Physical Chemistry: A Very Short Introduction (2014)
Chapter 2. Matter from the outside
In the early days of physical chemistry, maybe back in the 17th century but more realistically during the 19th century, its practitioners, lacking the extraordinary instruments we now possess, investigated appearances rather than burrowing into the then unknown inner structure of matter. That activity, when performed quantitatively rather than merely reporting on the way things looked, proved extraordinarily fruitful, especially in the field of study that came to be known as ‘thermodynamics’.
Thermodynamics is the science of energy and the transformations that it can undergo. It arose from considerations of the efficiencies of steam engines in the hands of engineers and physicists who were intent on extracting as much motive power as possible from a pound of coal or, as electricity developed, a pound of zinc. Those early pioneers had no conception, I suspect, that their discoveries would be imported into chemistry so effectively that thermodynamics would come to play a central role in understanding chemical reactions and acquire a reach far beyond what the phrase ‘transformation of energy’ suggests.
Modern chemical thermodynamics is still all about energy, but in the course of establishing the laws that govern energy it turns out that sometimes unsuspected relations are discovered between different properties of bulk matter. The usefulness of that discovery is that measurements of one property can be used to determine another that might not be readily accessible to measurement.
There are currently four laws of thermodynamics, which are slightly capriciously labelled 0, 1, 2, and 3. The Zeroth Law establishes the concept of temperature, and although temperature is of vital importance for the discussion of all forms of matter and their properties, I accept that its conceptual and logical basis is not of much concern to the general run of physical chemists and shall not discuss it further. (I touch on the significance of temperature again in Chapter 3.) The First Law concerns the hugely important conservation of energy, the Second Law that wonderfully illuminating property entropy, and the Third Law the seemingly frustrating inaccessibility of the absolute zero of temperature. As no chemistry happens at absolute zero, the Third Law might seem not to have much relevance to physical chemists, but in fact it plays an important role in the way they use data and I cannot ignore it. It is in the domain of the Second Law that most of the relations between measurable properties are found, and I shall explain what is involved in due course.
‘Matter from the outside’ is an appropriate title of a chapter on thermodynamics, for in its purest form, which is known as ‘classical thermodynamics’, no discussion or deduction draws on any model of the internal constitution of matter. Even if you don’t believe in atoms and molecules, you can be an effective (if blinkered) thermodynamicist. All the relations derived in classical thermodynamics refer to observable properties of bulk matter and do not draw on the properties of the constituent atoms and molecules. However, much insight into the origin of these bulk properties and the relations between them is obtained if the blinkers are removed and the atomic constitution of matter acknowledged. That is the role of the fearsomely named statistical thermodynamics, to which I give an introduction in Chapter 3.
Throughout this chapter I shall focus on the applications of thermodynamics in physical chemistry. It goes without saying that thermodynamics is also widely applicable in engineering and physics, where it originated. It is also a largely mathematical subject, especially when it comes to establishing relations between properties (and even defining those properties), but I shall do my best to present its content verbally.
The First Law
As far as physical chemists are concerned, the First Law is an elaboration of the law of the conservation of energy, the statement that energy can be neither created nor destroyed. The elaboration is that thermodynamics includes the transfer of energy as heat whereas the dynamics of Newton and his descendants, does not. The central concept of the First Law is the internal energy, U, the total energy of whatever region of the world we are interested in, the ‘system’, but not including its energy due to external causes, such as the motion of the entire system through space. The detailed experiments of James Joule (1818–89) in the middle of the 19th century established that the internal energy of a system could be changed either by doing work or by heating the system. Work, a concept from dynamics, involves moving against an opposing force; heat, the new concept in thermodynamics, is the transfer of energy that makes use of a temperature difference. The failure of often well-meaning but also innumerable charlatan-and greed-driven attempts to create a perpetual motion machine, a machine that can generate work without any input of energy (for instance, as heat) finally led to the conclusion that the internal energy of a system that is isolated from external influences does not change. That is the First Law of thermodynamics.
I need to interject a remark here. A system possesses energy, it does not possess work or heat (even if it is hot). Work and heat are two different modes for the transfer of energy into or out of a system. Work is a mode of transfer that is equivalent to raising a weight; heat is a mode of transfer that results from on a temperature difference (Figure 5). When we say ‘a system is hot’, we don’t mean that it has a lot of heat: it has a high temperature on account of the energy it currently stores.
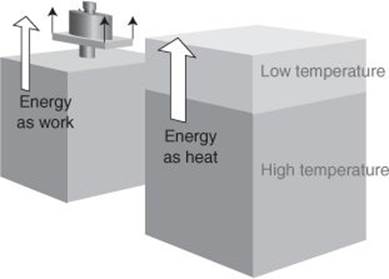
5. Work is a mode of transfer that is equivalent to raising a weight; heat is a mode of transfer that results from a temperature difference. These two modes of transfer decrease or increase the internal energy of a system
For practical reasons, in many instances physical chemists are less concerned with the internal energy than with a closely related property called the enthalpy, H. The name derives from the Greek words for ‘heat inside’; although there is no ‘heat’ as such inside a system; the name is quite sensible, as I shall shortly explain. For completeness, although it is immaterial to this verbal discussion, if you know the internal energy of a system, then you can calculate its enthalpy simply by adding to U the product of pressure and volume of the system (H = U + pV). The significance of the enthalpy—and this is the important point—is that a change in its value is equal to the output of energy as heat that can be obtained from the system provided it is kept at constant pressure. For instance, if the enthalpy of a system falls by 100 joules when it undergoes a certain change (such as a chemical reaction), then we know that 100 joules of energy can be extracted as heat from the system, provided the pressure is constant.
The relation between changes in enthalpy and heat output lies at the heart of the branch of physical chemistry, and more specifically of chemical thermodynamics, known as thermochemistry, the study of the heat transactions that accompany chemical reactions. This aspect of chemistry is of vital importance wherever fuels are deployed, where fuels include not only the gasoline of internal combustion engines but also the foods that power organisms. In the latter application, the role of energy deployment in organisms is studied in bioenergetics, the biochemically specific sub-section of thermochemistry.
The principal instrument used by thermochemists is a calorimeter, essentially a bucket fitted with a thermometer, but which, like so much scientific apparatus, has been refined considerably into an elaborate instrument of high precision subject to computer control and analysis. Broadly speaking, the reaction of interest is allowed to take place in the bucket and the resulting temperature rise is noted. That temperature rise (in rare cases, fall) is then converted to a heat output by comparing it with a known reaction or the effect of electrical heating. If the calorimeter is open to the atmosphere (and therefore at constant pressure), then the heat output is equal to the enthalpy change of the reaction mixture. Even if the reaction vessel is sealed and its contents undergo a change in pressure, chemists have ways of converting the data to constant-pressure conditions, and can deduce the enthalpy change from the observed change in temperature.
Thermochemists, by drawing on the First Law of thermodynamics know, with some relief, that they don’t have to examine every conceivable reaction. It may be that the reaction of interest can be thought of as taking place through a sequence of steps that have already been investigated. According to the First Law, the enthalpy change for the direct route must be the same as the sum of the enthalpy changes along the indirect route (just as two paths up a mountain between the same two points must lead to the same change in altitude), for otherwise you would have a neat way of creating energy: start with a compound, change it; change it back to the original compound by a different path, and extract the difference in energy. Therefore, they can tabulate data that can be used to predict enthalpy changes for any reaction of interest. For instance, if the enthalpy changes for the reactions A → B and B → C are known, then that of the reaction A → C is their sum. (Physical chemists, to the friendly scorn of inorganic and organic chemists, often don’t commit themselves to specific reactions. They argue that it is to retain generality that they use A, B, and C; their suspicious colleagues often wryly suspect that such generality actually conceals ignorance.)
In the old days of physical chemistry (well into the 20th century), the enthalpy changes were commonly estimated by noting which bonds are broken in the reactants and which are formed to make the products, so A → B might be the bond-breaking step and B → C the new bond-formation step, each with enthalpy changes calculated from knowledge of the strengths of the old and new bonds. That procedure, while often a useful rule of thumb, often gave wildly inaccurate results because bonds are sensitive entities with strengths that depend on the identities and locations of the other atoms present in molecules. Computation now plays a central role: it is now routine to be able to calculate the difference in energy between the products and reactants, especially if the molecules are isolated as a gas, and that difference easily converted to a change of enthalpy. The computation is less reliable for reactions that take place as liquids or in solution and progress there is still being made.
Enthalpy changes are very important for a rational discussion of changes in physical state (vaporization and freezing, for instance), and I shall return to them in Chapter 5. They are also essential for applications of the Second Law.
The Second Law
The Second Law of thermodynamics is absolutely central to the application of thermodynamics in chemistry, often appearing in disguised form, but always lurking beneath the surface of the discussion of chemical reactions and their applications in biology and technology.
The First Law circumscribes possible changes (they must conserve the total amount of energy, but allow for it to be shipped from place to place or transferred as heat or work). The Second Law identifies from among those possible changes the changes that can occur spontaneously. The everyday meaning of ‘spontaneously’ is ‘without external agency’; in physical chemistry it means without having to do work to bring the change about. It does not, as the word is sometimes coloured in common discourse, mean fast. Spontaneous changes can be very slow: the name simply indicates that they have a tendency to take place without intervention. The conversion of diamonds to graphite is spontaneous in this sense, but it is so slow that the change can be disregarded in practice. The expansion of a gas into a vacuum is both spontaneous and fast. Its compression into a smaller volume is not spontaneous: we have to do work and push on a piston to bring it about. In short: the First Law identifies possible changes, the Second Law identifies from among those possible changes the ones that are spontaneous.
The Second Law identifies spontaneous changes as those accompanied by an increase in entropy. Entropy, S, is a measure of the quality of energy: low entropy corresponds to high quality in a sense I shall explain; high entropy corresponds to low quality. The name is derived from the Greek words for ‘inward turning’, which gives an impression that it is concerned with changes taking place within a system. The increase in entropy therefore corresponds to the decline of the quality of energy, such as it becoming more widely dispersed and less useful for doing work.
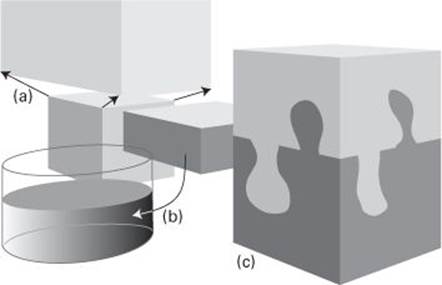
6. Entropy is a measure of ‘disorder’. Thus, it increases (a) when a gas disperses, (b) when a solid melts, and (c) when two substances mingle
Entropy is commonly associated as a measure of ‘disorder’, such as the dispersal of energy or the spreading of a gas (Figure 6). That is a very helpful interpretation to keep in mind. The quantitative expression for calculating the numerical value of the change in entropy was first proposed by the German physicist Rudolph Clausius (1822–88) in 1854: his formula implies that to calculate the entropy change we need to note the energy transferred carefully as heat during the change and divide it by the absolute temperature at which the change occurs. Thus, if 100 joules of heat enters a beaker of water at 20°C (293 K), the entropy of the water increases by 0.34 joules per kelvin. A very different but complementary and hugely insightful approach was developed by Ludwig Boltzmann, and I treat it in Chapter 3.
Physical chemists have used Clausius’s formula to compile tables of entropies of a wide range of substances (this is where they also have to use the Third Law, as I shall explain shortly), and can use them to assess the entropy change when reactants with one entropy change into products with a different entropy. This is essential information for judging whether a reaction is spontaneous at any given temperature (not necessarily fast, remember; just spontaneous). Here is another example of the utmost importance of the reliance of chemists on the achievements of physicists, in this case Clausius, for understanding why one reaction ‘goes’ but another doesn’t is central to chemistry and lies in the domain of physical chemistry, the interface of physics and chemistry.
Free energy
There is a catch. In all presentation of chemical thermodynamics it is emphasized that to use the Second Law it is necessary to consider the total entropy change, the sum of the changes within the system (the reaction mixture) and its surroundings. The former can be deduced from the tables of entropies that have been compiled, but how is the latter calculated?
This is where enthalpy comes back to play a role. If we know the enthalpy change taking place during a reaction, then provided the process takes place at constant pressure we know how much energy is released as heat into the surroundings. If we divide that heat transfer by the temperature, then we get the associated entropy change in the surroundings. Thus, if the enthalpy of a system falls by 100 joules during a reaction taking place at 25°C (298 K), that 100 joules leaves the system as heat and enters the surroundings and consequently their entropy increases by 0.34 joules per kelvin. All we have to do is to add the two entropy changes, that of the system and that of the surroundings, together, and identify whether the total change is positive (the reaction is spontaneous) or negative (the reaction is not spontaneous).
Physical chemists have found a smart way to do this fiddly part of the calculation, the assessment of the entropy change of the surroundings. Well, to be truthful, it was another theoretical physicist, the American Josiah Gibbs (1839–1903), who in the 1870s brought thermodynamics to the stage where it could be regarded as an aspect of physical chemistry and indeed effectively founded this branch of the subject. Once again, we see that physics provides the springboard for developments in physical chemistry.
Gibbs noted that the two changes in entropy, that of the system and that of the surroundings, could, provided the pressure and temperature are constant, be calculated by dealing solely in changes of what we now call the Gibbs energy, G, of the system alone and apparently ignoring the surroundings entirely. Remember how the enthalpy is calculated by adding pV to the internal energy (H = U + pV); similarly the Gibbs energy of a system is calculated simply by subtracting the product of the temperature and entropy from the enthalpy: G = H – TS. For instance, the entropy of 100 mL of water at 20°C (293 K) is 0.388 kilojoules per kelvin, so its Gibbs energy is about 114 kilojoules less than whatever its enthalpy happens to be.
Actual numbers, though, are not particularly interesting for our purposes: much more important is the presence of the negative sign in the definition G = H – TS. When this expression is analysed it turns out that, provided the pressure and temperature are constant, a spontaneous change corresponds to a decrease in Gibbs energy. This makes the Gibbs energy of the system alone a signpost of spontaneous change: all we need do is to compile tables of Gibbs energies for tables of enthalpies and entropies, work out how G changes in a reaction, and note whether the change is negative. If it is negative, then the reaction is spontaneous (Figure 7).
I need to make another interjection. Although it might seem to be more natural to think of a process as being natural if it corresponds to ‘down’ in some property, in this case the Gibbs energy, it must be remembered that the Gibbs energy is a disguised version of the total entropy. The spontaneous direction of change is invariably ‘up’ in total entropy. Gibbs showed chemists that, at the price of losing generality and dealing only with changes at constant temperature and pressure, they could deal with a property of the system alone, the Gibbs energy, and simply by virtue of the way it is defined with that negative sign, ‘down’ in Gibbs energy is actually ‘up’ in total entropy.
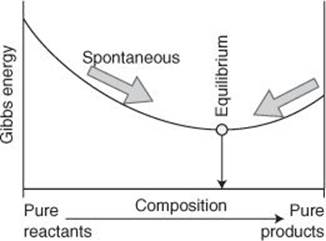
7. A reaction is spontaneous in the direction of decreasing Gibbs energy. When the Gibbs energy is a minimum, the reaction is at equilibrium with no tendency to change in either direction
There is a very important aspect of spontaneity that I now need to introduce. Chemists are very interested in the composition at which a chemical reaction has no further tendency to change: this is the state of chemical equilibrium. Neither the forward reaction nor its reverse is spontaneous when the mixture has reached equilibrium. In other words, at the composition corresponding to equilibrium, no change in composition, either the formation of more product or their decomposition, corresponds to a decrease in Gibbs energy: the Gibbs energy has reached a minimum, with a change in either direction corresponding to an increase in G. All a physical chemist needs to do to predict the composition at equilibrium is to identify the composition at which the Gibbs energy has reached a minimum. There are straightforward ways of doing that calculation, and the equilibrium composition of virtually any reaction at any temperature can be calculated provided the Gibbs energy data are available.
The ability to predict the equilibrium composition of a reaction and how it depends on the conditions is of immense importance in industry, for it is pointless to build a chemical factory if the yield of product is negligible, and questions of economy draw on finding the conditions of temperature and pressure that are likely to optimize the yield.
Finally, I need to explain the term ‘free energy’ that I have used as the title of this section and thereby open another important door on the biological and technological applications of the Second Law.
There are two kinds of work. One kind is the work of expansion that occurs when a reaction generates a gas and pushes back the atmosphere (perhaps by pressing out a piston). That type of work is called ‘expansion work’. However, a chemical reaction might do work other than by pushing out a piston or pushing back the atmosphere. For instance, it might do work by driving electrons through an electric circuit connected to a motor. This type of work is called ‘non-expansion work’. Now for the crucial point: a change in the Gibbs energy of a system at constant temperature and pressure is equal to the maximum non-expansion work that can be done by the reaction. Thus, a reaction for which the Gibbs energy decreases by 100 joules can do up to 100 joules of non-expansion work. That non-expansion work might be the electrical work of driving electrons through an external circuit. Thus, we arrive at a crucial link between thermodynamics and electrochemistry, which includes the generation of electricity by chemical reactions and specifically the operation of electric batteries and fuel cells (see Chapter 5).
There is another crucial link that this connection reveals: the link of thermodynamics with biology is that one chemical reaction might do the non-expansion work of building a protein from amino acids. Thus, a knowledge of the Gibbs energies changes accompanying metabolic processes is very important in bioenergetics, and much more important than knowing the enthalpy changes alone (which merely indicate a reaction’s ability to keep us warm). This connection is a major contribution of physical chemistry to biochemistry and biology in general.
The Third Law
I have cheated a little, but only in order to make fast progress with the very important Second Law. What I glossed over was the measurement of entropy. I mentioned that changes in entropy are measured by monitoring the heat supplied to a sample and noting the temperature. But that gives only the value of a change in entropy. What do we take for the initial value?
The Third Law of thermodynamics plays a role. Like the other two laws, there are ways of expressing the Third Law either in terms of direct observations or in terms of the thermodynamic properties I have mentioned, such as the internal energy or the entropy. One statement of the Third Law is of the first kind: it asserts that the absolute zero of temperature cannot be reached in a finite number of steps. That version is very important in the field of cryogenics, the attainment of very low temperatures, but has little directly to do with physical chemistry. The version that does have direct implications for physical chemistry is logically equivalent (it can be shown), but apparently quite different: the entropies of all perfect crystals are the same at the absolute zero of temperature. For convenience (and for reasons developed in Chapter 3) that common value is taken as zero.
Now we have a starting point for the measurement of absolute (or ‘Third-Law’) entropies. We lower the temperature of a substance to as close as possible to absolute zero, and measure the heat supplied at that temperature. We raise the temperature a little, and do the same. That sequence is continued until we reach the temperature at which we want to report the entropy, which is simply the sum of all those ‘heat divided by temperature’ quantities. This procedure still gives the change in entropy between zero temperature and the temperature of interest, but the former entropy is zero, by the Third Law, so the change gives us the absolute value of the entropy too.
Thus, the Third Law plays a role in physical chemistry by allowing us to compile tables of absolute entropies and to use those values to calculate Gibbs energies. It is a crucial part of the fabric of chemical thermodynamics, but is more of a technical detail than a contribution to insight. Or is that true? Can it be that the Third Law does provide insight? We shall see in Chapter 3 how it may be interpreted.
Relations between properties
I remarked at the beginning of this chapter that chemical thermodynamics provides relations, sometimes unexpected, between the properties of substances, those properties often being apparently unrelated to considerations of the transformations of energy. Thus, measurements of several properties might be stitched together using the guidance of thermodynamics to arrive at the value of a property that might be difficult to determine directly.
This section is difficult for me to generate not only because I want to avoid mathematics but also because you would be unlikely to find many of the relations in the least interesting and be unmoved by knowing that there are clever ways of measuring seemingly recondite properties! The best I can come up with to illustrate the kind of relation involved is that between different types of a moderately familiar property, heat capacity (commonly referred to as ‘specific heats’).
Let me establish the problem. A heat capacity, C, is the ratio of the heat supplied to a substance to the temperature rise produced. For instance, if 100 joules of energy is supplied as heat to 100 mL of water, its temperature rises by 0.24°C, so its heat capacity is 420 joules per degree. But when physical chemists wear their pernickety accountant’s hat, they stop to think. Did the water expand? If it did, then not all the energy supplied as heat remained in the sample because some was used to push back the atmosphere. On the other hand, suppose the water filled a rigid, sealed vessel and the same quantity of energy as heat was supplied to it. Now no expansion occurs, no work was done, and all that energy remains inside the water. In this case its temperature rises further than in the first case so the heat capacity is smaller (the heat supplied is the same but the temperature rise is greater, so their ratio is smaller). In other words, when doing precise calculations (and thermodynamics is all about circumspection and precision), we have to decide whether we are dealing with the heat capacity at constant pressure (when expansion is allowed) or at constant volume (when expansion is not allowed). The two heat capacities, which are denoted Cp and CV, respectively, are different.
What thermodynamics, and specifically a combination of the First and Second Laws, enables us to do, is to establish the relation between Cp and CV. I shall not give the final relation, which involves the compressibility (how its volume changes with pressure) and its expansivity (how its volume changes with temperature), but simply want to mention that by using these properties, which can be measure in separate experiments, Cp and CV can be related. There is one special case of the relation, however, that is worth presenting. For a perfect gas (a concept I develop in Chapter 4, but essentially an idealized gas in which all interactions between the molecules can be ignored, so they are flying around freely), the relation between the two heat capacities is Cp – CV = Nk, where N is the total number of molecules in the sample and k is the fundamental constant known as Boltzmann’s constant (once again, see Chapter 3). Because Nk is positive, we can infer that Cp is greater than CV, just as our discussion suggested should be the case.
This example might seem rather trivial and not particularly interesting (as I warned). However, it does reveal that physical chemists can deploy the laws of thermodynamics, laws relating to matter from the outside, to establish relations between properties and to make important connections.
The current challenge
Thermodynamics was developed for bulk matter. Attention now is shifting towards special forms of matter where ‘bulk’ might not be applicable: to very tiny nanosystems and to biological systems with their special functions and structures. One branch of thermodynamics that has made little progress since its formulation is ‘irreversible thermodynamics’, where the focus is on the rate at which energy is dissipated and entropy is generatedby systems that are not at equilibrium: a Fifth Law might be lurking currently undiscovered there.