March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 2. Delocalized Chemical Bonding
2.C. Molecules that have Delocalized Bonds
There are four main types of structure that exhibit delocalization:
1. Double (or Triple) Bonds in Conjugation.34 The double bonds in benzene are conjugated, of course, but the conjugation exists in acyclic molecules (e.g., butadiene, 6). In the MO picture (Fig. 2.4), the overlap of four orbitals gives two bonding orbitals that contain four electrons and two vacant antibonding orbitals. It can be seen that each orbital has one more node than the one of next lower energy. The energies of the four orbitals are (lowest to highest): α + 1.618β, α + 0.618β, α − 0.618β, and α − 1.618β; hence the total energy of the two occupied orbitals is 4α + 4.472β. Since the energy of two isolated double bonds is 4α + 4β, the resonance energy by this calculation is 0.472β.
In the resonance picture, structures 7 and 8 are considered to contribute.
![]()
Despite the invocation of structures 7 and 8 in the resonance picture, butadiene and similar conjugated systems are not considered to be resonance stabilized in the ground state. The bond order of the central bond should be > 1 and that of the other carbon–carbon bonds < 2, although neither predicts that the three bonds have equal electron density. Molecular orbital bond orders of 1.894 and 1.447 have been calculated.35
The existence of delocalization in butadiene and similar molecules has been questioned. The bond lengths in butadiene are 1.34 Å for the double bonds and 1.48 Å for the single bond.36 Since the typical single-bond distance of a bond that is not adjacent to an unsaturated group is 1.53 Å (Sec. 1.K), it has been argued that the shorter single bond in butadiene provides evidence for resonance. However, this shortening can also be explained by hybridization changes (See Sec. 1.K); and other explanations have also been offered.37 Resonance energies for butadienes, calculated from heats of combustion or hydrogenation, are only ~4 kcal mol−1 (17 kJ mol−1), and these values may not be entirely attributable to resonance.38 Thus, a calculation from heat of atomization data gives a resonance energy of 4.6 kcal mol−1 (19 kJ mol−1) for cis-1,3-pentadiene, and −0.2 kcal mol−1 (−0.8 kJ mol−1), for 1,4-pentadiene. These two compounds, each of which possesses two double bonds, two C–C single bonds, and eight C–H bonds, would seem to offer a direct comparison of a conjugated with a nonconjugated compound, but they are nevertheless not strictly comparable. The former has three sp3 C–H and five sp2 C–H bonds, while the latter has two and six, respectively. Also, the two single C–C bonds of the 1,4-diene are both sp2–sp3 bonds, while in the 1,3-diene, one is sp2–sp3 and the other is sp2–sp2. Therefore, it may be that some of the already small value of 4 kcal mol−1 (17 kJ mol−1) is not resonance energy, but arises from differing energies of bonds of different hybridization.39 As noted above, butadiene and related molecules are generally considered not to be resonance stabilized in the ground state.
Although bond distances fail to show it and the resonance energy is low, butadiene is planar.40 This has been taken as an indication that there is some delocalization. Similar effects are found in other conjugated systems (e.g., C=C–C=O41 and C=C–C=N), in longer systems with three or more multiple bonds in conjugation, and where double or triple bonds are conjugated with aromatic rings. Diynes (e.g., 1,3-butadiyne, 9) are also conjugated molecules. Based on calculations, Rogers et al.42 reported that the conjugation stabilization of 1,3-butadiyne is zero. Later calculations concluded that consideration of hyperconjugative interactions (Sec. 2.M) provides a more refined measure of conjugative stabilization.43 When this measure is used, the conjugation energies of the isomerization and hydrogenation reactions considered agree with a conjugative stabilization of 9.3 (0.5 kcal mol−1 for diynes and 8.2 0.1 kcal mol−1 for dienes).
![]()
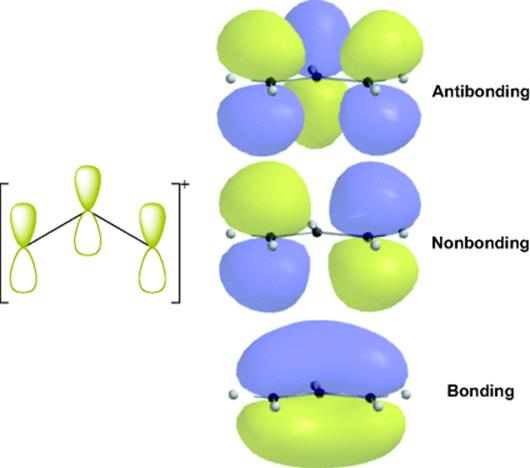
2. Double (or Triple) Bonds in Conjugation with a p Orbital on an Adjacent Atom. When a p orbital is on an atom adjacent to a double bond, there are three parallel p orbitals that overlap. As previously noted, it is a general rule that the overlap of n atomic orbitals creates n molecular orbitals, so overlap of a p orbital with an adjacent double bond gives rise to three new orbitals, as shown in Fig. 2.5. The middle orbital is a nonbonding orbital of zero-bonding energy. The central carbon atom does not participate in the nonbonding orbital.
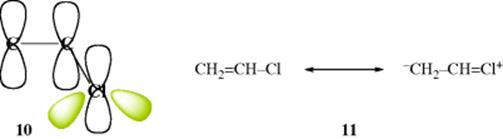
There are three cases: the original p orbital may have contained two, one, or no electrons. Since the original double bond contributes two electrons, the total number of electrons accommodated by the new orbitals is four, three, or two, respectively. A typical example of the first situation is vinyl chloride, (CH2=CH–Cl). Although the p orbital of the chlorine atom is filled, it still overlaps with the double bond (See 10). The four electrons occupy the two molecular orbitals of lowest energies, which is an example of resonance involving overlap between unfilled orbitals and a filled orbital. Canonical forms for vinyl chloride are shown in 11 (see Sec. 2.M).
Any system that contains an atom that has an unshared pair and is directly attached to a multiple-bond atom can show this type of delocalization. Resonance delocalization is more important with charged species (e.g., the carbonate ion) and true resonance contributors can be drawn:
![]()
The resonance delocalization in allylic carbanions, (e.g., CH2=CH–CH2−), is another example.
The other two cases have a p orbital that contains only one electron (radicals) or no electrons (cations). Allylic free radicals have one electron in the nonbonding orbital. In allylic cations, this orbital is vacant and only the bonding orbital is occupied. The orbital structures of the allylic carbanion, free radical, and cation differ from each other, only in that the nonbonding orbital is filled, half-filled, or empty. Since this is an orbital of zero bonding energy, it follows that the bonding π energies of the three species relative to electrons in the 2p orbitals of free atoms are the same. The electrons in the nonbonding orbital do not contribute to the bonding energy, positively or negatively.44
The resonance picture best describes three species that are charged or contain an unshared electron with double bonds in conjugation with, respectively, an unshared pair, an unpaired electron, and an empty orbital as in the allyl cation 12 (also see Chap 5).
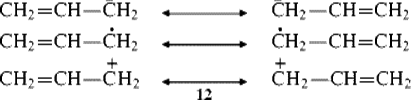
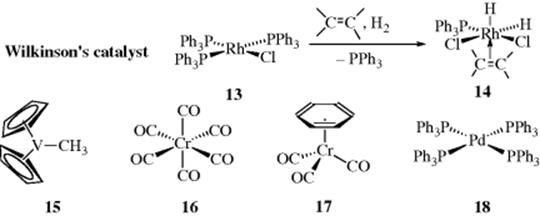
3. π-Allyl and Other η-Complexes. In the presence of transition metals, delocalized electrons in allylic cations may be donated to the metal, resulting in stabilization.45 In a carbon–metal bond (e.g., H3C–Fe), the carbon donates (shares) one electron with the metal, and is considered to be a one-electron donor. With a π bond (e.g., that found in ethylene), both electrons can be donated to the metal to form a complex (e.g., 14) by reaction of Wilkinson's catalyst (13) with an alkene and hydrogen gas,46 in addition, π bond is considered to be a two-electron donor. In these two cases, the electron donating ability of the group coordinated to the metal (the ligand) is indicated by terminology η1, η2, η3, and so on, for a one-, two-, and three-electron donor, respectively.
Ligands are categorized as η-ligands according to the ability to donate electrons to the metal. A hydrogen atom (as in 14) or a halogen ligand (as in 13) are η1 ligands. An amine (NR3), a phosphine (PR3, as in 13, 14, and 18), CO (as in 16 or 17), an ether (OR2) or a thioether (SR2) are η2 ligands. Hydrocarbon ligands include alkyl (as the methyl in 15) or aryl with a carbon-metal bond (η1), alkenes or carbenes (η2, see Sec. 3.C.i), π-allyl (η3), conjugated dienes (e.g., 1,3-butadiene) (η4), cyclopentadienyl (η5, as in 15 and see Sec. 2.I.ii), and arenes or benzene (η6).47 Note that in the formation of 14 from 13, the two-electron donor alkene displaces a two-electron donor phosphine. Other typical complexes include chromium hexacarbonyl (Cr(CO)6, 16), with six η2 CO ligands; η6-C6H6Cr(CO)3 (18), and tetrakis-triphenylphosphinopalladium (0), (17), with four η2 phosphine ligands.
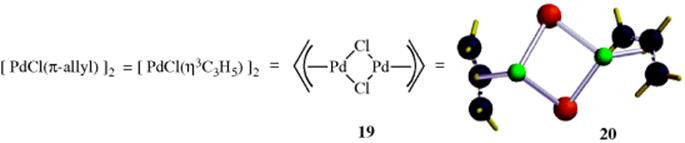
In the context of this section, the electron-delocalized ligand π-allyl (12) is an η3 donor and it is well known that allylic halides react with PdCl2 to form a bis-η3-complex 19 (see 20).48 Complexes (e.g., 19) react with nucleophiles to give the corresponding coupling product (10–60).49 The reaction of allylic acetates or carbons and a catalytic amount of Pd(0) compounds also lead to an η3-complex that can react with nucleophiles.50
4. Hyperconjugation. The type of delocalization called hyperconjugation is discussed in Section 2.M.
Note that there are examples of delocalization that cannot be strictly classified as any of these types.
Fig. 2.4 The four π orbitals of butadiene, formed by overlap of four p orbitals.
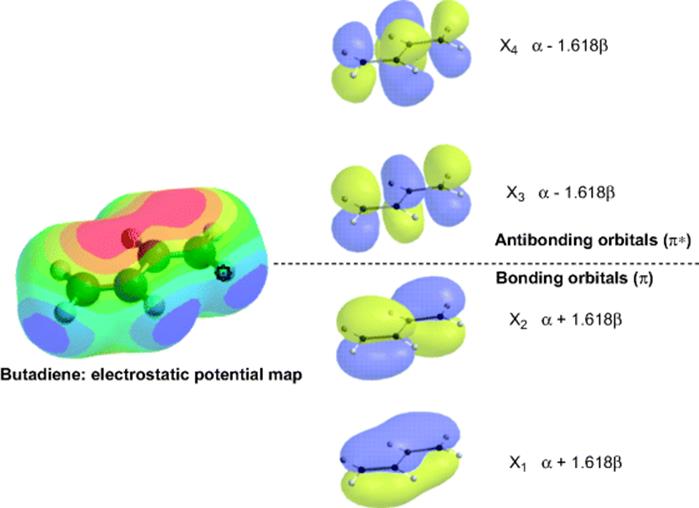
Fig. 2.5 Three orbitals of an allylic carbon, formed by overlap of three p orbitals.