THE LIVING WORLD
Unit Four. The Evolution and Diversity of Life
14. Evolution and Natural Selection
14.10. Sickle-Cell Anemia
Sickle-cell disease is a hereditary disease affecting hemoglobin molecules in the blood. It was first detected in 1904 in Chicago in a blood examination of an individual complaining of tiredness. You can see the original doctor’s report in figure 14.25. The disorder arises as a result of a single nucleotide change in the gene encoding b-hemoglobin, one of the key proteins used by red blood cells to transport oxygen. The sickle-cell mutation changes the sixth amino acid in the b-hemoglobin chain (position B6) from glutamic acid (very polar) to valine (nonpolar). The unhappy result of this change is that the nonpolar valine at position B6, protruding from a corner of the hemoglobin molecule, fits nicely into a nonpolar pocket on the opposite side of another hemoglobin molecule; the nonpolar regions associate with each other. As the two-molecule unit that forms still has both a B6 valine and an opposite nonpolar pocket, other hemoglobins clump on, and long chains form as in figure 14.26a. The result is the deformed “sickle-shaped” red blood cell you see in figure 14.26b. In normal everyday hemoglobin, by contrast, the polar amino acid glutamic acid occurs at position B6. This polar amino acid is not attracted to the nonpolar pocket, so no hemoglobin clumping occurs, and cells are normal shaped as in figure 14.26c.
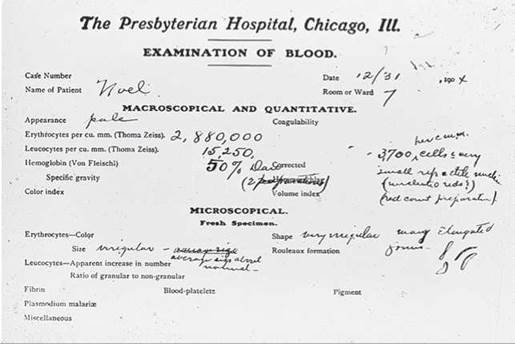
Figure 14.25. The first known sickle-cell disease patient.
Dr. Ernest Irons's blood examination report on his patient Walter Clement Noel, December 31, 1904, described his oddly shaped red blood cells.
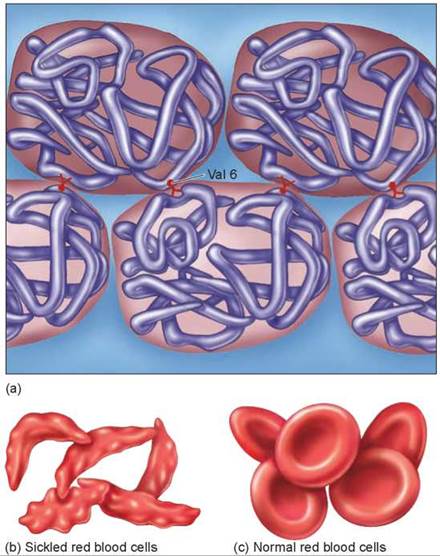
Figure 14.26. Why the sickle-cell mutation causes hemoglobin to clump.
Persons homozygous for the sickle-cell genetic mutation in the b-hemoglobin gene frequently have a reduced life span. This is because the sickled form of hemoglobin does not carry oxygen atoms well, and red blood cells that are sickled do not flow smoothly through the tiny capillaries but instead jam up and block blood flow. Heterozygous individuals, who have both a defective and a normal form of the gene, make enough functional hemoglobin to keep their red blood cells healthy.
The Puzzle: Why So Common?
The disorder is now known to have originated in Central Africa, where the frequency of the sickle-cell allele is about 0.12. One in 100 people is homozygous for the defective allele and develops the fatal disorder. Sickle-cell disease affects roughly two African Americans out of every thousand but is almost unknown among other racial groups.
If Darwin is right, and natural selection drives evolution, then why has natural selection not acted against the defective allele in Africa and eliminated it from the human population there? Why is this potentially fatal allele instead very common there?
The Answer: Stabilizing Selection
The defective allele has not been eliminated from Central Africa because people who are heterozygous for the sickle-cell allele are much less susceptible to malaria, one of the leading causes of death in Central Africa. Examine the maps in figure 14.27, and you will see the relationship between sickle-cell disease and malaria clearly. The map on the left shows the frequency of the sickle-cell allele, the darker green areas indicating a 10% to 20% frequency of the allele. The map on the right indicates the distribution of malaria in dark orange. Clearly, the areas that are colored in darker green on the left map overlap many of the dark orange areas in the map on the right. Even though the population pays a high price—the many individuals in each generation who are homozygous for the sickle-cell allele die—the deaths are far fewer than would occur due to malaria if the heterozygous individuals were not malaria resistant. One in 5 individuals (20%) are heterozygous and survive malaria, while only 1 in 100 (1%) are homozygous and die of sickle-cell disease. Similar inheritance patterns of the sickle-cell allele are found in other countries frequently exposed to malaria, such as areas around the Mediterranean, India, and Indonesia. Natural selection has favored the sickle-cell allele in Central Africa and other areas hit by malaria because the payoff in survival of heterozygotes more than makes up for the price in death of homozygotes. This phenomenon is an example of heterozygote advantage.
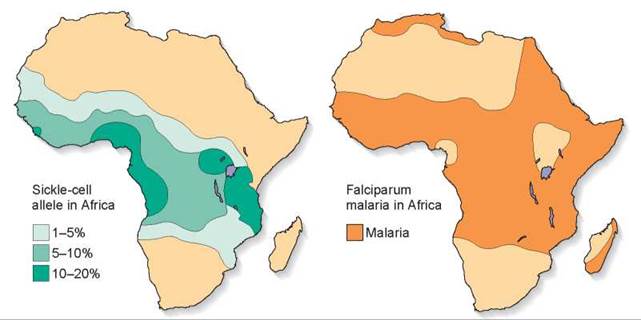
Figure 14.27. How stabilizing selection maintains sickle-cell disease.
The diagrams show the frequency of the sickle-cell allele (left) and the distribution of falciparum malaria (right). Falciparum malaria is one of the most devastating forms of the often fatal disease. As you can see, its distribution in Africa is closely correlated with that of the allele of the sickle-cell characteristic.
Stabilizing selection (also called balancing selection) is thus acting on the sickle-cell allele: (1) Selection tends to eliminate the sickle-cell allele because of its lethal effects on homozygous individuals, and (2) selection tends to favor the sicklecell allele because it protects heterozygotes from malaria. Like a manager balancing a store’s inventory, natural selection increases the frequency of an allele in a species as long as there is something to be gained by it, until the cost balances the benefit.
Stabilizing selection occurs because malarial resistance counterbalances lethal sickle-cell disease. Malaria is a tropical disease that has essentially been eradicated in the United States since the early 1950s, and stabilizing selection has not favored the sickle-cell allele here. Africans brought to America several centuries ago have not gained any evolutionary advantage in all that time from being heterozygous for the sickle-cell allele. There is no benefit to being resistant to malaria if there is no danger of getting malaria anyway. As a result, the selection against the sickle-cell allele in America is not counterbalanced by any advantage, and the allele has become far less common among African Americans than among native Africans in Central Africa.
Stabilizing selection is thought to have influenced many other human genes in a similar fashion. The recessive cf allele causing cystic fibrosis is unusually common in northwestern Europeans. People that are heterozygous for the cf allele are protected from the dehydration caused by cholera, and the cf allele may provide protection against typhoid fever too. Apparently, the bacterium causing typhoid fever uses the healthy version of the CFTR protein (see page 80) to enter the cells it infects, but it cannot use the cystic fibrosis version of the protein. As with sickle-cell disease, heterozygotes are protected.
Key Learning Outcome 14.10. The prevalence of sickle-cell disease in African populations is thought to reflect the action of natural selection. Natural selection favors individuals carrying one copy of the sickle-cell allele, because they are resistant to malaria, common in Africa.