March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 10. Aliphatic Substitution, Nucleophilic and Organometallic
10.H. Reactions
The reactions in this chapter are classified according to the attacking atom of the nucleophile in the order O, S, N, halogen, H, C. For a given nucleophile, reactions are classified by the substrate and leaving group. For the most part, only alkyl substrates are considered, since acyl substrates are considered in Chapter 16. Nucleophilic substitutions at a sulfur atom are treated at the end.
Not all the reactions in this chapter are actually nucleophilic substitutions. In some cases, the mechanisms are not known with enough certainty even to decide whether a nucleophile, an electrophile, or a free radical is attacking. In other cases, conversion of one compound to another can occur by two or even all three of these possibilities, depending on the reagent and reaction conditions. However, one or more of the nucleophilic mechanisms previously discussed do hold for the overwhelming majority of the reactions in this chapter. For the alkylations, the SN2 is by far the most common mechanism, as long as R is primary or secondary alkyl. For the acylations, the tetrahedral mechanism is the most common.
10.H.i. Oxygen Nucleophiles
A. Attack by OH at an Alkyl Carbon
10-1 Hydrolysis of Alkyl Halides
Hydroxy-de-halogenation
![]()
Alkyl halides can be converted to alcohols. Hydroxide ion is usually required, although particularly active substrates (e.g., allylic or benzylic alcohols) can be hydrolyzed by water. Ordinary halides can be hydrolyzed by water,530if the solvent is HMPA or N-methyl-2-pyrrolidinone,531 or if the reaction is done in an ionic solvent.532 If the hydrolysis (solvolysis) reaction proceeds via ionization, by an SN1 type mechanism, this reaction can be performed on tertiary substrates without significant interference from elimination side reactions. Tertiary alkyl α-halocarbonyl compounds can be converted to the corresponding alcohol with silver oxide in aq acetonitrile.533 The reaction is not frequently used for synthetic purposes, because alkyl halides are usually obtained from alcohols.
![]()
Vinylic halides are unreactive (Sec. 10.F), but they can be hydrolyzed to ketones at room temperature with mercuric trifluoroacetate, or with mercuric acetate in either trifluoroacetic acid or acetic acid containing BF3 etherate.534Primary bromides and iodides give alcohols when treated with bis(tributyltin)oxide (Bu3Sn–O–SnBu3) in the presence of silver salts.535
OS II, 408; III, 434; IV, 128; VI, 142, 1037.
10-2 Hydrolysis of gem-Dihalides
Oxo-de-dihalo-bisubstitution
![]()
gem-Dihalides can be hydrolyzed using either acid or basic catalysis to give aldehydes or ketones.536 Formally, the reaction may be regarded as giving R–C(OH)XR′, which is unstable and loses HX to give the carbonyl compound. For aldehydes derived from RCHX2, strong bases cannot be used, because the product undergoes the aldol reaction (16-34) or the Cannizzaro reaction (19-81). A mixture of calcium carbonate and sodium acetate is effective,537 and heating to 100 °C in DMSO gives good yields.538 A simple method heats a gem-dibromide with pyridine, and subsequent treatment with water gives the aldehyde.539 Heating 1,1-dihaloalkenes (C=CX2) with zinc and water leads to the corresponding methyl ketone.540
OS I, 95; II, 89, 133, 244, 549; III, 538, 788; IV, 110, 423, 807. Also see, OS III, 737.
10-3 Hydrolysis of 1,1,1-Trihalides
Hydroxy,oxo-de-trihalo-tersubstitution
![]()
This reaction is similar to 10-2. The utility of the method is limited by the lack of availability of trihalides, although these compounds can be prepared by addition of CCl4 and similar compounds to double bonds (Reaction 15-38) and by the free radical halogenation of methyl groups on aromatic rings (Reaction 14-1). When the reaction is carried out in the presence of an alcohol, a carboxylic ester can be obtained directly.541 1,1-Dichloroalkenes can also be hydrolyzed to carboxylic acids, by treatment with aq H2SO4. In general 1,1,1-trifluorides do not undergo this reaction,542 although exceptions are known.543
Aryl 1,1,1-trihalomethanes can be converted to acyl halides by treatment with sulfur trioxide.544 Hydrolysis of the acid chloride gives the carboxylic acid (Reaction 16-57).
![]()
Chloroform is more rapidly hydrolyzed with base than dichloromethane or carbon tetrachloride and gives not only formic acid, but also carbon monoxide.545 Hine546 showed that the mechanism of chloroform hydrolysis is quite different from that of dichloromethane or carbon tetrachloride, although superficially the three reactions appear similar. The first step is the loss of a proton to give CCl3−, which then loses Cl− to give dichlorocarbene (CCl2), which is hydrolyzed to formic acid or carbon monoxide.
![]()
This is an example of an SN1cB mechanism (Sec. 10.G.iii, category 1). The other two compounds react by the normal mechanisms. Carbon tetrachloride cannot give up a proton and dichloromethane is not acidic enough.
OS III, 270; V, 93. Also see, OS I, 327.
10-4 Hydrolysis of Alkyl Esters of Inorganic Acids
Hydroxy-de-sulfonyloxy-substitution, and so on
![]()
Esters of inorganic acids, including those given above and others, can be hydrolyzed to alcohols. The reactions are most successful when the ester is that of a strong acid, but it can be done for esters of weaker acids by the use of hydroxide ion (a more powerful nucleophile) or acidic conditions (which make the leaving group come off more easily). When vinylic substrates are hydrolyzed, the products are enols, which tautomerize to aldehydes or ketones (Sec. 2.N), as shown.
![]()
These reactions are all considered at one place because they are formally similar. Although some of them involve R–O cleavage and are thus nucleophilic substitutions at a saturated carbon, others involve cleavage of the bond between the inorganic atom and oxygen and are thus nucleophilic substitutions at a sulfur, nitrogen, and so on. It is even possible for the same ester to be cleaved at either position, depending on the conditions. Thus benzhydryl p-toluenesulfinate (Ph2CHOSOC6H4CH3) was found to undergo C–O cleavage in HClO4 solutions and S–O cleavage in alkaline media.547 In general, the weaker the corresponding acid, the less likely is C–O cleavage. Thus, sulfonic acid esters (ROSO2R′) generally give C–O cleavage,548 while nitrous acid esters (RONO) usually give N–O cleavage.549 Esters of sulfonic acids that are frequently hydrolyzed are mentioned in Section 10.G.iii. For hydrolysis of sulfonic acid esters (see also, Reaction 16-100).
OS VI, 852. See also, VIII, 50.
10-5 Hydrolysis of Diazoketones
Hydro, hydroxy-de-diazo-bisubstitution
![]()
Diazoketones are relatively easy to prepare (see Reaction 16-89). When treated with acid, they add a proton to give α-keto diazonium salts, which are hydrolyzed to the alcohols by the SN1 or SN2 mechanism.550 Relatively good yields of α-hydroxy ketones can be prepared in this way, since the diazonium ion is somewhat stabilized by the presence of the carbonyl group, which discourages N2 from leaving because that would result in an unstable α-carbonyl carbocation.
10-6 Hydrolysis of Acetals, Enol Ethers, and Similar Compounds551
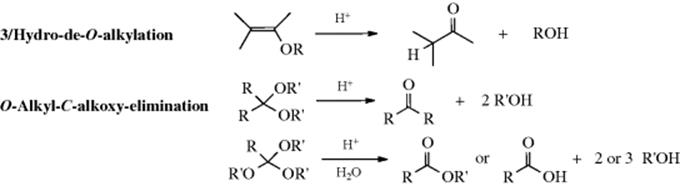
The alkoxyl group (OR) is not a leaving group in these reactions, so these compounds must be converted to the conjugate acids before they can be hydrolyzed. Although 100% sulfuric acid and other concentrated strong acids readily cleave simple ethers,552 the only acids used preparatively for this purpose are HBr and HI (Reaction 10-49). However, acetals, ketals, and ortho esters553 are easily cleaved by dilute acids. These compounds are hydrolyzed with greater facility because carbocations of type R2(RO)C+ are greatly stabilized by resonance (Sec. 5.A.ii). The reactions therefore proceed by the SN1 mechanism,554 as shown for acetals:555
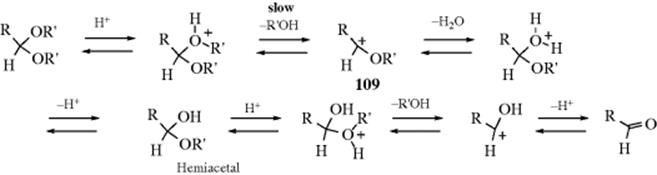
This mechanism, which is an SN1cA or A1 mechanism, is the reverse of that for acetal formation by reaction of an aldehyde and an alcohol (Reaction 16-5). Among the facts supporting the mechanism are556 (1) The reaction proceeds with specific H3O+ catalysis (see Sec. 8.D). (2) It is faster in D2O. (3) Optically active ROH are not racemized. (4) Even with tert-butyl alcohol the R–O bond does not cleave, as shown by 18O labeling.557 (5) In the case of acetophenone ketals, the intermediate corresponding to 109 [ArCMe(OR)2] could be trapped with sulfite ions (SO32−).558 (6) Trapping of this ion did not affect the hydrolysis rate,558 so the rate-determining step must come earlier. (7) In the case of 1,1-dialkoxyalkanes, intermediates corresponding to 109 were isolated as stable ions in superacid solution at −75 °C, where their spectra could be studied.559 (8) Hydrolysis rates greatly increase in the order CH2(OR′)2< RCH(OR′)2 < R2C(OR′)2 < RC(OR′)3, as would be expected for a carbocation intermediate.560 Formation of 109 is usually the rate-determining step (as marked above), but there is evidence that at least in some cases this step is fast, and the rate-determining step is loss of R′OH from the protonated hemiacetal.561 Rate-determining addition of water to 109 has also been reported.562
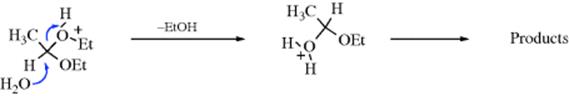
While the A1 mechanism shown above operates in most acetal hydrolyses, it has been shown that at least two other mechanisms can take place with suitable substrates.563 In one of these mechanisms, the second and third of the above steps are concerted, so that the mechanism is SN2cA (or A2). This has been shown, for example, in the hydrolysis of 1,1-diethoxyethane, by isotope effect studies:564
In the second mechanism, the first and second steps are concerted. In the case of hydrolysis of 2-(p-nitrophenoxy)tetrahydropyran, general acid catalysis was shown565 demonstrating that the substrate is protonated in the rate-determining step (Sec. 8.D). Reactions in which a substrate is protonated in the rate-determining step are called A-SE2 reactions.566 However, if protonation of the substrate were all that happens in the slow step, then the proton in the transition state would be expected to lie closer to the weaker base (Sec. 8.D). Because the substrate is a much weaker base than water, the proton should be largely transferred. Since the Br![]() nsted coefficient was found to be 0.5, the proton was actually transferred only about halfway. This can be explained if the basicity of the substrate is increased by partial breaking of the C–O bond. The conclusion drawn is that steps 1 and 2 are concerted. The hydrolysis of ortho esters in most cases is also subject to general acid catalysis.567
nsted coefficient was found to be 0.5, the proton was actually transferred only about halfway. This can be explained if the basicity of the substrate is increased by partial breaking of the C–O bond. The conclusion drawn is that steps 1 and 2 are concerted. The hydrolysis of ortho esters in most cases is also subject to general acid catalysis.567
The hydrolysis of acetals and ortho esters is governed by the stereoelectronic control factor discussed in Section 16.A.i, category 4,568 although the effect can generally be seen only in systems where conformational mobility is limited, especially in cyclic systems. There is evidence for synplanar stereoselection in the acid hydrolysis of acetals.569 The mechanism of Lewis acid mediated cleavage of chiral acetals is also known.570
Convenient reagents for the hydrolysis of acetals are wet silica gel571 and Amberlyst-15 (a sulfonic acid based polystyrene cation exchange resin).572 Both cyclic and acyclic acetals and ketals can be converted to aldehydes or ketones under nonaqueous conditions by treatment with TESOTf-2,6-lutidine (or 2,4,6-collidine) in dichloromethane followed by treatment with water,573 with Lewis acids e.g., 0.8% In(OTf)3 in acetone,574 ceric ammonium nitrate in aq acetonitrile,575 or Bi(OTf)3•xH2O.576
Although acetals, ketals, and ortho esters are easily hydrolyzed by acids, they are extremely resistant to hydrolysis by bases. An aldehyde or ketone can therefore be protected from attack by a base by conversion to the acetal or ketal (Reaction 16-5), and then can be cleaved with acid. Pyridine–HF has also been used for this conversion.577 Thioacetals, thioketals, gem-diamines, and other compounds that contain any two of the groups OR, OCOR, NR2, NHCOR, SR, and halogen on the same carbon can also be hydrolyzed to aldehydes or ketones, in most cases, by acid treatment. Thioacetals [RCH(SR′)2] and thioketals [R2C(SR′)2] are among those compounds generally resistant to acid hydrolysis.578 Because conversion to these compounds (Reaction 16-11) serves as an important method for protection of aldehydes and ketones, many methods have been devised to cleave them to the parent carbonyl compounds. Among reagents579 used for this purpose are HgCl2,580 FeCl2•6 H2O,581 cetyltrimethylammonium tribromide in dichloromethane,582m-chloroperoxybenzoic acid, the Dess–Martin periodinane583 (see Reaction 19-03), and sodium nitrite in aqueous acetyl chloride.584 Mixed acetals and ketals (RO–C–SR) can be hydrolyzed with most of the reagents mentioned above, including N-bromosuccinimide (NBS) in aq acetone,585 and glyoxylic acid on Amberlyst-15 with microwave irradiation.586
Enol ethers (vinyl ethers) are readily hydrolyzed by acids; the rate-determining step is protonation of the substrate.587 However, protonation does not take place at the oxygen, but at the β carbon,588 because that gives rise to the stable carbocation (110).589 After that, the mechanism is similar to the A1 mechanism given above for the hydrolysis of acetals.
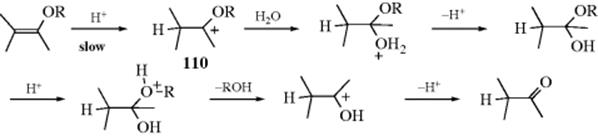
Among the facts supporting this mechanism, which is an A-SE2 mechanism because the substrate is protonated in the rate-determining step, are (1) the 18O labeling shows that in ROCH=CH2 it is the vinyl–oxygen bond and not the RO bond that cleaves;590 (2) the reaction is subject to general acid catalysis;591 (3) there is a solvent isotope effect when D2O is used.591 A method has been developed to determine primary kinetic isotope effects relating to proton transfer in the hydrolysis of enol ethers.592 Enantioselective protonation is possible in some cases. Cyclic silyl enol ethers are converted to chiral α-substituted ketones, for example, with high enantioselectivity using a chiral Br![]() nsted acid.593
nsted acid.593
Enamines are also hydrolyzed by acids (see Reaction 16-2); the mechanism is similar. Ketene dithioacetals [R2C=C(SR′)2] also hydrolyze by a similar mechanism, except that the initial protonation step is partially reversible.594Furans represent a special case of enol ethers that are cleaved by acid to give 1,4-diones.595 Thus oxonium ions are cleaved by water to give an alcohol and an ether:
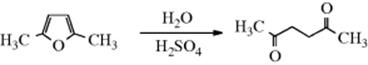
OS I, 67, 205; II, 302, 305, 323; III, 37, 127, 465, 470, 536, 541, 641, 701, 731, 800; IV, 302, 499, 660, 816, 903; V, 91, 292, 294, 703, 716, 937, 967, 1088; VI, 64, 109, 312, 316, 361, 448, 496, 683, 869, 893, 905, 996; VII, 12, 162, 241, 249, 251, 263, 271, 287, 381, 495; VIII, 19, 155, 241, 353, 373
10-7 Hydrolysis of Epoxides
(3) OC-seco-hydroxy-de-alkoxy substitution
![]()
The hydrolysis of epoxides is a convenient method for the preparation of vic-diols. The reaction is catalyzed by acids or bases. A basic reagent will attack the polarized carbon of the epoxide unit to open the ring, whereas an acid-catalyzed reaction leads to a protonated epoxide (an oxonium ion),596 which is opened by nucleophilic attack at an adjacent carbon. Among acid catalysts, perchloric acid leads to minimal side reactions,597 and 10% Bu4NHSO4 in water is effective.598 However, water reacts directly with epoxides at 60 °C.599 Dimethyl sulfoxide is a superior solvent for the alkaline hydrolysis of epoxides.600
Cobalt salen [salen = bis(salicylidene)ethylenediamine] catalysts, in the presence of water, open epoxides with high stereoselectivity.601 The enzyme epoxide hydrolase opens epoxides with high enantioselectivity.602
OS V, 414.
10.H.ii Attack by or at an Alkyl Carbon
10-8 Alkylation with Alkyl Halides: The Williamson Reaction
Alkoxy-de-halogenation
![]()
The Williamson reaction (Williamson ether synthesis), discovered in 1850, is still the best general method for the preparation of unsymmetrical or symmetrical ethers.603 The reaction can also be carried out with aromatic R′, although C-alkylation is sometimes a side reaction (see Sec. 10.G.vii).604 The normal method involves treatment of a primary or secondary alkyl halide with alkoxide or aroxide ion prepared from an alcohol or phenol by reaction with a suitable base, although methylation using dimethyl carbonate has been reported.605 The solvent is usually an aprotic solvent (THF, ether, etc.,) rather than an alcohol solvent, which typically promotes elimination reactions in the presence of alkoxides (see Chap. 17). It is also possible to mix the halide and alcohol or phenol directly with Cs2CO3 in acetonitrile,606 or with NaH in the presence of DMF.607 The reaction can also be carried out in a dry medium,608 neat,609 or in solvents using microwave irradiation.610 Williamson ether synthesis in ionic liquids has also been reported.611 The reaction is not successful for tertiary R (elimination predominates), and low yields are often obtained with secondary R. Monoethers can be formed from diols and alkyl halides.612 It is possible to selectively alkylate the primary hydroxyl in a diol [HOCH2CH(OH)R] using a tin complex.613
Many other functional groups can be present in the molecule without interference. Ethers with one tertiary group can be prepared by treatment of an alkyl halide or sulfate ester (Reaction 10-10) with a tertiary alkoxide (R′O−). Di-tert-butylether was prepared in high yield by direct attack by t-BuOH on reaction with the tert-butyl cation (at −80 °C in SO2ClF).614 Di-tert-alkyl ethers in general have proved difficult to make, but they can be prepared in low-to-moderate yields by treatment of a tertiary halide with Ag2CO3 or Ag2O.615 Alcohols react with Mg(ClO4)2 and an excess of Boc (Boc = t-butoxycarbonyl) anhydride (Boc2O) to give the tert-butyl ether.616
Active halides (e.g., Ar3CX) may react directly with the alcohol.617 Hindered alcohols may react as well.618 The mechanism for these cases is of course SN1. tert-Butyl halides can be converted to aryl tert-butyl ethers by treatment with phenols and an amine (e.g., pyridine).619 Aryl alkyl ethers can be prepared from alkyl halides by treatment with an aryl acetate (instead of a phenol) in the presence of K2CO3 and a crown ether.620 The Pd-catalyzed displacement of allylic acetates with aliphatic alcohols has been shown to give the corresponding alkyl allyl ether.621 A Rh-catalyst622 Ir catalyst,623 and an In-Si combined Lewis acid catalyst624 have been used in ether forming reactions. Aryl ethers have been prepared using Mitsunobu conditions (see Reaction 10-17).625
Vinyl ethers have been formed by coupling tetravinyl tin with phenols, in the presence of cupric acetate and oxygen.626 The Pd-catalyzed coupling of vinyl triflates and phenols has also been reported.627
Both aryl alkyl and dialkyl ethers can be efficiently prepared with the use of phase-transfer catalysis (Sec. 10.G.v)628 and with micellar catalysis.629 Symmetrical benzylic ethers have been prepared by reaction of benzylic alcohols with Mg/I2 followed by triflic anhydride.630
A slight variation of the Williamson ether synthesis has been used for the protection of hydroxy groups631 by reaction of their salts with chloromethyl methyl ether.
![]()
This protecting group is known as MOM (methoxymethyl) and such compounds are called MOM ethers. The resulting acetals are stable to bases and are easily cleaved with mild acid treatment (Reaction 10-7). Another protecting group, the 2-methoxyethoxymethyl group (the MEM group), is formed in a similar manner. Both MOM and MEM groups can be cleaved with dialkyl- and diarylboron halides (e.g., Me2BBr).632
Another common method for the protection of alcohols is conversion to the silyl ether (R–O–SiR'3). The alcohol is generally treated with a base (e.g., trimethylamine or imidazole) and then with a chlorotrialkylsilane (R3SiCl), or the analogous bromide.629 There are many variations of this basic procedure. Iodine promotes the reaction, for example.633 There are also many ways to remove the silyl group to regenerate the alcohol, although fluoride ion, including tetrabutylammonium fluoride in THF, is probably the most common method.629
Most Williamson reactions proceed by the SN2 mechanism, but there is evidence (see Sec. 10.C) that in some cases the SET mechanism can take place, especially with alkyl iodides.634 Secondary alcohols have been converted to the corresponding methyl ether by reaction with methanol in the presence of ferric nitrate nonahydrate.635
OS I, 75, 205, 258, 296, 435; II, 260; III, 127, 140, 209, 418, 432, 544; IV, 427, 457, 558, 590, 836; V, 251, 258, 266, 403, 424, 684; VI, 301, 361, 395, 683; VII, 34, 386, 435; VIII, 26, 161, 155, 373; 80, 227.
10-9 Epoxide Formation (Internal Williamson Ether Synthesis)
(3) OC-cyclo-Alkoxy-de-halogenation
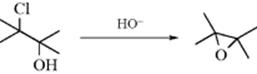
This is a special case of Reaction 10-8. The base removes the proton from the OH group of a halohydrin (chlorohydrin or bromohydrin), and the resulting alkoxide subsequently attacks in an internal SN2 reaction.636 Many epoxides have been made in this way.637 The course of the reaction can be influenced by neighboring group effects.638 Enantioselective epoxide-forming reactions are known, using chiral additives (e.g., dihydrocinchonidines).639Epoxidation of alkenes has also been accomplished using HOF–MeCN in a continuous flow system.640
Larger cyclic ethers can be prepared, including five-and six-membered rings (tetrahydrofurans and tetrahydropyrans, respectively).641 Additional treatment with base yields the glycol (Reaction 10-7). Thiiranes can be prepared by the reaction of α-chloro ketones with (EtO)2P(=O)-SH and NaBH4-Al2O3 with microwave irradiation.642
1,2-Diols can be converted to epoxides by treatment with DMF dimethyl acetal, [(MeO)2CHNMe2],643 with diethyl azodicarboxylate (Et2OCN=NCO2Et), and Ph3P,644 with a dialkoxytriphenylphosphorane,645 or with TsCl−Na−OHPhCH2NEt2+ Cl−.646
OS I, 185, 233; II, 256; III, 835; VI, 560; VII, 164, 356; VIII, 434.
10-10 Alkylation with Inorganic Esters
Alkoxy-de-sulfonyloxy substitution
![]()
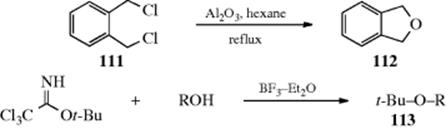
The reaction of alkyl sulfates with alkoxide ions is quite similar to Reaction 10-8 in mechanism and scope. Other inorganic esters can also be used. Methyl ethers of alcohols and phenols are commonly formed by treatment of alkoxides or aroxides with methyl sulfate. The alcohol or phenol can be methylated directly with dimethyl sulfate under various conditions.647 Carboxylic esters sometimes give ethers when treated with alkoxides (BAL2 mechanism, Reaction 16-59) in a very similar process (see also, Reaction 16-64). A related reaction heated 111 with alumina to give the corresponding benzofuran, (112).648 The reaction of aliphatic alcohols and potassium organotrifluoroborate salts also gives ethers.649
tert-Butyl ethers (113) can be prepared by treating the compound tert-butyl-2,2,2-trichloroacetimidate with an alcohol or phenol in the presence of boron trifluoride etherate.650 Trichloroimidates can be used to prepare other ethers as well.651tert-Butyl ethers can be cleaved by acid-catalyzed hydrolysis.652
OS I, 58, 537; II, 387, 619; III, 127, 564, 800; IV, 588; VI, 737, 859, VII, 41. Also see, OS V, 431.
10-11 Alkylation with Diazo Compounds
Hydro, alkoxy-de-diazo-bisubstitution
![]()
Alcohols react with diazo compounds to form ethers, but diazomethane and diazo ketones are most readily available, giving methyl ethers or α-keto ethers,653 respectively. With diazomethane654 the method is expensive and requires great caution, but the conditions are mild and high yields are obtained. Diazomethane is used chiefly to methylate alcohols and phenols that are expensive or available in small amounts. Hydroxy compounds react better as their acidity increases; ordinary alcohols do not react at all unless a catalyst (e.g., HBF4655 or silica gel)656 is present. The more acidic phenols react very well in the absence of a catalyst. The reaction of oximes, and ketones that have substantial enolic contributions, give O-alkylation to form, respectively, O-alkyl oximes and enol ethers. The mechanism657 is as in Reaction 10-5. Note that O-aryloximes are prepared from oximes and aryl halides, mediated by CuI.658
Diazoalkanes can also be converted to ethers by thermal or photochemical cleavage in the presence of an alcohol. These are carbene or carbenoid reactions.659 Enantioselective insertion into phenolic O–H bond leads to highly substituted ethers.660 Similar intermediates are involved when diazoalkanes react with alcohols in the presence of t-BuOCl to give acetals.661
![]()
OS V, 245. Also see, OS V, 1099.
10-12 Dehydration of Alcohols
Alkoxy-de-hydroxylation
![]()
The dehydration of alcohols to form symmetrical ethers662 is analogous to Reactions 10-8 and 10-10, but the species from which the leaving group departs is ROH2+ or ROSO2OH. The former is obtained directly on treatment of alcohols with sulfuric acid and may go, by an SN1 or SN2 pathway, directly to the ether if attacked by another molecule of alcohol. On the other hand, it may, again by either an SN1 or SN2 route, be attacked by the nucleophile HSO4−, in which case it is converted to ROSO2OH, which in turn may be attacked by an alcohol molecule to give ROR. Elimination is always a side reaction and, in the case of tertiary alkyl substrates, completely predominates. Good yields of ethers were obtained by heating diarylcarbinols [ArAr′CHOH → (ArAr′CH)2O] with TsOH in the solid state.663 Acids (e.g., Nafion-H with silyl ethers)664 can be used in this transformation, and Lewis acids can be used with alcohols in some cases.665
Mixed (unsymmetrical) ethers can be prepared if one group is tertiary alkyl and the other primary or secondary, since the latter group is not likely to compete with the tertiary group in the formation of the carbocation, while a tertiary alcohol is a very poor nucleophile.666 If one group is not tertiary, the reaction of a mixture of two alcohols leads to all three possible ethers. Unsymmetrical ethers have been formed by treatment of two different alcohols with MeReO3667 or with BiBr3.668 Unsymmetrical ethers have been prepared under Mitsunobu conditions (Reaction 10-17) with a polymer-supported phosphine and diethyl azadicarboxylate (DEAD).669 Symmetrical ethers are formed by heating benzylic alcohols with the polymer poly(3,4-ethylenedioxythiophene) in toluene or heptane (a two-phase system), with no other additives.670 Diols can be converted to cyclic ethers,671 although the reaction is most successful for five-membered rings, but five-, six-, and seven-membered rings have been prepared.672 Thus, 1,6-hexanediol gives mostly 2-ethyltetrahydrofuran. This reaction is also important in preparing furfural derivatives from aldoses, with concurrent elimination.
Phenols and primary alcohols form ethers when heated with dicyclohexylcarbodiimide673 (see Reaction 16-63).
OS I, 280; II, 126; IV, 25, 72, 266, 350, 393, 534; V, 539, 1024; VI, 887; VIII, 116. Also see, OS V, 721.
10-13 Transetherification
Hydroxy-de-alkoxylation and Alkoxy-de-hydroxylation
![]()
The exchange of one alkoxy group for another is rare for ethers without a reactive R group (e.g., diphenylmethyl),674 or by treatment of alkyl aryl ethers with alkoxide ions: ROAr + R′O− → ROR′ + ArO−.675 3-(2-Benzyloxyethyl)-3-methyl-oxetane was transformed into 3-benzyloxymethyl-3-methyltetrahydrofuran by an internal transetherification catalyzed by BF3•OEt2.676

Acetals and ortho esters undergo transetherification readily,677 as with the transformation of 114 to 115.678 As seen in Reaction 10-6, departure of the leaving group from an acetal gives a particularly stable carbocation. It is also possible to convert a dimethylketal directly to a dithiane by reaction with butane 1,4-dithiol on clay.679 These are equilibrium reactions, and most often the equilibrium is shifted by removing the lower-boiling alcohol by distillation. Enol ethers can be prepared by treating an alcohol with an enol ester or a different enol ether, with mercuric acetate as a catalyst,680 as shown in the example. N,N-Diethylaminoethylthiol reacts with aryl ethers to give the phenol derivative and the corresponding sulfide in what is effectively a transetherification.681
![]()
1,2-Diketones can be converted to α-keto enol ethers by treatment with an alkoxytrimethylsilane (ROSiMe3).682
OS VI, 298, 491, 584, 606, 869; VII, 334; VIII, 155, 173. Also see, OS V, 1080, 1096.
10-14 Alcoholysis of Epoxides
(3) OC-seco-alkoxy-de-alkoxylation
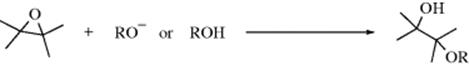
This reaction is analogous to 10-7. It may be acid (including Lewis acid683), base, or alumina684 catalyzed, and may occur by either an SN1 or SN2 mechanism. Catalysts (e.g., mesoporous aluminosilicate,685 Cu(BF4)2•nH2O,686Al(OTf)3,687 or BiCl3),688 have been used. β-Cyclodextrin has been used to promote the reaction with phenoxides in aqueous media.689 Many of the β-hydroxy ethers produced in this way are valuable solvents, [e.g., diethylene glycol and Cellosolve (2-ethoxyethanol)]. Reaction with thiols leads to hydroxy thioethers.690 Other nucleophilic oxygen or sulfur species have been shown to open epoxides, including thiols691 (catalyzed by Sc692 or In693). (Phenylseleno)silanes react with epoxides to give β-hydroxy selenides.694
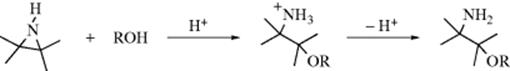
Opening an epoxide by an alkoxide moiety can be done intramolecularly, and a new cyclic ether is generated. Ethers of various ring sizes can be produced depending on the length of the tether between the alkoxide unit and the epoxide. Specialized conditions are common, as in the conversion of 116 to 117.695 Another variant of this transformation used a Co-salen catalyst.696 A specialized version has the alkoxide moiety on the carbon adjacent to the epoxide, leading to the Payne rearrangement where a 2,3-epoxy alcohol is converted to an isomeric one, by treatment with aqueous base, as shown in the example.697
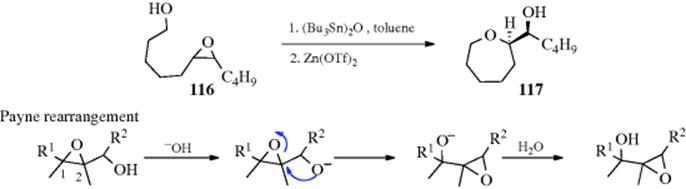
The reaction results in inverted configuration at C-2. Of course, the product can also revert to the starting material by the same pathway, so a mixture of epoxy alcohols is generally obtained.
The reaction of alcohols with aziridines leads to β-amino ethers,698 and reaction with thiols gives β-amino thioethers.699 It has been shown that ring opening of aziridines by phenols is promoted by tributylphosphine.700 Aldehydes open aziridines when catalyzed by nucleophilic carbenes.701 Metal catalysts [e.g., Cu(OTf)2] mediate the ring opening of N-tosylaziridines by alcohols.702 The reaction of N-tosyl aziridines with 10% ceric ammonium nitrate in aq methanol leads to N-tosylamino alcohols,703 and reaction with ethanol and 10% BF3•OEt2 gives N-tosyl ethers.704 In addition, N-tosylaziridines are opened by acetic acid in the presence of In(OTf)3 to give N-tosylamino acetates.705In the presence of Amberlyst-15, N-Boc (Boc = tert-butoxycarbonyl, –CO2t-Bu) aziridines react with LiBr to give the corresponding bromo amide.706 Aziridines are opened by potassium thiocyanate, catalyzed by LiClO4.707Catalytic enantioselective ring opening of N-acyl aziridines with TMSCN and a Gd catalyst leads to amino-nitriles.708aza-Payne rearrangements are known, based on reactions of aziridines rather than epoxides (see above).709
10-15 Alkylation with Onium Salts
Alkoxy-de-hydroxylation
![]()
Oxonium ions are excellent alkylating agents, and ethers can be conveniently prepared by treating them with alcohols or phenols.710 Quaternary ammonium salts can sometimes also be used.711
OS VIII, 536.
10-16 Hydroxylation via Silanes
Hydroxy-de-silylalkylation
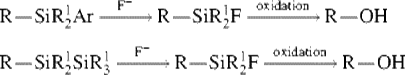
Alkylsilanes can be oxidized, with the silyl unit converted to a hydroxy unit. This usually requires either an aryl group712 or another silyl group713 attached to silicon. It has been shown that a strained four-membered ring silane (a siletane) also gives the corresponding alcohol upon oxidation.714 Treatment with a fluorinating agent (e.g., tetrabutylammonium fluoride or CsF) replaces Ar or SiR3 with F, which is oxidized with hydrogen peroxide or a peroxyacid to give the alcohol. This sequence is often called the Tamao–Fleming oxidation.712 There are several variations in substrate that allow versatility in the initial incorporation of the silyl unit.715 Hydroperoxide oxidation of a cyclic silane leads to a diol.716
C. Attack by OCOR at an Alkyl Carbon
10-17 Alkylation of Carboxylic Acid Salts
Acyloxy-de-halogenation
![]()
Sodium salts of carboxylic acids, including hindered acids (e.g., mesitoic), rapidly react with primary and secondary bromides and iodides at room temperature in dipolar aprotic solvents, especially HMPA, to give high yields of carboxylic esters.717 The mechanism is SN2. Several bases or basic media have been used to generate the carboxylate salt.718 Sodium salts are often used, but K, Ag, Cs,719 and substituted ammonium salts have also been used. An important variation uses phase transfer catalysis,720 and good yields of esters have been obtained from primary, secondary, benzylic, allylic, and phenacyl halides.721 Without phase-transfer catalysts and in protic solvents, the reaction is useful only for fairly active R [e.g., benzylic and allylic (SN1 mechanism)], but not for tertiary alkyl, since elimination occurs instead.722 Solid-state procedures are available. Addition of the dry carboxylate salt and the halide to alumina as a solid support, and microwave irradiation gives the ester in a procedure that is applicable to long-chain primary halides.723 A similar reaction of hexanoic acid and benzyl bromide on solid benzyltributylammonium chloride gave the ester with microwave irradiation.724 Ionic liquid solvents have been shown to facilitate this alkylation reaction.725
The reaction of an alcohol and a carboxylate anion with diethyl azodicarboxylate (EtOOCN=NCOOEt) and Ph3P726 is called the Mitsunobu reaction.727 Other azocarboxylates may be used in this reaction, including diisopropyl azodicarboxylate (DIAD), and di-2-methoxyethyl azodicarboxylate (DMEAD).728 Other Mitsunobu catalysts are available,729 including organocatalysts,730 and polymer-supported reagents have been used.731 A renewable phosphine ligand has been developed.732 Note that other functional groups, including azides733 and thiocyanates734 can be generated from alcohols using Mitsunobu conditions. This reaction can also be considered as an SN2 mechanism. Phenol esters can also be formed.735 Mitsunobu cyclodehydration of 1,2-diols leads to epoxides.736
Lactones can be prepared from halo acids by treatment with base (see Reaction 16-63). This has most often been accomplished with γ and δ lactones, but macrocyclic lactones (e.g., 11-17-members) have also been prepared in this way.737 An interesting variation treated 2-ethylbenzoic acid with hypervalent iodine and then I2/hν to give the five-membered ring lactone.738
Copper(I) carboxylates give esters with primary (including neopentyl without rearrangement), secondary, and tertiary alkyl, allylic, and vinylic halides.739 A simple SN mechanism is obviously precluded in this case. Vinylic halides can be converted to vinylic acetates by treatment with sodium acetate if palladium(II) chloride is present.740
A carboxylic acid (not the salt) can be the nucleophile if F− is present.741 Mesylates are readily displaced, for example, by benzoic acid/CsF.742 Dihalides have been converted to diesters by this method.741 A COOH group can be conveniently protected by reaction of its ion with a phenacyl bromide (ArCOCH2Br).743 The resulting ester is easily cleaved when desired with zinc and acetic acid. Dialkyl carbonates can be prepared without phosgene (see Reaction 16-61) by phase-transfer catalyzed treatment of primary alkyl halides with dry KHCO3 and K2CO3.744
Other leaving groups can also be replaced by OCOR. Alkyl chlorosulfites (ROSOCl) and other derivatives of sulfuric, sulfonic, and other inorganic acids can be treated with carboxylate ions to give the corresponding esters. Treatment with oxalyl chloride allows displacement by carboxylate salts.745 The use of dimethyl sulfate746 or trimethyl phosphate747 allows sterically hindered COOH groups to be methylated. The reaction of benzoic acid with aq lithium hydroxide, and then dimethyl sulfate gave methyl benzoate.748 Dimethyl carbonate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, see Reaction 17-13) has been used to prepare methyl esters.749 With certain substrates, carboxylic acids are strong enough nucleophiles for the reaction. Examples of such substrates are trialkyl phosphites [P(OR)3]750 and acetals of DMF.751
![]()
This is an SN2 process, since inversion is found at R. Another good leaving group is NTs2 and ditosylamines react quite well with acetate ion in dipolar aprotic solvents:752 RNTs2 + OAc− → ROAc. Ordinary primary amines have been converted to acetates and benzoates by the Katritzky pyrylium–pyridinium method (Sec. 10.G.iii).753 Quaternary ammonium salts can be cleaved by heating with AcO− in an aprotic solvent.754 Oxonium ions can also be used as substrates:755 R3O+ + R′COO− → R′COOR + R2O. The reaction of potassium thioacetate with alkyl halides give dithiocarboxylic esters.756
In a variation of this reaction, alkyl halides can be converted to carbamates, by treatment with a secondary amine and K2CO3 under phase transfer conditions.757 The reaction of alcohols and alkyl halides can lead to carbonates.758
![]()
OS II, 5; III, 650; IV, 582; V, 580; VI, 273, 576, 698.
10-18 Cleavage of Ethers with Acetic Anhydride or Acid Halides
Acyloxy-de-alkoxylation
![]()
Dialkyl ethers can be cleaved by treatment with anhydrous ferric chloride in acetic anhydride,759 or with Me3SiOTf in acetic anhydride.760 In this reaction, both R groups are converted to acetates and yields are moderate to high. Ethers can also be cleaved by the mixed, anhydride acetyl tosylate:761
![]()
Epoxides give β-hydroxyalkyl carboxylates when treated with a carboxylic acid or a carboxylate ion and a suitable catalyst.762 Tetrahydrofuran was opened to give O-acyl-4-iodo-1-butanol by treatment with acid chlorides and samarium halides763 or BCl3.764 In a highly specialized transformation, the reaction of an epoxide with CO2 and ZnCl2 in an ionic liquid leads to a cyclic carbonate.765 Epoxides react with CO and CH3OH in the presence of 10% of 3-hydroxypyridine and 5% of Co2(CO)8 to give a β-hydroxy methyl ester.766
OS VIII, 13.
10-19 Alkylation of Carboxylic Acids with Diazo Compounds
Hydro, acyloxy-de-diazo-bisubstitution
![]()
Carboxylic acids can be converted to esters with diazo compounds in a reaction essentially the same as 10-11. In contrast to alcohols, carboxylic acids undergo the reaction quite well at room temperature, since the reactivity of the reagent increases with acidity. The reaction is used where high yields are important or where the acid is sensitive to higher temperatures. Because of availability, diazomethane (CH2N2)654 is commonly used to prepare methyl esters, and diazo ketones are common. The mechanism is as shown in Reaction 10-11.
OS V, 797.
D. Other Oxygen Nucleophiles
10-20 Formation of Oxonium Salts
Dialkyloxonio-de-halogenation
![]()
Alkyl halides can be alkylated by ethers or ketones to give oxonium salts, if a very weak, negatively charged nucleophile is present to serve as a counterion and a Lewis acid is present to combine with X−.767 A typical procedure consists of treating the halide with the ether or the ketone in the presence of AgBF4 or AgSbF6. The Ag+ serves to remove X− and the BF4− or SbF6− acts as the counterion. Another method involves treatment of the halide with a complex formed between the oxygen compound and a Lewis acid (e.g., R2O•BF3 + RX → R3O+ BF4−), although this method is most satisfactory when the oxygen and halogen atoms are in the same molecule so that a cyclic oxonium ion is obtained. Ethers and oxonium ions also undergo exchange reactions:
![]()
OS V, 1080, 1096, 1099; VI, 1019.
10-21 Preparation of Peroxides and Hydroperoxides
Hydroperoxy-de-halogenation
![]()
Hydroperoxides can be prepared by treatment of alkyl halides, esters of sulfuric or sulfonic acids, or alcohols with hydrogen peroxide in basic solution, where it is actually HO2−.768 Sodium peroxide is similarly used to prepare dialkyl peroxides (2RX + Na2O2 → ROOR). Another method, which gives primary, secondary, or tertiary hydroperoxides and peroxides, involves treatment of the halide with H2O2 or a peroxide in the presence of silver trifluoroacetate.769 Peroxides can also be prepared770 by treatment of alkyl bromides or tosylates with potassium superoxide (KO2) in the presence of crown ethers (though alcohols may be side products771) and by the reaction between alkyl triflates and germanium or tin peroxide.772
Diacyl peroxides and acyl hydroperoxides can similarly be prepared773 from acyl halides or anhydrides and from carboxylic acids.774 Diacyl peroxides can also be prepared by the treatment of carboxylic acids with hydrogen peroxide in the presence of dicyclohexylcarbodiimide,775 Sulfuric acid, methanesulfonic acid, or some other dehydrating agent. Mixed alkyl–acyl peroxides (peresters) can be made from acyl halides and hydroperoxides.
![]()
OS III, 619, 649; V, 805, 904; VI, 276.
10-22 Preparation of Inorganic Esters
Nitrosooxy-de-hydroxylation, and so on.
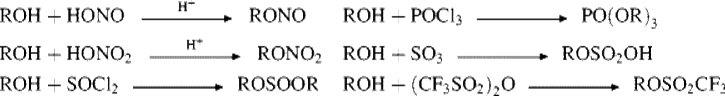
The above transformations show a few of the many inorganic esters that can be prepared by the reaction of an alcohol with an inorganic acid or, better, its acid halide or anhydride776 These similar reactions are grouped together for convenience, but not all involve nucleophilic substitutions at R. The other possible pathway is nucleophilic substitution at the inorganic central atom, such as the attack of the alcohol oxygen at the electrophilic sulfur atom in 118,777or a corresponding SN2 type process (see Sec. 16.B.v). In such cases, there
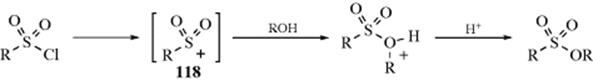
is no alkyl-O cleavage. Mono esters of sulfuric acid (alkylsulfuric acids), which are important industrially because their salts are used as detergents, can be prepared by treating alcohols with SO3, H2SO4, ClSO2OH, or SO3complexes.778 It is possible to prepare a primary sulfonate ester (e.g., tosylate), in the presence of a secondary alcohol unit when tosic acid reacts with a 1,2-diol in the presence of Fe3+-Montmorillonite.779 Polymer-bound reagents have been used to prepared sulfonate esters.780 Phenolic triflates have been prepared using N,N-ditrifylaniline and K2CO3 under microwave irradiation.781 Sulfinic esters are readily prepared from alcohols and sulfinyl chlorides, and in the presence of Cinchona alkaloids the reaction is enantioselective.782
Alkyl nitrites783 can be conveniently prepared by an exchange reaction ROH + R′ONO → RONO + R′OH, where R = t-Bu.784 Primary amines can be converted to alkyl nitrates (RNH2 → RONO2) by treatment with N2O4 at −78 °C in the presence of an excess of amidine base.785 Mitsunobu conditions (Reaction 10-17) can be used to prepare phosphate ester or phosphonate esters. The reaction can be done intramolecularly for prepare cyclic phosphonate esters.786
Alkyl halides are often used as substrates instead of alcohols. In such cases, the salt of the inorganic acid is usually used and the mechanism is nucleophilic substitution at the carbon atom. An important example is the treatment of alkyl halides with silver nitrate to form alkyl nitrates. This is used as a test for alkyl halides. In some cases, there is competition from the central atom. Thus nitrite ion is an ambident nucleophile that can give nitrites or nitro compounds (see Reaction 10-42).787 Dialkyl or aryl alkyl ethers can be cleaved with anhydrous sulfonic acids.788
![]()
Here R″ may be alkyl or aryl. For dialkyl ethers, the reaction does not end as indicated above, since R′OH is rapidly converted to R′OR′ by the sulfonic acid (Reaction 10-12), which in turn is further cleaved to R′OSO2R″ so that the product is a mixture of the two sulfonates. For aryl alkyl ethers, cleavage always takes place to give the phenol, which is not converted to the aryl ether under these conditions. Ethers can also be cleaved in a similar manner by mixed anhydrides of sulfonic and carboxylic acids789 (prepared as in Reaction 16-68). β-Hydroxyalkyl perchlorates790 and sulfonates can be obtained from epoxides.791 Epoxides and oxetanes give α,ω-dinitrates when treated with N2O5.792 Aziridines and azetidines react similarly, giving nitramine nitrates (e.g., N-butylazetidine gave NO2OCH2CH2CH2N(Bu)NO2).792
Phosphinate esters are prepared by transesterification-type reactions (16-64) from alcohols and other phosphinates.793
OS II, 106, 108, 109, 112, 204, 412; III, 148, 471; IV, 955; V, 839; VIII, 46, 50, 616. Also see, OS II, 111.
10-23 Alcohols from Amines
Hydroxy-de-amination
![]()
This transformation is rare. A rather direct method was reported whereby a primary amine reacted with KOH in diethylene glycol at 210 °C.794 The reaction of (S)-phenethylamine and the bis-(sulfonyl chloride) of 1,2-benzenesulfonic acid, followed by KNO2 and 18-crown-6 gave (R)-phenethyl alcohol in 70% yield and 40% enantiomeric excess (ee).795
10-24 Alkylation of Oximes796
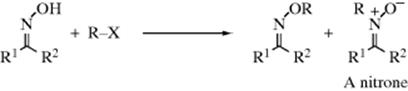
Oximes can be alkylated by alkyl halides or sulfates. N-Alkylation is a side reaction, yielding a nitrone.797 The relative yield of oxime ether and nitrone depends on the nature of the reagents, including the configuration of the oxime, and on the reaction conditions.798 For example, anti-benzaldoximes give nitrones, while the syn isomers give oxime ethers.799
OS III, 172; V, 1031. Also see, OS V, 269; VI, 199.
10.H.iii. Sulfur Nucleophiles
Sulfur compounds800 are better nucleophiles than their oxygen analogues (Sec. 10.G.ii), so in most cases these reactions take place faster and more smoothly than the corresponding reactions with oxygen nucleophiles. There is evidence that some of these reactions take place by SET mechanisms.801
10-25 Attack by SH at an Alkyl Carbon: Formation of Thiols802
Mercapto-de-halogenation
![]()
Sodium sulfhydride (NaSH) is a much better reagent for the formation of thiols (mercaptans) from alkyl halides than H2S and is used much more often. It is easily prepared by bubbling H2S into an alkaline solution, but hydrosulfide on a supported polymer resin has also been used.803 The reaction is most useful for primary halides. Secondary substrates give much lower yields, and the reaction fails completely for tertiary halides because elimination predominates. Sulfuric and sulfonic esters can be used instead of halides. Thioethers (RSR) are often side products.804 The conversion can also be accomplished under neutral conditions by treatment of a primary halide with F− and a tin sulfide (e.g., Ph3SnSSnPh3).805 An indirect method for the preparation of a thiol is the reaction of an alkyl halide with thiourea to give an isothiuronium salt (119), and subsequent treatment with alkali or a high-molecular-weight amine gives cleavage to the thiol.
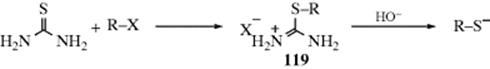
Other indirect methods are treatment of the halide with silyl-thiols and KH, followed by treatment with fluoride ion and water,806 and hydrolysis of Bunte salts (see Reaction 10-28) is another method.
Thiols have also been prepared from alcohols. One method involves treatment with H2S and a catalyst (e.g., Al2O3),807 but this is limited to primary alcohols. Another method involves treatment with Lawesson's reagent (see Reaction 16-10).808 Tertiary nitro compounds give thiols (RNO2 → RSH) when treated with sulfur and sodium sulfide, followed by amalgamated aluminum.809
OS III, 363, 440; IV, 401, 491; V, 1046; VIII, 592. Also see, OS II, 345, 411, 573; IV, 232; V, 223; VI, 620.
10-26 Attack by S at an Alkyl Carbon: Formation of Thioethers
Alkylthio-de-halogenation; Alkylthio-de-hydroxylation
![]()
Thioethers (sulfides) can be prepared by treatment of alkyl halides with salts of thiols (thiolate anions).810 The R′ group may be alkyl or aryl and organolithium bases can be used to deprotonate the thiol.811 As in Reaction 10-25, RX cannot be a tertiary halide, and sulfuric and sulfonic esters can be used instead of halides. As in the Williamson Reaction (10-8), yields are often improved by phase-transfer catalysis.812 Thiols react directly with alkyl halides in the presence of bases (e.g., DBU; see Reaction 17-13)813 or CsF.814 Leaving groups other than chloride can be used, as in the Ru catalyzed reaction of thiols with propargylic carbonates.815 Vinylic sulfides can be prepared by treating vinylic bromides with PhS− in the presence of a Ni complex,816 or in the presence of Pd(PPh3)4. Alternatively, the Ag salt of an enethiol reacts with iodomethane to give the corresponding methyl vinyl sulfide.817
In some cases, alcohols can be converted to thioethers by reaction with thiols. Tertiary alcohols react with thiols in the presence of sulfuric acid to give thioethers, and the reaction works best with tertiary substrates.818 This reaction is analogous to Reaction 10-12. Thiophenol reacts with propargylic alcohols in the presence of a Ru catalyst to give propargylic thioethers.819 Primary and secondary alcohols can be converted to alkyl aryl sulfides (ROH → RSAr) in high yields by treatment with Bu3P and an N- (arylthio)succinimide in benzene.820 Iodine catalyzes the allylic alkylation of thiols.821 Thioethers (RSR′) can be prepared from an alcohol ROH and a halide R′Cl by treatment with tetramethylthiourea Me2NC(=S)NMe2 followed by NaH.822
Thiolate ions are also useful for the demethylation of certain ethers,823 esters, amines, and quaternary ammonium salts. Aryl methyl ethers824 can be cleaved by heating with EtS− in the dipolar aprotic solvent DMF: ROAr + EtS−→ ArO− + EtSR.825 Allylic sulfides have been prepared by treating allylic carbonates ROCO2Me (R = an allylic group) with a thiol and a Pd(0) catalyst.826 A good method for the demethylation of quaternary ammonium salts consists of refluxing them with PhS− in 2-butanone to give the amine and methyl phenyl sulfide.827
A methyl group is cleaved more readily than other simple alkyl groups (e.g., ethyl), although loss of these groups competes. Benzylic and allylic groups cleave even more easily, and this is a useful procedure for the cleavage of benzylic and allylic groups from quaternary ammonium salts, even if methyl groups are also present.828
Symmetrical thioethers (R–S–R) can also be prepared by treatment of an alkyl halide (R–X) with sodium sulfide (Na2S).829 Symmetrical thioethers have also been prepared by the reaction of S(MgBr)2 with allylic halides.830 This reaction can be carried out internally, by treatment of sulfide ions with 1,4-, 1,5-, or 1,6-dihalides, to prepare five-, six-, and seven-membered831 sulfur-containing heterocyclic rings. Certain larger rings have also been closed in this way.832
gem-Dihalides can be converted to dithioacetals [RCH(SR′)2],833 and acetals have been converted to monothioacetals [R2C(OR′)(SR2)],834 and to dithioacetals.835 The combination of carbon disulfide and NaBH4 converted 1,3-dibromopropane to 1,3-dithiane.836
When epoxides are substrates,837 reaction with PhSeSnBu3/BF3•OEt2838 gives the corresponding β-hydroxy selenide in a manner analogous to that mentioned in Reaction 10-25. Reaction of an epoxide with Ph3SiSH followed by treatment with Bu4NF gives hydroxy-thiols.839
Epoxides can also be directly converted to episulfides (thiiranes)840 by treatment with a phosphine sulfide (e.g., Ph3PS),841 with thiourea and titanium tetraisopropoxide842 or thiourea and LiBF4 in acetonitrile,843, with NH4SCN and TiO (tfa)2 (tfa = trifluoacetyl),844 with (EtO)2P(=O)H/S/Al2O3,845 with KSCN and InBr3,846 and with KSCN in ionic liquids (Sec. 9.D.iii).847 2,4,6-Trichloro-1,3,5-triazine catalyzes this conversion under solvent-free conditions.848
![]()
Selenides (selenoethers) and tellurides can be prepared via RSe− and RTe− species,849 and Se and borohydride exchange resin followed by the halide give the selenoether.850 The La/I2 catalyzed reaction of diphenyl diselenide with primary alkyl iodides gave arylalkyl selenides,851 Indium has been used with alkyl halides.852 A Zn mediated synthesis of tertiary alkyl selenides from tertiary alkyl halides is known.853 Diaryl selenides (Ar–Se–Ar′) have been prepared by coupling aryl iodides with tin reagents (ArSeSnR3) with a Pd catalyst.854 α-Seleno aldehydes are prepared by the reaction of an aldehyde with PhSe(N(phthalimide).855
OS II, 31, 345, 547, 576; III, 332, 751, 763; IV, 396, 667, 892, 967; V, 562, 780, 1046; VI, 5, 31, 268, 364, 403, 482, 556, 601, 683, 704, 737, 833, 859; VII, 453; VIII, 592. See also, OS VI, 776.
10-27 Formation of Disulfides856
Dithio-de-dihalo- aggre -substitution
![]()
Disulfides can be prepared by treatment of alkyl halides with disulfide ions and also indirectly by the reaction of Bunte salts (see Reaction 10-28) with acid solutions of iodide, thiocyanate ion, or thiourea,857 or by pyrolysis or treatment with hydrogen peroxide. Alkyl halides also give disulfides when heated to reflux with sulfur and NaOH.858 Some molybdenum compounds convert alkyl halides to disulfides, including (BnNEt3)6Mo7S24.859
There are no OS references, but a similar preparation of a polysulfide may be found in OS IV, 295.
10-28 Formation of Bunte Salts
Sulfonatothio-de-halogenation
![]()
Primary and secondary, but not tertiary, alkyl halides are easily converted to Bunte salts (RSSO3−) by treatment with thiosulfate ion.860 Bunte salts can be hydrolyzed with acids to give the corresponding thiols861 or converted to disulfides, tetrasulfides, or pentasulfides.862
OS VI, 235.
10-29 Alkylation of Sulfinic Acid Salts
Alkylsulfonyl-de-halogenation
![]()
Alkyl halides or alkyl sulfates, treated with the salts of sulfinic acids, give sulfones.863 A Pd catalyzed reaction with a chiral complexing agent led to sulfones with modest asymmetric induction.864 Alkyl sulfinates (R′SO–OR) may be side products.865 Sodium tosylsulfinate reacted with allylic acetates in the presence of a Pd catalyst to give the corresponding sulfone.866 Sulfonic acids themselves can be used, if DBU (see Reaction 17-13) is present.867Sulfonyl halides react with allylic halides in the presence of AlCl3–Fe868 and with benzyl halides in the presence of Sm/HgCl2.869 Sulfones have also been prepared by treatment of alkyl halides with tosylhydrazide.870 The copper(II)-catalyzed cross-coupling of organoboronic acids and sulfinate salts leads to sulfones.871 Vinyl sulfones were prepared from PhSO2Na and vinyl iodinium salts C=C–I+Ph BF4−.872
OS IV, 674; IX, 497. See also, OS VI, 1016.
10-30 Formation of Alkyl Thiocyanates
Thiocyanato-de-halogenation
![]()
Alkyl halides873 or sulfuric or sulfonic esters can be heated with sodium or potassium thiocyanate to give alkyl thiocyanates,874 although the attack by the analogous cyanate ion (Reaction 10-44) gives exclusive N-alkylation. Primary amines can be converted to thiocyanates by the Katritzky pyrylium–pyridinium method (Sec. 10.G.iii).875 Tertiary chlorides are converted to tertiary thiocyanates with Zn(SCN)2 in pyridine and ultrasound.876
OS II, 366.
10.H.iv. Nitrogen Nucleophiles
A. Attack by NH2, NHR, or NR2 at an Alkyl Carbon
10-31 Alkylation of Amines
Amino-de-halogenation (alkyl)
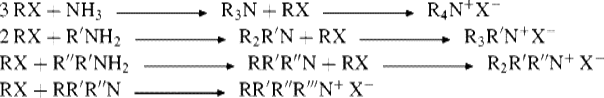
The reaction between alkyl halides and ammonia or primary amines is not usually a feasible method for the preparation of primary or secondary amines, since they are stronger bases than ammonia and preferentially attack the substrate. However, the reaction is very useful for the preparation of tertiary amines877 and quaternary ammonium salts. If ammonia is the nucleophile,878 the three or four alkyl groups on the nitrogen of the product must be identical. If a primary, secondary, or tertiary amine is used, then different alkyl groups can be placed on the same nitrogen atom. The conversion of tertiary amines to quaternary salts is called the Menshutkin reaction.879 It is sometimes possible to use this method for the preparation of a primary amine by the use of a large excess of ammonia or a secondary amine by the use of a large excess of primary amine. Metal-catalyzed methods are available to convert primary amines to secondary amines,880 and secondary amines can be converted to tertiary amines.881 Ionic liquids have been used to facilitate amination reactions.882 The use of ammonia in methanol with microwave irradiation has also been effective.883 Microwave irradiation has also been used in reactions of aniline with allyl iodides.884 Bromides react faster than chlorides, and secondary amines reaction with 3-chloro-1-bromopropane via the bromide, in the presence of Zn and THF.885N-Alkylation has been accomplished using alkyl halides in aqueous media.886
Bases other than amine can be used. Both sodium carbonate887 and lithium hydroxide888 have been used. Cesium hydroxide was successfully used as a base in the presence of molecular sieve 4 Å,889 and cesium fluoride has been used with benzylic halides.890 Potassium carbonate in DMSO has been used for the alkylation of aniline.891
The limitations of this approach can be seen in the reaction of a saturated solution of ammonia in 90% ethanol with ethyl bromide in a 16:1 molar ratio, which gave 34.2% of the primary amine (at a 1:1 ratio the yield was 11.3%).892 α-Halo acids are one type of substrate that give reasonable yields of primary amine (provided a large excess of NH3 is used) and are subsequently converted to amino acids. N-Chloromethyl lactams also react with amines to give good yields to the N-aminomethyl lactam.893 An indirect method to prepare primary amines from alkyl halides uses Reaction 10-43, followed by reduction of the azide (19-32),894 and the Gabriel synthesis (10-41) is effective.
The immediate product in any particular step is the protonated amine, but it rapidly loses a proton to another molecule of ammonia or amine in an equilibrium process, for example,
![]()
When a primary or secondary amine must be converted directly to the quaternary salt (exhaustive alkylation), the rate can be increased by the addition of a non-nucleophilic strong base that serves to remove the proton from RR′NH2+ or RR′R2NH+ and thus liberates the amine to attack another molecule of RX.895
The conjugate bases of ammonia and of primary and secondary amines (NH2−, RNH− R2N−) are generically known as amide bases, and are sometimes used as nucleophiles,896 including amide bases generated from organolithium reagents and amines (R2NLi).897 This is in contrast to analogous methods 10-1, 10-8, 10-25, and 10-26. Primary alkyl, allylic, and benzylic bromides, iodides, and tosylates react with sodium bis(trimethylsilyl)amide to give derivatives that are easily hydrolyzed to produce amine salts in high overall yields.898 Primary arylamines are easily alkylated, but diaryl- and triarylamines are very poor nucleophiles. However, the reaction has been carried out with diarylamines.899 Sulfates or sulfonates can be used instead of halides. N-Alkylation of heterocycles is sometimes problematic, but pyrrole is converted to N-methylpyrrole with KOH, iodomethane in ionic liquids.900
The reaction can be carried out intramolecularly to give cyclic amines, with three-, five-, and six-membered (but not four-membered) rings being easily prepared. Thus, 4-chloro-1-aminobutane treated with base gives pyrrolidine, and 2-chloroethylamine gives aziridine901 (analogous to Reaction 10-9):
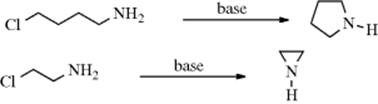
Reduction of N-(3-bromopropyl) imines gives a bromoamine in situ, which cyclizes to the aziridine.902 Five-membered ring amines (pyrrolidines) can be prepared from alkenyl amines via treatment with N-chlorosuccinimide (NCS) and then Bu3SnH.903 The Pd catalyzed internal addition of amine to allylic acetates leads to cyclic products via a SN2′ reaction.904 Three-membered cyclic amines (aziridines) can be prepared from chiral conjugated amides via bromination and reaction with an amine.905 Four-membered cyclic amines (azetidines) have been prepared from the ditosylate of 1,3-propanediol906 and from 1,3-dichloropropane.907 This reaction was also used to close five-, six-, and seven-membered rings.
![]()
As usual, tertiary substrates do not give the reaction at all, but undergo preferential elimination upon treatment with a basic amine. However, tertiary (but not primary or secondary) halides (e.g., R3CCl) can be converted to primary amines (R3CNH2) by treatment with NCl3 and AlCl3908 in a reaction related to Reaction 10-39. Ruthenium(II) complexes have been used for the alkylation of aryl amines.909
Primary amines can be prepared from alkyl halides by the use of hexamethylenetetramine910 followed by cleavage of the resulting salt with ethanolic HCl. The method called the Delépine reaction is most successful for active halides (e.g., allylic and benzylic halides and α-halo ketones).
A convenient way of obtaining secondary amines without contamination by primary or tertiary amines involves treatment of alkyl halides with the sodium or calcium salt of cyanamide (NH2–CN) to give disubstituted cyanamides, which are then hydrolyzed and decarboxylated to secondary amines. Good yields are obtained when the reaction is carried out under phase-transfer conditions.911 The R group may be primary, secondary, allylic, or benzylic. 1,ω-Dihalides give cyclic secondary amines. Aminoboranes react with sulfonate esters to give a derivative that can be hydrolyzed to a tertiary amine.912 An aminyl-radical cyclization process was used to prepare cyclic amines.913N-Silylalkyl amines are formed from amines by reaction with halotrialkylsilanes and a suitable base.914 Amines react directly with triarylsilanes in the presence of Yb catalysts.915
Palladium compounds react with allylic halides, acetates, or carbonate derivatives to generate π-allyl Pd intermediates that react with amines to give an allylic amine (see the reaction below).916 The same reaction is discussed in Reaction 10-60 with other nucleophiles. Propargylic amines can be prepared by similar methodology.917 Boronic acid derivatives leads to methylation of aniline derivatives in the presence of cupric acetate.918 tert-Butylamines can be prepared from isobutylene, HBr, and the amine by heating a sealed tube.919
![]()
Phosphines behave similarly to amines, and compounds, such as R3P and R4P+ X−, can be prepared.920 The reaction between triphenylphosphine and quaternary salts of nitrogen heterocycles in an aprotic solvent is probably the best way of dealkylating the heterocycles, for example,921
![]()
Other phosphorus compounds can be alkylated. Phosphinate esters, for example, react with a suitable base and then an alkyl halide to give the P-substituted product.922
OS I, 23, 48, 102, 300, 488; II, 85, 183, 290, 328, 374, 397, 419, 563; III, 50, 148, 254, 256, 495, 504, 523, 705, 753, 774, 813, 848; IV, 84, 98, 383, 433, 466, 582, 585, 980; V, 88, 124, 306, 361, 434, 499, 541, 555, 608, 736, 751, 758, 769, 825, 883, 985, 989, 1018, 1085, 1145; VI, 56, 75, 104, 106, 175, 552, 652, 704, 818, 967; VIII, 9, 152, 231, 358. Also see, OS II, 395; IV, 950; OS V, 121; OS I, 203.
For N-arylation of amines see Reaction 13-5.
10-32 Replacement of a Hydroxy or Alkoxy by an Amino Group
Amino-de-hydroxylation and Amino-de-alkoxylation
![]()
Alcohols can be converted to alkyl halides, which then react with amines (Reaction 10-43). Alcohols react with various amine reagents that give products convertible to the amine.923 The conversion ROH → RNH2 can be accomplished for primary and secondary alcohols by treatment with hydrazoic acid (HN3), diisopropyl azodicarboxylate (iPr–OOCN=NCOO–iPr), and excess Ph3P in THF, followed by water or aq acid.924 This is a type of Mitsunobu Reaction (see 10-17).925 Primary and secondary alcohols (ROH, but not methanol) can be converted to tertiary amines926 Primary amines can be generated directly from primary alcohols and ammonia.927 Formation of R′2NR required treatment with the secondary amine (R′2NH) with the (t-BuO)3Al compound in the presence of Raney nickel.928
Allylic alcohols (ROH) react with amines in the presence of Pt929 or Pd930 complexes, to give allylic amines.931 Amines can be N-alkylated by reaction with alcohols, in a sealed tube with microwave irradiation,932 and also by Ru-,933 Ir-,934 or Au catalyzed935 reactions, or by Ti mediated936 reactions. Copper–aluminum hydrotalcite can also be used to generate amines from alcohols.937 The use of aniline gives secondary amines (PhNHR). Phenols can be converted to aniline derivatives.938,939 Heating indoles with benzylic alcohols in the presence of Me3P=CH(CN) gives the N-benzylindole.940 Heating an alcohol on γ-Al2O3 leads to an amine,941 as does treatment with the amine, SnCl2, and Pd(PPh3)4.942 The Ru catalyzed reaction of amines and diols leads to cyclic amines.943
β-Amino alcohols give aziridines (120) when treated with triphenylphosphine dibromide in the presence of triethylamine.944 The fact that inversion takes place at the OH carbon indicates that an SN2 mechanism is involved, with OPPh3 as the leaving group.
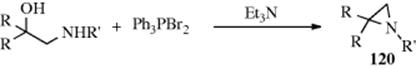
Alcohols can be converted to amines in an indirect manner.945 The alcohols are converted to alkyloxyphosphonium perchlorates, which in DMF successfully monoalkylate not only secondary but also primary amines.946
![]()
Thus by this means secondary as well as tertiary amines, can be prepared in good yields. Benzylic alcohols can be converted to an azide and then treated with triphenylphosphine to give the amine (Reaction 19-50).947
Cyanohydrins can be converted to amines by treatment with ammonia. The use of primary or secondary amines instead of ammonia leads to secondary and tertiary cyanoamines, respectively. It is more common to perform the conversion of an aldehyde or ketone directly to the cyanoamine without isolation of the cyanohydrin (see Reaction 16-52). α-Hydroxy ketones (acyloins and benzoins) behave similarly.948
![]()
A solution of the sodium salt of N-methylaniline in HMPA can be used to cleave the methyl group from aryl methyl ethers:949 ArOMe + PhNMe− → ArO− + PhNMe2. This reagent also cleaves benzylic groups. In a similar reaction, methyl groups of aryl methyl ethers can be cleaved with lithium diphenylphosphide (Ph2PLi).950 This reaction is specific for methyl ethers and can be carried out in the presence of ethyl ethers with high selectivity. Phenyl allyl ethers react with secondary amines in the presence of a Pd catalyst to give phenol and the tertiary allyl amine.951
OS II, 29, 231; IV, 91, 283; VI, 567, 788; VII, 501. Also see, OS I, 473; III, 272, 471.
10-33 Transamination
Alkylamino-de-amination
![]()
Where the nucleophile is the conjugate base of a primary amine, NH2 can be a leaving group. The method has been used to prepare secondary amines.952 In another process, primary amines are converted to secondary amines in which both R groups are the same (2 RNH2 → R2NH + NH3)953 by refluxing in xylene in the presence of Raney nickel.954 Quaternary salts can be dealkylated with ethanolamine.955
![]()
In this reaction, methyl groups are cleaved in preference to other saturated alkyl groups. A similar reaction takes place between a Mannich base (see Reaction 16-19) and a secondary amine, where the mechanism is elimination–addition (see Sec. 10.F). Transamination has been accomplished using yeast alcohol dehydrogenase.956
See also, Reaction 19-5.
OS V, 1018.
10-34 Alkylation of Amines With Diazo Compounds
Hydro, dialkylamino-de-diazo-bisubstitution
![]()
The reaction of diazo compounds with amines is similar to Reaction 10-11.957 The acidity of amines is not great enough for the reaction to proceed without a catalyst, but BF3, which converts the amine to the F3B–NHR′2 complex, enables the reaction to take place. Cuprous cyanide can also be used as a catalyst.958 Ammonia has been used rather than an amine but, as in the case of Reaction 10-31, mixtures of primary, secondary, and tertiary amines are obtained. However, a highly chemoselective reaction of amines in water has been reported.959 Primary aliphatic amines give mixtures of secondary and tertiary amines. Secondary amines give successful alkylation. Primary aromatic amines also give the reaction, but diaryl or arylalkylamines react very poorly.
10-35 Reaction of Epoxides with Nitrogen Reagents960
(3) OC-seco-Amino-de-alkoxylation
![]()
The reaction between epoxides and ammonia961 (or ammonium hydroxide)962 is a general and useful method for the preparation of β-hydroxyamines. With epoxides derived from terminal alkenes, the reaction with ammonia gives largely the primary amine, but secondary and tertiary amine products are possible from the appropriate epoxide. The reaction of 121 with ammonium hydroxide with microwave irradiation, for example,

gave 122.963 Ethanolamines, which are useful solvents, as well as synthetic precursors, are prepared by this reaction. Similar ring-opening occurs with alkyl and aromatic amines.964 For another way of accomplishing this conversion, see Reaction 10-40. Ring opening has been accomplished with aniline on silica gel,965 and with aromatic amines in the presence of heteropoly acids in water.966
![]()
Primary and secondary amines give, respectively, secondary and tertiary amines (121). Aniline reacts with epoxides in the presence of aq β-cyclodextrin967 in 5 M LiClO4 in ether,968 or in fluoro-alcohol solvents.969 Aniline reacts with epoxides in the presence of a VCl3 catalyst970 or a Cu(II) catalyst.971N-Boc-amine (H2N–CO2t-Bu) reacted with epoxides in the presence of a cobalt–salen catalyst to give the amido alcohol.972 Solvent-free reactions using a catalytic amount of SnCl4 are known.973 Other metal-catalyzed ring-opening reactions of epoxides with amines have been reported,974 often with high enantioselectivity.
Enantioselective ring-opening reactions typically use a metal catalyst in the presence of a chiral additive. Amines react with epoxides using a catalytic amount of a Nb complex, in the presence of a 1,1-bi-2-naphthol (BINOL) derivative, to give chiral amino alcohols.975 Other enantioselective ring-opening reactions include a V–salen-catalyzed reaction,976 and a Mg–BINOL complex.977
Tetrahydropyrimidones can be used to mediate the addition of indole to epoxides.978 Amide bases react differently with epoxides. Lithium 2,2,6,6-tetramethylpiperidide (LTMP), for example, reacted with epoxides, but the product was the corresponding enamine.979 This latter reaction follows a very different mechanism. Initial formation of the lithio-epoxide is followed by rearrangement to give the aldehyde,980 and subsequent reaction with the amine byproduct of the lithiation leads to the enamine.
An indirect method for generating an amino alcohol (124) is to open an epoxide with azide to give the azido-alcohol (123),981 and subsequent reduction (Reaction 19-50) gives the amine group.982 The cerium ammonium nitrate catalyzed reaction of epoxides and sodium azide, for example, gave the azido alcohol with selectivity for the azide group on the more substituted position.983 Cerium chloride has also been used, giving the azide on the less substituted carbon.984 Under Mitsunobu conditions (Reaction 10-17), epoxides are converted to 1,2-diazides with HN3.985 The reaction of trimethylsilyl azide and an epoxide was reported using an ionic solvent.986 In the presence of AlCl3 in water at pH 4, sodium azide reacts with epoxy acids to give the β-azido-α-hydroxycarboxylic acid.987 Silylazides can be used as well.988
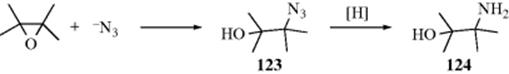
Sodium nitrate (NaNO2) reacts with epoxides in the presence of MgSO4 to give the nitro alcohol.989 The nitro group can also be reduced to give the amine (Reaction 19-45).990
Episulfides (thiiranes), which can be generated in situ in various ways, react similarly to give β-amino thiols,991 and aziridines react with amines to give 1,2-diamines (Reaction 10-38). Triphenylphosphine similarly reacts with epoxides to give an intermediate that undergoes elimination to give alkenes (see the Wittig Reaction, 16-44).
OS X, 29. See OS VI, 652 for a related reaction.
10-36 Formation of Aziridines from Epoxides
Amino-de-alkoxylation
![]()
It is possible to prepare aziridines, which are synthetically important molecules, directly from the corresponding epoxide. Reaction of Ph3P=NPh with an epoxide in the presence of ZnCl2 gives the N-phenyl aziridine.992Guanidines have also been used to prepare aziridines from epoxides.993 Tosylamines react with epoxides to give the N-tosylaziridine.994
Various methods are available to convert an aminomethyl epoxide to a hydroxymethyl aziridine (125).995

10-37 Amination of Oxetanes
(4) OC-homoseco-Amino-de-alkoxylation

Oxetanes are significantly less reactive with nucleophiles due to diminished ring strain. Under certain conditions, however, amines can open oxetanes to give amino alcohols. tert-Butyl amine reacts with oxetanes in the presence of Yb(OTf)3, for example, to give 3-hydroxy amines.996 Lithium tetrafluoroborate has also been used for this purpose.997
10-38 Reaction of Aziridines with Nitrogen
(3) NC-seco-Amino-de-aminoalkylation

Just as epoxides can be opened by amines to give hydroxy amines, aziridines can be opened to give diamines.998 With bicyclic aziridines, the major product is usually the trans diamine. N-Aryl or N-alkyl aziridines react with amines in the presence of T-Binolate,999 Sn(OTf)21000 or B(C6F5)31001 to give the diamine. Activated aziridines undergo regioselective ring opening with organoalanes.1002 Amines react with N-tosylaziridines, in the presence of various catalysts or additives to give the corresponding diamine derivative.1003 This reaction also takes place on activated silica.1004 The reaction of LiNTf2 and an amine, in the presence of an N-alkyl aziridine gives the diamine.1005
Tosyl-aziridines react with azide ion to generate azido tosylamines,1006 and a clay-catalyzed variation1007 has been reported. Reduction of the azide (Reaction 19-50) gives the diamine. Silylazides (e.g., Me3SiN3) also react with aziridine derivatives to give the azido-amine.1008 This latter reaction can be catalyzed by InCl3.1009
10-39 Amination of Alkanes
Amino-de-hydrogenation or Amination
![]()
Alkanes, arylalkanes, and cycloalkanes can be aminated, at tertiary positions only, by treatment with trichloroamine and aluminum chloride at 0–10 °C.1010 For example, p-MeC6H4CHMe2 gives p-MeC6H4CMe2NH2, methylcyclopentane gives 1-amino-1-methylcyclopentane, and adamantane gives 1-aminoadamantane, all in good yields. A Ag catalyzed reaction has also been reported.1011 There are not many other methods for the preparation of tert-alkyl amines. The mechanism has been rationalized as an SN1 process with H− as the leaving group:1010
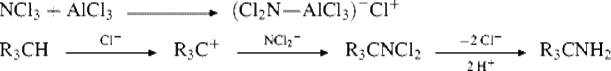
Note that under photochemical conditions, ammonia opens cyclopropane derivatives to give the corresponding alkyl amine.1012 See also Reaction, 12-12.
OS V, 35.
10-40 Formation of Isonitriles (Isocyanides)
Haloform-isocyanide transformation
![]()
There are several methods available for the preparation of isonitriles, otherwise known as isocyanides.1013 Reaction with chloroform under basic conditions is a common test for primary amines, both aliphatic and aromatic, since isonitriles (126) have very strong bad odors. The reaction probably proceeds by an SN1cB mechanism with dichlorocarbene (127) as an intermediate.
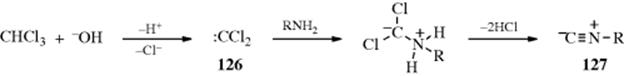
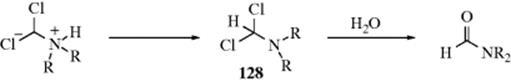
Yields are generally not high,1014 but an improved procedure has been reported.1015 When secondary amines are involved, the adduct 128 cannot lose two molar equivalents of HCl. Instead it is hydrolyzed to an N,N-disubstituted formamide.1016
A completely different way of preparing isocyanides involves the reaction of epoxides or oxetanes with trimethylsilyl cyanide and zinc iodide to give the isocyanide 129.1017
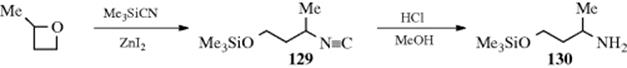
The products can be hydrolyzed to protected hydroxy-amines (e.g., 130).
OS VI, 232.
B. Attack by NHCOR
10-41 N-Alkylation or N-Arylation of Amides and Imides
Acylamino-de-halogenation
![]()
Amides are very weak nucleophiles,1018 far too weak to attack alkyl halides, so they first must be converted to their conjugate bases, the anion. By this method, unsubstituted amides can be converted to N-substituted, or N-substituted to N,N-disubstituted, amides.1019 Esters of sulfuric or sulfonic acids can also be substrates. Tertiary substrates give elimination and O-Alkylation is at times a side reaction.1020 Both amides and sulfonamides have been alkylated under phase-transfer conditions.1021 Metal-catalyzed amidations are known, including an Ir(I) catalyzed allylic amidation.1022
Lactams can be alkylated using similar procedures. Ethyl pyroglutamate (5-carboethoxy 2-pyrrolidinone) and related lactams were converted to N-alkyl derivatives via treatment with NaH (short contact time) followed by addition of the halide.1023 Other 2-pyrrolidinone derivatives can be alkylated using a similar procedure.1024N-Cyclopropyl lactams are prepared using a Bi reagent in the presence of cupric acetate.1025N-Aryl lactams can be prepared using Ph3Bi and Cu(OAc)2.1026N-Arylation of sulfonamides has been reported using a Pd catalyst,1027 and this method has been applied to an intramolecular arylation leading to bicyclic lactams.1028
N-Alkenyl amides have been prepared from vinyl iodides and primary amides, using 10% CuI and two molar equivalents of cesium carbonate.1029 A related Pd catalyzed vinylation of lactams was repeated using vinyl ethers as a substrate.1030 Oxazolidin-2-ones (a cyclic carbamate) can be N-alkylated using an alkyl halide with KF/Al2O3.1031
The Gabriel synthesis1032 for converting halides to primary amines is based on this reaction. The halide is treated with potassium phthalimide and the resulting product hydrolyzed (Reaction 16-60)
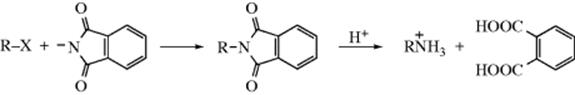
It is obvious that the primary amines formed in this reaction will be uncontaminated by secondary or tertiary amines (unlike Reaction 10-31). The reaction is usually rather slow, but the rate can be conveniently increased by the use of a dipolar aprotic solvent (e.g., DMF)1033 or with a crown ether.1034 Hydrolysis of the phthalimide, whether acid or base catalyzed (acid catalysis is used far more frequently), is also usually very slow, and better procedures are generally used. A common one is the Ing–Manske procedure,1035 in which the phthalimide is heated with hydrazine in an exchange reaction,1036 but other methods have been introduced, using Na2S in aq THF or acetone,1037 and 40% aq methylamine.1038 N-Aryl imides can be prepared from ArPb(OAc)3 and NaH.1039
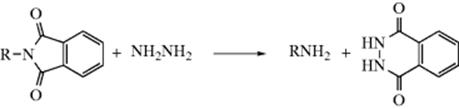
An alternative to the Gabriel synthesis, in which alkyl halides can be converted to primary amines in good yields, involves treatment of the halide with the strong base guanidine followed by alkaline hydrolysis.1040 There are several other alternative procedures.1041
N-Alkyl amides or imides can also be prepared starting from alcohols by treatment of the latter with equimolar amounts of the amide or imide (Ph3P) and diethyl azodicarboxylate (EtO2CN=NCO2Et) at room temperature (the Mitsunobu Reaction, 10-17).1042 A related reaction treats an alcohol with ClCH=NMe2+Cl−, followed by potassium phthalimide and treatment with hydrazine give the amine.1043 Metal-catalyzed syntheses of amides via oxidative coupling of alcohols and amines are known. Variations include the use of a Ru complex,1044 a RuCl3 catalyzed reaction,1045 an FeCl3 catalyzed reaction,1046 an Ir complex catalyzed reaction,1047 and an InCl3 catalyzed coupling of alcohols with ToSMIC (ToSMIC = toluene sulfonyl methyl cyanide).1048
Amides can also be alkylated with diazo compounds, as in Reaction 10-34. Salts of sulfonamides (ArSO2NH−) can be used to attack alkyl halides to prepare N-alkyl sulfonamides (ArSO2NHR) that can be further alkylated to ArSO2NRR′. Hydrolysis of the latter is a good method for the preparation of secondary amines. Secondary amines can also be made by crown-ether assisted alkylation of F3CCONHR (R = alkyl or aryl) and hydrolysis of the resulting F3CCONRR′.1049
The reaction of a primary amide and benzaldehyde, in the presence of a silane and trifluoroacetic acid, leads to the corresponding N-benzylamide.1050 This transformation is a reductive alkylation (Reaction 16-17). N-Alkynyl amides have been prepared by the copper-catalyzed reaction of 1-bromoalkynes and secondary amides.1051 1-Haloalkynes are typically prepared by base-induced elimination of 1,1-dihaloalkenes1052 or by direct halogenation of an alkyne with sodium or potassium hypohalite, prepared by reaction of the appropriate base with the halogen.1053
Internal N-alkylation has been used to prepare the highly strained compounds α-lactams.1054
OS I, 119, 203, 271; II, 25, 83, 208; III, 151; IV, 810; V, 1064; VI, 951; VII, 501.
C. Other Nitrogen Nucleophiles
10-42 Formation of Nitro Compounds1055
Nitro-de-halogenation
![]()
Sodium nitrite can be used to prepare nitro compounds from primary or secondary alkyl bromides or iodides, but the method is of limited scope. Silver nitrite gives nitro compounds only when RX is a primary bromide or iodide.1056 Nitrite esters are an important side product in all these cases (Reaction 10-22) and become the major product (by an SN1 mechanism) when secondary or tertiary halides are treated with silver nitrite. Alkyl nitro compounds can be prepared from the alkyl halide via the corresponding azide, by treatment with HOF in acetonitrile.1057
Nitro compounds can be prepared from alcohols using NaNO2/AcOH/HCl.1058
OS I, 410; IV, 368, 454, 724.
10-43 Formation of Azides
Azido-de-halogenation
![]()
Alkyl azides can be prepared by treatment of the appropriate halide with azide ion.1059 Phase-transfer catalysis,1060 ultrasound,1061 and the use of reactive clays1062 are important variations. Substrates with leaving groups other than halogen have been used,1063 including OMs (Ms = methanesulfonyl), OTs (Ts = tosyl),1064 and OAc (Ac = acetyl).1065 There are protocols for the conversion of alcohols to azides1066 Boronic acids are precursors to azides.1067 Aryl azides are prepared from aryl amines by reaction with t-BuONO and moist NaN3 in t-BuOH.1068
Ring-opening reactions of epoxides with nitrogen nucleophiles were discussed in Reaction 10-35. However, it is appropriate to discuss epoxide-opening reactions involving azides. Epoxides react with NaN3 (10-35), under various conditions and media, including in ionic liquids.1069 Other reagents include TMSN3 (TMS = trimethylsilyl) and Ph4SbOH1070 or SmI21071 or (i-Bu)2AlHN3Li1072 to give β-azido alcohols; these are easily converted to aziridines (131).1073
This conversion has been used as a key step in the preparation of optically active aziridines from optically active 1,2-diols (prepared by Reaction 15-48).1074 Even hydrogen can be the leaving group. Benzylic hydrogen atoms have been replaced and N3 and treatment with HN3 in CHCl3 in the presence of DDQ (see Reaction 19-01).1075
Tertiary alkyl azides can be prepared by stirring tertiary alkyl chlorides with NaN3 and ZnCl2 in CS21076 or by treating tertiary alcohols with NaN3 and CF3COOH1077 or with HN3 and TiCl41078 or BF3.1079 Aryl azides can be prepared from aniline and aniline derivatives.1080
Acyl azides, which can be used in the Curtius Reaction (18-14), are generally prepared from acyl halides, anhydrides,1081 esters,1082 or other acyl derivatives.1083 Acyl benzotriazoles are also precursors to acyl azides.1084 Acyl azides also can be prepared from aldehydes using SiCl4/NaN3–MnO2,1085 TMSN3/CrO31086 or the Dess–Martin periodinane (see Reaction 19-03, category 5) with NaN3.1087
OS III, 846; IV, 715; V, 273, 586; VI, 95, 207, 210, 910; VII, 433; VIII, 116; IX, 220; X, 378. See also, OS VII, 206.
10-44 Formation of Isocyanates and Isothiocyanates
Isocyanato-de-halogenation
![]()
When the reagent is the thiocyanate ion, S-alkylation is an important side reaction (10-30), but the cyanate ion practically always gives exclusive N-alkylation.509 Primary alkyl halides have been converted to isocyanates by treatment with sodium nitrocyanamide (NaNCNNO2) and m-chloroperoxybenzoic acid, followed by heating of the initially produced RN(NO2)CN.1088 When alkyl halides are treated with NCO− in the presence of ethanol, carbamates can be prepared directly (see Reaction 16-8).1089 Acyl halides give the corresponding acyl isocyanates and isothiocyanates.1090 For the formation of isocyanides (isonitriles), see Reaction 10-75. Isonitriles, in the presence of sulfur and a Rh catalyst, are converted to isothiocyanate,1091 as are amines.1092
OS III, 735.
10-45 Formation of Azoxy Compounds
Alkyl- NNO-azoxy-de-halogenation
![]()
The reaction between alkyl halides and alkanediazotates (132) gives azoxyalkanes.1093 The R and R′ groups may be the same or different, but neither may be aryl or tertiary alkyl. The reaction is regioselective; only the isomer shown is obtained.
10.H.v. Halogen Nucleophiles1094
10-46 Halide Exchange
Halo-de-halogenation
![]()
Halide exchange, sometimes call the Finkelstein reaction, is an equilibrium process, but it is often possible to shift the equilibrium.1095 The reaction is most often applied to the preparation of iodides and fluorides. Iodides can be prepared from chlorides or bromides by taking advantage of the fact that sodium iodide, but not the bromide or chloride, is soluble in acetone. When an alkyl chloride or bromide is treated with a solution of sodium iodide in acetone, the equilibrium is shifted by the precipitation of sodium chloride or bromide. Since the mechanism is SN2, the reaction is much more successful for primary halides than for secondary or tertiary halides; sodium iodide in acetone can be used as a test for primary bromides or chlorides. Tertiary chlorides can be converted to iodides by treatment with excess NaI in CS2, and ZnCl2 as catalyst.1096 Vinylic bromides give vinylic iodides with retention of configuration when treated with KI and a nickel bromide-zinc catalyst,1097 or with KI and CuI in hot HMPA.1098
Fluorides1099 are prepared by treatment of other alkyl halides with any of a number of fluorinating agents,1100 among them anhydrous HF (which is useful only for reactive substrates, e.g., benzylic or allylic), AgF, KF,1101 HgF2, Et3N•2HF,1102 4-Me–C6H4IF2,1103 and Me3SiF2Ph+![]() .1104 The Pd catalyzed conversion of chlorides to fluorides has also been reported.1105 The equilibria in these cases are shifted because the alkyl fluoride once formed has little tendency to react, owing to the extremely poor leaving-group ability of fluorine. Phase-transfer catalysis of the exchange reaction is a particularly effective way of preparing both fluorides and iodides.1106
.1104 The Pd catalyzed conversion of chlorides to fluorides has also been reported.1105 The equilibria in these cases are shifted because the alkyl fluoride once formed has little tendency to react, owing to the extremely poor leaving-group ability of fluorine. Phase-transfer catalysis of the exchange reaction is a particularly effective way of preparing both fluorides and iodides.1106
Primary alkyl chlorides can be converted to bromides with ethyl bromide, N-methyl-2-pyrrolidinone and a catalytic amount of NaBr,1107 with LiBr under phase-transfer conditions,1108 and with Bu4N+ Br−.1109 Primary bromides were converted to chlorides with TMSCl/imidazole in hot DMF.1110 For secondary and tertiary alkyl chlorides, treatment with excess gaseous HBr and an anhydrous FeBr3 catalyst in CH2Cl2 has given high yields1111 (this procedure is also successful for chloride-to-iodide conversions). Alkyl chlorides or bromides can be prepared from iodides by treatment with HCl or HBr in the presence of HNO3, making use of the fact that the leaving I− is oxidized to I2 by the HNO3.1112 Primary iodides give the chlorides when treated with PCl5 in POCl3.,1113 Primary alkyl halides are converted to the corresponding fluoride with tetrabutylammonium fluoride in tert-butanol.1114 Alkyl fluorides and chlorides are converted to the bromides and iodides (and alkyl fluorides to the chlorides) by heating with the corresponding HX in excess amounts.1115
OS II, 476; IV, 84, 525; VIII, 486; IX, 502.
10-47 Formation of Alkyl Halides from Esters of Sulfuric and Sulfonic Acids
Halo-de-sulfonyloxy-substitution, and so on
![]()
Alkyl sulfates, tosylates, and other esters of sulfuric and sulfonic acids can be converted to alkyl halides with any of the four halide ions.1116 Neopentyl tosylate, for example, reacts with Cl−, Br−, or I− without rearrangement in HMPA.1117 Similarly, allylic tosylates can be converted to chlorides without allylic rearrangement by reaction with LiCl in the same solvent.1118 Inorganic esters are intermediates in the conversion of alcohols to alkyl halides with SOCl2, PCl5, PCl3, and so on (Reaction 10-48), but those esters are seldom isolated.
OS I, 25; II, 111, 404; IV, 597, 753; V, 545.
10-48 Formation of Alkyl Halides from Alcohols
Halo-de-hydroxylation
![]()
Alcohols can be converted to alkyl halides with several reagents,1119 the most common of which are halogen acids (HX) and inorganic acid halides, (e.g., SOCl2,1120 PCl5, PCl3, and POCl3).1121 When the reagent is HX, the mechanism is SN1cA or SN2cA; that is, the leaving group is not ![]() , but OH2 (Sec. 10.G.iii). The leaving group is not
, but OH2 (Sec. 10.G.iii). The leaving group is not ![]() with the other reagents either, since in these cases the alcohol is first converted to an inorganic ester (e.g., ROSOCl) with SOCl2 (Reaction 10-22). The leaving group is therefore
with the other reagents either, since in these cases the alcohol is first converted to an inorganic ester (e.g., ROSOCl) with SOCl2 (Reaction 10-22). The leaving group is therefore ![]() or a similar group (Reaction 10-47). These may react by the SN1 or SN2 mechanism and, in the case of ROSOCl, by the SNi mechanism1122 (Sec. 10.D).
or a similar group (Reaction 10-47). These may react by the SN1 or SN2 mechanism and, in the case of ROSOCl, by the SNi mechanism1122 (Sec. 10.D).
The reagent HBr is usually used for alkyl bromides1123 and HI for alkyl iodides. These reagents are often generated in situ from the halide ion and an acid (e.g., phosphoric or sulfuric). The use of HI sometimes results in reduction of the alkyl iodide to the alkane (Reaction 19-53) and, if the substrate is unsaturated, can also reduce the double bond.1124 The reaction can be used to prepare primary, secondary, or tertiary halides, but alcohols of the isobutyl or neopentyl type often give large amounts of rearrangement products.1125 Tertiary chlorides are easily made with concentrated HCl, but primary and secondary alcohols react with HCl so slowly that a catalyst, usually zinc chloride, is required.1126 Primary alcohols give good yields of chlorides upon treatment with HCl in HMPA.1127
The inorganic acid chlorides (SOCl2,1128 PCl3, etc.) give primary, secondary, or tertiary alkyl chlorides with much less rearrangement than is observed with HCl. Inorganic bromides and iodides, especially PBr3, have also been used, but they are more expensive and used less often than HBr or HI, although some of them may also be generated in situ (e.g., PBr3 from phosphorous and bromine). Secondary alcohols always give some rearranged bromides if another secondary position is available, even with PBr3, PBr5, or SOBr2; thus 3-pentanol gives both 2- and 3-bromopentane. Such rearrangement can be avoided by converting the alcohol to a sulfonate and then using Reaction 10-47,1129 or by the use of phase-transfer catalysis.1130 Tertiary alcohols can be converted to the bromide with BBr3 at 0 °C.1131 Iodides have been prepared by simply heating the alcohol with iodine.1132 Trichloroisocyanuric acid (1,3,5-trichlorohexahydrotriazin-2,4,6-trione) and triphenylphosphine converts primary alcohols to the corresponding chloride.1133 Pivaloyl chloride–DMF has been used to convert alcohols to chlorides.1134 Sodium iodide and Amberlyst-151135 or tosic acid and KI with microwave irradiation1136 converts primary alcohols to the iodide.
The preparation of alkyl fluorides can be problematic, and specialized reagents are usually required. Hydrogen fluoride does not generally convert alcohols to alkyl fluorides.1137 The most important reagent for this purpose is the commercially available diethylaminosulfur trifluoride (Et2NSF3, DAST),1138 which converts primary, secondary, tertiary, allylic, and benzylic alcohols to fluorides in high yields under mild conditions.1139 Fluorides have also been prepared from alcohols by treatment with nonaflyl fluoride,1140 tetrabutylammonium difluoride,1141 CsI/BF3,1142 TMSI/ZnCl2,1143 and indirectly, by conversion to a sulfate or tosylate, and so on (Reaction 10-47). A mixture of IF5, NEt3, and excess KF1144 or (Cl3CO)2C=O [bis(trichloromethyl)carbonate] and KF (which gives COF2in situ) with 18-crown-61145 also converts primary alcohols to primary fluorides.
Primary, secondary, and tertiary alcohols can be converted to any of the four halides by treatment with the appropriate NaX, KX, or NH4X in polyhydrogen fluoride–pyridine solution.1146 This method is even successful for neopentyl halides. Ionic liquids can be used for halogenation, and bmim-Cl (1-n-butyl-3-methylimidazolium chloride) generates the chloride directly from the alcohol without any additional reagent.1147 Triphenylphosphine and iodine will convert alcohols to iodides in ionic liquids, under solvent-free conditions.1148tert-Butyl halides halogenate alcohols in the ionic liquid [pmim]Br with sonication.1149
Other reagents1150 have also been used, including ZrCl4/NaI,1151 Me3SiCl and BiCl3,1152 or Me3SiCl and InCl31153 or GaCl3–tartrate,1154 or simply Me3SiCl in DMSO.1155 1,2-Dipyridiniumditribromide ethane is an efficient brominating agent, and simply grinding the reagent and an alcohol in a porcelain mortar at room temperature with no solvent gives the product.1156 Other specialized reagents include (RO)3PRX1157 and R3PX21158, which give good yields for primary (including neopentyl), secondary, and tertiary halides without rearrangements,1159 Similarly, a mixture of PPh3 and CCl41160 (or CBr41161) give good results, and PPh3/Cl3CCONH2 is an efficient chlorinating reagent.1162 The compound PPh3–CCl3CN converts neopentyl alcohol to neopentyl chloride, in 95% yield.1163
![]()
The PPh3–CCl4 or CBr4 method converts allylic alcohols1164 to the corresponding halides without allylic rearrangements,1165 and also cyclopropylcarbinyl alcohols to the halides without ring opening.1166 A mixture of triphenylphosphine and iodine converts alcohols to iodides under solvent-free conditions, using microwave irradiation.1167 Hexabromoacetone–ethyltribromoacetate is an efficient brominating reagent.1168 N-Bromosaccharin and N-iodosaccharin in the presence of PPh3 gives the corresponding bromide or iodide.1169
Allylic and benzylic alcohols can also be converted to bromides or iodides with NaX–BF3 etherate,1170 and to iodides with AlI3.1171 A mixture of methanesulfonic acid and NaI also converts benzylic alcohols to benzylic iodides.1172 Allylic alcohols are converted to allylic halides in a procedure that uses acetyl halides, but the reaction proceeds with allylic rearrangement.1173 A simple method that is specific for benzylic and allylic alcohols (and does not give allylic rearrangement) involves reaction with NCS or NBS and methyl sulfide.1174 A mixture of NBS, Cu(OTf)2, and diisopropylcarbodiimide converted primary alcohols to the corresponding bromide.1175 The use of NCS gave the chloride and N-iodosuccinimide (NIS) gave the iodide under identical conditions. Thiols are converted to alkyl bromides by a similar procedure using PPh3 and NBS.1176
Trialkylsilyl ethers (e.g., ROSiMe3) are converted to the corresponding iodide with SiO2–Cl/NaI.1177 Hydroxy ketones are converted to the iodide with iodine and iodic acid.1178 Propargyilc fluorides can be prepared from allenylsilanes by treatment with Selectfluor.1179
OS I, 25, 36, 131, 142, 144, 292, 294, 533; II, 91, 136, 159, 246, 308, 322, 358, 399, 476; III, 11, 227, 370, 446, 698, 793, 841; IV, 106, 169, 323, 333, 576, 681; V, 1, 249, 608; VI, 75, 628, 634, 638, 781, 830, 835; VII, 210, 319, 356; VIII, 451. Also see, OS III, 818; IV, 278, 383, 597.
10-49 Formation of Alkyl Halides from Ethers
Halo-de-alkoxylation
![]()
Ethers can be cleaved by heating with concentrated HI or HBr.1180 Hydrogen chloride is seldom successful,1181 and HBr reacts more slowly than HI, but is often a superior reagent, since it causes fewer side reactions. Phase-transfer catalysis has also been used,1182 and 47% HBr in ionic liquids has proven effective.1183 Dialkyl ethers and alkyl aryl ethers can be cleaved. In the latter case, the alkyl–oxygen bond is the one broken. As in Reaction 10-48, the actual leaving group is not OR′−, but −OHR'. Although alkyl aryl ethers always cleave so as to give an alkyl halide and a phenol, there is no general rule for dialkyl ethers. Often cleavage occurs from both sides, and a mixture of two alcohols and two alkyl halides is obtained. However, methyl ethers are usually cleaved so that methyl iodide or bromide is a product. An excess of HI or HBr converts the alcohol product into alkyl halide, so that dialkyl ethers (but not alkyl aryl ethers) are converted to 2 equiv of alkyl halide. This procedure is often carried out so that a mixture of only two products is obtained instead of four.
O-Benzyl ethers are readily cleaved to the alcohol and the hydrocarbon via hydrogenolysis, and the most common methods are hydrogenation1184 or dissolving metal conditions (Na or K in ammonia).1185 Heating in anisole with 3% Sc(NTf2)31186 or with In metal in aq ethanol1187 also cleaves benzyl ethers. Isoprenyl alkyl ethers are cleaved using iodine in dichloromethane,1188 and allyl alkyl ethers are cleaved with Lewis acids under various conditions.1189The OCH2CH=CHPh unit of mixed allyl ethers (O–CH2CH=CH2 and OCH2CH=CHPh) can be cleaved selectively under electrolytic conditions.1190
Cyclic ethers (usually THF derivatives) can be similarly cleaved (see Reaction 10-50 for epoxides). Treatment of 2-methyltetrahydrofuran with acetyl chloride and ZnCl2 gave primarily O-acetyl-4-chloro-1-pentanol.1191 A mixture of Et2NSiMe3/2 MeI cleaved THF to give the O-trimethylsilyl ether of 4-iodo-1-butanol.1192 Ethers have also been cleaved with Lewis acids [e.g., BF3, Ce(OTf)4,1193 SiCl4/LiI/BF3,1194 BBr3,1195 or AlCl3].1196 In such cases, the departure of the OR is assisted by complex formation with the Lewis acid (see 133). The reagent NaI–BF3 etherate selectively cleaves ethers in the order benzylic ethers > alkyl methyl ethers > aryl methyl ethers.1197
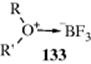
Dialkyl and alkyl aryl ethers are cleaved with Me3SiI1198: ROR′ + Me3SiI → RI + Me3SiOR.1199 A more convenient and less expensive alternative, which gives the same products, is a mixture of chlorotrimethylsilane and NaI.1200Triphenyldibromophosphorane (Ph3PBr2) cleaves dialkyl ethers to give 2 molar equivalents of alkyl bromide.1201 Alkyl aryl ethers can also be cleaved with LiI to give alkyl iodides and salts of phenols1202 in a reaction similar to Reaction 10-51. Allyl aryl ethers1203 are efficiently cleaved with NaI/Me3SiCl,1204 or NbCl5.1205 Aryl benzyl ethers are cleaved with BCl3 using pentamethylbenzene as a non-Lewis basic cation scavenger.1206 Cleavage in ionic liquids is also known.1207
A closely related reaction is cleavage of oxonium salts.
![]()
For these substrates, HX is not required, and X can be any of the four halide ions.
tert-Butyldimethylsilyl ethers (ROSiMe2CMe3) can be converted to bromides (RBr) by treatment with Ph3PBr2,1208 Ph3P–CBr4,1209 BBr3,1210 and CuBr2.1211 Alcohols are often protected by conversion to this kind of silyl ether.1212
OS I, 150; II, 571; III, 187, 432, 586, 692, 753, 774, 813; IV, 266, 321; V, 412; VI, 353. See also, OS VIII, 161, 556.
10-50 Formation of Halohydrins from Epoxides
(3)OC-seco -Halo-de-alkoxylation
This is a special case of Reaction 10-49 and is frequently used for the preparation of halohydrins.1213 In contrast to the situation with open-chain ethers and with larger rings, many epoxides react with all four hydrohalic acids, although with HF1214 the reaction is unsuccessful with simple aliphatic and cycloalkyl epoxides.1215 Hydrogen fluoride does react with more rigid epoxides, such as those in steroid systems. The reaction can be applied to simple epoxides1216 if polyhydrogen fluoride–pyridine is the reagent. The reagent NEt3•3HF converts epoxides to fluorohydrins with microwave irradiation.1217 Organocatalysts have been used to convert epoxides to fluorohydrins using an acetyl fluoride/fluorous alcohol combination.1218 Chloro-, bromo-, and iodohydrins can also be prepared1219 by treating epoxides with Ph3P and X2,1220 with 3/NaBr/H2O,1221 LiBr on Amberlyst-15 resin,1222 ceric ammonium nitrate/KBr,1223 I2 with a SmI2 catalyst,1224 and LiI on silica gel.1225 Epoxides can be converted directly to 1,2-dichloro compounds by treatment with SOCl2 and pyridine,1226 or with Ph3P and CCl4.1227 These are two-step reactions: a halohydrin is formed first and is then converted by the reagents to the dihalide (Reaction 10-48). As expected, inversion is found at both carbons. Meso epoxides were cleaved enantioselectively with the chiral B-halodiisopinocampheylboranes (see Reaction 15-16), where the halogen was Cl, Br, or I.1228 Diatomic iodine gives an iodohydrin with a 2,6-bis[2-(o-aminophenoxy)methyl]-4-bromo-1-methoxybenzene catalyst.1229 Epoxides are converted to the corresponding chlorohydrin upon treatment with the ionic liquid [AcMIm]Cl.1230
Bicyclic epoxides are usually opened to the trans-halohydrin. Unsymmetrical epoxides are usually opened to give mixtures of regioisomers. In a typical reaction, the halogen is delivered to the less sterically hindered carbon of the epoxide. In the absence of this structural feature, and in the absence of a directing group, relatively equal mixtures of regioisomeric halohydrins are expected. The phenyl is such a group, and in 1-phenyl-2-alkyl epoxides reaction with POCl3/DMAP (DMAP = 4-dimethylaminopyridine) leads to the chlorohydrin with the chlorine on the carbon bearing the phenyl.1231 When done in an ionic liquid with Me3SiCl, styrene epoxide gives 2-chloro-2-phenylethanol.1232 The reaction of thionyl chloride and poly(vinylpyrrolidinone) converts epoxides to the corresponding 2-chloro-1-carbinol.1233 Bromine with a phenylhydrazine catalyst, however, converts epoxides to the 1-bromo-2-carbinol.1234 An alkenyl group also leads to a halohydrin with the halogen on the carbon bearing the C=C unit.1235 Epoxy carboxylic acids are another example. When NaI reacts at pH 4, the major regioisomer is the 2-iodo-3-hydroxy compound, but when InCl3 is added, the major product is the 3-iodo-2-hydroxy carboxylic acid.1236
Acyl chlorides react with ethylene oxide in the presence of NaI to give 2-iodoethyl esters.1237
![]()
Acyl chlorides react with epoxides in the presence of a Eu(dpm)3 catalyst1238 [dpm = 1,1-bis(diphenylphosphino)methane] or a YCp2Cl catalyst1239 to give chloro esters.
A related reaction with episulfides leads to 2-chlorothio-esters.1240 Aziridines have been opened with PPh3 and halogenating agents,1241 and also by MgBr2 to give 2-haloamides in a related reaction1242N-Tosyl aziridines react with KF•2 H2O to give the 2-fluoro tosylamine product.1243 Aziridinium salts are opened by bromide ion.1244
OS I, 117; VI, 424; IX, 220.
10-51 Cleavage of Carboxylic Esters with Lithium Iodide
Iodo-de-acyloxy-substitution
![]()
Carboxylic esters, where R is methyl or ethyl, can be cleaved by heating with lithium iodide in refluxing pyridine or a higher-boiling amine.1245 The reaction is useful where a molecule is sensitive to acid and base (so that Reaction 16-59 cannot be used) or where it is desired to cleave selectively only one ester group in a molecule containing two or more. For example, refluxing O-acetyloleanolic acid methyl ester with LiI in s-collidine
cleaved only the 17-carbomethoxy group, not the 3-acetyl group.1246 Esters (RCO2R′) and lactones can also be cleaved with a mixture of Me3SiCl and NaI to give R′I and RCO2H.1247 The reaction of acetyl chloride and an allylic acetate leads to the allylic chloride.1248
10-52 Conversion of Diazo Ketones to α-halo Ketones
Hydro, halo-de-diazo bisubstitution
![]()
When diazo ketones are treated with HBr or HCl, they give the respective α-halo ketones. Hydrogeniodide does not give the reaction, since it reduces the product to a methyl ketone (Reaction 19-67). α-Fluoro ketones can be prepared by addition of the diazo ketone to polyhydrogen fluoride–pyridine.1249 This method is also successful for diazoalkanes.
Diazotization of α-amino acids in the above solvent at room temperature gives α-fluoro carboxylic acids.1250 If this reaction is run in the presence of excess KCl or KBr, the corresponding α-chloro or α-bromo acid is obtained instead.1251
OS III, 119.
10-53 Conversion of Amines to Halides
Halo-de-amination
![]()
Primary alkyl amines (RNH2) can be converted1252 to alkyl halides by (1) conversion to RNTs2 (Sec. 10.G.ii) and treatment of this with I− or Br− in DMF,415 or to N(Ts)–NH2 derivatives followed by treatment with NBS under photolysis conditions,1253 (2) diazotization with tert-butylnitrite and a metal halide (e.g., TiCl4 in DMF),1254 or (3) the Katritzky pyrylium–pyridinium method (Sec. 10.G.ii).1255 Alkyl groups can be cleaved from secondary and tertiary aromatic amines by concentrated HBr in a reaction similar to Reaction 10-49, for example,1256
![]()
Tertiary aliphatic amines are also cleaved by HI, but useful products are seldom obtained. Tertiary amines can be cleaved by reaction with phenyl chloroformate1257: R3N + ClCOOPh → RCl + R2NCOOPh. α-Chloroethyl chloroformate behaves similarly.1258 Alkyl halides may be formed when quaternary ammonium salts are heated: R4N+ X− → R3N + RX.1259
OS VIII, 119. See also, OS I, 428.
10-54 Conversion of Tertiary Amines to Cyanamides: The von Braun Reaction
Bromo-de-dialkylamino substitution
![]()
The von Braun reaction involves the cleavage of tertiary amines by cyanogen bromide to give an alkyl bromide and a disubstituted cyanamide, and can be applied to many tertiary amines.1260 Usually, the R group that cleaves is the one that gives the most reactive halide (e.g., benzyl or allyl). For simple alkyl groups, the smallest are the most readily cleaved. One or two of the groups on the amine may be aryl, but they do not cleave. Cyclic amines have been frequently cleaved by this reaction. Secondary amines also give the reaction, but the results are usually poor.1261
The mechanism consists of two successive nucleophilic substitutions, with the tertiary amine as the first nucleophile and the liberated bromide ion as the second:

The intermediate N-cyanoammonium bromide has been trapped, and its structure confirmed by chemical, analytical, and spectral data.1262 The BrCN in this reaction has been called a counterattack reagent; that is, a reagent that accomplishes, in one flask, two transformations designed to give the product.1263
OS III, 608.
10.H.vi. Carbon Nucleophiles
In any heterolytic reaction in which a new carbon–carbon bond is formed,1264 one carbon atom attacks as a nucleophile and the other as an electrophile. The classification of a given reaction as nucleophilic or electrophilic is a matter of convention and is usually based on analogy. Although not discussed in this chapter, Reactions 11-8–11-25 and 12-16–12-21 are nucleophilic substitutions with respect to one reactant, but following convention, we classify them with respect to the other. Similarly, all the reactions in this section would be called electrophilic substitution (aromatic or aliphatic) if we were to consider the reagent as the substrate.
In Reactions 10-56–10-65, the nucleophile is a “carbanion” part of an organometallic compound, often a Grignard reagent. There is much that is still not known about the mechanisms of these reactions and many of them are not nucleophilic substitutions at all. In those reactions that are nucleophilic substitutions, the attacking carbon brings a pair of electrons with it to the new C–C bond, whether or not free carbanions are actually involved. The connection of two alkyl or aryl groups is called coupling. Reactions 10-56–10-65 include both symmetrical and unsymmetrical coupling reactions. The latter are also called cross-coupling reactions. Other coupling reactions are considered in later chapters.
10-55 Coupling with Silanes
De-silylalkyl-coupling
![]()
Organosilanes (RSiMe3 or RSiMe2F, where R can be vinylic, allylic, or alkynyl) couple with vinylic, allylic, and aryl bromides and iodides (R′X), in the presence of certain catalysts, to give RR′ in good yields.1265 Allylsilanes react with allylic acetates in the presence of iodine.1266 The transition metal catalyzed coupling of silanes, particularly allyl silanes, is a mild method for incorporating alkyl fragments into a molecule.1267 Here PhSiMe2Cl couples to give biphenyl in the presence of CuI and Bu4NF,1268 and vinyl silanes react with allylic carbonates and a palladium catalyst to give dienes.1269 Allylsilanes have been coupled to substrates containing a benzotriazole unit, in the presence of BF3•etherate.1270 One variation used a silylmethyltin derivative in a palladium-catalyzed coupling with aryl iodides.1271 Homoallyl silanes coupled to Ph3BiF2 in the presence of BF3•OEt2 to give the phenyl coupling product.1272
α-Silyloxy methoxy derivatives [RCH(OMe)OSiR13], react with allyltrimethylsilane (Me3SiCH2CH=CH2) in the presence of TiX4 derivatives to give displacement of the OMe group and RCH(OSiR13)CH2CH=CH2).1273 A tertiary silyloxy group was displaced by allyl in the presence of ZnCl2.1274 Allylic acetates react with Me3SiSiMe3 and LiCl with a Pd catalyst to give the allyl silane.1275 The RSiF3 reagents can also be used in coupling reaction with aryl halides.1276 Allyl silanes react with epoxides, in the presence of BF3•OEt2 to give 2-allyl alcohols.1277 The reaction of α-bromo lactones and CH2=CHCH2Si(SiMe3)3 and azoisobutyronitrile (AIBN) leads to the α-allyl lactone.1278
Silyl epoxides have been prepared from epoxides via reaction with sec-butyllithium and chlorotrimethylsilane.1279 α-Silyl-N-Boc-amines were prepared in a similar manner from the N-Boc-amine.1280 Benzyl silanes coupled with allyl silanes to give ArCH2–R derivatives in the presence of VO(OEt)Cl21281 and allyltin compounds couple with allyl silanes in the presence of SnCl4.1282 Allyl silanes couple to the α-carbon of amines under photolysis conditions.1283
Arylsilanes were prepared by reaction of an aryllithium intermediate with TfOSi(OEt)3.1284 In the presence of BF3•etherate, allyl silane and α-methoxy N-carbobenzoxy (N-Cbz) amines were coupled.1285 Aryl cyanides have been converted to arylsilanes using a Rh catalyst and Me3SiSiMe3.1286
The reaction of a vinyl iodide with (EtO)3SiH and a Pd catalyst generated a good yield of the corresponding vinylsilane.1287
OSCV 10, 531.
10-56 Coupling of Alkyl Halides: The Wurtz Reaction
De-halogen-coupling
![]()
The coupling of alkyl halides by treatment with sodium to give a symmetrical product is called the Wurtz reaction. Side reactions (elimination and rearrangement) are so common that the reaction is seldom used. Mixed Wurtz reactions of two alkyl halides are even less feasible because of the number of products obtained. A somewhat more useful reaction (but still not very good) takes place when a mixture of an alkyl and an aryl halide is treated with sodium to give an alkylated aromatic compound (the Wurtz–Fittig reaction).1288 However, the coupling of two aryl halides with sodium is impractical (but see Reaction 13-11). Other metals have also been used to effect Wurtz reactions,1289 notably Ag, Zn,1290 Fe,1291 activated Cu,1292 In,1293 La,1294 and Mn compounds.1295 Lithium, under the influence of ultrasound, has been used to couple alkyl, aryl, and benzylic halides.1296
In a related reaction, Grignard reagents (Reaction 12-38) have been coupled in the presence of trifluorosulfonic anhydride.1297 Tosylates and other sulfonates and sulfates couple with Grignard reagents,1298 most often those prepared from aryl or benzylic halides.1299 Alkyl sulfates and sulfonates generally make better substrates in reactions with Grignard reagents than the corresponding halides (Reaction 10-57). The method is useful for primary and secondary R.
One type of Wurtz reaction that is quite useful is the closing of small rings, especially three-membered rings.1300 For example, 1,3-dibromopropane can be converted to cyclopropane by Zn and NaI.1301 Two highly strained molecules prepared this way are bicyclobutane1302 and tetracyclo[3.3.1.13,7.01,3]decane.1303 Three- and four-membered rings can also be closed in this manner with certain other reagents,1304 including benzoyl peroxide,1305t-BuLi,1306 and lithium amalgam,1307 as well as electrochemically.1308 The Pd or Ni catalyzed cross-coupling reaction of a Grignard reagent and an alkyl halide, is often called Kumada coupling.1309
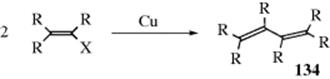
Vinylic halides can be coupled to give 1,3-butadienes (134) by treatment with activated Cu powder in a reaction analogous to the Ullmann Reaction (13-11).1310 This reaction is stereospecific, with retention of configuration at both carbons. Vinylic halides can also be coupled1311 with Zn–NiCl2,1312 and with n-BuLi in ether in the presence of MnCl2.1313 The coupling reaction with vinyltin reagents and vinyl halides occurs with a Pd catalyst.1314
It seems likely that the mechanism of the Wurtz reaction consists of two basic steps. The first is halogen–metal exchange to give an organometallic compound (RX + M → RM), which in many cases can be isolated (Reaction 12-38). Following this, the organometallic compound reacts with a second molecule of alkyl halide (RX + RM → RR). This reaction and its mechanism are considered in Section (Reaction 10-57).
OS III, 157; V, 328, 1058; VI, 133, 153.
A variation of the Wurtz coupling uses other metals to mediate or facilitate the coupling. In certain cases, such variations can be synthetically useful. Because of the presence of the 1,5-diene moiety in many naturally occurring compounds, methods that couple1315 allylic groups1316 are quite important. In one of these methods, allylic halides, tosylates, and acetates can be symmetrically coupled by treatment with nickel carbonyl1317 to give 1,5-dienes.1318The order of halide reactivity is I > Br > Cl. With unsymmetrical allylic substrates, coupling nearly always takes place at the less-substituted end.
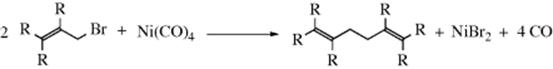
The reaction can be performed intramolecularly; large (11–20-membered) rings can be made in good yields (60–80%) by the use of high dilution.1319 The mechanism of coupling likely involves reaction of the allylic compound with Ni(CO)4 to give one or more π-allyl complexes, one of which may be the η3-complex 135. Loss of CO to give a π-allylnickel bromide (136) and ligand transfer leads to coupling and the final product. In some cases, the η3-complexes (136) can be isolated from the solution and crystallized as stable solids.
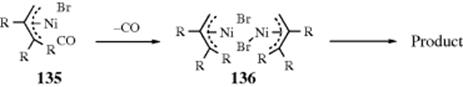
Unsymmetrical coupling can be achieved by treating an alkyl halide directly with 136, in a polar aprotic solvent,1320 and coupling occurs at the less substituted end. There is evidence that free radicals are involved in such couplings.1321 Hydroxy or carbonyl groups in the alkyl halide do not interfere. When 136 reacts with an allylic halide, a mixture of three products is obtained because of halogen–metal interchange. For example, allyl bromide treated with 136 prepared from methallyl bromide gave an approximately statistical mixture of 1,5-hexadiene, 2-methyl-1,5-hexadiene, and 2,5-dimethyl-1,5-hexadiene.1322 A symmetrical coupling of allylic tosylates used Ni(CO)4.

Symmetrical coupling of allylic halides occurs by heating with magnesium in ether.1323 The coupling of two different allylic groups has been achieved by treatment of an allylic bromide with an allylic Grignard reagent in THF containing HMPA,1324 or with an allylic tin reagent.1325 This type of coupling can be achieved with almost no allylic rearrangement in the substrate (and almost complete allylic rearrangement in the reagent) by treatment of allylic halides with lithium allylic boron ate complexes (RCH=CHCH2B–R23Li+).1326 The reaction between primary and secondary halides and allyltributylstannane provides another method for unsymmetrical coupling RX + CH2=CHCH2SnBu3 → RCH2CH=CH2.1327
In another method for the coupling of two different allylic groups,1328 a carbanion derived from a β,γ-unsaturated thioether couples with an allylic halide to give 137.1329 The product (137) contains an SPh group that must be removed (with Li in ethylamine) to give the 1,5-diene. Unlike most of the methods previously discussed, this method has the advantage that the coupling preserves the original positions and configurations of the two double bonds; no allylic rearrangements take place.
Treatment of conjugated ketones with SmI2 in HMPA gave the coupled diketone via Wurtz-type coupling.1330
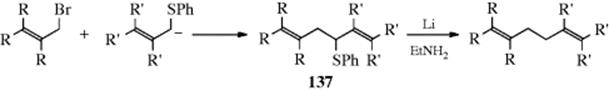
OS III, 121; IV, 748; VI, 722.
10-57 The Reaction of Alkyl Halides and Sulfonate Esters with Group 1 and 2 Organometallic Reagents1331
Alkyl-de-halogenation
![]()
A variety of Group 1 and 2 organometallic compounds1332 couple with alkyl halides.1333 Organosodium and organopotassium compounds are more reactive than Grignard reagents, and couple even with less reactive halides (see below). Coupling of organolithium compounds with alkyl halides1334 or aryl halides1335 is possible.1336 Unactivated aryl halides couple with alkyllithium reagents in THF.1337 The reaction of n-butyllithium/TMEDA with a homoallylic alcohol [CH2=C(Me)CH2CH2OH] leads to the allyllithium reagent, and subsequent reaction with an alkyl halide gives the substituted homoallylic alcohol [CH2=C(CH2R)CH2CH2OH].1338 Organolithium reagents exhibit an important side reaction: They react with ether solvents, and their half-life in such solvents is known.1339 With highly reactive organolithium reagents, preparing and keeping them long enough for the alkyl halide to be added is sometimes a problem (but usually not with simple primary organolithium reagents). Alkenes can be prepared by the coupling of vinylic lithium compounds with primary halides1340 or of vinylic halides with alkyllithium reagents in the presence of a Pd or Ru catalyst.1341 α-Lithioepoxides can also be formed, and reaction with an alkyl halide gives the substituted epoxide.1342 Arylsilanes (e.g., 2-trimethylsilylpyridine) undergo a deprotonation reaction of a silyl methyl group when treated with tert-butyllithium to give the corresponding ArMe2SiCH2Li reagent.1343 Subsequent reaction with an alkyl halide leads to the substituted silane. Organolithium reagents formed by Li–H exchange in the presence of (–)-sparteine couple with alkyl halides with high asymmetric induction.1344 Exchange of organotin compounds with organolithium reagents generates a new organolithium, and in one case intramolecular coupling in the presence of (–)-sparteine led to chiral pyrrolidine derivatives.1345 Propargyl lithium reagents formed in the presence of mercuric salts couple with halides.1346 Note that 1-lithioalkynes were coupled to alkyl halides in the presence of a palladium catalyst.1347
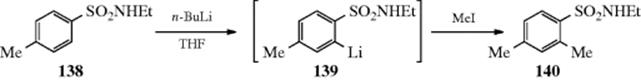
Aryllithium reagents are formed by metal–halogen exchange with aryl halides or H–metal exchange with various aromatic compounds, and they react with alkyl halides. The reaction of 138 with n-butyllithium, for example, generated the aryllithium (139), which reacted with iodomethane to give 140.1348 When an aromatic ring has an attached heteroatom or an heteroatom-containing substituent, reaction with a strong base (e.g., an organolithium reagent) usually leads to an ortho lithiated species.1349 Subsequent reaction with an electrophilic species gives the ortho substituted product. This phenomenon is known as directed ortho metalation (See 13-17). This selectivity was discovered independently by Gilman and by Wittig in 1939–1940, when anisole was found to give ortho deprotonation in the presence of butyllithium.1350 Alkylation ortho to a carbonyl is possible, and treatment of the acyl hydrazide [PhC(=O)NHNMe2] with sec-butyllithium and then iodoethane gave the ortho ethyl derivative.1351 Note that aminonaphthalene derivatives were reacted with tert-butyllithium and aryllithium formation occurred on the ring distal to the amino group, and subsequent reaction with iodomethane gave methylation on that ring.1352
In a method for propargylating an alkyl halide without allylic rearrangement, the halide is treated with lithio-1-trimethylsilylpropyne (141), which is a lithium compound protected by an SiMe3 group.1353 Reaction at its 1 position (which gives an allene) takes place only to a small extent, because of steric blockage by the large SiMe3 group. The SiMe3 group is easily removed by treatment with Ag+ followed by CN−. Propargyl derivative 141 is prepared by treating propynyllithium with Me3SiCl to give MeC![]() CSiMe3 from which a proton is removed with BuLi. The R group may be primary or allylic.1354 On the other hand, propargylic halides can be alkylated with essentially complete allylic rearrangement, to give allenes, by treatment with Grignard reagents and metallic salts,1355 or with dialkylcuprates (R2Cu).1356
CSiMe3 from which a proton is removed with BuLi. The R group may be primary or allylic.1354 On the other hand, propargylic halides can be alkylated with essentially complete allylic rearrangement, to give allenes, by treatment with Grignard reagents and metallic salts,1355 or with dialkylcuprates (R2Cu).1356
![]()
Grignard reagents are generally unreactive with alkyl halides unless allylic and benzylic reagents and substrates are used.1357 Grignard reagents have the advantage that they are usually simpler to prepare than the corresponding R'2CuLi (see Reaction 10-58), but the reaction is much narrower in scope. Grignard reagents couple only with active halides: allylic (though allylic rearrangements are common) and benzylic. They also couple with tertiary alkyl halides, but generally in low or moderate yields.1358
Allylic halides are more reactive than aliphatic alkyl halides, but Cu salts have been used to facilitate coupling with alkylmagnesium halides.1359 Indeed, Grignard reagents may react with alkyl halides in the presence of certain metal catalysts,1360 and stereocontrol is possible in these reactions.1361 These catalysts include Cu(I) compounds (see Reaction 10-58),1362 Ag compounds,1363 Pd1364 complexes, Co compounds,1365 Fe compounds,1366 and an Fe–amine complex was shown to catalyze Grignard coupling reactions.1367 Iron nanoparticles have also been employed to facilitate this type of coupling.1368 Alkyl triflates have been used rather than alkyl halides.1369 Chiral Cu complexes have been used with allylic halides to give rearranged alkylated products, with high enantioselectivity.1370 A similar reaction was reported using a Grignard reagent and a chiral imidazolium carbene complex.1371 As noted above, Grignard reagents react with allylic substrates, but if there is steric hindrance at the carbon bearing the leaving group, the reaction may proceed by an SN2' pathway (Sec. 10.E).1372
Aryl halides, even when activated, generally do not couple with Grignard reagents, although certain transition metal catalysts do effect this reaction in variable yields,1373 including V compounds.1374 The reaction with Grignard reagents proceeds better when OR can be the leaving group, providing that activating groups are present in the ring. Aryl triflates couple with arylmagnesium halides in the presence of a Pd catalyst,1375 as do vinyl halides with RMgX with a Pd1376 or Ni catalyst.1377 Alkyl halides are coupled to arylmagnesium bromides in the presence of a Co catalyst.1378 It is also possible to couple alkynylmagnesium halides with aryl iodides in the presence of Pd catalysts.1379 A silica-supported phosphine–Pd complex was used to couple arylmagnesium halides with aryl iodides.1380 Aryl Grignard reagents couple with alkyl halides, including neopentyl iodide, in the presence of ZnCl2 and a Ni catalyst.1381
Vinyl1382 and aryl halides1383 also couple with alkyl Grignard reagents in the presence of a catalytic amount of an Fe catalyst,1384 as do vinyl triflates with CuI1385 or vinyl halides with a Co catalyst.1386 Grignard reagents prepared from primary or secondary1387 alkyl or aryl halides can be coupled with vinylic or aryl halides (see Reaction 13-9) in high yields in the presence of a Ni(II) catalyst.1388 When a chiral Ni(II) catalyst is used, optically active hydrocarbons can be prepared from achiral reagents.1389 The Pd catalyzed coupling of arylmagnesium halides and vinyl bromides has also been reported.1390
Because Grignard reagents react with the C=O group (Reaction 16-24 and 16-82), they cannot be used to couple with halides containing ketone, CO2R, or amide functions. Although the coupling of Grignard reagents with ordinary alkyl halides is usually not useful for synthetic purposes, small amounts of symmetrical coupling product are commonly formed while Grignard reagents are being prepared.
For symmetrical coupling of organometallic reagents (2RM → RR), see Reaction 14-24 and 14-25.
Much study has been devoted to the mechanisms of these reactions,1391 but firm conclusions are still lacking, in part because the mechanisms vary depending on the metal, the R group, the catalyst, if any, and the reaction conditions. Two basic pathways can be envisioned: a nucleophilic substitution process (which might be SN1 or SN2) and a free radical mechanism. This could be an SET pathway, or some other route that provides radicals. In either case, the two radicals R• and R′• would be in a solvent cage:
![]()
It is necessary to postulate the solvent cage because, if the radicals were completely free, the products would be ~ 50% RR′, 25% RR, and 25% R′R′. This is generally not the case; in most of these reactions RR′ is the predominant or exclusive product.1392 An example where an SN2 mechanism has been demonstrated (by the finding of inversion of configuration at R) is the reaction between allylic or benzylic lithium reagents with secondary halides.1393 The fact that in some of these cases the reaction can be successfully applied to aryl and vinylic substrates indicates that a simple SN process cannot be the only mechanism. One possibility is that the reagents first undergo an exchange reaction: ArX + RM → RX + ArM, and then a nucleophilic substitution takes place. On the other hand, there is much evidence that many coupling reactions involving organometallic reagents with simple alkyl groups occur by free radical mechanisms. Among the evidence1394 is the observation of CIDNP in reactions of alkyl halides with simple organolithium reagents1395 (see Sec. 5.C.i), the detection of free radicals by ESR spectroscopy1396 (Sec. 5.C.i), and the formation of 2,3-dimethyl-2,3-diphenylbutane when the reaction was carried out in the presence of cumene1397 (this product is formed when a free radical abstracts a hydrogen from cumene to give PhCMe2, which dimerizes). Evidence for free radical mechanisms has also been found for the coupling of alkyl halides with simple organosodium compounds (Wurtz),1398 with Grignard reagents,1399 and with lithium dialkylcopper reagents (see Reaction 10-58).1400 Free radicals have also been implicated in the metal–ion catalyzed coupling of alkyl and aryl halides with Grignard reagents.1401
OS I, 186; III, 121; IV, 748; VI, 407; VII, 77, 172, 326, 485; VIII, 226, 396; IX, 530; X, 332, 396.
10-58 Reaction of Alkyl Halides and Sulfonate Esters with Organocuprates
Alkyl-de-halogenation
![]()
The reagents lithium dialkylcopper1402 (known as lithium dialkyl cuprates, also called Gilman reagents)1403 react with alkyl bromides, chlorides, and iodides in ether or THF to give good yields of the cross-coupling products.1404They are prepared (see Reaction 12-36) by the reaction of an organolithium compound with CuI or CuBr, but other Cu(I) compounds can be used.1405 They are usually generated at temperatures <0 °C due to the thermal instability of any dialkyl cuprate that has a hydrogen atom on a carbon that is β- to the Cu.
The reaction with alkyl halides is of wide scope1406 and R in R2CuLi may be primary alkyl, allylic, benzylic, aryl, vinylic, or allenic, and may contain keto, CO2H, CO2R, or CONR2 groups.1407 The mechanism of these reactions probably involves formation of a Cu(II) intermediate.1408 Reaction with allylic substrates usually proceeds with high selectivity for the γ-position,1409 in SN2'-type reactions.1410
Inversion of configuration has been shown in the reaction of 2-bromobutane with Ph2CuLi,1411 but the same reaction with 2-iodobutane was reported to proceed with racemization.1412 The reaction at a vinylic substrate occurs stereospecifically, with retention of configuration.1413 Many gem-dihalides do not react, but when the two halogens are on a carbon α to an aromatic ring1414 or on a cyclopropane ring,1415 both halogens can be replaced by R (e.g., PhCHCl2 → PhCHMe2). However, 1,2-dibromides give exclusive elimination (Reaction 17-22).1416 Vinylmagnesium halides, upon addition of a catalytic amount of Li2CuCl4, couple to alkyl halide.1416
Lithium dialkylcopper reagents couple with alkyl tosylates.1417 High yields are obtained with primary tosylates; secondary tosylates give lower yields,1418 but aryl tosylates do not react. Vinylic triflates1419 couple very well to give alkenes1420 and they also couple with allylic cuprates, to give 1,4-dienes.1421 Propargylic tosylates couple with vinylic cuprates to give vinylic allenes.1422
The R′ in R'2CuLi may be primary alkyl, vinylic, allylic, or aryl. Thus, in the reaction so far described, the alkyl groups on the organocuprate or the alkyl halide may not be secondary or tertiary alkyl. However, secondary and tertiary alkyl coupling can be achieved (on primary RX) by the use of R'2CuLi•PBu21423 (but this procedure introduces problems in the workup) or by the use of PhS(R′)CuLi,1424 which selectively couples a secondary or tertiary R′ with a primary iodide (RI) to give RR′.1425 It is possible to prepare mixed cuprates, where one ligand is tightly bound to the copper, allowing the other ligand to be transferred in a coupling reaction. A common example is adds a 2-thienyl group to the cuprate to give R(Th)CuLi, where the R group is transferred in lieu of the thienyl unit.1426 A lithium neopentyl aryl cuprate selectively transferred from an aryl group to an allylic halide.1427
Coupling to a secondary alkyl halide (R in RX above = secondary) can be achieved in high yield with the reagents R'2Cu(CN)Li2,1428 where R′ is primary alkyl or vinylic (but not aryl).1429 This modified reagent is commonly known as a higher order mixed cuprate. The reagents RCu(PPh2)Li, RCu(NR'2)Li, and RCu(PR'2)Li (R′ = cyclohexyl) are more stable than R2CuLi and can be used at higher temperatures.1430 These reagents are rather reactive. Unactivated aryl triflates1431 (ArOSO2CF3) react to give ArR in good yields when treated with R2Cu(CN)Li2,1432 with R3Al,1433 or with R3SnR and a Pd complex catalyst.1434 See Reaction 10-59 for other examples involving Al, Sn, and Pd coupling reactions. Both OTf units in RCH(OTf)2 can be replaced with Me2(CN)CuLi2.1435 With an allenic substrate, reaction with R(CN)CuLi can give ordinary displacement (with retention of configuration)1436 or an SN2′ reaction to produce an alkyne.1437 In the latter case, a chiral allene (see Sec. 4.C, category 5) gave a chiral alkyne. The structures of these “higher order mixed” cuprates has been called into question1438 by Bertz,1439 who suggested the reagent actually existed as R2CuLi•LiCN in THF. This was contradicted by Lipshutz and James.1440
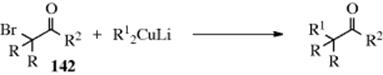
The fact that R'2CuLi do not react with ketones provides a method for the alkylation of ketones via the organocuprate coupling with α-haloketones (e.g., 1421441; see also, Reactions 10-68 and 10-73). Note that halogen–metal exchange (Reaction 12-39) is a side reaction and can become the main reaction.1442 When α,α′-dibromo ketones are treated with Me2CuLi in ether at −78 °C and the mixture quenched with methanol, monomethylation takes place1443(no dimethylation is observed). It has been suggested that the reaction involves cyclization (10-56) to a cyclopropanone followed by nucleophilic attack to give the enolate anion, which is protonated by the methanol. If iodomethane is added instead of methanol, an α,α′-dimethyl ketone is obtained, presumably from SN2 attack (Reaction 10-68). Primary, secondary, and tertiary monoalkylation can be achieved with a lithium tert-butoxy(alkyl)copper reagent1444instead of Me2CuLi, one of the few methods for introducing a tertiary alkyl group to a carbonyl group.
When dialkylcopperzinc reagents (R2CuZnCl) couple with allylic halides, allylic rearrangement occurs (SN2′) almost completely, and the reaction is diastereoselective if the allylic halide contains a δ alkoxy group.1445 Another type of copper reagent was prepared from RZnI/CuCN, and was shown to couple with alkenyl halides.1446 Diethylzinc in the presence of a catalytic amount of CuBr coupled to allylic chlorides.1447 When treated with organocopper compounds and Lewis acids (e.g., n-BuCu•BF3), allylic halides give substitution with almost complete allylic rearrangement, independently of the degree of substitution at the two ends of the allylic system.1448
OS IX, 502.
10-59 Reaction of Alkyl Halides and Sulfonate Esters with Other Organometallic Reagents
Alkyl-de-halogenation
![]()
In addition of Mg, Li, and Cu, other metals and metal complexes can be used to catalyze or mediate coupling reactions. Organoaluminum compounds couple very well with tertiary (to give products containing a quaternary carbon) and benzylic halides at −78 °C.1449 This reaction can also be applied to allylic, secondary, and some primary halides, but several days standing at room temperature is required (see also, Reaction 10-63). Vinylic aluminum compounds (in the presence of a suitable transition metal catalyst) couple with allylic halides, acetates, and alcohol derivatives to give 1,4-dienes,1450 and with vinylic and benzylic halides to give 1,3-dienes and allylic arenes, respectively.1451 Note that alkylboronic acids are coupled in the presence of Ag2O and a catalytic amount of CrCl2 to give the symmetrical alkyl derivative.1452
Products containing a quaternary carbon can also be obtained by treatment of tertiary halides with dialkyl or diaryl zinc reagents in CH2Cl2,1453 with Me3Si and AlCl3,1454 or with alkyltitanium reagents (RTiCl3 and R2TiCl2).1455Alkyl or aryl triflates (halides) couple with alkyl or ArZn(halide) reagents in the presence of a Pd catalyst.1456 This organozinc coupling reaction has been done in ionic liquids.1457 Vinyl halides can be coupled with vinyltin reagents in the presence of CuI,1458 and aryl tin compounds couple with vinyl halides1459 or vinyl triflates when a Pd catalyst is present.1460 When the vinyltin reagent is coupled with a vinyl triflate in the presence of a Pd catalyst, the reaction is known as the Stille reaction (Reaction 12-15). In the Stille reaction, vinylic triflates, in the presence of a Pd catalyst and LiCl, couple with organotin compounds (R′SnMe3), where R′ can be alkyl, allylic, vinylic, or alkynyl.1461The reaction has been performed intramolecularly, to prepare large-ring lactones.1462
The coupling of alkyl or alkenyl halides and an organozinc compound with a Ni complex has come to be known as Negishi coupling.1463 Several variations have been reported over the years. Arylzinc compounds1464 have been used, and also arylvinyl iodides.1465 The structure of bis(iodozincio)methane in THF solution has been reported.1466 Pyridylzinc compounds have been used in Negishi coupling.1467 Carbonylative cross coupling reactions have been reported.1468 Palladium-catalyzed variations are also known for alkyl or vinyl halides with organozinc compounds.1469 Dialkylzinc compounds can be coupled to alkyl halides in the presence of a nickel catalyst,1470 but with geminal diiodo compounds without a catalyst.1471 Asymmetric variations are known using various chiral additives or chiral catalysts,1472 including the example shown for an allylic chloride (DMA = dimethylactamide).1473 Coupling with propargylic substrates has also been reported.1474

Copper compounds can also be used as catalysts with dialkylzinc reagents.1475 The reaction of aryl halides with Me4ZnLi2 and then VO(OEt)Cl2 leads to the methylated aryl.1476 Isopropylzinc (iPrZn) displaces the iodide in γ-iodo ketones to give the alkyl substitution product, without reaction at the carbonyl.1477 Reactions of organozinc reagents with a carbonyl compound via acyl addition is presented in 16-31 (the Reformatsky reaction). Tertiary halides have also been coupled to allyltin reagents in the presence of AIBN.1478 Alkyl halides can be treated with SmI2 and then CuBr to give a reactive species that couples with other alkyl halides.1479 Trialkylindium compounds couple to allylic bromides in the presence of Cu(OTf)2•P(OEt)31480 and vinyl indium compounds are coupled to α-halo esters with a BEt3 catalyst.1481 Arylsulfonyl chlorides couple with allyl halides in the presence of bismuth to give allyl-aryls.1482Vinyl iodides couple with RMnCl with an iron catalyst1483 and Bu3MnMgBr reacted with a geminal dibromocyclopropane to give a dialkylated cyclopropane.1484 α-Haloketones are coupled with aryl halides using a Ni catalyst.1485Allylgallium reagents have been coupled to α-bromo esters in the presence of BEt3/O2.1486
Arylpalladium salts (ArPdX) prepared from arylmercury compounds and lithium palladium chloride couple with allylic chlorides in moderate yields, although allylic rearrangements can occur.1487 In most cases, better yields are obtained by addition of a Pd complex to the substrate, sometimes in conjunction with another metal, to facilitate coupling. Under these conditions, any arylpalladium species is generated in situ. Allylic, benzylic, vinylic, and aryl halides or triflates (trifluoromethylsulfonates) couple with organotin reagents in a reaction catalyzed by Pd complexes.1488 The advantage of this procedure is that the aryl group may contain nitro, ester, or aldehyde groups, and so on, which cannot be present in a Grignard reagent. Such functional groups as CO2R, CN, OH, and CHO may be present in either reagent, but the substrate may not bear a β hydrogen on an sp3 carbon, because that results in elimination. Indium metal has been used to mediate the coupling of an allylic halide and an arylpalladium complex.1489 Organoindium compounds were coupled to 1-iodonaphthalene with a Pd catalyst.1490 Aryl halides were also coupled to allylic silanes in the presence of a Pd catalyst.1491
Dimethylzinc was coupled to aryl halides with a Pd catalyst,1492 and Reformatsky-type zinc derivatives (see Reaction 16-28) have been coupled to aryl halides using a Pd catalyst and microwave irradiation.1493 Alkyl halides couple with ArMnCl or RMnCl in the presence of a Pd catalyst.1494 Cobalt-catalyzed coupling reactions are known.1495
In many cases, the organometallic reagent is prepared from the corresponding organolithium reagent (Reaction 10-57), as in the conversion of an aryllithium to an arylzirconium reagent, which was subsequently coupled to an aryl halide in the presence of a Pd catalyst.1496 Vinylzirconium reagents can be coupled to allylic halides in the presence of Cu(I) compounds.1497
Alkylboranes are coupled to alkyl halides in the presence of a Ni catalyst.1498
OS VII, 245; VIII, 295; X, 391.
10-60 Coupling of Organometallic Reagents with Carboxylic Esters
Alkyl-de-acyloxy-substitution
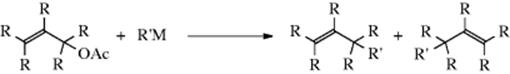
Several organometallic reagents react with allylic esters and carbonates to give the coupling product. Lithium dialkylcopper reagents couple with allylic acetates to give normal coupling products or those resulting from allylic rearrangement, depending on the substrate.1499 A mechanism involving a σ-allylic copper(III) complex has been suggested.1500 Silyl cuprates have also been used, with benzoate esters, to give allyl silanes.1501 Interestingly, allylic silanes have been coupled to acetates using B(C6F5)31502 or BF3.1503
![]()
Allenes are obtained when propargyl acetates are treated with methylmagnesium iodide.1504 Lithium dialkylcopper reagents give normal coupling products with enol acetates of β-dicarbonyl compounds.1505 It is also possible to carry out the coupling of allylic acetates with Grignard reagents, if catalytic amounts of cuprous salts are present.1506 Yields are better with this method, and regioselectivity can be controlled by the choice of cuprous salts.
Several metal-catalyzed coupling reactions are known. Allylic, benzylic, and cyclopropylmethyl acetates couple with trialkylaluminums,1507 and allylic acetates couple with aryl and vinylic tin reagents, in the presence of a Pd catalyst1508 (see below). Allylic acetates can be symmetrically coupled by treatment with Ni(CO)4 (Reaction 10-56) or with Zn and a Pd–complex catalyst,1509 or converted to unsymmetrical 1,5-dienes by treatment with an allylic stannane (R2C=CHCH2SnR3) in the presence of a Pd complex.1510 Other Ni(0) coupling reactions are known.1511 Titanium-mediated1512 coupling, Ir catalyzed,1513 and Fe catalyzed1514 reactions are also known. Aryl halides can be coupled to allylic acetates with CoBr2/Mn/FeBr2.1515 Allylic phosphonates have been used as substrates for displacement by higher order cuprates1516 (see Reaction 10-58) or dialkylzinc reagents.1517
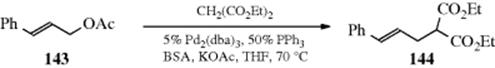
A common method is the reaction of η3-π-allyl palladium complexes1518 (see Sec 3.C.i) with various nucleophiles,1519 where the complex is obtained from allylic esters (acetate is the most common) or allylic carbonates (also see Reaction 10-31). This coupling reaction is often called the Tsuji–Trost reaction.1520 The mechanism of such π-allyl palladium reactions has been discussed.1521 The structure and nature of the ligands associated with the metal are important to the reaction, particularly with respect to stereoselectivity of a given reaction.1522 A typical transformation is shown for the reaction of 143 with diethyl malonate, BSA (N,O-bis(trimethylsilyl)acetamide), and potassium acetate, which gives coupling product 144 in the presence of the Pd catalyst.1523 This reaction is a variation of the basic transformation reported several years ago by Trost et al.1524 Enolate anions of active methylene compounds1525and also sulfone anions1526 have been used as nucleophiles most of the time. In most reported cases, the R′M species is the anion of an active methylene compound (e.g., sodium, potassium, or lithium dimethylmalonate) or Knoevenagel-type carbanions (see Reaction 16-38) or amino acid surrogates.1527 Enolate anions (see Reaction 10-68) have also been used.1528 Other nucleophiles can be used to displace allylic acetates.1529 The Pd catalyst used, the reaction conditions, and the nature of the organometallic compounds varies widely. Although two allylic coupling products are possible via the π-allyl intermediate, attack at the less substituted position is generally favored. This transformation has been done in ionic liquids1530 and ionic liquids have been used as additives in catalytic amounts in other solvents.1531 Palladium nanoparticles have been used to catalyze the reaction.1532 The SN2′ reactions with allylic substrates have been reported.1533 Benzoate esters have been used successfully in lieu of the acetate.1534 Catalyst metals other than Pd have been used for this reaction with allylic acetates.1535
The use of chiral ligands1536 or chiral additives that may act as ligands1537 lead to asymmetric induction in the coupling product.1538
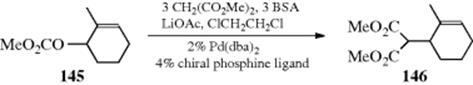
As mentioned above, a common variation is to replace the acetate leaving group with a carbonate (–OCO2R), where methyl carbonate (–OCO2Me) is most common.1539 A representative reaction is the transformation of 145 to 146,1540 where the use of a chiral ligand led to modest asymmetric induction. Indeed, as with allylic acetates, chiral ligands and chiral additives lead to asymmetric induction.1541 A variety of active methylene compounds can be used as nucleophiles,1542 including enolate anions.1543 Other nucleophiles can be used to displace allylic carbonates,1544 often in conjunction with chiral ligands to give the product with enantioselectivity. Polymer-supported phosphine ligands have been used successfully,1545 and catalyst systems other than Pd have been used for this reaction with allylic carbonates.1546. Potassium vinyltrifluoroborates (Reaction 10-73) have also been used in Pd catalyzed coupling reactions with allylic acetates.1547
Intramolecular cyclization is possible when the active methylene compound and an allylic acetate or carbonate is incorporated into the same molecule.1548 Propargylic esters have been used in Pd catalyzed coupling reactions, including a reaction with trialkylindium reagents.1549
10-61 Coupling of Organometallic Reagents with Sulfate Esters, Sulfoxides, Sulfones, Nitro, and Acetals
Alkyl-de-sulfonyl and de-sulfonyloxy-substitution, and so on;Alkyl-de-alkoxy-substitution, and so on;Alkyl-de-nitration, and so on
![]()
Leaving groups other than halide, esters or carbonate, or sulfonate esters are sometimes used. Sulfates, sulfonates, and epoxides give the expected products. The reactions of sodium sulfonates and alkyl halides in ionic liquids have been reported.1550 The SO2Ph group of allylic sulfones can be a leaving group if a Pd complex is present.1551 The NR2 group from Mannich bases (e.g., RCOCH2CH2NR2), can also act as a leaving group in this reaction (elimination–addition mechanism, Sec. 10.F). A nitro group can be displaced1552 from α-nitro esters, ketones, nitriles, and α,α-dinitro compounds,1553 and even from simple tertiary nitro compounds of the form R3CNO21554 or ArR2CNO21555 by salts of nitroalkanes, for example,
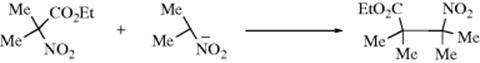
These reactions take place by SET mechanisms.1556 However, with α-nitro sulfones it is the sulfone group that is displaced, rather than the nitro group.1557 The SO2R group of allylic sulfones can be replaced by CHZZ′ (C=CCH2–SO2R → C=CCH2–CHZZ′) if a Mo(CO)6 catalyst is used.1558
tert-Butylsulfones react with organolithium reagents, in the presence of a catalytic amount of an iron complex, to give coupling.1559 In this case the t-BuSO2 unit becomes a “leaving group”. A sulfoxide was a “leaving group” in the cyclization of a carboxylic acid that contains a sulfoxide unit at C-4. Treatment with phenyliodonium bis(trifluoroacetate) gave the five-membered ring lactone.1560 Similar displacement of TolSO2 was observed with tolylsulfones and diethylzinc.1561
Phosphonic esters, ROPO(OR)2, react with allylic Grignard reagents to give the coupling product.1562
OS I, 471; II, 47, 360; VII, 351; VIII, 97, 471.
10-62 The Bruylants Reaction
Alkyl-de-cyanation
![]()
The Bruylants reaction is the reaction of an aminonitrile with a Grignard reagent to give a substituted amine.1563 This reaction is most often used for the preparation of aliphatic amines via aliphatic Grignard reagents. In a few cases, vinylic Grignard reagents can be used to prepare allylic amines.1564 The use of AgBF4 to convert amino nitriles to the corresponding iminium ion facilitates the Bruylants reaction with vinylic Grignard reagents.1565Replacement of the cyano group in a tertiary nitrile is also possible.1566
Displacement of a cyano group in α-cyanoketones is possible. Treatment of the α-cyanoketone with SmI2 followed by addition of an excess of allyl bromide gave the α-allyl ketone derivative.1567 α-Cyano amines react with allyl bromide and then zinc metal to give homoallylic amines after treatment with dilute acetic acid in THF.1568
10-63 Coupling Involving Alcohols
De-hydroxyl-coupling
![]()
In some cases, it is possible to couple an alcohol in the presence of an organometallic compound.1569 Allylic alcohols are coupled with alkylmagnesium bromides in the presence of Ti(OiPr)4, for example.1570 Allylic alcohols can be coupled with arylboronic acids in an ionic liquid solvent and a Rh catalyst.1571 The Pd catalyzed reaction of active methylene compounds with allylic alcohols1572 or benzylic alcohols1573 is also known. The coupling of an alcohol to the α carbon of a ketone (RCOMe + R′OH) to give a β-substituted alcohol [RCH(OH)CH2R′] is possible in the presence of a Ru catalyst.1574 Alcohols are coupled to allenes in the presence of an Ir catalyst.1575 Allylic carbonates are coupled to allylic alcohols with a Ni catalyst.1576
![]()
Allylic or benzylic alcohols can be symmetrically coupled1577 by treatment with methyllithium and titanium trichloride at −78 °C1578 or by refluxing with TiCl3 and LiAlH4 (as shown).1579 When the substrate is an allylic alcohol, the reaction is not regiospecific, but a mixture of normal coupling and allylic-rearranged products is found. A free radical mechanism is involved.1580 The TiCl3–LiAlH4 reagent can also convert 1,3-diols to cyclopropanes, provided that at least one phenyl group is present.1581
Tertiary alcohols (R3C–OH) react with trimethylaluminum at 80–200 °C to give methylation (R3C–Me).1582 The presence of side products from elimination and rearrangement, as well as the lack of stereospecificity,1583 indicate an SN1 mechanism. The reaction can also be applied to primary and secondary alcohols if these contain an aryl group in the α position. Higher trialkylaluminums are far less suitable, because reduction competes with alkylation [see also, reactions of Me3Al with ketones (16-24) and with carboxylic acids (16-82)]. The Me2TiCl2 compound reacts with tertiary alcohols in the same way.1584 β-Alkylation of secondary alcohols has been reported using alcohol substrates in the presence of an Ir complex.1585
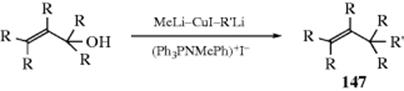
Allylic alcohols couple with a reagent prepared from MeLi, CuI, or R′Li in the presence of (Ph3PNMePh)+ I− to give alkenes (e.g., 147) that are products of allylic rearrangement.1586
The reaction gives good yields with primary, secondary, and tertiary alcohols, and with alkyl and aryllithium reagents.1587 Allylic alcohols also couple with certain Grignard reagents1588 in the presence of a nickel complex to give both normal products and the products of allylic rearrangement.
Allenic alcohols couple with allyl indium reagents at 140 °C to give allylic alcohol products.1589 Similarly, ω-hydroxy lactones couple with organoindium reagents.1590 Phenols react with vinyl boronates and a copper catalyst to give aryl vinyl ethers.1591
Alcohols react with allylsilanes, in the presence of an InCl31592 or InBr31593 catalyst to give the corresponding coupling product (R2CHOH → R2CH–CH2CH=CH2). Silyl ethers are also coupled to allylsilanes in the presence of InCl3.1594 Proparygylic alcohols have been coupled to allylic silanes using an Au catalyst1595 or a Rh catalyst.1596
10-64 Coupling of Organometallic Reagents with Compounds Containing the Ether Linkage1597
Alkyl-de-alkoxy-substitution
![]()
Acetals,1598 ketals, and ortho esters1599 react with Grignard reagents to give, respectively, ethers and acetals (or ketals). The latter can be hydrolyzed to aldehydes or ketones (Reaction 10-6). This procedure is a way of converting a halide (R″X, which may be alkyl, aryl, vinylic, or alkynyl) to an aldehyde (R″CHO), increasing the length of the carbon chain by one carbon (see also, Reaction 10-76). The ketone synthesis generally gives lower yields. Acetals, including allylic acetals, also give this reaction with organocopper compounds and BF3.1600 Dihydropyrans react with Grignard reagents in the presence of a Ni catalyst.1601 Acetals also undergo substitution when treated with silyl enol ethers or allylic silanes, with a Lewis acid catalyst,1602 for example,
![]()
ω-Ethoxy lactams react with Grignard regents to give ω-substituted lactams.1603 Tertiary amines can be prepared by the reaction of amino ethers with Grignard reagents,1604 (R2NCH2–OR′ + R2MgX → R2NCH2–R2) or with lithium dialkylcopper reagents.1605
Ordinary ethers are not cleaved by Grignard reagents (in fact, diethyl ether and THF are the most common solvents for Grignard reagents), although more active organometallic compounds often do cleave them.1606 However, methyl ethers have been replaced with a methyl group (MeMgX + ROMe → R–Me) via a Ni catalyzed coupling reaction with MeMgBr.1607 Oxetanes have been opened with organolithium reagents and BF3•OEt21608 and also with excess Li metal with a biphenyl catalyst.1609 Allylic ethers can be cleaved by Grignard reagents in THF if CuBr is present.1610 The reaction can take place either with or without allylic rearrangement.1611 Propargylic ethers give allenes.1612 Vinylic ethers can also be cleaved by Grignard reagents in the presence of a catalyst, in this case, a Ni complex.1613 Silyl enol ethers R2C=CROSiMe3 behave similarly.1614 Bicyclic benzofurans can be opened by dialkylzinc reagents in the presence of a Pd catalyst.1615
Certain acetals and ketals can be dimerized in a reaction similar to Reaction 10-56 by treatment with TiCl4–LiAlH4, for example,1616
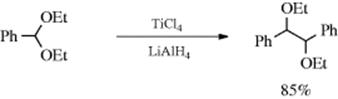
Also see, Reaction 10-65.
OS II, 323; III, 701. Also see, OS V, 431.
10-65 The Reaction of Organometallic Reagents with Epoxides
3(OC)-seco -Alkyl-de-alkoxy-substitution
![]()
The reaction between Grignard reagents or organolithium reagents and epoxides is very valuable and is often used to increase the length of a carbon chain by two carbons.1617 The Grignard reagent may be aromatic or aliphatic, although tertiary Grignard reagents give low yields. As expected for an SN2 process, attack is at the less substituted carbon. With allylic Grignard reagents, the addition of a catalytic amount of Yb(OTf)3 facilitated alkylation.1618Organolithium reagents,1619 in the presence of chiral additives, lead to the 2-substituted alcohol with good enantioselectivity. Similar reaction with a chiral Schiff base gave the same type of product, with excellent enantioselectivity.1620
Lithium dialkylcopper reagents also give the reaction,1621 as do higher order cuprates,1622 often producing higher yields. They have the additional advantage that they do not react with ester, ketone, or carboxyl groups so that the epoxide ring of epoxy esters, ketones, and carboxylic acids can be selectively attacked, often in a regioselective manner.1623 The use of BF3 increases the reactivity of R2CuLi, enabling it to be used with thermally unstable epoxides.1624 Lithium diaminocyano cuprates have also been used.1625
The reaction has also been performed with other organometallic compounds.1626 Trialkylaluminum reagents open epoxides with delivery of the alkyl group to carbon.1627 In the presence of a Lewis acid catalyst (e.g., BF3), alkylation can occur at the more substituted carbon.1628Friedel–Crafts type alkylation (see Reaction 11-11) is possible when an aromatic compound reacts with an epoxide and AlCl3.1629 Epoxides react with allyl bromide in the presence of In metal, with the expected delivery of allyl to the less substituted carbon.1630 When a substituted epoxide was treated with CO, BF3•OEt2, and a Co catalyst, carbonylation occurred and the final product was a β-lactone.1631 Similar β-lactone forming reactions were reported using substituted epoxides, CO, and a metal compound–BF3 complex.1632 A double carbonylation reaction was reported in the presence of an Al complex, generating an anhydride.1633 Five-membered ring lactams were formed from substituted epoxides using BF3•OEt2 followed by treatment with KHF2.1634 Ring opening of epoxides with Ti compounds has been shown to be selective for the more substituted carbon.1635 Epoxides react with Ag salts of alkynes, in the presence of Zr compounds, to give the rearrangement product, a propargylic alcohol.1636 A Ga/Sm induced ring opening with alkyl halides has been reported.1637
In the presence of a Sc catalyst, chiral allylic boranes open epoxides at the less substituted position to generate chiral, homoallylic alcohols.1638
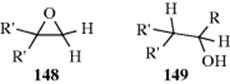
When gem-disubstituted epoxides (148), and sometimes other epoxides, are treated with Grignard reagents, the product may be 149, that is, the new alkyl group may appear on the same carbon as the OH. In such cases, the epoxide is isomerized to an aldehyde or a ketone before reacting with the Grignard reagent. Halohydrins are often side products.

When the substrate is a vinylic epoxide,1639 Grignard reagents generally give a mixture of the normal product and the product of allylic rearrangement (150).1640 Butyllithium reacted with a gem-difluoroalkylidene epoxide (F2C=CR–epoxide) and SN2′ displacement gave alkylation at the difluoro carbon and opened the epoxide.1641 The latter often predominates. In the case of R2CuLi,1642 acyclic substrates give mostly allylic rearrangement (SN2′).1639The double bond of the “vinylic” epoxide can be part of an enolate anion. In this case, R2CuLi give exclusive allylic rearrangement (SN2′) to 151 after hydrolysis, while Grignard and organolithium reagents opened the epoxide directly (SN2) to give 152 after hydrolysis.1643
An organometallic equivalent that opens epoxides is a hydrosilane, for example, Me3SiH, and CO, catalyzed by dicobalt octacarbonyl:1644 See Reaction 10-55 for other coupling reactions with organosilanes. Silyl enol ethers react with epoxides in a related reaction, but a Lewis acid (e.g., TiCl4) is required.1645
OS I, 306; VII, 501; VIII, 33, 516; X, 297.
10-66 Reaction of Organometallics with Aziridines
![]()
Aziridines have been opened by organometallic reagents to give amines.1646 It is also possible to open aziridines, although they are less reactive than epoxides,1647 with organometallic reagents particularly when there is an N-sulfonyl group (e.g., tosyl, formally making it a sulfonamide). Grignard reagents react with N-tosyl 2-phenylaziridine to give the corresponding N-tosylamine.1648 Organocuprates (10-58) reaction with N-alkylaziridines to give the corresponding amine.1649 In a Friedel–Crafts type reaction (11-11), aziridines react with benzene, in the presence of In(OTf)3, to give the β-aryl amine.1650
N-Tosyl aziridines have also been opened with enolate anions, which led to a pyrroline derivative,1651 and with Me2S=CHCO2Et (see Reaction 16-46) to generate a N-tosyl azetidine.1652 Allylic alcohols open N-tosylaziridines with KSF–Montmorillonite clay.1653 C-Arylation is possible with a Ag(I) catalyst.1654N-Sulfonyl aziridines react with the enolate anions of β-keto esters under phase-transfer conditions.1655N-Tosylaziridines react with InCl3 to give the chloro N-tosylamine.1656
Aziridines react with nucleophiles other than carbon nucleophiles. In the presence of tetrabutylammonium fluoride (TBAF), trimethylsilyl azide reacts with N-tosylaziridines to give the azido N-tosylamine.1657N-Benzylic aziridines are opened by trimethylsilyl azide in the presence of a Cr catalyst.1658 Acetic anhydride reacts with N-tosylaziridines, in the presence of PBu3, to give the N-tosylamino acetate.1659 Mediated by Lewis bases, aziridines react with silylated nucleophiles.1660
10-67 Alkylation at a Carbon Bearing an Active Hydrogen
Bis(ethoxycarbonyl)methyl-de-halogenation, and so on
![]()
The metal-catalyzed displacement of allylic acetates and carbonates (Reaction 10-60) clearly falls into this category. However, this section will focus on the more general reaction of active methylene compounds with substrates bearing a leaving group, not necessarily allylic substrates or metal catalyzed. When compounds contain two or three strong electron-withdrawing groups on a carbon atom bearing a proton (the so-called α-proton), that proton is more acidic than compounds without such groups (Sec. 5.B.i, category 1). Treatment with a suitable base (a base that has a conjugate acid with a pKa greater than the α-proton) removes the α-proton and generates the corresponding enolate anion (Reaction 10-68). These enolate anions react as carbon nucleophiles and attack alkyl halides, resulting in their alkylation.1661 Both Z and Z′ may be COOR′, CHO, COR′,1662 CONR'2, COO−, CN,1663 NO2, SOR′, SO2R′,1664 SO2OR′, SO2NR'2 or similar groups.1665 Some commonly used bases are sodium ethoxide and potassium tert-butoxide, each in its respective alcohol as solvent. With particularly acidic compounds (e.g., β-diketones–Z, Z′ = COR′), sodium hydroxide in water or aq alcohol or acetone, or even sodium carbonate,1666 is a strong enough base for the reaction. If at least one Z group is COOR′, saponification is a possible side reaction. In addition to the groups listed above, Z may also be phenyl, but if two phenyl groups are on the same carbon, the acidity is less than in the other cases and a stronger base must be used. However, the reaction can be successfully carried out with diphenylmethane with NaNH2 as the base.1667 If the solvent used in the reaction is acidic enough to protonate either the enolate anion or the base, an equilibrium will be established leading to only small amounts of the enolate anion (thermodynamic conditions). Such protic solvents include water, alcohols, or amines. To avoid this reaction, solvents that do not contain an acidic proton (aprotic solvents) are used, but protic solvents can be used in some cases. The use of polar aprotic solvents (e.g., DMF or DMSO), markedly increases the rate of alkylation1668 but also increases the extent of alkylation at the oxygen rather than the carbon with highly reactive species (e.g., iodomethane, Sec. 10.G.viii). In general, enolate anions, such as those described here, react with alkyl halides via C-alkylation, although trialkylsilyl halides and anhydrides tend to react via O-alkylation. Phase-transfer catalysis has also been used,1669and the use of chiral phase-transfer catalysts led to enantioselectivity in the alkylated product.1670 The reaction is successful for primary and secondary alkyl, allylic (with allylic rearrangement possible), and benzylic RX, but fails for tertiary halides, since these undergo elimination under the reaction conditions (see, however, Reaction 10-67). Various functional groups may be present in RX as long as they are not sensitive to base. Side reactions that may cause problems are the above-mentioned competing O-alkylation, elimination (if the enolate anion is a strong enough base), and dialkylation.
With substrates (e.g., ZCH2Z′) it is possible to alkylate twice. Initial removal of the proton with a base followed by alkylation of the resulting enolate anion with RX, can be followed by subsequent removal of the proton from ZCHRZ′ and then alkylation with the same or a different RX. An important example of this reaction is the malonic ester synthesis, in which both Z groups are COOEt. The product can be hydrolyzed and decarboxylated (Reaction 12-40) to give a carboxylic acid. An illustration is the preparation of 2-ethylpentanoic acid (153) from malonic ester. A variation of this alkylation sequence employs 1,2-dibromoethane as the alkylating agent, and subsequent treatment with 1, 8-diazabicycl [5.4.0]undec-7-ene (DBU) leads to incorporation of a vinyl group on the α carbon.1671 Another variation involved coupling of a dimalonate with an allylic carbonate (see Reaction 10-60), using a polymer-supported Pd catalyst.1672
It is obvious that many carboxylic acids of the formulas RCH2CO2H and RR′CHCO2H can be synthesized by this method [for some other ways of preparing such acids (see Reactions 10-70–10-73)]. Another important example is the acetoacetic ester synthesis, in which Z is CO2Et and Z′ is COCH3. In this case, the product can be decarboxylated with acid or dilute base (Reaction 12-40) to give a ketone (154) or cleaved with concentrated base (Reaction 12-43) to give a carboxylic ester (155) and a salt of acetic acid. This reaction has been done in tert-butanol in the presence of alumina, in vacuo, to give the alkylated keto acid directly from the keto ester.1673
Another way of preparing ketones involves alkylation1674 of β-keto sulfoxides1675 or sulfones,1676 to give 156.
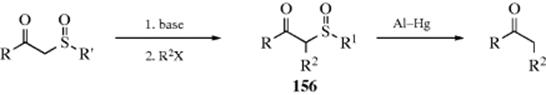
The sulfoxide group in the product (156) is easily reduced (desulfurized, see Reaction 19-70) to give the ketone in high yields using aluminum amalgam or by electrolysis.1677 β-Keto sulfoxides [e.g., 156 or sulfones (–SO2–)] are easily prepared (Reaction 16-86). When one group attached to the sulfur atom is chiral, the alkylation proceeds with reasonable enantioselectivity.1678
Other examples of the reaction are the cyanoacetic ester synthesis, in which Z is CO2Et and Z′ is CN (as in the malonic ester synthesis, the product here can be hydrolyzed and decarboxylated), and the Sorensen method of amino acid synthesis, using N-acetylaminomalonic esters, (EtO2C)2CHNHCOCH3. Hydrolysis and decarboxylation of the product in this case gives an α-amino acid. The amino group is also frequently protected by conversion to a phthalimido group.
The reaction is not limited to Z–CH2–Z′ compounds. Other compounds have acidic CH hydrogens. Some examples are the methyl hydrogens of α-aminopyridines, the methyl hydrogens of ynamines of the form CH3C![]() CNR21679(the product in this case can be hydrolyzed to an amide RCH2CH2CONR2), the CH2 hydrogens of cyclopentadiene and its derivatives (Sec. 2.I.ii), hydrogens connected to a triple-bond carbon (Reaction 10-74), and the hydrogen of HCN (Reaction 10-75) can also be removed with a base and the resulting ion alkylated (see also, Reactions 10-68–10-72). α-Imino esters have been used since treatment with a strong base with a titanium catalyst followed by an aldehyde leads to hydroxy amino esters.1680
CNR21679(the product in this case can be hydrolyzed to an amide RCH2CH2CONR2), the CH2 hydrogens of cyclopentadiene and its derivatives (Sec. 2.I.ii), hydrogens connected to a triple-bond carbon (Reaction 10-74), and the hydrogen of HCN (Reaction 10-75) can also be removed with a base and the resulting ion alkylated (see also, Reactions 10-68–10-72). α-Imino esters have been used since treatment with a strong base with a titanium catalyst followed by an aldehyde leads to hydroxy amino esters.1680
Alkylation takes place at the most acidic position of a reagent molecule; for example, acetoacetic ester (CH3COCH2COOEt) is alkylated at the methylene and not at the methyl group, because the former is more acidic than the latter, and hence gives up its proton to the base. However, if 2 molar equivalents of base are used, then not only is the most acidic proton removed but also the second most acidic. Alkylation of this doubly charged anion (a dianion) occurs at the less acidic position, in this case the second most acidic position1681 (see Sec. 10.G.vii). The first and second ion pair acidities of β-diketones has been studied.1682
When ω,ω′-dihalides are used, ring closures can be effected:1683
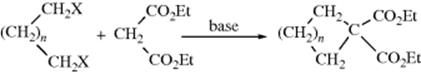
This method has been used to close rings of three (n = 0) to seven members, although five-membered ring closures proceed in highest yields. Another ring-closing method involves internal alkylation.1684
![]()
This method has been shown to be applicable to medium rings (10–14 members) without the use of high-dilution techniques.1685
The mechanism of these reactions is usually SN2 with inversion taking place at a chiral RX, although an SET1686 mechanism may be involved in certain cases,1687 especially where the nucleophile is an α-nitro carbanion1688 and/or the substrate contains a nitro or cyano1689 group. Tertiary alkyl groups can be introduced by an SN1 mechanism if the ZCH2Z′ compound (not the enolate anion) is treated with a tertiary carbocation generated in situ from an alcohol or alkyl halide and BF3 or AlCl3,1690 or with a tertiary alkyl perchlorate.1691
Alkylation α to a nitro group can be achieved with the Katritzky pyrylium–pyridinium reagents.1692 This reaction probably has a free radical mechanism.1693
OS I, 248, 250; II, 262, 279, 384, 474; III, 213, 219, 397, 405, 495, 705; IV, 10, 55, 288, 291, 623, 641, 962; V, 76, 187, 514, 523, 559, 743, 767, 785, 848, 1013; VI, 223, 320, 361, 482, 503, 587, 781, 991; VII, 339, 411; VIII, 5, 312, 381. See also, OS VIII, 235.
10-68 Alkylation of Ketones, Aldehydes, Nitriles, and Carboxylic Esters
α-Acylalkyl-de-halogenation, and so on
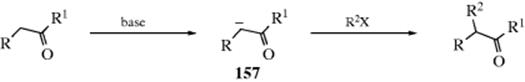
Ketones,1694 nitriles,1695 and carboxylic esters1696 can be alkylated in the α position in a reaction similar to 10-67.1652 The pKa of the proton α to the carbonyl or CN is in the range of 19–25 depending on the number of substituents (see Table 8.1), and a base that has a conjugate acid with a pKa greater than that proton must be employed. Note that since only one activating group is present, compared with two activating groups for the substrates in Reaction 10-67, the pKa of the α-proton is higher (a weaker acid) and a stronger base is required. Reaction of the α-proton with the base generates the key nucleophilic intermediate, an enolate anion (157). The most common bases1697 are lithium diethylamide (Et2NLi), lithium diisopropylamide[(Me2CH)2NLi, LDA],1698 lithium hexamethyldisilazide [LiN(SiMe3)2], t-BuOK, NaNH2, and KH. The base lithium N-isopropyl-N-cyclohexylamide (LICA) is particularly successful for carboxylic esters1699 and nitriles.1700 Enolate anion formation with lithium amides can also be regioselective (see Reaction 12-22).1701 Lithium enolate anions exist as aggregates in solution.1702 The mechanism for this deprotonation reaction has been studied,1703 as has the rate of deprotonation.1704
Solid KOH in Me2SO has been used to methylate ketones, in high yields.1705 Some of these bases are strong enough to convert the ketone, nitrile, or ester completely to its enolate anion conjugate base; others (especially t-BuOK) convert a significant fraction of the molecules. In the latter case, the aldol reaction (16-34) or Claisen condensation (16-85) may be side reactions, since under the thermodynamic conditions associated with this base both the free molecule and its conjugate base are present at the same time. Both lactones1706 and lactams are similarly alkylated.1707 Protic solvents are generally not suitable because they protonate the base (though of course this is not a problem with a conjugate pair, e.g., t-BuOK in t-BuOH). Some common solvents are DME, THF, DMF, and liquid NH3. Phase-transfer catalysis has been used to alkylate many nitriles, as well as some esters and ketones.1708 Amino acid surrogates (158, R = N derivative) can be alkylated, often under phase-transfer conditions.1709
Direct alkylation of aldehydes is difficult when bases (e.g., KOH and NaOMe) are used, due to rapid aldol reaction (16-34), but aldehydes bearing only one α hydrogen have been alkylated with allylic and benzylic halides in good yields using the base KH to prepare the potassium enolate,1710 or in moderate yields, by the use of a phase-transfer catalyst.1711 Even the use of amide bases (e.g., lithium diisopropylamide, LDA), lithium hexamethyl disilazide (LHMDS), or lithium tetramethylpiperidide (LTMP) to generate the enolate anion in an aprotic solvent (e.g., ether or THF) cannot completely suppress rapid aldol side reactions.
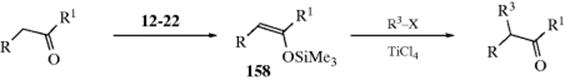
As in Reaction 10-67, the alkyl halide that reacts with the enolate anion may be primary or secondary. Tertiary halides give elimination. Even primary and secondary halides may give predominant elimination if the enolate anion is a strong enough base (e.g., the enolate anion from Me3CCOMe).1712 Tertiary alkyl groups, as well as other groups that normally give SN1 reactions, can be introduced if the reaction is performed on a silyl enol ether1713 of a ketone, aldehyde, or ester (see 158) with a Lewis acid catalyst.1714 Tertiary alkyl fluorides were coupled to silyl enol ethers with BF3•etherate.1715 Note that tin enolates (C=C–OSnR3) react with halides in the presence of a Zn catalyst.1716 A chiral variation of this latter reaction was reported involving generation of the enolate anion in the presence of Me3SnCl, a Pd catalyst, and a chiral ligand.1717
Metal-catalyzed alkylations that are related to this reaction are known. 1,3-Diketones are benzylated or allylated using Bi catalysts.1718 Monoalkylation is possible using Pd catalysts,1719 and Pd catalyzed asymmetric allylic alkylation is known1720 α-Alkylation of ketones with alcohols uses a Ru catalyst,1721 or Ni nanoparticles.1722 A recyclable Pd catalyst is available.1723 Zinc enolate anions have been used in enantioselective Pd catalyzed alkylation reactions.1724
Silyl enol ethers can be converted to the enolate anion, which can then be alkylated in the usual manner. The reaction of silyl enol ether (159) with KOEt followed by LiBr and a catalytic amount of n-butyllithium with allyl iodide gave 160.1725 Metal-catalyzed alkylation reactions are known with silyl enol ethers, including an In catalyzed1726 reaction. An Ir catalyzed regioselective and enantioselective alkylation of silyl enol ethers using allylic carbonates as a substrate has been reported.1727
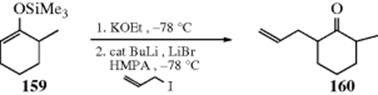
The metal-catalyzed (usually Ni) coupling reaction of an alkyl halide or an electrophilic substrate with a silane1728 is known as Hiyama coupling.1729 A Ni catalyzed Hiyama cross-coupling with a chiral additive leads to a chiral alkylated ketone.1730 The nature of the ligand has an important impact on the reaction.1731 There is a Pd catalyzed Hiyama cross-coupling.1732 Aryl siloxanes have been used in this reaction.1733
Enol carbonates react with alkylating agents in the presence of a Pd catalyst. The decarboxylative alkylation of allyl enol carbonates to the corresponding allylcyclohexanone derivatives is known.1734 An asymmetric version of this reaction has been reported.1735 The same reaction can be done using enolate anion and allylic acetates with a Pd catalyst.1736
Vinylic and aryl halides can be used to vinylate or arylate carboxylic esters (but not ketones) by the use of NiBr2 as a catalyst.1737 Ketones have been vinylated by treating their enol acetates with vinylic bromides in the presence of a Pd catalyst,1738 but direct reaction of a ketone, a vinyl halide, sodium tert-butoxide and a Pd catalyst also give the α-vinyl ketone.1739 Also as in Reaction 10-67, this reaction can be used to close rings.1740 Rings have been closed by treating a dianion of a dialkyl succinate with a 1,ω-dihalide or ditosylate.1741 This was applied to the synthesis of three-, four-, five-, and six-membered rings. When the attached groups were chiral (e.g., menthyl) the product was formed with > 90% ee.1740
Efficient enantioselective alkylations are known,1742 including the use a chiral base to form the enolate anion.1743 Alternatively, a chiral auxiliary can be attached. Many auxiliaries are based on the use of chiral amides1744 or esters.1745 Subsequent formation of the enolate anion allows alkylation to proceed with high enantioselectivity. A subsequent step is required to convert the chiral amide or ester to the corresponding carboxylic acid. Chiral additives can also be used,1746 and the influence of chelating ligands and hydrocarbon cosolvents has been studied for LiN(TMS)2 mediated enolization reactions.1747 The addition of triethylamine influences the (E/Z) selectivity of the enolate anion.1748 Dynamic kinetic resolution has been used for the asymmetric alkylation of malonate derivatives using allenyl acetates.1749
When the compound to be alkylated is an unsymmetrical ketone, the question arises as to which side will be alkylated (regioselectivity). If a phenyl or a vinylic group is present on one side, alkylation goes predominantly on that side. When only alkyl groups are present, the reaction is generally not regioselective; mixtures are obtained in which sometimes the more alkylated and sometimes the less alkylated side is predominantly alkylated. Which product is found in higher yield depends on the nature of the substrate, the base,1750 the cation, and the solvent. In any case, di- and trisubstitution are frequent1751 and it is often difficult to stop with the introduction of just one alkyl group.1752
Several methods have been developed for ensuring that alkylation takes place regioselectively on the desired side of a ketone.1753 Among these are the following:
1. Block one side of the ketone by introducing a removable group. Alkylation takes place on the other side; the blocking group is then removed. A common reaction for this purpose is formylation with ethyl formate (Reaction 16-86). This generally blocks the less hindered side. The formyl group is easily removed by alkaline hydrolysis (Reaction 12-43).
2. Introduce an activating group on one side. Alkylation then takes place on that side (Reaction 10-67). The activating group is then removed.
3. Prepare the desired one of the two possible enolate anions.1754 The two ions (e.g., 161 and 162) for
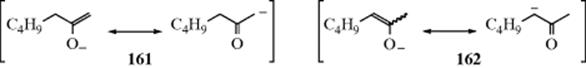
2-heptanone, interconvert rapidly only in the presence of the parent ketone or any stronger acid.1755 In the absence of such acids, it is possible to prepare either 161 or 162 and thus achieve selective alkylation on either side of the ketone.1756 The desired enolate anion can be obtained by treatment of the corresponding enol acetate with 2 equiv of methyllithium in 1,2-dimethoxyethane. Each enol acetate gives the corresponding enolate, for example,
![]()
The enol acetates, in turn, can be prepared by treatment of the parent ketone with an appropriate reagent.1745 Such treatment generally gives a mixture of the two enol acetates in which one or the other predominates, depending on the reagent. The mixtures are easily separable.1755 An alternate procedure involves conversion of a silyl enol ether1757 (see Reaction 12-17) or a dialkylboron enol ether1758 (an enol borinate, see Reaction 10-73) to the corresponding enolate anion. If the less hindered enolate anion is desired (e.g., 161), it can be prepared directly from the ketone by treatment with LDA in THF or DME at −78 °C.1759
4. Begin not with the ketone itself, but with an α,β-unsaturated ketone in which the double bond is present on the side where alkylation is desired. Upon treatment with lithium in liquid NH3, such a ketone is reduced to an enolate anion. When the alkyl halide is added, it must react with the enolate anion on the side where the double bond was.1760 Of course, this method is not actually an alkylation of the ketone, but of the α,β-unsaturated ketone, although the product is the same as if the saturated ketone had been alkylated on the desired side.
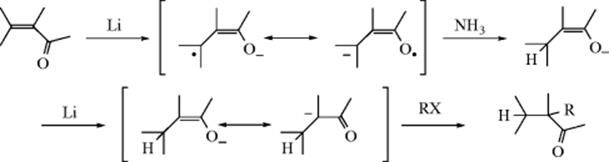
Both sides of acetone have been alkylated with different alkyl groups, in one operation, by treatment of the N,N-dimethylhydrazone of acetone with n-BuLi, followed by a primary alkyl, benzylic, or allylic bromide or iodide; then another mole of n-BuLi, a second halide, and finally hydrolysis of the hydrazone.1761 Alkylation of an unsymmetrical ketone at the more substituted position was reported using an alkyl bromide, NaOH, and a calix[n]arene catalyst (see Sec. 4.H. category 2 for calixarenes).1762
Among other methods for the preparation of alkylated ketones are (1) Alkylation of silyl enol ethers using various reagents as noted above, (2) the Stork enamine reaction (Reaction 10-69), (3) the acetoacetic ester synthesis (Reaction 10-67), (4) alkylation of β-keto sulfones or sulfoxides (Reaction 10-67), (5) acylation of CH3SOCH2− followed by reductive cleavage (Reaction 16-86), (6) treatment of α-halo ketones with lithium dialkylcopper reagents (Reaction 10-57), and (7) treatment of α-halo ketones with trialkylboranes (Reaction 10-73).
Aldehydes can be indirectly alkylated via an imine derivative of the aldehyde.1763 The derivative is easily prepared (Reaction 16-13) and the product easily hydrolyzed to the aldehyde (Reaction 16-2). Either or both R groups may be hydrogen, so that mono-, di-, and trisubstituted acetaldehydes can be prepared by this method. The R′ group may be primary alkyl, allylic, or benzylic. Imine alkylation can also be applied to the preparation of substituted amine derivatives. An amino acid surrogate (e.g., Ph2C=NCH2CO2R), when treated with KOH and an alkyl halide gives the C-alkylated product.1764 When a chiral additive is used, good enantioselectivity was observed. This reaction has also been done in the ionic liquid Bmim tetrafluoroborate (see Sec. 9.D.iii).1765 It is possible to alkylate α-amino amides directly.1766
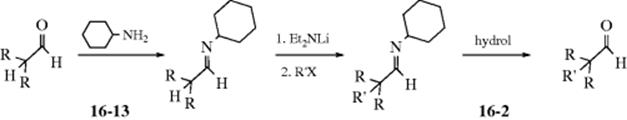
Hydrazones and other compounds with C=N bonds can be similarly alkylated.1743 The use of chiral amines or hydrazines1767 [followed by hydrolysis (Reaction 16-2) of the alkylated imine] can lead to chiral alkylated ketones in high optical yields1768 (for an example, see Sec. 4.J). Lithiated imines are generated by the treatment of an imine with a suitable base, and reaction with an alkyl halide gives the alkylated imine.1769 α-Magnesio imines also react with alkyl halides.1770
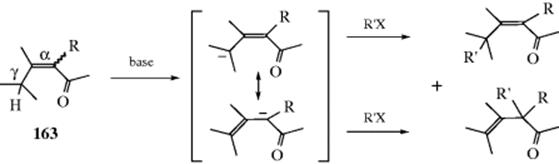
In α,β-unsaturated ketones, nitriles, and esters (e.g., 163), the γ hydrogen assumes the acidity normally held by the position α to the carbonyl group, especially when R is not hydrogen and so cannot compete. This principle, called vinylogy (see Sec. 6.B), operates because the resonance effect is transmitted through the double bond. However, because of the resonance, alkylation at the α position (with allylic rearrangement) competes with alkylation at the γ position and usually predominates.
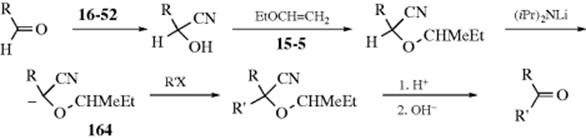
α-Hydroxynitriles (cyanohydrins), protected by conversion to acetals with ethyl vinyl ether (Reaction 15-5), can be easily alkylated with primary or secondary alkyl or allylic halides.1771 The R group can be aryl or a saturated or unsaturated alkyl. Since the cyanohydrins1772 are easily formed from aldehydes (Reaction 16-52) and the product is easily hydrolyzed to a ketone, this is a method for converting an aldehyde (RCHO) to a ketone (RCOR′)1773 (for other methods, see Reactions 10-71, 16-82, and 18-9).1774 In this procedure, the normal mode of reaction of a carbonyl carbon is reversed. The C atom of an aldehyde molecule is normally electrophilic and is attacked by nucleophiles (Chapter 16), but by conversion to the protected cyanohydrin this carbon atom has been induced to perform as a nucleophile.1775 The German word Umpolung1776 is used to describe this kind of reversal (another example is found in Reaction 10-71). Since the ion 164 serves as a substitute for the unavailable R(C=O)− anion, it is often called a “masked” R(C=O)− ion. This method fails for formaldehyde (R = H), but other masked formaldehydes have proved successful.1777 In an interesting variation of nitrile alkylation, a quaternary bromide [PhC(Br)(Me)CN] reacted with allyl bromide, in the presence of a Grignard reagent, to give the alkylated product [PhC(CN)(Me)CH2CH=CH2].1778
A coupling reaction of two ketones to form a 1,4-diketone has been reported, using ZnCl2/Et2NH.1779 An interesting allylic substitution at a 3° center involves the reaction of 3° 2-bromonitriles with iPrMgBr and allyl bromide to give the 2-allyl nitrile.1780
α-Alkylation of ketones with primary alcohols is possible using polymer-associated nanoparticulate Pd.1781
Protonation of enolate anions is discussed in Reaction 16-34, in connection with the aldol condensation.
OS III, 44, 219, 221, 223, 397; IV, 278, 597, 641, 962; V, 187, 514, 559, 848; VI, 51, 115, 121, 401, 818, 897, 958, 991; VII, 153, 208, 241, 424; VIII, 141, 173, 241, 403, 460, 479, 486; X, 59, 460; 80, 31.
10-69 The Stork Enamine Reaction
α-Acylalkyl-de-halogenation1782
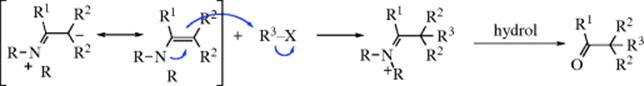
When enamines are treated with alkyl halides, an alkylation occurs to give an iminium salt via electron transfer from the electron pair on nitrogen, through the C=C to the electrophilic carbon of the alkyl halide.1783 In effect, an enamine behaves as a “nitrogen enolate anion” and generally reacts as carbon nucleophiles.1784 Hydrolysis of the iminium salt gives a ketone. Since the enamine is normally formed from a ketone (Reaction 16-13), the net result is alkylation of the ketone at the α position. The method, known as the Stork enamine reaction,1785 is an alternative to the ketone alkylation considered in Reaction 10-68, generally giving monoalkylation of the ketone. Alkylation usually takes place on the less substituted side of the original ketone. The most commonly used amines are the cyclic amines piperidine, morpholine, and pyrrolidine. There are metal-catalyzed enamine alkylation reactions, including the use of Ir catalysts.1786 There are reactions that are catalyzed by enamines, including asymmetric reactions.1787
The method is quite useful for particularly active alkyl halides, (e.g., allylic, benzylic, and propargylic halides), and for α-halo ethers and esters. Other primary and secondary halides can show sluggish reactivity. The reaction of enamines with benzotriazole derivatives has been reported.1788 Tertiary halides do not give the reaction at all since, with respect to the halide, this is nucleophilic substitution and elimination predominates. The reaction can also be applied to activated aryl halides (e.g., 2,4-dinitrochlorobenzene; see Chapter 13), to epoxides,1789 and to activated alkenes (e.g., acrylonitrile). The latter is a Michael-type reaction (15-24) with respect to the alkene.
Acylation1790 can be accomplished with acyl halides or with anhydrides. Hydrolysis of the resulting iminium salt leads to a 1,3-diketone. A COOEt group can be introduced by treatment of the enamine with ethyl chloroformate (ClCOOEt;1791 a CN group with cyanogen chloride1792 (not cyanogen bromide or iodide, which leads to halogenation of the enamine); a CHO group with the mixed anhydride of formic and acetic acids1791 or with DMF and phosgene;1793 and a C(R)=NR′ group with a nitrilium salt RC![]() N+R′.1794 The acylation of the enamine can take place by the same mechanism as alkylation, but another mechanism is also possible, if the acyl halide has an α hydrogen and if a tertiary amine is present, as it often is (it is added to neutralize the HX given off). In this mechanism, the acyl halide is dehydrohalogenated by the tertiary amine, producing a ketene (Reaction 17-14), which adds to the enamine to give a cyclobutanone (Reaction 15-63). This compound can be cleaved in the solution to form the same acylated imine salt (that would form by the more direct mechanism, or it can be isolated (in the case of enamines derived from aldehydes), or it may cleave in other ways.1795
N+R′.1794 The acylation of the enamine can take place by the same mechanism as alkylation, but another mechanism is also possible, if the acyl halide has an α hydrogen and if a tertiary amine is present, as it often is (it is added to neutralize the HX given off). In this mechanism, the acyl halide is dehydrohalogenated by the tertiary amine, producing a ketene (Reaction 17-14), which adds to the enamine to give a cyclobutanone (Reaction 15-63). This compound can be cleaved in the solution to form the same acylated imine salt (that would form by the more direct mechanism, or it can be isolated (in the case of enamines derived from aldehydes), or it may cleave in other ways.1795
N-Alkylation can be a problem, particularly with enamines derived from aldehydes. An alternative method, which gives good yields of alkylation with primary and secondary halides, is alkylation of enamine salts, which are prepared by treating an imine with ethylmagnesium bromide in THF:1796
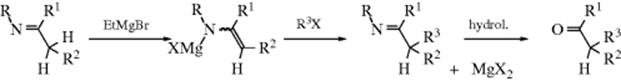
The imines are prepared by the reaction of secondary amines with aldehydes or ketones, mainly ketones (16-13). The enamine salt method has also been used to give good yields of mono α alkylation of α,β-unsaturated ketones.1797 Enamines prepared from aldehydes and butylisobutylamine can be alkylated by simple primary alkyl halides in good yields.1798 N-Alkylation in this case is presumably prevented by steric hindrance.
When the nitrogen of the substrate contains a chiral R group, both the Stork enamine synthesis and the enamine salt method can be used to perform enantioselective syntheses.1799 The use of S-proline can generate a chiral enamine in situ, thus allowing alkylation to occur, giving alkylated product with good enantioselectivity. The reaction has been done intramolecularly.1800
Conjugate addition (Michael addition) occurs when enamines react with conjugated ketones. This reaction is discussed in Reaction 15-24.
OS V, 533, 869; VI, 242, 496, 526; VII, 473.
10-70 Alkylation of Carboxylic Acid Salts
α-Carboxyalkyl-de-halogenation
![]()
Carboxylic acids can be alkylated in the α position by conversion of their salts to dianions, which have resonance contributors,1801 by treatment with a strong base (e.g., LDA).1802 The use of Li+ as the counterion increases the solubility of the dianionic salt. The reaction has been applied1803 to primary alkyl, allylic, and benzylic halides, and to carboxylic acids of the form RCH2CO2H and RR2CHCO2H.1695 Allkylation occurs at carbon, the more nucleophilic site relative to the carboxylate oxygen anion (see Sec. 10.G.vii). This procedure is an alternative to the malonic ester synthesis (Reaction 10-67) as a means of preparing carboxylic acids and has the advantage that acids of the form RR′R2CCO2H can also be prepared. In a related reaction, methylated aromatic acids can be alkylated at the methyl group by a similar procedure.1804
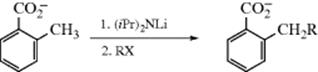
OS V, 526; VI, 517; VII, 249. See also, OS VII, 164.
10-71 Alkylation at a Position α to a Heteroatom
2-(2-Alkyl-thio) de-halogenation
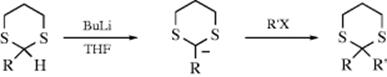
The presence of a sulfur atom on a carbon enhances the acidity of a proton on that carbon, and in dithioacetals and dithioketals that proton RSCH2SR is even more acidic. 1,3-Dithianes can be alkylated1805 if a proton is first removed by treatment with butyllithium in THF.1806 Since 1,3-dithianes can be prepared by treatment of an aldehyde or its acetal (see OS VI, 556) with 1,3-propanedithiol (Reaction 16-11) and can be hydrolyzed (Reaction 10-7), this is a method for the conversion of an aldehyde to a ketone1807 (see also, Reactions 10-68 and 18-9):
![]()
This is another example of Umpolung (see Reaction 10-68);1773 the normally electrophilic carbon of the aldehyde is made to behave as a nucleophile. The reaction can be applied to the unsubstituted dithiane (R = H) and one or two alkyl groups can be introduced, so a wide variety of aldehydes and ketones can be made starting with formaldehyde.1808 The R′ group may be a primary or secondary alkyl or benzylic. Iodides give the best results. The reaction has been used to close rings.1809 A similar synthesis of aldehydes can be performed starting with ethyl ethylthiomethyl sulfoxide (EtSOCH2SEt).1810
Group A may be regarded as a structural equivalent for carbonyl group B, since introduction of A into a molecule is actually an indirect means of introducing B. It is convenient to have a word for units within molecules. Such a word is synthon, introduced by Corey,1811 which is defined as a structural unit within a molecule that can be formed and/or assembled by known or conceivable synthetic operations. There are many other synthons equivalent to Aand B (e.g., C; by Reactions 19-36 and 19-3) and D (by Reactions 10-2 and 16-23).1812
Carbanions generated from 1,3-dithianes also react with epoxides1813 to give the expected products. Reaction with epoxides leads to intermediates that undergo the Brook rearrangement (Reaction 18-44), which is synthetically useful in what is known as anion relay chemistry.
Another useful application of this reaction stems from the fact that dithianes can be desulfurated with Raney nickel (Reaction 14-27). Aldehydes can therefore be converted to chain-extended hydrocarbons:1814
![]()
Similar reactions have been carried out with other thioacetals, as well as with compounds containing three thioether groups on a carbon.1815
If a stabilizing group other than sulfur is attached to the S–CH2 unit of a thioether (RSCH2X, where X is a stabilizing group), formation of the anion and alkylation can be facile. For example, benzylic and allylic thioethers (RSCH2Ar and RSCH2CH=CH2)1816 and thioethers of the form RSCH3 (R = tetrahydrofuranyl or 2-tetrahydropyranyl)1817 have been successfully alkylated at the carbon adjacent to the sulfur atom.1818 Stabilization by one thioether group has also been used in a method for the homologation of primary halides.1819 Thioanisole is treated with BuLi to give the corresponding anion,1820 which reacts with the halide to give the thioether, which is then refluxed with a mixture of methyl iodide and sodium iodide in DMF to give the alkyl iodide as the final product (via an intermediate sulfonium salt). By this sequence, an alkyl halide (RX) is converted to its homologue RCH2X by a pathway involving two laboratory steps (see also, Reaction 10-64).
Vinylic sulfides containing an α hydrogen can also be alkylated1821 by alkyl halides or epoxides. This method is for converting an alkyl halide (RX) to an α,β-unsaturated aldehyde, which is the synthetic equivalent of the unknown −HC=CH–CHO ion.1822 Even simple alkyl aryl sulfides (RCH2SAr and RR′CHSAr) have been alkylated α to the sulfur.1823
Sulfones1824 and sulfonic esters can also be alkylated in the α position if strong enough bases are used.1825 Alkylation at the α position of selenoxides allows the formation of alkenes, since selenoxides easily undergo elimination (Reaction 17-12).1826
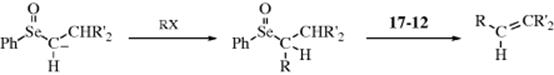
Alkylation can also be carried out, in certain compounds, at positions α to other heteroatoms,1827 for example, at a position α to the nitrogen of tertiary amines.1828 Alkylation α to the nitrogen of primary or secondary amines is not generally feasible because an NH hydrogen is usually more acidic than a CH hydrogen.
![]()
α-Lithiation of N-Boc amines has been accomplished and these react with halides in the presence of a Pd catalyst.1829 Alkylation α to the nitrogen atom of a carbamate occurs when the carbamate is treated with a Grignard reagentunder electrolysis conditions.1830 α-Methoxy amides also react with allyl halides and zinc metal to give alkylation via replacement of the OMe unit.1831 It has been accomplished, however, by replacing the NH hydrogens with other (removable) groups.1832 In one example, a secondary amine is converted to its N-nitroso derivative (Reaction 12-50).1833 The N-nitroso product is easily hydrolyzed to the product amine (Reaction 19-51).1834 Alkylation of secondary and primary amines has also been accomplished with > 10 other protecting groups, involving conversion of amines to amides, carbamates,1835 formamidines,1836 and phosphoramides.1831 In the case of formamidines (165), use of a chiral R′ leads to a chiral amine, in high ee, even when R is not chiral.1837 The reaction of hydrazones with aryl halides, in the presence of a Pd catalyst leads to replacement of H with an aryl group (R'NH–N=CRH → R'NHC=NRR”).1838
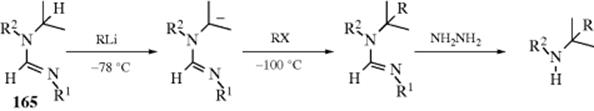
A proton can be removed from an allylic ether by treatment with an alkyllithium at about −70 °C (at higher temperatures the Wittig rearrangement, 18-22, takes place) to give the ion 166, which reacts with alkyl halides
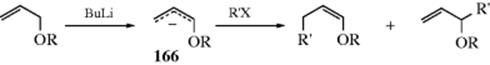
to give the two products shown.1839 Similar reactions1840 have been reported for allylic1841 and vinylic tertiary amines. In the latter case, enamines (167), treated with a strong base, are converted to anions that are then alkylated, generally at C-3.1842 (For direct alkylation of enamines at C-2, see Reaction 10-69.)
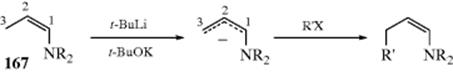
It is also possible to alkylate a methyl, ethyl, or other primary group of an aryl ester (ArCO2R), where Ar is a 2,4,6-trialkylphenyl group.1843 Since esters can be hydrolyzed to alcohols, this constitutes an indirect alkylation of primary alcohols. Methanol has also been alkylated by converting it to −CH2O−.1844
OS VI, 316, 364, 542, 704, 869; VIII, 573.
10-72 Alkylation of Dihydro-1,3-Oxazine. The Meyers Synthesis of Aldehydes, Ketones, and Carboxylic Acids
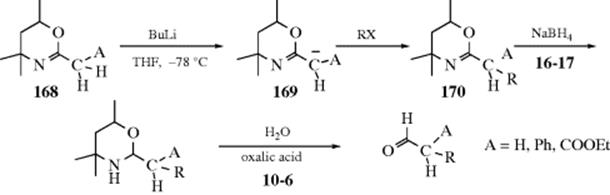
A synthesis of aldehydes1845 developed by Meyers et al.1846 begins with the commercially available dihydro-1,3-oxazine derivatives (168; A = H, Ph, or COOEt).1847 Removal of a proton from the indicated carbon in 168 leads to the resonance stabilized and bidentate anion (169). Alkylation occurs regioselectively at carbon using many alkyl bromides and iodides. The R group of RX can be primary or secondary alkyl, allylic, or benzylic and can carry another halogen or a CN group.1848 The alkylated oxazine (170) is then reduced and hydrolyzed to give an aldehyde containing two more carbons than the starting RX. This method thus complements Reaction 10-71, which converts RX to an aldehyde containing one more carbon. Since A can be H, mono- or disubstituted acetaldehydes can be produced by this method.
The ion 169 also reacts with epoxides, to form γ-hydroxy aldehydes after reduction and hydrolysis,1849 and with aldehydes and ketones (Reaction 16-38). Similar aldehyde synthesis has also been carried out with thiazoles1850 and thiazolines1851 (five-membered rings containing N and S in the 1 and 3 positions).
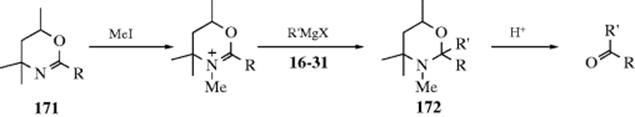
The reaction has been extended to the preparation of ketones:1852 Treatment of a dihydro-1,3-oxazine (171) with iodomethane forms the iminium salt (Reaction 10-31) which, when treated with a Grignard reagent or organolithium compound (16-31), produces 172, which can be hydrolyzed to a ketone. The R group can be alkyl, cycloalkyl, aryl, benzylic, and so on, and R′ of the Grignard reagent can be alkyl, aryl, benzylic, or allylic. Note that the heterocycles 168, 170, or 171 do not react directly with Grignard reagents. In another procedure, 2-oxazolines1853 (173) can be alkylated to give 174,1854 which are easily converted directly to the esters 175 by heating in 5–7% ethanolic sulfuric acid.
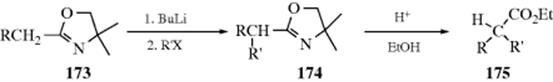
2-Oxazolines (173 and 174) are thus synthons for carboxylic acids; this is another indirect method for the α alkylation of a carboxylic acid,1855 representing an alternative to the malonic ester synthesis (10-67) and to Reactions 10-70 and 10-73. The method can be adapted to the preparation of optically active carboxylic acids by the use of a chiral reagent.1856 Note that, unlike 168, 173 can be alkylated even if R is alkyl. However, the C=N bond of 173 and 174cannot be effectively reduced, so that aldehyde synthesis is not feasible here.1857
OS VI, 905.
10-73 Alkylation with Boranes, Boronic Acids, and Boronates
Alkyl-de-halogenation
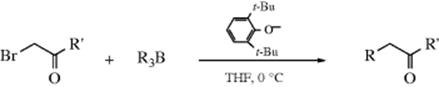
Trialkylboranes react rapidly and in high yields with α-halo ketones,1858 α-halo esters,1859 α-halo nitriles,1860 and α-halo sulfonyl derivatives (sulfones, sulfonic esters, sulfonamides)1861 in the presence of a base to give, respectively, alkylated ketones, esters, nitriles, and sulfonyl derivatives.1862 Potassium tert-butoxide is often a suitable base, but potassium 2,6-di-tert-butylphenoxide at 0 °C in THF gives better results in most cases, possibly because the large bulk of the two tert-butyl groups prevents the base from coordinating with the R3B.1863 The trialkylboranes are prepared by treatment of 3 equiv of an alkene with 1 equiv of BH3 (Reaction 15-16).1864 With appropriate boranes, the R group transferred to α-halo ketones, nitriles, and esters can be vinylic,1865 or (for α-halo ketones and esters) aryl.1866
The reaction can be extended to α,α-dihalo esters1867 and α,α-dihalo nitriles.1868 It is possible to replace just one halogen or both. In the latter case, the two alkyl groups can be the same or different. When dialkylation is applied to dihalo nitriles, the two alkyl groups can be primary or secondary, but with dihalo esters, dialkylation is limited to primary R. Another extension is the reaction of boranes (BR3) with γ-halo-α,β-unsaturated esters.1869 Alkylation takes place in the γ position, but the double bond migrates out of conjugation with the CO2Et unit [BrCH2CH=CHCO2Et → RCH=CHCH2CO2Et]. In this case, however, double-bond migration is an advantage, because nonconjugated β,γ-unsaturated esters are usually much more difficult to prepare than their α,β-unsaturated isomers.
The alkylation of activated halogen compounds is one of several reactions of trialkylboranes developed by H.C. Brown1870 (see also, Reactions 15-16, 15-27, 18-31–18-40, etc.). These compounds are extremely versatile and can be used for the preparation of many types of compounds. In this reaction, for example, an alkene (via the BR3) can be coupled to a ketone, a nitrile, a carboxylic ester, or a sulfonyl derivative. Note that this is still another indirect way to alkylate a ketone (see Reaction 10-68) or a carboxylic acid (see Reaction 10-70), and provides an additional alternative to the malonic ester and acetoacetic ester syntheses (Reaction 10-67).
Although superficially this reaction resembles Reaction 10-57, it is likely that the mechanism is quite different, involving migration of an R group from boron to carbon (see also, Reactions 18-23–18-26). The mechanism is not known with certainty,1871 but it may be tentatively shown as (illustrated for an α-halo ketone):
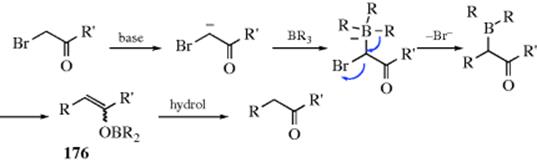
The first step is removal of the acidic proton by the base to give an enolate anion that combines with the borane (Lewis acid–base reaction). An R group then migrates, displacing the halogen leaving group.1872 Another migration follows, this time of BR2 from carbon to oxygen to give the enol borinate (176),1873 which is hydrolyzed. Configuration at the alkyl group R is retained.1874
Alkenylboranes (R'2C=CHBZ2; Z = various groups) couple in high yields with vinylic,1875 alkynyl, aryl, benzylic, and allylic halides or triflates in the presence of a Pd catalyst and a base to give R'2C=CHR.1876 9-Alkyl-9-BBN compounds (Reaction 15-16) also couple with vinylic and aryl halides,1877 as well as with α-halo ketones, nitriles, and esters.1878
The reaction has also been applied to compounds with other leaving groups. Diazo ketones, diazo esters, diazo nitriles, and diazo aldehydes (177)1879 react with trialkylboranes in a similar manner.
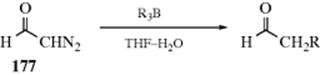
The mechanism is probably also similar. In this case, a base is not needed, since the carbon already has an available pair of electrons. The reaction with diazo aldehydes1880 is especially notable, since successful reactions cannot be obtained with α-halo aldehydes.1881
Alkyl1882 and aryl1883 boronic acids [RB(OH)2] react with allylic acetates to give the alkylated product in the presence of a Pd catalyst.1884 A cyclopropylboronic acid was coupled to an allylic bromide with silver oxide/KOH and a Pd catalyst.1885 Arylboronic acids undergo a coupling reaction with epoxides in the presence of a Pd catalyst.1886 Alkylboronic acids can also be coupled to aromatic compounds in the presence of Cu(OAc)2 and a Pd catalyst.1887 The Pd catalyzed coupling of vinyl halides and alkylboronic acids,1888 which gives substituted alkenes, is related to the Suzuki coupling (Reaction 13-12). Vinyl zirconium reagents were coupled to alkyl halides with a Pd catalyst.1889Arylation of sp3 C–H positions can be done using arylboronates and a Rh catalyst.1890 In a related but metal-free reaction, activated epoxides and aziridines are opened by borates.1891
Potassium aryl- and 1-alkenyltrifluoroborates (ArBF3K and RBF3K) are easily prepared from organoboronic acids or esters. In general, the trifluoroborates have greater air stability and greater nucleophilicity1892 when compared to the corresponding organoboranes and organoboronic acid derivatives. Potassium alkyltrifluoroborates undergo the Pd catalyzed coupling reaction with arenediazonium tetrafluoroborates,1893 diaryliodonium salts,1894 aryl halides,1895 as well as with aryl triflates. An example of the latter reaction converted 178 to diphenylmethane via coupling with phenyl triflate.1896 Alkenyltrifluoroborates can be coupled to aryl halides.1897
![]()
where dppf = bis(diphenylphosphino)ferrocene.
OS VI, 919; IX, 107.
10-74 Alkylation at an Alkynyl Carbon
Alkynyl-de-halogenation
![]()
The reaction between alkyl halides and acetylide ions is useful, but of limited scope.1898 Only primary halides unbranched in the β-position give good yields, although allylic halides can be used if CuI is present.1899 If acetylene is the reagent, two different groups can be successively attached. Sulfates, sulfonates, and epoxides1900 are sometimes used as substrates. The acetylide ion is often prepared by treatment of an alkyne with a strong base (e.g., NaNH2). Magnesium acetylides (prepared as in Reaction 12-22) are also frequently used, although they react only with active substrates (e.g., allylic, benzylic, and propargylic halides) and not with primary alkyl halides. Alternatively, the alkyl halide can be treated with a lithium acetylide–ethylenediamine complex.1901 If 2 equiv of a very strong base are used, alkylation can be effected at a carbon α to a terminal triple bond: RCH2C![]() CH + 2BuLi → RCHC(C:− + R′Br → RR′CHC
CH + 2BuLi → RCHC(C:− + R′Br → RR′CHC![]() C:−.1902 For another method of alkylating at an alkynyl carbon, see Reaction 18-26. An alternative method for generating an alkyne anion treated a trialkylsilyl alkyne with potassium carbonate in methanol, and then methyllithium/LiBr.1903 In the presence of an alkyl iodide, alkylation at the alkynyl carbon occurred. Terminal alkynes react with alkylzinc reagents in the presence of a Pd catalyst.1904
C:−.1902 For another method of alkylating at an alkynyl carbon, see Reaction 18-26. An alternative method for generating an alkyne anion treated a trialkylsilyl alkyne with potassium carbonate in methanol, and then methyllithium/LiBr.1903 In the presence of an alkyl iodide, alkylation at the alkynyl carbon occurred. Terminal alkynes react with alkylzinc reagents in the presence of a Pd catalyst.1904
Other metalated terminal alkynes can be coupled to substrates with a leaving group, and even with other organometallics. In the presence of a Pd catalysts, an alkynyltin reagent reacts with an alkylzinc compound to give the corresponding alkyne.1905 Terminal alkynes react with allylic bromides in the presence of a Ni catalyst.1906 The reaction of a terminal alkyne with Zn(II) compounds allows reaction with silanes to give the 1-silylalkyne.1907 A Re catalyzed C–H insertion reaction is known using terminal alkynes and an active methylene compound.1908 Alkynylzinc compounds undergo Pd catalyzed cross-coupling reactions.1909
1-Haloalkynes react with various substrates in the presence of a metal catalyst. 1-Haloalkynes (e.g., R–C![]() C–X react with ArSnBu3 and CuI to give R–C
C–X react with ArSnBu3 and CuI to give R–C![]() C–Ar.1910 Organozirconium compounds react in a similar manner.1911Acetylene reacts with 2 equiv of iodobenzene, in the presence of a Pd catalyst and CuI, to give 1,2-diphenylethyne.1912 1-Trialkylsilyl alkynes react with 1-haloalkynes, in the presence of a CuCl catalyst, to give diynes1913 and with aryl triflates to give 1-aryl alkynes.1914 The enolate anion derived from a β-keto ester couples with 1-bromoalkynes to give the corresponding substitution product.1915 1-Bromoalkynes react with nitrogen compounds (e.g., imidazole) in the presence of a Cu catalyst to give the corresponding alkyne.1916 1-Bromoalkynes react with Grignard derived reagents in the presence of an Fe catalyst.1917
C–Ar.1910 Organozirconium compounds react in a similar manner.1911Acetylene reacts with 2 equiv of iodobenzene, in the presence of a Pd catalyst and CuI, to give 1,2-diphenylethyne.1912 1-Trialkylsilyl alkynes react with 1-haloalkynes, in the presence of a CuCl catalyst, to give diynes1913 and with aryl triflates to give 1-aryl alkynes.1914 The enolate anion derived from a β-keto ester couples with 1-bromoalkynes to give the corresponding substitution product.1915 1-Bromoalkynes react with nitrogen compounds (e.g., imidazole) in the presence of a Cu catalyst to give the corresponding alkyne.1916 1-Bromoalkynes react with Grignard derived reagents in the presence of an Fe catalyst.1917
Alkynes couple with alkyl halides in the presence of SmI2/Sm,1918 or a copper catalyst.1919 Alkynes react with hypervalent iodine compounds1920 and with reactive alkanes (e.g., adamantane) in the presence of AIBN.1921 The reaction of benzylic amines with terminal alkynes, in the presence of copper triflate and tert-butylhydroperoxide, leads to incorporation of the alkyne group α to the nitrogen.1922 A similar reaction occurs at a methyl group of N,N-dimethylaniline.1923 α-Methoxycarbamates (MeO–CHR–NR1– CO2R2) react with terminal alkynes and CuBr to give the alkynylamine.1924 In the presence of GaCl3, ClC![]() CSiMe3 reacts with silyl enol ethers to give, after treatment with methanolic acid, an α-ethynyl ketone.1925
CSiMe3 reacts with silyl enol ethers to give, after treatment with methanolic acid, an α-ethynyl ketone.1925
In a related reaction, terminal alkynes react with silanes (R3SiH) in the presence of an Ir catalyst1926 or zinc triflate1927 to give the 1-trialkylsilyl alkyne. Similar products are obtained when terminal alkynes react with N-trialkylsilylamines and ZnCl2.1928
OS IV, 117; VI, 273, 564, 595; VIII, 415; IX, 117, 477, 688; 76, 263. Also see, OS IV, 801; VI, 925.
10-75 Preparation of Nitriles
Cyano-de-halogenation
![]()
The reaction between cyanide ion and alkyl halides is a convenient method for the preparation of nitriles,1929 The reaction proceeds by a SN2 mechanism,1930 so primary, benzylic, and allylic halides give good yields of nitriles; secondary halides give moderate yields. The reaction fails for tertiary halides, which give elimination under these conditions. Many other groups on the molecule do not interfere. A number of solvents have been used, but the high yields and short reaction times observed with DMSO make it a very good solvent for this reaction.1931 In general, polar aprotic solvents are the best choice. Other ways to obtain high yields under mild conditions are to use a phase-transfer catalyst,1932 in alternative solvents (e.g., PEG 400 (a polyethylene glycol)],1933 or with ultrasound.1934 This is an important way of increasing the length of a carbon chain by one carbon, since nitriles are easily hydrolyzed to carboxylic acids (Reaction 16-4).
The cyanide ion is an ambident nucleophile (it can react via N or via C) and isonitriles (also called isocyanides, R–N![]() C) may be side products.1935 If the preparation of isocyanides is desired (see Reaction 10-40), they can be made the main products by the use of reagents with more covalent metal–carbon bonds [e.g., silver or copper(I) cyanide,1936 Sec. 10.G.vii, category 3]. However, the use of an excess of LiCN in acetone/THF gave the nitrile as the major product.1937 Tosyl cyanide (TolSO2CN) has been used in some cases.1938 A radical cyanation of alkyl iodides has been reported using diethylphosphoryl cyanide1939 (see Chap. 14).
C) may be side products.1935 If the preparation of isocyanides is desired (see Reaction 10-40), they can be made the main products by the use of reagents with more covalent metal–carbon bonds [e.g., silver or copper(I) cyanide,1936 Sec. 10.G.vii, category 3]. However, the use of an excess of LiCN in acetone/THF gave the nitrile as the major product.1937 Tosyl cyanide (TolSO2CN) has been used in some cases.1938 A radical cyanation of alkyl iodides has been reported using diethylphosphoryl cyanide1939 (see Chap. 14).
Vinylic bromides can be converted to vinylic cyanides with CuCN,1940 with KCN, a crown ether, and a Pd complex,1941 or with KCN and a Ni(0) catalyst.1942 Halides can be converted to the corresponding nitriles by treatment with trimethylsilyl cyanide in the presence of catalytic amounts of SnCl4: R3CCl + Me3SiCN → R3CCN.1943 Primary, secondary, and tertiary alcohols are converted to nitriles in good yields by treatment with NaCN, Me3SiCl, and a catalytic amount of NaI in DMF–MeCN.1944 Lewis acids have been used in conjunction with NaCN or KCN.1945 α,β-Epoxy amides were opened to the β-cyano-α-hydroxyamide with Et2AlCN.1946 Cyanohydrins react with alkyl halides in some cases to give the nitrile.1947
Substrates that react with cyanide may contain leaving groups other than halides (e.g., esters of sulfuric and sulfonic acids, sulfates and sulfonates, respectively). Vinylic triflates give vinylic cyanides when treated with LiCN, a crown ether, and a Pd catalyst.1948 Epoxides give β-hydroxy nitriles. The C-2-Selectivity was observed when NaCN and B(OMe)3 were reacted with a disubstituted epoxide.1949 The use of trimethylsilyl cyanide (Me3SiCN) and a Lewis acid generates the O-TMS β-hydroxy nitrile, and the use of YbCl3 and a salen complex gave good enantioselectivity.1950 Tetrabutylammonium cyanide converted a primary alcohol to the corresponding nitrile in the presence of PPh3/DDQ.1951 Alcohols are converted to cyanides by reaction with triphenylphosphine and cyanogen bromide.1952
Sodium cyanide in HMPA selectively cleaves methyl esters in the presence of ethyl esters:1953
![]()
OS I, 46, 107, 156, 181, 254, 256, 536; II, 292, 376; III, 174, 372, 557; IV, 438, 496, 576; V, 578, 614.
10-76 Direct Conversion of Alkyl Halides to Aldehydes and Ketones
Formyl-de-halogenation
![]()
The direct conversion of alkyl bromides to aldehydes, with an increase in the chain length by one carbon, can be accomplished1954 by treatment with sodium tetracarbonylferrate(-2)1955 (Collman's reagent) in the presence of triphenylphosphine and subsequent quenching of 179 with acetic acid. The reagent Na2Fe(CO)4 can be prepared by treatment of iron pentacarbonyl [Fe(CO)5] with sodium amalgam in THF. Good yields are obtained from primary alkyl bromides; secondary bromides give lower yields. The reaction is generally not satisfactory for benzylic bromides, but a good yield of the ketone was obtained using benzyl chloride and aryl iodides.1956 The initial species produced from RX and Na2Fe(CO)4 is the ion RFe(CO)4−, which can be isolated;1957 it then reacts with Ph3P to give 179.1958
The synthesis can be extended to the preparation of ketones in six distinct ways.1959 These include quenching 179 with a second alkyl halide (R′X) rather than acetic acid; omitting PPh3 with first RX and then adding the second, R′X; treatment with RX in the presence of CO,1955 followed by treatment with R′X′; treatment with an acyl halide followed by treatment with an alkyl halide or an epoxide, gives an α,β-unsaturated ketone.1960 The final variations involve reaction of alkyl halides or tosylates with Na2Fe(CO)4 in the presence of ethylene to give alkyl ethyl ketones;1961 when 1,4-dihalides are used, five-membered cyclic ketones are prepared.1962
Yet another approach uses electrolysis conditions with the alkyl chloride, Fe(CO)5, and a Ni catalyst, which gives the ketone directly, in one step.1963 In the first stage of methods 1, 2, and 3, primary bromides, iodides, and tosylates and secondary tosylates can be used. The second stage of the first-four methods requires more active substrates (e.g., primary iodides or tosylates or benzylic halides). Method 5 has been applied to primary and secondary substrates.
Other acyl organometallic reagents are known. An acyl zirconium reagent [e.g., RCOZr(Cl)Cp2] reacted with allylic bromide in the presence of CuI to give the corresponding ketone, but with allylic rearrangement.1964
Symmetrical ketones (R2CO) can be prepared by treatment of a primary alkyl or benzylic halide with Fe(CO)5 and a phase-transfer catalyst,1965 or from a halide RX (R = primary alkyl, aryl, allylic, or benzylic) and CO by an electrochemical method involving a nickel complex.1966 Aryl, benzylic, vinylic, and allylic halides have been converted to aldehydes by treatment with CO and Bu3SnH, with a Pd catalyst.1967 Various other groups do not interfere. Several procedures for the preparation of ketones are catalyzed by Pd complexes. Alkyl aryl ketones are formed in good yields by treatment of a mixture of an aryl iodide, an alkyl iodide, and a Zn–Cu couple with CO (ArI + RI + CO → RCOAr).1968 Vinylic halides react with vinylic tin reagents in the presence of CO to give unsymmetrical divinyl ketones.1969 Aryl, vinylic, and benzylic halides can be converted to methyl ketones (RX → RCOMe) by reaction with (α-ethoxyvinyl)tributyltin [Bu3SnC(OEt)=CH2].1970 In addition, SmI2 can be used to convert alkyl chlorides to ketones, in the presence of 50 atm of CO.1971
The conversion of alkyl halides to aldehydes and ketones can also be accomplished indirectly (Reaction 10-71). See also, Reaction 12-33.
OS VI, 807.
10-77 Carbonylation of Alkyl Halides, Alcohols, or Alkanes
Alkoxycarbonyl-de-halogenation
![]()
A direct method for preparing a carboxylic acid treats an alkyl halide with NaNO2 in acetic acid and DMSO.1972 Reaction of an alkyl halide with ClCOCO2Me and (Bu3Sn)2 under photochemical conditions leads to the corresponding methyl ester.1973
Several methods, all based on carbon monoxide or metal carbonyls, have been developed for converting an alkyl halide to a carboxylic acid or an acid derivative with the chain extended by one carbon.1974 When an alkyl halide is treated with SbCl5–SO2 at −70 °C, it dissociates into the corresponding carbocation (Sec. 5.A.ii). If carbon monoxide and an alcohol are present, a carboxylic ester is formed by the following route:1975
This has also been accomplished with concentrated H2SO4 saturated with CO.1976 Not surprisingly, only tertiary halides perform satisfactorily; secondary halides give mostly rearrangement products. An analogous reaction takes place with alkanes possessing a tertiary hydrogen, using HF–SbF5–CO.1977
Carboxylic acids or esters are the products, depending on whether the reaction mixture is solvolyzed with water or an alcohol. Alcohols with more than seven carbons are cleaved into smaller fragments by this procedure.1978Similarly, tertiary alcohols1979 react with H2SO4 and CO (which is often generated from HCOOH and the H2SO4 in the solution) to give trisubstituted acetic acids in a process called the Koch–Haaf reaction (see also, 15-35).1980 If a primary or secondary alcohol is the substrate, the carbocation initially formed rearranges to a tertiary ion before reacting with the CO. Better results are obtained if trifluoromethanesulfonic acid (F3CSO2OH) is used instead of H2SO4.1981 Iodo alcohols were transformed into lactones under radical conditions (AIBN, allylSnBu3) and 45 atm of CO.1982
Another method1983 for the conversion of alkyl halides to carboxylic esters is treatment of a halide with nickel carbonyl [Ni(CO)4] in the presence of an alcohol and its conjugate base.1984 When R′ is primary, RX may only be a vinylic or an aryl halide; retention of configuration is observed at a vinylic R. Consequently, a carbocation intermediate is not involved here. When R′ is tertiary, R may be primary alkyl as well as vinylic or aryl. This is thus one of the few methods for preparing esters of tertiary alcohols. Alkyl iodides give the best results, then bromides. In the presence of an amine, an amide can be isolated directly, at least in some instances.
![]()
Still another method for the conversion of halides to acid derivatives makes use of Na2Fe(CO)4. As described in Reaction 10-76, primary and secondary alkyl halides and tosylates react with this reagent to give the ion RFe(CO)4−or, if CO is present, the ion RCOFe(CO)4−. Treatment of RFe(CO)4− or RCOFe(CO)4− with oxygen or sodium hypochlorite gives, after hydrolysis, a carboxylic acid.1985 Alternatively, RFe(CO)4− or RCOFe(CO)4− reacts with a halogen (e.g., I2) in the presence of an alcohol to give a carboxylic ester,1986 or in the presence of a secondary amine or water to give, respectively, the corresponding amide or free acid. Both RFe(CO)4− and RCOFe(CO)4−, which are prepared from primary R, give high yields. With secondary R, the best results are obtained in the solvent THF by the use of RCOFe(CO)4− prepared from secondary tosylates. Ester and keto groups may be present in R without being affected. Carboxylic esters (RCO2R′) have also been prepared by treating primary alkyl halides RX with alkoxides R′O− in the presence of Fe(CO)5.1987 Here RCOFe(CO)4− is presumably an intermediate.
Palladium complexes also catalyze the carbonylation of halides.1988 Aryl (see Reaction 13-15),1989 vinylic,1990 benzylic, and allylic halides (especially iodides) can be converted to carboxylic esters with CO, an alcohol or alkoxide, and a Pd complex.1991 The Pd catalyzed carbonylation of organoindium compounds in the presence of methanol gives methyl esters.1992 Similar reactivity was reported with vinyl triflates.1993 α-Halo ketones are converted to β-keto esters with CO, an alcohol, NBu3 and a palladium catalyst at 110 °C.1994 Use of an amine instead of the alcohol or alkoxide leads to an amide.1995 Reaction with an amine, AIBN, CO, and a tetraalkyltin catalyst also leads to an amide.1996 Benzylic and allylic halides were converted to carboxylic acids electrocatalytically, with CO and a Co–imine complex.1997 Vinylic halides were similarly converted with CO and nickel cyanide, under phase-transfer conditions.1998 Allylic O-phosphates were converted to allylic amides with CO and ClTi=NTMS, in the presence of a Pd catalyst.1999 Terminal alkynes were converted to the alkynyl ester using CO, PdBr2, CuBr2 in methanol, and sodium bicarbonate.2000
Other organometallic reagents can be used to convert alkyl halides to carboxylic acid derivatives. Benzylic halides were converted to carboxylic esters with CO in the presence of a rhodium complex.2001 Variations introduce the R′ group via an ether (R'2O),2002 or an Al, Ti, or Zr alkoxide.2003 The reaction of an alkene, a primary alcohol, and CO, in the presence of a Rh catalyst, led to carbonylation of the alkene and formation of the corresponding ester.2004Vinyl triflates were converted to the conjugated carboxylic acid with CO2 and a Ni catalyst.2005 Reaction with an α,ω-diiodide, Bu4NF, and Mo(CO)6 gave the corresponding lactone.2006
A number of double carbonylations have been reported. In these reactions, two molecules of CO are incorporated in the product, leading to α-keto acids or their derivatives.2007 When the catalyst is a Pd complex, best results are obtained in the formation of α-keto amides.2008 The R group is usually aryl or vinylic.2009 The formation of α-keto acids2010 or esters2011 requires more severe conditions. α-Hydroxy acids were obtained from aryl iodides when the reaction was carried out in the presence of an alcohol, which functioned as a reducing agent.2012 Cobalt catalysts have also been used and require lower CO pressures.2007
OS V, 20, 739.
Notes
1. See Hartshorn, S.R. Aliphatic Nucleophilic Substitution, Cambridge University Press, Cambridge, 1973; Katritzky, A.R.; Brycki, B.E. Chem. Soc. Rev. 1990, 19, 83; Richard, J.P. Adv. Carbocation Chem. 1989, 1, 121; Streitwieser, A. Solvolytic Displacement Reactions, McGraw-Hill, NY, 1962.
2. See Sun, L.; Hase, W. L.; Song, K. J. Am. Chem. Soc. 2001, 123, 5753. Nucleophlicity and leaving group ability for frontside and backside attack have been studied. See Bento, A.P.; Bickelhaupt, F.M. J. Org. Chem. 2008, 73,7290.
3. Hasanayn, F.; Streitwieser, A.; Al-Rifai, R. J. Am. Chem. Soc. 2005, 127, 2249. See also Cruickshank, F.R.; Hyde, A.J.; Pugh, D. J. Chem. Ed. 1977, 54, 288.
4. For a theoretical investigation of a kinetic isotope effect, see Matsson, O.; Dybala-Defratyka, A.; Rostkowski, M.; Paneth, P.; Westaway, K.C. J. Org. Chem. 2005, 70, 4022.
5. For a discussion of this type of solvent effect, see Arnaut, L.G.; Formosinho, S.J. Chemistry: European J. 2007, 13, 8018.
6. Cowdrey, W.A.; Hughes, E.D.; Ingold, C.K.; Masterman, S.; Scott, A.D. J. Chem. Soc. 1937, 1252. The idea that the addition of one group and removal of the other are simultaneous was first suggested by Lewis, G.N. in Valence and the Structure of Atoms and Molecules, Chemical Catalog Company, NY, 1923, p. 113. The idea that a one-step substitution leads to inversion was proposed by Olsen, A.R. J. Chem. Phys. 1933, 1, 418.
7. Walden, P. Ber. 1893, 26, 210; 1896, 29, 133; 1899, 32, 1855.
8. For a discussion of these cycles, see Kryger, L.; Rasmussen, S.E. Acta Chem. Scand. 1972, 26, 2349.
9. Phillips, H. J. Chem. Soc. 1923, 123, 44. See Garwood, D.C.; Cram, D.J. J. Am. Chem. Soc. 1970, 92, 4575; Cram, D.J.; Cram, J.M. Fortschr. Chem. Forsch. 1972, 31, 1.
10. See Kenyon, J.; Phillips, H.; Shutt, G.R. J. Chem. Soc. 1935, 1663 and references cited therein.
11. Streitwieser, Jr., A. J. Am. Chem. Soc. 1953, 75, 5014.
12. Speranza, M.; Angelini, G. J. Am. Chem. Soc. 1980, 102, 3115 and references cited therein; Kempf, B.; Hampel, N.; Ofial, A.R.; Mayr, H. Chem. Eur. J. 2003, 9, 2209. See Riveros, J.M.; José, S.M.; Takashima, K. Adv. Phys. Org. Chem. 1985, 21, 197.
13. Li, C.; Ross, P.; Szulejko, J.E.; McMahon, T.B. J. Am. Chem. Soc. 1996, 118, 9360.
14. See Müller, P.; Mareda, J. in Olah, G.A. Cage Hydrocarbons, Wiley, NY, 1990, pp. 189–217; Fort, Jr., R.C.; Schleyer, P.v.R. Adv. Alicyclic Chem. 1966, 1, 283.
15. Doering, W. von E.; Levitz, M.; Sayigh, A.; Sprecher, M.; Whelan, Jr., W.P. J. Am. Chem. Soc. 1953, 75, 1008. Actually, a slow substitution was observed in this case, but not by an SN2 mechanism.
16. Cope, A.C.; Synerholm, M.E. J. Am. Chem. Soc. 1950, 72, 5228.
17. Hughes, E.D.; Juliusburger, F.; Masterman, S.; Topley, B.; Weiss, J. J. Chem. Soc. 1935, 1525.
18. Tenud, L.; Farooq, S.; Seibl, J.; Eschenmoser, A. Helv. Chim. Acta 1970, 53, 2059. See also, King, J.F.; McGarrity, M.J. J. Chem. Soc., Chem. Commun. 1979, 1140.
19. See Angel, L.A; Ervin, K.M. J. Am. Chem. Soc. 2003, 125, 1014.
20. Taken from Chandrasekhar, J.; Smith, S.F.; Jorgensen, W.L. J. Am. Chem. Soc. 1985, 107, 154.
21. Gao, J.; Xia, X. J. Am. Chem. Soc. 1993, 115, 9667.
22. Lee, I.; Kim, C.K.; Chung, D.S.; Lee, B.-S. J. Org. Chem. 1994, 59, 4490.
23. Evanseck, J.D.; Blake, J.F.; Jorgensen, W.L. J. Am. Chem. Soc. 1987, 109, 2349; Kozaki, T.; Morihashi, K.; Kikuchi, O. J. Am. Chem. Soc. 1989, 111, 1547; Jorgensen, W.L. Acc. Chem. Res. 1989, 22, 184.
24. Chandrasekhar, J.; Jorgensen, W.L. J. Am. Chem. Soc. 1985, 107, 2974.
25. For a discussion of environmentally benign substitution reactions, see Vogel, P.; Figueira, S.; Muthukrishnan, S.; Mack, J. Tetrahedron Lett. 2009, 50, 55.
26. See Reactions 10-60–10-68 and Bachrach, S.M.; Gailbreath, B.D. J. Org. Chem. 2001, 66, 2005.
27. Hoz, S.; Basch, H.; Wolk, J.L.; Hoz, T.; Rozental, E. J. Am. Chem. Soc. 1999, 121, 7724.
28. Yi, R.; Basch, H.; Hoz, S. J. Org. Chem. 2002, 67, 5891.
29. Grinblat, J.; Ben-Zion, M.; Hoz, S. J. Am. Chem. Soc., 2001, 123, 10738.
30. Pearson, R.G.; Dillon, R.L. J. Am. Chem. Soc. 1953, 75, 2439.
31. Yamataka, H.; Mustanir; Mishima, M. J. Am. Chem. Soc. 1999, 121, 10223.
32. See Mayr, H.; Minegishi, S. Angew. Chem. Int. Ed. 2002, 41, 4493. For a discussion of dynamic processes associated with the SN1 mechanism, see Peters, K.S. Chem. Rev. 2007, 107, 859.
33. For a related computational study, see Ruff, F.; Farkas, Ö; Kucsman, Á Eur. J. Org. Chem. 2006, 5570.
34. Parr, R. G.; Szentpály, L.V.; Liu, S. J. Am. Chem. Soc. 1999, 121, 1922. Also see Denekamp, C.; Sandlers, Y. Angew. Chem. Int. Ed. 2006, 45, 2093.
35. See Pérez, P. J. Org. Chem. 2004, 69, 5048.
36. Pérez, P.; Toro-Labbé, A.; Contreras, R. J. Am. Chem. Soc. 2001, 123, 5527.
37. Pérez, P.; Toro-Labbé, A.; Aizman, A.; Contreras, R. J. Org. Chem. 2002, 67, 4747.
38. Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Chem. Rev. 2006, 106, 2065.
39. Schaller, H.F.; Tishkov, A.A.; Feng, X.; Mayr, H. J. Am. Chem. Soc. 2008, 130, 3012.
40. See Okamoto, K. Adv. Carbocation Chem. 1989, 1, 171; Blandamer, M.J.; Scott, J.M.W.; Robertson, R.E. Prog. Phys. Org. Chem. 1985, 15, 149. Also see Dvorko, G.F.; Ponomareva, E.A.; Kulik, N.I. Russ. Chem. Rev. 1984, 53, 547.
41. Blandamer, M.J.; Burgess, J.; Duce, P.P.; Symons, M.C.R.; Robertson, R.E.; Scott, J.M.W. J. Chem. Res. (S) 1982, 130.
42. Benfey, O.T.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1952, 2488.
43. Bateman, L.C.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1940, 960.
44. In the experiments mentioned, the solvent was actually "70" or "80%" aq acetone. The "80%" aq acetone consists of 4 vol of dry acetone and 1 vol of water.
45. Creary, X.; Wang, Y.-X. J. Org. Chem. 1992, 57, 4761. Also see, Farcasiu, D.; Marino, G.; Harris, J.M.; Hovanes, B.A.; Hsu, C.S. J. Org. Chem. 1994, 59, 154.
46. Bateman, L.C.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1940, 1011.
47. McClelland, R.A.; Kanagasabapathy, V.M.; Steenken, S. J. Am. Chem. Soc. 1988, 110, 6913.
48. Schaller, H.F.; Mayr, H. Angew. Chem. Int. Ed. 2008, 47, 3958.
49. Fort, Jr., R.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 4, Wiley, NY, 1973, pp. 1783–1835.
50. Bartlett, P.D.; Knox, L.H. J. Am. Chem. Soc. 1939, 61, 3184.
51. For synthetic examples, see Kraus, G.A.; Hon, Y. J. Org. Chem. 1985, 50, 4605.
52. Olah, G.A.; Liang, G.; Wiseman, J.R.; Chong, J.A. J. Am. Chem. Soc. 1972, 74, 4927.
53. Della, E.W.; Pigou, P.E.; Tsanaktsidis, J. J. Chem. Soc., Chem. Commun. 1987, 833.
54. Eaton, P.E.; Yang, C.; Xiong, Y. J. Am. Chem. Soc. 1990, 112, 3225; Moriarty, R.M.; Tuladhar, S.M.; Penmasta, R.; Awasthi, A.K. J. Am. Chem. Soc. 1990, 112, 3228.
55. Hrovat, D.A.; Borden, W.T. J. Am. Chem. Soc. 1990, 112, 3227.
56. Lee, I.; Kim, N.D.; Kim, C.K. Tetrahedron Lett. 1992, 33, 7881.
57. White, E.H.; McGirk, R.H.; Aufdermarsh, Jr., C.A.; Tiwari, H.P.; Todd, M.J. J. Am. Chem. Soc. 1973, 95, 8107; Beak, P.; Harris, B.R. J. Am. Chem. Soc. 1974, 96, 6363.
58. For a review of reactions with the OCOCl leaving group, see Beak, P. Acc. Chem. Res. 1976, 9, 230.
59. See Arnett, E.M.; Molter, K.E. Acc. Chem. Res. 1985, 18, 339.
60. Lee, I.; Lee, Y.S.; Lee, B.-S.; Lee, H.W. J. Chem. Soc. Perkin Trans. 2 1993, 1441.
61. See Beletskaya, I.P. Russ. Chem. Rev. 1975, 44, 1067; Harris, J.M. Prog. Phys. Org. Chem. 1974, 11, 89; Raber, D.J.; Harris, J.M.; Schleyer, P.v.R. in Szwarc, M. Ions and Ion Pairs in Organic Reactions, Vol. 2, Wiley, NY, 1974, pp. 247–374.
62. For an alternative view, See Uggerud, E. J. Org. Chem. 2001, 66, 7084.
63. Proposed by Winstein, S.; Clippinger, E.; Fainberg, A.H.; Heck, R.; Robinson, G.C. J. Am. Chem. Soc. 1956, 78, 328.
64. Marcus, Y.; Hefter, G. Chem. Rev. 2006, 106, 4585.
65. See Kessler, H.; Feigel, M. Acc. Chem. Res. 1982, 15, 2.
66. Fry, J.L.; Lancelot, C.J.; Lam, L.K.M.; Harris, J.M.; Bingham, R.C.; Raber, D.J.; Hall, R.E.; Schleyer, P.v.R. J. Am. Chem. Soc. 1970, 92, 2538.
67. Savéant, J.-M. J. Am. Chem. Soc. 2008, 130, 4732.
68. See Richard, J.P.; Toteva, M.M.; Amyes, T.L. Org. Lett. 2001, 3, 2225.
69. Shiner Jr., V.J.; Fisher, R.D. J. Am. Chem. Soc. 1971, 93, 2553.
70. See McManus, S.P.; Safavy, K.K.; Roberts, F.E. J. Org. Chem. 1982, 47, 4388; Kinoshita, T.; Komatsu, K.; Ikai, K.; Kashimura, K.; Tanikawa, S.; Hatanaka, A.; Okamoto, K. J. Chem. Soc. Perkin Trans. 2 1988, 1875; Ronco, G.; Petit, J.; Guyon, R.; Villa, P. Helv. Chim. Acta 1988, 71, 648; Kevill, D.N.; Kyong, J.B.; Weitl, F.L. J. Org. Chem. 1990, 55, 4304.
71. Jia, Z.S.; Ottosson, H.; Zeng, X.; Thibblin, A. J. Org. Chem. 2002, 67, 182.
72. Diaz, A.F.; Lazdins, I.; Winstein, S. J. Am. Chem. Soc. 1968, 90, 1904.
73. Paradisi, C.; Bunnett, J.F. J. Am. Chem. Soc. 1985, 107, 8223; Fujio, M.; Sanematsu, F.; Tsuno, Y.; Sawada, M.; Takai, Y. Tetrahedron Lett. 1988, 29, 93.
74. Goering, H.L.; Hopf, H. J. Am. Chem. Soc. 1971, 93, 1224 and references cited therein.
75. Dietze, P.E.; Wojciechowski, M. J. Am. Chem. Soc. 1990, 112, 5240.
76. Cristol, S.J.; Noreen, A.L.; Nachtigall, G.W. J. Am. Chem. Soc. 1972, 94, 2187.
77. Simon, J.D.; Peters, K.S. J. Am. Chem. Soc. 1982, 104, 6142.
78. Goering, H.L.; Briody, R.G.; Sandrock, G. J. Am. Chem. Soc. 1970, 92, 7401.
79. Goering, H.L.; Briody, R.G.; Levy, J.F. J. Am. Chem. Soc. 1963, 85, 3059.
80. Winstein, S.; Gall, J.S.; Hojo, M.; Smith, S. J. Am. Chem. Soc. 1960, 82, 1010. See also, Shiner, Jr., V.J.; Hartshorn, S.R.; Vogel, P.C. J. Org. Chem. 1973, 38, 3604.
81. Jorgensen, W.L.; Buckner, J.K.; Huston, S.E.; Rossky, P.J. J. Am. Chem. Soc. 1987, 109, 1891.
82. Okamoto, K. Pure Appl. Chem. 1984, 56, 1797. Also see Lee, I.; Kim, H.Y.; Kang, H.K.; Lee, H.W. J. Org. Chem. 1988, 53, 2678; Lee, I.; Kim, H.Y.; Lee, H.W.; Kim, I.C. J. Phys. Org. Chem. 1989, 2, 35.
83. Okamoto, K.; Kinoshita, T.; Shingu, H. Bull. Chem. Soc. Jpn. 1970, 43, 1545.
84. Kinoshita, T.; Ueno, T.; Ikai, K.; Fujiwara, M.; Okamoto, K. Bull. Chem. Soc. Jpn. 1988, 61, 3273; Kinoshita, T.; Komatsu, K.; Ikai, K.; Kashimura, K.; Tanikawa, S.; Hatanaka, A.; Okamoto, K. J. Chem. Soc. Perkin Trans. 21988, 1875.
85. For an essay on borderline mechanisms in general, see Jencks, W.P. Chem. Soc. Rev. 1982, 10, 345.
86. Sneen, R.A.; Felt, G.R.; Dickason, W.C. J. Am. Chem. Soc. 1973, 95, 638 and references cited therein; Sneen, R.A. Acc. Chem. Res. 1973, 6, 46.
87. See Kevill, D.N.; Degenhardt, C.R. J. Am. Chem. Soc. 1979, 101, 1465.
88. See Sneen, R.A.; Felt, G.R.; Dickason, W.C. J. Am. Chem. Soc. 1973, 95, 638 and references cited therein; Sneen, R.A. Acc. Chem. Res. 1973, 6, 46; Blandamer, M.J.; Robertson, R.E.; Scott, J.M.W.; Vrielink, A. J. Am. Chem. Soc. 1980, 102, 2585; Stein, A.R. Can. J. Chem. 1987, 65, 363.
89. See Raber, D.J.; Harris, J.C.; Hall, R.E.; Schleyer, P.v.R. J. Am. Chem. Soc. 1971, 93, 4821; McLennan, D.J. Acc. Chem. Res. 1976, 9, 281; Stein, A.R. J. Org. Chem. 1976, 41, 519; Katritzky, A.R.; Musumarra, G.; Sakizadeh, K. J. Org. Chem. 1981, 46, 3831. For a reply, see Sneen, R.A.; Robbins, H.M. J. Am. Chem. Soc. 1972, 94, 7868. See Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982, pp. 442–450.
90. See Thibblin, A. J. Chem. Soc. Perkin Trans. 2 1987, 1629.
91. Katritzky, A.R.; Sakizadeh, K.; Gabrielsen, B.; le Noble, W.J. J. Am. Chem. Soc. 1984, 106, 1879.
92. Bentley, T.W.; Bowen, C.T.; Morten, D.H.; Schleyer, P.v.R. J. Am. Chem. Soc. 1981, 103, 5466.
93. Also see Laureillard, J.; Casadevall, A.; Casadevall, E. Tetrahedron 1984, 40, 4921; Helv. Chim. Acta 1984, 67, 352. For evidence against the SN2 (intermediate) mechanism, see Richard, J.P.; Amyes, T.L.; Vontor, T. J. Am. Chem. Soc. 1991, 113, 5871.
94. Amyes, T.L.; Richard, J.P. J. Am. Chem. Soc. 1990, 112, 9507. Also see Richard, J.P.; Rothenberg, M.E.; Jencks, W.P. J. Am. Chem. Soc. 1984, 106, 1361; Richard, J.P.; Jencks, W.P. J. Am. Chem. Soc. 1984, 106, 1373, 1383; Katritzky, A.R.; Brycki, B.E. J. Phys. Org. Chem. 1988, 1, 1; Stein, A.R. Can. J. Chem. 1989, 67, 297.
95. The relationship between electropilicity and rate coefficients is discussed in Aizman, A.; Contreras, R.; Pérez, P. Tetrahedron 2005, 61, 889.
96. See, however, Sneen, R.A.; Larsen, J.W. J. Am. Chem. Soc. 1969, 91, 6031.
97. Weiner, H.; Sneen, R.A. J. Am. Chem. Soc. 1965, 87, 287.
98. According to this scheme, the configuration of the isolated RN3 should be retained. It was, however, largely inverted, owing to a competing SN2 reaction where N3− directly attacks ROBs.
99. See Streitwieser, Jr., A.; Walsh, T.D.; Wolfe, Jr., J.R. J. Am. Chem. Soc. 1965, 87, 3682; Streitwieser, Jr., A.; Walsh, T.D. J. Am. Chem. Soc. 1965, 87, 3686; Beronius, P.; Nilsson, A.; Holmgren, A. Acta Chem. Scand. 1972, 26, 3173. See also, Knier, B.L.; Jencks, W.P. J. Am. Chem. Soc. 1980, 102, 6789.
100. Bank, S.; Noyd, D.A. J. Am. Chem. Soc. 1973, 95, 8203; Ashby, E.C.; Goel, A.B.; Park, W.S. Tetrahedron Lett. 1981, 22, 4209. For discussions of the relationship between SN2 and SET mechanisms, see Lewis, E.S. J. Am. Chem. Soc. 1989, 111, 7576; Shaik, S.S. Acta Chem. Scand. 1990, 44, 205.
101. See Savéant, J. Adv. Phys. Org. Chem. 1990, 26, 1; Ashby, E.C. Acc. Chem. Res. 1988, 21, 414. See also, Pross, A. Acc. Chem. Res. 1985, 18, 212; Chanon, M. Acc. Chem. Res. 1987, 20, 214. See Rossi, R.A.; Pierini, A.B.; Peñéñory, A.B. Chem. Rev. 2003, 103, 71.
102. Daasbjerg, K.; Lund, T.; Lund, H. Tetrahedron Lett. 1989, 30, 493.
103. See also, Fuhlendorff, R.; Lund, T.; Lund, H.; Pedersen, J.A. Tetrahedron Lett. 1987, 28, 5335.
104. See, for example, Russell, J.A.; Pecoraro, J.M. J. Am. Chem. Soc. 1979, 101, 3331.
105. Santiago, A.N.; Morris, D.G.; Rossi, R.A. J. Chem. Soc., Chem. Commun. 1988, 220.
106. See Newcomb, M.; Curran, D.P. Acc. Chem. Res. 1988, 21, 206; Newcomb, M. Acta Chem. Scand. 1990, 44, 299. For replies to this criticism, see Ashby, E.C. Acc. Chem. Res. 1988, 21, 414; Ashby, E.C.; Pham, T.N.; Amrollah-Madjdabadi, A.A. J. Org. Chem. 1991, 56, 1596.
107. In this book, there is a distinction between the SET and SRN1 mechanisms. However, many workers use the designation SET to refer to the SRN1, the chain version of the SET, or both.
108. Nazareno, M.A.; Rossi, R.A. J. Org. Chem. 1996, 61, 1645.
109. Ashby, E.C.; Sun, X.; Duff, J.L. J. Org. Chem. 1994, 59, 1270.
110. Haberfield, P. J. Am. Chem. Soc.1995, 117, 3314.
111. Shaik, S.S. Acta Chem. Scand.1990, 44, 205.
112. Ashby, E.C.; Park, B.; Patil, G.S.; Gadru, K.; Gurumurthy, R. J. Org. Chem. 1993, 58, 424.
113. See Capon, B.; McManus, S. Neighboring Group Participation, Vol. 1, Plenum, NY, 1976.
114. See McCortney, B.A.; Jacobson, B.M.; Vreeke, M.; Lewis, E.S. J. Am. Chem. Soc. 1990, 112, 3554.
115. See Page, M.I. Chem. Soc. Rev. 1973, 2, 295.
116. Winstein, S.; Lucas, H.J. J. Am. Chem. Soc. 1939, 61, 1576, 2845.
117. For a theoretical treatment of strain energy release and intrinsic barriers for internal SN2 reactions, see Wolk, J.L.; Rozental, E.; Basch, H.; Hoz, S. J. Org. Chem. 2006, 71, 3876.
118. Allred, E.L.; Winstein, S. J. Am. Chem. Soc. 1967, 89, 3991, 3998.
119. Allred, E.L.; Winstein, S. J. Am. Chem. Soc. 1967, 89, 4012.
120. Eliel, E.L.; Clawson, L.; Knox, D.E. J. Org. Chem. 1985, 50, 2707; Eliel, E.L.; Knox, D.E. J. Am. Chem. Soc. 1985, 107, 2946.
121. See Wilen, S.H.; Delguzzo, L.; Saferstein, R. Tetrahedron 1987, 43, 5089.
122. See Perst, H. Oxonium Ions in Organic Chemistry, Verlag Chemie, Deerfield Beach, FL, 1971, pp. 100–127. Also see Francl, M.M.; Hansell, G.; Patel, B.P.; Swindell, C.S. J. Am. Chem. Soc. 1990, 112, 3535.
123. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 141–145.
124. Lambert, J.B.; Beadle, B.M.; Kuang, K. J. Org. Chem. 1999, 64, 9241.
125. Peterson, P.E. Acc. Chem. Res. 1971, 4, 407, and references cited therein.
126. Peterson, P.E.; Bopp, R.J.; Chevli, D.M.; Curran, E.L.; Dillard, D.E.; Kamat, R.J. J. Am. Chem. Soc. 1967, 89, 5902. See also, Reich, I.L.; Reich, H.J. J. Am. Chem. Soc. 1974, 96, 2654.
127. See Olah, G.A. Halonium Ions, Wiley, NY, 1975; Koster, G.F. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, pp. 1265–1351.
128. See Henrichs, P.M.; Peterson, P.E. J. Org. Chem. 1976, 41, 362; Vancik, H.; Percac, K.; Sunko, D.E. J. Chem. Soc., Chem. Commun. 1991, 807.
129. Olah, G.A.; Bollinger, J.M.; Mo, Y.K.; Brinich, J.M. J. Am. Chem. Soc. 1972, 94, 1164.
130. Haupt, F.C.; Smith, M.R. Tetrahedron Lett. 1974, 4141.
131. See Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972; Bartlett, P.D. Nonclassical Ions, W.A. Benjamin, NY, 1965. Barkhash, V.A. Top. Curr. Chem. 1984, 116/117, 1; McManus, S.P.; Pittman Jr., C.U. in McManus, S.P. Organic Reactive Intermediates, Academic Press, NY, 1973, pp. 302–321.
132. Olah, G.A. J. Org. Chem. 2005, 70, 2413.
133. Sieber, S.; Schleyer, P.v.R.; Vancik, H.; Mesic, M.; Sunko, D.E. Angew. Chem. Int. Ed. 1993, 32, 1604; Schleyer, P.v.R.; Sieber, S. Angew. Chem. Int. Ed. 1993, 32, 1606.
134. Herrmann, R.; Kirmse, W. Liebigs Ann. Chem. 1995, 703.
135. Bartlett, P.D.; Bank, S.; Crawford, R.J.; Schmid, G.H. J. Am. Chem. Soc. 1965, 88, 1288.
136. Winstein, S.; Carter, P. J. Am. Chem. Soc. 1961, 83, 4485.
137. For example, see Brunelle, P.; Sorensen, T.S.; Taeschler, C. J. Org. Chem. 2001, 66, 7294.
138. Okazaki, T.; Terakawa, E.; Kitagawa, T.; Takeuchi, K. J. Org. Chem. 2000, 65, 1680.
139. Smith, W. B. J. Org. Chem. 2001, 66, 376.
140. This was pointed out by Cram, D.J. J. Am. Chem. Soc. 1964, 86, 3767.
141. See Brown, H.C. The Nonclassical Ion Problem, Plenum, NY, 1977. This book also includes rebuttals by Schleyer, P.v.R. See also, Brown, H.C. Pure Appl. Chem. 1982, 54, 1783.
142. See Story, P.R.; Clark, Jr., B.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, pp. 1007–1060; Richey, Jr., H.G. in Zabicky, J. The Chemistry of Alkenes, Vol. 2, Wiley, NY, 1970, pp. 77–101.
143. Winstein, S.; Shatavsky, M. J. Am. Chem. Soc. 1956, 78, 592.
144. Story, P.R.; Snyder, L.C.; Douglass, D.C.; Anderson, E.W.; Kornegay, R.L. J. Am. Chem. Soc. 1963, 85, 3630. See Story, P.R.; Clark, Jr., B.C. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, pp. 1026–1041; Lustgarten, R.K.; Brookhart, M.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2347.
145. See Gassman, P.G.; Doherty, M.M. J. Am. Chem. Soc. 1982, 104, 3742 and references cited therein; Laube, T. J. Am. Chem. Soc. 1989, 111, 9224.
146. See Schleyer, P.v.R.; Bentley, T.W.; Koch, W.; Kos, A.J.; Schwarz, H. J. Am. Chem. Soc. 1987, 109, 6953; Fernández-Mateos, A.; Rentzsch, M.; Sánchez, L.R.; González, R.R. Tetrahedron 2001, 57, 4873.
147. See Ferber, P.H.; Gream, G.E. Aust. J. Chem. 1981, 34, 1051; Orlovic, M.; Borcic, S.; Humski, K.; Kronja, O.; Imper, V.; Polla, E.; Shiner, Jr., V.J. J. Org. Chem. 1991, 56, 1874.
148. Bly, R.S.; Bly, R.K.; Bedenbaugh, A.O.; Vail, O.R. J. Am. Chem. Soc. 1967, 89, 880.
149. See Peterson, P.E.; Vidrine, D.W. J. Org. Chem. 1979, 44, 891; Rappoport, Z. React. Intermed. (Plenum) 1983, 3, 440.
150. Von Lehman, T.; Macomber, R. J. Am. Chem. Soc. 1975, 97, 1531.
151. Gassman, P.G.; Zeller, J.; Lamb, J.T. Chem. Commun. 1968, 69.
152. See Olah, G.A.; Berrier, A.L.; Arvanaghi, M.; Prakash, G.K.S. J. Am. Chem. Soc. 1981, 103, 1122.
153. Gassman, P.G.; Fentiman, Jr., A.F. J. Am. Chem. Soc. 1969, 91, 1545; 1970, 92, 2549.
154. See Lambert, J.B.; Mark, H.W.; Holcomb, A.G.; Magyar, E.S. Acc. Chem. Res. 1979, 12, 317.
155. Malnar, I.; Juric, S.; Vrcek, V.; Gjuranovic, Z.; Mihalic, Z.; Kronja, O. J. Org. Chem. 2002, 67, 1490.
156. Gassman, P.G.; Hall, J.B. J. Am. Chem. Soc. 1984, 106, 4267.
157. In this section, systems are considered in which at least one carbon separates the cyclopropyl ring from the carbon bearing the leaving group. For a discussion of systems in which the cyclopropyl group is directly attached to the leaving-group carbon, see below, category 4.b.
158. For a review, see Haywood-Farmer, J. Chem. Rev. 1974, 74, 315.
159. Tanida, H.; Tsuji, T.; Irie, T. J. Am. Chem. Soc. 1967, 89, 1953; Battiste, M.A.; Deyrup, C.L.; Pincock, R.E.; Haywood-Farmer, J. J. Am. Chem. Soc. 1967, 89, 1954.
160. For a competitive study of cyclopropyl versus double-bond participation, see Lambert, J.B.; Jovanovich, A.P.; Hamersma, J.W.; Koeng, F.R.; Oliver, S.S. J. Am. Chem. Soc. 1973, 95, 1570.
161. Also see Gassman, P.G.; Creary, X. J. Am. Chem. Soc. 1973, 95, 2729; Takakis, I.M.; Rhodes, Y.E. Tetrahedron Lett. 1983, 24, 4959.
162. Haywood-Farmer, J. Chem. Rev. 1974, 74, 315.
163. Haywood-Farmer, J.; Pincock, R.E. J. Am. Chem. Soc. 1969, 91, 3020. Also see Rhodes, Y.E.; Takino, T. J. Am. Chem. Soc. 1970, 92, 4469; Hanack, M.; Krause, P. Liebigs Ann. Chem. 1972, 760, 17.
164. Gassman, P.G.; Seter, J.; Williams, F.J. J. Am. Chem. Soc. 1971, 93, 1673. See Haywood-Farmer, J.; Pincock, R.E. J. Am. Chem. Soc. 1969, 91, 3020; Chenier, P.J.; Jenson, T.M.; Wulff, W.D. J. Org. Chem. 1982, 47, 770.
165. See Schipper, P.; Driessen, P.B.J.; de Haan, J.W.; Buck, H.M. J. Am. Chem. Soc. 1974, 96, 4706; Ohkata, K.; Doecke, C.W.; Klein, G.; Paquette, L.A. Tetrahedron Lett. 1980, 21, 3253.
166. See Lancelot, L.A.; Cram, D.J.; Schleyer, P.v.R. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, pp. 1347–1483.
167. Kevill, D.N.; D'Souza, M.J. J. Chem. Soc. Perkin Trans. 2 1997, 257.
168. Cram, D.J. J. Am. Chem. Soc.1949, 71, 3863; 1952, 74, 2129.
169. Brookhart, M.; Anet, F.A.L.; Cram, D.J.; Winstein, S. J. Am. Chem. Soc. 1966, 88, 5659; Lee, C.C.; Unger, D.; Vassie, S. Can. J. Chem. 1972, 50, 1371.
170. Brown, H.C.; Kim, C.J. J. Am. Chem. Soc. 1971, 93, 5765.
171. Diaz, A.; Winstein, S. J. Am. Chem. Soc. 1969, 91, 4300. See also, Schadt, F.L.; Lancelot, C.J.; Schleyer, P.v.R. J. Am. Chem. Soc. 1978, 100, 228.
172. Nordlander, J.E.; Kelly, W.J. J. Am. Chem. Soc. 1969, 91, 996.
173. Jablonski, R.J.; Snyder, E.I. J. Am. Chem. Soc. 1969, 91, 4445.
174. Thompson, J.A.; Cram, D.J. J. Am. Chem. Soc. 1969, 91, 1778. See also Kingsbury, C.A.; Best, D.C. Bull. Chem. Soc. Jpn. 1972, 45, 3440.
175. Coke, J.L.; McFarlane, F.E.; Mourning, M.C.; Jones, M.G. J. Am. Chem. Soc. 1969, 91, 1154; Jones, M.G.; Coke, J.L. J. Am. Chem. Soc. 1969, 91, 4284. See also, Harris, J.M.; Schadt, F.L.; Schleyer, P.v.R.; Lancelot, C.J. J. Am. Chem. Soc. 1969, 91, 7508.
176. See Ando, T.; Shimizu, N.; Kim, S.; Tsuno, Y.; Yukawa, Y. Tetrahedron Lett. 1973, 117.
177. Lancelot, C.J.; Schleyer, P.v.R. J. Am. Chem. Soc. 1969, 91, 4291, 4296; Lancelot, C.J.; Harper, J.J.; Schleyer, P.v.R. J. Am. Chem. Soc. 1969, 91, 4294; Schleyer, P.v.R.; Lancelot, C.J. J. Am. Chem. Soc. 1969, 91, 4297.
178. Ramsey, B.; Cook Jr., J.A.; Manner, J.A. J. Org. Chem. 1972, 37, 3310.
179. Olah, G.A.; Comisarow, M.B.; Kim, C.J. J. Am. Chem. Soc. 1969, 91, 1458. See, however, Ramsey, B.; Cook, Jr., J.A.; Manner, J.A. J. Org. Chem. 1972, 37, 3310.
180. Olah, G.A.; Spear, R.J.; Forsyth, D.A. J. Am. Chem. Soc. 1976, 98, 6284.
181. See Olah, G.A.; Singh, B.P.; Liang, G. J. Org. Chem. 1984, 49, 2922; Olah, G.A.; Singh, B.P. J. Am. Chem. Soc. 1984, 106, 3265.
182. Mishima, M.; Tsuno, Y.; Fujio, M. Chem. Lett. 1990, 2277.
183. See Tanida, H. Acc. Chem. Res. 1968, 1, 239; Shiner, Jr., V.J.; Seib, R.C. J. Am. Chem. Soc. 1976, 98, 862; Ferber, P.H.; Gream, G.E. Aust. J. Chem. 1981, 34, 2217; Fujio, M.; Goto, M.; Seki, Y.; Mishima, M.; Tsuno, Y.; Sawada, M.; Takai, Y. Bull. Chem. Soc. Jpn. 1987, 60, 1097. For a discussion of evidence obtained from isotope effects, see Scheppele, S.E. Chem. Rev. 1972, 72, 511, p. 522.
184. Jackman, L.M.; Haddon, V.R. J. Am. Chem. Soc. 1974, 96, 5130; Gates, M.; Frank, D.L.; von Felten, W.C. J. Am. Chem. Soc. 1974, 96, 5138; Ando, T.; Yamawaki, J.; Saito, Y. Bull. Chem. Soc. Jpn. 1978, 51, 219.
185. See Olah, G.A. Angew. Chem. Int. Ed. 1973, 12, 173, pp. 192–198.
186. See Olah, G.A.; Prakash, G.K.S.; Williams, R.E. Hypercarbon Chemistry, Wiley, NY, 1987, pp. 157–170; Grob, C.A. Angew. Chem. Int. Ed. 1982, 21, 87; Sargent, G.D. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, pp. 1099–1200; Sargent, G.D. Q. Rev. Chem. Soc. 1966, 20, 301; Gream, G.E. Rev. Pure Appl. Chem. 1966, 16, 25. Also see Kirmse, W. Acc. Chem. Res. 1986, 19, 36. See also, Ref. 190.
187. Winstein, S.; Clippinger, E.; Howe, R.; Vogelfanger, E. J. Am. Chem. Soc. 1965, 87, 376.
188. For another view, see Bielmann, R.; Fuso, F.; Grob, C.A. Helv. Chim. Acta 1988, 71, 312; Flury, P.; Grob, C.A.; Wang, G.Y.; Lennartz, H.; Roth, W.R. Helv. Chim. Acta 1988, 71, 1017.
189. See Menger, F.M.; Perinis, M.; Jerkunica, J.M.; Glass, L.E. J. Am. Chem. Soc. 1978, 100, 1503.
190. See Lenoir, D.; Apeloig, Y.; Arad, D.; Schleyer, P.v.R. J. Org. Chem. 1988, 53, 661; Grob, C.A. Acc. Chem. Res. 1983, 16, 426; Brown, H.C. Acc. Chem. Res. 1983, 16, 432; Walling, C. Acc. Chem. Res. 1983, 16, 448. Also see Arnett, E.M.; Hofelich, T.C.; Schriver, G.W. React. Intermed. (Wiley) 1985, 3, 189, pp. 193–202.
191. See Lajunen, M. Acc. Chem. Res. 1985, 18, 254; Apeloig, Y.; Arad, D.; Schleyer, P.v.R. J. Org. Chem. 1988, 53, 661.
192. Also see Werstiuk, N.H.; Dhanoa, D.; Timmins, G. Can. J. Chem. 1983, 61, 2403; Brown, H.C.; Ikegami, S.; Vander Jagt, D.L. J. Org. Chem. 1985, 50, 1165; Nickon, A.; Swartz, T.D.; Sainsbury, D.M.; Toth, B.R. J. Org. Chem. 1986, 51, 3736.
193. The presence of hydride shifts (Reaction 18-01) under solvolysis conditions has complicated the interpretation of the data.
194. Olah, G.A. Acc. Chem. Res. 1976, 9, 41; Saunders, M. Acc. Chem. Res. 1983, 16, 440. See also, Johnson, S.A.; Clark, D.T. J. Am. Chem. Soc. 1988, 110, 4112.
195. See Kramer, G.M.; Scouten, C.G. Adv. Carbocation Chem. 1989, 1, 93. See, however, Olah, G.A.; Prakash, G.K.S.; Farnum, D.G.; Clausen, T.P. J. Org. Chem. 1983, 48, 2146.
196. Myhre, P.C.; Webb, G.G.; Yannoni, C.S. J. Am. Chem. Soc. 1990, 112, 8991.
197. See Lossing, F.P.; Holmes, J.L. J. Am. Chem. Soc. 1984, 106, 6917 and references cited therein.
198. Koch, W.; Liu, B.; DeFrees, D.J.; Sunko, D.E.; Vancik, H. Angew. Chem. Int. Ed. 1990, 29, 183.
199. See, for example, Koch, W.; Liu, B.; DeFrees, D.J. J. Am. Chem. Soc. 1989, 111, 1527.
200. Olah, G.A.; DeMember, J.R.; Lui, C.Y.; White, A.M. J. Am. Chem. Soc. 1969, 91, 3958. See also, Forsyth, D.A.; Panyachotipun, C. J. Chem. Soc., Chem. Commun. 1988, 1564.
201. Olah, G.A. Acc. Chem. Res. 1976, 9, 41. See also, Farnum, D.G.; Wolf, A.D. J. Am. Chem. Soc. 1974, 96, 5166.
202. Nickon, A.; Lin, Y. J. Am. Chem. Soc. 1969, 91, 6861. See also, Montgomery, L.K.; Grendze, M.P.; Huffman, J.C. J. Am. Chem. Soc. 1987, 109, 4749.
203. Fry, A.J.; Farnham, W.B. J. Org. Chem. 1969, 34, 2314.
204. Farnum, W.B.; Botto, R.E.; Chambers, W.T.; Lam, B. J. Am. Chem. Soc. 1978, 100, 3847. See also, Olah, G.A.; Berrier, A.L.; Prakash, G.K.S. J. Org. Chem. 1982, 47, 3903.
205. See in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, the articles by Richey, Jr., H.G. pp. 1201–1294, and by Wiberg, K.B.; Hess, Jr., B.A.; Ashe III, A.J. pp. 1295–1345; Sarel, S.; Yovell, J.; Sarel-Imber, M. Angew. Chem. Int. Ed. 1968, 7, 577.
206. Roberts, D.D.; Mazur, R.H. J. Am. Chem. Soc. 1951, 73, 2509.
207. See Roberts, D.D.; Snyder, Jr., R.C. J. Org. Chem. 1979, 44, 2860, and references cited therein.
208. Wiberg, K.B.; Ashe, III, A.J. J. Am. Chem. Soc. 1968, 90, 63.
209. Schleyer, P.v.R.; Van Dine, G.W. J. Am. Chem. Soc. 1966, 88, 2321. See also, Kevill, D.N.; Abduljaber, M.H. J. Org. Chem. 2000, 65, 2548.
210. See Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 3, Wiley, NY, 1972, the article by Wiberg, K.B.; Hess, Jr., B.A.; Ashe, III, A.J. pp. 1300–1303.
211. See Rhodes, Y.E.; DiFate, V.G. J. Am. Chem. Soc. 1972, 94, 7582. See, however, Brown, H.C.; Peters, E.N. J. Am. Chem. Soc. 1975, 97, 1927.
212. Majerski, Z.; Schleyer, P.v.R. J. Am. Chem. Soc. 1971, 93, 665.
213. Koch, W.; Liu, B.; DeFrees, D.J. J. Am. Chem. Soc. 1988, 110, 7325; Saunders, M.; Laidig, K.E.; Wiberg, K.B.; Schleyer, P.v.R. J. Am. Chem. Soc. 1988, 110, 7652.
214. Staral, J.S.; Yavari, I.; Roberts, J.D.; Prakash, G.K.S.; Donovan, D.J.; Olah, G.A. J. Am. Chem. Soc. 1978, 100, 8016. See also, Prakash, G.K.S.; Arvanaghi, M.; Olah, G.A. J. Am. Chem. Soc. 1985, 107, 6017; Myhre, P.C.; Webb, G.G.; Yannoni, C.S. J. Am. Chem. Soc. 1990, 112, 8992.
215. See Yamataka, H.; Ando, T.; Nagase, S.; Hanamura, M.; Morokuma, K. J. Org. Chem. 1984, 49, 631. For an opposing view, see Zamashchikov, V.V.; Rudakov, E.S.; Bezbozhnaya, T.V.; Matveev, A.A. J. Org. Chem. USSR1984, 20, 11.
216. Silver, M.S.; Meek, A.G. Tetrahedron Lett. 1971, 3579; Dupuy, W.E.; Hudson, H.R. J. Chem. Soc. Perkin Trans. 2 1972, 1715.
217. For further discussions of protonated cyclopropanes, see Sec. 15.B.iv, 18.A.ii.
218. Olah, G.A.; DeMember, J.R.; Commeyras, A.; Bribes, J.L. J. Am. Chem. Soc. 1971, 93, 459.
219. Johnson, S.A.; Clark, D.T. J. Am. Chem. Soc. 1988, 110, 4112. See also, Carneiro, J.W.; Schleyer, P.v.R.; Koch, W.; Raghavachari, K. J. Am. Chem. Soc. 1990, 112, 4064.
220. Fujiyama, R.; Munechika, T. Tetrahedron Lett. 1993, 34, 5907.
221. See Buzek, P.; Schleyer, P.v.R.; Sieber, S.; Koch, W.; Carneiro, J.W. de M.; Vancik, H.; Sunko, D.E. J. Chem. Soc., Chem. Commun. 1991, 671; Imhoff, M.A.; Ragain, R.M.; Moore, K.; Shiner, V.J. J. Org. Chem. 1991, 56, 3542.
222. Dannenberg, J.J.; Barton, J.K.; Bunch, B.; Goldberg, B.J.; Kowalski, T. J. Org. Chem. 1983, 48, 4524; Allen, A.D.; Ambidge, I.C.; Tidwell, T.T. J. Org. Chem. 1983, 48, 4527.
223. Lee, C.C.; Clayton, J.W.; Lee, C.C.; Finlayson, A.J. Tetrahedron 1962, 18, 1395.
224. Lewis, E.S.; Boozer, C.E. J. Am. Chem. Soc. 1952, 74, 308.
225. Lewis, E.S.; Witte, K. J. Chem. Soc. B 1968, 1198. Also see Kice, J.L.; Hanson, G.C. J. Org. Chem. 1973, 38, 1410; Cohen, T.; Solash, J. Tetrahedron Lett. 1973, 2513; Verrinder, D.J.; Hourigan, M.J.; Prokipcak, J.M. Can. J. Chem. 1978, 56, 2582.
226. See DeWolfe, R.H. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 417–437. For comprehensive older reviews, see DeWolfe, R.H.; Young, W.G. Chem. Rev. 1956, 56, 753; in Patai, S. The Chemistry of Alkenes, Wiley, NY, 1964, the sections by Mackenzie, K. pp. 436–453 and DeWolfe, R.H.; Young, W.G. pp. 681–738.
227. DeWolfe, R.H.; Young, W.G. Chem. Rev. 1956, 56, 753 give several dozen such examples.
228. Katritzky, A.R.; Fara, D.C.; Yang, H.; Tämm, K.; Tamm, T.; Karelson, M. Chem. Rev. 2004, 104, 175.
229. Young, W.G.; Winstein, S.; Goering, H.L. J. Am. Chem. Soc. 1951, 73, 1958.
230. See Kantner, S.S.; Humski, K.; Goering, H.L. J. Am. Chem. Soc. 1982, 104, 1693; Thibblin, A. J. Chem. Soc. Perkin Trans. 2 1986, 313.
231. See Magid, R.M. Tetrahedron 1980, 36, 1901, see pp. 1901–1910.
232. Streitwieser, A.; Jayasree, E.G.; Leung, S.S.-H.; Choy, G.S.-C. J. Org. Chem. 2005, 70, 8486.
233. Bordwell, F.G.; Clemens, A.H.; Cheng, J. J. Am. Chem. Soc. 1987, 109, 1773. Also see, Young, J.-j; Jung, L.-j.; Cheng, K.-m. Tetrahedron Lett. 2000, 41, 3411.
234. Hirab, T.; Nojima, M.; Kusabayashi, S. J. Org. Chem. 1984, 49, 4084.
235. Hirashita, T.; Hayashi, Y.; Mitsui, K.; Araki, S. Tetrahedron Lett. 2004, 45, 3225.
236. Bordwell, F.G.; Mecca, T.G. J. Am. Chem. Soc. 1972, 94, 5829. See also Dewar, M.J.S. J. Am. Chem. Soc. 1984, 106, 209.
237. See Uebel, J.J.; Milaszewski, R.F.; Arlt, R.E. J. Org. Chem. 1977, 42, 585.
238. See Fry, A. Pure Appl. Chem. 1964, 8, 409; Georgoulis, C.; Ville, G. Bull. Soc. Chim. Fr. 1985, 485; Meislich, H.; Jasne, S.J. J. Org. Chem. 1982, 47, 2517.
239. Paquette, L.A.; Stirling, C.J.M. Tetrahedron 1992, 48, 7383.
240. See Magid, R.M.; Fruchey, O.S. J. Am. Chem. Soc. 1979, 101, 2107; Bäckvall, J.E.; Vågberg, J.O.; Genêt, J.P. J. Chem. Soc., Chem. Commun. 1987, 159.
241. See Stork, G.; Schoofs, A.R. J. Am. Chem. Soc. 1979, 101, 5081.
242. Bach, R.D.; Wolber, G.J. J. Am. Chem. Soc. 1985, 107, 1352; Stohrer, W. Angew. Chem. Int. Ed. 1983, 22, 613.
243. Young, W.G. J. Chem. Educ. 1962, 39, 456. See Corey, E.J.; Boaz, N.W. Tetrahedron Lett. 1984, 25, 3055.
244. For a theoretical study, see Streitwieser, A.; Jayasree, E.G.; Hasanayn, F.; Leung, S.S.-H. J. Org. Chem. 2008, 73, 9426.
245. Vermeer, P.; Meijer, J.; Brandsma, L. Recl. Trav. Chim. Pays-Bas 1975, 94, 112.
246. See Schuster, H.F.; Coppola, G.M. Allenes in Organic Synthesis, Wiley, NY, 1984, pp. 12–19, 26–30; Taylor, D.R. Chem. Rev. 1967, 67, 317, pp. 324–328. See Larock, R.C.; Reddy, Ch.K. Org. Lett. 2000, 2, 3325.
247. See Swaminathan, S.; Narayanan, K.V. Chem. Rev. 1971, 71, 429; Andres, J.; Cardenas, R.; Silla, E.; Tapi, O. J. Am. Chem. Soc. 1988, 110, 666.
248. Corey, E.J.; Boaz, N.W. Tetrahedron Lett. 1984, 25, 3059, 3063.
249. Ibuka, T.; Taga, T.; Habashita, H.; Nakai, K.; Tamamura, H.; Fujii, N.; Chounan, Y.; Nemoto, H.; Yamamoto, Y. J. Org. Chem. 1993, 58, 1207.
250. Wipf, P.; Fritch, P.C. J. Org. Chem. 1994, 59, 4875.
251. Smith, M.B.; Hrubiec, R.T. Tetrahedron 1984, 40, 1457; Hrubiec, R.T.; Smith, M.B. J. Org. Chem. 1984, 49, 385.
252. See Rappoport, Z. Recl. Trav. Chim. Pays-Bas 1986, 104, 309; Shainyan, B.A. Russ. Chem. Rev. 1986, 55, 511; Modena, G. Acc. Chem. Res. 1971, 4, 73.
253. Truce, W.E.; Boudakian, M.M. J. Am. Chem. Soc. 1956, 78, 2748.
254. Hancock, J.W.; Morrell, C.E.; Rhom, D. Tetrahedron Lett. 1962, 987.
255. Bernasconi, C.F.; Fassberg, J.; Killion, Jr., R.B.; Rappoport, Z. J. Org. Chem. 1990, 55, 4568.
256. See Rappoport, Z.; Peled, P. J. Am. Chem. Soc. 1979, 101, 2682, and references cited therein.
257. Avramovitch, B.; Weyerstahl, P.; Rappoport, Z. J. Am. Chem. Soc. 1987, 109, 6687.
258. See Rybinskaya, M.I.; Nesmeyanov, A.N.; Kochetkov, N.K. Russ. Chem. Rev. 1969, 38, 433.
259. Rappoport, Z. Adv. Phys. Org. Chem. 1969, 7, see pp. 31–62; Shainyan, B.A. Russ. Chem. Rev. 1986, 55, 516. See also, Rappoport, Z.; Gazit, A. J. Am. Chem. Soc. 1987, 109, 6698.
260. See Rappoport, Z.; Gazit, A. J. Am. Chem. Soc. 1986, 51, 4112; Park, K.P.; Ha, H. Bull. Chem. Soc. Jpn. 1990, 63, 3006.
261. Shiers, J.J.; Shipman, M.; Hayes, J.-F.; Slawin, A.M.Z. J. Am. Chem. Soc. 2004, 126, 6868.
262. Apeloig, Y.; Rappoport, Z. J. Am. Chem. Soc. 1979, 101, 5095.
263. See Stang, P.J.; Rappoport, Z.; Hanack, H.; Subramanian, L.R. Vinyl Cations, Chapter 5, Academic Press, NY, 1979; Stang, P.J. Acc. Chem. Res. 1978, 11, 107; Rappoport, Z. Acc. Chem. Res. 1976, 9, 265.
264. See Stang, P.J.; Rappoport, Z.; Hanack, H.; Subramanian, L.R. Vinyl Cations, Chap. 6, Academic Press, NY, 1979.
265. Hanack, M.; Bässler, T.; Eymann, W.; Heyd, W.E.; Kopp, R. J. Am. Chem. Soc. 1974, 96, 6686.
266. Grob, C.A.; Spaar, R. Helv. Chim. Acta 1970, 53, 2119.
267. Hassdenteufel, J.R.; Hanack, M. Tetrahedron Lett. 1980, 503. See also, Kobayashi, S.; Nishi, T.; Koyama, I.; Taniguchi, H. J. Chem. Soc., Chem. Commun. 1980, 103.
268. Schiavelli, M.D.; Gilbert, R.P.; Boynton, W.A.; Boswell, C.J. J. Am. Chem. Soc. 1972, 94, 5061.
269. See Hanack, M.; Märkl, R.; Martinez, A.G. Chem. Ber. 1982, 115, 772.
270. Kelsey, D.R.; Bergman, R.G. J. Am. Chem. Soc. 1971, 93, 1941.
271. Pfeifer, W.D.; Bahn, C.A.; Schleyer, P.v.R.; Bocher, S.; Harding, C.E.; Hummel, K.; Hanack, M.; Stang, P.J. J. Am. Chem. Soc. 1971, 93, 1513.
272. See Clarke, T.C.; Bergman, R.G. J. Am. Chem. Soc. 1974, 96, 7934; Summerville, R.H.; Schleyer, P.v.R. J. Am. Chem. Soc. 1972, 94, 3629; 1974, 96, 1110
273. Maroni, R.; Melloni, G.; Modena, G. J. Chem. Soc., Chem. Commun. 1972, 857.
274. Angelini, G.; Hanack, M.; Vermehren, J.; Speranza, M. J. Am. Chem. Soc. 1988, 110, 1298.
275. See Cacace, F. Adv. Phys. Org. Chem. 1970, 8, 79. See also, Fornarini, S.; Speranza, M. J. Am. Chem. Soc. 1985, 107, 5358.
276. Flynn Jr., J.; Badiger, V.V.; Truce, W.E. J. Org. Chem. 1963, 28, 2298. See also, Shainyan, B.A.; Mirskova, A.N. J. Org. Chem. USSR 1984, 20, 885, 1989; 1985, 21, 283.
277. Bottini, A.T.; Corson, F.P.; Fitzgerald, R.; Frost II, K.A. Tetrahedron 1972, 28, 4883.
278. See Popov, A.F.; Piskunova, Z.; Matvienko, V.N. J. Org. Chem. USSR 1986, 22, 1299.
279. See Andrisano, R.; Angeloni, A.S.; De Maria, P.; Tramontini, M. J. Chem. Soc. C 1967, 2307.
280. See Rappoport, Z. Acc. Chem. Res. 1981, 14, 7; Rappoport, Z.; Avramovitch, B. J. Org. Chem. 1982, 47, 1397.
281. Galli, C.; Gentili, P.; Rappoport, Z. J. Org. Chem. 1994, 59, 6786.
282. See DePuy, C.H.; Gronert, S.; Mullin, A.; Bierbaum, V.M. J. Am. Chem. Soc. 1990, 112, 8650.
283. For a reported example, see Edwards, O.E.; Grieco, C. Can. J. Chem. 1974, 52, 3561.
284. See Anderson, P.H.; Stephenson, B.; Mosher, H.S. J. Am. Chem. Soc. 1974, 96, 3171.
285. See Streitwieser, A. Solvolytic Displacement Reactions, McGraw-Hill, NY, 1962, p. 13.
286. For evidence, see Caldwell, G.; Magnera, T.F.; Kebarle, P. J. Am. Chem. Soc. 1984, 106, 959.
287. For a discussion of the interplay between steric and electronic effects, see Fernández, I.; Frenking, G.; Uggerud, E. Chemistry: Eur. J. 2009, 15, 2166.
288. Liu, K.-T.; Hou, S.-J.; Tsao, K.-L. J. Org. Chem. 1998, 63, 1360.
289. Fujio, M.; Nomura, H.; Nakata, K.; Saeki, Y.; Mishima, M.; Kobayashi, S.; Matsushita, T.; Nishimoto, K.; Tsuno, Y. Tetrahedron Lett. 1994, 35, 5005.
290. Fujio, M.; Nakata, K.; Kuwamura, T.; Nakamura, H.; Saeki, Y.; Mishima, M.; Kobayashi, S.; Tsuno, Y. Tetrahedron Lett. 1992, 34, 8309.
291. Liu, K.T.; Tsao, M.-L.; Chao, I. Tetrahedron Lett. 1996, 37, 4173.
292. See DeTar, D.F.; Binzet, S.; Darba, P. J. Org. Chem. 1987, 52, 2074.
293. See Streitwieser, A. Solvolytic Displacement Reactions, McGraw-Hill, NY, 1962, p. 43.
294. See Zamashchikov, V.V.; Bezbozhnaya, T.V.; Chanysheva, I.R. J. Org. Chem. USSR 1986, 22, 1029.
295. See Dietze, P.E.; Jencks, W.P. J. Am. Chem. Soc. 1986, 108, 4549; Dietze, P.E.; Hariri, R.; Khattak, J. J. Org. Chem. 1989, 54, 3317.
296. Fry, J.L.; Harris, J.M.; Bingham, R.C.; Schleyer, P.v.R. J. Am. Chem. Soc. 1970, 92, 2540; Schleyer, P.v.R.; Fry, J.L.; Lam, L.K.M.; Lancelot, C.J. J. Am. Chem. Soc. 1970, 92, 2542. Also see Dutler, R.; Rauk, A.; Sorensen, T.S.; Whitworth, S.M. J. Am. Chem. Soc. 1989, 111, 9024.
297. Fry, J.L.; Engler, E.M.; Schleyer, P.v.R. J. Am. Chem. Soc. 1972, 94, 4628. See also, Gassman, P.G.; Pascone, J.M. J. Am. Chem. Soc. 1973, 95, 7801.
298. See Minegishi, S.; Kobayashi, S.; Mayr, H. J. Am. Chem. Soc. 2004, 126, 5174; Kevill, D.N. in Charton, M. Advances in Quantitative Structure–Property Relationships, Vol. 1, JAI Press, Greenwich, CT, 1996, pp 81–115; Schadt, F.L.; Bentley, T.W.; Schleyer, P.v.R. J. Am. Chem. Soc. 1976, 98, 7667.
299. Dafforn, G.A.; Streitwieser, Jr., A. Tetrahedron Lett. 1970, 3159.
300. Cafferata, L.F.R.; Desvard, O.E.; Sicre, J.E. J. Chem. Soc. Perkin Trans. 2 1981, 940.
301. Kornblum, N.; Cheng, L.; Davies, T.M.; Earl, G.W.; Holy, N.L.; Kerber, R.C.; Kestner, M.M.; Manthey, J.W.; Musser, M.T.; Pinnick, H.W.; Snow, D.H.; Stuchal, F.W.; Swiger, R.T. J. Org. Chem. 1987, 52, 196.
302. See Miller, S.I.; Dickstein, J.I. Acc. Chem. Res. 1976, 9, 358.
303. Streitwieser, A. Solvolytic Displacement Reactions, McGraw-Hill, NY, 1962, p. 75.
304. For a Grunwald–Winstein correlation analysis of the solvolysis of benzyl bromide see Liu, K.-T.; Hou, I.-J. Tetrahedron 2001, 57, 3343.
305. See DeWolfe, R.H.; Young, W.G. in Patai, S. The Chemistry of Alkenes, Wiley, NY, 1964, pp. 683–688, 695–697.
306. King, J.F.; Tsang, G.T.Y.; Abdel-Malik, M.M.; Payne, N.C. J. Am. Chem. Soc. 1985, 107, 3224.
307. Jacobs, T.L.; Brill, W.F. J. Am. Chem. Soc. 1953, 75, 1314.
308. See Gross, H.; Höft, E. Angew. Chem. Int. Ed. 1967, 6, 335.
309. See De Kimpe, N.; Verhé, R. The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines, Wiley, NY, 1988, pp. 225–368.
310. Allen, A.D.; Kanagasabapathy, V.M.; Richard, J.P. J. Am. Chem. Soc. 1989, 111, 1455.
311. For reviews of such carbocations, see Bégué, J.; Charpentier-Morize, M. Acc. Chem. Res. 1980, 13, 207; Charpentier-Morize, M. Bull. Soc. Chim. Fr. 1974, 343.
312. For reviews, see Creary, X. Acc. Chem. Res. 1985, 18, 3; Creary, X.; Hopkinson, A.C.; Lee-Ruff, E. Adv. Carbocation Chem. 1989, 1, 45; Charpentier-Morize, M.; Bonnet-Delpon, D. Adv. Carbocation Chem. 1989, 1, 219.
313. Creary, X. J. Org. Chem. 1979, 44, 3938.
314. The resonance contributor that has the positive charge on the more electronegative atom is less stable, according to rule c in Section 2.E, but it nevertheless seems to be contributing in this case.
315. Creary, X. J. Am. Chem. Soc. 1984, 106, 5568. See, however, Takeuchi, K.; Yoshida, M.; Ohga,Y.; Tsugeno, A.; Kitagawa, T. J. Org. Chem. 1990, 55, 6063.
316. Gassman, P.G.; Saito, K.; Talley, J.J. J. Am. Chem. Soc. 1980, 102, 7613.
317. Takeuchi, K.; Kitagawa, T.; Okamoto, K. J. Chem. Soc., Chem. Commun. 1983, 7. See also, Dao, L.H.; Maleki, M.; Hopkinson, A.C.; Lee-Ruff, E. J. Am. Chem. Soc. 1986, 108, 5237.
318. Halvorsen, A.; Songstad, J. J. Chem. Soc., Chem. Commun. 1978, 327.
319. Bordwell, F.G.; Brannen, Jr., W.T. J. Am. Chem. Soc. 1964, 86, 4645. Sisti, A.J.; Lowell, S. Can. J. Chem. 1964, 42, 1896.
320. See McLLee, I.; Shim, C.S.; Chung, S.Y.; Lee, I. J. Chem. Soc. Perkin Trans. 2 1988, 975; Yoh, S.; Lee, H.W. Tetrahedron Lett. 1988, 29, 4431.
321. Cinquini, M.; Colonna, S.; Landini, D.; Maia, A.M. J. Chem. Soc. Perkin Trans. 2 1976, 996.
322. See Creary, X.; Mehrsheikh-Mohammadi, M.E.; Eggers, M.D. J. Am. Chem. Soc. 1987, 109, 2435.
323. See Gronert, S.; Pratt, L.M.; Mogali, S. J. Am. Chem. Soc. 2001, 123, 3081.
324. See Sedaghat-Herati, M.R.; McManus, S.P.; Harris, J.M. J. Org. Chem. 1988, 53, 2539.
325. See, for example, Okamoto, K.; Kita, T.; Araki, K.; Shingu, H. Bull. Chem. Soc. Jpn. 1967, 40, 1913.
326. Sugawara, M.; Yoshida, J.-i. Bull. Chem. Soc. Jpn. 2000, 73, 1253.
327. Nakashima, T.; Fujiyama, R.; Kim, H.-J.; Fujio, M.; Tsuno, Y. Bull. Chem. Soc. Jpn. 2000, 73, 429
328. Jorge, J.A.L.; Kiyan, N.Z.; Miyata, Y.; Miller, J. J. Chem. Soc. Perkin Trans. 2 1981, 100; Vitullo, V.P.; Grabowski, J.; Sridharan, S. J. Chem. Soc., Chem. Commun. 1981, 737.
329. See Fernández, I.; Frenking, G. J. Org. Chem. 2006, 71, 2251.
330. See Sugden, S.; Willis, J.B. J. Chem. Soc. 1951, 1360; Baker, J.W.; Nathan, W.S. J. Chem. Soc. 1935, 1840; Lee, I.; Sohn, S.C.; Oh, Y.J.; Lee, B.C. Tetrahedron 1986, 42, 4713.
331. Holtz, H.D.; Stock, L.M. J. Am. Chem. Soc. 1965, 87, 2404.
332. See Friedrich, E.C. in Rappoport, Z. The Chemistry of the Cyclopropyl Group, pt. 1, Wiley, NY, 1987, pp. 633–700; Aksenov, V.S.; Terent'eva, G.A.; Savinykh, Yu.V. Russ. Chem. Rev. 1980, 49, 549.
333. Roberts, J.D.; Chambers, V.C. J. Am. Chem. Soc. 1951, 73, 5034.
334. See Banert, K. Chem. Ber. 1985, 118, 1564; Vilsmaier, E.; Weber, S.; Weidner, J. J. Org. Chem. 1987, 52, 4921.
335. See Jefford, C.W.; Wojnarowski, W. Tetrahedron 1969, 25, 2089; Hausser, J.W.; Uchic, J.T. J. Org. Chem. 1972, 37, 4087.
336. See Wolk, J. L.; Hoz, T.; Basch, H.; Hoz, S. J. Org. Chem. 2001, 66, 915.
337. Brown, H.C.; Rao, C.G.; Ravindranathan, M. J. Am. Chem. Soc. 1978, 100, 7946.
338. Sella, A.; Basch, H.; Hoz, S. Tetrahedron Lett. 1996, 37, 5573.
339. See Kraus, G.A.; Hon, Y.; Thomas, P.J.; Laramay, S.; Liras, S.; Hanson, J. Chem. Rev. 1989, 89, 1591.
340. Adcock. J.L.; Gakh, A.A. Tetrahedron Lett. 1992, 33, 4875.
341. Wiberg, K.B.; McMurdie, N. J. Org. Chem. 1993, 58, 5603.
342. Bentley, T.W.; Roberts, K. J. Org. Chem. 1988, 50, 5852.
343. Bentley, T.W.; Roberts, K. J. Org. Chem. 1988, 50, 5852.
344. Shiner, Jr., V.J.; Fisher, R.D. J. Am. Chem. Soc. 1971, 93, 2553. For a review of secondary isotope effects in SN2 reactions, see Westaway, K.C. Isot. Org. Chem. 1987, 7, 275.
345. Harris, J.M.; McManus, S.P. Nucleophilicity, American Chememical Society, Washington, 1987; Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982, pp. 145–167, 181–186; Hudson, R.F. in Klopman, G. Chemical Reactivity and Reaction Paths, Wiley, NY, 1974, pp. 167–252.
346. See Ritchie, C.D.; Minasz, R.J.; Kamego, A.A.; Sawada, M. J. Am. Chem. Soc. 1977, 99, 3747; McClelland, R.A.; Banait, N.; Steenken, S. J. Am. Chem. Soc. 1986, 108, 7023.
347. Bateman, L.C.; Cooper, K.A.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1940, 925.
348. See Wei, Y.; Sastry, G.N.; Zipse, H. J. Am. Chem. Soc. 2008, 130, 3473.
349. Uggerud, E. Chem. Eur. J.2006, 12, 1127.
350. See Bordwell, F.G.; Hughes, D.L. J. Am. Chem. Soc. 1984, 106, 3234.
351. Parker, A.J. J. Chem. Soc. 1961, 1328 has a list of ~ 20 such reactions.
352. Weaver, W.M.; Hutchison, J.D. J. Am. Chem. Soc. 1964, 86, 261; See also, Bordwell, F.G.; Hughes, D.L. J. Org. Chem. 1981, 46, 3570. For a contrary result in liquid sulfur dioxide, see Lichtin, N.N.; Puar, M.S.; Wasserman, B. J. Am. Chem. Soc. 1967, 89, 6677.
353. Winstein, S.; Savedoff, L.G.; Smith, S.G.; Stevens, I.D.R.; Gall, J.S. Tetrahedron Lett. 1960, no. 9, 24.
354. Gordon, J.E.; Varughese, P. Chem. Commun. 1971, 1160. See also, Ford, W.T.; Hauri, R.J.; Smith, S.G. J. Am. Chem. Soc. 1974, 96, 4316.
355. Olmstead, W.N.; Brauman, J.I. J. Am. Chem. Soc. 1977, 99, 4219. See also, Tanaka, K.; Mackay, G.I.; Payzant, J.D.; Bohme, D.K. Can. J. Chem. 1976, 54, 1643.
356. See Kormos, B.L.; Cramer, C.J. J. Org. Chem. 2003, 68, 6375.
357. Parker, A.J. J. Chem. Soc. 1961, 4398.
358. Pearson, R.G. Surv. Prog. Chem. 1969, 5, 1, pp. 21-38.
359. See Guibe, F.; Bram, G. Bull. Soc. Chim. Fr. 1975, 933.
360. Zaugg, H.E.; Leonard, J.E. J. Org. Chem. 1972, 37, 2253. See also, Solov'yanov, A.A.; Ahmed, E.A.A.; Beletskaya, I.P.; Reutov, O.A. J. Org. Chem. USSR 1987, 23, 1243; Jackman, L.M.; Lange, B.C. J. Am. Chem. Soc.1981, 103, 4494.
361. See, for example Williard, P.G.; Carpenter, G.B. J. Am. Chem. Soc. 1986, 108, 462.
362. Zook, H.D.; Gumby, W.L. J. Am. Chem. Soc. 1960, 82, 1386. See also, Cacciapaglia, R.; Mandolini, L. J. Org. Chem. 1988, 53, 2579.
363. See Barlow, S.E.; Van Doren, J.M.; Bierbaum, V.M. J. Am. Chem. Soc. 1988, 110, 7240; Merkel, A.; Havlas, Z.; Zahradník, R. J. Am. Chem. Soc. 1988, 110, 8355.
364. Olmstead, W.N.; Brauman, J.I. J. Am. Chem. Soc. 1977, 99, 4219.
365. Bohme, D.K.; Raksit, A.B. J. Am. Chem. Soc. 1984, 106, 3447. See also, Hierl, P.M.; Ahrens, A.F.; Henchman, M.; Viggiano, A.A.; Paulson, J.F.; Clary, D.C. J. Am. Chem. Soc. 1986, 108, 3142.
366. Bohme, D.K.; Raksit, A.B. Can. J. Chem. 1985, 63, 3007.
367. Chen, X.; Brauman, J.I. J. Am. Chem. Soc. 2008, 130, 15038.
368. Landini, D.; Maia, A.; Rampoldi, A. J. Org. Chem. 1989, 54, 328.
369. Edwards, J.O.; Pearson, R.G. J. Am. Chem. Soc. 1962, 84, 16.
370. Swain, C.G.; Scott, C.B. J. Am. Chem. Soc. 1953, 75, 141.
371. Albery, W.J.; Kreevoy, M.M. Adv. Phys. Org. Chem. 1978, 16, 87, pp. 113–115.
372. Also see Ritchie, C.D. Pure Appl. Chem. 1978, 50, 1281; Duboc, C. in Chapman, N.B.; Shorter, J. Correlation Analysis in Chemistry, Recent Advances, Plenum, NY, 1978, pp. 313–355; Ibne-Rasa, K.M. J. Chem. Educ. 1967, 44, 89; Kawazoe, Y.; Ninomiya, S.; Kohda, K.; Kimoto, H. Tetrahedron Lett. 1986, 27, 2897; Kevill, D.N.; Fujimoto, E.K. J. Chem. Res. (S) 1988, 408.
373. From Wells, P.R. Chem. Rev. 1963, 63, 171, p. 212. See also, Koskikallio, J. Acta Chem. Scand. 1969, 23, 1477, 1490.
374. See Pellerite, M.J.; Brauman, J.I. J. Am. Chem. Soc. 1983, 105, 2672.
375. For reference scales for the characterization of cationic electrophiles and neutral nucleophiles see Mayr, H.; Bug, T.; Gotta, M.F.; Hering, N.; Irrgang, B.; Janker, B.; Kempf, B.; Loos, R.; Ofial, A.R.; Remennikov, G.; Schimmel, H. J. Am. Chem. Soc. 2001, 123, 9500.
376. For discussions, see Dietze, P.; Jencks, W.P. J. Am. Chem. Soc. 1989, 111, 5880.
377. Jencks, W.P.; Gilchrist, M. J. Am. Chem. Soc. 1968, 90, 2622.
378. For theoretical treatments of nucleophilicity at a carbonyl carbon, see Buncel, E.; Shaik, S.S.; Um, I.; Wolfe, S. J. Am. Chem. Soc. 1988, 110, 1275, and references cited therein.
379. Baer, S.; Stoutland, P.O.; Brauman, J.I. J. Am. Chem. Soc. 1989, 111, 4097.
380. Definition in the Glossary of Terms used in Physical Organic Chemistry, Pure Appl. Chem. 1979, 51, 1731.
381. See Ren, Y.; Yamataka, H. J. Org. Chem. 2007, 72, 5660; Org. Lett. 2006, 8, 119; Chemistry: European J. 2007, 13, 677.
382. Hoz, S.; Buncel, E. Israel J. Chem. 1985, 26, 313.
383. Grekov, A.P.; Veselov, V.Ya. Russ. Chem. Rev. 1978, 47, 631; Jencks, W.P. Catalysis in Chemistry and Enzymology, McGraw-Hill, New York, 1969; pp 107–111.
384. See Ho, S.; Buncel, E. Isr. J. Chem. 1985, 26, 313.
385. Buncel, E.; Hoz, S. Tetrahedron Lett. 1983, 24, 4777. For evidence that this is not the sole cause, see Oae, S.; Kadoma, Y. Can. J. Chem. 1986, 64, 1184.
386. See Hoz, S. J. Org. Chem. 1982, 47, 3545; Laloi-Diard, M.; Verchere, J.; Gosselin, P.; Terrier, F. Tetrahedron Lett. 1984, 25, 1267.
387. Also see Hudson, R.F.; Hansell, D.P.; Wolfe, S.; Mitchell, D.J. J. Chem. Soc., Chem. Commun. 1985, 1406. For a discussion, see Herschlag, D.; Jencks, W.P. J. Am. Chem. Soc. 1990, 112, 1951.
388. Buncel, E.; Um, I. J. Chem. Soc., Chem. Commun. 1986, 595; Terrier, F.; Degorre, F.; Kiffer, D.; Laloi, M. Bull. Soc. Chim. Fr. 1988, 415. For some evidence against this explanation, see Moss, R.A.; Swarup, S.; Ganguli, S. J. Chem. Soc., Chem. Commun. 1987, 860.
389. Buncel, E.; Um, I.-H. Tetrahedron 2004, 60, 7801.
390. For example, see Kice, J.L.; Legan, E. J. Am. Chem. Soc. 1973, 95, 3912.
391. Dixon, J.E.; Bruice, T.C. J. Am. Chem. Soc. 1971, 93, 3248, 6592.
392. McIsaac, Jr., J.E.; Subbaraman, L.R.; Subbaraman, J.; Mulhausen, H.A.; Behrman, E.J. J. Org. Chem. 1972, 37, 1037. See, however, Buncel, E.; Wilson, H.; Chuaqui, C. J. Am. Chem. Soc. 1982, 104, 4896; Int. J. Chem. Kinet. 1982, 14, 823.
393. Phan, T.B.; Breugst, M.; Mayr, H. Angew. Chem. Int. Ed. 2006, 45, 3869.
394. Phan, T.B.; Mayr, H. Can. J. Chem. 2005, 83, 1554.
395. Phan, T.B.; Mayr, H. Eur. J. Org. Chem. 2006, 2530.
396. Brotzel, F.; Chu, Y.C.; Mayr, H. J. Org. Chem. 2007, 72, 3679; Korzhenevskaya, N.G. Russ. J. Org. Chem. 2008, 44, 1255.
397. Brotzel, F.; Kempf, B.; Singer, T.; Zipse, H.; Mayr, H. Chemistry: European J. 2007, 13, 336.
398. Nigst, T.A.; Westermaier, M.; Ofial, A.R.; Mayr, H. Eur. J. Org. Chem. 2008, 2369.
399. Lakhdar, S.; Westermaier, M.; Terrier, F.; Goumont, R.; Boubaker, T.; Ofial, A.R.; Mayr, H. J. Org. Chem. 2006, 71, 9088.
400. Breugst, M.; Tokuyasu, T.; Mayr, H. J. Org. Chem. 2010, 75, 5250.
401. Brotzel, F.; Mayr, H. Org. Biomol. Chem. 2007, 5, 3814.
402. Appel, R.; Mayr, H. Chemistry: Eur. J. 2010, 16, 8610.
403. See Staude, E.; Patat, F. in Patai, S. The Chemistry of the Ether Linkage, Wiley, NY, 1967, pp. 22–46.
404. Olah, G.A.; O'Brien, D.H. J. Am. Chem. Soc. 1967, 89, 1725; Olah, G.A.; Sommer, J.; Namanworth, E. J. Am. Chem. Soc. 1967, 89, 3576; Olah, J.A.; Olah, G.A. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, pp. 743–747.
405. See Okada, S.; Abe, Y.; Taniguchi, S.; Yamabe, S. J. Chem. Soc., Chem. Commun. 1989, 610.
406. See Smith, J.G. Synthesis 1984, 629; Bartók, M.; Láng, K.L.in Patai, S. The Chemistry of Functional Groups, Supplement E, Wiley, NY, 1980, pp. 609–681.
407. See Hu, X.E. Tetrahedron 2004, 60, 2701.
408. See Di Vona, M.L.; Illuminati, G.; Lillocci, C. J. Chem. Soc. Perkin Trans. 2 1985, 1943; Bury, A.; Earl, H.A.; Stirling, C.J.M. J. Chem. Soc., Chem. Commun. 1985, 393.
409. Bentley, T.W.; Christl, M.; Kemmer, R.; Llewellyn, G.; Oakley, J.E. J. Chem. Soc. Perkin Trans. 2 1994, 2531.
410. Perst, H. Oxonium Ions in Organic Chemistry, Verlag Chemie, Deerfield Beach, FL, 1971, pp. 100–127; Perst, H. in Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 5, Wiley, NY, 1976, pp. 1961–2047; Granik, V.G.; Pyatin, B.M.; Glushkov, R.G. Russ. Chem. Rev. 1971, 40, 747; See Curphey, T.J. Org. Synth. VI, 1021.
411. See Stang, P.J.; Hanack, M.; Subramanian, L.R. Synthesis 1982, 85; Howells, R.D.; McCown, J.D. Chem. Rev. 1977, 77, 69, pp. 85–87.
412. Crossland, R.K.; Wells, W.E.; Shiner, Jr., V.J. J. Am. Chem. Soc. 1971, 93, 4217.
413. Olah, G.A.; Mo, Y.K. J. Am. Chem. Soc. 1974, 96, 3560.
414. See Baumgarten, R.J.; Curtis, V.A. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 2, Wiley, NY, 1982, pp. 929–997.
415. See Müller, P.; Thi, M.P.N. Helv. Chim. Acta 1980, 63, 2168; Curtis, V.A.; Knutson, F.J.; Baumgarten, R.J. Tetrahedron Lett. 1981, 22, 199.
416. See Katritzky, A.R.; Marson, C.M. Angew. Chem. Int. Ed. 1984, 23, 420; Katritzky, A.R.; Sakizadeh, K.; Musumarra, G. Heterocycles 1985, 23, 1765; Katritzky, A.R.; Musumarra, G. Chem. Soc. Rev. 1984, 13, 47.
417. See Katritzky, A.R.; Brycki, B. J. Am. Chem. Soc. 1986, 108, 7295, and other papers in this series.
418. For a review of Mannich bases, see Tramontini, M. Synthesis 1973, 703.
419. See Kirmse, W. Angew. Chem. Int. Ed. 1976, 15, 251; Collins, C.J. Acc. Chem. Res. 1971, 4, 315.
420. See Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986; Hegarty, A.F. in Patai, S. The Chemistry of Diazonium and Diazo Groups, pt. 2, Wiley, NY, 1978, pp. 511–591, pp. 571–575; More O'Ferrall, R.A. Adv. Phys. Org. Chem. 1967, 5, 331; Studzinskii, O.P.; Korobitsyna, I.K. Russ. Chem. Rev. 1970, 39, 834.
421. For aromatic diazoium salts, see Weiss, R.; Wagner, K.; Priesner, C.; Macheleid, J. J. Am. Chem. Soc. 1985, 107, 4491); Laali, K.; Olah, G.A. Rev. Chem. Intermed. 1985, 6, 237; Bott, K. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 1, Wiley, NY, 1983, pp. 671–697.
422. See Mohrig, J.R.; Keegstra, K.; Maverick, A.; Roberts, R.; Wells, S. J. Chem. Soc., Chem. Commun. 1974, 780.
423. Berner, D.; McGarrity, J.F. J. Am. Chem. Soc. 1979, 101, 3135
424. See Manuilov, A.V.; Barkhash, V.A. Russ. Chem. Rev. 1990, 59, 179; Saunders, Jr., W.H.; Cockerill, A.F. Mechanisms of Elimination Reactions, Wiley, NY, 1973, pp. 280–317.
425. Semenow, D.; Shih, C.; Young, W.G. J. Am. Chem. Soc. 1958, 80, 5472. See Olah, G.A.; Schleyer, P.v.R. Carbonium Ions, Vol. 2, Wiley, NY, 1970, the articles by Keating, J.T.; Skell, P.S. pp. 573–653.
426. Maskill, H.; Thompson, J.T.; Wilson, A.A. J. Chem. Soc. Perkin Trans. 2 1984, 1693; Connor, J.K.; Maskill, H. Bull. Soc. Chim. Fr. 1988, 342.
427. Weiss, R.; Wagner, K.; Priesner, C.; Macheleid, J. J. Am. Chem. Soc. 1985, 107, 4491.
428. Streitwieser, Jr., A.; Schaeffer, W.D. J. Am. Chem. Soc. 1957, 79, 2888.
429. Pearson, R.G.; Edgington, D.N. J. Am. Chem. Soc. 1962, 84, 4607.
430. See Knipe, A.C. in Stirling, C.J.M. The Chemistry of the Sulphonium Group, pt. 1, Wiley, NY, 1981, pp. 313–385. See also, Badet, B.; Julia, M.; Lefebvre, C. Bull. Soc. Chim. Fr. 1984, II-431.
431. See McMurry, J.E. Org. React. 1976, 24, 187.
432. For the effect of nitro substitution, see Sinnott, M.L.; Whiting, M.C. J. Chem. Soc. B 1971, 965. See also, Page, I.D.; Pritt, J.R.; Whiting, M.C. J. Chem. Soc. Perkin Trans. 2 1972, 906.
433. See Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 2nd ed., VCH, NY, 1988; Klumpp, G.W. Reactivity in Organic Chemistry, Wiley, NY, 1982, pp. 186–203; Bentley, T.W.; Schleyer, P.v.R. Adv. Phys. Org. Chem. 1977, 14, 1.
434. Mu, L.; Drago, R.S.; Richardson, D.E. J. Chem. Soc. Perkin Trans 2, 1998, 159; Fujio, M.; Saeki, Y.; Nakamoto, K.; Kim, S.H.; Rappoport, Z.; Tsuno, Y. Bull. Chem. Soc. Jpn. 1996, 69, 751.
435. Bentley, T.W.; Llewellyn, G.; Ryu, Z.H. J. Org. Chem. 1998, 63, 4654.
436. This analysis is due to Ingold, C.K. Structure and Mechanism in Organic Chemistry, 2d ed., Cornell University Press, Ithaca, NY, 1969, pp. 457–463.
437. See Ponomareva, E.A.; Dvorko, G.F.; Kulik, N.I.; Evtushenko, N.Yu. Doklad. Chem. 1983, 272, 291.
438. See Bug, T.; Mayr, H. J. Am. Chem. Soc. 2003, 125, 12980; Brinchi, L.; DiProfio, P.; Germani, R.; Savelli, G.; Spreti, N.; Bunton, L.A. Eur. J. Org. Chem. 2000, 3849.
439. See Buncel, E.; Wilson, H. Adv. Phys. Org. Chem. 1977, 14, 133; Martin, D.; Weise, A.; Niclas, H. Angew. Chem. Int. Ed. 1967, 6, 318.
440. See Normant, H. Russ. Chem. Rev. 1970, 39, 457; Angew. Chem. Int. Ed. 1967, 6, 1046.
441. Klamt, A.; Schüürmann, G. J. Chem. Soc. Perkin Trans. 2 1993, 799.
442. Kim, D.W.; Song, C.E.; Chi, D.Y. J. Org. Chem. 2003, 68, 4281; Chiappe, C.; Pieraccini, D.; Saullo, P. J. Org. Chem. 2003, 68, 6710.
443. DeSimone, J.; Selva, M.; Tundo, P. J. Org. Chem. 2001, 66, 4047.
444. See Craig, S.L.; Brauman, J.I. J. Am. Chem. Soc. 1999, 121, 6690.
445. Westaway, K.C.; Lai, Z. Can. J. Chem. 1989, 67, 345.
446. For reviews of the effects of protic and aprotic solvents, see Parker, A.J. Chem. Rev. 1969, 69, 1; Madaule-Aubry, F. Bull. Soc. Chim. Fr. 1966, 1456.
447. See Magnera, T.F.; Caldwell, G.; Sunner, J.; Ikuta, S.; Kebarle, P. J. Am. Chem. Soc. 1984, 106, 6140.
448. See, for example, Fuchs, R.; Cole, L.L. J. Am. Chem. Soc. 1973, 95, 3194.
449. See, however, Haberfield, P.; Clayman, L.; Cooper, J.S. J. Am. Chem. Soc. 1969, 91, 787.
450. Smith, S.G.; Fainberg, A.H.; Winstein, S. J. Am. Chem. Soc. 1961, 83, 618.
451. Capon, B.; McManus, S. Neighboring Group Participation, Vol. 1, Plenum, NY, 1976; Haywood–Farmer, J. Chem. Rev. 1974, 74, 315.
452. Schadt, F.L.; Schleyer, P.v.R.; Bentley, T.W. Tetrahedron Lett. 1974, 2335.
453. See Bunton, C.A.; Robinson, L. J. Am. Chem. Soc. 1968, 90, 5965.
454. See Kevill, D.N. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, pp. 933–984.
455. See Rudakov, E.S.; Kozhevnikov, I.V.; Zamashchikov, V.V. Russ. Chem. Rev. 1974, 43, 305. For an example of assistance in removal of F by H+, see Coverdale, A.K.; Kohnstam, G. J. Chem. Soc. 1960, 3906.
456. Zamashchikov, V.V.; Rudakov, E.S.; Bezbozhnaya, T.V.; Matveev, A.A. J. Org. Chem. USSR 1984, 20, 424. See, however, Kevill, D.N.; Fujimoto, E.K. J. Chem. Soc., Chem. Commun. 1983, 1149.
457. Kornblum, N.; Jones, W.J.; Hardies, D.E. J. Am. Chem. Soc. 1966, 88, 1704; Kornblum, N.; Hardies, D.E. J. Am. Chem. Soc. 1966, 88, 1707.
458. Grunwald, E.; Winstein, S. J. Am. Chem. Soc. 1948, 70, 846.
459. See Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 2nd ed., VCH, NY, 1988, pp. 339–405; Langhals, H. Angew. Chem. Int. Ed. 1982, 21, 724.
460. For a criticism of the Y scale, see Abraham, M.H.; Doherty, R.M.; Kamlet, M.J.; Harris, J.M.; Taft, R.W. J. Chem. Soc. Perkin Trans. 2 1987, 1097.
461. Y values are from Fainberg, A.H.; Winstein, S. J. Am. Chem. Soc. 1956, 78, 2770, except for the value for CF3CH2OH which is from Shiner, Jr., V.J.; Dowd, W.; Fisher, R.D.; Hartshorn, S.R.; Kessick, M.A.; Milakofsky, L.; Rapp, M.W. J. Am. Chem. Soc. 1969, 91, 4838. YOTs values are from Bentley, T.W.; Llewellyn, G. Prog. Phys. Org. Chem. 1990, 17, pp. 143–144. Z values are from Kosower, E.M.; Wu, G.; Sorensen, T.S. J. Am. Chem. Soc. 1961, 83, 3147. See also, Larsen, J.W.; Edwards, A.G.; Dobi, P. J. Am. Chem. Soc. 1980, 102, 6780. ET(30) values are from Reichardt, C.; Dimroth, K. Fortschr. Chem. Forsch. 1969, 11, 1; Reichardt, C. Angew. Chem. Int. Ed. 1979, 18, 98; Laurence, C.; Nicolet, P.; Reichardt, C. Bull. Soc. Chim. Fr. 1987, 125; Laurence, C.; Nicolet, P.; Lucon, M.; Reichardt, C. Bull. Soc. Chim. Fr. 1987, 1001; Reichardt, C.; Eschner, M.; Schäfer, G. Liebigs Ann. Chem. 1990, 57. Also see Reichardt, C. Solvents and Solvent Effects in Organic Chemistry, 2nd ed., VCH, NY, 1988.
462. A scale of solvent nucleophilicity (as opposed to ionizing power), called the NT scale, has been developed: Kevill, D.N.; Anderson, S.W. J. Org. Chem. 1991, 56, 1845.
463. See Kevill, D.N.; Anderson, S.W. J. Am. Chem. Soc. 1986, 108, 1579; McManus, S.P.; Neamati-Mazreah, N.; Karaman, R.; Harris, J.M. J. Org. Chem. 1986, 51, 4876; Abraham, M.H.; Doherty, R.M.; Kamlet, M.J.; Harris, J.M.; Taft, R.W. J. Chem. Soc. Perkin Trans. 2 1987, 913.
464. Schadt, F.L.; Bentley, T.W.; Schleyer, P.v.R. J. Am. Chem. Soc. 1976, 98, 7667.
465. Bentley, T.W.; Carter, G.E. J. Org. Chem. 1983, 48, 579.
466. For a review of these scales, see Bentley, T.W.; Llewellyn, G. Prog. Phys. Org. Chem. 1990, 17, 121.
467. Kevill, D.N.; Anderson, S.W. J. Org. Chem. 1985, 50, 3330. See also, Creary, X.; McDonald, S.R. J. Org. Chem. 1985, 50, 474.
468. Bentley, T.W.; Carter, G.E. J. Am. Chem. Soc. 1982, 104, 5741. See also, Liu, K.; Sheu, H. J. Org. Chem. 1991, 56, 3021.
469. Bentley, T.W.; Carter, G.E.; Roberts, K. J. Org. Chem. 1984, 49, 5183.
470. See Kevill, D.N.; Hawkinson, D.C. J. Org. Chem. 1990, 55, 5394 and references cited therein.
471. Fujio, M.; Saeki, Y.; Nakamoto, K.; Yatsugi, K.-i.; Goto, N.; Kim, S.H.; Tsuji, Y.; Rappoport, Z.; Tsuno, Y. Bull. Chem. Soc. Jpn. 1995, 68, 2603; Liu, K.-T.; Chin, C.-P.; Lin, Y.-S.; Tsao, M.-L. J. Chem. Res. (S) 1997, 18.
472. Fujio, M.; Susuki, T.; Goto, M.; Tsuji, Y.; Yatsugi, K.; Saeki, Y.; Kim, S.H.; Tsuno, Y. Bull. Chem. Soc. Jpn. 1994, 67, 2233.
473. Tsuji, Y.; Fujio, M.; Tsuno, Y. Tetrahedron Lett. 1992, 33, 349.
474. See Abraham, M.H.; Grellier, P.L.; Abboud, J.M.; Doherty, R.M.; Taft, R.W. Can. J. Chem. 1988, 66, 2673; Shorter, J. Correlation Analysis of Organic Reactivity, Wiley, NY, 1982, pp. 127–172; Reichardt, C. Angew. Chem. Int. Ed. 1979, 18, 98; Abraham, M.H. Prog. Phys. Org. Chem. 1974, 11, 1. See also, Chastrette, M.; Rajzmann, M.; Chanon, M.; Purcell, K.F. J. Am. Chem. Soc. 1985, 107, 1.
475. Kosower, E.M.; Wu, G.; Sorensen, T.S. J. Am. Chem. Soc. 1961, 83, 3147. See also, Larsen, J.W.; Edwards, A.G.; Dobi, P. J. Am. Chem. Soc. 1980, 102, 6780.
476. Dimroth, K.; Reichardt, C. Liebigs Ann. Chem. 1969, 727, 93. See also, Haak, J.R.; Engberts, J.B.F.N. Recl. Trav. Chim. Pays-Bas 1986, 105, 307.
477. The symbol ET comes from energy, transition. The (30) is used because the ion 100 bore this number in Dimroth, K.; Reichardt, C. Liebigs Ann. Chem. 1969, 727, 93. Values based on other ions have also been reported: See, for example, Reichardt, C.; Harbusch-Görnert, E.; Schäfer, G. Liebigs Ann. Chem. 1988, 839.
478. Reichardt, C.; Dimroth, K. Fortschr. Chem. Forsch. 1969, 11, p. 32.
479. Doherty, R.M.; Abraham, M.H.; Harris, J.M.; Taft, R.W.; Kamlet, M.J. J. Org. Chem. 1986, 51, 4872. See also, Bekárek, V. J. Chem. Soc. Perkin Trans. 2 1986, 1425; Abe, T. Bull. Chem. Soc. Jpn. 1990, 63, 2328.
480. Buncel, E.; Rajagopal, S. J. Org. Chem. 1989, 54, 798.
481. Dong, D.C.; Winnik, M.A. Can. J. Chem. 1984, 62, 2560.
482. For a review of such scales, see Buncel, E.; Rajagopal, S. Acc. Chem. Res. 1990, 23, 226.
483. Kaupp, G. Angew. Chem. Int. Ed.1994, 33, 1452.
484. Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 2nd ed., Verlag Chemie, Deerfield Beach, FL, 1983; Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Academic Press, NY, 1978; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977; Makosza, M. Pure Appl. Chem. 2000, 72, 1399; Montanari, F.; Landini, D.; Rolla, F. Top. Curr. Chem. 1982, 101, 147; Alper, H. Adv. Organomet. Chem. 1981, 19, 183; Sjöberg, K. Aldrichimica Acta 1980, 13, 55.
485. Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Academic Press, NY, 1978, p. 2.
486. See Lissel, M.; Feldman, D.; Nir, M.; Rabinovitz, M. Tetrahedron Lett. 1989, 30, 1683.
487. Landini, D.; Maia, A.; Montanari, F.J. Am. Chem. Soc. 1978, 100, 2796.
488. For a review, see Rabinovitz, M.; Cohen, Y.; Halpern, M. Angew. Chem. Int. Ed. 1986, 25, 960.
489. See Makosza, M. Pure Appl. Chem. 1975, 43, 439. See also, Dehmlow, E.V.; Thieser, R.; Sasson, Y.; Pross, E. Tetrahedron 1985, 41, 2927; Mason, D.; Magdassi, S.; Sasson, Y. J. Org. Chem. 1990, 55, 2714.
490. Bhalerao, U.T.; Mathur, S.N.; Rao, S.N. Synth. Commun. 1992, 22, 1645.
491. Badri, M.; Brunet, J.-J.; Perron R. Tetrahedron Lett. 1992, 33, 4435.
492. See Liotta, C. in Patai, S. The Chemistry of Functional Groups, Supplement E, Wiley, NY, 1980, pp. 157–174.
493. See Liotta, C.; Harris, H.P.; McDermott, M.; Gonzalez, T.; Smith, K. Tetrahedron Lett. 1974, 2417; Sam, D.J.; Simmons, H.E. J. Am. Chem. Soc. 1974, 96, 2252; Durst, H.D. Tetrahedron Lett. 1974, 2421.
494. Soula, G. J. Org. Chem.1985, 50, 3717.
495. Furukawa, N.; Ogawa, S.; Kawai, T.; Oae, S. J. Chem. Soc. Perkin Trans. 1 1984, 1833. See also, Fujihara, H.; Imaoka, K.; Furukawa, N.; Oae, S. J. Chem. Soc. Perkin Trans. 1 1986, 333.
496. See Iwamoto, H.; Yoshimura, M.; Sonoda, T.; Kobayashi, H. Bull. Chem. Soc. Jpn. 1983, 56, 796.
497. See, for example, Dehmlow, E.V.; Slopianka, M. Chem. Ber. 1979, 112, 2765.
498. Fife, W.K.; Xin, Y. J. Am. Chem. Soc. 1987, 109, 1278.
499. Deshayes, S.; Liagre, M.; Loupy, A.; Luche, J.-L.; Petit, A. Tetrahedron 1999, 55, 10851.
500. Quici, S.; Regen, S.L. J. Org. Chem. 1979, 44, 3436.
501. See Regen, S.L. Nouv. J. Chim. 1982, 6, 629; Angew. Chem. Int. Ed. 1979, 18, 421. See also, Bogatskii, A.V.; Luk'yanenko, N.G.; Pastushok, V.N.; Parfenova, M.N. Doklad. Chem. 1985, 283, 210; Pugia, M.J.; Czech, B.P.; Czech, B.P.; Bartsch, R.A. J. Org. Chem. 1986, 51, 2945.
502. See Mingos, D.M.P.; Baghurst, D.R. Chem. Soc. Rev. 1991, 20, 1; Giguere, R.J. Org. Synth. Theory Appl. 1989, 1, 103.
503. Keusenkothen, P.F.; Smith, M.B. Tetrahedron Lett. 1989, 30, 3369.
504. See Einhorn, C.; Einhorn, J.; Dickens, M.J.; Luche, J. Tetrahedron Lett. 1990, 31, 4129.
505. Kad, G.-L.; Singh, V.; Kuar, K.P.; Singh, J. Tetrahedron Lett. 1997, 38, 1079.
506. Matsumoto, K.; Morris, A.R. Organic Synthesis at High Pressure, Wiley, NY, 1991; Matsumoto, K.; Sera, A.; Uchida, T. Synthesis 1985, 1, 999.
507. Isaacs, N.S. Liquid Phase High Pressure Chemistry, Wiley, Chichester, 1981; Asano, T.; le Noble, W.J. Chem. Rev. 1978, 78, 407.
508. Firestone, R.A.; Vitale, M.A. J. Org. Chem. 1981, 46, 2160.
509. See Reutov, O.A.; Beletskaya, I.P.; Kurts, A.L. Ambident Anions, Plenum, NY, 1983. For a review, see Black, T.H. Org. Prep. Proced. Int. 1989, 21, 179.
510. See Holm, A.; Wentrup, C. Acta Chem. Scand. 1966, 20, 2123.
511. This term was introduced by Hassner, A. J. Org. Chem. 1968, 33, 2684.
512. For an exception, see Trimitsis, G.B.; Hinkley, J.M.; TenBrink, R.; Faburada, A.L.; Anderson, R.; Poli, M.; Christian, B.; Gustafson, G.; Erdman, J.; Rop, D. J. Org. Chem. 1983, 48, 2957.
513. See Hauser, C.R.; Harris, C.M. J. Am. Chem. Soc. 1958, 80, 6360. For reviews, see Thompson, C.M.; Green, D.L.C. Tetrahedron 1991, 47, 4223; Harris, T.M.; Harris, C.M. Org. React. 1969, 17, 155.
514. For a review, see Erashko, V.I.; Shevelev, S.A.; Fainzil'berg, A.A. Russ. Chem. Rev. 1966, 35, 719.
515. See Jackman, L.M.; Lange, B.C. Tetrahedron 1977, 33, 2737; Reutov, O.A.; Kurts, A.L. Russ. Chem. Rev. 1977, 46, 1040; Gompper, R.; Wagner, H. Angew. Chem. Int. Ed. 1976, 15, 321.
516. See Bégué, J.; Charpentier-Morize, M.; Née, G. J. Chem. Soc., Chem. Commun. 1989, 83.
517. This principle, sometimes called Kornblum's rule, was first stated by Kornblum, N.; Smiley, R.A.; Blackwood, R.K.; Iffland, D.C. J. Am. Chem. Soc. 1955, 77, 6269.
518. See Austad, T.; Songstad, J.; Stangeland, L.J. Acta Chem. Scand. 1971, 25, 2327; Carretero, J.C.; García Ruano, J.L. Tetrahedron Lett. 1985, 26, 3381.
519. Kornblum, N.; Berrigan, P.J.; le Noble, W.J. J. Chem. Soc. 1963, 85, 1141; Kornblum, N.; Seltzer, R.; Haberfield, P. J. Am. Chem. Soc. 1963, 85, 1148. For other examples, see le Noble, W.J.; Puerta, J.E. Tetrahedron Lett.1966, 1087; Schick, H.; Schwarz, H.; Finger, A.; Schwarz, S. Tetrahedron 1982, 38, 1279.
520. Kornblum, N.; Seltzer, R.; Haberfield, P. J. Am. Chem. Soc. 1963, 85, 1148; Kurts, A.L.; Beletskaya, I.P.; Masias, A.; Reutov, O.A. Tetrahedron Lett. 1968, 3679. See, however, Sarthou, P.; Bram, G.; Guibe, F. Can. J. Chem.1980, 58, 786.
521. Smith, S.G.; Hanson, M.P. J. Org. Chem. 1971, 36, 1931; Akabori, S.; Tuji, H. Bull. Chem. Soc. Jpn. 1978, 51, 1197. See also, le Noble, W.J.; Palit, S.K. Tetrahedron Lett. 1972, 493.
522. Jones, M.E.; Kass, S.R.; Filley, J.; Barkley, R.M.; Ellison, G.B. J. Am. Chem. Soc. 1985, 107, 109.
523. See, for example O'Neill, P.; Hegarty, A.F. J. Org. Chem. 1987, 52, 2113.
524. Chechik, V.O.; Bobylev, V.A. Acta Chem. Scand. B, 1994, 48, 837.
525. Rao, A.S.; Paknikar, S.K.; Kirtane, J.G. Tetrahedron 1983, 39, 2323; Behrens, C.H.; Sharpless, K.B. Aldrichimica Acta 1983, 16, 67; Enikolopiyan, N.S. Pure Appl. Chem. 1976, 48, 317; Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines, Academic Press, NY, 1969, pp. 206–273.
526. Biggs, J.; Chapman, N.B.; Finch, A.F.; Wray, V. J. Chem. Soc. B 1971, 55.
527. Caron M.; Sharpless, K.B. J. Org. Chem. 1985, 50, 1557. See also, Chong, J.M.; Sharpless, K.B. J. Org. Chem. 1985, 50, 1560; Behrens, C.H.; Sharpless, K.B. J. Org. Chem. 1985, 50, 5696.
528. Murphy, D.K.; Alumbaugh, R.L.; Rickborn, B. J. Am. Chem. Soc. 1969, 91, 2649. For a method of overriding this preference, see McKittrick, B.A.; Ganem, B. J. Org. Chem. 1985, 50, 5897.
529. Gao, Y.; Sharpless, K.B. J. Am. Chem. Soc. 1988, 110, 7538; Kim, B.M.; Sharpless, K.B. Tetrahedron Lett. 1989, 30, 655.
530. See, however, Kurz, J.L.; Lee, J.; Love, M.E.; Rhodes, S. J. Am. Chem. Soc. 1986, 108, 2960.
531. Hutchins, R.O.; Taffer, I.M. J. Org. Chem. 1983, 48, 1360.
532. Kim, D.W.; Hong, D.J.; Seo, J.W.; Kim, H.S.; Kim, H.K.; Song, C.E.; Chi, D.Y. J. Org. Chem. 2004, 69, 3186.
533. Cavicchioni, G. Synth. Commun. 1994, 24, 2223.
534. Martin, S.F.; Chou, T. Tetrahedron Lett. 1978, 1943; Yoshioka, H.; Takasaki, K.; Kobayashi, M.; Matsumoto, T. Tetrahedron Lett. 1979, 3489.
535. Gingras, M.; Chan, T.H. Tetrahedron Lett. 1989, 30, 279.
536. Salomaa, P. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 177–210.
537. Mataka, S.; Liu, G.-B.; Sawada, T.; Tori-i, A.; Tashiro, M. J. Chem. Res. (S) 1995, 410.
538. Li, W.; Li, J.; DeVincentis, D.; Masour, T.S. Tetrahedron Lett. 2004, 45, 1071.
539. Augustine, J.K.; Naik, Y.A.; Mandal, A.B.; Chowdappa, N.; Praveen, V.B. Tetrahedron 2008, 64, 688.
540. Wang, L.; Li, P.; Yan, J.; Wu, Z. Tetrahedron Lett. 2003, 44, 4685.
541. See, for example, Le Fave, G.M.; Scheurer, P.G. J. Am. Chem. Soc. 1950, 72, 2464.
542. Sheppard, W.A.; Sharts, C.M. Organic Fluorine Chemistry, W.A. Benjamin, NY, 1969, pp. 410–411; Hudlicky, M. Chemistry of Organic Fluorine Compounds, 2nd ed., Ellis Horwood, Chichester, 1976, pp. 273–274.
543. See, for example, Kobayashi, Y.; Kumadaki, I. Acc. Chem. Res. 1978, 11, 197.
544. Rondestvedt, Jr., C.S. J. Org. Chem. 1976, 41, 3569, 3574, 3576. For another method, see Nakano, T.; Ohkawa, K.; Matsumoto, H.; Nagai, Y. J. Chem. Soc., Chem. Commun. 1977, 808.
545. See Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 129–141.
546. Hine, J. J. Am. Chem. Soc. 1950, 72, 2438. Also see, le Noble, W.J. J. Am. Chem. Soc. 1965, 87, 2434.
547. Batts, B.D. J. Chem. Soc. B 1966, 551.
548. Barnard, P.W.C.; Robertson, R.E. Can. J. Chem. 1961, 39, 881. See also, Drabicky, M.J.; Myhre, P.C.; Reich, C.J.; Schmittou, E.R. J. Org. Chem. 1976, 41, 1472.
549. SeeWilliams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 162–163.
550. Dahn, H.; Gold, H. Helv. Chim. Acta 1963, 46, 983; Thomas, C.W.; Leveson, L.L. Int. J. Chem. Kinet. 1983, 15, 25. See Smith, III, A.B; Dieter, R.K. Tetrahedron 1981, 37, 2407.
551. Bergstrom, R.G. in Patai, S. The Chemistry of Functional Groups, Supplement E, Wiley, NY, 1980, pp. 881–902; Cockerill, A.F.; Harrison, R.G. in Patai, S. The Chemistry of Functional Groups, Supplement A, pt. 1, Wiley, NY, 1977, pp. 149–329; Cordes, E.H.; Bull, H.G. Chem. Rev. 1974, 74, 581; Pindur, U.; Müller, J.; Flo, C.; Witzel, H. Chem. Soc. Rev. 1987, 16, 75 (ortho esters); DeWolfe, R.H. Carboxylic Ortho Acid Derivatives, Academic Press, NY, 1970, pp. 134–146 (ortho esters); Rekasheva, A.F. Russ. Chem. Rev. 1968, 37, 1009 (enol ethers).
552. Jaques, D.; Leisten, J.A. J. Chem. Soc. 1964, 2683. See also, Olah, G.A.; O'Brien, D.H. J. Am. Chem. Soc. 1967, 89, 1725.
553. See Pavlova, L.A.; Davidovich, Yu.A.; Rogozhin, S.V. Russ. Chem. Rev. 1986, 55, 1026.
554. See Satchell, D.P.N.; Satchell, R.S. Chem. Soc. Rev. 1990, 19, 55.
555. Kreevoy, M.M.; Taft, R.W. J. Am. Chem. Soc. 1955, 77, 3146, 5590.
556. For a discussion of these, and of other evidence, see Cordes, E.H. Prog. Phys. Org. Chem. 1967, 4, 1.
557. Cawley, J.J.; Westheimer, F.H. Chem. Ind. (London) 1960, 656.
558. Young, P.R.; Jencks, W.P. J. Am. Chem. Soc. 1977, 99, 8238. See also, Jencks, W.P. Acc. Chem. Res. 1980, 13, 161; Young, P.R.; Bogseth, R.C.; Rietz, E.G. J. Am. Chem. Soc. 1980, 102, 6268. However, see Amyes, T.L.; Jencks, W.P. J. Am. Chem. Soc. 1988, 110, 3677.
559. See White, A.M.; Olah, G.A. J. Am. Chem. Soc. 1969, 91, 2943; Akhmatdinov, R.T.; Kantor, E.A.; Imashev, U.B.; Yasman, Ya.B.; Rakhmankulov, D.L. J. Org. Chem. USSR 1981, 17, 626.
560. See Belarmino, A.T.N.; Froehner, S.; Zanette, D.; Farah, J.P.S.; Bunton, C.A.; Romsted, L.S. J. Org. Chem. 2003, 68, 706.
561. Fife, T.H.; Natarajan, R. J. Am. Chem. Soc. 1986, 108, 2425, 8050; McClelland, R.A.; S![]() rensen, P.E. Acta Chem. Scand. 1990, 44, 1082.
rensen, P.E. Acta Chem. Scand. 1990, 44, 1082.
562. Fife, T.H.; Natarajan, R. J. Am. Chem. Soc. 1986, 108, 2425, 8050.
563. See Fife, T.H. Acc. Chem. Res. 1972, 5, 264; Wann, S.R.; Kreevoy, M.M. J. Org. Chem. 1981, 46, 419.
564. Kresge, A.J.; Weeks, D.P. J. Am. Chem. Soc. 1984, 106, 7140. See also, Amyes, T.L.; Jencks, W.P. J. Am. Chem. Soc. 1989, 111, 7888, 7900.
565. Fife, T.H.; Brod, L.H. J. Am. Chem. Soc. 1970, 92, 1681; Jensen, J.L.; Herold, L.R.; Lenz, P.A.; Trusty, S.; Sergi, V.; Bell, K.; Rogers, P. J. Am. Chem. Soc. 1979, 101, 4672.
566. See Williams Jr., J.M.; Kreevoy, M.M. Adv. Phys. Org. Chem. 1968, 6, 63.
567. Chiang, Y.; Kresge, A.J.; Lahti, M.O.; Weeks, D.P. J. Am. Chem. Soc. 1983, 105, 6852 and references cited therein; Fife, T.H.; Przystas, T.J. J. Chem. Soc. Perkin Trans. 2 1987, 143.
568. See, for example, Kirby, A.J. Acc. Chem. Res. 1984, 17, 305; Bouab, O.; Lamaty, G.; Moreau, C. Can. J. Chem. 1985, 63, 816. See, however, Ratcliffe, A.J.; Mootoo, D.R.; Andrews, C.W.; Fraser-Reid, B. J. Am. Chem. Soc.1989, 111, 7661.
569. Li, S.; Kirby, A.J.; Deslongchamps, P. Tetrahedron Lett. 1993, 34, 7757.
570. Sammakia, T.; Smith, R.S. J. Org. Chem. 1992, 57, 2997.
571. Huet, F.; Lechevallier, A.; Pellet, M.; Conia, J.M. Synthesis 1978, 63. See Caballero, G.M.; Gros, E.G. Synth. Commun. 1995, 25, 395.
572. Coppola, G.M. Synthesis 1984, 1021.
573. Fujioka, H.; Okitsu, T,; Sawama, Y.; Murata, N.; Li, R.; Kita, Y. J. Am. Chem. Soc. 2006, 128, 5930.
574. Gregg, B.T.; Golden, K.C.; Quinn, J.F. J. Org. Chem. 2007, 72, 5890.
575. Ates, A.; Gautier, A.; Leroy, B.; Plancher, J.M.; Quesnel, Y.; Markó, I.E. Tetrahedron Lett. 1999, 40, 1799.
576. Carringan, M.D.; Sarapa, D.; Smith, R.C.; Wieland, L.C.; Mohan, R.S. J. Org. Chem. 2002, 67, 1027.
577. Watanabe, Y.; Kiyosawa, Y.; Tatsukawa, A.; Hayashi, M. Tetrahedron Lett. 2001, 42, 4641.
578. Ali, M.; Satchell, D.P.N. J. Chem. Soc. Perkin Trans. 2 1992, 219; 1993, 1825; Ali, M.; Satchell, D.P.N.; Le, V.T. J. Chem. Soc. Perkin Trans. 2 1993, 917.
579. See Gröbel, B.; Seebach, D. Synthesis 1977, 357, see pp. 359–367; Cussans, N.J.; Ley, S.V.; Barton, D.H.R. J. Chem. Soc. Perkin Trans. 1 1980, 1654.
580. Corey, E.J.; Erickson, B.W. J. Org. Chem. 1971, 36, 3553; Satchell, D.P.N.; Satchell, R.S. J. Chem. Soc. Perkin Trans. 2 1987, 513.
581. Kamal, A.; Laxman, E.; Reddy, P.S.M.M. Synlett 2000, 1476.
582. Mondal, E.; Bose, G.; Khan, A.T. Synlett 2001, 785.
583. Langille, N.F.; Dakin, L.A.; Panek, J.S. Org. Lett. 2003, 5, 575. See also, Stork, G.; Zhao, K. Tetrahedron Lett. 1989, 30, 287.
584. Khan, A.T.; Mondal, E.; Sahu, P.R. Synlett 2003, 377.
585. Karimi, B.; Seradj, H.; Tabaei, M.H. Synlett 2000, 1798.
586. Chavan, S.P.; Soni, P.; Kamat, S.K. Synlett 2001, 1251.
587. Jones, J.; Kresge, A.J. Can. J. Chem. 1993, 71, 38.
588. See Burt, R.A.; Chiang, Y.; Kresge, A.J.; Szilagyi, S. Can. J. Chem. 1984, 62, 74.
589. See Chwang, W.K.; Kresge, A.J.; Wiseman, J.R. J. Am. Chem. Soc. 1979, 101, 6972.
590. Kiprianova, L.A.; Rekasheva, A.F. Dokl. Akad. Nauk SSSR, 1962, 142, 589.
591. Fife, T.H. J. Am. Chem. Soc. 1965, 87, 1084; Kresge, A.J.; Yin, Y. Can. J. Chem. 1987, 65, 1753.
592. Tsang, W.-Y.; Richard, J.P. J. Am. Chem. Soc. 2007, 129, 10330.
593. Cheon, C.H.; Yamamoto, H. J. Am. Chem. Soc 2008, 130, 9246. For a different example, see Nakashima, D.; Yamamoto, H. Synlett 2006, 150.
594. For a review, see Okuyama, T. Acc. Chem. Res. 1986, 19, 370.
595. See Finlay, J.; McKervey, M.A.; Gunaratne, H.Q.N. Tetrahedron Lett. 1998, 39, 5651.
596. For a density functional analysis of this ion, see Zhao, Y.; Truhlar, D.G. J. Org. Chem. 2007, 72, 295.
597. Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis Vol. 1, Wiley, NY, 1967, p. 796.
598. Fan, R.-H.; Hou, X.-L. Org. Biomol. Chem. 2003, 1, 1565. For a reaction with NaHSO4, see Cavdar, H.; Saracoglu, N. Tetrahedron 2009, 65, 985.
599. Wang, Z.; Cui, Y.-T.; Xu, Z.-B.; Qu, J. J. Org. Chem. 2008, 73, 2270.
600. Berti, G.; Macchia, B.; Macchia, F. Tetrahedron Lett. 1965, 3421.
601. Ready, J.M.; Jacobsen, E.N. J. Am. Chem. Soc. 2001, 123, 2687.
602. See Zhao, L.; Han, B.; Huang, Z.; Miller, M.; Huang, H.; Malashock, D.S.; Zhu, Z.; Milan, A.; Robertson, D.E.; Weiner, D.P.; Burk, M. J. J. Am. Chem. Soc. 2004, 126, 11156.
603. See Feuer, H.; Hooz, J. in Patai, S. The Chemistry of the Ether Linkage, Wiley, NY, 1967, pp. 446–450, 460–468.
604. For a list of reagents used to convert alcohols and phenols to ethers, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 890–893.
605. Ouk, S.; Thiebaud, S.; Borredon, E.; Legars, P.; Lecomte, L. Tetrahedron Lett. 2002, 43, 2661.
606. Lee, J.C.; Yuk, J.Y.; Cho, S.H. Synth. Commun. 1995, 25, 1367.
607. Jin, C.H.; Lee, H.Y.; Lee, S.H.; Kim, I.S.; Jung, Y.H. Synlett 2007, 2695.
608. Bogdal, D.; Pielichowski, J.; Jaskot, K. Org. Prep. Proceed. Int. 1998, 30, 427.
609. Yuncheng, Y.; Yulin, J.; Jun, P.; Xiaohui, Z.; Conggui, Y. Gazz. Chim. Ital. 1993, 123, 519.
610. Paul, S.; Gupta, M. Tetrahedron Lett. 2004, 45, 8825.
611. Xu, Z.Y.; Xu, D.Q.; Liu, B.Y. Org. Prep. Proceed. Int. 2004, 36, 156. Also see More, S.V.; Ardhapure, S.S.; Naik, N.H.; Bhusare, S.R.; Jadhav, W.N.; Pawar, R.P. Synth. Commun. 2005, 35, 3113.
612. See Jha, S.C.; Joshi, N.N. J. Org. Chem. 2002, 67, 3897.
613. Boons, G.-J.; Castle, G.H.; Clase, J.A.; Grice, P.; Ley, S.V.; Pinel, C. Synlett, 1993, 913.
614. Olah, G.A.; Halpern, Y.; Lin, H.C. Synthesis 1975, 315. Also see Masada, H.; Yonemitsu, T.; Hirota, K. Tetrahedron Lett. 1979, 1315.
615. Masada, H.; Sakajiri, T. Bull. Chem. Soc. Jpn. 1978, 51, 866.
616. Bartoli, G.; Bosco, M.; Locatelli, M.; Marcantoni, E.; Melchiorre, P.; Sambri, L. Org. Lett. 2005, 7, 427.
617. See Salomaa, P.; Kankaanperä, A.; Pihlaja, K. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 454–466. Also see Biordi, J.; Moelwyn-Hughes, E.A. J. Chem. Soc. 1962, 4291.
618. Aspinall, H.C.; Greeves, N.; Lee, W.-M.; McIver, E.G.; Smith, P.M. Tetrahedron Lett. 1997, 38, 4679.
619. Masada, H.; Oishi, Y. Chem. Lett. 1978, 57; Camps, F.; Coll, J.; Moretó, J.M. Synthesis 1982, 186.
620. Banerjee, S.K.; Gupta, B.D.; Singh, K. J. Chem. Soc., Chem. Commun. 1982, 815.
621. Nakagawa, H.; Hirabayashi, T.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2004, 69, 3474; Haight, A.R.; Stoner, E.J.; Peterson, M.J.; Grover, V.K. J. Org. Chem. 2003, 68, 8092.
622. Evans, P.A.; Leahy, D.K. J. Am. Chem. Soc. 2002, 124, 7882.
623. Ueno, S.; Hartwig, J.F. Angew. Chem. Int. Ed. 2008, 47, 1928.
624. Saito, T.; Yasuda, M.; Baba, A. Synlett 2005, 1737.
625. Lepore, S.D.; He, Y. J. Org. Chem. 2003, 68, 8261.
626. Blouin, M.; Frenette, R. J. Org. Chem. 2001, 66, 9043.
627. Willis, M.C.; Taylor, D.; Gillmore, A.T. Chem. Commun. 2003, 2222.
628. Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Springer, NY, 1978, pp. 128–138; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 73–84. See also, Eynde, J.J.V.; Mailleux, I. Synth. Commun. 2001, 31, 1; de la Zerda, J.; Barak, G.; Sasson, Y. Tetrahedron 1989, 45, 1533.
629. Jursic, B. Tetrahedron 1988, 44, 6677.
630. Nishiyama, T.; Kameyama, H.; Maekawa, H.; Watanuki, K. Can. J. Chem. 1999, 77, 258.
631. See Greene, T.W. Protective Groups in Organic Synthesis Wiley, New York, 1980; Wuts, P.G.M.; Greene, T.W. ProtectiveGroups in Organic Synthesis 2nd ed., Wiley, New York, 1991; Wuts, P.G.M.; Greene, T.W. Protective Groups in OrganicSynthesis 3rd ed., Wiley, New York, 1999; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis 4th ed., Wiley, New Jersey, 2006.
632. Guindon, Y.; Yoakim, C.; Morton, H.E. J. Org. Chem. 1984, 49, 3912. For other methods, see Hanessian, S.; Delorme, D.; Dufresne,Y. Tetrahedron Lett. 1984, 25, 2515; Rigby, J.H.; Wilson, J.Z. Tetrahedron Lett. 1984, 25, 1429.
633. Bartoszewicz, A.; Kalek, M.; Stawinski, J. Tetrahedron 2008, 64, 8843.
634. Ashby, E.C.; Bae, D.; Park, W.; Depriest, R.N.; Su, W. Tetrahedron Lett. 1984, 25, 5107.
635. Namboodiri, V.V.; Varma, R.S. Tetrahedron Lett. 2002, 43, 4593.
636. See Knipe, A.C. J. Chem. Soc. Perkin Trans. 2 1973, 589.
637. See Berti, G. Top. Stereochem. 1973, 7, 93, pp. 187.
638. Lang, F.; Kassab, D.J.; Ganem, B. Tetrahedron Lett. 1998, 39, 5903.
639. Lygo, B.; Gardiner, S.D.; McLeod, M.C.; To, D.C.M. Org. Biomol. Chem. 2007, 5, 2283.
640. McPake, C.B.; Murray, C.B.; Sandford, G. Tetrahedron Lett. 2009, 50, 1674.
641. See Kim, K.M.; Jeon, D.J.; Ryu, E.K. Synthesis 1998, 835. Also see Marek, I.; Lefrancois, J.-M.; Normant, J.-F. Tetrahedron Lett. 1992, 33, 1747.
642. Yadav, L.D.S.; Kapoor, R. Synthesis 2002, 2344.
643. Neumann, H. Chimia, 1969, 23, 267.
644. Guthrie, R.D.; Jenkins, I.D.; Yamasaki, R.; Skelton, B.W.; White, A.H. J. Chem. Soc. Perkin Trans. 1 1981, 2328 and references cited therein. For a review of diethyl azodicarboxylate–Ph3P, see Mitsunobu, O. Synthesis 1981, 1.
645. Kelly, J.W.; Evans, Jr., S.A. J. Org. Chem. 1986, 51, 5490. See also, Hendrickson, J.B.; Hussoin, M.S. Synlett, 1990, 423.
646. Szeja, W. Synthesis 1985, 983.
647. Cao, Y.-Q.; Pei, B.-G. Synth. Commun. 2000, 30, 1759.
648. Mihara, M.; Ishino, Y.; Minakata, S.; Komatsu, M. Synlett 2002, 1526.
649. Quach, T.D.; Batey, R.A. Org. Lett. 2003, 5, 1381.
650. Armstrong, A.; Brackenridge, I.; Jackson, R.F.W.; Kirk, J.M. Tetrahedron Lett. 1988, 29, 2483.
651. Rai, A.N.; Basu, A. Tetrahedron Lett. 2003, 44, 2267.
652. Lajunen, M.; Ianskanen-Lehti, K. Acta Chem. Scand. B, 1994, 48, 861.
653. Pansare, S.V.; Jain, R.P.; Bhattacharyya, A. Tetrahedron Lett. 1999, 40, 5255.
654. For a review of diazomethane, see Pizey, J.S. Synthetic Reagents, Vol. 2; Wiley, NY, 1974, pp. 65–142.
655. Neeman, M.; Caserio, M.C.; Roberts, J.D.; Johnson, W.S. Tetrahedron 1959, 6, 36.
656. Ogawa, H.; Hagiwara, H.; Chihara, T.; Teratani, S.; Taya, K. Bull. Chem. Soc. Jpn. 1987, 60, 627.
657. Kreevoy, M.M.; Thomas, S.J. J. Org. Chem. 1977, 42, 3979. See also, McGarrity, J.F.; Smyth, T. J. Am. Chem. Soc. 1980, 102, 7303.
658. De, P.; Nonappa; Pandurangan, K.; Maitra, U.; Wailes, S. Org. Lett. 2007, 9, 2767.
659. Noels, A.F.; Demonceau, A.; Petiniot, N.; Hubert, A.J.; Teyssié, P. Tetrahedron 1982, 38, 2733.
660. Chen, C.; Zhu, S.-F.; Liu, B.; Wang, L.-X.; Zhou, Q.-L. J. Am. Chem. Soc. 2007, 129, 12616.
661. Baganz, H.; May, H. Angew. Chem. Int. Ed. 1966, 5, 420.
662. See Feuer, H.; Hooz, J. in Patai, S. The Chemistry of the Ether Linkage, Wiley, NY, 1967, pp.457–460, 468–470.
663. Toda, F.; Takumi, H.; Akehi, M. J. Chem. Soc. Perkin Trans. 2 1990, 1270.
664. Zolfigol, M.A.; Mohammadpoor-Baltork, I.; Habibi, D.; Mirjalili, B.B.F.; Bamoniri, A. Tetrahedron Lett. 2003, 44, 8165.
665. See Ooi, T.; Ichikawa, H.; Itagaki, Y.; Maruoka, K. Heterocycles 2000, 52, 575.
666. See, for example, Jenner, G. Tetrahedron Lett. 1988, 29, 2445.
667. Zhu, Z.; Espenson, J.H. J. Org. Chem. 1996, 61, 324.
668. Boyer, B.; Keramane, E.-M.; Roque, J.-P.; Pavia, A.A. Tetrahedron Lett. 2000, 41, 2891.
669. Lizarzaburu, M.E.; Shuttleworth, S. Tetrahedron Lett. 2002, 43, 2157.
670. D'Angelo, J.G.; Sawyer, R.; Kumar, A.; Onorato, A.; McCluskey, C.; Delude, C.; Vollenweider, L.; Reyes, N.; French, R.; Warner, S.; Chou, J.; Stenzel, J.; Sotzing, G.A.; Smith, M.B. J. Polymer Sci., Part A 2007, 45, 2328.
671. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 893–894.
672. See Olah, G.A.; Fung, A.P.; Malhotra, R. Synthesis 1981, 474.
673. Vowinkel, E. Chem. Ber. 1962, 95, 2997; 1963, 96, 1702; 1966, 99, 42.
674. Pratt, E.F.; Draper, J.D. J. Am. Chem. Soc. 1949, 71, 2846. See Salehi, P.; Irandoost, M.; Seddighi, B.; Behbahani, F.K.; Tahmasebi, D.P. Synth. Commun. 2000, 30, 1743.
675. Zoltewicz, J.A.; Sale, A.A. J. Org. Chem. 1970, 35, 3462.
676. Itoh, A.; Hirose, Y.; Kashiwagi, H.; Masaki, Y. Heterocycles 1994, 38, 2165.
677. See Salomaa, P.; Kankaanperä, A.; Pihlaja, K. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 458–463; DeWolfe, R.H. Carboxylic Ortho Acid Derivatives, Academic Press, NY, 1970, pp. 18–29, 146–148.
678. McElvain, S.M.; Curry, M.J. J. Am. Chem. Soc. 1948, 70, 3781.
679. Jnaneshwara, G.K.; Barahate, N.B.; Sudalai, A.; Deshpande, V.H.; Wakharkar, R.D.; Gajare, A.S.; Shingare, M.S.; Sukumar, R. J. Chem. Soc. Perkin Trans. 1 1998, 965.
680. Watanabe, W.H.; Conlon, L.E. J. Am. Chem. Soc. 1957, 79, 2828; Shostakovskii, M.F.; Trofimov, B.A.; Atavin, A.S.; Lavrov, V.I. Russ. Chem. Rev. 1968, 37, 907; Gareev, G.A. J. Org. Chem. USSR 1982, 18, 36.
681. Magano, J.; Chen, M.H.; Clark, J.D.; Nussbaumer, T. J. Org. Chem. 2006, 71, 7103.
682. Ponaras, A.A.; Meah, M.Y. Tetrahedron Lett. 1986, 27, 4953.
683. Iranpoor, N.; Tarrian, T.; Movahedi, Z. Synthesis 1996, 1473. See Moberg, C.; Rákos, L.; Tottie, L. Tetrahedron Lett. 1992, 33, 2191 for an example that generates a hydroxy ether with high enantioselectivity. Also see, Chini, M.; Crotti, P.; Gardelli, C.; Macchia, F. Synlett, 1992, 673.
684. See Posner, G.H.; Rogers, D.Z. J. Am. Chem. Soc. 1977, 99, 8208, 8214.
685. Robinson, M.W.C.; Buckle, R.; Mabbett, I.; Grant, G.M.; Graham, A.E. Tetrahedron Lett. 2007, 48, 4723.
686. Barluenga, J.; Vázquez-Villa, H.; Ballesteros, A.; González, J.M. Org. Lett. 2002, 4, 2817.
687. Williams, D.B.G.; Lawton, M. Org. Biomol. Chem. 2005, 3, 3269.
688. Mohammadpoor-Baltork, I.; Tangestaninejad, S.; Aliyan, H.; Mirkhani, V. Synth. Commun., 2000, 30, 2365.
689. Surendra, K.; Krishnaveni, N.; Nageswar, Y.V.D.; Rao, K.R. J. Org. Chem. 2003, 68, 4994.
690. Fringuelli, F.; Pizzo, F.; Tortoioli, S.; Vaccaro, L. J. Org. Chem. 2003, 68, 8248; Amantini, D.; Friguelli, F.; Pizzo, F.; Tortoioli, S.; Vaccaro, L. Synlett 2003, 2292.
691. See also Degl'Innocenti, A.; Capperucci, A.; Cerreti, A.; Pollicino, S.; Scapecchi, S.; Malesci, I.; Castagnoli, G. Synlett 2005, 3063.
692. Ogawa, C.; Wang, N.; Kobayashi, S. Chem. Lett. 2007, 36, 34.
693. Nandakumar, M.V.; Tschöp, A.; Krautscheid, H.; Schneider, C. Chem. Commun. 2007, 2756.
694. Tiecco, M.; Testaferri, L.; Marini, F.; Sternativo, S.; Del Verme, F.; Santi, C.; Bagnoli, L.; Temperini, A. Tetrahedron 2008, 64, 3337.
695. Matsumura, R.; Suzuki, T.; Sato, K.; Oku, K.-i.; Hagiwara, H.; Hoshi, T.; Ando, M.; Kamat, V.P. Tetrahedron Lett. 2000, 41, 7701. See also, Karikomi, M.; Watanabe, S.; Kimura, Y.; Uyehara, T. Tetrahedron Lett. 2002, 43,1495.
696. Wu, M.H.; Hansen, K.B.; Jacobsen, E.N. Angew. Chem. Int. Ed. 1999, 38, 2012.
697. Behrens, C.H.; Ko, S.Y.; Sharpless, K.B.; Walker, F.J. J. Org. Chem. 1985, 50, 5687. See Yamazaki, T.; Ichige, T.; Kitazume, T. Org. Lett. 2004, 6, 4073.
698. See Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines, Academic Press, NY, 1969, pp. 224–227, 256–257.
699. Wu, J.; Hou, X.-L.; Dai, L.-X. J. Chem. Soc., Perkin Trans. 1 2001, 1314.
700. Hou, X.-L.; Fan, R.-H.; Dai, L.-X. J. Org. Chem. 2002, 67, 5295.
701. Liu, Y.-K.; Li, R.; Yue, L.; Li, B.-J.; Chen, Y.-C.; Wu, Y.; Ding, L.-S. Org. Lett. 2006, 8, 1521.
702. Ghorai, M.K.; Das, K.; Shukla, D. J. Org. Chem. 2007, 72, 5859.
703. Chandrasekhar, S.; Narsihmulu, Ch.; Sultana, S.S. Tetrahedron Lett. 2002, 43, 7361.
704. Prasad, B.A.B.; Sekar, G.; Singh, V.K. Tetrahedron Lett. 2000, 41, 4677.
705. Yadav, J.S.; Reddy, B.V.S.; Sadashiv, K.; Harikishan, K. Tetrahedron Lett. 2002, 43, 2099.
706. Righi, G.; Potini, C.; Bovicelli, P. Tetrahedron Lett. 2002, 43, 5867.
707. Yadav, J.S.; Subba Reddy, B.V.; Jyothirmai, B.; Murty, M.S.R. Tetrahedron Lett. 2005, 46, 6385. Also in water, with β-cyclodextrin, see Reddy, M.S.; Narender, M.; Nageswar, Y.V.D.; Rao, K.R. Tetrahedron Lett. 2005, 46,6437.
708. Mita, T.; Fujimori, I.; Wada, R.; Wen, J.; Kanai, M.; Shibasaki, M. J. Am. Chem. Soc. 2005, 127, 11252.
709. Xichun, F.; Guofu, Q.; Shucai, L.; Hanbing, T.; Lamei, W.; Xianming, H. Tetrahedron Asymmetry 2006, 17, 1394.
710. Granik, V.G.; Pyatin, B.M.; Glushkov, R.G.Russ. Chem. Rev. 1971, 40, 747, see p. 749.
711. See Vogel, D.E.; Büchi, G.H. Org. Synth., 66, 29. With pyridinium salts, see Poon, K.W.C.; Dudley, G.B. J. Org. Chem. 2006, 71, 3923. See also Saitoh, T.; Ichikawa, J. J. Am. Chem. Soc. 2005, 127, 9696.
712. Tamao, K.; Kakui, T.; Akita, M.; Iwahara, T.; Kanatani, R.; Yoshida, J.; Kumada, M. Tetrahedron 1983, 39, 983; Fleming, I.; Henning, R.; Plaut, H. J. Chem. Soc., Chem. Commun. 1984, 29. For the protodesilylation step see Häbich, D.; Effenberger, F. Synthesis 1979, 841. Also see Buncel, E.; Davies, A.G. J. Chem. Soc. 1958, 1550.
713. Suginome, M.; Matsunaga, S.; Ito, Y. Synlett, 1995, 941.
714. Sunderhaus, J.D.; Lam, H.; Dudley, G.B. Org. Lett. 2003, 5, 4571.
715. See Matsumoto, Y.; Hayashi, T.; Ito, Y. Tetrahedron 1994, 50, 335; Uozumi, Y.; Kitayama, K.; Hayashi, T.; Yanagi, K.; Fukuyo, E. Bull. Chem. Soc. Jpn. 1995, 68, 713.
716. Liu, D.; Kozmin, S.A. Angew. Chem. Int. Ed. 2001, 40, 4757.
717. Shaw, J.E.; Kunerth, D.C. J. Org. Chem. 1974, 39, 1968; Larock, R.C. J. Org. Chem. 1974, 39, 3721; Pfeffer, P.E.; Silbert, L.S. J. Org. Chem. 1976, 41, 1373.
718. Bases include DBU (see Reaction 17-13): See Mal, D. Synth. Commun. 1986, 16, 331. Cs2CO3: Lee, J.C.; Oh, Y.S.; Cho, S.H.; Lee, J.I. Org. Prep. Proceed. Int. 1996, 28, 480. CsF-Celite: Lee, J.C.; Choi, Y. Synth. Commun.1998, 28, 2021.
719. See Dijkstra, G.; Kruizinga, W.H.; Kellogg, R.M. J. Org. Chem. 1987, 52, 4230.
720. See Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Acaemic Press, NY, 1978, pp. 140–155; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 85–95.
721. See Clark, J.H.; Miller, J.M. Tetrahedron Lett. 1977, 599.
722. See, however, Moore, G.G.; Foglia, T.A.; McGahan, T.J. J. Org. Chem. 1979, 44, 2425.
723. Bram, G.; Loupy, A.; Majdoub, M.; Gutierrez, E.; Ruiz-Hitzky, E. Tetrahedron 1990, 46, 5167. See Arrad, O.; Sasson, Y. J. Am. Chem. Soc. 1988, 110, 185; Dakka, J.; Sasson, Y.; Khawaled, K.; Bram, G.; Loupy, A. J. Chem. Soc., Chem. Commun. 1991, 853.
724. Yuncheng, Y.; Yulin, J.; Dabin, G. Synth. Commun. 1992, 22, 3109.
725. Brinchi, L.; Germani, R.; Savelli, G. Tetraheron Lett. 2003, 44, 2027, 6583; Liu, Z.; Chen, Z.-C.; Zheng, Q.-G. Synthesis 2004, 33.
726. Mitsunobu, O.; Yamada, M. Bull. Chem. Soc. Jpn. 1967, 40, 2380; Camp, D.; Jenkins, I.D. Aust. J. Chem. 1988, 41, 1835.
727. But, T.Y.S.; Toy, P.H. Chemistry: Asian J. 2007, 2, 1340. See Ahn, C.; Correia, R.; DeShong, P. J. Org. Chem. 2002, 67, 1751 and references cited therein. See also, Hughes, D.L. Org. Prep. Proceed. Int. 1996, 28, 127; Dembinski, R. Eur. J. Org. Chem. 2004, 2763; Dandapani, S.; Curran, D.P. Chem. Eur. J. 2004, 10, 3131. Also see Steinreiber, A.; Stadler, A.; Mayer, S.F.; Faber, K.; Kappe, C.O. Tetrahedron Lett. 2001, 42, 6283. For a chromatography-free product separation, see Proctor, A.J.; Beautement, K.; Clough, J.M.; Knight, D.W.; Li, Y. Tetrahedron Lett. 2006, 47, 5151.
728. Sugimura, T.; Hagiya, K. Chem. Lett. 2007, 36, 566.
729. See Tsunoda, T.; Yamamiya, Y.; Kawamura, Y.; Itô, S. Tetrahedron Lett. 1995, 36, 2529; Tsunoda, T.; Nagaku, M.; Nagino, C.; Kawamura, Y.; Ozaki, F.; Hioki, H.; Itô, S. Tetrahedron Lett. 1995, 36, 2531. For fluorous reactions and reagents see Dandapani, S.; Curran, D.P. Tetrahedron 2002, 58, 3855.
730. But, T.Y.S.; Toy, P.H. J. Am. Chem. Soc. 2006, 128, 9636.
731. Harned, A.M.; He, H.S.; Toy, P.H.; Flynn, D.L.; Hanson, P.R. J. Am. Chem. Soc. 2005, 127, 52.
732. Yoakim, C.; Guse, I.; O'Meara, J.A.; Thavonokham, B. Synlett 2003, 473.
733. See Papeo, G.; Poster, H.; Vianello, P.; Varasi, M. Synthesis 2004, 2886.
734. Iranpoor, N.; Firouzabadi, H.; Akhlaghinia, B.; Azadi, R. Synthesis 2004, 92.
735. Fitzjarrald, V.P.; Pongdee, R. Tetrahedron Lett. 2007, 48, 3553.
736. García-Delgado, N.; Riera, A.; Verdaguer, X. Org. Lett. 2007, 9, 635.
737. See Galli, C.; Mandolini, L. Org. Synth. VI, 698; Kimura, Y.; Regen, S.L. J. Org. Chem. 1983, 48, 1533.
738. Togo, H.; Muraki, T.; Yokoyama, M. Tetrahedron Lett. 1995, 36, 7089.
739. Klumpp, G.W.; Bos, H.; Schakel, M.; Schmitz, R.F.; Vrielink, J.J. Tetrahedron Lett. 1975, 3429.
740. Yamaji, M.; Fujiwara, Y.; Asano, R.; Teranishi, S. Bull. Chem. Soc. Jpn. 1973, 46, 90.
741. Ooi, T.; Sugimoto, H.; Doda, K.; Maruoka, K. Tetrahedron Lett. 2001, 42, 9245.
742. Sato, T.; Otera, J. Synlett, 1995, 336.
743. Hendrickson, J.B.; Kandall, L.C. Tetrahedron Lett. 1970, 343.
744. Verdecchia, M.; Frochi, M.; Palombi, L.; Rossi, L. J. Org. Chem. 2002, 67, 8287. See also, Kadokawa, J.-i.; Habu, H.; Fukamachi, S.; Karasu, M.; Tagaya, H.; Chiba, K. J. Chem. Soc., Perkin Trans. 1 1999, 2205.
745. Barrett, A.G.M.; Braddock, D.C.; James, R.A.; Koike, N.; Procopiou, P.A. J. Org. Chem. 1998, 63, 6273.
746. Grundy, J.; James, B.G.; Pattenden, G. Tetrahedron Lett. 1972, 757.
747. Harris, M.M.; Patel, P.K. Chem. Ind. (London) 1973, 1002.
748. Chakraborti, A.K.; Basak, A.; Grover, V. J. Org. Chem. 1999, 64, 8014. See also, Avila–Zárraga, J.G.; Martínez, R. Synth. Commun. 2001, 31, 2177.
749. Shieh, W.-C.; Dell, S.; Repic, O. Tetrahedron Lett. 2002, 43, 5607.
750. Szmuszkovicz, J. Org. Prep. Proceed. Int. 1972, 4, 51.
751. Vorbrüggen, H. Angew. Chem. Int. Ed. 1963, 2, 211; Brechbühler, H.; Büchi, H.; Hatz, E.; Schreiber, J.; Eschenmoser, A. Angew. Chem. Int. Ed. 1963, 2, 212.
752. Curtis, V.A.; Schwartz, H.S.; Hartman, A.F.; Pick, R.M.; Kolar, L.W.; Baumgarten, R.J. Tetrahedron Lett. 1977, 1969.
753. See Katritzky, A.R.; Gruntz, U.; Kenny, D.H.; Rezende, M.C.; Sheikh, H. J. Chem. Soc. Perkin Trans. 1 1979, 430.
754. Wilson, N.D.V.; Joule, J.A. Tetrahedron 1968, 24, 5493.
755. Raber, D.J.; Gariano, Jr., P.; Brod, A.O.; Gariano, A.; Guida, W.C.; Guida, A.R.; Herbst, M.D. J. Org. Chem. 1979, 44, 1149.
756. Zheng, T.-C.; Burkart, M.; Richardson, D.E. Tetrahedron Lett. 1999, 40, 603.
757. Gómez-Parra, V.; Sánchez, F.; Torres, T. J. Chem. Soc. Perkin Trans. 2 1987, 695.
758. Dueno, E.E.; Chu, F.; Kim, S.-I.; Jung, K.W. Tetrahedron Lett. 1999, 40, 1843. Also see Yoshida, M.; Fujita, M.; Ishii, T.; Ihara, M. J. Am. Chem. Soc. 2003, 125, 4874.
759. Ganem, B.; Small, Jr., V.M. J. Org. Chem. 1974, 39, 3728.
760. Procopiou, P.A.; Baugh, S.P.D.; Flack, S.S.; Inglis, G.G.A. Chem. Commun. 1996, 2625.
761. Karger, M.H.; Mazur, Y. J. Am. Chem. Soc. 1968, 90, 3878. See also, Coffi-Nketsia, S.; Kergomard, A.; Tautou, H. Bull. Soc. Chim. Fr. 1967, 2788.
762. See Otera, J.; Matsuzaki, S. Synthesis 1986, 1019; Deardorff, D.R.; Myles, D.C. Org. Synth., 67, 114.
763. Kwon, D.W.; Kim, Y.H.; Lee, K. J. Org. Chem. 2002, 67, 9488.
764. Malladi, R.R.; Kabalka, G.W. Synth. Commun. 2002, 32, 1997.
765. Li, F.; Xiao, L.; Xia, C.; Hu, B. Tetrahedron Lett. 2004, 45, 8307.
766. Denmark, S.E.; Ahmad, M. J. Org. Chem. 2007, 72, 9630.
767. Meerwein, H.; Hederich, V.; Wunderlich, K. Arch. Pharm. 1958, 291/63, 541. See Perst, H. Oxonium Ions in Organic Chemistry, Verlag Chemie, Deerfield Beach, FL, 1971, pp. 22–39.
768. See Hiatt, R. in Swern, D. Organic Peroxides, Vol. 2, Wiley, NY, 1971, pp. 1–151; Pandiarajan, K. in Pizey, J.S. Synthetic Reagents, Vol. 6, Wiley, NY, 1985, pp. 60–155.
769. Cookson, P.G.; Davies, A.G.; Roberts, B.P. J. Chem. Soc., Chem. Commun. 1976, 1022. Also see Bourgeois, M.; Montaudon, E.; Maillard, B. Synthesis 1989, 700.
770. Johnson, R.A.; Nidy, E.G.; Merritt, M.V. J. Am. Chem. Soc. 1978, 100, 7960.
771. See San Filippo, Jr., J.; Chern, C.; Valentine, J.S. J. Org. Chem. 1975, 40, 1678; Corey, E.J.; Nicolaou, K.C.; Shibasaki, M.; Machida, Y.; Shiner, C.S. Tetrahedron Lett. 1975, 3183.
772. Salomon, M.F.; Salomon, R.G. J. Am. Chem. Soc. 1979, 101, 4290.
773. See Bouillon, G.; Lick, C.; Schank, K. in Patai, S. The Chemistry of Peroxides, Wiley, NY, 1983, pp. 279–309; Hiatt, R.; Swern, D. Organic Peroxides, Vol. 2, Wiley, NY, 1971, pp. 799–929.
774. See Silbert, L.S.; Siegel, E.; Swern, D. J. Org. Chem. 1962, 27, 1336.
775. Greene, F.D.; Kazan, J. J. Org. Chem. 1963, 28, 2168.
776. See Salomaa, P.; Kankaanperä, A.; Pihlaja, K. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pp. 481–497.
777. See Aldred, S.E.; Williams, D.L.H.; Garley, M. J. Chem. Soc. Perkin Trans. 2 1982, 777.
778. Sandler, S.R.; Karo, W. Organic Functional Group Preparations, 2nd ed., Vol 3, Academic Press, NY, 1989, pp. 129–151.
779. Choudary, B.M.; Chowdari, N.S.; Kantam, M.L. Tetraheron 2000, 56, 7291.
780. Vignola, N.; Dahmen, S.; Enders, D.; Bräse, S. Tetrahedron Lett. 2001, 42, 7833.
781. Bengtson, A.; Hallberg, A.; Larhed, M. Org. Lett. 2002, 4, 1231.
782. Shibata, N.; Matsunaga, M.; Fukuzumi, T.; Nakamura, S.; Toru, T. Synlett 2005, 1699.
783. See Williams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 150–172.
784. Doyle, M.P.; Terpstra, J.W.; Pickering, R.A.; LePoire, D.M. J. Org. Chem. 1983, 48, 3379. See Williams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 150–156.
785. Barton, D.H.R.; Narang, S.C. J. Chem. Soc. Perkin Trans. 1 1977, 1114.
786. Pungente, M.D.; Weiler, L. Org. Lett. 2001, 3, 643.
787. See Boguslavskaya, L.S.; Chuvatkin, N.N.; Kartashov, A.V. Russ. Chem. Rev. 1988, 57, 760.
788. Klamann, D.; Weyerstahl, P. Chem. Ber. 1965, 98, 2070.
789. Karger, M.H.; Mazur, Y. J. Org. Chem. 1971, 36, 532, 540.
790. See Zefirov, N.S.; Zhdankin, V.V.; Koz'min, A.S. Russ. Chem. Rev. 1988, 57, 1041.
791. Zefirov, N.S.; Kirin, V.N.; Yur'eva, N.M.; Zhdankin, V.V.; Kozmin, A.S. J. Org. Chem. USSR 1987, 23, 1264.
792. Golding, P.; Millar, R.W.; Paul, N.C.; Richards, D.H. Tetrahedron Lett. 1988, 29, 2731, 2735.
793. Han, L.-B.; Zhao, C.-Q. J. Org. Chem. 2005, 70, 10121.
794. Rahman, S.M.A.; Ohno, H.; Tanaka, T. Tetrahedron Lett. 2001, 42, 8007.
795. S![]() rbye, K.; Tautermann, C.; Carlsen, P.; Fiksdahl, A. Tetraheron Asymmetry, 1998, 9, 681.
rbye, K.; Tautermann, C.; Carlsen, P.; Fiksdahl, A. Tetraheron Asymmetry, 1998, 9, 681.
796. See Abele, E.; Lukevics, E. Org. Prep. Proceed. Int. 2000, 32, 235.
797. See Torssell, K.B.G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, VCH, NY, 1988, pp. 75–93; Katritzky, A.R.; Cui, X.; Long, Q.; Yanga, B.; Wilcox, A.L.; Zhang, Y.-K. Org. Prep. Proceed. Int. 2000, 32, 175.
798. See Reutov, O.A.; Beletskaya, I.P.; Kurts, A.L. Ambident Anions, Plenum, NY, 1983, pp. 262–272.
799. Buehler, E. J. Org. Chem. 1967, 32, 261.
800. See Bernardi, F.; Csizmadia, I.G.; Mangini, A. Organic Sulfur Chemistry, Elsevier, NY, 1985; Oae, S. Organic Chemistry of Sulfur, Plenum, NY, 1977. For selenium compounds, see Krief, A.; Hevesi, L. Organoselenium Chemistry I, Springer, NY, 1988; Liotta, D. Organoselenium Chemistry, Wiley, NY, 1987.
801. See Ashby, E.C.; Park, W.S.; Goel, A.B.; Su, W. J. Org. Chem. 1985, 50, 5184.
802. See Wardell, J.L. in Patai, S. The Chemistry of the Thiol Group, pt. 1, Wiley, NY, 1974, pp. 179–211.
803. Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. Chem. Lett. 2000, 1304.
804. See Vasil'tsov, A.M.; Trofimov, B.A.; Amosova, S.V. J. Org. Chem. USSR 1983, 19, 1197.
805. Gingras, M.; Harpp, D.N. Tetrahedron Lett. 1990, 31, 1397.
806. Miranda, E.I.; Díaz, M.J.; Rosado, I.; Soderquist, J.A. Tetrahedron Lett. 1994, 35, 3221; Rane, A.M.; Miranda, E.I.; Soderquist, J. Tetrahedron Lett. 1994, 35, 3225.
807. Lucien, J.; Barrault, J.; Guisnet, M.; Maurel, R. Nouv. J. Chim. 1979, 3, 15.
808. Nishio, T. J. Chem. Soc. Perkin Trans. 1 1993, 1113.
809. Kornblum, N.; Widmer, J. J. Am. Chem. Soc. 1978, 100, 7086.
810. See Peach, M.E. in Patai, S. The Chemistry of the Thiol Groups, pt. 2, Wiley, NY, 1974, pp. 721–735.
811. Yin, J.; Pidgeon, C. Tetrahedron Lett. 1997, 38, 5953.
812. See Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 221–233. Also see Salvatore, R.N.; Smith, R.A.; Nischwitz, A.K.; Gavi, T. Tetrahedron Lett. 2005, 46, 8931.
813. Ono, N.; Miyake, H.; Saito, T.; Kaji, A. Synthesis 1980, 952. See also, Ferreira, J.T.B.; Comasseto, J.V.; Braga, A.L. Synth. Commun. 1982, 12, 595; Ando, W.; Furuhata, T.; Tsumaki, H.; Sekiguchi, A. Synth. Commun. 1982, 12, 627.
814. Shah, S.T.A.; Khan, K.M.; Heinich, A.M.; Voelter, W. Tetrahedron Lett. 2002, 43, 8281.
815. Kondo, T.; Kanda, Y.; Baba, A.; Fukuda, K.; Nakamura, A.; Wada, K.; Morisaki, Y.; Mitsudo, T.-a. J. Am. Chem. Soc. 2002, 124, 12960.
816. Cristau, H.J.; Chabaud, B.; Labaudiniere, R.; Christol, H. J. Org. Chem. 1986, 51, 875.
817. Ochiai, M.; Hirobe, M.; Miyamoto, K. J. Am. Chem. Soc. 2006, 128, 9046.
818. See Cain, M.E.; Evans, M.B.; Lee, D.F. J. Chem. Soc. 1962, 1694.
819. Inada, Y.; Nishibayashi, Y.; Hidai, M.; Uemura, S. J. Am. Chem. Soc. 2002, 124, 15172.
820. Walker, K.A.M. Tetrahedron Lett. 1977, 4475. See the references in this paper for other methods of converting alcohols to sulfides. See also, Cleary, D.G. Synth. Commun. 1989, 19, 737.
821. Zhang, X.; Rao, W.; Chan, P.W.H. Synlett 2008, 2204.
822. Fujisaki, S.; Fujiwara, I.; Norisue, Y.; Kajigaeshi, S. Bull. Chem. Soc. Jpn. 1985, 58, 2429.
823. See Evers, M. Chem. Scr. 1986, 26, 585.
824. Also see Hanessian, S.; Guindon, Y. Tetrahedron Lett. 1980, 21, 2305; Williard, P.G.; Fryhle, C.B. Tetrahedron Lett. 1980, 21, 3731; Node, M.; Nishide, K.; Fuji, K.; Fujita, E. J. Org. Chem. 1980, 45, 4275; Evers, M.; Christiaens, L. Tetrahedron Lett. 1983, 24, 377; Tiecco, M. Synthesis 1988, 749.
825. Feutrill, G.I.; Mirrington, R.N. Tetrahedron Lett. 1970, 1327, Aust. J. Chem. 1972, 25, 1719, 1731.
826. Goux, C.; Lhoste, P.; Sinou, D. Tetrahedron 1994, 50, 10321.
827. Shamma, M.; Deno, N.C.; Remar, J.F. Tetrahedron Lett. 1966, 1375. For alternative procedures, see Hutchins, R.O.; Dux, F.J. J. Org. Chem. 1973, 38, 1961; Posner, G.H.; Ting, J. Synth. Commun. 1974, 4, 355.
828. Kametani, T.; Kigasawa, T.; Hiiragi, M.; Wagatsuma, N.; Wakisaka, K. Tetrahedron Lett. 1969, 635.
829. For another reagent, see Harpp, D.N.; Gingras, M.; Aida, T.; Chan, T.H. Synthesis 1987, 1122.
830. Nedugov, A.N.; Pavlova, N.N. Zhur. Org. Khim., 1992, 28, 1401 (Engl. 1103).
831. Tan, L.C.; Pagni, R.M.; Kabalka, G.W.; Hillmyer, M.; Woosley, J. Tetrahedron Lett. 1992, 33, 7709.
832. See Singh, A.; Mehrotra, A.; Regen, S.L. Synth. Commun. 1981, 11, 409.
833. See, for example Wähälä, K.; Ojanperä, I.; Häyri, L.; Hase, T.A. Synth. Commun. 1987, 17, 137.
834. Sato, T.; Kobayashi, T.; Gojo, T.; Yoshida, E.; Otera, J.; Nozaki, H. Chem. Lett. 1987, 1661.
835. Firouzabadi, H.; Iranpoor, N.; Hazarkhani, H. J. Org. Chem. 2001, 66, 7527 and references cited therein; Ranu, B.C.; Das, A.; Samanta, S. Synlett. 2002, 727.
836. Wan, Y.; Kurchan, A.N.; Barnhurst, L.A.; Kutateladze, A.G. Org. Lett. 2000, 2, 1133.
837. Chini, M.; Crotti, P.; Giovani, E.; Macchia, F.; Pineschi, M. Synlett, 1992, 303.
838. Nishiyama, Y.; Ohashi, H.; Itoh, K.; Sonoda, N. Chem. Lett. 1998, 159.
839. Brittain, J.; Gareau, Y. Tetrahedron Lett. 1993, 34, 3363.
840. See Fokin, A.V.; Kolomiets, A.F. Russ. Chem. Rev. 1975, 44, 138. Key intermediates have been isolated: Kleiner, C.M.; Horst, L.; Würtele, C.; Wende, R.; Schreiner, P.R. Org. Biomol. Chem. 2009, 7, 1397. See Das, B.; Reddy, V.S.; Krishnaiah, M. Tetrahedron Lett. 2006, 47, 8471.
841. Chan, T.H.; Finkenbine, J.R. J. Am. Chem. Soc. 1972, 94, 2880.
842. Gao, Y.; Sharpless, K.B. J. Org. Chem. 1988, 53, 4114. Also see Bouda, H.; Borredon, M.E.; Delmas, M.; Gaset, A. Synth. Commun. 1987, 17; 943, 1989, 19, 491.
843. Kazemi, F.; Kiasat, A.R.; Ebrahimi, S. Synth. Commun. 2003, 33, 595.
844. Iranpoor, N.; Zeynizadeh, B. Synth. Commun. 1998, 28, 3913. See also, Tamami, B.; Kolahdoozan, M. Tetrahedron Lett. 2004, 45, 1535.
845. Kaboudin, B.; Norouzi, H. Tetrahedron Lett. 2004, 45, 1283.
846. Yadav, J.S.; Reddy, B.V.S.; Baishya, G. Synlett. 2003, 396.
847. Yadav, J.S.; Reddy, B.V.S.; Reddy, Ch.S.; Rajasekhar, K. J. Org. Chem. 2003, 68, 2525.
848. Bandgar, B.P.; Joshi, N.S.; Kamble, V.T. Tetrahedron Lett. 2006, 47, 4775.
849. Cohen, R.J.; Fox, D.L.; Salvatore, R.N. J. Og. Chem. 2004, 69, 4265. Also see Monahan, R.; Brown, D.; Waykole, L.; Liotta, D. in Liotta, D.C. Organoselenium Chemistry, Wiley, NY, 1987, pp. 207–241.
850. Yanada, K.; Fujita, T.; Yanada, R. Synlett, 1998, 971.
851. Nishino, T.; Okada, M.; Kuroki, T.; Watanabe, T.; Nishiyama, Y.; Sonoda, N. J. Org. Chem. 2002, 67, 8696. Zinc in aqueous media has also been used: see Bieber, L.W.; de Sá, A.C.P.F.; Menezes, P.H.; Goncalves, S.M.C. Tetrahedron Lett. 2001, 42, 4597.
852. Munbunjong, W.; Lee, E.H.; Chavasiri, W.; Jang, D.O. Tetrahedron Lett. 2005, 46, 8769.
853. Krief, A.; Derock, M.; Lacroix, D. Synlett 2005, 2832.
854. Nishiyama, Y.; Tokunaga, K.; Sonoda, N. Org. Lett. 1999, 1, 1725.
855. Wang, J.; Li, H.; Mei, Y.; Lou, B.; Xu, D.; Xie, D.; Guo, H.; Wang, W. J. Org. Chem. 2005, 70, 5678.
856. See Arisawa, M.; Yamaguchi, M. J. Am. Chem. Soc. 2004, 125, 6624.
857. Milligan, B.; Swan, J.M. J. Chem. Soc. 1962, 2712.
858. Chorbadjiev, S.; Roumian, C.; Markov, P. J. Prakt. Chem. 1977, 319, 1036. For an example using microwave irradiation, see Wang, J.-X.; Gao, L.; Huang, D. Synth. Commun. 2002, 32, 963.
859. See Polshettiwar, V.; Nivsarkar, M.; Acharya, J.; Kaushik, M.P. Tetrahedron Lett. 2003, 44, 887.
860. For a review of Bunte salts, see Distler, H. Angew. Chem. Int. Ed. 1967, 6, 544–553.
861. Kice, J.L. J. Org. Chem. 1963, 28, 957.
862. Milligan, B.; Saville, B.; Swan, J.M. J. Chem. Soc. 1963, 3608.
863. See Schank, K. in Patai, S.; Rappoport, Z.; Stirling, C. The Chemistry of Sulphones and Sulphoxides, Wiley, NY, 1988, pp. 165–231, pp. 177–188. For a reaction using the MgBr salt of an aryl sulfinate, see Wu, J.-P.; Emeigh, J.; Su, X.-P. Org. Lett. 2005, 7, 1223.
864. Eichelmann, H.; Gais, H.-J. Tetrahedron Asymmetry, 1995, 6, 643.
865. See Kielbasinski, P.; Zurawinski, R.; Drabowicz, J.; Mikolajczyk, M. Tetrahedron 1988, 44, 6687.
866. Felpin, F.-X.; Landais, Y. J. Org. Chem. 2005, 70, 6441. Also see Chandrasekhar, S.; Jagadeshwar, V.; Saritha, B.; Narsihmulu, C. J. Org. Chem. 2005, 70, 6506.
867. Biswas, G.; Mal, D. J. Chem. Res. (S) 1988, 308.
868. Saikia, P.; Laskar, D.D.; Prajapati, D.; Sandhu, J.S. Chem. Lett. 2001, 512.
869. Zhang, J.; Zhang, Y. J. Chem. Res. (S) 2001, 516.
870. Ballini, R.; Marcantoni, E.; Petrini, M. Tetrahedron 1989, 45, 6791.
871. Huang, F.; Batey, R.A. Tetrahedron 2007, 63, 7667.
872. Ochiai, M.; Oshima, K.; Masaki, Y.; Kunishima, M.; Tani, S. Tetrahedron Lett. 1993, 34, 4829.
873. Renard, P.-Y.; Schwebel, H.; Vayron, P.; Leclerc, E.; Dias, S.; Mioskowski, C. Tetrahedron Lett. 2001, 42, 8479. The reagent Ph3P(SCN)2 has also been used: see Iranpoor, N.; Firouzabadi, H.; Shaterian, H.R. Tetrahedron Lett. 2002, 43, 3439. Also see Mohanazadeh, F.; Aghvami, M. Tetrahedron Lett. 2007, 48, 7240.
874. See Guy, R.G. in Patai, S. The Chemistry of Cyanates and Their Thio Derivatives, pt. 2, pp. 819–886, Wiley, NY, 1977, pp. 819–886.
875. Katritzky, A.R.; Gruntz, U.; Mongelli, N.; Rezende, M.C. J. Chem. Soc. Perkin Trans. 1 1979, 1953. See Tamura, Y.; Kawasaki, T.; Adachi, M.; Tanio, M.; Kita, Y. Tetrahedron Lett. 1977, 4417.
876. Bettadaiah, B.K.; Gurudutt, K.N.; Srinivas, P. Synth. Commun. 2003, 33, 2293.
877. See Gibson, M.S. in Patai, S. The Chemistry of the Amino Group, Wiley, NY, 1968, pp. 45–55; Spialter, L.; Pappalardo, J.A. The Acyclic Aliphatic Tertiary Amines, Macmillan, NY, 1965, pp. 14–29.
878. See Jeyaraman, R. in Pizey, J.S. Synthetic Reagents, Vol. 5, Wiley, NY, 1983, pp. 9–83.
879. For a discussion of solvent effects see Sola, M.; Lledos, A.; Duran, M.; Bertran, J.; Abboud, J.L.M. J. Am. Chem. Soc. 1991, 113, 2873. For other parameters, see Bottini, A.T. Sel. Org. Transform. 1970, 1, 89; Persson, J.; Berg, U.; Matsson, O. J. Org. Chem. 1995, 60, 5037; Zoltewicz, J.A.; Deady, L.W. Adv. Heterocycl. Chem. 1978, 22, 71; Shaik, S.; Ioffe, A.; Reddy, A.C.; Pross, A. J. Am. Chem. Soc. 1994, 116, 262.
880. Lorentz-Petersen, L.L.R.; Jensen, P.; Madsen, R. Synthesis 2009, 4110.
881. Kurosu, M.; Dey, S.S.; Crick, D.C. Tetrahedron Lett. 2006, 47, 4871.
882. Lyubimov, S.E.; Davankov, V.A.; Gavrilov, K.N. Tetrahedron Lett. 2006, 47, 2721.
883. Saulnier, M.G.; Zimmermann, K.; Struzynski, C.P.; Sang, X.; Velaparthi, U.; Wittman, M.; Frennesson, D.B. Tetrahedron Lett. 2004, 45, 397.
884. Romera, J.L.; Cid, J.M.; Trabanco, A.A. Tetrahedron Lett. 2004, 45, 8797.
885. Murty, M.S.R.; Jyothirmai, B.; Krishna, P.R.; Yadav, J.S. Synth. Commun. 2003, 33, 2483.
886. Singh, C.B.; Kavala, V.; Samal, A.K.; Patel, B.K. Eur. J. Org. Chem. 2007, 1369; Simion, A.M.; Arimura, T.; Miyazawa, A.; Simion, C.; Prakash, G.K.S.; Olah, G.A.; Tashiro, M. Synth. Commun. 2009, 39, 2859.
887. Faul, M.M.; Kobierski, M.E.; Kopach, M.E. J. Org. Chem. 2003, 68, 5739.
888. Cho, J.H.; Kim, B.M. Tetrahedron Lett. 2002, 43, 1273.
889. Salvatore, R.N.; Schmidt, S.E.; Shin, S.I.; Nagle, A.S.; Worrell, J.H.; Jung, K.W. Tetrahedron Lett. 2000, 41, 9705.
890. Hayat, S.; Rahman, A.-u.; Choudhary, M.I.; Khan, K.M.; Schumann, W.; Bayer, E. Tetrahedron 2001, 57, 9951.
891. Salvatore, R.N.; Nagle, A.S.; Jung, K.W. J. Org. Chem. 2002, 67, 674.
892. Werner, E.A. J. Chem. Soc. 1918, 113, 899.
893. Chen, P.; Suh, D.J.; Smith, M.B. J. Chem. Soc. Perkin Trans. 1 1995, 1317; Deskus, J.; Fan, D.-p.; Smith. M.B. Synth. Commun. 1998, 28, 1649.
894. See Kumar, H.M.S.; Anjaneyulu, S.; Reddy, B.V.S.; Yadav, J.S. Synlett. 1999, 551.
895. Sommer, H.Z.; Lipp, H.I.; Jackson, L.L. J. Org. Chem. 1971, 36, 824. See also, Chuang, T.-H.; Sharpless, K.B. Org. Lett. 2000, 2, 3555.
896. See DePue, J.S.; Collum, D.B. J. Am. Chem. Soc. 1988, 110, 5524.
897. Vitale, A.A.; Chiocconi, A.A. J. Chem. Res. (S) 1996, 336.
898. Bestmann, H.J.; Wölfel, G. Chem. Ber. 1984, 117, 1250.
899. Patai, S.; Weiss, S. J. Chem. Soc. 1959, 1035.
900. Le, Z.-G.; Chen, Z.-C.; Hu, Y.; Zheng, Q.-G. Synthesis 2004, 1951.
901. See Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines, Academc Press, NY, 1969, pp. 1–59.
902. DeKimpe, N.; DeSmaele, D. Tetrahedron Lett. 1994, 35, 8023. Also see, De Kimpe, N.; Boelens, M.; Piqueur, J.; Baele, J. Tetrahedron Lett. 1994, 35, 1925.
903. Tokuda, M.; Fujita, H.; Suginome, H. J. Chem. Soc. Perkin Trans. 1 1994, 777.
904. Grellier, M.; Pfeffer, M.; van Koten, G. Tetrahedron Lett. 1994, 35, 2877.
905. Garner, P.; Dogan, O.; Pillai, S. Tetrahedron Lett.,1994, 35, 1653.
906. Juaristi, E.; Madrigal, D. Tetrahedron 1989, 45, 629.
907. Ju, Y.; Varma, R.S. J. Org. Chem. 2006, 71, 135.
908. Strand, J.W.; Kovacic, M.K. J. Am. Chem. Soc. 1973, 95, 2977.
909. Naskar, S.; Bhattacharjee, M. Tetrahedron Lett. 2007, 48, 3367; Hollmann, D.; Bähn, S.; Tillack, A.; Parton, R.; Altink, R.; Beller, M. Tetrahedron Lett. 2008, 49, 5742.
910. See Blazevic, N.; Kolbah, D.; Belin, B.; Sunjic, V.; Kajfez, F. Synthesis 1979, 161.
911. Jonczyk, A.; Ochal, Z.; Makosza, M. Synthesis 1978, 882.
912. Thomas, S.; Huynh, T.; Enriquez-Rios, V.; Singaram, B. Org. Lett. 2001, 3, 3915.
913. Crich, D.; Shirai, M.; Rumthao, S. Org. Lett. 2003, 5, 3767.
914. Greene, T.W. Protective Groups in Organic Synthesis Wiley, New York, 1980; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis 2nd ed., Wiley, New York, 1991; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis 3rd ed., Wiley, New York, 1999; Wuts, P.G.M.; Greene, T.W. Protective Groups in Organic Synthesis 4th ed., Wiley, New Jersey, 2006.
915. Takaki, K.; Kamata, T.; Miura, Y.; Shishido, T.; Takehira, K. J. Org. Chem. 1999, 64, 3891.
916. Faller, J.W.; Wilt, J.C. Org. Lett. 2005, 7, 633; Nagano, T.; Kobayashi, S. J. Am. Chem. Soc. 2009, 131, 4200; Watson, I.D.G.; Styler, S.A.; Yudin, A.K. J. Am. Chem. Soc. 2004, 126, 5086. See also, Evans, P.A.; Robinson, J.E.; Moffett, K.K. Org. Lett. 2001, 3, 3269; Mahrwald, R.; Quint, S. Tetrahedron Lett. 2001, 42, 1655. For mechanistic insights, see Watson, I.D.G.; Yudin, A.K. J. Am. Chem. Soc. 2005, 127, 17516.
917. Detz, R.J.; Delville, M.M.E.; Hiemstra, H.; van Maarseveen, J.H. Angew. Chem. Int. Ed. 2008, 47, 3777. A Cu(I) mediated reaction in ionic liquids is known, see Park, S.B.; Alper, H. Chem. Commun. 2005, 1315.
918. González, I.; Mosquera, J.; Guerrero, C.; Rodríguez, R.; Cruces, J. Org. Lett. 2009, 11, 1677. See also, Bariwal, J.B.; Ermolat'ev, D.S.; Van der Eycken, E.V. Chemistry: Eur. J. 2010, 16, 3281.
919. Gage, J.R.; Wagner, J.M. J. Org. Chem. 1995, 60, 2613.
920. See Honaker, M.T.; Sandefur, B.J.; Hargett, J.L.; McDaniel, A.L.; Salvatore, R.N. Tetrahedron Lett. 2003, 44, 8373.
921. See Deady, L.W.; Finlayson, W.L.; Korytsky, O.L. Aust. J. Chem. 1979, 32, 1735.
922. Abrunhosa-Thomas, I.; Sellers, C.E.; Montchamp, J.-L. J. Org. Chem. 2007, 72, 2851.
923. See Katritzky, A.R.; Huang, T.-B.; Voronkov, M.V. J. Org. Chem. 2001, 66, 1043; Cami-Kobeci, G.; Williams, J.M.J. Chem. Commun. 2004, 1072. See also, Salehi, P.; Motlagh, A.R. Synth. Commun. 2000, 30, 671; Lakouraj. M.M.; Movassagh, B.; Fasihi, J. Synth. Commun. 2000, 30, 821.
924. Fabiano, E.; Golding, B.T.; Sadeghi, M.M. Synthesis 1987, 190. See also, Klepacz, A.; Zwierzak, A. Synth. Commun. 2001, 31, 1683.
925. See Edwards, M.L.; Stemerick, D.M.; McCarthy, J.R. Tetrahedron Lett. 1990, 31, 3417.
926. See Arcelli, A.; Evans, Jr., S.A. J. Org. Chem. 1986, 51, 95; Huh, K.; Tsuji, Y.; Kobayashi, M.; Okuda, F.; Watanabe, Y. Chem. Lett. 1988, 449.
927. Gunanathan, C.; Milstein, D. Angew. Chem. Int. Ed. 2008, 47, 8661. For a Ru-catalyzed reaction of primary and secondary alcohols with ammonia, see Imm, S.; Bähn, S.; Neubert, L.; Neumann, H.; Beller, M. Angew. Chem. Int. Ed. 2010, 49, 8126.
928. Botta, M.; De Angelis, F.; Nicoletti, R. Synthesis 1977, 722.
929. Utsunomiya, M.; Miyamoto, Y.; Ipposhi, J.; Ohshima, T.; Mashima, K. Org. Lett. 2007, 9, 3371.
930. Yamashita, Y.; Gopalarathnam, A.; Hartwig, J.F. J. Am. Chem. Soc. 2007, 129, 7508; Yang, S.-C.; Hsu, Y.-C.; Gan, K.-H. Tetrahedron 2006, 62, 3949.
931. Tsuji, Y.; Takeuchi, R.; Ogawa, H.; Watanabe, Y. Chem. Lett. 1986, 293.
932. Jiang, Y.-L.; Hu, Y.-Q.; Feng, S.-Q.; Wu, J.-S.; Wu, Z.-W.; Yuan, Y.-C.; Liu, J.-M.; Hao, Q.-S.; Li, D.-P. Synth. Commun. 1996, 26, 161.
933. Hamid, M.H.S.A.; Williams, J.M.J. Tetrahedron Lett. 2007, 48, 8263; Malai Haniti S.A.; Hamid, M.H.S.A.; Williams, J.M.J. Chem. Commun. 2007, 725; Tillack, A.; Hollmann, D.; Mevius, K.; Michalik, D.; Bähn, S.; Beller, M. Eur. J. Org. Chem. 2008, 4745.
934. Fujita, K.; Enoki, Y.; Yamaguchi, R. Tetrahedron 2008, 64, 1943; Defieber, C.; Ariger, M.A.; Moriel, P.; Carreira, E.M. Angew. Chem. Int. Ed. 2007, 46, 3139.
935. Guo, S.; Song, F.; Liu, Y. Synlett 2007, 964.
936. Ramanathan, B.; Odom, A.L. J. Am. Chem. Soc. 2006, 128, 9344.
937. Likhar, P.R.; Arundhathi, R.; Kantam, M.L.; Prathima, P.S. Eur. J. Org. Chem. 2009, 5383.
938. Mizuno, M.; Yamano, M. Org. Lett. 2005, 7, 3629.
939. Kaboudin, B. Tetrahedron Lett. 2003, 44, 1051.
940. Bombrun, A.; Casi, G. Tetrahedron Lett. 2002, 43, 2187.
941. Valot, F.; Fache, F.; Jacquot, R.; Spagnol, M.; Lemaire, M. Tetrahedron Lett. 1999, 40, 3689. See Selva, M.; Tundo, P.; Perosa, A. J. Org. Chem. 2003, 68, 7374.
942. Masuyama, Y.; Kagawa, M.; Kurusu, Y. Chem. Lett. 1995, 1121.
943. Fujita, K.-i.; Fujii, T.; Yamaguchi, R. Org. Lett. 2004, 6, 3525.
944. Okada, I.; Ichimura, K.; Sudo, R. Bull. Chem. Soc. Jpn. 1970, 43, 1185. See also, Pfister, J.R. Synthesis 1984, 969; Suzuki, H.; Tani, H. Chem. Lett. 1984, 2129; Marsella, J.A. J. Org. Chem. 1987, 52, 467.
945. Also see Hendrickson, J.B.; Joffee, I. J. Am. Chem. Soc. 1973, 95, 4083; Trost, B.M.; Keinan, E. J. Org. Chem. 1979, 44, 3451; Koziara, A.; Osowska-Pacewicka, K.; Zawadzki, S.; Zwierzak, A. Synthesis 1985, 202; 1987, 487.
946. Castro, B.; Selve, C. Bull. Soc. Chim. Fr. 1971, 4368. For a similar method, see Tanigawa, Y.; Murahashi, S.; Moritani, I. Tetrahedron Lett. 1975, 471.
947. Reddy, G.V.S.; Rao, G.V.; Subrmanyam, R.V.K.; Iyengar, D.S. Synth. Commun. 2000, 30, 2233.
948. See Klemmensen, P.; Schroll, G.; Lawesson, S. Ark. Kemi, 1968, 28, 405.
949. Loubinoux, B.; Coudert, G.; Guillaumet, G. Synthesis 1980, 638.
950. Ireland, R.E.; Walba, D.M. Org. Synth. VI, 567.
951. Widehem, R.; Lacroix, T.; Bricout, H.; Monflier, E. Synlett 2000, 722.
952. Baltzly, R.; Blackman, S.W. J. Org. Chem. 1963, 28, 1158.
953. See Geller, B.A. Russ. Chem. Rev. 1978, 47, 297.
954. De Angelis, F.; Grgurina, I.; Nicoletti, R. Synthesis 1979, 70; See also, Tsuji, Y.; Shida, J.; Takeuchi, R.; Watanabe, Y. Chem. Lett. 1984, 889; Bank, S.; Jewett, R. Tetrahedron Lett. 1991, 32, 303.
955. Hünig, S.; Baron W. Chem. Ber. 1957, 90, 395, 403.
956. Cassimjee, K.E.; Branneby, C.; Abedi, V.; Wells, A.; Berglund, P. Chem. Commun. 2010, 5569.
957. Müller, E.; Huber-Emden, H.; Rundel, W. Liebigs Ann. Chem. 1959, 623, 34.
958. Saegusa, T.; Ito, Y.; Kobayashi, S.; Hirota, K.; Shimizu, T. Tetrahedron Lett. 1966, 6131.
959. Azizi, N.; Saidi, M.R. Org. Lett. 2005, 7, 3649.
960. Lu, P. Tetrahedron 2010, 66, 2549.
961. See Charrada, B.; Hedhli, A.; Baklouti, A. Tetrahedron Lett. 2000, 41, 7347.
962. Pastó, M.; Rodríguez, B.; Riera, A.; Pericàs, M.A. Tetrahedron Lett. 2003, 44, 8369.
963. Lindström, U.M.; Olofsson, B.; Somfai, P. Tetrahedron Lett. 1999, 40, 9273.
964. See Harrack, Y.; Pujol, M.D. Tetrahedron Lett. 2002, 43, 819; Steiner, D.; Sethofer, S.G.; Goralski, C.T.; Singaram, B. Tetrahedron Asymmetry 2002, 13, 1477. For a reaction catalyzed by LiBr, see Chakraborti, A.K.; Rudrawar, S.; Kondaskar, A. Eur. J. Org. Chem. 2004, 3597.
965. Chakraborti, A.K.; Rudrawar, S.; Kondaskar, A. Org. Biomol. Chem. 2004, 2, 1277.
966. Azizi, N.; Saidi, M.R. Tetrahedron 2007, 63, 888.
967. Reddy, L.R.; Reddy, M.A.; Chanumathi, N.; Rao, K.R. Synlett 2000, 339.
968. Heydari, A.; Mehrdad, M.; Malecki, A.; Ahmadi, N. Synthesis 2004, 1563.
969. Das, U.; Crousse, B.; Kesavan, V.; Bonnet-Delpon, D.; Bégue, J.P. J. Org. Chem. 2000, 65, 6749.
970. Sabitha, G.; Reddy, G.S.K.K.; Reddy, K.B.; Yadav, J.S. Synthesis 2003, 2298.
971. Kamal, A.; Ramu, R.; Azhar, M.A.; Khanna, G.B.R. Tetrahedron Lett. 2005, 46, 2675.
972. Bartoli, G.; Bosco, M.; Carlone, A.; Locatelli, M.; Mechiorre, P.; Sambri, L. Org. Lett. 2004, 6, 3973.
973. Zhao, P.-Q.; Xu, L.-W.; Xia, C.-G. Synlett 2004, 846.
974. Examples include Al compounds: Williams, D.B.G.; Lawton, M. Tetrahedron Lett. 2006, 47, 6557; Robinson, M.W.C.; Timms, D.A.; Williams, S.M.; Graham, A.E. Tetrahedron Lett. 2007, 48, 6249. Bi compounds: McCluskey, A.; Leitch, S.K.; Garner, J.; Caden, C.E.; Hill, T.A.; Odell, L.R.; Stewart, S.G. Tetrahedron Lett. 2005, 46, 8229; Ollevier, T.; Nadeau, E. Tetrahedron Lett. 2008, 49, 1546. Ce compounds: Reddy, L.R.; Reddy, M.A.; Bhanumathi, N.; Rao, K.R. Synthesis 2001, 831. Co compounds: Sundararajan, G.; Viyayakrishna, K.; Varghese, B. Tetrahedron Lett. 2004, 45, 8253. Er compounds: Procopio, A.; Gaspari, M.; Nardi, M.; Oliverio, M.; Rosati, O. Tetrahedron Lett. 2008, 49, 2289. In compounds: Rodríguez, J.R.; Navarro, A. Tetrahedron Lett. 2004, 45, 7495. Sc cmpounds: Azoulay, S.; Manabe, K.; Kobayashi, S. Org. Lett. 2005, 7, 4593; Placzek, A.T.; Donelson, J.L.; Trivedi, R.; Gibbs, R.A.; De, S.K. Tetrahedron Lett. 2005, 46, 9029. Sm compounds: Carrée, F.; Gil, R.; Collin, J. Org. Lett. 2005, 7, 1023;. Sn compounds: Sekar, G.; Singh, V.K. J. Org. Chem. 1999, 64, 287. Zn compounds:Bonollo, S.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. Synlett 2008, 1574. Zr compounds: Charkraborti, A.K.; Kondaskar, A. Tetrahedron Lett. 2003, 44, 8315.
975. Arai, K.; Lucarini, S.; Salter, M.M.; Ohta, K.; Yamashita, Y.; Kobayashi, S. J. Am. Chem. Soc. 2007, 129, 8103; Arai, K; Salter, K.M.; Yamashita, Y.; Kobayashi, S. Angew. Chem. Int. Ed. 2007, 46, 955.
976. Sun, J.; Dai, Z.; Yang, M.; Pan, X.; Zhu, C. Synthesis 2008, 2100.
977. Bao, H.; Wu, J.; Li, H.; Wang, Z.; You; T.; Ding, K. Eur. J. Org. Chem. 2010, 6722.
978. Fink, D.M. Synlett 2004, 2394.
979. Hodgson, D.M.; Bray, C.D.; Kindon, N.D. J. Am. Chem. Soc. 2004, 126, 6870. For a similar reaction with LiNTf2, see Cossy, J.; Bellosta, V.; Hamoir, C.; Desmurs, J.-R. Tetrahedron Lett. 2002, 43, 7083.
980. Yanagisawa, A.; Yasue, K.; Yamamoto, H. J. Chem. Soc., Chem. Commun. 1994, 2103.
981. Kazemi, F.; Kiasat, A.R.; Ebrahimi, S. Synth. Commun. 2003, 33, 999. For a reaction done under phase transfer conditions, see Tamami, B.; Mahdavi, H. Tetrahedron Lett. 2001, 42, 8721.
982. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 815.
983. Iranpoor, N.; Kazemi, F. Synth. Commun. 1999, 29, 561.
984. Sabitha, G.; Babu, R.S.; Rajkumar, M.; Yadav, J.S. Org. Lett. 2002, 4, 343.
985. Göksu, S.; Socen, H.; Sütbeyaz, Y. Synthesis 2002, 2373.
986. Song, C.E.; Oh, C.R.; Roh, E.J.; Choo, D.J. Chem. Commun. 2000, 1743.
987. Fringuelli, F.; Pizzo, F.; Vaccaro, L. Tetrahedron Lett. 2001, 42, 1131.
988. Schneider, C. Synlett 2000, 1840.
989. Kalita, B.; Barua, N.C.; Bezbarua, M.; Bez, G. Synlett 2001, 1411.
990. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 821.
991. Dong, Q.; Fang, X.; Schroeder, J.D.; Garvey, D.S. Synthesis 1999, 1106.
992. Kühnau, D.; Thomsen, I.; J![]() rgensen, K.A. J. Chem. Soc. Perkin Trans. 1, 1996, 1167.
rgensen, K.A. J. Chem. Soc. Perkin Trans. 1, 1996, 1167.
993. Tsuchiya, Y.; Kumamoto, T.; Ishikawa, T. J. Org. Chem. 2004, 69, 8504.
994. Albanese, D.; Landini, D.; Penso, M.; Petricci, S. Tetrahedron 1999, 55, 6387.
995. Najime, R.; Pilard, S.; Vaultier, M. Tetrahedron Lett. 1992, 33, 5351; Moulines, J.; Bats, J.-P.; Hautefaye, P.; Nuhrich, A.; Lamidey, A.-M. Tetrahedron Lett. 1993, 34, 2315.
996. Crotti, P.; Favero, L.; Macchia, F.; Pineschi, M. Tetrahedron Lett. 1994, 35, 7089.
997. Chini, M.; Crotti, P.; Favero, L.; Macchia, F. Tetrahedron Lett. 1994, 35, 761.
998. See Dermer, O.C.; Ham, G.E. Ethylenimine and Other Aziridines, Academic Press, NY, 1969, pp. 262–268. See also, Scheuermann, J.E.W.; Ilyashenko, G.; Griffiths, D.V.; Watkinson, M. Tetrahedron Asymmetry 2002, 13,269.
999. Peruncheralathan, S.; Teller, H.; Schneider, C. Angew. Chem. Int. Ed. 2009, 48, 4849.
1000. Sekar, G.; Singh, V.K. J. Org. Chem. 1999, 64, 2537.
1001. Watson, I.D.G.; Yudin, A.K. J. Org. Chem. 2003, 68, 5160.
1002. Bertolini, F.; Woodward, S.; Crotti, S.; Pineschi, M. Tetrahedron Lett. 2009, 50, 4515.
1003. Examples include Aqueous media with β-cyclodextrin: Reddy, M.A.; Reddy, L.R.; Bhanamathi, N.; Rao, K.R. Chem. Lett. 2001, 246. BiCl3: Swamy, N.R.; Venkateswarlu, Y. Synth. Commun. 2003, 33, 547. InBr3: Yadav, J.S.; Reddy, B.V.S.; Rao, K.; Raj, K.S.; Prasad, A.R. Synthesis 2002, 1061. InCl3: Yadav, J.S.; Reddy, B.V.S.; Abraham, S.; Sabitha, G. Tetrahedron Lett. 2002, 43, 1565; LiClO4: Yadav, J.S.; Reddy, B.V.S.; Jyothirmai, B.; Murty, M.S.R. Synlett 2002, 53; Yadav, J.S.; Reddy, B.V.S.; Parimala, G.; Reddy, P.V. Synthesis 2002, 2383. PBu3: Fan, R.-H.; Hou, X.-L. J. Org. Chem. 2003, 68, 726. TaCl5/SiO2: Chandrasekhar, S.; Prakash, S.J.; Shyamsunder, T.; Ramachandar, T. Synth. Commun. 2004, 34, 3865. Yb(OTf)3: Meguro, M.; Yamamoto, Y. Heterocycles 1996, 43, 2473.
1004. Kumar, G.D.K.; Baskaran, S. Synlett 2004, 1719.
1005. Cossy, J.; Bellosta, V.; Alauze, V.; Desmurs, J.-R. Synthesis 2002, 2211.
1006. Bisai, A.; Pandey, G.; Pandey, M.K.; Singh, V.K. Tetrahedron Lett. 2003, 44, 5839.
1007. Nadir, U.K.; Singh, A. Tetrahedron Lett. 2005, 46, 2083.
1008. Rowland, E.B.; Rowland, G.B.; Rivera-Otero, E.; Antilla, J.C. J. Am. Chem. Soc. 2007, 129, 12084; Chandrasekhar, M.; Sekar, G.; Singh, V.K. Tetrahedron Lett. 2000, 41, 10079.
1009. Yadav, J.S.; Reddy, B.V.S.; Kumar, G.M.; Murthy, Ch.V.S.R. Synth. Commun. 2002, 32, 1797.
1010. Wnuk, T.A.; Chaudhary, S.S.; Kovacic, P. J. Am. Chem. Soc. 1976, 98, 5678, and references cited therein.
1011. Li, Z.; Capretto, D.A.; Rahaman, R.; He, C. Angew. Chem. Int. Ed. 2007, 46, 5184.
1012. Yasuda, M.; Kojima, R.; Tsutsui, H.; Utsunomiya, D.; Ishii, K.; Jinnouchi, K.; Shiragami, T.; Yamashita, T. J. Org. Chem. 2003, 68, 7618.
1013. For a method for the preparation of benzyl isocyanides, see Kitano, Y.; Manoda, T.; Miura, T.; Chiba, K.; Tada, M. Synthesis 2006, 405.
1014. See Periasamy, M.P.; Walborsky, H.M. Org. Prep. Proced. Int. 1979, 11, 293.
1015. Weber, W.P.; Gokel, G.W. Tetrahedron Lett. 1972, 1637; Weber, W.P.; Gokel, G.W.; Ugi, I. Angew. Chem. Int. Ed. 1972, 11, 530.
1016. Saunders, M.; Murray, R.W. Tetrahedron 1959, 6, 88; Frankel, M.B.; Feuer, H.; Bank, J. Tetrahedron Lett. 1959, no. 7, 5.
1017. Gassman, P.G.; Haberman, L.M. Tetrahedron Lett. 1985, 26, 4971, and references cited therein.
1018. Brace, N.O. J. Org. Chem. 1993, 58, 1804.
1019. For procedures, see Yamawaki, J.; Ando, T.; Hanafusa, T. Chem. Lett. 1981, 1143; Sukata, K. Bull. Chem. Soc. Jpn. 1985, 58, 838.
1020. See Challis, B.C.; Challis, J.A., in Zabicky, J. The Chemistry of Amides, Wiley, NY, 1970, pp. 734–754.
1021. Salvatore, R.N.; Shin, S.I.; Flanders, V.L.; Jung, K.w. Tetrahedron Lett. 2001, 42, 1799.
1022. Singh, O.V.; Han, H. Tetrahedron Lett. 2007, 48, 7094.
1023. Simandan, T.; Smith, M.B. Synth. Commun. 1996, 26, 1827.
1024. Liu, H.; Ko, S.-B.; Josien, H.; Curran, D.P. Tetrahedron Lett. 1995, 36, 8917.
1025. Gagnon, A.; St-Onge, M.; Little, K.; Duplessis, M.; Barabé, F. J. Am. Chem. Soc. 2007, 129, 44.
1026. Chan, D.M.T. Tetrahedron Lett. 1996, 37, 9013.
1027. Ikawa, T.; Barder, T.E.; Biscoe, M.R.; Buchwald, S.L. J. Am. Chem. Soc. 2007, 129, 13001.
1028. Wasa, M.; Yu, J.-Q. J. Am. Chem. Soc. 2008, 130, 14058. See also, Poondra, R.R.; Turner, N.J. Org. Lett. 2005, 7, 863.
1029. Pan, X.; Cai, Q.; Ma, D. Org. Lett. 2004, 6, 1809.
1030. Brice, J.L.; Meerdink, J.E.; Stahl, S.S. Org. Lett. 2004, 6, 1845.
1031. Blass, B.E.; Drowns, M.; Harris, C.L.; Liu, S.; Portlock, D.E. Tetrahedron Lett. 1999, 40, 6545.
1032. For a review, see Gibson, M.S.; Bradshaw, R.W. Angew. Chem. Int. Ed. 1968, 7, 919.
1033. See Sheehan, J.C.; Bolhofer, W.A. J. Am. Chem. Soc. 1950, 72, 2786. See also, Landini, D.; Rolla, F. Synthesis 1976, 389.
1034. Soai, K.; Ookawa, A.; Kato, K. Bull. Chem. Soc. Jpn. 1982, 55, 1671.
1035. Ing, H.R.; Manske, R.H.F. J. Chem. Soc. 1926, 2348.
1036. See Khan, M.N. J. Org. Chem. 1995, 60, 4536 for the kinetics of hydrazinolysis of phthalimides.
1037. Kukolja, S.; Lammert, S.R. J. Am. Chem. Soc. 1975, 97, 5582.
1038. Wolfe, S.; Hasan, S.K. Can. J. Chem. 1970, 48, 3572.
1039. López-Alvarado, P.; Avendaño, C.; Menéndez, J.C. Tetrahedron Lett. 1992, 33, 6875.
1040. Hebrard, P.; Olomucki, M. Bull. Soc. Chim. Fr. 1970, 1938.
1041. See Grehn, L.; Ragnarsson, U. Synthesis 1987, 275; Dalla Croce, P.; La Rosa, C.; Ritieni, A. J. Chem. Res. (S) 1988, 346; Yinglin, H.; Hongwen, H. Synthesis 1990, 122.
1042. Mitsunobu, O.; Wada, M.; Sano, T. J. Am. Chem. Soc. 1972, 94, 679; Grunewald, G.L.; Kolasa, T.; Miller, M.J. J. Org. Chem. 1987, 52, 4978; Sammes, P.G.; Thetford, D. J. Chem. Soc. Perkin Trans. 1 1989, 655.
1043. Barrett, A.G.M.; Braddock, D.C.; James, R.A.; Procopiou, P.A. Chem. Commun. 1997, 433.
1044. Nordstr![]() m, L.U.; Vogt, H.; Madsen, R. J. Am. Chem. Soc. 2008, 130, 17672. See also, Watson, A.J.A.; Maxwell, A.C.; Williams, J.M.J. Org. Lett. 2009, 11, 2667; Dam, J.H.; Osztrovszky, G.; Nordstr
m, L.U.; Vogt, H.; Madsen, R. J. Am. Chem. Soc. 2008, 130, 17672. See also, Watson, A.J.A.; Maxwell, A.C.; Williams, J.M.J. Org. Lett. 2009, 11, 2667; Dam, J.H.; Osztrovszky, G.; Nordstr![]() m, L.U.; Madsen, R. Chemistry: Eur. J. 2010, 16, 6820.
m, L.U.; Madsen, R. Chemistry: Eur. J. 2010, 16, 6820.
1045. Ghosh, S.C.; Hong, S.-H. Eur. J. Org. Chem. 2010, 4266.
1046. Jana, U.; Maiti, S.; Biswas, S. Tetrahedron Lett. 2008, 49, 858.
1047. Fujita, K.; Komatsubara, A.; Yamaguchi, R. Tetrahedron 2009, 65, 3624.
1048. Krishna, P.R.; Sekhar, E.R.; Prapurna, Y.L. Tetrahedron Lett. 2007, 48, 9048.
1049. Nordlander, J.E.; Catalane, D.B.; Eberlein, T.H.; Farkas, L.V.; Howe, R.S.; Stevens, R.M.; Tripoulas, N.A. Tetrahedron Lett. 1978, 4987. For other methods, see Briggs, E.M.; Brown, G.W.; Jiricny, J.; Meidine, M.F. Synthesis 1980, 295; Zwierzak, A.; Brylikowska-Piotrowicz, J. Synthesis 1982, 922
1050. Dubé, D.; Scholte, A.A. Tetrahedron Lett. 1999, 40, 2295.
1051. Zhang, Y.; Hsung, R.P.; Tracey, M.R.; Kurtz, K.C.M.; Vera, E.L. Org. Lett. 2004, 6, 1151; Frederick, M.O.; Mulder, J.A.; Tracey, M.R.; Hsung, R.P.; Huang, J.; Kurtz, K.C.M.; Shen, L.; Douglas, C.J. J. Am. Chem. Soc.2003, 125, 2368.
1052. For an example involving bromine see Besstmann, H.-J.; Frey, H. Liebigs Ann. Chem. 1980, 12, 2061.
1053. For examples with hypobromite, see Mozuraitis, R.; Buda, V.; Liblikas, I.; Unelius, C.R.; Borg-Karlson, A.-K. J. Chem. Ecol. 2002, 28, 1191; Barbu, E.; Tsibouklis, J. Tetrahedron Lett. 1996, 37, 5023.
1054. See Quast, H.; Leybach, H. Chem. Ber. 1991, 124, 849. For a review of α-lactams, see Lengyel, I.; Sheehan, J.C. Angew. Chem. Int. Ed. 1968, 7, 25.
1055. See Larson, H.O. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 1, Wiley, NY, 1969, pp. 325–339; Kornblum, N. Org. React. 1962, 12, 101.
1056. See Ballini, R.; Barboni, L.; Giarlo, G. J. Org. Chem. 2004, 69, 6907.
1057. Rozen, S.; Carmeli, M. J. Am. Chem. Soc. 2003, 125, 8118.
1058. Baruah, A.; Kalita, B.; Barua, N.C. Synlett 2000, 1064.
1059. See Scriven, E.F.V.; Turnbull, K. Chem. Rev. 1988, 88, 297; Biffin, M.E.C.; Miller, J.; Paul, D.B. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 57–119; Alvarez, S.G.; Alvarez, M.T. Synthesis 1997, 413. Also see Kim, J.-G.; Jang, D.O. Synlett 2008, 2075.
1060. See Reeves, W.P.; Bahr, M.L. Synthesis 1979, 823; Marti, M.J.; Rico, I.; Ader, J.C.; de Savignac, A.; Lattes, A. Tetrahedron Lett. 1989, 30, 1245.
1061. Priebe, H. Acta Chem. Scand. Ser. B, 1984, 38, 895.
1062. See, for example, Varma, R.S.; Naicker, K.P.; Aschberger, J. Synth. Commun. 1999, 29, 2823.
1063. See Murahashi, T.; Tanigawa, Y.; Imada, Y.; Taniguchi, Y. Tetrahedron Lett. 1986, 27, 227.
1064. Scriven, E.F.V.; Turnbull, K. Chem. Rev. 1988, 88, 297, see p. 306.
1065. Murahashi, S.; Taniguchi, Y.; Imada, Y.; Tanigawa, Y. J. Org. Chem. 1989, 54, 3292.
1066. Rad, M.N.S.; Behrouz, S.; Khalafi-Nezhad, A. Tetrahedron Lett. 2007, 48, 3445; Hajipour, A.R.; Rajaei, A.; Ruoho, A.E. Tetrahedron Lett. 2009, 50, 708.
1067. Tao, C.-Z.; Cui, X.; Li, J.; Liu, A.-X.; Liu, L.; Guo, Q.-X. Tetrahedron Lett. 2007, 48, 3525.
1068. Das, J.; Patil, S.N.; Awasthi, R.; Narasimhulu, C.P.; Trehan, S. Synthesis 2005, 1801.
1069. Yadav, J.S.; Reddy, B.V.S.; Jyothirmai, B.; Murty, M.S.R. Tetrahedron Lett. 2005, 46, 6559.
1070. Fujiwara, M.; Tanaka, M.; Baba, A.; Ando, H.; Souma, Y. Tetrahedron Lett. 1995, 36, 4849.
1071. Van de Weghe, P.; Collin, J. Tetrahedron Lett. 1995, 36, 1649.
1072. Youn, Y.S.; Cho, I.S.; Chung, B.Y. Tetrahedron Lett. 1998, 39, 4337.
1073. See Ittah, Y.; Sasson, Y.; Shahak, I.; Tsaroom, S.; Blum, J. J. Org. Chem. 1978, 43, 4271. For the mechanism of the conversion to aziridines, see Pöchlauer, P.; Müller, E.P.; Peringer, P. Helv. Chim. Acta 1984, 67, 1238.
1074. Lohray, B.B.; Gao, Y.; Sharpless, K.B. Tetrahedron Lett. 1989, 30, 2623.
1075. Guy, A.; Lemor, A.; Doussot, J.; Lemaire, M. Synthesis 1988, 900.
1076. Miller, J.A. Tetrahedron Lett. 1975, 2959. See also, Koziara, A.; Zwierzak, A. Tetrahedron Lett. 1987, 28, 6513.
1077. Balderman, D.; Kalir, A. Synthesis 1978, 24.
1078. Hassner, A.; Fibiger, R.; Andisik, D. J. Org. Chem. 1984, 49, 4237.
1079. See, for example, Adam, G.; Andrieux, J.; Plat, M. Tetrahedron 1985, 41, 399.
1080. Liu, Q.; Tor, Y. Org. Lett. 2003, 5, 2571.
1081. See Lwowski, W. in Patai, S. The Chemistry of the Azido Group, Wiley, NY, 1971, pp. 503–554.
1082. Rawal, V.H.; Zhong, H.M. Tetrahedron Lett. 1994, 35, 4947.
1083. Affandi, H.; Bayquen, A.V.; Read, R.W. Tetrahedron Lett. 1994, 35, 2729. For a preparation using triphosgene see Gumaste, V.K.; Bhawal, B.M.; Deshmukh, A.R.A.S. Tetrahedron Lett. 2002, 43, 1345.
1084. Katritzky, A.R.; Widyan, K.; Kirichenko, K. J. Org. Chem. 2007, 72, 5802.
1085. Elmorsy, S.S. Tetrahedron Lett. 1995, 36, 1341.
1086. Lee, J.G.; Kwak, K.H. Tetrahedron Lett. 1992, 33, 3165.
1087. Bose, D.S.; Reddy, A.V.N. Tetrahedron Lett. 2003, 44, 3543.
1088. Manimaran, T.; Wolford, L.T.; Boyer, J.H. J. Chem. Res. (S) 1989, 331.
1089. Effenberger, F.; Drauz, K.; Förster, S.; Müller, W. Chem. Ber. 1981, 114, 173.
1090. See Tsuge, O. in Patai, S. The Chemistry of Cyanates and Their Thio Derivatives, pt. 1, Wiley, NY, 1977, pp. 445–506; Nuridzhanyan, K.A. Russ. Chem. Rev. 1970, 39, 130; Lozinskii, M.O.; Pel'kis, P.S. Russ. Chem. Rev.1968, 37, 363.
1091. Arisawa, M.; Ashikawa, M.; Suwa, A.; Yamaguchi, M. Tetrahedron Lett. 2005, 46, 1727.
1092. Munch, H.; Hansen, J.S.; Pittelkow, M.; Christensen, J.B.; Boas, U. Tetrahedron Lett. 2008, 49, 3117.
1093. See Yandovskii, V.N.; Gidaspov, B.V.; Tselinskii, I.V. Russ. Chem. Rev. 1980, 49, 237; Moss, R.A. Acc. Chem. Res. 1974, 7, 421.
1094. See Hudlicky, M.; Hudlicky, T. in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, pp. 1021–1172.
1095. For a list of reagents for alkyl halide interconversion, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 667–671.
1096. Miller, J.A.; Nunn, M.J. J. Chem. Soc. Perkin Trans. 1 1976, 416.
1097. Takagi, K.; Hayama, N.; Inokawa, S. Chem. Lett. 1978, 1435.
1098. Suzuki, H.; Aihara, M.; Yamamoto, H.; Takamoto, Y.; Ogawa, T. Synthesis 1988, 236.
1099. See Mann, J. Chem. Soc. Rev. 1987, 16, 381; Rozen, S.; Filler, R. Tetrahedron 1985, 41, 1111; Hudlicky, M. Chemistry of Organic Fluorine Compounds, pt. 2, Ellis Horwood, Chichester, 1976, pp. 24–169; Sheppard, W.A.; Sharts, C.M. Organic Fluorine Chemistry, W.A. Benjamin, NY, 1969, pp. 52–184, 409–430.
1100. See Sharts, C.M.; Sheppard, W.A. Org. React. 1974, 21, 125; Hudlicky, M. Chemistry of Organic Fluorine Compunds, pt. 2, Ellis Horwood, Chichester, 1976, pp. 91–136.
1101. See Makosza, M.; Bujok, R. Tetrahedron Lett. 2002, 43, 2761.
1102. Giudicelli, M.B.; Picq, D.; Veyron B. Tetrahedron Lett. 1990, 31, 6527. Also see Sawaguchi, M.; Ayuba, S.; Nakamura, Y.; Fukuhara, J.; Hara, S.; Yoneda, N. Synlett 2000, 999.
1103. Sawaguchi, M.; Hara, S.; Nakamura, Y.; Ayuba, S.; Kukuhara, T.; Yoneda, N. Tetrahedron 2001, 57, 3315.
1104. Kvícala, J.; Mysík, P.; Paleta, O. Synlett 2001, 547.
1105. Katcher, M.H.; Doyle, A.G. J. Am. Chem. Soc. 2010, 132, 17402.
1106. See Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Academic Press, NY, 1978, pp. 112–125; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis. Springer, NY, 1977, pp. 117–124. See also, Bram, G.; Loupy, A.; Pigeon, P. Synth. Commun. 1988, 18, 1661.
1107. Willy, W.E.; McKean, D.R.; Garcia, B.A. Bull. Chem. Soc. Jpn. 1976, 49, 1989. See also, Babler, J.H.; Spina, K.P. Synth. Commun. 1984, 14, 1313.
1108. Loupy, A.; Pardo, C. Synth. Commun. 1988, 18, 1275.
1109. Bidd, I.; Whiting, M.C. Tetrahedron Lett. 1984, 25, 5949.
1110. Peyrat, J.-F.; Figadère, B.; Cavé, A. Synth. Commun. 1996, 26, 4563.
1111. Yoon, K.B.; Kochi, J.K. J. Org. Chem. 1989, 54, 3028.
1112. Svetlakov, N.V.; Moisak, I.E.; Averko-Antonovich, I.G. J. Org. Chem. USSR 1969, 5, 971.
1113. Bartley, J.P.; Carman, R.M.; Russell-Maynard, J.K.L. Aust. J. Chem. 1985, 38, 1879.
1114. Kim, D.W.; Jeong, H.-J.; Lim, S.T.; Sohn, M.-H. Tetrahedron Lett. 2010, 51, 432.
1115. Namavari, M.; Satyamurthy, N.; Phelps, M.E.; Barrio, J.R. Tetrahedron Lett. 1990, 31, 4973.
1116. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 697–700.
1117. Stephenson, B.; Solladié, G.; Mosher, H.S. J. Am. Chem. Soc. 1974, 96, 3171.
1118. Stork, G.; Grieco, P.A.; Gregson, M. Tetrahedron Lett. 1969, 1393.
1119. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 689–697.
1120. See Pizey, J.S. Synthetic Reagents, Vol. 1, Wiley, NY, 1974, pp. 321–357. See Mohanazadeh, F.; Momeni, A.R. Org. Prep. Proceed. Int. 1996, 28, 492 for the use of SOCl2 on silica gel.
1121. See Salomaa, P.; Kankaanperä, A.; Pihlaja, K. in Patai, S. The Chemistry of the Hydroxyl Group, pt. 1, Wiley, NY, 1971, pt. 1, pp. 595–622.
1122. Schreiner, P.R.; Schleyer, P.v.R.; Hill, R.K. J. Org. Chem. 1993, 58, 2822.
1123. Chong, J.M.; Heuft, M.A.; Rabbat, P. J. Org. Chem. 2000, 65, 5837.
1124. Jones, R.; Pattison, J.B. J. Chem. Soc. C 1969, 1046.
1125. See Di Deo, M.; Marcantoni, E.; Torregiani, E.; Bartoli, G.; Bellucci, M.C.; Bosco, M.; Sambri, L. J. Org. Chem. 2000, 65, 2830.
1126. Other phase-transfer catalysts have been used: Landini, D.; Montanari, F.; Rolla, F. Synthesis 1974, 37.
1127. Fuchs, R.; Cole, L.L. Can. J. Chem. 1975, 53, 3620.
1128. See Chaudhari, S.S.; Akamanchi, K.G. Synlett 1999, 1763.
1129. Cason, J.; Correia, J.S. J. Org. Chem. 1961, 26, 3645.
1130. Dakka, G.; Sasson, Y. Tetrahedron Lett. 1987, 28, 1223.
1131. Pelletier, J.D.; Poirier, D. Tetrahedron Lett. 1994, 35, 1051.
1132. Joseph, R.; Pallan, P.S.; Sudalai, A.; Ravindranathan, T. Tetrahedron Lett. 1995, 36, 609.
1133. Hiegel, G.A.; Rubino, M. Synth. Commmun. 2002, 32, 2691.
1134. Dubey, A.; Upadhyay, A.K.; Kumar, P. Tetrahedron Lett. 2010, 51, 744.
1135. Tajbakhsh, M.; Hosseinzadeh, R.; Lasemi, Z. Synlett 2004, 635.
1136. Lee, J.C.; Park, J.Y.; Yoo, E.S. Synth. Commun. 2004, 34, 2095.
1137. For an exception, see Hanack, M.; Eggensperger, H.; Hähnle, R. Liebigs Ann. Chem. 1962, 652, 96; See also, Politanskii, S.F.; Ivanyk, G.D.; Sarancha, V.N.; Shevchuk, V.U. J. Org. Chem. USSR 1974, 10, 697.
1138. See Hudlicky, M. Org. React. 1988, 35, 513.
1139. Middleton, W.J. J. Org. Chem. 1975, 40, 574.
1140. Vorbrüggen, H. Synthesis 2008, 1165.
1141. Kim, K.-Y.; Kim, B.C.; Lee, H.B.; Shin, H. J. Org. Chem. 2008, 73, 8106. See also Zhao, X.; Zhuang, W.; Fang, D.; Xue, X.; Zhou, J. Synlett 2009, 779.
1142. Hayat, S.; Atta-ur-Rahman; Khan, K.M.; Choudhary, M.I.; Maharvi, G.M.; Zia-Ullah; Bayer, E. Synth. Commun. 2003, 33, 2531.
1143. Manickam, GF.; Siddappa, U.; Li, Y. Tetrahedron Lett. 2006, 47, 5867.
1144. Yoneda, N.; Fukuhara, T. Chem. Lett. 2001, 222.
1145. Flosser, D.A.; Olofson, R.A. Tetrahedron Lett. 2002, 43, 4275.
1146. Olah, G.A.; Welch, J.; Vankar, Y.D.; Nojima, M.; Kerekes, I.; Olah, J.A. J. Org. Chem. 1979, 44, 3872. See also, Yin, J.; Zarkowsky, D.S.; Thomas, D.W.; Zhao, M.W.; Huffman, M.A. Org. Lett. 2004, 6, 1465.
1147. Ren, R. X.; Wu, J. X. Org. Lett. 2001, 3, 3727.
1148. Hajipour, A.R.; Mostafavi, M.; Ruoho, A.E. Org. Prep. Proceed. Int. 2009, 41, 87.
1149. Ranu, B.C.; Jana, R. Eur. J. Org. Chem. 2005, 755.
1150. Also see Classon, B.; Liu, Z.; Samuelsson, B. J. Org. Chem. 1988, 53, 6126; Munyemana, F.; Frisque-Hesbain, A.; Devos, A.; Ghosez, L. Tetrahedron Lett. 1989, 30, 3077; Ernst, B.; Winkler, T. Tetrahedron Lett. 1989, 30, 3081.
1151. Firouzabadi, H.; Iranpoor, N.; Jafarpour, M. Tetrahedron Lett. 2004, 45, 7451.
1152. Labrouillère, M.; LeRoux, C.; Oussaid, A.; Gaspard-Iloughmane, H.; Dubac, J. Bull. Soc. Chim. Fr. 1995132, 522.
1153. Yasuda, M.; Yamasaki, S.; Onishi, Y.; Baba, A. J. Am. Chem. Soc. 2004, 126, 7186.
1154. Yasuda, M.; Shimizu, K.; Yamasaki, S.; Baba, A. Org. Biomol. Chem. 2008, 6, 2790.
1155. Snyder, D.C. J. Org. Chem. 1995, 60, 2638.
1156. Kavala, V.; Naik, S.; Patel, B.K. J. Org. Chem. 2005, 70, 4267.
1157. Rydon, H.N. Org. Synth. VI, 830.
1158. Sandri, J.; Viala, J. Synth. Commun. 1992, 22, 2945.
1159. See Castro, B.R. Org. React. 1983, 29, 1; Mackie, R.K. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979; pp. 433–466.
1160. See Appel, R. Angew. Chem. Int. Ed. 1975, 14, 801; Appel, R.; Halstenberg, M. in Cadogan, J.I.G. Organophosphorus Reagents in Organic Synthesis, Academic Press, NY, 1979, pp. 387–431. Also see, Slagle, J.D.; Huang, T.T.; Franzus, B. J. Org. Chem. 1981, 46, 3526; Pollasatri, M.P.; Sagal, J.F.; Chang, G. Tetrahedron Lett. 2001, 42, 2459.
1161. Wagner, A.; Heitz, M.; Mioskowski, C. Tetrahedron Lett. 1989, 30, 557. See also, Desmaris, L.; Percina, N.; Cottier, L.; Sinou, D. Tetrahedron Lett. 2003, 44, 7589.
1162. Pluempanupat, W.; Chavasiri, W. Tetrahedron Lett. 2006, 47, 6821. Also see Pluempanupat, W.; Chantarasriwong, O.; Taboonpong, P.; Jang, D.O.; Chavasiri, W. Tetrahedron Lett. 2007, 48, 223.
1163. Matveeva, E.D.; Yalovskaya, A.I.; Cherepanov, I.A.; Kurts, A.L.; Bundel', Yu.G. J. Org. Chem. USSR 1989, 25, 587.
1164. See Magid, R.M. Tetrahedron 1980, 36, 1901, pp. 1924–1926.
1165. Axelrod, E.H.; Milne, G.M.; van Tamelen, E.E. J. Am. Chem. Soc. 1973, 92, 2139.
1166. Hrubiec, R.T.; Smith, M.B. Synth. Commun. 1983, 13, 593.
1167. Hajipour, A.R.; Falahati, A.R.; Ruoho, A.E. Tetrahedron Lett. 2006, 47, 4191.
1168. Tongkate, P.; Pluempanupat, W.; Chavasiri, W. Tetrahedron Lett. 2008, 49, 1146.
1169. Firouzabadi, H.; Iranpoor, N.; Ebrahimzadeh, F. Tetrahedron Lett. 2006, 47, 1771.
1170. Bandgar, B.P.; Sadavarte, V.S.; Uppalla, L.S. Tetrahedron Lett. 2001, 42, 951.
1171. Sarmah, P.; Barua, N.C. Tetrahedron 1989, 45, 3569.
1172. Kamal, A.; Ramesh, G.; Laxman, N. Synth. Commun. 2001, 31, 827.
1173. Kishali, N.; Polat, M.F.; Altundas, R.; Kara, Y. Helv. Chim. Acta 2008, 91, 67.
1174. Corey, E.J.; Kim, C.U.; Takeda, M. Tetrahedron Lett. 1972, 4339.
1175. Li, Z.; Crosignani, S.; Linclau, B. Tetrahedron Lett. 2003, 44, 8143; Crosignani, S.; Nadal, B.; Li, Z.; Linclau, B. Chem. Commun. 2003, 260.
1176. Iranpoor, N.; Firouzabadi, H.; Aghapour, G. Synlett 2001, 1176.
1177. Firouzabadi, H.; Iranpoor, N.; Hazarkhani, H. Tetrahedron Lett. 2002, 43, 7139.
1178. Patil, B.R.; Bhusare, S.R.; Pawar, R.P.; Vibhute, Y.R. Tetrahedron Lett. 2005, 46, 7179.
1179. Carroll, L.; Pacheco, Ma C.; Garcia, L.; Gouverneur, V. Chem. Commun. 2006, 4113.
1180. See Bhatt, M.V.; Kulkarni, S.U. Synthesis 1983, 249; Staude, E.; Patat, F. in Patai, S. The Chemistry of the Ether Linkage, Wiley, NY, 1967, p. 22; Tiecco, M. Synthesis 1988, 749.
1181. Also see Jursic, B. J. Chem. Res. (S) 1989, 284.
1182. Landini, D.; Montanari, F.; Rolla, F. Synthesis 1978, 771.
1183. Boovanahalli, S.K.; Kim, D.W.; Chi, D.Y. J. Org. Chem. 2004, 69, 3340.
1184. Heathcock, C.H.; Ratcliffe, R. J. Am. Chem. Soc. 1971, 93, 1746.
1185. Reist, E.J.; Bartuska, V.J.; Goodman, L. J. Org. Chem. 1964, 29, 3725.
1186. Ishihara, K.; Hiraiwa, Y.; Yamamoto, H. Synlett 2000, 80.
1187. Moody, C.J.; Pitts, M.R. Synlett 1999, 1575.
1188. Vatèle, J.-M. Synlett 2001, 1989. For a procedure using DDQ see Vatèle, J.-M. Synlett 2002, 507
1189. See Dahlen, A.; Sundgren, A.; Lahmann, M.; Oscarson, S.; Hilmersson, G. Org. Lett. 2003, 5, 4085; Bartoli, G.; Cupone, G.; Dalpozzo, R.; DeNino, A.; Maiuolo, L.; Marcantoni, E.; Procopio, A. Synlett 2001, 1897; Chandrasekhar, S.; Reddy, Ch.R.; Rao, R.J. Tetrahedron 2001, 57, 3435; Tanaka, S.; Saburi, H.; Ishibashi, Y.; Kitamura, M. Org. Lett. 2004, 6, 1873. See also, Murakami, H.; Minami, T.; Ozawa, F. J. Org. Chem. 2004, 69, 4482.
1190. Solis-Oba, A.; Hudlicky, T.; Koroniak, L.; Frey, D. Tetrahedron Lett. 2001, 42, 1241.
1191. Mimero, P.; Saluzzo, C.; Amouroux, R. Tetrahedron Lett. 1994, 35, 1553.
1192. Ohshita, J.; Iwata, A.; Kanetani, F.; Kunai, A.; Yamamoto, Y.; Matui, C. J. Org. Chem. 1999, 64, 8024.
1193. Khalafi-Nezhad, A.; Alamdari, R.F. Tetrahedron 2001, 57, 6805.
1194. Zewge, D.; King, A.; Weissman, S.; Tschaen, D. Tetrahedron Lett. 2004, 45, 3729.
1195. Niwa, H.; Hida, T.; Yamada, K. Tetrahedron Lett. 1981, 22, 4239.
1196. Johnson, F. in Olah, G.A. Friedel–Crafts and Related Reactions, Vol. 4, Wiley, NY, 1965, pp. 1–109.
1197. Vankar, Y.D.; Rao, C.T. J. Chem. Res. (S) 1985, 232. See also, Sharma, G.V.M.; Reddy, Ch.G.; Krishna, P.R. J. Org. Chem. 2003, 68, 4574.
1198. See Olah, G.A.; Prakash, G.K.S.; Krishnamurti, R. Adv. Silicon Chem. 1991, 1, 1.
1199. Jung, M.E.; Lyster, M.A. J. Org. Chem. 1977, 42, 3761; Org. Synth. VI, 353.
1200. Olah, G.A.; Narang, S.C.; Gupta, B.G.B.; Malhotra, R. J. Org. Chem. 1979, 44, 1247; Amouroux, R.; Jatczak, M.; Chastrette, M. Bull. Soc. Chim. Fr. 1987, 505.
1201. Anderson, Jr., A.G.; Freenor, F.J. J. Org. Chem. 1972, 37, 626.
1202. Harrison, I.T. Chem. Commun. 1969, 616.
1203. See Ishizaki, M.; Yamada, M.; Watanabe, S.-i.; Hoshino, O.; Nishitani, K.; Hayashida, M.; Tanaka, A.; Hara, H. Tetrahedron 2004, 60, 7973.
1204. Kamal, A.; Laxman, E.; Rao, N.V. Tetrahedron Lett. 1999, 40, 371.
1205. Yadav, J.S.; Ganganna, B.; Bhunia, D.C.; Srihari, P. Tetrahedron Lett. 2009, 50, 4318.
1206. Okano, K.; Okuyama, K.; Fukuyama, Y.; Tokuyama, H. Synlett 2008, 1977. See also, Konieczny, M.T.; Maciejewski, G.; Konieczny, W. Synthesis 2005, 1575.
1207. Park, J.; Chae, J. Synlett 2010, 1651; Cheng, L.; Aw, C.; Ong, S.S.; Lu, Y. Bull. Chem. Soc. Jpn. 2007, 80, 2008.
1208. Aizpurua, J.M.; Cossío, F.P.; Palomo, C. J. Org. Chem. 1986, 51, 4941.
1209. Mattes, H.; Benezra, C. Tetrahedron Lett. 1987, 28, 1697.
1210. Kim, S.; Park, J.H. J. Org. Chem. 1988, 53, 3111.
1211. Bhatt, S.; Nayak, S.K. Tetrahedron Lett. 2006, 47, 8395.
1212. See Corey, E.J.; Venkateswarlu, A. J. Am. Chem. Soc. 1972, 94, 6190.
1213. Wang, T.; Ji, W.-H.; Xu, Z.-Y.; Zeng, B.-B. Synlett 2009, 1511.
1214. See Sharts, C.M.; Sheppard, W.A. Organic Fluorine Chemistry, W.A. Benjamin, NY, 1969, pp. 52–184, 409–430. For a related review, see Yoneda, N. Tetrahedron 1991, 47, 5329.
1215. Shahak, I.; Manor, S.; Bergmann, E.D. J. Chem. Soc. C 1968, 2129.
1216. Olah, G.A.; Meidar, D. Isr. J. Chem. 1978, 17, 148.
1217. Inagaki, T.; Fukuhara, T.; Hara, S. Synthesis 2003, 1157.
1218. Kalow, J.A.; Doyle, A.G. J. Am. Chem. Soc. 2010, 132, 3268.
1219. Einhorn, C.; Luche, J. J. Chem. Soc., Chem. Commun. 1986, 1368; Ciaccio, J.A.; Addess, K.J.; Bell, T.W. Tetrahedron Lett. 1986, 27, 3697; Spawn, C.; Drtina, G.J.; Wiemer, D.F. Synthesis 1986, 315. For reviews, see Bonini, C.; Righi, G. Synthesis 1994, 225; Chini, M.; Crotti, P.; Gardelli, C.; Macchia, F. Tetrahedron 1992, 48, 3805.
1220. Palumbo, G.; Ferreri, C.; Caputo, R. Tetrahedron Lett. 1983, 24, 1307. See Afonso, C.A.M.; Vieira, N.M.L.; Motherwell, W.B. Synlett 2000, 382.
1221. Amantini, D.; Fringuelli, F.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2001, 66, 4463.
1222. Bonini, C.; Giuliano, C.; Righi, G.; Rossi, L. Synth. Commun. 1992, 22, 1863.
1223. Lu, Z.; Wu, W.; Peng, L.; Wu, L. Can. J. Chem. 2008, 86, 142.
1224. Kwon, D.W.; Cho, M.S.; Kim, Y.H. Synlett 2003, 959. Thiophenol promotes ring openeing by iodine, see Wu, J.; Sun, X.; Sun, W.; Ye, S. Synlett 2006, 2489.
1225. Kotsuki, H.; Shimanouchi, T. Tetrahedron Lett. 1996, 37, 1845.
1226. Campbell, J.R.; Jones, J.K.N.; Wolfe, S. Can. J. Chem. 1966, 44, 2339.
1227. Isaacs, N.S.; Kirkpatrick, D. Tetrahedron Lett. 1972, 3869.
1228. Srebnik, M.; Joshi, N.N.; Brown, H.C. Isr. J. Chem. 1989, 29, 229.
1229. Nikam, K.; Nashi, T. Tetrahedron, 2002, 58, 10259. Also see Sharghi, H.; Niknam, K.; Pooyan, M. Tetrahedron 2001, 57, 6057; Sharghi, H.; Naeimi, H. Bull. Chem. Soc. Jpn. 1999, 72, 1525.
1230. Ranu, B.C.; Banerjee, S. J. Org. Chem. 2005, 70, 4517.
1231. Sartillo-Piscil, F.; Quinero, L.; Villegas, C.; Santacruz-Juárez, E.; de Parrodi, C.A. Tetrahedron Lett. 2002, 43, 15.
1232. Xu, L.-W.; Li, L.; Xia, C.-G.; Zhao, P.-Q. Tetrahedron Lett. 2004, 45, 2435.
1233. Tamami, B.; Ghazi, I.; Mahdavi, H. Synth. Commun. 2002, 32, 3725.
1234. Sharghi, H.; Eskandari, M.M. Synthesis 2002, 1519.
1235. Ha, J.D.; Kim, S.Y.; Lee, S.J.; Kang, S.K.; Ahn, J.H.; Kim, S.S.; Choi, J.-K. Tetrahedron Lett. 2004, 45, 5969.
1236. Fringuelli, F.; Pizzo, F.; Vaccaro, L. J. Org. Chem. 2001, 66, 4719. Also see Concellón, J.M.; Bardales, E.; Concellón, C.; García-Granda, S.; Díaz, M.R. J. Org. Chem. 2004, 69, 6923.
1237. Belsner, K.; Hoffmann, H.M.R. Synthesis 1982, 239. See also, Iqbal, J.; Khan, M.A.; Srivastava, R.R. Tetrahedron Lett. 1988, 29, 4985.
1238. Taniguchi, Y.; Tanaka, S.; Kitamura, T.; Fujiwara, Y. Tetrahedron Lett. 1998, 39, 4559.
1239. Qian, C.; Zhu, D. Synth. Commun. 1994, 24, 2203.
1240. Kameyama, A.; Kiyota, M.; Nishikubo, T. Tetrahedron Lett. 1994, 35, 4571.
1241. Kumar, M.; Pandey, S.K.; Gandhi, S.; Singh, V.K. Tetrahedron Lett. 2009, 50, 363.
1242. Righi, G.; D'Achille, R.; Bonini, C. Tetrahedron Lett. 1996, 37, 6893.
1243. Fan, R.-H.; Zhou, Y.-G.; Zhang, W.-X.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2004, 69, 335.
1244. D'hooghe, M.; Speybroeck, V.V.; Waroquier, M.; De Kimpe, N. Chem. Commun. 2006, 1554.
1245. See McMurry, J. Org. React. 1976, 24, 187–224.
1246. Elsinger, F.; Schreiber, J.; Eschenmoser, A. Helv. Chim. Acta 1960, 43, 113.
1247. Olah, G.A.; Narang, S.C.; Gupta, B.G.B.; Malhotra, R.J. Org. Chem. 1979, 44, 1247. See also, Kolb, M.; Barth, J. Synth. Commun. 1981, 11, 763.
1248. Yadav, V.K.; Babu, K.G. Tetrahedron 2003, 59, 9111.
1249. Olah, G.A.; Welch, J.; Vankar, Y.D.; Nojima, M.; Kerekes, I.; Olah, J.A. J. Org. Chem. 1979, 44, 3872.
1250. Olah, G.A.; Prakash, G.K.S.; Chao, Y.L. Helv. Chim. Acta 1981, 64, 2528; Barber, J.; Keck, R.; Rétey, J. Tetrahedron Lett. 1982, 23, 1549.
1251. Olah, G.A.; Shih, J.; Prakash, G.K.S. Helv. Chim. Acta 1983, 66, 1028.
1252. For another method, see Lorenzo, A.; Molina, P.; Vilaplana, M.J. Synthesis 1980, 853.
1253. Collazo, L.R.; Guziec Jr., F.S.; Hu, W.-X.; Pankayatselvan, R. Tetrahedron Lett. 1994, 35, 7911.
1254. Doyle, M.P.; Bosch, R.J.; Seites, P.G. J. Org. Chem. 1978, 43, 4120.
1255. Katritzky, A.R.; Chermprapai, A.; Patel, R.C. J. Chem. Soc. Perkin Trans. 1 1980, 2901.
1256. Chambers, R.A.; Pearson, D.E. J. Org. Chem. 1963, 28, 3144.
1257. See Cooley, J.H.; Evain, E.J. Synthesis 1989, 1.
1258. Olofson, R.A.; Martz, J.T.; Senet, J.; Piteau, M.; Malfroot, T. J. Org. Chem. 1984, 49, 2081; Olofson, R.A.; Abbott, D.E. J. Org. Chem. 1984, 49, 2795. See also, Campbell, A.L.; Pilipauskas, D.R.; Khanna, I.K.; Rhodes, R.A. Tetrahedron Lett. 1987, 28, 2331.
1259. See Ko, E.C.F.; Leffek, K.T. 1971, 49, 129; Deady, L.W.; Korytsky, O.L. Tetrahedron Lett. 1979, 451.
1260. See Cooley, J.H.; Evain, E.J. Synthesis 1989, 1. See Vaccari, D.; Davoli, P.; Spaggiari, A.; Prati, F. Synlett 2008, 1317.
1261. See Hageman, H.A. Org. React. 1953, 205.
1262. Fodor, G.; Abidi, S.; Carpenter, T.C. J. Org. Chem. 1974, 39, 1507. See also, Paukstelis, J.V.; Kim, M. J. Org. Chem. 1974, 39, 1494.
1263. See Hwu, J.R.; Gilbert, B.A. Tetrahedron 1989, 45, 1233.
1264. See Stowell, J.C. Carbanions in Organic Synthesis, Wiley, NY, 1979; Noyori, R. in Alper, H. Transition Metal Organometallics in Organic Synthesis Vol. 1, Academic Press, NY, 1976, pp. 83–187.
1265. Cho, Y.S.; Kang, S.-H.; Han, J.-S.; Yoo, B.R.; Jung, I.N. J. Am. Chem. Soc. 2001, 123, 5584.
1266. Yadav, J.S.; Reddy, B.V.S.; Rao, K.V.; Raj, K.S.; Rao, P.P.; Prasad, A.R.; Gunasekar, D. Tetrahedron Lett. 2004, 45, 6505.
1267. See Kakiuchi, F.; Tsuchiya, K.; Matsumoto, M.; Mizushima, E.; Chatani, N. J. Am. Chem. Soc. 2004, 126, 12792; Nii, S.; Terao, J.; Kambe, N. Tetrahedron Lett. 2004, 45, 1699.
1268. Kang, S.-K.; Kim, T.H.; Pyun, S.-J. J. Chem. Soc. Perkin Trans. 1 1997, 797.
1269. Matsuhashi, H.; Asai, S.; Hirabayashi, K.; Hatanaka, Y.; Mori, A.; Hiyama, T. Bull. Chem. Soc. Jpn. 1997, 70, 1943.
1270. Katritzky, A.R.; Mehta, S.; He, H.-Y.; Cui, X. J. Org. Chem. 2000, 65, 4364.
1271. Itami, K.; Kamei, T.; Yoshida, J.-i. J. Am. Chem. Soc. 2001, 123, 8773.
1272. Matano, Y.; Yoshimune, M.; Suzuki, H. Tetrahedron Lett. 1995, 36, 7475.
1273. Maeda, K.; Shinokubo, H.; Oshima, K. J. Org. Chem. 1997, 62, 6429.
1274. Yokozawa, T.; Furuhashi, K.; Natsume, H. Tetrahedron Lett. 1995, 36, 5243.
1275. Tsuji, Y.; Funato, M.; Ozawa, M.; Ogiyama, H.; Kajita, S.; Kawamura, T. J. Org. Chem. 1996, 61, 5779.
1276. Hatanaka, Y.; Goda, K.; Hiyama, T. Tetrahedron Lett. 1994, 35, 6511; Matsuhashi, H.; Kuroboshi, M.; Hatanaka, Y.; Hiyama, T. Tetrahedron Lett. 1994, 35, 6507.
1277. Prestat, G.; Baylon, C.; Heck, M.-P.; Mioskowski, C. Tetrahedron Lett. 2000, 41, 3829.
1278. Chatgilialoglu, C.; Ferreri, C.; Ballestri, M.; Curran, D.P. Tetrahedron Lett. 1996, 37, 6387; Chatgilialoglu, C.; Alberti, A.; Ballestri, M.; Macciantelli, D.; Curran, D.P. Tetrahedron Lett. 1996, 37, 6391.
1279. Hodgson, D.M.; Norsikian, S.L.M. Org. Lett. 2001, 3, 461.
1280. Harrison, J.R.; O'Brien, P.; Porter, D.W.; Smith, N.W. Chem. Commun. 2001, 1202.
1281. Hirao, T.; Fujii, T.; Ohshiro, Y. Tetrahedron Lett. 1994, 35, 8005.
1282. Takeda, T.; Takagi, Y.; Takano, H.; Fujiwara, T. Tetrahedron Lett. 1992, 33, 5381.
1283. Pandey, G.; Rani, K.S.; Lakshimaiah, G. Tetrahedron Lett. 1992, 33, 5107. See Gelas-Mialhe, Y.; Gramain, J.-C.; Louvet, A.; Remuson, R. Tetrahedron Lett. 1992, 33, 73.
1284. Seganish, W.M.; DeShong, P. J. Org. Chem. 2004, 69, 6790.
1285. Matos, M.R.P.N.; Afonso, C.A.M.; Batey, R.A. Tetrahedron Lett. 2001, 42, 7007.
1286. Tobisu, M.; Kita, Y.; Ano, Y.; Chatani, N. J. Am. Chem. Soc. 2008, 130, 15982.
1287. Murata, M.; Watanabe, S.; Masuda, Y. Tetrahedron Lett. 1999, 40, 9255.
1288. For an example, see Kwa, T.L.; Boelhouwer, C. Tetrahedron 1970, 25, 5771.
1289. For a list of reagents, including metals and other reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 83–84.
1290. See, for example, Nosek, J. Collect. Czech. Chem. Commun. 1964, 29, 597.
1291. Onsager, O. Acta Chem. Scand. Ser. B, 1978, 32, 15.
1292. Ginah, F.O.; Donovan, T.A.; Suchan, S.D.; Pfennig, D.R.; Ebert, G.W. J. Org. Chem. 1990, 55, 584.
1293. Ranu, B.C.; Dutta, P.; Sarkar, A. Tetrahedron Lett. 1998, 39, 9557.
1294. Nishino, T.; Watanabe, T.; Okada, M.; Nishiyama, Y.; Sonoda, N. J. Org. Chem. 2002, 67, 966.
1295. See Ma, J.; Chan, T.-H. Tetrahedron Lett. 1998, 39, 2499; Gilbert, B.C.; Lindsay, C.I.; McGrail, P.T.; Parsons, A.F.; Whittaker, D.T.E. Synth. Commun. 1999, 29, 2711.
1296. Han, B.H.; Boudjouk, P. Tetrahedron Lett. 1981, 22, 2757.
1297. Nishiyama, T.; Seshita, T.; Shodai, H.; Aoki, K.; Kameyama, H.; Komura, K. Chem. Lett. 1996, 549.
1298. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 1277–1286.
1299. See Danheiser, R.L.; Tsai, Y.; Fink, D.M. Org. Synth. 66, 1.
1300. See Freidlina, R.Kh.; Kamyshova, A.A.; Chukovskaya, E.Ts. Russ. Chem. Rev. 1982, 51, 368; in Rappoport, Z. The Chemistry of the Cyclopropyl Group, pt. 1, Wiley, NY, 1987, the reviews by Tsuji, T.; Nishida, S. pp. 307–373, and Verhé, R.; De Kimpe, N. pp. 445–564.
1301. For a discussion of the mechanism, see Applequist, D.E.; Pfohl, W.F. J. Org. Chem. 1978, 43, 867.
1302. Wiberg, K.B.; Lampman, G.M. Tetrahedron Lett. 1963, 2173; Lampman, G.M.; Aumiller, J.C. Org. Synth. VI, 133.
1303. Pincock, R.E.; Schmidt, J.; Scott, W.B.; Torupka, E.J. Can. J. Chem. 1972, 50, 3958.
1304. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 175–184.
1305. Kaplan, L. J. Am. Chem. Soc.1967, 89, 1753; J. Org. Chem. 1967, 32, 4059.
1306. Bailey, W.F.; Gagnier, R.P. Tetrahedron Lett. 1982, 23, 5123.
1307. Connor, D.S.; Wilson, E.R. Tetrahedron Lett. 1967, 4925.
1308. Rifi, M.R. J. Am. Chem. Soc.1967, 89, 4442; Org. Synth. VI, 153.
1309. Tamao, K.; Sumitani, K.; Kumada, M. J. Am. Chem. Soc. 1972, 94, 4374; Terao, J.; Kambe, N. Acc. Chem. Res. 2008, 41, 1545; Lopez-Perez, A.; Adrio, J.; Carretero, J.C. Org. Lett. 2009, 11, 5514; Frisch, A.C.; Shaikh, N.; Zapf, A.; Beller, M. Angew. Chem., 2002, 114, 4218. See Chen, X.; Wang, L.; Liu, J. Synthesis 2009, 2408; Limmert, M.E.; Roy, A.H.; Hartwig, J.F. J. Org. Chem. 2005, 70, 9364; Tsai, F.-Y.; Lin, B.-N.; Chen, M.-J.; Mou, C.-Y.; Liu, S.T. Tetrahedron 2007, 63, 4304; Organ, M.G.; Abdel-Hadi, M.; Avola, S.; Hadei, N.; Nasielski, J.; O'Brien, C.J.; Valente, C. Chemistry: European J. 2007, 13, 150; Gauthier, D.; Beckendorf, S.; G![]() gsig, T.M.; Lindhardt, A.T.; Skrydstrup, T. J. Org. Chem. 2009, 74, 3536. For a coupling reaction of aryl iodides with heterocyclic Grignard reagents, see Ruben Martin, R.; Buchwald, S.L. J. Am. Chem. Soc. 2007, 129, 3844.
gsig, T.M.; Lindhardt, A.T.; Skrydstrup, T. J. Org. Chem. 2009, 74, 3536. For a coupling reaction of aryl iodides with heterocyclic Grignard reagents, see Ruben Martin, R.; Buchwald, S.L. J. Am. Chem. Soc. 2007, 129, 3844.
1310. Cohen, T.; Poeth, T. J. Am. Chem. Soc. 1972, 94, 4363.
1311. See Grigg, R.; Stevenson, P.; Worakun, T. J. Chem. Soc., Chem. Commun. 1985, 971; Vanderesse, R.; Fort, Y.; Becker, S.; Caubere, P. Tetrahedron Lett. 1986, 27, 3517.
1312. Takagi, K.; Mimura, H.; Inokawa, S. Bull. Chem. Soc. Jpn. 1984, 57, 3517.
1313. Cahiez, G.; Bernard, D.; Normant, J.F. J. Organomet. Chem. 1976, 113, 99.
1314. Paley, R.S.; de Dios, A.; de la Pradilla, R.F. Tetrahedron Lett. 1993, 34, 2429.
1315. See Magid, R.M. Tetrahedron 1980, 36, 1901, see pp. 1910–1924.
1316. In this section, methods are discussed in which one molecule is a halide. For other allylic coupling reactions, see 10-57, 10-63, and 10-60.
1317. See Tamao, K.; Kumada, M. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond Vol. 4, Wiley, NY, 1987, pp. 819–887.
1318. Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R. Principles and Applications of Organotransition Metal Chemsitry, 2nd ed., University Science Books, Mill Valley, CA, 1987, pp. 739–748; Billington, D.C. Chem. Soc. Rev. 1985, 14, 93; Kochi, J.K. Organometallic Mechanisms and Catalysis, Academic Press, NY, 1978, pp. 398–408; Semmelhack, M.F. Org. React. 1972, 19, 115, see pp. 162–170; Baker, R. Chem. Rev. 1973, 73, 487, see pp. 512–517.
1319. Corey, E.J.; Wat, E.K.W. J. Am. Chem. Soc. 1967, 89, 2757. See also, Reijnders, P.J.M.; Blankert, J.F.; Buck, H.M. Recl. Trav. Chim. Pays-Bas 1978, 97, 30.
1320. See Semmelhack, M.F. Org. React. 1972, 19, 115, see pp. 147–162; Semmelhack, M.F. Org. React. 1972,199, 115, see pp. 144–146.
1321. Hegedus, L.S.; Thompson, D.H.P. J. Am. Chem. Soc. 1985, 107, 5663.
1322. Corey, E.J.; Semmelhack, M.F.; Hegedus, L.S. J. Am. Chem. Soc. 1968, 90, 2416.
1323. Turk, A.; Chanan, H. Org. Synth. III, 121.
1324. Stork, G.; Grieco, P.A.; Gregson, M. Tetrahedron Lett. 1969, 1393; Grieco, P.A. J. Am. Chem. Soc. 1969, 91, 5660.
1325. Hosomi, A.; Imai, T.; Endo, M.; Sakurai, H. J. Organomet. Chem. 1985, 285, 95. See also, Yanagisawa, A.; Norikate, Y.; Yamamoto, H. Chem. Lett. 1988, 1899.
1326. Yamamoto, Y.; Yatagai, H.; Maruyama, K. J. Am. Chem. Soc. 1981, 103, 1969.
1327. See Keck, G.E.; Yates, J.B. J. Am. Chem. Soc. 1982, 104, 5829; Migita, T.; Nagai, K.; Kosugi, M. Bull. Chem. Soc. Jpn 1983, 56, 2480.
1328. See Axelrod, E.H.; Milne, G.M.; van Tamelen, E.E. J. Am. Chem. Soc. 1970, 92, 2139; Morizawa, Y.; Kanemoto, S.; Oshima, K.; Nozaki, H. Tetrahedron Lett. 1982, 23, 2953.
1329. Biellmann, J.F.; Ducep, J.B. Tetrahedron Lett. 1969, 3707.
1330. Cabrera, A.; Rosas, N.; Sharma, P.; LeLagadec, R.; Velasco, L.; Salmón, M. Synth. Commun. 1998, 28, 1103.
1331. See Naso, F.; Marchese, G. in Patai, S.; Rappoport, Z. The Chemstry of Functional Groups, Supplement D, pt. 2, Wiley, NY, 1983, pp. 1353–1449.
1332. For lists of reagents and substrates, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 101–127.
1333. See Beletskaya, I.P. J. Organomet. Chem. 1983, 250, 551; Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 249–262.
1334. Snieckus, V.; Rogers-Evans, M.; Beak, P.; Lee, W.K.; Yum, E.K.; Freskos, J. Tetrahedron Lett. 1994, 35, 4067.
1335. Dieter, R.K.; Li, S.J. J. Org. Chem. 1997, 62, 7726. Also see Beak, P.; Wu, S.; Yum, E.K.; Jun, Y.M. J. Org. Chem. 1994, 59, 276.
1336. For example, see Brimble, M.A.; Gorsuch, S. Aust. J. Chem. 1999, 52, 965.
1337. Merrill, R.E.; Negishi, E. J. Org. Chem., 1974, 39, 3452. For another method, see Hallberg, A.; Westerlund, C. Chem. Lett., 1982, 1993.
1338. Yong, K.H.; Lotoski, J.A.; Chong, J.M. J. Org. Chem. 2001, 66, 8248.
1339. Stanetty, P.; Mihovilovic, M.D. J. Org. Chem. 1997, 62, 1514.
1340. Duhamel, L.; Poirier, J. J. Am. Chem. Soc. 1977, 99, 8356.
1341. Murahashi, S.; Yamamura, M.; Yanagisawa, K.; Mita, N.; Kondo, K. J. Org. Chem. 1979, 44, 2408.
1342. Marié, J.-C.; Curillon, C.; Malacria, M. Synlett 2002, 553.
1343. Itami, K.; Kamei, T.; Mitsudo, K.; Nokami, T.; Yoshida, J.-i. J. Org. Chem. 2001, 66, 3970.
1344. Basu, A.; Beak, P. J. Am. Chem. Soc. 1996, 118, 1575; Wu, S.; Lee, S.; Beak, P. J. Am. Chem. Soc. 1996, 118, 715; Dieter, R.K.; Sharma, R.R. Tetrahedron Lett. 1997, 38, 5937.
1345. Serino, C.; Stehle, N.; Park, Y.S.; Florio, S.; Beak, P. J. Org. Chem. 1999, 64,1160.
1346. Ma, S.; Wang, L. J. Org. Chem. 1998, 63, 3497.
1347. Yang, L.-M.; Huang, L.-F.; Luh, T.-Y. Org. Lett. 2004, 6, 1461.
1348. MacNeil, S.L.; Familoni, O.B.; Snieckus, V. J. Org. Chem. 2001, 66, 3662.
1349. See Snieckus, V. Chem. Rev. 1990, 90, 879; Gschwend, H.W.; Rodriguez, H.R. Org. React. 1979, 26, 1. See also, Green, L.; Chauder, B.; Snieckus, V. J. Heterocyclic Chem. 1999, 36, 1453.
1350. Gilman, H.; Bebb, R.L. J. Am. Chem. Soc. 1939, 61, 109; Wittig, G.; Fuhrman, G. Chem. Ber. 1940, 73, 1197.
1351. McCombie, S.W.; Lin, S.-I.; Vice, S.F. Tetrahedron Lett. 1999, 40, 8767.
1352. Kraus, G.A.; Kim, J. J. Org. Chem. 2002, 67, 2358.
1353. Corey, E.J.; Kirst, H.A.; Katzenellenbogen, J.A. J. Am. Chem. Soc. 1970, 92, 6314.
1354. See Ireland, R.E.; Dawson, M.I.; Lipinski, C.A. Tetrahedron Lett. 1970, 2247.
1355. Pasto, D.J.; Chou, S.; Waterhouse, A.; Shults, R.H.; Hennion, G.F. J. Org. Chem. 1978, 43, 1385; Jeffery-Luong, T.; Linstrumelle, G. Tetrahedron Lett. 1980, 21, 5019.
1356. Pasto, D.J.; Chou, S.; Fritzen, E.; Shults, R.H.; Waterhouse, A.; Hennion, G.F. J. Org. Chem. 1978, 43, 1389. See also, Tanigawa, Y.; Murahashi, S. J. Org. Chem. 1980, 45, 4536.
1357. See Raston, C.L.; Salem, G. in Hartley, F.R. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, 1987, pp. 161–306, 269–283; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 1046–1165.
1358. See Ohno, M.; Shimizu, K.; Ishizaki, K.; Sasaki, T.; Eguchi, S. J. Org. Chem. 1988, 53, 729.
1359. Tissot-Croset, K.; Alexakis, A. Tetrahedron Lett. 2004, 45, 7375; Tissot-Croset, K.; Polet, D.; Alexakis, A. Angew. Chem. Int. Ed. 2004, 43, 2426.
1360. See Erdik, E. Tetrahedron 1984, 40, 641; Kochi, J.K. Organometallic Mechanisms and Catalysis, Academic Press, NY, 1978, pp. 374–398.
1361. Bäckvall, J.-E.; Persson, E.S.M.; Bombrun, A. J. Org. Chem. 1994, 59, 4126.
1362. Terao, J.; Ikumi, A.; Kuniyasu, H.; Kambe, N. J. Am. Chem. Soc. 2003, 125, 5646. See also, Hintermann, L.; Xiao, L.; Labonne, A. Angew. Chem. Int. Ed. 2008, 47, 8246; Cahiez, G.; Gager, O.; Buendia, J. Synlett 2010, 299.
1363. Someya, H.; Ohmiya, H.; Yorimitsu, H.; Oshima, K. Org. Lett. 2008, 10, 969.
1364. López-Pérez, A.; Adrio, J.; Carretero, J.C. Org. Lett. 2009, 11, 5514. For other references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 386–392.
1365. Hamaguchi, H.; Uemura, M.; Yasui, H.; Yorimitsu, H.; Koichiro Oshima, K. Chem. Lett. 2008, 37, 1178.
1366. Cahiez, G.; Habiak, V.; Duplais, C.; Moyeux, A. Angew. Chem. Int. Ed. 2007, 46, 4364; Molander, G.A.; Rahn, B.J.; Shubert, D.C.; Bonde, S.E. Tetrahedron Lett. 1983, 24, 5449. See Bedford, R.B.; Bruce, D.W.; Frost, R.M.; Goodby, J.W.; Hird, M. Chem. Commun. 2004, 2822.
1367. Bedford, R.B.; Bruce, D.W.; Frost, R.M.; Hird, M. Chem. Commun. 2005, 4161.
1368. Bedford, R.B.; Betham, M.; Bruce, D.W.; Davis, S.A.; Frost, R.M.; Hird, M. Chem. Commun. 2006, 1398.
1369. Wang, S.; Zhang, A. Org. Prep. Proceed. Int. 2008, 40, 293.
1370. Geurts, K.; Fletcher, S.P.; Feringa, B.L. J. Am. Chem. Soc. 2006, 128, 15572.
1371. Lee, Y.; Hoveyda, A.H. J. Am. Chem. Soc. 2006, 128, 15604.
1372. Kar, A.; Argade, N.P. Synthesis 2005, 2995. For an example, see Sen, S.; Singh, S.; Sieburth, S.McN. J. Org. Chem. 2009, 74, 2884.
1373. See Bell, T.W.; Hu, L.; Patel, S.V. J. Org. Chem., 1987, 52, 3847; Ozawa, F.; Kurihara, K.; Fujimori, M.; Hidaka, T.; Toyoshima, T.; Yamamoto, A. Organometallics 1989, 8, 180.
1374. Yasuda, S.; Yorimitsu, H.; Oshima, K. Bull. Chem. Soc. Jpn. 2008, 81, 287.
1375. Kamikawa, T.; Hayashi, T. Synlett, 1997, 163.
1376. Hoffmann, R.W.; Gieson, V.; Fuest, M. Liebigs Ann. Chem. 1993, 629.
1377. Babudri, F.; Fiandanese, V.; Mazzone, L.; Naso, F. Tetrahedron Lett. 1994, 35, 8847.
1378. Ohmiya, H.; Yorimitsu, H.; Oshima, K. J. Am. Chem. Soc. 2006, 128, 1886.
1379. Negishi, E.; Kotora, M.; Xu, C. J. Org. Chem. 1997, 62, 8957.
1380. Cai, M.-Z.; Song, C.-S.; Huang, X. J. Chem. Res. (S) 1998, 264.
1381. Kondo, S.; Ohira, M.; Kawasoe, S.; Kunisada, H.; Yuki, Y. J. Org. Chem. 1993, 58, 5003.
1382. Nagano, T.; Hayashi, T. Org. Lett. 2004, 6, 1297; Terao, J.; Watabe, H.; Kambe, N. J. Am. Chem. Soc. 2005, 127, 3656.
1383. Martin, R.; Fürstner, A. Angew. Chem. Int. Ed.2004, 43, 3955.
1384. Dohle, W.; Kopp, F.; Cahiez, G.; Knochel, P. Synlett 2001, 1901. See Scheiper, B.; Bonnekessel, M.; Krause, H.; Fürstner, A. J. Org. Chem. 2004, 69, 3943.
1385. Karlström, A.S.E.; Rönn, M.; Thorarensen, A.; Bäckvall, J.-E. J. Org. Chem. 1998, 63, 2517.
1386. Cahiez, G.; Avedissian, H. Tetrahedron Lett. 1998, 39, 6159.
1387. Hayashi, T.; Konishi, M.; Kobori, Y.; Kumada, M.; Higuchi, T.; Hirotsu, K. J. Am. Chem. Soc. 1984, 106, 158.
1388. Böhm, V.P.W.; Gstöttmayr, C.W.K.; Weskamp, T.; Hermann, W.A. Angew. Chem. Int. Ed. 2001, 40, 3387; Terao, J.; Watanabe, H.; Ikumi, A.; Kuniyasu, H.; Kambe, N. J. Am. Chem. Soc. 2002, 124, 4222. See Kumada, M. Pure Appl. Chem. 1980, 52, 669.
1389. See Hayashi, T.; Kumada, M. in Morrison, J.D. Asymmetic Synthesis Vol. 5, Academic Press, NY, 1985, pp. 147–169. See also, Iida, A.; Yamashita, M. Bull. Chem. Soc. Jpn. 1988, 61, 2365.
1390. Rathore, R.; Deselnicu, M.I.; Burns, C.L. J. Am. Chem. Soc. 2002, 124, 14832.
1391. See Beletskaya, I.P.; Artamkina, G.A.; Reutov, O.A. Russ. Chem. Rev. 1976, 45, 330.
1392. When a symmetrical distribution of products is found, this is evidence for a free-radical mechanism: the solvent cage is not efficient and breaks down.
1393. Sommer, L.H.; Korte, W.D. J. Org. Chem. 1970, 35, 22; Korte, W.D.; Kinner, L.; Kaska, W.C. Tetrahedron Lett. 1970, 603. See also, Schlosser, M.; Fouquet, G. Chem. Ber. 1974, 107, 1162, 1171.
1394. See Muraoka, K.; Nojima, M.; Kusabayashi, S.; Nagase, S. J. Chem. Soc. Perkin Trans. 2 1986, 761.
1395. Podoplelov, A.V.; Leshina, T.V.; Sagdeev, R.Z.; Kamkha, M.A.; Shein, S.M. J. Org. Chem. USSR 1976, 12, 488; Ward, H.R.; Lawler, R.G.; Cooper, R.A. in Lepley, A.R.; Closs, G.L. Chemically Induced Magnetic Polarization, Wiley, NY, 1973, pp. 281–322.
1396. Russell, G.A.; Lamson, D.W. J. Am. Chem. Soc. 1969, 91, 3967.
1397. Bryce-Smith, D. Bull. Soc. Chim. Fr. 1963, 1418.
1398. Garst, J.F.; Hart, P.W. J. Chem Soc. Chem. Commun. 1975, 215.
1399. Kasukhin, L.F.; Ponomarchuk, M.P.; Buteiko, Zh.F. J. Org. Chem. USSR 1972, 8, 673; Singh, P.R.; Tayal, S.R.; Nigam, A. J. Organomet. Chem. 1972, 42, C9.
1400. Bertz, S.H.; Dabbagh, G.; Mujsce, A.M. J. Am. Chem. Soc. 1991, 113, 631.
1401. Tamura, M.; Kochi, J.K. J. Am. Chem. Soc. 1971, 93, 1483, 1485, 1487; J. Organomet. Chem. 1971, 31, 289; 1972, 42, 205; Lehr, G.F.; Lawler, R.G. J. Am. Chem. Soc. 1986, 106, 4048.
1402. See Pearson, R.G.; Gregory, C.D. J. Am. Chem. Soc. 1976, 98, 4098. See also, Lipshutz, B.H.; Kozlowski, J.A.; Breneman, C.M. Tetrahedron Lett. 1985, 26, 5911; Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, 2nd ed., University Science Books, Mill Valley, CA, 1987, pp. 682–698.
1403. See Stemmler, T.L.; Barnhart, T.M.; Penner-Hahn, J.E.; Tucker, C.E.; Knochel, P.; Böhme, M.; Frenking, G. J. Am. Chem. Soc. 1995, 117, 12489. Solution compositions of Gilman reagents have also been studied. See Lipshutz, B.H.; Kayser, F.; Siegmann, K. Tetrahedron Lett. 1993, 34, 6693.
1404. Bergbreiter, D.E.; Whitesides, G.M. J. Org. Chem. 1975, 40, 779. See Bertz, S.H.; Eriksson, M.; Miao, G.; Snyder, J.P. J. Am. Chem. Soc. 1998, 118, 10906 for the reactivity of β-silyl organocuprates.
1405. For an example using a Cu(II) salt, see Nguyen, T.T.; Chevallier, F.; Jouikov, V.; Mongin, F. Tetrahedron Lett. 2009, 50, 6787.
1406. See Posner, G.H. Org. React. 1975, 22, 253; Lipshutz, B.H. Accts. Chem. Res. 1997, 30, 277; Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, NY, 1980. For lists of substrates and reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 392–399, 599–604, 1564.
1407. See Mori, S.; Nakamura, E.; Morokuma, K. J. Am. Chem. Soc. 2000, 122, 7294.
1408. For an extensive discussion of the mechanism of reaction between organocuprates and alkyl haldies or epoxides, see Mori, S.; Nakamura, E.; Morokuma, K. J. Am. Chem. Soc. 2000, 122, 7294; Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, NY, 1980. See Bertz, S.H.; Cope, S.; Dorton, D.; Murphy, M.; Ogle, C.A. Angew. Chem. Int. Ed. 2007, 46, 7082.
1409. Yoshikai, N.; Zhang, S.-L.; Nakamura, E. J. Am. Chem. Soc. 2008, 130, 12862.
1410. Some intermediates in this reaction have been prepared, see Bartholomew, E.R.; Bertz, S.H.; Cope, S.; Murphy, M.; Ogle, C.A. J. Am. Chem. Soc. 2008, 130, 11244. For a review, see Falciola, C.A.; Alexakis, A. Eur. J. Org. Chem. 2008, 3765.
1411. Posner, G.H.; Ting, J. Synth. Commun. 1973, 3, 281.
1412. Lipshutz, B.H.; Wilhelm, R.S.; Nugent, S.T.; Little, R.D.; Baizer, M.M. J. Org. Chem. 1983, 48, 3306.
1413. Klein, J.; Levene, R. J. Am. Chem. Soc. 1972, 94, 2520. For a discussion of the mechanism, see Yoshikai, N.; Nakamura, E. J. Am. Chem. Soc. 2004, 126, 12264.
1414. Posner, G.H.; Brunelle, D.J. Tetrahedron Lett. 1972, 293.
1415. See Kitatani, K.; Hiyama, T.; Nozaki, H. Bull. Chem. Soc. Jpn. 1977, 50, 1600.
1416. Cahiez, G.; Chaboche, C.; Jézéquel, M. Tetrahedron 2000, 56, 2733.
1417. Johnson, C.R.; Dutra, G.A. J. Am. Chem. Soc. 1973, 95, 7777, 7783. See Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, NY, 1980, pp. 85–90.
1418. Secondary tosylates give higher yields when they contain an O or S atom: Hanessian, S.; Thavonekham, B.; DeHoff, B. J. Org. Chem. 1989, 54, 5831.
1419. See Scott, W.J.; McMurry, J.E. Acc. Chem. Res. 1988, 21, 47.
1420. Tsushima, K.; Araki, K.; Murai, A. Chem. Lett. 1989, 1313.
1421. Lipshutz, B.H.; Elworthy, T.R. J. Org. Chem. 1990, 55, 1695.
1422. Baudouy, R.; Goré, J. J. Chem. Res. (S) 1981, 278. See also, Elsevier, C.J.; Vermeer, P. J. Org. Chem. 1989, 54, 3726.
1423. Whitesides, G.M.; Fischer, Jr., W.F.; San Filippo, Jr., J.; Bashe, R.W.; House, H.O., J. Am. Chem. Soc. 1969, 91, 4871.
1424. Prepared as in Ref. 1444 or treatment of PhSCu with RLi: Posner, G.H.; Brunelle, D.J.; Sinoway, L. Synthesis 1974, 662.
1425. Posner, G.H.; Whitten, C.E.; Sterling, J.J. J. Am. Chem. Soc. 1973, 95, 7788.
1426. See Malmberg, H.; Nilsson, M.; Ullenius, C. Tetrahedron Lett. 1982, 23, 3823; Lipshutz, B.H.; Kozlowski, J.A.; Parker, D.A.; Nguyen, S.L.; McCarthy, K.E. J. Organomet. Chem. 1985, 285, 437.
1427. Piazza, C.; Knochel, P. Angew. Chem. Int. Ed. 2002, 41, 3263.
1428. See Lipshutz, B.H. Synthesis 1987, 325; Synlett 1990, 119. See also, Bertz, S.H. J. Am. Chem. Soc. 1990, 112, 4031; Lipshutz, B.H.; Sharma, S.; Ellsworth, E.L. J. Am. Chem. Soc. 1990, 112, 4032.
1429. Lipshutz, B.H.; Wilhelm, R.S.; Floyd, D.M. J. Am. Chem. Soc. 1981, 103, 7672.
1430. Bertz, S.H.; Dabbagh, G. J. Org. Chem. 1984, 49, 1119.
1431. See Aoki, S.; Fujimura, T.; Nakamura, E.; Kuwajima, I. J. Am. Chem. Soc., 1988, 110, 3296.
1432. McMurry, J.E.; Mohanraj, S. Tetrahedron Lett., 1983, 24, 2723.
1433. Hirota, K.; Isobe, Y.; Maki, Y. J. Chem. Soc., Perkin Trans. 1, 1989, 2513.
1434. Echevarren, E.M.; Stille, J.K. J. Am. Chem. Soc., 1987, 109, 5478. For a similar reaction with aryl fluorosulfonates, see Roth, G.P.; Fuller, C.E. J. Org. Chem., 1991, 56, 3493.
1435. Martínez, A.G.; Barcina, J.O.; Díez, B.R.; Subramanian, L.R. Tetrahedron 1994, 50, 13231.
1436. Mooiweer, H.H.; Elsevier, C.J.; Wijkens, P.; Vermeer, P. Tetrahedron Lett. 1985, 26, 65.
1437. Corey, E.J.; Boaz, N.W. Tetrahedron Lett. 1984, 25, 3059, 3063. For the reaction of these reagents with haloalkynes, see Yeh, M.C.P.; Knochel, P. Tetrahedron Lett. 1989, 30, 4799.
1438. Bertz, S.H.; Miao, G.; Eriksson, M. Chem. Commun. 1996, 815; Snyder, J.P.; Bertz, S.H. J. Org. Chem. 1995, 60, 4312. Also see, Snyder, J.P.; Tipsword, G.E.; Spangler, D.P. J. Am. Chem. Soc. 1992, 114, 1507.
1439. Bertz, S.H. J. Am. Chem. Soc. 1990, 112, 4031.
1440. Lipshutz, B.H.; James, B. J. Org. Chem. 1994, 59, 7585 and references cited therein.
1441. Dubois, J.E.; Fournier, P.; Lion, C. Bull. Soc. Chim. Fr. 1976, 1871.
1442. See Corey, E.J.; Posner, G.H. J. Am. Chem. Soc. 1967, 89, 3911; Wakselman, C.; Mondon, M. Tetrahedron Lett. 1973, 4285.
1443. Posner, G.H.; Sterling, J.J. J. Am. Chem. Soc. 1973, 95, 3076. See also, Posner, G.H.; Sterling, J.J.; Whitten, C.E.; Lentz, C.M.; Brunelle, D.J. J. Am. Chem. Soc. 1975, 97, 107; Lion, C.; Dubois, J.E. Tetrahedron 1975, 31, 1223. See Lei, X.; Doubleday, Jr., C.; Turro, N.J. Tetrahedron Lett. 1986, 27, 4671.
1444. Prepared by treating CuI with t-BuOLi in THF at 0 °C and adding RLi to this solution.
1445. Nakamura, E.; Sekiya, K.; Arai, M.; Aoki, S. J. Am. Chem. Soc. 1989, 111, 3091.
1446. Marquais, S.; Cahiez, G.; Knochel, P. Synlett, 1994, 849.
1447. Malda, H.; van Zijl, A.W.; Arnold, L.A.; Feringa, B.L. Org. Lett. 2001, 3, 1169.
1448. Yamamoto, Y.; Yamamoto, S.; Yatagai, H.; Maruyama, K. J. Am. Chem. Soc. 1980, 102, 2318. See also, Lipshutz, B.H.; Ellsworth, E.L.; Dimock, S.H. J. Am. Chem. Soc. 1990, 112, 5869.
1449. Kennedy, J.P. J. Org. Chem. 1970, 35, 532. See also, Sato, F.; Kodama, H.; Sato, M. J. Organomet. Chem. 1978, 157, C30.
1450. See Lee, Y.; Akiyama, K.; Gillingham, D.G.; Brown, M.K.; Hoveyda, A.H. J. Am. Chem. Soc. 2008,130, 446.
1451. Negishi, E.; Takahashi, T.; Baba, S.; Van Horn, D.E.; Okukado, N. J. Am. Chem. Soc. 1987, 109, 2393.
1452. Falck, J.R.; Mohaptra, S.; Bondlela, M.; Venkataraman, S.K. Tetrahedron Lett. 2002, 43, 8149.
1453. Reetz, M.T.; Wenderoth, B.; Peter, R.; Steinbach, R.; Westermann, J. J. Chem. Soc., Chem. Commun. 1980, 1202. See also, Klingstedt, T.; Frejd, T. Organometallics 1983, 2, 598.
1454. Bolestova, G.I.; Parnes, Z.N.; Latypova, F.M.; Kursanov, D.N. J. Org. Chem. USSR 1981, 17, 1203.
1455. Reetz, M.T.; Westermann, J.; Steinbach, R. Angew. Chem. Int. Ed. 1980, 19, 900, 901.
1456. Takahashi, H.; Hossain, K.M.; Nishihara, Y.; Shibata, T.; Takagi, K. J. Org. Chem. 2006, 71, 671. See also Sase, S.; Jaric, M.; Metzger, A.; Malakhov, V.; Knochel, P. J. Org. Chem. 2008, 73, 7380.
1457. Sirieix, J.; Oßberger, M.; Betzemeier, B.; Knochel, P. Synlett 2000, 1613.
1458. Kang, S.-K.; Kim, J.-S.; Choi, S.-C. J. Org. Chem. 1997, 62, 4208.
1459. Shen, W.; Wang, L. J. Org. Chem. 1999, 64, 8873.
1460. Fouquet, E.; Rodriguez, A.L. Synlett 1998, 1323.
1461. Kwon, H.B.; McKee, B.H.; Stille, J.K. J. Org. Chem. 1990, 55, 3114. See Stang, P.J.; Kowalski, M.H.; Schiavelli, M.D.; Longford, D. J. Am. Chem. Soc. 1989, 111, 3347; Stang, P.J.; Kowalski, M.H. J. Am. Chem. Soc. 1989, 111, 3356.
1462. Stille, J.K.; Tanaka, M. J. Am. Chem. Soc. 1987, 109, 3785.
1463. Negishi, E. Acc. Chem. Res. 1982, 15, 340; Negishi, E.; Baba, S. J. Chem. Soc., Chem. Commun. 1976, 597; Baba, S.; Negishi, E. J. Am. Chem. Soc. 1976, 98, 6729; King, A.O.; Okukado, N.; Negishi, E. J. Chem. Soc., Chem. Commun. 1977, 683; Negishi, E.; Okukado, N.; King, A. O.; van Horn, D. E.; Spiegel, B. I. J. Am. Chem. Soc. 1978, 100, 2254; Schade, M.A.; Metzger, A.; Hug, S.; Knochel, P. Chem. Commun. 2008, 3046; Phapale, V.B.; Guisán-Ceinos, M.; Buñuel, E.; Cárdenas, D.J. Chemistry: Eur. J. 2009, 15, 12681; Dong, Z.-B.; Manolikakes, G.; Shi, L.; Knochel, P.; Mayr, H. Chemistry: Eur. J. 2010, 16, 248. For insights into the mechanism, see Casares, J.A.; Espinet, P.; Fuentes, B.; Salas, G. J. Am. Chem. Soc. 2007, 129, 3508. For coupling with nonactivated secondary halides, see Glorius, F. Angew. Chem. Int. Ed. 2008, 47, 8347.
1464. Mutule, I.; Suna, E. Tetrahedron 2005, 61, 11168.
1465. Kabir, M.S.; Monte, A.; Cook, J.M. Tetrahedron Lett. 2007, 48, 7269.
1466. Matsubara, S.; Oshima, K.; Matsuoka, H.; Matsumoto, K.; Ishikawa, K.; Matsubara, E. Chem. Lett. 2005, 34, 952.
1467. Coleridge, B.M.; Bello, C.S.; Ellenberger, D.H.; Leitner, A. Tetrahedron Lett. 2010, 51, 357.
1468. Wang, Q.; Chen, C. Tetrahedron Lett. 2008, 49, 2916.
1469. See Hadei, N.; Kantchev, E.A.B.; O'Brien, C.J.; Organ, M.G. J. Org. Chem. 2005, 70, 8503; Andrei, D.; Wnuk, S.F. J. Org. Chem. 2006, 71, 405. For a variation using Pd-nanoparticles, see Liu, J.; Deng, Y.; Wang, H.; Zhang, H.; Yu, G.; Wu, B.; Zhang, H.; Li, Q.; Marder, T.B.; Yang, Z.; Lei, A. Org. Lett. 2008, 10, 2661.
1470. Giovannini, R.; Stüdemann, T.; Devasagayaraj, A.; Dussin, G.; Knochel, P. J. Org. Chem. 1999, 64, 3544; Jensen, A.E.; Knochel, P. J. Org. Chem. 2002, 67, 79; Zhou, J.; Fu, G.C. J. Am. Chem. Soc. 2003, 125, 14726; Terao, J.; Todo, H.; Watanabe, H.; Ikumi, A.; Kambe, N. Angew. Chem. Int. Ed. 2004, 43, 6180.
1471. Shibli, A.; Varghese, J.P.; Knochel, P.; Marek, I. Synlett 2001, 818.
1472. See Fischer, C.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 4594; Arp, F.O.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 10482.
1473. Son, S.; Fu, G.C. J. Am. Chem. Soc. 2008, 130, 2756.
1474. Smith, S.W.; Fu, G.C. Angew. Chem. Int. Ed. 2008, 47, 9334.
1475. Shi, W.J.; Wang, L.-X.; Fu, Y.; Zhu, S.-F.; Zhou, Q.-L. Tetrahedron Asymmetry 2003, 14, 3867.
1476. Hu, J.-b.; Zhao, G.; Yang, G.-s.; Ding, Z.-d. J. Org. Chem. 2001, 66, 303.
1477. Jensen, A.E.; Knochel, P. J. Org. Chem. 2002, 67, 79.
1478. Kraus, G.A.; Ansersh, B.; Su, Q.; Shi, J. Tetrahedron Lett. 1993, 34, 1741.
1479. Berkowitz, W.F.; Wu, Y. Tetrahedron Lett. 1997, 38, 3171.
1480. Rodríguez, D.; Sestelo, J.P.; Sarandeses, L.A. J. Org. Chem. 2003, 68, 2518.
1481. Takami, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2004, 6, 4555.
1482. Baruah, M.; Boruah, A.; Prajapati, D.; Sandu, J.S. Synlett, 1998, 1083.
1483. Cahiez, G.; Marquais, S. Tetrahedron Lett. 1996, 37, 1773.
1484. Kakiya, H.; Inoue, R.; Shinokubo, H.; Oshima, K. Tetrahedron 2000, 56, 2131.
1485. Durandetti, M.; Sibille, S.; Nédélec, J.-Y.; Périchon, J. Synth. Commun. 1994, 24, 145.
1486. Usugi, S.-i.; Yorimitsu, H.; Oshima, K. Tetrahedron Lett. 2001, 42, 4535.
1487. Heck, R.F. J. Am. Chem. Soc. 1968, 90, 5531. See Reaction 13-10. See Heck, R.F. Palladium Reagents in Organic Syntheses, Academic Press, NY, 1985, pp. 208–214, 242–249. Also see Yin, L.; Liebscher, J. Chem. Rev.2007, 107, 133.
1488. See Stille, J.K. Angew. Chem. Int. Ed. 1986, 25, 508; Bumagin, N.A.; Beletskaya, I.P. Russ. Chem. Rev. 1990, 59, 1174. See Martínez, A.G.; Barcina, J.O.; Heras, Md.R.C.; Cerezo, A.d.F. Org. Lett. 2000, 2, 1377
1489. Lee, P.H.; Sung, S.-y.; Lee, K. Org. Lett. 2001, 3, 3201.
1490. Rodríguez, D.; Sestelo, J.P.; Sarandeses, L.A. J. Org. Chem. 2004, 69, 8136.
1491. Denmark, S.E.; Werner, N.S. J. Am. Chem. Soc. 2008, 130, 16382.
1492. Herbert, J.M. Tetrahedron Lett. 2004, 45, 817.
1493. Bentz, E.; Moloney, M.G.; Westaway, S.M. Tetrahedron Lett. 2004, 45, 7395.
1494. Riquet, E.; Alami, M.; Cahiez, G. Tetrahedron Lett. 1997, 38, 4397.
1495. Czaplik, W.M.; Mayer, M.; Jacobi von Wangelin, A. Synlett 2009, 2931.
1496. Frid, M.; Pérez, D.; Peat, A.J.; Buchwald, S.L. J. Am. Chem. Soc. 1999, 121, 9469. See also, Villiers, P.; Vicart, N.; Ramondenc, Y.; Plé, G. Tetrahedron Lett. 1999, 40, 8781.
1497. Sato, A.; Ito, H.; Taguchi, T. J. Org. Chem. 2005, 70, 709.
1498. Saito, B.; Fu, G.C. J. Am. Chem. Soc. 2008, 130, 6694.
1499. Purpura, M.; Krause, N. Eur. J. Org. Chem. 1999, 267.
1500. Goering, H.L.; Kantner, S.S.; Seitz, Jr., E.P. J. Org. Chem. 1985, 50, 5495.
1501. Fleming, I.; Higgins, D.; Lawrence, N.J.; Thomas, A.P. J. Chem. Soc. Perkin Trans. 1 1992, 3331.
1502. Rubin, M.; Gevorgyan, V. Org. Lett. 2001, 3, 2705. See Schwier, T.; Rubin, M.; Gevorgyan, V. Org. Lett. 2004, 6, 1999.
1503. Smith, D.M.; Tran, M.B.; Woerpel, K.A. J. Am. Chem. Soc. 2003, 125, 14149; Ayala, L.; Lucero, C.G.; Romero, J.A.C.; Tabacco, S.A.; Woerpel, K.A. J. Am. Chem. Soc. 2003, 125, 15521.
1504. Roumestant, M.; Gore, J. Bull. Soc. Chim. Fr. 1972, 591, 598; Crabbé, P.; Barreiro, E.; Dollat, J.; Luche, J. J. Chem. Soc., Chem. Commun. 1976, 183, and references cited therein.
1505. Casey, C.P.; Marten, D.F. Tetrahedron Lett. 1974, 925. See also, Posner, G.H.; Brunelle, D.J. J. Chem. Soc., Chem. Commun. 1973, 907; Kobayashi, S.; Takei, H.; Mukaiyama, T. Chem. Lett. 1973, 1097.
1506. Karlström, A.S.E.; Huerta, F.F.; Muezelaar, G.J.; Bäckvall, J.-E. Synlett 2001, 923; Alexakis, A.; Malan, C.; Lea, L.; Benhaim, C.; Fournioux, X. Synlett 2001, 927.
1507. van Klaveren, M.; Persson, E.S.M.; del Villar, A.; Grove, D.M.; Bäckvall, J.-E.; van Koten, G. Tetrahedron Lett. 1995, 36, 3059.
1508. Del Valle, L.; Stille, J.K.; Hegedus, L.S. J. Org. Chem. 1990, 55, 3019. For another method, see Legros, J.; Fiaud, J. Tetrahedron Lett. 1990, 31, 7453.
1509. Sasaoka, S.; Yamamoto, T.; Kinoshita, H.; Inomata, K.; Kotake, H. Chem. Lett. 1985, 315.
1510. Trost, B.M.; Keinan, E. Tetrahedron Lett. 1980, 21, 2595.
1511. Yatsumonji, Y.; Ishida, Y.; Tsubouchi, A.; Takeda, T. Org. Lett. 2007, 9, 4603.
1512. Mandal, S.K.; Paira, M.; Roy, S.C. J. Org. Chem. 2008, 73, 3823.
1513. Spiess, S.; Welter, C.; Franck, G.; Taquet, J.-P.; Helmchen, G. Angew. Chem. Int. Ed. 2008, 47, 7652.
1514. Plietker, B. Angew. Chem. Int. Ed. 2006, 45, 6053.
1515. Gomes, P.; Gosmini, C.; Périchon, J. Org. Lett. 2003, 5, 1043.
1516. Belelie, J.L.; Chong, J.M. J. Org. Chem. 2001, 66, 5552.
1517. Kacprzynski, M.A.; Hoveyda, A.H. J. Am. Chem. Soc. 2004, 126, 10676.
1518. See Tsuji, J. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 3, Wiley, NY, 1985, pp. 163–199.
1519. See Trost, B.M.; Crawley, M.L. Chem. Rev. 2003, 103, 2921. For a discussion of the mechanism see Tsurugi, K.; Nomura, N.; Aoi, K. Tetrahedron Lett. 2002, 43, 469.
1520. See Mohr, J.T.; Stoltz, B.M. Chemistry: Asian J. 2007, 2, 1476; Trost, B.M. Angew. Chem. Int. Ed. 1989, 28, 1173; Acc. Chem. Res. 1980, 13, 385; Tsuji, J.; Minami, I. Acc. Chem. Res. 1987, 20, 140, Organic Synthesis with Palladium Compounds, Springer, Berlin, 1981, pp. 45, 125; Heck, R.F. Palladium Reagents in Organic Synthesis, Academic Press, NY, 1985, pp. 130–166; Hegedus, L.S. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, Vol. 5, pt. B, Elsevier, NY, 1984, pp. 30-44. See Liao, M.-c.; Duan, X.-h.; Liang, Y.-m. Tetrahedron Lett. 2005, 46, 3469.
1521. Trost, B.M.; Toste, F.D. J. Am. Chem. Soc. 1999, 121, 4545.
1522. For example, see Liu, D.; Xie, F.; Zhang, W. Tetrahedron Lett. 2007, 48, 585; Guimet, E.; Diéguez, M.; Ruiz, A.; Claver, C. Tetrahedron Asymmetry 2005, 16, 959; Mikhael, I.; Goux-Henry, C.; Sinou, D. Tetrahedron Asymmetry 2006, 17, 1853; Ruzziconi, R.; Santi, C.; Spizzichino, S. Tetrahedron Asymmetry 2007, 18, 1742; Wang, Q.-F.; He, W.; Liu, X.-Y.; Chen, H.; Qin, X.-Y.; Zhang, S.-Y. Tetrahedron Asymmetry 2008, 19, 2447; Polet, D.; Alexakis, A.; Tissot-Croset, K.; Corminboeuf, C.; Ditrich, K. Chem. Eur. J. 2006, 12, 3596. Also see Mino, T.; Sato, Y.; Saito, A.; Tanaka, Y.; Saotome, H.; Sakamoto, M.; Fujita, T. J. Org. Chem. 2005, 70, 7979.
1523. Poli, G.; Giambastiani, G.; Mordini, A. J. Org. Chem. 1999, 64, 2962.
1524. Trost, B.M.; Weber, L.; Strege, P.E.; Fullerton, T.J.; Dietsche, T.J. J. Am. Chem. Soc. 1978, 100, 3416, 3426. These papers include a discussion of the mechanism of this reaction.
1525. Braun, M.; Meier, T. Angew. Chem. Int. Ed. 2006, 45, 6952.
1526. Manchand, P.S.; Wong, H.S.; Blount, J.F. J. Org. Chem. 1978, 43, 4769.
1527. Nakoji, M.; Kanayama, T.; Okino, T.; Takemoto, Y. Org. Lett. 2001, 3, 3329.
1528. Braun, M.; Laicher, F.; Meier, T. Angew. Chem. Int. Ed. 2000, 39, 3494.
1529. NaN(CHO)2: Wang, Y.; Ding, K. J. Org. Chem. 2001, 66, 3238. Indene: Hayashi, T.; Suzuka, T.; Okada, A.; Kawatsura, M. Tetrahedron Asymmetry 2004, 15, 545.
1530. See Chen, W.; Xu, L.; Chatterton, C.; Xiao, J. Chem. Commun. 1999, 1247.
1531. Sato, Y.; Yoshino, T.; Mori, M. Org. Lett. 2003, 5, 31.
1532. Jansat, S.; Gómez, M.; Philippot, K.; Muller, G.; Guiu, E.; Claver, C.; Castillón, S.; Chaudret, B. J. Am. Chem. Soc., 2004, 126, 1592.
1533. Falciola, C.A.; Tissot-Croset, K.; Alexakis, A. Angew. Chem. Int. Ed. 2006, 45, 5995.
1534. Krafft, M.E.; Sugiura, M.; Abboud, K.A. J. Am. Chem. Soc. 2001, 123, 9174.
1535. Ir: Kinoshita, N.; Marx, K.H.; Tanaka, K.; Tsubaki, K.; Kawabata, T.; Yoshikai, N.; Nakamura, E.; Fuji, K. J. Org. Chem. 2004, 69, 7960. Pt: Blacker, A.J.; Clarke, M.L.; Loft, M.S.; Mahon, M.F.; Humphries, M.E.; Williams, J.M.J. Chem. Eur. J. 2000, 6, 353. Ru: Renaud, J.-L.; Bruneau, C.; Demerseman, B. Synlett 2003, 408.
1536. See Boaz, N.W.; Ponaskik Jr., J.A.; Large, S.E.; Debenham, S.D. Tetrahedron Asymmetry 2004, 15, 2151.
1537. Molander, G.A.; Burke, J.P.; Carroll, P.J. J. Org. Chem. 2004, 69, 8062; Kloetzing, R.J.; Lotz, M.; Knochel, P. Tetrahedron Asymmetry 2003, 14, 255; Nakano, H.; Yokayama, J.-i.; Koiyama, Y.; Fjita, R.; Hongo, H. Tetrahedron Asymmetry 2003, 14, 2361; Mercier, F.; Brebion, F.; Dupont, R.; Mathey, F. Tetrahedron Asymmetry 2003, 14, 3137.
1538. See Consiglio, G.; Waymouth, R.M. Chem. Rev. 1989, 89, 257.
1539. See Ito, K.; Kashiwagi, R.; Hayashi, S.; Uchida, T.; Katsuki, T. Synlett 2001, 284.
1540. Hamada, Y.; Sakaguchi, K.-e.; Hatano, K.; Hara, O. Tetrahedron Lett. 2001, 42, 1297.
1541. Kuwano, R.; Kondo, Y.; Matsuyama, Y. J. Am. Chem. Soc. 2003, 125, 12104; Faller, J.W.; Wilt, J.C. Tetrahedron Lett. 2004, 45, 7613.
1542. Amide esters: Kazmaieer, U.; Zumpe, F.L. Angew. Chem. Int. Ed. 1999, 38, 1468.
1543. Evans, P.A.; Lawler, M.J. J. Am. Chem. Soc. 2004, 126, 8642. For a reaction is a silyl enol ether see Muraoka, T.; Matsuda, I.; Itoh, K. Tetrahedron Lett. 2000, 41, 8807.
1544. Aryllithium reagents: Evans, P.A.; Uraguchi, D. J. Am. Chem. Soc. 2003, 125, 7158. Alkoxides: Evans, P.A.; Leahy, D.K.; Slieker, L.M. Tetrahedron Asymmetry 2003, 14, 3613. Phenoxide anions: Evans, P.A.; Leahy, D.K. J. Am. Chem. Soc. 2000, 122, 5012; López, F.; Ohmura, T.; Hartwig, J.F. J. Am. Chem. Soc. 2003, 125, 3426. Secondary amines: Matsushima, Y.; Onitsuka, K.; Kondo, T.; Mitsudo, T.-a.; Takahashi, S. J. Am. Chem. Soc.2001, 123, 10405. Primary amines: Ohmura, T.; Hartwig, J.F. J. Am. Chem. Soc. 2002, 124, 15164. N-Lithio-sulfonamides: Evans, P.A.; Robinson, J.E.; Baum, E.W.; Fazal, A.N. J. Am. Chem. Soc. 2002, 124, 8782. C-Alkylation with an indole: Bandini, M.; Melloni, A.; Umani-Ronchi, A. Org. Lett. 2004, 6, 3199. Michael addition of conjugated esters: Muraoka, T.; Matsuda, I.; Itoh, K. J. Am. Chem. Soc. 2000, 12, 9552.
1545. Uozumi, Y.; Shibatmoi, K. J. Am. Chem. Soc. 2001, 123, 2919.
1546. Ru: Trost, B.M.; Fraisse, P.L.; Ball, Z.T. Angew. Chem. Int. Ed. 2002, 41, 1059. Mo: Glorius, F.; Pfaltz, A. Org. Lett. 1999, 1, 141; Malkov, A.V.; Spoor, P.; Vinader, V.; Kocovsky, P. Tetrahedron Lett. 2001, 42, 509. Ir: Alexakis, A.; Polet, D. Org. Lett. 2004, 6, 3529; Lee, P.H.; Sung, S.-y.; Lee, K.; Chang, S. Synlett 2002, 146.
1547. Kabalka, G.W.; Al-Masum, M. Org. Lett. 2006, 8, 11.
1548. Castaño, A.M.; Méndez, M.; Ruano, M.; Echavarren, A.M. J. Org. Chem. 2001, 66, 589. See also, Zhang, Q.; Lu, X.; Han, X. J. Org. Chem. 2001, 66, 7676.
1549. Riveiros, R.; Rodríguez, D.; Sestelo, J.P.; Sarandeses, L.A. Org. Lett. 2006, 8, 1403.
1550. Hu, Y.; Chen, Z.-C.; Le, Z.-G.; Zheng, Q.G. Synth. Commun. 2004, 34, 4031.
1551. Trost, B.M.; Schmuff, N.R.; Miller, M.J. J. Am. Chem. Soc. 1980, 102, 5979.
1552. See Kornblum, N. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 1, Wiley, NY, 1982, pp. 361–393; Kornblum, N. Angew. Chem. Int. Ed. 1975, 14, 734; Tamura, R.; Kamimura, A.; Ono, N. Synthesis1991, 423; Kornblum, N. in Feuer, H.; Nielsen, A.T. Nitro Compounds: Recent Advances in Synthesis and Chemisry, VCH, NY, 1990, pp. 46–85.
1553. Kornblum, N.; Kelly, W.J.; Kestner, M.M. J. Org. Chem. 1985, 50, 4720.
1554. Kornblum, N.; Erickson, A.S. J. Org. Chem. 1981, 46, 1037.
1555. Kornblum, N.; Carlson, S.C.; Widmer, J.; Fifolt, M.J.; Newton, B.N.; Smith, R.G. J. Org. Chem. 1978, 43, 1394.
1556. For a review of the mechanism, see Beletskaya, I.P.; Drozd, V.N. Russ. Chem. Rev. 1979, 48, 431. See also, Kornblum, N.; Wade, P.A. J. Org. Chem. 1987, 52, 5301; Bowman, W.R. Chem. Soc. Rev. 1988, 17, 283; Ref. 1479.
1557. Kornblum, N.; Boyd, S.D.; Ono, N. J. Am. Chem. Soc. 1974, 96, 2580.
1558. Trost, B.M.; Merlic, C.A. J. Org. Chem. 1990, 55, 1127.
1559. Jin, L.; Julia, M.; Verpeaux, J.N. Synlett 1994, 215.
1560. Casey, M.; Manage, A.C.; Murphy, P.J. Tetrahedron Lett. 1992, 33, 965.
1561. Dahmen, S.; Bräse, S. J. Am. Chem. Soc. 2002, 124, 5940.
1562. Yanagisawa, A.; Hibino, H.; Nomura, N.; Yamamoto, H. J. Am. Chem. Soc. 1993, 115, 5879.
1563. Bruylants, P. Bull. Soc. Chem. Belg. 1924, 33, 467.
1564. Trost, B.M.; Spagnol, M.D. J. Chem. Soc., Perkin Trans. 1 1995, 2083.
1565. Agami, C.; Couty, F.; Evano, G. Org. Lett. 2000, 2, 2085.
1566. Katritzky, A.R.; Yang, H.; Singh, S.K. J. Org. Chem. 2005, 70, 286.
1567. Zhu, J.-L.; Shia, K.-S.; Liu, H.-J. Tetrahedron Lett. 1999, 40, 7055.
1568. Bernardi, L.; Bonini, B.F.; Capitò, E.; Dessole, G.; Fochi, M.; Comes-Franchini, M.; Ricci, A. Synlett 2003, 1778.
1569. For a review of Pd catalyzed reactions, see Muzart, J. Tetrahedron 2005, 61, 4179.
1570. Kulinkovich, O.G.; Epstein, O.L.; Isakov, V.E.; Khmel'nitskaya, E.A. Synlett 2001, 49.
1571. Kabalka, G.W.; Dong, G.; Venkataiah, B. Org. Lett. 2003, 5, 893.
1572. Kinoshita, H.; Shinokubo, H.; Oshima, K. Org. Lett. 2004, 6, 4085.
1573. Bisaro, F.; Prestat, G.; Vitale, M.; Poli, G. Synlett 2002, 1823.
1574. Cho, C.S.; Kim, B.T.; Kim. T.-J.; Shim. S.C. J. Org. Chem. 2001, 66, 9020. See also, Morita, M.; Obora, Y.; Ishii, Y. Chem. Commun. 2007, 2850.
1575. Bower, J.F.; Skucas, E.; Patman, R.L.; Krische, M.J. J. Am. Chem. Soc. 2007, 129, 15134.
1576. Sumida, Y.; Hayashi, S.; Hirano, K.; Hideki, H.; Oshima, K. Org. Lett. 2008, 10, 1629.
1577. See Lai, Y. Org. Prep. Proceed. Int. 1980, 12, 363, pp. 377–388.
1578. Sharpless, K.B.; Hanzlik, R.P.; van Tamelen, E.E. J. Am. Chem. Soc. 1968, 90, 209.
1579. McMurry, J.E.; Silvestri, M.G.; Fleming, M.P.; Hoz, T.; Grayston, M.W. J. Org. Chem. 1978, 43, 3249. For another method, see Nakanishi, S.; Shundo, T.; Nishibuchi, T.; Otsuji, Y. Chem. Lett. 1979, 955.
1580. van Tamelen, E.E.; Åkermark, B.; Sharpless, K.B. J. Am. Chem. Soc. 1969, 91, 1552.
1581. Walborsky, H.M.; Murati, M.P. J. Am. Chem. Soc. 1980, 102, 426.
1582. Harney, D.W.; Meisters, A.; Mole, T. Aust. J. Chem. 1974, 27, 1639.
1583. Salomon, R.G.; Kochi, J.K. J. Org. Chem. 1973, 38, 3715.
1584. Reetz, M.T.; Westermann, J.; Steinbach, R. J. Chem. Soc., Chem. Commun. 1981, 237.
1585. Fujita, K.; Asai, C.; Yamaguchi, T.; Hanasaka, F.; Yamaguchi, R. Org. Lett. 2005, 7, 4017.
1586. Goering, H.L.; Tseng, C.C. J. Org. Chem. 1985, 50, 1597. For another procedure, see Yamamoto, Y.; Maruyama, K. J. Organomet. Chem. 1978, 156, C9.
1587. See Cella, J.A. J. Org. Chem. 1982, 47, 2125.
1588. Consiglio, G.; Morandini, F.; Piccolo, O. J. Am. Chem. Soc. 1981, 103, 1846, and references cited therein. See Felkin, H.; Swierczewski, G. Tetrahedron 1975, 31, 2735; Fujisawa, T.; Iida, S.; Yukizaki, H.; Sato, T. Tetrahedron Lett. 1983, 24, 5745.
1589. Araki, S.; Usui, H.; Kato, M.; Butsugan, Y. J. Am. Chem. Soc, 1996, 118, 4699.
1590. Bernardelli, P.; Paquette, L.A. J. Org. Chem. 1997, 62, 8284.
1591. McKinley, N.F.; O'Shea, D.F. J. Org. Chem. 2004, 69, 5087.
1592. Saito, T.; Nishimoto, Y.; Yasuda, M.; Baba, A. J. Org. Chem. 2006, 71, 8516; Yasuda, M.; Somyo, T.; Baba, A. Angew. Chem. Int. Ed. 2006, 45, 793.
1593. Kim, S.H.; Shin, C.; Pae, A.N.; Koh, H.Y.; Chang, M.H.; Chung, B.Y.; Cho, Y.S. Synthesis 2004, 1581.
1594. Saito, T.; Nishimoto, Y.; Yasuda, M.; Baba, A. J. Org. Chem. 2007, 72, 8588.
1595. Georgy, M.; Boucard, V.; Campagne, J.-M. J. Am. Chem. Soc. 2005, 127, 14180.
1596. Funayama, A.; Satoh, T.; Miura, M. J. Am. Chem. Soc. 2005, 127, 15354.
1597. See Trofimov, B.A.; Korostova, S.E. Russ. Chem. Rev. 1975, 44, 41.
1598. See Mukaiyama, T.; Murakami, M. Synthesis 1987, 1043; Abell, A.D.; Massy-Westropp, R.A. Aust. J. Chem. 1985, 38, 1031. For a list of substrates and reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 934–942.
1599. See DeWolfe, R.H. Carboxylic Ortho Acid Derivatives, Academic Press, NY, 1970, pp. 44–45, 224–230.
1600. Normant, J.F.; Alexakis, A.; Ghribi, A.; Mangeney, P. Tetrahedron 1989, 45, 507; Alexakis, A.; Mangeney, P.; Ghribi, A.; Marek, I.; Sedrani, R.; Guir, C.; Normant, J.F. Pure Appl. Chem. 1988, 60, 49.
1601. Ducoux, J.-P.; LeMénez, P.; Kunesch, N.; Wenkert, E. J. Org. Chem. 1993, 58, 1290.
1602. See Mori, I.; Ishihara, K.; Flippin, L.A.; Nozaki, K.; Yamamoto, H.; Bartlett, P.A.; Heathcock, C.H. J. Org. Chem. 1990, 55, 6107, and references cited therein.
1603. Wei, Z.Y.; Knaus, E.E. Org. Prep. Proceed. Int. 1993, 25, 255.
1604. See Mesnard, D.; Miginiac, L. J. Organomet. Chem. 1989, 373, 1. See also, Bourhis, M.; Bosc, J.; Golse, R. J. Organomet. Chem. 1983, 256, 193.
1605. Germon, C.; Alexakis, A.; Normant, J.F. Bull. Soc. Chim. Fr. 1984, II-377.
1606. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 1013–1045.
1607. Guan, B.-T.; Xiang, S.-K.; Wang, B.-Q.; Sun, Z.-P.; Wang, Y.; Zhao, K.-Q.; Shi, Z.-J. J. Am. Chem. Soc. 2008,130, 3268.
1608. Bach, T.; Eilers, F. Eur. J. Org. Chem. 1998, 2161.
1609. Rama, K.; Pasha, M.A. Tetrahedron Lett. 2000, 41, 1073.
1610. Commercon, A.; Bourgain, M.; Delaumeny, M.; Normant, J.F.; Villieras, J. Tetrahedron Lett. 1975, 3837; Claesson, A.; Olsson, L. J. Chem. Soc., Chem. Commun. 1987, 621.
1611. Calo, V.; Lopez, L.; Pesce, G. J. Chem. Soc. Perkin Trans. 1 1988, 1301. See also, Valverde, S.; Bernabé, M.; Garcia-Ochoa, S.; Gómez, A.M. J. Org. Chem. 1990, 55, 2294.
1612. Alexakis, A.; Marek, I.; Mangeney, P.; Normant, J.F. J. Am. Chem. Soc. 1990, 112, 8042.
1613. Kocienski, P.; Dixon, N.J.; Wadman, S. Tetrahedron Lett. 1988, 29, 2353.
1614. Hayashi, T.; Katsuro, Y.; Kumada, M. Tetrahedron Lett. 1980, 21, 3915.
1615. Lauens, M.; Renaud, J.-L.; Hiebert, S. J. Am. Chem. Soc. 200, 122, 1804.
1616. Ishikawa, H.; Mukaiyama, T. Bull. Chem. Soc. Jpn. 1978, 51, 2059.
1617. See Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 961–1012; Schaap, A.; Arens, J.F. Recl. Trav. Chim. Pays-Bas 1968, 87, 1249. Also see Schrumpf, G.; Grätz, W.; Meinecke, A.; Fellenberger, K. J. Chem. Res. (S) 1982, 162.
1618. Likhar, P.R.; Kumar, M.P.; Bandyopadhyay, A.K. Tetrahedron Lett. 2002, 43, 3333.
1619. Hodgson, D.M.; Stent, M.A.H.; Stefane, B.; Wilson, F.X. Org. Biomol. Chem. 2003, 1, 1139; Hodgson, D.M.; Maxwell, C.R.; Miles, T.J.; Paruch, E.; Stent, M.A.H.; Matthews, I.R.; Wilson, F.X.; Witherington, J. Angew. Chem. Int. Ed. 2002, 41, 4313.
1620. Oguni, N.; Miyagi, Y.; Itoh, K. Tetrahedron Lett. 1998, 39, 9023.
1621. See Posner, G.H. An Introduction to Synthesis Using Organocopper Reagents, Wiley, NY, 1980, pp. 103–113. See also, Lipshutz, B.H.; Kozlowski, J.; Wilhelm, R.S. J. Am. Chem. Soc. 1982, 104, 2305; Blanchot-Courtois, V.; Hanna, I. Tetrahedron Lett. 1992, 33, 8087.
1622. Chauret, D.C.; Chong, J.M. Tetrahedron Lett. 1993, 34, 3695.
1623. Chong, J.M.; Cyr, D.R.; Mar, E.K. Tetrahedron Lett. 1987, 28, 5009; Larchevêque, M.; Petit, Y. Tetrahedron Lett. 1987, 28, 1993.
1624. See Alexakis, A.; Jachiet, D.; Normant, J.F. Tetrahedron 1986, 42, 5607.
1625. Yamamoto, Y.; Asao, N.; Meguro, M.; Tsukada, N.; Nemoto, H.; Sadayori, N.; Wilson, J.G.; Nakamura, H. J. Chem. Soc., Chem. Commun. 1993, 1201.
1626. See Wardell, J.L.; Paterson, E.S. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 307–310; Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1045–1063. Ba: Yasue, K.; Yanagisawa, A.; Yamamoto, H. Bull. Chem. Soc. Jpn. 1997, 70, 493. Mn: Tang, J.; Yorimitsu, H.; Kakiya, H.; Inoue, R.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 1997, 38, 9019. Sn: Yadav, J.S.; Reddy, B.V.S.; Satheesh, G. Tetahedron Lett. 2003, 44, 6501. Zn: Equey, O.; Vrancken, E.; Alexakis, A. Eur. J. Org. Chem. 2004, 2151
1627. Schneider, C.; Brauner, J. Eur. J. Org. Chem. 2001, 4445; Sasaki, M.; Tanino, K.; Miyashita, M. J. Org. Chem. 2001, 66, 5388; Sasaki, M.; Tanino, K.; Miyashita, M. Org. Lett. 2001, 3, 1765; Shanmugam, P.; Miyashita, M. Org. Lett. 2003, 5, 3265 (formation of O-silyl ether product). For the reaction in an ionic liquid see Zhou, H.; Campbell, E.J.; Nguyen, S.T. Org. Lett. 2001, 3, 2229.
1628. See Zhao, H.; Pagenkopf, B.L. Chem. Commun. 2003, 2592.
1629. Lin, J.; Kanazaki, S.; Kashino, S.; Tsuboi, S. Synlett 2002, 899.
1630. Hirashita, T.; Mitsui, K.; Hayashi, Y.; Araki, S. Tetrahedron Lett. 2004, 45, 9189. For a reaction using Pd nanoparticles, see Jiang, N.; Hu, Q.; Reid, C.S.; Ou, Y.; Li, C.J. Chem. Commun. 2003, 2318.
1631. Lee, J.T.; Thomas, P.J.; Apler, H. J. Org. Chem. 2001, 66, 5424.
1632. Schmidt, J.A.R.; Mahadevan, V.; Getzler, Y.D.Y.L.; Coates, G.W. Org. Lett. 2004, 6, 373.
1633. Rowley, J.M.; Lobkovsky, E.B.; Coates, G.W. J. Am. Chem. Soc. 2007, 129, 4948.
1634. Movassaghi, M.; Jacobsen, E.N. J. Am. Chem. Soc. 2002, 124, 2456.
1635. Tanaka, T.; Hiramatsu, K.; Kobayashi, Y.; Ohno, H. Tetrahedron 2005, 61, 6726.
1636. Albert, B.J.; Koide, K. J. Org. Chem. 2008, 73, 1093.
1637. Gohain, M.; Prajapati, D. Chem. Lett. 2005, 34, 90.
1638. Lautens, M.; Maddess, M.L.; Sauer, E.L.O.; Oullet, S.G. Org. Lett. 2002, 4, 83.
1639. For a list of organometallic reagents that react with vinylic epoxides, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 244–250.
1640. Marshall, J.A.; Trometer, J.D.; Cleary, D.G. Tetrahedron 1989, 45, 391.
1641. Ueki, H.; Chiba, T.; Yamazaki, T.; Kitazume, T. J. Org. Chem. 2004, 69, 7616.
1642. See Marshall, J.A. Chem. Rev. 1989, 89, 1503.
1643. Wender, P.A.; Erhardt, J.M.; Letendre, L.J. J. Am. Chem. Soc. 1981, 103, 2114.
1644. Murai, T.; Kato, S.; Murai, T.; Toki, T.; Suzuki, S.; Sonoda, N. J. Am. Chem. Soc. 1984, 106, 6093.
1645. Lalic, G.; Petrovski, Z.; Galonic, D.; Matovic, R.; Saicic, R.N. Tetrahedron 2001, 57, 583.
1646. See Onistschenko, A.; Buchholz, B.; Stamm, H. Tetrahedron 1987, 43, 565.
1647. Crotti, P.; Favero, L.; Gardelli, C.; Macchia, F.; Pineschi, M. J. Org. Chem. 1995, 60, 2514.
1648. Müller, P.; Nury, P. Org. Lett. 1999, 1, 439; Müller, P.; Nury, P. Helv. Chim. Acta 2001, 84, 662.
1649. Penkett, C.S.; Simpson, I.D. Tetrahedron Lett. 2001, 42, 1179.
1650. Saidi, M.R.; Azizi, N.; Naimi-Jamal, M.R. Tetrahedron Lett. 2001, 42, 8111.
1651. Lygo, B. Synlett 1993, 764.
1652. Nadir, U.K.; Arora, A. J. Chem. Soc. Perkin Trans. 1 1995, 2605.
1653. Yadav, J.S.; Reddy, B.V.S.; Balanarsaiah, E.; Raghavendra, S. Tetrahedron Lett. 2002, 43, 5105.
1654. Bera, M.; Roy, S. Tetrahedron Lett. 2007, 48, 7144.
1655. Moss, T.A.; Fenwick, D.R.; Dixon, D.J. J. Am. Chem. Soc. 2008, 130, 10076.
1656. Yadav, J.S.; Subba Reddy, B.V.; Kumar, G.M. Synlett 2001, 1417.
1657. Wu, J.; Hou, X.-L.; Dai, L.-X. J. Org. Chem. 2000, 65, 1344.
1658. Li, Z.; Fernández, M.; Jacobsen, E.N. Org. Lett. 1999, 1, 1611.
1659. Fan, R.-H.; Hou, X.-L. Tetrahedron Lett. 2003, 44, 4411.
1660. Minakata, S.; Okada, Y.; Oderaotoshi, Y.; Komatsu, M. Org. Lett. 2005, 7, 3509. See also Matsukawa, S.; Tsukamoto, K. Org. Biomol. Chem. 2009, 7, 3792.
1661. For dicussions of Reactions 10-67 and 10-68, see House, H.O. Modern Synthetic Reactions, 2nd ed., W. A. Benjamin, NY, 1972, pp. 492–570, 586–595; Carruthers, W. Some Modern Methods of Organic Synthesis 3rd ed., Cambridge University Press, Cambridge, 1986, pp. 1–26.
1662. See Christoffers, J. Synth. Commun. 1999, 29, 117.
1663. See Fatiadi, A.J. Synthesis 1978, 165, 241; Freeman, F. Chem. Rev. 1969, 69, 591.
1664. See Neplyuev, V.M.; Bazarova, I.M.; Lozinskii, M.O. Russ. Chem. Rev. 1986, 55, 883.
1665. For lists of examples, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1522–1527 ff, 1765–1769.
1666. See Fedorynski, M.; Wojciechowski, K.; Matacz, Z.; Makosza, M. J. Org. Chem. 1978, 43, 4682.
1667. Murphy, W.S.; Hamrick, Jr., P.J.; Hauser, C.R. Org. Synth. V. 523.
1668. Zaugg, H.E.; Dunnigan, D.A.; Michaels, R.J.; Swett, L.R.; Wang, T.S.; Sommers, A.H.; DeNet, R.W. J. Org. Chem. 1961, 26, 644; Johnstone, R.A.W.; Tuli, D.; Rose, M.E. J. Chem. Res. (S) 1980, 283.
1669. See Tundo, P.; Venturello, P.; Angeletti, E. J. Chem. Soc. Perkin Trans. 1 1987, 2159.
1670. Park, E.J.; Kim, M.H.; Kim, D.Y. J. Org. Chem. 2004, 69, 6897.
1671. Bunce, R.A.; Burns, S.E. Org. Prep. Proceed. Int. 1999, 31, 99.
1672. Akiyama, R.; Kobayashi, S. J. Am. Chem. Soc. 2003, 125, 3412.
1673. Bhar, S.; Chaudhuri, S.K.; Sahu, S.G.; Panja, C. Tetrahedron 2001, 57, 9011.
1674. See Trost, B.M. Chem. Rev. 1978, 78, 363; Solladié, G. Synthesis 1981, 185.
1675. Gassman, P.G.; Richmond, G.D. J. Org. Chem. 1966, 31, 2355; Kuwajima, I.; Iwasawa, H. Tetrahedron Lett. 1974, 107.
1676. Kurth, M.J.; O'Brien, M.J. J. Org. Chem. 1985, 3846.
1677. Lamm, B.; Samuelsson, B. Acta Chem. Scand. 1969, 23, 691.
1678. Enders, D.; Harnying, W.; Vignola, N. Eur. J. Org. Chem. 2003, 3939.
1679. Corey, E.J.; Cane, D.E. J. Org. Chem. 1970, 35, 3405.
1680. Kanemasa, S.; Mori, T.; Wada, E.; Tatsukawa, A. Tetrahedron Lett. 1993, 34, 677. See Kotha, S.; Kuki, A. Tetrahedron Lett. 1992, 33, 1565 for a related reaction.
1681. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1540–1541. Also see, Lu, Y.-Q.; Li, C.-J. Tetrahedron Lett. 1996, 37, 471.
1682. Facchetti, A.; Streitwieser, A. J. Org. Chem. 2004, 69, 8345.
1683. Zefirov, N.S.; Kuznetsova, T.S.; Kozhushkov, S.I.; Surmina, L.S.; Rashchupkina, Z.A. J. Org. Chem. USSR 1983, 19, 474.
1684. See Walborsky, H.M.; Murari, M.P. Can. J. Chem. 1984, 62, 2464; Bose, A.K.; Manhas, M.S.; Chatterjee, B.G.; Abdulla, R.F. Synth. Commun. 1971, 1, 51. For a list of examples, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 156–157, 165–166.
1685. Deslongchamps, P.; Lamothe, S.; Lin, H. Can. J. Chem. 1987, 65, 1298; Brillon, D.; Deslongchamps, P. Can. J. Chem. 1987, 65, 43, 56.
1686. These SET mechanisms are often called SRN1 mechanisms. See also, Ref. 107.
1687. Bordwell, F.G.; Harrelson, Jr., J.A. J. Am. Chem. Soc. 1989, 111, 1052.
1688. For a review of mechanisms with these nucleophiles, see Bowman, W.R. Chem. Soc. Rev. 1988, 17, 283.
1689. Kornblum, N.; Fifolt, M. Tetrahedron 1989, 45, 1311.
1690. See Crimmins, T.F.; Hauser, C.R. J. Org. Chem. 1967, 32, 2615; Boldt, P.; Militzer, H.; Thielecke, W.; Schulz, L. Liebigs Ann. Chem. 1968, 718, 101.
1691. Boldt, P.; Ludwieg, A.; Militzer, H. Chem. Ber. 1970, 103, 1312.
1692. Katritzky, A.R.; Kashmiri, M.A.; Wittmann, D.K. Tetrahedron 1984, 40, 1501.
1693. Katritzky, A.R.; Chen, J.; Marson, C.M.; Maia, A.; Kashmiri, M.A. Tetrahedron 1986, 42, 101.
1694. See Caine, D. in Augustine, R.L. Carbon–Carbon Bond Formation, Vol. 1; Marcel Dekker, NY, 1979, pp. 85–352.
1695. See Arseniyadis, S.; Kyler, K.S.; Watt, D.S. Org. React. 1984, 31, 1. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1801–1808. See Taber, D.F.; Kong, S. J. Org. Chem. 1997, 62, 8575; Rojas, G.; Baughman, T.W.; Wagener, K.B. Synth. Commun. 2007, 37, 3923.
1696. See Petragnani, N.; Yonashiro, M. Synthesis 1982, 521. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1724–1758ff.
1697. For a list of some bases, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 1476–1479.
1698. See Klusener, P.A.A.; Brandsma, L.; Verkruijsse, H.D.; Schleyer, P.v.R.; Friedl, T.; Pi, R. Angew. Chem. Int. Ed. 1986, 25, 465.
1699. Rathke, M.W.; Lindert, A. J. Am. Chem. Soc. 1971, 93, 2319; Bos, W.; Pabon, H.J.J. Recl. Trav. Chim. Pays-Bas 1980, 99, 141. See also, Cregge, R.J.; Herrmann, J.L.; Lee, C.S.; Richman, J.E.; Schlessinger, R.H. Tetrahedron Lett. 1973, 2425.
1700. Watt, D.S. Tetrahedron Lett. 1974, 707.
1701. See Comins, D.L.; Killpack, M.O. J. Org. Chem. 1987, 52, 104. See Xie, L.; Isenberger, K.M.; Held, G.; Dahl, M. J. Org. Chem. 1997, 62, 7516 for steric versus electronic effects in kinetic enolate formation.
1702. Liou, L.R.; McNeil, A.J.; Toombes, G.E.S.; Collum, D.B. J. Am. Chem. Soc. 2008, 130, 17334; Khartabil, H.K.; Gros, P.C.; Fort, Y.; Ruiz-López, M.F. J. Org. Chem. 2008, 73, 9393; Pratt, L.M.; Mu, R.; Carter, C.; Woodford, B. Tetrahedron 2007, 63, 1331. See also Pratt, L.M.; Nguyen, S.C.; Thanh, B.T. J. Org. Chem. 2008, 73, 6086.
1703. Sun, X.; Kenkre, S.L.; Remenar, J.F.; Gilchrist, J.H. J. Am. Chem. Soc. 1997, 119, 4765.
1704. Majewski, M.; Nowak, P. Tetrahedron Lett. 1998, 39, 1661.
1705. Langhals, E.; Langhals, H. Tetrahedron Lett. 1990, 31, 859.
1706. See Ibrahim-Ouali, M.; Parrain, J.-L.; Santelli, M. Org. Prep. Proceed. Int. 1999, 31, 467. Enolate anions of β-lactones are subject to ring opening: see Mori, S.; Shindo, M. Org. Lett. 2004, 6, 3945.
1707. Matsuo, J.-i.; Kobayashi, S.; Koga, K. Tetrahedron Lett. 1998, 39, 9723.
1708. See Makosza, M. Russ. Chem. Rev. 1977, 46, 1151; Pure Appl. Chem. 1975, 43, 439; Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Acaemic Press, NY, 1978, pp. 170–217; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 136–204.
1709. Ooi, T.; Takeuchi, M.; Kato, D.; Uematsu, Y.; Tayama, E.; Sakai, D.; Maruoka, K. J. Am. Chem. Soc. 2005, 127, 5073.
1710. Artaud, I.; Torossian, G.; Viout, P. Tetrahedron 1985, 41, 5031.
1711. Purohit, V.G.; Subramanian, R. Chem. Ind. (London) 1978, 731; Buschmann, E.; Zeeh, B. Liebigs Ann. Chem. 1979, 1585.
1712. Zook, H.D.; Kelly, W.L.; Posey, I.Y. J. Org. Chem. 1968, 33, 3477.
1713. For a list of alkylations of silyl enol ethers, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1494–1505.
1714. Kang, S.-K.; Ryu, H.-C.; Hong, Y.-T. J. Chem. Soc., Perkin Trans. 1 2000, 3350. For a review, see Reetz, M.T. Angew. Chem. Int. Ed. 1982, 21, 96.
1715. Hirano, K.; Fujita, K.; Yorimitsu, H.; Shinokubo, H.; Oshima, K. Tetrahedron Lett. 2004, 45, 2555.
1716. Yasuda, M.; Tsuji, S.; Shigeyoshi, Y.; Baba, A. J. Am. Chem Soc. 2002, 124, 7440.
1717. Trost, B.M.; Schroeder, G.M. J. Am. Chem. Soc. 1999, 121, 6759.
1718. Rueping, M.; Nachtsheim, B.J.; Kuenkel, A. Org. Lett. 2007, 9, 825.
1719. Ranu, B.C.; Chattopadhyay, K.; Adak, L. Org. Lett. 2007, 9, 4595. See Zheng, W.-H.; Zheng, B.-H.; Zhang, Y.; Hou, X.-L. J. Am. Chem. Soc. 2007, 129, 7718.
1720. Trost, B.M.; Schroeder, G.M. Chemistry: European J. 2005, 11, 174.
1721. Martínez, R.; Ramón, D.J.; Yus, M. Tetrahedron 2006, 62, 8988.
1722. Alonso, F.; Riente, P.; Yus, M. Synlett 2007, 1872.
1723. Kwon, M.S.; Kim, N.; Seo, S.H.; Park, I.S.; Cheedrala, R.K.; Park, J. Angew. Chem. Int. Ed. 2005, 44, 6913.
1724. Kinoshita, N.; Kawabata, T.; Tsubaki, K.; Bando, M.; Fuji, K. Tetrahedron 2006, 62, 1756.
1725. Yu, W.; Jin, Z. Tetrahedron Lett. 2001, 42, 369.
1726. Nishimoto, Y.; Saito, T.; Yasuda, M.; Baba, A. Tetrahedron 2009, 65, 5462.
1727. Graening, T.; Hartwig, J.F. J. Am. Chem. Soc. 2005, 127, 17192.
1728. For a reaction with a vinyl silane substrate, see Wang, Z.; Pitteloud, J.-P.; Montes, L.; Rapp, M.; Derane, D.; Wnuk, S.F. Tetrahedron 2008, 64, 5322.
1729. Hiyama, T.; Shirakawa, E. Top. Curr. Chem. 2002, 219, 61; Denmark, S. E.; Sweis, R. F. In Metal-Catalyzed Cross-Coupling Reactions, de Meijere, A.; Diederich, F., Eds., Wiley–VCH, New York, 2004; Chap. 4.
1730. Dai, X.; Strotman, N.A.; Fu, G.C. J. Am. Chem. Soc. 2008,130, 3302.
1731. Raders, S.M.; Kingston, J.V.; Verkade, J.G. J. Org. Chem. 2010, 75, 1744.
1732. Zhang, L.; Wu, J. J. Am. Chem. Soc. 2008, 130, 12250; Zhang, L.; Qing, J.; Yang, P.; Wu, J. Org. Lett. 2008, 10, 4971; Ranu, B.C.; Dey, R.; Chattopadhyay, K. Tetrahedron Lett. 2008, 49, 3430; Li, J.-H.; Deng, C.-L.; Liu, W.-J.; Xie, Y.-X. Synthesis 2005, 3039; Li, J.-H.; Deng, C.-L.; Xie, Y.-X. Synthesis 2006, 969.
1733. Chen, S.-N.; Wu, W.-Y.; Tsai, F.-Y. Tetrahedron 2008, 64, 8164.
1734. Tsuji, J.; Minami, I. Acc. Chem. Res. 1987, 20, 140. See also, Nicolaou, K.C.; Vassilikogiannakis, G.; Mägerlein, W.; Kranich, R. Angew. Chem. Int. Ed. 2001, 40, 2482.
1735. Behenna, D.C.; Stoltz, B.M. J. Am. Chem. Soc. 2004, 126, 15044.
1736. Trost, B.M.; Schroeder, G.M.; Kristensen, J. Angew. Chem. Int. Ed. 2002, 41, 3492.
1737. Millard, A.A.; Rathke, M.W. J. Am. Chem. Soc. 1977, 99, 4833.
1738. Kosugi, M.; Hagiwara, I.; Migita, T. Chem. Lett. 1983, 839. For other methods, see Negishi, E.; Akiyoshi, K. Chem. Lett. 1987, 1007; Chang, T.C.T.; Rosenblum, M.; Simms, N. Org. Synth. 66, 95.
1739. Chieffi, A.; Kamikawa, K.; Åhman, J.; Fox, J.M.; Buchwald, S.L Org. Lett. 2001, 3, 1897.
1740. See Stork, G.; Boeckman, Jr., R.K. J. Am. Chem. Soc. 1973, 95, 2016; Stork, G.; Cohen, J.F. J. Am. Chem. Soc. 1974, 96, 5270. In the last case, the substrate moiety is an epoxide function.
1741. Misumi, A.; Iwanaga, K.; Furuta, K.; Yamamoto, H. J. Am. Chem. Soc. 1985, 107, 3343; Furuta, K.; Iwanaga, K.; Yamamoto, H. Org. Synth. 67, 76.
1742. See Nógrádi, M. Stereoselective Synthesis, VCH, NY, 1986, pp. 236–245; Evans, D.A. in Morrison, J.D. Asymmetric Synthesis Vol. 3, Academic Press, NY, 1984, pp. 1–110.
1743. See Murakata, M.; Nakajima, N.; Koga, K. J. Chem. Soc., Chem. Commun. 1990, 1657. For a review, see Cox. P.J.; Simpkins, N.S. Tetrahedron: Asymmetry 1991, 2, 1, pp. 6–13.
1744. See Lafontaine, J.A.; Provencal, D.P.; Gardelli, C.; Leahy, J.W. J. Org. Chem. 2003, 68, 4215. See Evans, D.A.; Chapman, K.T.; Bisaha, J. Tetrahedron Lett. 1984, 25, 4071; Evans, D.A.; Chapman, K.T.; Bisaha, J. J. Am. Chem. Soc. 1984, 106, 4261; Oppolzer, W.; Chapuis, C.; Dupuis, D.; Guo, M. Helv. Chim. Acta 1985, 68, 2100; Schmierer, R.; Grotemeier, G.; Helmchen, G.; Selim, A. Angew. Chem. Int. Ed. 1981, 20, 207.
1745. Oppolzer, W.; Dudfield, P.; Stevenson, T.; Godel, T. Helv. Chim. Acta 1985, 68, 212.
1746. Denmark, S.E.; Stavenger, R.A. Acc. Chem. Res. 2000, 33, 432; Machajewski, T.D.; Wong, C.–H. Angew. Chem. Int. Ed. 2000, 39, 1352.
1747. Godenschwager, P.F.; Collum, D.B. J. Am. Chem. Soc. 2007, 129, 12023.
1748. Godenschwager, P.F.; Collum, D.B. J. Am. Chem. Soc. 2008, 130, 8726.
1749. Trost, B.M.; Fandrick, D.R.; Dinh, D.C. J. Am. Chem. Soc. 2005, 127, 14186.
1750. See, for example, Prieto, J.A.; Suarez, J.; Larson, G.L. Synth. Commun. 1988, 18, 253; Gaudemar, M.; Bellassoued, M. Tetrahedron Lett. 1989, 30, 2779.
1751. See Lissel, M.; Neumann, B.; Schmidt, S. Liebigs Ann. Chem. 1987, 263.
1752. See Morita, J.; Suzuki, M.; Noyori, R. J. Org. Chem. 1989, 54, 1785.
1753. See House, H.O. Rec. Chem. Prog. 1968, 28, 99; Podraza, K.F. Org. Prep. Proced. Int. 1991, 23, 217.
1754. See d'Angelo, J. Tetrahedron 1976, 32, 2979; Stork, G. Pure Appl. Chem. 1975, 43, 553.
1755. House, H.O.; Trost, B.M. J. Org. Chem. 1965, 30, 1341.
1756. House, H.O.; Gall, M.; Olmstead, H.D. J. Org. Chem. 1971, 36, 2361. For an improved procedure, see Liotta, C.L.; Caruso, T.C. Tetrahedron Lett. 1985, 26, 1599.
1757. See Kuwajima, I.; Nakamura, E. Acc. Chem. Res. 1985, 18, 181; Rasmussen, J.K. Synthesis 1977, 91. See Bélanger, É.; Cantin, K.; Messe, O.; Tremblay, M.; Paquin, J.-F. J. Am. Chem. Soc. 2007, 129, 1034.
1758. Pasto, D.J.; Wojtkowski, P.W. J. Org. Chem. 1971, 36, 1790.
1759. House, H.O.; Gall, M.; Olmstead, H.D. J. Org. Chem. 1971, 36, 2361. See also, Corey, E.J.; Gross, A.W. Tetrahedron Lett. 1984, 25, 495.
1760. See Caine, D. Org. React. 1976, 23, 1. Also see Näf, F.; Decorzant, R. Helv. Chim. Acta 1974, 57, 1317; Wender, P.A.; Eissenstat, M.A. J. Am. Chem. Soc. 1978, 100, 292.
1761. Yamashita, M.; Matsuyama, K.; Tanabe, M.; Suemitsu, R. Bull. Chem. Soc. Jpn. 1985, 58, 407.
1762. Shimizu, S.; Suzuki, T.; Sasaki, Y.; Hirai, C. Synlett 2000, 1664.
1763. See Fraser, R.R. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, Vol. 5, pt. B, Elsevier, NY, 1984, pp. 65–105; Whitesell, J.K.; Whitesell, M.A. Synthesis 1983, 517. For a list of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1513–1518. Also see Goering, H.L.; Tseng, C.C. J. Org. Chem. 1981, 46, 5250.
1764. Park, H.-g.; Jeong, B.-s.; Yoo, M.-s.; Park, M.-k.; Huh, H.; Jew, S.-s. Tetrahedron Lett. 2001, 42, 4645; Jew, S.-s.; Jeong, B.-s.; Yoo, M.-s.; Huh, H.; Park, H.-g. Chem. Commun. 2001, 1244.
1765. Pellissier, H.; Santelli, M. Tetrahedron 2003, 59, 701.
1766. Myers, A.G.; Schnider, P.; Kwon, S.; Kung, D.W. J. Org. Chem. 1999, 64, 3322.
1767. See Enders, D. in Morrison, J.D. Asymmetric Synthesis, Vol. 3, Academic Press, NY, 1984, pp. 275–339.
1768. Meyers, A.I.; Williams, D.R.; White, S.; Erickson, G.W. J. Am. Chem. Soc. 1981, 103, 3088; Enders, D.; Bockstiegel, B. Synthesis 1989, 493; Enders, D.; Kipphardt, H.; Fey, P. Org. Synth. 65, 183.
1769. Zuend, S.J.; Ramirez, A.; Lobkovsky, E.; Collum, D.B. J. Am. Chem. Soc. 2006, 128, 5939.
1770. Hatakeyama, T.; Ito, S.; Nakamura, M.; Nakamura, E. J. Am. Chem. Soc. 2005, 127, 14192.
1771. Stork, G.; Depezay, J.C.; D'Angelo, J. Tetrahedron Lett. 1975, 389. See also, Hünig, S.; Marschner, C.; Peters, K.; von Schnering, H.G. Chem. Ber. 1989, 122, 2131, and other papers in this series.
1772. For a review of 164, see Albright, J.D. Tetrahedron 1983, 39, 3207.
1773. Also see Stetter, H.; Schmitz, P.H.; Schreckenberg, M. Chem. Ber. 1977, 110, 1971; Hünig, S. Chimia, 1982, 36, 1.
1774. See Martin, S.F. Synthesis 1979, 633.
1775. See Block, E. Reactions of Organosulfur Compounds, Academic Press, NY, 1978, pp. 56–67; Gröbel, B.; Seebach, D. Synthesis1977, 357; Lever, Jr., O.W. Tetrahedron 1976, 32, 1943; Seebach, D. Angew. Chem. Int. Ed.1969, 8, 639. Also see Hase, T.A.; Koskimies, J.K. Aldrichimica Acta 1981, 14, 73; Hase, T.A. Umpoled Synthons, Wiley, NY, 1987, pp. xiii-xiv, 7-18, 219-317. For lists of references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1435–1438.
1776. See Hase, T.A. Umpoled Synthons, Wiley, NY, 1987; Seebach, D. Angew. Chem. Int. Ed. 1979, 18, 239.
1777. Stork, G.; Ozorio, A.A.; Leong, A.Y.W. Tetrahedron Lett. 1978, 5175.
1778. Fleming, F.F.; Zhang, Z.; Knochel, P. Org. Lett. 2004, 6, 501.
1779. Nevar, N.M.; Kel'in, A.V.; Kulinkovich, O.G. Synthesis 2000, 1259.
1780. Fleming, F.F.; Zhang, Z.; Liu, W.; Knochel, P. J. Org. Chem. 2005, 70, 2200.
1781. Yamada, Y.M.A.; Uozumi, Y. Org. Lett. 2006, 8, 1375.
1782. This is the IUPAC name with respect to the halide as substrate.
1783. See Adams, J.P. J. Chem. Soc., Perkin Trans. 1 2000, 125.
1784. See Kempf, B.; Hampel, N.; Ofial, A.R.; Mayr, H. Chem. Eur. J. 2003, 9, 2209.
1785. See Hickmott, P.W. Tetrahedron, 1984, 40, 2989; 1982, 38, 1975, 3363; Granik, V.G. Russ. Chem. Rev., 1984, 53, 383. Also see in Cook, A.G. Enamines, 2nd ed.; Marcel Dekker, NY, 1988, the articles by Alt, G.H.; Cook, A.G. pp. 181–246, and Gadamasetti, G.; Kuehne, M.E. pp. 531–689; Whitesell, J.K.; Whitesell, M.A. Synthesis, 1983, 517; House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 570–582, 766–772; Bláha, K.; Cervinka, O. Adv. Heterocycl. Chem., 1966, 6, 147, p. 186.
1786. Weix, D.J.; Hartwig, J.F. J. Am. Chem. Soc. 2007, 129, 7720.
1787. Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Chem. Rev. 2007, 107, 5471.
1788. Katritzky, A.R.; Fang, Y.; Silina, A. J. Org. Chem. 1999, 64, 7622; Katritzky, A.R.; Huang, Z.; Fang, Y. J. Org. Chem. 1999, 64, 7625.
1789. Britten, A.Z.; Owen, W.S.; Went, C.W. Tetrahedron 1969, 25, 3157.
1790. See Hickmott, P.W. Chem. Ind. (London) 1974, 731; Hünig, S.; Hoch, H. Fortschr. Chem. Forsch. 1970, 14, 235.
1791. Stork, G.; Brizzolara, A.; Landesman, H.; Szmuszkovicz, J.; Terrell, R. J. Am. Chem. Soc. 1963, 85, 207.
1792. Kuehne, M.E. J. Am. Chem. Soc., 1959, 81, 5400.
1793. Ziegenbein, W. Angew. Chem. Int. Ed. Engl. 1965, 4, 358.
1794. Baudoux, D.; Fuks, R. Bull. Soc. Chim. Belg. 1984, 93, 1009.
1795. See Alt, G.H.; Cook, A.G. in Cook, A.G. Enamines, 2nd ed., Marcel Dekker, NY, 1988, pp. 204–215.
1796. Stork, G.; Dowd, S.R. J. Am. Chem. Soc., 1963, 85, 2178.
1797. Stork, G.; Benaim, J. J. Am. Chem. Soc., 1971, 93, 5938.
1798. Curphey, T.J.; Hung, J.C.; Chu, C.C.C. J. Org. Chem., 1975, 40, 607. See also, Ho, T.; Wong, C.M. Synth. Commun., 1974, 4, 147.
1799. See Nógrádi, M. Stereoselective Synthesis, VCH, NY, 1986, pp. 248–255; Whitesell, J.K. Acc. Chem. Res. 1985, 18, 280; Bergbreiter, D.E.; Newcomb, M. in Morrison, J.D. Asymmetric Synthesis, Vol. 2, Academic Press, NY, 1983, pp. 243–273.
1800. Vignola, N.; List, B. J. Am. Chem. Soc. 2004, 126, 450.
1801. Mladenova, M.; Blagoev, B.; Gaudemar, M.; Dardoize, F.; Lallemand, J.Y. Tetrahedron 1981, 37, 2153.
1802. Pfeffer, P.E.; Silbert, L.S.; Chirinko, Jr., J.M. J. Org. Chem. 1972, 37, 451.
1803. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1717–1720ff.
1804. Cregar, P.L. J. Am. Chem. Soc.1970, 92, 1396.
1805. Seebach, D.; Corey, E.J. J. Org. Chem. 1975, 40, 231. See Page, P.C.B.; van Niel, M.B.; Prodger, J.C. Tetrahedron 1989, 45, 7643; Ager, D.J. in Hase, T.A. Umpoled Synthons, Wiley, NY, 1987, pp. 19–37; Seebach, D. Synthesis 1969, 17, especially pp. 24–27; Olsen, R.K.; Curriev, Jr., Y.O. in Patai, S. The Chemistry of the Thiol Group, pt. 2, Wiley, NY, 1974, pp. 536–547.
1806. See Lipshutz, B.H.; Garcia, E. Tetrahedron Lett. 1990, 31, 7261.
1807. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1451–1454.
1808. For a direct conversion of RX to RCHO, see Reaction 10-76.
1809. See Seebach, D.; Jones, N.R.; Corey, E.J. J. Org. Chem. 1968, 33, 300; Hylton, T.; Boekelheide, V. J. Am. Chem. Soc. 1968, 90, 6887; Ogura, K.; Yamashita, M.; Suzuki, M.; Tsuchihashi, G. Tetrahedron Lett. 1974, 3653.
1810. Richman, J.E.; Herrmann, J.L.; Schlessinger, R.H. Tetrahedron Lett. 1973, 3267. See also, Schill, G.; Jones, P.R. Synthesis 1974, 117; Hori, I.; Hayashi, T.; Midorikawa, H. Synthesis 1974, 705.
1811. Corey, E.J. Pure Appl. Chem. 1967, 14, 19, pp. 20-23.
1812. See Hase, T.A.; Koskimies, J.K. Aldrichimica Acta 1982, 15, 35.
1813. See Corey, E.J.; Seebach, D. J. Org. Chem. 1975, 40, 231.
1814. See Hylton, T.; Boekelheide, V. J. Am. Chem. Soc. 1968, 90, 6887; Jones, J.B.; Grayshan, R. Chem. Commun. 1970, 141, 741.
1815. See Lissel, M. Liebigs Ann. Chem. 1982, 1589.
1816. Uemoto, K.; Kawahito, A.; Matsushita, N.; Skamoto, I.; Kaku, H.; Tsunoda, T. Tetrahedron Lett. 2001, 42, 905.
1817. Block, E.; Aslam, M. J. Am. Chem. Soc. 1985, 107, 6729.
1818. Biellmann, J.F.; Ducep, J.B. Tetrahedron 1971, 27, 5861. See also, Narasaka, K.; Hayashi, M.; Mukaiyama, T. Chem. Lett. 1972, 259.
1819. Corey, E.J.; Jautelat, M. Tetrahedron Lett. 1968, 5787.
1820. Corey, E.J.; Seebach, D. J. Org. Chem. 1966, 31, 4097.
1821. Oshima, K.; Shimoji, K.; Takahashi, H.; Yamamoto, H.; Nozaki, H. J. Am. Chem. Soc. 1973, 95, 2694.
1822. See Funk, R.L.; Bolton, G.L. J. Am. Chem. Soc. 1988, 110, 1290.
1823. Dolak, T.M.; Bryson, T.A. Tetrahedron Lett. 1977, 1961.
1824. See Magnus, P.D. Tetrahedron 1977, 33, 2019, pp. 2022–2025; Hendrickson, J.B.; Sternbach, D.D.; Bair, K.W. Acc. Chem. Res. 1977, 10, 306.
1825. See Truce, W.E.; Hollister, K.R.; Lindy, L.B.; Parr, J.E. J. Org. Chem. 1968, 33, 43; Julia, M.; Arnould, D. Bull. Soc. Chim. Fr. 1973, 743, 746.
1826. Reich, H.J.; Shah, S.K. J. Am. Chem. Soc. 1975, 97, 3250.
1827. See Krief, A. Top. Curr. Chem. 1987, 135, 1. Also see Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, pp. 336–341.
1828. Lepley, A.R.; Khan, W.A. Chem. Commun. 1967, 1198; Lepley, A.R.; Giumanini, A.G. J. Org. Chem. 1966, 31, 2055; Ahlbrecht, H.; Dollinger, H. Tetrahedron Lett. 1984, 25, 1353.
1829. Dieter, R.K.; Li, S. Tetrahedron Lett. 1995, 36, 3613.
1830. Suga, S.; Okajima, M.; Yoshida, J.-i. Tetrahedron Lett. 2001, 42, 2173.
1831. Kise, N.; Yamazaki, H.; Mabuchi, T.; Shono, T. Tetrahedron Lett. 1994, 35, 1561.
1832. For a review, see Beak, P.; Zajdel, W.J.; Reitz, D.B. Chem. Rev. 1984, 84, 471.
1833. Seebach, D.; Enders, D.; Renger, B. Chem. Ber. 1977, 110, 1852; Renger, B.; Kalinowski, H.; Seebach, D. Chem. Ber. 1977, 110, 1866. For a review, see Seebach, D.; Enders, D. Angew. Chem. Int. Ed. 1975, 14, 15.
1834. Fridman, A.L.; Mukhametshin, F.M.; Novikov, S.S. Russ. Chem. Rev. 1971, 40, 34, pp. 41–42.
1835. For the use of tert-butyl carbamates, see Beak, P.; Lee, W. Tetrahedron Lett. 1989, 30, 1197.
1836. For a review, see Meyers, A.I. Aldrichimica Acta 1985, 18, 59.
1837. Meyers, A.I.; Miller, D.B.; White, F. J. Am. Chem. Soc. 1988, 110, 4778; Gonzalez, M.A.; Meyers, A.I. Tetrahedron Lett. 1989, 30, 43, 47 and references cited therein.
1838. Takemiya, A.; Hartwig, J.F. J. Am. Chem. Soc. 2006, 128, 14800.
1839. Funk, R.L.; Bolton, G.L. J. Am. Chem. Soc. 1988, 110, 1290. See Hommes, H.; Verkruijsse, H.D.; Brandsma, L. Recl. Trav. Chim. Pays-Bas 1980, 99, 113, and references cited therein.
1840. See Biellmann, J.F.; Ducep, J. Org. React. 1982, 27, 1.
1841. Martin, S.F.; DuPriest, M.T. Tetrahedron Lett. 1977, 3925 and references cited therein.
1842. For a review, see Ahlbrecht, H. Chimia 1977, 31, 391.
1843. Beak, P.; Carter. L.G. J. Org. Chem. 1981, 46, 2363.
1844. Seebach, D.; Meyer, N. Angew. Chem. Int. Ed. 1976, 15, 438.
1845. Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1461–1465.
1846. Meyers, A.I.; Nabeya, A.; Adickes, H.W.; Politzer, I.R.; Malone, G.R.; Kovelesky, A.C.; Nolen, R.L.; Portnoy, R.C. J. Org. Chem. 1973, 38, 36.
1847. See Schmidt, R.R. Synthesis 1972, 333; Collington, E.W. Chem. Ind. (London) 1973, 987.
1848. Meyers, A.I.; Malone, G.R.; Adickes, H.W. Tetrahedron Lett. 1970, 3715.
1849. Adickes, H.W.; Politzer, I.R.; Meyers, A.I. J. Am. Chem. Soc. 1969, 91, 2155.
1850. Altman, L.J.; Richheimer, S.L. Tetrahedron Lett. 1971, 4709.
1851. Meyers, A.I.; Durandetta, J.L. J. Org. Chem. 1975, 40, 2021.
1852. Meyers, A.I.; Smith, E.M. J. Org. Chem. 1972, 37, 4289.
1853. For a review, see Meyers, A.I.; Mihelich, E.D. Angew. Chem. Int. Ed. 1976, 15, 270.
1854. Meyers, A.I.; Temple, Jr., D.L.; Nolen, R.L.; Mihelich, E.D. J. Org. Chem. 1974, 39, 2778; Meyers, A.I.; Mihelich, E.D.; Nolen, R.L. J. Org. Chem. 1974, 39, 2783; Meyers, A.I.; Mihelich, E.D.; Kamata, K. J. Chem. Soc., Chem. Commun. 1974, 768.
1855. See Meyers, A.I. Pure Appl. Chem. 1979, 51, 1255; Acc. Chem. Res. 1978, 11, 375. See also, Hoobler, M.A.; Bergbreiter, D.E.; Newcomb, M. J. Am. Chem. Soc. 1978, 100, 8182; Meyers, A.I.; Snyder, E.S.; Ackerman, J.J.H. J. Am. Chem. Soc. 1978, 100, 8186.
1856. See Lutomski, K.A.; Meyers, A.I. in Morrison, J.D. Asymmetric Synthesis, Vol. 3, Academic Press, NY, 1984, pp. 213–274.
1857. Meyers, A.I.; Temple Jr., D.L. J. Am. Chem. Soc. 1970, 92, 6644, 6646.
1858. Brown, H.C.; Rogic, M.M.; Rathke, M.W. J. Am. Chem. Soc. 1968, 90, 6218.
1859. Brown, H.C.; Rogic, M.M.; Rathke, M.W.; Kabalka, G.W. J. Am. Chem. Soc. 1968, 90, 818.
1860. Brown, H.C.; Nambu, H.; Rogic, M.M. J. Am. Chem. Soc. 1969, 91, 6854.
1861. Truce, W.E.; Mura, L.A.; Smith, P.J.; Young, F. J. Org. Chem. 1974, 39, 1449.
1862. See Negishi, E.; Idacavage, M.J. Org. React. 1985, 33, 1, pp. 42–43, 143–150; Weill-Raynal, J. Synthesis 1976, 633; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 372–391, 404–409; Cragg, G.M.L. Organoboranes in Organic Synthesis, Marcel Dekker, NY, 1973, pp. 275–278, 283–287.
1863. Brown, H.C.; Nambu, H.; Rogic, M.M. J. Am. Chem. Soc. 1969, 91, 6852, 6854, 6855.
1864. For an improved procedure, with 9-BBN (see Reaction 15-16), see Brown, H.C.; Rogic, M.M. J. Am. Chem. Soc. 1969, 91, 2146; Brown, H.C.; Rogic, M.M.; Nambu, H.; Rathke, M.W. J. Am. Chem. Soc. 1969, 91, 2147; Katz, J.; Dubois, J.E.; Lion, C. Bull. Soc. Chim. Fr. 1977, 683.
1865. Brown, H.C.; Bhat, N.G.; Campbell, Jr., J.B. J. Org. Chem. 1986, 51, 3398.
1866. Brown, H.C.; Rogic, M.M. J. Am. Chem. Soc. 1969, 91, 4304.
1867. Brown, H.C.; Rogic, M.M.; Rathke, M.W.; Kabalka, G.W. J. Am. Chem. Soc. 1968, 90, 1911.
1868. Nambu, H.; Brown, H.C. J. Am. Chem. Soc. 1970, 92, 5790.
1869. Brown, H.C.; Nambu, H. J. Am. Chem. Soc. 1970, 92, 1761.
1870. Brown, H.C. Organic Syntheses via Boranes, Wiley, NY, 1975; Hydroboration, W.A. Benjamin, NY, 1962; Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972; Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988.
1871. See Prager, R.H.; Reece, P.A. Aust. J. Chem. 1975, 28, 1775.
1872. See Midland, M.M.; Zolopa, A.R.; Halterman, R.I. J. Am. Chem. Soc. 1979, 101, 248. See also, Midland, M.M.; Preston, S.B. J. Org. Chem. 1980, 45, 747.
1873. Pasto, D.J.; Wojtkowski, P.W. J. Org. Chem. 1971, 36, 1790.
1874. Brown, H.C.; Rogic, M.M.; Rathke, M.W.; Kabalka, G.W. J. Am. Chem. Soc. 1969, 91, 2150.
1875. Occhiato, E.G.; Trabocchi, A.; Guarna, A. Org. Lett. 2000, 2, 1241.
1876. Sato, M.; Miyaura, N.; Suzuki, A. Chem. Lett. 1989, 1405; Rivera, I.; Soderquist, J.A. Tetrahedron Lett. 1991, 32, 2311; and references cited therein. For a review, see Matteson, D.S. Tetrahedron 1989, 45, 1859.
1877. Miyaura, N.; Ishiyama, T.; Sasaki, H.; Ishikawa, M.; Satoh, M.; Suzuki, A. J. Am. Chem. Soc. 1989, 111, 314. See also, Soderquist, J.A.; Santiago, B. Tetrahedron Lett. 1990, 31, 5541.
1878. Ishiyama, T.; Abe, S.; Miyaura, N.; Suzuki, A. Chem. Lett. 1992, 691; Brown, H.C.; Joshi, N.N.; Pyun, C.; Singaram, B. J. Am. Chem. Soc. 1989, 111, 1754. For another such coupling, see Matteson, D.S.; Tripathy, P.B.; Sarkar, A.; Sadhu, K.M. J. Am. Chem. Soc. 1989, 111, 4399.
1879. Mikhailov, B.M.; Gurskii, M.E. Bull. Acad. Sci. USSR Div. Chem. Sci. 1973, 22, 2588.
1880. Hooz, J.; Morrison, G.F. Can J. Chem. 1970, 48, 868.
1881. See Hooz, J.; Bridson, J.N.; Calzada, J.G.; Brown, H.C.; Midland, M.M.; Levy, A.B. J. Org. Chem. 1973, 38, 2574.
1882. Kondolff, I.; Doucet, H.; Santelli, M. Tetrahedron 2004, 60, 3813. For a variation involving a borate complex, see Zou, G.; Falck, J.R. Tetrahedron Lett. 2001, 42, 5817.
1883. Nobre, S.M.; Monteiro, A.L. Tetrahedron Lett. 2004, 45, 8225; Langle, S.; Abarbri, M.; Duchêne, A. Tetrahedron Lett. 2003, 44, 9255.
1884. Ohmiya, H.; Makida, Y.; Tanaka, T.; Sawamura, M. J. Am. Chem. Soc. 2008, 130, 17276.
1885. Chen, H.; Deng, M.-Z. J. Org. Chem. 2000,65, 4444.
1886. Yoshida, M.; Ueda, H.; Ihara, M. Tetrahedron Lett. 2005, 46, 6705.
1887. Chen, X.; Goodhue, C.E.; Yu, J.-Q. J. Am. Chem. Soc. 2006, 128, 12634.
1888. Bellina, F.; Anselmi, C.; Rossi, R. Tetrahedron Lett. 2001, 42, 3851. See also, Yoshida, H.; Yamaryo, Y.; Oshita, J.; Kunai, A. Tetrahedron Lett. 2003, 44, 1541.
1889. Wiskur, S.L.; Lorte, A.; Fu, G.C. J. Am. Chem. Soc. 2004, 126, 82.
1890. Pastine, S.J.; Gribkov, D.V.; Sames, D. J. Am. Chem. Soc. 2006, 128, 14220.
1891. Pineschi, M.; Bertolini, F.; Haak, R.M.; Crotti, P.; Macchia, F. Chem. Commun. 2005, 1426.
1892. Batey, R.A.; Thadani, A.N.; Smil, D.V.; Lough, A.J. Synthesis 2000, 990.
1893. Darses, S.; Michaud, G.; Genêt, J.-P. Eur. J. Org. Chem. 1999, 1875.
1894. Xia, M.; Chen, Z.-C. Synth. Commun. 1999, 29, 2457.
1895. Molander, G.A.; Gormisky, P.E. J. Org. Chem. 2008, 73, 7481.
1896. Molander, G.A.; Ito, T. Org. Lett. 2001, 3, 393.
1897. Molander, G.A.; Rivero, M.R. Org. Lett. 2002, 4, 107.
1898. See Ben-Efraim, D.A. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, Wiley, NY, 1978, pp. 790–800; Ziegenbein, W. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 185–206, 241–244. Also see Bernadou, F.; Mesnard, D.; Miginiac, L. J. Chem. Res. (S) 1978, 106; 1979, 190.
1899. Jeffery, T. Tetrahedron Lett. 1989, 30, 2225.
1900. See Krause, N.; Seebach, D. Chem. Ber. 1988, 121, 1315.
1901. Smith, W.N.; Beumel Jr., O.F. Synthesis 1974, 441.
1902. Bhanu, S.; Scheinmann, F. J. Chem. Soc. Perkin Trans.1, 1979, 1218; Quillinan, A.J.; Scheinmann, F. Org. Synth. VI, 595.
1903. Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. Tetrahedron Lett. 2003, 44, 9087.
1904. Chen, M.; Zheng, X.; Li, W.; He, J.; Lei, A. J. Am. Chem. Soc. 2010, 132, 4101.
1905. Zhao, Y.; Wang, H.; Hou, X.; Hu, Y.; Lei, A.; Zhang, H.; Zhu, L. J. Am. Chem. Soc. 2006, 128, 15048.
1906. Nadal, M.L.; Bosch, J.; Vila, J.M.; Klein, G.; Ricart, S.; Moretó, J.M. J. Am. Chem. Soc. 2005, 127, 10476.
1907. Rahaim, Jr., R.J.; Shaw, J.T. J. Org. Chem. 2008, 73, 2912.
1908. Kuninobu, Y.; Kawata, A.; Takai, K. Org. Lett. 2005, 7, 4823.
1909. Qian, M.; Negishi, E. Tetrahedron Lett. 2005, 46, 2927.
1910. Kang, S.-K.; Kim, W.-Y.; Jiao, X. Synthesis 1998, 1252.
1911. Liu, Y.; Xi, C.; Hara, R.; Nakajima, K.; Yamazaki, A.; Kotora, M.; Takahashi, T. J. Org. Chem. 2000, 65, 6951.
1912. Pal, M.; Kundu, N.G. J. Chem. Soc. Perkin Trans 1, 1996, 449. Also see, Nguefack, J.-F.; Bolitt, V.; Sinou, D. Tetrahedron Lett, 1996, 37, 5527.
1913. Nishihara, Y.; Ikegashira, K.; Mori, A.; Hiyama, T. Tetrahedron Lett. 1998, 39, 4075.
1914. See Nishihara, Y.; Ikegashira, K.; Mori, A.; Hiyama, T. Chem. Lett. 1997, 1233.
1915. Poulsen, T.B.; Bernardi, L.; Alemán, J.; Overgaard, J.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2007, 129, 441.
rgensen, K.A. J. Am. Chem. Soc. 2007, 129, 441.
1916. Laroche, C.; Li, J.; Freyer, M.W.; Kerwin, S.M. J. Org. Chem. 2008, 73, 6462.
1917. Castagnolo, D.; Botta, M. Eur. J. Org. Chem. 2010, 3224.
1918. Murakami, M.; Hayashi, M.; Ito, Y. Synlett, 1994, 179.
1919. Bieber, L.W.; da Silva, M.F. Tetrahedron Lett. 2007, 48, 7088.
1920. Kang, S.-K.; Lim, K.-H.; Ho, P.-S.; Kim, W.-Y. Synthesis 1997, 874.
1921. Xiang, J.; Jiang, W.; Fuchs, P.L. Tetrahedron Lett. 1997, 38, 6635.
1922. Li, Z.; Li, C.-J. Org. Lett. 2004, 6, 4997.
1923. Li, Z.; Li, C.-J. J. Am. Chem. Soc. 2004, 126, 11810.
1924. Zhang, J.; Wei, C.; Lei, C.-J. Tetrahedron Lett. 2002, 43, 5731.
1925. Arisawa, M.; Amemiya, R.; Yamaguchi, M. Org. Lett. 2002, 4, 2209.
1926. Shimizu, R; Fuchikami, T. Tetrahedron Lett. 2000, 41, 907.
1927. Jiang, H.; Zhu, S. Tetrahedron Lett. 2005, 46, 517.
1928. Andreev, A.A.; Konshin, V.V.; Komarov, N.V.; Rubin, M.; Brouwer, C.; Gevorgyan, V. Org. Lett. 2004, 6, 421.
1929. See in Patai, S.; Rappoport, Z. The Chemistry of Functional Groups, Supplement C, pt. 1, Wiley, NY, 1983, the articles by Fatiadi, A.J. pt. 2, pp. 1057–1303, and Friedrich, K. pt. 2, pp. 1343–1390; Friedrich, K.; Wallenfels, K. in Rappoport, Z. The Chemistry of the Cyano Group, Wiley, NY, 1970, pp. 77–86.
1930. For a discusion about the influence of solvent on this transition state, see Fang, Y.-r.; MacMillar, S.; Eriksson, J.; Kołodziejska-Huben, M.; Dybała-Defratyka, A.; Paneth, P.; Matsson, O.; Westaway, K.C. J. Org. Chem. 2006, 71, 4742.
1931. Smiley, R.A.; Arnold, C. J. Org. Chem. 1960, 25, 257; Friedman, L.; Shechter, H. J. Org. Chem. 1960, 25, 877.
1932. Starks, C.M.; Liotta, C. Phase Transfer Catalysis, Acaemic Press, NY, 1978, pp. 94–112; Weber, W.P.; Gokel, G.W. Phase Transfer Catalysis in Organic Synthesis, Springer, NY, 1977, pp. 96–108. See also, Bram, G.; Loupy, A.; Pedoussaut, M. Tetrahedron Lett. 1986, 27, 4171; Bull. Soc. Chim. Fr. 1986, 124.
1933. Cao, Y.-Q.; Che, B.-H.; Pei, B.-G. Synth. Commun. 2001, 31, 2203.
1934. Ando, T.; Kawate, T.; Ichihara, J.; Hanafusa, T. Chem. Lett. 1984, 725.
1935. See Luanay, D.; Booth, S.; Clemens, I.; Merritt, A.; Bradley, M. Tetrahedron Lett. 2002, 43, 7201.
1936. See Jackson, H.L.; McKusick, B.C. Org. Synth. IV, 438.
1937. Ciaccio, J.A.; Smrtka, M.; Maio, W.A.; Rucando, D. Tetrahedron Lett. 2004, 45, 7201.
1938. Kim, S.; Song, H.-J. Synlett 2002, 2110.
1939. Cho, C.H.; Lee, J.Y.; Kim, S. Synlett 2009, 81.
1940. See Lapouyade, R.; Daney, M.; Lapenue, M.; Bouas-Laurent, H. Bull. Soc. Chim. Fr. 1973, 720.
1941. Yamamura, K.; Murahashi, S. Tetrahedron Lett. 1977, 4429.
1942. Procházka, M.; Siroky, M. Collect. Czech. Chem. Commun. 1983, 48, 1765.
1943. Zieger, H.E.; Wo, S. J. Org. Chem. 1994, 59, 3838. See Tsuji, Y.; Yamada, N.; Tanaka, S. J. Org. Chem. 1993, 58, 16 for a similar reaction with allylic acetates. See Hayashi, M.; Tamura, M.; Oguni, N. Synlett, 1992, 663 for a similar reaction with epoxides using a Ti catalyst.
1944. Camps, F.; Gasol, V.; Guerrero, A. Synth. Commun. 1988, 18, 445.
1945. See Iranpoor, N.; Shekarriz, M. Synth. Commun. 1999, 29, 2249.
1946. Ruano, J.L.G.; Fernández-Ibáñez, M.Á.; Castro, A.M.M.; Ramos, J.H.R.; Flamarique, A.C.R. Tetrahedron Asymmetry 2002, 13, 1321.
1947. Dowd, P.; Wilk, B.K.; Wlostowski, M. Synth. Commun. 1993, 23, 2323; Wilk, B.K. Synth. Commun. 1993, 23, 2481 and see Ohno, H.; Mori, A.; Inoue, S. Chem. Lett. 1993, 975 for similar reactions with epoxides.
1948. Piers, E.; Fleming, F.F. J. Chem. Soc., Chem. Commun. 1989, 756.
1949. Sasaki, M.; Tanino, K.; Hirai, A.; Miyashita, M. Org. Lett. 2003, 5, 1789.
1950. Schaus, S.E.; Jacobsen, E.N. Org. Lett. 2000, 2, 1001.
1951. Iranpoor, N.; Firouzabadi, H.; Akhlaghinia, B.; Nowrouzi, N. J. Org. Chem. 2004, 69, 2562.
1952. Tarrade-Matha, A.; Pillon, F.; Doris, E. Synth. Commun. 2010, 40, 1646.
1953. Müller, P.; Siegfried, B. Helv. Chim. Acta 1974, 57, 987.
1954. Cooke, Jr., M.P. J. Am. Chem. Soc. 1970, 92, 6080.
1955. See Collman, J.P. Acc. Chem. Res. 1975, 8, 342. Also see Brunet, J. Chem. Rev. 1990, 90, 1041.
1956. Dolhem, E.; Barhdadi, R.; Folest, J.C.; Nédélec, J.Y.; Troupel, M. Tetrahedron 2001, 57, 525.
1957. Siegl, W.O.; Collman, J.P. J. Am. Chem. Soc. 1972, 94, 2516.
1958. See Collman, J.P.; Finke, R.G.; Cawse, J.N.; Brauman, J.I. J. Am. Chem. Soc. 1978, 100, 4766.
1959. See Collman, J.P.; Hoffman, N.W. J. Am. Chem. Soc. 1973, 95, 2689.
1960. Yamashita, M.; Yamamura, S.; Kurimoto, M.; Suemitsu, R. Chem. Lett. 1979, 1067.
1961. Cooke Jr., M.P.; Parlman, R.M. J. Am. Chem. Soc. 1975, 97, 6863. However, see McMurry, J.E.; Andrus, A. Tetrahedron Lett. 1980, 21, 4687, and references cited therein.
1962. Yamashita, M.; Uchida, M.; Tashika, H.; Suemitsu, R. Bull. Chem. Soc. Jpn. 1989, 62, 2728.
1963. Dolhem, E.; Ocafrain, M.; Nédélec, J.Y.; Troupel, M. Tetrahedron 1997, 53, 17089.
1964. Hanzawa, Y.; Narita, K.; Taguchi, T. Tetrahedron Lett. 2000, 41, 109.
1965. des Abbayes, H.; Clément, J.; Laurent, P.; Tanguy, G.; Thilmont, N. Organometallics 1988, 7, 2293.
1966. Garnier, L.; Rollin, Y.; Périchon, J. J. Organomet. Chem. 1989, 367, 347.
1967. Baillargeon, V.P.; Stille, J.K. J. Am. Chem. Soc. 1986, 108, 452. See also, Ben-David, Y.; Portnoy, M.; Milstein, D. J. Chem. Soc., Chem. Commun. 1989, 1816.
1968. Tamaru, Y.; Ochiai, H.; Yamada, Y.; Yoshida, Z. Tetrahedron Lett. 1983, 24, 3869.
1969. Goure, W.F.; Wright, M.E.; Davis, P.D.; Labadie, S.S.; Stille, J.K. J. Am. Chem. Soc. 1984, 106, 6417. See Merrifield, J.H.; Godschalx, J.P.; Stille, J.K. Organometallics 1984, 3, 1108.
1970. Kosugi, M.; Sumiya, T.; Obara, Y.; Suzuki, M.; Sano, H.; Migita, T. Bull. Chem. Soc. Jpn. 1987, 60, 767.
1971. Ogawa, A.; Sumino, Y.; Nanke, T.; Ohya, S.; Sonoda, N.; Hirao, T. J. Am. Chem. Soc., 1997, 119, 2745.
1972. Matt, C.; Wagner, A.; Mioskowski, C. J. Org. Chem. 1997, 62, 234.
1973. Kim, S.; Jon, S.Y. Tetrahedron Lett. 1998, 39, 7317.
1974. See Colquhoun, H.M.; Holton, J.; Thompson, D.J.; Twigg, M.V. New Pathways for Organic Synthesis, Plenum, NY, 1984, pp. 199–204, 212–220, 234–235. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley-VCH, NY, 1999, pp. 1684–1685, 1694–1698, 1702–1704.
1975. Puzitskii, K.V.; Pirozhkov, S.D.; Ryabova, K.G.; Myshenkova, T.N.; Éidus, Ya.T. Bull. Acad. Sci. USSR Div. Chem. Sci. 1974, 23, 192.
1976. Takahashi, Y.; Yoneda, N. Synth. Commun. 1989, 19, 1945.
1977. Paatz, R.; Weisgerber, G. Chem. Ber. 1967, 100, 984. See Akhrem, I.; Afanas'eva, L.; Petrovskii, P.; Vitt, S.; Orlinkov, A. Tetrahedron Lett. 2000, 41, 9903.
1978. Yoneda, N.; Takahashi, Y.; Fukuhara, T.; Suzuki, A. Bull. Chem. Soc. Jpn. 1986, 59, 2819.
1979. See Bahrmann, H.; Cornils, B. in Falbe, J. New Syntheses with Carbon Monoxide, Springer, NY, 1980, pp. 226–241; Piacenti, F.; Bianchi, M. in Wender, I.; Pino, P. Organic Syntheses via Metal Carbonyls, Vol. 2, Wiley, NY, 1977, pp. 1–42.
1980. See Bahrmann, H. in Falbe, J. New Syntheses with Carbon Monoxide, Springer, NY, 1980, pp. 372–413.
1981. Booth, B.L.; El-Fekky, T.A. J. Chem. Soc. Perkin Trans. 1 1979, 2441.
1982. Kreimerman, S.; Ryu, I.; Minakata, S.; Komatsu, M. Org. Lett. 2000, 2, 389.
1983. See Collman, J.P.; Hegedus, L.S.; Norton, J.R.; Finke, R.G. Principles and Applications of Organotransition Metal Chemistry, 2nd ed.; University Science Books: Mill Valley, CA, 1987, pp. 749–768; Anderson, G.K.; Davies, J.A. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 3, Wiley, NY, pp. 335–359, pp. 348–356; Heck, R.F. Adv. Catal., 1977, 26, 323, p. 323; Cassar, L.; Chiusoli, G.P.; Guerrieri, F. Synthesis 1973, 509.
1984. Corey, E.J.; Hegedus, L.S. J. Am. Chem. Soc. 1969, 91, 1233. See also, Crandall, J.K.; Michaely, W.J. J. Organomet. Chem. 1973, 51, 375.
1985. Collman, J.P.; Winter, S.R.; Komoto, R.G. J. Am. Chem. Soc. 1973, 95, 249.
1986. Collman, J.P.; Winter, S.R.; Komoto, R.G. J. Am. Chem. Soc. 1973, 95, 249.
1987. Yamashita, M.; Mizushima, K.; Watanabe, Y.; Mitsudo, T.; Takegami, Y. Chem. Lett. 1977, 1355. See also, Tanguy, G.; Weinberger, B.; des Abbayes, H. Tetrahedron Lett. 1983, 24, 4005.
1988. See Gulevich, Yu.V.; Bumagin, N.A.; Beletskaya, I.P. Russ. Chem. Rev. 1988, 57, 299, pp. 303–309; Heck, R.F. Palladium Reagents in Organic Synthesis, Academic Press, NY, 1985, pp. 348–356, 366–370. See Kormos, C.M.; Leadbeater, N.E. Synlett 2007, 2006.
1989. See Bessard, Y; Crettaz, R. Heterocycles 1999, 51, 2589.
1990. See Cacchi, S.; Morera, E.; Ortar, G. Tetrahedron Lett. 1985, 26, 1109.
1991. Kiji, J.; Okano, T.; Higashimae, Y.; Kukui, Y. Bull. Chem. Soc. Jpn. 1996, 69, 1029.
1992. Zhao, Y.; Jin, L.; Li, P.; Lei, A. J. Am. Chem. Soc. 2008, 130, 9429.
1993. Jutand, A.; Négri, S. Synlett, 1997, 719.
1994. Lapidus, A.L.; Eliseev, O.L.; Bondarenko, T.N.; Sizan, O.E.; Ostapenko, A.G.; Beletskaya, I.P. Synthesis 2002, 317.
1995. Schoenberg, A.; Heck, R.F. J. Org. Chem. 1974, 39, 3327. See also, Cai, M.-Z.; Song, C.-S.; Huang, X. Synth. Commun. 1997, 27, 361; Screttas, C.G.; Steele, B.R. Org. Prep. Proceed. Int. 1990, 22, 271, pp. 288–314; Satoh, T.; Ikeda, M.; Kushino, Y.; Miura, M.; Nomura, M. J. Org. Chem. 1997, 62, 2662.
1996. Ryu, I.; Nagahara, K.; Kambe, N.; Sonoda, N.; Kreimerman, S.; Komatsu, M. Chem. Commun. 1998, 1953.
1997. Isse, A.A.; Gennaro, A. Chem. Commun. 2002, 2798.
1998. Alper, H.; Amer, I.; Vasapollo, G. Tetrahedron Lett. 1989, 30, 2615. See also, Amer, I.; Alper, H. J. Am. Chem. Soc. 1989, 111, 927.
1999. Ueda, K.; Mori, M. Tetrahedron Lett. 2004, 45, 2907. For an intramolecular carbonylation to generate a cyclic amide, see Trost, B.M.; Ameriks, M.K. Org. Lett. 2004, 6, 1745.
2000. Li, J.; Jiang, H.; Chen, M. Synth. Commun. 2001, 31, 199.
2001. For an example, see Giroux, A.; Nadeau, C.; Han, Y. Tetrahedron Lett. 2000, 41, 7601.
2002. Buchan, C.; Hamel, N.; Woell, J.B.; Alper, H. Tetrahedron Lett. 1985, 26, 5743.
2003. Woell, J.B.; Fergusson, S.B.; Alper, H. J. Org. Chem. 1985, 50, 2134.
2004. Yokoa, K.; Tatamidani, H.; Fukumoto, Y.; Chatani, N. Org. Lett. 2003, 5, 4329.
2005. Senboku, H.; Kanaya, H.; Tokuda, M. Synlett 2002, 140.
2006. Imbeaux, M.; Mestdagh, H.; Moughamir, K.; Rolando, C. J. Chem. Soc., Chem. Commun. 1992, 1678.
2007. For a review, see Collin, J. Bull. Soc. Chim. Fr. 1988, 976.
2008. Kobayashi, T.; Tanaka, M. J. Organomet. Chem. 1982, 233, C64; Ozawa, F.; Sugimoto, T.; Yuasa, Y.; Santra, M.; Yamamoto, T.; Yamamoto, A. Organometallics 1984, 3, 683.
2009. Son, T.; Yanagihara, H.; Ozawa, F.; Yamamoto, A. Bull. Chem. Soc. Jpn. 1988, 61, 1251.
2010. Tanaka, M.; Kobayashi, T.; Sakakura, T. J. Chem. Soc., Chem. Commun. 1985, 837.
2011. See Ozawa, F.; Kawasaki, N.; Okamoto, H.; Yamamoto, T.; Yamamoto, A. Organometallics 1987, 6, 1640.
2012. Kobayashi, T.; Sakakura, T.; Tanaka, M. Tetrahedron Lett. 1987, 28, 2721.