March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 18. Rearrangements
In a rearrangement reaction, a group moves from one atom to another in the same molecule.1 Most are migrations from an atom to an adjacent one (called 1,2-shifts), but some are over longer distances. The group
![]()
that migrates (W) may move with its electron pair (these can be called nucleophilic or anionotropic rearrangements; the migrating group can be regarded as a nucleophile), without its electron pair (electrophilic or cationotropicrearrangements; in the case of migrating hydrogen, prototropic rearrangements), or with just one electron (radical rearrangements). The atom A is called the migration origin and B is the migration terminus. However, there are some rearrangements that do not lend themselves to neat categorization in this manner. Among these are those with cyclic transition states (Reactions 18-27–18-36).
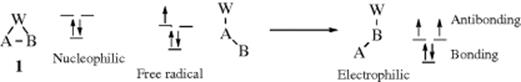
As will be seen, nucleophilic 1,2-shifts are much more common than electrophilic or free radical 1,2-shifts. The reason for this can be seen by a consideration of the transition states (or in some cases intermediates) involved. The transition state or intermediate for all three cases can be represented by 1, in which the two-electron A–W bond overlaps with the orbital on atom B, which contains zero, one, and two electrons, in the case of nucleophilic, free radical, and electrophilic migration, respectively. The overlap of these orbitals gives rise to three new orbitals, which have an energy relationship similar to those in Section 2.K.i (one bonding and two degenerate antibonding orbitals). In a nucleophilic migration, where only two electrons are involved, both can go into the bonding orbital and 1 is a low-energy transition state, In a free radical or electrophilic migration, there are, respectively, three or four electrons that must be accommodated, and antibonding orbitals must be occupied. It is not surprising therefore that, when 1,2-electrophilic or free radical shifts are found, the migrating group W is usually aryl or some other group that can accommodate the extra one or two electrons and thus effectively remove them from the three-membered transition state or intermediate (see 41).
In any rearrangement two possible modes of reaction can, in principle, be distinguished. In one of these, the group W becomes completely detached from A and may end up on the B atom of a different molecule (intermolecularrearrangement). In the other, W goes from A to B in the same molecule (intramolecular rearrangement), in which case there must be some continuing tie holding W to the A–B system, preventing it from coming completely free. Strictly speaking, only the intramolecular type fits our definition of a rearrangement, but the general practice, which is followed here, is to include under the title “rearrangement” all net rearrangements whether they are inter- or intramolecular. It is usually not difficult to tell whether a given rearrangement is inter- or intramolecular. The most common method involves the use of cross over experiments. In this type of experiment, rearrangement is carried out on a mixture of W–A–B and V–A–C, where V is closely related to W (say, methyl vs ethyl) and B to C. In an intramolecular process, only A–B–W and A–C–V are recovered, but if the reaction is intermolecular, then not only will these two be found, but also A–B–V and A–C–W.
18.A. Mechanisms
18.A.i. Nucleophilic Rearrangements2
Broadly speaking, such rearrangements consist of three steps, of which the actual migration is the second:
![]()
This process has been called the Whitmore 1,2-shift.3 Since the migrating group carries the electron pair with it, the migration terminus B must be an atom with only six electrons in its outer shell (an open sextet). The first step therefore is creation of a system with an open sextet. Such a system can arise in various ways, but two of these are the most important:
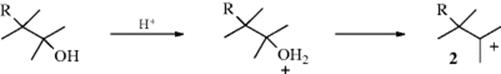
1. Formation of a Carbocation: These can be formed in a number of ways (see Sec. 5.A.iii), but one of the most common methods is the acid treatment of an alcohol to give 2 from an intermediate oxonium ion. Such carbocations are subject to rearrangement to a more stable carbocation. These two steps are of course the same as the first two steps of the SN1cA or the E1 reactions of alcohols.
2. Formation of a Nitrene: The decomposition of acyl azides is one of several ways in which acyl nitrenes (3) are formed (see Sec. 5.E). After the migration has taken place, the atom at the migration origin (A) must necessarily have an open sextet. In the third step, this atom acquires an octet. In the case of carbocations, combinations with a nucleophile (rearrangement with substitution) and loss of H+ (rearrangement with elimination). Constitute the most common third stop.
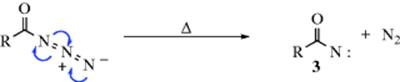
Although this mechanism is presented as taking place in three steps, and some reactions do take place in this way, in others the transformation is simultaneous. For example, in the nitrene example above, as the R migrates, an electron pair from the nitrogen moves into the C–N bond to give a stable isocyanate, (4). In this example, what are shown as the second and third steps are simultaneous. It is also possible for the second and third steps to be simultaneous even when the “third” step involves more than just a simple motion of a pair of electrons. Similarly, there are many reactions in which the first two steps are simultaneous; that is, there is no actual formation of a species (e.g., 2 or 3). In these instances, it may be said that R assists in the removal of the leaving group, with migration of R and the removal of the leaving group taking place simultaneously. Many investigations have been carried out in attempts to determine, in various reactions, whether such intermediates as (2) or (3) actually form, or whether the steps are simultaneous (see, e.g., the discussions in Reaction 16-45 and Sec. 18.A.ii), but the difference between the two possibilities is often subtle, and the question is not always easily answered.4

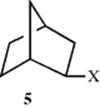
Evidence for this mechanism is that rearrangements of this sort occur under conditions where previously carbocations have been encountered: SN1 conditions, Friedel–Crafts alkylation, and so on. Solvolysis of neopentyl bromide leads to rearrangement products, and the rate increases with increasing ionizing power of the solvent, but is unaffected by concentration of base,5 so that the first step is carbocation formation. The same compound under SN2 conditions gave no rearrangement, but slowly gave only ordinary substitution. Thus with neopentyl bromide, formation of a carbocation leads only to rearrangement. Carbocations usually rearrange to more stable carbocations. Thus the direction of rearrangement is usually primary → secondary → tertiary. Neopentyl (Me3CCH2), neophyl (PhCMe2CH2), and norbornyl (e.g., 5) type systems are especially prone to carbocation rearrangement reactions. It has been shown that the rate of migration increases with the degree of electron deficiency at the migration terminus.6
It was previously mentioned (Sec. 5.A.ii) that stable tertiary carbocations could be obtained, in solution, at very low temperatures. The NMR studies have shown that when these solutions are warmed, rapid migrations of hydride and of alkyl groups take place, resulting in an equilibrium mixture of structures.7 For example, the tert-pentyl cation (5)8 equilibrates as follows:
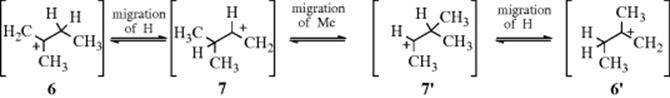
Carbocations that rearrange to give products of identical structure ![]() are called degenerate carbocations. Such rearrangements are degenerate rearrangements and many examples are known.9
are called degenerate carbocations. Such rearrangements are degenerate rearrangements and many examples are known.9
18.A.ii. The Actual Nature of the Migration
Most nucleophilic 1,2-shifts are intramolecular. The W group does not become free, but always remains connected in some way to the substrate. Apart from the evidence from cross-over experiments, the strongest evidence is that when the W group is chiral, the configuration is retained in the product. For example, (+)-PhCHMeCO2H was converted to (−)-PhCHMeNH2 by the Curtius (18-14), Hofmann (18-13), Lossen (18-15), and Schmidt (18-16) reactions.10 In these reactions, the extent of retention varied from 95.8 to 99.6%. Retention of configuration in the migrating group has been shown many times since.11 Another experiment demonstrating retention was the easy conversion of 8 to 9.11 Neither inversion nor racemization could take place at a bridgehead.
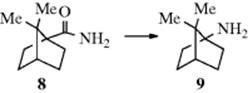
Using the simple example W-A-B, there is much evidence that retention of configuration usually occurs in W, and inversion never occurs.12 However, this is not the state of affairs at A and B. In many reactions, of course, the structure of W–A–B is such that the product has only one steric possibility at A or B or both, and in most of these cases nothing can be learned. But in cases where the steric nature of A or B can be investigated, the results are mixed. It has been shown that either inversion or racemization can occur at A or B. One example is 10, where the conversion proceeded with inversion (equivalent to inversion at B.13 Inversion at A has been
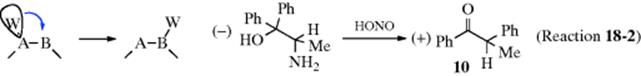
shown in other cases.14 However, in many other cases, racemization occurs at A or B or both.15 It is not always necessary for the product to have two steric possibilities in order to investigate the stereochemistry at A or B. Thus, in most Beckmann rearrangements (18-17), only the group trans (usually called anti) to the hydroxyl group migrates, as illustrated by formation of 11, which shows inversion at B.

This information tells us about the degree of concertedness of the three steps of the rearrangement. First, consider the migration terminus B in R–A–B–X. If racemization is found at B, it is probable that the first step takes place before the second and that a positively charged carbon (or other sextet atom) is present at B:
![]()
With respect to B this is an SN1 type process. If inversion occurs at B, it is likely that the first two steps are concerted, that a carbocation is not an intermediate, and that the process is SN2 like:
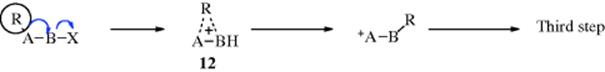
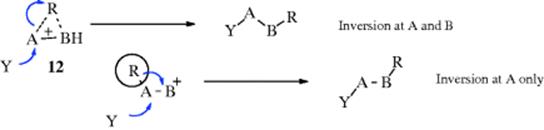
In this case, participation by R assists in removal of X in the same way that neighboring groups do (Sec. 10.C). Indeed, R is a neighboring group here. The only difference is that, in the case of the neighboring-group mechanism of nucleophilic substitution, R never becomes detached from A, while in a rearrangement the bond between R and A is broken. In either case, the anchimeric assistance results in an increased rate of reaction. Of course, for such a process to take place, R must be in a favorable geometrical position (R and X antiperiplanar). Intermediate 12 may be a true intermediate, or there may only be a transition state, depending on what migrates. In certain cases of the SN1 type process, it is possible for migration to take place with net retention of configuration at the migrating terminus because of conformational effects in the carbocation.16
A few conclusions may be summarized:
1. The SN1 type process occurs mostly when B is a tertiary atom or has one aryl group and at least one other alkyl or aryl group. In other cases, the SN2 type process is more likely. Inversion of configuration (indicating an SN2 type process) has been shown for a neopentyl substrate by the use of the chiral neopentyl-1-d alcohol.17 There is other evidence that neopentyl systems undergo rearrangement by a carbocation (SN1 type) mechanism.18
2. The question as to whether 12 is an intermediate or a transition state has been much debated. When R is aryl or vinyl, then 12 is probably an intermediate and the migrating group lends anchimeric assistance19 (see Sec. 10.C.i, category 3-preceding category 4 for resonance stabilization of this intermediate, when R is aryl). When R is alkyl, 12 is a protonated cyclopropane (edge- or corner protonated; see Sec. 15.B.iv). There is much evidence that in simple migrations of a methyl group, the bulk of the products formed do not arise from protonated cyclopropane intermediates. Evidence for this statement has already been given (Sec. 10.C.i, category 4c). Further evidence was obtained from experiments involving labeling.
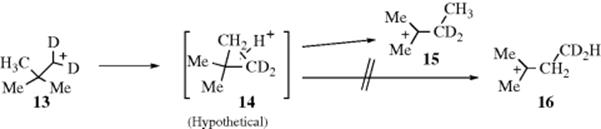
Rearrangement of the neopentyl cation labeled with deuterium in the 1 position (13) gave only tert-pentyl products with the label in the 3 position (derived from 15), though if 14 were an intermediate, the cyclopropane ring could just as well cleave the other way to give tert-pentyl derivatives labeled in the 4 position (derived from 16).20 Another experiment that led to the same conclusion was the generation, in several ways, of Me3C13CH2+. The only tert-pentyl products isolated were labeled in C-3, that is, Me2C+–13CH2CH3 derivatives; no derivatives of Me2C+–CH213CH3 were found.21
Although the bulk of the products are not formed from protonated cyclopropane intermediates, there is considerable evidence that at least in 1-propyl systems, a small part of the product can in fact arise from such intermediates.22 Among this evidence is the isolation of 10–15% cyclopropanes (mentioned in Sec. 10.C.i, category 4c). Additional evidence comes from propyl cations generated by diazotization of labeled amines (CH3CH2CD2+, CH3CD2CH2+, CH3CH214CH2+), where isotopic distribution in the products indicated that a small amount (~5%) of the product had to be formed from protonated cyclopropane intermediates, for example,23
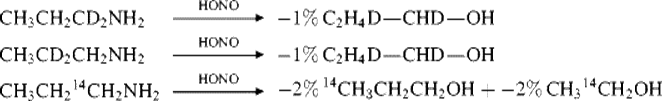
Even more scrambling was found in trifluoroacetolysis of 1-propyl-1-![]() -mercuric perchlorate.24 However, protonated cyclopropane intermediates accounted for <1% of the products from diazotization of labeled isobutylamine25 and from formolysis of labeled 1-propyl tosylate.26
-mercuric perchlorate.24 However, protonated cyclopropane intermediates accounted for <1% of the products from diazotization of labeled isobutylamine25 and from formolysis of labeled 1-propyl tosylate.26
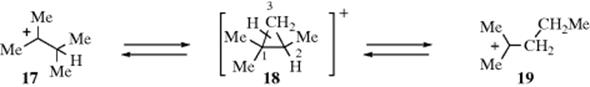
It is likely that protonated cyclopropane transition states or intermediates are also responsible for certain non-1,2 rearrangements. For example, in superacid solution, the ions 17 and 19 are in equilibrium. It is not possible for these to interconvert solely by 1,2-alkyl or hydride shifts unless primary carbocations, which are highly unlikely, are intermediates. However, the reaction can be explained27 by postulating that (in the forward reaction) it is the 1,2-bond of the intermediate or transition state 18 that opens up rather than the 2,3-bond, which is the one that would open if the reaction were a normal 1,2-shift of a methyl group. In this case, opening of the 1,2-bond produces a tertiary cation, while opening of the 2,3-bond would give a secondary cation. (In the reaction 19 → 17, it is of course the 1,3-bond that opens).
3. There has been much discussion of H as the migrating group. There is no conclusive evidence that 10 in this case is or is not a true intermediate, although both positions have been argued (see Sec. 10.C.i, category 4c).
The stereochemistry at the migration origin A is less often involved, since in most cases it does not end up as a tetrahedral atom; but when there is inversion here, there is an SN2 type process at the beginning of the migration. This may or may not be accompanied by an SN2 process at the migration terminus B:
In some cases, it has been found that, when H is the migrating species, the configuration at A may be retained.28
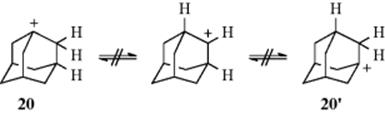
There is evidence that the configuration of the molecule may be important even where the leaving group is gone long before migration takes place. For example, the 1-adamantyl cation (20) does not equilibrate intramolecularly, even at temperatures up to 130 °C,29 though open-chain (e.g., 6-6′) and cyclic tertiary carbocations undergo such equilibration at 0 °C or below. On the basis of this and other evidence, it has been concluded that for a 1,2-shift of hydrogen or methyl to proceed as smoothly as possible, the vacant p orbital of the carbon bearing the positive charge and the sp3 orbital carrying the migrating group must be coplanar,29 which is not possible for 20.
18.A.iii. Migratory Aptitudes30
In many reactions, there is no question about which group migrates. For example, in the Hofmann, Curtius, and similar reactions there is only one possible migrating group in each molecule, and one can measure migratory aptitudes only by comparing the relative rearrangement rates of different compounds. In other instances, there are two or more potential migrating groups, but which migrates is settled by the geometry of the molecule. The Beckmann rearrangement (Reaction 18-17) provides an example. As seen in the formation of 11, only the group trans to the OH migrates. In compounds whose geometry is not restricted in this manner, there still may be eclipsing effects (see Sec. 17.C), so that the choice of migrating group is largely determined by which group is in the right place in the most stable conformation of the molecule.31 However, in some reactions, especially the Wagner–Meerwein (18-1) and the pinacol (18-2) rearrangements, the molecule may contain several groups that, geometrically at least, have approximately equal chances of migrating, and these reactions have often been used for the direct study of relative migratory aptitudes. In the pinacol rearrangement, there is the additional question of which OH group leaves and which does not, since a group can migrate only if the OH group on the other carbon is lost.
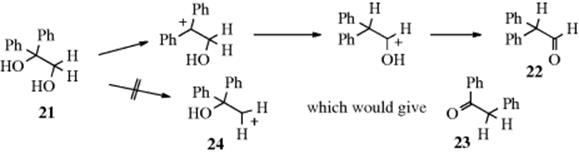
We deal with the second question first. To study this question, the best type of substrate to use is one of the form R2C(OH)–C(OH)R′2, since the only thing that determines migratory aptitude is which OH group comes off. Once the OH group is gone, the migrating group is determined. As might be expected, the OH that leaves is the one whose loss gives rise to the more stable carbocation. Thus 1,1-diphenylethanediol (21) gives diphenylacetaldehyde (22), not phenylacetophenone (23). Obviously, it does not matter in this case whether phenyl has a greater inherent migratory aptitude than hydrogen or not. Only the hydrogen can migrate because 24 is not formed. Remember that carbocation stability is enhanced by groups in the order aryl > alkyl > hydrogen, and this normally determines which side loses the OH group. However, exceptions are known, and which group is lost may depend on the reaction conditions (e.g., see the reaction of 59).
In order to answer the question about inherent migratory aptitudes, the obvious type of substrate to use (in the pinacol rearrangement) is R′RC(OH)–COH)RR′, since the same carbocation is formed no matter which OH leaves, and it would seem that a direct comparison of the migratory tendencies of R and R′ is possible. On closer inspection, however, it is clear that several factors are operating. Apart from the question of possible conformational effects, already mentioned, there is also the fact that whether the group R or R′ migrates is determined not only by the relative inherent migrating abilities of R and R′, but also by whether the group that does not migrate is better at stabilizing the positive charge that will now be found at the migration origin.32 Thus, migration of R gives rise to the cation R′C+(OH)CR2R2′, while migration of R′ gives the cation R+C(OH)CRR2′ and these cations have different stabilities. It is possible that in a given case R might be found to migrate less than R′, not because it actually has a lower inherent migrating tendency, but because it is much
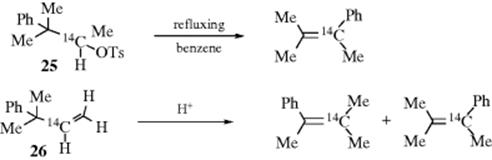
better at stabilizing the positive charge. In addition to this factor, the migrating ability of a group is also related to its capacity to render anchimeric assistance to the departure of the nucleofuge. An example of this effect is the decomposition of tosylate (25) where only the phenyl group migrates, while in acid treatment of the corresponding alkene (26), there is competitive migration of both methyl and phenyl (in these reactions ![]() labeling is necessary to determine which group has migrated).33 Both 25 and 26 give the same carbocation; the differing results must be caused by the fact that in 25 the phenyl group can assist the leaving group, while no such process is possible for 26. This example clearly illustrates the difference between migration to a relatively free terminus and one that proceeds with the migrating group lending anchimeric assistance.34
labeling is necessary to determine which group has migrated).33 Both 25 and 26 give the same carbocation; the differing results must be caused by the fact that in 25 the phenyl group can assist the leaving group, while no such process is possible for 26. This example clearly illustrates the difference between migration to a relatively free terminus and one that proceeds with the migrating group lending anchimeric assistance.34
Therefore, it is not surprising that clear-cut answers as to relative migrating tendencies are not available. More often than not migratory aptitudes are in the order aryl > alkyl, but exceptions are known, and the position of hydrogen in this series is often unpredictable. In some cases, migration of hydrogen is preferred to aryl migration; in other cases, migration of alkyl is preferred to that of hydrogen. Mixtures are often found, and the isomer that predominates often depends on conditions. For example, the comparison between methyl and ethyl has been made many times in various systems, and in some cases methyl migration and in others ethyl migration has been found to predominate.35 However, it can be said that among aryl migrating groups, electron-donating substituents in the para and meta positions increase the migratory aptitudes, while the same substituents in the ortho positions decrease them. Electron-withdrawing groups decrease migrating ability in all positions. The following are a few of the relative migratory aptitudes determined for aryl groups:36 p-anisyl, 500; p-tolyl, 15.7; m-tolyl, 1.95; phenyl, 1.00; p-chlorophenyl, 0.7; o-anisyl, 0.3. For the o-anisyl group, the poor migrating ability probably has a steric cause, while for the others there is a fair correlation with activation or deactivation of electrophilic aromatic substitution, which is what the process is with respect to the benzene ring. It has been reported that at least in certain systems acyl groups have a greater migratory aptitude than alkyl groups.37
18.A.iv. Memory Effects38
Solvolysis of the endo bicyclic compound (27, X = ONs, see Sec. 10.G.iii, or Br) gave mostly the bicyclic allylic alcohol (30), along with a smaller amount of the tricyclic alcohol (34), while solvolysis of the exo isomers, (31), gave mostly 34, with smaller amounts of 30.39 Note that endo and exo here refers to the position of the XCH2 group over the C=C unit or opposite the C=C unit, respectively. The two isomers gave entirely different ratios of products, although the carbocation initially formed seems to be the same for each (marked 28 and 32 for convenience). With 28, a second rearrangement (a shift of the 1,7 bond) follows to give 29, while with 32
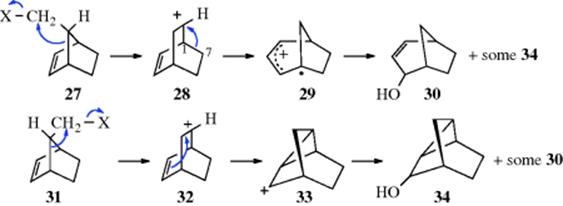
an intramolecular addition of the positive carbon to the double bond gives 33. It seems as if 28 and 32 “remember” how they were formed before they go on to give the second step. Such effects are called memory effects and other such cases are known.40 The causes of these effects are not well understood, although there has been much discussion. One possible cause is differential solvation of the apparently identical ions 28 and 32. Other possibilities are (1) that the ions have geometrical structures that are twisted in opposite senses (e.g., a twisted 32 might have its positive
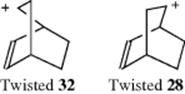
carbon closer to the double bond than a twisted 28); (2) that ion pairing is responsible41; and (3) that nonclassical carbocations are involved.42 One possibility that has been ruled out is that steps 27 → 28 → 29 and 31 → 32 → 33 are concerted, so that 28/32 never exist at all. This possibility has been excluded by several kinds of evidence, including the fact that 27 gives not only 30, but also some 34; and 31 gives some 30 along with 34. This means that some of the 28 and 32 ions interconvert, a phenomenon known as leakage.