March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part I. Introduction
Chapter 8. Acids and Bases
8.G. The Effects of the Medium on Acid and Base Strength
Structural features are not the only factors that affect acidity or basicity. The same compound can have its acidity or basicity changed when the reaction conditions are changed. The effect of temperature (Sec. 8.A) has already been mentioned. More important is the effect of the solvent, which can exert considerable influence on acid and base strengths by differential solvation.234 If a base is more solvated than its conjugate acid, its stability is increased relative to the conjugate acid. For example, Table 8.6 shows that in reactions with a proton, where steric effects are absent, methylamine is a stronger base than ammonia and dimethylamine is stronger still.235 These results are easily explainable if one assumes that methyl groups are electron donating. However, trimethylamine, which should be even stronger, is a weaker base than dimethylamine or methylamine. This apparently anomalous behavior can be explained by differential hydration.236 Thus, NH4+ is much better hydrated (by hydrogen bonding to the water solvent) than NH3 because of its positive charge.237 It has been estimated that this effect contributes ~11 pK units to the base strength of ammonia.238 When methyl groups replace hydrogen, this difference in hydration decreases239 until, for trimethylamine, it contributes only ~6 pK units to the base strength.195 Thus two effects act in opposite directions, the field effect increasing the basicity as the number of methyl groups increases and the hydration effect decreasing it. Taken together, the strongest base is dimethylamine and the weakest is ammonia in solution. If alkyl groups are electron donating, one would expect that in the gas phase,240 where the solvation effect does not exist, the basicity order of amines toward the proton should be R3N > R2NH > RNH2 > NH3, and this has indeed been confirmed, for R = Me, as well as R = Et and Pr.241 Aniline too, in the gas phase, is a stronger base than NH3,242 so its much lower basicity in aqueous solution (pKa of PhNH3+ 4.60 compared with 9.24 for aq NH4+) is caused by similar solvation effects and not by resonance and field electron-withdrawing effects of a phenyl group. Similarly, pyridine243 and pyrrole244 are both much less basic than NH3 in aqueous solution (pyrrole245 is neutral in aqueous solution), but more basic in the gas phase. Care must be taken in attributing relative acidities or basicities to any particular effect. Solvent has a significant influence on the Hammett reaction constant (Sec. 11.D), which influences the acidity of substituted benzoic acids.246
In the case of Lewis acids, protic solvents (e.g., water or alcohol) can strongly influence their reactivity, cause it to react via an alternative path to the one desired, or even cause decomposition. Rare earth metal triflates have been used to develop water tolerant Lewis acids that can be used in many organic reactions.247
For simple alcohols, the order of gas-phase acidity is completely reversed from that in aqueous solution. In solution, the acidity is in the order H2O > MeCH2OH > Me2CHOH > Me3COH, but in the gas phase the order is precisely the opposite.248 Once again solvation effects can be invoked to explain the differences. Comparing the two extremes, H2O and Me3COH, we see that the OH− ion is very well solvated by water while the bulky Me3CO− is much more poorly solvated because the water molecules cannot get as close to the oxygen. Thus in solution H2O gives up its proton more readily. When solvent effects are absent, however, the intrinsic acidity is revealed and Me3COH is a stronger acid than H2O. This result demonstrates that simple alkyl groups cannot be simply regarded as electron donating. If methyl is an electron-donating group, then Me3COH should be an intrinsically weaker acid than H2O, yet it is stronger. A similar pattern is found with carboxylic acids, where simple aliphatic acids (e.g., propanoic) are stronger than acetic acid in the gas phase,249 although weaker in aqueous solution (Table 8.5). The evidence in these and other cases250 is that alkyl groups can be electron donating when connected to unsaturated systems, but may have either no effect or may actually be electron withdrawing in other systems. It appears that the intrinsic gas-phase acidity order of alcohols as well as the basicity order of amines is due to the effect of alkyl groups, because of their polarizability, which can spread both positive and negative charges.251 It has been calculated that even in the case of alcohols the field effects of the alkyl groups are still operating normally, but are swamped by the greater polarizability effects.252 Polarizability effects on anionic centers are a major factor in gas-phase acid–base reactions.253 It has been shown (by running reactions on ions that are solvated in the gas phase) that solvation by even one molecule of solvent can substantially affect the order of basicities.254
The effect on the orientation of solvent molecules when an acid or base is converted to its conjugate is an important aspect of solvent effects. For example, consider an acid (RCOOH) converted to RCOO− in aqueous solution. The solvent molecules, by hydrogen bonding, arrange themselves around the COO− group in a much more orderly fashion than they had been arranged around the COOH group (because they are more strongly attracted to the negative charge). This leads to a considerable loss of freedom and a decrease in entropy. Thermodynamic measurements show that for simple aliphatic and halogenated aliphatic acids in aqueous solution at room temperature, the entropy (TΔS) usually contributes much more to the total free energy change ΔG than does the enthalpy ΔH.255 Two examples are shown in Table 8.7.256 Resonance and field effects of functional groups therefore affect the acidity of RCOOH in two distinct ways. They affect the enthalpy (electron-withdrawing groups increase acidity by stabilizing RCOO− by charge dispersal), but they also affect the entropy (by lowering the charge on the COO− group and by changing the electron-density distribution in the COOH group, electron-withdrawing groups alter the solvent orientation patterns around both the acid and the ion, and consequently change ΔS).
Table 8.7 Thermodynamic Values for the Ionizations of Acetic and Chloroacetic Acids in H2O at 25°Ca
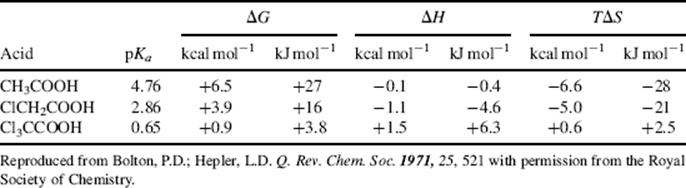
a. See Ref. 256.
A change from a protic to an aprotic solvent can also affect the acidity or basicity, since there is a difference in solvation of anions by a protic solvent (which can form hydrogen bonds) and an aprotic one.257 The effect can be extreme: In DMF, picric acid is stronger than HBr,258 though in water HBr is far stronger. This particular result can be attributed to size. That is, the large ion (O2N)3C6H2O− is better solvated by DMF than the smaller ion Br−.259 The ionic strength of the solvent also influences acidity or basicity, since it has an influence on activity coefficients.
In summary, solvation can have powerful effects on acidity and basicity. In the gas phase, the effects discussed in Section 8.F, especially resonance and field effects, operate unhindered by solvent molecules. Electron-withdrawing groups generally increase acidity (and decrease basicity); electron-donating groups act in the opposite way. In solution, especially aqueous solution, these effects still largely persist (which is why pK values in Table 8.5 do largely correlate with resonance and field effects), but in general are much weakened, and occasionally reversed.179
Notes
1. For monographs on acids and bases, see Stewart, R. The Proton: Applications to Organic Chemistry, Academic Press, NY, 1985; Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell University Press, Ithaca, NY, 1973; Finston, H.L.; Rychtman, A.C. A New View of Current Acid–Base Theories, Wiley, NY, 1982.
2. For discussion of the historical development of acid–base theory, see Bell, R.P. Q. Rev. Chem. Soc. 1947, 1, 113; Bell, R.P. The Proton in Chemistry, 1st ed., Cornell University Press, Ithaca, NY, 1959, pp. 7–17.
3. According to IUPAC terminology (Bunnett, J.F.; Jones, R.A.Y. Pure Appl. Chem. 1988, 60, 1115), an acid is a hydron donor. The IUPAC recommends that the term proton be restricted to the nucleus of the hydrogen isotope of mass 1, while the nucleus of the naturally occurring element, which contains ~0.015% deuterium, be called the hydron (the nucleus of mass 2 has always been known as the deuteron). This accords with the naturally occurring negative ion, which has long been called the hydride ion. In this book, however, we will continue to use proton for the naturally occurring form, because most of the literature uses this term.
4. Although equilibrium is reached in most acid–base reactions extremely rapidly (see Sec. 8.B), some are slow (especially those in which the proton is lost from a carbon) and in these cases time must be allowed for the system to come to equilibrium.
5. For a review of stronger Bronsted acids, see Akiyama, T. Chem. Rev. 2007, 107, 5744.
6. Table 8.1 is a thermodynamic acidity scale and applies only to positions of equilibria. For the distinction between thermodynamic and kinetic acidity (see Sec. 8.B).
7. For a first principles calculation of pK values in nonaqueous solution, see Ding, F.; Smith, J.M.; Wang, H. J. Org. Chem. 2009, 74, 2679.
8. Gold,V.; Laali, K.; Morris, K.P.; Zdunek, L.Z. J. Chem. Soc. Chem. Commun. 1981, 769; Sommer, J.; Canivet, P.; Schwartz, S.; Rimmelin, P. Nouv. J. Chim. 1981, 5, 45.
9. Arnett, E.M. Prog. Phys. Org. Chem. 1963, 1, 223, pp. 324–325.
10. Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell University Press, Ithaca, NY, 1973.
11. Deno, N.C.; Gaugler, R.W.; Wisotsky, M.J. J. Org. Chem. 1966, 31, 1967.
12. Levy, G.C.; Cargioli, J.D.; Racela, W. J. Am. Chem. Soc. 1970, 92, 6238. See, however, Brouwer, D.M.; van Doorn, J.A. Recl. Trav. Chim. Pays-Bas 1971, 90, 1010.
13. Carboxylic acids, esters, and amides are shown in this table to be protonated on the carbonyl oxygen. See Smith, C.R.; Yates, K. Can. J. Chem. 1972, 50, 771; Benedetti, E.; Di Blasio, B.; Baine, P. J. Chem. Soc. Perkin Trans. 2 1980, 500; Homer, R.B.; Johnson, C.D. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 188–197. It has been shown that some amides protonate at nitrogen: see Perrin, C.L. Acc. Chem. Res. 1989, 22, 268. For a review of alternative proton sites, see Liler, M. Adv. Phys. Org. Chem. 1975, 11, 267.
14. Stewart, R.; Granger, M.R. Can. J. Chem. 1961, 39, 2508.
15. Yates, K.; Stewart, R. Can. J. Chem. 1959, 37, 664; Stewart, R.; Yates, K. J. Am. Chem. Soc. 1958, 80, 6355.
16. Lee, D.G. Can. J. Chem. 1970, 48, 1919.
17. Cerfontain, H.; Koeberg-Telder, A.; Kruk, C. Tetrahedron Lett. 1975, 3639.
18. Arnett, E.M.; Wu, C.Y. J. Am. Chem. Soc. 1960, 82, 5660; Koeberg-Telder, A.; Lambrechts, H.J.A.; Cerfontain, H. Recl. Trav. Chim. Pays-Bas 1983, 102, 293.
19. Fischer, A.; Grigor, B.A.; Packer, J.; Vaughan, J. J. Am. Chem. Soc. 1961, 83, 4208.
20. Arnett, E.M.; Wu, C.Y. J. Am. Chem. Soc. 1960, 82, 4999.
21. Boyd, R.H. J. Phys. Chem. 1963, 67, 737.
22. Arnett, E.M.; Quirk, R.P.; Burke, J.J. J. Am. Chem. Soc. 1970, 92, 1260.
23. McTigue. P.T.; Sime, J.M. Aust. J. Chem. 1963, 16, 592.
24. Deno, N.C.; Turner, J.O. J. Org. Chem. 1966, 31, 1969.
25. Chandler, W.D.; Lee, D.G. Can. J. Chem. 1990, 68, 1757.
26. For a discussion, see Campbell, M.L.; Waite, B.A. J. Chem. Educ. 1990, 67, 386.
27. Grant, H.M.; McTigue, P.; Ward, D.G. Aust. J. Chem. 1983, 36, 2211.
28. Bruckenstein, S.; Kolthoff, I.M. in Kolthoff, I.M.; Elving, P.J. Treatise on Analytical Chemistry, Vol. 1, pt. 1, Wiley, NY, 1959, pp. 432–433.
29. Brown, H.C.; McDaniel, D.H.; Häflinger, O. in Braude, E.A.; Nachod, F.C. Determination of Organic Structures by Physical Methods, Vol. 1, Academic Press, NY, 1955, pp. 567–662.
30. Pearson, R.G.; Dillon, R.L. J. Am. Chem. Soc. 1953, 75, 2439.
31. This value includes the CO2 usually present. The value for H2CO3 alone is 3.9 in Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell University Press, Ithaca, NY, 1973.
32. Crampton, M.R. in Patai, S. The Chemistry of the Thiol Group, pt. 1, Wiley, NY, 1974, pp. 396–410.
33. See Bunting, J.W.; Kanter, J.P. J. Am. Chem. Soc. 1993, 115, 11705.
34. Perrin, D.D. Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution, 2nd ed., Pergamon, Elmsford, NY, 1982.
35. Rochester, C.H. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, p. 374.
36. Cram, D.J. Chem. Eng. News 1963, 41 (No. 33, Aug. 19), 94.
37. Bowden, K.; Stewart, R. Tetrahedron 1965, 21, 261.
38. Hine, J.; Philips, J.C.; Maxwel, J.I. J. Org. Chem. 1970, 35, 3943. See also, Ang, K.P.; Lee, T.W.S. Aust. J. Chem. 1977, 30, 521.
39. Reeve, W.; Erikson, C.M.; Aluotto, P.F. Can. J. Chem. 1979, 57, 2747.
40. See also, Olmstead, W.N.; Margolin, Z.; Bordwell, F.G. J. Org. Chem. 1980, 45, 3295.
41. Harned, H.S.; Robinson, R.A. Trans. Faraday Soc. 1940, 36, 973.
42. Streitwieser, Jr., A.; Nebenzahl, L. J. Am. Chem. Soc. 1976, 98, 2188.
43. Guthrie, J.P.; Cossar, J. Can. J. Chem. 1986, 64, 2470.
44. Homer, R.B.; Johnson, C.D. in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 238–240.
45. The pKa of acetone in DMSO is reported to be 26.5. See Bordwell, F.G.; Zhang, X.-M. Accts. Chem. Res. 1997, 26, 510.
46. Guthrie, J.P.; Cossar, J.; Klym, A. J. Am. Chem. Soc. 1984, 106, 1351; Chiang, Y.; Kresge, A.J.; Tang, Y.S.; Wirz, J. J. Am. Chem. Soc. 1984, 106, 460.
47. Streitwieser, Jr., A.; Ciuffarin, E.; Hammons, J.H. J. Am. Chem. Soc. 1967, 89, 63.
48. Streitwieser, Jr., A.; Hollyhead, W.B.; Pudjaatmaka, H.; Owens, P.H.; Kruger, T.L.; Rubenstein, P.A.; MacQuarrie, R.A.; Brokaw, M.L.; Chu, W.K.C.; Niemeyer, H.M. J. Am. Chem. Soc. 1971, 93, 5088.
49. Streitwieser, A.; Wang, G.P.; Bors, D.A. Tetrahedron 1997, 53, 10103.
50. For a review of the acidity of cyano compounds, see Hibbert, F. in Patai, S.; Rappoport, Z. The Chemistry of Triple-bonded Functional Groups, pt. 1; Wiley, NY, 1983, pp. 699–736.
51. Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, p. 19. See also, Dessy, R.E.; Kitching, W.; Psarras, T.; Salinger, R.; Chen, A.; Chivers, T. J. Am. Chem. Soc. 1966, 88, 460.
52. Amyes, T.L.; Richard, J.P. J. Am. Chem. Soc. 1996, 118, 3129.
53. Streitwieser, Jr., A.; Hollyhead, W.B.; Sonnichsen, G.; Pudjaatmaka, H.; Chang, C.J.; Kruger, T.L. J. Am. Chem. Soc. 1971, 93, 5096.
54. Buncel, E.; Menon, B. J. Am. Chem. Soc. 1977, 99, 4457.
55. Buncel, E.; Menon, B. J. Organomet. Chem. 1977, 141, 1.
56. Albrech, H.; Schneider, G. Tetrahedron 1986, 42, 4729.
57. Boerth, D.W.; Streitwieser, Jr., A. J. Am. Chem. Soc. 1981, 103, 6443.
58. Streitwieser, Jr., A.; Scannon, P.J.; Niemeyer, H.M. J. Am. Chem. Soc. 1972, 94, 7936.
59. Streitwieser, Jr., A.; Boerth, D.W. J. Am. Chem. Soc. 1978, 100, 755.
60. This value is calculated from results given in Streitwieser, Jr., A.; Caldwell, R.A.; Young, W.R. J. Am. Chem. Soc. 1969, 91, 529. For a review of acidity and basicity of cyclopropanes, see Battiste, M.A.; Coxon, J.M. in Rappoport, Z. The Chemistry of the Cyclopropyl Group, pt. 1, Wiley, NY, 1987, pp. 255–305.
61. See Daasbjerg, K. Acta Chem. Scand. B 1995, 49, 878 for pKa values of various hydrocarbons in DMF.
62. This value is calculated from results given in Streitwieser, Jr., A.; Taylor, D.R. J. Chem. Soc. D 1970, 1248.
63. These values are based on those given in Cram, D.J. Chem. Eng. News 1963, 41 (No. 33, Aug. 19), 94, but are corrected to the newer scale of Streitwieser, A.; Streitwieser, Jr., A.; Scannon, P.J.; Niemeyer, H.M. J. Am. Chem. Soc. 1972, 94, 7936; Streitwieser, Jr., A.; Boerth, D.W. J. Am. Chem. Soc. 1978, 100, 755.
64. Breslow, R. and co-workers report a value of 71 (Breslow, R.; Grant, J.L. J. Am. Chem. Soc. 1977, 99, 7745), but this was obtained by a different method, and is not comparable to the other values in Table 8.1. A more comparable value is 53. See also, Juan, B.; Schwarz, J.; Breslow, R. J. Am. Chem. Soc. 1980, 102, 5741.
65. This table gives average values for functional groups. See Brown, H.C.; McDaniel, D.H.; Häflinger, O. in Braude, E.A.; Nachod, F.C. Determination of Organic Structures by Physical Methods, Vol. 1, Academic Press, NY, 1955; Serjeant, E.P.; Dempsey, B. Ionisation Constants of Organic Acids in Aqueous Solution, Pergamon, Elmsford NY, 1979; Kortüm, G.; Vogel, W.; Andrussow, K. Dissociation Constants of Organic Acids in Aqueous Solution, Butterworth, London, 1961. The index in the 1979 volume covers both volumes. Kortüm, G.; Vogel, W.; Andrussow, K. Pure Appl. Chem. 1960, 1, 190; Arnett, E.M. Prog. Phys. Org. Chem. 1963, 1, 223; Perrin, D.D. Dissociation Constants of Organic Bases in Aqueous Solution, Butterworth, London, 1965, and Supplement, 1972; Collumeau, A. Bull. Soc. Chim. Fr. 1968, 5087; Bordwell, F.G. Acc. Chem. Res. 1988, 21, 456; Perrin, D.D. Ionisation Constants of Inorganic Acids and Bases in Aqueous Solution, 2nd ed., Pergamon, Elmsford NY, 1982; Pure Appl. Chem. 1969, 20, 133.
66. Gillespie, R.J. Acc. Chem. Res. 1968, 1, 202.
67. For discussions of pKa determinations for the conjugate acids of ketones, see Bagno, A.; Lucchini,V.; Scorrano, G. Bull. Soc. Chim. Fr. 1987, 563; Toullec, J. Tetrahedron Lett. 1988, 29, 5541.
68. For a review of methods of determining pKa values, see Cookson, R.F. Chem. Rev. 1974, 74, 5.
69. Kolthoff, I.M.; Bruckenstein, S. in Kolthoff, I.M.; Elving, P.J. Treatise on Analytical Chemistry, Vol. 1, pt. 1, Wiley, NY, 1959, pp. 475–542, p. 479.
70. For reviews of organic compounds protonated at O, N, or S, see Olah, G.A.; White, A.M.; O'Brien, D.H. Chem. Rev. 1970, 70, 561; Olah, G.A.; White, A.M.; O'Brien, D.H. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1973, pp. 1697–1781.
71. Rochester, C.H. Acidity Functions, Academic Press, NY, 1970. For discussion of the basicity of such compounds, see Liler, M. Reaction Mechanisms in Sulfuric Acid, Academic Press, NY, 1971, pp. 118–139.
72. Pedireddi, V.R.; Desiraju, G.R. J. Chem. Soc. Chem. Commun. 1992, 988.
73. Alkorta, I.; Campillo, N.; Rozas, I.; Elguero, J. J. Org. Chem. 1998, 63, 7759.
74. Streitwieser, A.; Keevil, T.A.; Taylor, D.R.; Dart, E.C. J. Am. Chem. Soc. 2005, 127, 9290.
75. See Reutov, O.A.; Beletskaya, I.P.; Butin, K.P. CH-Acids, Pergamon, NY, 1978; Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 1–45; Streitwieser, Jr., A.; Hammons, J.H. Prog. Phys. Org. Chem. 1965, 3, 41; Wiberg, K.B. J. Org. Chem. 2002, 67, 1613.
76. See Jones, J.R. Q. Rev. Chem. Soc. 1971, 25, 365; Fischer, H.; Rewicki, D. Prog. Org. Chem. 1968, 7, 116; Reutov, O.A.; Beletskaya, I.P.; Butin, K.P. CH-Acids, Chapter 1, Pergamon, NY, 1978 (an earlier version of this chapter appeared in Russ. Chem. Rev. 1974, 43, 17); Gau, G.; Assadourian, L.; Veracini, S. Prog. Phys. Org. Chem. 1987, 16, 237; in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, pt. A, Elsevier, NY, 1980, the reviews by Pellerite, M.J.; Brauman, J.I. pp. 55–96 (gas-phase acidities); and Streitwieser, Jr., A.; Juaristi, E.; Nebenzahl, L. pp. 323–381.
77. See Olah, G.A.; Prakash, G.K.S.; Sommer, J. Superacids, Wiley, NY, 1985; Gillespie, R.J.; Peel, T.E. Adv. Phys. Org. Chem. 1971, 9, 1; Arata, K. Adv. Catal. 1990, 37, 165. For a review of methods of measuring superacidity, see Jost, R.; Sommer, J. Rev. Chem. Intermed. 1988, 9, 171.
78. Prakash, G.K.S. J. Org. Chem. 2006, 71, 3661.
79. These reactions are equilibria. What the rule actually says is that the position of equilibrium will be such that the weaker acid predominates. However, this needs to be taken into account only when the acid and base are close to each other in the table (within ~2 pK units).
80. See Gal, J.; Maria, P. Prog. Phys. Org. Chem. 1990, 17, 159.
81. Bohme, D.K.; Lee-Ruff, E.; Young, L.B. J. Am. Chem. Soc. 1972, 94, 4608, 5153.
82. Gerrard, W.; Macklen, E.D. Chem. Rev. 1959, 59, 1105. For other examples, see Calder, G.V.; Barton, T.J. J. Chem. Educ. 1971, 48, 338; Hambly, A.N. Rev. Pure Appl. Chem. 1965, 15, 87, p. 88.
83. Tal'Rose, V.L.; Frankevitch, E.L. J. Am. Chem. Soc. 1958, 80, 2344; DeKock, R.L. J. Am. Chem. Soc. 1975, 97, 5592; McDaniel, D.H.; Coffman, N.B.; Strong, J.M. J. Am. Chem. Soc. 1970, 92, 6697. For a computational study of the proton affinities of ketones, vicinal diketones and α-keto esters, see Taskinen, A.; Nieminen, V.; Toukoniitty, E.; Murzin, D.Yu.; Hotokka, M. Tetrahedron 2005, 61, 8109.
84. For measnurement of amine basicity via ion pair statibity in ionic liquids (Sec. 9.D.iii), see D'Anna, F.; Renato Noto, R. Tetrahedron 2007, 63, 11681.
85. Caskey, D.C.; Damrauer, R.; McGoff, D. J. Org. Chem. 2002, 67, 5098.
86. Streitwieser, A.; Kim, H.-J. J. Am. Chem. Soc. 2000, 122, 11783; Garrido, G.; Koort, E.; Ràfols, C.; Bosch, E.; Rodima, T.; Leito, I.; Rosés, M. J. Org. Chem. 2006, 71, 9062.
87. Canle, L.M.; Demirtas, I.; Freire, A.; Maskill, H.; Mishima, M. Eur. J. Org. Chem. 2004, 5031.
88. Chmurzynski, L. J. Heterocyclic Chem. 2000, 37, 71.
89. Perrin, C.L.; Ohta, B.K.; Kuperman, J.; Liberman, J.; Erdélyi, M. J. Am. Chem. Soc. 2005, 127, 9641.
97. Carrasco, N.; González-Nilo, F.; Rezende, M.C. Tetrahedron 2002, 58, 5141.
98. A new scale of π-basicity is proposed. See Stoyanov, E.S.; Stoyanova, I.V.; Reed, C.A. Chemistry: European J. 2008, 14, 7880.
99. Ly, T.; Krout, M.; Pham, D.K.; Tani, K.; Stoltz, B.M.; Julian, R.R. J. Am. Chem. Soc. 2007, 129, 1864.
90. Le Questel, J.-Y.; Laurence, C.; Lachkar, A.; Helbert, M.; Berthelot, M. J. Chem. Soc. Perkin Trans. 2 1992, 2091.
91. Berthelot, M.; Besseau, F.; Laurence, C. Eur. J. Org. Chem. 1998, 925.
92. Korzhenevskaya, N.G.; Rybachenko, V.I.; Kovalenko, V.V.; Lyashchuk, S.N.; Red'ko, A.N. Russ. J. Org. Chem. 2007, 43, 1475.
93. Berthelot, M.; Helbert, M.; Laurence, C.; LeQuestel, J.-Y.; Anvia, F.; Taft, R.W. J. Chem. Soc. Perkin Trans. 2 1993, 625.
94. Besseau, F.; Laurence, C.; Berthelot, M. J. Chem. Soc. Perkin Trans. 2 1994, 485.
95. Besseau, F.; Luçon, M.; Laurence, C.; Berthelot, M. J. Chem. Soc. Perkin Trans. 2 1998, 101.
96. Laurence, C.; Berthelot, M.; Luçon, M.; Morris, D.G. J. Chem. Soc. Perkin Trans. 2 1994, 491.
100. For calculated basicities of super bases see Glasovac, Z.; Eckert-Maksi![]() , M.; Maksi
, M.; Maksi![]() , Z.B. New J. Chem., 2009, 33, 588.
, Z.B. New J. Chem., 2009, 33, 588.
101. Schwesinger, R.; Mißfeldt, M.; Peters, K.; von Schnering, H.G. Angew. Chem. Int. Ed. 1987, 26, 1165; Schwesinger, R.; Schlemper, H.; Hasenfratz, Ch.; Willaredt, J.; Dimbacher, T.; Breuer, Th.; Ottaway, C.; Fletschinger, M.; Boele, J.; Fritz, H.; Putzas, D.; Rotter, H.W.; Bordwell, F.G.; Satish, A.V.; Ji, G.Z.; Peters, E.-M.; Peters, K.; von Schnering, H.G. Liebigs Ann. 1996, 1055.
102. Kova![]() evi
evi![]() , B.; Maksi
, B.; Maksi![]() , Z.B. Org. Lett. 2001, 3, 1523.
, Z.B. Org. Lett. 2001, 3, 1523.
103. Alder, R.W.; Bowman, P.S.; Steele, W.R.S.; Winterman, D.R. Chem. Commun. 1968, 723; Alder, R.W. Chem. Rev. 1989, 89, 1215.
104. Raczynska, E.D.; Darowska, M.; Dabkowska, I.; Decouzon, M.; Gal, J.-F.; Maria, P.-C.; Poliart, C.D. J. Org. Chem. 2004, 69, 4023.
105. Raab, V.; Kipke, J.; Gschwind, R.M.; Sundermeyer, J. Chem. Eur. J. 2002, 8, 1682.
106. Krieger, C.; Newsom, I.; Zirnstein, M.A.; Staab, H.A. Angew. Chem. Int. Ed. 1989, 28, 84.
107. Gorecka-Kobylinska, J.; Schlosser, M. J. Org. Chem. 2009, 74, 222.
108. For reviews of such proton transfers, see Hibbert, F. Adv. Phys. Org. Chem. 1986, 22, 113; Crooks, J.E. in Bamford, C.H.; Tipper, C.F.H. Chemical Kinetics, Vol. 8; Elsevier, NY, 1977, pp. 197–250. See Bernasconi, C.F.; Fairchild, D.E.; Montañez, R.L.; Aleshi, P.; Zheng, H.; Lorance, E. J. Org. Chem. 2005, 70, 7721.
109. See Eigen, M. Angew. Chem. Int. Ed. 1964, 3, 1.
110. See, for example, Hojatti, M.; Kresge, A.J.; Wang, W. J. Am. Chem. Soc. 1987, 109, 4023.
111. See Ritchie, C.D.; Lu, S. J. Am. Chem. Soc. 1989, 111, 8542.
112. Chen, X.; Walthall, D.A.; Brauman, J.I. J. Am. Chem. Soc. 2004, 126, 12614.
113. Abraham, M. H.; Enomoto, K.; Clarke, E. D.; Sexton, G. J. Org. Chem. 2002, 67, 4782.
114. See Hibbert, F. in Bamford, C.H.; Tipper, C.F.H. Chemical Kinetics, Vol. 8, Elsevier, NY, 1977, pp. 97–196; Kreevoy, M.M. Isot. Org. Chem. 1976, 2, 1; Leffek, K.T. Isot. Org. Chem. 1976, 2, 89.
115. See Bernasconi, C.F. Tetrahedron 1985, 41, 3219.
116. Houk, R.J.T.; Anslyn, E.V.; Stanton, J.F. Org. Lett. 2006, 8, 3461.
117. Kresge, A.J.; Powell, M.F. J. Org. Chem. 1986, 51, 822; Formosinho, S.J.; Gal, V.M.S. J. Chem. Soc. Perkin Trans. 2 1987, 1655.
118. Not all 1-alkynes behave as normal acids; see Aroella, T.; Arrowsmith, C.H.; Hojatti, M.; Kresge, A.J.; Powell, M.F.; Tang, Y.S.; Wang, W. J. Am. Chem. Soc. 1987, 109, 7198.
119. Juhasz, M.; Hoffmann, S.; Stoyanov, E.; Kim, K.-C.; Reed, C.A. Angew. Chem. Int. Ed. 2004, 43, 5352.
120. See Kurz, J.L. J. Am. Chem. Soc. 1989, 111, 8631.
121. Wang, W.; Cheng, P.; Huang, C.; Jong, Y. Bull. Chem. Soc. Jpn. 1992, 65, 562.
122. Wang, W.-h.; Cheng, C.-c. Bull. Chem. Soc. Jpn. 1994, 67, 1054.
123. Lambert, C.; Hampel, F.; Schleyer, P.v.R. Angew. Chem. Int. Ed. 1992, 31, 1209.
124. For fuller treatments, see Hammett, L.P. Physical Organic Chemistry, 2nd ed., McGraw-Hill, NY, 1970, pp. 263–313; Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge University Press, Cambridge, 1984, pp. 83–93; Arnett, E.M.; Scorrano, G. Adv. Phys. Org. Chem. 1976, 13, 83.
125. Holt, J.; Karty, J.M. J. Am. Chem. Soc. 2003, 125, 2797.
126. Thomazeau, C.; Olivier-Bourbigou, H.; Magna, L.; Luts, S.; Gilbert, B. J. Am. Chem. Soc. 2003, 125, 5264.
127. Yang, Y.-l.; Kou, Y. Chem. Commun. 2004, 226.
128. Hammett, L.P.; Deyrup, A.J. J. Am. Chem. Soc. 1932, 54, 2721.
129. See Rochester, C.H. Acidity Functions, Academic Press, NY, 1970; Cox, R.A.; Yates, K. Can. J. Chem. 1983, 61, 2225; Boyd R.H. in Coetzee, J.F.; Ritchie, C.D. Solute–Solvent Interactions, Marcel Dekker, NY, 1969, pp. 97–218.
130. See Yates, K.; McClelland, R.A. Prog. Phys. Org. Chem. 1974, 11, 323.
131. See Kreevoy, M.M.; Baughman, E.H. J. Am. Chem. Soc. 1973, 95, 8178; García, B.; Leal, J.M.; Herrero, L.A.; Palacios, J.C. J. Chem. Soc. Perkin Trans. 2 1988, 1759; Arnett, E.M.; Quirk, R.P.; Burke, J.J. J. Am. Chem. Soc.1970, 92, 1260.
132. For lengthy tables of many acidity scales, with references, see Cox, R.A.; Yates, K. Can. J. Chem. 1983, 61, 2225. For an equation that is said to combine the vast majority of acidity functions, see Zalewski, R.I.; Sarkice, A.Y.; Geltz, Z. J. Chem. Soc. Perkin Trans. 2 1983, 1059.
133. See Vianello, R.; Maksi![]() , Z.B. Eur. J. Org. Chem. 2004, 5003.
, Z.B. Eur. J. Org. Chem. 2004, 5003.
134. Deno, N.C.; Berkheimer, H.E.; Evans, W.L.; Peterson, H.J. J. Am. Chem. Soc. 1959, 81, 2344.
135. Reagan, M.T. J. Am. Chem. Soc. 1969, 91, 5506.
136. Edward, J.T.; Wong, S.C. Can. J. Chem. 1977, 55, 2492; Liler, M.; Markovi![]() , D. J. Chem. Soc. Perkin Trans. 2 1982, 551.
, D. J. Chem. Soc. Perkin Trans. 2 1982, 551.
137. Hammett, L.P. Physical Organic Chemistry, 2nd ed., McGraw-Hill, NY, 1970, p. 278; Rochester, C.H. Acidity Functions, Academic Press, NY, 1970, p. 21.
138. For another approach to solvent basicity scales, see Catalán, J.; Gómez, J.; Couto, A.; Laynez, J. J. Am. Chem. Soc. 1990, 112, 1678.
139. See Rochester, C.H. Q. Rev. Chem. Soc. 1966, 20, 511; Rochester, C.H. Acidity Functions, Academic Press, NY, 1970, pp. 234–264; Bowden, K. Chem. Rev. 1966, 66, 119.
140. Bunnett, J.F.; McDonald, R.L.; Olsen, F.P. J. Am. Chem. Soc. 1974, 96, 2855.
141. More O'Ferrall, R.A. J. Chem. Soc. Perkin Trans. 2 1972, 976.
142. Bagno, A.; Scorrano, G.; More O'Ferrall, R.A. Rev. Chem. Intermed. 1987, 7, 313. See also, Cox, R.A. Acc. Chem. Res. 1987, 20, 27.
143. Bunnett, J.F. J. Am. Chem. Soc. 1961, 83, 4956, 4968, 4973, 4978.
144. Catalán, J.; Couto, A.; Gomez, J.; Saiz, J.L.; Laynez, J. J. Chem. Soc. Perkin Trans. 2 1992, 1181.
145. Abraham, M.H.; Taft, R.W. J. Chem. Soc. Perkin Trans. 2 1993, 305.
146. Liu, P.C.; Hoz, S.; Buncel, E. Gazz. Chim. Ital. 1996, 126, 31. See also, Abraham, M.H.; Zhao, Y.J. J. Org. Chem. 2004, 69, 4677.
147. Fattahi, A.; Kass, S.R. J. Org. Chem. 2004, 69, 9176.
148. See Stewart, R. The Proton: Applications to Organic Chemistry, Academic Press, NY, 1985, pp. 251–305; Willi, A.V. in Bamford, C.H.; Tipper, C.F.H. Chemical Kinetics, Vol. 8, Elsevier, NY, 1977, pp. 1–95; Jones, R.A.Y. Physical and Mechanistic Organic Chemistry, 2nd ed., Cambridge University Press, Cambridge, 1984, pp. 72–82; Bender, M.L. Mechanisms of Homogeneous Catalysis from Protons to Proteins, Wiley, NY, 1971, pp. 19–144.
149. See Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982, pp. 167–179; Bell, R.P. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry: Recent Advances, Plenum Press, 1978, pp. 55–84; Kresge, A.J. Chem. Soc. Rev. 1973, 2, 475.
150. See Silva, P.J. J. Org. Chem. 2009, 74, 914.
151. See Marcus, R.A. J. Phys. Chem. 1968, 72, 891; Kresge, A.J. Chem. Soc. Rev. 1973, 2, 475.
152. Omitting the work terms.
153. Albery, W.J. Annu. Rev. Phys. Chem. 1980, 31, 227, p. 244.
154. See Jencks, W.P. Acc. Chem. Res. 1976, 9, 425; Stewart, R.; Srinivasan, R. Acc. Chem. Res. 1978, 11, 271; Guthrie, J.P. J. Am. Chem. Soc. 1980, 102, 5286.
155. See Agmon, N. J. Am. Chem. Soc. 1980, 102, 2164; Murray, C.J.; Jencks, W.P. J. Am. Chem. Soc. 1988, 110, 7561.
156. Pross, A.; Shaik, S.S. New J. Chem. 1989, 13, 427; Lewis, E.S. J. Phys. Org. Chem. 1990, 3, 1.
157. Lewis bases are useful catalysts in organic synthesis. See Denmark, S.E.; Beutner, G.L. Angew. Chem. Int. Ed. 2008, 47, 1560.
158. For a monograph on Lewis acid–base theory, see Jensen, W.B. The Lewis Acid–Base Concept, Wiley, NY, 1980. For a discussion of the definitions of Lewis acid and base, see Jensen, W.B. Chem. Rev. 1978, 78, 1.
159. Rauk, A.; Hunt, I.R.; Keay, B.A. J. Org. Chem. 1994, 59, 6808.
160. For a review of ate complexes, see Wittig, G. Q. Rev. Chem. Soc. 1966, 20, 191.
161. See Satchell, D.P.N.; Satchell, R.S. Q. Rev. Chem. Soc. 1971, 25, 171; Chem. Rev. 1969, 69, 251. See also, Sandström, M.; Persson, I.; Persson, P. Acta Chem. Scand. 1990, 44, 653; Laszlo, P.; Teston-Henry, M. Tetrahedron Lett. 1991, 32, 3837.
162. Springer, G.; Elam, C.; Edwards, A.; Bowe, C.; Boyles, D.; Bartmess, J.; Chandler, M.; West, K.; Williams, J.; Green, J.; Pagni, R.M.; Kabalka, G.W. J. Org. Chem. 1999, 64, 2202.
163. See Ayers, P.W.; Parr, R.G. J. Am. Chem. Soc. 2000, 122, 2010.
164. Pearson, R.G.; Songstad, J. J. Am. Chem. Soc. 1967, 89, 1827. For a monograph on the concept, see Ho, T. Hard and Soft Acids and Bases Principle in Organic Chemistry, Academic Press, NY, 1977; Pearson, R.G. J. Chem. Educ. 1987, 64, 561; Ho, T. Tetrahedron 1985, 41, 1; Pearson, R.G. in Chapman, N.B.; Shorter, J. Advances in Linear Free-Energy Relationships, Plenum Press, NY, 1972, pp. 281–319. For a collection of papers, see Pearson, R.G. Hard and Soft Acids and Bases, Dowden, Hutchinson, and Ross, Stroudsberg, PA, 1973.
165. Taken from Pearson, R.G. J. Chem. Ed. 1968, 45, 581, 643.
166. Pearson, R.G. Inorg. Chem. 1988, 27, 734; J. Org. Chem. 1989, 54, 1423. See also, Orsky, A.R.; Whitehead M.A. Can. J. Chem. 1987, 65, 1970.
167. See Sauers, R.R. Tetrahedron 1999, 55, 10013.
168. Parr, R.G.; Pearson, R.G. J. Am. Chem. Soc. 1983, 105, 7512. Note that there is not always a strict correlation between the values in Table 8.4 and the categories of Table 8.3.
169. Pearson, R.G. J. Am. Chem. Soc. 1988, 110, 7684.
170. For proofs of this principle, see Chattaraj, P.K.; Lee, H.; Parr, R.G. J. Am. Chem. Soc. 1991, 113, 1855.
171. Wolman, Y. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, p. 677; Maskill, H. The Physical Basis of Organic Chemistry, Oxford University Press, Oxford 1985, p. 159.
172. See also, Bochkov, A.F. J. Org. Chem. USSR 1986, 22, 1830, 1837.
173. Méndez, F.; Gázguez, J.L. J. Am. Chem. Soc. 1994, 116, 9298.
174. Woodward, S. Tetrahedron 2002, 58, 1017.
175. See Hine, J. Structural Effects on Equilibria in Organic Chemistry, Wiley, NY, 1975; Taft, R.W. Prog. Phys. Org. Chem. 1983, 14, 247; Petrov, E.S. Russ. Chem. Rev. 1983, 52, 1144 (NH acids); Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell Univ. Press, Ithaca, NY, 1973, pp. 86–110. For a monograph on methods of estimating pK values by analogy, extrapolation, and so on, see Perrin, D.D.; Dempsey, B.; Serjeant, E.P. pKa Prediction for Organic Acids and Bases, Chapman and Hall, NY, 1981.
176. The varying degrees by which the different factors that affect gas-phase acidities of 25 acids has been calculated: Taft, R.W.; Koppel, I.A.; Topsom, R.D.; Anvia, F. J. Am. Chem. Soc. 1990, 112, 2047.
177. For a review of the enhancement of acidity by NO2, see Lewis, E.S. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 2, Wiley, NY, 1982, pp. 715–729.
178. Filler, R.; Wang, C. Chem. Commun. 1968, 287.
179. See Edward, J.T. J. Chem. Educ. 1982, 59, 354; Schwartz, L.M. J. Chem. Educ. 1981, 58, 778.
180. Headley, A.D.; McMurry, M.E.; Starnes, S.D. J. Org. Chem. 1994, 59, 1863.
181. Wiberg, K.B. J. Org. Chem. 2003, 68, 875.
182. For calculated gas-phase acidities of substituted benzoic acids see Wiberg, K.B. J. Org. Chem. 2002, 67, 4787. Also see Gupta, K.; Giri, S.; Chattaraj, P.K. New J. Chem. 2008, 32, 1945.
183. DeMaria, P.; Fontana, A.; Spinelli, D.; Dell'Erba, C.; Novi, M.; Petrillo, G.; Sancassan, F. J. Chem. Soc. Perkin Trans. 2 1993, 649.
184. Chen, C.-T.; Siegel, J.S. J. Am. Chem. Soc. 1994, 116, 5959. See also, Sotomatsu, T.; Shigemura, M.; Murata, Y.; Fujita, T. Bull. Chem. Soc. Jpn. 1992, 65, 3157.
185. See Exner, O.; ![]() ársky, P. J. Am. Chem. Soc. 2001, 123, 9564. See also, Liptak, M.D.; Shields, G.C. J. Am. Chem. Soc. 2001, 123, 7314.
ársky, P. J. Am. Chem. Soc. 2001, 123, 9564. See also, Liptak, M.D.; Shields, G.C. J. Am. Chem. Soc. 2001, 123, 7314.
186. It has been contended that resonance delocalization plays only a minor role in the increased strength of carboxylic acids compared to alcohols, and the “...higher acidity of acids arises principally because the electrostatic potential of the acidic hydrogens is more positive in the neutral acid molecule...”: Siggel, M.R.; Streitwieser, Jr., A.; Thomas, T.D. J. Am. Chem. Soc. 1988, 110, 8022; Thomas, T.D.; Carroll, T.X.; Siggel, M.R. J. Org. Chem. 1988, 53, 1812. For contrary views, see Exner, O. J. Org. Chem. 1988, 53, 1810; Perrin, D.D. J. Am. Chem. Soc. 1991, 113, 2865. See also, Godfrey, M. Tetrahedron Lett. 1990, 31, 5181.
187. Copper complexes of active methylene compounds show large pKa shifts. See Zhong, Z.; Postnikova, B.J.; Hanes, R.E.; Lynch, V.M.; Anslyn, E.V. Chemistry: European J. 2005, 11, 2385.
188. Goumont, R.; Magnier, E.; Kizilian, E.; Terrier, F. J. Org. Chem. 2003, 68, 6566.
189. See, however, Krygowski, T.M.; Maurin, J. J. Chem. Soc. Perkin Trans. 2 1989, 695.
190. Smith, J.W. in Patai, S. The Chemistry of the Amino Group; Wiley, NY, 1968, pp. 161–204.
191. Liptak, M.D.; Gross, K.C.; Seybold, P.G.; Feldus, S.; Shields, G.C. J. Am. Chem. Soc. 2002, 124, 6421.
192. Taft, R.W. Prog. Phys. Org. Chem. 1983, 14, 247, see Sec. 5.B.i.
193. Note that Lewis acidity decreases, whereas Br![]() nsted acidity increases, going down the table. There is no contradiction here when we remember that in the Lewis picture the actual acid in all Br
nsted acidity increases, going down the table. There is no contradiction here when we remember that in the Lewis picture the actual acid in all Br![]() nsted acids is the same, namely, the proton. In comparing, say, HI and HF, we are not comparing different Lewis acids but only how easily F– and I– give up the proton.
nsted acids is the same, namely, the proton. In comparing, say, HI and HF, we are not comparing different Lewis acids but only how easily F– and I– give up the proton.
194. The effect discussed here is an example of a symmetry factor. For an extended discussion, see Eberson, L. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 211–293.
195. Brown, H.C. J. Am. Chem. Soc. 1945, 67, 378, 1452, Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 53–64. See also, Brown, H.C.; Krishnamurthy, S.; Hubbard, J.L. J. Am. Chem. Soc. 1978, 100, 3343.
196. Hibbert, F.; Simpson, G.R. J. Chem. Soc. Perkin Trans. 2 1987, 243, 613.
197. For a review of the effect of strain on amine basicities, see Alder, R.W. Chem. Rev. 1989, 89, 1215.
198. Staab, H.A.; Saupe, T.; Krieger, C. Angew. Chem. Int. Ed. 1983, 22, 731.
199. Saupe, T.; Krieger, C.; Staab, H.A. Angew. Chem. Int. Ed. 1986, 25, 451.
200. For a review, see Staab, H.A.; Saupe, T. Angew. Chem. Int. Ed. 1988, 27, 865.
201. Howard, S.T. J. Am. Chem. Soc. 2000, 122, 8238.
202. Krieger, C.; Newsom, I.; Zirnstein, M.A.; Staab, H.A. Angew. Chem. Int. Ed. 1989, 28, 84. See also, Staab, H.A.; Zirnstein, M.A.; Krieger, C. Angew. Chem. Int. Ed. 1989, 28, 86.
203. Miyahara, Y.; Goto, K.; Inazu, T. Tetrahedron Lett. 2001, 42, 3097.
204. Ueno, M.; Ishitani, H.; Kobayashi, S. Org. Lett. 2002, 4, 3395.
205. Mathieu, B.; de Fays, L.; Ghosez, L. Tetrahedron Lett 2000, 41, 9651
206. Brown, H.C.; Kanner, B. J. Am. Chem. Soc. 1953, 75, 3865; 1966, 88, 986.
207. Meot-Ner, M.; Smith, S.C. J. Am. Chem. Soc. 1991, 113, 862, and references cited therein. See also, Benoit, R.L.; Fréchette, M.; Lefebvre, D. Can. J. Chem. 1988, 66, 1159.
208. Arnett, E.M.; Harrelson, Jr., J.A. J. Am. Chem. Soc. 1987, 109, 809.
209. For a discussion of why esters and amides are weaker acids than ketones, see Fersner, A.; Karty, J.M.; Mo, Y. J. Org. Chem. 2009, 74, 7245.
210. Wang, X.; Houk, K.N. J. Am. Chem. Soc. 1988, 110, 1870; Wiberg, K.B.; Laidig, K.E. J. Am. Chem. Soc. 1988, 110, 1872.
211. Majewski, M.; Wang, F. Tetrahedron 2002, 58, 4567.
212. Amedjkouh, M. Tetrahedron Asymm. 2004, 15, 577.
213. Gung, B.W.; Yanik, M.M. J. Org. Chem. 1996, 61, 947.
214. Grotjahn, D.B.; Sheridan, P.M.; Al Jihad, I.; Ziurys, L.M. J. Am. Chem. Soc. 2001, 123, 5489.
215. Fressigné, C.; Maddaluno, J.; Giessner-Prettre, C.; Silvi, B. J. Org. Chem. 2001, 66, 6476.
216. Williard, P.G.; Salvino, J.M. J. Org. Chem. 1993, 58, 1. For a study of the oligomer structure of LDA at low ligand concentrations, see Rutherford, J.L.; Collum, D.B. J. Am. Chem. Soc. 2001, 123, 199.
217. Ito, H.; Nakamura, T.; Taguchi, T.; Hanzawa, Y. Tetrahedron Lett. 1992, 33, 3769.
218. Aubrecht, K.B.; Collum, D.B. J. Org. Chem. 1996, 61, 8674.
219. Romesberg, F.E.; Collum, D.B. J. Am.Chem. Soc. 1994, 116, 9198, 9187. For a study of other mixed aggregates, see Thomas, R.D.; Huang, J. J. Am. Chem. Soc. 1999, 121, 11239.
220. Sakuma, K.; Gilchrist, J.H.; Romesberg, F.E.; Cajthami, C.E.; Collum, D.B. Tetrahedron Lett. 1993, 34, 5213.
221. Lucht, B.L.; Collum, D.B. J. Am. Chem. Soc. 1994, 116, 7949.
222. Lucht, B.L.; Collum, D.B. J. Am. Chem. Soc. 1995, 117, 9863. See also, Lucht, B.L.; Collum, D.B. J. Am. Chem. Soc. 1996, 118, 2217, 3529. See Romesberg, F.E.; Bernstein, M.P.; Gilchrist, J.H.; Harrison, A.T.; Fuller, D.J.; Collum, D.B. J. Am. Chem. Soc. 1993, 115, 3475 for the structure in HMPA.
223. Lucht, B.L.; Collum, D.B. J. Am. Chem. Soc. 1994, 116, 6009.
224. Collum, D.B. Acc. Chem. Res. 1993, 26, 227. For NMR studies of LiNEt2 and ring laddering see Rutherford, J.L.; Collum, D.B. J. Am. Chem. Soc. 1999, 121, 10198.
225. Hilmersson, G.; Davidsson, Ö. J. Org. Chem. 1995, 60, 7660. See O'Brien, P. J. Chem. Soc. Perkin Trans. 1 1998, 1439; Sott, R.; Grandander, J.; Dinér, P.; Hilmersson, G. Tetrahedron Asymm. 2004, 15, 267.
226. Arvidsson, P.I.; Hilmersson, G.; Ahlberg, P. J. Am. Chem. Soc. 1999, 121, 183.
227. Carlier, P.R.; Madura, J.D. J. Org. Chem. 2002, 67, 3832.
228. Williard, P. G.; Jacobson, M. A. Org. Lett. 2000, 2, 2753. For the structure and bonding of dilithiodiamines see Pratt, L.M.; Mu, R. J. Org. Chem. 2004, 69, 7519.
229. Sun, C.; Williard, P.G. J. Am. Chem. Soc. 2000, 122, 7829. See also, Pratt, L.M.; Streitwieser, A. J. Org. Chem. 2003, 68, 2830.
230. Jackman, L.M.; Çizmeciyan, D.; Williard, P.G.; Nichols, M.A. J. Am. Chem. Soc. 1993, 115, 6262.
231. Jackman, L.M.; Chen, X. J. Am. Chem. Soc. 1992, 114, 403.
232. Streitwieser, A.; Juaristi, E.; Kim, Y.-J.; Pugh, J.K. Org. Lett. 2000, 2, 3839.
233. Alconcel, L.S.; Deyerl, H.-J.; Continetti, R.E. J. Am. Chem. Soc. 2001, 123, 12675.
234. See Epshtein, L.M.; Iogansen, A.V. Russ. Chem. Rev. 1990, 59, 134; Dyumaev, K.M.; Korolev, B.A. Russ. Chem. Rev. 1980, 49, 1021; Taft, R.W.; Bordwell, F.G. Acc. Chem. Res. 1988, 21, 463; Heemstra, J.M.; Moore, J.S. Tetrahedron 2004, 60, 7287.
235. See Smith, J.W. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 161–204.
236. Aue, D.H.; Webb, H.M.; Bowers, M.T. J. Am. Chem. Soc. 1972, 94, 4726; 1976, 98, 311, 318; Mucci, A.; Domain, R.; Benoit, R.L. Can. J. Chem. 1980, 58, 953. See also, Drago, R.S.; Cundari, T.R.; Ferris, D.C. J. Org. Chem. 1989, 54, 1042.
237. For discussions of the solvation of ammonia and amines, see Jones, III, F.M.; Arnett, E.M. Prog. Phys. Org. Chem. 1974, 11, 263; Grunwald, E.; Ralph, E.K. Acc. Chem. Res. 1971, 4, 107.
238. Condon, F.E. J. Am. Chem. Soc. 1965, 87, 4481, 4485.
239. For two reasons: (1) the alkyl groups are poorly solvated by the water molecules, and (2) the strength of the hydrogen bonds of the BH+ ions decreases as the basicity of B increases: Lau, Y.K.; Kebarle, P. Can. J. Chem. 1981, 59, 151.
240. See Liebman, J.F. Mol. Struct. Energ. 1987, 4, 49; Dixon, D.A.; Lias, S.G. Mol. Struct. Energ. 1987, 2, 269; Bohme, D.K. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 2, Wiley, NY, 1982, pp. 731–762; Arnett, E.M. Acc. Chem. Res. 1973, 6, 404. See Lias, S.G.; Liebman, J.F.; Levin, R.D. J. Phys. Chem. Ref. Data, 1984, 13, 695. See also, the tables of gas-phase acidities and basicities in the following articles, and their cited references: Meot-Ner, M.; Kafafi, S.A. J. Am. Chem. Soc. 1988, 110, 6297; Headley, A.D. J. Am. Chem. Soc. 1987, 109, 2347; Fujio, M.; McIver, Jr., R.T.; Taft, R.W. J. Am. Chem. Soc. 1981, 103, 4017; Lau, Y.K.; Nishizawa, K.; Tse, A.; Brown, R.S.; Kebarle, P. J. Am. Chem. Soc. 1981, 103, 6291.
241. Briggs, J.P.; Yamdagni, R.; Kebarle, P. J. Am. Chem. Soc. 1972, 94, 5128; Aue, D.H.; Webb H.M.; Bowers, M.T. J. Am. Chem. Soc. 1972, 94, 4726; 1976, 98, 311, 318.
242. Ikuta, S.; Kebarle, P. Can. J. Chem. 1983, 61, 97.
243. Taft, R.W.; Taagepera, M.; Summerhays, K.D.; Mitsky, J. J. Am. Chem. Soc. 1973, 95, 3811.
244. Yamdagni, R.; Kebarle, P. J. Am. Chem. Soc. 1973, 95, 3504.
245. See Catalan, J.; Abboud, J.L.M.; Elguero, J. Adv. Heterocycl. Chem. 1987, 41, 187.
246. Bartnicka, H.; Bojanowska, I.; Kalinowski, M.K. Aust. J. Chem. 1993, 46, 31.
247. Kobayashi, S. Synlett, 1994, 689.
248. Arnett, E.M.; Small, L.E.; McIver, Jr., R.T.; Miller, J.S. J. Am. Chem. Soc. 1974, 96, 5638; Blair, L.K.; Isolani, P.C.; Riveros, J.M. J. Am. Chem. Soc. 1973, 95, 1057; McIver, Jr., R.T.; Scott, J.A.; Riveros, J.M. J. Am. Chem. Soc. 1973, 95, 2706. Also see Bartmess, J.E.; McIver, Jr., R.T. J. Am. Chem. Soc. 1977, 99, 4163.
249. See Caldwell, G.; Renneboog, R.; Kebarle, P. Can. J. Chem. 1989, 67, 611.
250. Brauman, J.I.; Blair, L.K. J. Am. Chem. Soc. 1971, 93, 4315; Laurie, V.W.; Muenter, J.S. J. Am. Chem. Soc. 1966, 88, 2883.
251. Brauman, J.I.; Riveros, J.M.; Blair, L.K. J. Am. Chem. Soc. 1971, 93, 3914; Huheey, J.E. J. Org. Chem. 1971, 36, 204; Radom, L. Aust. J. Chem. 1975, 28, 1; Aitken, E.J.; Bahl, M.K.; Bomben, K.D.; Gimzewski, J.K.; Nolan, G.S.; Thomas, T.D. J. Am. Chem. Soc. 1980, 102, 4873.
252. Taft, R.W.; Taagepera, M.; Abboud, J.M.; Wolf, J.F.; DeFrees, D.J.; Hehre, W.J.; Bartmess, J.E.; McIver, Jr., R.T. J. Am. Chem. Soc. 1978, 100, 7765. For a scale of polarizability parameters, see Hehre, W.J.; Pau, C.; Headley, A.D.; Taft, R.W.; Topsom, R.D. J. Am. Chem. Soc. 1986, 108, 1711.
253. Bartmess, J.E.; Scott, J.A.; McIver, Jr., R.T. J. Am. Chem. Soc. 1979, 101, 6056.
254. Bohme, D.K.; Rakshit, A.B.; Mackay, G.I. J. Am. Chem. Soc. 1982, 104, 1100.
255. Bolton, P.D.; Hepler, L.G. Q. Rev. Chem. Soc. 1971, 25, 521; Gerrard, W.; Macklen, E.D. Chem. Rev. 1959, 59, 1105. See also, Wilson, B.; Georgiadis, R.; Bartmess, J.E. J. Am. Chem. Soc. 1991, 113, 1762.
256. Bolton, P.D.; Hepler, L.G. Q. Rev. Chem. Soc. 1971, 25, 521; p. 529.
257. For a review, see Parker, A.J. Q. Rev. Chem. Soc. 1962, 16, 163.
258. Sears, P.G.; Wolford, R.K.; Dawson, L.R. J. Electrochem. Soc. 1956, 103, 633.
259. Miller, J.; Parker, A.J. J. Am. Chem. Soc. 1961, 83, 117.