Medical Microbiology
Section 3 The conflicts
16 Parasite survival strategies and persistent infections
Introduction
The most common infectious organisms have developed ‘answers’ to host defences
So far, we have concentrated on the battery of mechanisms available to the host, both natural and adaptive, to keep out and destroy the parasite. Powerful as these are, they are obviously not 100% effective; otherwise healthy people would never have infections. In fact, most of the common infectious organisms described in this book have developed ‘answers’ to host defences because their ability to survive as human parasites has depended upon this. They successfully infect humans and are of concern to the physician precisely because they have developed strategies for evading or actively interfering with host defences.
Strategies to evade natural non-adaptive defences such as the phagocyte
These include the following:
• Killing or avoiding being killed by phagocytes. Successful parasites have evolved numerous ingenious antiphagocytic devices. Antiphagocytic devices (Fig. 16.1) range from killing or inhibiting the phagocyte itself, via more subtle ways of eluding contact, to protection against intracellular death allowing the microorganism to survive within the phagocyte – a very serious challenge to the host.
• Interfering with ciliary action (see Fig. 13.3).
• Interfering with the activation of complement. Microorganisms can acquire or mimic complement regulators, actively inhibit complement components, or enzymatically destroy complement components. A variety of microbes can bind complement regulators, including E. coli, streptococci and Candida albicans. The smallpox and vaccinia viruses produce proteins that mimic host complement regulators. Staph. aureus, streptococci, herpes simplex virus, Schistosoma and Trypanosoma express complement inhibitors. Proteases that destroy complement components are produced by Pseudomonas, Serratia marcescans and Schistosoma mansoni. Other strategies are to physically block complement lysis – the insertion of the C567 complex is prevented by the long side chains of the cell wall polysaccharides of smooth strains of Salmonellae and by the capsules of staphylococci, which do not activate complement, and the cell wall of Gram-positive bacteria prevents lysis by the complement membrane attack complex (Fig. 16.2). However, some pathogens take the opposite approach, choosing to enter host cells by exploiting opsonization with complement components – HIV-1 and Mycobacterium tuberculosis exploit the CR3 receptor in this way.
• Producing iron-binding molecules. Nearly all bacteria need iron, but the host’s iron-binding proteins such as transferrin limit the availability of this element. Accordingly, certain bacteria (e.g. Neisseria) produce their own powerful iron-binding proteins to circumvent the shortage
• Blocking interferons. Host cells respond to double-stranded DNA (dsRNA) from infecting microbes (including all viruses), by forming interferons alpha and beta. These are produced rapidly, within 24–h, after infection and are part of the non-adaptive response. Certain viruses are either poor inducers of interferons (hepatitis B) or produce molecules that block the action of interferons in cells (hepatitis B, HIV, adenoviruses, Epstein–Barr virus, vaccinia virus). Interferon gamma (IFNγ), an essential part of the adaptive response, is also affected.
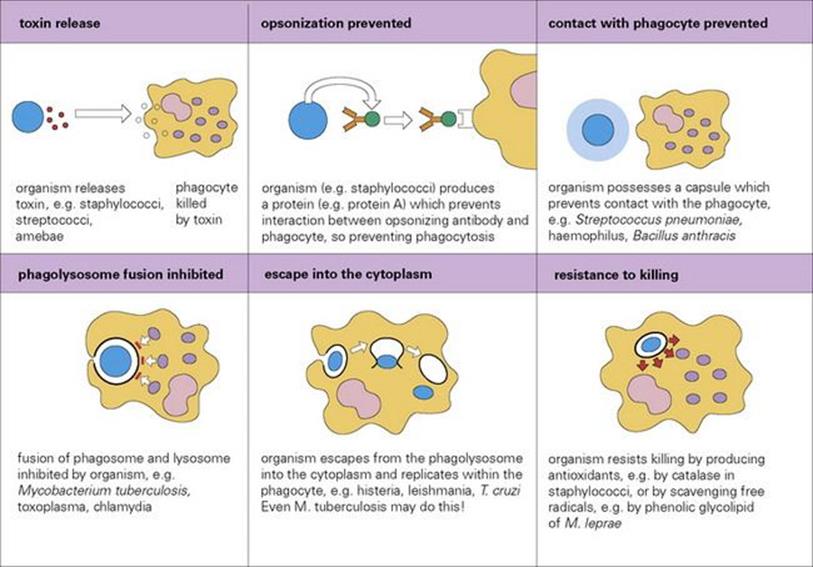
Figure 16.1 Various mechanisms adopted by microorganisms to avoid phagocytosis.
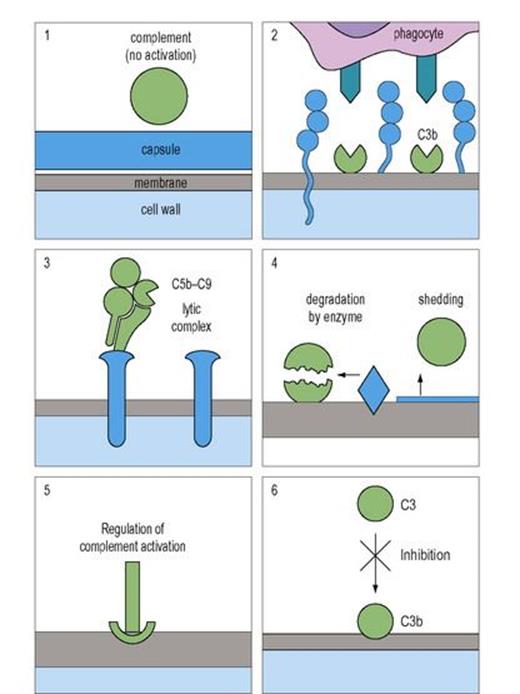
Figure 16.2 Bacteria avoid complement-mediated damage by a variety of strategies. (1) An outer capsule or coat prevents complement activation. (2) An outer surface can be configured so that complement receptors on phagocytes cannot obtain access to fixed C3b. (3) Surface structures can be expressed that divert attachment of the lytic complex (MAC) from the cell membrane. (4) Membrane-bound enzyme can degrade fixed complement or cause it to be shed. (5) Complement inhibitors can be captured onto the surface. (6) Direct inhibition of the C3 and C5 convertases blocks complement activation.
(Panels 1–4 reproduced from: Male, D., Brostoff, J., Roth, D.B., Roitt, I. (2006) Immunology. Mosby Elsevier, with permission.)
Strategies to evade adaptive defences
Strategies to evade adaptive defences are more sophisticated than those for evading innate defences
The success of microbes in evading or interfering with adaptive (immune) defences is discussed in this chapter. The strategies involved are more sophisticated than those for evading innate defences, because lymphocytes are programmed so that their cell receptors can recognize virtually any shape (B cells) or amino acid sequence (T cells), provided it is not identical to self. For example:
• The polysaccharide capsules of bacteria prevent non-immune contact between phagocytes and the bacterial cell wall, but are quickly recognized as foreign by B-cell surface receptors (immunoglobulin), leading to the formation of antibody with consequent opsonization and phagocytosis of the bacteria
• Many microorganisms such as bacteria and fungi can resist intracellular destruction by macrophages, but their peptides are presented in association with major histocompatibility complex (MHC) molecules on the macrophage surface, and their presence is detected by T cells. A new set of cytotoxic and other immune mechanisms is then brought into action.
In both these examples, the lymphocytes are behaving like a highly specialized and sharply observant secret police force in contrast to the everyday activities of the more pedestrian macrophages.
Parasite survival strategies
Parasite survival strategies can take as many forms as there are parasites, but they can be usefully classified according to the immune component that is evaded and the means selected to do this (see Ch. 12). As a result, the microbe is able to undergo what are often quite lengthy periods of growth and spread during the incubation period before being shed and transmitted to the next host, as occurs in hepatitis B and tuberculosis. Shedding of the microbe for just a few extra days after clinical recovery gives more extensive transmission in the community, and this is a worthwhile result for the microbe.
Viruses are particularly good at thwarting immune defences
Viruses are able to thwart immune defences for a number of reasons:
• Their invasion of tissues and cells is often ‘silent’. Unlike most bacteria, they do not form toxins, and as long as they do not cause extensive cell destruction there is no sign of illness until the onset of immune and inflammatory responses, sometimes several weeks after infection, as occurs in hepatitis B virus and EBV infections.
• Viruses such as rubella virus, wart viruses, hepatitis B virus and EBV can infect cells for long periods without adverse effects on cell viability.
Some microbes are able to persist in the host
Certain microbes are able to remain (persist) in the host for many years, often for life. From the microbe’s point of view, persistence is worthwhile only if shedding occurs during the persistence. Persistent microbes fall into two categories:
• those that are shed more or less continuously, such as the Epstein–Barr virus (EBV) into saliva, hepatitis B virus into blood, and eggs into faeces in various helminth infections
• those that are shed intermittently, such as herpes simplex virus (HSV), polyomaviruses, typhoid bacilli, tubercle bacilli and malaria parasites.
Virus latency is a type of persistence and is based on an intimate molecular relationship with the infected cell. The viral genome continues to be present in the host without producing antigens or infectious material, and only does so very occasionally, when the virus reactivates (becomes patent) (Box 16.1).
![]()
Box 16.1  Lessons in Microbiology
Lessons in Microbiology
Trail of illness from a slippery cook
In 1901, Mary Mallon, from Long Island, New York, took a job as cook with a family in New York City. Soon afterwards, the family washerwoman and a visitor to the house became ill with enteric fever (typhoid). Mary moved to another job and a few weeks later all seven family members plus two of the servants went down with enteric fever. Similar infections followed her movements as a cook and in 1906 the authorities tried to dissuade her from such work. She was indignant at the suggestion that she was carrying a dangerous germ, knowing that she was healthy, and failed to keep promises to have regular checks and give up work. She was suspicious of officials and aggressive, on one occasion advancing towards the questioner brandishing a carving knife. She was later arrested and put in an isolation hospital. After appealing to the US Supreme Court, she was released in 1910 with promises not to work as a cook. Then in 1914 typhoid epidemics broke out in a hospital and in a sanatorium where she had worked as a cook. She was traced, living under a false name, and in the interests of public safety, she was detained permanently on North Brother Island, where she died in 1938. In her cooking career she had been responsible for about 200 cases of typhoid in eight different families and had started seven epidemics of the disease. Her favourite recipe, an iced peaches dessert, may have been a good source of infection.
Mary had recovered fully from an attack of typhoid earlier in life, but she had gallstones and this enabled the bacteria to persist in her gallbladder for many years, appearing intermittently in the faeces. About 5% of cases become carriers, either in gallbladder or urinary bladder, and they play a central role as foci of infection. Nowadays, Mary would have to have acquired her original infection in a region such as the Indian subcontinent where typhoid is endemic. Each year, there are up to 22 million typhoid cases worldwide, 300 of which are in the USA, most being travellers to the Indian subcontinent (see also Ch. 22).
![]()
Strategies for evading host defences cause a rapid ‘hit-and-run’ infection
One evasion strategy for microorganisms is to cause a rapid ‘hit-and-run’ infection. The microbe invades, multiplies and is shed within a few days, before adaptive immune defences have had time to come into action. Infections of the body surfaces (rhinoviruses, rotaviruses) come into this category. Otherwise, the principal strategies employed by parasites to elude the lymphocyte (as discussed in the following pages) are:
• concealment of antigens
• antigenic variation
• immunosuppression.
Concealment of antigens
A spy in a foreign country can conceal his presence from the police by hiding, by never venturing out of doors, or by adopting the disguise of a native. Parasites have the same choices. Places to hide include the interior of host cells (though the MHC molecules act as ‘informers’ for this compartment, picking up and transporting microbial peptides to the cell surface where they will be recognized) and particular sites in the body where lymphocytes do not normally circulate (‘privileged sites’, the equivalent of ‘no-go’ areas).
Remaining inside cells without their antigens being displayed on the surface prevents recognition
If a microbe can remain inside cells without allowing its antigens to be displayed on the cell surface, it will remain unrecognized (‘incognito’) as far as immune defences are concerned. Even if specific antibody and T-cell responses have been induced, the microbe inside such a cell is unaffected. Persistent latent viruses such as HSV in sensory neurones behave in this way. During reactivation, of course, re-exposure and boosting of immune defences is inevitable.
Other strategies are possible. Several viruses (HIV in macrophages, coronaviruses) display their proteins ‘secretly’ on the walls of intracellular vacuoles instead of at the cell surface, and bud into these vacuoles. Adenoviruses have taken more active steps to avoid antigen display. One of the adenoviral proteins (E19) combines with class I MHC molecules and prevents their passage to the cell surface so that infected cells are not recognized by cytotoxic T cells.
Colonizing privileged sites keeps the microbe out of reach of circulating lymphocytes
The vast numbers of microbes that colonize the skin and the intestinal lumen, together with those that are shed directly into external secretions, are effectively out of reach of circulating lymphocytes. They are exposed to secretory antibodies, which although able to bind to the microbe (e.g. influenza virus) and render it less infectious, are generally unable to kill the microbe or control its replication in or on the epithelial surface (Figs 16.3, 16.4). A local inflammatory response, however, can enhance host defences.
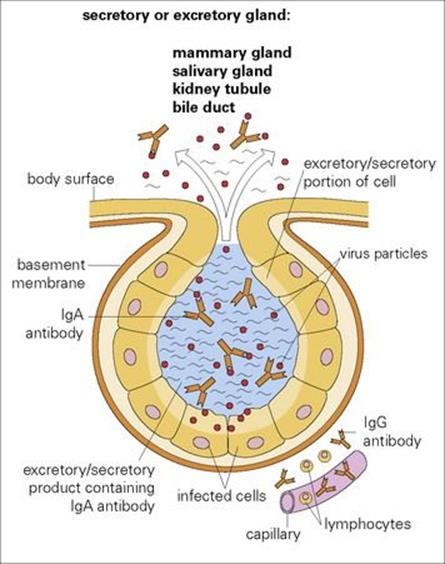
Figure 16.3 Viral infection of cell surfaces facing the external world. Infection of the surface epithelium of, for instance, a secretory or excretory gland allows direct shedding of the virus to the exterior, as well as avoidance of host immune defences.
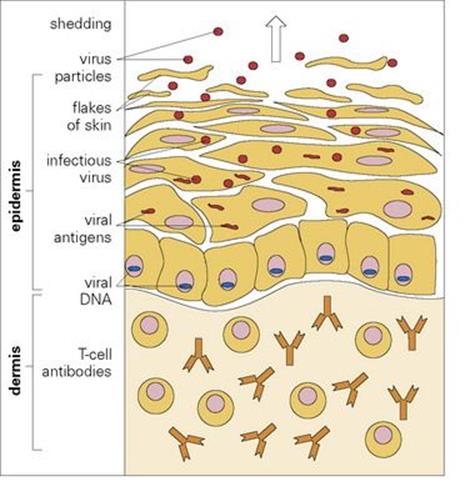
Figure 16.4 Wart virus replication in epidermis – a privileged site? Cell differentiation such as keratinization controls virus replication, and as a result virus matures when it is physically removed from immune defences.
Within the body, it is more difficult to avoid lymphocytes and antibodies, but certain sites are safer than others. These include the central nervous system, joints, testes and placenta. Here, lymphocyte circulation is less intense, and access of antibodies and complement is more restricted. However, as soon as inflammatory responses are induced, then lymphocytes, monocytes and antibodies are rapidly delivered and the site loses its privilege.
Additional privileged sites can be created by the infectious organism itself. A good example is the hydatid cyst that develops in liver, lung or brain around growing colonies of the tapeworm Echinococcus granulosus (Fig. 16.5), inside which the worms can survive even though the blood of the host contains protective levels of antibody.

Figure 16.5 Hydatid cysts. Multiple, thin-walled, fluid-filled cysts in a surgical specimen. The lung is a common site. Growing within a cyst is a survival strategy for Echinococcus granulosus.
(Courtesy of J.A. Innes.)
Perhaps the most highly privileged site of all is host DNA, and this is occupied by the retroviruses. Retroviral RNA is transcribed by the reverse transcriptase into DNA as a necessary part of the replicative cycle, and this then becomes integrated into the DNA of the host cell (see Ch. 21). Once integrated, and as long as there is no cell damage and viral products are not expressed on the cell surface where they can be recognized by immune defences, the virus enjoys total anonymity. This is what makes complete cure and complete removal of virus from a patient infected with HIV such a daunting task. The intragenomic site becomes even more privileged if the egg or sperm is infected. The viral genome will then be present in all embryonic cells and transferred from one generation to another as if it were the host’s own DNA. Luckily, this does not happen with HIV or with human T-cell lymphotropic virus (HTLV) 1 and 2. However, the ‘endogenous’ retroviruses of humans present in profusion as DNA sequences in our genome, but not expressed as antigens, come into this category. They are part of our inheritance. This surely represents the ultimate, the final logical step in parasitism, at the borderline between infection and heredity.
Mimicry sounds like a useful strategy, but does not prevent the host from making an antimicrobial response
If the microbe can in some way avoid inducing an immune response, this can be regarded as a ‘concealment’ of its antigens. One method is by mimicking host antigens, as such self antigens are not recognized as foreign. Numerous examples are known of parasite-derived molecules that resemble those of the host (Table 16.1). In the case of viral proteins, mimicry based on amino acid sequence homology (sharing of 8–10 consecutive amino acids) is seen to be common when computer comparisons are made between viral and host proteins. Perhaps the most celebrated example, however, is the cross-reaction between group A beta-haemolytic streptococci and human myocardium. This cross-reaction underlies the development of rheumatic heart disease, following repeated streptococcal infection because of antibody made against the cross-reacting determinant meromyosin (Fig. 16.6). The fact that the host makes such autoantibodies shows that in this case mimicry does not protect the bacteria. The conclusion is that, although mimicry sounds like a useful strategy for microbes and occurs quite frequently, it is probably an accident rather than a sinister microbial strategy. It does not prevent the host from making an antimicrobial and autoimmune response.
Table 16.1 Some examples of mimicry or uptake of host antigens by parasites
|
Microbe’s strategy |
Parasite |
Corresponding host antigen |
|
Mimicry |
Epstein–Barr virus |
Human fetal thymusa |
|
Streptococci |
Cardiac muscle (meromyosin) |
|
|
Mycobacterium tuberculosis |
65 kDa heat shock protein |
|
|
Neisseria meningitidis |
Embryonic brain, vitronectin |
|
|
Treponema |
Cardiolipinb |
|
|
Mycoplasma pneumoniae |
Erythrocytesc |
|
|
Plasmodium falciparum |
Spondind |
|
|
Trypanosoma cruzi |
Heart myosin, nerve |
|
|
Schistosoma |
Glutathione transferase |
|
|
Antigen uptake |
Neisseria meningitides |
Vitronectin |
|
Haemophilus influenzae |
Vitronectin |
|
|
Cytomegalovirus |
β2-microglobulin |
|
|
Schistosoma |
Glycolipids, HLA I, HLA II |
|
|
Filarial nematodes |
Albumin |
|
|
Ascaris |
Blood group antigens |
a Also cross-reacts with erythrocytes of certain species and is the basis for the Paul Bunnell (heterophil antibody) test.
b Basis for Wasserman-type antibody test for syphilis.
c Basis for cold agglutinin test, but may result from damage to erythrocytes rather than direct mimicry.
d The homologous malaria protein thrombospondin-related anonymous protein (TRAP) shares sequence with the circumsporozoite protein that mediates binding to hepatocytes. HLA, human leukocyte antigen; Ig, immunoglobulin.
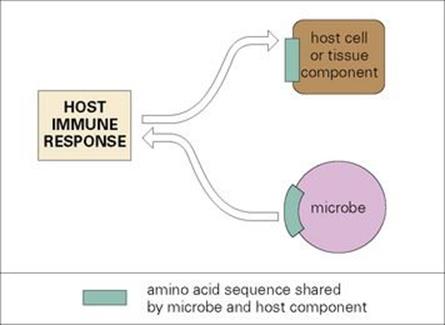
Figure 16.6 Molecular mimicry by the microbe can induce host cell damage, for example rheumatic heart disease following streptococcal infection is caused by antibodies reacting with meromyosin, the cross-reacting determinant.
Microbes can conceal themselves by taking up host molecules to cover their surface
This is illustrated in Table 16.1. A superb example of this is the blood fluke Schistosoma, as the schistosomula acquire a surface coat of host blood group glycolipids and MHC antigens from the plasma. Such a parasite must indeed be virtually invisible even to a lymphocyte. For unknown reasons, however, this strategy is essentially restricted to worms.
The uptake of immunoglobulin molecules by the microbe seems to be a more widespread phenomenon. A number of viruses and bacteria produce Fc receptors, which are displayed on their surface and bind immunoglobulin molecules of all specificities in an immunologically useless upside-down position (Fig. 16.7, and see below). This prevents the access of specific antibodies or T cells to the microbe or the infected cell.
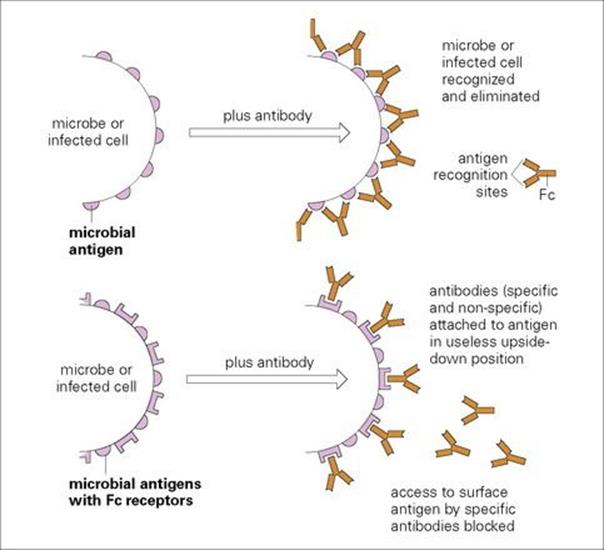
Figure 16.7 The production of Fc receptors is of some benefit to microbes, for example staphylococci, streptococci, herpes simplex virus, varicella-zoster virus and cytomegalovirus.
Immune modulation
Modulation of the host immune response by the microbe can prevent this response being an effective one. An alternative strategy for the microbe is to avoid inducing an immune response or to induce a poor response. There are five possible methods:
• infection during early embryonic life
• the production of large quantities of the microbial antigen or of antigen–antibody complexes
• exploiting ‘gaps’ in the host’s immune repertoire
• upsetting the balance between antibody and cell-mediated immune responses – between T-helper cell (Th) 1 and 2 responses
• inducing immune responses that down-regulate or suppress protective immunity.
Infection during early embryonic life
Before development of the immune system, a time when antigens present are regarded as ‘self’, infection could possibly result in immune tolerance. However, in the case of intrauterine infection with cytomegalovirus (CMV), rubella virus and syphilis, the fetus does eventually produce IgM antibody, which is detectable in umbilical cord blood, but cell-mediated responses are more seriously impaired. Children with congenital CMV or rubella fail to develop lymphoproliferative responses to CMV or rubella antigens and consequently take years to clear the virus from the body (see Ch. 23). In some cases, infection in the neonatal period is more likely to result in tolerance than infection in later life. Therefore, neonatal infection with hepatitis B virus frequently results in permanent carriage of the virus, though the mechanism is unknown.
Production of large quantities of microbial antigen or antigen–antibody complexes
Large quantities of microbial antigen or antigen–antibody complexes circulating in the body can cause immune tolerance to that antigen. Anergy, as evidenced by normal antibody but depressed cell-mediated immune responses to the invading microbe, is seen in disseminated coccidioidomycosis and cryptococcosis, and in visceral and diffuse cutaneous leishmaniasis, in each case associated with large amounts of microbial antigen in the circulation.
Exploiting ‘gaps’ in the host’s immune repertoire
There are likely to be certain peptides to which the host makes a poor immune response, based on the nature of the host’s MHC class II molecules. These represent genetically determined ‘gaps’ in the host’s immune repertoire, and microbes, as they evolve, might be expected to match these peptides. In other words, microbes may be constantly ‘probing’ the immune repertoire of the host, seeking out weaknesses. European populations today may have more resistant immune repertoires selected following the deaths of so many young adults from tuberculosis in the nineteenth century.
Upsetting the balance between Th1 and Th2 responses
Resistance to infection often depends upon a suitable balance between Th1 and Th2 responses (see Ch. 10). Good defence against tuberculosis and herpesviruses needs cell-mediated immunity, whereas antibody is required for good defence against polioviruses or Streptococcus pneumoniae. In active tuberculosis, T cells making IL-4 can be detected, with a reduction in the beneficial Th1 cytokine response. By inducing an ineffective type of response a microbe can promote its own survival.
An altered Th1:Th2 balance can also alter the phenotype of macrophages. In helminth infections, as well as allergy, Th2 cytokines such as IL-4 and IL-13 induce alternatively activated macrophages (classically activated macrophages are those activated by IFNγ). Alternatively activated macrophages are thought to play a role in worm expulsion from the gut.
Regulatory T cells
Some bacteria evade protective Th1 responses by inducing antigen-specific regulatory T cells. Bordetella pertussis infection induces regulatory T cells specific for its filamentous haemagglutinin and pertactin. These regulatory T cells produce IL-10, and thus suppress Th1 immunity to two vital bacterial components which help the bacteria attach to host cells. Regulatory T cells are induced by many other bacteria and even by some helminth antigens. (Other types of immunomodulation caused by microbes, e.g. superantigen production, are described below, see ‘Immunosuppression’.)
Antigenic variation
Reverting to the metaphor of a spy in foreign territory, there is another way to confuse the enemy and that is by repeated changes in appearance. The African trypanosome, the causative organism of sleeping sickness, does this, and so do a wide range of viruses, bacteria and protozoa. Antigenic variation can occur:
• during the course of infection in a given individual
• during spread of the microbe through the host community (Fig. 16.8).
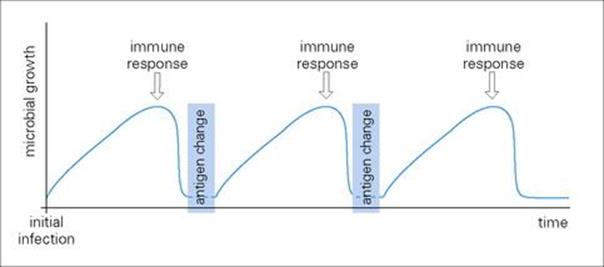
Figure 16.8 Antigenic variation as a microbial strategy. The change in antigens may take place in the originally infected individual, enabling the microbe to undergo renewed growth (e.g. trypanosomiasis, relapsing fever – see Ch. 27) or it may take place as the microbe passes through the host population, enabling it to re-infect a given individual (e.g. influenza).
As a strategy for evading host immune responses, antigenic variation depends upon variation occurring in antigens whose recognition is involved in protection. Antigenic variation is common as the microbe passes through the host community and it tends to be more important in longer-lived hosts, such as humans in whom microbial survival is favoured by multiple re-infections during the lifetime of a given individual. Also, it is more common in infections limited to respiratory or intestinal epithelium where the incubation period is < 1 week and the microbe can commonly infect, multiply and be shed from the body before a significant secondary immune response is generated. During systemic infections (e.g. measles, mumps, typhoid), the incubation period is longer and secondary responses have more opportunity to come into action and control an infection by an antigenic variant. Accordingly, antigenic variation is not an important feature of these systemic infections.
At the molecular level, there are three main mechanisms for antigenic variation:
• mutation
• recombination
• gene switching.
The best known example of mutation is the influenza virus
As the influenza virus spreads through the community there are repeated mutations in the genes coding for haemagglutinin and neuraminidase (see Ch. 19), causing small antigenic changes that are sufficient to reduce the effectiveness of B- and T-cell memory built up in response to earlier infections. This is called ‘antigenic drift’. Human rhinoviruses and enteroviruses are evolving rapidly and show a similar drift. Antigenic drift could account for the wealth of antigenic types of staphylococci, streptococci and pneumococci. During earlier poliovirus epidemics, mutations occurred at the rate of about two base substitutions per week, some of them involving the main antigenic sites on the virus. HIV (see Ch. 21) undergoes antigenic drift, but in this case it occurs during infection of a given individual, which helps to explain the difficulties experienced by the immune system in controlling this infection. Mutations affecting the epitopes recognized by cytotoxic T (Tc) cells are the source of ‘escape mutants’.
The classic example of recombination involves influenza A virus
More extensive and sudden alterations in antigens can take place by the exchange of genetic material between two different microbes. The classic example is genetic ‘shift’ in influenza A virus (Fig. 16.9; and see Ch. 19). When human and avian virus strains recombine, a completely new strain of influenza A virus suddenly emerges, brandishing a haemagglutinin or neuraminidase of avian origin. This new virus, not previously experienced by the present population, gives rise to an influenza pandemic. The 2009/2010 ‘swine flu’ epidemic which originated in Mexico spread rapidly and was caused by a H1N1 virus – segments of its genome were identified in flu isolates almost 20 years earlier but reassortment led to the new pandemic strain (Fig. 16.9). Surprisingly, new evidence suggests that the 1918 Spanish flu pandemic may not have been caused by antigenic shift, but by an avian flu that became able to infect humans.
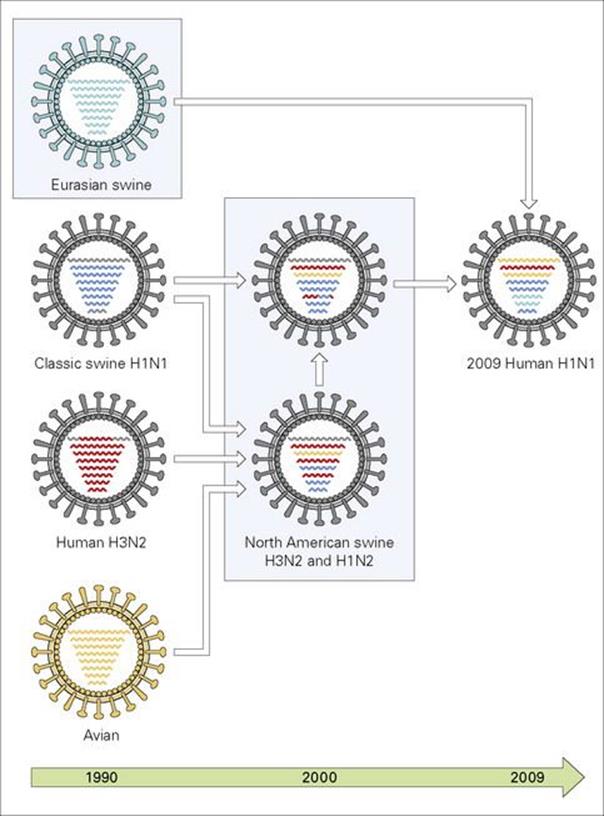
Figure 16.9 The major surface antigens of influenza virus are haemagglutinin and neuraminidase. Haemagglutinin is involved in attachment to cells, and antibodies to haemagglutinin are protective. Antibodies to neuraminidase are much less effective. The influenza virus can change its antigenic properties slightly (antigenic drift) or radically (antigenic shift). Pandemics can arise when there is antigenic shift with reassortment of genes. The diagram shows the origins of the 2009 pandemic influenza A (H1N1). The official influenza antigen nomenclature is based on the type of haemagglutinin (H1, H2, etc.) and neuraminidase (N1, N2, etc) expressed on the surface of the virion. Note that, although new strains replace old strains, the internal antigens remain largely unchanged.
(Redrawn from Trifonof V. et al., N Engl J Med (2009) 361: 115–119.)
Gene switching was first demonstrated in African trypanosomes
Gene switching represents the most dramatic form of antigenic variation and was first demonstrated in the African trypanosomes, Trypanosoma gambiense and T. rhodesiense (see Ch. 27). These organisms carry genes for about 1000 quite distinct surface molecules known as variant-specific glycoproteins, which cover almost the entire surface and are immunodominant. The trypanosome can switch from the use of one gene to another, much as a B cell does with the immunoglobulin heavy chain-constant genes. The effect on the host is a sequence of unrelated infections at approximately weekly intervals. This enables the trypanosome to persist while the immune system is constantly trying to catch up with it. The main stimulus for each gene switch is possibly the antibody response itself, but the exact mechanism is not clear. About 10% of the trypanosome genome consists of surface coat genes, but this is a worthwhile investment for the parasite.
Gene switching is thought to result in the relapsing persistent course of certain infections
Gene switching is also thought to be responsible for the relapsing persistent course of certain other infections, including that by Borrelia recurrentis (relapsing fever) and brucellosis. It is also important in gonorrhoea, not because of antigenic variation, but because changes in bacterial properties are desirable at different stages of the infection. For instance, attachment to urethral epithelium is vital early in infection by Neisseria gonorrhoeae, but attachment to phagocytes is less desirable. Hence, there is a switching of genes coding for the pilin and outer membrane proteins that mediate attachment. However, gonococci also show great antigenic variation as they circulate through the host community, and this is achieved by genetic rearrangements and recombinations in the repertoire of pilin genes.
Immunosuppression
Many virus infections cause a general temporary immunosuppression
A large variety of microorganisms cause immunosuppression in the infected host. The mechanism is generally not understood, but it often involves invasion of the immune system by the microbe – in other words, ‘to evade, invade’. As a subversive strategy this makes sense, but the extent to which the microbe benefits is often debatable. The host shows a depressed immune response to antigens of the infecting microbe (antigen-specific suppression) or, more commonly, both to antigens of the infecting microbe and unrelated antigens. HIV is one of the most spectacular, but by no means the only microbe that interferes with the immune system in this way (Table 16.2). HIV causes death of CD4+ T cells, resulting in a disastrous loss of T-cell function.
Table 16.2 Depressed immune responses in microbial infections
|
Parasite |
Feature of immunosuppression |
Mechanism |
|
Viruses |
||
|
HIV |
↓Ab ↓CMI, long-lasting |
↓CD4a T cells |
|
Immunosuppressive molecule (gp41) |
||
|
↓antigen presentation by infected APC |
||
|
Polyclonal activation of B cells |
||
|
Epstein–Barr virus |
↓CMI, temporary |
Includes polyclonal activation of infected B cellsa |
|
Measles |
↓CMI, temporaryb |
Differentiation blocked in infected T and B cells |
|
Cytomegalovirus |
↓CMI, temporary |
Unknown; very occasional infection of mononuclear cells |
|
Varicella-zoster virus, mumps |
↓CMI, temporary |
Infection of T cells |
|
Bacteria |
||
|
M. leprae (lepromatous leprosy) |
↓CMI |
Polyclonal activation of B cells, production of IL-4 and IL-10 |
|
Protozoa |
||
|
Trypanosoma |
↓Ab ↓CMI |
Regulatory T cells, production of IL-10, ↓T cell proliferation |
|
Plasmodia |
Regulatory T cells, ↓antigen presentation |
|
|
Toxoplasma |
↓CD4 T cells, IFNγ |
|
|
Leishmania |
Production of IL-10 and TGFβ |
In most cases, the mechanisms are unclear, but possible important factors are listed. aFor HIV, the depressed responses are seen later, after initial neutralizing antibody and cytotoxic cell responses. There are at least nine possible mechanisms involved in HIV immunosuppression, but decreased numbers of CD4+ T cells is probably the most important.
b Also, the BCRF-1 gene of the virus codes for an IL-10-like molecule that enhances antibody rather than protective CMI responses.
c Patients with a positive tuberculin skin test become temporarily negative during measles infection. Measles also stops macrophages producing IL-12, a molecule needed for the Th1-type (protective) immune response. Ab, antibody; CMI, cell mediated immunity; APC, antigen-presenting cell.
Clearly it would benefit the microbe if most responses to its own but not to other antigens were suppressed, but this is uncommon. However, a general immunosuppression, as long as it is temporary, might give the microbe enough time to grow, spread and be shed before being eliminated. This is what happens in many virus infections. A lasting general immunosuppression would be detrimental to the microbe because susceptibility to other infections would cause unnecessary damage to the host species. From this point of view, HIV has certainly overstepped the mark.
Different microbes have different immunosuppressive effects
Immunosuppression by microbes often involves actual infection of immune cells:
• T cells (HIV, measles)
• B cells (EBV)
• macrophages (HIV, CMV, leishmania)
• dendritic cells (HIV).
This may result in impaired cell function, such as blocking of cell division, blocking of release of interleukin 2 (IL-2) or other cytokines, or in cell death.
Additional immunosuppressive actions taken by microbes include the release of immunosuppressive molecules. For instance, the gp41 polypeptide formed by HIV acts as an ‘immunologic anaesthetic’, temporarily blocking T-cell function. Other microbes (poxviruses, herpesviruses, T. cruzi) release molecules that interfere with the action of complement or with immunologically important cytokines such as IL-2, IFNs (see above) or tumour necrosis factor (TNF).
Certain microbe toxins are immunomodulators
A particularly dramatic form of immune interference is practised by the staphylococci. Many strains liberate exotoxins (staphylococcal enterotoxin, epidermolytic toxin and toxic shock syndrome toxin) that are responsible for disease. At first sight, producing these toxins seems to be of no advantage to the staphylococci, but it is now recognized that they have extremely powerful immunomodulatory actions – they are the most potent T-cell mitogens known, and act at picomolar concentrations. They function as ‘superantigens’ and, after binding to class II MHC molecules on antigen-presenting cells, act as polyclonal activators of T cells (Fig. 16.10). A large proportion (2–20%) of all T cells then respond by dividing and releasing cytokines; only 0.001–0.01% are capable of doing this in response to a regular antigen.
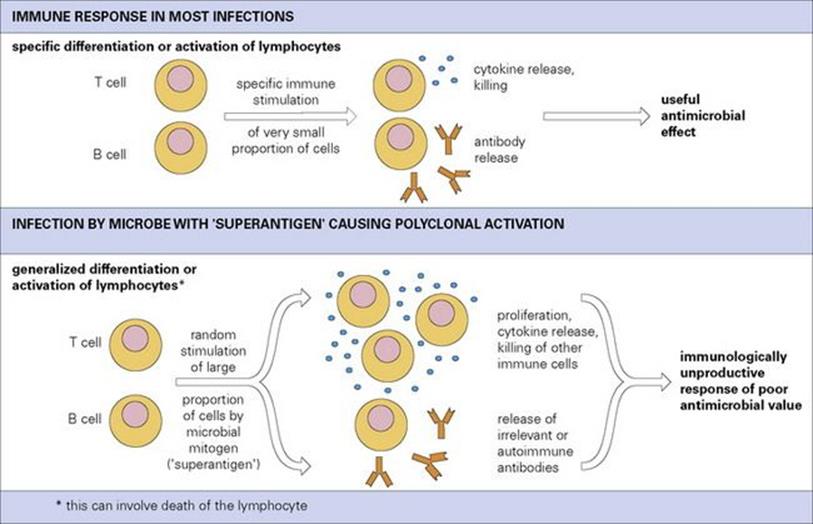
Figure 16.10 Microbial interference with the immune system by production of T- or B-cell superantigens (polyclonal activators).
It would be logical to presume that these toxins, which are coded for by plasmids, were acquired by the parasite to upset immune responses and therefore to help in the eternal battle with host defences. As if to confirm this, it has been found that similar molecules are produced by certain streptococci and mycoplasmas.
Possible mechanisms by which the staphylococcal toxins may interfere with immune defences include:
• excessive local liberation of cytokines by activated cells, upsetting the delicate balance of immune regulation
• killing of T cells or other immune cells
• diversion of T cells of all specificities into immunologically unproductive activity by polyclonal activation.
Less dramatic polyclonal activation is seen in many other infections. Microbes may cause polyclonal activation of B cells as well as T cells, e.g. in EBV and HIV infections, and this can be interpreted as an ‘immunodiversion’ by the infecting microbe, or in the case of EBV as production of a supply of B cells in which the virus can grow. One consequence is that a range of ‘irrelevant’, sometimes autoimmune, antibodies are formed (e.g. heterophil antibodies in EBV infection).
Successful microbes often interfere with signalling between immune cells, with cytotoxic T-cell recognition or with host apoptotic responses
Many microbes interfere with host molecules such as cytokines, chemokines, MHC, and apoptotic and complement receptors, all of which are essential components of host defence. Many DNA viruses code for fake molecules or fake cell receptors for the host molecules, and this disrupts the antimicrobial response. Herpes simplex virus (HSV) produces a molecule, gC (glycoprotein C), that functions as a receptor for C3b. It is present on the virus particle and on the infected cell and interferes with complement activation, protecting both the virus and the infected cell from destruction by antibody and complement.
EB virus produces a homologue of IL-10, which favours a humoral rather than the more protective CMI response induced by IL-12. Virulent strains of Mycobacterium tuberculosis induce IL-10 production by infected macrophages, which again favours the infecting microbe. Furthermore, M. tuberculosis, as well as other intracellular organisms (Leishmania major, Histoplasma capsulatum) inhibit IL-12 production by the infected macrophages. T cells are therefore not activated by IL-12 to form IFNγ, and the immune response is again pushed away from the protective Th1 pattern.
Adenoviruses and herpesviruses reduce MHC class I expression on infected cells so that cytotoxic T cells fail to recognize such cells. Other viruses (rotaviruses, adenoviruses) interfere with the production or action of interferons.
A strategy useful for one microbe is not necessarily good for others. For example, a local cell infected with a virus can commit suicide by undergoing apoptosis, a useful defence if it takes place before virus replication is complete. Accordingly, certain viruses (HSV, EBV, HIV) code for proteins that interfere with apoptosis, permitting long-term infection of the cell. Other viruses, however, such as measles, induce apoptosis, as do certain bacteria (Shigella flexneri, Salmonella) after encountering macrophages, enabling them to escape destruction. It may be useful to induce apoptosis in one cell but not in another. Thus, HIV inhibits apoptosis in the infected immune cell, but induces apoptosis in neighbouring uninfected cells.
Some microbes interfere with the local expression of the immune response in tissues
Some microbes do not interfere with the development of an immune response, but actively interfere with its expression in tissues. For instance N. gonorrhoeae, Strep. pneumoniae and many strains of Haemophilus influenzae liberate a protease that cleaves human IgA antibody. These bacteria are residents or invaders of mucosal surfaces where IgA antibodies operate, and the ability to produce such an enzyme seems unlikely to be mere coincidence.
An equally worthwhile local interference, practised by so many different infectious agents that it is likely to be significant, is the production by the microbe of Fc receptor molecules (see Fig. 16.7). The best-known example is protein A, a cell wall protein excreted from virulent staphylococci that inhibits the phagocytosis of antibody-coated bacteria. Certain herpesviruses (HSV, varicella-zoster virus (VZV), CMV) code for molecules that act as Fc receptors for IgG, and streptococci produce an Fc receptor for IgA, as shown in Figure 16.7.
Other examples include the production by Pseudomonas of an elastase that inactivates the C3b and C5a components of complement and hence tends to inhibit opsonic and other host defence functions of complement.
Unfortunately, although the above phenomena look convincingly like microbial adaptations for upsetting host defences, it is not always easy to prove that this is the case.
Persistent infections
Persistent infections represent a failure of host defences
One way of looking at persistent infections (Table 16.3) is to regard them as failures of host defences. Host defences are designed to control microbial growth and spread and to eliminate the microbe from the body. The microbe may persist:
• in a flagrantly defiant infectious form, as with hepatitis B in the blood or the schistosome in the blood vessels of the alimentary tract or bladder
• in a form with low or partial infectivity, for instance adenoviruses in the tonsils and adenoids
• in a metabolically altered state, such as M. tuberculosis
• in a completely non-infectious form, often without producing any microbial antigens. Latent virus infections are classic examples of this type of persistence. In the case of HSV, viral DNA persists for many years, probably for life, in sensory neurones in the dorsal root ganglia.
Table 16.3 Examples of persistent infections in humans
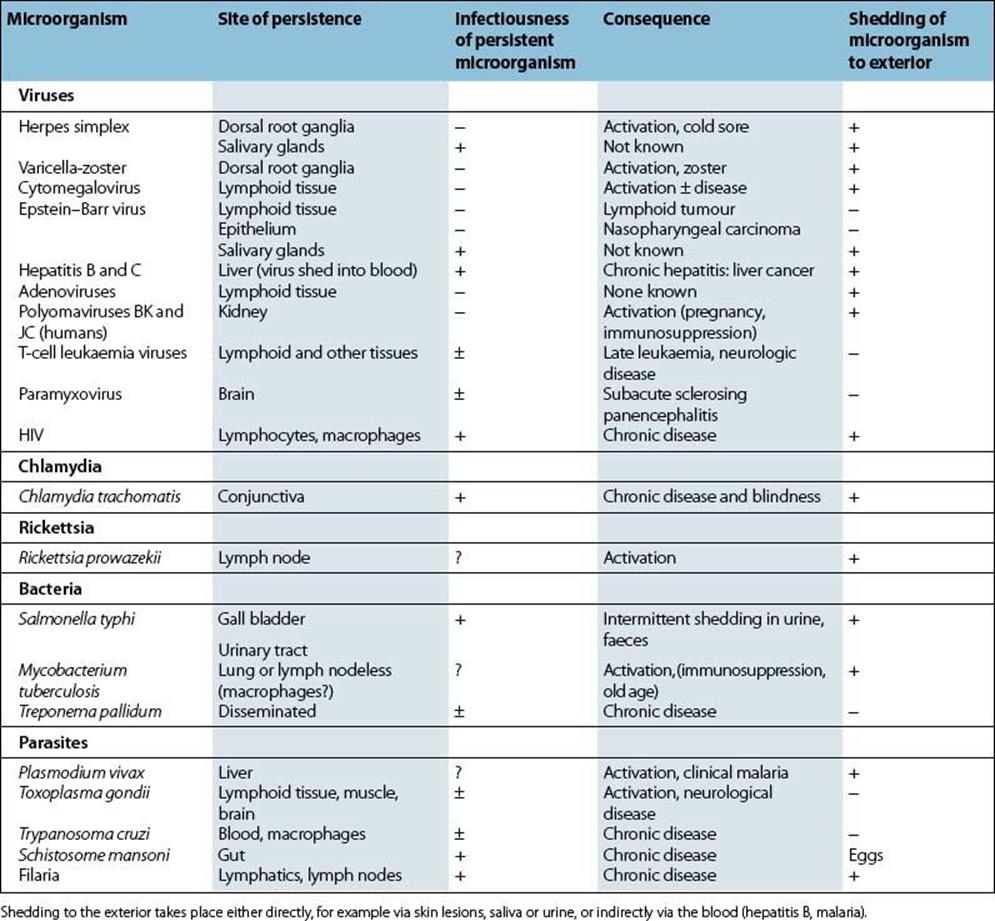
The molecular basis for viral latency has still not been elucidated. It involves special adaptations by the virus to the state of latency – in the case of HSV and VZV, there is very limited transcription of viral RNA in infected neurones, known as ‘latency-associated transcripts’. The viral genome is not integrated with host DNA, and instead of being linear it is circular, and exists in free episomal form.
Latent infections can become patent
Latent infections are so-called because they can become patent. This is where they become of immense medical interest, and the legacy of latent herpesvirus infections in humans is described in Chapter 26. Different patterns of persistent infections are illustrated in Figure 16.11, and they are important for four main reasons:
1. They can be reactivated.
2. They are sometimes associated with chronic disease, as in the case of chronic hepatitis B infections, subacute sclerosing panencephalitis following measles, and AIDS.
3. They are sometimes associated with cancers, such as hepatocellular carcinoma with hepatitis B virus, and Burkitt’s lymphoma and nasopharyngeal carcinoma with EBV.
4. From the microbial viewpoint, they enable the infectious agent to persist in the host community (Box 16.2).
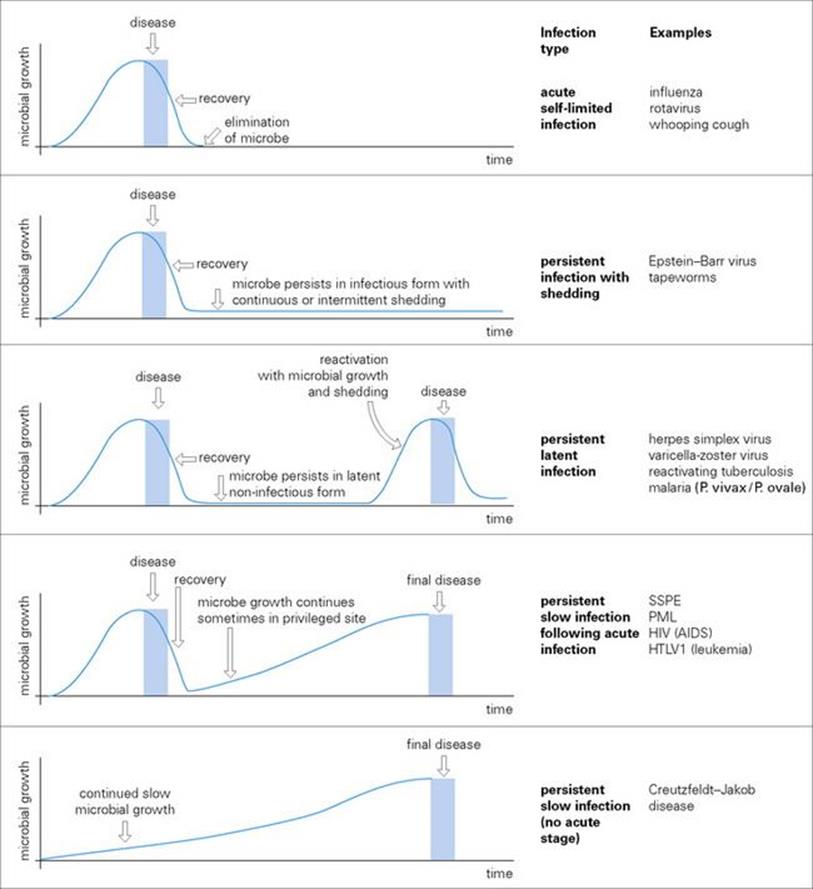
Figure 16.11 Patterns of acute and persistent infections. For some microbes (e.g. CMV, tuberculosis), the distinction between persistence in infectious form and true latency is not clear. HTLV1, human T-cell leukaemia virus 1; PML, progressive multifocal leukoencephalopathy; SSPE, subacute sclerosing panencephalitis.
![]()
Box 16.2 Lessons in Microbiology
Persistence is of survival value for the microbe
Persistence without any further shedding, as occurs in subacute sclerosing panencephalitis and progressive multifocal leukoencephalopathy (see Ch. 24), is of no survival value, but there are obvious advantages if the microbe is also shed, either continuously or intermittently. This is especially true when the host species consists of small isolated groups of individuals (Fig. 16.12). Measles, for instance, is not normally a persistent infection. It only infects humans, does not survive for long outside the body and has nowhere else to go (i.e. there is no animal reservoir). Without a continued supply of fresh susceptible humans, the virus could not maintain itself and would become extinct. There has to be, at all times, someone acutely infected with measles. From studies of island communities it is clear that you need a minimum of about 500 000 humans to maintain measles without reintroduction from outside. In Palaeolithic times, when humans lived in small, isolated groups, measles could not have existed in its present form.
In contrast, persistent and latent infections are admirably adapted for survival under these circumstances. VZV can maintain itself in a community of < 1000 individuals. Children get chickenpox, the virus persists in latent form in sensory neurones, and later in life the virus reactivates to cause shingles. By this time, a new generation of susceptible individuals has appeared and the shingles vesicles provide a fresh source of virus.
Serologic studies show that the viral infections prevalent in small, completely isolated Indian communities in the Amazon basin are persistent or latent (e.g. due to adenoviruses, polyomaviruses, papillomaviruses, herpesviruses) rather than non-persistent (e.g. due to influenza, measles, poliovirus). The same principles apply to non-viral infections. Those present in small communities are either persistent/latent (typhoid, respiratory tuberculosis) or have an animal reservoir for maintenance of the microbe.
![]()
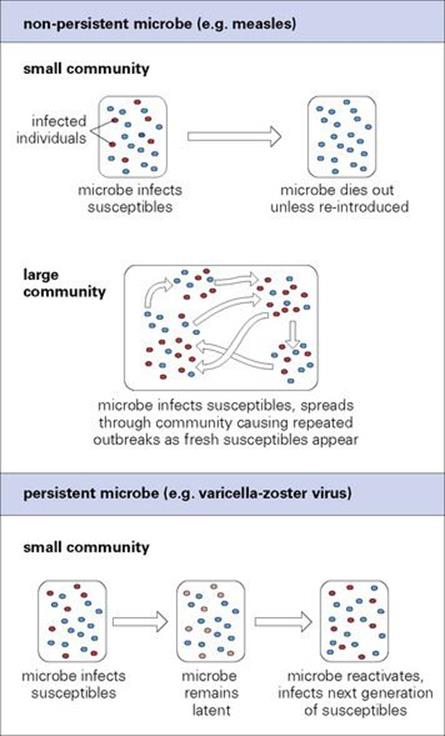
Figure 16.12 Persistence is a microbial survival strategy.
Reactivation of latent infections
Reactivation is clinically important in immunosuppressed individuals
Reactivation occurs in immunocompromised patients, and is of major clinical importance in those immunosuppressed as a result of chronic disease or infection (AIDS), tumours (leukaemias, lymphomas), or in those immunosuppressed by the physician following transplantation (Table 16.4). Reactivation also occurs during naturally occurring periods of immunocompromise, the most important of these being pregnancy and old age. From the microbe’s point of view, latency is an adaptation that allows reactivation with renewed growth and shedding of the infectious agent during these naturally occurring periods.
Table 16.4 Reactivation of persistent infections
|
Circumstance |
Infectious agent |
Site of shedding |
|
Old age |
Varicella-zoster virus |
Skin vesicles |
|
Mycobacterium tuberculosis |
Lung |
|
|
Pregnancy |
Polyomaviruses (BK, JC) |
Urine |
|
Cytomegalovirus |
Cervix |
|
|
Herpes simplex virus 2 |
Cervix |
|
|
Epstein–Barr virus |
Saliva |
|
|
Leukaemias, lymphomas (e.g. Hodgkin’s disease) |
Varicella-zoster virus |
Skin vesicles |
|
Polyomavirus |
CNS (PML)a |
|
|
Post-transplant immunosuppression |
Herpes simplex virus |
Skin/mucosal lesions |
|
Varicella-zoster virus |
Skin vesicles |
|
|
Wart viruses |
Skin |
|
|
Cytomegaloviruses |
Viraemia, pneumonitisa |
|
|
Epstein–Barr virus |
Saliva |
|
|
Hepatitis B,C |
Blood |
|
|
Polyomavirus (BK) |
Urine |
|
|
HIV infection |
Pneumocystis jiroveciib |
Lunga |
|
Toxoplasma gondii |
CNSa |
|
|
Varicella-zoster virus |
Skin vesicles |
|
|
Herpes simplex virus |
Skin/mucosal lesions |
|
|
Mycobacterium tuberculosis |
Lung |
|
|
Polyomavirus (JC) |
CNS (PML)a |
a No shedding from these sites.
b Formerly P. carinii. PML, progressive multifocal leukoencephalopathy.
Features of reactivation in herpesvirus infections are described in Chapters 21 and 26. We still know very little about reactivation mechanisms at the molecular level, as might be expected in view of our ignorance about the latent state itself. Latency is often thought of as a period when the microbe is in deep sleep. However, recent experiments suggest it may be a more active state. For example, M. tuberculosis needs to make certain proteins to keep itself in a latent state. Other products – called resuscitation promoting factors – may be needed to wake up latent M. tuberculosis.
It is useful to distinguish two stages in reactivation
The first event (stage A) in reactivation (Fig. 16.13), the resumption of viral activity in the latently infected cell, is the most mysterious stage. In the case of HSV, this can be triggered by sensory stimuli arriving in the neurone (from skin areas responding to sunlight) and also by certain fevers (i.e. during other infections) or by hormonal influences. Little more than this is known.
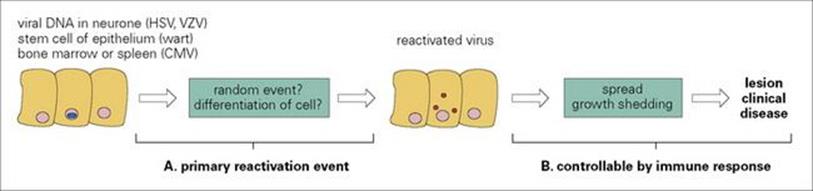
Figure 16.13 Two stages in reactivation of latent viruses. CMV, cytomegalovirus.
The second event (stage B) involves the spread and replication of the reactivated virus. HSV must travel down the sensory axon to the skin or mucosal surface, infect and spread in subepithelial tissues and then in the epithelium, finally forming a virus-rich vesicle (> 1 million infectious units/mL of vesicle fluid). All this takes at least 3–4 days. Stage B is less mysterious than stage A and can be controlled by the immune system. Therefore, cold sores may be associated with poor lymphocyte responses to HSV antigens, and zoster with declining cell-mediated responses (specifically to VZV antigens) in old people.
Stage A probably occurs more frequently than stage B, because immune defences often arrest the process during stage B before final production of the lesion. Hence, as many as 10–20% of HSV reactivation episodes are thought to be ‘non-lesional’ with burning, tingling and itching at the site, but no signs of a cold sore. Also, zoster may involve no more than the sensory prodrome associated with virus reactivation and replication in sensory neurones; skin lesions are prevented by host defences.
Reactivation of EBV and CMV with appearance of the virus in saliva (EBV) or blood (CMV) is generally asymptomatic. In immunologically deficient individuals, however, reactivation may progress to cause clinical disease: either hepatitis and pneumonitis in the case of CMV or the rarer hairy tongue leukoplakia due to EBV (see Ch. 30).
![]()
 Key Facts
Key Facts
• Many successful parasites have adopted strategies for evading immune responses. These enable them to stay in the body long enough to complete their business of infection and shedding to fresh hosts. Some parasites persist indefinitely in the body.
• Mechanisms of immune evasion include:
• concealing parasite antigens from the host (staying inside host cells, infecting ‘privileged sites’)
• changing parasite antigens, either in the infected individual (trypanosomiasis) or during spread through the host population (influenza)
• direct action on immune cells (e.g. HIV on CD4+ T cells) or on immune signalling systems (e.g. production of fake cytokine molecules)
• local interference with immune defences (production of IgA proteases, Fc receptors).
• During persistent infections, the microbe may continue to multiply and be able to infect others (HIV, hepatitis B).
• Alternatively, during persistent infections, the microbe enters into a latent state and later in life reactivates with renewed multiplication and the ability to infect others (herpesviruses, M. tuberculosis).
![]()