March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 2. Delocalized Chemical Bonding
2.M. Hyperconjugation
Conjugation in molecules (e.g., 1,3-butadiene and benzene) was well known to organic chemists in the 19th century. For example, it became understood that one could view 1,3-butadiene as two ethylene units. The overlap of the end p orbitals of the two ethylenes that are to be attached leads to conjugation, which in turn leads to a lowering of the energy of the system, a change in geometry in several respects, and most obviously, to changes in chemical and physical properties of the molecule. In this case, two p orbitals overlap. Mulliken suggested that if one has a sigma orbital overlapping with a p orbital, then one has “hyperconjugation.” Qualitatively hyperconjugation is similar to conjugation, but smaller. When a methyl group is attached to ethylene, for example, there is a shift of the UV absorption spectrum to longer wavelength, an increase in the reactivity of the molecule, and a lowering of energy, similar to the changes that occur in the ethylene to butadiene case, but to a lesser extent. Hyperconjugation shows the same effects, but to a lesser amount, because the sigma orbital lies at a lower energy than the π orbital. Hence the electrons delocalize out of the sigma orbital in hyperconjugation to a lesser extent than from a π orbital in conjugation. The term hyperconjugation therefore arises from the hyperconjugative forms that make small but definite contributions to the ground state of a molecule.409
Another delocalization phenomenon has been discussed that involves σ electrons.410 Baker and Nathan411 observed that the rates of reaction of p-substituted benzyl bromides with pyridine (see Reaction 10-31) were opposite from the results expected from electron release. That is, the methyl-substituted compound reacted fastest and the tert-butyl substituted compounded reacted slowest. This appeared to be an anomalous electron-release pattern for alkyl groups because the field effect predicted the order of electron release for simple alkyl groups connected to an unsaturated system should be tert-butyl > isopropyl > ethyl > methyl. At least one hydrogen atom should be attached to the α-carbon that is connected to the sp2 carbon for the Baker–Nathan effect. The Baker–Nathan effect has clouded the issue because some interpreted that it indicates hyperconjugation occurs with hydrogen and a double bond, but either did not occur or occurred to a very small amount with carbon. In the 1930s, Baker and Nathan did not have the tools to experimentally or theoretically understand hyperconjugation to any great extent. Those chemists looked for lowering of energy that could be detected, but these experiments do not necessarily help to explain hyperconjugation. Indeed, it is now known that the Baker–Nathan effect is a result of changes in solvation energy412 and has very little to do with hyperconjugation. It was recently reported that hyperconjugation is an important factor determining alkane C–H bond dissociation energies.413 In certain instances where the Baker–Nathan effect was found to apply in solution, the order was completely reversed in the gas phase.414 Since the molecular structures are unchanged in going from the gas phase into solution, it appears that each alkyl group is solvated to a different extent.415 However, this only demonstrates that the Baker–Nathan effect is not the same as hyperconjugation. The structural changes that occur upon hyperconjugation can be determined from quantum mechanics and from experiment. Such changes can be qualitatively predicted by looking at the resonance structures involved.
Hyperconjugation is probably important for carbocations, as well as for free radicals416 and for excited states of molecules.417 In free radicals and carbocations, the canonical forms display no more charge separation than the main form. Muller and Mulliken call this isovalent hyperconjugation.
Apart from contributions to aromaticity, hyperconjugation has been used to explain the stability of intermediates (e.g., carbocations, see Chapter 5 for an introduction to carbocations). It was stated that C–C hyperconjugation is most important in stabilizing carbocations when the C–C bond(s) involved have more than 75% p character.418 This effect can be illustrated by a typical case (e.g., carbocation 140), where hyperconjugation is invoked to explain the relative stability of the ion as attached groups are varied. If the orbitals of an adjacent C–H bond align with the empty orbital of the positive center; a canonical form can be
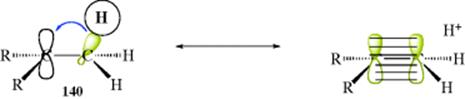
drawn by electron donation to C+ to form a canonical form that is formally an alkene and H+. Note that the H in 140 could be any atom and C could be any sp2-hybridized atom and hyperconjugation would still occur. The key part is that a σ-bond overlaps a π-bond. For 140, an alkene and a closely bound proton constitute a canonical form that helps stabilize the carbocation. Each of the three methyl hydrogen atoms in 140 can contribute to the hyperconjugative stabilization. In other words, resonance contributors involving the C–H bonds represent the bond elongation due to hyperconjugation, and provide stabilization of a carbocation.419 To determine whether hyperconjugation is important in a given situation using molecular modeling, one must ask if the localized model is adequate for that situation at the particular level of precision, or whether the model must be corrected by including some delocalization.420 To a first approximation, delocalization can be neglected, but it is needed for better approximations. The effect of the alkene canonical form on 140 is that the electrons in the C–H bond are closer to the carbon than if hyperconjugation did not contribute at all.
In neutral molecules, the structural elements noted above should be present for hyperconjugation. There is usually at least one sp2-hybridized atom, usually carbon, but for hyperconjugation in general, all that is needed is a σ-bond. Resonating structures due to hyperconjugation may be written involving “no bonds” between the alpha carbon and hydrogen atoms, as shown below for propene.
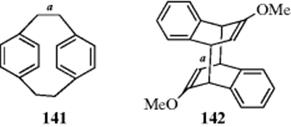
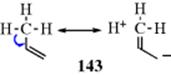
Contributions of this type are seen by a comparison of the X-ray data for 141 and 142 with the calculated data (MM40). The bond length for bond a in 141 is 1.571 Å by analysis of the X-ray data, but analysis by MM40 calculations gave a value of 1.565 A.421 Similar analysis of bond a in 142 gave an experimental value of 1.627 Å, but MM40 calculations gave a value of 1.589 Å. The calculated values are shorter than the actual values. When hyperconjugation was included in the MM4 calculations, the calculated values were 1.574 Å for 141 and 1.623 Å for 142.421 The hyperconjugative stretching effect was calculated to be 0.009 Å for 141 and 0.034 Å for 142. This work suggests that hyperconjugation is actually a bond stretching effect,421 and it has been represented as resonance contributors several times in this chapter. If the bond elongation found in propene, due to hyperconjugation, is represented as the canonical forms shown for 143, the charge separation illustrates bond elongation. In a different example using toluene, there is evidence that the main interaction between methyl groups and the ring system in the positive ions of aromatic hydrocarbons is due to hyperconjugation rather than an inductive effect.422
There is evidence that bond length effects were the result of the s character of the saturated carbon rather than of neutral hyperconjugation.423 These experimental results seem to follow from hyperconjugation in the ground states of neutral molecules, and there is evidence in favor of hyperconjugation.424 Indeed, hyperconjugation appears to operate for both carbon and hydrogen in various systems (supported by quantum mechanics).425 These works tie together experimental and computational results into a unified picture that supports hyperconjugation.424,425 A study of the one-bond coupling constants for the aromatic system 144
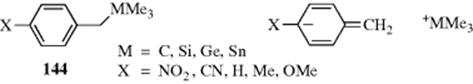
appears to provide structural evidence for hyperconjugation in a neutral ground state.426 Hyperconjugation in the ground state of neutral molecules has been called sacrificial hyperconjugation by Muller and Mulliken.427
Another way to view this phenomenon is to say that electron release is permitted by a mechanism that is essentially a type of tautomeric effect (Sec. 2.N). Dewar suggested that the delocalization of electrons of single bonds (hyperconjugation) and of p or π electrons (conjugation) should be included as part of the electronic description only if the localized bond picture fails.428 This result is a good first approximation, but modern molecular mechanics allows a much better analysis.
Hyperconjugation has been invoked to explain various aspects of aromaticity, as utilized early in this chapter. It is known that 5,5-disubstituted cyclopentadienes, where the substituents are electropositive groups, show enhanced cyclic conjugation in comparison with cyclopentadiene itself.429 5,5-Distannylcyclopentadiene, for example, was found to be nearly as aromatic as furan. This is explained by hyperconjugative electron donation by the substituents, resulting in a partially anionic ring.424 Another effect is the so-called C∗-aromaticity, which is a hyperconjugative effect found in small disubstituted rings that leads to lowering of ring-strain energies for the unsaturated rings, particularly when electronegative substituents are attached.430