Medical Microbiology
Section 2 The adversaries–host defences
10 Adaptive responses provide a ‘quantum leap’ in effective defence
Introduction
Infectious agents frequently find ways around the innate defences
In Chapter 9, we discussed the many ways in which the primary or innate defences of the body may counteract microbial infection. However, infectious agents frequently find ways around these defences, as there is a huge number of different microorganisms surrounding us and they have a powerful ability to mutate, e.g.:
• The surface of some microbes fails to activate the alternative complement pathway.
• Other microbes can activate the alternative complement pathway, but do so at the end of flagella, so that the membrane attack complex builds up at a site distant from the body of the organism and therefore causes no damage.
• In other cases, microorganisms taken into the body of the macrophage develop subterfuges that prevent the development of the awesome battery of microbicidal mechanisms that the macrophage normally expresses (see Ch. 16).
• Cells infected with certain viruses may prove to be resistant to the cytotoxic action of natural killer cells, or the viruses may be only weak stimulators of interferon, so that cell-to-cell transmission of the virus proceeds unchecked.
• Yet another microbial subterfuge is the production of bacterial toxins that can kill the phagocyte if not neutralized.
Adaptive responses act against microorganisms that overcome the innate defences
It is clear that the body needs to provide immune defences that can be ‘tailor-made’ to each individual variant of the different species of microorganisms. Ideally, these should link the organism directly into the various killing mechanisms of the innate system. In this chapter, we shall see how evolution has achieved this by inserting specific recognition sites on antibody molecules and on T cells. When an infectious agent enters the body, the lymphocytes respond to it and produce a reaction that is specific for that particular microorganism. Furthermore, the magnitude of this response increases with time, often to quite high levels, so that we speak of it as an ‘adaptive’ or ‘acquired’ response. We know that the body produces millions of different antibodies, which as a population are capable of recognizing virtually any pathogen that has arisen or might arise.
The role of antibodies
The acute inflammatory response
Antibodies act as adaptors to focus acute inflammatory reactions
Antibodies are immunoglobulin molecules (Fig. 10.1, Table 10.1) which are synthesized by host B lymphocytes (so-called because they mature in the bone marrow; see Fig. 11.2) when they make contact with an infectious microbe, which acts as a foreign antigen (i.e. it generatesantibodies). Each antibody has two identical recognition sites that are complementary in shape to the surface of the foreign antigen and which enable it to bind with varying degrees of strength to that antigen. The recognition site is hypervariable in that antibodies of different antigen specificities each have a unique amino acid sequence in this region. This hypervariability is confined to three loops on the heavy and three on the light peptide chains, which make up the antibody molecule (Fig. 10.1) and are referred to as complementarity determining regions (CDRs) because they make complementary contact with the antigen. Thus, the amino acid sequences of these CDRs determine which antigen is recognized by a given antibody. Other sites on the antibody molecule are specialized for functions such as activating the complement system and inducing phagocytosis by macrophages and polymorphs (Fig. 10.2). Therefore, when a microbial antigen is coated with several of these adaptor antibody molecules, they induce complement fixation and phagocytosis, processes that the microbe may well have evolved to try and avoid. In this way, the reluctant microorganism becomes drawn into the innate defence mechanism of the acute inflammatory response. We will now examine the ways in which antibody can mediate these different phenomena.
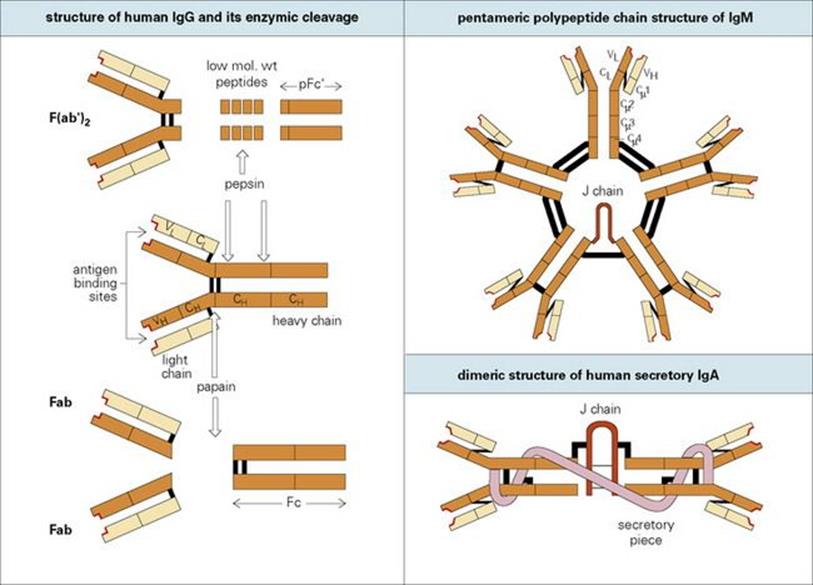
Figure 10.1 The structure of immunoglobulins. The basic structure of immunoglobulins is a unit consisting of two identical light polypeptide chains and two identical heavy polypeptide chains linked together by disulfide bonds (black bars). Each chain is made up of individual globular domains. Different antibodies have different VL and VH domains, the highly variable regions of the light and heavy chains, respectively. This hypervariability is confined to three loops on the VL and three on the VH domains. These make up the antigen-binding site (highlighted in red). In contrast, the remaining domains (CL, CH1, etc.) are relatively constant in amino acid structure. Cleavage of human immunoglobulin G (IgG) by pepsin induces a divalent antigen-binding fragment, F(ab′)2 and a pFc′ fragment composed of two terminal CH3 domains. Papain produces two univalent antigen binding fragments, Fab, and an Fc portion containing the CH2 and CH3 heavy chain domains. Polymerization of the basic immunoglobulin units to form IgM and IgA is catalysed by the J (joining) chain. The portion of the transporter (which transfers IgA across the mucosal cell to the lumen) which remains attached to the IgA is termed ‘secretory piece’.
Table 10.1 Biologic properties of major immunoglobulin (Ig) classes in the human
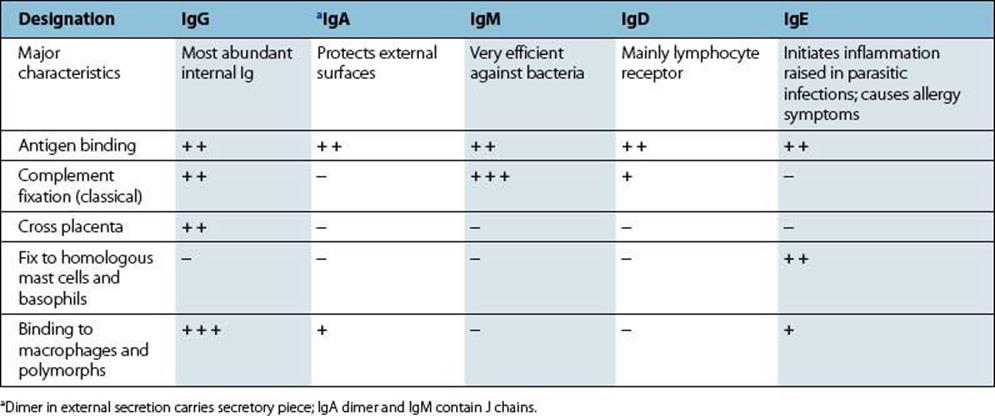
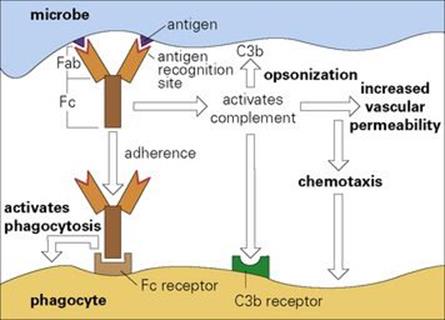
Figure 10.2 The antibody adaptor molecule. Antibodies (anti-foreign bodies) are produced by host lymphocytes on contact with invading microbes, which act as antigens (i.e. generate antibodies). Each antibody (see Fig. 10.1) has a recognition site (Fab) enabling it to bind antigen, and a backbone structure (Fc) capable of some secondary biologic action such as activating complement and phagocytosis. Thus, in the present case, antibody bound to the microbe activates complement and initiates an acute inflammatory reaction (cf. Fig. 9.14). The C3b generated fixes to the microbe and, together with the antibody molecules, facilitates adherence to Fc and C3b receptors on the phagocyte and thence microbial ingestion.
Antibody complexed with antigen activates complement through the ‘classical’ pathway
When antibody molecules bind an antigen, the resulting complex activates the first component of complement, C1, converting it into an esterase (![]() ). This initiates a second route of complement activation (Fig. 10.3) termed the ‘classical’ pathway, mainly because scientists discovered it before the ‘alternative’ pathway (see Ch. 9), although the evidence indicates that the alternative pathway is of greater antiquity in evolutionary terms. The activated first component splits off a small peptide from each of the succeeding components C4 and C2, the residual fragments forming a composite, the
). This initiates a second route of complement activation (Fig. 10.3) termed the ‘classical’ pathway, mainly because scientists discovered it before the ‘alternative’ pathway (see Ch. 9), although the evidence indicates that the alternative pathway is of greater antiquity in evolutionary terms. The activated first component splits off a small peptide from each of the succeeding components C4 and C2, the residual fragments forming a composite, the ![]() , complex. The
, complex. The ![]() complex has the enzymatic ability or property of a C3 convertase. Being an enzyme, the
complex has the enzymatic ability or property of a C3 convertase. Being an enzyme, the ![]() protease creates large numbers of the
protease creates large numbers of the ![]() convertase which itself also having proteolytic activity, cleaves many C3 molecules, this so-called enzyme cascade providing a mechanism for the striking amplification of the relatively few initial complement activation events.
convertase which itself also having proteolytic activity, cleaves many C3 molecules, this so-called enzyme cascade providing a mechanism for the striking amplification of the relatively few initial complement activation events. ![]() has a similar function to the alternative pathway C3 convertase,
has a similar function to the alternative pathway C3 convertase, ![]() , and the sequence of events following the splitting of C3 which generates an acute inflammatory response is indistinguishable from that occurring in the alternative pathway. C3a and C5a anaphylatoxins are formed, and C3b binds to the surface of the microbe–antibody complex (Fig. 10.4; compare Fig. 9.14). Subsequently, the later components are assembled into a membrane attack complex (MAC) (see Fig. 9.18), which may help to kill the microorganism if it has been focused onto a vulnerable site.
, and the sequence of events following the splitting of C3 which generates an acute inflammatory response is indistinguishable from that occurring in the alternative pathway. C3a and C5a anaphylatoxins are formed, and C3b binds to the surface of the microbe–antibody complex (Fig. 10.4; compare Fig. 9.14). Subsequently, the later components are assembled into a membrane attack complex (MAC) (see Fig. 9.18), which may help to kill the microorganism if it has been focused onto a vulnerable site.
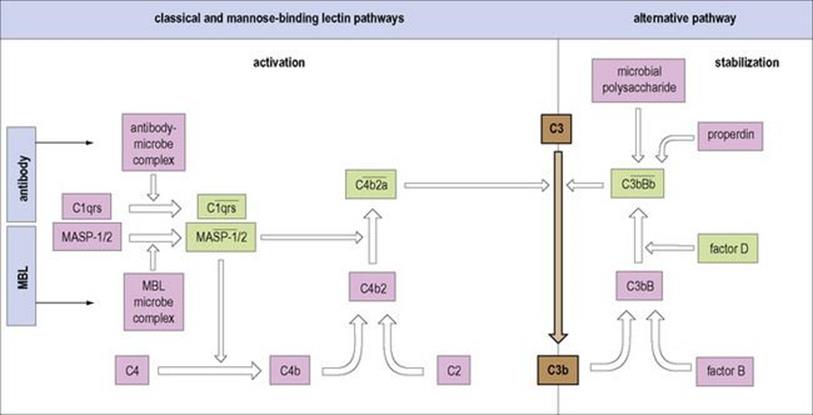
Figure 10.3 Comparison of the alternative classical and mannose binding lectin complement pathways. All converge with the formation of C3 convertase enzymes (in heavily outlined boxes) which split the dominant protein C3 into the C3b fragment, an event which is at the heart of the complement interactions. The complex of antibody with microbial antigen activates the first component of the ‘classical’ pathway (step 1) leading to cleavage of C3 through the ![]() , C3 convertase. Mannose binding lectin, when combined with microbial surface carbohydrate, associates with serine proteases MASP-1 and -2 which split C4 and C2, just like
, C3 convertase. Mannose binding lectin, when combined with microbial surface carbohydrate, associates with serine proteases MASP-1 and -2 which split C4 and C2, just like ![]() . In contrast, the activation of the ‘alternative’ pathway depends upon stabilization of the C3 convertase (
. In contrast, the activation of the ‘alternative’ pathway depends upon stabilization of the C3 convertase (![]() ) on the microbial surface produced by the feedback loop (cf. Fig. 9.13). The molecular units with protease activity are highlighted in green, the enzymatic domains showing considerable homology. Note that the acute phase protein, C-reactive protein, on binding to microbial phosphorylcholine, can trigger the classical pathway. An upper bar (—) indicates an active complex.
) on the microbial surface produced by the feedback loop (cf. Fig. 9.13). The molecular units with protease activity are highlighted in green, the enzymatic domains showing considerable homology. Note that the acute phase protein, C-reactive protein, on binding to microbial phosphorylcholine, can trigger the classical pathway. An upper bar (—) indicates an active complex.
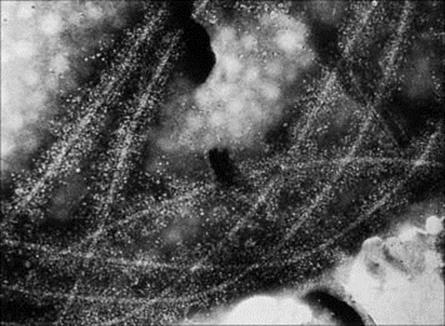
Figure 10.4 Electron microscopy of C3-coated salmonella flagella. The flagella have been incubated with anti-flagellum antibody and complement. The electron-dense material extending 30 nm on either side of each flagellum is believed to be C3b. The interpretation of this is that complement fixation by antibody results in a heavy macromolecular coating of C3b on biologic membranes to which complement has been fixed (× 700 000).
(Courtesy of A. Feinstein and E. Munn.)
It is appropriate at this stage to recall the activation of complement by innate immune mechanisms involving the binding of mannose binding lectin (MBL) and C-reactive protein to carbohydrates on microbial surfaces (cf. Fig. 9.20), and it is noteworthy that both acute phase proteins activate the classical pathway albeit through different routes (Fig. 10.3).
The acute inflammatory reaction can also be initiated by antibody bound to mast cells
A specialized antibody, immunoglobulin E (IgE), has a backbone site with a high affinity for specific receptors on the surface of mast cells. When microbial antigen attaches to these cell-bound antibodies the surface receptors are cross-linked and transduce a signal to the interior of the cell. This signal leads to the release of mediators capable of increasing vascular permeability and inducing polymorph chemotaxis (Fig. 10.5).
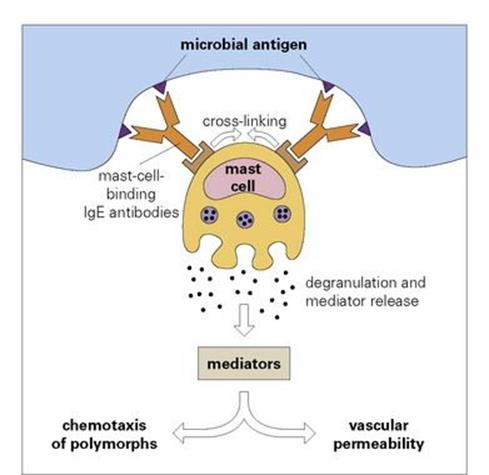
Figure 10.5 Degranulation of mast cells by interaction of microbial antigen with specific antibodies of the IgE class, which bind to special receptors on the mast cell surface. The cross-linking of receptors caused by this interaction leads to the release of mediators, which induce an increase in vascular permeability and attract polymorphs, i.e. they provoke an acute inflammatory reaction at the site of the microbial antigen.
Activation of phagocytic cells
Antigen–antibody complexes activate phagocytic cells
Other sites on the Fc backbone of certain types of antibody molecule bind to specialized Fc receptors on the surface of phagocytic cells. If there is more than one antibody in the antigen–antibody complex, these receptors are cross-linked, so inducing the cell to put out arms of cytoplasm, which enclose the complex in a phagocytic vacuole (Fig. 10.6). Note also that there is a ‘bonus effect’ of multivalent binding of reversible ligand-receptor links; for example, the association constant for a complex binding through two antibody molecules to the phagocyte is the product rather than the sum of the individual association constants.
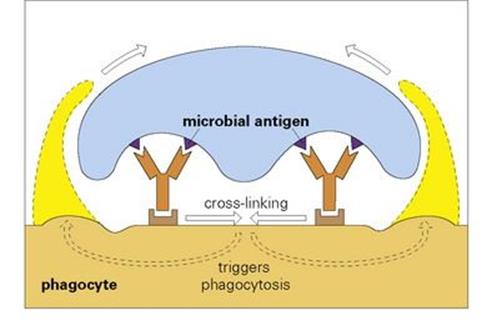
Figure 10.6 The binding of a microbe to a phagocyte by more than one antibody cross-links the antibody receptors on the phagocyte surface and triggers phagocytosis of the microorganism, which is engulfed by the extending cytoplasmic projections.
Blocking microbial reactions
Antibodies block microbial interactions by combining with one of the reacting molecules
For example, an antibody directed against the influenza haemagglutinin will prevent the virus from attaching to its specific receptor on a cell, making it unable to infect that cell (Fig. 10.7). Likewise, antibodies to an essential transport molecule on a bacterial surface can prevent the uptake of that nutrient and cause a metabolic block. As a final example, an antibody to a bacterial toxin will prevent damage to the cells with which the toxin would otherwise interact.
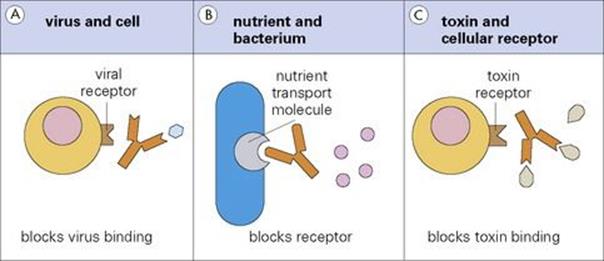
Figure 10.7 Because of its size, antibody can block interactions between (A) a virus and a cell, (B) a nutrient and a bacterium and (C) a toxin and a cellular receptor.
The role of T lymphocytes
Defence against intracellular organisms
The body has evolved an adaptive immune defence system based upon the T lymphocyte, so-called because it matures in the thymus gland. There are several specialized subsets of T cells (Table 10.2) but here we are concerned with those which provide a defence against viruses and many different species of microorganisms which can live within cells, where they are shielded from attack by antibody.
Table 10.2 Subpopulations of T cells
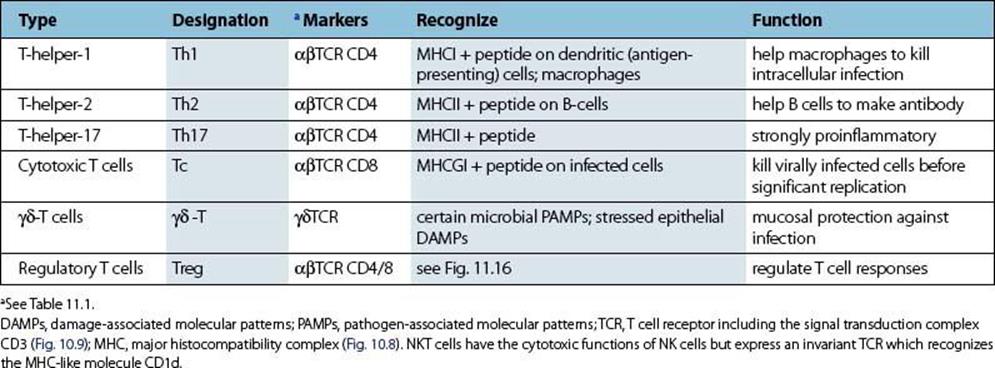
Most T lymphocytes bind to peptide derived from intracellular organisms complexed with major histocompatibility complex
As microorganisms go through their various life cycles they sometimes die within the cells they infect. The proteins derived from these dead organisms and also newly synthesized viral proteins are fragmented by intracellular cytosolic enzymes (‘processing’) and the peptides are incorporated into cytoplasmic vacuoles where they bind to a molecule of the major histocompatibility complex (MHC) (Fig. 10.8). (MHC molecules were originally discovered because of their ability to bring about the most violent rejection of grafts interchanged between members of the same species.) We now know that one of their important functions is to act as cellular surface markers. Class I MHC molecules are present on virtually every cell in the body and can therefore be used as a marker indicating an instance of ‘cell’. Class II MHC molecules appear mainly on macrophages and B cells.
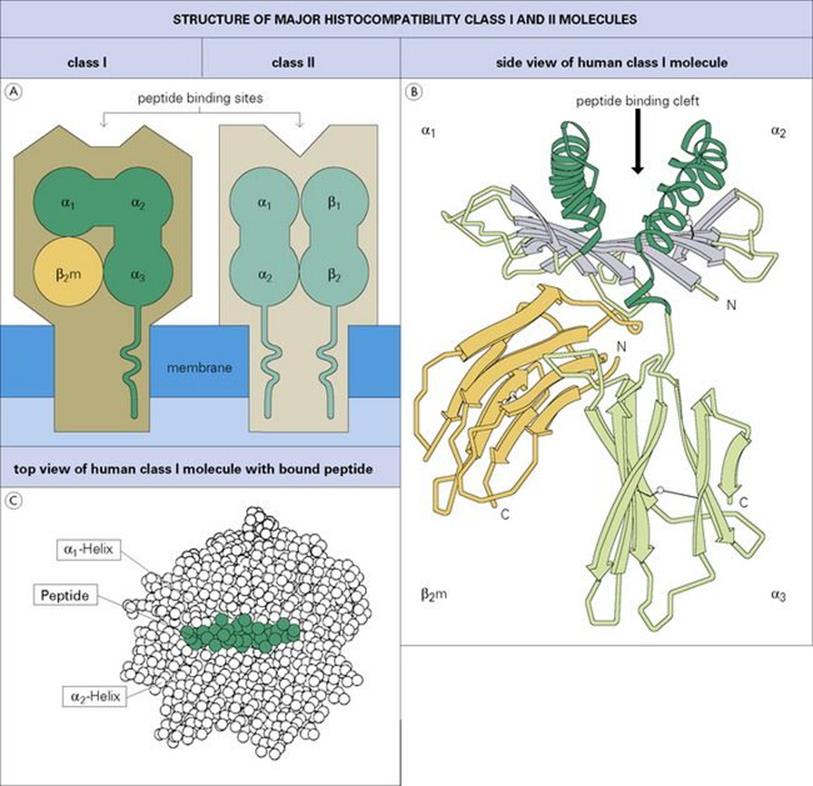
Figure 10.8 Class I and class II major histocompatibility complex molecules. (A) Diagram showing domains and transmembrane segments; the α-helices and β-pleated sheets are viewed end-on. (B) Side view of human class I molecule (HLA-A2) based on X-ray crystallographic structure showing the cleft and the typical immunoglobulin folding of the α3 and β2-microglobulin (β2m) domains (four antiparallel β-strands on one face and three on the other). The strands making the β-pleated sheet are shown as thick gray arrows in the amino to carboxyl direction, α-helices are represented as helical ribbons. The inside facing surfaces of the two helices and the upper surface of the β-pleated sheet form a cleft which binds the peptide. (Adapted from: Bjorkman, P. I. et al. (1987) Nature; 329:512, with permission.) (C) Top view of a peptide bound tightly within the MHC class I cleft, in this case peptide 309–317 from HIV-1 reverse transcriptase bound to HLA-A2. This is the ‘view’ seen by the combining site of the T-cell receptor described below.
(Based on Vignali, D. A. A. and Strominger, J. L. (1994) The Immunologist; 2:112, with permission.)
A specialized T-cell receptor (TCR) on the T-lymphocyte surface (Fig. 10.9) is analogous to an antibody molecule in its ability to recognize foreign antigen. It resembles the immunoglobulin Fab portion in structure, with α and β chains instead of heavy and light chains, again with hypervariable loops to contact the antigen. However, unlike the antibody recognition site which interacts directly with a foreign antigen, the T-cell surface receptor is specialized for binding to the complex of MHC molecule and peptide derived from the processed intracellular organism. Thus, not only is the MHC a molecular signal for ‘cell’, but the foreign peptide is a signal that the cell has an intracellular microbe. Therefore, when the TCR recognizes these two moieties together, the T lymphocyte must be binding to an infected cell of a type indicated by the class of the MHC (see Table 10.2). The T lymphocyte then becomes activated and, depending upon its particular characteristics, sets off an effector mechanism to deal with the intracellular microorganisms, as explained below.
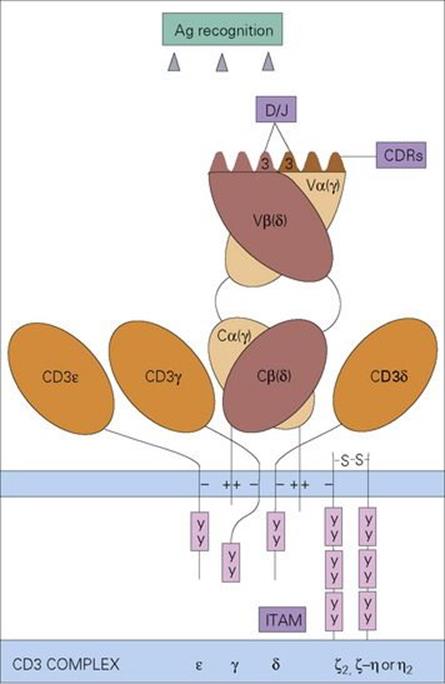
Figure 10.9 The T-cell receptor on αβ-T cells consists of an α and a β chain each composed of a variable (V) and a constant (C) domain resembling the immunoglobulin Fab antigen-binding fragment in structure. The highly variable (complementarity determining) regions (CDRs) on the variable domains contact the MHC-peptide antigen complex. This produces a signal which is transduced by the invariant CD3 complex composed of γ, δ, ɛ and ζ or η chains, through their cytoplasmic immune receptor tyrosine-based activation motifs (ITAM) which contact protein tyrosine kinases. γδ T cells (see below) have receptors composed of γ and δ chains as indicated in the figure.
T lymphocytes help macrophages kill intracellular parasites
The task of recognizing macrophages that have unwelcome guests, such as listeria or tubercle bacilli living within them, falls mainly to a subset of lymphocytes called the Th1 T-helper cells (see Table 10.2). When a specific Th1 cell combines with a complex of class II MHC molecule and microbial peptide on the surface of an infected macrophage, the T cell is triggered to release macrophage activating factors, notably interferon gamma (IFNγ) (see Ch. 9). This unleashes previously suppressed microbicidal mechanisms within the macrophage, in particular the generation of NO radicals, so leading to the death of the intracellular parasites (Fig. 10.10). In general, Th1 cells evoke a chronic inflammatory response dominated by macrophages but, in addition, recent studies have revealed the powerful pro-inflammatory role of another subset, the Th17 cell.
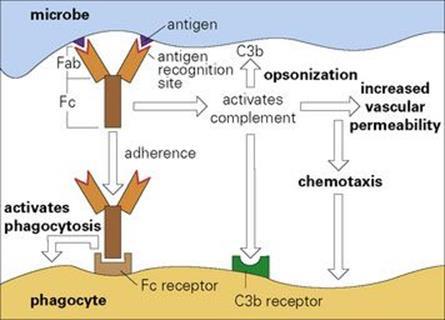
Figure 10.10 T-helper (Th1) cells trigger the killing of intracellular parasites within macrophages (Mφ). Recognition of the infected macrophage by the Th1 cell TCR results in lymphocyte activation with release of IFNγ. This then activates the macrophage, which turns on its microbicidal mechanisms to kill the intracellular parasite. Th17 helper cells subserve a similar role and are also thought to be a prominent factor in the pathogenesis of certain autoimmune disorders.
T lymphocytes inhibit intracellular replication of viruses
Cells infected with virus express complexes consisting of class I MHC and a virally derived peptide on their surface. These are recognized by the specific receptors on cytotoxic T (Tc) cells (Fig. 10.11), which are therefore led into close proximity to their virally infected target. The target cell is then killed by similar extracellular mechanisms to those described in Chapter 9. Since the virally derived peptides appear on the cell surface at a very early stage of infection, if the Tc cells kill the cell before the virus has had an opportunity to replicate significantly, the host has won an important battle. The natural killer (NK) cell fulfils a similar function to that of the Tc cell, but because it lacks the specialized receptors for recognizing the particular viral peptide in association with class I MHC, its chances of binding strongly to the surface of the infected target cell are much less than those of the Tc cell. However, it is of interest that not only can antibody binding to the target cell enhance NK cell potency (see Fig. 10.13) but both the Tc cell and the Th cell are capable of releasing IFNs, particularly IFNγ, which markedly improve the performance of the NK cell, so making a useful integrated system. An important additional responsibility of these IFNs is to render adjacent cells resistant to replication of viral particles, which gain entrance through intercellular transport mechanisms (Fig. 10.12).
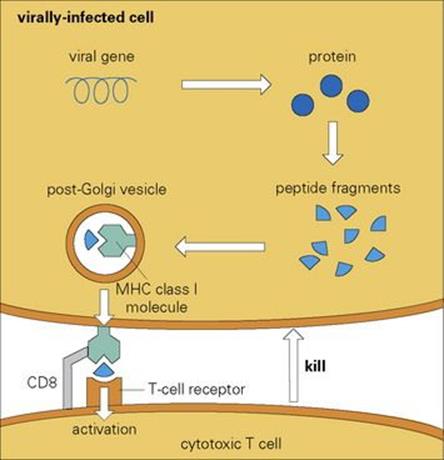
Figure 10.11 The cytotoxic T lymphocytes are activated when their specific cell surface receptors recognize an infected cell by binding to a surface MHC class I molecule that is associated with a peptide fragment derived from a degraded intracellular viral protein.
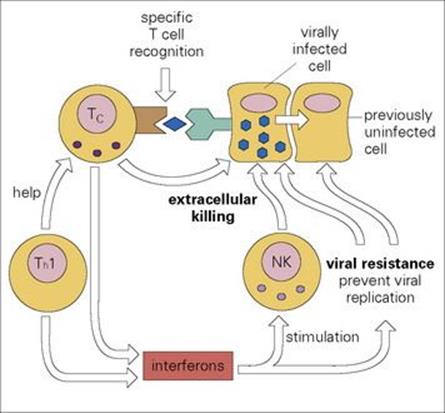
Figure 10.12 Cellular defences against viral infection. Cytotoxic T (Tc) cells specifically recognize surface MHC class I plus peptide derived from degraded viral protein and kill the infected cells before the virus replicates. Natural killer (NK) cells can do the same, though far less effectively; however, their activity is enhanced by interferons (IFNs) produced by Tc and Th1 cells. Local production of IFNs also prevents adjacent cells from becoming infected by intercellular viral transport.
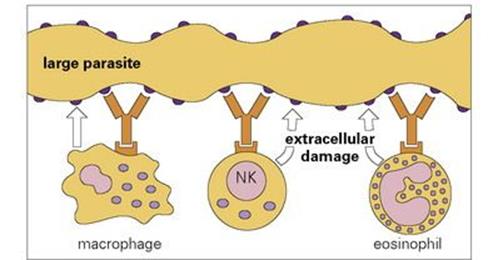
Figure 10.13 Antibody-dependent cellular cytotoxicity. Different effector cells bind to the parasite surface through their receptor for antibody and damage the parasite target. Macrophages burn the target cell surface by a stream of reactive oxygen intermediates generated by the respiratory oxygen burst, NK cells induce apoptosis by granzyme, TNF and Fas/FasL mechanisms, while eosinophils damage the target cell membrane by release of major basic protein, a perforin-like molecule and copious reactive oxygen metabolites. The antibodies mostly belong to the IgG class (seeFig. 10.1, Table 10.1).
Extracellular attack on large infectious agents
Defensive cells attack the antibody-coated surfaces of parasites
Where a parasite is demonstrably larger than a phagocytic cell, it is physically impossible for phagocytosis to occur. However, it is still possible for the defensive cells to deliver an extracellular attack on the surface of the parasite. This can occur through the phenomenon of ‘antibody-dependent cellular cytotoxicity’ (ADCC) in which effector cells bind through their surface receptors to antibody molecules coating the target cell (Fig. 10.13). The result of this interaction is to induce activation of the effector cell and the release of materials to damage the parasite target. Major cell types that indulge in this type of activity are:
• macrophages
• eosinophils
• NK cells.
Local defences at mucosal surfaces
The immune mechanisms involving the acute inflammatory response and T-cell-mediated systems operate well within the milieu of the body. It is worth examining, however, the special nature of the defences required to protect the body at the mucosal surfaces which face the exterior, for example in the lung and the gastrointestinal tract (Fig. 10.14).
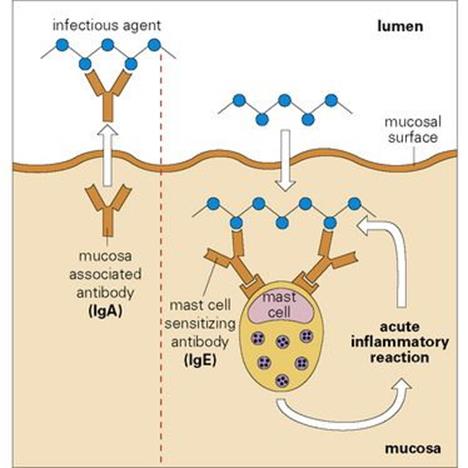
Figure 10.14 Defence of body mucosal surfaces. A specialized antibody associated with the mucosal surface – secretory immunoglobulin A (IgA) – is generated when mucosal IgA held as a dimer by J-chains (red in Fig. 10.1) is transported by the poly-Ig receptor across mucosal epithelium to the lumen where the major portion of the receptor remains bound to the IgA dimer as secretory piece and evidently protects the secretory IgA molecule from local adverse conditions. Secretory IgA blocks adherence of the microbe to the mucosa and hence entry into the body. An infectious agent gaining entrance to the body will fire IgE-sensitized mast cells, which cluster beneath the surface and generate a protective local acute inflammatory response by attracting complement-fixing antibodies, complement and polymorphs from the blood.
The first line of defence aims to prevent the microbe from adhering to the mucosal surface. Adhesion to the mucosal surface is a prerequisite for penetrating the body. To prevent this, there is the innate mechanism of mucus production. In addition, a special antibody, IgA, is synthesized by the lymphoid aggregates, some of which are organized (adenoids, tonsil, Peyer’s patches), while others are less organized (lamina propria, lung, urinogenital tract). Together, these lymphoid aggregates constitute the mucosal-associated lymphoid tissue (MALT). The IgA is then actively transported by a carrier molecule, the so-called poly-Ig receptor, into the lumen and is associated with the mucosal surface in a high concentration where it continues to bear a portion of the carrier called ‘secretory piece’ (see Fig. 10.1). When coated with such IgA antibodies, the adhesion of infectious agents to the mucosa is greatly diminished, but they can still be captured by local macrophages with surface receptors for IgA. Mast cells tend to cluster in the submucosal region and, should a microorganism break through the mucosal barrier, it could encounter a mast cell that has bound the specialized IgE antibody to its surface; on reaction with this surface antibody, the mast cell is triggered to release mediators of the acute inflammatory reaction. By increasing vascular permeability, these mediators will bring about the flooding of the site with plasma proteins, including other classes of antibody and complement, while chemotactic agents will attract polymorphonuclear leukocytes.
Larger parasites, such as nematodes, within the lumen of the gut pose special problems
It is thought that antigens derived from the nematode may penetrate the submucosal space and activate T and B cells and degranulate sensitized mast cells. The latter will produce an acute inflammation at the mucosal surface and almost certainly lead to an outflow of antibody, complement and probably effectors of ADCC into the lumen. In the lumen, the antibody, complement and effectors of ADCC can then interact with the parasite and inflict metabolic damage. In the meantime, the interaction with sensitized Th cells will lead to the release of soluble factors termed cytokines, which include a mediator capable of stimulating the goblet cells lining the intestinal villi. The goblet cells then release their mucins into the lumen where they coat the damaged parasite and facilitate expulsion from the body (Fig. 10.15).
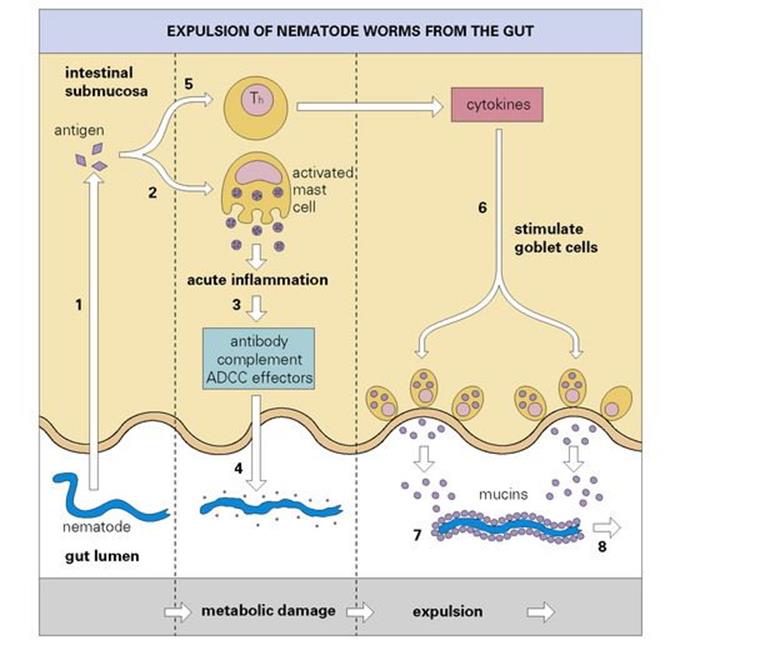
Figure 10.15 Expulsion of nematode worms from the gut. Worm antigen (1) is thought to trigger an acute inflammatory reaction in the submucosa (2). This facilitates the recruitment of complement and possibly antibody-dependent cellular cytotoxicity (ADCC) effectors (3), which damage the parasite (4). Soluble factors (cytokines), released by antigen-specific triggering of T-helper cells (Th) (5), stimulate the secretion of mucins by goblet cells (6), which coat the worm (7) and aid its expulsion (8).
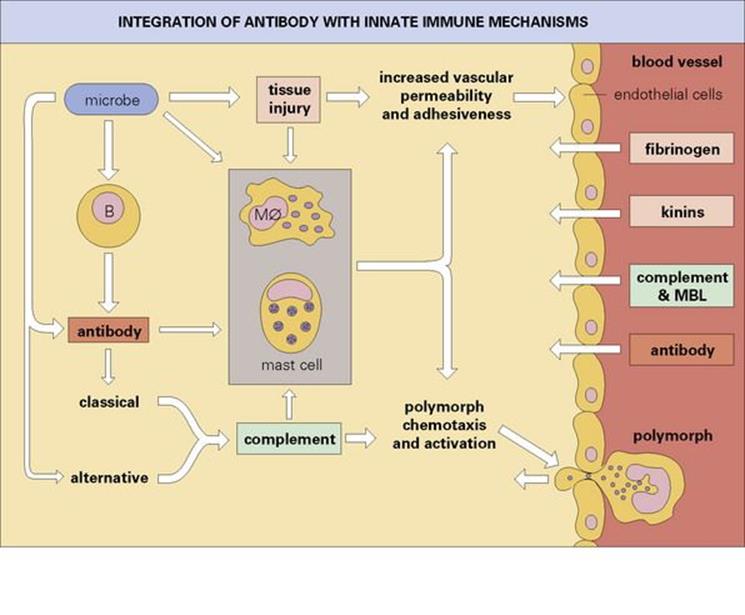
Figure 10.16 Integration of antibody with the innate immune mechanisms, leading to the production of a protective acute inflammatory reaction. The activated endothelial cells allow exudation of soluble proteins from the circulation and express accessory molecules, which aid the binding of the polymorphs to the capillary wall and their subsequent escape into the infected site. Mφ, macrophage.
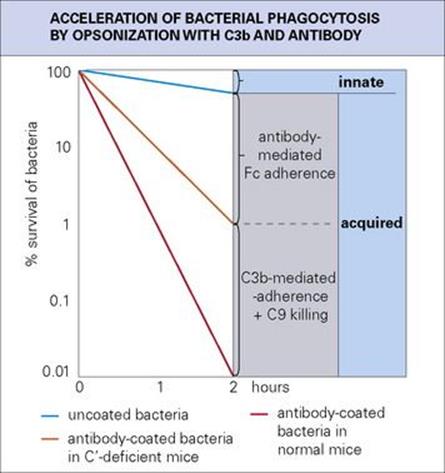
Figure 10.17 The slow rate of phagocytosis of uncoated bacteria (innate immunity) is increased many times by acquired immunity through coating with antibody and then C3b (opsonization). Killing may also take place through the C5–9 terminal complement components. This is a hypothetical but realistic situation; the natural proliferation of the bacteria has been ignored.
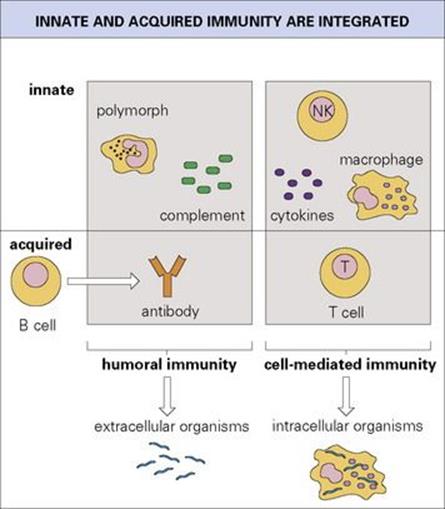
Figure 10.18 The mechanisms of innate and acquired immunity are integrated to provide the basis for humoral and cell-mediated immunity. Deficiencies of humoral immunity predispose to infection with extracellular organisms, and deficiencies of T-cell-mediated responses are associated primarily with intracellular infections.
A subset of T cells bearing γδ receptors dominates the mucosal epithelium
In the human, T lymphocytes expressing αβ receptors represent the large majority of T cells in the blood, but a subset composed of γδ chains dominates the intestinal epithelium and skin. Unlike αβ T cells, the γδ subset can recognize antigen directly without the need for antigen processing. Heat shock proteins released from stressed or damaged cells are potent stimulators of γδ T cells as are low molecular weight phosphate-containing non-proteinaceous antigens such as isopentenyl pyrophosphate and alkylamines which occur in a wide range of pathogens. γδ T cells can also collaborate with mucosal epithelial cells expressing surface CD1 molecules (containing β2 microglobulin but non-classical MHC-like chains) which have a hydrophobic cleft enabling them to present lipid and glycolipid microbial antigens such as lipoarabinomannan, the mycobacterial cell wall component.
![]()
 Key Facts
Key Facts
• The evolution of the adaptive response has provided the body with a powerful series of mechanisms that extend and exploit the innate mechanisms of defence. Thus, this lymphocyte-mediated response greatly augments the innate defence against each particular infecting organism.
• In most cases, the effector mechanisms involve the innate systems of defence such as phagocytosis, complement activation and macrophage intracellular killing.
• Taking an overall view of the adaptive responses, humoral immunity mediated by antibody produced by B lymphocytes is effective in neutralizing bacterial toxins, and, by interacting with complement, mast cells and polymorphs, produces the acute inflammatory reaction (Fig. 10.16). This response is especially effective against extracellular microbes, and the ‘quantum leap’ provided by antibody in the clearance of extracellular bacteria from the blood is clearly shown in the example in Figure 10.17. The IgE-mediated acute inflammatory response and secreted IgA defend the mucosal surfaces against extracellular infections.
• In contrast, most T-cell-mediated responses are directed to intracellular organisms. The receptors on the majority of T cells are composed of α and β chains, each with a variable and constant domain resembling antibody Fab fragments. However, they recognize an infected cell as a target by binding to the surface major histocompatibility complex (MHC) molecule, which is a marker for a cell, linked to a peptide derived by degradation of intracellular microbial protein.
• αβ T cells are divided into T-helper Th1, T-helper Th2, T-helper Th17, cytotoxic Tc cells and regulatory T cells. CD4 Th1 and Th17 cells recognize class II MHC on macrophages and produce soluble factors (cytokines), first chemotactic factors to attract, and second IFNγ, to activate phagocytic cells to switch on their intracellular antimicrobial mechanisms. CD4 Th2 cells recognize class II MHC on B cells and help them to produce antibody. CD8 Tc cells recognize MHC class I on most cells and are effective against viruses, killing virally infected targets and preventing the spread of virus through the local production of interferons. Tc cells can also be divided into subsets expressing either Th1- or Th2-type cytokine patterns. The different classes of regulatory cells which provide overall control of T-cell proliferation and, in particular, police the activity of any self-reacting T cells which have escaped elimination in the thymus are discussed in the next chapter (see Fig. 11.15 and Fig. 11.16). A subset expressing γδ receptors dominates in the intestinal epithelium and skin and recognizes heat shock stress proteins and microbial non-proteinaceous phosphate-containing antigens without the need for antigen processing.
• Figure 10.18 emphasizes the close interactions between innate and acquired mechanisms leading to defence against extracellular microorganisms, on the one hand, and intracellular infections, on the other. In keeping with these concepts, deficiencies in humoral immunity from whatever cause, predispose the individual to infection by extracellular organisms, whereas defects in T-cell-mediated responses are primarily associated with intracellular infections.
• The first contact with antigen evokes a response that leaves behind a memory of the encounter so that the subsequent response to a second contact with antigen is more powerful and evolves more rapidly than on the first occasion. The cellular bases for these phenomena are explained inChapter 11. The production of memory by a primary interaction with antigen provides the basis for vaccination, where the first contact is with an avirulent form of the microorganism or its component antigens.
• The other point to stress at this stage is the specificity of memory – infection with measles, for example, produces a subsequent immunity to that virus, but does not afford protection against an unrelated virus such as mumps.
![]()