen, P. Acta Chem. Scand. Ser. B 1974, 28, 586.
yen, P. Acta Chem. Scand. Ser. B 1974, 28, 586.
1400. See Kayser, M.M.; Breau, L. Can. J. Chem. 1989, 67, 1401. For a study of the mechanism, see Abell, A.D.; Clark, B.M.; Robinson, W.T. Aust. J. Chem. 1988, 41, 1243.
1401. With microwave irradiation, see Sabitha, G.; Reddy, M.M.; Srinivas, D.; Yadov, J.S. Tetrahedron Lett. 1999, 40, 165.
1402. Murphy, P.J.; Brennan, J. Chem. Soc. Rev. 1988, 17, 1; Flitsch, W.; Schindler, S.R. Synthesis 1975, 685.
1403. Bestmann, H.J.; Seng, F. Tetrahedron 1965, 21, 1373.
1404. Bestmann, H.J.; Denzel, T.; Salbaum, H. Tetrahedron Lett. 1974, 1275.
1405. Lawrence, N.J.; Beynek, H. Synlett 1998, 497.
1406. Kano, N.; Hua, X.J.; Kawa, S.; Kawashima, T. Tetrahedron Lett. 2000, 41, 5237.
1407. For a review, see Palacios, F.; Alonso, C.; Aparicio, D.; Rubiales, G.; de los Santos, J.M. Tetrahedron 2007, 63, 523.
1408. Singh, P.N.D.; Klima, R.F.; Muthukrishnan, S.; Murthy, R.S.; Sankaranarayanan, J.; Stahlecker, H.M.; Patel, B.; Gudmundsdóttir, A.D. Tetrahedron Lett. 2005, 46, 4213.
1409. For in situ generation of this reagent, see Cannizzo, L.F.; Grubbs, R.H. J. Org. Chem. 1985, 50, 2386.
1410. Tebbe, F.N.; Parshall, G.W.; Reddy, G.S. J. Am. Chem. Soc. 1978, 100, 3611; Pine, S.H.; Pettit, R.J.; Geib, G.D.; Cruz, S.G.; Gallego, C.H.; Tijerina, T.; Pine, R.D. J. Org. Chem. 1985, 50, 1212. See also, Clawson, L.; Buchwald, S.L.; Grubbs, R.H. Tetrahedron Lett. 1984, 25, 5733; Clift, S.M.; Schwartz, J. J. Am. Chem. Soc. 1984, 106, 8300.
1411. Petasis, N.A.; Bzowej, E.I. J. Am. Chem. Soc. 1990, 112, 6392.
1412. Meurer, E.C.; Santos, L.S.; Pilli, R.A.; Eberlin, M.N. Org. Lett. 2003, 5, 1391.
1413. Pine, S.H.; Shen, G.S.; Hoang, H. Synthesis 1991, 165.
1414. Martínez, I.; Andrews, A.E.; Emch, J.D.; Ndakala, A.J.; Wang, J.; Howell, A.R. Org. Lett. 2003, 5, 399.
1415. Petasis, N.A.; Lu, S.-P. Tetrahedron Lett. 1995, 36, 2393.
1416. Tehrani, K.A.; De Kimpe, N. Tetrahedron Lett. 2000, 41, 1975. See Martínez, I.; Howell, A.R. Tetrahedron Lett. 2000, 41, 5607.
1417. Petasis, N.A.; Browej, E.I. Tetrahedron Lett. 1993, 34, 943.
1418. Petasis, N.A.; Akritopoulou, I. Synlett 1992, 665.
1419. Petasis, N.A.; Hu, Y.-H. J. Org. Chem,. 1997, 62, 782. Also see, Petasis, N.A.; Browej, E.I. J. Org. Chem. 1992, 57, 1327.
1420. Fujiwara, T.; Iwasaki, N.; Takeda, T. Chem. Lett. 1998, 741. For an example using a gem-dichloride, see Takeda, T.; Sasaki, R.; Fujiwara, T. J. Org. Chem. 1998, 63, 7286.
1421. Yan, T.H.; Tsai, C.-C.; Chien, C.-T.; Cho, C.-C.; Huang, P.-C. Org. Lett. 2004, 6, 4961.
1422. Yan, T.-H.; Chien, C.-T.; Tsai, C.-C.; Lin, K.-W.; Wu, Y.-H. Org. Lett. 2004, 6, 4965.
1423. Takeda, T.; Shimane, K.; Ito, K.; Saeki, N.; Tsubouchi, A. Chem. Communn. 2002, 1974.
1424. Ishino, Y.; Mihara, M.; Nishihama, S.; Nishiguchi, I. Bull. Chem. Soc. Jpn. 1998, 71, 2669.
1425. Okazoe, T.; Takai, K.; Oshima, K.; Utimoto, K. J. Org. Chem. 1987, 52, 4410; Matsubra, S.; Ukai, K.; Mizuno, T.; Utimoto, K. Chem. Lett. 1999, 825; Takai, K.; Kataoka, Y.; Okazoe, T.; Utimoto, K. Tetrahedron Lett. 1988, 29, 1065.
1426. For a review, see Aguero, A.; Osborn, J.A. New J. Chem. 1988, 12, 111.
1427. See Hartner Jr., F.W.; Schwartz, J.; Clift, S.M. J. Am. Chem. Soc. 1990, 105, 640.
1428. Hartley, R.C.; Li, J.; Main, C.A.; McKiernan, G.J. Tetrahedron 2007, 63, 4825.
1429. See List, B.; Doehring, A.; Fonseca, M.T.H.; Job, A.; Torres, R.R. Tetrahedron 2006, 62, 476.
1430. Sada, M.; Komagawa, S.; Uchiyama, M.; Kobata, M.; Mizuno, T.; Utimoto, K.; Oshima, K.; Matsubara, S. J. Am. Chem. Soc. 2010, 132, 17452.
1431. Matsubara, S.; Horiuchi, M.; Takai, K.; Utimoto, K. Chem. Lett. 1995, 259. See also, Concellón, J.M.; Concellón, C. J. Org. Chem. 2006, 71, 1728.
1432. Barma, D.K.; Kundu, A.; Zhang, H.; Mioskowski, C.; Falck, J.R. J. Am. Chem. Soc. 2003, 125, 3218.
1433. Peng, Z.-Y.; Ma, F.-F.; Zhu, L.-F.; Xie, X.-M.; Zhang, Z. J. Org. Chem. 2009, 74, 6855.
1434. Chen, Y.; Huang, L.; Zhang, X.P. Org. Lett. 2003, 5, 2493; Aggarwal, V.K.; Fulton, J.R.; Sheldon, C.G.; de Vincente, J. J. Am. Chem. Soc. 2003, 125, 6034.
1435. Lebel, H.; Guay, D.; Paquet, V.; Huard, K. Org. Lett. 2004, 6, 3047. For a synthesis of dienes from conjugated aldehydes, see Lebel, H.; Paquet, V. J. Am. Chem. Soc. 2004, 126, 320.
1436. Pedro, F.M.; Hirner, S.; Kühn, F.E. Tetrahedron Lett. 2005, 46, 7777.
1437. Lebrun, M.-E.; Le Marquand, P.; Berthelette, C. J. Org. Chem. 2006, 71, 2009.
1438. See Block, E. Reactions of Organosulfur Compounds Academic Press, NY, 1978, pp. 101–105; Berti, G. Top. Stereochem. 1973, 7, 93, pp. 218–232. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 944–951.
1439. The bond enthalpies for S and Se ylids has been determined. See Stoffregen, S.A.; McCulla, R.D.; Wilson, R.; Cercone, S.; Miller, J.; Jenks, W.S. J. Org. Chem. 2007, 72, 8235.
1440. See Paxton, R.J.; Taylor, R.J.K. Synlett 2007, 633.
1441. See Kavanagh, S.A.; Piccinini, A.; Fleming, E.M.; Connon, S.J. Org. Biomol. Chem. 2008, 6, 1339. For reviews, see House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972,, pp. 709–733; Durst, T. Adv. Org. Chem. 1969, 6, 285, see pp. 321–330. For a monograph on sulfur ylids, see Trost, B.M.; Melvin, Jr., L.S. Sulfur Ylids, Academic Press, NY, 1975.
1442. Corey, E.J.; Chaykovsky, M. J. Am. Chem. Soc. 1965, 87, 1353.
1443. Adams, J.; Hoffman, Jr., L.; Trost, B.M. J. Org. Chem. 1970, 35, 1600; Braun, H.; Huber, G.; Kresze, G. Tetrahedron Lett. 1973, 4033; Corey, E.J.; Jautelat, M.; Oppolzer, W. Tetrahedron Lett. 1967, 2325.
1444. See Forbes, D.C.; Amin, S.R.; Bean, C.J.; Standen, M.C. J. Org. Chem. 2006, 71, 8287.
1445. Farrall, M.J.; Durst, T.; Fréchet, J.M.J. Tetrahedron Lett. 1979, 203.
1446. Toda, F.; Kanemoto, K. Heterocycles 1997, 46, 185.
1447. Alcaraz, L.; Harnett, J.J.; Mioskowski, C.; Martel, J.P.; Le Gall, T.; Shin, D.-S.; Falck, J.R. Tetrahedron Lett. 1994, 35, 5449. Also see, Alcaraz, L.; Harnett, J.J.; Mioskowski, C.; Martel, J.P.; Le Gall, T.; Shin, D.-S.; Falck, J.R. Tetrahedron Lett. 1994, 35, 5453.
1448. See Aggarwal, V.K.; Angelaud, R.; Bihan, D.; Blackburn, P.; Fieldhouse, R.; Fonguerna, S.J.; Ford, G.D.; Hynd, G.; Jones, E.; Jones, R.V.H.; Jubault, P.; Palmer, M.J.; Ratcliffe, P.D.; Adams, H. J. Chem. Soc., Perkin Trans. 1 2001, 2604.
1449. See Sone, T.; Yamaguchi, A.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2008, 130, 10078.
1450. Hansch, M.; Illa, O.; McGarrigle, E.M.; Aggarwal, V.K. Chemistry: Asian J. 2008, 3, 1657.
1451. See Takada, H.; Metzner, P.; Philouze, C. Chem. Commun. 2001, 2350.
1452. See Aggarwal, V.K.; Harvey, J.N.; Richardson, J. J. Am. Chem. Soc. 2002, 124, 5747.
1453. See Johnson, C.R.; Schroeck, C.W.; Shanklin, J.R. J. Am. Chem. Soc. 1973, 95, 7424.
1454. Johnson, C.R.; Schroeck, C.W.; Shanklin, J.R. J. Am. Chem. Soc. 1973, 95, 7424.
1455. See Gutsche, C.D. Org. React. 1954, 8, 364.
1456. See Davies, H.M.L.; De Meese, J. Tetrahedron Lett. 2001, 42, 6803.
1457. M![]() kosza, M.; Urba
kosza, M.; Urba![]() ska, N.; Chesnokov, A.A. Tetrahedron Lett. 2003, 44, 1473.
ska, N.; Chesnokov, A.A. Tetrahedron Lett. 2003, 44, 1473.
1458. Arai, S.; Shioiri, T. Tetrahedron 2002, 58, 1407.
1459. For exceptions, see Sadhu, K.M.; Matteson, D.S. Tetrahedron Lett. 1986, 27, 795; Araki, S.; Butsugan, Y. J. Chem. Soc., Chem. Commun. 1989, 1286.
1460. Alex, A.; Larmanjat, B.; Marrot, J.; Couty, F.; David, O. Chem. Commun. 2007, 2500.
1461. Liao, W.-W.; Deng, X.-M.; Tang, Y. Chem. Commun. 2004, 1516.
1462. Zhu, S.; Liao, Y.; Zhu, S. Synlett 2005, 1429.
1463. For a review, see Muller, L.L.; Hamer, J. 1,2-Cycloaddition Reactions, Wiley, NY, 1967, pp. 57–86.
1464. Iranpoor, N.; Kazemi, F. Synthesis 1996, 821.
1465. Schönberg, A.; Frese, E. Chem. Ber. 1962, 95, 2810.
1466. For example, see Beiner, J.M.; Lecadet, D.; Paquer, D.; Thuillier, A. Bull. Soc. Chim. Fr. 1973, 1983.
1467. Mlosto![]() , G.; Roma
, G.; Roma![]() ski, J.; Swiatek, A.; Heimgartner, H. Helv. Chim. Acta 1999, 82, 946.
ski, J.; Swiatek, A.; Heimgartner, H. Helv. Chim. Acta 1999, 82, 946.
1468. Opitz, G.; Fischer, K. Angew. Chem. Int. Ed. 1965, 4, 70.
1469. For a review of this process, see Fischer, N.S. Synthesis1970, 393.
1470. Trost, B.M.; Melvin, Jr., L.S. Sulfur Ylids, Academic Press, NY, 1975. For reviews, see Fava, A. in Bernardi, F.; Csizmadia, I.G.; Mangini, A. Organic Sulfur Chemistry, Elsevier, NY, 1985, pp. 299–354; Belkin, Yu.V.; Polezhaeva, N.A. Russ. Chem. Rev. 1981, 50, 481; Block, E. in Stirling, C.J.M. The Chemistry of the Sulphonium Group, part 2, Wiley, NY, 1981, pp. 680–702; Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 91–127.
1471. See Gololobov, Yu.G.; Nesmeyanov, A.N.; Lysenko, V.P.; Boldeskul, I.E. Tetrahedron 1987, 43, 2609.
1472. Chandrasekhar, S.; Jagadeshwar, N.V.; Reddy, K.V. Tetrahedron Lett. 2003, 44, 3629.
1473. See Kennewell, P.D.; Taylor, J.B. Chem. Soc. Rev. 1980, 9, 477.
1474. Johnson, C.R. Aldrichimica Acta 1985, 18, 1; Acc. Chem. Res. 1973, 6, 341; Kennewell, P.D.; Taylor, J.B. Chem. Soc. Rev. 1975, 4, 189; Trost, B.M. Acc. Chem. Res. 1974, 7, 85.
1475. Oswald, M.F.; Raw, S.A.; Taylor, R.J.K. Org. Lett. 2004, 6, 3997.
1476. Bestmann, H.J.; Seng, F. Angew. Chem. Int. Ed. 1962, 1, 116; Grieco, P.A.; Finkelhor, R.S. Tetrahedron Lett. 1972, 3781.
1477. Shestopalov, A.M.; Sharanin, Yu.A.; Litvinov, V.P.; Nefedov, O.M. J. Org. Chem. USSR 1989, 25, 1000.
1478. Cohen, T.; Myers, M. J. Org. Chem. 1988, 53, 457.
1479. Kunz, R.K.; MacMillan, D.W.C. J. Am. Chem. Soc. 2005, 127, 3240.
1480. Kakei, H.; Sone, T.; Sohtome, Y.; Matsunaga, S.; Shibasaki, M. J. Am. Chem. Soc. 2007, 129, 13410.
1481. Zheng, J.-C.; Liao, W.-W.; Tang, Y.; Sun, X.-L.; Daim L.-X.J. Am. Chem. Soc. 2005, 127, 12222.
1482. Bertus, P.; Gandon, V.; Szymoniak, J. Chem. Commun. 2000, 171.
1483. Taylor, E.C.; McKillop, A. The Chemistry of Cyclic Enaminonitriles and ortho-Amino Nitriles, Wiley, NY, 1970; Schaefer, J.P.; Bloomfield, J.J. Org. React. 1967, 15, 1.
1484. See Page, P.C.B.; van Niel, M.B.; Westwood, D. J. Chem. Soc. Perkin Trans. 1 1988, 269.
1485. Panek, J.S.; Liu, P. Tetrahedron Lett. 1997, 38, 5127.
1486. Fukuzumi, S.; Okamoto, T.; Otera, J. J. Am. Chem. Soc. 1994, 116, 5503.
1487. Braddock, D.C.; Badine, D.M.; Gottschalk, T. Synlett 2001, 1909.
1488. Chataigner, I.; Piarulli, U.; Gennari, C. Tetrahedron Lett. 1999, 40, 3633.
1489. Mayr, H.; Gabriel, A.O.; Schumacher, R. Liebigs Ann. Chem. 1995, 1583.
1490. Ishihara, K.; Mouri, M.; Gao, Q.; Maruyama, T.; Furuta, K.; Yamamoto, H. J. Am. Chem. Soc. 1993, 115, 11490. See Malkov, A.V.; Liddon, A.J.P.S.; Ramírez-López, P.; Bendová, L.; Haigh, D.; Ko![]() ovský, P. Angew. Chem. Int. Ed. 2006, 45, 1432.
ovský, P. Angew. Chem. Int. Ed. 2006, 45, 1432.
1491. Kercher, T.; Livinghouse, T. J. Am. Chem. Soc. 1996, 118, 4200.
1492. Maruyama, T.; Mizuno, Y.; Shimizu, I.; Suga, S.; Yoshida, J. J. Am. Chem. Soc. 2007, 129, 1902.
1493. Friedrich, K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 2, Wiley, NY, 1983, pp. 1345–1390; Friedrich, K.; Wallenfels, K. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 72–77.
1494. Rasmussen, J.K.; Heilmann, S.M.; Krepski, L. Adv. Silicon Chem. 1991, 1, 65; Yoneda, R.; Santo, K.; Harusawa, S.; Kurihara, T. Synthesis 1986, 1054; Sukata, K. Bull. Chem. Soc. Jpn. 1987, 60, 3820.
1495. See Kanai, M.; Hamashima, Y.; Shibasaki, M. Tetrahedron Lett. 2000, 41, 2405. The reaction works in some cases without a Lewis acid, see Manju, K.; Trehan, S. J. Chem. Soc. Perkin Trans. 1 1995, 2383.
1496. See Gröger, H.; Capan, E.; Barthuber, A.; Vorlop, K.-D. Org. Lett. 2001, 3, 1969, and references cited therein; Lundgren, S.; Wingstrand, E.; Penhoat, M.; Moberg, C. J. Am. Chem. Soc. 2005, 127, 11592; Kim, S.S.; Kwak, J.M. Tetrahedron 2006, 62, 48; Shen, K.; Liu, X.; Li, Q.; Feng, X. Tetrahedron 2008, 64, 147; Kim, S.S.; Song. D.H. Eur. J. Org. Chem. 2005, 1777. See also, North, M.; Omedes-Pujol, M.; Williamson, C. Chemistry: Eur. J. 2010, 16, 11367. See Bruneh, J.-M.; Holmes, I.P. Angew. Chem. Int. Ed. 2004, 43, 2752.
1497. Ryu, D.H.; Corey, E.J. J. Am. Chem. Soc. 2005, 127, 5384; Douglas E.; Fuerst, D.E.; Jacobsen, E.N. J. Am. Chem. Soc. 2005, 127, 8964; Liu, X.; Qin, B.; Zhou, X.; He, B.; Feng, X. J. Am. Chem. Soc. 2005, 127, 12224; Wen, Y.; Huang, X.; Huang, J.; Xiong, Y.; Qin, B.; Feng, X. Synlett 2005, 2445.
1498. Lv, C.; Wu, M.; Wang, S.; Xia, C.; Sun, W. Tetrahedron Asymm. 2010, 21, 1869.
1499. van Langen, L.M.; Selassa, R.P.; van Rantwijk, F.; Sheldon, R.A. Org. Lett. 2005, 7, 327.
1500. Gerrits, P.J.; Marcus, J.; Birikaki, L.; van der Gen, A. Tetrahedron Asymmetry 2001, 12, 971.
1501. Gaisberger, R.P.; Fechter, M.H.; Griengl, H. Tetrahedron Asymmetry 2004, 15, 2959.
1502. He, B.; Li, Y.; Feng, X.; Zhang, G. Synlett 2004, 1776.
1503. Shen, Y.; Feng, X.; Li, Y.; Zhang, G.; Jiang, Y. Tetrahedron 2003, 59, 5667. See Shen, Y.; Feng, X.; Li, Y.; Zhang, G.; Jiang, Y. Eur. J. Org. Chem. 2004, 129.
1504. Amurrio, I.; Córdoba, R.; Csákÿ, A.G.; Plumet, J. Tetrahedron 2004, 60, 10521.
1505. Jenner, G. Tetrahedron Lett. 1999, 40, 491.
1506. Kurono, N.; Yamaguchi, M.; Suzuki, K.; Ohkuma, T. J. Org. Chem. 2005, 70, 6530.
1507. Song, J.J.; Gallou, F.; Reeves, J.T.; Tan, Z.; Yee, N.K.; Senanayake, C.H. J. Org. Chem. 2006, 71, 1273; Suzuki, Y.; Abu Bakar M.D.; Muramatsu, K.; Sato, M. Tetrahedron 2006, 62, 4227.
1508. Shen, Z.-L.; Ji, S.-J.; Loh, T.-P. Tetrahedron Lett. 2005, 46, 3137.
1509. Ward, D.E.; Hrapchak, M.J.; Sales, M. Org. Lett. 2000, 2, 57.
1510. Fossey, J.S.; Richards, C.J. Tetrahedron Lett. 2003, 44, 8773.
1511. Cho, W.K.; Kang, S.M.; Medda, A.K.; Lee, J.K.; Choi, I.S.; Lee, H.-S. Synthesis 2008, 50.
1512. Huang, W.; Song, Y.; Bai, C.; Cao, G.; Zheng, Z. Tetrahedron Lett. 2004, 45, 4763; He, B.; Chen, F.-X.; Li, Y.; Feng, X.; Zhang, G. Tetrahedron Lett. 2004, 45, 5465.
1513. Bandini, M.; Cozzi, P.G.; Melchiorre, P.; Umani-Ronchi, A. Tetrahedron Lett 2001, 42, 3041.
1514. See Ryu, D.H.; Corey, E.J. J. Am. Chem. Soc. 2004, 126, 8106.
1515. See He, B.; Qin, B.; Feng, X.; Zhang, G. J. Org. Chem. 2004, 69, 7910; Chen, F.-X.; Qin, B.; Feng, X.; Zhang, G.; Jiang, Y. Tetrahedron 2004, 60, 10449; Uang, B.-J.; Fu, I.-P.; Hwang, C.-D.; Chang, C.-W.; Yang, C.-T.; Hwang, D.-R. Tetrahedron 2004, 60, 10479. Also see Aspinall, H.C.; Greeves, N.; Smith, P.M. Tetrahedron Lett. 1999, 40, 1763; Deng, H.; Isler, M.P.; Snapper, M.L.; Hoveyda, A.H. Angew. Chem. Int. Ed. 2002, 41, 1009; Karimi, B.; Ma'Mani, L. Org. Lett. 2004, 6, 4813.
1516. Baleizão, C.; Gigante, B.; Garcia, H.; Corma, A. Tetrahedron Lett. 2003, 44, 6813.
1517. De, S.K.; Gibbs, R.A. Tetrahedron Lett. 2004, 45, 7407.
1518. Belokon, Y.N.; Gutnov, A.V.; Moskalenko, M.A.; Yashkina, L.V.; Lesovoy, D.E.; Ikonnikov, N.S.; Larichev, V.S.; North, M. Chem. Commun. 2002, 244; Kawasaki, Y.; Fujii, A.; Nakano, Y.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 1999, 64, 4214.
1519. Ogata, Y.; Kawasaki, A. in Zabicky, J. The Chemistry of the Carbonyl Group, Vol. 2, Wiley, NY, 1970, pp. 21–32. See also, Ching, W.; Kallen, R.G. J. Am. Chem. Soc. 1978, 100, 6119.
1520. Lapworth, A. J. Chem. Soc. 1903, 83, 998.
1521. See Shafran, Yu.M.; Bakulev, V.A.; Mokrushin, V.S. Russ. Chem. Rev. 1989, 58, 148.
1522. See Williams, R.M. Synthesis of Optically Active α-Amino Acids Pergamon, Elmsford, NY, 1989, pp. 208–229; Yet, L. Angew. Chem. Int. Ed. 2001, 40, 875; Gröger, H. Chem. Rev. 2003, 103, 2795.
1523. See Zhang, G.-W.; Zheng, D.-H.; Nie, J.; Wang, T.; Ma, J.-A. Org. Biomol. Chem. 2010, 8, 1399. See also, Yazaki, R.; Kumagai, N.; Shibasaki, M. J. Am. Chem. Soc. 2010, 132, 5522.
1524. See Mai, K.; Patil, G. Tetrahedron Lett. 1984, 25, 4583; Synth. Commun. 1985, 15, 157.
1525. Jenner, G.; Salem, R.B.; Kim, J.C.; Matsumoto, K. Tetrahedron Lett. 2003, 44, 447.
1526. Martínez, R.; Ramón, D.J.; Yus, M. Tetrahedron Lett. 2005, 46, 8471.
1527. Shen, Z.-L.; Ji, S.-J.; Loh, T.-P. Tetrahedron 2008, 64, 8159.
1528. Ooi, T.; Uematsu, Y.; Maruoka, K. J. Am. Chem. Soc. 2006, 128, 2548.
1529. Hou, Z.; Wang, J.; Liu, X.; Feng, X. Chemistry: European J. 2008, 14, 4484.
1530. Rueping, M.; Sugiono, E.; Azap, C. Angew. Chem. Int. Ed. 2006, 45, 2617.
1531. Blacker, J.; Clutterbuck, L.A.; Crampton, M.R.; Grosjean, C.; North, M. Tetrahedron Asymmetry 2006, 17, 1449.
1532. Cannella, R.; Clerici, A.; Panzeri, W.; Pastori, N.; Punta, C.; Porta, O. J. Am. Chem. Soc. 2006, 128, 5358.
1533. Saito, K.; Harada, K. Tetrahedron Lett. 1989, 30, 4535.
1534. Huang, J.; Corey, E.J. Org. Lett. 2004, 6, 5027.
1535. Prasad, B.A.B.; Bisai, A.; Singh, V.K. Tetrahedron Lett. 2004, 45, 9565.
1536. Ishitani, H.; Komiyama, S.; Hasegawa, Y.; Kobayashi, S. J. Am. Chem. Soc. 2000, 122, 762.
1537. Nakamura, S.; Sato, N.; Sugimoto, M.; Toru, T. Tetrahedron Asymmetry 2004, 15, 1513.
1538. Royer, L.; De, S.K.; Gibbs, R.A. Tetrahedron Lett. 2005, 46, 4595.
1539. Chavarot, M.; Byrne, J.J.; Chavant, P.Y.; Vallée, Y. Tetrahedron Asymmetry 2001, 12, 1147.
1540. Krueger, C.A.; Kuntz, K.W.; Dzierba, C.D.; Wirschun, W.G.; Gleason, J.D.; Snapper, M.L.; Hoveyda, A.H. J. Am. Chem. Soc. 1999, 121, 4284.
1541. Jiricny, J.; Orere, D.M.; Reese, C.B. J. Chem. Soc. Perkin Trans. 1 1980, 1487. Also see Okimoto, M.; Chiba, T. J. Org. Chem. 1990, 55, 1070.
1542. See Ferris, J.P.; Sanchez, R.A. Org. Synth. V, 344.
1543. Pan, S.C.; Zhou, J.; List, B. Angew. Chem. Int. Ed. 2007, 46, 612.
1544. With basic catalysts: Griengl, H.; Sieber, W. Monatsh. Chem. 1973, 104, 1008, 1027.
1545. See Adams, D.R.; Bhatnagar, S.P. Synthesis 1977, 661; Isagulyants, V.I.; Khaimova, T.G.; Melikyan, V.R.; Pokrovskaya, S.V. Russ. Chem. Rev. 1968, 37, 17. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 248.
1546. See Safarov, M.G.; Nigmatullin, N.G.; Ibatullin, U.G.; Rafikov, S.R. Doklad. Chem. 1977, 236, 507.
1547. Hellin, M.; Davidson, M.; Coussemant, F. Bull. Soc. Chim. Fr. 1966, 1890, 3217.
1548. Blomquist, A.T.; Wolinsky, J. J. Am. Chem. Soc. 1957, 79, 6025; Schowen, K.B.; Smissman, E.E.; Schowen, R.L. J. Org. Chem. 1968, 33, 1873.
1549. See Safarov, M.G.; Isagulyants, V.I.; Nigmatullin, N.G. J. Org. Chem. USSR 1974, 10, 1378.
1550. Fremaux, B.; Davidson, M.; Hellin, M.; Coussemant, F. Bull. Soc. Chim. Fr. 1967, 4250.
1551. Meresz, O.; Leung, K.P.; Denes, A.S. Tetrahedron Lett. 1972, 2797.
1552. See Wilkins, C.L.; Marianelli, R.S. Tetrahedron 1970, 26, 4131; Karpaty, M.; Hellin, M.; Davidson, M.; Coussemant, F. Bull. Soc. Chim. Fr. 1971, 1736; Coryn, M.; Anteunis, M. Bull. Soc. Chim. Belg. 1974, 83, 83.
1553. See Isagulyants, V.I.; Isagulyants, G.V.; Khairudinov, I.R.; Rakhmankulov, D.L. Bull. Acad. Sci. USSR. Div. Chem. Sci., 1973, 22, 1810; Sharf, V.Z.; Kheifets, V.I.; Freidlin, V.I. Bull. Acad. Sci. USSR Div. Chem. Sci., 1974, 23, 1681.
1554. Jasti, R.; Rychnovsky, S.D. J. Am. Chem. Soc. 2006, 128, 13640.
1555. Yadav, J.S.; Subba Reddy, B.V.; Hara Gopal, A.V.; Narayana Kumar, G.G.K.S.; Madavi, C.; Kunwar, A.C. Tetrahedron Lett. 2008, 49, 4420; Reddy, S.; Krishna, V.H.; Swamy, T.; Narayana Kumar, G.G.K.S. Can. J. Chem.2007, 85, 412.
1556. Yang, D.H.; Yang, N.C.; Ross, C.B. J. Am. Chem. Soc. 1959, 81, 133.
1557. Ellis, W.W.; Odenkirk, W.; Bosnich, B. Chem. Commun. 1998, 1311.
1558. Sarkar, T.K.; Nandy, S.K. Tetrahedron Lett. 1996, 37, 5195.
1559. Cossy, J.; Gille, B.; Bellosta, V. J. Org. Chem. 1998, 63, 3141.
1560. Kimura, M.; Ezoe, A.; Mori, M.; Iwata, K.; Tamaru, Y. J. Am. Chem. Soc. 2006, 128, 8559; Cho, H.Y.; Morken, J.P. J. Am. Chem. Soc. 2008, 130, 16140. See Yang, Y.; Zhu, S.-F.; Duan, H.-F.; Zhou, C.-Y.; Wang, L.-X.; Zhou, Q.-L. J. Am. Chem. Soc. 2007, 129, 2248.
1561. Song, M.; Montgomery, J. Tetrahedron 2005, 61, 11440.
1562. Jiménez-Núñez, E.; Claverie, C.K.; Nieto-Oberhuber, C.; Echavarren, A.M. Angew. Chem. Int. Ed. 2006, 45, 5452.
1563. Yadav, J.S.; Reddy, B.V.S.; Kumar, G.M.; Murthy, Ch.V.S.R. Tetrahedron Lett. 2001, 42, 89.
1564. Yang, J.; Viswanathan, G.S.; Li, C.-J. Tetrahedron Lett. 1999, 40, 1627.
1565. Jaber, J.J.; Mitsui, K.; Rychnovsky, S.D. J. Org. Chem. 2001, 66, 4679.
1566. Dobbs, A.P.; Martinovi![]() , S. Tetrahedron Lett. 2002, 43, 7055.
, S. Tetrahedron Lett. 2002, 43, 7055.
1567. Arnold, R.T.; Veeravagu, P. J. Am. Chem. Soc. 1960, 82, 5411; Klimova, E.I.; Abramov, A.I.; Antonova, N.D.; Arbuzov, Yu.A. J. Org. Chem. USSR 1969, 5, 1308; Klimova, E.I.; Antonova, N.D.; Arbuzov, Yu.A. J. Org. Chem. USSR 1969, 5, 1312, 1315.
1568. See Ben Salem, R.; Jenner, G. Tetrahedron Lett. 1986, 27, 1575. There is evidence that the mechanism is somewhat more complicated than shown here: Kwart, H.; Brechbiel, M. J. Org. Chem. 1982, 47, 3353.
1569. Also see Achmatowicz, Jr., O.; Szymoniak, J. J. Org. Chem. 1980, 45, 1228; Ben Salem, R.; Jenner, G. Tetrahedron Lett. 1986, 27, 1575; Papadopoulos, M.; Jenner, G. Tetrahedron Lett. 1981, 22, 2773.
1570. See Cartaya-Marin, C.P.; Jackson, A.C.; Snider, B.B. J. Org. Chem. 1984, 49, 2443.
1571. Jackson, A.C.; Goldman, B.E.; Snider, B.B. J. Org. Chem. 1984, 49, 3988.
1572. See Song, Z.; Beak, P. J. Org. Chem. 1990, 112, 8126.
1573. Benner, J.P.; Gill, G.B.; Parrott, S.J.; Wallace, B. J. Chem. Soc. Perkin Trans. 1 1984, 291, 315, 331.
1574. Mikami, K.; Terada, M.; Nakai, T. J. Am. Chem. Soc. 1990, 112, 3949.
1575. See Ujikawa, O.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1989, 30, 2837; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1178–1179.
1576. Ujikawa, O.; Inanaga, J.; Yamaguchi, M. Tetrahedron Lett. 1989, 30, 2837.
1577. Corey, E.J.; Pyne, S.G. Tetrahedron Lett. 1983, 24, 2821.
1578. See Shono, T.; Kashimura, S.; Mori, Y.; Hayashi, T.; Soejima, T.; Yamaguchi, Y. J. Org. Chem. 1989, 54, 6001.
1579. See Belotti, D.; Cossy, J.; Pete, J.P.; Portella, C. J. Org. Chem. 1986, 51, 4196.
1580. Shimizu, M.; Baba, T.; Toudou, S.; Hachiya, I. Chem. Lett. 2007, 36, 12.
1581. For a review, see Ide, W.S.; Buck, J.S. Org. React. 1948, 4, 269.
1582. Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Tetrahedron Lett. 2005, 46, 5317. See also, Iwamoto, K.; Kimura, H.; Oike, M.; Sato, M. Org. Biomol. Chem. 2008, 6, 912; Iwamoto, K.; Hamaya, M.; Hashimoto, N.; Kimura, H.; Suzuki, Y.; Sato, M. Tetrahedron Lett. 2006, 47, 7175;
1583. Storey, J.M.D.; Williamson, C. Tetrahedron Lett. 2005, 46, 7337.
1584. See Kuebrich, J.P.; Schowen, R.L.; Wang, M.; Lupes, M.E. J. Am. Chem. Soc. 1971, 93, 1214.
1585. Lapworth, A. J. Chem. Soc. 1903, 83, 995; 1904, 85, 1206.
1586. See Diederich, F.; Lutter, H. J. Am. Chem. Soc. 1989, 111, 8438. Also see Lappert, M.F.; Maskell, R.K. J. Chem. Soc., Chem. Commun. 1982, 580.
1587. Kuhlmann, H. Org. Synth. VII, 95; Matsumoto, T.; Ohishi, M.; Inoue, S. J. Org. Chem. 1985, 50, 603.
1588. Stetter, H.; Dämbkes, G. Synthesis 1977, 403.
1589. Rozwadowska, M.D. Tetrahedron 1985, 41, 3135.
1590. Demir, A.S.; Dünnwald, T.; Iding, H.; Pohl, M.; Müller, M. Tetrahedron Asymmetry 1999, 10, 4769.
1591. Enders, D.; Han, J. Tetrahedron Asymmetry 2008, 19, 1367; O'Toole, S.E.; Connon, S.J. Org. Biomol. Chem. 2009, 7, 3584; Baragwanath, L.; Rose, C.A.; Zeitler, K.; Connon, S.J. J. Org. Chem. 2009, 74, 9214.
1592. Enders, D.; Niemeier, O.; Balensiefer, T. Angew. Chem. Int. Ed. 2006, 45, 1463. See also, Mavis, M.E.; Yolacan, C.; Aydoga, F. Tetrahedron Lett. 2010, 51, 4509.
1593. Bausch, C.C.; Johnson, J.S. J. Org. Chem. 2004, 69, 4283.
1594. Linghu, X.; Bausch, C.C.; Jeffrey S.; Johnson, J.S. J. Am. Chem. Soc. 2005, 127, 1833.
1595. Tormo, J.; Hays, D.S.; Fu, G.C. J. Org. Chem. 1998, 63, 201.
1596. Ooi, T.; Doda, K.; Sakai, D.; Maruoka, K. Tetrahedron Lett. 1999, 40, 2133.
1597. Li, C.-Y.; Wang, X.-B.; Sun, X.-L.; Tang, Y.; Zheng, J.-C.; Xu, Z.-H.; Zhou, Y.-G.; Dai, L.-X. J. Am. Chem. Soc. 2007, 129, 1494.
1598. Boivin, J.; Fouquet, E.; Zard, S.Z. Tetrahedron 1994, 50, 1745.
1599. Boivin, J.; Schiano, A.-M.; Zard, S.Z. Tetrahedron Lett. 1994, 35, 249.
1600. Bowman, W.R.; Stephenson, P.T.; Terrett, N.K.; Young, A.R. Tetrahedron Lett. 1994, 35, 6369.
1601. Kim, S.; Jon, S.Y. Chem. Commun. 1996, 1335.
1602. For a review, see Friestad, G.K. Tetrahedron 2001, 57, 5461.
1603. Miyabe, H.; Ueda, M.; Naito, T. J. Org. Chem. 2000, 65, 5043.
1604. Miyabe, H.; Ueda, M.; Yoshioka, N.; Yamakawa, K.; Naito, T. Tetrahedron 2000, 56, 2413.
1605. Jin, M.; Zhang, D.; Yang, L.; Liu, Y.; Liu, Z. Tetrahedron Lett. 2000, 41, 7357.
1606. McNabb, S.B.; Ueda, M.; Naito, T. Org. Lett. 2004, 6, 1911.
1607. Halland, N.; J![]() rgensen, K.A. J. Chem. Soc., Perkin Trans. 1 2001, 1290.
rgensen, K.A. J. Chem. Soc., Perkin Trans. 1 2001, 1290.
1608. Clive, D.L.J.; Pham, M.P.; Subedi, R. J. Am. Chem. Soc. 2007, 129, 2713.
1609. Jang, D.O.; Kim, S.Y. J. Am. Chem. Soc. 2008, 130, 16152.
1610. Friestad, G.K.; Marié, J.-C.; Suh, Y.S.; Qin, J. J. Org. Chem. 2006, 71, 7016.
1611. Kim, S.S.; Mah, Y.J.; Kim, A.R. Tetrahedron Lett. 2001, 42, 8315.
1612. See Bentley, T.W.; Shim, C.S. J. Chem. Soc. Perkin Trans. 2 1993, 1659.
1613. Talbot, R.J.E. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 10; Elsevier, NY, 1972, pp. 226–257. See Kivinen, A. in Patai, S. The Chemistry of Acyl Halides, Wiley, NY, 1972, pp. 177–230.
1614. Bender, M.L.; Chen, M.C. J. Am. Chem. Soc. 1963, 85, 30. See also, Song, B.D.; Jencks, W.P. J. Am. Chem. Soc. 1989, 111, 8470; Bentley, T.W.; Koo, I.S.; Norman, S.J. J. Org. Chem. 1991, 56, 1604.
1615. Guthrie, J.P.; Pike, D.C. Can. J. Chem. 1987, 65, 1951. See also, Lee, I.; Sung, D.D.; Uhm, T.S.; Ryu, Z.H. J. Chem. Soc. Perkin Trans. 2 1989, 1697.
1616. Bevan, C.W.L.; Hudson, R.F. J. Chem. Soc. 1953, 2187; Satchell, D.P.N. J. Chem. Soc. 1963, 555.
1617. Motie, R.E.; Satchell, D.P.N.; Wassef, W.N. J. Chem. Soc. Perkin Trans. 2 1992, 859; 1993, 1087.
1618. See Satchell, D.P.N.; Wassef, W.N.; Bhatti, Z.A. J. Chem. Soc. Perkin Trans. 2 1993, 2373.
1619. Satchell, D.P.N. Q. Rev. Chem. Soc. 1963, 17, 160, see pp. 172–173. See Talbot, R.J.E. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 10, Elsevier, NY, 1972, pp. 280–287.
1620. See Deady, L.W.; Finlayson, W.L. Aust. J. Chem. 1983, 36, 1951.
1621. Wilk, B.K. Synth. Commun. 1996, 26, 3859.
1622. For a list of catalysts and reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1959–1968
1623. See Bender, M.L.; Komiyama, M. Cyclodextrin Chemistry; Springer, NY, 1978, pp. 34–41. The mechanism is shown in Saenger, W. Angew. Chem. Int. Ed. 1980, 19, 344.
1624. For reviews of ester hydrolysis catalyzed by pig liver esterase, see Zhu, L.; Tedford, M.C. Tetrahedron 1990, 46, 6587; Ohno, M.; Otsuka, M. Org. React. 1989, 37, 1. See Wong, C. Science 1989, 244, 1145; Whitesides, G.M.; Wong, C. Angew. Chem. Int. Ed. 1985, 24, 617; Barbayianni, E.; Fotakopoulou, I.; Schmidt, M.; Constantinou-Kokotou, V.; Bornscheuer, U.T.; Kokotos, G. J. Org. Chem. 2005, 70, 8730; Fotakopoulou, I.; Barbayianni, E.; Constantinou-Kokotou, V.; Bornscheuer, U.T.; Kokotos, G. J. Org. Chem. 2007, 72, 782.
1625. Basu, M.K.; Sarkar, D.C.; Ranu, B.C. Synth. Commun. 1989, 19, 627.
1626. See Olah, G.A.; Husain, A.; Singh, B.P.; Mehrotra, A.K. J. Org. Chem. 1983, 48, 3667.
1627. Ranu, B.C.; Dutta, P.; Sarkar, A. Synth. Commun. 2000, 30, 4167.
1628. Yanada, R.; Negoro, N.; Bessho, K.; Yanada, K. Synlett 1995, 1261.
1629. Ramesh, C.; Mahender, G.; Ravindranath, N.; Das, B. Tetrahedron 2003, 59, 1049.
1630. Das, B.; Banerjee, J.; Ramu, R.; Pal, R.; Ravindranath, N.; Ramesh, C. Tetrahedron Lett. 2003, 44, 5465.
1631. Varma, R.S.; Varma, M.; Chatterjee, A.K. J. Chem. Soc. Perkin Trans. 1 1993, 999.
1632. Taksande, K.N.; Sakate, S.S.; Lokhande, P.D. Tetrahedron Lett. 2006, 47, 643.
1633. Sharma, L.; Nayak, M.K.; Chakraborti, A.K. Tetrahedron 1999, 55, 9595.
1634. Nishizawa, M.; Yamamoto, H.; Seo, K.; Imagawa, H.; Sugihara, T. Org. Lett. 2002, 4, 1947.
1635. Yadav, J.S.; Reddy, B.V.S.; Rao, C.V.; Chand, P.K.; Prasad, A.R. Synlett 2002, 137.
1636. Ramesh, C.; Mahender, G.; Ravindranath, N.; Das, B. Tetrahedron Lett. 2003, 44, 1465.
1637. See Kaiser, E.T.; Kézdy, F.J. Prog. Bioorg. Chem. 1976, 4, 239, pp. 254–265.
1638. Wallace, O.B.; Springer, D.M. Tetrahedron Lett. 1998, 39, 2693.
1639. Choi, J.; Yoon, N.M. Synth. Commun. 1995, 25, 2655.
1640. Jin, C.K.; Jeong, H.J.; Kim, M.K.; Kim, J.Y.; Yoon, Y.-J.; Lee, S.-G. Synlett 2001, 1956,
1641. Gassman, P.G.; Schenk, W.N. J. Org. Chem. 1977, 42, 918.
1642. Wu, Y.-g.; Limburg, D.C.; Wilkinson, D.E.; Vaal, M.J.; Hamilton, G.S. Tetrahedron Lett. 2000, 41, 2847.
1643. Jackson, R.W. Tetrahedron Lett. 2001, 42, 5163.
1644. Ley, S.V.; Mynett, D.M. Synlett 1993, 793.
1645. Moon, S.; Duchin, L.; Cooney, J.V. Tetrahedron Lett. 1979, 3917.
1646. Loupy, A.; Pedoussaut, M.; Sansoulet, J. J. Org. Chem. 1986, 51, 740.
1647. See Nair, R.V.; Shukla, M.R.; Patil, P.N.; Salunkhe, M.M. Synth. Commun. 1999, 29, 1671.
1648. Niwayama, S. J. Org. Chem. 2000, 65, 5834.
1649. Hirata, T.; Shimoda, K.; Kawano, T. Tetrahedron Asymmetry 2000, 11, 1063.
1650. Kajiro, H.; Mitamura, S.; Mori, A.; Hiyama, T. Bull. Chem. Soc. Jpn. 1999, 72, 1553.
1651. Ingold, C.K. Structure and Mechanism in Organic Chemistry, 2d ed., Cornell University Press, Ithaca, NY, 1969, pp. 1129–1131.
1652. As given here, the IUPAC designations for BAC1 and BAL1 are the same, but Rule A.2 adds further symbols so that they can be distinguished: Su–AL for BAL1 and Su-AC for BAC1. See the IUPAC rules: Guthrie, R.D. Pure Appl. Chem. 1989, 61, 23, see p. 49.
1653. Kirby, A.J. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 10, 1972, pp. 57–207; Euranto, E.K. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 505–588.
1654. Gunaydin, H.; Houk, K.N. J. Am. Chem. Soc. 2008, 130, 15232.
1655. This is an SN1 mechanism with OR' as leaving group, which does not happen.
1656. See Zimmermann, H.; Rudolph, J. Angew. Chem. Int. Ed. 1965, 4, 40.
1657. See Polanyi, M.; Szabo, A.L. Trans. Faraday Soc. 1934, 30, 508.
1658. Holmberg, B. Ber. 1912, 45, 2997.
1659. Ingold, C.K.; Ingold, E.H. J. Chem. Soc. 1932, 758.
1660. Norton, H.M.; Quayle, O.R. J. Am. Chem. Soc. 1940, 62, 1170.
1661. Martin, R.B. J. Am. Chem. Soc. 1962, 84, 4130. See also Yates, K. Acc. Chem. Res. 1971, 6, 136; Huskey, W.P.; Warren, C.T.; Hogg, J.L. J. Org. Chem. 1981, 46, 59.
1662. See Euranto, E.K.; Kanerva, L.T. Acta Chem. Scand. Ser. B 1988, 42 717.
1663. Treffers, H.P.; Hammett, L.P. J. Am. Chem. Soc. 1937, 59, 1708. For other evidence for this mechanism, see Bender, M.L.; Chen, M.C. J. Am. Chem. Soc. 1963, 85, 37.
1664. Yates, K. Acc. Chem. Res. 1971, 6, 136; Al-Shalchi, W.; Selwood, T.; Tillett J.G. J. Chem. Res. (S) 1985, 10.
1665. For discussions, see Kirby, A.J. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 86–101; Ingold, C.K. Structure and Mechanism in Organic Chemistry, 2nd ed., Cornell University Press, Ithica, NY, 1969, pp. 1137–1142, 1157–1163.
1666. Douglas, J.E.; Campbell, G.; Wigfield, D.C. Can. J. Chem. 1993, 71, 1841.
1667. Cowdrey, W.A.; Hughes, E.D.; Ingold, C.K.; Masterman, S.; Scott, A.D. J. Chem. Soc. 1937, 1264; Long, F.A.; Purchase, M. J. Am. Chem. Soc. 1950, 73, 3267.
1668. Barclay, L.R.C.; Hall, N.D.; Cooke, G.A. Can. J. Chem. 1962, 40, 1981.
1669. Sneen, R.A.; Rosenberg, A.M. J. Org. Chem. 1961, 26, 2099. See also, Müller, P.; Siegfried, B. Helv. Chim. Acta 1974, 57, 987.
1670. Moore, J.A.; Schwab, J.W. Tetrahedron Lett. 1991, 32, 2331.
1671. Takashima, K.; José, S.M.; do Amaral, A.T.; Riveros, J.M. J. Chem. Soc., Chem. Commun. 1983, 1255.
1672. Comisarow, M. Can. J. Chem. 1977, 55, 171.
1673. Fukuda, E.K.; McIver, Jr., R.T. J. Am. Chem. Soc. 1979, 101, 2498.
1674. See Williams, A.; Douglas, K.T. Chem. Rev. 1975, 75, 627.
1675. See Broxton, T.J.; Chung, R.P. J. Org. Chem. 1986, 51, 3112.
1676. Inoue, T.C.; Bruice, T.C. J. Org. Chem. 1986, 51, 959; Isaacs, N.S.; Najem, T.S. Can. J. Chem. 1986, 64, 1140; J. Chem. Soc. Perkin Trans. 2 1988, 557.
1677. Allen, A.D.; Kitamura, T.; Roberts, K.A.; Stang, P.J.; Tidwell, T.T. J. Am. Chem. Soc. 1988, 110, 622.
1678. See Euranto, E.K. Pure Appl. Chem. 1977, 49, 1009.
1679. Mujika, J.I.; Mercero, J.M.; Lopez, X. J. Am. Chem. Soc. 2005, 127, 4445.
1680. See Zahn, D. Eur. J. Org. Chem. 2004, 4020.
1681. See Kahne, D.; Still, W.C. J. Am. Chem. Soc. 1988, 110, 7529.
1682. For a list of catalysts and, with references, see Larock, R.C. Comprehensive Organic Transformatinos, 2nd ed., Wiley–VCH, NY, 1999, pp. 1976–1977. Also see, Bagno, A.; Lovato, G.; Scorrano, G. J. Chem. Soc. Perkin Trans. 2 1993, 1091.
1683. Chemat, F. Tetrahedron Lett. 2000, 41, 3855.
1684. Schwyzer, R.; Costopanagiotis, A.; Sieber, P. Helv. Chim. Acta 1963, 46, 870.
1685. Bose, D.S.; Lakshminarayana, V. Synthesis 1999, 66.
1686. Tom, N.J.; Simon, W.M.; Frost, H.N.; Ewing, M. Tetrahedron Lett. 2004, 45, 905.
1687. Kim, Y.H.; Kim, K.; Park, Y.J. Tetrahedron Lett. 1990, 31, 3893.
1688. See Flynn, D.L.; Zelle, R.E.; Grieco, P.A. J. Org. Chem. 1983, 48, 2424.
1689. Ladenheim, H.; Bender, M.L. J. Am. Chem. Soc. 1960, 82, 1895.
1690. Vaughan, H.L.; Robbins, M.D. J. Org. Chem. 1975, 40, 1187.
1691. Gassman, P.G.; Hodgson, P.K.G.; Balchunis, R.J. J. Am. Chem. Soc. 1976, 98, 1275.
1692. Hibbert, F.; Malana, M.A. J. Chem. Soc. Perkin Trans. 2 1992, 755.
1693. O'Connor, C. Q. Rev. Chem. Soc. 1970, 24, 553; Talbot, R.J.E. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 257–280; Challis, B.C.; Challis, J.C. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 731–857.
1694. See DeWolfe, R.H.; Newcomb, R.C. J. Org. Chem. 1971, 36, 3870.
1695. Hori, K.; Kamimura, A.; Ando, K.; Mizumura, M.; Ihara, Y. Tetrahedron 1997, 53, 4317. See Marlier, J.F.; Campbell, E.; Lai, C.; Weber, M.; Reinhardt, L.A.; Cleland, W.W. J. Org. Chem. 2006, 71, 3829.
1696. Khan, M.N.; Olagbemiro, T.O. J. Org. Chem. 1982, 47, 3695.
1697. Eriksson, S.O. Acta Chem. Scand. 1968, 22, 892; Acta Pharm. Suec., 1969, 6, 139.
1698. Menger, F.M.; Donohue, J.A. J. Am. Chem. Soc. 1973, 95, 432; Pollack, R.M.; Dumsha, T.C. J. Am. Chem. Soc. 1973, 95, 4463; Kijima, A.; Sekiguchi, S. J. Chem. Soc. Perkin Trans. 2 1987, 1203.
1699. Schowen, R.L.; Jayaraman, H.; Kershner, L. J. Am. Chem. Soc. 1966, 88, 3373. See also, Bowden, K.; Bromley, K. J. Chem. Soc. Perkin Trans. 2 1990, 2103.
1700. Bennet, A.J.; Slebocka-Tilk, H.; Brown, R.S.; Guthrie, J.P.; Jodhan, A. J. Am. Chem. Soc. 1990, 112, 8497.
1701. See Slebocka-Tilk, H.; Bennet, A.J.; Hogg, H.J.; Brown, R.S. J. Am. Chem. Soc. 1991, 113, 1288; Bennet, A.J.; Slebocka-Tilk, H.; Brown, R.S.; Guthrie, J.P.; Jodhan, A. J. Am. Chem. Soc. 1990, 112, 8497.
1702. See Yates, K.; Stevens, J.B. Can. J. Chem. 1965, 43, 529; Yates, K.; Riordan, J.C. Can. J. Chem. 1965, 43, 2328.
1703. Lacey, R.N. J. Chem. Soc. 1960, 1633; Druet, L.M.; Yates, K. Can. J. Chem. 1984, 62, 2401.
1704. Stodola, F.H. J. Org. Chem. 1972, 37, 178.
1705. See Barnett, J.W.; O'Connor, C.J. J. Chem. Soc., Chem. Commun. 1972, 525; J. Chem. Soc. Perkin Trans. 2 1972, 2378.
1706. Bentley, T.W.; Llewellyn, G.; McAlister, J.A. J. Org. Chem. 1996, 61, 7927.
1707. Bandgar, B.P.; Kamble, V.T.; Sadavarte, V.S.; Uppalla, L.S. Synlett 2002, 735.
1708. See Kaiser, E.M.; Woodruff, R.A. J. Org. Chem. 1970, 35, 1198.
1709. Nagasawa, K.; Yoshitake, S.; Amiya, T.; Ito, K. Synth. Commun. 1990, 20, 2033.
1710. Taylor, E.C.; McLay, G.W.; McKillop, A. J. Am. Chem. Soc. 1968, 90, 2422.
1711. Ghosh, R.; Maiti, S.; Chakraborty, A. Tetrahedron Lett. 2004, 45, 6775.
1712. Illi, V.O. Tetrahedron Lett. 1979, 2431. For another method, see Nekhoroshev, M.V.; Ivakhnenko, E.P.; Okhlobystin, O.Yu. J. Org. Chem. USSR 1977, 13, 608.
1713. Yadav, J.S.; Reddy, G.S.; Svinivas, D.; Himabindu, K. Synth. Commun. 1998, 28, 2337.
1714. Ghosh, R.; Maiti, S.; Chakraborty, A. Tetrahedron Lett. 2005, 46, 147.
1715. Sirkecioglu, O.; Karliga, B.; Talinli, N. Tetrahedron Lett. 2003, 44, 8483.
1716. Srivastava, V.; Tandon, A.; Ray, S. Synth. Commun. 1992, 22, 2703.
1717. Meshram, H.M.; Reddy, G.S.; Bindu, K.H.; Yadav, J.S. Synlett 1998, 877.
1718. Chen, R.; Zhang, Y. Synth. Commun. 2000, 30, 1331.
1719. Liu, Y.; Zhang, Y. Synth. Commun. 1999, 29, 4043.
1720. See Ahmad, S.; Iqbal, J. Chem. Lett. 1987, 953, and references cited therein.
1721. Lakouraj, M.; Movassagh, B.; Fasihi, J. J. Chem. Res. (S) 2001, 378.
1722. Nafion-H has been used: Kumareswaran, R.; Pachamuthu, K.; Vankar, Y.D. Synlett 2000, 1652.
1723. Ce: Dalpozzo, R.; DeNino, A.; Maiuolo, L.; Procopio, A.; Nardi, M.; Bartoli, G.; Romeo, R. Tetrahedron Lett. 2003, 44, 5621. Cu: Saravanan, P.; Singh, V.K. Tetrahedron Lett. 1999, 40, 2611. In: Chakraborti, A.K.; Gulhane, R. Tetrahedron Lett. 2003, 44, 6749. Li: Nakae, Y.; Kusaki, I.; Sato, T. Synlett 2001, 1584. Mg: Bartoli, G.; Bosco, M.; Dalpozzo, R.; Marcantoni, E.; Massaccesi, M.; Sambri, L. Eur. J. Org. Chem. 2003, 4611. Ru: De, S.K. Tetrahedron Lett. 2004, 45, 2919. Ti: Chandrasekhar, S.; Ramachandar, T.; Reddy, M.V.; Takhi, M. J. Org. Chem. 2000, 65, 4729. Yb: Dumeunier, R.; Markó, I.E. Tetrahedron Lett. 2004, 45, 825.
1724. For a list of catalysts, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1955–1957.
1725. Constantinou-Kokotou, V.; Peristeraki, A. Synth. Commun. 2004, 34, 4227. Also see Bandgar, B.P.; Kasture, S.P.; Kamble, V.T. Synth. Commun. 2001, 31, 2255.
1726. Clarke, P.A.; Kayaleh, N.E.; Smith, M.A.; Baker, J.R.; Bird, S.J.; Chan, C. J. Org. Chem. 2002, 67, 5226; Clarke, P.A. Tetrahedron Lett. 2002, 43, 4761.
1727. Sakakura, A.; Kawajiri, K.; Ohkubo, T.; Kosugi, Y.; Ishihara, K. J. Am. Chem. Soc. 2007, 129, 14775. See Scriven, E.F.V. Chem. Soc. Rev. 1983, 12, 129; Höfle, G.; Steglich, W.; Vorbrüggen, H. Angew. Chem. Int. Ed. 1978, 17, 569.
1728. Birman, V.B.; Li, X.; Han, Z. Org. Lett. 2007, 9, 37.
1729. See van Es, A.; Stevens, W. Recl. Trav. Chim. Pays-Bas 1965, 84, 704.
1730. See Sofuku, S.; Muramatsu, I.; Hagitani, A. Bull. Chem. Soc. Jpn. 1967, 40, 2942.
1731. See Chen, Y.; Tian, S.-K.; Deng, L. J. Am. Chem. Soc. 2000, 122, 9542.
1732. Chen, Y.; McDaid, P.; Deng, L. Chem. Rev. 2003, 103, 2965.
1733. Fatiadi, A.J. Carbohydr. Res. 1968, 6, 237.
1734. Temperini, A.; Annesi, D.; Testaferri, L.; Tiecco, M. Tetrahedron Lett. 2010, 51, 5368.
1735. Ayers, J.T.; Anderson, S.R. Synth. Commun 1999, 29, 351. See Movassagh, B.; Lakouraj, M.M.; Fadaei, Z. J. Chem. Res. (S) 2001, 22.
1736. For a review of some methods, see Haslam, E. Tetrahedron 1980, 36, 2409.
1737. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1932–1941.
1738. Nascimento, M.de G.; Zanotto, S.P.; Scremin, M.; Rezende, M.C. Synth. Commun. 1996, 26, 2715.
1739. Newman, M.S. An Advanced Organic Laboratory Course, Macmillan, NY, 1972, pp. 8–10.
1740. See Werner, W. J. Chem. Res. (S) 1980, 196; Hill, D.R.; Hsiao, C.-N.; Kurukulasuriya, R.; Wittenberger, S.J. Org. Lett. 2002, 4, 111.
1741. Johnston, B.H.; Knipe, A.C.; Watts, W.E. Tetrahedron Lett. 1979, 4225.
1742. See Ishihara, K.; Nakagawa, S.; Sakakura, A. J. Am. Chem. Soc. 2005, 127, 4168.
1743. Houston, T.A.; Wilkinson, B.L.; Blanchfield, J.T. Org. Lett. 2004, 6, 679.
1744. Das, B.; Venkataiah, B.; Madhsudhan, P. Synlett 2000, 59.
1745. Wakasugi, K.; Misaki, T.; Yamada, K.; Tanabe, Y. Tetrahedron Lett. 2000, 41, 5249.
1746. Lee, A.S.-Y.; Yang, H.-C.; Su, F.-Y. Tetrahedron Lett. 200142, 301.
1747. Hwu, J.R.; Hsu, C.-Y.; Jain, M.L. Tetrahedron Lett. 200445, 5151.
1748. McNulty, J.; Cheekoori, S.; Nair, J.J.; Larichev, V.; Capretta, A.; Robertson, A.J. Tetrahedron Lett. 2005, 46, 3641; Yoshino, T.; Imori, S.; Togo, H. Tetrahedron 2006, 62, 1309.
1749. Eshghi, H.; Rafei, M.; Karimi, M.H. Synth. Commun. 2001, 31, 771.
1750. Srinivas, K.V.N.S.; Mahender, I.; Das, B. Synlett 2003, 2419.
1751. Bosco, J.W.J.; Agrahari, A.; Saikia, A.K. Tetrahedron Lett. 2006, 47, 4065.
1752. Crosignani, S.; White, P.D.; Linclau, B. Org. Lett. 2002, 4, 2961.
1753. See Crosignani, S.; White, P.D.; Linclau, B. J. Org. Chem. 2004, 69, 5897.
1754. Chen, C.-T.; Munot, Y.S. J. Org. Chem. 2005, 70, 8625.
1755. Velusamy, S.; Borpuzari, S.; Punniyamurthy, T. Tetrahedron 2005, 61, 2011.
1756. Sedighi, M.; Çalimsiz, S.; Lipton, M.A. J. Org. Chem. 2006, 71, 9517.
1757. Salomé, C.; Kohn, H. Tetrahedron 2009, 65, 456.
1758. Vorbrüggen, H. Synlett 2008, 1603.
1759. Iranpoor, N.; Firouzabadi, H.; Khalili, D.; Motevalli, S. J. Org. Chem. 2008, 73, 4882.
1760. Wolfe, J.F.; Ogliaruso, M.A. in Patai, S. The Chemistry of Acid Derivatives, pt. 2, Wiley, NY, 1979, pp. 1062–1330. For a list of methods for converting hydroxy acids to lactones, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1989, pp. 1861–1867.
1761. Adam, W.; Baeza, J.; Liu, J. J. Am. Chem. Soc. 1972, 94, 2000. Also see Merger, F. Chem. Ber. 1968, 101, 2413; Blume, R.C. Tetrahedron Lett. 1969, 1047.
1762. Lardelli, G.; Lamberti,V.; Weller, W.T.; de Jonge, A.P. Recl. Trav. Chim. Pays-Bas 1967, 86, 481.
1763. See Parenty, A.; Moreau, X.; Campagne, J.-M. Chem. Rev. 2006, 106, 911.
1764. Wollenberg, R.H.; Nimitz, J.S.; Gokcek, D.Y. Tetrahedron Lett. 1980, 21, 2791; Thalmann, A.; Oertle, K.; Gerlach, H. Org. Synth. VII, 470. See also, Schmidt, U.; Heermann, D. Angew. Chem. Int. Ed. 1979, 18, 308; Trost, B.M.; Chisholm, J.D. Org. Lett. 2002, 4, 3743.
1765. See Mukaiyama, T. Angew. Chem. Int. Ed. 1979, 18, 707; Convers, E.; Tye, H.; Whittaker, M. Tetrahedron Lett. 2004, 45, 3401. For a microwave-assisted reaction, see Donati, D.; Morelli, C.; Taddei, M. Tetrahedron Lett.2005, 46, 2817.
1766. Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc. Jpn. 1979, 52, 1989; Mundy, B.P.; Ellerd, M.G.; Favaloro, Jr., F.G. Name Reactions and Reagents in Organic Synthesis, 2nd ed., Wiley–Interscience, New Jersey, 2005, pp. 710–711. For a discussion of the mechanism, see Dhimitruka, I.; SantaLucia, Jr., J. Org. Lett. 2006, 8, 47.
1767. See Salomaa, P.; Kankaanperä, A.; Pihlaja, K. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 466–481.
1768. Balcom, B.J.; Petersen, N.O. J. Org. Chem. 1989, 54, 1922.
1769. Doleschall, G.; Lempert, K. Tetrahedron Lett. 1963, 1195.
1770. Shimizu, T.; Hiramoto, K.; Nakata, T. Synthesis 2001, 1027.
1771. Keck, G.E.; Sanchez, C.; Wager, C.A. Tetrahedron Lett. 2000, 41, 8673.
1772. Nahmany, M.; Melman, A. Org. Lett. 2001, 3, 3733.
1773. See Arrieta, A.; García, T.; Lago, J.M.; Palomo, C. Synth. Commun. 1983, 13, 471.
1774. Boden, E.P.; Keck, G.E. J. Org. Chem. 1985, 50, 2394.
1775. Brook, M.A.; Chan, T.H. Synthesis 1983, 201.
1776. See Staab, H.A.; Rohr, W. Newer Methods Prep. Org. Chem. 1968, 5, 61. See also, Morton, R.C.; Mangroo, D.; Gerber, G.E. Can. J. Chem. 1988, 66, 1701.
1777. See Kadaba, P.K. Synth. Commun. 1974, 4, 167.
1778. Bi: Carrigan, D.; Freiberg, D.A.; Smith, R.C.; Zerth, H.M.; Mohan, R.S. Synthesis 2001, 2091; Mohammadpoor-Baltork, I.; Khosropour, A.R.; Aliyan, H. J. Chem. Res. 2001, 280. Ce: Pan, W.-B.; Chang, F.-R.; Wei, L.-M.; Wu, M.J.; Wu, Y.-C. Tetrahedron Lett. 2003, 44, 331. Fe: Sharma, G.V.M.; Mahalingam, A.K.; Nagarajan, M.; Ilangovan, P.; Radhakrishna, P. Synlett 1999, 1200; Zhang, G.-S. Synth. Commun. 1999, 29, 607.. Hf: Ishihara, K.; Nakayama, M.; Ohara, S.; Yamamoto, H. Tetrahedron 2002, 58, 8179;
1779. Derevitskaya, V.A.; Klimov, E.M.; Kochetkov, N.K. Tetrahedron Lett. 1970, 4269. See also, Mohacsi, E. Synth. Commun. 1982, 12, 453.
1780. Sudalai, A.; Kanagasabapathy, S.; Benicewicz, B.C. Org. Lett. 2000, 2, 3213.
1781. Curphey, T.J. Tetrahedron Lett. 2002, 43, 371.
1782. Iimura, S.; Manabe, K.; Kobayashi, S. Chem. Commun. 2002, 94.
1783. Otera, J. Chem. Rev. 1993, 93, 1449.
1784. For a list of catalysts, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1969–1973.
1785. See Chavan, S.P.; Subbarao, Y.T.; Dantale, S.W.; Sivappa, R. Synth. Commun. 2001, 31, 289.
1786. Stanton, M.G.; Gagné, M.R. J. Org. Chem. 1997, 62, 8240; Vasin, V.A.; Razin, V.V. Synlett 2001, 658.
1787. See Imwinkelried, R.; Schiess, M.; Seebach, D. Org. Synth., 65, 230; Bandgar, B.P.; Uppalla, L.S.; Sadavarte, V.S. Synlett 2001, 1715.
1788. See Bose, D.S.; Satyender, A.; Rudra Das, A.P.; Mereyala, H.B. Synthesis 2006, 2392.
1789. For a review see Grasa, G.A.; Singh, R.; Nolan, S.P. Synthesis 2004, 971.
1790. Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. Synlett 2001, 1338; Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. Synth. Commun. 2001, 31, 2063; Štefane, B.; Ko![]() evar, M.; Polanc, S. Synth. Commun. 2002, 32, 1703.
evar, M.; Polanc, S. Synth. Commun. 2002, 32, 1703.
1791. Ishihara, K.; Niwa, M.; Kosugi, Y. Org. Lett. 2008, 10, 2187.
1792. Hagiwara, H.; Koseki, A.; Isobe, K.; Shimizu, K.-i.; Hoshi, T.; Suzuki, T. Synlett 2004, 2188.
1793. See Shirae, Y.; Mino, T.; Hasegawa, T.; Sakamoto, M.; Fujita, T. Tetrahedron Lett. 2005, 46, 5877.
1794. Yamada, S. Tetrahedron Lett. 1992, 33, 2171. See also, Costa, A.; Riego, J.M. Can. J. Chem. 1987, 65, 2327.
1795. Wong, C.H.; Whitesides, G. M. in Baldwin, J.E. Enzymes in Synthetic Organic Chemistry, Tetrahedron Organic Chemistry Series Vol. 12, Pergamon Press, NY, 1994; Faber, K. Biotransformations in Organic Chemistry. A Textbook, 2nd ed, Springer–Verlag, NY, 1995; Córdova, A.; Janda, K.D. J. Org. Chem. 2001, 66, 1906; Ciuffreda, P.; Casati, S.; Santaniello, E. Tetrahedron Lett. 2003, 44, 3663.
1796. Anand, R.C.; Sevlapalam, N. Synth. Commun. 1994, 24, 2743.
1797. Barry, J.; Bram, G.; Petit, A. Tetrahedron Lett. 1988, 29, 4567. See also, Nishiguchi, T.; Taya, H. J. Chem. Soc. Perkin Trans. 1 1990, 172.
1798. Ilankumaran, P.; Verkade, J.G. J. Org. Chem. 1999, 64, 3086.
1799. Ooi, T.; Sugimoto, H.; Maruoka, K. Heterocycles 2001, 54, 593.
1800. Joshi, U.M.; Patkar, L.N.; Rajappa, S. Synth. Commun. 2004, 34, 33.
1801. See Koskikallio, E.A. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 103–136.
1802. Ilankumaran, P.; Verkade, J.G. J. Org. Chem. 1999, 64, 9063.
1803. Grasa, G.A.; Kissling, R.M.; Nolan, S.P. Org. Lett. 2002, 4, 3583.
1804. Bosco, J.W.J.; Saikia, A.K. Chem. Commun. 2004, 1116.
1805. See House, H.O.; Trost, B.M. J. Org. Chem. 1965, 30, 2502.
1806. See Mondal, M.A.S.; van der Meer, R.; German, A.L.; Heikens, D. Tetrahedron 1974, 30, 4205.
1807. Henry, P.M. J. Am. Chem. Soc. 1971, 93, 3853; Acc. Chem. Res. 1973, 6, 16.
1808. Das, B.; Venkataiah, B. Synthesis 2000, 1671.
1809. Chavan, S.P.; Kale, R.R.; Shivasankar, K.; Chandake, S.I.; Benjamin, S.B. Synthesis 2003, 2695.
1810. For example, see Czarnik, A.W. Tetrahedron Lett. 1984, 25, 4875. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 197–1978.
1811. DeLuca, L.; Giacomelli, G.; Porcheddu, A. J. Org. Chem. 2002, 67, 5152.
1812. Charette, A.B.; Chua, P. Synlett 1998, 163.
1813. Anelli, P.L.; Brocchetta, M.; Palano, D.; Visigalli, M. Tetrahedron Lett. 1997, 38, 2367.
1814. Kiessling, A.J.; McClure, C.K. Synth. Commun. 1997, 27, 923.
1815. Glatzhofer, D.T.; Roy, R.R.; Cossey, K.N. Org. Lett. 2002, 4, 2349. See Naik, R.; Pasha, M.A. Synth. Commun. 2005, 35, 2823.
1816. See Yamaguchi, J.-i.; Aoyagi, T.; Fujikura, R.; Suyama, T. Chem. Lett. 2001, 466.
1817. Fisher, L.E.; Caroon, J.M.; Stabler, S.R.; Lundberg, S.; Zaidi, S.; Sorensen, C.M.; Sparacino, M.L.; Muchowski, J.M. Can. J. Chem. 1994, 72, 142.
1818. Orita, A.; Nagano, Y.; Hirano, J.; Otera, J. Synlett 2001, 637.
1819. Serieys, A.; Botuha, C.; Chemla, F.; Ferreira, F.; Pérez-Luna, A. Tetrahedron Lett. 2008, 49, 5322.
1820. Srivastava, R.R.; Kabalka, G.W. Tetrahedron Lett. 1992, 33, 593.
1821. Fife, W.K.; Zhang, Z. J. Org. Chem. 1986, 51, 3744. For a review of acetic formic anhydride see Strazzolini, P.; Giumanini, A.G.; Cauci, S. Tetrahedron 1990, 46 1081.
1822. Plusquellec, D.; Roulleau, F.; Lefeuvre, M.; Brown, E. Tetrahedron 1988, 44, 2471; Wang, J.; Hu, Y.; Cui, W. J. Chem. Res. (S) 1990, 84.
1823. Hu, Y.; Wang, J.-X.; Li, S. Synth. Commun. 1997, 27, 243.
1824. For lists of other dehydrating agents with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1930–1932; Ogliaruso, M.A.; Wolfe, J.F. in Patai, S. The Chemistry of Acid Derivatives, pt.1, Wiley, NY, 1979, pp. 437–438.
1825. See Rammler, D.H.; Khorana, H.G. J. Am. Chem. Soc. 1963, 85, 1997. See also, Hata, T.; Tajima, K.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 1968, 41, 2746.
1826. Kim, J.; Jang, D.O. Synth. Commun. 2001, 31, 395.
1827. Villemin, D.; Labiad, B.; Loupy, A. Synth. Commun. 1993, 23, 419.
1828. Rinderknecht, H.; Ma, V. Helv. Chim. Acta 1964, 47, 152. See also, Nangia, A.; Chandrasekaran, S. J. Chem. Res. (S) 1984, 100.
1829. Weiss, J.; Havelka, F.; Nefedov, B.K. Bull. Acad. Sci. USSR Div. Chem. Sci. 1978, 27, 193.
1830. Avison, A.W.D. J. Chem. Soc. 1955, 732.
1831. Karger, M.H.; Mazur, Y. J. Org. Chem. 1971, 36, 528.
1832. See Satchell, D.P.N. Q. Rev. Chem. Soc. 1963, 17, 160, pp. 182–184.
1833. See Scheithauer, S.; Mayer, R. Top. Sulfur Chem. 1979, 4, 1.
1834. Ahmad, S.; Iqbal, J. Tetrahedron Lett. 1986, 27, 3791.
1835. Hirabayashi, Y.; Mizuta, M.; Mazume, T. Bull. Chem. Soc. Jpn. 1965, 38, 320.
1836. Nomura, R.; Miyazaki, S.; Nakano, T.; Matsuda, H. Chem. Ber. 1990, 123, 2081.
1837. Imamoto, T.; Kodera, M.; Yokoyama, M. Synthesis 1982, 134. See also, Dellaria, Jr., F.F.; Nordeen, C.; Swett, L.R. Synth. Commun. 1986, 16, 1043.
1838. Rao, Y.; Li, X.; Nagorny, P.; Hayashida, J.; Danishefsky, S.J. Tetrahedron Lett. 2009, 50, 6684.
1839. Mukaiyama, T.; Takeda, T.; Atsumi, K. Chem. Lett. 1974, 187. See also, Hatch, R.P.; Weinreb, S.M. J. Org. Chem. 1977, 42, 3960; Cohen, T.; Gapinski, R.E. Tetrahedron Lett. 1978, 4319.
1840. Gauthier, J.Y.; Bourdon, F.; Young, R.N. Tetrahedron Lett. 1986, 27, 15.
1841. See Knipe, A.C. J. Chem. Soc. Perkin Trans. 2 1973, 589.
1842. Eldred, S.E.; Stone, D.A.; Gellman, S.H.; Stahl, S.S. J. Am. Chem. Soc. 2003, 125, 3422.
1843. Shapiro, G.; Marzi, M. J. Org. Chem. 1997, 62, 7096.
1844. Roos, E.C.; Bernabé, P.; Hiemstra, H.; Speckamp, W.N.; Kaptein, B.; Boesten, W.H.J. J. Org. Chem. 1995, 60, 1733.
1845. El Kaim, L.; Grimaud, L.; Lee, A.; Perroux, Y.; Tiria, C. Org. Lett. 2004, 6, 381.
1846. Bon, E.; Bigg, D.C.H.; Bertrand, G. J. Org. Chem. 1994, 59, 4035.
1847. Sibi, M.P.; Hasegawa, H.; Ghorpade, S.R. Org. Lett. 2002, 4, 3343.
1848. Ley, S.V.; Leach, A.G.; Storer, R.I. J. Chem. Soc. Perkin Trans. 1 2001, 358.
1849. Mohammadpoor-Baltork, I.; Khodaei, M.M.; Nikoofar, K. Tetrahedron Lett. 2003, 44, 591.
1850. Inamoto, K.; Shiraishi, M.; Hiroya, K.; Doi, T. Synthesis 2010, 3087.
1851. Saravanan, V.; Mukherjee, C.; Das, S.; Chandrasekaran, S. Tetrahedron Lett. 2004, 45, 681.
1852. You, H.-W.; Lee, K.-J. Synlett 2001, 105.
1853. Telvekar, V.N.; Chettiar, S.N. Tetrahedron Lett. 2007, 48, 4529.
1854. Cho, C.-G.; Park, J.-S.; Jung, I.-H.; Lee, H. Tetrahedron Lett. 2001, 42, 1065.
1855. See Challis, M.S.; Butler, A.R. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 279–290.
1856. See Beckwith, A.L.J. in Zabicky, J.The Chemistry of Amides, Wiley, NY, 1970, pp. 73–185; Jedrzejczak, M.; Motie, R.E.; Satchell, D.P.N. J. Chem. Soc. Perkin Trans. 2 1993, 599.
1857. See Bhattacharyya, S.; Gooding, O.W.; Labadie, J. Tetrahedron Lett. 2003, 44, 6099.
1858. Reddy, A.S.; Kumar, M.S.; Reddy, G.R. Tetrahedron Lett. 2000, 41, 6285.
1859. Meshram, H.M.; Reddy, G.S.; Reddy, M.M.; Yadav, J.S. Tetrahedron Lett. 1998, 39, 4103.
1860. Hajipour, A.R.; Mazloumi, Gh. Synth. Commun. 2002, 32, 23.
1861. Cho, D.H.; Jang, D.O. Tetrahedron Lett. 2004, 45, 2285.
1862. Shi, F.; Li, J.; Li, C.; Jia, X. Tetrahedron Lett. 2010, 51, 6049.
1863. Ghosh, R.; Maiti, S.; Chakraborty, A. Tetrahedron Lett. 2004, 45, 6775.
1864. Lee, W.S.; Park, K.H.; Yoon, Y.-J. Synth. Commun. 2000, 30, 4241.
1865. Kim, J.-G.; Jang, D.O. Synlett 2010, 2093. For other formylation reactions, see Shekhar, A.C.; Kumar, A.R.; Sathaiah, G.; Paul, V.L.; Sridhar, M.; Rao, P.S. Tetrahedron Lett. 2009, 50, 7099; Brahmachari, G.; Laskar, S. Tetrahedron Lett. 2010, 51, 2319; Rahman, M.; Kundu, D.; Hajra, A.; Majee, A. Tetrahedron Lett. 2010, 51, 2896; Deutsch, J.; Eckelt, R.; Köckritz, A.; Martin, A. Tetrahedron 2009, 65, 10365.
1866. See Paulsen, H.; Stoye, D. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 515–600.
1867. For an improved method, see Ando, W.; Tsumaki, H. Synth. Commun. 1983, 13, 1053.
1868. Richter, R.; Ulrich, H. pp. 619–818, and Drobnica, L.; Kristián, P.; Augustín, J. pp. 1003–1221, in Patai, S. The Chemistry of Cyanates and Their Thio Derivatives, pt. 2, Wiley, NY, 1977.
1869. See Ozaki, S. Chem. Rev. 1972, 72, 457, see pp. 457–460. For a review of the industrial preparation of isocyanates by this reaction, see Twitchett, H.J. Chem. Soc. Rev. 1974, 3, 209.
1870. For a review of thiophosgene, see Sharma, S. Sulfur Rep. 1986, 5, 1.
1871. Kurita, K.; Iwakura, Y. Org. Synth. VI, 715.
1872. Heydari, A.; Shiroodi, R.K.; Hamadi, H.; Esfandyari, M.; Pourayoubi, M. Tetrahedron Lett. 2007, 48, 5865; Upadhyaya, D.J.; Barge, A.; Stefania, R.; Cravotto, G. Tetrahedron Lett. 2007, 48, 8318; Shrikhande, J.J.; Gawande, M.B.; Jayaram, R.V. Tetrahedron Lett. 2008, 49, 4799. See Vilaivan, T. Tetrahedron Lett. 2006, 47, 6739.
1873. See Yasuhara, T.; Nagaoka, Y.; Tomioka, K. J. Chem. Soc. Perkin Trans. 1 1999, 2233.
1874. Greene, T.W. Protective Groups in Organic Synthesis Wiley, NY, 1980, pp 222–248, 324–326; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis, 2nd ed., Wiley, NY, 1991, pp 327–330; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis, 3rd ed., Wiley, NY, 1999, pp 518–525; 737–739.
1875. Kivinen, A. in Patai, S. The Chemistry of Acyl Halides, Wiley, NY, 1972; Bender, M.L.; Jones, M.J. J. Org. Chem. 1962, 27, 3771. See also, Song, B.D.; Jencks, W.P. J. Am. Chem. Soc. 1989, 111, 8479.
1876. Lemoucheux, L.; Seitz, T.; Rouden, J.; Lasne, M.-C. Org. Lett. 2004, 6, 3703.
1877. For a discussion of the mechanism, see Kluger, R.; Hunt, J.C. J. Am. Chem. Soc. 1989, 111, 3325.
1878. See Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 86–96. See also, Naik, S.; Bhattacharjya, G.; Talukdar, B.; Patel, B.K. Eur. J. Org. Chem. 2004, 1254.
1879. Dave, P.R.; Kumar, K.A.; Duddu, R.; Axenrod, T.; Dai, R.; Das, K.K.; Guan, X.-P.; Sun, J.; Trivedi, N.J.; Gilardi, R.D. J. Org. Chem. 2000, 65, 1207.
1880. Anuradha, M.V.; Ravindranath, B. Tetrahedron 1997, 53, 1123.
1881. See Wheeler, O.H.; Rosado, O. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 335–381; Hargreaves, M.K.; Pritchard, J.G.; Dave, H.R. Chem. Rev. 1970, 70, 439 (cyclic imides).
1882. Tsubouchi, H.; Tsuji, K.; Ishikawa, H. Synlett 1994, 63.
1883. Kacprzak, K. Synth. Commun. 2003, 33, 1499.
1884. Le, Z.-G.; Chen, Z.-C.; Hu, Y.; Zheng, Q.-G. Synthesis 2004, 995.
1885. Martin, B.; Sekljic, H.; Chassaing, C. Org. Lett. 2003, 5, 1851.
1886. Eaton, J.T.; Rounds, W.D.; Urbanowicz, J.H.; Gribble, G.W. Tetrahedron Lett. 1988, 29, 6553.
1887. Vlietstra, E.J.; Zwikker, J.W.; Nolte, R.J.M.; Drenth, W. Recl. Trav. Chim. Pays-Bas 1982, 101, 460.
1888. Barrett, A.G.M.; Lana, J.C.A. J. Chem. Soc., Chem. Commun. 1978, 471.
1889. Vauthey, I.; Valot, F.; Gozzi, C.; Fache, F.; Lemaire, M. Tetrahedron Lett. 2000, 41, 6347.
1890. Singh, D.U.; Singh, P.R.; Samant, S.D. Tetahedron Lett. 2004, 45, 4805.
1891. Martin, M.T.; Roschangar, F.; Eaddy, J.F. Tetrahedron Lett. 2003, 44, 5461.
1892. Arseniyadis, S.; Subhash, P.V.; Valleix, A.; Mathew, S.P.; Blackmond, D.G.; Wagner, A.; Mioskowski, C. J. Am. Chem. Soc. 2005, 127, 6138.
1893. See Gooßen, L.J.; Ohlmann, D.M.; Lange, P.P. Synthesis 2009, 160.
1894. See Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 105–109.
1895. Chou, W.-C.; Chou, M.-C.; Lu, Y.-Y.; Chen, S.-F. Tetrahedron Lett. 1999, 40, 3419. Also see White, J.M.; Tunoori, A.R.; Turunen, B.J.; Georg, G.I J. Org. Chem. 2004, 69, 2573.
1896. Ishihara, K.; Kondo, S.; Yamamoto, H. Synlett 2001, 1371.
1897. Crosignani, S.; Gonzalez, J.; Swinnen, D. Org. Lett. 2004, 6, 4579; Chichilla, R.; Dodsworth, D.J.; Nájera, C.; Soriano, J.M. Tetrahedron Lett. 2003, 44, 463.
1898. da C. Rodrigues, R.; Barros, I.M.A.; Lima, E.L.S. Tetrahedron Lett. 2005, 46, 5945.
1899. Wodka, D.; Robbins, M.; Lan, P.; Martinez, R.L.; Athanasopoulos, J.; Makara, G.M. Tetrahedron Lett. 2006, 47, 1825.
1900. Khalafi-Nezhad, A.; Mokhtari, B.; Rad, M.N.S. Tetrahedron Lett. 2003, 44, 7325; Perreux, L.; Loupy, A.; Volatron, F. Tetrahedron 2002, 58, 2155. See also, Bose, A.K.; Ganguly, S.N.; Manhas, M.S.; Guha, A.; Pombo-Villars, E. Tetrahedron Lett. 2006, 47, 4605.
1901. De Luca, L.; Giacomelli, G.; Porcheddu, A.; Salaris, M. Synlett 2004, 2570.
1902. Peng, Y.; Song, G. Org. Prep. Proceed. Int. 2002, 34, 95.
1903. Hosseini-Sarvari, M.; Sharghi, H. J. Org. Chem. 2006, 71, 6652.
1904. See Bladé-Font, A. Tetrahedron Lett. 1980, 21, 2443. Also see Wei, Z.-Y.; Knaus, E.E. Tetrahedron Lett. 1993, 34, 4439 for a variation of this reaction.
1905. Gutman, A.L.; Meyer, E.; Yue, X.; Abell, C. Tetrahedron Lett. 1992, 33, 3943.
1906. Bosch, I.; Romea, P.; Urpí, F.; Vilarrasa, J. Tetrahedron Lett. 1993, 34, 4671. See Bai, D.; Shi, Y. Tetrahedron Lett. 1992, 33, 943 for the preparation of lactam units in p-cyclophanes.
1907. See Klausner, Y.S.; Bodansky, M. Synthesis 1972, 453.
1908. It was first used this way by Sheehan, J.C.; Hess, G.P. J. Am. Chem. Soc. 1955, 77, 1067.
1909. See Gross, E.; Meienhofer, J. The Peptides, 3 Vols., Academic Press, NY, 1979–1981. See Bodanszky, M.; Bodanszky, A. The Practice of Peptide Synthesis, Springer, NY, 1984.
1910. Feuerstein, M.; Doucet, H.; Santelli, M. Tetrahedron Lett. 2001, 42, 6667.
1911. See Rebek, J.; Feitler, D. J. Am. Chem. Soc. 1974, 96, 1606. Also see Rebek, J.; Feitler, D. J. Am. Chem. Soc. 1973, 95, 4052.
1912. Banwell, M.; Smith, J. Synth. Commun. 2001, 31, 2011. For another procedure, see Kim, M.; Lee, H.; Han, K.-J.; Kay, K.-Y. Synth. Commun. 2003, 33, 4013.
1913. Shiina, I.; Suenaga, Y.; Nakano, M.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 2000, 73, 2811.
1914. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1941–1949.
1915. Ishihara, K.; Ohara, S.; Yamamoto, H. J. Org. Chem. 1996, 61, 4196.
1916. See Vaidyanathan, R.; Kalthod, V.G.; Ngo, D.; Manley, J.M.; Lapekas, S.P. J. Org. Chem. 2004, 69, 2565. Also see Grzyb, J.A.; Batey, R.A. Tetrahedron Lett. 2003, 44, 7485.
1917. Klosa, J. J. Prakt. Chem. 1963, [4] 19, 45.
1918. Wilson, J.D.; Weingarten, H. Can. J. Chem. 1970, 48, 983.
1919. Cossy, J.; Pale-Grosdemange, C. Tetrahedron Lett. 1989, 30, 2771.
1920. Thorsen, M.; Andersen, T.P.; Pedersen, U.; Yde, B.; Lawesson, S. Tetrahedron 1985, 41, 5633.
1921. Jászay, Z.M.; Petneházy, I.; Töke, L. Synth. Commun. 1998, 28, 2761.
1922. Higuchi, T.; Miki, T.; Shah, A.C.; Herd, A.K. J. Am. Chem. Soc. 1963, 85, 3655.
1923. For example, see Schindbauer, H. Monatsh. Chem. 1968, 99, 1799.
1924. Zhmurova, I.N.; Voitsekhovskaya, I.Yu.; Kirsanov, A.V. J. Gen. Chem. USSR 1959, 29, 2052. See also, Liu, H.; Chan, W.H.; Lee, S.P. Synth. Commun. 1979, 9, 31.
1925. Pelter, A.; Levitt, T.E.; Nelson, P. Tetrahedron 1970, 26, 1539; Pelter, A.; Levitt, T.E. Tetrahedron 1970, 26, 1545, 1899.
1926. Sanchez, R.; Vest, G.; Despres, L. Synth. Commun. 1989, 19, 2909.
1927. Park, S.-D.; Oh, J.-H.; Lim, D. Tetrahedron Lett. 2002, 43, 6309.
1928. Merrifield, R.B. J. Am. Chem. Soc. 1963, 85, 2149.
1929. Birr, C. Aspects of the Merrifield Peptide Synthesis, Springer, NY, 1978. For reviews, see Bayer, E. Angew. Chem. Int. Ed. 1991, 30, 113; Kaiser, E.T. Acc. Chem. Res. 1989, 22, 47; Jacquier, R. Bull. Soc. Chim. Fr. 1989, 220; Barany, G.; Kneib-Cordonier, N.; Mullen, D.G. Int. J. Pept. Protein Res. 1987, 30, 705; Andreev, S.M.; Samoilova, N.A.; Davidovich, Yu.A.; Rogozhin, S.V. Russ. Chem. Rev. 1987, 56, 366; Gross, E.; Meienhofer, J. The Peptides, Vol. 2, Academic Press, NY, 1980, the articles by Barany, G.; Merrifield, R.B. pp. 1–184; Fridkin, M. pp. 333–363; Erickson, B.W.; Merrifield, R.B. in Neurath, H.; Hill, R.L.; Boeder, C.-L. The Proteins, 3rd ed., Vol. 2, Academic Press, NY, 1976, pp. 255–527. For R. B. Merrifield's Nobel Prize lecture, see Merrifield, R.B. Angew. Chem. Int. Ed. 1985, 24, 799.
1930. Laszlo, P. Preparative Organic Chemistry Using Supported Reagents, Academic Press, NY, 1987; Mathur, N.K.; Narang, C.K.; Williams, R.E. Polymers as Aids in Organic Chemistry, Academic Press, NY 1980; Hodge, P.; Sherrington, D.C. Polymer-Supported Reactions in Organic Synthesis, Wiley, NY, 1980. For reviews, see Pillai, V.N.R.; Mutter, M. Top. Curr. Chem. 1982, 106, 119; Akelah, A.; Sherrington, D.C. Chem. Rev. 1981, 81, 557; Akelah, A. Synthesis 1981, 413; Rebek, J. Tetrahedron 1979, 35, 723; McKillop, A.; Young, D.W. Synthesis 1979, 401, 481; Crowley, J.I.; Rapoport, H. Acc. Chem. Res. 1976, 9, 135; Patchornik, A.; Kraus, M.A. Pure Appl. Chem.1975, 43, 503.
1931. See Whitney, D.B.; Tam, J.P.; Merrifield, R.B. Tetrahedron 1984, 40, 4237.
1932. Merrifield, R.B.; Stewart, J.M.; Jernberg, N. Anal. Chem. 1966, 38, 1905.
1933. See Schnorrenberg, G.; Gerhardt, H. Tetrahedron 1989, 45, 7759.
1934. For a review, see Bannwarth, W. Chimia 1987, 41, 302.
1935. Fréchet, J.M.J. Tetrahedron 1981, 37, 663; Fréchet, J.M.J. in Hodge, P.; Sherrington, D.C. Polymer-Supported Reactions in Organic Synthesis, Wiley, NY, 1980, pp. 293–342, Leznoff, C.C. Acc. Chem. Res. 1978, 11, 327; Chem. Soc. Rev. 1974, 3, 64.
1936. Czarnik, A.W.; DeWitt, S.H. A Practical Guide to Combinatorial Chemistry, American Chemical Society, Washington, D.C., 1997; Chaiken, I.N.; Janda, K.D. Molecular Diversity and Combinatorial Chemistry: Libraries and Drug Discovery, American Chemical Society, Washington, D.C; 1996; Balkenhol, F.; von dem Bussche-Hünnefeld, C.; Lansky, A.; Zechel, C. Angew. Chem. Int. Ed. 1996, 35, 2289; Thompson, L.A.; Ellman, J.A. Chem. Rev.1996, 96, 555; Crowley, J.I.; Rapoport, H. Acc. Chem. Res. 1976, 9, 135; Leznoff, C.C. Acc. Chem. Res. 1978, 11, 327.
1937. Beckwith, A.L.J. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 96–105. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1973–1976. See Sabot, C.; Kumar, K.A.; Meunier, S.; Mioskowski, C. Tetrahedron Lett. 2007, 48, 3863.
1938. See Mizuhara, T.; Hioki, K.; Yamada, M.; Sasaki, H.; Morisaki, D.; Kunishima, M. Chem. Lett. 2008, 37, 1190. Magnesium nitride is a useful source of ammonia in this reaction. See Veitch, G.E.; Bridgwood, K.L.; Ley, S.V. Org. Lett. 2008, 10, 3623.
1939. Xu, D.; Prasad, K.; Repic, O.; Blacklock, T.J. Tetrahedron Lett. 1995, 36, 7357.
1940. See Matsumoto, K.; Hashimoto, S.; Uchida, T.; Okamoto, T.; Otani, S. Chem. Ber. 1989, 122, 1357.
1941. Högberg, T.; Ström, P.; Ebner, M.; Rämsby, S. J. Org. Chem. 1987, 52, 2033.
1942. Guo, Z.; Dowdy, E.D.; Li, W.-S.; Polniaszek, R.; Delaney, E. Tetrahedron Lett. 2001, 42, 1843.
1943. Ranu, B.C.; Dutta, P. Synth. Commun. 2003, 33, 297.
1944. Matsumoto, K.; Hashimoto, S.; Uchida, T.; Okamoto, T.; Otani, S. Chem. Ber. 1989, 122, 1357.
1945. Varma, R.S.; Naicker, K.P. Tetrahedron Lett. 1999, 40, 6177.
1946. Zradni, F.-Z.; Hamelin, J.; Derdour, A. Synth. Commun. 2002, 32, 3525.
1947. See Wang, J.; Rosingana, M.; Discordia, R.P.; Soundararajan, N.; Polniaszek, R. Synlett 2001, 1485.
1948. Labelle, M.; Gravel, D. J. Chem. Soc., Chem. Commun. 1985, 105.
1949. Ooi, T.; Tayama, E.; Yamada, M.; Maruoka, K. Synlett. 1999, 729.
1950. Youshko, M.I.; van Rantwijk, F.; Sheldon, R.A. Tetrahedron Asymmetry 2001, 12, 3267.
1951. Distaso, M.; Quaranta, E. Tetrahedron 2004, 60, 1531.
1952. Orrling, K.M.; Wu, X.; Russo, F.; Larhed, M. J. Org. Chem. 2008, 73, 8627.
1953. Ho, C.Y.; Strobel, E.; Ralbovsky, J.; Galemmo, Jr., R.A. J. Org. Chem. 2005, 70, 4873.
1954. van Melick, J.E.W.; Wolters, E.T.M. Synth. Commun. 1972, 2, 83.
1955. Satchell, D.P.N.; Satchell, R.S. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 410–431; Ilieva, S.; Galabov, B.; Musaev, D.G.; Morokuma, K.; Schaefer, III, H.F. J. Org. Chem. 2003, 68, 1496.
1956. Bruice, T.C.; Donzel, A.; Huffman, R.W.; Butler, A.R. J. Am. Chem. Soc. 1967, 89, 2106.
1957. Bunnett, J.F.; Davis, G.T. J. Am. Chem. Soc. 1960, 82, 665, Jencks, W.P.; Carriuolo, J. J. Am. Chem. Soc. 1960, 82, 675; Bruice, T.C.; Mayahi, M.F. J. Am. Chem. Soc. 1960, 82, 3067.
1958. Bruice, T.C.; Felton, S.M. J. Am. Chem. Soc. 1969, 91, 2799; Felton, S.M.; Bruice, T.C. J. Am. Chem. Soc. 1969, 91, 6721; Nagy, O.B.; Reuliaux,V.; Bertrand, N.; Van Der Mensbrugghe, A.; Leseul, J.; Nagy, J.B. Bull. Soc. Chim. Belg. 1985, 94, 1055.
1959. Gresser, M.J.; Jencks, W.P. J. Am. Chem. Soc. 1977, 99, 6963, 6970. See also, Um, I.-H.; Lee, J.-Y.; Lee, H.W.; Nagano, Y.; Fujio, M.; Tsuno, Y. J. Org. Chem. 2005, 70, 4980.
1960. Blackburn, G.M.; Jencks, W.P. J. Am. Chem. Soc. 1968, 90, 2638.
1961. Bunnett, J.F.; Davis, G.T. J. Am. Chem. Soc. 1960, 82, 665.
1962. Zaugg, H.E.; Helgren, P.F.; Schaefer, A.D. J. Org. Chem. 1963, 28, 2617. See also, Weintraub, L.; Terrell, R. J. Org. Chem. 1965, 30, 2470; Harada, R.; Kinoshita, Y. Bull. Chem. Soc. Jpn. 1967, 40, 2706.
1963. Huang, P.-Q.; Zheng, X.; Deng, X.-M. Tetrahedron Lett. 2001, 42, 9039. See also, Taylor, S.K.; Ide, N.D.; Silver, M.E.; Stephan, M. Synth. Commun. 2001, 31, 2391.
1964. For a list of procedures, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1978–1982.
1965. Murakami, Y.; Kondo, K.; Miki, K.; Akiyama, Y.; Watanabe, T.; Yokoyama, Y. Tetrahedron Lett. 1997, 38, 3751.
1966. Hoerter, J.M.; Otte, K.M.; Gellman, S.H.; Stahl, S.S. J. Am. Chem. Soc. 2006, 128, 5177.
1967. See Chimishkyan, A.L.; Snagovskii, Yu.S.; Gulyaev, N.D.; Leonova, T.V.; Kusakin, M.S. J. Org. Chem. USSR 1985, 21, 1955.
1968. Yang, Y.; Lu, S. Org. Prep. Proceed. Int. 1999, 31, 559.
1969. Garcia, J.; Vilarrasa, J. Tetrahedron Lett. 1982, 23, 1127.
1970. Askitoglu, E.; Guggisberg, A.; Hesse, M. Helv. Chim. Acta 1985, 68, 750 and references cited therein. For a carbon analog, see Süsse, M.; Hájicek, J.; Hesse, M. Helv. Chim. Acta 1985, 68, 1986.
1971. See Stach, H.; Hesse, M. Tetrahedron 1988, 44, 1573.
1972. Kotsuki, H.; Iwasaki, M.; Nishizawa, H. Tetrahedron Lett. 1992, 33, 4945.
1973. See Douglas, K.T. Acc. Chem. Res. 1986, 19, 186.
1974. See Collum, D.B.; Chen, S.; Ganem, B. J. Org. Chem. 1978, 43, 4393.
1975. Coniglio, S.; Aramini, A.; Cesta, M.C.; Colagioia, S.; Curti, R.; D'Alessandro, F.; D'anniballe, G.; D'Elia, V.; Nano, G.; Orlando, V.; Allegretti, M. Tetrahedron Lett. 2004, 45, 5375.
1976. Salvatore, R.N.; Shin, S.I.; Nagle, A.S.; Jung, K.W. J. Org. Chem. 2001, 66, 1035.
1977. For a review, see Challis, B.C.; Challis, J.A. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 759–773.
1978. Baburao, K.; Costello, A.M.; Petterson, R.C.; Sander, G.E. J. Chem. Soc. C 1968, 2779; Davidson, D.; Skovronek, H. J. Am. Chem. Soc. 1958, 80, 376.
1979. See LaLonde, R.T.; Davis, C.B. J. Org. Chem. 1970, 35, 771.
1980. See Bojarski, J.T.; Mokrosz, J.L.; Barton, H.J.; Paluchowska, M.H. Adv. Heterocycl. Chem. 1985, 38, 229.
1981. Speziale, A.J.; Smith, L.R.; Fedder, J.E. J. Org. Chem. 1965, 30, 4306.
1982. Marinescu, L.G.; Pedersen, C.M.; Bols, M. Tetrahedron 2005, 61, 123. See Marinescu, L.; Thinggaard, J.; Thomsen, I. B.; Bols, M. J. Org. Chem. 2003, 68, 9453; Hünig, S.; Schaller, R. Angew. Chem. Int. Ed. 1982, 21, 36.
1983. Arote, N.D.; Akamanchi, K.G. Tetrahedron Lett. 2007, 48, 5661.
1984. See Ansell, M.F. in Patai, S. The Chemistry of Acyl Halides, Wiley, NY, 1972, pp. 35–68.
1985. See Keinan, E.; Sahai, M. J. Org. Chem. 1990, 55, 3922.
1986. Bains, S.; Green, J.; Tan, L.C.; Pagni, R.M.; Kabalka, G.W. Tetrahedron Lett. 1992, 33, 7475.
1987. Kang, D.H.; Joo, T.Y.; Lee, E.H.; Chaysripongkul, S.; Chavasiri, W.; Jang, D.O. Tetrahedron Lett. 2006, 47, 5693.
1988. See Pizey, J.S. Synthetic Reaagents, Vol. 1, Wiley, NY, 1974, pp. 321–357. See Mohanazadeh, F.; Momeni, A.R. Org. Prep. Proceed. Int. 1996, 28, 492.
1989. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1929–1930.
1990. Lee, J.B. J. Am. Chem. Soc. 1966, 88, 3440. See Venkataraman, K.; Wagle, D.R. Tetrahedron Lett. 1979, 3037; Devos, A.; Remion, J.; Frisque-Hesbain, A.; Colens, A.; Ghosez, L. J. Chem. Soc., Chem. Commun. 1979, 1180.
1991. Adams, R.; Ulich, L.H. J. Am. Chem. Soc. 1920, 42, 599; Wood, T.R.; Jackson, F.L.; Baldwin, A.R.; Longenecker, H.E. J. Am. Chem. Soc. 1944, 66, 287; Zhang, A.; Nie, J. J. Agric. Food Chem. 2005, 53, 2451.
1992. Olah, G.A.; Nojima, M.; Kerekes, I. Synthesis 1973, 487. For other methods of preparing acyl fluorides, see Mukaiyama, T.; Tanaka, T. Chem. Lett. 1976, 303; Ishikawa, N.; Sasaki, S. Chem. Lett. 1976, 1407.
1993. Švec, P.; Eisner, A.; Kolá![]() ová, L.; Weidlich, T.; Pejchal, V.; R
ová, L.; Weidlich, T.; Pejchal, V.; R![]() ži
ži![]() ka, A. Tetrahedron Lett. 2008, 49, 6320.
ka, A. Tetrahedron Lett. 2008, 49, 6320.
1994. For lists of reagents converting acid derivatives to acyl halides, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1950–1951, 1955, 1968.
1995. Olah, G.A.; Welch, J.; Vankar, Y.D.; Nojima, M.; Kerekes, I.; Olah, J.A. J. Org. Chem. 1979, 44, 3872. See also, Yin, J.; Zarkowsky, D.S.; Thomas, D.W.; Zhao, M.W.; Huffman, M.A. Org. Lett. 2004, 6, 1465.
1996. Olah, G.A.; Kuhn, S.J. J. Org. Chem. 1961, 26, 237.
1997. Olah, G.A.; Kuhn, S.J. J. Am. Chem. Soc. 1960, 82, 2380.
1998. Emsley, J.; Gold, V.; Hibbert, F.; Szeto, W.T.A. J. Chem. Soc. Perkin Trans. 2 1988, 923.
1999. Markovski, L.N.; Pashinnik, V.E. Synthesis 1975, 801.
2000. Burton, D.J.; Koppes, W.M. J. Chem. Soc., Chem. Commun. 1973, 425; J. Org. Chem. 1975, 40, 3026; Anderson Jr., A.G.; Kono, D.H. Tetrahedron Lett. 1973, 5121.
2001. See Schmidt, A.H.; Russ, M.; Grosse, D. Synthesis 1981, 216; Hoffmann, H.M.R.; Haase, K. Synthesis 1981, 715.
2002. House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 691–694, 734–765.
2003. For a review, see Cais, M.; Mandelbaum, A. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, Vol. 1, pp. 303–330.
2004. See Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents,Wiley, NY, 1980, pp. 81–85. Ryu, I.; Ikebe, M.; Sonoda, N.; Yamamoto, S.-y.; Yamamura, G.-h.; Komatsu, M. Tetrahedron Lett. 2002, 43, 1257.
2005. Posner, G.H.; Whitten, C.E.; McFarland, P.E. J. Am. Chem. Soc. 1972, 94, 5106; Luong-Thi, N.; Rivière, H. J. Organomet. Chem. 1974, 77, C52.
2006. See Bennett, G.B.; Nadelson, J.; Alden, L.; Jani, A. Org. Prep. Proced. Int. 1976, 8, 13.
2007. Johnson, C.R.; Dhanoa, D.S. J. Org. Chem. 1987, 52, 1885.
2008. Bergbreiter, D.E.; Killough, J.M. J. Org. Chem. 1976, 41, 2750.
2009. Knochel, P.; Yeh, M.C.P.; Berk, S.C.; Talbert, J. J. Org. Chem. 1988, 53, 2390.
2010. Castro, C.E.; Havlin, R.; Honwad, V.K.; Malte, A.; Mojé, S. J. Am. Chem. Soc. 1969, 91, 6464. See Verkruijsse, H.D.; Heus-Kloos, Y.A.; Brandsma, L. J. Organomet. Chem. 1988, 338, 289.
2011. Stack, D.E.; Dawson, B.T.; Rieke, R.D. J. Am. Chem. Soc. 1992, 114, 5110.
2012. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood, NJ, 1954, pp. 712–724. See Wang, X.-j.; Zhang, L.; Sun, X.; Xu, Y.; Krishnamurthy, D.; Senanayake, C.H. Org. Lett. 2005, 7, 5593.
2013. Föhlisch, B.; Flogaus, R. Synthesis 1984, 734.
2014. Maeda, H.; Okamoto, J.; Ohmori, H. Tetrahedron Lett. 1996, 37, 5381.
2015. Babudri, F.; Fiandanese, V.; Marchese, G.; Punzi, A. Tetrahedron 1996, 52, 13513.
2016. Malanga, C.; Aronica, L.A.; Lardicci, L. Tetrahedron Lett. 1995, 36, 9185. See Lemoucheux, L.; Rouden, J.; Lasne, M.-C. Tetrahedron Lett. 2000, 41, 9997.
2017. See Dubois, J.E.; Lion, C.; Arouisse, A. Bull. Soc. Chim. Belg. 1984, 93, 1083.
2018. Cooke, Jr., M.P. J. Org. Chem. 1986, 51, 951.
2019. Fehr, C.; Galindo, J.; Perret, R. Helv. Chim. Acta 1987, 70, 1745.
2020. See Fujisawa, T.; Sato, T. Org. Synth. 66, 116; Babudri, F.; D'Ettole, A.; Fiandanese, V.; Marchese, G.; Naso, F. J. Organomet. Chem. 1991, 405, 53.
2021. See MacPhee, J.A.; Boussu, M.; Dubois, J.E. J. Chem. Soc. Perkin Trans. 2 1974, 1525.
2022. Bandgar, B.P.; Patil, A.V. Tetrahedron Lett. 2005, 46, 7627; Ekoue-Kovi, K.; Xu, H.; Wolf, C. Tetrahedron Lett. 2008, 49, 5773. For a Cu-mediated reaction, see Nishihara, Y.; Inoue, Y.; Fujisawa, M.; Takagi, K. Synlett2005, 2309.
2023. Thimmaiah, M.; Zhang, X.; Fang, S. Tetrahedron Lett. 2008, 49, 5605.
2024. Xin, B.; Zhang, Y.; Cheng, K. Synthesis 2007, 1970.
2025. Lysén, M.; Kelleher, S.; Begtrup, M.; Kristensen, J.L. J. Org. Chem. 2005, 70, 5342.
2026. Xin, B.; Zhang, Y.; Cheng, K. J. Org. Chem. 2006, 71, 5725.
2027. Wang, J.-X.; Wei, B.; Hu, Y.; Liu, Z.; Yang, Y. Synth. Commun. 2001, 31, 3885.
2028. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1389–1400.
2029. Cason, J.; Fessenden, R. J. Org. Chem. 1960, 25, 477.
2030. Baruah, B.; Boruah. A.; Prajapati, D.; Sandhu, J.S. Tetrahedron Lett. 1996, 37, 9087.
2031. See Grey, R.A. J. Org. Chem. 1984, 49, 2288; Tamaru, Y.; Ochiai, H.; Nakamura, T.; Yoshida, Z. Org. Synth. 67, 98.
2032. Labadie, J.W.; Stille, J.K. J. Am. Chem. Soc. 1983, 105, 669, 6129; Labadie, J.W.; Tueting, D.; Stille, J.K. J. Org. Chem. 1983, 48, 4634. See Inoue, K.; Shimizu, Y.; Shibata, I.; Baba, A. Synlett 2001, 1659.
2033. Yamada, J.; Yamamoto, Y. J. Chem. Soc., Chem. Commun. 1987, 1302.
2034. Yadav, J.S.; Srinivas, D.; Reddy, G.S.; Bindu, K.H. Tetrahedron Lett. 1997, 38, 8745. Also see, Bryan, V.J.; Chan, T.-H. Tetrahedron Lett. 1997, 38, 6493 for a similar reaction with an acyl imidazole.
2035. Kim, S.-H.; Rieke, R.D. J. Org. Chem. 1998, 63, 6766; Cahiez, G.; Martin, A.; Delacroix, T. Tetrahedron Lett. 1999, 40, 6407.
2036. Filon, H.; Gosmini, C.; Périchon, J. Tetrahedron 2003, 59, 8199.
2037. Rao, M.L.N.; Venkatesh, V.; Banerjee, D. Tetrahedron 2007, 63, 12917.
2038. Markó, I.E.; Southern, J.M. J. Org. Chem. 1990, 55, 3368.
2039. Ying, T.; Bao, W.; Zhang, Y.; Xu, W. Tetrahedron Lett. 1996, 37, 3885.
2040. Kakusawa, N.; Yamaguchi, K.; Kurita, J.; Tsuchiya, T. Tetraehdron Lett. 2000, 41, 4143.
2041. Chowdhury, C.; Kundu, N.G. Tetrahedron 1999, 55, 7011; Wang, J.-X.; Wei, B.; Hu, Y.; Liua, Z.; Kang, L. J. Chem. Res. (S) 2001, 146.
2042. Karpov, A.S.; Müller, T.J.J. Org. Lett. 2003, 5, 3451.
2043. Wang, B.; Wang, S.; Li, P.; Wang, L. Chem. Commun. 2010, 5891.
2044. Iwai, T.; Fujihara, T.; Terao, J.; Tsuji, Y. J. Am. Chem. Soc. 2009, 131, 6668.
2045. Böttcher, A.; Becker, H.; Brunner, M.; Preiss, T.; Henkelmann, J.; De Bakker, C.; Gleiter, R. J. Chem. Soc., Perkin Trans.1 1999, 3555.
2046. Wang, J.-x.; Wei, B.; Huang, D.; Hu, Y.; Bai, L. Synth. Commun. 2001, 31, 3337.
2047. Wang, J.-X.; Wei, B.; Hu, Y.; Liu, Z.; Fu, Y. Synth. Commun. 2001, 31, 3527.
2048. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood, NJ, 1954, pp. 561–562, 846–908.
2049. Deskus, J.; Fan, D.; Smith, M.B. Synth. Commun. 1998, 28, 1649.
2050. Hallouis, S.; Saluzzo, C.; Amouroux, R. Synth. Commun. 2000, 30, 313.
2051. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 549–766, 846–869.
2052. Canonne, P.; Bernatchez, M. J. Org. Chem. 1986, 51, 2147; 1987, 52, 4025.
2053. Kresge, A.J.; Weeks, D.P. J. Am. Chem. Soc. 1984, 106, 7140. See also, Amyes, T.L.; Jencks, W.P. J. Am. Chem. Soc. 1989, 111, 7888, 7900.
2054. See Newman, M.S.; Smith, A.S. J. Org. Chem. 1948, 13, 592; Edwards, Jr., W.R.; Kammann, Jr., K.P. J. Org. Chem. 1964, 29, 913; Araki, M.; Sakat, S.; Takei, H.; Mukaiyama, T. Chem. Lett. 1974, 687.
2055. Huet, F.; Pellet, M.; Conia, J.M. Tetrahedron Lett. 1976, 3579.
2056. For a list of reactions with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1386–1389, 1400–1419.
2057. See Olah, G.S.; Prakash, G.K.S.; Arvanaghi, M. Synthesis 1984, 228; Martín, R.; Romea, P.; Tey, C.; Urpí, F.; Vilarrasa, J. Synlett 1997, 1414. Also see Kashima, C.; Kita, I.; Takahashi, K.; Hosomi, A. J. Heterocyclic Chem.1995, 32, 25 for a related reaction.
2058. Shintani, R.; Fu, G.C. Angew. Chem. Int. Ed. 2002, 41, 1057.
2059. Bogavac, M.; Arsenijevic, L.; Pavlov, S.; Arsenijevic, V. Tetrahedron Lett. 1984, 25, 1843.
2060. Petrov, A.D.; Kaplan, E.P.; Tsir, Ya. J. Gen. Chem. USSR 1962, 32, 691.
2061. Evans, E.A. J. Chem. Soc. 1956, 4691. See Clark, C.T.; Milgram, B.C.; Scheidt, K.A. Org. Lett. 2004, 6, 3977. See Wakefield, B.J. Organolithium Methods; Academic Press, NY, 1988, pp. 82–88.
2062. Mueller-Westerhoff, U.T.; Zhou, M. Synlett 1994, 975.
2063. Zhang, P.; Terefenko, E.A.; Slavin, J. Tetrahedron Lett. 2001, 42, 2097.
2064. DeLuca, L.; Giacomelli, G.; Porcheddu, A. Org. Lett. 2001, 3, 1519.
2065. Spialtr, L.; Pappalardo, J.A. The Acyclic Aliphatic Tertiary Amines, Macmillan, NY, 1965, pp. 59–63.
2066. Prakash, G.K.S.; York, C.; Liao, Q.; Kotian, K.; Olah, G.A. Heterocycles 1995, 40, 79.
2067. Jones, K.; Storey, J.M.D. J. Chem. Soc., Perkin Trans. 1 2000, 769.
2068. Tominaga, Y.; Kohra, S.; Hosomi, A. Tetrahedron Lett. 1987, 28, 1529.
2069. Calderwood, D.J.; Davies, R.V.; Rafferty, P.; Twigger, H.L.; Whelan, H.M. Tetrahedron Lett. 1997, 38, 1241.
2070. Ahn, Y.; Cohen, T. Tetrahedron Lett. 1994, 35, 203.
2071. Comins, D.L.; Dernell, W. Tetrahedron Lett. 1981, 22, 1085.
2072. Mitchell, R.H.; Iyer, V.S. Tetrahedron Lett. 1993, 34, 3683. Also see, Sibi, M.P.; Sharma, R.; Paulson, K.L. Tetrahedron Lett. 1992, 33, 1941.
2073. Baird, M.S.; Huber, F.A.M.; Tverezovsky, V.V.; Bolesov, I.G. Tetrahedron 2001, 57, 1593.
2074. Nahm, S.; Weinreb, S.M. Tetrahedron Lett. 1981, 22, 3815. For a review, see Balasubramaniam, S.; Aidhen, I.S. Synthesis 2008, 3707.
2075. See Tallier, C.; Bellosta, V.; Meyer, C.; Cossy, J. Org. Lett. 2004, 6, 2145.
2076. Xie, W.; Zou, B.; Pei, D.; Ma, D. Org. Lett. 2005, 7, 2775. For other exmaples see Andrés, J.M.; Pedrosa, R.; Pérez-Encabo, A. Tetrahedron 2000, 56, 1217.
2077. Lee, N.R.; Lee, J.I. Synth. Commun. 1999, 29, 1249.
2078. Ruiz, J.; Sotomayor, N.; Lete, E. Org. Lett. 2003, 5, 1115.
2079. See Hansford, K.A.; Dettwiler, J.E.; Lubell, W.D. Org. Lett. 2003, 5, 4887.
2080. Hlasta, D.J.; Court, J.J. Tetrahedron Lett. 1989, 30, 1773. See also, Nahm, S.; Weinreb, S.M. Tetrahedron Lett. 1981, 22, 3815.
2081. Kojima, S.; Hidaka, T.; Yamakaw, A. Chem. Lett. 2005, 34, 470.
2082. Yamaguchi, M.; Waseda, T.; Hirao, I. Chem. Lett. 1983, 35.
2083. Bubnov, Y.N.; Pastukhov, F.V.; Yampolsky, I.V.; Ignatenko, A.V. Eur. J. Org. Chem. 2000, 1503; Li, Z.; Zhang, Y. Tetrahedron Lett. 2001, 42, 8507.
2084. Bubnov, Yu.N.; Klimkina, E.V.; Zhun', I.V.; Pastukhov, F.V.; Yampolsky, I.V. Pure Appl. Chem. 2000, 72, 1641.
2085. Michael, U.; Hörnfeldt, A. Tetrahedron Lett. 1970, 5219; Scilly, N.F. Synthesis 1973, 160.
2086. Collins, S.; Hong, Y. Tetrahedron Lett. 1987, 28, 4391.
2087. Periasamy, M.; Reddy, M.R.; Bharathi, P. Synth. Commun. 1999, 29, 677.
2088. Meisters, A.; Mole, T. Aust. J. Chem. 1974, 27, 1665.
2089. Chung, E.-A.; Cho, C.-W.; Ahn, K.H. J. Org. Chem. 1998, 63, 7590.
2090. Yu, Y.; Liebeskind, L.S. J. Org. Chem. 2004, 69, 3554.
2091. Wittenberg, R.; Srogl, J.; Egi, M.; Liebeskind, L.S. Org. Lett. 2003, 5, 3033.
2092. Tatamidani, H.; Kakiuchi, F.; Chatani, N. Org. Lett. 2004, 6, 3597.
2093. Tatamidani, H.; Yokota, K.; Kakiuchi, F.; Chatani, N. J. Org. Chem. 2004, 69, 5615.
2094. Frost, C.G.; Wadsworth, K.J. Chem. Commun. 2001, 2316.
2095. Gooßen, L.J.; Ghosh, K. Eur. J. Org. Chem. 2002, 3254.
2096. Fausett, B.W.; Liebeskind, L.S. J. Org. Chem. 2005, 70, 4851.
2097. Anderson, R.J.; Henrick, C.A.; Rosenblum, L.D. J. Am. Chem. Soc. 1974, 96, 3654. See also, Kim, S.; Lee, J.I. J. Org. Chem. 1983, 48, 2608.
2098. Shimizu, T.; Seki, M. Tetrahedron Lett. 2002, 43, 1039.
2099. Bercot, E.A.; Rovis, T. J. Am. Chem. Soc. 2004, 126, 10248.
2100. O'Brien, E.M.; Bercot, E.A.; Rovis, T. J. Am. Chem. Soc. 2003, 125, 10498.
2101. Shimizu, T.; Seki, M. Tetrahedron Lett. 2001, 42, 429.
2102. See Mori, Y.; Seki, M. Tetrahedron Lett. 2004, 45, 7343. For a different but related cross-coupling, see Zhang, Y.; Rovis, T. J. Am. Chem. Soc . 2004, 126, 15964.
2103. Lannou, M.-I.; Hélion, F.; Namy, J.-L. Tetrahedron Lett. 2002, 43, 8007.
2104. Cacchi, S.; Fabrizi, G.; Gavazza, F.; Goggiamani, A. Org. Lett. 2003, 5, 289.
2105. Kowalski, C.J.; Haque, M.S.; Fields, K.W. J. Am. Chem. Soc. 1985, 107, 1429; Kowalski, C.J.; Haque, M.S. J. Org. Chem. 1985, 50, 5140.
2106. Kowalski, C.J.; Haque, M.S. J. Am. Chem. Soc. 1986, 108, 1325.
2107. Katritzky, A.R.; Wang, Z.; Wang, M.; Wilkerson, C.R.; Hall, C.D.; Akhmedov, N.G. J. Org. Chem. 2004, 69, 6617.
2108. Yoo, B.W.; Choi, K.H.; Lee, S.J.; Nam, G.S.; Chang, K.Y.; Kim, S.H.; Kim, J.H. Synth. Commun. 2002, 32, 839.
2109. Wang, X.; Zhang, Y. Tetrahedron Lett. 2002, 43, 5431.
2110. Saikia, P.; Laskar, D.D.; Prajapati, D.; Sandhu, J.S. Tetahedron Lett. 2002, 43, 7525.
2111. Takahashi, T.; Xi, C.; Ura, Y.; Nakajima, K. J. Am. Chem. Soc. 2000, 122, 3228.
2112. Mészáros, L. Tetrahedron Lett. 1967, 4951.
2113. Souppe, J.; Namy, J.; Kagan, H.B. Tetrahedron Lett. 1984, 25, 2869. See also, Collin, J.; Namy, J.; Dallemer, F.; Kagan, H.B. J. Org. Chem. 1991, 56, 3118.
2114. Voronkov, M.G.; Belousova, L.I.; Vlasov, A.V.; Vlasova, N.N. Russ. J. Org. Chem 2008, 44, 929.
2115. Han, B.H.; Boudjouk, P. Tetrahedron Lett. 1981, 22, 2757.
2116. Verlhac, J.; Chanson, E.; Jousseaume, B.; Quintard, J. Tetrahedron Lett. 1985, 26, 6075. For another procedure, see Olah, G.A.; Wu, A. J. Org. Chem. 1991, 56, 902.
2117. For examples of reactions in this section, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1484–1485, 1522–1527.
2118. For an improved procedure, see Rathke, M.W.; Cowan, P.J. J. Org. Chem. 1985, 50, 2622.
2119. When phase-transfer catalysts are used, O-acylation becomes the main reaction: Jones, R.A.; Nokkeo, S.; Singh, S. Synth. Commun. 1977, 7, 195.
2120. Taylor, E.C.; Hawks, III, G.H.; McKillop, A. J. Am. Chem. Soc. 1968, 90, 2421.
2121. See Skarzewski, J. Tetrahedron 1989, 45, 4593; Newkome, G.R.; Baker, G.R. Org. Prep. Proced. Int. 1986, 19, 117.
2122. Hegedus, L.S.; Williams, R.E.; McGuire, M.A.; Hayashi, T. J. Am. Chem. Soc. 1980, 102, 4973.
2123. See House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 762–765; House, H.O.; Auerbach, R.A.; Gall, M.; Peet, N.P. J. Org. Chem. 1973, 38, 514.
2124. Seebach, D.; Weller, T.; Protschuk, G.; Beck, A.K.; Hoekstra, M.S. Helv. Chim. Acta 1981, 64, 716.
2125. Tirpak, R.E.; Rathke, M.W. J. Org. Chem. 1982, 47, 5099.
2126. See Hauser, C.R.; Swamer, F.W.; Adams, J.T. Org. React. 1954, 8, 59, pp. 98–106.
2127. See Hayden, W.; Pucher, R.; Griengl, H. Monatsh. Chem. 1987, 118, 415.
2128. Mermerian, A.H.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 5604.
2129. See Rablen, P.R.; Bentrup, KL.H. J. Am. Chem. Soc. 2003, 125, 2142.
2130. See Tanabe, Y. Bull. Chem. Soc. Jpn. 1989, 62, 1917.
2131. Tanabe, Y.; Hamasaki, R.; Funakoshi, S. Chem. Commun. 2001, 1674.
2132. Misaki, T.; Nagase, R.; Matsumoto, K.; Tanabe, Y. J. Am. Chem. Soc. 2005, 127, 2854.
2133. Maki, T.; Ishihara, K.; Yamamoto, H. Tetrahedron 2007, 63, 8645.
2134. Sullivan, D.F.; Woodbury, R.P.; Rathke, M.W. J. Org. Chem. 1977, 42, 2038.
2135. Rathke, M.W.; Lindert, A. J. Am. Chem. Soc. 1971, 93, 2318.
2136. Yoshizawa, K.; Toyota, S.; Toda, F. Tetrahedron Lett. 2001, 42, 7983.
2137. Kawata, A.; Takata, K.; Kuninobu, Y.; Takai, K. Angew. Chem. Int. Ed. 2007, 46, 7793.
2138. See Schaefer, J.P.; Bloomfield, J.J. Org. React. 1967, 15, 1.
2139. Toda, F.; Suzuki, T.; Higa, S. J. Chem. Soc. Perkin Trans. 1 1998, 3521.
2140. Crowley, J.I.; Rapoport, H. J. Org. Chem. 1980, 45, 3215. For another method, see Yamada, Y.; Ishii, T.; Kimura, M.; Hosaka, K. Tetrahedron Lett. 1981, 22, 1353.
2141. Yoshida, Y.; Hayashi, R.; Sumihara, H.; Tanabe, Y. Tetrahedron Lett. 1997, 38, 8727.
2142. Marquié, J.; Laporterie, A.; Dubac, J.; Roques, N. Synlett 2001, 493.
2143. In some cases, an SET mechanism may be involved: Ashby, E.C.; Park, W. Tetrahedron Lett. 1983, 1667. See Nishimura, T.; Sunagawa, M.; Okajima, T.; Fukazawa, Y. Tetrahedron Lett. 1997, 38, 7063.
2144. Brown, C.A. Synthesis 1975, 326.
2145. See Garst, J.F. J. Chem. Educ. 1979, 56, 721.
2146. Popik, V.V.; Nikolaev, V.A. J. Org. Chem. USSR 1989, 25, 1636.
2147. Mander, L.N.; Sethi, P. Tetrahedron Lett. 1983, 24, 5425.
2148. Hellou, J.; Kingston, J.F.; Fallis, A.G. Synthesis 1984, 1014.
2149. See Durst, T. Adv. Org. Chem. 1969, 6, 285, pp. 296–301.
2150. Schank, K.; Hasenfratz, H.; Weber, A. Chem. Ber. 1973, 106, 1107, House, H.O.; Larson, J.K. J. Org. Chem. 1968, 33, 61.
2151. Corey, E.J.; Seebach, D. J. Org. Chem. 1975, 40, 231
2152. See Corey, E.J.; Erickson, B.W. J. Org. Chem. 1971, 36, 3553.
2153. Miles, M.L.; Harris, T.M.; Hauser, C.R. J. Org. Chem. 1965, 30, 1007.
2154. Hill, D.G.; Burkus, T.; Hauser, C.R. J. Am. Chem. Soc. 1959, 81, 602.
2155. Kuo, Y.; Yahner, J.A.; Ainsworth, C. J. Am. Chem. Soc. 1971, 93, 6321; Angelo, B. C.R. Seances Acad. Sci. Ser. C 1973, 276, 293.
2156. Koch, G.K.; Kop, J.M.M. Tetrahedron Lett. 1974, 603.
2157. Krapcho, A.P.; Kashdan, D.S.; Jahngen, Jr., E.G.E.; Lovey, A.J. J. Org. Chem. 1977, 42, 1189; Lion, C.; Dubois, J.E. J. Chem. Res. (S) 1980, 44.
2158. See Hünig, S.; Schaller, R. Angew. Chem. Int. Ed. 1982, 21, 36.
2159. Taylor, E.C.; Andrade, J.G.; John, K.C.; McKillop, A. J. Org. Chem. 1978, 43, 2280.
2160. Olah, G.A.; Arvanaghi, M.; Prakash, G.K.S. Synthesis 1983, 636.
2161. Tanaka, M. Tetrahedron Lett. 1980, 21, 2959. See also, Tanaka, M.; Koyanagi, M. Synthesis 1981, 973.
2162. Ando, T.; Kawate, T.; Yamawaki, J.; Hanafusa, T. Synthesis 1983, 637.
2163. Koenig, K.E.; Weber, W.P. Tetrahedron Lett. 1974, 2275. See also, Sukata, K. Bull. Chem. Soc. Jpn. 1987, 60, 1085.
2164. See Fridman, A.L.; Ismagilova, G.S.; Zalesov, V.S.; Novikov, S.S. Russ. Chem. Rev. 1972, 41, 371; Ried, W.; Mengler, H. Fortshr. Chem. Forsch., 1965, 5, 1.
2165. Hodson, D.; Holt, G.; Wall, D.K. J. Chem. Soc. C 1970, 971.
2166. See Kwart, H.; King, K. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 362–370.
2167. Granito, C.; Schultz, H.P. J. Org. Chem. 1963, 28, 879.
2168. See Ruzicka, L.; Stoll, M.; Schinz, H. Helv. Chim. Acta 1926, 9, 249; 1928, 11, 1174; Ruzicka, L.; Brugger, W.; Seidel, C.F.; Schinz, H. Helv. Chim. Acta 1928, 11, 496.
2169. Hites, R.A.; Biemann, K. J. Am. Chem. Soc. 1972, 94, 5772. See also, Bouchoule, C.; Blanchard, M.; Thomassin, R. Bull. Soc. Chim. Fr. 1973, 1773.
2170. Ritter, J.J.; Minieri, P.P. J. Am. Chem. Soc. 1948, 70, 4045. See Krimen, L.I.; Cota, D.J. Org. React. 1969, 17, 213; Johnson, F.; Madroñero, R. Adv. Heterocycl. Chem. 1966, 6, 95; Tongco, E.C.; Prakash, G.K.S.; Olah, G.A. Synlett 1997, 1193.
2171. Salehi, P.; Khodaei, M.M.; Zolfigol, M.A.; Keyvan, A. Synth. Commun. 2001, 31, 1947.
2172. Lebedev, M.Y.; Erman, M.B. Tetrahedron Lett. 2002, 43, 1397.
2173. Chen, H.G.; Goel, O.P.; Kesten, S.; Knobelsdorf, J. Tetrahedron Lett. 1996, 37, 8129.
2174. Martinez, A.G.; Alvarez, R.M.; Vilar, E.T.; Fraile, A.G.; Hanack, M.; Subramanian, L.R. Tetrahedron Lett. 1989, 30, 581.
2175. Barton, D.H.R.; Magnus, P.D.; Garbarino, J.A.; Young, R.N. J. Chem. Soc. Perkin Trans. 1 1974, 2101. See also, Top, S.; Jaouen, G. J. Org. Chem. 1981, 46, 78.
2176. Tamaddon, F.; Khoobi, M.; Keshavarz, E. Tetrahedron Lett. 2007, 48, 3643.
2177. Polshettiwar, V.; Varma, R.S. Tetrahedron Lett. 2008, 49, 2661.
2178. Anxionnat, B.; Guérinot, A.; Reymond, S.; Cossy, J. Tetrahedron Lett. 2009, 50, 3470.
2179. Theerthagiri, P.; Lalitha, A.; Arunachalam, P.N. Tetrahedron Lett. 2010, 51, 2813.
2180. See Fry, A.J.; Simon, J.A. J. Org. Chem. 1982, 47, 5032.
2181. Sakaguchi, S.; Hirabayashi, T.; Ishii, Y. Chem. Commun. 2002, 516.
2182. Anatol, J.; Berecoechea, J. Bull. Soc. Chim. Fr. 1975, 395; Synthesis 1975, 111.
2183. See Bevington, J.C. Q. Rev. Chem. Soc. 1952, 6, 141.
2184. See Camarena, R.; Cano, A.C.; Delgado, F.; Zúñiga, N.; Alvarez, C. Tetrahedron Lett. 1993, 34, 6857.
2185. Barón, M.; de Manderola, O.B.; Westerkamp, J.F. Can. J. Chem. 1963, 41, 1893.
2186. See Martin, D.; Bauer, M.; Pankratov, V.A. Russ. Chem. Rev. 1978, 47, 975. See Pankratov, V.A.; Chesnokova, A.E. Russ. Chem. Rev. 1989, 58, 879.
2187. Williams, A.; Ibrahim, I.T. Chem. Rev. 1981, 81, 589; Mikolajczyk, M.; Kielbasinski, P. Tetrahedron 1981, 37, 233; Kurzer, F.; Douraghi-Zadeh, K. Chem. Rev. 1967, 67, 107.
2188. Campbell, T.W.; Monagle, J.J.; Foldi, V.S. J. Am. Chem. Soc. 1962, 84, 3673.
2189. Monagle, J.J.; Campbell, T.W.; McShane Jr., H.F. J. Am. Chem. Soc. 1962, 84, 4288.
2190. Monagle, J.J.; Mengenhauser, J.V. J. Org. Chem. 1966, 31, 2321.
2191. See Ostrogovich, G.; Kerek, F.; Buzás, A.; Doca, N. Tetrahedron 1969, 25, 1875.
2192. Zhang, M.; Vedantham, P.; Flynn, D.L.; Hanson, P.R. J. Org. Chem. 2004, 69, 8340.
2193. Mlinari![]() -Majerski, K.; Margeta, R.; Veljkovi
-Majerski, K.; Margeta, R.; Veljkovi![]() , J. Synlett 2005, 2089.
, J. Synlett 2005, 2089.
2194. Douglas, D.E.; Burditt, A.M. Can. J. Chem. 1958, 36, 1256.
2195. Barltrop, J.A.; Day, A.C.; Bigley, D.B. J. Chem. Soc. 1961, 3185.
2196. See Calter, M.A.; Tretyak, O.A.; Flaschenriem, C. Org. Lett. 2005, 7, 1809.
2197. Muller, L.L.; Hamer, J. 1,2-Cycloaddition Reactions, Wiley, NY, 1967, pp. 139–168; Ulrich, H. Cycloaddition Reactions of Heterocumulenes, Academic Press, NY, 1967, pp. 39–45, 64–74.
2198. Wynberg, H.; Staring, E.G.J. J. Am. Chem. Soc. 1982, 104, 166; J. Chem. Soc., Chem. Commun. 1984, 1181.
2199. Gnanadesikan, V.; Corey, E.J. Org. Lett. 2006, 8, 4943.
2200. Nelson, S.G.; Zhu, C.; Shen, X. J. Am. Chem. Soc. 2004, 126, 14.
2201. Wynberg, H.; Staring, E.G.J. J. Org. Chem. 1985, 50, 1977.
2202. Elam, E.U. J. Org. Chem. 1967, 32, 215.
2203. Aben, R.W.; Hofstraat, R.; Scheeren, J.W. Recl. Trav. Chim. Pays-Bas 1981, 100, 355. For a discussion of oxetane cycloreversion, see Miranda, M.A.; Izquierdo, M.A.; Galindo, F. Org. Lett. 2001, 3, 1965.
2204. Ciufolini, M.A.; Rivera-Fortin, M.A.; Byrne, N.E. Tetrahedron Lett. 1993, 34, 3505.
2205. Ninomiya, I.; Naito, T. Photochemical Synthesis, Academic Press, NY, 1989, pp. 138–152; Carless, H.A.J. in Coyle, J.D. Photochemistry in Organic Synthesis, Royal Society of Chemistry, London, 1986, pp. 95–117; Carless, H.A.J. in Horspool, W.M. Synthetic Organic Photochemistry, Plenum, NY, 1984, pp. 425–487; Jones II, M. Org. Photochem. 1981, 5, 1; Arnold, D.R. Adv. PhotoChem. 1968, 6, 301–423; Chapman, O.L.; Lenz, G. Org. Photochem. 1967, 1, 283, pp. 283–294; Muller, L.L.; Hamer, J. 1,2-Cycloaddition Reactions, Wiley, NY, 1967, pp. 111–139. Also see, Bosch, E.; Hubig, S.M.; Kochi, J.K. J. Am. Chem. Soc. 1998, 120, 386.
2206. Turro, N.J. Pure Appl. Chem. 1971, 27, 679; Yang, N.C.; Kimura, M.; Eisenhardt, W. J. Am. Chem. Soc. 1973, 95, 5058; Barltrop, J.A.; Carless, H.A.J. J. Am. Chem. Soc. 1972, 94, 1951, 8761.
2207. Arnold, D.R.; Hinman, R.L.; Glick, A.H. Tetrahedron Lett. 1964, 1425; Yang, N.C.; Nussim, M.; Jorgenson, M.J.; Murov, S. Tetrahedron Lett. 1964, 3657.
2208. See references cited in Griesbeck, A.G.; Stadmüller, S. J. Am. Chem. Soc. 1990, 112, 1281. See also, Kutateladze, A.G. J. Am. Chem. Soc. 2001, 123, 9279.
2209. Freilich, S.C.; Peters, K.S. J. Am. Chem. Soc. 1985, 107, 3819; Griesbeck, A.G.; Mauder, H.; Stadmüller, S. Accts. Chem. Res. 1994, 27, 70.
2210. Adam, W.; Stegmann, V.R. J. Am. Chem. Soc. 2002, 124, 3600. See Ciufolini, M.A.; Rivera-Fortin, M.A.; Zuzukin, V.; Whitmire, K.H. J. Am. Chem. Soc. 1994, 116, 1272.
2211. Greisbeck, A.G.; Bondock, S. J. Am. Chem. Soc. 2001, 123, 6191. See also, Adam, W.; Stegmann, V.R. Synthesis 2001, 1203.
2212. For a spin-directed reaction, see Griesbeck, A.G.; Fiege, M.; Bondock, S.; Gudipati, M.S. Org. Lett. 2000, 2, 3623.
2213. Howell, A.R.; Fan, R.; Truong, A. Tetrahedron Lett. 1996, 37, 8651. See Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis, Wiley, NY, 1984, pp. 317–326.
2214. Abe, M.; Tachibana, K.; Fujimoto, K.; Nojima, M. Synthesis 2001, 1243.
2215. Kang, T.; Scheffer, J.R. Org. Lett. 2001, 3, 3361.
2216. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 1919–1921. See Fu, N.; Tidwell, T.T. Tetrahedron 2008, 64, 10465; Brown, M.J. Heterocycles 1989, 29, 2225; Isaacs, N.S. Chem. Soc. Rev. 1976, 5, 181; Mukerjee, A.K.; Srivastava, R.C. Synthesis 1973, 327; Muller, L.L.; Hamer, J. 1,2-Cycloaddition Reactions, Wiley, NY, 1967, pp. 173–206; Anselme, J. in Patai, S. The Chemistry of the Carbon–Nitrogen Double Bond, Wiley, NY, 1970, pp. 305–309; Sandhu, J.S.; Sain, B. Heterocycles 1987, 26, 777.
2217. See Schaumann, E. Tetrahedron 1988, 44, 1827.
2218. Schaumann, E. Chem. Ber. 1976, 109, 906.
2219. With indium: Banik, B.K.; Ghatak, A.; Becker, F.F. J. Chem. Soc., Perkin Trans. 1 2000, 2179.
2220. For a review, see Hart, D.J.; Ha, D. Chem. Rev. 1989, 89, 1447.
2221. Hegedus, L.S.; McGuire, M.A.; Schultze, L.M.; Yijun, C.; Anderson, O.P. J. Am. Chem. Soc. 1984, 106, 2680; Hegedus, L.S.; McGuire, M.A.; Schultze, L.M. Org. Synth. 65, 140.
2222. Hegedus, L.S.; Imwinkelried, R.; Alarid-Sargent, M.; Dvorak, D.; Satoh, Y. J. Am. Chem. Soc. 1990, 112, 1109.
2223. Sharma, S.D.; Pandhi, S.B. J. Org. Chem. 1990, 55, 2196.
2224. Donati, D.; Morelli, C.; Porcheddu, A.; Taddei, M. J. Org. Chem. 2004, 69, 9316.
2225. D'hooghe, M.; Brabandt, W.V.; Dekeukeleire, S.; Dejaegher, Y.; De Kimpe, N. Chemistry: European J. 2008, 14, 6336.
2226. Taggi, A.E.; Hafez, A.M.; Wack, H.; Young, B.; Drury III, W.J.; Lectka, T. J. Am. Chem. Soc. 2000, 122, 7831.
2227. Hodous, B.L.; Fu, G.C. J. Am. Chem. Soc. 2002, 124, 1578.
2228. Taggi, A.E.; Hafez, A.M.; Wack, H.; Young, B.; Ferraris, D.; Lectka, T. J. Am. Chem. Soc. 2002, 124, 6626.
2229. France, S.; Shah, M.H.; Weatherwax, A.; Wack, H.; Roth, J.P.; Lectka, T. J. Am. Chem. Soc. 2005, 127, 1206.
2230. Shah, M.H.; France, S.; Lectka, T. Synlett 2003, 1937.
2231. Clark, A.J.; Battle, G.M.; Bridge, A. Tetrahedron Lett. 2001, 42, 4409.
2232. See Brady, W.T.; Shieh, C.H. J. Org. Chem. 1983, 48, 2499.
2233. See Opitz, G.; Koch, J. Angew. Chem. Int. Ed. 1963, 2, 152.
2234. Kamal, A.; Sattur, P.B. Heterocycles 1987, 26, 1051; Szabo, W.A. Aldrichimica Acta 1977, 10, 23; Rasmussen, J.K.; Hassner, A. Chem. Rev. 1976, 76, 389; Graf, R. Angew. Chem. Int. Ed. 1968, 7, 172.
2235. Bestian, H. Pure Appl. Chem. 1971, 27, 611. See also, Barrett, A.G.M.; Betts, M.J.; Fenwick, A. J. Org. Chem. 1985, 50, 169.
2236. See McAllister, M.A.; Tidwell, T.T. J. Chem. Soc. Perkin Trans. 2 1994, 2239.
2237. Moriconi, E.J.; Kelly, J.F. J. Org. Chem. 1968, 33, 3036. See also, Martin, J.C.; Carter, P.L.; Chitwood, J.L. J. Org. Chem. 1971, 36, 2225.
2238. Malpass, J.R.; Tweddle, N.J. J. Chem. Soc. Perkin Trans. 1 1977, 874.
2239. Moriconi, E.J.; Kelly, J.F.; Salomone, R.A. J. Org. Chem. 1968, 33, 3448.
2240. Taguchi, Y.; Tsuchiya, T.; Oishi, A.; Shibuya, I. Bull. Chem. Soc. Jpn. 1996, 69, 1667.
2241. Linder, M.R.; Podlech, J. Org. Lett. 2001, 3, 1849.
2242. Torii, S.; Okumoto, H.; Sadakane, M.; Hai, A.K.M.A.; Tanaka, H. Tetrahedron Lett. 1993, 34, 6553.
2243. Shindo, M.; Oya, S.; Sato, Y.; Shishido, K. Heterocycles 1998, 49, 113.
2244. Cho, C.S.; Jiang, L.H.; Shim, S.C. Synth. Commun. 1999, 29, 2695.
2245. Naskar, D.; Roy, S. J. Chem. Soc., Perkin Trans. 1 1999, 2435.
2246. See Davoli, P.; Forni, A.; Moretti, I.; Prati, F.; Torre, G. Tetrahedron 2001, 57, 1801.
2247. Lu, S.-M.; Alper, H. J. Org. Chem. 2004, 69, 3558.
2248. Awasthi, C.; Yadav, L.D.S. Synlett 2010, 1783.
2249. Ugi, I. Isonitrile Chemistry, Academic Press, NY, 1971; Walborsky, H.M.; Periasamy, M.P. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 2, Wiley, NY, 1983, pp. 835–887; Hoffmann, P.; Marquarding, D.; Kliimann, H.; Ugi, I. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 853–883.
2250. Lim, Y.Y.; Stein, A.R. Can. J. Chem. 1971, 49, 2455.
2251. Cunningham, I.D.; Buist, G.J.; Arkle, S.R. J. Chem. Soc. Perkin Trans. 2 1991, 589.
2252. Suginome, M.; Fukuda, T.; Ito, Y. Org. Lett. 1999, 1, 1977.
2253. Ugi, I. Angew. Chem. Int. Ed. 1982, 21, 810; Marquarding, D.; Gokel, G.W.; Hoffmann, P.; Ugi, I. in Ugi, I. Isonitrile Chemistry, Academic Press, NY, 1971, pp. 133–143, Gokel, G.W.; Lüdke, G.; Ugi, I. in Ugi, I. Ref. 936, pp. 145–199, 252–254.
2254. Denmark, S.E.; Fan, Y. J. Org. Chem. 2005, 70, 9667.
2255. Koszelewski, D.; Szymanski, W.; Krysiak, J.; Ostaszewski, R. Synth. Commun. 2008, 38, 1120.
2256. Fan, X.; Li, Y.; Zhang, X.; Qu, G.; Wang, J. Can. J. Chem. 2006, 84, 794.
2257. See Jenner, G. Tetrahedron Lett. 2000, 43, 1235.
2258. Pan, S.C.; List, B. Angew. Chem. Int. Ed. 2008, 47, 3622.
2259. Constabel, F.; Ugi, I. Tetrahedron 2001, 57, 5785.
2260. El Kaïm, L.; Grimaud, L.; Schiltz, A. Org. Biomol. Chem. 2009, 7, 3024.
2261. Godet, T.; Bovin, Y.; Vincent, G.; Merle, D.; Thozet, A.; Ciufolini, M.A. Org. Lett. 2004, 6, 3281.
2262. Ugi, I. in Gross, E.; Meienhofer, J. The Peptides, Vol. 2, Academic Press, NY, 1980, pp. 365–381, Intra-Sci. Chem. Rep. 1971, 5, 229; Gokel, G.W.; Hoffmann, P.; Kleimann, H.; Klusacek, H.; Lüdke, G.; Marquarding, D.; Ugi, I. in Ugi, I. Isonitrile Chemistry, Academic Press, NY, 1971, pp. 201–215. See also, Kunz, H.; Pfrengle, W. J. Am. Chem. Soc. 1988, 110, 651.
2263. Okandeji, B.O.; Gordon, J.R.; Sello, J.K. J. Org. Chem. 2008, 73, 5595.
2264. See Ito, Y.; Murakami, M. Synlett 1990, 245.
2265. Walborsky, H.M. J. Org. Chem. 1981, 46, 5405; 1982, 47, 52. See also, Murakami, H.; Ito, H.; Ito, Y. J. Org. Chem. 1988, 53, 4158.
2266. See Cooper, A.J.L.; Ginos, J.Z.; Meister, A. Chem. Rev. 1983, 83, 321.
2267. Jones, W.D.; Foster, G.P.; Putinas, J.M. J. Am. Chem. Soc. 1987, 109, 5047.
2268. See Ciuffarin, E.; Fava, A. Prog. Phys. Org. Chem. 1968, 6, 81.
2269. For a comparative reactivity study, see Hirata, R.; Kiyan, N.Z.; Miller, J. Bull. Soc. Chim. Fr. 1988, 694.
2270. See Gordon, I.M.; Maskill, H.; Ruasse, M. Chem. Soc. Rev. 1989, 18, 123.
2271. Sabol, M.A.; Andersen, K.K. J. Am. Chem. Soc. 1969, 91, 3603. See also, Jones, M.R.; Cram, D.J. J. Am. Chem. Soc. 1974, 96, 2183.
2272. Kaiser, E.T.; Zaborsky, O.R. J. Am. Chem. Soc. 1968, 90, 4626.
2273. Lee, I.; Kang, H.K.; Lee, H.W. J. Am. Chem. Soc. 1987, 109, 7472; Arcoria, A.; Ballistreri, F.P.; Spina, E.; Tomaselli, G.A.; Maccarone, E. J. Chem. Soc. Perkin Trans. 2 1988, 1793; Gnedin, B.G.; Ivanov, S.N.; Shchukina, M.V. J. Org. Chem. USSR 1988, 24, 731.
2274. Ciuffarin, E.; Senatore, L.; Isola, M. J. Chem. Soc. Perkin Trans. 2 1972, 468.
2275. Ciuffarin, E.; Senatore, L. Tetrahedron Lett. 1974, 1635.
2276. Opitz, G. Angew. Chem. Int. Ed. 1967, 6, 107. See Thea, S.; Guanti, G.; Hopkins, A.; Williams, A. J. Org. Chem. 1985, 50, 5592; Skonieczny, S. Tetrahedron Lett. 1987, 28, 5001; Pregel, M.J.; Buncel, E. J. Chem. Soc. Perkin Trans. 2 1991, 307.
2277. King, J.F. Acc. Chem. Res. 1975, 8, 10; Nagai, T.; Tokura, N. Int. J. Sulfur Chem. Part B 1972, 207; Opitz, G. Angew. Chem. Int. Ed. 1967, 6, 107; Wallace, T.J. Q. Rev. Chem. Soc. 1966, 20, 67.
2278. See Netscher, T.; Prinzbach, H. Synthesis 1987, 683.
2279. See Tagaki, W.; Kurusu, T.; Oae, S. Bull. Chem. Soc. Jpn. 1969, 42, 2894.
2280. Kice, J.L.; Legan, E. J. Am. Chem. Soc. 1973, 95, 3912.
2281. Chang, F.C. Tetrahedron Lett. 1964, 305.
2282. Cuvigny, T.; Larchevêque, M. J. Organomet. Chem. 1974, 64, 315.
2283. Sridhar, M.; Kumar, B.A.; Narender, R. Tetrahedron Lett. 1998, 39, 2847.
2284. Reddy, G.S.; Mohan, G.H.; Iyengar, D.S. Synth. Commun. 2000, 30, 3829.
2285. See Simpson, L.S.; Widlanski, T.S. J. Am. Chem. Soc. 2006, 128, 1605.
2286. Rogne, O. J. Chem. Soc. B 1971, 1334. See also, Litvinenko, M.; Shatskaya, V.A.; Savelova, V.A. Doklad. Chem. 1982, 265, 199.
2287. Klamann, D.; Fabienke, E. Chem. Ber. 1960, 93, 252.
2288. Padmapriya, A.A.; Just, G.; Lewis, N.G. Synth. Commun. 1985, 15, 1057.
2289. Karaman, R.; Leader, H.; Goldblum, A.; Breuer, E. Chem. Ind. (London) 1987, 857.
2290. Martinelli, M.J.; Vaidyanathan, R.; Khau, V.V. Tetrahedron Lett. 2000, 41, 3773; Bucher, B.; Curran, D.P. Tetrahedron Lett. 2000, 41, 9617.
2291. De Luca, L.; Giacomelli, G. J. Org. Chem. 2008, 73, 3967.
2292. See Kamal, A.; Reddy, J.S.; Bharathi, E.V.; Dastagiri, D. Tetrahedron Lett. 2008, 49, 348.
2293. See Gambill, C.R.; Roberts, T.D.; Shechter, H. J. Chem. Educ. 1972, 49, 287.
2294. Fanta, P.E.; Wang, C.S. J. Chem. Educ. 1964, 41, 280.
2295. Hendrickson, J.B.; Bergeron, R. Tetrahedron Lett. 1970, 345.
2296. For an example, see Regitz, M.; Hocker, J.; Liedhegener, A. Org. Synth. V, 179.
2297. Milan, D.S.; Prager, R.H. Aust. J. Chem. 1999, 52, 841.
2298. Kim, D.Y.; Kim. H.S.; Choi, Y.J.; Mang, J.Y.; Lee, K. Synth. Commun. 2001, 31, 2463.
2299. Bouchez, L.C.; Dubbaka, S.R.; Urks, M.; Vogel, P. J. Org. Chem. 2004, 69, 6413.
2300. Blotny, G. Tetrahedron Lett. 2003, 44, 1499.
2301. Poshkus, A.C.; Herweh, J.E.; Magnotta, F.A. J. Org. Chem. 1963, 28, 2766; Litvinenko, L.M.; Dadali, V.A.; Savelova, V.A.; Krichevtsova, T.I. J. Gen. Chem. USSR 1964, 34, 3780.
2302. See Bianchi, T.A.; Cate, L.A. J. Org. Chem. 1977, 42, 2031, and references cited therein.
2303. Frye, L.L.; Sullivan, E.L.; Cusack, K.P.; Funaro, J.M. J. Org. Chem, 1992, 57, 697.
2304. Baarschers, W.H. Can. J. Chem. 1976, 54, 3056.
2305. Neumann, W.P.; Wicenec, C. Chem. Ber. 1993, 126, 763.
2306. Volla, C.M.R.; Vogel, P. Angew. Chem. Int. Ed. 2008, 47, 1305.
2307. Sun, X.; Wang, L.; Zhang, Y. Synth. Commun. 1998, 28, 1785.
2308. Labadie, S.S. J. Org. Chem. 1989, 54, 2496.
2309. See Waykole, L.; Paquette, L.A. Org. Synth. 67, 149.
2310. Steensma, R.W.; Galabi, S.; Tagat, J.R.; McCombie, S.W. Tetrahedron Lett. 2001, 42, 2281.
2311. Bandgar, B.P.; Bettigeri, S.V.; Phopase, J. Org.Lett. 2004, 6, 2105.
2312. Beaulieu, C.; Guay, D.; Wang, Z.; Evans, D.A. Tetrahedron Lett. 2004, 45, 3233.