March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 10. Aliphatic Substitution, Nucleophilic and Organometallic
In nucleophilic aliphatic substitution the attacking (electron donating) reagent (the nucleophile) brings an electron pair to the substrate, using this pair to form the new bond, and the leaving group (the nucleofuge) comes away with an electron pair:
![]()
As written, this equation says nothing about charges. Nucleophile Y may be neutral or negatively charged; RX may be neutral or positively charged; so there are four charge types, examples of which follows:
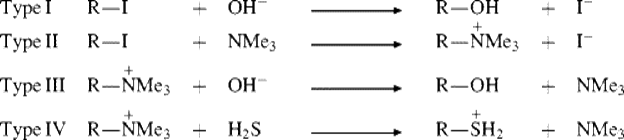
In all cases, Y must have an unshared pair of electrons, so that all nucleophiles are Lewis bases. When Y is the solvent, the reaction is called solvolysis. Nucleophilic substitution at an aromatic carbon is considered in Chapter 13.
Nucleophilic substitution at an alkyl carbon is said to alkylate the nucleophile. For example, the above reaction between RI and NMe3 is an alkylation of trimethylamine. Nucleophilic substitution at an acyl carbon is an acylationof the nucleophile, and such reactions are found in Chapter 16.
10.A. Mechanisms
Several distinct mechanisms are possible for aliphatic nucleophilic substitution reactions, depending on the substrate, nucleophile, leaving group, and reaction conditions. In all of them, however, the attacking reagent carries the electron pair with it. Mechanisms that occur at a saturated carbon atom are considered first.1 By far the most common are the SN1 and SN2 mechanisms.
10.A.i. The SN2 Mechanism
The designation SN2 stands for substitution nucleophilic bimolecular. The IUPAC designation (Sec. 9.F) is ANDN. In this mechanism, there is backside attack,2 which means that the nucleophile approaches the substrate from a position 180° away from the leaving group. This approach will minimize steric and electronic repulsion of the substrate and the incoming nucleophile. The reaction is a one-step process with no intermediate (see below, and Sec. 10.A.iv). The C–Y bond is formed as the C–X bond is broken to generate pentacoordinate transition state 1.
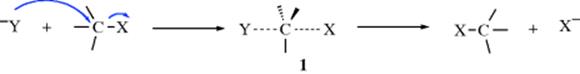
The energy necessary to break the C–X bond is supplied by the collision of the nucleophile (Y) with the carbon bearing the leaving group (X). The top of the curve of free energy of activation is taken to be the transition state, and the position of the atoms for this reaction are shown in transition state 1. The transition state is not a real structure, of course, but the energetic midpoint of the reaction. There are various computational methods to ascertain characteristics of a given transition state, and the experimental examination of kinetic isotope effects has been used to infer information about the transition state.3 The group X must leave as the group Y comes in, because at no time can the carbon have more than eight electrons in its outer shell. When the transition state is reached, the central carbon atom has gone from its initial sp3 hybridization to essentially sp2 with an approximately perpendicular p orbital. One lobe of this p orbital overlaps with the nucleophile and the other with the leaving group. This is the reason a frontside SN2 mechanism has never been observed. In a hypothetical frontside transition state, both the nucleophile and the leaving group would have to overlap with the same lobe of the p orbital. The backside mechanism involves the maximum amount of overlap throughout the course of the reaction. At the energy point of the transition state, the three nonreacting substituents and the central carbon are approximately coplanar. They will be exactly coplanar if both the entering and leaving group are the same.
There is a large amount of evidence for the SN2 mechanism. First, there is the kinetic evidence.4 Since both the nucleophile and the substrate are involved in the rate-determining step (the only step, in this case), the reaction should be first order in each component, second order overall, and satisfy the rate expression shown in Eq. (10-1).
(10-1) ![]()
This rate law has been found to apply. Note that the 2 in SN2 stands for bimolecular. It must be remembered that this is not always the same as second order (see Sec. 6.J.vi). If a large excess of nucleophile is present (e.g., if it is the solvent5) the mechanism may still be bimolecular, although the experimentally determined kinetics will be first order, Eq. (10-2)).
(10-2) ![]()
As previously mentioned (Sec. 6.J.vi), such kinetics are called pseudo-first order.
The kinetic evidence is a necessary but not a sufficient condition; other mechanisms will be encountered that are also consistent with these data. Much more convincing evidence is obtained from the fact that the mechanism predicts inversion of configuration when substitution occurs at a chiral carbon and this has been observed many times. This inversion of configuration (see Sec. 4.E.ii) that proceeds through transition state 1 is called the Walden inversion and was observed long before the SN2 mechanism was formulated by Hughes and Ingold.6
At this point, it is useful to see just how it was originally proved that a given substitution reaction proceeds with inversion of configuration, even before the mechanism was known. Walden presented a number of examples7 in which inversion must have taken place. For example, (+)-malic acid (2) could be converted to (+)-chlorosuccinic acid by thionyl chloride and to (−)-chlorosuccinic acid by phosphorus pentachloride.
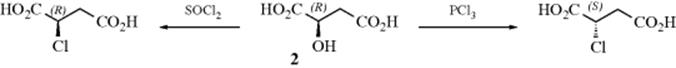
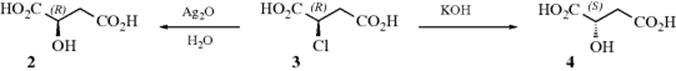
One was an inversion and the other a retention of configuration, but the question was which was which? The signs of rotation are of no help in answering this question since rotation need not be related to configuration (Sec. 4.F). Another example discovered by Walden is formation of 3 from 4.8
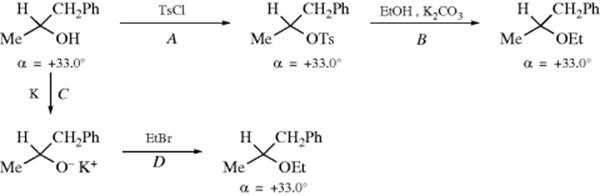
A series of experiments designed to settle the matter of exactly where inversion takes place was performed by Phillips,9 Kenyon,10 and co-workers. In 1923, Phillips and co-workers9 carried out the following cycle based on (+)-1-phenyl-2-propanol. In this cycle, (+)-1-phenyl-2-propanol is converted to its ethyl ether by two routes, path AB giving the (−) ether, and path CD giving the (+) ether. Therefore, at least one of the four steps must be an inversion. It is extremely unlikely that there is inversion in step A, C, or D, since in all these steps the C–O bond is unbroken, and in none of them could the oxygen of the bond have come from the reagent. There is a high probability that A, C, and D proceeded with retention, leaving B as the inversion. A number of other such cycles were carried out, always with nonconflicting results.10 These experiments not only definitely showed that certain specific reactions proceed with inversion, but also established the configurations of many compounds.
Walden inversion has been found at a primary carbon atom by the use of a chiral substrate containing a deuterium and a hydrogen atom at the carbon bearing the leaving group.11 Inversion of configuration has also been found for SN2 reactions proceeding in the gas phase.12 High-pressure mass spectrometry has been used to probe the energy surface for gas-phase SN2 reactions, which have two transition states (a “loose” and a “tight” transition state.13
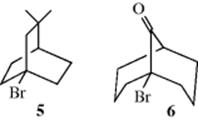
Another kind of evidence for the SN2 mechanism comes from compounds with potential leaving groups at bridgehead carbons. If the SN2 mechanism is correct, these compounds should not be able to react by this mechanism, since the nucleophile cannot approach from the rear. Among the many known examples of unsuccessful reaction attempts at bridgeheads under SN2 conditions14 are treatment of the [2.2.2] system (5) with ethoxide ion15 and treatment of the [3.3.1] system (6) with sodium iodide in acetone.16 In these cases, open-chain analogues underwent the reactions readily. As a final example of evidence for the SN2 mechanism, the reaction between optically active 2-octyl iodide and radioactive iodide ion may be mentioned:
![]()
Racemization is expected in this reaction, since if the pure R isomer is the starting material each exchange will produce an (S) isomer. With increasing concentration of (S) isomer, it will begin to compete for I− with the R isomer, until at the end a racemic mixture is left. The point investigated was a comparison of the rate of inversion with the rate of uptake of radioactive ![]() . It was found17 that the rates were identical within experimental error:
. It was found17 that the rates were identical within experimental error:
![]()
The rate of racemization was the parameter actually measured, which is twice the rate of inversion, since each inversion creates, in effect, two racemic molecules. The significance of this result is that it shows that every act of exchange is an act of inversion.
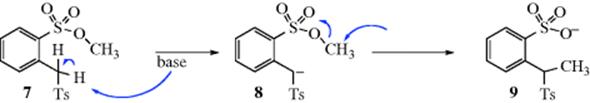
Eschenmoser and co-workers18 provided strong evidence that the transition state in an SN2 reaction must be linear. Base treatment of methyl α-tosyl-o-toluenesulfonate (7) gives the o-(l-tosylethyl)benzenesulfonate ion (9). The role of the base is to remove the benzylic proton α- to the tosyl group to give the ion 8. It might be supposed that the negatively charged carbon of 8 attacks the methyl group in an internal SN2 process, but this is not the case. Crossover experiments18 (See 11-27) have shown that the negatively charged carbon attacks the methyl group of another molecule rather than the nearby one in the same molecule; that is, the reaction is intermolecular (see 8) and not intramolecular, despite the more favorable entropy of the latter pathway (Sec. 6.D). It is likely that intramolecular attack does not take place because complete linearity cannot be attained. This behavior is in sharp contrast to that in cases in which the leaving group is not constrained (Sec. 10.C), where intramolecular SN2 mechanisms operate freely.
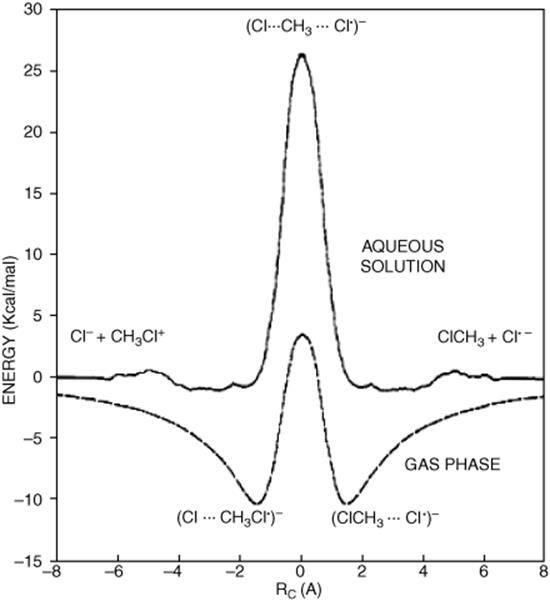
There is evidence, both experimental and theoretical, that there are intermediates in at least some SN2 reactions in the gas phase, in charge-type I reactions, where a negative ion nucleophile attacks a neutral substrate.19 Two energy minima, one before and one after the transition state, appear in the reaction coordinate Fig. 10.1).20 The energy surface for the SN2 Menshutkin reaction (10-31) has been examined. It was shown that charge separation was promoted by the solvent.21 An ab initio study of the SN2 reaction at primary and secondary carbon centers has looked at the energy barrier (at the transition state) to the reaction.22 These minima correspond to unsymmetrical ion–dipole complexes.23 Theoretical calculations also show such minima in certain solvents (e.g., N, N-dimethylformamide DMF), but not in water.24 In general, polar aprotic solvents (those that do not have an acidic hydrogen X–H, where X–O, S, N, etc.), favor polarized transition state 1.25 The rate of the reaction is generally slower in protic solvents (e.g., alcohol or water).
Fig. 10.1 Free energy profile for the gas phase (solid line) and aqueous solution (dashed line) SN2 reaction between CH3Cl and Cl-, from molecular orbital calculations.20 [Reprinted with permission from Chandrasekhar, J.; Smith, S.F.; Jorgensen, W.L. J. Am. Chem. Soc. 1985, 107,154. Copyright © 1985 American Chemical Society.
The SN2 reactions can occur at atoms other than carbon, X (e.g., nitrogen or sulfur26), and analogous to the phenomenon observed for SN2 reactions at carbon.27 the valence of the element X, controls the intrinsic barrier for the reaction in accord with the properties seen in the periodic table.28
For a list of some of the more important reactions that operate by the SN2 mechanism, see Table 10.7.
Note that in some reactions, (e.g., bromine transfer between carbanions via nucleophilic attack on bromine), anomalous kinetic behavior is observed. The largest rate constants are associated with bromine transfer between cyano-activated carbanions and the smallest relate to the removal of bromine from the nitromethane and nitroethane moieties.29 The Br![]() nsted plot (log k vs ΔpKa) for this reaction shows that unlike any normal Br
nsted plot (log k vs ΔpKa) for this reaction shows that unlike any normal Br![]() nsted plot, which by definition displays a positive slope, the plot for MeNO2 and EtNO2 is negative. In deprotonation reactions of carbon compounds, the reactivity of nitroethane and nitromethane were shown to be anomalous.30 In the series nitromethane, ethane, and isopropane, compounds with higher acidity undergo slower deprotonation (i.e., the Br
nsted plot, which by definition displays a positive slope, the plot for MeNO2 and EtNO2 is negative. In deprotonation reactions of carbon compounds, the reactivity of nitroethane and nitromethane were shown to be anomalous.30 In the series nitromethane, ethane, and isopropane, compounds with higher acidity undergo slower deprotonation (i.e., the Br![]() nsted plot displays a negative slope), contrary to expectations.31
nsted plot displays a negative slope), contrary to expectations.31
10.A.ii. The SN1 Mechanism
The most ideal version of the SN1 mechanism (substitutional nucleophilic unimolecular) consists of two steps32 (once again, possible charges on the substrate and nucleophile are not shown):
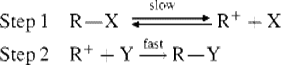
The first step is a slow ionization of the substrate and is the rate-determining step. The second is a rapid reaction between the intermediate carbocation and the nucleophile. There are, of course, transition states for both step 1 (R![]() X) and step 2 (R+
X) and step 2 (R+![]() Y).33 The reactive nature of the carbocation can be expressed by its electrophilic character, or electrophilicity. A theoretical discussion concerning the origin of the electrophilicity concept was proposed by Parr et al.34 In general, a good electrophile was characterized by having a high value of electronegativity (or a high value of electronic chemical potential), and a low value of chemical hardness (Sec. 8.E.i). The effect of substitution has been studied35 in the context of superelectrophilicity (where carbocations are generated in superacidic media). Solvent effects have also been studied.36 Electrophilicity scales have been proposed using other carbocations,37 and there is an electrophilicity index.38 Carbocation intermediates have been studied for the reaction Ar2CH–O2CR → Ar2CH+, and the relative ionization rates with the same anionic leaving group does not correlate with the corresponding relative reactivities of the carbocation toward a common nucleophile.39
Y).33 The reactive nature of the carbocation can be expressed by its electrophilic character, or electrophilicity. A theoretical discussion concerning the origin of the electrophilicity concept was proposed by Parr et al.34 In general, a good electrophile was characterized by having a high value of electronegativity (or a high value of electronic chemical potential), and a low value of chemical hardness (Sec. 8.E.i). The effect of substitution has been studied35 in the context of superelectrophilicity (where carbocations are generated in superacidic media). Solvent effects have also been studied.36 Electrophilicity scales have been proposed using other carbocations,37 and there is an electrophilicity index.38 Carbocation intermediates have been studied for the reaction Ar2CH–O2CR → Ar2CH+, and the relative ionization rates with the same anionic leaving group does not correlate with the corresponding relative reactivities of the carbocation toward a common nucleophile.39
Returning to the SN1 mechanism, ionization of a leaving group to form the carbocation is always assisted by the solvent,40 since the energy necessary to break the bond is largely recovered by solvation of R+ and of X. For example, the ionization of t-BuCl to t-Bu+ and Cl− in the gas phase without a solvent requires 150 kcal mol−1 (630 kJ mol−1). In the absence of a solvent, such a process simply would not take place, except at very high temperatures. In water, this ionization requires only 20 kcal mol−1 (84 kJ mol−1). The difference is solvation energy. This means that the water is effectively “pulling” the leaving group away from the substrate. In cases where the role of the solvent is solely to assist in departure of the leaving group from the frontside the mechanism is called limiting SN1. In other words, there is a complete absence of backside (SN2) participation by solvent molecules. There is kinetic and other evidence41 that two molecules of a protic solvent form weak hydrogen bonds with X in order to pull the leaving group X away from RX.
![]()
In the IUPAC system, the SN1 mechanism is DN + AN or DN‡ + AN (where ‡ denotes the rate-determining step). The IUPAC designations for the SN1 and SN2 mechanisms thus clearly show the essential differences between them: ANDN indicates that bond breaking is concurrent with bond formation; DN + AN shows that the former happens first.
In looking for evidence for the SN1 mechanism, the first thought is that it should be a first-order reaction following the rate law:
(10-3) ![]()
Since the slow step involves only the substrate, the rate should be dependent only on the concentration of that. Although the solvent is necessary to assist in the process of ionization, it does not enter the rate expression, because it is present in large excess. However, the simple rate law given in Eq. (10-3)) is not sufficient to account for all the data. Many cases are known where pure first-order kinetics are followed, but in many other cases more complicated kinetics are found. This fact can be explained by taking into account the reversibility of the first step. The X formed in this step competes with Y for the cation and the rate law must be modified as shown (see Chap 6).
(10-4) 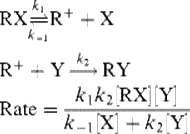
At the beginning of the reaction, when the concentration of X is very small, k-1[X] is negligible compared with k2[Y] and the rate law is reduced to Eq. (10-3). Indeed, SN1 reactions generally do display simple first-order kinetics in their initial stages. Most kinetic studies of SN1 reactions fall into this category. In the later stages of SN1 solvolyses, [X] becomes large and Eq. (10-4) predicts that the rate should decrease. This is found to be the case for diarylmethyl halides,42 although not for tert-butyl halides, which follow Eq. (10-3) for the entire reaction.43 An explanation for this difference is that tert-butyl cations are less selective than the relatively stable diarylmethyl type (Sec. 5.A.ii). Although halide ion is a much more powerful nucleophile than water, there is much more water available since it is the solvent.44 The selective diphenylmethyl cation survives many collisions with solvent molecules before combining with a reactive halide. However, some of the collisions with solvent lead to product, so despite the fact that the halide ion is more reactive, the slower reaction with solvents leads to product because of the overwhelming number of solvent molecules.
If the X formed during the reaction can decrease the rate, at least in some cases, it should be possible to add X from the outside and further decrease the rate in that way. This retardation of rate by addition of X is called common-ion effect or the mass-law effect. Once again, addition of halide ions decreases the rate for diphenylmethyl, but not for tert-butyl halides.
One factor that complicates the kinetic picture is the salt effect. An increase in ionic strength of the solution usually increases the rate of an SN1 reaction (Sec. 10.G.iv). But when the reaction is of charge type II, where both Y and RX are neutral, so that X is negatively charged (and most solvolyses are of this charge type), the ionic strength increases as the reaction proceeds and this increases the rate. This effect must be taken into account in studying the kinetics. Incidentally, the fact that the addition of outside ions increases the rate of most SN1 reactions makes especially impressive the decrease in rate caused by the common ion.
Note that the pseudo-first-order rate law for an SN2 reaction in the presence of a large excess of Y [Eq. (10-1)] is the same as that for an ordinary SN1 reaction [Eq. (10-3)]. It is thus not possible to tell these cases apart by simple kinetic measurements. However, they can often be distinguished by the common-ion effect mentioned above. Addition of a common ion will not markedly affect the rate of an SN2 reaction beyond the effect caused by other ions. Unfortunately, as seen above, not all SN1 reactions show the common-ion effect, and this test fails for tert-butyl and similar cases.
Kinetic studies also provide other evidence for the SN1 mechanism. One technique used ![]() NMR to follow the solvolysis of trifluoroacetyl esters.45 If this mechanism operates essentially as shown above, the rate should be the same for a given substrate under a given set of conditions, regardless of the identity of the nucleophile or its concentration. In one experiment, benzhydryl chloride (Ph2CHCl) was treated in SO2 with the nucleophiles fluoride ion, pyridine, and triethylamine at several concentrations of each nucleophile.46 In each case, the initial rate of the reaction was approximately the same when corrections were made for the salt effect. The same type of behavior has been shown in a number of other cases, even when the reagents are as different in their nucleophilicities (see Sec. 10.G.ii) as H2O and HO−.
NMR to follow the solvolysis of trifluoroacetyl esters.45 If this mechanism operates essentially as shown above, the rate should be the same for a given substrate under a given set of conditions, regardless of the identity of the nucleophile or its concentration. In one experiment, benzhydryl chloride (Ph2CHCl) was treated in SO2 with the nucleophiles fluoride ion, pyridine, and triethylamine at several concentrations of each nucleophile.46 In each case, the initial rate of the reaction was approximately the same when corrections were made for the salt effect. The same type of behavior has been shown in a number of other cases, even when the reagents are as different in their nucleophilicities (see Sec. 10.G.ii) as H2O and HO−.
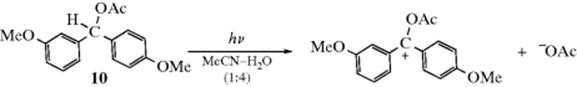
It is normally not possible to detect the carbocation intermediate of an SN1 reaction directly, because its lifetime is very short. However, in the case of 3,4-dimethoxydiphenylmethyl acetate (10), and certain other substrates in polar solvents, it was possible to initiate the reaction photolytically, and under these conditions the UV spectra of the intermediate carbocations could be obtained,47 providing additional evidence for the SN1 mechanism. Further, addition of water to a colorless solution of Ar2CH–OAc (Ar = morpholinophenyl) in acetone, leads to direct observation of the intermediate carbocation.48
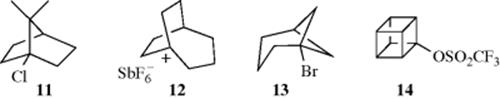
Further evidence for the SN1 mechanism is that reactions run under SN1 conditions fail or proceed very slowly at the bridgehead positions13 of [2.2.1] (norbornyl) systems49 (e.g., 1-chloroapocamphane, 8). If SN1 reactions require carbocations and if carbocations must be planar or nearly planar, then it is no surprise that bridgehead 1-norbornyl carbon atoms, which cannot assume planarity, do not become the seat of carbocations. As an example, 11, boiled 21 h with 30% KOH in 80% ethanol or 48 h with aq ethanolic silver nitrate, gave no reaction in either case,50 although analogous open-chain systems reacted readily. According to this theory, SN1 reactions should be possible with larger rings, since near-planar carbocations might be expected there. This turns out to be the case. For example, [2.2.2] bicyclic systems undergo SN1 reactions much faster than smaller bicyclic systems, although the reaction is still slower than with open-chain systems.51 Proceeding to a still larger system, the bridgehead [3.2.2] cation (12) is actually stable enough to be kept in solution in SbF5-SO3ClF at temperatures below −50 °C52 (see also, Sec. 10.G, category 6). Other small bridgehead systems that undergo SN1 reactions are the [3.1.1] (e.g., 13)53 and the cubyl (e.g., 14)54 systems. Ab initio calculations show that the cubyl cation, although it cannot be planar, requires less energy to form than the 1-norbornyl cation.55 There are reactions where the cationic carbon is not coplanar with conjugating substituents (e.g., phenyl), and formation of the carbocation is more difficult but the reaction proceeds.56
Certain nucleophilic substitution reactions that normally involve carbocations can take place at norbornyl bridgeheads57 (though it is not certain that carbocations are actually involved in all cases) if the leaving group used is of the type that cannot function as a nucleophile (and thus come back) once it has gone, and in the displacement of ClCO2 in 15. In this example,58 chlorobenzene is the nucleophile (see Reaction 11-10).
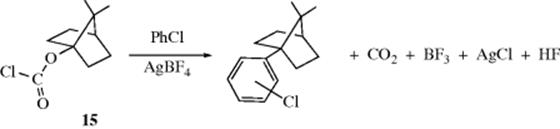
Additional evidence for the SN1 mechanism, in particular, for the intermediacy of carbocations, is that solvolysis rates of alkyl chlorides in ethanol parallel carbocation stabilities, as determined by heats of ionization measured in superacid solutions (Sec. 5.A.ii).59 It is important to note that some solvolysis reactions proceed by an SN2 mechanism.60
10.A.iii. Ion Pairs in the SN1 Mechanism61
Like the kinetic evidence, the stereochemical evidence for the SN1 mechanism is less clear-cut than it is for the SN2 mechanism.62 If there is a free carbocation, it is planar (Sec. 5.A.ii), and the nucleophile should attack with equal facility from either side of the plane, resulting in complete racemization. Although many first-order substitutions do give complete racemization, many others do not. Typically there is 5–20% inversion, although in a few cases, a small amount of retention of configuration has been found. These and other results have led to the conclusion that in many SN1 reactions at least some of the products are not formed from free carbocations, but rather from ion pairs. According to this concept,63 SN1 reactions proceed in this manner:
![]()
where 16 (an intimate, contact, or tight ion pair), 17 (a loose, or solvent-separated ion pair),64 or 18 (the dissociated ions, which means that each is surrounded by molecules of solvent).65 The reaction in which the intimate ion pair recombines to give the original substrate is referred to as internal return. The reaction products can result from attack by the nucleophile at any stage. In the intimate ion pair (16), R+ does not behave like the free cation of 18. There is probably significant bonding between R+ and X− and asymmetry may well be maintained.66 Here, X− “solvates” the cation on the side from which it departed, while solvent molecules near 16 can only solvate it from the opposite side. Nucleophilic attack by a solvent molecule on 16 thus leads to inversion. Note that there is evidence for concerted pathways in some ion pairing reactions.67
Ignoring the possibilities of elimination or rearrangement (see Chapters 17 and 18), a complete picture of the possibilities for solvolysis reactions68 in a solvent SH is represented by the following scheme,69 although in any particular case it is unlikely that all these reactions occur:
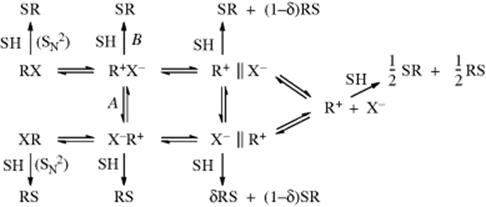
In this scheme, (RS) and (SR) represent enantiomers, and so on, and δ represents some fraction. The following are the possibilities: (1) Direct attack by SH on RX gives SR (complete inversion) in a straight SN2 process. (2) If the intimate ion pair R+ X− is formed, the solvent can attack at this stage. This can lead to total inversion if reaction A does not take place or to a combination of inversion and racemization if there is competition between A and B. (3) If the solvent-separated ion pair is formed, SH can attack here. The stereochemistry is not maintained as tightly and more racemization (perhaps total) is expected. (4) Finally, if free R+ is formed, it is planar, and attack by SH gives complete racemization.
The ion-pair concept thus predicts that SN1 reactions can display either complete racemization or partial inversion. The fact that this behavior is generally found is evidence that ion pairs are involved in many SN1 reactions. There is much other evidence for the intervention of ion pairs,70 including ion–molecule pairs.71
1. The compound 2-octyl brosylate was labeled at the sulfone oxygen with ![]() and solvolyzed. The unreacted brosylate recovered at various stages of solvolysis had the
and solvolyzed. The unreacted brosylate recovered at various stages of solvolysis had the ![]() considerably, although not completely, scrambled:72
considerably, although not completely, scrambled:72
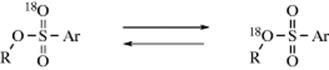
In an intimate ion pair, the three oxygen atoms become equivalent:
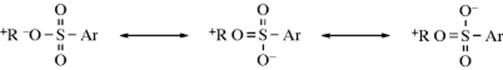
Similar results were obtained with several other sulfonate esters.73 The possibility must be considered that the scrambling resulted from ionization of one molecule of ROSO2Ar to R+ and ArSO2O− followed by attack by the ArSO2O− ion on another carbocation or perhaps on a molecule of ROSO2Ar in an SN2 process. However, this was ruled out by solvolyzing unlabeled substrate in the presence of labeled HOSO2Ar. These experiments showed that there was some intermolecular exchange (3–20%), but not nearly enough to account for the amount of scrambling found in the original experiments. Similar scrambling was found in solvolysis of labeled carboxylic esters (R–18O–COR′), where the leaving group is R′COO−74. Also in this case, the external addition of RCOO− did not result in significant exchange. However, it has been proposed that the scrambling could result from a concerted process, not involving ion-pair intermediates, and there is some evidence for this view.75
2. The special salt effect. The addition of LiClO4 or LiBr in the acetolysis of certain tosylates produced an initial steep rate acceleration that then decreased to the normal linear acceleration (caused by the ordinary salt effect).76This result is interpreted as: the ClO4− (or Br−) traps the solvent-separated ion pair to give ![]() which, being unstable under these conditions, goes to product. Hence, the amount of solvent-separated ion pair that would have returned to the starting material is reduced, and the rate of the overall reaction is increased. The special salt effect has been directly observed by the use of picosecond absorption spectroscopy.77
which, being unstable under these conditions, goes to product. Hence, the amount of solvent-separated ion pair that would have returned to the starting material is reduced, and the rate of the overall reaction is increased. The special salt effect has been directly observed by the use of picosecond absorption spectroscopy.77
3. The possibilities of racemization or inversion of the product (RS) of a solvolysis reaction were discussed previously. However, the formation of an ion pair followed by internal return can also affect the stereochemistry of the substrate molecule RX. Cases have been found where internal return racemizes an original optically active RX, an example being solvolysis in aq acetone of α-p-anisylethyl p-nitrobenzoate,78 while in other cases partial or complete retention is found (e.g., solvolysis in aq acetone of p-chlorobenzhydryl p-nitrobenzoate).79 Racemization of RX is presumably caused by the equilibrium pathway:
![]()
Evidence for ion pairs includes some cases where internal return involves racemization, it has been shown that such racemization is faster than solvolysis. For example, optically active p-chlorobenzhydryl chloride racemizes ~ 30 times faster than it solvolyzes in acetic acid.80
Molecular orbital calculations81 on tert-BuCl show that the C–Cl distance in the intimate ion pair is 2.9 Å and the onset of the solvent-separated ion pair takes place at ~ 5.5 Å (cf. the C–Cl bond length of 1.8 Å).
In a few cases, SN1 reactions have been found to proceed with partial retention (20–50%) of configuration. Ion pairs have been invoked to explain some of these.82 For example, it has been proposed that the phenolysis of optically active α-phenylethyl chloride, in which the ether of net retained configuration is obtained, involves a four-center mechanism:

This conclusion is strengthened by the fact that partial retention was obtained in this system only with chloride or other neutral leaving groups; with leaving groups bearing a positive charge, which are much less likely to form hydrogen bonds with the solvent, no retention was found.83 Partial retention can also arise when the ion pair is shielded at the backside by an additive (e.g., acetonitrile, acetone, or aniline).84
The difference between the SN1 and SN2 mechanisms is in the timing of the steps. In the SN1 mechanism, first X leaves, then Y attacks. In the SN2 case, the two things happen simultaneously. One could imagine a third possibility: first the attack of Y and then the removal of X. This is not possible at a saturated carbon, since it would mean that there are more than eight electrons in the outer shell of carbon. However, this type of mechanism is possible and indeed occurs at other types of substrate (Sec. 10.F and Chap 13).
10.A.iv. Mixed SN1 and SN2 Mechanisms
Some reactions of a given substrate under a given set of conditions display all the characteristics of SN2 mechanisms; other reactions seem to proceed by SN1 mechanisms, but cases are found that cannot be characterized so easily. There seems to be something in between, a mechanistic “borderline” region.85 At least two broad theories have been devised to explain these phenomena. One theory holds that intermediate behavior is caused by a mechanism that is neither “pure” SN1 nor “pure” SN2, but some “in-between” type. According to the second theory, there is no intermediate mechanism at all, and borderline behavior is caused by simultaneous operation, in the same flask, of both the SN1 and SN2 mechanisms; that is, some molecules react by the SN1, while others react by the SN2 mechanism.
One formulation of the intermediate-mechanism theory is that of Sneen et al.86 The formulation is in fact very broad and applies not only to borderline behavior, but to all nucleophilic substitutions at a saturated carbon.87According to Sneen, et al.88 all SN1 and SN2 reactions can be accommodated by one basic mechanism (the ion-pair mechanism). The substrate first ionizes to an intermediate ion pair that is then converted to products:
![]()
The difference between the SN1 and SN2 mechanisms is that in the former case the formation of the ion pair (k1) is rate determining, while in the SN2 mechanism its destruction (k2) is rate determining. Borderline behavior is found where the rates of formation and destruction of the ion pair are of the same order of magnitude.88 However, a number of investigators have asserted that these results could also be explained in other ways.89
There is evidence for the Sneen formulation where the leaving group has a positive charge. In this case, there is a cation–molecule pair (RX+ → R+ X−).90 instead of the ion pair that would be present if the leaving group were uncharged. Katritzky et al.91 found that when such a reaction was run at varying high pressures, there was a minimum in the plot of rate constant versus pressure. A minimum of this sort usually indicates a change in mechanism, and the interpretation in this case was that the normal SN2 mechanism operates at higher pressures and the cation–molecule mechanism at lower pressures.
An alternative view that also favors an intermediate mechanism is that of Schleyer and co-workers,92 who believe that the key to the problem is varying degrees of nucleophilic solvent assistance to ion-pair formation. They have proposed an SN2 (intermediate) mechanism.93
Among the experiments that have been cited for the viewpoint that borderline behavior results from simultaneous SN1 and SN2 mechanisms is the behavior of 4-methoxybenzyl chloride in 70% aq acetone.94 In this solvent, hydrolysis95 (i.e., conversion to 4-methoxybenzyl alcohol) occurs by an SN1 mechanism. When azide ions are added, the alcohol is still a product, but now 4-methoxybenzyl azide is another product. Addition of azide ions increases the rate of ionization (by the salt effect), but decreases the rate of hydrolysis. If more carbocations are produced, but fewer go to the alcohol, then some azide must be formed by reaction with carbocations: an SN1 process. However, the rate of ionization is always less than the total rate of reaction, so some azide must also form by an SN2 mechanism.94 Thus, the conclusion is that SN1 and SN2 mechanisms operate simultaneously.96
Some nucleophilic substitution reactions that seem to involve a “borderline” mechanism actually do not. Thus, one of the principal indications that a “borderline” mechanism is taking place has been the finding of partial racemization and partial inversion. However, this type of stereochemical behavior is quite consistent with a strictly SN2 process.97 The reaction of optically active 2-octyl brosylate in 75% aq dioxane, gave inverted 2-octanol in 77% optical purity.97 When sodium azide was added, 2-octyl azide was obtained along with the 2-octanol, but the latter was now 100% inverted. It is apparent that, in the original case, 2-octanol was produced by two different processes: An SN2 reaction leading to inverted product, and another process in which some intermediate leads to racemization or retention. When azide ions were added, they scavenged this intermediate, so that the entire second process now went to produce azide, while the SN2 reaction, unaffected by addition of azide, still went on to give inverted 2-octanol. What is the nature of the intermediate in the second process? At first thought, it is a carbocation, so that this would be another example of simultaneous SN1 and SN2 reactions. However, solvolysis of 2-octyl brosylate in pure methanol or of 2-octyl methanesulfonate in pure water, in the absence of azide ions, gave methyl 2-octyl ether or 2-octanol, respectively, with 100% inversion of configuration, indicating that the mechanism in these solvents was pure SN2. Since methanol and water are more polar than 75% aq dioxane and since an increase in polarity of solvent increases the rate of SN1 reactions at the expense of SN2 (Sec. 10.G.iii), it is extremely unlikely that any SN1 process could occur in 75% aq dioxane. The intermediate in the second process is thus not a carbocation. It's nature is suggested by the fact that, in the absence of azide ions, the amount of inverted 2-octanol decreased with an increasing percentage of dioxane in the solvent. Thus the intermediate is an oxonium ion (19) formed by an SN2 attack by dioxane. This ion is not a stable product, but reacts with water in another SN2 process to produce 2-octanol with retained configuration.
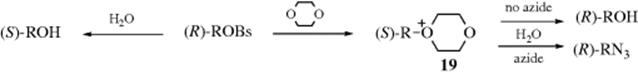
That part of the original reaction that resulted in retention of configuration98 is thus seen to stem from two successive SN2 reactions and not from any “borderline” behavior.99