THE LIVING WORLD
Unit Three. The Continuity of Life
10. Foundations of Genetics
10.10. The Role of Mutation
Hemophilia: A Sex-Linked Trait
Blood in a cut clots as a result of the polymerization of protein fibers circulating in the blood. A dozen proteins are involved in this process, and all must function properly for a blood clot to form. A mutation causing any of these proteins to lose their activity leads to a form of hemophilia, a hereditary condition in which the blood clots slowly or not at all.
Hemophilias are recessive disorders, expressed only when an individual does not possess any copy of the normal allele and so cannot produce one of the proteins necessary for clotting. Most of the genes that encode the blood-clotting proteins are on autosomes, but two (designated VIII and IX) are on the X chromosome. These two genes are sex-linked (see section 10.7): Any male who inherits a mutant allele will develop hemophilia because his other sex chromosome is a Y chromosome that lacks any alleles of those genes.

The most famous instance of hemophilia, often called the Royal hemophilia, is a sex-linked form that arose in the royal family of England. This hemophilia was caused by a mutation in gene IX that occurred in one of the parents of Queen Victoria of England (1819-1901). The pedigree in figure 10.30 shows that in the six generations since Queen Victoria, 10 of her male descendants have had hemophilia (the solid squares). The present British royal family has escaped the disorder because Queen Victoria’s son, King Edward VII, did not inherit the defective allele, and all the subsequent rulers of England are his descendants. Three of Victoria’s nine children did receive the defective allele, however, and they carried it by marriage into many of the other royal families of Europe. In this photo, Queen Victoria of England is surrounded by some of her descendants in 1894. Standing behind Victoria and wearing feathered boas are two of Victoria’s granddaughters, Alice’s daughter’s: Princess Irene of Prussia (right), and Alexandra (left), who would soon become Czarina of Russia. Both Irene and Alexandra were also carriers of hemophilia.
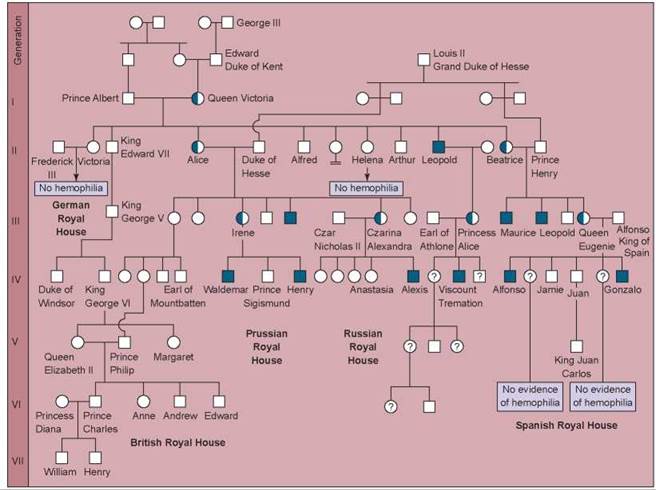
Figure 10.30. The Royal hemophilia pedigree.
Queen Victoria's daughter Alice introduced hemophilia into the Russian and Prussian royal houses, and her daughter Beatrice introduced it into the Spanish royal house. Victoria's son Leopold, himself a victim, also transmitted the disorder in a third line of descent. Half-shaded symbols represent carriers with one normal allele and one defective allele; fully shaded symbols represent affected individuals. Squares represent males; circles represent females.
Sickle-Cell Disease: A Recessive Trait
Sickle-cell disease is a recessive hereditary disorder. Its inheritance is shown in the pedigree in figure 10.31, where affected individuals are homozygous, carrying two copies of the mutated gene. Affected individuals have defective molecules of hemoglobin, the protein within red blood cells that carries oxygen. Consequently, these individuals are unable to properly transport oxygen to their tissues. The defective hemoglobin molecules stick to one another, forming stiff, rodlike structures and resulting in the formation of sickle-shaped red blood cells (figure 10.31). As a result of their stiffness and irregular shape, these cells have difficulty moving through the smallest blood vessels; they tend to accumulate in those vessels and form clots. People who have large proportions of sickle-shaped red blood cells tend to have intermittent illness and a shortened life span.
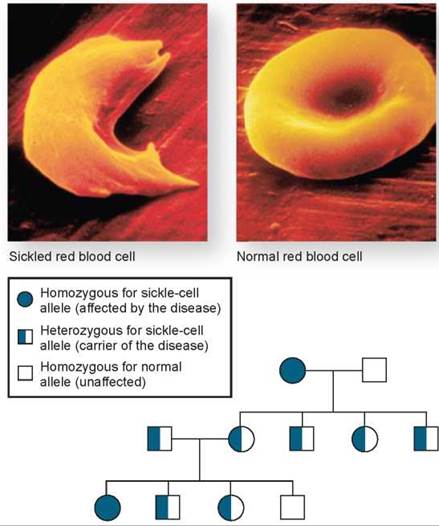
Figure 10.31. Inheritance of sickle-cell disease.
Sickle-cell disease is a recessive autosomal disorder. If one parent is homozygous for the recessive trait, all of the offspring will be carriers (heterozygotes), like the F1 generation of Mendel's testcross. A normal red blood cell is shaped like a flattened sphere. In individuals homozygous for the sickle-cell trait, many of the red blood cells have sickle shapes.
The hemoglobin in the defective red blood cells differs from that in normal red blood cells in only one of hemoglobin’s 574 amino acid subunits. In the defective hemoglobin, the amino acid valine replaces a glutamic acid at a single position in the protein. Interestingly, the position of the change is far from the active site of hemoglobin where the iron-bearing heme group binds oxygen. Instead, the change occurs on the outer edge of the protein. Why then is the result so catastrophic? The sickle-cell mutation puts a very nonpolar amino acid on the surface of the hemoglobin protein, creating a “sticky patch” that sticks to other such patches—nonpolar amino acids tend to associate with one another in polar environments like water. As one hemoglobin adheres to another, chains of hemoglobin molecules form.
Individuals heterozygous for the sickle-cell allele are generally indistinguishable from normal persons. However, some of their red blood cells show the sickling characteristic when they are exposed to low levels of oxygen. The allele responsible for sickle-cell disease is particularly common among people of African descent because the sickle-cell allele is more common in Africa. About 9% of African Americans are heterozygous for this allele, and about 0.2% are homozygous and therefore have the disorder. In some groups of people in Africa, up to 45% of all individuals are heterozygous for this allele, and fully 6% are homozygous and express the disorder. What factors determine the high frequency of sickle-cell disease in Africa? It turns out that heterozygosity for the sickle-cell allele increases resistance to malaria, a common and serious disease in Central Africa. Comparing the two maps shown in figure 10.32, you can see that the area of the sickle-cell trait matches well with the incidence of malaria. The interactions of sickle-cell disease and malaria are discussed further in chapter 14.
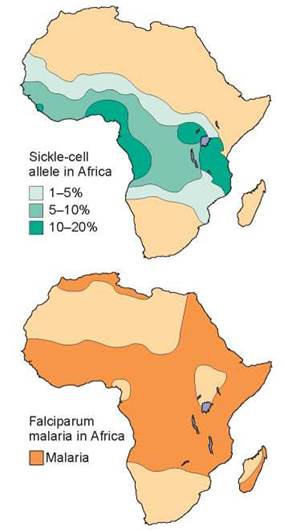
Figure 10.32. The sickle-cell allele confers resistance to malaria.
The distribution of sickle-cell disease closely matches the occurrence of malaria in central Africa. This is not a coincidence. The sicklecell allele, when heterozygous, confers resistance to malaria, a very serious disease.
Tay-Sachs Disease: A Recessive Trait
Tay-Sachs disease is an incurable hereditary disorder in which the brain deteriorates. Affected children appear normal at birth and usually do not develop symptoms until about the eighth month, when signs of mental deterioration appear. The children are blind within a year after birth, and they rarely live past five years of age.
The Tay-Sachs allele produces the disease by encoding a nonfunctional form of the enzyme hexosaminidase A. This enzyme breaks down gangliosides, a class of lipids occurring within the lysosomes of brain cells. As a result, the lysosomes fill with gangliosides, swell, and eventually burst, releasing oxidative enzymes that kill the cells. There is no known cure for this disorder.
Tay-Sachs disease is rare in most human populations, occurring in only 1 in 300,000 births in the United States. However, the disease has a high incidence among Jews of Eastern and Central Europe (Ashkenazi) and among American Jews, 90% of whom trace their ancestry to Eastern and Central Europe. In these populations, it is estimated that 1 in 28 individuals is a heterozygous carrier of the disease, and approximately 1 in 3,500 infants has the disease. Because the disease is caused by a recessive allele, most of the people who carry the defective allele do not themselves develop symptoms of the disease because, as shown by the middle bar in figure 10.33, their one normal gene produces enough enzyme activity (50%) to keep the body functioning normally.
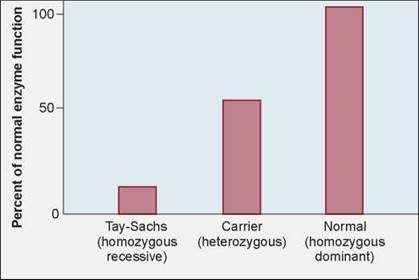
Figure 10.33. Tay-Sachs disease.
Homozygous individuals (left bar) typically have less than 10% of the normal level of hexosaminidase A (right bar), while heterozygous individuals (middle bar) have about 50% of the normal level— enough to prevent deterioration of the central nervous system.
Huntington's Disease: A Dominant Trait
Not all hereditary disorders are recessive. Huntington’s disease is a hereditary condition caused by a dominant allele that causes the progressive deterioration of brain cells. Perhaps 1 in 24,000 individuals develops the disorder. Because the allele is dominant, every individual who carries the allele expresses the disorder. Nevertheless, the disorder persists in human populations because its symptoms usually do not develop until the affected individuals are more than 30 years old, and by that time most of those individuals have already had children. Consequently, as illustrated by the pedigree in figure 10.34, the allele is often transmitted before the lethal condition develops.
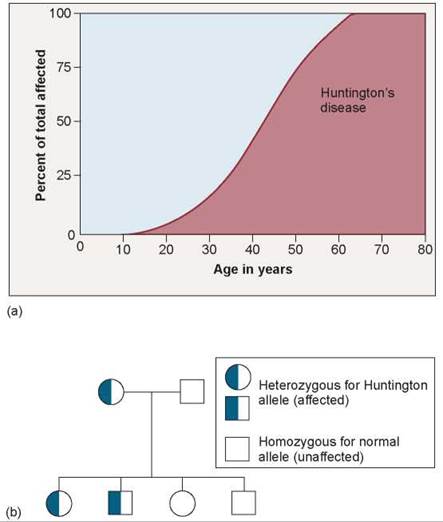
Figure 10.34. Huntington's disease is a dominant genetic disorder.
(a) Because of the late age of onset of Huntington's disease, the allele causing it persists despite being both dominant and fatal. (b) The pedigree illustrates how a dominant lethal allele can be passed from one generation to the next. Although the mother was affected, we can tell that she was heterozygous because if she were homozygous dominant, all of her children would have been affected. However, by the time she found out that she had the disease, she had probably already given birth to her children. In this way the trait passes on to the next generation even though it is fatal.
Key Learning Outcome 10.10. Many human hereditary disorders reflect the presence of rare (and sometimes not so rare) mutations within human populations.