March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 4. Stereochemistry and Conformation
4.N. Conformational Analysis
For acyclic molecules with single covalent bonds, there is rotation about those bonds. As a practical matter, such rotation leads to different arrangements of the atoms with respect to a given bond, but all arrangements constitute the same molecule. The different arrangements for a molecule due to such rotation are called rotamers. In principle, there is rotation about every single bond and a near infinite number of rotamers. If two different 3D spatial arrangements of the atoms in an acyclic molecule are interconvertible merely by free rotation about bonds, they are called conformations.269 If they are not interconvertible, they are called configurations.270 Configurations represent isomers that can be separated, as previously discussed in this chapter. Conformations represent conformers, which are rapidly interconvertible and thus nonseparable. The terms “conformational isomer” or more commonly “rotamer”271 are used to identify one of many structures that result from rotation about single covalent bonds. Typically, the conformation is the average of the collection of lower energy rotatmers for an acyclic compound. A number of methods have been used to determine conformations.272 These include X-ray and electron diffraction, IR, Raman, UV, NMR,273 and microwave spectra,274 PES,275 supersonic molecular jet spectroscopy,276 and ORD and CD measurements.277 Ring current NMR anisotropy has been applied to conformational analysis,278 as has chemical shift simulation.279 Some of these methods are useful only for solids. It must be kept in mind that the conformation of a molecule in the solid state is not necessarily the same as in solution.280 Conformations can be calculated by a method called molecular mechanics (Sec. 4.P). A method was reported that characterized six-membered ring conformations as a linear combination of ideal basic conformations.281 The term absolute conformation has been introduced for molecules where one conformation is optically inactive but, by internal rotation about a C(sp3)–C(sp3) bond, optically active conformers are produced.282
Note that “free” rotation about single bonds is not possible in cyclic molecules, but rather pseudorotation that leads to different conformations. This discussion will therefore separate rotation in acyclic molecules from pseudorotation in cyclic molecules.
4.N.i. Conformation in Open-Chain Systems283
For any open-chain molecule with a single bond that connects two sp3 carbon atoms, an infinite number of rotatmers are possible, each of which has a certain energy associated with it, which leads to an infinite number of conformations. As a practical matter, the number of conformations is much less. If one ignores duplications due to symmetry, the number of conformations can be estimated as being > 3n, where n = the number of internal C–C bonds. For example, n-pentane, has 11, n-hexane 35, n-heptane 109, n-octane 347, n-nonane 1101, and n-decane 3263.284 For ethane, there are two important rotamers that are taken as the extremes, a conformation of highest (marked eclipsed) and one of lowest (marked staggered) potential energy, depicted in two ways as sawhorse diagrams or Newman projections. In Newman projection formulas, the observer looks at the C–C bond head on. The three lines emanating from the center of the circle represent the bonds coming from the front carbon, with respect to the observer.
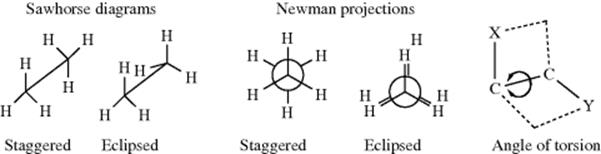
The staggered conformation is the conformation of lowest potential energy for ethane. As rotation about the bond occurs, the energy gradually increases until the eclipsed conformation is reached, when the energy is at a maximum. Further rotation decreases the energy again. Figure 4.4 illustrates this finding. The angle of torsion, which is a dihedral angle, is the angle between the X–C–C and the C–C–Y planes, as shown in the diagram. For ethane, the difference in energy is ~2.9 kcal mol−1 (12 kJ mol−1).285 This difference is called the energy barrier or rotational barrier,286 since in free rotation about a single bond there must be enough rotational energy present to cross the barrier every time two hydrogen atoms are opposite each other. There was much speculation about the cause of the barriers and many explanations have been suggested.287 It has been concluded from MO calculations (see Sec. 4.P) that the barrier is caused by repulsion between overlapping filled molecular orbitals.288 The staggered conformation of ethane is lowest in energy because the orbitals of the C–H bonds in this conformation have the least amount of overlap with the C–H orbitals of the adjacent carbon.
Fig. 4.4 Conformational energy diagram for ethane.
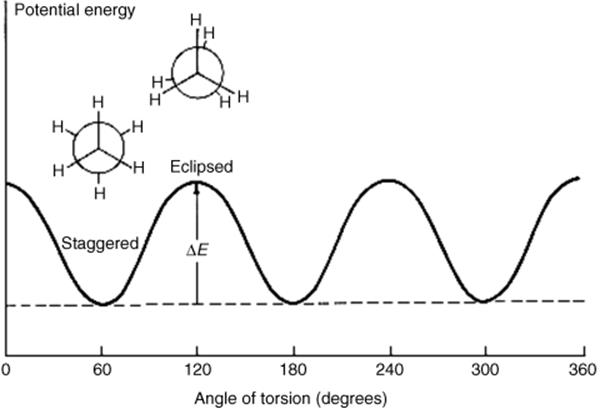
At ordinary temperatures, enough rotational energy is present for the ethane molecule to rotate rapidly, but it spends most of its time at or near the energy minimum. Groups larger than hydrogen cause larger barriers, presumably due to steric interactions between the larger units.289 When the barriers are large enough, as in the case of suitably substituted biphenyls (Sec. 4.C, category 5) or the diadamantyl compound mentioned (see 107 and 108) rotation at room temperature is completely prevented, which is described as a q configuration not a conformations. Even for compounds with small barriers, cooling to low temperatures may remove enough rotational energy for what would otherwise be conformational isomers to become configurational isomers.
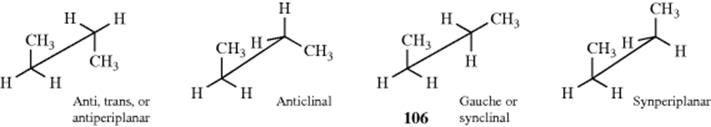
A 1,2-disubstituted ethane (YCH2–CH2Y or YCH2–CH2X,290 e.g., n-butane),291 is somewhat more complicated. There are four extremes: a fully staggered conformation, called anti, trans, or antiperiplanar; another staggered conformation, called gauche or synclinal; and two types of eclipsed conformations, called synperiplanar and anticlinal.
An energy diagram for this system is given in Fig. 4.5. Although there is constant rotation about the central bond, it is possible to estimate what percentage of the molecules are in each conformation at a given time. For example, a consideration of dipole moment and polarizability measurements led to the conclusion that for 1,2-dichloroethane in CCl4 solution at 25°C ~ 70% of the molecules are in the anti and ~ 30% in the gauche conformation.292 The corresponding figures for 1,2-dibromoethane are 89% anti and 11% gauche.293 The eclipsed conformations are unpopulated and serve only as pathways from one staggered conformation to another. Solids normally consist of a single conformer.
Fig. 4.5 Conformational energy for YCH2–CH2Y or YCH2–CH2X. For n-butane, ΔE1 = 4–6, ΔE2 = 0.9, and ΔE3 = 3.4 kcal mol−1 (17–25, 3.8, 14 kJ mol−1, respectively).
It may be observed that the gauche conformation of butane (see 106), or any other similar molecule, appears to be chiral. It is not. The lack of optical activity in such compounds arises from the fact that 106 is not a static molecule, but is in dynamic equilibrium with many other conformations, including its mirror image. In effect, they interconvert too rapidly for separation.
For butane and for most other molecules of the forms YCH2–CH2Y and YCH2– CH2X, the anti conformer is the most stable, but exceptions are known. One group of exceptions consists of molecules containing small electronegative atoms, especially fluorine and oxygen. Thus 2-fluoroethanol,294 1,2-difluoroethane,295 and 2-fluoroethyl trichloroacetate (FCH2CH2OCOCCl3)296 exist predominantly in the gauche form and compounds, such as, 2-chloroethanol and 2-bromoethanol,294 also prefer the gauche form. It has been proposed that the preference for the gauche conformation in these molecules is an example of a more general phenomenon, known as the gauche effect; that is, a tendency to adopt that structure that has the maximum number of gauche interactions between adjacent electron pairs or polar bonds.297 It was believed that the favorable gauche conformation of 2-fluoroethanol was the result of intramolecular hydrogen bonding, but this explanation does not do for molecules like 2-fluoroethyl trichloroacetate. It has in fact been ruled out for 2-fluoroethanol as well.298 The effect of β-substituents in Y–C–C–OX systems where Y = F or SiR3 has been examined and there is a small bond shortening effect on C–OX that is greatest when OX is a good leaving group. Bond lengthening was also observed with the β-silyl substituent.299 Other exceptions are known, where small electronegative atoms are absent. For example 1,1,2,2-tetrachloroethane and 1,1,2,2-tetrabromoethane both prefer the gauche conformation,300 even though 1,1,2,2-tetrafluoroethane prefers the anti.301 Also, both 2,3-dimethylpentane and 3,4-dimethylhexane prefer the gauche conformation,302 and 2,3-dimethylbutane shows no preference for either.303 Furthermore, the solvent can exert a powerful effect. For example, the compound 2,3-dinitro-2,3-dimethylbutane exists entirely in the gauche conformation in the solid state, but in benzene, the gauche/anti ratio is 79:21; while in CCl4 the anti form is actually favored (gauche/anti ratio 42:58).304 In many cases, there are differences in the conformation of these molecules between the gas and the liquid phase (as when X = Y = OMe) because of polar interactions with the solvent.305
In one case, two conformational isomers of a single aliphatic hydrocarbon, 3,4-di(1-adamantyl)-2,2,5,5-tetramethylhexane, have proven stable enough for isolation at room temperature.306 The two isomers 107 and 108 were separately crystallized, and the structures were proven by X-ray crystallography. The actual dihedral angles are distorted from the 60° angles shown in the drawings, due to steric hindrance between the large adamantyl and tert-butyl groups.
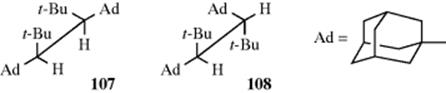
All the conformations so far discussed have involved rotation about sp3–sp3 bonds. Many studies have also been made of compounds with sp3–sp2 bonds.307 For example, propanal (or any similar molecule) has four extreme conformations, two of which are called eclipsing and the other two bisecting. For propanal, the eclipsing conformations have lower energy than the other two, with 109 favored over 110 by ~1 kcal mol−1 (4 kJ mol−1).308 As already pointed out (Sec. 4.K.i), for a few of these compounds, rotation is slow enough to permit cis-trans isomerism, although for simple compounds rotation is rapid. The cis conformer of acetic acid was produced in solid Ar,309 and it was reported that acetaldehyde has a lower rotational barrier (~1 kcal mol−1 or 4 kJ mol−1) than ethane.310 Calculations have examined the rotational barriers around the CO and CC bonds in formic acid, ethanedial, and glycolaldehyde molecules.311
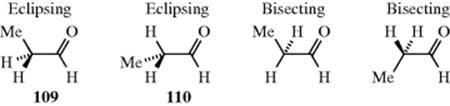
Other carbonyl compounds exhibit rotation about sp3–sp3 bonds, including amides.312 In N-acetyl-N-methylaniline, the cis- conformation (111) is more stable than the trans- (112) by 3.5 kcal mol−1 (14.6 kJ mol−1).313 This is due to destabilization of (S) due to steric hindrance between two methyl groups, and to electronic repulsion between the carbonyl lone-pair electrons and the phenyl π-electrons in the twisted phenyl orientation.313
A similar conformational analysis has been done with formamide derivatives,314 with secondary amides,315 and for hydroxamide acids.316 It is known that thioformamide has a larger rotational barrier than formamide, which can be explained by a traditional picture of amide “resonance” that is more appropriate for the thioformamide than formamide itself.317 Torsional barriers in α-keto amides have been reported,318 and the C–N bond of acetamides,319thioamides,320 enamides321 carbamates (R2N–CO2R′),322 and enolate anions derived from amides323 have been examined. It is known that substituents influence rotational barriers.324
In Section 4.C, category 5, atropisomerism was possible when ortho substituents on biphenyl derivatives and certain other aromatic compounds prevented rotation about the ![]() bond. The presence of ortho-substituents can also influence the conformation of certain groups.325 In 113, R = alkyl and the carbonyl unit is planar, with the trans C=O
bond. The presence of ortho-substituents can also influence the conformation of certain groups.325 In 113, R = alkyl and the carbonyl unit is planar, with the trans C=O![]() F conformer is more stable when X = F. When X = CF3, the cis and trans are planar and the trans predominates.326 When R = alkyl, there is one orthogonal conformation, but there are two interconverting nonplanar conformations when R = O-alkyl.326 In 1,2-diacylbenzenes, the carbonyl units tend to adopt a twisted conformation to minimize steric interactions.327
F conformer is more stable when X = F. When X = CF3, the cis and trans are planar and the trans predominates.326 When R = alkyl, there is one orthogonal conformation, but there are two interconverting nonplanar conformations when R = O-alkyl.326 In 1,2-diacylbenzenes, the carbonyl units tend to adopt a twisted conformation to minimize steric interactions.327
4.N.ii. Conformation in Six-Membered Rings328
For cyclic compounds, complete rotation (360°) about a single bond is impossible. However, repulsion between atoms and groups leads to motion about each bond called pseudorotation. Pseudorotation leads to a variety of different conformations, depending on the size of the ring. In many such conformations, the ring is said to be puckered. For cyclohexane, there are two extreme conformations in which all the angles are tetrahedral (the C–C–C angles in cyclohexane are actually 111.5°).329 These are called the boat and the chair conformations. The chair conformation is the low-energy structure that participates in a dynamic equilibrium (there are two chair conformations that are equivalent in energy for cyclohexane), and the boat form is a higher energy form330 in equilibrium with a somewhat more stable form

known as the twist conformation. The twist form is ~1.5 kcal mol−1 (6.3 kJ mol−1) more stable than the boat because it has less eclipsing interaction (see below).331 The chair form is more stable than the twist form by ~5 kcal mol−1(21 kJ mol−1).332 In the vast majority of compounds containing a cyclohexane ring, the molecules exist almost entirely as equilibrating chair forms.333 It is known that the boat or twist form exists transiently. In some cases, chair and twist–boat conformations have actually been observed (cis-1,4-di-tert-butylcyclohexane, e.g.).334
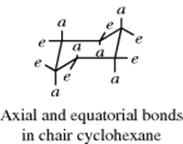
An inspection of the chair form shows that six of its bonds are directed differently from the other six. On each carbon, one bond is directed up or down and the other more or less in the “plane” of the ring. The up or down bonds are called axial and the others are equatorial. The axial bonds point alternately up and down. If a molecule were frozen into a chair form, there would be isomerism in monosubstituted cyclohexanes. For example, there would be an equatorial methylcyclohexane and an axial isomer. This result is incorrect, however, as it has never been possible to isolate isomers of this type at room temperature.335 In order for the two types of methylcyclohexane to be nonseparable, there must be rapid interconversion of one chair form to another (in which all axial bonds become equatorial and vice versa) and this is possible only if the boat or twist conformations are transient species. Conversion of one chair form to another requires an activation energy of ~10 kcal mol−1 (42 kJ mol−1)336 and is very rapid at room temperature.337 However, by working at low temperatures, Jensen and Bushweller338 were able to obtain the pure equatorial conformers of chlorocyclohexane and trideuteriomethoxycyclohexane as solids and in solution. Equatorial chlorocyclohexane has a half-life of 22 years in solution at −160°C.
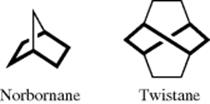
In some molecules, the twist conformation is actually preferred.339 Of course, in certain bicyclic compounds, the six-membered ring is forced to maintain a boat or twist conformation, as in norbornane or twistane.
In monosubstituted cyclohexanes, the substituent normally prefers the equatorial position because there is an interaction between the substituent and the axial hydrogens in the axial 3 and 5 positions, but the extent of this preference depends greatly on the nature of the group.340 Alkyl groups have a greater preference for the equatorial position than polar groups. For alkyl groups, the preference increases with size, although size seems to be unimportant for polar groups. Both the large HgBr341 and HgCl342 groups and the small F group have been reported to have little or no conformational preference (the HgCl group actually shows a slight preference for the axial position). Table 4.3 gives approximate values of the free energy required for various groups to go from the equatorial position to the axial (these are called A values),343 although it must be kept in mind that they vary somewhat with physical state, temperature, and solvent.344 Values for other groups in kcal mol−1 include D345 (0.008), NH2346 (1.4), CH=CH2347 (1.7), CH3348 (1.74), C6H11349 (2.15), Si(CH3),350 (2.4–2.6), OCH3351 (0.75), C6H5352 (2.7), and t-(CH3)3C353 (4.9).
Table 4.3 Free-Energy Differences between Equatorial and Axial Substituents on a Cyclohexane Ringa,b
[Reprinted with permission from Corey, E.J.; Feiner, N.F. J. Org. Chem. 1980, 45, 765. Copyright © 1980 American Chemical Society.]
a. See Ref. 343.
b. The A values or A1,3-strain.
For alkyl groups in disubstituted compounds, the conformation is such that as many groups as possible adopt the equatorial position. This conformation will minimize the axial interactions (known as A1,3-strain), and will be the lower energy conformation. The preference for one chair conformation over the other depends on the groups attached to the cyclohexane ring, and their relative positions on that ring. In a cis-1,2-disubstituted cyclohexane, one substituent must be axial and the other equatorial. In a trans-1,2, compound both may be equatorial or both axial. This finding is also true for 1,4-disubstituted cyclohexanes, but the reverse holds for 1,3-compounds: the trans isomer must have the age conformation and the cis isomer may be a or ee. For alkyl groups, the ee conformation predominates over the a, but for other groups this is not necessarily so. For example, both trans-1,4-dibromocyclohexane and the corresponding dichloro compound have the ee and a conformations about equally populated354 and most trans-1,2-dihalocyclohexanes exist predominantly in the a conformation.355 Note that in the latter case the two halogen atoms are anti in the a conformation, but gauche in the ee conformation.356
Since compounds with alkyl equatorial substituents are generally more stable, trans-1,2 compounds, which can adopt the ee conformation, are thermodynamically more stable than their cis-1,2 isomers, which must exist in the ageconformation. For the 1,2-dimethylcyclohexanes, the difference in stability is ~2 kcal mol−1 (8 kJ mol−1). Similarly, trans-1,4 and cis-1,3 compounds are more stable than their stereoisomers.
An interesting anomaly is all-trans-1,2,3,4,5,6-hexaisopropylcyclohexane, in which the six isopropyl groups prefer the axial position, although the six ethyl groups of the corresponding hexaethyl compound prefer the equatorial position.357 The alkyl groups of these compounds can of course only be all axial or all equatorial, and it is likely that the molecule prefers the all-axial conformation because of unavoidable strain in the other conformation.
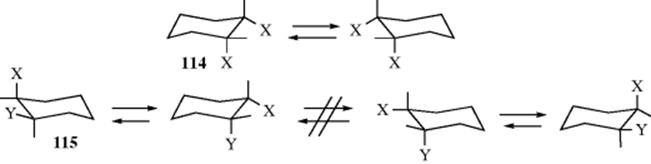
Incidentally, it is now apparent, at least in one case, why the correct number of stereoisomers could be predicted by assuming planar rings, even though they are not planar (Sec. 4.K.ii). In the case of both a cis-1,2-X,X-disubstituted and a cis-1,2-X,Y-disubstituted cyclohexane, the molecule is nonsuperimposable on its mirror image; neither has a plane of symmetry. However, in the former case (114) conversion of one chair form to the other which of course happens rapidly, turns the molecule into its mirror image, while in the latter case (115) rapid interconversion does not give the mirror image, but merely the conformer in which the original axial and equatorial substituents exchange places. Thus the optical inactivity of 114 is not due to a plane of symmetry, but to a rapid interconversion of the molecule and its mirror image. A similar situation holds for cis-1,3 compounds. However, for cis-1,4 isomers (both X,X and X,Y) optical inactivity arises from a plane of symmetry in both conformations. All trans-1,2- and trans-1,3-disubstituted cyclohexanes are chiral (whether X,X or X,Y), while trans-1,4 compounds (both X,X and X,Y) are achiral, since all conformations have a plane of symmetry. It has been shown that the equilibrium is very dependent on both the solvent and the concentration of the disubstituted cyclohexane.358 A theoretical study of the 1,2-dihalides showed a preference for the diaxial form with X = Cl, but predicted that the energy difference between diaxial and diequatorial was small when X = F.359
The conformation of a group can be frozen into a desired position by putting a large alkyl group into the ring (most often tert-butyl), which introduces significant A1,3-strain and leads to a preference for the chair with the groups in the equatorial position.360 It is known that silylated derivatives of trans-1,4- and trans-1,2-dihydroxycyclohexane, some monosilyloxycyclohexanes and some silylated sugars have unusually large populations of chair conformations with axial substituents.361 Adjacent silyl groups in the 1,2-disubstituted series show a stabilizing interaction in all conformations, generally leading to unusually large axial populations.
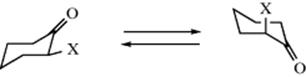
The principles involved in the conformational analysis of six-membered rings containing one or two trigonal atoms. For example, cyclohexanone and cyclohexene, are similar.362–364 The barrier to interconversion in cyclohexane has been calculated to be 8.4–12.1 kcal mol−1(35.2–50.7 kJ mol−1).365 Cyclohexanone derivatives also assume a chair–conformation. Substituents at C-2 can assume an axial or equatorial position depending on steric and electronic influences. The proportion of the conformation with an axial X group is shown in Table 4.4 for a variety of substituents (X) in 2-substituted cyclohexanones.366
Table 4.4 Proportion of Axial Conformation in 2-Substituted Cyclohexanones, in CDCl3a
Reprinted with permission from Basso, E.A.; Kaiser, C.; Rittner, R.; Lambert, J.B. J. Org. Chem. 1993, 58, 7865. Copyright © 1993 American Chemical Society.
X
% Axial Conformation
F
17 ± 3
Cl
45 ± 4
Br
71 ± 4
I
88 ± 5
MeO
28 ± 4
MeS
85 ± 7
MeSe
(92)
Me2N
44 ± 3
Me
(26)
a. See Ref. 366.
4.N.iii. Conformation in Six-Membered Rings Containing Heteroatoms
In six-membered rings containing heteroatoms,367 the basic principles are the same; that is, there are chair, twist, and boat forms, axial, and equatorial groups. The conformational equilibrium for tetrahydropyridines, for example, has been studied.368 In certain compounds, a number of new factors enter the picture. Only two of these will be examined.369
1. In 5-alkyl-substituted 1,3-dioxanes, the 5-substituent has a much smaller preference for the equatorial position than in cyclohexane derivatives;370 the A1,3-strain is much lower. This fact indicates that the lone pairs on the oxygens have a smaller steric requirement than the C–H bonds in the corresponding cyclohexane derivatives. There is some evidence of a homoanomeric interaction in these systems.371 Similar behavior is found in the 1,3-dithianes,372 and 2,3-disubstituted-1,4-dithianes have also been examined.373 With certain non-alkyl substituents (e.g., F, NO2, SOMe,374 NMe3+) the axial position is actually preferred.375
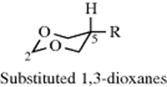
2. An alkyl group located on a carbon α to a heteroatom prefers the equatorial position, which is of course the normally expected behavior, but a polar group in such a location prefers the axial position. An example of this phenomenon, known as the anomeric effect,376 is the greater stability of α-glucosides over β-glucosides. A number of explanations have been offered for the anomeric
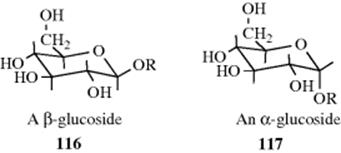
effect.377 The one378 that has received the most acceptance379 is that one of the lone pairs of the polar atom connected to the carbon (an oxygen atom in the case of 117) can be stabilized by overlapping with an antibonding orbital of the bond between the carbon and the other polar atom:

This can happen only if the two orbitals are in the positions shown. The situation can also be represented by this type of hyperconjugation (called “negative hyperconjugation,” see Sec. 2.M):
![]()
It is possible that simple repulsion between parallel dipoles in 116 also plays a part in the greater stability of 117. It has been shown that aqueous solvation effects reduce anomeric stabilization in many systems, particularly for tetrahydropyranosyls.380 In contrast to cyclic acetals, simple acyclic acetals rarely adopt the anomeric conformation, apparently because the eclipsed conformation better accommodates steric interactions of groups linked by relatively short carbon–oxygen bonds.381 In all cis-2,5-di-tert-butyl-1,4-cyclohexanediol, hydrogen bonding stabilizes the otherwise high-energy form382 and 1,3-dioxane (118) exists largely as the twist conformation shown.383The conformational preference of 1-methyl-1-silacyclohexane (121) has been studied.384 A strongly decreased activation barrier in silacyclohexane was observed, as compared to that in the parent ring, and is explained by the longer endocyclic Si–C bonds.
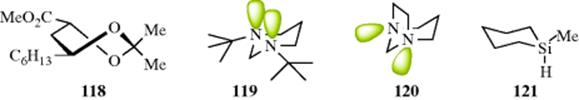
Second-row heteroatoms are known to show a substantial anomeric effect.385 There appears to be evidence for a reverse anomeric effect in 2-aminotetrahydropyrans,386 but it has been called into question whether a reverse anomeric effect exists at all.387 In 119, the lone-pair electrons assume an axial conformation and there is an anomeric effect.388 In 120, however, the lone-pair electron orbitals are oriented gauche to both the axial and equatorial α-CH bond and there is no anomeric effect.388
4.N.iv. Conformation in Other Rings389
Three-membered saturated rings are usually planar, but other small rings can have some flexibility. Cyclobutane390 is not planar, but exists as in 122, with an angle between the planes of ~35°.391 The deviation from planarity is presumably caused by eclipsing in the planar form (see Sec. 4.Q.i). Oxetane is closer to
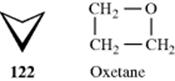
planarity because there is less eclipsing, with an angle between the planes of ~10°.392 Cyclopentane might be expected to be planar, since the angles of a regular pentagon are 108°, but it is not so, also because of eclipsing effects.393There are two puckered conformations for cyclopentane, the envelope and the half-chair. There is little energy difference between these two forms and many five-membered ring systems have conformations somewhere in between them.394 Although in the envelope conformation one carbon is shown above the others,
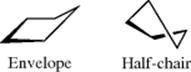
ring motions cause each of the carbons in rapid succession to assume this position. The puckering rotates around the ring in what is called a pseudorotation395 (see Sec. 4.O.ii). In substituted cyclopentanes and five-membered rings in which at least one atom does not contain two substituents [e.g., tetrahydrofuran (THF), cyclopentanone, C3- and C7-monosubstituted and disubstituted hexahydroazepin-2-ones (caprolactams),396 tetrahydrothiophene S-oxide397], one conformer may be more stable than the others. The barrier to planarity in cyclopentane has been reported to be 5.2 kcal mol−1 (22 kJ mol−1).398 Contrary to previous reports, there is only weak stabilization (<2 kcal mol−1; <8 kJ mol−1) of three-, four-, and five-membered rings by gem-dialkoxycarbonyl substituents (e.g., COOR).399
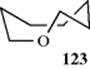
Rings larger than six-membered are always puckered400 unless they contain a large number of sp2 atoms (see the section on strain in medium rings, Sec. 4.Q.ii). The energy and conformations of the alkane series cycloheptane to cyclodecane has been reported.401 The conformation shown for oxacyclooctane (123), for example, appears to be the most abundant one.402 The conformations of other large-ring compounds have been studied, including cycloundecane,403 11-membered ring lactones,404 10- and 11-membered ring ketones,405 and 11- and 14-membered ring lactams.406 Dynamic NMR was used to determine the conformation large-ring cycloalkenes and lactones,407and C–H coupling constants have been used for conformational analysis.408 Strain estimates have been made for small-ring cyclic allenes and butatrienes.409 Note that axial and equatorial hydrogens are found only in the chair conformations of six-membered rings. In rings of other sizes, the hydrogens protrude at angles that generally do not lend themselves to classification in this way,410 although in some cases the terms “pseudo-axial” and “pseudo-equatorial” have been used to classify hydrogens in rings of other sizes.411