March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 7th Edition (2013)
Part II. Introduction
Chapter 12. Aliphatic, Alkenyl, and Alkynyl Substitution, Electrophilic and Organometallic
12.C. Reactions
The reactions in this chapter are arranged in order of leaving group: hydrogen, metals, halogen, and carbon. Electrophilic substitutions at a nitrogen atom are treated last.
12.C.i. Hydrogen as Leaving Group
A. Hydrogen as the Electrophile
12-1 Hydrogen Exchange
Deuterio-de-hydrogenation or Deuteriation
![]()
Hydrogen exchange can be accomplished by treatment with acids or bases. As with Reaction 11-1, the exchange reaction is mostly used to study mechanistic questions (e.g., relative acidities), but it can be used synthetically to prepare deuterated or tritiated molecules. When ordinary strong acids (e.g., H2SO4) are used, only fairly acidic protons on carbon can exchange (e.g., acetylenic and allylic). However, primary, secondary, and tertiary hydrogen atoms of alkanes can be exchanged by treatment with superacids (Sec. 5.A.ii).48 The order of hydrogen reactivity is tertiary > secondary > primary. Where C–C bonds are present, they may be cleaved also (Reaction 12-47). The mechanism of the exchange (illustrated for methane) has been formulated as involving attack of H+ on the C–H bond to give the pentavalent methanonium ion, which loses
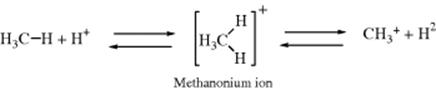
H2 to give a tervalent carbocation.49 The methanonium ion (CH5+) has a three-center, two-electron bond.50 It is not known whether the methanonium ion is a transition state or a true intermediate, but an ion (CH5+) has been detected in the mass spectrum.51 The IR spectrum of the ethanonium ion (C2H7+) has been measured in the gas phase.52 Note that the two electrons in the three-center, two-electron bond can move in three directions, in accord with the threefold symmetry of such a structure. The electrons can move to unite the two hydrogen atoms, leaving the CH3+ free (the forward reaction), or they can unite the CH3 with either of the two hydrogen atoms, leaving the other hydrogen as a free H+ ion (the reverse reaction). Actually, the methyl cation is not stable under these conditions. It can go back to CH4 by the route shown (leading to H+ exchange), or it can react with additional CH4 molecules (Reaction 12-20) to eventually yield the tert-butyl cation, which is stable in these superacid solutions. Hydride ion can also be removed from alkanes (producing tervalent carbocations) by treatment with pure SbF5 in the absence of any source of H+.53 Complete or almost complete perdeuteriation of cyclic alkenes has been achieved by treatment with dilute DCl/D2O in sealed Pyrex tubes at 165–280 °C.54
Exchange with bases involves an SE1 mechanism.
![]()
Of course, such exchange is most successful for relatively acidic protons (e.g., those α to a carbonyl group), but even weakly acidic protons can exchange with bases if the bases are strong enough (see Sec. 5.B.i).
Alkanes and cycloalkanes, of both low and high molecular weight, can be fully perdeuterated treatment with D2 gas and a catalyst (e.g., Rh, Pt, or Pd).55
OS VI, 432.
12-2 Migration of Double Bonds
3/Hydro-de-hydrogenation
![]()
The double bonds of many unsaturated compounds may be isomerized56 upon treatment with strong bases.57 In many cases, equilibrium mixtures are obtained and the thermodynamically most stable isomer predominates.58 If the new double bond can be in conjugation with one already present or with an aromatic ring, the conjugated compound is favored.59 If the choice is between an exocyclic and an endocyclic double bond (particularly with six-membered rings), endocyclic is usually preferred. In the absence of such considerations, Zaitsev's rule (Sec. 17.B) applies and the double bond goes to the carbon with the fewest hydrogen atoms. All these considerations lead to predictions that terminal alkenes can be isomerized to internal ones, nonconjugated alkenes to conjugated, exo six-membered ring alkenes to endo, and so on, and not the other way around.
The term prototropic rearrangement is sometimes used as an example of electrophilic substitution with accompanying allylic rearrangement. The mechanism involves abstraction by a base to give a resonance-stabilized carbanion, and reaction with a proton is at the position that will give the more stable alkene:60
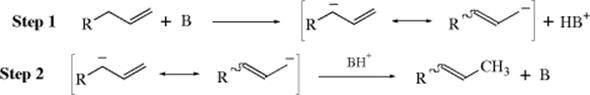
This mechanism is exactly analogous to the allylic-rearrangement mechanism for nucleophilic substitution (Sec. 10.D). Ultraviolet spectra of allylbenzene and 1-propenylbenzene in solutions containing NH2+ are identical, showing that the same carbanion is present in both cases, as required by this mechanism.61 The acid BH+ protonates the position that will give the more stable product, although the ratio of the two possible products can vary with the identity of BH+.62 It has been shown that base-catalyzed double-bond shifts are partially intramolecular, at least in some cases.63 The intramolecular nature has been ascribed to a conducted tour mechanism (Sec. 12.A.iii) in which the base leads the proton from one carbanionic site to the other (13 → 14).64
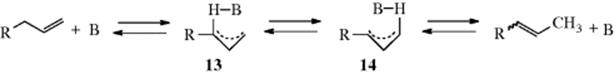
Double-bond rearrangements can also take place on treatment with acids. Both proton and Lewis65 acids can be used. The mechanism in the case of proton acids is the reverse of the previous one; first a proton is gained, giving a carbocation and then another is lost:
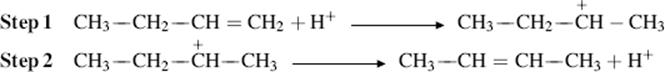
As in the case of the base-catalyzed reaction, the thermodynamically most stable alkene is the one predominantly formed. However, the acid-catalyzed reaction is much less synthetically useful because carbocations give rise to many side products. If the substrate has several possible locations for a double bond, mixtures of all possible isomers are usually obtained. Isomerization of 1-decene, for example, gives a mixture that contains not only 1-decene and cis-and trans-2-decene, but also the cis and trans isomers of 3-, 4-, and 5-decene as well as branched alkenes resulting from rearrangement of carbocations. It is true that the most stable alkenes predominate, but many of them have stabilities that are close together.
Double-bond isomerization can take place in other ways. Nucleophilic allylic rearrangements were discussed in Chapter 10 (Sec. 10.E). Electrocyclic and sigmatropic rearrangements are treated at Reactions 18-27 to 18-35. Double-bond migrations have also been accomplished photochemically,66 and by means of metallic ion (most often complex ions containing Pt, Rh, or Ru) or metal carbonyl catalysts.67 With metal compounds there are at least two possible mechanisms. One of these, which requires external hydrogen, is called the metal hydride addition–elimination mechanism:
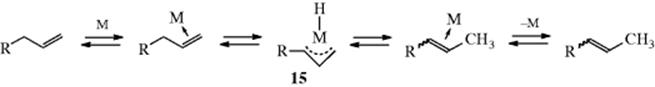
![]()
The other mechanism, called the π-allyl complex mechanism, does not require external hydrogen and proceeds by hydrogen abstraction to form the η3-π-allyl complex 15 (see Sec. 3.C.i, category 1 and Reaction 10-60). Another difference between the two mechanisms is that the former involves 1,2- and the latter 1,3-shifts. The isomerization of 1-butene Rh catalyzed reaction is an example that takes place by the metal hydride mechanism,68 while an example of the π-allyl complex mechanism is found in the Fe3(CO)12 catalyzed isomerization of 3-ethyl-1-pentene.69 A Pd catalyst was used to convert alkynones (RCOC![]() CCH2CH2R′) to 2,4-alkadien-1-ones (RCOCH=CHCH=CHCHR′).70 The reaction of an en-yne with HSiCl3 and a Pd catalyst generated an allene with moderate enantioselectivity (see Sec. 4.C, category 5 for chiral allenes).71
CCH2CH2R′) to 2,4-alkadien-1-ones (RCOCH=CHCH=CHCHR′).70 The reaction of an en-yne with HSiCl3 and a Pd catalyst generated an allene with moderate enantioselectivity (see Sec. 4.C, category 5 for chiral allenes).71
The metal-catalysis method has been used for the preparation of simple enols, by isomerization of allylic alcohols, for example.72 Some enols are stable enough for isolation (see Sec. 4.Q.iv), but slowly tautomerize to the aldehyde or ketone, with half-lives ranging from 40 to 50 min to several days.72
No matter which of the electrophilic methods of double-bond shifting is employed, the thermodynamically most stable alkene is usually formed in the largest amount, although a few anomalies are known. An indirect method of double-bond isomerization is known, leading to migration in the other direction. This involves conversion of the alkene to a borane (Reaction 15-16), rearrangement of the borane (Reaction 18-11), oxidation and hydrolysis of the newly formed borane to the alcohol (17) (see Reaction 12-31), and dehydration of the alcohol (Reaction 17-1) to the alkene. The reaction is driven by the fact that with heating the addition of borane is reversible, and the equilibrium favors formation of the less sterically hindered borane, 16 in this case.
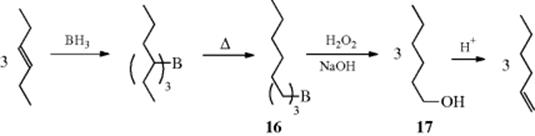
Since the migration reaction is always toward the end of a chain, terminal alkenes can be produced from internal ones, so the migration is often opposite to that with the other methods. Alternatively, the rearranged borane can be converted directly to the alkene by heating with an alkene of molecular weight higher than that of the product (Reaction 17-15). Photochemical isomerization can also lead to the thermodynamically less stable isomer.73
See Reaction 15-1 for related reactions in which double bonds migrate or isomerize.
![]()
Triple bonds can also migrate in the presence of bases,74 but through an allene intermediate:75 In general, strong bases (e.g., NaNH2) convert internal alkynes to terminal alkynes (a particularly good base for this purpose is potassium 3-aminopropylamide, NH2CH2CH2CH2NHK76), because the equilibrium is shifted by formation of the acetylide ion. With weaker bases (e.g., NaOH), which are not strong enough to remove the acetylenic proton, the internal alkynes are favored because of their greater thermodynamic stability. In some cases, the reaction can be stopped at the allene stage.77 The reaction then becomes a method for the preparation of allenes.78 The reaction of propargylic alcohols with tosylhydrazine (PPh3) and DEAD also generates allenes.79 In a related reaction, base induced isomerization of propargylic alcohols leads to conjugated ketones in some cases.80 Acid-catalyzed migration of triple bonds (with allene intermediates) can be accomplished if very strong acids (e.g., HF–PF5) are used.81 If the mechanism is the same as that for double bonds, vinyl cations are intermediates.
OS II, 140; III, 207; IV, 189, 192, 195, 234, 398, 683; VI, 68, 87, 815, 925; VII, 249; VIII, 146, 196, 251, 396, 553; X, 156, 165; 81, 147
12-3 Keto–Enol Tautomerization
3/ O-Hydro-de-hydrogenation
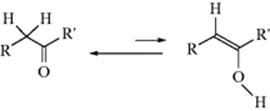
The tautomeric equilibrium between enols and ketones or aldehydes (keto–enol tautomerism) is a form of prototropy,82 but is not normally a preparative reaction. For some ketones, however, both forms can be prepared (see Sec. 2.N.i, category 3 for a discussion of this and other types of tautomerism). Keto–enol tautomerism occurs in systems containing one or more carbonyl groups linked to sp3 carbons bearing one or more hydrogen atoms. The keto is generally more stable than the enol tautomer for neutral systems, and for most ketones and aldehydes only the keto form is detectable under ordinary conditions. The availability of additional intramolecular stabilization through hydrogen bonding or complete electron delocalization (as in phenol), may cause the enol tautomer to be favored.
Keto–enol tautomerism is usally a slow process, but it can be catalyzed by a trace of acid or base.83 In this equilibrium, the heteroatom is the basic site and the proton is the acidic site. For tautomerism in general (see Sec. 2.N.i),84the presence of an acid or a base is not necessary to initiate the isomerization since each tautomeric substance possesses amphiprotic properties.84 Polar protic solvents (e.g., water or alcohol) may participate in the proton transfer by forming a cyclic or a linear complex with the tautomers.85 Whether the complex formed is cyclic or linear depends on the conformation and configuration of the tautomers. In a strongly polar aprotic solvent and in the presence of an acid or a base, the tautomeric molecule may lose or gain a proton and form the corresponding mesomeric anion or cation, which, in turn, may gain or lose a proton, respectively, and yield a new tautomeric form.86 The structural features of the carbonyl compound influences the equilibrium.87 Differing conjugative stabilization by CH-π orbital overlap does not directly influence stereoselectivity, and steric effects are generally not large enough to cause the several kilocalorie per mole (kcal mol−1) energy difference seen between transition structures unless there is exceptional crowding.88 Note that sterically stabilized enols are known,89 including arylacetaldehydes.90 Torsional strain involving vicinal bonds does contribute significantly to stereoselectivity in enolate formation.88
The acid base catalyzed mechanisms are identical to those in Reaction 12-2.91
Acid catalyzed
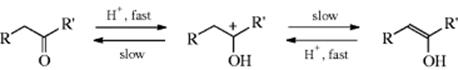
Base catalyzed92
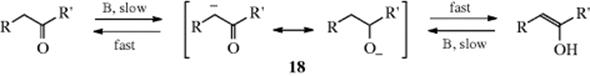
For each catalyst, the mechanism for one direction is the exact reverse of the other, by the principle of microscopic reversibility.93 As expected from mechanisms in which the C–H bond is broken in the rate-determining step, substrates of the type RCD2COR show deuterium isotope effects (of ~5) in both the basic-94 and the acid95-catalyzed processes. The keto–enol/enolate anion equilibrium has been studied in terms of the influence of β-oxygen96 or β-nitrogen97 substituents. The stereochemistry of enol protonation can be controlled by varying the proximal group and by changing the acidity of the medium.98
The base induced reaction generates an enolate anion rather than an enol, and the formation of and reactions of enolate anions are discussed further in Reactions 10-60, 10-67, 16-24, and 16-34. Note that ring strain plays no significant role on the rate of base-catalyzed enolization.99 In certain cases (e.g., benzofuranones), base-induced enolate anion formation may give a transition state in which aromaticity can play a role. One study showed that aromatic stabilization of the transition state is ahead of proton transfer, and aromaticity appears to lower the intrinsic barrier to this reaction.100 Enolizable hydrogen atoms can be replaced by deuterium (and ![]() by
by ![]() ) by passage of a sample through a deuterated (or
) by passage of a sample through a deuterated (or ![]() containing) gas-chromatography column.101
containing) gas-chromatography column.101
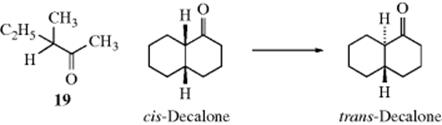
Although the conversion of an aldehyde or a ketone to its enol tautomer is not generally a preparative procedure, the reactions do have their preparative aspects. When enol ethers or esters are hydrolyzed, the initially formed enols immediately tautomerize to the aldehydes or ketones. In addition, the overall processes (forward plus reverse reactions) are often used for equilibration purposes. When an optically active compound in which the chirality is due to a stereogenic carbon α to a carbonyl group (as in 19) is treated with acid or base, racemization results.102 If there is another stereogenic center in the molecule, the less stable diastereomer can be converted to the more stable one in this manner. For example, cis-decalone can be equilibrated to the trans isomer. Isotopic exchange can similarly be accomplished at the α position of an aldehyde or ketone. In cyclic compounds, cis- to trans- isomerization can occur via the enol.103 The role of additives (e.g., ZnCl2) on the stereogenic enolization reactions using chiral cases has been discussed.104
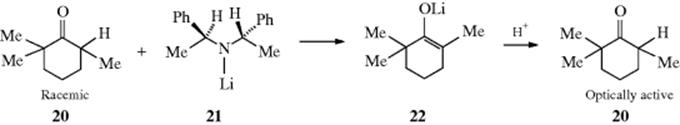
If a full equivalent of base per equivalent of ketone is used, the enolate ion (18) is formed and can be isolated105 (see, e.g., the alkylation reaction in Reaction 10-68).106 Enantioselective enolate anion protonation reactions have been studied.107 Enolate protonation is discussed in section Reaction 16-34. For the acid-catalyzed process, exchange or equilibration is accomplished only if the carbonyl compound is completely converted to the enol and then back, but in the base-catalyzed process exchange or equilibration can take place if only the first step (conversion to the enolate ion) takes place. The difference is usually academic. Aggregation behavior of stereoselective enolizations mediated by Mg and Ca bis(amides) have been studied.108
In the case of the ketone (20), a racemic mixture was converted to an optically active mixture (optical yield 46%) by treatment with the chiral base (21).109 This happened because 21 reacted with one enantiomer of 20 faster than with the other (an example of kinetic resolution). The enolate (22) must remain coordinated with the chiral amine, and it is the amine that reprotonate 22, not an added proton donor.
There are many enol–keto interconversions and acidification reactions of enolate ions to the keto forms listed in Organic Syntheses. No attempt is made to list them here.
B. Halogen Electrophiles
Halogenation of unactivated hydrocarbons is discussed in Reaction 14-1.
12-4 Halogenation of Aldehydes and Ketones
Halogenation or Halo-de-hydrogenation
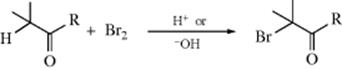
Aldehydes and ketones can be halogenated in the α position with bromine, chlorine, or iodine,110 although the reaction is less successful with fluorine.111 Sulfuryl chloride,112 Me3SiCl–Me2SO,113 and NCS114 have been used as reagents for chlorination. α-Chloroaldehydes are formed with Cl2 and a catalytic amount of tetraethylammonium chloride.115 Bromination methods include NBS (see Reaction 14-3),116 Me3SiBr–DMSO,117 tetrabutylammonium tribromide,118 in situ generated ZnBr2 in water,119 and bromine•dioxane on silica with microwave irradiation.120 α-Chlorination121 and also bromination122 have been reported in ionic liquids. Enantioselective chlorination123 and bromination124 methods are known, including methods that use enolate anions as intermediates.125 Organocatalyzed asymmetric α-halogenation methods are known that can be applied to incorporation of virtually any halogen.126 β-Keto esters and 1,3-diketones are α-brominated using bromodimethylsulfonium bromide.127 1,3-Diketones, β-ketoesters, and malonates are chlorinated using sodium hypochlorite or brominated using sodium hypobromite.128
Iodination has been accomplished by the direct reaction of ketones with molecular iodine,129 with I2-cerium(IV) ammonium nitrate,130 NCS/NaI,131 ICl/NaI/FeCl3,132 and with iodine using 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) in methanol.133 Methyl ketones react with NIS and tosic acid with microwave irradiation without solvent to give the α-iodoketone.134 An asymmetric iodination of aldehydes used NIS, with a catalytic amount of benzoic acid and a chiral biaryl amine.135
Although less prevalent than those noted above, several methods have been reported for the preparation of α-fluoro aldehydes and ketones,136 including enantioselective fluorination protocols.137 Organocatalytic α-fluorination is known for aldehydes and ketones.138 Selectfluor, [F–TEDA–BF4. 1-Fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)] has been used for the monofluorination of ketones,139 as has a mixture of KI–KIO3–H2SO4.140 Active compounds (e.g., β-keto esters and β-diketones) have been fluorinated with an N-fluoro-N-alkylsulfonamide141 (this can result in enantioselective fluorination, if an optically active N-fluorosulfonamide is used142), with F2/N2–HCO2H,143 and with NF3O/Bu4NOH.144 Acetyl hypofluorite fluorinates simple ketones in the form of their lithium enolate anions.145 Aldehydes have been α-fluorinated using N-fluorobenzenesulfonimide as an electrophilic source of fluorine and an imidazolidinone as an organocatalyst.146 The enantioselective α-fluorination of oxindoles has been reported using N-fluorobenzenesulfonimide, a Pd catalyst, and a chiral ligand,147 and also with an organocatalyst.148
For unsymmetrical ketones, the preferred position of halogenation is usually the more substituted: a CH group, then a CH2 group, and then CH3149; however, mixtures are frequent. With aldehydes the aldehydic hydrogen is sometimes replaced, but only when there is no α-hydrogen and the reaction is generally not very useful (see Reaction 14-4). It is also possible to prepare di- and polyhalides. When basic catalysts are used, one α position of a ketone is completely halogenated before the other is attacked, and the reaction cannot be stopped until all the hydrogen atoms of the first carbon have been replaced (see below). If one of the groups is methyl, the haloform reaction (12-44) takes place. With acid catalysts, it is usually possible to stop the reaction after only one halogen has been incorporated, although a second halogen can be introduced by the use of excess reagent. In chlorination, the second halogen generally appears on the same side as the first,150 while in bromination the α,α′-dibromo product is found.151 Actually, with both halogens it is the α,α-dihalo ketone that is formed first, but in the case of bromination this compound isomerizes under the reaction conditions to the α,α′-isomer.150 α,α′-Dichloro ketones are formed by reaction of a methyl ketone with an excess of CuCl2 and LiCl in DMF152 or with HCl and H2O2 in methanol.153 Aryl methyl ketones can be dibrominated in high yields with benzyltrimethylammonium tribromide.154 Active methylene compounds are chlorinated with NCS and Mg(ClO4)2.155 Similar chlorination in the presence of a chiral copper catalyst led to α-chlorination with modest enantioselectivity.156
It is not the aldehyde or ketone itself that is halogenated, but the corresponding enol or enolate ion. The purpose of the catalyst is to provide a small amount of enol or enolate (Reaction 12-3). The reaction is often done without addition of acid or base, but traces of acid or base are always present, and these are enough to catalyze formation of the enol or enolate. With acid catalysis the mechanism is
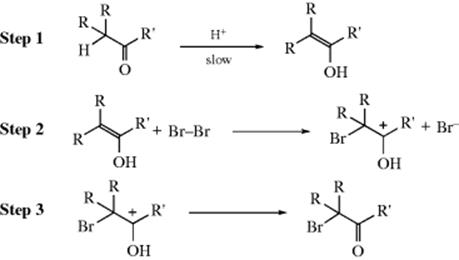
The first step, as seen in Reaction 12-3, actually consists of two steps. The second step is very similar to the first step in electrophilic addition to double bonds (Sec. 15.A.i). There is a great deal of evidence for this mechanism: (1) the rate is first order in substrate; (2) bromine does not appear in the rate expression at all,157 a fact consistent with a rate-determining first step;158 (3) the reaction rate is the same for bromination, chlorination, and iodination under the same conditions;159 (4) the reaction shows an isotope effect; and (5) the rate of the step 2–step 3 sequence has been independently measured (by starting with the enol) and found to be very fast.160
With basic catalysts the mechanism may be the same as that given above (since bases also catalyze formation of the enol), or the reaction may go directly through the enolate ion without formation of the enol:
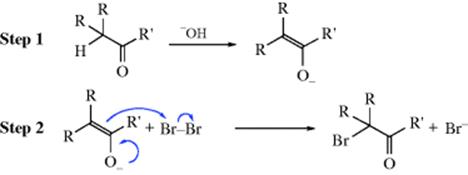
It is difficult to distinguish the two possibilities. It was mentioned above that in the base-catalyzed reaction, if the substrate has two or three α halogens on the same side of the C=O group, it is not possible to stop the reaction after just one halogen atom has entered. The reason is that the electron-withdrawing field effect of the first halogen increases the acidity of the remaining hydrogen atoms; that is, a CHX group is more acidic than a CH2 group, so that the initially formed halo ketone is converted to enolate ion (and hence halogenated) more rapidly than the original substrate. Other halogenating agents can be used in this reaction.
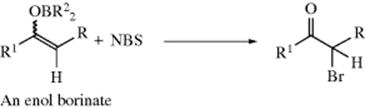
Regioselectivity in the halogenation of unsymmetrical ketones can be attained by treatment of the appropriate enol borinate of the ketone with NBS or NCS.161 The desired halo ketone is formed in high yield. The appropriate lithium enolate can be brominated at a low temperature162 (see Reaction 10-68, category 4 for the regioselective formation of enolate ions). α-Halo aldehydes have been prepared in good yield by treatment of silyl enol ethers (R2C=CHOSiMe3) with Br2 or Cl2,163 with sulfuryl chloride (SO2Cl2),164 or with I2 and silver acetate.165 Silyl enol ethers generate α-chloroketones with good enantioselectivity using ZrCl4 in conjunction with an α,α-dichloromalonate ester.166 Silyl enol ethers can also be fluorinated, with XeF2167 or with 5% F2 in N2 at −78 °C in FCCl3.168 Enol acetates have been regioselectively iodinated with I2 and either Th(I) acetate169 or Cu(II) acetate.170
α,β-Unsaturated ketones can be converted to α-halo-α,β-unsaturated ketones by treatment with phenylselenium bromide or chloride,171 and to α-halo-β,γ-unsaturated ketones by two-phase treatment with HOCl.172 Conjugated ketones were converted to the α-bromo conjugated ketone (a vinyl bromide) using the Dess–Martin periodinane (see Reaction 19-3, category 5) and tetraethylammonium bromide.173
OS I, 127; II, 87, 88, 244, 480; III, 188, 343, 538; IV, 110, 162, 590; V, 514; VI, 175, 193, 368, 401, 512, 520, 711, 991; VII, 271; VIII, 286. See also, OS VI, 1033; VIII, 192.
12-5 Halogenation of Carboxylic Acids and Acyl Halides
Halogenation or Halo-de-hydrogenation
![]()
The α hydrogen atoms of carboxylic acids are replaced by bromine or chlorine using a phosphorus halide as catalyst.174 The reaction, known as the Hell–Volhard–Zelinskii reaction, is not applicable to iodine or fluorine. When there are two α hydrogen atoms, one or both may be replaced, although it is often hard to stop with just one. The reaction actually takes place on the acyl halide formed initially from the carboxylic acid and the halogenating reagent. This means that each molecule of acid is α halogenated while it is in the acyl halide stage. The acids alone are inactive, except for those with relatively high enol content (e.g., malonic acid). Less than one full molar equivalent of catalyst (per molar equivalent of substrate) is required, because of the exchange reaction between carboxylic acids and acyl halides (see Reaction 16-79). The halogen from the catalyst is not transferred to the α position. For example, the use of Cl2 and PBr3 results in α-chlorination, not bromination. As expected from the foregoing, acyl halides undergo a halogenation without a catalyst. An enantioselective α-halogenation was reported to give chiral α-haloesters via an alkaloid-catalyzed reaction of acyl halides with perhaloquinone-derived reagents.175 So do anhydrides and many compounds that enolize easily (e.g., malonic ester and aliphatic nitro compounds). The mechanism is usually regarded as proceeding through the enol as in Reaction 12-4.176 If chlorosulfuric acid (ClSO2OH) is used as a catalyst, carboxylic acids can be α-iodinated,177 as well as chlorinated or brominated.178 N-Bromosuccinimide in a mixture of sulfuric acid–trifluoroacetic acid can monobrominate simple carboxylic acids.179
A number of other methods exist for the α halogenation of carboxylic acids or their derivatives.180 Under electrolytic conditions with NaCl, malonates are converted to 2-chloro malonates.181 Acyl halides can be a brominated or chlorinated by use of NBS or NCS and HBr or HCl.182 The latter is an ionic, not a free radical halogenation (see Reaction 14-3). Direct iodination of carboxylic acids has been achieved with I2-Cu(II) acetate in HOAc.183 Acyl chlorides can be α iodinated with I2 and a trace of HI.184 Carboxylic acids, esters, and amides have been α-fluorinated at −78 °C with F2 diluted in N2.185 Amides have been α-iodinated using iodine and s-collidine.186
OS I, 115, 245; II, 74, 93; III, 347, 381, 495, 523, 623, 705, 848; IV, 254, 348, 398, 608, 616; V, 255; VI, 90, 190, 403; IX, 526. Also see, OS IV, 877; VI, 427.
12-6 Halogenation of Sulfoxides and Sulfones
Halogenation or Halo-de-hydrogenation
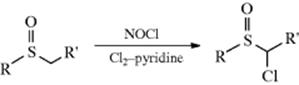
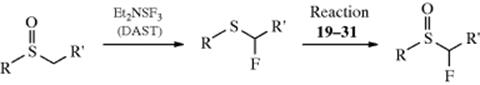
Sulfoxides can be chlorinated in the α position187 by treatment with Cl2188 or NCS,189 in the presence of pyridine. These methods involve basic conditions. The reaction can also be accomplished in the absence of base with SO2Cl2in CH2Cl2,190 or with TsNCl2.191 The bromination of sulfoxides with bromine192 and with NBS–bromine193 have also been reported. Sulfones have been chlorinated by treatment of their conjugate bases (RSO2C−HR′) with various reagents, among them SO2Cl2, CCl4,194 or NCS.195 The α-fluorination of sulfoxides was reported via treatment with diethylaminosulfur trifluoride (Et2NSF3, DAST) to give an α-fluoro thioether, usually in high yield. Oxidation of this compound with m-chloroperoxybenzoic acid gave the sulfoxide.196
C. Nitrogen Electrophiles
12-7 Aliphatic Diazonium Coupling
Arylhydrazono-de-dihydro-bisubstitution
![]()
If a C–H unit is acidic enough, that carbon couples with diazonium salts in the presence of a base (via the enolate anion), most often aq sodium acetate.197 The reaction is commonly carried out on compounds of the form Z–CH2–Z′, where Z and Z′ are as defined in Section 16-38 (e.g., β-keto esters, β-keto amides, malonic ester).
The mechanism is probably of the simple SE1 type:
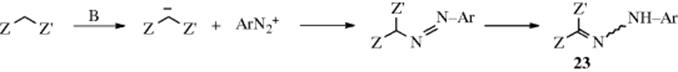
Aliphatic azo compounds in which the carbon containing the azo group is attached to a hydrogen are unstable and tautomerize to the isomeric hydrazones (23), which are the products of the reaction.
When the reaction is carried out on a compound of the form Z–CHR–Z′, the azo compound does not have a hydrogen that can lead to tautomerism, and at least one Z is acyl or carboxyl, this group usually cleaves:
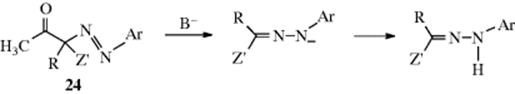
so the product in this case is also the hydrazone, and not the azo compound. In fact, compounds of the type 24 are seldom isolable from the reaction, although this has been accomplished.198 The cleavage step shown is an example of Reaction 12-43 and, when a carboxyl group cleaves, of Reaction 12-40. The overall process in this case is called the Japp–Klingemann reaction199 and involves conversion of a ketone (25) or a carboxylic acid (26) to a hydrazone (27). When an acyl and a carboxyl group are both present, the leaving group order has been reported to be MeCO > COOH > PhCO.200 When there is no acyl or carboxyl group present, the aliphatic azo compound is stable.

OS III, 660; IV, 633.
12-8 Nitrosation at a Carbon Bearing an Active Hydrogen

Carbons adjacent to a Z group (as defined in Reaction 10-67) can be nitrosated with nitrous acid or alkyl nitrites.201 The initial product is the C-nitroso compound, but these are stable only when there is no hydrogen that can undergo tautomerism. When there is, the product is the more stable oxime. The situation is analogous to that with azo compounds and hydrazones (Reaction 12-7). The mechanism is similar to that in Reaction 12-7:202 R–H → R− + +N=O → R–N=O. The reactive species is either NO+ or a carrier of it. When the substrate is a simple ketone, the mechanism goes through the enol (as in halogenation Reaction 12-4):
Evidence is that the reaction, in the presence of X− (Br−, Cl−, or SCN−) was first order in ketone and in H+, but zero order in HNO2 and X−.203 Furthermore, the rate of the nitrosation was about the same as that for enolization of the same ketones. The species NOX is formed by HONO + X− + H+ → HOX + H2O. In the cases of F3CCOCH2COCF3 and malononitrile, the nitrosation went entirely through the enolate ion rather than the enol.204
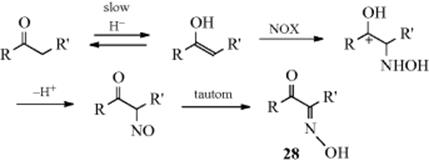
As in the Japp–Klingemann reaction, when Z is an acyl or carboxyl group (in the case of R2CH–Z), it can be cleaved. Since oximes and nitroso compounds can be reduced to primary amines, this reaction often provides a route to amino acids. As in the case of Reaction 12-4, the silyl enol ether of a ketone can be used instead of the ketone itself.205 Good yields of α-oximinoketones (28) can be obtained by treating ketones with tert-butyl thionitrate.206
Imines can be prepared in a similar manner by treatment of an active hydrogen compound with a nitroso compound:

Alkanes can be nitrosated photochemically, by treatment with NOCl and UV light.207 For nitration at an activated carbon, see Reaction 12-9. Trialkyltin enol ethers (C=C–O–SnR3) react with PhNO to give α-(N-hydroxylamino)ketones.208
OS II, 202, 204, 223, 363; III, 191, 513; V, 32, 373; VI, 199, 840. Also see, OS V, 650.
12-9 Nitration of Alkanes
Nitration or Nitro-de-hydrogenation
![]()
Nitration of alkanes209 can be carried out in the gas phase at ~400 °C or in the liquid phase. The reaction is not practical for the production of pure products for any alkane except methane. For other alkanes, not only does the reaction produce mixtures of the mono-, di-, and polynitrated alkanes at every combination of positions, but extensive chain cleavage occurs.210 A free radical mechanism is involved.211
![]()
Activated positions (e.g., ZCH2Z′ compounds) can be nitrated by fuming nitric acid in acetic acid, by acetyl nitrate and an acid catalyst,212 or by alkyl nitrates under alkaline conditions.213 In the latter case, it is the carbanionic form of the substrate that is actually nitrated. The conjugate base of the nitro compound is isolated under these alkaline conditions, but yields are not high. Of course, the mechanism in this case is not of the free radical type, but is electrophilic substitution with respect to the carbon (similar to the mechanisms of Reactions 12-7 and 12-8). Positions activated by only one electron-withdrawing group (e.g., α positions of simple ketones, nitriles, sulfones, or N,N-dialkyl amides) can be nitrated with alkyl nitrates if a very strong base (e.g., t-BuOK or NaNH2,) is present to convert the substrate to the carbanionic form.214
Electrophilic nitration of alkanes has been performed with nitronium salts (e.g., NO2+ PF6− and with HNO3–H2SO4 mixtures), but mixtures of nitration and cleavage products are obtained and yields are generally low.215 The reaction of alkanes with nitric acid and N-hydroxysuccinimide (NHS), however, gave moderate-to-good yields of the corresponding nitroalkane.216 Similar nitration was accomplished with NO2, NHS and air.217 Aliphatic nitro compounds can be a nitrated [R2C-NO2 → R2C(NO2)2] by treatment of their conjugate bases RCNO2 with NO2− and K3Fe(CN)6.218
OS I, 390; II, 440, 512.
12-10 Direct Formation of Diazo Compounds
Diazo-de-dihydro-bisubstitution
![]()
Compounds containing a CH2 bonded to two Z groups (active methylene compounds, with Z as defined in Reaction 10-67) can be converted to diazo compounds on treatment with tosyl azide in the presence of a base.219 The use of phase-transfer catalysis increases the convenience of the method.220 Sulfonyl azides also give the reaction.221 The diazo-transfer reaction can also be applied to other reactive positions (e.g., the 5 position of cyclopentadiene).222The mechanism is probably as follows:
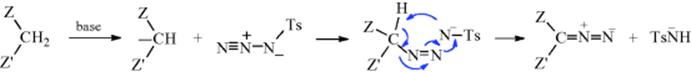
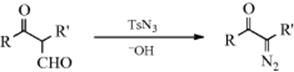
A diazo group can be introduced adjacent to a single carbonyl group indirectly by first converting the ketone to an α-formyl ketone (Reaction 16-85) and then treating it with tosyl azide. As in the similar cases of Reactions 12-7and 12-8, the formyl group is cleaved during the reaction.223
OS V, 179; VI, 389, 414.
12-11 Conversion of Amides to α-Azido Amides
Azidation or Azido-de-hydrogenation
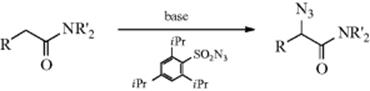
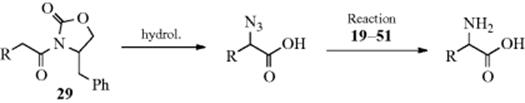
In Reaction 12-10, treatment of Z–CH2–Z′ with tosyl azide gave the α-diazo compound via diazo transfer. When this reaction is performed on a compound with a single Z group (e.g., an amide), formation of the azide becomes a competing process via the enolate anion.224 Factors favoring azide formation rather than diazo transfer include K+ as the enolate counterion rather than Na+ or Li+ and the use of 2,4,6-triisopropylbenzenesulfonyl azide rather than TsN3. When the reaction was applied to amides with a chiral R′ (e.g., the oxazolidinone derivative 29), it was highly stereoselective, and the product could be converted to an optically active amino acid.224
12-12 Direct Amination at an Activated Position
Alkyamino-de-hydrogenation, and so on
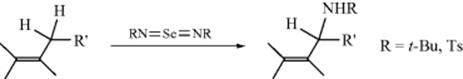
Alkenes can be aminated225 in the allylic position by treatment with solutions of imido selenium compounds (R–N=Se=N–R).226 The reaction, which is similar to the allylic oxidation of alkenes with SeO2 (see Reaction 19-14), has been performed with R = t-Bu and R = Ts. The imido sulfur compound TsN=S=NTs has also been used,227 as well as PhNHOH–FeCl2/FeCl3.228 Benzylic positions can be aminated with t-BuOOCONHTs in the presence of a catalytic amount of Cu(OTf)2.229 Enantioselective allylic amination has been reported using organocatalyts.230 A Rh catalyzed amination of benzylic positions has also been reported.231
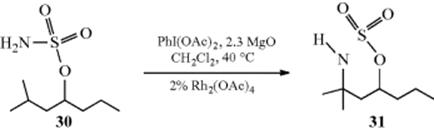
Tertiary alkyl hydrogen can be replaced in some cases via C–H nitrogen insertion. The reaction of sulfamate ester (30) with PhI(OAc)2, MgO, and a dinuclear Rh carboxylate catalyst, for example, generated oxathiazinane (31).232This transformation is a formal oxidation, and primary carbamates have been similarly converted to oxazolidin-2-ones.233
Amination of 1,3-dicarbonyl compounds can be done using functionalized dimides and an appropriate catalyst, generating the corresponding hydrazone. Enantioselective amination using this method has been reported, using a chiral guanidine catalyst.234
See also, Reaction 10-39.
12-13 Insertion by Nitrenes
CH-[Acylimino]-insertion, and so on

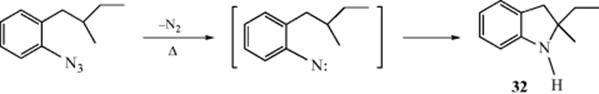
Carbonylnitrenes (:NCOW, W = R′, Ar, or OR′) are very reactive species (Sec. 5.E) and insert into the C–H bonds of alkanes to give amides (W = R′ or Ar) or carbamates (W = OR′).235 The nitrenes are generated as discussed in Section 5.E. The order of reactivity among alkane C–H bonds is tertiary > secondary > primary.236 Nitrenes are much more selective (and less reactive) in this reaction than carbenes (Reaction 12-17).237 It is likely that only singlet and not triplet nitrenes insert.238 Retention of configuration is found at a stereogenic carbon.239 The mechanism is presumably similar to the simple one-step mechanism for insertion of carbenes (Reaction 12-21). Other nitrenes [e.g., cyanonitrene (NCN)240 and arylnitrenes (NAr)241] can also insert into C–H bonds, but alkylnitrenes usually undergo rearrangement before they can react with the alkane. The Au(III) catalyzed insertion of nitrenes into aromatic and benzylic C–H groups has been reported.242 N-Carbamoyl nitrenes undergo insertion reactions that often lead to mixtures of products, but exceptions are known,243 chiefly in cyclizations.244 For example, heating of 2-(2-methylbutyl)phenyl azide gave ~60% 2-ethyl-2-methylindoline (32).239 Enantioselective nitrene insertion reactions are known.245
D. Sulfur Electrophiles
12-14 Sulfenylation, Sulfonation, and Selenylation of Ketones and Carboxylic Esters
Alkylthio-de-hydrogenation, and so on
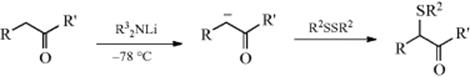
Sulfonation or Sulfo-de-hydrogenation
Ketones, carboxylic esters (including lactones),246 and amides (including lactams)247 can be sulfenylated248 in the α position by conversion to the enolate anion (see Sec. 8.F, part 7), and subsequent treatment with a disulfide.249The reaction, shown above for ketones, involves nucleophilic substitution at sulfur. α-Phenylseleno ketones [RCH(SePh)COR′] and α-phenylseleno esters [RCH(SePh)COOR′] can be similarly prepared250 by treatment of the corresponding enolate anions with PhSeBr,251 PhSeSePh,252 or benzeneseleninic anhydride [PhSe(O)OSe(O)Ph].253 Another method for the introduction of a phenylseleno group into the α position of a ketone involves simple treatment of an ethyl acetate solution of the ketone with PhSeCl (but not PhSeBr) at room temperature.254 This procedure is also successful for aldehydes but not for carboxylic esters. N-Phenylselenophthalimide has been used to convert ketones255 and aldehydes256 to the α- PhSe derivative. Silyl enol ethers are converted to α-alklylthio and α-arylthio ketones via a sulfenylation method, driven by aromatization of an added quinone mono-O,S-acetal in the presence of Me3SiOTf.257
The α-seleno and α-sulfenyl carbonyl compounds prepared by this reaction can be converted to α,β-unsaturated carbonyl compounds (Reaction 17-12). The sulfenylation reaction has also been used258 as a key step in a sequence for moving the position of a carbonyl group to an adjacent carbon.259

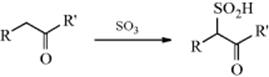
Aldehydes, ketones, and carboxylic acids containing α hydrogen atoms can be sulfonated with sulfur trioxide.260 The mechanism is presumably similar to that of Reaction 12-4. Sulfonation has also been accomplished at vinylic hydrogen.
OS VI, 23, 109; VIII, 550. OS IV, 846, 862.
E. Carbon Reagents
12-15 Alkylation and Alkenylation of Alkenes
Alkylation or Alkyl-de-oxysulfonation (de-halogenation), Arylation or Aryl-de-oxysulfonation (de-halogenation), and so on
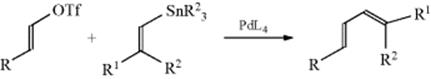
Vinyl triflates (C=C–OSO2CF3) react with vinyl tin derivatives in the presence of Pd catalysts to form dienes, in what is known as Stille coupling.261 Phosphine or bis(phosphine) ligands are most commonly used with the Pd catalyst,262 but other ligands have been used,263 including triphenylarsine.264 Vinyl triflates can be prepared from the enolate anion by reaction with N-phenyl triflimide.265 Vinyltin compounds are generally prepared by the reaction of an alkyne with an trialkyltin halide (see Reactions 15-17 and 15-21).266 Stille cross-coupling reactions are an important variation of the basic reaction,267 including cross-coupling reactions of unactivated secondary halides and monoorganotin reagents.268 Stille reactions are compatible with many functional groups. Vinyl halides can be used,269 and allenic tin compounds have been used.270 Intramolecular reactions are possible.271 Stille coupling has been done using microwave irradiation,272 in fluorous solvents,273 and in supercritical carbon dioxide (see Sec. 9.D.ii).274 Stille coupling using alkynes as a substrate are known.275
This reaction is highly stereoselective, and proceeds with a retention of geometry of the C=C units, and are usually regiospecific with respect to the newly formed C–C σ-bond. Cine substitution is known with this reaction, and its mechanism has been studied.276 Using ArSnCl3 derivatives, Stille coupling can be done in aq KOH.277
Aryl halides,278 heteroaryl halides,279 and heteroaryl triflates280 can be coupled to vinyltin reagents281 using a Pd catalyst. A Mo catalyzed variation is known.282 A Cu catalyzed cross coupling variation283 has been reported in ionic liquids.284 Vinyl halides can be coupled to alkenes to form dienes.285 The reaction of dihydrofurans with vinyl triflates and a Pd catalyst leads to a nonconjugated diene,286 illustrating that the product is formed by an elimination step, as with the Heck reaction (13-10), and double-bond migration can occur resulting in allylic rearrangement.
The accepted mechanism for the Stille reaction involves a catalytic cycle287 in which an oxidative addition288 and a reductive elimination step289 are fast, relative to Sn/Pd transmetalation (the rate-determining step).290 It appears that the greater the coordinating ability of the unsaturated species is important, and a coordinated solvent molecule is likely involved in the electrophilic substitution at tin. Another mechanism has been proposed, in which oxidative addition of the vinyl triflate to the ligated Pd gives a cis-Pd complex that isomerizes rapidly to a trans-Pd complex, which then reacts with the organotin compound following an SE2 (cyclic) mechanism, with release of a ligand.291This pathway gives a bridged intermediate, and subsequent elimination of XSnBu3 yields a three-coordinate species cis-Pd complex, which readily gives the coupling product.291 Most of the major intermediates have been intercepted, isolated, and characterized using electrospray ionization mass spectrometry.292
Cyclopropylboronic acids (Reaction 12-28) couple with vinylic halides293 or vinyl triflates294 to give vinylcyclopropanes, using a Pd catalyst. Vinyl borates (Reaction 12-28) were coupled to vinyl triflates using a Pd catalyst.295Vinyltrifluoroborates can be coupled to allylic chlorides using microwave irradiation296 and vinyl halides react with vinyltrifluoroborates to give dienes with high stereoselectivity.297 Stille coupling to enols has been reported.298 The coupling of vinyl silanes to give the symmetrically conjugated diene using CuCl and air has also been reported.299
Other methods are available to give Stille-like products. 1-Lithioalkynes were coupled to vinyl tellurium compounds (C=C–TeBu) using a Ni300 or a Pd catalyst301 to give a conjugated en-yne. 2-Alkynes (R–C![]() C–Me) react with HgCl2, n-butyllithium, and ZnBr2, sequentially, and then with vinyl iodides and a Pd catalyst to give the nonconjugated en-yne.302 Alkynyl groups can be coupled to vinyl groups to give ene-ynes, via reaction of silver alkynes (Ag–C
C–Me) react with HgCl2, n-butyllithium, and ZnBr2, sequentially, and then with vinyl iodides and a Pd catalyst to give the nonconjugated en-yne.302 Alkynyl groups can be coupled to vinyl groups to give ene-ynes, via reaction of silver alkynes (Ag–C![]() C–R) with vinyl triflates and a Pd catalyst.303 In the presence of CuI and a Pd catalyst, vinyl triflates304 or vinyl halides305 couple to terminal alkynes. Alkynyl zinc reagents (R–C
C–R) with vinyl triflates and a Pd catalyst.303 In the presence of CuI and a Pd catalyst, vinyl triflates304 or vinyl halides305 couple to terminal alkynes. Alkynyl zinc reagents (R–C![]() C–ZnBr) can be coupled to vinyl halides with a Pd catalyst to give the conjugated en-yne.306
C–ZnBr) can be coupled to vinyl halides with a Pd catalyst to give the conjugated en-yne.306
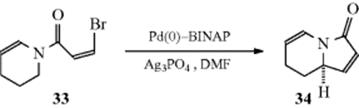
Alkyl groups can be coupled to a vinyl unit to give substituted alkenes. The reaction of vinyl iodides and EtZnBr, with a Pd catalyst, gave the ethylated alkene (C=C–Et).307 Aliphatic alkyl bromides reacted with vinyltin compounds to give the alkylated alkene using a Pd catalyst.308 Allylic tosylates were coupled to conjugated alkenes to give a non-conjugated diene using a Pd catalyst.309 An internal coupling reaction was reported in which an alkenyl enamide (33) reacted with Ag3PO4 and a chiral palladium catalyst to give 34 enantioselectively.310
For the related coupling reaction of alkenes and aryl compounds (arylation of alkenes), see Reaction 13-10.
12-16 Acylation at an Aliphatic Carbon
Acylation or Acyl-de-hydrogenation
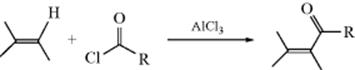
Alkenes can be acylated with an acyl halide and a Lewis acid catalyst in what is essentially a Friedel–Crafts Reaction (11-17) at an aliphatic carbon.311 The product can arise by two paths. The initial attack is by the π bond of the alkene unit on the acyl cation (RCO+; or on the acyl halide free or complexed; see Reaction 11-17) to give a carbocation, (35).
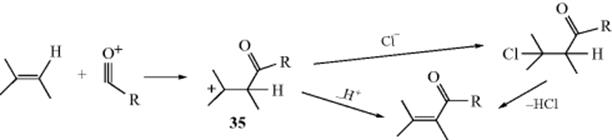
Ion 35 can either lose a proton or combine with chloride ion. If it loses a proton, the product is an unsaturated ketone. The mechanism is similar to the tetrahedral mechanism in Section 16.A.i, but with the charges reversed. If it combines with chloride, the product is a β-halo ketone, which can be isolated, so that the result is addition to the double bond (see Reaction 15-47). On the other hand, the β-halo ketone may, under the conditions of the reaction, lose HCl to give the unsaturated ketone, this time by an addition–elimination mechanism. In the case of unsymmetrical alkenes, the more stable alkene is formed (the more highly substituted and/or conjugated alkene, following Markovnikov's rule, see Sec. 15.B.ii). Anhydrides and carboxylic acids (the latter with a proton acid e.g., anhydrous HF, H2SO4, or polyphosphoric acid as a catalyst) are sometimes used instead of acyl halides. With some substrates and catalysts, double-bond migrations are occasionally encountered so that, for example, when 1-methylcyclohexene was acylated with acetic anhydride and zinc chloride, the major product was 6-acetyl-1-methylcyclohexene.312
Conjugated dienes can be acylated by treatment with acyl- or alkylcobalt tetracarbonyls, followed by base-catalyzed cleavage of the resulting π-allyl carbonyl derivatives313 (π-allyl metal complexes were discussed in Sec. 3.C.i. The reaction is very general. With unsymmetrical dienes, the acyl group generally substitutes most readily at a cis double bond, next at a terminal alkenyl group, and least readily at a trans double bond. The most useful bases are strongly basic, hindered amines (e.g., dicyclohexylethylamine). Acylation of vinylic ethers has been accomplished with aromatic acyl chlorides, a base, and a Pd catalyst: ROCH=CH2 → ROCH=CHCOAr.314
Formylation of alkenes can be accomplished with N-disubstituted formamides and POCl3.315 This is an aliphatic Vilsmeier reaction (see Reaction 11-18). Vilsmeier formylation can also be performed on the α position of acetals and ketals, so that hydrolysis of the products gives keto aldehydes or dialdehydes:316 A variation heated a 1,1-dibromoalkene with a secondary amine in aq DMF to give the corresponding amide.317
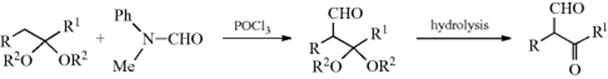
Acetylation of acetals or ketals can be accomplished with acetic anhydride and BF3–etherate.318 The mechanism with acetals or ketals also involves attack at an alkenyl carbon, since enol ethers are intermediates.318 Ketones can be formylated in the α position by treatment with CO and a strong base.319
OS IV, 555, 560; VI, 744. Also see, OS VI, 28.
12-17 Conversion of Enolates to Silyl Enol Ethers, Silyl Enol Esters, and Silyl Enol Sulfonate Esters
3/O-Trimethylsilyl-de-hydrogenation
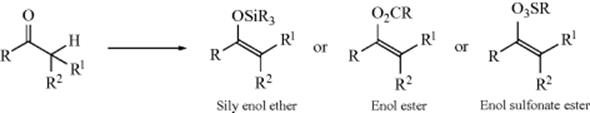
Silyl enol ethers,320 important reagents with a number of synthetic uses (see, e.g., Reactions 10-68, 12-4, 15-24, 15-64, and 16-36), can be prepared by base treatment of a ketone (converting it to its enolate anion) followed by addition of a trialkylchlorosilane. Other silylating agents have also been used.321 Both strong bases (e.g., LDA), and weaker bases (Et3N) have been used for this purpose.322 In some cases, the base and the silylating agent can be present at the same time.323 Enolate anions prepared in other ways (e.g., as shown in Reaction 10-58) also give the reaction.324 The reaction can be applied to aldehydes by the use of the base KH in 1,2-dimethoxyethane.325 A particularly mild method for conversion of ketones or aldehydes to silyl enol ethers uses Me3SiI and the base hexamethyldisilazane [(Me3Si)2NH.]326 Cyclic ketones can be converted to silyl enol ethers in the presence of acyclic ketones, by treatment with Me3SiBr, tetraphenylstibonium bromide (Ph4SbBr), and an aziridine.327 bis(Trimethylsilyl)acetamide is an effective reagent for the conversion of ketones to the silyl enol ether, typically giving the thermodynamic product (see below).328 Silyl enol ethers have also been prepared by the direct reaction of a ketone and a silane (R3SiH) with a Pt catalyst.329
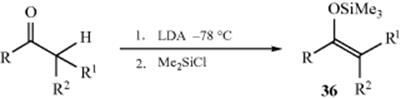
For substituted ketones, (E) and (Z) isomers are usually formed. For 36, the enol is (Z) when R1 is the priority group, but (E) when R2 is the priority group. In some cases, it is possible to control the selectivity to favor more of one isomer than the other. Treatment of 2-methyl-3-pentanone with LDA (THF, -78 °C), for example, gave a 60:40 mixture of the (Z) and (E) enolates.330 The base used to generate an enolate anion, the solvent and temperature, the conjugate acid of the base used, and the nature of the carbonyl substrate will all play a role in the selectivity. In general, equilibrating (thermodynamic) conditions [protic solvents (e.g., ethanol, water, or ammonia), a base generating a conjugate acid stronger than the starting ketone, more ionic counterions (e.g., K or Na), higher temperatures and longer reaction times] are expected to give more of the (E)-isomer. Conversely, kinetic conditions [aprotic solvents (e.g., ether or THF), a base generating a conjugate acid weaker than the starting ketone, more covalent counterions (e.g., Li, lower temperatures), and relatively short reaction times] usually give more of the (Z)-isomer. It is not always easy to predict the ratio, however. Either isomer is possible from aldehydes using the proper Rh catalyst.331
Magnesium diisopropylamide has been used to prepare kinetic silyl enol ethers in virtual quantitative yield.332 Reaction with Me3SiCl/KI in DMF gives primarily the thermodynamic silyl enol ether.333
An interesting synthesis of silyl enol ethers involves chain extension of an aldehyde. Aldehydes are converted to the silyl enol ether of a ketone upon reaction with lithium (trimethylsilyl)diazomethane and then a dirhodium catalyst.334 For example, initial reaction of lithium(trimethylsilyl)diazomethane [LTMSD, prepared in situ by reaction of butyllithium with (trimethylsilyl)diazomethane] to the aldehyde (e.g., 37) gave the alkoxide addition product. Protonation and then capture by a transition metal catalyst, and a 1,2-hydride migration gave the silyl enol ether, (38). Silyl enol ethers can be prepared from acyloin derivatives (see Reaction 19-78).335
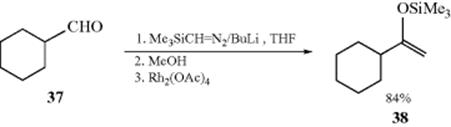
Enol acetates are generally prepared by the reaction of an enolate anion with a suitable acylating reagent.336 Enolate anions react with acyl halides and with anhydrides to give the acylated product. Both C- and O-acylation are possible, but in general O-acylation predominates.337 Note that the extent of O- versus C-acylation is very dependent on the local environment and electronic effects within the enolate anion.338 O-Benzoate enols are formed in good yield from aldehydes or 1,3-diketones in the presence of CuBr and tert-butylhydroperoxide.339 Silyl sulfonate esters can be prepared by similar methods, using sulfonic acid anhydrides rather than carboxylic anhydrides. A polymer-supported triflating agent was used to prepare silyl enol triflate from ketones, in the presence of diisopropylethylamine.340
When a silyl enol ether is the trimethylsilyl derivative (Me3Si–O-C=C), treatment with methyllithium will regenerate the lithium enolate anion and the volatile trimethylsilane (Me3SiH).341
OS VI, 327, 445; VII, 282, 312, 424, 512; VIII, 1, 286, 460; IX, 573. See also, OS VII, 66, 266. For the conversion of ketones to vinylic triflates,342 see OS VIII, 97, 126.
12-18 Conversion of Aldehydes to β-Keto Esters or Ketones
Alkoxycarbonylalkylation or Alkoxycarbonylalkyl-de-hydrogenation
![]()
β-Keto esters have been prepared in moderate to high yields by treatment of aldehydes with diethyl diazoacetate in the presence of a catalytic amount of a Lewis acid (e.g., SnCl2, BF3, or GeCl2).343 The reaction was successful for both aliphatic and aromatic aldehydes, but the former react more rapidly than the latter, and the difference is great enough to allow selective reactivity. In a similar process, aldehydes react with certain carbanions stabilized by boron, in the presence of (F3CCO)2O or NCS, to give ketones.344
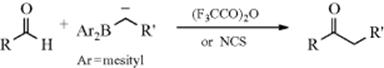
Ketones can be prepared from aryl aldehydes (ArCHO) by treatment with a Rh complex [(Ph3P)2Rh(CO)Ar′], whereby the Ar group is transferred to the aldehyde, producing the ketone (Ar–CO–Ar′).345 In another Rh catalyzed reaction, aryl aldehydes (ArCHO) react with Me3SnAr′ to give the diaryl ketone (Ar–CO–Ar′).346
Acylation of aryl halides with aldehydes gives arylketones in the presence of a Pd catalyst.347
12-19 Cyanation or Cyano-de-hydrogenation
![]()
There are several reactions in which a C–H unit is replaced by C–CN. In virtually all cases, the hydrogen being replaced is on a carbon α to a heteroatom or functional group. There are several examples.
Introduction of a cyano group α to the carbonyl group of a ketone can be accomplished by prior formation of the enolate anion with LDA in THF and addition of this solution to p-TsCN at −78 °C.348 The products are formed in moderate to high yields but the reaction is not applicable to methyl ketones. Treatment of TMSCH2N(Me)C=Nt-Bu with sec-butyllithium and R2C=O, followed by iodomethane and NaOMe leads to the nitrile (R2CH–CN).349

Cyanation has been shown to occur α to a nitrogen, specifically in N,N-dimethylaniline derivatives. Treatment with a catalytic amount of RuCl3 in the presence of oxygen and NaCN leads to the corresponding cyanomethylamine.350 Conversion of tertiary amines to the α-cyanoamine has been reported in the presence of FeCl2 and t-BuOOH.351
In a different kind of reaction, nitro compounds are α-cyanated by treatment with −CN and K3Fe(CN)6.352 The mechanism probably involves ion radicals. In still another reaction, secondary amines are converted to α-cyanoamines by treatment with phenylseleninic anhydride and NaCN or Me3SiCN.353,354
Another specialized reaction converts the methyl group of arenes (e.g., toluene) into a cyano group: toluene → benzonitrile, for example.355
12-20 Alkylation of Alkanes
Alkylation or Alkyl-de-hydrogenation
![]()
Alkanes can be alkylated by treatment with solutions of stable carbocations356 (Sec. 5.A.ii), but the availability of such carbocations is limited and mixtures are usually obtained. In a typical experiment, the treatment of propane with isopropyl fluoroantimonate (Me2HC+ SbF6−) gave 26% 2,3-dimethylbutane, 28% 2-methylpentane, 14% 3-methylpentane, and 32% n-hexane, as well as some butanes, pentanes (formed by Reaction 12-47), and higher alkanes. Mixtures arise in part because intermolecular hydrogen exchange (RH + R′+ → R+ + R′H) is much faster than alkylation, so that alkylation products are also derived from the new alkanes and carbocations formed in the exchange reaction. Furthermore, the carbocations present are subject to rearrangement (Chapter 18), giving rise to new carbocations. Products result from all the hydrocarbons and carbocations present in the system. As expected from their relative stabilities, secondary alkyl cations alkylate alkanes more readily than tertiary alkyl cations (the tert-butyl cation does not alkylate methane or ethane). Stable primary alkyl cations are not available, but alkylation has been achieved with complexes formed between CH3F or C2H5F and SbF5.357 The mechanism of alkylation can be formulated (similar to that shown in hydrogen exchange with superacids, Reaction 12-1) as
It is by means of successive reactions of this sort that simple alkanes like methane and ethane give tert-butyl cations in superacid solutions (Sec. 5.A.ii).358
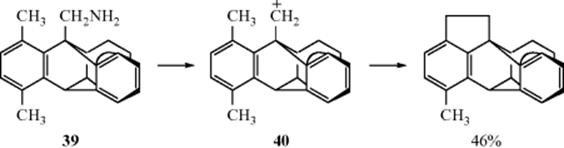
Intramolecular insertion has been reported. The positively charged carbon of the carbocation (40), generated from the diazonium salt of the triptycene compound (39), reacted with the CH3 group in close proximity with it.359
12-21 Insertion by Carbenes
CH-Methylene-insertion
![]()
The highly reactive species methylene (:CH2) inserts into C–H bonds,360 both aliphatic and aromatic,361 although with aromatic compounds subsequent ring expansion is also possible (see Reaction 15-64). This is effectively a homologation reaction.362 The methylene insertion reaction has limited utility because of its nonselectivity (see Sec. 5.D.i). The insertion reaction of carbenes has been used for synthetic purposes.363
The carbenes can be generated in any of the ways mentioned in Chapter 5 (Sec. 5.D.ii). Alkylcarbenes usually rearrange rather than give insertion (Sec. 5.D.ii, category 4), but, when this is impossible, intramolecular insertion364 is found rather than intermolecular.365 Methylene (:CH2) generated by photolysis of diazomethane (CH2N2) in the liquid phase is indiscriminate (totally nonselective) in its reactivity (Sec. 5.D.ii, category 2). Methylene (:CH2) generated in other ways and monoalkyl and dialkyl carbenes are less reactive and insert in the order tertiary > secondary > primary.366 Carbene insertion with certain allylic systems can proceed with rearrangement of the double bond.367 Carbenes have been generated using ultrasound.368 Halocarbenes (:CCl2,:CBr2, etc.) insert much less readily, although a number of instances have been reported.369
Insertion at an allylic carbon of alkenes has been reported.370 Dirhodium catalyzed insertion into H–Csp2 bonds is known,371 and also H–Csp bonds.372 Note that cyclopropanation may compete with C–H insertion with electron-rich highly substituted alkenes.373 Palladacycles formed by C–H insertion reactions with biphenylene have been intercepted.374 Such species have been impicated in the Heck reaction (Reaction 13-10). Insertion of diazoalkane and diazocarbonyl compounds can be catalyzed by copper compounds375 and silver compounds376 as well. Insertion into the α-C–H bond of an aldehyde gives an α-substituted aldehyde.377 Intramolecular insertion at the α carbon of a ketone by a diazoketone, using TiCl4, gives a bicyclic 1,3-diketone.378 The reaction in which aldehydes are converted to methyl ketones, RCHO + CH2N2 → RCOCH3, while apparently similar, does not involve a free carbene intermediate and is considered in Reaction 18-9. Note that aryl ketenes react with Me3SiCHN2 and then silica to give 2-indanone derivatives.379 A three component coupling reaction of vinyl iodides, secondary amines, and diazo(trimethylsilyl)methane gives allylic amines.380 A gold-catalyzed reaction is known that uses alkynes as an α-diazo ketone equivalent.381
Insertion into the O–H bond of alcohols, to produce ethers, has been reported using a diazocarbonyl compound and an In(OTf)3 catalyst.382 The Cu catalyzed insertion of a diazo ester into an oxetane gives the ring-expanded THF derivative.383 Insertion is also possible with other ethers, including silyl ethers.384 Metal-catalyzed silylene insertion into allylic ethers leads to allylic silanes.385 Similar insertion at the α carbon of an ether leads to cyclic ethers, with high enantioselectivity when a chiral ligand is used with a Rh catalyst.386
The insertion of the diazocarbonyl unit into the C–H bond of an α-diazo amide gives the lactam shown in the reaction.387 Insertion into a 2-pyrrolidinone derivative using Me3SiCH2N2 followed by AgCO2Ph with ultrasound gave the ring-expanded 2-piperidone derivative.388 Intramolecular insertion reactions are well known,389 and tolerate a variety of functional groups.390
The metal carbene insertion reaction, in contrast to the methylene insertion reaction can be highly selective391 and useful in synthesis.392 There are numerous examples, usually requiring a transition metal catalyst.393 The catalyst typically converts a diazoalkane or diazocarbonyl compound to the metal carbene in situ, allowing the subsequent insertion reaction. Intermolecular reactions are known, including diazoalkane insertion reaction with a dirhodium catalyst.394 When chiral ligands are present good enantioselectivity is observed in the insertion product.395
The mechanism396 of the insertion reaction is not known with certainty, but there seem to be at least two possible pathways.
1. A simple one-step process involving a three-center cyclic transition state:

The most convincing evidence for this mechanism is that in the reaction between isobutene-1![]() and carbene the product 2-methyl-1-butene was labeled only in the 1 position.397 This rules out a free radical or a carbocation or carbanion intermediate. If 41 (or a corresponding ion) were an intermediate, resonance would ensure that some carbene attacked at the 1 position:
and carbene the product 2-methyl-1-butene was labeled only in the 1 position.397 This rules out a free radical or a carbocation or carbanion intermediate. If 41 (or a corresponding ion) were an intermediate, resonance would ensure that some carbene attacked at the 1 position:
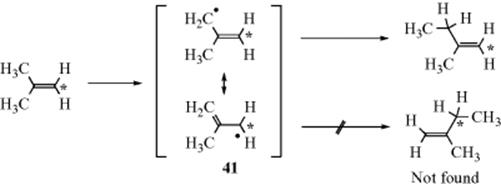
Other evidence is that retention of configuration, which is predicted by this mechanism, has been found in a number of instances.398 An ylid intermediate was trapped in the reaction of :CH2 with allyl alcohol.399
2. A free radical process in which the carbene directly abstracts a hydrogen from the substrate to generate a pair of free radicals:
![]()
One fact supporting this mechanism is that among the products obtained (beside butane and isobutane) on treatment of propane with CH2 (generated by photolysis of diazomethane and ketene) were propene and ethane,400which could arise, respectively, by
![]()
and
![]()
That this mechanism can take place under suitable conditions has been demonstrated by isotopic labeling401 and by other means.402 However, the formation of disproportionation and dimerization products does not always mean that the free radical abstraction process takes place. In some cases, these products arise in a different manner.403 The product of the reaction between a carbene and a molecule may have excess energy (see Sec. 5.D.ii). Therefore it is possible for the substrate and the carbene to react by mechanism 1 (the direct-insertion process) and for the excess energy to cause the compound thus formed to cleave to free radicals. When this pathway is in operation, the free radicals are formed after the actual insertion reaction.
The mechanism of cyclopropylcarbene reactions has also been discussed.404
It has been suggested405 that singlet carbenes insert by the one-step direct-insertion process and triplets (which, being free radicals, are more likely to abstract hydrogen) by the free radical process. In support of this suggestion, CIDNP signals406 (Sec. 5.C.i) were observed in the ethylbenzene produced from toluene and triplet CH2, but not from the same reaction with singlet CH2.407 Carbenoids (e.g., compounds of the form R2CMCl, see Reaction 12-39) can insert into a C–H bond by a different mechanism, similar to pathway 2, but involving abstraction of a hydride ion rather than a hydrogen atom.408
For the similar insertion reaction of nitrenes, see Reaction 12-13.
OS VII, 200.
F. Metal Electrophiles
12-22 Metalation with Organometallic Compounds
Metalation or Metalo-de-hydrogenation
![]()
Many organic compounds can be metalated by treatment with an organometallic compound.409 Since the reaction involves a proton transfer, the equilibrium lies on the side of the weaker acid.410 For example, fluorene reacts with n-butyllithium to give butane and 9-fluorenyllithium. Since aromatic hydrocarbons are usually stronger acids than aliphatic ones, R is most often aryl. The most common reagent is probably butyllithium.411 Reductive lithiation is an important method for the preparation of organolithium reagents.412 Normally, only active aromatic rings react with butyllithium. Benzene itself reacts very slowly and in low yield, although benzene can be metalated by butyllithium either in the presence of tert-BuOK413 or by n-butyllithium that is coordinated with various diamines.414 Metalation of aliphatic RH is most successful when the carbanions are stabilized by resonance (allylic, benzylic, propargylic,415 etc.) or when the negative charge is at an sp carbon (at triple bonds). Trimethylsilylmethyl potassium (Me3SiCH2K)416 and also a combination of an organolithium compound with a bulky alkoxide (LICKOR superbase)417 are very good reagents for allylic metalation. The former is also useful for benzylic positions. A combination of BuLi, t-BuOK, and tetramethylethylenediamine has been used to convert ethylene to vinylpotassium.418The reaction can be used to determine relative acidities of very weak acids by allowing two R–H compounds to compete for the same R′M and to determine which proton in a molecule is the most acidic.419
Note that organolithium compounds are aggregated species and can form hetero-aggregates containing different organic groups.420 N-Lithio-N-(trialkylsilyl)allylamines are deprotonated in ether solvents at the cis-vinylic position to give 3,N-dilithio-N-(trialkylsilyl)allylamines.421
In general, the reaction can be performed only with organometallics of active metals (e.g., Li, Na, and K), but Grignard reagents abstract protons from a sufficiently acidic C–H bond, as in R–C![]() C–H → R–C
C–H → R–C![]() C–MgX. This is the best method for the preparation of alkynyl Grignard reagents.422 Lewis acids have been used to promote α-lithiation of amines.423 Triethylgallium has been used to generate enolate anions from ketones.424
C–MgX. This is the best method for the preparation of alkynyl Grignard reagents.422 Lewis acids have been used to promote α-lithiation of amines.423 Triethylgallium has been used to generate enolate anions from ketones.424
When a heteroatom (e.g., N, O, S,425 or a halogen),426 is present in a molecule containing an aromatic ring or a double bond, lithiation is usually quite regioselective.427 It has been shown that fluorine is more effective for stabilization of carbanions when compared to the heavier halogens.428 In such compounds, the lithium usually bonds with the sp2 carbon closest to the heteroatom, probably because the attacking species coordinates with the heteroatom.429 This type of reaction with compounds such as anisole are often called directed metalations.430 In the case of aromatic rings, this means attack at the ortho position,431 but this is considered in Reaction 13-17.
![]()
Ref. 432
In the case of γ,δ-unsaturated disubstituted amides (42), the lithium does not go to the closest position, but in this case too the regiochemistry is controlled by coordination to the oxygen.433 Cyclopropyllithium reagents are rather stable.434

The mechanism involves an attack by R′− (or a polar R′) on the hydrogen435 (an acid–base reaction) Evidence is that resonance effects of substituents in R seem to make little difference. When R is aryl, OMe and CF3 both direct ortho, while isopropyl directs meta and para (mostly meta).436 These results are exactly what would be expected from pure field effects, with no contribution from resonance effects, which implies that attack occurs at the hydrogen and not at R. Other evidence for the involvement of H in the rate-determining step is that there are large isotope effects.437 The nature of R′ also has an effect on the rate. In the reaction between triphenylmethane and R′Li, the rate decreased in the order R′ = allyl > Bu > Ph > vinyl > Me, although this order changed with changing concentration of R′Li, because of varying degrees of aggregation of the R′Li.438 With respect to the reagent, this reaction is a special case of Reaction 12-24.
Enantioselective reactions are known. The preparation of chlorodeuteriomethylithium proceeds with inversion from the corresponding enantiopure stannyl derivative.439 Although highly reactive chemically, it is configurationally stable at temperatures up to −78 °C. Enantioselective catalytic deprotonation with chiral ligands has been used for the deprotonation of N-Boc amines to give chiral α-trimethylsilyl derivatives.440 A barrier to enantiomerization has been observed for unstablized, chelated, and dipole-stabilized organolithium compounds. Studies of lithiopyrrolidines show free energies for enantiomerization in the range of 19–22 kcal mol−1 (79.5–92.1 kJ mol−1) at 0 °C.441
A closely related reaction is formation of nitrogen ylids442 from quaternary ammonium salts (see Reaction 17-8):
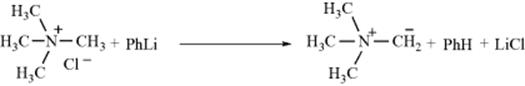
Phosphonium salts undergo a similar reaction (see Reaction 16-44).
OS II, 198; III, 413, 757; IV, 792; V, 751; VI, 436, 478, 737, 979; VII, 172, 334, 456, 524; VIII, 19, 391, 396, 606.
12-23 Metalation with Metals and Strong Bases
Metalation or Metalo-de-hydrogenation
![]()
Organic compounds can be metalated at suitably acidic positions by active metals and by strong bases.443 The reaction has been used to study the acidities of very weak acids (see Sec. 5.B.i). The conversion of terminal alkynes to acetylide ions is one important application.444 A gold-catalyst conversion of trimethylsilyl substituted esters and carbonates to the corresponding enolate anion has been reported445. Synthetically, an important use of the method is to convert aldehydes and ketones,446 carboxylic esters, and similar compounds to their enolate forms,447 for example, for use in nucleophilic substitutions (Reactions 10-67, 10-68, and 13-14) and in additions to multiple bonds (Reactions 15-24 and 16-53). Note that the reaction of carbonyl compounds with lithium dialkylamides leads to the corresponding enolate anion. This reaction was discussed in Reaction 10-68, in connection with the alkylation reaction of enolate anions.
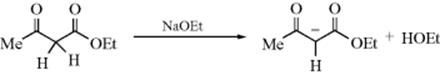
OS I, 70, 161, 490; IV, 473; VI, 468, 542, 611, 683, 709; VII, 229, 339. Conversions of ketones or esters to enolates are not listed.
12.C.ii. Metals as Leaving Groups
A. Hydrogen as the Electrophile
12-24 Replacement of Metals by Hydrogen
Hydro-de-metalation or Demetalation
![]()
Organometallic compounds, including enolate anions, react with acids in reactions that replace the metal with hydrogen.448 The R group may be aryl (see Reaction 11-41). The reaction is often used to introduce deuterium or tritium into susceptible positions. For Grignard reagents, water is usually a strong enough acid, but stronger acids are also used. An important method for the reduction of alkyl halides consists of the process RX → RMgX → RH.
The organometallic compounds that are hydrolyzed by water are the ones high in the electromotive series: Na, K, Li, Zn, and so on. Enantioselective protonation of lithium enolates449 and cyclopropyllithium compounds450 have been reported. When the metal is less active, stronger acids are required. For example, R2Zn compounds react explosively with water, R2Cd slowly, and R2Hg not at all, although the latter can be cleaved with concentrated HCl. However, this general statement has many exceptions, some hard to explain. For example, BR3 compounds are completely inert to water, and GaR3 at room temperature cleave just one R group, but AlR3 reacts violently with water. However, BR3 can be converted to RH with carboxylic acids.451 For less active metals, it is often possible to cleave just one R group from a multivalent metal. For example,
![]()
Organometallic compounds of less active metals and metalloids (e.g., Si,452 Sb, and Bi), are quite inert to water. Organomercury compounds (RHgX or R2Hg) can be reduced to RH by H2, NaBH4, or other reducing agents.453 The reduction with NaBH4 takes place by a free radical mechanism.454 Alkyl-Si bonds are cleaved by H2SO4 [e.g., HOOCCH2CH2SiMe3 → 2CH2 + (HOOCCH2CH2SiMe3)2O].455
When the hydrogen of the HA is attached to carbon, this reaction is the same as 12-22.
This section does not list the many hydrolyses of Na or K enolates, and so on found in Organic Syntheses. The hydrolysis of a Grignard reagent to give an alkane is found at OS II, 478; the reduction of a vinylic tin compound at OS VIII, 381; and the reduction of an alkynylsilane at OS VIII, 281.
B. Oxygen Electrophiles
12-25 The Reaction between Organometallic Reagents and Oxygen456
Hydroperoxy-de-metalation; Hydroxy-de-metalation
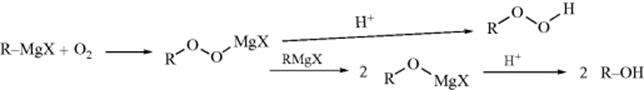
Oxygen reacts with Grignard reagents to give either hydroperoxides457 or alcohols. The reaction can be used to convert alkyl halides to alcohols without side reactions. With aryl Grignard reagents, yields are lower and only phenols are obtained, not hydroperoxides. Because of this reaction, oxygen should be excluded when Grignard reagents are prepared and used in various reactions.
Most other organometallic compounds also react with oxygen. Trialkylboranes and alkyldichloroboranes (RBCl2) can be conveniently converted to hydroperoxides by treatment with oxygen followed by hydrolysis.458 Dilithiated carboxylic acids (see Reaction 10-70) react with oxygen to give (after hydrolysis) α-hydroxy carboxylic acids.459 There is evidence that the reaction between Grignard reagents and oxygen involves a free radical mechanism.460
OS V, 918. See also, OS VIII, 315.
12-26 Reaction between Organometallic Reagents and Peroxides
tert-Butoxy-de-metalation
![]()
A convenient method of preparation of tert-butyl ethers consists of treating Grignard reagents with tert-butyl acyl peroxides.461 Both alkyl and aryl Grignard reagents can be used. The application of this reaction to Grignard reagents prepared from cyclopropyl halides permits cyclopropyl halides to be converted to tert-butyl ethers of cyclopropanols,462 which can then be easily hydrolyzed to the cyclopropanols. The direct conversion of cyclopropyl halides to cyclopropanols by Reaction 10-1 is not generally feasible, because cyclopropyl halides do not generally undergo nucleophilic substitutions without ring opening.
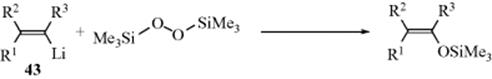
Vinyllithium reagents (43) react with silyl peroxides to give high yields of silyl enol ethers with retention of configuration.463 Since the preparation of 43 from vinylic halides (Reaction 12-39) also proceeds with retention, the overall procedure is a method for the stereospecific conversion of a vinylic halide to a silyl enol ether. Dialky ethers have been prepared from organotrifluoroborates and acetals464.
OS V, 642, 924.
12-27 Oxidation of Trialkylboranes to Borates
![]()
The reaction of alkenes with borane, monoalkyl, and dialkylboranes leads to a new organoborane (see Reaction 15-16). Treatment of organoboranes with alkaline H2O2 oxidizes trialkylboranes to esters of boric acid.465 This reaction does not affect double or triple bonds, aldehydes, ketones, halides, or nitriles that may be present elsewhere in the molecule. There is no rearrangement of the R group itself, and this reaction is a step in the hydroboration method of converting alkenes to alcohols (Reaction 15-16). The mechanism has been formulated as involving initial formation of an ate complex when the hydroperoxide anion attacks the electrophilic boron atom. Subsequent rearrangement from boron to oxygen,465 as shown, leads to the B–O–R unit.
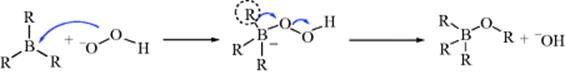
Similar migration of the other two R groups and hydrolysis of the B–O bonds leads to the alcohol and boric acid. Retention of configuration is observed in R. Boranes can also be oxidized to borates in good yields with oxygen,466with sodium perborate (NaBO3)467 and with trimethylamine oxide, either anhydrous468 or in the form of the dihydrate.469 The reaction with oxygen is free radical in nature.470
OS V, 918; VI, 719, 852, 919.
12-28 Preparation of Borates and Boronic Acids
Alkylboronic and arylboronic acids [RB(OH)2, and ArB(OH)2], respectively, are increasingly important in organic chemistry. The Pd catalyzed coupling reaction of aryl halides and aryl triflates with arylboronic acids (the Suzuki–Miyaura reaction, 13-12) is probably the most notable example. A simple synthesis involves the reaction of a Grignard reagent (e.g., phenylmagnesium bromide) with an alkyl borate to give phenylboronic acid.471 Alkylboronic acids are similarly prepared.472 Note that boronic acids are subject to cyclic trimerization with loss of water to form boroxines. Tetrahydroxydiboron has been used to prepare allylboronic acids, as well as potassium trifluoro(allyl)borates.473
Trimethylborate [B(OMe)3] can be used in place of tri-n-butyl borate.474 Newer methods involve the Pd mediated borylation of alcohols with bis(pinacolato)diboron475 or pinacolborane,476 but deprotection of the boronate esters can be a problem. Diolboranes (e.g., catecholborane 44)477 are prepared by the reaction of a diol with borane. Cedranediolborane (45, prepared from the cedrane-8,9-diol478 by treatment with borane•dimethyl sulfide) can be coupled to aryl iodides with a palladium catalyst, and generates the free boronic acid by treatment with diethanolamine and then aq acid.479 Boronate esters are often prepared as a means to purify the organoboron species, but some of these esters are hydrolytically unstable and difficult to deal with upon completion of the reaction.480
Alkeneboronic esters and acids are also readily available, as in the addition of vinylmagnesium chloride481 to trimethyl borate below −50 °C, followed by hydrolysis.482 A nonaqueous workup procedure has been reported for the preparation of arylboronic esters [ArB(OR′2)].483 Uncontrollable polymerization or oxidation of much of the boronic acid occurred during the final stages of the isolation procedure, but could be avoided by in situ conversion to the dibutyl ester by adding the crude product to 1-butanol. The Sm(III) catalyzed hydroboration of olefins with catecholborane is a good synthesis of boronate esters.484
Trialkyl borates (sometimes called orthoborates) can be prepared by heating the appropriate alcohol with boron trichloride in a sealed tube, but the procedure works well only for relatively simple alkyl groups.485 Heating alcohols with boron trioxide (B2O3) in an autoclave at 110–170 °C give the trialkyl borate.486 Boric acid can be used for the preparation of orthoborates487 by heating with alcohols in the presence of either hydrogen chloride or concentrated sulfuric acid. Removal of water as an azeotrope with excess alcohol improves the yield,488 and good yields can be obtained. for trialkyl borates489 and even for triphenyl borate.490 This method is unsuccessful for those borates whose parent alcohols do not form azeotropes with water and for the tertiary alkyl borates,489 impure samples are usually obtained.491
Potassium organotrifluoroborates (RBF3K) are readily prepared by the addition of inexpensive KHF2 to a variety of organoboron intermediates.492 They are monomeric, crystalline solids that are readily isolated and indefinitely stable in the air. These reagents can be used in several of the applications where boronic acids or esters are used (Reactions 13-10–13-13).493 Note that vinylboronic acid and even vinylboronate esters are unstable to polymerization,494 whereas the analogous vinyltrifluoroborate is readily synthesized and completely stable.495
OS 13, 16; 81, 134.
12-29 Oxygenation of Organometallic Reagents and Other Substrates to O-Esters and Related Compounds
![]()
In some cases, it is possible to oxygenate a nonaromatic carbon atom using various reagents, where the product is an O- ester rather than an alcohol. In one example, a vinyl iodonium salt was heated with DMF to produce the corresponding formate ester.496
![]()
C. Sulfur Electrophiles
12-30 Conversion of Organometallic Reagents to Sulfur Compounds
Thiols and sulfides are occasionally prepared by treatment of Grignard reagents with sulfur.497 Analogous reactions are known for selenium and tellurium compounds. Grignard reagents and other organometallic compounds498react with sulfuryl chloride to give sulfonyl chlorides,499 with esters of sulfinic acids to give (stereospecifically) sulfoxides,500 with disulfides to give sulfides,501 and with SO2 to give sulfinic acid salts,502 which can be hydrolyzed to sulfinic acids or treated with halogens to give sulfonyl halides.503
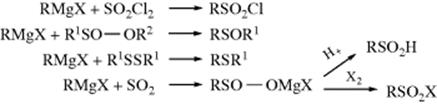
OS III, 771; IV, 667; VI, 533, 979.
D. Halogen Electrophiles
12-31 Halo-de-metalation
![]()
Grignard reagents react with halogens to give alkyl halides. The reaction is useful for the preparation of iodo compounds from the corresponding chloro or bromo compounds. The reaction is not useful for preparing chlorides, since the reagents RMgBr and RMgI react with Cl2 to give mostly RBr and RI, respectively.504
Most organometallic compounds, both alkyl and aryl, also react with halogens to give alkyl or aryl halides.505 The reaction can be used to convert acetylide ions to 1-haloalkynes.506 Vinyliodonium tetrafluoroborates were converted to vinyl fluorides by heating.507 Similarly, vinyl trifluoroborates were converted to the vinyl iodide with NaI and chloramine-T in aq THF.508 The reaction of an alkene with CuO·BF4, iodine and triethylsilane gave the 2-iodoalkane.509 Vinylzirconate reagents react with I2 to give the corresponding vinyl iodide.510
Enolate anions can be converted to the corresponding vinyl phosphate, and subsequent reaction with triphenylphosphine dihalide leads to the vinyl halide.511
Trialkylboranes react rapidly with I2512 or Br2513 in the presence of NaOMe in methanol, or with FeCl3 or other reagents514 to give alkyl iodides, bromides, or chlorides, respectively. Combined with the hydroboration reaction (Reaction 15-16), this is an indirect way of adding HBr, HI, or HCl to a double bond to give products with an anti-Markovnikov orientation (see Reaction 15-1). Trialkylboranes can also be converted to alkyl iodides by treatment with allyl iodide and air in a free radical process.515 trans-1-Alkenylboronic acids (47), prepared by hydroboration of terminal alkynes with catecholborane to give 46516 (Reaction 15-16), followed by hydrolysis, react with I2 in the presence of NaOH at 0 °C in ethereal solvents to give trans-vinylic iodides.517
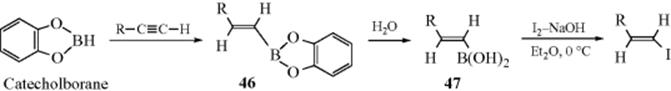
Treatment with ICl also gives the vinyl iodide.518 This is an indirect way of accomplishing the anti-Markovnikov addition of HI to a terminal triple bond. The reaction cannot be applied to alkenylboronic acids prepared from internal alkynes. However, alkenylboronic acids prepared from both internal and terminal alkynes react with Br2 (2 molar equivalents of Br2 must be used) followed by base to give the corresponding vinylic bromide, but in this case with inversion of configuration; so the product is the cis-vinylic bromide.519 Alkenylboronic acids also give vinylic bromides and iodides when treated with a mild oxidizing agent and NaBr or NaI, respectively.520 Treatment of 47(prepared from terminal alkynes) with Cl2 gave vinylic chlorides with inversion.521 Vinylic boranes can be converted to the corresponding vinylic halide by treatment with NCS or NBS.522 Vinylic halides can also be prepared from vinylic silanes523 and from vinylic copper reagents. The latter react with I2 to give iodides,524 and with NCS or NBS at −45 °C to give chlorides or bromides.525 The reaction of an aryl alkyne with HInCl2/BEt3 and then iodine leads to a (Z)-vinyl iodide with respect to the aryl group and the iodine atom.526 Boronic acids can be fluorinated in a reaction mediated by Ag(I) triflate.527
For the reaction of lithium enolate anions of esters with I2 or CX4, see Reaction 12-5.
The conversion of terminal alkynes to 1-iodo-1-alkynes was reported using NaI under electrochemical conditions.528 1-Bromo-1-alkynes were converted to the 1-iodo-1-alkyne with CuI.529 1-Trialkyldisilylalkynes were converted to the corresponding 1-bromoalkyne via reaction with NBS and AgF.530 Terminal alkynes react with (diacetoxyiodo)benzene, KI, and CuI to give 1-iodo-alkynes.531 Trichloroisocyanuric acid has been used to convert terminal alkynes to 1-chloroalkynes.532
It is unlikely that a single mechanism suffices to cover all conversions of organometallic compounds to alkyl halides.533 In a number of cases, the reaction has been shown to involve inversion of configuration (see Sec. 12.A.i), indicating an SE2 (back) mechanism, while in other cases retention of configuration has been shown,534 implicating an SE2 (front) or SEi mechanism. In still other cases, complete loss of configuration as well as other evidence demonstrated the presence of a free radical mechanism.534,535
OS I, 125, 325, 326; III, 774, 813; V, 921; VI, 709; VII, 290; VIII, 586; IX, 573. Also see, OS II, 150.
E. Nitrogen Electrophiles
12-32 The Conversion of Organometallic Compounds to Amines
Amino-de-metalation
![]()
There are several methods for conversion of alkyl- or aryllithium compounds to primary amines.536 The two most important are treatment with hydroxylamine derivatives and with certain azides.537 In the first of these methods, treatment of RLi with methoxyamine and MeLi in ether at −78 °C gives RNH2.538 Grignard reagents from aliphatic halides give lower yields. The reaction can be extended to give secondary amines by the use of N-substituted methoxyamines (CH3ONHR′).539 There is evidence540 that the mechanism involves the direct displacement of OCH3 by R on an intermediate CH2ONR′− (CH2ONR′− Li+ + RLi → CH3OLi + RNR′− Li+). Tosyl azide (TsN3) is a highly useful azide.541 The initial product is usually RN3, but this is easily reduced to the amine (Reaction 19-51). With some azides (e.g., azidomethyl phenyl sulfide, PhSCH2N3), the group attached to the N3 is a poor leaving group, so the initial product is a triazene (in this case ArNHN=NCH2SPh from ArMgX), which can be hydrolyzed to the amine.542
![]()
Organoboranes react with a mixture of aq NH3 and NaOCl to produce primary amines.543 It is likely that the actual reagent is chloramine (NH2Cl). Chloramine itself,544 hydroxylamine-O-sulfonic acid in diglyme,545 and trimethylsilyl azide546 also give the reaction. Since the boranes can be prepared by the hydroboration of alkenes (Reaction 15-16), this is an indirect method for the addition of NH3 to a double bond with anti-Markovnikovorientation. Secondary amines can be prepared547 by the treatment of alkyl- or aryldichloroboranes or dialkylchloroboranes with alkyl or aryl azides.
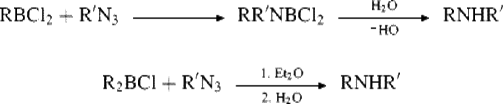
The use of an optically active R∗BCl2 gave secondary amines of essentially 100% optical purity.548 Aryllead triacetates [ArPb(OAc)3] give secondary amines (ArNHAr′) when treated with primary aromatic amines Ar′NH2 and Cu(OAc)2.549
Secondary amines have been converted to tertiary amines by treatment with lithium dialkylcuprate reagents: R2CuLi + NHR → RNR′2.550 The reaction was also used to convert primary amines to secondary, but yields were lower.551
Terminal alkynes reacted with chlorodiphenylphosphine (Ph3PCl) and a Ni catalyst to give the 1-diphenylphosphino alkyne (R-C![]() C-PPh2).552 Alkynyl halides can be used for a similar reaction. Treatment of methyl carbamates with KHMDS and CuI, followed by 2 equiv of 1-bromophenylacetylene gave the N-substituted alkyne, Ph-C
C-PPh2).552 Alkynyl halides can be used for a similar reaction. Treatment of methyl carbamates with KHMDS and CuI, followed by 2 equiv of 1-bromophenylacetylene gave the N-substituted alkyne, Ph-C![]() C-N(CO2Me)R.553
C-N(CO2Me)R.553
Metal-catalyzed amination reactions are increasingly important in organic methodology. In a typical reaction, an amine is coupled to an alkyl, vinyl, or aryl halide (or with a different leaving group) in the presence of a transition metal, usually Pd. Presumably, the amination occurs via reaction with a transient organometallic species. Amination of aromatic compounds via this approach is discussed in section Reaction 13-5. Aliphatic and vinyl substrates are treated here. In one example, a vinyl triflate is converted to an enamine via reaction with pyrrole in the presence of a Pd catalyst.554
OS VI, 943.
F. Carbon Electrophiles
12-33 The Conversion of Organometallic Compounds to Ketones, Aldehydes, Carboxylic Esters, or Amides
Acyl-de-metalation, and so on
![]()
Symmetrical ketones555 can be prepared in good yields by the reaction of organomercuric halides556 with dicobalt octacarbonyl in THF,557 or with nickel carbonyl in DMF or certain other solvents.558 The R group may be aryl or alkyl. However, when R is alkyl, rearrangements may intervene in the CO2(CO)8 reaction, although the Ni(CO)4 reaction seems to be free from such rearrangements.559 Divinylic ketones (useful in the Nazarov cyclization, 15-20) have been prepared in high yields by treatment of vinylic mercuric halides with CO and a Rh catalyst.559 In a more general synthesis of unsymmetrical ketones, tetraalkyltin compounds (R4Sn) are treated with a halide R′X (R′ = aryl, vinylic, benzylic), CO, and a Pd complex catalyst.560 Similar reactions use Grignard reagents, Fe(CO)5, and an alkyl halide.561
Grignard reagents react with formic acid to give good yields of aldehydes. Two molar equivalents of RMgX are used; the first converts HCO2H to HCOO−, which reacts with the second equivalent to give RCHO.562 Alkyllithium reagents and Grignard reagents react with CO to give symmetrical ketones.563 An interesting variation reacts CO2 with an organolithium, which is then treated with a different organolithium reagent to give the unsymmetrical ketone.564 α,β-Unsaturated aldehydes can be prepared by treatment of vinylic silanes with dichloromethyl methyl ether and TiCl4 at −90 °C.565
α,β-Unsaturated esters can be prepared by treating boronic esters (27) with CO, PdCl2, and NaOAc in MeOH.566 The synthesis of α,β-unsaturated esters has also been accomplished by treatment of vinylic mercuric chlorides with CO at atmospheric pressure and a Pd catalyst in an alcohol as solvent, for example,567
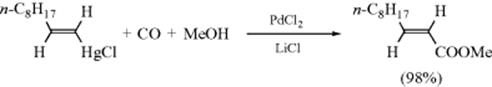
Alkyl and aryl Grignard reagents can be converted to carboxylic esters with Fe(CO)5 instead of CO.568
Amides have been prepared by the treatment of trialkyl or triarylboranes with CO and an imine, in the presence of catalytic amounts of cobalt carbonyl569:
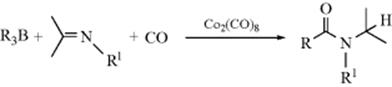
In another method for the conversion RM → RCONR, Grignard reagents and organolithium compounds are treated with a formamide (HCONR′2) to give the intermediate RCH(OM)NR′2, which is not isolated, but treated with PhCHO or Ph2CO to give the product RCONR′2.570
Direct conversion of a hydrocarbon to an aldehyde (R–H → R–CHO) was reported by treatment of the hydrocarbon with GaCl3 and CO.571
For carbonylation reactions of aryl halides, see Reaction 13-15.
See also, Reactions 10-76, 15-32, and 18-23–18-24.
OS VIII, 97.
12-34 Cyano-de-metalation
![]()
Vinylic copper reagents react with ClCN to give vinyl cyanides, although BrCN and ICN give the vinylic halide instead.572 Vinylic cyanides have also been prepared by the reaction between vinylic lithium compounds and phenyl cyanate (PhOCN).573 Alkyl nitriles (RCN) have been prepared, in varying yields, by treatment of sodium trialkylcyanoborates with NaCN and lead tetraacetate.574 Vinyl bromides reacted with KCN, in the presence of a Ni complex and Zn metal to give the vinyl nitrile.575 Vinyl triflates react with LiCN, in the presence of a Pd catalyst, to give the vinyl nitrile.576
For other electrophilic substitutions of the type RM → RC, which are discussed under nucleophilic substitutions in Chapter 10, see also, Reactions 16-81–16-85 and 16-99.
OS IX, 548
G. Metal Electrophiles
12-35 Transmetalation with a Metal
Metalo-de-metalation
![]()
Many organometallic compounds are best prepared by this reaction, which involves replacement of a metal in an organometallic compound by another metal. The RM′ compound can be successfully prepared only when M′ is above M in the electromotive series, unless some other way is found to shift the equilibrium. That is, RM is usually an unreactive compound and M′ is a metal more active than M. Most often, RM is R2Hg, since mercury alkyls556are easy to prepare and mercury is far down in the electromotive series.577 Alkyls of Li, Na, K, Be, Mg, Al, Ga, Zn, Cd, Te, Sn, and so on, have been prepared this way. An important advantage of this method over Reaction 12-38 is that it ensures that the organometallic compound will be prepared free of any possible halide. This method can be used for the isolation of solid sodium and potassium alkyls.578 If the metals lie too close together in the series, it may not be possible to shift the equilibrium. For example, alkylbismuth compounds cannot be prepared in this way from alkylmercury compounds.
OS V, 1116.
12-36 Transmetalation with a Metal Halide
Metalo-de-metalation
![]()
In contrast to Reaction 12-35, the reaction between an organometallic compound and a metal halide is successful only when M′ is below M in the electromotive series.579 The two reactions considered together therefore constitute a powerful tool for preparing all kinds of organometallic compounds. In this reaction, the most common substrates are Grignard reagents and organolithium compounds.580
The MgX of Grignard reagents581 can migrate to terminal positions in the presence of small amounts of TiCl4.582 The proposed mechanism consists of metal exchange (Reaction 12-36), elimination–addition, and metal exchange:
The addition step is similar to Reactions 15-16 or 15-17 and follows Markovnikov's rule, so the positive titanium goes to the terminal carbon.
Among others, alkyls of Be, Zn,583 Cd, Hg, Al, Sn, Pb, Co, Pt, and Au have been prepared by treatment of Grignard reagents with the appropriate halide.584 The reaction has been used to prepare alkyls of almost all nontransition metals and even of some transition metals. Alkyls of metalloids and of nonmetals, including Si, B,585 Ge, P, As, Sb, and Bi, can also be prepared in this manner.586 Except for alkali-metal alkyls and Grignard reagents, the reaction between RM and M′X is the most common method for the preparation of organometallic compounds.587 In the presence of Ir,588 or Pd catalysts,589 aromatic compunds react with boranes to give the corresponding arylborane.
Lithium dialkylcopper reagents are prepared from 2 molar equivalents of RLi with 1 molar equivalent of a cuprous halide in ether at low temperatures:590 The formation of organocuprates of this type are discussed in more detail in Reaction 10-58, in connection with the coupling reaction of organocuprates with alkyl halides.
![]()
Another way is to dissolve an alkylcopper compound in an alkyllithium solution. Higher order cuprates can also be prepared, as well as “non-ate” copper reagents.591
Metallocenes (48, see Sec. 2.I.ii) are usually made by this method. Among others, metallocenes of Sc, Ti, V, Cr, Mn, Fe, Co, and Ni have been prepared in this manner.592
![]()
In a related reaction, sulfurated boranes (R2B–SSiR′2) react with Grignard reagents (e.g., methylmagneisum bromide) to give the β-alkyl borane (e.g., R2B–Me) upon heating in vacuo.593
OS I, 231, 550; III, 601; IV, 258, 473, 881; V, 211, 496, 727, 918, 1001; VI, 776, 875, 1033; VII, 236, 290, 524; VIII, 23, 57, 268, 474, 586, 606, 609. Also see, OS IV, 476
12-37 Transmetalation with an Organometallic Compound
Metalo-de-metalation
![]()
This type of metallic exchange is used much less often than Reactions 12-35 and 12-36. It is an equilibrium reaction and is useful only if the equilibrium lies in the desired direction. Usually the goal is to prepare a lithium compound that is not prepared easily in other ways,594 for example, a vinylic or an allylic lithium, most commonly from an organotin substrate. Examples are the preparation of vinyllithium from phenyllithium and tetravinyltin and the formation of α-dialkylamino organolithium compounds from the corresponding organotin compounds595
![]()
The reaction has also been used to prepare 1,3-dilithiopropanes596 and 1,1-dilithiomethylenecyclohexane597 from the corresponding mercury compounds. In general, the equilibrium lies in the direction in which the more electropositive metal is bonded to that alkyl or aryl group that is the more stable carbanion (Sec. 5.B.i). The reaction proceeds with retention of configuration;598 an SEi mechanism is likely.599
“Higher order” cuprates600 (see Reaction 10-58) have been produced by this reaction starting with a vinylic tin compound:601
![]()
These compounds are not isolated, but used directly in situ for conjugate addition reactions (Reaction 15-25). Another method for the preparation of such reagents (but with Zn instead of Li) allows them to be made from α-acetoxy halides:602
![]()
OS V, 452; VI, 815; VIII, 97.
12.C.iii. Halogen as Leaving Group
The reduction of alkyl halides can proceed by an electrophilic substitution mechanism, but it is considered in Chapter 19 (Reaction 19-53).
12-38 Metalo-de-halogenation
![]()
Alkyl halides react directly with certain metals to give organometallic compounds.603 The most common metal is Mg, and of course this is by far the most common method for the preparation of Grignard reagents.604 The Grignard reaction with aldehydes or ketones is discussed in Reaction 16-24. The order of halide activity is I > Br > Cl. This reaction can be applied to many alkyl halides primary, secondary, and tertiary and to aryl halides, although aryl chlorides require the use of THF or another higher-boiling solvent instead of the usual ether, or special entrainment methods.605 Aryl iodides and bromides can be treated in the usual manner. Allylic Grignard reagents can also be prepared in the usual manner (or in THF),606 although in the presence of excess halide these may give Wurtz-type coupling products (see Reaction 10-56).607 Like aryl chlorides, vinylic halides require higher-boiling solvents (see OS IV, 258). A good procedure for benzylic and allylic halides is to use magnesium anthracene (prepared from Mg and anthracene in THF)608 instead of ordinary magnesium,609 although activated magnesium turnings have also been used.610 Alkynyl Grignard reagents are generally prepared by the method in Reaction 12-22.
Dihalides611 can be converted to Grignard reagents if the halogens are different and are at least three carbons apart. If the halogens are the same, it is possible to obtain dimagnesium compounds [e.g., BrMg(CH2)4MgBr].612 1,2-Dihalides give elimination613 rather than Grignard reagent formation (Reaction 17-22), and the reaction is seldom successful with 1,1-dihalides, although the preparation of gem-disubstituted compounds [e.g., CH2(MgBr)2], has been accomplished with these substrates.614 α-Halo Grignard reagents and α-halolithium reagents can be prepared by the method given in Reaction 12-39.615 Alkylmagnesium fluorides can be prepared by refluxing alkyl fluorides with Mg in the presence of appropriate catalysts (e.g., I2 or EtBr) in THF for several days.616 Nitrogen-containing Grignard reagents have been prepared.617
The presence of other functional groups in the halide usually affects the preparation of the Grignard reagent. Groups that contain active hydrogen (defined as any hydrogen that will react with a Grignard reagent, e.g., OH, NH2, and CO2H), can be present in the molecule, but only if they are converted to the salt form (O−, NH−, COO−, respectively). Groups that react with Grignard reagents (e.g., C=O, C![]() N, NO2, CO2R) inhibit Grignard formation entirely. In general, the only functional groups that may be present in the halide molecule without any interference at all are double and triple bonds (except terminal triple bonds) and OR and NR2 groups. However, β-halo ethers generally give β elimination when treated with Mg (see Reaction 17-24), and Grignard reagents from α-halo ethers618 can only be formed in THF or dimethoxymethane at a low temperature, for example,619 because such reagents immediately undergo α elimination (see Reaction 12-39) at room temperature in ether solution.
N, NO2, CO2R) inhibit Grignard formation entirely. In general, the only functional groups that may be present in the halide molecule without any interference at all are double and triple bonds (except terminal triple bonds) and OR and NR2 groups. However, β-halo ethers generally give β elimination when treated with Mg (see Reaction 17-24), and Grignard reagents from α-halo ethers618 can only be formed in THF or dimethoxymethane at a low temperature, for example,619 because such reagents immediately undergo α elimination (see Reaction 12-39) at room temperature in ether solution.
![]()
Because Grignard reagents react with water (Reaction 12-24) and with oxygen (Reaction 12-25), it is generally best to prepare them in an anhydrous nitrogen atmosphere. Grignard reagents are generally neither isolated nor stored; solutions of Grignard reagents are used directly for the required synthesis. Grignard reagents can also be prepared in benzene or toluene, if a tertiary amine is added to complex with the RMgX.620 This method eliminates the need for an ether solvent. With certain primary alkyl halides it is even possible to prepare alkylmagnesium compounds in hydrocarbon solvents in the absence of an organic base.621 It is also possible to obtain Grignard reagents in powdered form, by complexing them with the chelating agent tris(3,6-dioxaheptyl)amine [N(CH2CH2OCH2CH2OCH3)3].622
Next to the formation of Grignard reagents, the most important application of this reaction is the conversion of alkyl and aryl halides to organolithium compounds,623 but it has also been carried out with many other metals (e.g., Na, Be, Zn, Hg, As, Sb, and Sn). With Na, the Wurtz Reaction (10-56) is an important side reaction. In some cases, where the reaction between a halide and a metal is too slow, an alloy of the metal with K or Na can be used instead. The most important example is the preparation of tetraethyl-lead from ethyl bromide and a Pb–Na alloy.
The efficiency of the reaction can often be improved by use of the metal in its powdered624 or vapor625 form. These techniques have permitted the preparation of some organometallic compounds that cannot be prepared by the standard procedures. Among the metals produced in an activated form are Mg,626 Ca,627 Zn,628 Al, Sn, Cd,629 Ni, Fe, Ti, Cu,630 Pd, and Pt.631
The mechanism of Grignard reagent formation involves free radicals,632 and there is much evidence for this, from CIDNP633 (Sec. 5.C.i) and from stereochemical, rate, and product studies.634 Further evidence is that free radicals have been trapped,635 and that experiments that studied the intrinsic reactivity of MeBr on a magnesium single-crystal surface showed that Grignard reagent formation does not take place by a single-step insertion mechanism.636 The following SET mechanism has been proposed:633
Other evidence has been offered to support a SET initiated radical process for the second step of this mechanism.637 The species R–X√− and Mg√+ are radical ions.638 The subscript “s” is meant to indicate that the species so marked are bound to the surface of the magnesium. It is known that this is a surface reaction.639 It has been suggested that some of the R√ radicals diffuse from the magnesium surface into the solution and then return to the surface to react with the XMg√. There is evidence both for640 and against641 this suggestion. Another proposal is that the fourth step is not the one shown here, but that the R√ is reduced by Mg+ to the carbanion R−, which combines with MgX+ to give RMgX.642
There are too many preparations of Grignard reagents in Organic Syntheses for us to list here. Chiral Grignard reagents are rare, since they are configurationally unstable in most cases. However, a few chiral Grignard reagentsare known.643 Use of the reaction to prepare other organometallic compounds can be found in OS I, 228; II, 184, 517, 607; III, 413, 757; VI, 240; VII, 346; VIII, 505. The preparation of unsolvated butylmagnesium bromide is described at OS V, 1141. The preparation of highly reactive (powdered) magnesium is given at OS VI, 845.
12-39 Replacement of a Halogen by a Metal from an Organometallic Compound
Metalo-de-halogenation
![]()
The exchange reaction between halides and organometallic compounds occurs most readily when M is Li and X is Br or I,644 although it has been shown to occur with Mg.645 The R′ group is usually, although not always, alkyl, and often butyl; R is usually aromatic.646 Alkyl halides are generally not reactive enough, while allylic and benzylic halides usually give Wurtz coupling. Of course, the R that becomes bonded to the halogen is the one for which RH is the weaker acid. Despite the preponderance of reactions with bromides and iodides, it is noted that the reaction of 1-fluorooctane with 4–10 equiv of Li powder and 2–4 equiv of DTBB (4,4′-di-tert-butylbiphenyl) in THP (THP=tetrahydropyran) at 0 °C for 5 min, was shown to give a solution of the corresponding 1-octyllithium.647 Vinylic halides react with retention of configuration.648 The reaction can be used to prepare α-halo organolithium and α-halo organomagnesium compounds649 Carbon tetrachloride reacts with butyllithium to give lithiotrichloromethane (Cl3C–Li), for example.650 Such compounds can also be prepared by hydrogen–metal exchange, for example,651
![]()
This is an example of Reaction 12-22. However, these α-halo organometallic compounds are stable (and configurationally stable as well652) only at low temperatures (ca. -100 °C) and only in THF or mixtures of THF and other solvents (e.g., HMPA). At ordinary temperatures, they lose MX (α elimination) to give carbenes, which then react further, or carbenoid reactions. The α-chloro-α-magnesio sulfones [ArSO2CH(Cl)MgBr] are exceptions, being stable in solution at room temperature and even under reflux.653 Compounds in which a halogen and a transition metal are on the same carbon can be more stable than the ones with lithium.654
There is evidence that the mechanism655 of the reaction of alkyllithium compounds with alkyl and aryl iodides involves free radicals.656
![]()
Among the evidence is the fact that coupling and disproportionation products are obtained from R√ and R′√ and the observation of CIDNP.656,657 However, in the degenerate exchange between PhI and PhLi the ate complex Ph2I− Li+has been shown to be an intermediate,658 and there is other evidence that radicals are not involved in all instances of this reaction.659
In a completely different kind of process, alkyl halides can be converted to certain organometallic compounds by treatment with organometalate ions, for example,
![]()
Most of the evidence is in accord with a free radical mechanism involving electron transfer, although an SN2 mechanism can compete under some conditions.660 Electrochemically genrated zinc has been used to prepare organozinc bomide from the corresponding alkyl halide.661
OS VI, 82; VII, 271, 326, 495; VIII, 430. See also, OS VII, 512; VIII, 479.
12.C.iv. Carbon Leaving Groups
In these reactions (12-40–12-48), a carbon–carbon bond cleaves. The substrate is the side that retains the electron pair; hence the reactions are considered electrophilic substitutions. The incoming group is hydrogen in all but one (Reaction 12-42) of the cases. The reactions in groups A and B are sometimes called anionic cleavages,662 although they do not always occur by mechanisms involving free carbanions (SE1). When they do, the reactions are facilitated by increasing stability of the carbanion.
A. Carbonyl-Forming Cleavages
These reactions follow the pattern:
![]()
The leaving group is stabilized because the electron deficiency at its carbon is satisfied by a pair of electrons from the oxygen. With respect to the leaving group the reaction is elimination to form a C=O bond. Retrograde aldol reactions (16-34) and cleavage of cyanohydrins (16-52) belong to this classification but are treated in Chapter 16 under their more important reverse reactions. Other eliminations to form C=O bonds are discussed in Reaction 17-32.
12-40 Decarboxylation of Aliphatic Acids
Hydro-de-carboxylation
![]()
Many carboxylic acids can be successfully decarboxylated, either as the free acid or in the salt form, but not simple aliphatic acids.663 An exception is acetic acid, which as the acetate, heated with base, gives good yields of methane. Malonic acid derivatives are the most common substrates for decarboxylation, giving the corresponding mono-carboxylic acid. Decarboxylation of 2-substituted malonic acids has been reported using microwave irradiation.664 Aliphatic acids that do undergo successful decarboxylation have certain functional groups or double or triple bonds in the α or β position. Some of these are shown in Table 12.2.
Table 12.2 Some Acids That Undergo Decarboxylation Fairly Readilya
Acid Type
Decarboxylation Product
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
a. Others are described in the text.
For decarboxylation of aromatic acids, see Reaction 11-35. Decarboxylation of an α-cyano acid can give a nitrile or a carboxylic acid, since the cyano group may or may not be hydrolyzed in the course of the reaction. In addition to the compounds listed in Table 12.2, decarboxylation can be carried out on α,β-unsaturated665 and α,β-acetylenic acids. Glycidic acids give aldehydes on decarboxylation. The following mechanism has been suggested:666
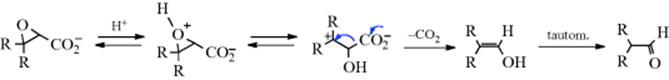
The direct product is an enol that tautomerizes to the aldehyde.667 This is the usual last step in the Darzens Reaction (16-40).
Decarboxylations can be regarded as reversal of the addition of carbanions to carbon dioxide (Reaction 16-82), but free carbanions are not always involved.668 When the carboxylate ion is decarboxylated, the mechanism can be either SE1 or SE2. In the case of the SE1 mechanism, the reaction is of course aided by the presence of electron-withdrawing groups, which stabilize the carbanion.669 Decarboxylation of carboxylate ions can be accelerated by the addition of a suitable crown ether, which in effect removes the metallic ion.670 The reaction without the metallic ion has also been performed in the gas phase.671 Some acids can also be decarboxylated directly and, in most of these cases, there is a cyclic, six-center mechanism:
Here too there is an enol that tautomerizes to the product. The mechanism is illustrated for the case of β-keto acids,672 but it is likely that malonic, α-cyano, α-nitro, and β,γ-unsaturated acids673 behave similarly, since similar six-membered transition states can be written for them. Some α,β-unsaturated acids are also decarboxylated by this mechanism by isomerizing to the β,γ-isomers before they actually decarboxylate.674 Evidence is that 49 and similar bicyclic β-keto acids resist decarboxylation.675 In such compounds, the six-membered cyclic transition state cannot form for steric reasons,676 and if it could, formation of the intermediate enol would violate Bredt's rule (Sec. 4.Q.iii).
Some carboxylic acids that cannot form a six-membered transition state can still be decarboxylated, and these presumably react through an SE1 or SE2 mechanism.677 Further evidence for the cyclic mechanism is that the reaction rate varies very little with a change from a nonpolar to a polar solvent (even from benzene to water678), and is not subject to acid catalysis.679 The rate of decarboxylation of a β,γ-unsaturated acid was increased ~105–106 times by introduction of a β-methoxy group, indicating that the cyclic transition state has dipolar character.680 Rate constants for decarboxylation reactions have been calculated using no barrier theory.681
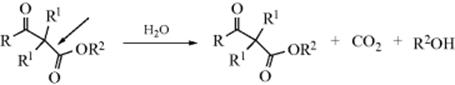
β-Keto acids682 are easily decarboxylated, but such acids are usually prepared from β-keto esters, and the esters are easily decarboxylated themselves on hydrolysis without isolation of the acids.683 This decarboxylation of β-keto esters involving cleavage on the carboxyl side of the substituted methylene group (arrow) is carried out under acidic, neutral, or slightly basic conditions to yield a ketone. When strongly basic conditions are used, cleavage occurs on the other side of the CR2 group (Reaction 12-43). β-Keto esters can be decarbalkoxylated without passing through the free-acid stage by treatment with boric anhydride (B2O3) at 150 °C.684 The alkyl portion of the ester (R′) is converted to an alkene or, if it lacks a β hydrogen, to an ether (R′OR′). Another method for the decarbalkoxylation of β-keto esters, malonic esters, and α-cyano esters consists of heating the substrate in wet DMSO containing NaCl, Na3PO4, or some other simple salt.685 In this method too, the free acid is probably not an intermediate, but here the alkyl portion of the substrate is converted to the corresponding alcohol. α-Amino acids have been decarboxylated by treatment with a catalytic amount of 2-cyclohexenone.686 Amino acids are decarboxylated by sequential treatment with NBS at pH 5 followed by NaBH4 and NiCl2.687 Certain decarboxylations can also be accomplished photochemically.688 See also, the decarbonylation of acyl halides, mentioned in Reaction 14-32. In some cases, decarboxylations can give organometallic compounds: RCOOM → RM + CO2.689 The Cu catalyzed decarboxylation of 2-alkynoic acids to terminal alkynes has been reported.690
Decarboxylative alkylation and arylation reactions are known. In the presence of a Ru catalyst and a B-phenyl borinate, decarboxylation of proline esters leads to 2-phenylpyrrolidine derivatives.691 In the presence of a Pd catalyst, esters undergo decarboxylation with coupling between the alkyl groups on the carbonyl and the ester oxygen to give the corresponding hydrocarbon fragment.692
Some of the decarboxylations listed in Organic Syntheses are performed with concomitant ester or nitrile hydrolysis and others are simple decarboxylations.
With ester or nitrile hydrolysis: OS I, 290, 451, 523; II, 200, 391; III, 281, 286, 313, 326, 510, 513, 591; IV, 55, 93, 176, 441, 664, 708, 790, 804; V, 76, 288, 572, 687, 989; VI, 615, 781, 873, 932; VII, 50, 210, 319; VIII, 263.
Simple decarboxylations: OS I, 351, 401, 440, 473, 475; II, 21, 61, 93, 229, 302, 333, 368, 416, 474, 512, 523; III, 213, 425, 495, 705, 733, 783; IV, 234, 254, 278, 337, 555, 560, 597, 630, 731, 857; V, 251, 585; VI, 271, 965; VII, 249, 359; VIII, 235, 444, 536; 75, 195. Also see, OS IV, 633.
12-41 Cleavage of Alkoxides
Hydro-de-(α-oxidoalkyl)-substitution
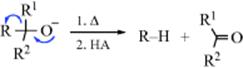
Alkoxides of tertiary alcohols can be cleaved in a reaction that is essentially the reverse of addition of carbanions to ketones (Reaction 16-24).693 The reaction is unsuccessful when the R groups are simple unbranched alkyl groups (e.g., the alkoxide of triethylcarbinol). Cleavage is accomplished with branched alkoxides (e.g., the alkoxides of diisopropylneopentylcarbinol or tri-tert-butylcarbinol).694 Allylic,695 benzylic,696 and aryl groups also cleave (e.g., the alkoxide of triphenylcarbinol gives benzene and benzophenone). Studies in the gas phase show that the cleavage is a simple one, giving the carbanion and ketone directly in one step.697 However, with some substrates in solution, substantial amounts of dimer R–R have been found, indicating a radical pathway.698 Hindered alcohols (not the alkoxides) also lose one R group by cleavage, also by a radical pathway.699 The so-called retro-aldol (see Reaction 16-34) is another example.
The reaction has been used for extensive mechanistic studies (see Sec. 12.A.ii).
OS VI, 268.
12-42 Replacement of a Carboxyl Group by an Acyl Group
Acyl-de-carboxylation
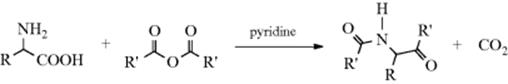
When an α-amino acid is treated with an anhydride in the presence of pyridine, the carboxyl group is replaced by an acyl group and the NH2 becomes acylated. This is called the Dakin–West reaction.700 The mechanism involves formation of an oxazolone.701 The reaction sometimes takes place on carboxylic acids even when an a amino group is not present. A number of N-substituted amino acids [RCH(NHR′)COOH] give the corresponding N-alkylated products.
OS IV, 5; V, 27.
B. Acyl Cleavages
In these reactions (12-43–2-46), a carbonyl group is attacked by a hydroxide ion (or an amide ion), giving an intermediate that undergoes cleavage to a carboxylic acid (or an amide). With respect to the leaving group, this is nucleophilic substitution at a carbonyl group and the mechanism is the tetrahedral one discussed in Section 16.A.i.
![]()
With respect to R this is of course electrophilic substitution. The mechanism is usually SE1.
12-43 Basic Cleavage of β-Keto Esters and β-Diketones
Hydro-de-acylation
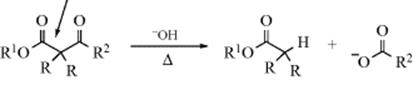
When β-keto esters are treated with concentrated base, cleavage occurs, but on the keto side of the CR2 group (arrow) in contrast to the acid cleavage mentioned in Reaction 12-40. The products are a carboxylic ester and the salt of an acid. However, the utility of the reaction is somewhat limited by the fact that decarboxylation is a side reaction, even under basic conditions. β-Diketones behave similarly to give a ketone and the salt of a carboxylic acid. With both β-keto esters and β-diketones, ![]() can be used instead of
can be used instead of ![]() , in which case the ethyl esters of the corresponding acids are obtained instead of the salts. In the case of β-keto esters, this is the reverse of Claisen condensation(Reaction 16-85). The related cleavage of cyclic α-cyano ketones, in an intramolecular fashion, has been used in a synthesis of macrocyclic lactones (e.g., 50).702
, in which case the ethyl esters of the corresponding acids are obtained instead of the salts. In the case of β-keto esters, this is the reverse of Claisen condensation(Reaction 16-85). The related cleavage of cyclic α-cyano ketones, in an intramolecular fashion, has been used in a synthesis of macrocyclic lactones (e.g., 50).702
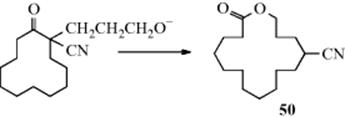
Activated F− (from KF and a crown ether) has been used as the base to cleave an α-cyano ketone.703 Treatment with ceric ammonium nitrate led to cleavage of β-diketones to give a carboxylic acid.704
OS II, 266, 531; III, 379; IV, 415, 957; V, 179, 187, 277, 533, 747, 767.
12-44 Haloform Reaction
![]()
In the haloform reaction, methyl ketones (and the only methyl aldehyde, acetaldehyde) are cleaved with halogen and a base.705 The halogen can be bromine, chlorine, or iodine. What takes place is actually a combination of two reactions. The first is an example of Reaction 12-4, in which, under the basic conditions employed, the methyl group is trihalogenated. Then the resulting trihalo ketone is attacked by hydroxide ion to give tetrahedral intermediate (51).706 The X3C− group is a sufficiently good leaving group (not HX2C− or H2XC−) that a carboxylic acid is formed, which quickly reacts with the carbanion to give the final products. Primary or secondary methylcarbinols also give the reaction, because they are oxidized to the carbonyl compounds under the conditions employed.
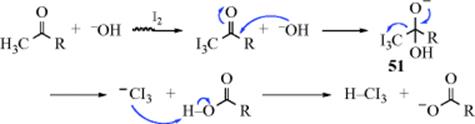
As with Reaction 12-4, the rate-determining step is the preliminary enolization of the methyl ketone.707 A side reaction is α halogenation of the non-methyl R group. Sometimes these groups are also cleaved.708 The reaction cannot be applied to F2, but ketones of the form RCOCF3 (R = alkyl or aryl) give fluoroform and RCOO− when treated with base.709 Rate constants for cleavage of X3CCOPh (X = F, Cl, Br) were found to be in the ratio 1 : 5.3 × 1010 : 2.2 × 1013, showing that an F3C− group cleaves much more slowly than the others.710 In the past, the haloform reaction was used as a test for methylcarbinols and methyl ketones. Iodine was most often used as the test reagent, since iodoform (HCI3) is an easily identifiable yellow solid. The reaction can be used for synthetic purposes. Methyl ketones (RCOCH3) can be converted directly to methyl esters (RCO2CH3) by an electrochemical reaction.711Trifluoromethyl ketones have been converted to ethyl esters via treatment with NaH in aq DMF followed by reaction with bromoethane.712
OS I, 526; II, 428; III, 302; IV, 345; V, 8. Also see, OS VI, 618.
12-45 Cleavage of Nonenolizable Ketones
Hydro-de-acylation
![]()
Ordinary ketones are generally much more difficult to cleave than trihalo ketones or β-diketones. However, nonenolizable ketones can be cleaved by treatment with a 10:3 mixture of t-BuOK–H2O in an aprotic solvent [e.g., ether, DMSO, 1,2-dimethoxyethane (glyme),713 or with solid t-BuOK in the absence of a solvent].714 When the reaction is applied to monosubstituted diaryl ketones, that aryl group preferentially cleaves that comes off as the more stable carbanion, except that aryl groups substituted in the ortho position are more readily cleaved than otherwise because of the steric effect (relief of strain).714,715 In certain cases, cyclic ketones can be cleaved by base treatment, even if they are enolizable.716
OS VI, 625. See also, OS VII, 297.
12-46 The Haller–Bauer Reaction
Hydro-de-acylation
![]()
Cleavage of ketones with sodium amide is called the Haller–Bauer reaction.717 As with Reaction 12-45, which is exactly analogous, the reaction is usually applied only to non-enolizable ketones, most often to ketones of the form ArCOCR3, where the products R3CCONH2 (after hydrolysis) are not easily attainable by other methods. However, many other ketones have been used, although benzophenone is virtually unaffected. It has been shown that the configuration of optically active alkyl groups (R) is retained.718 The NH2 loses its proton from the tetrahedral intermediate (52) before the R group is cleaved.719

An extension of this cleavage process involves the reaction of α-nitro ketones (O=C–CHRNO2) with a primary amine, neat, to give the corresponding amide, O=C–NHR′.720
OS V, 384, 1074.
C. Other Cleavages
12-47 The Cleavage of Alkanes
Hydro-de- tert- butylation, and so on
![]()
The C–C bonds of alkanes can be cleaved by treatment with superacids (Sec. 5.A.ii). For example, neopentane in FSO3H–SbF5 can cleave to give methane and the tert-butyl cation. The C–H cleavage (see Reaction 12-1) is a competing reaction and, for example, neopentane can give H2 and the tert-pentyl carbocation (formed by rearrangement of the initially formed neopentyl cation) by this pathway. In general, the order of reactivity is tertiary C–H > C–C > secondary C–H ![]() primary C–H, although steric factors cause a shift in favor of C–C cleavage in such a hindered compound as tri-tert-butylmethane. The mechanism is similar to that shown in Reactions 12-1 and 12-20 and involves attack by H+ on the C–C bond to give a pentavalent cation.
primary C–H, although steric factors cause a shift in favor of C–C cleavage in such a hindered compound as tri-tert-butylmethane. The mechanism is similar to that shown in Reactions 12-1 and 12-20 and involves attack by H+ on the C–C bond to give a pentavalent cation.
Catalytic hydrogenation seldom breaks unactivated C–C bonds (i.e., R–R′ + H2 → RH + R′H), but methyl and ethyl groups have been cleaved from substituted adamantanes by hydrogenation with a Ni-Al2O3 catalyst at ~250 °C.721 Certain C–C bonds have been cleaved by alkali metals.722
The C–C bond of 2-allyl-2-arylmalonate derivatives was cleaved, with loss of the allylic group to give the 2-arylmalonate, by treatment with a Ni catalyst.723
12-48 Decyanation or Hydro-de-cyanation
![]()
The cyano group of alkyl nitriles can be removed724 by treatment with metallic Na, either in liquid ammonia,725 or together with tris(acetylacetonato)iron(III) [Fe(acac)3]726 or, with lower yields, titanocene. The two procedures are complementary. Although both can be used to decyanate many kinds of nitriles, the Na–NH3 method gives high yields with R groups (e.g., trityl, benzyl, phenyl, and tertiary alkyl), but lower yields (~35–50%) when R = primary or secondary alkyl. On the other hand, primary and secondary alkyl nitriles are decyanated in high yields by the Na–Fe(acac)3 procedure. Sodium in liquid ammonia is known to be a source of solvated electrons, and the reaction may proceed through the free radical R√ that would then be reduced to the carbanion R−, which by abstraction of a proton from the solvent, would give RH. The mechanism with Fe(acac)3 is presumably different. Another procedure,727which is successful for R = primary, secondary, or tertiary, involves the use of potassium metal and the crown ether dicyclohexano-18-crown-6 in toluene.728
α-Amino and α-amido nitriles RCH(CN)NR′2 and RCH(CN)NHCOR′ can be decyanated in high yield by treatment with NaBH4.729
12.C.v. Electrophilic Substitution at Nitrogen
In most of the reactions in this section, an electrophile bonds with the unshared pair of a nitrogen atom. The electrophile may be a free positive ion or a positive species attached to a carrier that breaks off in the course of the attack or shortly after:
![]()
Further reaction of 53 depends on the nature of Y and of the other groups attached to the nitrogen.
12-49 The Conversion of Hydrazines to Azides
Hydrazine-azide transformation
![]()
Monosubstituted hydrazines treated with nitrous acid give azides in a reaction exactly analogous to the formation of aliphatic diazo compounds mentioned in Reaction 13-19. Among other reagents used for this conversion have been N2O4730 and nitrosyl tetrafluoroborate (NOBF4).731
OS III, 710; IV, 819; V, 157.
12-50 N-Nitrosation
N-Nitroso-de-hydrogenation
![]()
When secondary amines are treated with nitrous acid (typically formed from sodium nitrite and a mineral acid),732 N-nitroso compounds (also called nitrosamines) are formed.733 The reaction can be accomplished with dialkyl-, diaryl-, or alkylarylamines, and even with mono-N-substituted amides: RCONHR′ + HONO → RCON(NO)R′.734 Tertiary amines have also been N-nitrosated, but in these cases one group cleaves, so that the product is the nitroso derivative of a secondary amine.735 The group that cleaves appears as an aldehyde or ketone product. Other reagents have also been used (e.g., NOCl), which is useful for amines or amides that are not soluble in an acidic aqueous solution or where the N-nitroso compounds are highly reactive. N-Nitroso compounds can be prepared in basic solution by treatment of secondary amines with gaseous N2O3, N2O4,736 or alkyl nitrites,737 and, in aqueous or organic solvents, by treatment with BrCH2NO2.738 Secondary amines are converted to the N-nitroso compound with H5IO6 on wet silica.739
The mechanism of nitrosation is essentially the same as in Reaction 13-19 up to the point where 54 is formed. Since this species cannot lose a proton, it is stable and the reaction ends there. The attacking entity can be any of those mentioned in Reaction 13-19. The following has been suggested as the mechanism for the reaction with tertiary amines:740
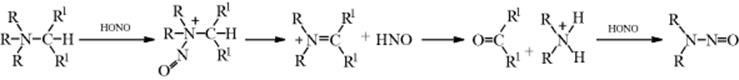
The evidence for this mechanism includes the facts that nitrous oxide is a product (formed by 2HNO → H2O + N2O) and that quinuclidine, where the nitrogen is at a bridgehead and cannot give elimination, does not react. Tertiary amines have also been converted to nitrosamines with nitric acid in Ac2O741 and with N2O4.742
Amines and amides can be N-nitrated743 with nitric acid,744 or NO2+,745 and aromatic amines can be converted to triazenes with diazonium salts. Aliphatic primary amines can also be converted to triazenes if the diazonium salts contain electron-withdrawing groups.746 C-Nitrosation is discussed at Reactions 11-3 and 12-8.
OS I, 177, 399, 417; II, 163, 211, 290, 460, 461, 462, 464 (Also see, V, 842); III, 106, 244; IV, 718, 780, 943; V, 336, 650, 797, 839, 962; VI, 542, 981. Also see, OS III, 711.
12-51 Conversion of Nitroso Compounds to Azoxy Compounds

In a reaction similar to 13-24, azoxy compounds can be prepared by the condensation of a nitroso compound with a hydroxylamine.747 The position of the oxygen in the final product is determined by the nature of the R groups, not by which R groups came from which starting compound. Both R and R′ can be alkyl or aryl, but when two different aryl groups are involved, mixtures of azoxy compounds (ArNONAr, ArNONAr′, and Ar′NONAr′) are obtained748 and the unsymmetrical product (ArNONAr′) is likely to be formed in the smallest amount. This behavior is probably caused by an equilibration between the starting compounds prior to the actual reaction (ArNO + Ar′NHOH → Ar′NO + ArNHOH).749 The mechanism750 has been investigated in the presence of base. Under these conditions both reactants are converted to radical anions, which couple:
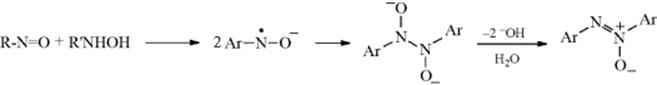
These radical anions have been detected by ESR.751 This mechanism is consistent with the following result: When nitrosobenzene and phenylhydroxylamine are coupled, ![]() and
and ![]() labeling show that the two nitrogen atoms and the two oxygen atoms become equivalent.752 Unsymmetrical azoxy compounds can be prepared753 by combination of a nitroso compound with an N,N-dibromoamine. Symmetrical and unsymmetrical azo and azoxy compounds are produced when aromatic nitro compounds react with aryliminodimagnesium reagents [ArN(MgBr)2].754
labeling show that the two nitrogen atoms and the two oxygen atoms become equivalent.752 Unsymmetrical azoxy compounds can be prepared753 by combination of a nitroso compound with an N,N-dibromoamine. Symmetrical and unsymmetrical azo and azoxy compounds are produced when aromatic nitro compounds react with aryliminodimagnesium reagents [ArN(MgBr)2].754
12-52 N-Halogenation
N-Halo-de-hydrogenation
![]()
Treatment with sodium hypochlorite or hypobromite converts primary amines into N-halo- or N,N-dihaloamines. Secondary amines can be converted to N-halo secondary amines. Similar reactions can be carried out on unsubstituted and N-substituted amides and on sulfonamides. With unsubstituted amides the N-halogen product is seldom isolated but usually rearranges (see Reaction 18-13); however, N-halo-N-alkyl amides and N-halo imides are quite stable. The important reagents NBS and NCS are made in this manner. N-Halogenation has also been accomplished with other reagents (e.g., sodium bromite, NaBrO2),755 benzyltrimethylammonium tribromide (PhCH2NMe3+Br3−),756 NaCl with Oxone,757 and NCS.758 Sodium hypohalite in the presence of tert-butanol and acetic acid is an efficient method for the preparation of N-haloamines.759 Amides are N-chlorinated with trichloroisocyanuric acid.760 The mechanisms of these reactions761 involve attack by a positive halogen and are probably similar to those of Reactions 13-19 and 12-50.762 N-Fluorination can be accomplished by direct treatment of amines763 or amides764 with F2. Fluorination of N-alkyl-N-fluoroamides [RRN(F)COR′] results in cleavage to N,N-difluoroamines (RNF2).764,765 Trichloroisocyanuric acid converts primary amines to the N,N-dichloroamine.766
OS III, 159; IV, 104, 157; V, 208, 663, 909; VI, 968; VII, 223; VIII, 167, 427.
12-53 The Reaction of Amines With Carbon Monoxide or Carbon Dioxide
N-Formylation or N-Formyl-de-hydrogenation, and so on
![]()
Three types of product can be obtained from the reaction of amines with CO, depending on the catalyst. (1) Both primary and secondary amines react with CO in the presence of various catalysts [e.g., Cu(CN)2, Me3N-H2Se, and Rh or Ru complexes] to give N-substituted and N,N-disubstituted formamides, respectively.767 Primary aromatic amines react with ammonium formate to give the formamide.768 Tertiary amines react with CO and a Pd catalyst to give an amide.769 (2) Symmetrically substituted ureas can be prepared by treatment of a primary amine (or ammonia) with CO770 in the presence of Se771 or S.772 The R source can be alkyl or aryl. The same thing can be done with secondary amines, using Pd(OAc)2–I2–K2CO3.773 Primary aromatic amines react with β-keto esters and a Mo–ZrO2 catalyst to give the symmetrical urea.774 Treatment of a secondary amine with nitrobenzene, selenium and carbon monoxide leads to the unsymmetrical urea.775 (3) When PdCl2 is the catalyst, primary amines yield isocyanates.776 Isocyanates can also be obtained by treatment of CO with azides: RN3 + CO → RNCO,777 or with an aromatic nitroso or nitro compound and a Rh complex catalyst.778 Primary amines react with di-tert-butyltricarbonate to give the isocyanate.779
Lactams are converted to the corresponding N-chloro lactam with Ca(OCl)2 with moist alumina in dichloromethane.780 Ring-expanded lactams are obtained from cyclic amines via a similar reaction781 (see also, Reaction 16-22). Intramolecular carbonylation of amines also leads to lactams.782
A fourth type of product, a carbamate (RNHCOOR′), can be obtained from primary or secondary amines, if these are treated with CO, O2, and an alcohol (R′OH) in the presence of a catalyst.783 Primary amines react with dimethyl carbonate in supercritical CO2 (see Sec. 9.D.ii) to give a carbamate.784 Carbamates can also be obtained from nitroso compounds, by treatment with CO, R′OH, Pd(OAc)2, and Cu(OAc)2,785 and from nitro compounds.786 When allylic amines (R2C=CHRCHRNR′2) are treated with CO and a Pd–phosphine catalyst, the CO inserts to produce the β,γ-unsaturated amides (R2C=CHRCHRCONR′2) in good yields.787 Silyloxy carbamates (RNHCO2SiR′3) can be prepared by the reaction of a primary amine with carbon dioxide and triethylamine, followed by reaction with triisopropylsilyl triflate and tetrabutylammonium fluoride.788
Carbon dioxide reacts with amines (ArNH2) and alkyl halides, under electrolysis conditions, to give the corresponding carbamate (ArNHCO2Et).789 Secondary amines react with all halides and an onium salt in supercritical CO2(see Sec. 9.D.ii) to give the carbamate.790 N-Phenylthioamines react with CO and a palladium catalyst to give a thiocarbamate (ArSCO2NR′2).791 Urea derivatives were obtained from amines, CO2, and an antimony catalyst.792
Aziridines can be converted to cyclic carbamates (oxazolidinones) by heating with carbon dioxide and a chromium–salen catalyst.793 The reaction of aziridines with LiI, and then CO2 also generates oxazolidinones.794
Notes
1. See Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, 5 Vols., Wiley, NY, 1984–1990; Haiduc, I.; Zuckerman, J.J. Basic Organometallic Chemistry, Walter de Gruyter, NY, 1985; Negishi, E. Organometallics in Organic Synthesis, Wiley, NY, 1980; Aylett, B.J. Organometallic Compounds, 4th ed., Vol. 1, pt. 2; Chapman and Hall, NY, 1979; Maslowsky, Jr., E. Chem. Soc. Rev. 1980, 9, 25, and in Tsutsui, M. Characterization of Organometallic Compounds, Wiley, NY, 1969–1971, the articles by Cartledge, F.K.; Gilman, H. pt. 1, pp. 1–33, and by Reichle, W.T. pt. 2, pp. 653–826.
2. See Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H. Eds., Vol. 12, Elsevier, NY, 1973; Reutov, O.A.; Beletskaya, I.P. Reaction Mechanisms of Organometallic Compounds, North-Holland Publishing Company, Amsterdam, The Netherlands, 1968; Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, pp. 25–149; Reutov, O.A. Pure Appl. Chem. 1978, 50, 717; Tetrahedron 1978, 34, 2827.
3. Gawley, R.E. Tetrahedron Lett. 1999, 40, 4297.
4. Buckle, M.J.C.; Fleming, I.; Gil, S. Tetrahedron Lett. 1992, 33, 4479.
5. The names for these mechanisms vary throughout the literature. For example, the SEi mechanism has also been called the SE2, the SE2 (closed), and the SE2 (cyclic) mechanism. The original designations, SE1, SE2, and so on, were devised by the Hughes–Ingold school.
6. It has been contended that the SEi mechanism violates the principle of conservation of orbital symmetry (sec Reaction 15-60, A), and that the SE2 (back) mechanism partially violates it: Slack, D.A.; Baird, M.C. J. Am. Chem. Soc. 1976, 98, 5539.
7. See Flood, T.C. Top. Stereochem. 1981, 12, 37. See also, Jensen, F.R.; Davis, D.D. J. Am. Chem. Soc. 1971, 93, 4048.
8. Winstein, S.; Traylor, T.G.; Garner, C.S. J. Am. Chem. Soc. 1955, 77, 3741.
9. Schöllkopf, U. Angew. Chem. 1960, 72, 147. See Fort, Jr., R.C.; Schleyer, P.v.R. Adv. Alicyclic Chem. 1966, 1, 283, pp. 353–370.
10. Hughes, E.D.; Volger, H.C. J. Chem. Soc. 1961, 2359.
11. Jensen, F.R. J. Am. Chem. Soc. 1960, 82, 2469; Ingold, C.K. Helv. Chim. Acta 1964, 47, 1191.
12. Jensen, F.R.; Davis, D.D. J. Am. Chem. Soc. 1971, 93, 4048. See Fukuto, J.M.; Jensen, F.R. Acc. Chem. Res. 1983, 16, 177.
13. See Magnuso, R.H.; Halpern, J.; Levitin, I.Ya.; Vol'pin, M.E. J. Chem. Soc. Chem. Commun. 1978, 44.
14. See Rahm, A.; Pereyre, M. J. Am. Chem. Soc. 1977, 99, 1672; McGahey, L.F.; Jensen, F.R. J. Am. Chem. Soc. 1979, 101, 4397; Olszowy, H.A.; Kitching, W. Organometallics 1984, 3, 1676. Also see Rahm, A.; Grimeau, J.; Pereyre, M. J. Organomet. Chem. 1985, 286, 305.
15. See Bergbreiter, D.E.; Rainville, D.P. J. Organomet. Chem. 1976, 121, 19.
16. See Sokolov, V.I. Chirality and Optical Activity in Organometallic Compounds, Gordon and Breach, NY, 1990.
17. See Jensen, F.R.; Whipple, L.D.; Wedegaertner, D.K.; Landgrebe, J.A. J. Am. Chem. Soc. 1959, 81, 1262; Charman, H.B.; Hughes, E.D.; Ingold, C.K. J. Chem. Soc. 1959, 2523, 2530.
18. This was done first by Walborsky, H.M.; Young, A.E. J. Am. Chem. Soc. 1964, 86, 3288.
19. Jensen, F.R.; Nakamaye, K.L. J. Am. Chem. Soc. 1966, 88, 3437.
20. Abraham, M.H.; Johnston, G.F. J. Chem. Soc. A, 1970, 188.
21. See Abraham, M.H.; Dorrell, F.J. J. Chem. Soc. Perkin Trans. 2 1973, 444.
22. Fukuto, J.M.; Newman, D.A.; Jensen, F.R. Organometallics 1987, 6, 415.
23. Abraham, M.H.; Hill, J.A. J. Organomet. Chem. 1967, 7, 11.
24. Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H., Eds., Vol. 12, Elsevier, NY, 1973, p. 15.
25. Fukuzumi, S.; Kochi, J.K. J. Am. Chem. Soc. 1980, 102, 2141, 7290.
26. Hsu, S.K.; Ingold, C.K.; Wilson, C.L. J. Chem. Soc. 1938, 78.
27. Wilson, C.L. J. Chem. Soc. 1936, 1550.
28. See Hoffman, T.D.; Cram, D.J. J. Am. Chem. Soc. 1969, 91, 1009. For a discussion, see Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 138–158.
29. See Roitman, J.N.; Cram, D.J. J. Am. Chem. Soc. 1971, 93, 2225, 2231 and references cited therein; Cram, J.M.; Cram, D.J. Intra-Sci. Chem. Rep. 1973, 7(3), 1; Cram, D.J. Fundamentals of Carbanion Chemistry, Academic Press, NY, 1965, pp. 85–105.
30. Cram, D.J.; Ford, W.T.; Gosser, L. J. Am. Chem. Soc. 1968, 90, 2598; Ford, W.T.; Cram, D.J. J. Am. Chem. Soc. 1968, 90, 2606, 2612. See also, Buchholz, S.; Harms, K.; Massa, W.; Boche, G. Angew. Chem. Int. Ed. 1989, 28,73.
31. Almy, J.; Hoffman, D.H.; Chu, K.C.; Cram, D.J. J. Am. Chem. Soc. 1973, 95, 1185.
32. Dreiding, A.S.; Pratt, R.J. J. Am. Chem. Soc. 1954, 76, 1902. See also, Walborsky, H.M.; Turner, L.M. J. Am. Chem. Soc. 1972, 94, 2273.
33. See Courtois, G.; Miginiac, L. J. Organomet. Chem. 1974, 69, 1.
34. Sleezer, P.D.; Winstein, S.; Young, W.G. J. Am. Chem. Soc. 1963, 85, 1890. See also, Kashin, A.N.; Bakunin, V.N.; Khutoryanskii, V.A.; Beletskaya, I.P.; Reutov, O.A. J. Organomet. Chem. 1979, 171, 309.
35. Matassa, V.G.; Jenkins, P.R.; Kümin, A.; Damm, L.; Schreiber, J.; Felix, D.; Zass, E.; Eschenmoser, A. Isr. J. Chem. 1989, 29, 321.
36. Young, D.; Kitching, W. J. Org. Chem. 1983, 48, 614; Tetrahedron Lett. 1983, 24, 5793.
37. Kashin, A.N.; Bakunin, V.N.; Beletskaya, I.P.; Reutov, O.A. J. Org. Chem. USSR 1982, 18, 1973. See also, Wickham, G.; Young, D.; Kitching, W. Organometallics 1988, 7, 1187.
38. See Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H., Eds., Vol. 12, Elsevier, NY, 1973, pp. 211–241.
39. Also see Isaacs, N.S.; Laila, A.H. Tetrahedron Lett. 1984, 25, 2407.
40. See Abraham, M.H.; Broadhurst, A.T.; Clark, I.D.; Koenigsberger, R.U.; Dadjour, D.F. J. Organomet. Chem. 1981, 209, 37.
41. Sayre, L.M.; Jensen, F.R. J. Am. Chem. Soc. 1979, 101, 6001.
42. Also see Nugent, W.A.; Kochi, J.K. J. Am. Chem. Soc. 1976, 98, 5979.
43. Reutov, O.A. J. Organomet. Chem. 1983, 250, 145. See also, Butin, K.P.; Magdesieva, T.V. J. Organomet. Chem. 1985, 292, 47.
44. Richard, J.P. J. Org. Chem. 1992, 57, 625.
45. See Reutov, O.A. Bull. Acad. Sci. USSR Div. Chem. Sci. 1980, 29, 1461. See also, Dembech, P.; Eaborn, C.; Seconi, G. J. Chem. Soc. Chem. Commun. 1985, 1289.
46. See Kitching, W. Rev. Pure Appl. Chem. 1969, 19, 1.
47. See Petrosyan, V.S. J. Organomet. Chem. 1983, 250, 157.
48. See Olah, G.A.; Prakash, G.K.S.; Sommer, J. Superacids, Wiley, NY, 1985, pp. 244–249; Olah, G.A. Angew. Chem. Int. Ed. 1973, 12, 173.
49. See McMurry, J.E.; Lectka, T. J. Am. Chem. Soc. 1990, 112, 869; Culmann, J.; Sommer, J. J. Am. Chem. Soc. 1990, 112, 4057.
50. See Olah, G.A.; Prakash, G.K.S.; Williams, R.E.; Field, L.D.; Wade, K. Hypercarbon Chemistry, Wiley, NY, 1987.
51. See Sefcik, M.D.; Henis, J.M.S.; Gaspar, P.P. J. Chem. Phys. 1974, 61, 4321.
52. Yeh, L.I.; Pric, J.M.; Lee, Y.T. J. Am. Chem. Soc. 1989, 111, 5597.
53. Lukas, J.; Kramer, P.A.; Kouwenhoven, A.P. Recl. Trav. Chim. Pays-Bas 1973, 92, 44.
54. Werstiuk, N.H.; Timmins, G. Can. J. Chem. 1985, 63, 530; 1986, 64, 1564.
55. See Atkinson, J.G.; Luke, M.O.; Stuart, R.S. Can. J. Chem. 1967, 45, 1511.
56. For a list of methods used to shift double and triple bonds, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 220–226, 567–568.
57. See Pines, H.; Stalick, W.M. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds, Academic Press, NY, 1977, pp. 25–123; DeWolfe, R.H. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 9, Elsevier, NY, 1973, pp. 437–449; Hubert, A.J.; Reimlinger, H. 1970, 405; Mackenzie, K. in The Chemistry of Alkenes, Vol. 1, Patai, S., pp. 416–436, Vol. 2, Zabicky, J., pp. 132–148; Wiley, NY, 1964, 1970; Broaddus, C.D. Acc. Chem. Res. 1968, 1, 231.
58. See Hine, J.; Skoglund, M.J. J. Org. Chem. 1982, 47, 4766. See also, Hine, J.; Linden, S. J. Org. Chem. 1983, 48, 584.
59. For a review of conversions of β,γ enones to α,β enones, see Pollack, R.M.; Bounds, P.L.; Bevins, C.L. in Patai, S.; Rappoport, Z. The Chemistry of Enones, pt. 1, Wiley, NY, 1989, pp. 559–597.
60. See Pollack, R.M.; Mack, J.P.G.; Eldin, S. J. Am. Chem. Soc. 1987, 109, 5048.
61. Rabinovich, E.A.; Astaf'ev, I.V.; Shatenshtein, A.I. J. Gen. Chem. USSR 1962, 32, 746.
62. Hünig, S.; Klaunzer, N.; Schlund, R. Angew. Chem. Int. Ed. 1987, 26, 1281.
63. See Cram, D.J.; Uyeda, R.T. J. Am. Chem. Soc. 1964, 86, 5466; Ohlsson, L.; Wold, S.; Bergson, G. Ark. Kemi., 1968, 29, 351.
64. Hussénius, A.; Matsson, O.; Bergson, G. J. Chem. Soc. Perkin Trans. 2 1989, 851.
65. See Cameron G.S.; Stimson, V.R. Aust. J. Chem. 1977, 30, 923.
66. Schönberg, A. Preparative Organic Photochemistry, Springer, NY, 1968, pp. 22–24.
67. See Rodriguez, J.; Brun, P.; Waegell, B. Bull. Soc. Chim. Fr. 1989, 799–823; Otsuka, S.; Tani, K. in Morrison, J.D. Asymmetric Synthesis Vol. 5, Academic Press, NY, 1985, pp. 171–191 (enantioselective); Colquhoun, H.M.; Holton, J.; Thompson, D.J.; Twigg, M.V. New Pathways for Organic Synthesis, Plenum, NY, 1984, pp. 173–193; Khan, M.M.T.; Martell, A.E. Homogeneous Catalysis by Metal Complexes, Academic Press, NY, 1974, pp. 9–37; Heck, R.F. Organotransition Metal Chemistry, Academic Press, NY, 1974, pp. 76–82; Jira, R.; Freiesleben, W. Organomet. React. 1972, 3, 1, pp. 133–149.
68. Cramer, R. J. Am. Chem. Soc. 1966, 88, 2272.
69. Casey, C.P.; Cyr, C.R. J. Am. Chem. Soc. 1973, 95, 2248.
70. Trost, B.M.; Schmidt, T. J. Am. Chem. Soc. 1988, 110, 2301.
71. Han, J.W.; Tokunaga, N.; Hayashi, T. J. Am. Chem. Soc. 2001, 123, 12915.
72. Bergens, S.H.; Bosnich, B. J. Am. Chem. Soc. 1991, 113, 958.
73. See Duhaime, R.M.; Lombardo, D.A.; Skinner, I.A.; Weedon, A.C. J. Org. Chem. 1985, 50, 873.
74. See Pines, H.; Stalick, W.M. Base-Catalyzed Reactions of Hydrocarbons and Related Compounds, Academic Press, NY, 1977, pp. 124–204; Théron F.; Verny, M.; Vessière, R. in Patai, S. The Chemistry of Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 381–445; Bushby, R.J. Q. Rev. Chem. Soc. 1970, 24, 585; Iwai, I. Mech. Mol. Migr. 1969, 2, 73.
75. See Huntsman, W.D. in Patai, S. The Chemistry of Ketenes, Allenes, and Related Compounds, pt. 2, Wiley, NY, 1980, pp. 521–667.
76. Macaulay, S.R. J. Org. Chem. 1980, 45, 734; Abrams, S.R. Can. J. Chem. 1984, 62, 1333.
77. See Oku, M.; Arai, S.; Katayama, K.; Shioiri, T. Synlett 2000, 493.
78. See Cunico, R.F.; Zaporowski, L.F.; Rogers, M. J. Org. Chem. 1999, 64, 9307.
79. Myers, A.G.; Zheng, B. J. Am. Chem. Soc. 1996, 118, 4492. See Moghaddam, F.M.; Emami, R. Synth. Commun. 1997, 27, 4073 for the formation of alkoxy allenes from propargyl ethers.
80. Sonye, J.P.; Koide, K. J. Org. Chem. 2006, 71, 6254.
81. Barry, B.J.; Beale, W.J.; Carr, M.D.; Hei, S.; Reid, I. J. Chem. Soc. Chem. Commun. 1973, 177.
82. Patai, S. The Chemistry of the Carbonyl Group, Wiley, London, 1966; Rappoport, Z. The Chemistry of Enols, Wiley, NY, 1990; Rappoport, Z.; Frey, J.; Sigalov, M.; Rochlin, E. Pure Appl. Chem. 1997, 69, 1933; Fontana, A.; De Maria, P.; Siani, G.; Pierini, M.; Cerritelli, S.; Ballini, R. Eur. J. Org. Chem. 2000, 1641; Iglesias, E. Curr. Org. Chem. 2004, 8, 1.
83. See Jones, J.R. The Ionisation of Carbon Acids, Academic Press, London, 1973; Toullec, J. Adv. Phys. Org. Chem. 1982, 18, 1; Chiang, Y.; Kresge, A.J.; Santaballa, J.A.; Wirz, J. J. Am. Chem. Soc. 1988, 110, 5506.
84. See Raczynska, E.D.; Kosinska, W.; Osmialowski, B.; Gawinecki, R. Chem. Rev. 2005, 105, 3561 for a general discussion of tautomerism. Also see Rappoport, Z. The Chemistry of Enols, Wiley, NY, 1990; Zabicky, J. The Chemistry of Amides, Wiley, London, 1970; Boyer, J.H. The Chemistry of the Nitro and Nitroso Groups, Interscience Publishers, NY, 1969; Patai, S. The Chemistry of Amino, Nitroso, Nitro Compounds and their Derivatives, Wiley, NY, 1982; Patai, S. The Chemistry of Amino, Nitroso, Nitro and Related Groups, Supplement F2, Wiley, Chichester, 1996; Cook, A.G. Enamines, 2nd ed., Marcel Dekker, NY, 1998.
85. Gorb, L.; Leszczynski, J. J. Am. Chem. Soc. 1998, 120, 5024; Guo, J. X.; Ho, J. J. J. Phys. Chem. A 1999, 103, 6433.
86. Briegleb, G.; Strohmeier, W. Angew. Chem. 1952, 64, 409; Baddar, F. G.; Iskander, Z. J. Chem. Soc. 1954, 203.
87. Hegarty, A.F.; Dowling, J.P.; Eustace, S.J.; McGarraghy, M. J. Am. Chem. Soc. 1998, 120, 2290.
88. Behnam, S.M.; Behnam, S.E.; Ando, K.; Green, N.S.; Houk, K.N. J. Org. Chem. 2000, 65, 8970.
89. Miller, A.R. J. Org. Chem., 1976, 41,3599.
90. Fuson, R.C.; Tan, T.-L. J. Am. Chem. Soc. 1948, 70, 602.
91. See Keeffe, J.R.; Kresge, A.J. in Rappoport, Z. The Chemistry of Enols, Wiley, NY, 1990, pp. 399–480; Toullec, J. Adv. Phys. Org. Chem. 1982, 18, 1. Also see Bell, R.P. The Proton in Chemistry, 2nd ed., Cornell Univ. Press, Ithaca, NY, 1973, pp. 171–181; Shelly, K.P.; Venimadhavan, S.; Nagarajan, K.; Stewart, R. Can. J. Chem. 1989, 67, 1274. Also see Pollack, R.M. Tetrahedron 1989, 45, 4913.
92. Another mechanism for base-catalyzed enolization has been reported when the base is a tertiary amine: See Bruice, P.Y. J. Am. Chem. Soc. 1990, 112, 7361 and references cited therein.
93. For a proposed concerrted mechanism, see Capon, B.; Siddhanta, A.K.; Zucco, C. J. Org. Chem. 1985, 50, 3580. For evidence against it, see Chiang, Y.; Hojatti, M.; Keeffe, J.R.; Kresge, A.J.; Schepp, N.P.; Wirz, J. 1987, 109,4000 and references cited therein.
94. Xie, L.; Saunders, Jr., W.H. J. Am. Chem. Soc. 1991, 113, 3123.
95. Lienhard, G.E.; Wang, T. J. Am. Chem. Soc. 1969, 91, 1146. See also, Toullec, J.; Dubois, J.E. J. Am. Chem. Soc. 1974, 96, 3524.
96. Chiang, Y.; Kresge, A.J.; Meng, Q.; More O'Ferrall, R.A.; Zhu, Y. J. Am. Chem. Soc. 2001, 123, 11562.
97. Chiang, Y.; Griesbeck, A.G.; Heckroth, H.; Hellrung, B.; Kresge, A.J.; Meng, Q.; O'Donoghue, A.C.; Richard, J.P.; Wirz, J. J. Am. Chem. Soc. 2001, 123, 8979.
98. Zimmerman, H.E.; Cheng, J. J. Org. Chem. 2006, 71, 873.
99. Cantlin, R.J.; Drake, J.; Nagorski, R.W. Org. Lett. 2002, 4, 2433.
100. Bernasconi, C.F.; Pérez-Lorenzo, M. J. Am. Chem. Soc. 2007, 129, 2704.
101. Senn, M.; Richter, W.J.; Burlingame, A.L. J. Am. Chem. Soc. 1965, 87, 680; Richter, W.J.; Senn, M.; Burlingame, A.L. Tetrahedron Lett. 1965, 1235.
102. For an exception, see Guthrie, R.D.; Nicolas, E.C. J. Am. Chem. Soc. 1981, 103, 4637.
103. Dechoux, L.; Doris, E. Tetrahedron Lett. 1994, 35, 2017.
104. Coggins, P.; Gaur, S.; Simpkins, N.S. Tetrahedron Lett. 1995, 36, 1545.
105. See Wen, J.Q.; Grutzner, J.B. J. Org. Chem. 1986, 51, 4220.
106. See d'Angelo, J. Tetrahedron 1976, 32, 2979. Also see Fruchart, J.-S.; Lippens, G.; Kuhn, C.; Gran-Masse, H.; Melnyk, O. J. Org. Chem. 2002, 67, 526.
107. Vedejs, E.; Kruger, A.W.; Suna, E. J. Org. Chem. 1999, 64, 7863.
108. He, X.; Allan, J.F.; Noll, B.C.; Kennedy, A.R.; Henderson, K.W. J. Am. Chem. Soc. 2005, 127, 6920.
109. Eleveld, M.B.; Hogeveen, H. Tetrahedron Lett. 1986, 27, 631. See also, Cain, C.M.; Cousins, R.P.C.; Coumbarides, G.; Simpkins, N.S. Tetrahedron 1990, 46, 523.
110. See House, H.O. Modern Synthetic Reactions, 2nd ed., W.A. Benjamin, NY, 1972, pp. 459–478; De Kimpe, N.; Verhé, R. The Chemistry of α-Haloketones, α-Haloaldehydes, and α-Haloimines, Wiley, NY, 1988. For lists of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp.709–719.
111. See Rozen, S.; Filler, R. Tetrahedron 1985, 41, 1111; German, L.; Zemskov, S. New Fluorinating Agents in Organic Chemistry; Springer, NY, 1989.
112. Tabushi, I.; Kitaguchi, H. in Pizey, J.S. Synthetic Reagents, Vol. 4; Wiley, NY, 1981, pp. 336–396.
113. Fraser, R.R.; Kong, F. Synth. Commun. 1988, 18, 1071.
114. See Mei, Y.; Bentley, P.A.; Du, J. Tetrahedron Lett. 2008, 49, 3802. Alse see Pravst, I.; Zupan, M.; Stavber, S. Tetrahedron 2008, 64, 5191.
115. Bellesia, F.; DeBuyck, L.; Ghelfi, F.; Pagnoni, U.M.; Parson, A.F.; Pinetti, A. Synthesis 2003, 2173.
116. Tanemura, K.; Suzuki, T.; Nishida, Y.; Satsumabayashi, K.; Horaguchi, T. Chem. Commun. 2004, 470. See Guha, S.K.; Wu, B.; Kim, B.S.; Baik, W.; Koo, S. Tetrahedron Lett. 2006, 47, 291; Arbuj, S.S.; Waghmode, S.B.; Ramaswamy, A.V. Tetrahedron Lett. 2007, 48, 1411. See also Sreedhar, B.; Reddy, P.S.; Madhavi, M. Synth. Commun. 2007, 37, 4149.
117. Bellesia, F.; Ghelfi, F.; Grandi, R.; Pagnoni, U.M. J. Chem. Res. (S) 1986, 428.
118. Kajigaeshi, S.; Kakinami, T.; Okamoto, T.; Fujisaki, S. Bull. Chem. Soc. Jpn. 1987, 60, 1159.
119. Juneja, S.K. Choudhary, D.; Paul, S.; Gupta, R. Synth. Commun. 2006, 36, 2877.
120. Paul, S.; Gupta, V.; Gupta, R.; Loupy, A. Tetrahedron Lett. 2003, 44, 439.
121. Lee, J.C.; Park, H.J. Synth. Commun. 2006, 36, 777.
122. Pingali, S.R.K.; Madhav, M.; Jursic, B.S. Tetrahedron Lett. 2010, 51, 1383.
123. Brochu, M.P.; Brown, S.P.; MacMillan, D.W.C. J. Am. Chem. Soc. 2004, 126, 4108; Halland, N.; Braunton, A.; Bachmann, S.; Marigo, M.; Jorgensen, K.A. J. Am. Chem. Soc. 2004, 126, 4790. See Wang, L.; Cai, C.; Curran, D.P.; Zhang, W. Synlett 2010, 433.
124. See Bertelsen, S.; Halland, N.; Bachmann, S.; Marigo, M.; Braunton, A.; J![]() rgensen, K.A. Chem. Commun. 2005, 4821.
rgensen, K.A. Chem. Commun. 2005, 4821.
125. See France, S.; Weatherwax, A.; Lectka, T. Eur. J. Org. Chem. 2005, 475.
126. See Ueda, M.; Kano, T.; Maruoka, K. Org. Biomol. Chem. 2009, 7, 2005.
127. Khan, A.T.; Ali, Md.A.; Goswami, P.; Choudhury, L.H. J. Org. Chem. 2006, 71, 8961.
128. Meketa, M.L.; Mahajan, Y.R.; Weinreb, S.M. Tetrahedron Lett. 2005, 46, 4749.
129. Rao, M.L.N.; Jadhav, D.N. Tetrahedron Lett. 2006, 47, 6883. Also see Yadav, J.S.; Kondaji, G.; Reddy, M.S.R.; Srihari, P. Tetrahedron Lett. 2008, 49, 3810.
130. Horiuchi, C.A.; Kiji, S. Bull. Chem. Soc. Jpn. 1997, 70, 421. For another reagent, see Sket, B.; Zupet, P.; Zupan, M.; Dolenc, D. Bull. Chem. Soc. Jpn. 1989, 62, 3406.
131. Yamamoto, T.; Toyota, K.; Morita, N. Tetrahedron Lett. 2010, 51, 1364.
132. Mohanakrishnan, A.K.; Prakash, C.; Ramesh, N. Tetrahedron 2006, 62, 3242.
133. Jereb, M.; Stavber, S.; Zupan, M. Tetrahedron 2003, 59, 5935.
134. Lee, J.C.; Bae, Y.H. Synlett 2003, 507.
135. Kano, T.; Ueda, M.; Maruoka, K. J. Am. Chem. Soc. 2008, 130, 3728.
136. Davis, F.A.; Kasu, P.V.N. Org. Prep. Proceed. Int. 1999, 31, 125.
137. See Pihko, P.M. Angew. Chem. Int. Ed. 2006, 45, 544.
138. Enders, D.; Hüttl, M.R.M. Synlett 2005, 991.
139. See Loghmani-Khouzani, H.; Poorheravi, M.R.; Sadeghi, M.M.M.; Caggiano, L.; Jackson, R.F.W. Tetrahedron 2008, 64, 7419.
140. Okamoto, T.; Kakinami, T.; Nishimura, T.; Hermawan, I.; Kajigaeshi, S. Bull. Chem. Soc. Jpn. 1992, 65, 1731.
141. Barnette, W.E. J. Am. Chem. Soc. 1984, 106, 452; Ma, J.-A. For an example with asymmetric induction, see Cahard, D. Tetrahedron Asymm 2004, 15, 1007.
142. Differding, E.; Lang, R.W. Tetrahedron 1988, 29, 6087.
143. Chambers, R.D.; Greenhall, M.P.; Hutchinson, J. J. Chem. Soc. Chem. Commun. 1995, 21.
144. Gupta, O.D.; Shreeve, J.M. Tetrahedron Lett. 2003, 44, 2799.
145. Rozen, S.; Brand, M. Synthesis 1985, 665. For another reagent, see Davis, F.A.; Han, W. Tetrahedron Lett. 1991, 32, 1631.
146. Beeson, T.D.; MacMillan, D.W.C. J. Am. Chem. Soc. 2005, 127, 8826.
147. Hamashima, Y.; Suzuki, T.; Takano, H.; Shimura, Y.; Sodeoka, M. J. Am. Chem. Soc. 2005, 127, 10164.
148. Steiner, D.D.; Mase, N.; Barbas III, C.F. Angew. Chem. Int. Ed. 2005, 44, 3706.
149. For chlorination this is reversed if the solvent is methanol: Gallucci, R.R.; Going, R. J. Org. Chem. 1981, 46, 2532.
150. Rappe, C. Ark. Kemi 1965, 24, 321. But see also, Teo, K.E.; Warnhoff, E.W. J. Am. Chem. Soc. 1973, 95, 2728.
151. Garbisch Jr., E.W. J. Org. Chem. 1965, 30, 2109.
152. Nobrega, J.A.; Goncalves, S.M.C.; Reppe, C. Synth. Commun. 2002, 32, 3711.
153. Terent'ev, A.O.; Khodykin, S.V.; Troitskii, N.A.; Ogibin, Y.N.; Nikishin, G.I. Synthesis 2004, 2845.
154. Kajigaeshi, S.; Kakinami, T.; Tokiyama, H.; Hirakawa, T.; Okamoto, T. Bull. Chem. Soc. Jpn. 1987, 60, 2667.
155. Yang, D.; Yan, Y.-L.; Lui, B. J. Org. Chem. 2002, 67, 7429.
156. Marigo, M.; Kumaragurubaran, N.; J![]() rgensen, K.A. Chem. Eur. J. 2004, 10, 2133.
rgensen, K.A. Chem. Eur. J. 2004, 10, 2133.
157. See Tapuhi, E.; Jencks, W.P. J. Am. Chem. Soc. 1982, 104, 5758. Also see Pinkus, A.G.; Gopalan, R. J. Am. Chem. Soc. 1984, 106, 2630; Pinkus, A.G.; Gopalan, R. Tetrahedron 1986, 42, 3411.
158. See, however, Deno, N.C.; Fishbein, R. J. Am. Chem. Soc. 1973, 95, 7445.
159. Bell, R.P.; Yates, K. J. Chem. Soc. 1962, 1927.
160. Hochstrasser, R.; Kresge, A.J.; Schepp, N.P.; Wirz, J. J. Am. Chem. Soc. 1988, 110, 7875.
161. Hooz, J.; Bridson, J.N. Can. J. Chem. 1972, 50, 2387.
162. Stotter, P.L.; Hill, K.A. J. Org. Chem. 1973, 38, 2576.
163. Blanco, L.; Amice, P.; Conia, J.M. Synthesis 1976, 194.
164. Olah, G.A.; Ohannesian, L.; Arvanaghi, M.; Prakash, G.K.S. J. Org. Chem. 1984, 49, 2032.
165. Rubottom, G.M.; Mott, R.C. J. Org. Chem. 1979, 44, 1731.
166. Zhang, Y.; Shibatomi, K.; Yamamoto, H. J. Am. Chem. Soc. 2004, 126, 15038.
167. Tsushima, T.; Kawada, K.; Tsuji, T. Tetrahedron Lett. 1982, 23, 1165.
168. Purrington, S.T.; Bumgardner, C.L.; Lazaridis, N.V.; Singh, P. J. Org. Chem. 1987, 52, 4307.
169. Cambie, R.C.; Hayward, R.C.; Jurlina, J.L.; Rutledge, P.S.; Woodgate, P.D. J. Chem. Soc. Perkin Trans. 1 1978, 126.
170. Horiuchi, C.A.; Satoh, J.Y. Synthesis 1981, 312.
171. Ley, S.V.; Whittle, A.J. Tetrahedron Lett. 1981, 22, 3301.
172. Hegde, S.G.; Wolinsky, J. Tetrahedron Lett. 1981, 22, 5019.
173. Fache, F.; Piva, O. Synlett 2002, 2035.
174. See Harwood, H.J. Chem. Rev. 1962, 62, 99, pp. 102-103.
175. Wack, H.; Taggi, A.E.; Hafez, A.M.; Drury III, W.J.; Lectka, T. J. Am. Chem. Soc. 2001, 123, 1531. See also, France, S.; Wack, H.; Taggi, A.E.; Hafez, A.M.; Wagerle, Ty.R.; Shah, M.H.; Dusich, C.L.; Lectka, T. J. Am. Chem. Soc. 2004, 126, 4245.
176. See, however, Kwart, H.; Scalzi, F.V. J. Am. Chem. Soc. 1964, 86, 5496.
177. Ogata, Y.; Watanabe, S. J. Org. Chem. 1979, 44, 2768; 1980, 45, 2831.
178. Ogata, Y.; Adachi, K. J. Org. Chem. 1982, 47, 1182.
179. Zhang, L.H.; Duan, J.; Xu, Y.; Dolbier, Jr., W.R. Tetrahedron Lett. 1998, 39, 9621.
180. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 730–738.
181. Okimoto, M.; Takahashi, Y. Synthesis 2002, 2215.
182. Harpp, D.N.; Bao, L.Q.; Black, C.J.; Gleason, J.G.; Smith, R.A. J. Org. Chem. 1975, 40, 3420.
183. Horiuchi, C.A.; Satoh, J.Y. Chem. Lett. 1984, 1509.
184. Rathke, M.W.; Lindert, A. Tetrahedron Lett. 1971, 3995.
185. Purrington, S.T.; Woodard, D.L. J. Org. Chem. 1990, 55, 3423.
186. Kitagawa, O.; Hanano, T.; Hirata, T.; Inoue, T.; Taguchi, T. Tetrahedron Lett. 1992, 33, 1299.
187. For a review, see Venier, C.G.; Barager, III, H.J. Org. Prep. Proced. Int. 1974, 6, 77, pp. 81–84.
188. Tsuchihashi, G.; Iriuchijima, S. Bull. Chem. Soc. Jpn. 1970, 43, 2271.
189. Ogura, K.; Imaizumi, J.; Iida, H.; Tsuchihashi, G. Chem. Lett. 1980, 1587.
190. Tin, K.; Durst, T. Tetrahedron Lett. 1970, 4643.
191. Kim, Y.H.; Lim, S.C.; Kim, H.R.; Yoon, D.C. Chem. Lett. 1990, 79.
192. Cinquini, M.; Colonna, S. J. Chem. Soc. Perkin Trans. 11972, 1883. See also, Cinquini, M.; Colonna, S. Synthesis1972, 259.
193. Iriuchijima, S.; Tsuchihashi, G. Synthesis 1970, 588.
194. Regis, R.R.; Doweyko, A.M. Tetrahedron Lett. 1982, 23, 2539.
195. Paquette, L.A.; Houser, R.W. J. Org. Chem. 1971, 36, 1015.
196. McCarthy, J.R.; Pee, N.P.; LeTourneau, M.E.; Inbasekaran, M. J. Am. Chem. Soc. 1985, 107, 735. See also, Umemoto, T.; Tomizawa, G. Bull. Chem. Soc. Jpn. 1986, 59, 3625.
197. See Parmerter, S.M. Org. React. 1959, 10, 1.
198. See Yao, H.C.; Resnick, P. J. Am. Chem. Soc. 1962, 84, 3514.
199. For a review, see Phillips, R.R. Org. React. 1959, 10, 143.
200. Neplyuev, V.M.; Bazarova, I.M.; Lozinskii, M.O. J. Org. Chem. USSR 1989, 25, 2011. This paper also includes a sequence of leaving group ability for other Z groups.
201. For a review, see Williams, D.L.H. Nitrosation, Cambridge Univ. Press, Cambridge, 1988, pp. 1–45.
202. For a review, see Williams, D.L.H. Adv. Phys. Org. Chem. 1983, 19, 381. See also, Williams, D.L.H. Nitrosation, Cambridge Univ. Press, Cambridge, 1988.
203. Leis, J.R.; Peña, M.E.; Williams, D.L.H.; Mawson, S.D. J. Chem. Soc. Perkin Trans. 2 1988, 157.
204. Iglesias, E.; Williams, D.L.H. J. Chem. Soc. Perkin Trans. 2 1989, 343; Crookes, M.J.; Roy, P.; Williams, D.L.H. J. Chem. Soc. Perkin Trans. 2 1989, 1015. See also, Graham, A.; Williams, D.L.H. J. Chem. Soc. Chem. Commun. 1991, 407.
205. Rasmussen, J.K.; Hassner, A. J. Org. Chem. 1974, 39, 2558.
206. Kim, Y.H.; Park, Y.J.; Kim, K. Tetrahedron Lett. 1989, 30, 2833.
207. See Pape, M. Fortschr. Chem. Forsch. 1967, 7, 559.
208. Momiyama, N.; Yamamoto, H. Org. Lett. 2002, 4, 3579.
209. See Olah, G.A.; Malhotra, R.; Narang, S.C. Nitration, VCH, NY, 1989, pp. 219–295; Ogata, Y. in Trahanovsky, W.S. Oxidation in Organic Chemisry, part C, Academic Press, NY, 1978, pp. 295–342; Ballod, A.P.; Shtern, V.Ya. Russ. Chem. Rev. 1976, 45, 721.
210. See Matasa, C.; Hass, H.B. Can. J. Chem. 1971, 49, 1284.
211. Titov, A.I. Tetrahedron 1963, 19, 557.
212. Sifniades, S. J. Org. Chem. 1975, 40, 3562.
213. See Larson, H.O. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, Vol. 1, Wiley, NY, 1969, pp. 310–316.
214. See Feuer, H.; Van Buren, II, W.D.; Grutzner, J.B. J. Org. Chem. 1978, 43, 4676.
215. Olah, G.A.; Lin, H.C. J. Am. Chem. Soc. 1973, 93, 1259. See also, Bach, R.D.; Holubka, J.W.; Badger, R.C.; Rajan, S. J. Am. Chem. Soc. 1979, 101, 4416.
216. Isozaki, S.; Nishiwaki, Y.; Sakaguchi, S.; Ishii, Y. Chem. Commun. 2001, 1352.
217. Nishiwaki, Y.; Sakaguchi, S.; Ishii, Y. J. Org. Chem. 2002, 67, 5663.
218. Garver, L.C.; Grakauskas, V.; Baum, K. J. Org. Chem. 1985, 50, 1699.
219. See Regitz, M.; Maas, G. Diazo Compounds, Academic Press, NY, 1986, pp. 326–435; Regitz, M. Synthesis 1972, 351. See also, Koskinen, A.M.P.; Muñoz, L. J. Chem. Soc. Chem. Commun. 1990, 652.
220. Ledon, H. Synthesis 1974, 347, Org. Synth. VI, 414; Also see Ghosh, S.; Datta, I. Synth. Commun. 1991, 21, 191.
221. Taber, D.F.; Ruckle, Jr., R.E.; Hennessy, M.J. J. Org. Chem. 1986, 51, 4077; Baum, J.S.; Shook, D.A.; Davies, H.M.L.; Smith, H.D. Synth. Commun. 1987, 17, 1709.
222. Doering, W. von E.; DePuy, C.H. J. Am. Chem. Soc. 1953, 75, 5955.
223. See also Danheiser, R.L.; Miller, R.F.; Brisbois, R.G.; Park, S.Z. J. Org. Chem. 1990, 55, 1959.
224. Evans, D.A.; Britton, T.C. J. Am. Chem. Soc. 1987, 109, 6881, and references cited therein.
225. See Sheradsky, T. in Patai, S. The Chemistry of Functional Groups, Supplement F, pt. 1, Wiley, NY, 1982, pp. 395–416.
226. Sharpless, K.B.; Hori, T.; Truesdale, L.K.; Dietrich, C.O. J. Am. Chem. Soc. 1976, 98, 269; Kresze, G.; Münsterer, H. J. Org. Chem. 1983, 48, 3561. For a review, see Cheikh, R.B.; Chaabouni, R.; Laurent, A.; Mison, P.; Nafti, A. Synthesis 1983, 685, pp. 691–696.
227. Sharpless, K.B.; Hori, T. J. Org. Chem. 1979, 41, 176. For other reagents, see Tsushima, S.; Yamada, Y.; Onami, T.; Oshima, K.; Chaney, M.O.; Jones, N.D.; Swartzendruber, J.K. Bull. Chem. Soc. Jpn. 1989, 62, 1167.
228. Srivastava, R.S.; Nicholas, K.M. Tetrahedron Lett. 1994, 35, 8739.
229. Kohmura, Y.; Kawasaki, K.; Katsuki, T. Synlett, 1997, 1456.
230. Poulsen, T.B.; Alemparte, C.; J![]() rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 11614.
rgensen, K.A. J. Am. Chem. Soc. 2005, 127, 11614.
231. Fiori, J.W.; Du Bois, J. J. Am. Chem. Soc. 2007, 129, 562.
232. Espino, C. G.; Wehn, P. M.; Chow, J.; Du Bois, J. J. Am. Chem.Soc. 2001, 123, 6935.
233. Espino, C.G.; Du Bois, J. Angew. Chem. Int. Ed. 2001, 40, 598.
234. Terada, M.; Nakano, M.; Ube, H. J. Am. Chem. Soc. 2006, 128, 16044.
235. See Lwowski, W. in Lwowski, W. Nitrenes, Wiley, NY, 1970, pp. 199–207.
236. See Maslak, P. J. Am. Chem. Soc. 1989, 111, 8201.
237. See Alewood, P.F.; Kazmaier, P.M.; Rauk, A. J. Am. Chem. Soc. 1973, 95, 5466.
238. See Inagaki, M.; Shingaki, T.; Nagai, T. Chem. Lett. 1981, 1419.
239. Smolinsky, G.; Feuer, B.I. J. Am. Chem. Soc. 1964, 86, 3085.
240. See Anastassiou, A.G.; Shepelavy, J.N.; Simmons, H.E.; Marsh, F.D. in Lwowski, W. Nitrenes, Wiley, NY, 1970, pp. 305–344.
241. See Scriven, E.F.V. Azides and Nitrenes, Academic Press, NY, 1984, pp. 95–204.
242. Li, Z.; Capretto, D.A.; Rahaman, R.O.; He, C. J. Am. Chem. Soc. 2007, 129, 12058.
243. See also, Meinwald, J.; Aue, D.H. Tetrahedron Lett. 1967, 2317.
244. For a list of examples, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1148–1149.
245. See Müller, P.; Fruit, C. Chem. Rev. 2003, 103, 2905.
246. See Trost, B.M. Pure Appl. Chem. 1975, 43, 563, pp. 572–578; Caine, D. in Augustine, R.L. Carbon–Carbon Bond Formation, Vol. 1, Marcel Dekker, NY, 1979, pp. 278–282.
247. Gassman, P.G.; Balchunis, R.J. J. Org. Chem. 1977, 42, 3236.
248. See Sandrinelli, F.; Fontaine, G.; Perrio, S.; Beslin, P. J. Org. Chem. 2004, 69, 6916.
249. For another reagent, see Scholz, D. Synthesis 1983, 944.
250. See Back, T.G. in Liotta, D.C. Organoselenium Chemistry, Wiley, NY, 1987, pp. 1–125; Paulmier, C. Selenium Reagents and Intermediates in Organic Synthesis, Pergamon, Elmsford, NY, 1986, pp. 95–98.
251. Brocksom, T.J.; Petragnani, N.; Rodrigues, R. J. Org. Chem. 1974, 39, 2114. See also, Liotta, D. Acc. Chem. Res. 1984, 17, 28.
252. Grieco, P.A.; Miyashita, M. J. Org. Chem. 1974, 39, 120. See Miyoshi, N.; Yamamoto, T.; Kambe, N.; Murai, S.; Sonoda, N. Tetrahedron Lett. 1982, 23, 4813.
253. Barton, D.H.R.; Morzycki, J.W.; Motherwell, W.B.; Ley, S.V. J. Chem. Soc. Chem. Commun. 1981, 1044.
254. Sharpless, K.B.; Lauer, R.F.; Teranishi, A.Y. J. Am. Chem. Soc. 1973, 95, 6137.
255. Cossy, J.; Furet, N. Tetrahedron Lett. 1993, 34, 7755.
256. Wang, W.; Wang, K.; Li, H. Org. Lett. 2004, 6, 2817.
257. Matsugi, M.; Murata, K.; Gotanda, K.; Nambu, H.; Anilkumar, G.; Matsumoto, K.; Kita, Y. J. Org. Chem., 2001, 66, 2434.
258. Trost, B.M.; Hiroi, K.; Kurozumi, S. J. Am. Chem. Soc. 1975, 97, 438.
259. See OS VI, 23, 109; 68, 8. See also Morris, D.G. Chem. Soc. Rev. 1982, 11, 397; Kane, V.V.; Singh, V.; Martin, A.; Doyle, D.L. Tetrahedron 1983, 39, 345.
260. See Gilbert, E.E. Sulfonation and Related Reactions, Wiley, NY, 1965, pp. 33–61.
261. Scott, W.J.; Crisp, G.T.; Stille, J.K. J. Am. Chem. Soc. 1984, 106, 4630. See Roth, G.P.; Farina, V.; Liebeskind, L.S.; Peña-Cabrera, E. Tetrahedron Lett. 1995, 36, 2191 for an optimized version of this reaction. Also see Echavarren, A.M. Angew. Chem. Int. Ed. 2005, 44, 3962; Reiser, O. Angew. Chem. Int. Ed. 2006, 45, 2838.
262. See Zhou, W.-J.; Wang, K.-H.; Wang, J.-X. J. Org. Chem. 2009, 74, 5599.
263. Gajare, A.S.; Jensen, R.S.; Toyota, K.; Yoshifuji, M.; Ozawa, F. Synlett 2005, 144. For a ligand free reaction, see Yabe, Y.; Maegawa, T.; Monguchi, Y.; Sajiki, H. Tetrahedron 2010, 66, 8654.
264. Lau, K.C.Y.; Chiu, P. Tetrahedron Lett. 2007, 48, 1813.
265. McMurry, J.E.; Scott, W.J. Tetrahedron Lett. 1983, 24, 979.
266. See Maleczka Jr., R.E.; Lavis, J.M.; Clark, D.H.; Gallagher, W.P. Org. Lett. 2000, 2, 3655.
267. Farina, V.; Krishnamurthy, V.; Scott, W.J. Org. React. 1997, 50, 1; Li, J.-H.; Liang, Y.; Wang, D.-P.; Liu, W.-J.; Xie, Y.-X.; Yin, D.-L. J. Org. Chem. 2005, 70, 2832.
268. Powell, D.A.; Maki, T.; Fu, G.C. J. Am. Chem. Soc. 2005, 127, 510.
269. Johnson, C.R.; Adams, J.P.; Braun, M.P.; Senanayake, C.B.W. Tetrahedron Lett. 1992, 33, 919.
270. Badone, D.; Cardamone, R.; Guzzi, U. Tetrahedron Lett. 1994, 35, 5477.
271. Segorbe, M.M.; Adrio, J.; Carretero, J.C. Tetrahedron Lett. 2000, 41, 1983.
272. Larhed, M.; Hoshino, M.; Hadida, S.; Curran, D.P. J. Org. Chem. 1997, 62, 5583.
273. Olofsson, K.; Kim, S.-Y.; Larhed, M.; Curran, D.P.; Hallberg, A. J. Org. Chem. 1999, 64, 4539.
274. Jessop, P. G.; Ikariya, T.; Noyori, R. Chem. Rev. 1999, 99, 475.
275. Shi, Y.; Peterson, S.M.; Haberaecker III, W.W.; Blum, S.A. J. Am. Chem. Soc. 2008, 130, 2168.
276. Farina, V.; Hossain, M.A. Tetrahedron Lett. 1996, 37, 6997.
277. Rai, R.; Aubrecht, K.B.; Collum, D.B. Tetrahedron Lett. 1995, 36, 3111.
278. Littke, A.F.; Fu, G.C. Angew. Chem. Int. Ed. 1999, 38, 2411.
279. Clapham, B.; Sutherland, A.J. J. Org. Chem. 2001, 66, 9033.
280. Schaus, J.V.; Panek, J.S. Org. Lett. 2000, 2, 469.
281. See Rousset, S.; Abarbri, M.; Thibonnet, J.; Duchêne, A.; Parrain, J.-L. Org. Lett. 1999, 1, 701. Also see Minière, S.; Cintrat, J.-C. J. Org. Chem. 2001, 66, 7385.
282. Lindh, J.; Fardost, A.; Almeida, M.; Nilsson, P. Tetrahedron Lett. 2010, 51, 2470; Sävmarker, J.; Lindh, J.; Nilsson, P. Tetrahedron Lett. 2010, 51, 6886.
283. Mee, S.P.H.; Lee, V.; Baldwin, J.E. Chemistry: European J. 2005, 11, 3294.
284. Li, J.-H.; Tang, B.-X.; Tao, L.-M.; Xie, Y.-X.; Liang, Y.; Zhang, M.-B. J. Org. Chem. 2006, 71, 7488.
285. Voigt, K.; Schick, U.; Meyer, F.E.; de Meijere, A. Synlett 1994, 189.
286. Gilbertson, S.R.; Fu, Z.; Xie, D. Tetrahedron Lett. 2001, 42, 365.
287. Scott, W.J.; Stille, J.K. J. Am. Chem. Soc. 1986, 108, 3033; Stille, J.K. Angew. Chem., Int. Ed. 1986, 25, 508; Farina, V. in Abel, E.W.; Stone, F.G.A.; Wilkinson, G. Comprehensive Organometallic Chemistry II, Vol. 12, Pergamon, Oxford, U.K., 1995, Chapter 3.4.; Brown, J.M.; Cooley, N.A. Chem. Rev. 1988, 88, 1031.
288. Amatore, C.; Jutand, A.; Suarez, A. J. Am. Chem. Soc. 1993, 115, 9531 and references cited therein.
289. Ozawa, F.; Fujimori, M.; Yamamoto, T.; Yamamoto, A. Organometallics 1986, 5, 2144; Tatsumi, K.; Hoffmann, R.; Moravski, A.; Stille, J.K. J. Am. Chem. Soc. 1981, 103, 4182; Loar, M.K.; Stille, J.K. J. Am. Chem. Soc.1981, 103, 4174.
290. Deacon, G.B.; Gatehouse, B.M.; Nelson-Reed, K.T. J. Organomet. Chem. 1989, 359, 267.
291. Casado, A.L.; Espinet, P.; Gallego, A.M. J. Am. Chem. Soc. 2000, 122, 11771.
292. Santos, L.S.; Rosso, G.B.; Pilli, R.A.; Eberlin, M.N. J. Org. Chem. 2007, 72, 5809.
293. Zhou, S.-m.; Deng, M.-z. Tetrahedron Lett. 2000, 41, 3951.
294. Yao, M.-L.; Deng, M.-Z. J. Org. Chem. 2000, 65, 5034; Yao, M.-L.; Deng, M.-Z. Tetrahedron Lett. 2000, 41, 9083.
295. Occhiato, E.G.; Trabocchi, A.; Guarna, A. J. Org. Chem. 2001, 66, 2459.
296. Kabalka, G.W.; Dadush, E.; Al-Masum, M. Tetrahedron Lett. 2006, 47, 7459.
297. Molander, G.A.; Felix, L.A. J. Org. Chem. 2005, 70, 3950.
298. Fu, X.; Zhang, S.; Yin, J.; McAllister, T.L.; Jiang, S.A.; Tann, C.-H.; Thiruvengadam, T.K.; Zhang, F. Tetrahedron Lett. 2002, 43, 573. See Vallin, K.S.A.; Larhed, M.; Johansson, K.; Hallberg, A. J. Org. Chem. 2000, 65, 4537.
299. Nishihara, Y.; Ikegashira, K.; Toriyama, F.; Mori, A.; Hiyama, T. Bull. Chem. Soc. Jpn. 2000, 73, 985.
300. Raminelli, C.; Gargalak Jr., J.; Silveira, C.C.; Comasseto, J.V. Tetrahedron Lett. 2004, 45, 4927; Silveira, C.C.; Braga, A.L.; Vieira, A.S.; Zeni, G. J. Org. Chem. 2003, 68, 662.
301. Zeni, G.; Comasseto, J.V. Tetrahedron Lett. 1999, 40, 4619.
302. Ma, S.; Zhang, A.; Yu, Y.; Xia, W. J. Org. Chem. 2000, 65, 2287.
303. Dillinger, S.; Bertus, P.; Pale, P. Org. Lett. 2001, 3, 1661. See Halbes, U.; Bertus, P.; Pale, P. Tetrahedron Lett. 2001, 42, 8641; Bertus, P.; Halbes, U.; Pale, P. Eur. J. Org. Chem. 2001, 4391.
304. Braga, A.L.; Emmerich, D.J.; Silveira, C.C.; Martins, T.L.C.; Rodrigues, O.E.D. Synlett 2001, 369.
305. Lee, J.-H.; Park, J.-S.; Cho, C.-G. Org. Lett. 2002, 4, 1171. Also see Bates, C.G.; Saejueng, P.; Venkataraman, D. Org. Lett. 2004, 6, 1441.
306. Negishi, E.; Qian, M.; Zeng, F.; Anastasia, L.; Babinski, D. Org. Lett. 2003, 5, 1597.
307. Abarbri, M.; Parrain, J.-L.; Kitamura, M.; Noyori, R.; Duchêne, A. J. Org. Chem. 2000, 65, 7475.
308. Menzel, K.; Fu, G.C. J. Am. Chem. Soc. 2003, 125, 3718.
309. Tsukada, N.; Sato, T.; Inoue, Y. Chem. Commun. 2003, 2404.
310. Kiewel, K.; Tallant, M.; Sulikowski, G.A. Tetrahedron Lett. 2001, 42, 6621.
311. See Groves, E.E. Chem. Soc. Rev. 1972, 1, 73; Satchell, D.P.N.; Satchell, R.S. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 259–266, 270–273; Nenitzescu, C.D.; Balaban, A.T. in Olah, G.A. Friedel–Crafts and Related Reactions, Vol. 3, Wiley, NY, 1964, pp. 1033–1152.
312. Deno, N.C.; Chafetz, H. J. Am. Chem. Soc. 1952, 74, 3940. For other examples, see Grignon-Dubois, M.; Cazaux, M. Bull. Soc. Chim. Fr. 1986, 332.
313. See Heck, R.F. in Wender, I.; Pino, P. Organic Syntheses via Metal Carbonyls, Vol. 1, Wiley, NY, 1968, pp. 388–397.
314. Andersson, C.; Hallberg, A. J. Org. Chem. 1988, 53, 4257.
315. See Burn, D. Chem. Ind. (London) 1973, 870; Satchell, D.P.N.; Satchell, R.S. in Patai, S. The Chemistry of the Carbonyl Group, Vol. 1, Wiley, NY, 1966, pp. 281–282.
316. Youssefyeh, R.D. Tetrahedron Lett. 1964, 2161.
317. Shen, W.; Kunzer, A. Org. Lett. 2002, 4, 1315.
318. Youssefyeh, R.D. J. Am. Chem. Soc. 1963, 85, 3901.
319. See van der Zeeuw, A.J.; Gersmann, H.R. Recl. Trav. Chim. Pays-Bas 1965, 84, 1535.
320. See Poirier, J. Org. Prep. Proced. Int. 1988, 20, 319; Brownbridge, P. Synthesis 1983, 1, 85; Colvin, E.W. Silicon Reagents in Organic Synthesis, Academic Press, NY, 1988; Colvin, E.W. in Hartley, C.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, pp. 539–621; Ager, D.J. Chem. Soc. Rev. 1982, 11, 493.
321. See Mizhiritskii, M.D.; Yuzhelevskii, Yu.A. Russ. Chem. Rev. 1987, 56, 355. For a list, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1488–1491.
322. Di-tert-butylmagnesium has also been used. See Kerr, W.J.; Watson, A.J.B.; Hayes, D. Synlett 2008, 1386.
323. Corey, E.J.; Gross, A.W. Tetrahedron Lett. 1984, 25, 495. Also see Lipshutz, B.H.; Wood, M.R.; Lindsley, C.W. Tetrahedron Lett. 1995, 36, 4385.
324. See Cahiez, G.; Figadère, B.; Cléry, P. Tetrahedron Lett. 1994, 35, 6295.
325. Ladjama, D.; Riehl, J.J. Synthesis 1979, 504. See Orban, J.; Turner, J.V.; Twitchin, B. Tetrahedron Lett. 1984, 25, 5099.
326. Miller, R.D.; McKean, D.R. Synth. Commun. 1982, 12, 319. See also, Ahmad, S.; Khan, M.A.; Iqbal, J. Synth. Commun. 1988, 18, 1679.
327. Fujiwara, M.; Baba, A.; Matsuda, H. Chem. Lett. 1989, 1247.
328. Smietana, M.; Mioskowski, C. Org. Lett. 2001, 3, 1037. See also, Tanabe, Y.; Misaki, T.; Kurihara, M.; Iida, A.; Nishii, Y. Chem. Commun. 2002, 1628.
329. Ozawa, F.; Yamamoto, S.; Kayagishi, S.; Hiraoka, M.; Ideda, S.; Minami, T.; Ito, S.; Yoshifuji, M. Chem. Lett. 2001, 972. See Blackwell, J.M.; Morrison, D.J.; Piers, W.E. Tetahedron 2002, 58, 8247; Mori, A.; Kato, T. Synlett2002, 1167.
330. Heathcock, C.H.; Buse, C.T.; Kleschick, W.A.; Pirrung, M.A.; Sohn, J.E.; Lampe, J. J. Org. Chem. 1980, 45, 1066.
331. Vitale, M.; Lecourt, T.; Sheldon, C.G.; Aggarwal, V.K. J. Am. Chem. Soc. 2006, 128, 2524.
332. Lessène, G.; Tripoli, R.; Cazeau, P.; Biran, C.; Bordeau, M. Tetrahedron Lett. 1999, 40, 4037. Also see Patonay, T.; Hajdu, C.; Jeko, J.; Lévai, A.; Micskei, K.; Zucchi, C. Tetrahedron Lett. 1999, 40, 1373.
333. Lin, J.-M.; Liu, B.-S. Synth. Commun. 1997, 27, 739.
334. Aggarwal, V.K.; Sheldon, C.G.; Macdonald, G.J.; Martin, W.P. J. Am. Chem. Soc. 2002, 124, 10300.
335. Robertson, B.D.; Hartel, A.M. Tetrahedron Lett. 2008, 49, 2088.
336. For the synthesis of enol acetates, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, 1484–1485.
337. See Krapcho, A.P.; Diamanti, J.; Cayen, C.; Bingham, R. Org. Synth. Coll. Vol. V 1973, 198.
338. See Honda, T.; Namiki, H.; Kudoh, M.; Watanabe, N.; Nagase, H.; Mizutani, H. Tetrahedron Lett. 2000, 41, 5927.
339. Yoo, W.-J.; Li, C.-J. J. Org. Chem. 2006, 71, 6266.
340. Wentworth, A.D.; Wentworth, Jr., P.; Mansoor, U.F.; Janda, K.D. Org. Lett. 2000, 2, 477.
341. House, H.O.; Czuba, L.J.; Gall, M.; Olmstead, H.D. J. Org. Chem. 1969, 34, 2324.
342. Comins, D.L.; Dehghani, A. Tetrahedron Lett. 1992, 33, 6299.
343. Holmquist, C.R.; Roskamp, E.J. J. Org. Chem. 1989, 54, 3258.
344. Pelter, A.; Smith, K.; Elgendy, S.; Rowlands, M. Tetrahedron Lett. 1989, 30, 5643.
345. Krug, C.; Hartwig, J.F. J. Am. Chem. Soc. 2002, 124, 1674.
346. Pucheault, M.; Darses, S.; Genet, J.-P. J. Am. Chem. Soc. 2004, 126, 15356.
347. Ruan, J.; Saidi, O.; Iggo, J.A.; Xiao, J. J. Am. Chem. Soc. 2008, 130, 10510.
348. Kahne, D.; Collum, D.B. Tetrahedron Lett. 1981, 22, 5011.
349. Santiago, B.; Meyers, A.I. Tetrahedron Lett. 1993, 34, 5839.
350. North, M. Angew. Chem. Int. Ed. 2004, 43, 4126.
351. Han, W.; Ofial, A.R. Chem. Commun. 2009, 5024.
352. Kornblum, N.; Singh, N.K.; Kelly, W.J. J. Org. Chem. 1983, 48, 332.
353. Barton, D.H.R.; Billion, A.; Boivin, J. Tetrahedron Lett. 1985, 26, 1229.
354. Lemaire, M.; Doussot, J.; Guy, A. Chem. Lett. 1988, 1581. See also, Hayashi, Y.; Mukaiyama, T. Chem. Lett. 1987, 1811.
355. Zhou, W.; Zhang, L.; Jiao, N. Angew. Chem. Int. Ed 2009, 48, 7094.
356. Olah, G.A.; Mo, Y.K.; Olah, J.A. J. Am. Chem. Soc. 1973, 95, 4939. See Olah, G.A.; Farooq, O.; Prakash, G.K.S. in Hill, C.L. Activation and Functionalization of Alkanes, Wiley, NY, 1989, pp. 27–78; Ref. 48; Fabre, P.; Devynck, J.; Trémillon, B. Chem. Rev. 1982, 82, 591. See also, Olah, G.A.; Prakash, G.K.S.; Williams, R.E.; Field, L.D.; Wade, K. Hypercarbon Chemistry, Wiley, NY, 1987.
357. Olah, G.A.; DeMember, J.R.; Shen, J. J. Am. Chem. Soc. 1973, 95, 4952. See also, Sommer, J.; Muller, M.; Laali, K. Nouv. J. Chem. 1982, 6, 3.
358. For example, see Hogeveen, H.; Roobeek, C.F. Recl. Trav. Chim. Pays-Bas 1972, 91, 137.
359. Yamamoto, G.; Oki, M. Chem. Lett. 1987, 1163.
360. First reported by Meerwein, H.; Rathjen, H.; Werner, H. Ber. 1942, 75, 1610. See Doyle, M.P.; Duffy, R.; Ratnikov, M.; Zhou, L. Chem. Rev. 2010, 110, 704; Bethell, D. in McManus, S.P. Organic Reactive Intermediates, Academic Press, NY, 1973, pp. 92–101; Kirmse, W. Carbene Chemistry, 2nd ed., Academic Press, NY, 1971, pp. 209–266.
361. Terao, T.; Shida, S. Bull. Chem. Soc. Jpn. 1964, 37, 687. See also, Moss, R.A.; Fedé, J.-M.; Yan, S. J. Am. Chem. Soc. 2000, 122, 9878.
362. See Marek, I. Tetrahedron 2002, 58, 9463.
363. See Paquette, L.A.; Kobayashi, T.; Gallucci, J.C. J. Am. Chem. Soc. 1988, 110, 1305; Doyle, M.P.; Bagheri, V.; Pearson, M.M.; Edwards, J.D. Tetrahedron Lett. 1989, 30, 7001.
364. Friedman, L.; Berger, J.G. J. Am. Chem. Soc. 1961, 83, 492, 500. See Padwa, A.; Krumpe, K.E. Tetrahedron 1992, 48, 5385.
365. See Burke, S.D.; Grieco, P.A. Org. React. 1979, 26, 361.
366. Doering, W. von E.; Knox, L.H. J. Am. Chem. Soc. 1961, 83, 1989.
367. Carter, D.S.; Van Vranken, D.L. Org. Lett. 2000, 2, 1303; Doyle, M.P.; McKervey, M.A.; Ye, T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides, Wiley, NY, 1998.
368. Bertram, A.K.; Liu, M.T.H. J. Chem. Soc. Chem. Commun. 1993, 467.
369. See Steinbeck, K. Tetrahedron Lett. 1978, 1103; Boev, V.I. J. Org. Chem. USSR 1981, 17, 1190.
370. Davies, H.M.L.; Ren, P.; Jin, Q. Org. Lett. 2001, 3, 3587.
371. Gibe, R.; Kerr, M.A. J. Org. Chem. 2002, 67, 6247.
372. Arduengo, III, A.J.; Calabrese, J.C.; Davidson, F.; Dias, H.V.R.; Goerlich, J.R.; Krafczyk, R.; Marshall, W.J.; Tamm, M.; Schmutzler, R. Helv. Chim. Acta. 1999, 82, 2348.
373. Ventura, D.L.; Li, Z.; Coleman, M.G.; Davies, H.M.L. Tetrahedron 2009, 65, 3052.
374. Masselot, D.; Charmant, J.P.H.; Gallagher, T. J. Am. Chem. Soc. 2006, 128, 694.
375. See Caballero, A.; Díaz-Requejo, M.M.; Belderraín, T.R.; Nicasio, M.C.; Trofimenko, S.; Pérez, P.J. J. Am. Chem. Soc. 2003, 125, 1446.
376. Dias, H.V.R.; Browning, R.G.; Polach, S.A.; Diyabalanage, H.V.K.; Lovely, C.J. J. Am. Chem. Soc. 2003, 125, 9270.
377. Hashimoto, T.; Naganawa, Y.; Maruoka, K. J. Am. Chem. Soc. 2008,130, 2434.
378. Wee, A.G.H.; Duncan, S.C. J. Org. Chem. 2005, 70, 8372.
379. Dalton, A.M.; Zhang, Y.; Davie, C.P.; Danheiser, R.L. Org. Lett. 2002, 4, 2465.
380. Devine, S.K.J.; Van Vranken, D.L. Org. Lett. 2007, 9, 2047.
381. Ye, L.; Cui, L.; Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2010, 132, 3258.
382. Matusamy, S.; Arulananda, S.; Babu, A.; Gunanathan, C. Tetrahedron Lett. 200243, 3133.
383. Lo, M.M.-C.; Fu, G.C. Tetrahedron 2001, 57, 2621.
384. Davies, H.M.L.; Hedley, S.J.; Brooks R.; Bohall, B.R. J. Org. Chem. 2005, 70, 10737.
385. Bourque, L.E.; Cleary, P.A.; Woerpel, K.A. J. Am. Chem. Soc. 2007, 129, 12602.
386. Davies, H.M.L.; Grazini, M.V.A.; Aouad, E. Org. Lett. 2001, 3, 1475.
387. Doyle, M.P.; Protopopova, M.N.; Winchester, W.R.; Daniel, K.L. Tetrahedron Lett. 1992, 33, 7819. See also, Clark, J.S.; Hodgson, P.B.; Goldsmith, M.D.; Street, L.J. J. Chem. Soc., Perkin Trans. 1 2001, 3312.
388. Coutts, I.G.C.; Saint, R.E.; Saint, S.L.; Chambers-Asman, D.M. Synthesis 2001, 247.
389. See Shi, W.; Zhang, B.; Zhang, J.; Liu, B.; Zhang, S.; Wang, J. Org. Lett. 2005, 7, 3103.
390. See Doyle, M.P.; Kalinin, A.V. Synlett, 1995, 1075; Watanabe, N.; Ohtake, Y.; Hashimoto, S.; Shiro, M.; Ikegami, S. Tetrahedron Lett. 1995, 36, 1491; Maruoka, K.; Concepcion, A.B.; Yamamoto, H. J. Org. Chem. 1994, 59, 4725.
391. See Sulikowski, G.A.; Cha, K.L.; Sulikowski, M.M. Tetrahedron Asymmetry, 1998, 9, 3145.
392. Ye, T.; McKervey, M.A. Chem. Rev. 1994, 94, 1091.
393. Doyle, M.P. Pure Appl. Chem. 1998, 70, 1123. See Taber, D.F.; Malcolm, S.C. J. Org. Chem. 1998, 63, 3717 for a discussion of transition state geometry in rhodium mediated C–H insertion.
394. Davies, H.M.L.; Jin, Q. Org. Lett. 2004, 6, 1769; Davies, H.M.L.; Loe, ∅. Synthesis 2004, 2595.
395. See Davies, H.M.L.; Beckwith, R.E.J. Chem. Rev. 2003, 103, 2861. See also Davies, H.M.L.; Nikolai, J. Org. Biomol. Chem. 2005, 3, 4176; Suematsu, H.; Katsuki, T. J. Am. Chem. Soc. 2009, 131, 14218.
396. See Bethell, D. Adv. Phys. Org. Chem. 1969, 7, 153, pp. 190–194.
397. Doering, W. von E.; Prinzbach, H. Tetrahedron 1959, 6, 24.
398. See Seyferth, D.; Cheng, Y.M. J. Am. Chem. Soc. 1971, 93, 4072.
399. Sobery, W.; DeLucca, J.P. Tetrahedron Lett. 1995, 36, 3315.
400. Frey, H.M. Proc. Chem. Soc. 1959, 318.
401. McNesby, J.R.; Kelly, R.V. Int. J. Chem. Kinet. 1971, 3, 293.
402. Ring, D.F.; Rabinovitch, B.S. J. Am. Chem. Soc. 1966, 88, 4285; Can J. Chem. 1968, 46, 2435.
403. Bell, J.A. Prog. Phys. Org. Chem. 1964, 2, 1, pp. 30–43.
404. Cummins, J.M.; Porter, T.A.; Jones, Jr., M. J. Am. Chem. Soc. 1998, 120, 6473.
405. Richardson, D.B.; Simmons, M.C.; Dvoretzky, I. J. Am. Chem. Soc. 1961, 83, 1934.
406. See Roth, H.D. Acc. Chem. Res. 1977, 10, 85.
407. Roth, H.D. J. Am. Chem. Soc. 1972, 94, 1761. See also, Bethell, D.; McDonald, K. J. Chem. Soc. Perkin Trans. 2 1977, 671.
408. See Oku, A.; Yamaura, Y.; Harada, T. J. Org. Chem. 1986, 51, 3730; Ritter, R.H.; Cohen, T. J. Am. Chem. Soc. 1986, 108, 3718.
409. See Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 44–107; Wardell, J.L. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 4, Wiley, NY, pp. 1–157, pp. 27–71; Narasimhan, M.S.; Mali, R.S. Synthesis 1983, 957; Biellmann, J.F.; Ducep, J. Org. React. 1982, 27, 1; Gschwend, H.W.; Rodriguez, H.R. Org. React. 1979, 26, 1; Mallan, J.M.; Bebb, R.L. Chem. Rev. 1969, 69, 693.
410. See Saá, J.M.; Martorell, G.; Frontera, A. J. Org. Chem. 1996, 61, 5194.
411. See Durst, T. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, Vol. 5, pt. B, Elsevier, NY, 1984, pp. 239–291, pp. 265–279. For an article on the safe handling of RLi compounds, see Anderson, R. Chem. Ind. (London) 1984, 205.
412. Ivanov, R.; Marek, I.; Cohen, T. Tetrahedron Lett. 2010, 51, 174.
413. Schlosser, M. J. Organomet. Chem. 1967, 8, 9. See also, Schlosser, M.; Katsoulos, G.; Takagishi, S. Synlett, 1990, 747.
414. Rausch, M.D.; Ciappenelli, D.J. J. Organomet. Chem. 1967, 10, 127.
415. See Klein, J. Tetrahedron 1983, 39, 2733; Klein, J. in Patai, S. The Chemistry of the Carbon–Carbon Triple Bond, pt. 1, Wiley, NY, 1978, pp. 343–379.
416. Hartmann, J.; Schlosser, M. Helv. Chim. Acta 1976, 59, 453.
417. Schlosser, M. Pure Appl. Chem. 1988, 60, 1627. For sodium analogues, see Schlosser, M.; Hartmann, J.; Stähle, M.; Kramar, J.; Walde, A.; Mordini, A. Chimia, 1986, 40, 306.
418. Brandsma, L.; Verkruijsse, H.D.; Schade, C.; Schleyer, P.v.R. J. Chem. Soc. Chem. Commun. 1986, 260.
419. See Shirley, D.A.; Hendrix, J.P. J. Organomet. Chem. 1968, 11, 217.
420. Gossage, R.A.; Jastrzebski, J.T.B.H.; van Koten, G. Angew. Chem. Int. Ed. 2005, 44, 1448.
421. Jacobson, M.A.; Keresztes, I.; Williard, P.G. J. Am. Chem. Soc. 2005, 127, 4965. For a computational study of mixed aggregates of chloromethyllithium and lithium dialkylamides, see Pratt, L.M.; Lê, L.T.; Truong, T.N. J. Org. Chem. 2005, 70, 8298. Also see Gupta, L.; Hoepker, A.C.; Singh, K.J.; Collum, D.B. J. Org. Chem. 2009, 74, 2231 for a LiCl catalyzed reaction.
422. See Blagoev, B.; Ivanov, D. Synthesis 1970, 615.
423. Kessar, S.V.; Singh, P.; Singh, K.N.; Venugopalan, P.; Kaur, A.; Bharatam, P.V.; Sharma, A.K. J. Am. Chem. Soc. 2007, 129, 4506.
424. Nishimura, Y.; Miyake, Y.; Amemiya, R.; Yamaguchi, M. Org. Lett. 2006, 8, 5077.
425. See Figuly, G.D.; Loop, C.K.; Martin, J.C. J. Am. Chem. Soc. 1989, 111, 654; Block, E.; Eswarakrishnan, V.; Gernon, M.; Ofori-Okai, G.; Saha, C.; Tang, K.; Zubieta, J. J. Am. Chem. Soc. 1989, 111, 658; Smith, K.; Lindsay, C.M.; Pritchard, G.J. J. Am. Chem. Soc. 1989, 111, 665.
426. See Gilday, J.P.; Negri, J.T.; Widdowson, D.A. Tetrahedron 1989, 45, 4605.
427. See Katritzky, A.R.; Lam, J.N.; Sengupta, S. Prog. Heterocycl. Chem. 1989, 1, 1.
428. Bickelhaupt, F.M.; Hermann, H.L.; Boche, G. Angew. Chem. Int. Ed. 2006, 45, 823.
429. See Beak, P.; Meyers, A.I. Acc. Chem. Res. 1986, 19, 356; Beak, P.; Snieckus, V. Acc. Chem. Res. 1982, 15, 306; Narasimhan, N.S.; Mali, R.S. Top. Curr. Chem. 1987, 138, 63; Reuman, M.; Meyers, A.I. Tetrahedron 1985, 41, 837.
430. Slocum, D.W.; Coffey, D.S.; Siegel, A.; Grimes, P. Tetrahedron Lett. 1994, 35, 389.
431. See Snieckus, V. Chem. Rev. 1990, 90, 879; Pure Appl. Chem. 1990, 62, 2047. For a discussion of the mechanism, see Bauer, W.; Schleyer, P. v.R. J. Am. Chem. Soc. 1989, 111, 7191.
432. Baldwin, J.E.; Höfle, G.A.; Lever Jr., O.W. J. Am. Chem. Soc. 1974, 96, 7125.
433. Beak, P.; Hunter, J.E.; Jun, Y.M.; Wallin, A.P. J. Am. Chem. Soc. 1987, 109, 5403. See also, Stork, G.; Polt, R.L.; Li, Y.; Houk, K.N. J. Am. Chem. Soc. 1988, 110, 8360; Barluenga, J.; Foubelo, F.; Fañanas, F.J.; Yus, M. J. Chem. Res. (S) 1989, 200.
434. Peñafiel, I.; Pastor, I.M.; Yus, M. Tetrahedron 2010, 66, 2928.
435. Benkeser, R.A.; Trevillyan, E.A.; Hooz, J. J. Am. Chem. Soc. 1962, 84, 4971.
436. Bryce-Smith, D. J. Chem. Soc. 1963, 5983; Benkeser, R.A.; Hooz, J.; Liston, T.V.; Trevillyan, E.A. J. Am. Chem. Soc. 1963, 85, 3984.
437. Pocker, Y.; Exner, J.H. J. Am. Chem. Soc. 1968, 90, 6764.
438. West, P.; Waack, R.; Purmort, J.I. J. Am. Chem. Soc. 1970, 92, 840.
439. Kapeller, D.C.; Hammerschmidt, F. J. Am. Chem. Soc. 2008, 130, 2329.
440. McGrath, M.J.; O'Brien, P. J. Am. Chem. Soc. 2005, 127, 16378.
441. Ashweek, J.; Brandt, P.; Coldham, I.; Dufour, S.; Gawley, R.E.; Hæffner, F.; Klein, R.; Sanchez-Jimenezy, G. J. Am. Chem. Soc. 2005, 127, 449.
442. Zugravescu, I.; Petrovanu, M. Nitrogen–Ylid Chemistry, McGraw Hill, NY, 1976, pp. 251–283; Wittig, G.; Rieber, M. Ann. 1949, 562, 177; Wittig, G.; Polster, R. Ann. 1956, 599, 1.
443. See Durst, T. in Buncel, E.; Durst, T. Comprehensive Carbanion Chemistry, Vol. 5, pt. B, Elsevier, NY, 1984, pp. 239–291; Wardell, J.L. Ref. 388; Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988, pp. 32–44.
444. See Ziegenbein, W. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 170–185. For an improved method, see Fisch, A.; Coisne, J.M.; Figeys, H.P. Synthesis 1982, 211.
445. Wang, S.; Zhang, L. Org. Lett. 2006, 8, 4585.
446. Hegarty, A.F.; Dowling, J.P.; Eustace, S.J.; McGarraghy, M. J. Am. Chem. Soc. 1998, 120, 2290.
447. See Caine, D. in Augustine, R.L. Carbon–Carbon Bond Formation, Vol. 1, Marcel Dekker, NY, 1979, pp. 95–145, 284–291.
448. See Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, pp. 25–149, pp. 105–136; Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H. Eds., Vol. 12, Elsevier, NY, 1973, pp. 107–134; Schlosser, M. Angew. Chem. Int. Ed. 1964, 3, 287, 362; Newer Methods Prep. Org. Chem. 1968, 5, 238.
449. Mitsuhashi, K.; Ito, R.; Arai, T.; Yanagisawa, A. Org. Lett. 2006, 8, 1721.
450. Walborsky, H.M.; Ollman, J.; Hamdouchi, C.; Topolski, M. Tetrahedron Lett. 1992, 33, 761.
451. Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Academic Press, NY, 1988, see pp. 242–244.
452. See Fleming, I.; Dunoguès, J.; Smithers, R. Org. React. 1989, 37, 57, pp. 89–97, 194–243.
453. See Makarova, L.G. Organomet. React. 1970, 1, 119, see pp. 251–270, 275–300.
454. See Barluenga, J.; Yus, M. Chem. Rev. 1988, 88, 487.
455. Sommer, L.H.; Marans, N.S.; Goldberg, G.M.; Rockett, J.; Pioch, R.P. J. Am. Chem. Soc. 1951, 73, 882. See also, Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, p. 117.
456. See Brilkina, T.G.; Shushunov, V.A. Reactions of Organometallic Compounds with Oxygen and Peroxides, CRC Press, Boca Raton, FL, 1969; Wardell, J.L.; Paterson, E.S. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, see pp. 219–338, pp. 311–316.
457. See Harada, T.; Kutsuwa, E. J. Org. Chem. 2003, 68, 6716.
458. Brown, H.C.; Midland, M.M. Tetrahedron 1987, 43, 4059.
459. Adam, W.; Cueto, O. J. Org. Chem. 1977, 42, 38.
460. Garst, J.F.; Smith, C.D.; Farrar, A.C. J. Am. Chem. Soc. 1972, 94, 7707. See Davies, A.G. J. Organomet. Chem. 1980, 200, 87.
461. Lawesson, S.; Frisell, C.; Denney, D.B.; Denney, D.Z. Tetrahedron 1963, 19, 1229. See Brilkina, T.G.; Shushunov, V.A. Reactions of Organometallic Compounds with Oxygen and Peroxides, CRC Press, Boca Raton, FL, 1969; Razuvaev, G.A.; Shushunov, V.A.; Dodonov, V.A.; Brilkina, T.G. in Swern, D. Organic Peroxides, Vol. 3, Wiley, NY, 1972, pp. 141–270.
462. Longone, D.T.; Miller, A.H. Tetrahedron Lett. 1967, 4941.
463. Davis, F.A.; Lal, G.S.; Wei, J. Tetrahedron Lett. 1988, 29, 4269.
464. Mitchell, T.A.; Bode, J.W. J. Am. Chem. Soc. 2009, 131, 18057.
465. See Pelter, A.; Smith, K.; Brown, H.C. Borane Reagents, Aademic Press, NY, 1988, pp. 244–249; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithaca, NY, 1972, pp. 321–325. See also, Brown, H.C.; Snyder, C.; Subba Rao, B.C.; Zweifel, G. Tetrahedron 1986, 42, 5505.
466. Brown, H.C.; Midland, M.M.; Kabalka, G.W. Tetrahedron 1986, 42, 5523.
467. Kabalka, G.W.; Shoup, T.M.; Goudgaon, N.M. J. Org. Chem. 1989, 54, 5930.
468. Köster, R.; Arora, S.; Binger, P. Angew. Chem. Int. Ed. 1969, 8, 205.
469. Kabalka, G.W.; Slayden, S.W. J. Organomet. Chem. 1977, 125, 273.
470. Midland, M.M.; Brown, H.C. J. Am. Chem. Soc. 1971, 93, 1506.
471. Bean, F.R.; Johnson, J.R. J. Am. Chem Soc. 1932, 54, 4415; Lappert, M.F. Chem. Rev. 1956, 56, 959.
472. Khotinsky, E.; Melamed, M. Chem. Ber. 1909, 42, 3090.
473. Sebelius, S.; Olsson, V.J.; Szabó, K.J. J. Am. Chem. Soc. 2005, 127, 10478.
474. Soloway, A.H. J. Am. Chem. Soc. 1959, 81, 3017.
475. Ishiyama, T.; Murata, M.; Miyaura, N. J. Org. Chem. 1995, 60, 7508.
476. Murata, M.; Oyama, T.; Watanabe, S.; Masuda, Y. J. Org. Chem. 2000, 65, 164; Song, Y.L. Synlett 2000, 1210.
477. Kanth, J.V.B.; Periasamy, M.; Brown, H.C. Org. Process Res. Dev. 2000, 4, 550.
478. Song, Y.; Ding, Z.; Wang, Q.; Tao, F. Synth. Commun. 1998, 28, 3757.
479. Song, Y.-L.; Morin, C. Synlett 2001, 266.
480. Lightfoot, A.P.; Maw, G.; Thirsk, C.; Twiddle, S.J.R.; Whiting, A. Tetrahedron Lett. 2003, 44, 7645.
481. Ramsden, H.E.; Leebrick, J.R.; Rosenberg, S.D.; Miller, E.H.; Walburn, J.J.; Balint, A.E.; Cserr, R. J. Org, Chem., 1957, 22, 1602.
482. Matteson, D.S. Acc. Chem. Res. 1970, 3, 186; Matteson, D.S. Progr. Boron Chem. 1970, 3, 117.
483. Wong, K.-T.; Chien, Y.-Y.; Liao, Y.-L.; Lin, C.-C.; Chou, M.-Y.; Leung, M.-K. J. Org. Chem. 2002, 67, 1041.
484. Evans, D.A.; Muci, A.R.; Stuermer, R. J. Org. Chem., 1993, 58, 5307.
485. Councler, C. Ber. 1876, 9, 485; 1877, 10, 1655; 1878, 11, 1106.
486. Schiff, H. Ann. Suppl. 1867, 6, 158; Councler, C. J. Prakt. Chem. 1871, 16, 371.
487. Cohn, G. Pharm. Zentr. 1911, 62, 479.
488. Bannister, W.J. U.S. Patent 1,668,797 (Chem. Abstr. 1928, 22:2172).
489. Haider, S.Z.; Khundhar, M.H.; Siddiqulah, Md. J. Appl. Chem. 1954, 4, 93.
490. Colclough, T.; Gerrard, W.; Lappert, M.F. J. Chem. Soc. 1955, 907.
491. Ahmad, T.; Khundkar, M.H. Chem. Ind. 1954, 248.
492. Vedejs, E.; Fields, S.C.; Hayashi, R.; Hitchcock, S.R.; Powell, D.R.; Schrimpf, M.R. J. Am. Chem. Soc. 1999, 121, 2460.
493. Molander, G.A.; Biolatto, B. J. Org. Chem. 2003, 68, 4302; Molander, G.A.; Yun, C.; Ribagorda, M.; Biolatto, B. J. Org. Chem. 2003, 68, 5534; Molander, G.A.; Ribagorda, M. J. Am. Chem. Soc. 2003, 125, 11148.
494. Matteson, D. S. J. Am. Chem. Soc. 1960, 82, 4228.
495. Molander, G. A.; Felix, L. A. J. Org. Chem. 2005, 70, 3950.
496. Ochiai, M.; Yamamoto, S.; Sato, K. Chem. Commun. 1999, 1363.
497. See Wardell, J.L.; Paterson, E.S. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, 1985, pp. 316–323; Wardell, J.L. in Patai, S. The Chemistry of the Thiol Group, pt. 1, Wiley, NY, 1974, pp. 211–215; Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988, pp. 135–142.
498. Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 210–216.
499. Bhattacharya, S.N.; Eaborn, C.; Walton, D.R.M. J. Chem. Soc. C 1968, 1265. For similar reactions with organolithiums, see Hamada, T.; Yonemitsu, O. Synthesis 1986, 852.
500. Harpp, D.N.; Vines, S.M.; Montillier, J.P.; Chan, T.H. J. Org. Chem. 1976, 41, 3987.
501. See Negishi, E. Organometallics in Organic Synthesis, Wiley, NY, 1980, pp. 243–247.
502. See Kitching, W.; Fong, C.W. Organomet. Chem. Rev. Sect. A 1970, 5, 281.
503. Asinger, F.; Laue, P.; Fell, B.; Gubelt, C. Chem. Ber. 1967, 100, 1696.
504. Zakharkin, L.I.; Gavrilenko, V.V.; Paley, B.A. J. Organomet. Chem. 1970, 21, 269.
505. See Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Metal–Carbon Bond, Vol. 2, Wiley, NY, pp. 72–105; Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 158–178; Makarova, L.G. Organomet. React. 1970, 1, 119, pp. 325–348.
506. See Delavarenne, S.Y.; Viehe, H.G. in Viehe, H.G. Acetylenes, Marcel Dekker, NY, 1969, pp. 665–688. For a list of reagents, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 655–656. For an improved procedure, see Brandsma, L.; Verkruijsse, H.D. Synthesis 1990, 984.
507. Okuyama, T.; Fujita, M.; Gronheid, R.; Lodder, G. Tetrahedron Lett. 2000, 41. 5125.
508. Kabalka, G.W.; Mereddy, A.R. Tetrahedron Lett. 2004, 45, 1417.
509. Campos, P.J.; García, B.; Rodríguez, M.A. Tetrahedron Lett. 2002, 43, 6111.
510. Zhang, D.; Ready, J.M. J. Am. Chem. Soc. 2007, 129, 12088.
511. Kamei, K.; Maeda, N.; Tatsuoka, T. Tetrahedron Lett. 2005, 46, 229.
512. Brown, H.C.; Rathke, M.W.; Rogic, M.M.; De Lue, N.R. Tetrahedron 1988, 44, 2751.
513. Brown, H.C.; Lane, C.F. Tetrahedron 1988, 44, 2763; Brown, H.C.; Lane, C.F.; De Lue, N.R. Tetrahedron 1988, 44, 2273. Also see Nelson, D.J.; Soundararajan, R. J. Org. Chem. 1989, 54, 340.
514. Nelson, D.J.; Soundararajan, R. J. Org. Chem. 1988, 53, 5664. For other reagents, see Jigajinni, V.B.; Brown, H.C.; De Lue, N.R. Tetrahedron 1988, 44, 2785.
515. Suzuki, A.; Nozawa, S.; Harada, M.; Itoh, M.; Brown, H.C.; Midland, M.M. J. Am. Chem. Soc. 1971, 93, 1508; Brown, H.C.; Midland, M.M. Angew. Chem. Int. Ed. 1972, 11, 692, pp. 699–700; Brown, H.C. Boranes in Organic Chemistry, Cornell University Press, Ithica, NY, 1972, pp. 442–446.
516. See Kabalka, G.W. Org. Prep. Proced. Int. 1977, 9, 131.
517. Brown, H.C.; Hamaoka, T.; Ravindran, N.; Subrahmanyam, C.; Somayaji, V.; Bhat, N.G. J. Org. Chem. 1989, 54, 6075. See also, Kabalka, G.W.; Gooch, E.E.; Hsu, H.C. Synth. Commun. 1981, 11, 247.
518. Stewart, S.K.; Whiting, A. Tetrahedron Lett. 1995, 36, 3929.
519. Brown, H.C.; Hamaoka, T.; Ravindran, N. J. Am. Chem. Soc. 1973, 95, 6456. See also, Brown, H.C.; Bhat, N.G. Tetrahedron Lett. 1988, 29, 21.
520. See Kabalka, G.W.; Sastry, K.A.R.; Knapp, F.F.; Srivastava, P.C. Synth. Commun. 1983, 13, 1027.
521. Kunda, S.A.; Smith, T.L.; Hylarides, M.D.; Kabalka, G.W. Tetrahedron Lett. 1985, 26, 279.
522. Hoshi, M.; Shirakawa, K. Tetrahedron Lett. 2000, 41, 2595.
523. See Chou, S.P.; Kuo, H.; Wang, C.; Tsai, C.; Sun, C. J. Org. Chem. 1989, 54, 868.
524. Normant, J.F.; Chaiez, G.; Chuit, C.; Villieras, J. J. Organomet. Chem. 1974, 77, 269; Synthesis 1974, 803.
525. Westmijze, H.; Meijer, J.; Vermeer, P. Recl. Trav. Chim. Pays-Bas 1977, 96, 168; Levy, A.B.; Talley, P.; Dunford, J.A. Tetrahedron Lett. 1977, 3545.
526. Takami, K.; Yorimitsu, H.; Oshima, K. Org. Lett. 2002, 4, 2993.
527. Furuya, T.; Ritter, T. Org. Lett. 2009, 11, 2860.
528. Nishiguchi, I.; Kanbe, O.; Itoh, K.; Maekawa, H. Synlett 2000, 89.
529. Abe, H.; Suzuki, H. Bull. Chem. Soc. Jpn. 1999, 72, 787.
530. Lee, T.; Kang, H.R.; Kim, S.; Kim, S. Tetrahedron 2006, 62, 4081.
531. Yan, J.; Li, J.; Cheng, D. Synlett 2007, 2442.
532. Vilhelmsen, M.H.; Andersson, A.S.; Nielsen, M.B. Synthesis 2009, 1469.
533. See Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Carbon–Metal Bond, Vol. 2, Wiley, NY, p. 72; Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H., Eds., Vol. 12; Elsevier, NY, 1973, pp. 135–177; Jensen, F.R.; Rickborn, B. Electrophilic Substitution of Organomercurials, McGraw-Hill, NY, 1968, pp. 75–97.
534. See Jensen, F.R.; Gale, L.H. J. Am. Chem. Soc. 1960, 82, 148.
535. See de Ryck, P.H.; Verdonck, L.; Van der Kelen, G.P. Bull. Soc. Chim. Belg., 1985, 94, 621.
536. See Erdik, E.; Ay, M. Chem. Rev. 1989, 89, 1947.
537. See Genet, J.P.; Mallart, S.; Greck, C.; Piveteau, E. Tetrahedron Lett. 1991, 32, 2359.
538. Beak, P.; Kokko, B.J. J. Org. Chem. 1982, 47, 2822; Colvin, E.W.; Kirby, G.W.; Wilson, A.C. Tetrahedron Lett. 1982, 23, 3835; Boche, G.; Bernheim, M.; Schrott, W. Tetrahedron Lett. 1982, 23, 5399; Boche, G.; Schrott, W. Tetrahedron Lett. 1982, 23, 5403.
539. Kokko, B.J.; Beak, P. Tetrahedron Lett. 1983, 24, 561.
540. Beak, P.; Basha, A.; Kokko, B.; Loo, D. J. Am. Chem. Soc. 1986, 108, 6016.
541. Spagnolo, P.; Zanirato, P.; Gronowitz, S. J. Org. Chem. 1982, 47, 3177; Reed, J.N.; Snieckus, V. Tetrahedron Lett. 1983, 24, 3795; Mori, S.; Aoyama, T.; Shioiri, T. Tetrahedron Lett. 1984, 25, 429.
542. Trost, B.M.; Pearson, W.H. J. Am. Chem. Soc. 1981, 103, 2483; 1983, 105, 1054.
543. Kabalka, G.W.; Wang, Z.; Goudgaon, N.M. Synth. Commun. 1989, 19, 2409. See Kabalka, G.W.; Wang, Z. Organometallics 1989, 8, 1093; Synth. Commun. 1990, 20, 231.
544. Brown, H.C.; Heydkamp, W.R.; Breuer, E.; Murphy, W.S. J. Am. Chem. Soc. 1964, 86, 3565.
545. Brown, H.C.; Kim, K.; Srebnik, M.; Singaram, B. Tetrahedron 1987, 43, 4071. See Brown, H.C.; Kim, K.; Cole, T.E.; Singaram, B. J. Am. Chem. Soc. 1986, 106, 6761.
546. Kabalka, G.W.; Goudgaon, N.M.; Liang, Y. Synth. Commun. 1988, 18, 1363.
547. Carboni, B.; Vaultier, M.; Courgeon, T.; Carrié, R. Bull. Soc. Chim. Fr. 1989, 844.
548. Brown, H.C.; Salunkhe, A.M.; Singaram, B. J. Org. Chem. 1991, 56, 1170.
549. Barton, D.H.R.; Donnelly, D.M.X.; Finet, J.; Guiry, P.J. Tetrahedron Lett. 1989, 30, 1377.
550. Yamamoto, H.; Maruoka, K. J. Org. Chem. 1980, 45, 2739.
551. Merkushev, E.B. Synthesis 1988, 923
552. Beletskaya, I.P.; Affanasiev, V.V.; Kazankova, M.A.; Efimova, I.V. Org. Lett. 2003, 5, 4309.
553. Dunetz, J.R.; Danheiser, R.L. Org. Lett. 2003, 5, 4011.
554. Movassaghi, M.; Ondrus, A.E. J. Org. Chem. 2005, 70, 8638.
555. See Narayana, C.; Periasamy, M. Synthesis 1985, 253; Gulevich, Yu.V.; Bumagin, N.A.; Beletskaya, I.P. Russ. Chem. Rev. 1988, 57, 299.
556. See Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985; Larock, R.C. Tetrahedron 1982, 38, 1713; Angew. Chem. Int. Ed. 1978, 17, 27.
557. Seyferth, D.; Spohn, R.J. J. Am. Chem. Soc. 1969, 91, 3037.
558. Ryu, I.; Ryang, M.; Rhee, I.; Omura, H.; Murai, S.; Sonoda, N. Synth. Commun. 1984, 14, 1175 and references cited therein. For another method, see Hatanaka, Y.; Hiyama, T. Chem. Lett. 1989, 2049.
559. Larock, R.C.; Hershberger, S.S. J. Org. Chem. 1980, 45, 3840.
560. Tanaka, M. Tetrahedron Lett. 1979, 2601.
561. Yamashita, M.; Suemitsu, R. Tetrahedron Lett. 1978, 761. See also, Vitale, A.A.; Doctorovich, F.; Nudelman, N.S. J. Organomet. Chem. 1987, 332, 9.
562. Sato, F.; Oguro, K.; Watanabe, H.; Sato, M. Tetrahedron Lett. 1980, 21, 2869. See Amaratunga, W.; Fréchet, J.M.J. Tetrahedron Lett. 1983, 24, 1143.
563. Trzupek, L.S.; Newirth, T.L.; Kelly, E.G.; Sbarbati, N.E.; Whitesides, G.M. J. Am. Chem. Soc. 1973, 95, 8118.
564. Zadel, G.; Breitmaier, E. Angew. Chem. Int. Ed. 1992, 31, 1035.
565. Yamamoto, K.; Yohitake, J.; Qui, N.T.; Tsuji, J. Chem. Lett. 1978, 859.
566. Miyaura, N.; Suzuki, A. Chem. Lett. 1981, 879. See also, Yamashina, N.; Hyuga, S.; Hara, S.; Suzuki, A. Tetrahedron Lett. 1989, 30, 6555.
567. Larock, R.C. J. Org. Chem. 1975, 40, 3237.
568. Yamashita, M.; Suemitsu, R. Tetrahedron Lett. 1978, 1477.
569. Alper, H.; Amaratunga, S. J. Org. Chem. 1982, 47, 3593.
570. Screttas, C.G.; Steele, B.R. J. Org. Chem. 1988, 53, 5151.
571. Oshita, M.; Chatani, N. Org. Lett. 2004, 6, 4323.
572. Westmijze, H.; Vermeer, P. Synthesis 1977, 784.
573. Murray, R.E.; Zweifel, G. Synthesis 1980, 150.
574. Masuda, Y.; Hoshi, M.; Yamada, T.; Arase, A. J. Chem. Soc. Chem. Commun. 1984, 398.
575. Sakakibara, Y.; Enami, H.; Ogawa, H.; Fujimoto, S.; Kato, H.; Kunitake, K.; Sasaki, K.; Sakai, M. Bull. Chem. Soc. Jpn. 1995, 68, 3137.
576. Piers, E.; Fleming, F.F. Can. J. Chem. 1993, 71, 1867.
577. See Makarova, L.G. Organomet. React. 1970, 1, 119, pp. 190–226; Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 31–44.
578. See Pi, R.; Bauer, W.; Brix, B.; Schade, C.; Schleyer, P.v.R. J. Organomet. Chem. 1986, 306, C1.
579. See Abraham, M.H.; Grellier, P.L. in Hartley, F.R.; Patai, S. The Chemistry of the Carbon–Metal Bond, Vol. 2, Wiley, NY, pp. 25–149; Abraham, M.H. Comprehensive Chemical Kinetics, Bamford, C.H.; Tipper, C.F.H., Eds., Vol. 12; Elsevier, NY, 1973, pp. 39–106; Jensen, F.R.; Rickborn, B. Electrophilic Substituton of Organomercurials, McGraw-Hill, NY, 1968, pp. 100–192. Also see, Schlosser, M. Angew. Chem. Int. Ed. 1964, 3, 287, 362; Newer Methods Prep. Org. Chem. 1968, 5, 238.
580. See Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988; Wakefield, B.J. The Chemistry of Organolithium Compounds, Pergamon, Elmsford, NY, 1974.
581. See Hill, E.A. Adv. Organomet. Chem. 1977, 16, 131; J. Organomet. Chem. 1975, 91, 123.
582. Fell, B.; Asinger, F.; Sulzbach, R.A. Chem. Ber. 1970, 103, 3830. See also, Ashby, E.C.; Ainslie, R.D. J. Organomet. Chem. 1983, 250, 1.
583. See Erdik, E. Tetrahedron 1987, 43, 2203.
584. See Noltes, J.G. Bull. Soc. Chim. Fr. 1972, 2151.
585. See Brown, H.C.; Racherla, U.S. Tetrahedron Lett. 1985, 26, 4311.
586. See Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988, pp. 149–158; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Nonmetallic Substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 1306–1345.
587. See Mole, T. Organomet. React. 1970, 1, 1, pp. 31–43; Larock, R.C. Organomercury Compounds in Organic Synthesis, Springer, NY, 1985, pp. 9–26; Makarova, L.G. Organomet. React. 1970, 1, 119, pp. 129–178, 227–240; van Koten, G. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 219–232; Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 248–270.
588. Chotana, G.A.; Rak, M.A.; Smith III, M.R. J. Am. Chem. Soc. 2005, 127, 10539; Harrisson, P.; Morris, J.; Marder, T.B.; Steel, P.G. Org. Lett. 2009, 11, 3586.
589. Billingsley, K.L.; Buchwald, S.L. J. Org. Chem. 2008, 73, 5589.
590. House, H.O.; Chu, C.; Wilkins, J.M.; Umen, M.J. J. Org. Chem. 1975, 40, 1460. But see also, Lipshutz, B.H.; Whitney, S.; Kozlowski, J.A.; Breneman, C.M. Tetrahedron Lett. 1986, 27, 4273; Bertz, S.H.; Dabbagh, G. Tetrahedron 1989, 45, 425.
591. Stack, D.E.; Klein, W.R.; Rieke, R.D. Tetrahedron Lett. 1993, 34, 3063.
592. See Bublitz, D.E.; Rinehart Jr., K.L. Org. React. 1969, 17, 1; Birmingham, J.M. Adv. Organomet. Chem. 1965, 2, 365, pp. 375.
593. Soderquist, J.A.; DePomar, J.C.J. Tetrahedron Lett. 2000, 41, 3537.
594. See Wardell, J.L. in Hartley, F.R.; Patai, S. The Chemistry of the Carbon–Metal Bond, Vol. 4, Wiley, NY, pp. 1–157, see pp. 81–89; Kauffmann, T. Top. Curr. Chem. 1980, 92, 109, pp. 130.
595. Pearson, W.H.; Lindbeck, A.C. J. Org. Chem. 1989, 54, 5651.
596. Seetz, J.W.F.L.; Schat, G.; Akkerman, O.S.; Bickelhaupt, F. J. Am. Chem. Soc. 1982, 104, 6848.
597. Maercker, A.; Dujardin, R. Angew. Chem. Int. Ed. 1984, 23, 224.
598. Sawyer, J.S.; Kucerovy, A.; Macdonald, T.L.; McGarvey, G.J. J. Am. Chem. Soc. 1988, 110, 842.
599. Dessy, R.E.; Kaplan, F.; Coe, G.R.; Salinger, R.M. J. Am. Chem. Soc. 1963, 85, 1191.
600. See Lipshutz, B.H. Synlett, 1990, 119. See also, Bertz, S.H. J. Am. Chem. Soc. 1990, 112, 4031; Lipshutz, B.H.; Sharma, S.; Ellsworth, E.L. J. Am. Chem. Soc. 1990, 112, 4032.
601. Behling, J.R.; Babiak, K.A.; Ng, J.S.; Campbell, A.L.; Moretti, R.; Koerner, M.; Lipshutz, B.H. J. Am. Chem. Soc. 1988, 110, 2641.
602. Chou, T.; Knochel, P. J. Org. Chem. 1990, 55, 4791.
603. See Massey, A.G.; Humphries, R.E. Aldrichimica Acta 1989, 22, 31; Negishi, E. Organometallics in Organic Synthesis, Wiley, NY, 1980, pp. 30–37
604. See Raston, C.L.; Salem, G. in Hartley, F.R.; Patai, S. The Chemistry of the Carbon–Metal Bond, Vol. 4, Wiley, NY, pp. 159–306, 162–175; Kharasch, M.S.; Reinmuth, O. Grignard Reactions of Monmetallic substances, Prentice-Hall, Englewood Cliffs, NJ, 1954, pp. 5–91.
605. Pearson, D.E.; Cowan, D.; Beckler, J.D. J. Org. Chem. 1959, 24, 504.
606. See Benkeser, R.A. Synthesis 1971, 347.
607. See Oppolzer, W.; Schneider, P. Tetrahedron Lett. 1984, 25, 3305.
608. Bogdanovic, B.; Janke, N.; Kinzelmann, H. Chem. Ber. 1990, 123, 1507, and other papers in this series.
609. Gallagher, M.J.; Harvey, S.; Raston, C.L.; Sue, R.E. J. Chem. Soc. Chem. Commun. 1988, 289.
610. Baker, K.V.; Brown, J.M.; Hughes, N.; Skarnulis, A.J.; Sexton, A. J. Org. Chem. 1991, 56, 698. See Lai, Y. Synthesis 1981, 585.
611. See Raston, C.L.; Salem, G. in Hartley, F.R.; Patai, S. The Chemistry of the Carbon–Metal Bond, Vol. 4, Wiley, NY, pp. 187–193; Heaney, H. Organomet. Chem. Rev. 1966, 1, 27. For a review of di-Grignard reagents, see Bickelhaupt, F. Angew. Chem. Int. Ed. 1987, 26, 990.
612. See Seetz, J.W.F.L.; Hartog, F.A.; Böhm, H.P.; Blomberg, C.; Akkerman, O.S.; Bickelhaupt, F. Tetrahedron Lett. 1982, 23, 1497.
613. See van Eikkema Hommes, N.J.R.; Bickelhaupt, F.; Klumpp, G.W. Angew. Chem. Int. Ed. 1988, 27, 1083.
614. See Bruin, J.W.; Schat, G.; Akkerman, O.S.; Bickelhaupt, F. J. Organomet. Chem. 1985, 288, 13.
615. See Chivers, T. Organomet. Chem. Rev. Sect. A 1970, 6, 1.
616. Yu, S.H.; Ashby, E.C. J. Org. Chem. 1971, 36, 2123.
617. Sugimoto, O.; Yamada, S.; Tanji, K. J. Org. Chem. 2003, 68, 2054.
618. See Peterson, D.J. Organomet. Chem. Rev. Sect. A 1972, 7, 295.
619. See Castro, B. Bull. Soc. Chim. Fr. 1967, 1533, 1540, 1547.
620. Gitlitz, M.H.; Considine, W.J. J. Organomet. Chem. 1970, 23, 291.
621. Smith, Jr., W.N. J. Organomet. Chem. 1974, 64, 25.
622. Boudin, A.; Cerveau, G.; Chuit, C.; Corriu, R.J.P.; Reye, C. Tetrahedron 1989, 45, 171.
623. See Wakefield, B.J. Organolithium Methods, Academic Press, NY, 1988, pp. 21–32; Wardell, J.L. in Hartley, F.R.; Patai, S. Vol. 4, pp. 1–157, 5–27; Newcomb, M.E. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 3–14. For a study of halogen–lithium exhange in hydrocarbon solvents, see Slocum, D.W.; Kusmic, D.; Raber, J.C.; Reinscheld, T.K.; Whitley, P.E. Tetrahedron Lett. 2010, 51, 4793.
624. See Rieke, R.D. Science 1989, 246, 1260.
625. See Klabunde, K.J. React. Intermed. (Plenum) 1980, 1, 37; Acc. Chem. Res. 1975, 8, 393; Skell, P.S. Havel, J.J.; McGlinchey, M.J. Acc. Chem. Res. 1973, 6, 97.
626. Ebert, G.W.; Rieke, R.D. J. Org. Chem. 1988, 53, 4482. See also, Baker, K.V.; Brown, J.M.; Hughes, N.; Skarnulis, A.J.; Sexton, A. J. Org. Chem. 1991, 56, 698.
627. Wu, T.; Xiong, H.; Rieke, R.D. J. Org. Chem. 1990, 55, 5045.
628. Rieke, R.D.; Li, P.T.; Burns, T.P.; Uhm, S.T. J. Org. Chem. 1981, 46, 4323. See also, Zhu, L.; Wehmeyer, R.M.; Rieke, R.D. J. Org. Chem. 1991, 56, 1445.
629. Burkhardt, E.R.; Rieke, R.D. J. Org. Chem. 1985, 50, 416.
630. Stack, D.E.; Dawson, B.T.; Rieke, R.D. J. Am. Chem. Soc. 1991, 113, 4672, and references cited therein.
631. See Lai, Y. Synthesis 1981, 585; Rieke, R.D. Acc. Chem. Res. 1977, 10, 301.
632. See Blomberg, C. Bull. Soc. Chim. Fr. 1972, 2143.
633. Bodewitz, H.W.H.J.; Blomberg, C.; Bickelhaupt, F. Tetrahedron 1975, 31, 1053. See also, Schaart, B.J.; Blomberg, C.; Akkerman, O.S.; Bickelhaupt, F. Can. J. Chem. 1980, 58, 932.
634. See Rogers, H.R.; Hill, C.L.; Fujiwara, Y.; Rogers, R.J.; Mitchell, H.L.; Whitesides, G.M. J. Am. Chem. Soc. 1980, 102, 217; Barber, J.J.; Whitesides, G.M. J. Am. Chem. Soc. 1980, 102, 239.
635. Root, K.S.; Hill, C.L.; Lawrence, L.M.; Whitesides, G.M. J. Am. Chem. Soc. 1989, 111, 5405.
636. Nuzzo, R.G.; Dubois, L.H. J. Am. Chem. Soc. 1986, 108, 2881.
637. Hoffmann, R.W.; Brönstrup, M.; Müller, M. Org. Lett. 2003, 5, 313.
638. See Sergeev, G.B.; Zagorsky, V.V.; Badaev, F.Z. J. Organomet. Chem. 1983, 243, 123. See, however, de Souza-Barboza, J.C.; Luche, J.; Pétrier, C. Tetrahedron Lett. 1987, 28, 2013.
639. Walborsky, H.M.; Topolski, M. J. Am. Chem. Soc. 1992, 114, 3455; Walborsky, H.M.; Zimmermann, C. J. Am. Chem. Soc. 1992, 114, 4996; Walborsky, H.M. Accts. Chem. Res. 1990, 23, 286.
640. Garst, J.F. Acc. Chem. Res. 1991, 24, 95; Garst, J.F.; Ungváry, F.; Batlaw, R.; Lawrence, K.E. J. Am. Chem. Soc. 1991, 113, 5392.
641. Walborsky, H.M. Acc. Chem. Res. 1990, 23, 286.
642. de Boer, H.J.R.; Akkerman, O.S.; Bickelhaupt, F. Angew. Chem. Int. Ed. 1988, 27, 687.
643. See Hölzer, B.; Hoffmann, R.W. Chem. Commun. 2003, 732; Dakternieks, D.; Dunn, K.; Henry, D.J.; Schiesser, C.H.; Tiekink, E.R. Organometallics 1999, 18, 3342.
644. See Wardell, J.L. in Zuckerman, J.J. Inorganic Reactions and Methods, Vol. 11, VCH, NY, 1988, pp. 107–129; Parham, W.E.; Bradsher, C.K. Acc. Chem. Res. 1982, 15, 300.
645. See Tamborski, C.; Moore, G.J. J. Organomet. Chem. 1971, 26, 153.
646. See Bailey, W.F.; Punzalan, E.R. J. Org. Chem. 1990, 55, 5404; Negishi, E.; Swanson, D.R.; Rousset, C.J. J. Org. Chem. 1990, 55, 5406.
647. Yus, M.; Herrera, R.P.; Guijarro, A. Tetrahedron Lett., 2003, 44, 5025.
648. For examples of exchange, R = vinylic, see Miller, R.B.; McGarvey, G. Synth. Commun. 1979, 9, 831; Sugita, T.; Sakabe, Y.; Sasahara, T.; Tsukuda, M.; Ichikawa, K. Bull. Chem. Soc. Jpn. 1984, 57, 2319.
649. See Siegel, H. Top. Curr. Chem. 1982, 106, 55; Negishi, E. Organometallics in Organic Synthesis, Wiley, NY, 1980, pp. 136–151; Köbrich, G. Angew. Chem. Int. Ed. 1972, 11, 473. Also see Krief, A. Tetrahedron 1980, 36, 2531; Normant, H. J. Organomet. Chem. 1975, 100, 189.
650. Hoeg, D.F.; Lusk, D.I.; Crumbliss, A.L. J. Am. Chem. Soc. 1965, 87, 4147. See also, Villieras, J.; Tarhouni, R.; Kirschleger, B.; Rambaud, M. Bull. Soc. Chim. Fr. 1985, 825.
651. Villieras, J. Bull. Soc. Chim. Fr. 1967, 1520.
652. Schmidt, A.; Köbrich, G.; Hoffmann, R.W. Chem. Ber. 1991, 124, 1253; Hoffmann, R.W.; Bewersdorf, M. Chem. Ber. 1991, 124, 1259.
653. Stetter, H.; Steinbeck, K. Liebigs Ann. Chem. 1972, 766, 89.
654. Kauffmann, T.; Fobker, R.; Wensing, M. Angew. Chem. Int. Ed. 1988, 27, 943.
655. For reviews of the mechanism, see Bailey, W.F.; Patricia, J.J. J. Organomet. Chem. 1988, 352, 1; Beletskaya, I.P.; Artamkina, G.A.; Reutov, O.A. Russ. Chem. Rev. 1976, 45, 330.
656. Ashby, E.C.; Pham, T.N. J. Org. Chem. 1987, 52, 1291. See also, Bailey, W.F.; Patricia, J.J.; Nurmi, T.T.; Wang, W. Tetrahedron Lett. 1986, 27, 1861.
657. Ward, H.R.; Lawler, R.G.; Loken, H.Y. J. Am. Chem. Soc. 1968, 90, 7359.
658. See Reich, H.J.; Green, D.P.; Phillips, N.H. J. Am. Chem. Soc. 1989, 111, 3444.
659. Beak, P.; Allen, D.J.; Lee, W.K. J. Am. Chem. Soc. 1990, 112, 1629.
660. See Ashby, E.C.; Su, W.; Pham, T.N. Organometallics 1985, 4, 1493; Alnajjar, M.S.; Kuivila, H.G. J. Am. Chem. Soc. 1985, 107, 416.
661. Kurono, N.; Inoue, T.; Tokuda, M. Tetrahedron 2005, 61, 11125.
662. See Artamkina, G.A.; Beletskaya, I.P. Russ. Chem. Rev. 1987, 56, 983.
663. March, J. J. Chem. Educ. 1963, 40, 212.
664. Zara, C.L.; Jin, T.; Giguere, R.J. Synth. Commun. 2000, 30, 2099.
665. See Roy, S.C.; Guin, C.; Maiti, G. Tetrahedron Lett. 2001, 42, 9253.
666. Singh, S.P.; Kagan, J. J. Org. Chem. 1970, 35, 2203.
667. Shiner, Jr., V.J.; Martin, B. J. Am. Chem. Soc. 1962, 84, 4824.
668. See Richardson, W.H.; O'Neal, H.E. in Bamford, C.H.; Tipper, C.F.H. Comprehensive Chemical Kinetics, Vol. 5, Elsevier, NY, 1972, pp. 447–482; Clark, L.W. in Patai, S. The Chemistry of Carboxylic Acids and Esters, Wiley, NY, 1969, pp. 589–622. See Dunn, G.E. Isot. Org. Chem. 1977, 3, 1.
669. See Buncel, E.; Venkatachalam, T.K.; Menon, B.C. J. Org. Chem. 1984, 49, 413.
670. Hunter, D.H.; Patel, V.; Perry, R.A. Can. J. Chem. 1980, 58, 2271, and references cited therein.
671. Graul, S.T.; Squires, R.R. J. Am. Chem. Soc. 1988, 110, 607.
672. See Jencks, W.P. Catalysis in Chemistry and Enzmology, McGraw-Hill, NY, 1969, pp. 116–120.
673. Bigley, D.B.; Clarke, M.J. J. Chem. Soc. Perkin Trans. 2 1982, 1, and references cited therein. For a review, see Smith, G.G.; Kelly, F.W. Prog. Phys. Org. Chem. 1971, 8, 75, pp. 150–153.
674. Bigley, D.B. J. Chem. Soc. 1964, 3897.
675. Wasserman, H.H. in Newman Steric Effects in Organic Chemistry, Wiley, NY, 1956, p. 352. See also, Buchanan, G.L.; Kean, N.B.; Taylor, R. Tetrahedron 1975, 31, 1583.
676. Sterically hindered β-keto acids decarboxylate more slowly: Meier, H.; Wengenroth, H.; Lauer, W.; Krause, V. Tetrahedron Lett. 1989, 30, 5253.
677. See Ferris, J.P.; Miller, N.C. J. Am. Chem. Soc. 1966, 88, 3522.
678. Swain, C.G.; Bader, R.F.W.; Esteve Jr., R.M.; Griffin, R.N. J. Am. Chem. Soc. 1961, 83, 1951.
679. Noyce, D.S.; Metesich, M.A. J. Org. Chem. 1967, 32, 3243.
680. Bigley, D.B.; Al-Borno, A. J. Chem. Soc. Perkin Trans. 2 1982, 15.
681. Guthrie, J.P.; Peiris, S.; Simkin, M.; Wang, Y. Can. J. Chem. 2010, 88, 79.
682. See Oshry, L.; Rosenfeld, S.M. Org. Prep. Proced. Int. 1982, 14, 249.
683. For a list of examples, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, pp. 1542–1543. See Yu, Y.; Zhang, Y. Synth. Commun. 1999, 29, 243.
684. Lalancette, J.M.; Lachance, A. Tetrahedron Lett. 1970, 3903.
685. See Krapcho, A.P. Synthesis 1982, 805, 893. For other methods, see Dehmlow, E.V.; Kunesch, E. Synthesis 1985, 320; Taber, D.F.; Amedio, Jr., J.C.; Gulino, F. J. Org. Chem. 1989, 54, 3474.
686. Hashimoto, M.; Eda, Y.; Osanai, Y.; Iwai, T.; Aoki, S. Chem. Lett. 1986, 893.
687. Laval, G.; Golding, B.T. Synlett 2003, 542.
688. See Okada, K.; Okubo, K.; Oda, M. Tetrahedron Lett. 1989, 30, 6733.
689. See Deacon, G.B. Organomet. Chem. Rev. A 1970, 355; Deacon, G.B.; Faulks, S.J.; Pain, G.N. Adv. Organomet. Chem. 1986, 25, 237.
690. Kolarovi, A.; Fberov, Z. J. Org. Chem. 2009, 74, 7199.
691. Gribkov, D.V.; Pastine, S.J.; Schnürch, M.; Sames, D. J. Am. Chem. Soc. 2007, 129, 11750.
692. Waetzig, S.R.; Tunge, J.A. J. Am. Chem. Soc. 2007, 129, 14860.
693. Benkeser, R.A.; Siklosi, M.P.; Mozdzen, E.C. J. Am. Chem. Soc. 1978, 100, 2134.
694. Arnett, E.M.; Small, L.E.; McIver Jr., R.T.; Miller, J.S. J. Org. Chem. 1978, 43, 815. See also, Lomas, J.S.; Dubois, J.E. J. Org. Chem. 1984, 49, 2067.
695. See Snowden, R.L.; Linder, S.M.; Muller, B.L.; Schulte-Elte, K.H. Helv. Chim. Acta 1987, 70, 1858, 1879.
696. Partington, S.M.; Watt, C.I.F. J. Chem. Soc. Perkin Trans. 2 1988, 983.
697. Tumas, W.; Foster, R.F.; Brauman, J.I. J. Am. Chem. Soc. 1988, 110, 2714; Ibrahim, S.; Watt, C.I.F.; Wilson, J.M.; Moore, C. J. Chem. Soc. Chem. Commun. 1989, 161.
698. Paquette, L.A.; Gilday, J.P.; Maynard, G.D. J. Org. Chem. 1989, 54, 5044; Paquette, L.A.; Maynard, G.D. J. Org. Chem. 1989, 54, 5054.
699. See Lomas, J.S.; Fain, D.; Briand, S. J. Org. Chem. 1990, 55, 1052, and references cited therein.
700. See Buchanan, G.L. Chem. Soc. Rev. 1988, 17, 91.
701. Allinger, N.L.; Wang, G.L.; Dewhurst, B.B. J. Org. Chem. 1974, 39, 1730.
702. Milenkov, B.; Hesse, M. Helv. Chim. Acta 1987, 70, 308. For a similar preparation of lactams, see Wälchli, R.; Bienz, S.; Hesse, M. Helv. Chim. Acta 1985, 68, 484.
703. Beletskaya, I.P.; Gulyukina, N.S.; Borodkin, V.S.; Solov'yanov, A.A.; Reutov, O.A. Doklad. Chem. 1984, 276, 202. See also, Mignani, G.; Morel, D.; Grass, F. Tetrahedron Lett. 1987, 28, 5505.
704. Zhang, Y.; Jiao, J.; Flowers II, R.A. J. Org. Chem. 2006, 71, 4516.
705. See Chakrabartty, S.K. in Trahanovsky, W.S. Oxidation in Organic Chemistry, pt. C, Academic Press, NY, 1978, pp. 343–370.
706. See Guthrie, J.P.; Cossar, J. Can. J. Chem. 1986, 64, 1250; Zucco, C.; Lima, C.F.; Rezende, M.C.; Vianna, J.F.; Nome, F. J. Org. Chem. 1987, 52, 5356.
707. Pocker, Y. Chem. Ind. (London) 1959, 1383.
708. Levine, R.; Stephens, J.R. J. Am. Chem. Soc. 1950, 72, 1642.
709. See Hudlicky, M. Chemistry of Organic Fluorine Compounds, 2nd ed., Ellis Horwood, Chichester, 1976, pp. 276–278.
710. Guthrie, J.P.; Cossar, J. Can. J. Chem. 1990, 68, 1640.
711. Nikishin, G.I.; Elinson, M.N.; Makhova, I.V. Tetrahedron 1991, 47, 895.
712. Delgado, A.; Clardy, J. Tetrahedron Lett. 1992, 33, 2789.
713. Gassman, P.G.; Lumb, J.T.; Zalar, F.V. J. Am. Chem. Soc. 1967, 89, 946.
714. March, J.; Plankl, W. J. Chem. Soc. Perkin Trans. 1 1977, 460.
715. Davies, D.G.; Derenberg, M.; Hodge, P. J. Chem. Soc. C 1971, 455.
716. See Hoffman, T.D.; Cram, D.J. J. Am. Chem. Soc. 1969, 91, 1009.
717. See Gilday, J.P.; Paquette, L.A. Org. Prep. Proced. Int. 1990, 22, 167.
718. Paquette, L.A.; Gilday, J.P. J. Org. Chem. 1988, 53, 4972; Paquette, L.A.; Ra, C.S. J. Org. Chem. 1988, 53, 4978.
719. Bunnett, J.F.; Hrutfiord, B.F. J. Org. Chem. 1962, 27, 4152.
720. Ballini, R.; Bosica, G.; Fiorini, D. Tetrahedron 2003, 59, 1143.
721. Grubmüller, P.; Schleyer, P.v.R.; McKervey, M.A. Tetrahedron Lett. 1979, 181.
722. See Grovenstein Jr., E.; Bhatti, A.M.; Quest, D.E.; Sengupta, D.; VanDerveer, D. J. Am. Chem. Soc. 1983, 105, 6290.
723. Necas, D.; Tursky, M.; Kotora, M. J. Am. Chem. Soc. 2004, 126, 10222.
724. For a list of procedures, with references, see Larock, R.C. Comprehensive Organic Transformations, 2nd ed., Wiley–VCH, NY, 1999, p. 75.
725. Birch, A.J.; Hutchinson, E.G. J. Chem. Soc. Perkin Trans. 1 1972, 1546; Yamada, S.; Tomioka, K.; Koga, K. Tetrahedron Lett. 1976, 61.
726. Van Tamelen, E.E.; Rudler, H.; Bjorklund, C. J. Am. Chem. Soc. 1971, 93, 7113.
727. See Berkoff, C.E.; Rivard, D.E.; Kirkpatrick, D.; Ives, J.L. Synth. Commun. 1980, 10, 939; Savoia, D.; Tagliavini, E.; Trombini, C.; Umani-Ronchi, A. J. Org. Chem. 1980, 45, 3227; Ozawa, F.; Iri, K.; Yamamoto, A. Chem. Lett. 1982, 1707.
728. Ohsawa, T.; Kobayashi, T.; Mizuguchi, Y.; Saitoh, T.; Oishi, T. Tetrahedron Lett. 1985, 26, 6103.
729. Fabre, C.; Hadj Ali Salem, M.; Welvart, Z. Bull. Soc. Chim. Fr. 1975, 178. See also, Ogura, K.; Shimamura, Y.; Fujita, M. J. Org. Chem. 1991, 56, 2920.
730. Kim, Y.H.; Kim, K.; Shim, S.B. Tetrahedron Lett. 1986, 27, 4749.
731. Pozsgay, V.; Jennings, H.J. Tetrahedron Lett. 1987, 28, 5091.
732. See Zolfigol, M.A. Synth. Commun. 1999, 29, 905; Zolfigol, M.A.; Ghaemi, E.; Madrikian, E.; Kiany-Burazjani, M. Synth. Commun. 2000, 30, 2057.
733. See Williams, D.L.H. Nitrosation, Cambridge University Press, Cambridge, 1988, pp. 95–109; Kostyukovskii, Ya.L.; Melamed, D.B. Russ. Chem. Rev. 1988, 57, 350; Saavedra, J.E. Org. Prep. Proced. Int. 1987, 19, 83; Challis, B.C.; Challis, J.A. in Patai, S.; Rappoport, Z. The Chemistry of the Functional Groups Supplement F, pt. 2, Wiley, NY, 1982, pp. 1151–1223. Also see Zyranov, G.V.; Rudkevich, D.M. Org. Lett. 2003, 5, 1253.
734. Castro, A.; Iglesias, E.; Leis, J.R.; Peña, M.E.; Tato, J.V. J. Chem. Soc. Perkin Trans. 2 1986, 1725.
735. Hein, G.E. J. Chem. Educ. 1963, 40, 181. See also, Verardo, G.; Giumanini, A.G.; Strazzolini, P. Tetrahedron 1990, 46, 4303.
736. Challis, B.C.; Kyrtopoulos, S.A. J. Chem. Soc. Perkin Trans. 1 1979, 299.
737. Casado, J.; Castro, A.; Lorenzo, F.M.; Meijide, F. Monatsh. Chem. 1986, 117, 335.
738. Challis, B.C.; Yousaf, T.I. J. Chem. Soc. Chem. Commun. 1990, 1598.
739. Zolfigol, M.A.; Choghamarani, A.G.; Shivini, F.; Keypour, H.; Salehzadeh, S. Synth. Commun. 2001, 31, 359. See Zolfigol, M.A.; Bagherzadeh, M.; Choghamarani, A.G.; Keypour, H.; Salehzadeh, S. Synth. Commun. 2001, 31, 1161.
740. Gowenlock, B.G.; Hutchison, R.J.; Little, J.; Pfab, J. J. Chem. Soc. Perkin Trans. 2 1979, 1110. See also, Loeppky, R.N.; Outram, J.R.; Tomasik, W.; Faulconer, J.M. Tetrahedron Lett. 1983, 24, 4271.
741. Boyer, J.H.; Pillai, T.P.; Ramakrishnan, V.T. Synthesis 1985, 677.
742. Boyer, J.H.; Kumar, G.; Pillai, T.P. J. Chem. Soc. Perkin Trans. 1 1986, 1751.
743. See Bottaro, J.C.; Schmitt, R.J.; Bedford, C.D. J. Org. Chem. 1987, 52, 2292; Suri, S.C.; Chapman, R.D. Synthesis 1988, 743; Carvalho, E.; Iley, J.; Norberto, F.; Rosa, E. J. Chem. Res. (S) 1989, 260.
744. Cherednichenko, L.V.; Dmitrieva, L.G.; Kuznetsov, L.L.; Gidaspov, B.V. J. Org. Chem. USSR 1976, 12, 2101, 2105.
745. Andreev, S.A.; Lededev, B.A.; Tselinskii, I.V. J. Org. Chem. USSR 1980, 16, 1166, 1170, 1175, 1179.
746. See Vaughan, K.; Stevens, M.F.G. Chem. Soc. Rev. 1978, 7, 377.
747. Boyer, J.H. in Feuer, H. The Chemistry of the Nitro and Nitroso Groups, pt. 1, Wiley, NY, 1969, pp. 278–283.
748. See Ogata, Y.; Tsuchida, M.; Takagi, Y. J. Am. Chem. Soc. 1957, 79, 3397.
749. Knight, G.T.; Saville, B. J. Chem. Soc. Perkin Trans. 2 1973, 1550.
750. For discussions of the mechanism in the absence of base, see Becker, A.R.; Sternson, L.A. J. Org. Chem. 1980, 45, 1708. See also, Pizzolatti, M.G.; Yunes, R.A. J. Chem. Soc. Perkin Trans. 1 1990, 759.
751. Russell, G.A.; Geels, E.J.; Smentowski, F.J.; Chang, K.; Reynolds, J.; Kaupp, G. J. Am. Chem. Soc. 1967, 89, 3821.
752. Oae, S.; Fukumoto, T.; Yamagami, M. Bull. Chem. Soc. Jpn. 1963, 36, 728.
753. Zawalski, R.C.; Kovacic, P. J. Org. Chem. 1979, 44, 2130. Also see Moriarty, R.M.; Hopkins, T.E.; Prakash, I.; Vaid, B.K.; Vaid, R.K. Synth. Commun. 1990, 20, 2353.
754. Okubo, M.; Matsuo, K.; Yamauchi, A. Bull. Chem. Soc. Jpn. 1989, 62, 915, and other papers in this series.
755. Kajigaeshi, S.; Nakagawa, T.; Fujisaki, S. Chem. Lett. 1984, 2045.
756. Kajigaeshi, S.; Murakawa, K.; Asano, K.; Fujisaki, S.; Kakinami, T. J. Chem. Soc. Perkin Trans. 1 1989, 1702.
757. Curini, M.; Epifano, F.; Marcotullio, M.C.; Rosati, O.; Tsadjout, A. Synlett 2000, 813.
758. See Guillemin, J.; Denis, J.N. Synthesis 1985, 1131.
759. Zhong, Y.-L.; Zhou, H.; Gauthier, D.R.; Lee, J.; Askin, D.; Dolling, U.H.; Volante, R.P. Tetrahedron Lett. 2005, 46, 1099.
760. De Luca, L.; Giacomelli, G.; Nieddu, G. Synlett 2005, 223.
761. See Matte, D.; Solastiouk, B.; Merlin, A.; Deglise, X. Can. J. Chem. 1989, 67, 786.
762. See Thomm, E.W.C.W.; Wayman, M. Can. J. Chem. 1969, 47, 3289; Higuchi, T.; Hussain, A.; Pitman, I.H. J. Chem. Soc. B, 1969, 626.
763. Sharts, C.M. J. Org. Chem. 1968, 33, 1008.
764. Grakauskas, V.; Baum, K. J. Org. Chem. 1969, 34, 2840; 1970, 35, 1545.
765. See Barton, D.H.R.; Hesse, R.H.; Klose, T.R.; Pechet, M.M. J. Chem. Soc. Chem. Commun. 1975, 97.
766. DeLuca, L.; Giacomelli, G. Synlett 2004, 2180.
767. See Bitsi, G.; Jenner, G. J. Organomet. Chem. 1987, 330, 429.
768. Reddy, P.G.; Kumar, .D.K.; Baskaran, S. Tetrahedron Lett. 2000, 41, 9149.
769. Troisi, L.; Granito, C.; Rosato, F.; Videtta, V. Tetrahedron Lett. 2010, 51, 371; Wu, X.-F.; Neumann, H.; Beller, M. Chemistry: Eur. J. 2010, 16, 9750.
770. See Gabriele, B.; Salerno, G.; Mancuso, R.; Costa, M. J. Org. Chem. 2004, 69, 4741.
771. Sonoda, N.; Yasuhara, T.; Kondo, K.; Ikeda, T.; Tsutsumi, S. J. Am. Chem. Soc. 1971, 93, 6344.
772. Franz, R.A.; Applegath, F.; Morriss, F.V.; Baiocchi, F.; Bolze, C. J. Org. Chem. 1961, 26, 3309.
773. Pri-Bar, I.; Alper, H. Can. J. Chem. 1990, 68, 1544.
774. Reddy, B.M.; Reddy, V.R. Synth. Commun. 1999, 29, 2789.
775. Yang, Y.; Lu, S. Tetrahedron Lett. 1999, 40, 4845.
776. Stern, E.W.; Spector, M.L. J. Org. Chem. 1966, 31, 596.
777. Bennett, R.P.; Hardy, W.B. J. Am. Chem. Soc. 1968, 90, 3295.
778. Unverferth, K.; Tietz, H.; Schwetlick, K. J. Prakt. Chem. 1985, 327, 932. See also, Kunin, A.J.; Noirot, M.D.; Gladfelter, W.L. J. Am. Chem. Soc. 1989, 111, 2739.
779. Peerlings, H.W.I.; Meijer, E.W. Tetrahedron Lett. 1999, 40, 1021.
780. Larionov, O.V.; Kozhushkov, S.I.; de Meijere, A. Synthesis 2003, 1916.
781. Wang, M.D.; Alper, H. J. Am. Chem. Soc. 1992, 114, 7018.
782. Lu, S.-M.; Alper, H. J. Am. Chem. Soc. 2005, 127, 14776.
783. Feroci, M.; Inesi, A.; Rossi, L. Tetrahedron Lett. 2000, 41, 963.
784. Selva, M.; Tundo, P.; Perosa, A. Tetrahedron Lett. 2002, 43, 1217. Also see Selva, M.; Tundo, P.; Perosa, A.; Dall'Acqua, F. J. Org. Chem. 2005, 70, 2771.
785. Alper, H.; Vasapollo, G. Tetrahedron Lett. 1987, 28, 6411.
786. Cenini, S.; Crotti, C.; Pizzotti, M.; Porta, F. J. Org. Chem. 1988, 53, 1243; Reddy, N.P.; Masdeu, A.M.; El Ali, B.; Alper, H. J. Chem. Soc. Chem. Commun. 1994, 863.
787. Murahashi, S.; Imada, Y.; Nishimura, K. J. Chem. Soc. Chem. Commun.1988, 1578.
788. Lipshutz, B.H.; Papa, P.; Keith, J.M. J. Org. Chem. 1999, 64, 3792.
789. Feroci, M.; Casadei, M.A.; Orsini, M.; Palombi, L.; Inesi, A. J. Org. Chem. 2003, 68, 1548.
790. Yoshida, M.; Hara, N.; Okuyama, S. Chem. Commun. 2000, 151.
791. Kuniyasu, H.; Hiraike, H.; Morita, M.; Tanaka, A.; Sugoh, K.; Kurosawa, H. J. Org. Chem. 1999, 64, 7305.
792. Nomura, R.; Hasegawa, Y.; Ishimoto, M.; Toyosaki, T.; Matsuda, H. J. Org. Chem. 1992, 57, 7339.
793. Miller, A.W.; Nguyen, S.T. Org. Lett. 2004, 6, 2301.
794. Hancock, M.T.; Pinhas, A.R. Tetrahedron Lett. 2003, 44, 5457.