March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 5. Carbocations, Carbanions, Free Radicals, Carbenes, and Nitrenes
There are four types of organic species in which a carbon atom has a valence of only 2 or 3.1 They are usually very short-lived, and most exist only as intermediates that are quickly converted to more stable molecules. However, some are more stable than others and fairly stable examples have been prepared for three of the four types. The four types of species are carbocations (A), carbon radicals (B), carbanions (C), and carbenes (D). Of the four, only carbanions have a complete octet around the carbon. There are many other organic ions and radicals with charges and unpaired electrons on atoms other than carbon, but only nitrenes (E), the nitrogen analogues of carbenes, will be discussed. Each of these five types is discussed in a separate section, which in each case includes brief summaries of the ways in which the species form and react. These summaries are short and schematic. The generation and fate of the five types are more fully treated for the appropriate specific reactions in Part II.
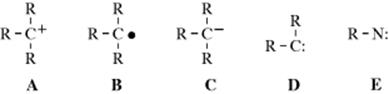
5.A. Carbocations2
5.A.i. Nomenclature
First, the nomenclature of carbocations (A) is discussed. For many years, these species were called “carbonium ions,” although it was suggested3 as long ago as 1902 that this was inappropriate because “-onium” usually refers to a covalency higher than that of the neutral atom. Nevertheless, the name “carbonium ion” was well established and created few problems4 until some years ago, when Olah and co-workers2,5 found evidence for another type of intermediate in which there is a positive charge at a carbon atom, but in which the formal covalency of the carbon atom is five rather than three. The simplest example is the methanonium ion (CH5+; see Reaction 12-01). Olah5proposed that the name “carbonium ion” be henceforth reserved for pentacoordinated positive ions, and that A be called a “carbenium ions.” He also proposed the term “carbocation” to encompass both types. IUPAC has accepted these definitions.6 For the most part, intermediates such as A are called carbenium ions or carbocations, but the latter term will be used more often in this book.
5.A.ii. Stability and Structure of Carbocations
[Reprinted with permission from Kato, T.; Reed, C.A. Angew. Chem. Int. Ed. 2004, 43, 2908, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 2004 by Wiley–VCH Verlag]
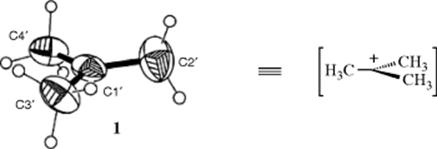
Carbocations are intermediates in several kinds of reactions.7 The more stable ones have been prepared in solution and in some cases even as solid salts. X-ray crystallographic structures also have been obtained in some cases.8The X-ray of the tert-butyl cation complexed with dichloromethane was reported,9 for example, and is presented as 1 with the solvent molecules removed for clarity. The IR spectrum of the tert-butyl cation has been recorded in the gas phase.10 An isolable dioxa-stabilized pentadienylium ion was isolated and its structure was determined by ![]() NMR,
NMR, ![]() NMR, mass spectrometry, and IR.11 A β-fluoro substituted 4-methoxyphenethyl cation has been observed directly by laser flash photolysis.12 In solution, the carbocation may be free (this is more likely in polar solvents, in which it is solvated) or it may exist as an ion pair,13 which means that it is closely associated with a negative ion, called a counterion or gegenion. Ion pairs are more likely in nonpolar solvents.
NMR, mass spectrometry, and IR.11 A β-fluoro substituted 4-methoxyphenethyl cation has been observed directly by laser flash photolysis.12 In solution, the carbocation may be free (this is more likely in polar solvents, in which it is solvated) or it may exist as an ion pair,13 which means that it is closely associated with a negative ion, called a counterion or gegenion. Ion pairs are more likely in nonpolar solvents.
Among simple alkyl carbocations,14 the order of stability is tertiary > secondary > primary. There are many known examples of rearrangements of primary or secondary carbocations to tertiary, both in solution and in the gas phase (see Sec. 18.A.ii). Since simple alkyl cations are unstable in ordinary strong-acid solutions (e.g., H2SO4), the study of these species was greatly facilitated by the discovery that many of them could be kept indefinitely as stable solutions in mixtures of fluorosulfuric acid and antimony pentafluoride. Such mixtures, usually dissolved in SO2 or SO2ClF, are among the strongest acidic solutions known and are often called superacids.15 The original experiments involved the addition of alkyl fluorides to SbF5.16
![]()
Subsequently, it was found that the same carbocations could also be generated from alcohols in superacid–SO2 at −60 °C17 and from alkenes by the addition of a proton from superacid or HF–SbF5 in SO2 or SO2ClF at low temperatures.18 Even alkanes give carbocations in superacid by loss of H−. For example,19 2-methylpropane gives the tert-butyl cation.
![]()
No matter how they are generated, study of the simple alkyl carbocations has provided dramatic evidence for the stability order.20 Both propyl fluorides gave the isopropyl cation; all four butyl fluorides21 gave the tert-butyl cation, and all seven of the pentyl fluorides examined gave the tert-pentyl cation. n-Butane, in superacid, gave only the tert-butyl cation. To date, no primary cation has survived long enough for detection. Neither methyl nor ethyl fluoride gave the corresponding carbocations when treated with SbF5. At low temperatures, methyl fluoride gave chiefly the methylated sulfur dioxide salt [(CH3OSO)+SbF6−],22 while ethyl fluoride rapidly formed the tert-butyl and tert-hexyl cations by addition of the initially formed ethyl cation to ethylene molecules also formed.23 At room temperature, methyl fluoride also gave the tert-butyl cation.24 In accord with the stability order, hydride ion is abstracted from alkanes by superacid most readily from tertiary and least readily from primary positions.
![]()
The stability order can be explained by the polar effect and by hyperconjugation (Sec. 2.M). In the polar effect, nonconjugated substituents exert an influence on stability through bonds (inductive effect) or through space (field effect). Since a tertiary carbocation has more carbon substituents on the positively charged carbon, relative to a primary, there is a greater polar effect that leads to great stability. In the hyperconjugation explanation,25 a primary carbocation is compared with a tertiary, and “the hyperconjugation concept arises from model-building procedures (see Sec. 2.M). In general, this means that the model must be corrected by including some delocalization in order to get a good enough description.”26 Evidence used to support the hyperconjugation explanation is that the equilibrium constant for this reaction involving 2 and 3 is 1.97, showing that 3 is more stable than 2.27 Due to a β secondary isotope effect, there is less hyperconjugation in 2 than in 3 (see Sec. 6.J.V.ii for isotope effects).28 The field effect explanation is that the electron-donating effect of alkyl groups increases the electron density at the charge-bearing carbon, reducing the net charge on the carbon, and in effect spreading the charge over the α carbons. It is a general rule that the more concentrated any charge is, the less stable the species bearing it will be. There are several structural types of delocalization, as summarized in Table 5.1.29
Table 5.1 Structural Types of Delocalizationa
Valence Structures
Abbreviation
Name
![]()
ππ
Simple conjugation
![]()
σπ
Hyperconjugation
![]()
πσ
Homoconjugation
![]()
σσ
Homohyperconjugation
![]()
σπ/ππ
Hyperconjugation/conjugation
![]()
σπ/σπ
Double hyperconjugation
[Reprinted with permission from Radom, L.; Poppinger, D.; Haddon, R.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 2303–2426, Wiley–VCH Verlag GmbH & Co. KGaA, Weinheim. Copyright © 1976 by Wiley–VCH Verlag].
a. See Ref. 25.
The most stable of the simple alkyl cations is the tert-butyl cation. Even the relatively stable tert-pentyl and tert-hexyl cations fragment at higher temperatures to produce the tert-butyl cation, as do all other alkyl cations with four or more carbons to date studied.30 Methane,31 ethane, and propane, in superacid, also yield tert-butyl cations as the main product (see Reaction 12-20). Even paraffin wax and polyethylene give a tert-butyl cation. Solid salts of tert-butyl and tert-pentyl cations, [e.g., Me3C+ SbF6−], have been prepared from superacid solutions and are stable below −20 °C.32
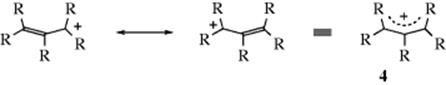
In carbocations, where the positive carbon is in conjugation with a double bond, as in allylic cations (the allyl cation is 4, R = H), the stability is greater because of increased delocalization due to resonance33 where the positive charge is spread over several atoms instead of being concentrated on one (see the molecular orbital picture of 4 in Sec. 2.C, category 2). Each of the terminal atoms in 4 has a charge of ![]() (the charge is exactly
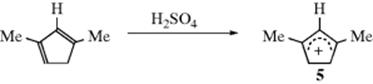
(the charge is exactly ![]() if all of the R groups are the same). Stable cyclic and acyclic allylic-type carbocations34 have been prepared by dissolving conjugated dienes in concentrated sulfuric acid; the cyclopentadienyl cation, (5) is an example.35
if all of the R groups are the same). Stable cyclic and acyclic allylic-type carbocations34 have been prepared by dissolving conjugated dienes in concentrated sulfuric acid; the cyclopentadienyl cation, (5) is an example.35
Stable allylic carbocations have also been obtained by the reaction between alkyl halides, alcohols, or alkenes (by hydride extraction) and SbF5 in SO2 or SO2ClF.36 Bis(allylic) cations37 are more stable than the simple allylic type, and some of these have been prepared in concentrated sulfuric acid.38 Arenium ions (Sec. 11.A.i) are familiar examples of this type. Propargyl cations (RC![]() CC+R2) have also been prepared.39
CC+R2) have also been prepared.39
Canonical forms can be drawn for benzylic carbocations, as shown,40 and they are similar to those shown above for allylic cations.
![]()
A number of benzylic carbocations have been obtained in solution as SbF6− salts.41 Diarylmethyl and triarylmethyl cations are even more stable because more canonical forms are possible (i.e., there is more extensive delocalization, hence greater stability). Chlorotriphenylmethane ionizes in polar solvents to give the stable triphenylmethyl cation (trityl cation, see 18), for example, because the solvent does not react with the ion,
![]()
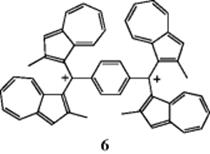
whereas water does react with the ion. In liquid SO2, for example, the ion remains stable for many years. Both triphenylmethyl and diphenylmethyl cations have been isolated as solid salts.42 In fact, Ph3C+ BF4− and related salts are available commercially. Arylmethyl cations are further stabilized if they have electron-donating substituents in ortho or para positions.43 Dications44 and trications are also possible, including the particularly stable dication (6), where each positively charged benzylic carbon is stabilized by two azulene rings.45 A related trication is known where two azulene rings stabilize each benzylic cationic center.46
Cyclopropylmethyl carbocations47 are even more stable than benzylic carbocations. Carbocations 7, 8, and similar ions have been prepared by dissolution of the alcohols in FSO3H–SO2–SbF5,48 and 9 has been prepared from the corresponding alcohol in 96% H2SO4.49 This special stability, which increases with each additional cyclopropyl group, is
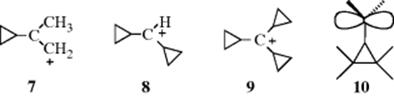
a result of conjugation between the bent orbitals of the cyclopropyl rings (Sec. 4.Q.i) and the vacant p orbital of the cationic carbon (see 10). Nuclear magnetic resonance and other studies have shown that the vacant p orbital lies parallel to the C-2–C-3 bond of the cyclopropane ring and not perpendicular to it.50 In this respect, the geometry is similar to that of a cyclopropane ring conjugated with a double bond (Sec. 4.Q.i). Cyclopropylmethyl cations are further discussed in Section 10.C.i, category 4. The stabilizing effect just discussed is unique to cyclopropyl groups. Cyclobutyl and larger cyclic groups are about as effective at stabilizing a carbocation as ordinary alkyl groups.51
Another structural feature that increases carbocation stability is the presence, adjacent to the cationic center, of a heteroatom bearing an unshared pair52 (e.g., oxygen,53 nitrogen,54 or halogen).55 Such ions are stabilized by resonance, as with the oxocarbenium ion (R2C=O+Me).
![]()
This methoxymethyl cation can be obtained as a stable solid, MeOCH2+ SbF6−.56 Carbocations containing either α, β, or γ silicon atom are also stabilized,57 relative to similar ions without the silicon atom. γ-Silyl cyclobutylcarbocations are known.58 In superacid solution, ions [e.g., CX3+ (X = Cl, Br, I)] have been prepared.59 Vinyl-stabilized halonium ions are also known.60
Simple acyl cations (RCO+) have been prepared61 in solution and the solid state.62 The acetyl cation (CH3CO+) is about as stable as the tert-butyl cation (see Table 5.1). The 2,4,6-trimethylbenzoyl and 2,3,4,5,6-pentamethylbenzoyl cations are especially stable (for steric reasons) and are easily formed in 96% H2SO4.63 These ions, often referred to as acylium ions, are stabilized by a canonical form containing a triple bond (12), although the positive charge is principally located on the carbon,64 so that 11 contributes more than 12.
![]()
The stabilities of many other stable carbocations can also be attributed to resonance. Among these are the tropylium, cyclopropenium,65 and other aromatic cations discussed in Chapter 2. Where resonance stability is completely lacking, as in the phenyl (C6H5+) or vinyl cations,66 the ion, if formed at all, is usually very short lived.67 Neither a vinyl68 nor a phenyl cation has as yet been prepared as a stable species in solution.69 However, stable alkenyl carbocations have been generated on Zeolite Y,70 and the phenyl cation has been observed in cryogenic argon matrices.71
Various quantitative methods have been developed to express the relative stabilities of carbocations.72 One of the most common of these, although useful only for relatively stable carbocations that are formed by ionization of alcohols in acidic solutions, is based on the equation73
![]()
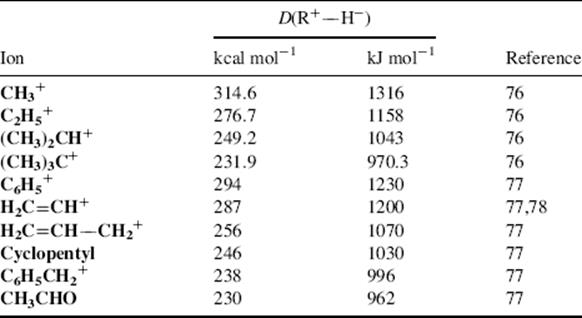
pKR+ is the pK value for the reaction ![]() and is a measure of the stability of the carbocation. The HR parameter is an easily obtainable measurement of the stability of a solvent (Sec. 8.C) and approaches pH at low concentrations of acid. In order to obtain pKR+, for a cation R+, one dissolves the alcohol ROH in an acidic solution of known HR. Then, the concentration of R+ and ROH are obtained, generally from spectra, and pKR+ is easily calculated.74 A measure of carbocation stability that applies to less stable ions is the dissociation energy [D(R+–H−)] for the cleavage reaction R–H → R+ + H−, which can be obtained from PES (Sec. 1.E) and other measurements. Some values of D(R+–H−) are shown in Table 5.2.75–7875767778 Within a given class of ion, (primary, secondary, allylic, aryl, etc.), D(R+–H−) has been shown to be a linear function of the logarithm of the number of atoms in R+, with larger ions being more stable.77
and is a measure of the stability of the carbocation. The HR parameter is an easily obtainable measurement of the stability of a solvent (Sec. 8.C) and approaches pH at low concentrations of acid. In order to obtain pKR+, for a cation R+, one dissolves the alcohol ROH in an acidic solution of known HR. Then, the concentration of R+ and ROH are obtained, generally from spectra, and pKR+ is easily calculated.74 A measure of carbocation stability that applies to less stable ions is the dissociation energy [D(R+–H−)] for the cleavage reaction R–H → R+ + H−, which can be obtained from PES (Sec. 1.E) and other measurements. Some values of D(R+–H−) are shown in Table 5.2.75–7875767778 Within a given class of ion, (primary, secondary, allylic, aryl, etc.), D(R+–H−) has been shown to be a linear function of the logarithm of the number of atoms in R+, with larger ions being more stable.77
Table 5.2 R–H → R+ + H− Dissociation Energies in the Gas Phase.
Since the central carbon of tricoordinated carbocations has only three bonds and no other valence electrons, the bonds are sp2 and should be planar.79 Raman, IR, and NMR spectroscopic data on simple alkyl cations show this to be so.80 In methylcycohexyl cations, there are two chair conformations where the carbon bearing the positive charge is planar (13 and 14), and there is evidence that 14 is more stable due to a difference in hyperconjugation.81Arenonium ions (15) are also known, and are relatively stable.82 Other evidence is that carbocations are difficult to form at bridgehead atoms in [2.2.1] systems,83 where they cannot be planar (see Sec. 10.A.ii).84 Bridgehead carbocations are known, however, as in [2.1.1]hexanes85 and cubyl carbocations.86 However, larger bridgehead ions can exist. For example, the adamantyl cation (15) has been synthesized, as the SF6− salt.87 The relative stability of 1-adamantyl cations is influenced by the number and nature of substituents. For example, the stability of the 1-adamantyl cation increases with the number of isopropyl substituents at C-3, C-5, and C-7.88 Among other bridgehead carbocations that have been prepared in superacid solution at –78 °C are the dodecahydryl cation (16)89 and the 1-trishomobarrelyl cation (17).90 In the latter case, the instability of the bridgehead position is balanced by the extra stability gained from the conjugation with the three cyclopropyl groups.
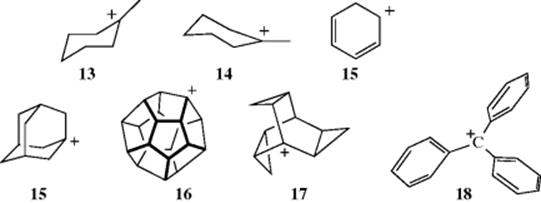
Triarylmethyl cations (e.g., the triphenylmethyl carbocation, 18),91 are propeller-shaped, although the central carbon and the three ring carbons connected to it are in a plane:92 The three benzene rings cannot be all in the same plane due to steric hindrance, although increased resonance energy would be gained if they could.
An important tool for the investigation of carbocation structure is measurement of the ![]() NMR chemical shift of the carbon atom bearing the positive charge.93 This shift approximately correlates with electron density on the carbon. The
NMR chemical shift of the carbon atom bearing the positive charge.93 This shift approximately correlates with electron density on the carbon. The ![]() chemical shifts for a number of ions are given in Table 5.3.94 As shown in Table 5.3, the substitution of an ethyl for a methyl or a methyl for a hydrogen atom causes a downfield shift, indicating that the central carbon becomes somewhat more positive. On the other hand, the presence of hydroxy or phenyl groups decreases the positive character of the central carbon. The
chemical shifts for a number of ions are given in Table 5.3.94 As shown in Table 5.3, the substitution of an ethyl for a methyl or a methyl for a hydrogen atom causes a downfield shift, indicating that the central carbon becomes somewhat more positive. On the other hand, the presence of hydroxy or phenyl groups decreases the positive character of the central carbon. The ![]() chemical shifts are not always in exact order of carbocation stabilities, as determined in other ways. Thus the chemical shift shows that the triphenylmethyl cation has a more positive central carbon than diphenylmethyl cation, although the former is more stable. Also, the 2-cyclopropylpropyl and 2-phenylpropyl cations have shifts of −86.8 and −61.1, respectively, although we have seen that according to other criteria a cyclopropyl group is better than a phenyl group at stabilizing a carbocation.95 The reasons for this discrepancy are not fully understood.88,96
chemical shifts are not always in exact order of carbocation stabilities, as determined in other ways. Thus the chemical shift shows that the triphenylmethyl cation has a more positive central carbon than diphenylmethyl cation, although the former is more stable. Also, the 2-cyclopropylpropyl and 2-phenylpropyl cations have shifts of −86.8 and −61.1, respectively, although we have seen that according to other criteria a cyclopropyl group is better than a phenyl group at stabilizing a carbocation.95 The reasons for this discrepancy are not fully understood.88,96
Table 5.3 The ![]() NMRChemical-Shift Values, in Parts per Million from
NMRChemical-Shift Values, in Parts per Million from ![]() S2, for the Charged Carbon Atom of Some Carbocations in SO2ClF–SbF5, SO2–FSO3H–SbF6, or SO2–SbF5a
S2, for the Charged Carbon Atom of Some Carbocations in SO2ClF–SbF5, SO2–FSO3H–SbF6, or SO2–SbF5a
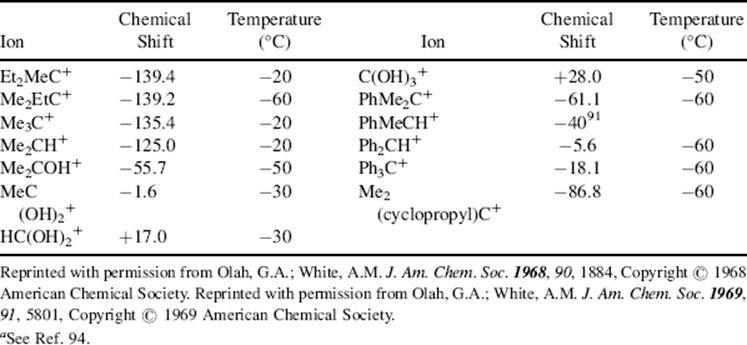
Nonclassical carbocations are discussed in section 10.C.i.
5.A.iii. The Generation and Fate of Carbocations
A number of methods are available to generate carbocations, stable or unstable.
1. A direct ionization, in which a leaving group attached to a carbon atom leaves with its pair of electrons, as in solvolysis reactions of alkyl halides (see Sec. 10.G.i) or sulfonate esters (Reaction 10-04):
![]()
2. Ionization after an initial reaction that converts one functional group into a leaving group, as in protonation of an alcohol to give an oxonium ion (ROH2+) or conversion of a primary amine to a diazonium salt, both of which ionize to the corresponding carbocation:
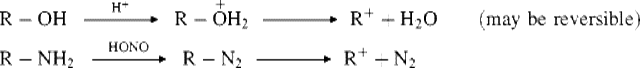
Oxonium ions are also generated by protonation of ethers,97 including epoxides.98 However, these ions do not always lead to carbocations via ionization, but often undergo substitution reactions (see Chap 10). Oxatriquinane, (19) is a fused, tricyclic alkyl oxonium ion that is remarkably stable. Oxonium ion 19 has been heated to reflux in water, can be chromatographed, and does not react with alcohols or alkyl thiols.99 The X-ray crystal structure shows longer C–O bond distances and more acute C–O–C bond angles than any reported alkyloxonium salt. Oxatriquinene (20) has also been synthesized.
3. A proton or other positive species adds to one atom of an alkene or alkyne, leaving the adjacent carbon atom with a positive charge (see Chapters 11 and 15).
![]()
4. A proton or other positive species adds to one atom of a C=X bond, where X = O, S, N in most cases, leaving the adjacent carbon atom with a positive charge (see Chapter 16). When X = O, S, this ion is a resonance stabilized oxocarbenium ion (X = O) or thiacarbenium ion (X = S), as shown. When X = NR, protonation leads to an iminium ion (X = N), with the charge localized on the nitrogen. A silylated carboxonium ion (e.g., 21) has been reported.100
![]()
When formed by any of the processes 1–3, carbocations are most often short-lived transient species and react further without being isolated. Oxocarbenium ions are more stable and may be longer lived, but even oxocarbenium ions are transient intermediates. The intrinsic barriers to formation and reaction of carbocations have been studied.101
There are two principal pathways by which carbocations react to give stable products that are effectively the reverse of the two pathways just described.
1. A carbocation may combine with a species possessing an electron pair (essentially a Lewis acid–base reaction, see Chapter 8). This reaction occurs by an atom or group donating electrons to the positive carbon of the carbocation. The atom or group that donates the electrons to carbon is called a nucleophile (see Chapter 10):
![]()
Any reasonable nucleophile will react with the carbocation, but the nucleophile may also be a neutral species with a pair to donate, in which case, of course, the immediate product must bear a positive charge (see Chapters 10, 13, 15, and 16). These reactions are very fast. A recent study measured ks (the rate constant for reaction of a simple tertiary carbocation) to be 3.5 × 1012 s−1.102
2. The carbocation may have a proton (or much less often, another positive ion) removed from the adjacent atom (see Chapters 11 and 17):
![]()
Carbocations can also adopt two other pathways that lead not to stable products, but to other carbocations:
3. Rearrangement. An alkyl or aryl group or a hydrogen atom (sometimes another group) migrates with its electron pair to the positive center, leaving another positive charge behind (see Chap 18):
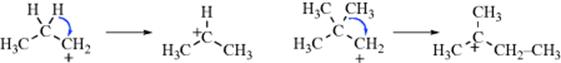
A novel rearrangement has been observed. The 2-methyl-2-butyl-1-13C cation (13C-labeled tert-amyl cation) shows an interchange of the inside and outside carbons with a barrier of 19.5 kcal mol−1 (81.6 kJ mol−1) (±2.0 kcal mol−1, 8.4 kJ mol−1).103 Another unusual migratory process has been observed for the nonamethylcyclopentyl cation. It has been shown that “four methyl groups undergo rapid circumambulatory migration with a barrier <2 kcal mol−1 (8.4 kJ mol−1) while five methyl groups are fixed to ring carbons. The process that equalizes the two sets of methyls has a barrier of 7.0 kcal mol−1 (29.3 kJ mol−1).”104
4. Addition. A carbocation may add to a double bond, generating a positive charge at a new position (see Chaps 11 and 15). This means that the π bond donates two electrons to a positive atom, generating positive charge on the carbon as shown:

Whether formed by pathway 3 or 4, the new carbocation normally reacts further in an effort to stabilize itself, usually by pathway 1 or 2. However, 22 can add to another alkene molecule, and this product can add to still another, and so on. This is one of the mechanisms for vinyl polymerization.