Medical Microbiology
Section 4 Clinical manifestation and diagnosis of infections by body system
27 Vector-borne infections
Introduction
A number of important human diseases, caused by organisms ranging from viruses to worms, are transmitted by blood-feeding arthropods. These vectors inject the organisms into humans as they take a blood meal. Two classes of arthropods make the major contribution to disease transmission, the six-legged insects and the eight-legged ticks and mites. Arthropod-transmitted infections are commonest in warmer countries, but occur worldwide. Of these, malaria is undoubtedly the most important. This chapter will also cover schistosomiasis, a major tropical disease, which is often described as vector-transmitted, but the aquatic snail ‘vectors’ are more accurately referred to as intermediate hosts.
Transmission of disease by vectors
In sparsely populated areas, transmission by insects is an effective means of spread
Disease transmission by insects has major implications for the host, the vector and the parasite. To consider the parasite first, it requires the organism to be present in the right place (in the blood) and at the right time (some insects, for example, bite only at night). Blood is an inhospitable environment, and this may require quite subtle evasion mechanisms for parasite survival. In addition, the conditions found in the vector are likely to be very different from those in the human host, and the parasite may have to make a remarkably complex transition in a short time. With the larger protozoal and helminth parasites, this transition often involves clearly visible changes in appearance and is responsible for much of the complicated nomenclature of parasite life cycles. Since some insect vectors have lifespans hardly longer than those of their parasites, there is considerable wastage due to death of the vector before the parasite has matured to the infective stage for humans. A difference of a few days in a mosquito’s lifespan can make an enormous difference to the effectiveness of malaria transmission, and indeed this simple factor is believed to underlie much of the difference between the African pattern of endemic infection and the Indian pattern of sporadic epidemics. However, what may be lost from wastage is more than compensated for by the increased distances over which spread of the parasite can occur.
Vector transmission of disease means that the disease may be controlled by controlling the vector and is, for instance, a major reason why malaria is not endemic in many European countries, where it used to be common.
A potential advantage of this type of transmission for the host is that it is sometimes possible to immunize specifically against the stages infective to humans or those responsible for infecting the vector transmission stages of the parasite. Again, malaria can serve as an example – vaccines against the sporozoites, gametocytes and gametes having been clearly shown to block transmission in animal models. Once transmission is blocked, there is a mathematically calculable possibility that the disease will die out. A vaccine against sporozoites has shown promising activity in protecting African children from falciparum malaria.
Arbovirus infections
Arboviruses are arthropod-borne viruses
A wide range of about 500 different viruses is transmitted by arthropods such as ticks, mosquitoes and sandflies. These arboviruses multiply in the arthropod vector, and for each virus there is a natural cycle involving vertebrates (various birds or mammals) and arthropods. The virus enters the arthropod when the latter takes a blood meal from the infected vertebrate, and passes through the gut wall to reach the salivary gland where replication takes place. Once this has occurred, 1–2 weeks after ingesting the virus, the arthropod becomes infectious, and can transmit the virus to another vertebrate during a blood meal. Certain arboviruses that infect ticks are also transmitted directly from adult tick to egg (transovarial transmission), so that future generations of ticks are infected without the need for a vertebrate host.
Only a small number of arboviruses are important causes of human disease
Arboviruses tend to replicate in vascular endothelium, the central nervous system (CNS), skin and muscle, and are therefore multisystem infections. They are generally named after the clinical disease (e.g. yellow fever) or the place where they were first discovered (e.g. Rift Valley fever, Japanese encephalitis). A few (e.g. Ross River virus in Australia and the Pacific and Chikungunya virus in Africa and Asia) cause arthritis.
The human stage of the virus cycle may be essential (urban yellow fever, dengue), there being no other vertebrate host, or it may be ‘accidental’ from the virus’s point of view, with humans acting as ‘dead end’ hosts who do not form a necessary part of the natural cycle (e.g. equine encephalitides, West Nile virus).
Yellow fever
Yellow fever virus is transmitted by mosquitoes and is restricted to Africa, Central and South America and the Caribbean
Yellow fever virus is a flavivirus, and there is only one antigenic type. It was taken to the Americas by the early slave traders, and the first recorded case was in Yucatan in 1640. Yellow fever virus is transmitted by two different cycles:
• from human to human by the mosquito Aedes aegypti; which is well-adapted to breeding around human habitations; the infection can be maintained in this way as ‘urban’ yellow fever
• from infected monkeys to humans by mosquitoes such as Haemagogus. This is ‘jungle’ yellow fever and is seen in Africa and South America.
Yellow fever is not transmitted directly from human to human by day-to-day contact, but transmission from ill patients to healthcare workers has been reported, notably after needlestick injury.
Clinical features of yellow fever may be mild, but in 10% to 20% of cases classic yellow fever with liver damage occurs, which can prove fatal
The virus enters dermal tissues or blood vessels at the site of a mosquito bite and spreads through the body, infecting vascular endothelium and liver. After an incubation period of 3–6 days, there is a sudden onset of fever, headache and muscular aches. Although mild cases occur, prostration and shock are not uncommon, and severe liver damage may result in death. Coagulation defects (largely due to prothrombin deficiency) cause haemorrhage into the gastrointestinal tract (manifest as haematemesis and melena) and elsewhere. Renal damage is also seen, with proteinuria and occasionally tubular necrosis. The case mortality rate is in the range of 5–50%.
The diagnosis is usually clinical; there is no specific treatment, but there is a vaccine
The virus can be isolated from blood during the acute stage, and a post-mortem diagnosis can be made from the severe mid-zonal changes and acidophilic inclusion bodies seen in the liver. Virus-specific IgM antibodies are detectable after a week.
The best prevention is to give the live attenuated 17D yellow fever vaccine to those who may be exposed. Protection lasts at least 10 years, and vaccination is necessary for entry into and travel through endemic areas. The international health regulations permit a state to require a valid certificate of vaccination from a traveller from an endemic area to another country where the right mosquitoes are present but the disease does not occur (e.g. from tropical Africa to India). Vaccines based on recombinant DNA technology have been developed. As with all arthropod-borne infections, control of arthropod vectors (insecticides, attention to breeding sites) and reduced exposure (insect repellents, mosquito nets) are also important.
Dengue fever
Dengue virus is transmitted by mosquitoes and occurs in SE Asia, the Pacific area, India, South and Central America
Dengue is one of the most rapidly re-emerging arbovirus diseases, with between 50 and 100 million cases each year. Dengue virus is a flavivirus with four antigenic subtypes. The mosquito A. aegypti is the principal human vector. The virus also circulates in monkeys and can be transmitted by mosquitoes to cause ‘jungle’ dengue in humans, a disease analogous to jungle yellow fever.
Dengue fever may be complicated by dengue haemorrhagic fever/dengue shock syndrome
Dengue virus replicates in monocytes and possibly in vascular endothelium. After an incubation period of 4–8 days, there is malaise, fever, headache, arthralgia, nausea and vomiting, and sometimes a maculopapular or erythematous rash. Recovery may be followed by prolonged fatigue and/or depression.
Dengue haemorrhagic fever/dengue shock syndrome (DHF/DSS) is a particularly severe form of the disease. In the past, mortality rates were high, but with prompt access to expert hospital care, a fatality rate of below 1% can be achieved. The pathogenesis of this syndrome is shown inFigure 27.1. After an earlier attack of dengue, antibodies are formed that are specific for that serotype. On subsequent infection with a different serotype, the antibodies bind to the virus and not only fail to neutralize it (as might be expected for a different subtype), but actually enhance its ability to infect monocytes. The Fc portion of the virus-bound immunoglobulin molecule attaches to Fc receptors on monocytes, and entry into the cell by this route increases the efficiency of infection. Infection of increased numbers of monocytes results in an increased release of cytokines into the circulation (see Ch. 17) and this leads to vascular damage, shock and haemorrhage, especially into the gastrointestinal tract and skin. Similar ‘enhancing’ antibodies are formed in many other virus infections, but it is only in dengue haemorrhagic fever that they are known to play a pathogenic role. A number of other factors can influence the course of dengue infection, including age and dengue virus strain virulence.
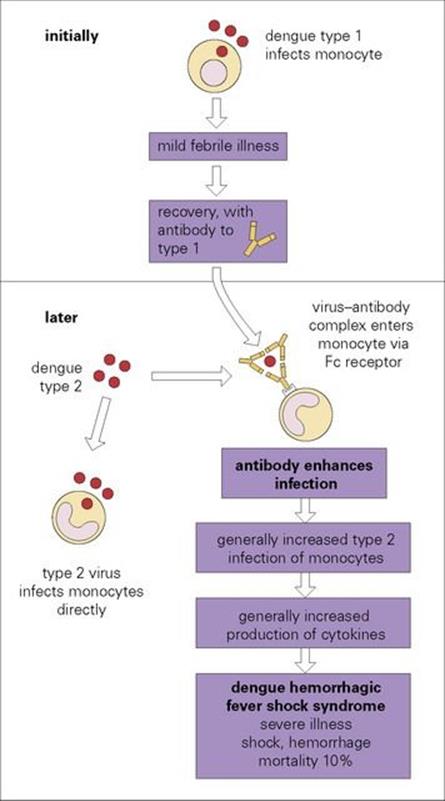
Figure 27.1 The pathogenesis of dengue haemorrhagic shock syndrome. There are four serotypes of dengue virus. Types 1 and 2 are illustrated as examples. Antibody to type 1 binds to type 2 without preventing infection with type 2.
There is no antiviral therapy for dengue fever. Treatment is supportive. The World Health Organization has published a revised dengue case classification based on the presence or absence of warning signs in order to improve patient care (see bibliography).
There is no currently licensed dengue vaccine. A suitable vaccine must be tetravalent to avoid the danger that a vaccine could induce the type of antibody associated with DHF/DSS. Candidate live attenuated tetravalent vaccines are undergoing field testing.
Chikungunya virus infection (CHIKV)
Chikungunya is an arbovirus transmitted mainly by Aedes aegypti. The disease is present in Africa and Asia and resulted in an outbreak in Italy in 2007. The illness is similar to dengue but polyarthritis is very common and retro-orbital pain rare in CHIKV.
Arbovirus encephalitis
The encephalitic arboviruses only occasionally cause encephalitis
Six of the ten encephalitic arboviruses listed in Table 27.1 cause disease in the USA, and although most infections are subclinical or mild, fatal encephalitis can occur. The viruses replicate in the CNS, but a cell-mediated immune response to infection makes a major contribution to the encephalitis. Vaccines against Western equine encephalitis (WEE), Eastern equine encephalitis (EEE) and Venezuelan equine encephalitis (VEE), each of which may cause disease in horses, have been used for laboratory workers. A Japanese encephalitis vaccine is also available and is used in the UK for the occasional at-risk traveller. Laboratory diagnosis is carried out in special centres, occasionally by virus isolation, but more commonly by demonstrating a rise in specific antibody.
Table 27.1 Arboviruses causing encephalitis
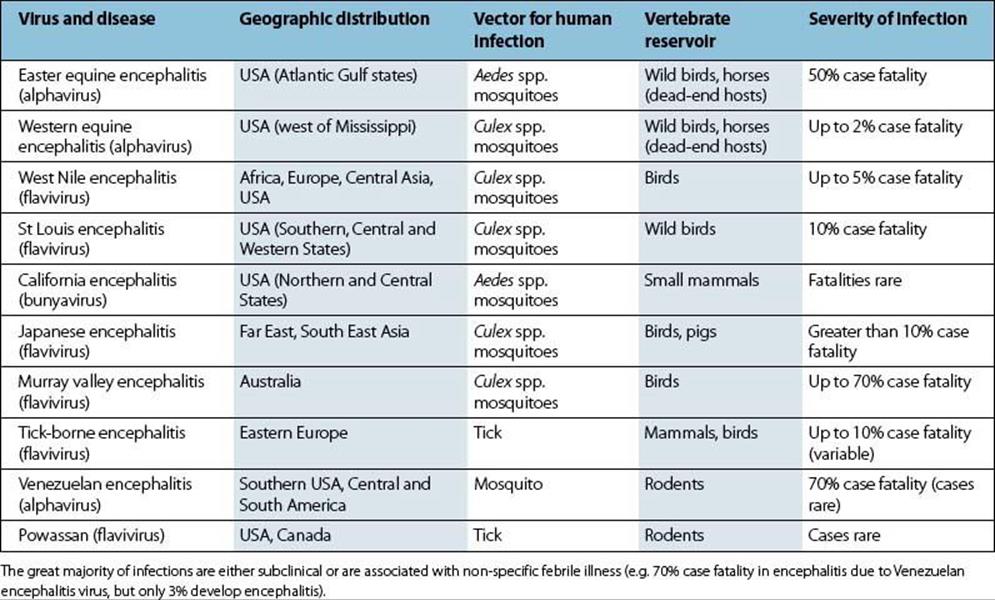
Prior to the mid-1990s, West Nile virus, which is transmitted from infected birds by Culex mosquitoes and for which humans are considered to be dead-end hosts, was not considered a major public health problem, but viral changes then resulted in cases with severe neurologic disease. The virus, which had not previously been reported from the Western hemisphere, was recorded in New York in 1999 and since then has spread widely in the USA, Canada, Mexico and the Caribbean. In 2006, the CDC reported a total of more than 1500 human cases in the USA and more than 150 blood donors with the virus. By 2010, it was reported to have caused more than 25 000 cases, 12 000 of whom had severe neurologic disease with more than 1100 deaths. Quite how the virus crossed the Atlantic is unknown though it has been suggested that it was probably imported in a live bird. Infection is diagnosed clinically (fever > 38.8°C, neurologic symptoms, elevated CSF cell count and protein, possible muscular weakness) and serologically. Vaccines and immunotherapeutic agents are being developed.
Arboviruses and haemorrhagic fevers
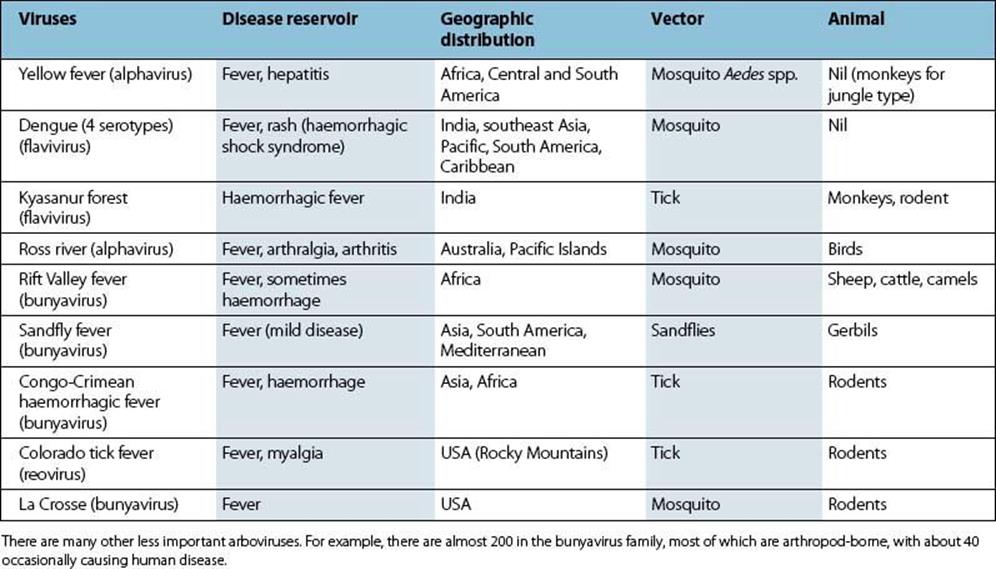
Arboviruses are major causes of fever in endemic areas of the world
Arbovirus infections are often subclinical or mild, but occasionally there is a severe haemorrhagic illness. Some of the best known of these infections are listed in Table 27.2. Laboratory diagnosis by isolation of virus, by detection of viral genome or by demonstration of a rise in antibody is possible in special centres.
Table 27.2 Arboviruses causing fevers and haemorrhagic disease
Infections caused by rickettsiae
The rickettsiae are a group of intracellular, arthropod-transmitted Gram-negative aerobic rods (see Ch. 2 and Appendix). Previously the group included, among others, the genera Rickettsia, Bartonella, Coxiella, Ehrlichia and Orientia. Genomic-based analysis has resulted in a complete reclassification of the group, but for convenience, these genera are all included here. These organisms are ‘debilitated’ in the sense that all, except Bartonella, are obligate intracellular parasites. All are carried in arthropod or animal reservoirs (Fig. 27.2). All are transmitted to humans by arthropods except Coxiella, which appears to infect following inhalation from environmental sources; thus person-to-person transmission does not occur.
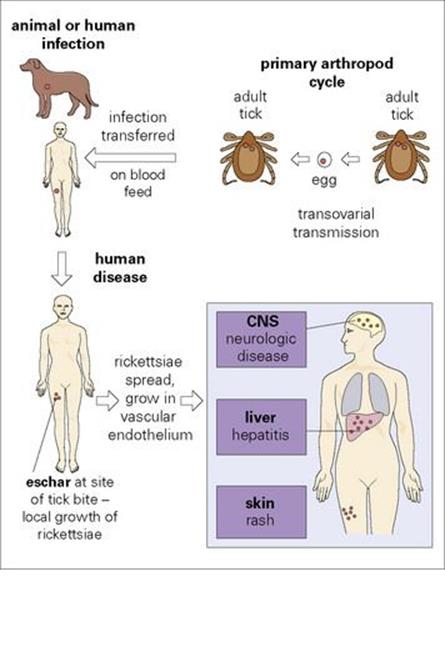
Figure 27.2 Typical events in rickettsial infection. There is no direct person-to-person spread. Q fever is atypical (see text). Typhus is unusual because the infected arthropod transmits from person to person, eventually dies and there is no eschar. CNS, central nervous system.
The rickettsiae are small bacteria and infections tend to be persistent or become latent
Howard T. Ricketts identified ‘Rocky Mountain spotted fever’ in 1906 and showed that it was transmitted transovarially in ticks. Rickettsiae probably arose as parasites of blood-sucking or other arthropods in which they were maintained by vertical transmission, transfer to the arthropod’s vertebrate host being initially ‘accidental’ and not necessary for rickettsial survival. The infected arthropod does not appear to be adversely affected. Rickettsia prowazekii is perhaps a more recent parasite of the human body louse, because the louse dies 1–3 weeks after infection. As with most arthropod-borne infections, transmission from person to person does not occur.
Typical clinical symptoms of rickettsial infection are fever, headache and rash
Rickettsiae multiply in vascular endothelium to cause vasculitis in skin, CNS and liver, and hence are multisystem infections (Table 27.3). In spite of immune responses, there is a tendency for rickettsial infections to persist in the body for long periods or become latent.
Table 27.3 The principal rickettsial diseases in humans
Except for Q fever (caused by Coxiella burnetii), the typical clinical features are fever, headache and rash. A history suggesting contact with rickettsial vectors or reservoir animals may suggest a diagnosis (e.g. camping, working, engaging in military activities in endemic areas).
Laboratory diagnosis is based on serologic tests
Complement fixation tests are specific for different rickettsiae. However, microimmunofluorescence methods are the most common serological approach, and demonstration of a fourfold or greater rise in titre is considered to be a positive result. Infected patients also make antibody to the rickettsiae that cross-react with the O antigen polysaccharide of various strains of Proteus vulgaris, as detected by agglutination in the Weil–Felix test. The agglutination pattern with three strains of Proteus can be used to identify the rickettsiae. Although the phenomenon is of interest, the test is not of great value, because of false-positive and false-negative results. Earlier diagnosis can often be made by fluorescent antibody staining of skin biopsy material. PCR diagnostic tests are now available. Isolation of rickettsiae is difficult and dangerous, and laboratory infections have occurred.
All rickettsiae are susceptible to tetracyclines
Prevention is based on reducing exposure to the vector (e.g. ticks). A killed R. prowazekii vaccine is available for the military, and a killed C. burnetii vaccine is available for those at high risk (e.g. military, veterinarians, etc.) with no demonstrated sensitivity to the antigen.
Rocky Mountain spotted fever
Rocky Mountain spotted fever is transmitted by dog ticks and has a mortality of up to 10%
The rickettsiae causing this disease are carried by the dog tick (Dermacentor variabilis) or by wood ticks (Dermacentor andersoni) and are transmitted vertically from adult tick to egg. Human infection occurs in the warm months of the year as ticks become active. Children are most commonly infected, but their disease is milder.
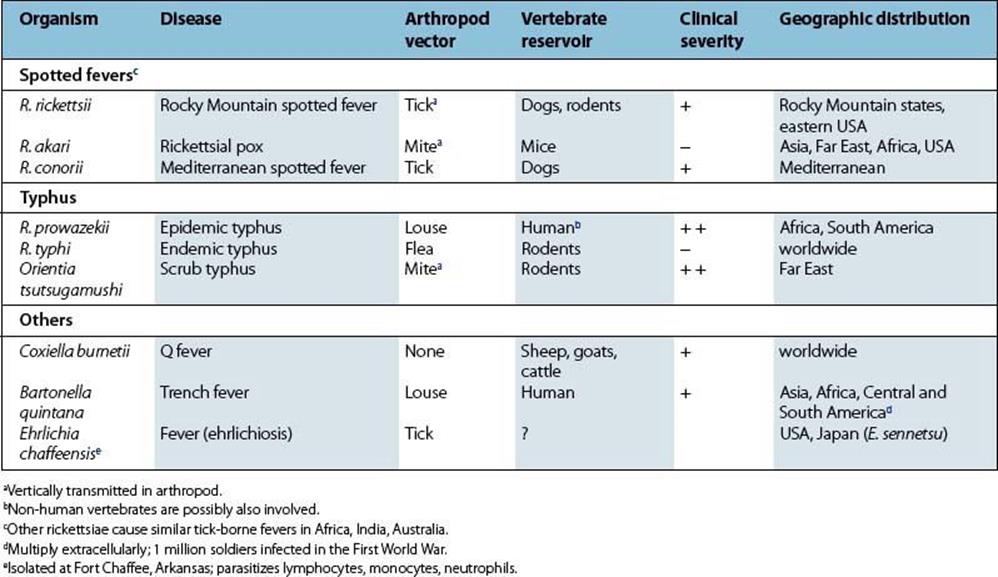
The rickettsiae multiply in the skin at the site of the tick bite, then spread to blood and infect vascular endothelium in the lung, spleen, brain and skin. After an incubation period of about 1 week, there is onset of fever, severe headache and myalgia, and often respiratory symptoms. A generalized maculopapular rash appears a few days later, often becoming petechial or purpuric (Fig. 27.3). There is splenomegaly, and neurologic involvement is frequent, with later onset of clotting defects (disseminated intravascular coagulation), shock and death. Fatal cases are usually those with a delayed diagnosis. Peak mortality is seen in 40–60-year-olds.
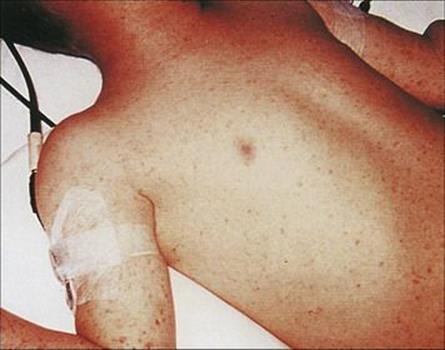
Figure 27.3 Generalized maculopapular rash with petechiae in Rocky Mountain spotted fever.
(Courtesy of T.F. Sellers, Jr.)
Mediterranean spotted fever
Mediterranean spotted fever is transmitted by dog ticks
Mediterranean spotted fever is caused by Rickettsia conorii, carried by the dog tick Rhipicephalus sanguineus. Human infection, which occurs mainly in October, is known in all Mediterranean countries and can occur in urban as well as rural areas. After an incubation period of about 1 week, 50% of cases develop fever, headache and myalgia, and 2–4 days later a rash, especially on the palms and soles. The bite usually goes unnoticed as it is caused by immature ticks and is painless; a local lesion is not usually seen.
African tick-bite fever
Four rickettsial species are found in Africa. R. africae is found mainly in urban areas and R. conorii in semi-rural and rural areas. African tick-bite fever is regularly seen in travellers returning from Africa to the temperate zone.
Rickettsialpox
Rickettsialpox is a mild infection
About 5 days after the bite of a rodent-associated mite (Allodermanyssus sanguineus) infected with R. akari, a local eschar develops with fever and headache occurring approximately 1 week later. After a few more days, a generalized papulovesicular rash then appears. The disease is, however, mild and usually lasts for about 1 week.
Epidemic typhus
Epidemic typhus is transmitted by the human body louse
Epidemic typhus is transmitted from person to person by Pediculus humanus. The rickettsiae (R. prowazekii) multiply in the gut epithelium of the louse and are excreted in faeces during the act of biting. The rickettsiae enter the skin when the bite is scratched. The disease cannot maintain itself unless enough people are infested with lice. Epidemic typhus is therefore classically associated with poverty and war, when clothes and bodies are washed less frequently. There were 30 million cases in Eastern Europe and the Soviet Union from 1918 to 1922. The disease is seen in Africa, Central and South America, and sporadically (as a sylvatic form) in the USA. As there is no direct person-to-person spread, outbreaks can be terminated by delousing campaigns.
Untreated epidemic typhus has a mortality as high as 60%
Rickettsiae proliferate at the site of the bite and then spread in the blood to infect vascular endothelium in skin, heart, CNS, muscle and kidney. About 1 week after the louse bite (there is no eschar) the infected person develops fever, headache and flu-like symptoms. The generalized maculopapular rash appears 5–9 days later and sometimes there is severe meningoencephalitis with delirium and coma. In untreated cases, mortality can range from 20% in healthy individuals to as high as 60% in elderly or compromised patients, due to peripheral vascular collapse or secondary bacterial pneumonia.
Convalescence may take months. In some individuals, the rickettsiae are not eliminated from the body on clinical recovery and remain in the lymph nodes. As much as 50 years later, the infection can reactivate to cause Brill–Zinsser disease, and the patient once again acts as a source of infection for any lice that may be present.
Endemic (murine) typhus
Endemic typhus is caused by Rickettsia typhi and is transmitted to humans by the rat flea. The disease is similar to epidemic typhus, but is less severe.
Scrub typhus
Scrub typhus is a severe illness caused by Orientia tsutsugamushi and is transmitted to humans by trombiculid mites (chiggers). It occurs only in the Far East; cases were seen in American soldiers in Vietnam. The rickettsiae are maintained in the mites by transovarial transfer and are transmitted to humans or rodents during feeding. There is fever and an eschar, and a macular rash appears after about 5 days of illness. Pneumonitis, meningitis, disseminated intravascular coagulation and circulatory collapse may ensue. Treatment is with tetracycline or doxycycline and must be given early. Vaccines based on recombinant antigens or on DNA are being developed.
Borrelia infections
Relapsing fever
The epidemic form of relapsing fever is caused by Borrelia recurrentis, which is transmitted by human body lice
Borrelia recurrentis is a Gram-negative spirochete consisting of an irregular spiral, 10–30 μm long, and is highly flexible, moving by rotation and twisting.
Epidemics of relapsing fever (Fig. 27.4) are due to transmission of infection by the human body louse. Bacteria multiply in the louse, and when louse bites are rubbed, the lice are crushed and the bacteria are introduced into the bite wound. Lice are essential for person-to-person transmission of louse-borne relapsing fever. As with other louse-borne infections (e.g. typhus), spread of the disease in humans is favoured when people rarely wash and when clothes are not changed (e.g. in wars, natural disasters). The last great epidemic in North Africa and Europe during the Second World War caused 50 000 deaths.
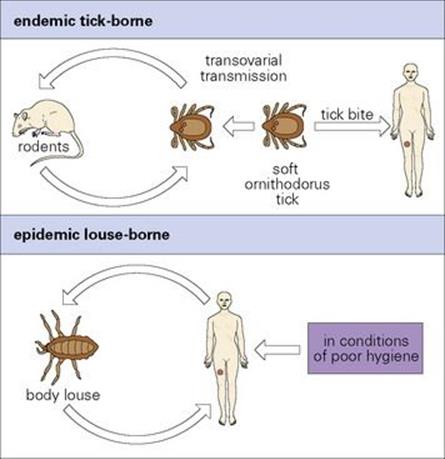
Figure 27.4 Transmission in relapsing fever.
The endemic form of relapsing fever in humans is transmitted by tick bites
Infections with other species of Borrelia are endemic in rodents in many parts of the world, including western USA, and are transmitted by soft ticks of the genus Ornithodoros. In the tick, the bacteria are transmitted transovarially from generation to generation, which, together with their ability to survive for up to 15 years between feeds, helps maintain the endemic cycle of this form of relapsing fever.
Relapsing fever is characterized by repeated febrile episodes due to antigenic variation in the spirochetes
The bacteria multiply locally and enter the blood. After an incubation period of 3–10 days, there is a sudden onset of illness with chills and fever, lasting for 3–5 days (Fig. 27.5). The afebrile period lasts about a week before there is a second attack of fever, which is followed by another afebrile period. Generally, there are 3–10 such episodes, of diminishing severity. More serious illness can occur if there is extensive growth of bacteria in the spleen, liver and kidneys.
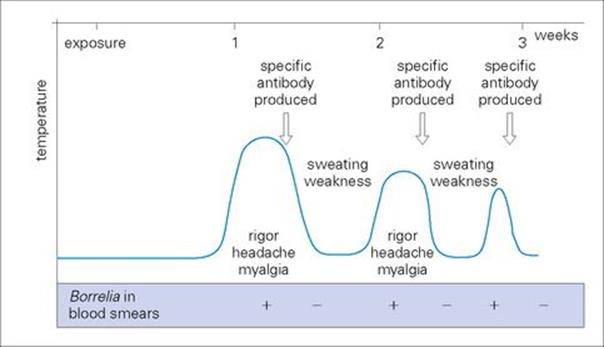
Figure 27.5 Course of events in relapsing fever.
Agglutinating and lytic antibodies are formed against the infecting bacteria, which are cleared from the blood. Under the ‘pressure’ of this immune response, a new antigenic type emerges and is free to multiply and cause a fresh febrile episode.
Antigenic variation involves switching of variable proteins on the bacterial surface. The Borrelia have arrays of genes (variable large proteins (Vlp) and variable small proteins (Vsp)) that are altered and activated by gene conversion involving plasmids carrying collections of these genes. The result is that a single cloned bacterium can give rise spontaneously to approximately 30 serotypes and switching occurs at a rate of 1:1000 to 1:10 000 per cell generation. Similar phenomena are seen in trypanosomes. Direct person-to-person transmission does not occur. Mortality with endemic (tick-borne) relapsing fever is < 5%, but may be up to 40% in epidemic (louse-borne) relapsing fever.
Relapsing fever is diagnosed in the laboratory and treated with tetracycline
The bacteria can be cultivated in the laboratory and can be seen in Giemsa-stained smears of blood taken during the febrile period (Fig. 27.6). Complement fixation antibody tests are available, but are rarely useful because of the problem of antigenic variation.
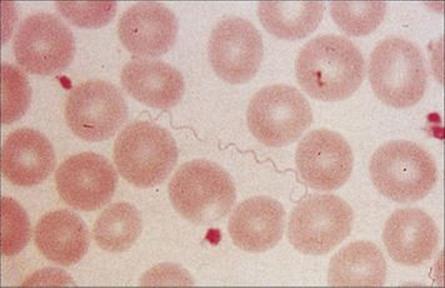
Figure 27.6 Tightly coiled helical spirochetes of Borrelia recurrentis in the blood of a patient with relapsing fever.
(Courtesy of T.F. Sellers.)
Tetracycline is used in treatment and to prevent relapses. The best preventative measure is avoidance of arthropod vectors.
Lyme disease
Lyme disease is caused by Borrelia spp. and is transmitted by Ixodes ticks
Lyme disease (or Lyme borreliosis) occurs in Europe, the USA and most continents of the world, and is named after the town in Connecticut, USA, where the first cases were recognized in 1975. It is caused by Borrelia burgdorferi in the USA; in Europe by B. garinii, B. afzelii predominate, with B. burgdorferi less common. The natural cycle of infection takes place in small mammals in which it is transmitted by hard ticks of the genus Ixodes (Fig. 27.7). Human infection follows the bite of an infected tick (most commonly the nymph). In Europe and the USA, infection is more common in summer months when recreational exposure to infected ticks is more likely. Person-to-person transmission does not occur.
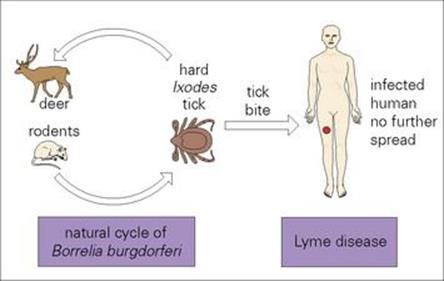
Figure 27.7 Transmission of Lyme disease.
Erythema migrans is a characteristic feature of Lyme disease
The bacteria multiply locally, and after an incubation period of about 1 week, fever, headache, myalgia, lymphadenopathy and a characteristic lesion at the site of the tick bite develop. The skin lesion is called erythema migrans (Fig. 27.8), its name describing its main features. It begins as a macule and enlarges over the next few weeks, remaining red and flat, but with the centre clearing, until it is several centimeters in diameter. In 50% of patients, fresh transient lesions appear on the skin elsewhere in the body. Immunologic findings include circulating immune complexes and sometimes elevated serum IgM levels and cryoglobulins that contain IgM. Borrelia is capable of evading the human immune response and mechanisms include antigenic variation and the ability to evade complement-mediated killing.
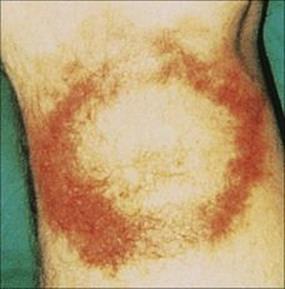
Figure 27.8 Rash of erythema chronicum migrans on the leg in Lyme disease.
(Courtesy of E. Sahn.)
Lyme disease commonly causes additional disease 1 week to 2 years after the initial illness
In 75% of untreated patients, in spite of antibody and T-cell responses to the Borrelia, there are additional later manifestations of disease. These are seen from 1 week to > 2 years after the onset of illness. The first of these manifestations to appear are neurologic (meningitis, encephalitis, peripheral neuropathy) and cardiologic (heart block, myocarditis). The second of these manifestations to appear are arthralgia and arthritis, which may persist for months or years. Immune complexes are found in affected joints. These late manifestations are immunologic in origin and are probably due to antigenic cross-reactivity between Borrelia and host tissues. The Borrelia themselves are rarely detectable at this stage.
Lyme disease is diagnosed serologically and treated with antibiotic
Borrelia can be cultured (in NSK medium) from early-stage cutaneous tissues but is rarely seen at later stages; culture may take several weeks. Thus, Lyme disease is primarily diagnosed on clinical presentation and known exposure. When indicated, serologic tests such as enzyme-linked immunosorbent assay (ELISA) are useful, with Western blot confirmation of all positive and equivocal results. Specific IgM antibodies are detected 3–6 weeks after infection and IgG antibodies at a later stage. PCR diagnosis has been disappointing.
Doxycycline or amoxicillin are effective in treatment of early disease. Late disease, especially with neurologic complications, may require more aggressive therapy, for example, with intravenous ceftriaxone for 30 days.
Prevention of Lyme disease is by avoidance of tick bites.
Protozoal infections
Malaria
Malaria is initiated by the bite of an infected female anopheline mosquito
Malaria is restricted to areas where these mosquitoes can breed, i.e. the tropics between 60°N and 40°S (except areas higher than about 2000 m). It is of major importance in Africa, India, the Far East and South America. Drug and insecticide resistance present major challenges to malaria control. About 35% of the world’s population is estimated to be infected, with some 10 million new cases annually and perhaps 2 million deaths. Increased air travel means malaria is regularly seen as an imported disease in non-malarious countries, and unless the possibility of malaria is constantly borne in mind, the diagnosis may be delayed or missed altogether, with fatal results. Malaria can also be transmitted by blood transfusion, needlestick accidents or from mother-to-fetus or neonate.
The life cycle of the malaria parasite comprises three stages
Five species of Plasmodium cause malaria in humans, of which P. falciparum and P. knowlesi are the most virulent (Table 27.4). All have similar life cycles, which are the most complex of any human infection, comprising three quite distinct stages and characterized by alternating extracellular and intracellular forms (Figs 27.9, 27.10).
Table 27.4 Human malaria parasites
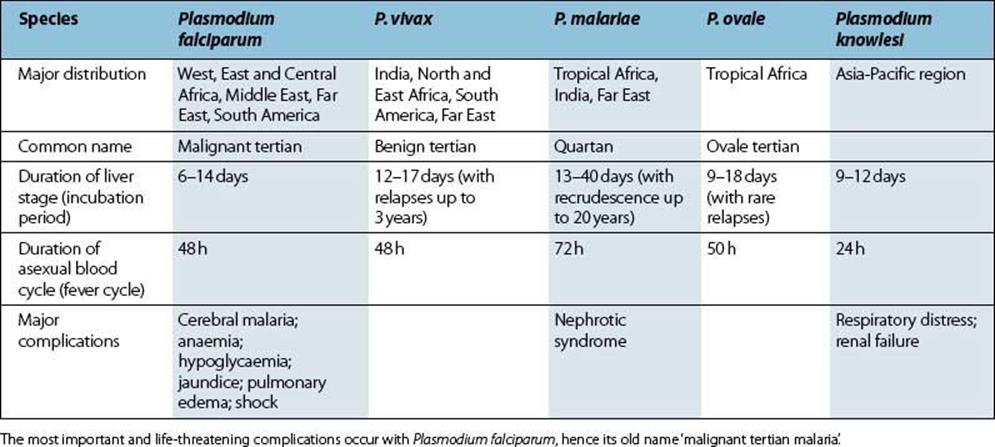
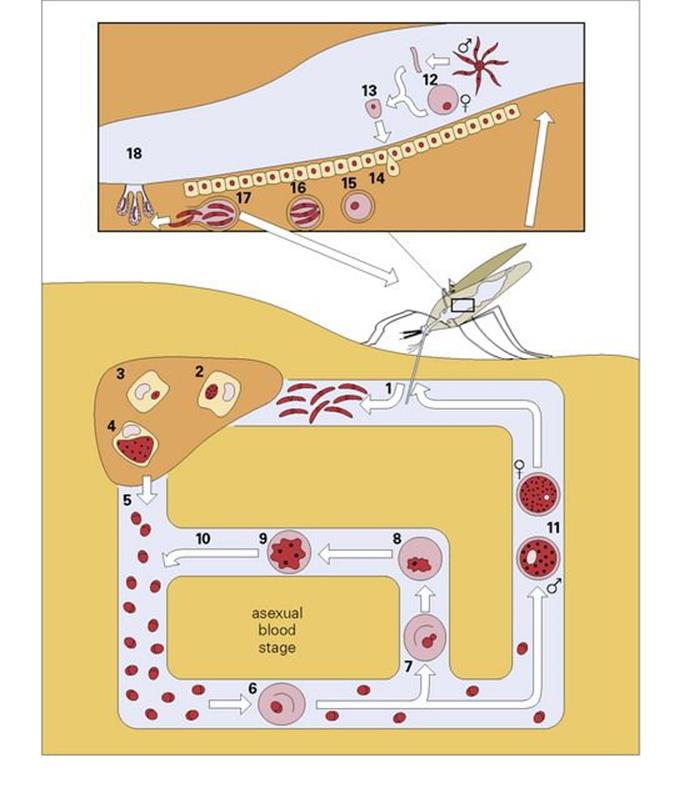
Figure 27.9 The life cycle of malaria in human and mosquito. In the symptomless pre-erythrocytic stage, sporozoites from the saliva of an infected Anopheles mosquito are injected into the human bloodstream when the mosquito bites (1). They then enter the parenchymal cells of the liver (2), where they mature in approximately 2 weeks into tissue schizonts (4), finally rupturing to produce 10 000– 40 000 merozoites (5). These circulate in the blood for a few minutes before entering the red blood cells (6) to initiate the asexual blood stage. For P. vivax and P. ovale only, some parasites, however, remain within the liver to lie dormant as hypnozoites (3), which are the cause of relapses. Once in the red blood cells, the merozoites mature into the ring form (7), trophozoite (8) and schizont (9), which complete the cycle by maturing to release merozoites back into the circulation (10). This cycle may last for months or even years. Some merozoites, however, go on to initiate the sexual stage, maturing within the red blood cells to form male and female gametocytes (11), which can be taken up by the Anopheles mosquito on feeding. On entering the gut of the insect, the male gametocyte exflagellates (12) to form male microgametes, which fertilize the female gamete to form the zygote (13). This then invades the gut mucosa (14), where it develops into an oocyst (15). This develops to produce thousands of sporozoites (16), which are released (17) and migrate to the salivary glands of the insect (18), whence the cycle begins again.
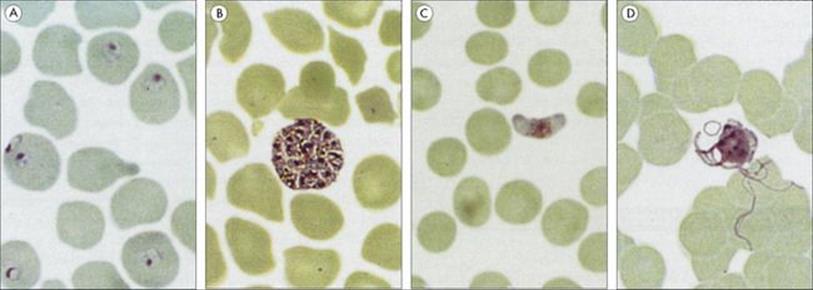
Figure 27.10 Different stages of the malaria parasites. (A) Plasmodium falciparum ring forms in red blood cells. (B) Plasmodium vivax erythrocytic schizont. (C) P. falciparum female gametocyte. (D) P. vivax male gametocytes exflagellating to form microgametes 20–25 μm long.
Invasion of red cells requires at least two separate receptor–ligand interactions; the lack of one red cell surface receptor, the Duffy antigen, explains the resistance to P. vivax of most West Africans. Other genetic traits that contribute to resistance to malaria include haemoglobin S (sickle cell), β-thalassaemia, and glucose-6-phosphate dehydrogenase deficiency.
Clinical features of malaria include a fluctuating fever and drenching sweats
Symptoms range from fever to fatal cerebral disease or multi-organ failure and are associated exclusively with the asexual blood stage (Fig. 27.9). The clinical picture depends upon the age and immune status of the patient, as well as the species of parasite. The most characteristic feature is fever, which follows rupture of erythrocytic schizonts and is mainly due to the induction of cytokines such as interleukin 1 (IL-1) and tumour necrosis factor (TNF). The synchronous cycle in red cells means that the different species of malaria give characteristic patterns of fever, with either a 48-h (tertian: days 1 and 3) or 72-h (quartan: days 1 and 4) periodicity (Fig. 27.11). However, this classical pattern of fever is seldom seen in clinical practice where a chaotic fever pattern is common. Furthermore, it is possible to be afebrile yet obviously very unwell with malaria. A typical paroxysm starts with a feeling of intense cold with shivering, followed by a hot dry stage and finally a period of drenching sweats. Headache, muscle pains and vomiting are common. The symptoms of malaria closely resemble those of influenza, which is a common misdiagnosis. Jaundice may be present and lead to an erroneous diagnosis of viral hepatitis. Fever may be the only physical sign in early malaria, but later enlargement of the spleen and liver is common and anaemia almost invariable.
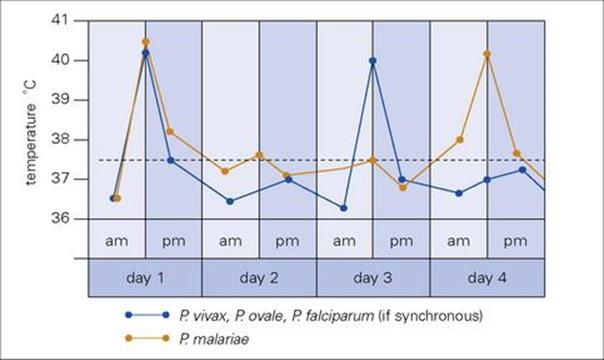
Figure 27.11 Malaria fever charts showing cyclical fluctuations in temperature. The peaks coincide with the maturation and rupture of the intraerythrocytic schizonts, occurring every 48 h (Plasmodium falciparum, Plasmodium vivax and Plasmodium ovale) or every 72 h (Plasmodium malariae), when the cycles are synchronized.
In the absence of reinfection, P. vivax, P. ovale and P. malariae malarias are normally self-limiting though debilitating infections. P. malariae may persist in the blood at a low level for decades and recrudesce to cause symptoms from time to time. Relapses (defined as hypnozoite-induced) may occur with P. vivax and P. ovale months or even 1–2 years after the initial malarial illness.
P. falciparum malaria is frequently fatal during the first 2 weeks due to a variety of complications (Table 27.4). In hyperendemic areas, complicated falciparum malaria is most common in children aged between 6 months and 5 years, and in pregnant, particularly primigravid, women. However, it can occur at any age in the non-immune (e.g. tourists). The most dangerous complication is cerebral malaria, with convulsions and diminished level of consciousness progressing to coma. Possible causes include binding of parasitized red cells in cerebral capillaries, increased permeability of the blood–brain barrier, and excessive induction of cytokines such as TNF. If successfully treated, there is usually little or no impairment of cerebral function, although neurologic and psychiatric sequelae may occur in 5–10% of childhood cases.
Severe anaemia is also common, due partly to red cell destruction and partly to dyserythropoiesis in the bone marrow. Of the other complications, hypoglycaemia and lactic acidosis are thought to be important contributors to mortality. Acute renal failure due to acute tubular necrosis is an important complication of falciparum malaria and nephrotic syndrome may occur with P. malariae (quartan malarial nephropathy).
Malaria has an immunosuppressive effect and interacts with HIV infection
The strong epidemiologic correlation between malaria and endemic Burkitt’s lymphoma probably reflects reduced T-cell cytotoxicity against Epstein–Barr virus (EBV)-infected cells. Malaria may also interfere with the efficacy of vaccines against common viral or bacterial infections.
In pregnant females, HIV-1 infection is associated with more peripheral blood malaria, more placental malaria, higher parasite densities, more fever, and increased risk of adverse birth outcomes. In semi-immune non-pregnant adults, HIV-1 infection is associated with higher rates of malaria infection and higher rates of clinical disease. In non-immune, non-pregnant adults, HIV-1 infection is associated with higher rates of severe malaria and death.
Immunity to malaria develops gradually and seems to need repeated boosting
Immunity to malaria develops in stages, and in endemic areas, children who survive early attacks become resistant to severe disease by about 5 years. Parasite levels fall progressively until adulthood when they are low or absent most of the time. However, 1 year spent away from exposure is sufficient for most of this immunity to wane, i.e. repeated boosting is needed to maintain it. The actual mechanisms are still controversial and seem to involve both antibody and cell-mediated immunity (Fig. 27.12).
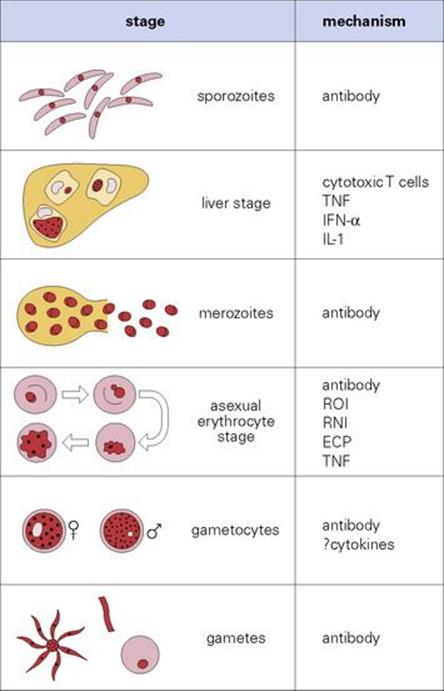
Figure 27.12 Immunity to malaria. The principal mechanisms thought to be responsible for immunity at each stage of the cycle. IFN, interferon; IL, interleukin; TNF, tumour necrosis factor; ROI, reactive oxygen intermediates; RNI, reactive nitrogen intermediates; ECP, eosinophil cationic proteins.
Malaria is diagnosed by finding parasitized red cells in thin and thick blood films
Later (schizont) stages may be sequestered in deep tissues, so parasites may be deceptively scarce in, or even absent from, the peripheral blood. A single negative blood film does not exclude malaria. Further samples should be taken 12–24, and 48 h later. Any case of fever, especially with anaemia, splenomegaly or cerebral signs, in a patient who conceivably could have malaria is therefore best treated as malaria, while continuing to look for other diagnoses and seeking expert help. However, the presence of parasites in the blood of an ill patient from an endemic area does not mean malaria is the cause of the illness, so other causes of fever should still be borne in mind while they receive treatment for malaria. For example, they might have lobar pneumonia and coincidental malarial parasitaemia, since low-grade parasitaemia may be asymptomatic in those with partial malarial immunity.
Where available, intravenous artesunate (in combination with other antimalarials to avoid the development of drug resistance) is the drug of choice for severe malaria. Intravenous quinine is used if artesunate cannot be obtained without delay.
Chloroquine is no longer of value for the treatment of falciparum malaria, but remains the drug of choice for the blood stages of P. vivax, P. ovale and P. malariae infections. Primaquine (contraindicated in G6PD deficiency) is used to kill hypnozoites of P. vivax or P. ovale in the liver and thus prevent relapses of these infections.
In endemic areas, the most promising method of prevention is the use of bed nets impregnated with an insecticide, e.g. permethrin. DDT is again being recommended for indoor residual spraying, where it does not affect the environment or get into food chains. The prospects for a malaria vaccine are discussed in Chapter 34.
Trypanosomiasis
Three species of the protozoan Trypanosoma cause human disease
Trypanosoma brucei gambiense and T. b. rhodesiense cause African trypanosomiasis or sleeping sickness, and T. cruzi causes South American trypanosomiasis or Chagas disease. The diseases differ markedly in:
• the insect vector
• the localization of the parasite
• the effects on the immune system.
African trypanosomiasis
African trypanosomiasis is transmitted by the tsetse fly and restricted to equatorial Africa
The vector of African trypanosomiasis is the tsetse fly Glossina, and there is a reservoir of infection in several domestic and wild animals (cattle, pigs, deer). In humans, T. brucei remains extracellular, first in the tissues near the insect bite and then in the blood, where it divides rapidly and continuously.
Clinical features of African trypanosomiasis include lymphadenopathy and sleeping sickness
Following an infected bite, a swollen chancre develops at the site (T. b. rhodesiense only), with widespread lymph node enlargement, especially in the back of the neck (Winterbottom’s sign; Fig. 27.13A). The parasite establishes in the blood and multiplies rapidly, with fever, splenomegaly and, often, signs of myocardial involvement. The CNS may become involved (more acutely in the East African T. b. rhodesiense than the West African T. b. gambiense), with the gradual development of headache, psychologic changes (‘silent grief’), voracious appetite and weight loss, and finally coma (sleeping sickness; Fig. 27.13B) and death. Unlike malaria, parasitologically cured trypanosomiasis can leave the patient with severe residual neurologic and mental disability.
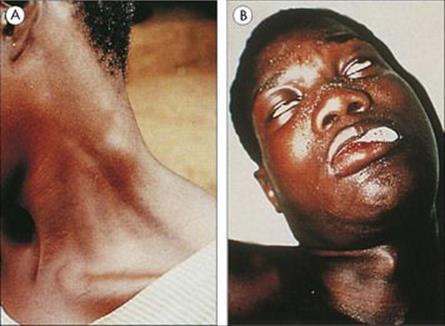
Figure 27.13 African trypanosomiasis. (A) Enlargement of the lymph nodes in the neck (Winterbottom’s sign). (Courtesy of P.G. Janssens.) (B) Coma (sleeping sickness) due to generalized encephalitis.
(Courtesy of M.E. Krampitz and P. de Raadt.)
T. brucei evades host defences by varying the antigens in its glycoprotein coat
T. brucei survives freely in the blood because of its remarkable degree of antigenic variation, based on switching between some 1000 different genes for the glycoprotein coat. A high concentration of IgM is found in the blood, and later in the cerebrospinal fluid (CSF), and this is manufactured by the plasma cells (Mott cells), which are a feature of the lymphocytic infiltrate seen as perivascular cuffing around blood vessels in the brain (Fig. 27.14).
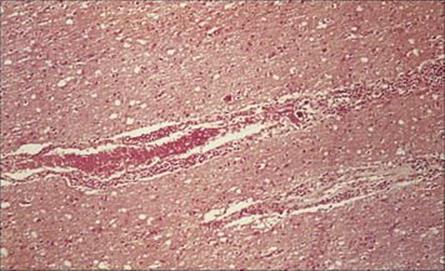
Figure 27.14 Lymphocytic infiltration around a blood vessel in the brain in Trypanosoma brucei infection (H&E stain).
(Courtesy of R. Muller and J.R. Baker.)
African trypanosomiasis is diagnosed by demonstrating parasites microscopically in blood, lymph nodes (by puncture) or in late cases in CSF. Detection of antitrypanosomal antibody is used to screen populations, with further parasitological examination of those who are seropositive.
East African trypanosomiasis is treated with suramin intravenously for the haemolymphatic stage, followed by melarsoprol intravenously (very toxic) if the CNS is involved. West African trypanosomiasis is treated with pentamidine intramuscularly or eflornithine intravenously for the haemolymphatic stage. CNS involvement is treated with eflornithine intravenously or nifurtimox orally plus eflornithine intravenously (NECT).
Pentamidine prophylaxis is no longer deployed
Control of the tsetse fly vector is difficult, though insecticides are widely used. Bed nets are ineffective, as the flies feed during daylight hours.
Chagas disease
T. cruzi is transmitted by the reduviid (‘kissing’) bug
T. cruzi is transmitted by reduviid (kissing) bugs, which readily inhabit poor housing, so Chagas disease is characteristically a disease of the rural poor. Almost all species of mammal can act as reservoirs of infection. The parasite invades host cells, notably macrophages and cardiac muscle cells.
Chagas disease has serious long-term effects, which include fatal heart disease
Chancres (‘chagomas’) may develop at the site of infection, with a transient febrile illness that may rarely lead to death by heart failure. Following invasion of host cells, the disease pursues an extremely slow and chronic course. Some infected individuals remain in the indeterminate phase of the disease and do not develop complications. In cases where the disease does progress, the major complications, which can take years to appear, involve the heart and the intestinal tract. The major cause of death is myocarditis, with progressive weakening and dilation of the ventricles due to destruction of cardiac muscle by the parasite (Fig. 27.15) and probably also to autoimmune mechanisms induced by cross-reacting antigens. Cardiac aneurysm and heart block are particularly serious features. Dilatation of the intestinal tract is due to similar processes in nerve cells, and the organs become incapable of proper peristalsis; megaoesophagus and megacolon are the two commonest manifestations.
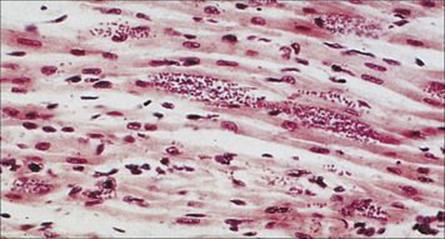
Figure 27.15 Amastigote forms of Trypanosoma cruzi in cardiac muscle in Chagas disease (H&E stain).
(Courtesy of H. Tubbs.)
Chronic Chagas disease is usually diagnosed serologically
In the acute phase, parasites may be seen in a blood film, but the chronic disease is usually diagnosed serologically or by xenodiagnosis. Clean reduviid bugs are fed on the patient and their rectal contents examined 1–2 months later, or homogenized and injected into mice, in which even a single trypanosome will produce a patent infection. Polymerase chain reaction (PCR) techniques are now in use and may replace or be used in combination with xenodiagnosis.
Chagas disease is very difficult to cure. Children respond better to antitrypanosomal drugs than do adults. Nifurtimox orally or benznidazole orally are the drugs of choice. Side effects are common with both of them. Posaconazole shows promising activity and has been used clinically.
Prevention is achieved by improved housing and living standards, vector control plus active case finding and treatment. However, vector control by insecticides is difficult. Trypanosoma cruzi is adept at evading the immune response and thus cannot be eliminated by the human host, a major challenge to vaccine development.
Leishmaniasis
Leishmania parasites are transmitted by sandflies and cause New World and Old World leishmaniasis
Several species of Leishmania parasites cause disease in both the New World and the Old World (Table 27.5). In the latter areas especially, dogs can act as an important reservoir of infection. All are transmitted by sandflies.
Table 27.5 Leishmania species – their distribution and clinical syndromes
|
Species |
Distribution |
Diseases |
|
L. donovani |
|
|
|
L. chagasi |
South America |
|
|
L. major |
|
|
|
L. tropica |
||
|
L. aethiopica |
Africa |
|
|
L. mexicana |
Mexico and Central America |
|
|
L. braziliensis |
South America |
Leishmania is an intracellular parasite and inhabits macrophages
Leishmania evades the killing mechanisms of macrophages (Fig. 27.16) unless they are strongly activated, e.g. by interferon-gamma (IFNγ). The two principal sites of parasite growth are:
• the liver and spleen (visceral leishmaniasis)
• the skin (cutaneous leishmaniasis).
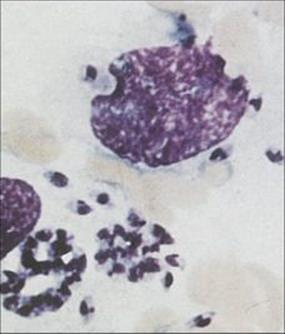
Figure 27.16 Leishmania within macrophages in aspirate from a lesion of New World leishmaniasis.
(Courtesy of M.J.Wood.)
Untreated visceral leishmaniasis (‘kala-azar’) is fatal in 80–90% of cases
Visceral leishmaniasis, or kala-azar, usually develops slowly, with fever and weight loss, followed months or years later by hepatomegaly and, especially, splenomegaly. Skin lesions may appear following treatment; these contain massive numbers of parasites, and the syndrome is known as post-kala-azar dermal leishmaniasis (PKDL). With appropriate treatment, only those who are very ill at diagnosis die.
Cutaneous leishmaniasis is characterized by plaques, nodules or ulcers
Classic cutaneous leishmaniasis progresses insidiously, from a small papule at the site of infection to a large ulcer. This may eventually heal with considerable scarring (Fig. 27.17), leaving the patient relatively immune to reinfection. Old World leishmanial lesions are known as Oriental sores (also ‘Baghdad boil’ and ‘Delhi sore’) and New World leishmaniasis as espundia (mucosal leishmaniasis due to Leishmania (Viannia) braziliensis) and chiclero ulcer (Leishmania mexicana infection of the pinna).
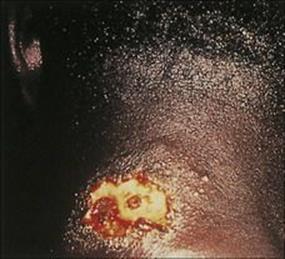
Figure 27.17 Cutaneous lesion on the neck in Leishmania braziliensis infection.
(Courtesy of P.J. Cooper.)
Immunodeficient patients may suffer more severe leishmaniasis
In immunodeficient patients, widespread chronic skin lesions can occur – diffuse cutaneous leishmaniasis – analogous to lepromatous leprosy. Visceral leishmaniasis is a major complication of HIV infection not only in the tropics but also around the Mediterranean, though it is now easier to manage with antileishmanial drugs since the advent of highly active antiretroviral therapy (HAART).
Leishmaniasis is diagnosed by demonstrating the organism microscopically and is treated with antimonials
Demonstration of the organism by microscopy of splenic aspirate or biopsies of bone marrow or skin lesions (depending upon the clinical picture) is definitive proof of leishmaniasis.
Detection of antileishmanial antibody by the Leishmania direct agglutination test is valuable in the diagnosis of visceral leishmaniasis.
A positive delayed hypersensitivity reaction to leishmanial antigens (Montenegro or leishmanin skin test) can be useful in the diagnosis of cutaneous leishmaniasis if parasites are not found.
Where available, the polymerase chain reaction is now the method of choice for the detection and species identification of Leishmania in skin biopsies.
Cutaneous leishmaniasis is treated by local injection of the edge of the ulcer with sodium stibogluconate (an antimonial). Intravenous sodium stibogluconate is used to treat multiple or potentially disfiguring lesions. The agent of choice for the treatment of visceral leishmaniasis is intravenous liposomal amphotericin B. Intravenous sodium stibogluconate is an alternative, though there is now significant antimony-resistant visceral leishmaniasis in parts of India.
Impregnated bed nets are effective against the sandfly vector, and the animal reservoir can be eliminated by dog control.
The prospects for vaccination against the cutaneous disease are quite promising.
Helminth infections
Schistosomiasis
Schistosomiasis is transmitted through a snail vector
All digeneans (flukes) must pass through a mollusc intermediate host in order to complete their larval development. However, schistosomes are the only group in which larvae penetrate directly into the final host after release from the snail.
The life cycle of schistosomes is illustrated in Figure 27.18. Infected snails, which are always aquatic, release fork-tailed larvae into the surrounding water. These penetrate the host’s skin, enter the dermis and pass via the blood, through the lungs to the liver, where they mature and form permanent male and female pairs before relocating to their final site:
• the veins surrounding the bladder for Schistosoma haematobium
• the mesenteric veins around the colon for Schistosoma japonicum and Schistosoma mansoni.
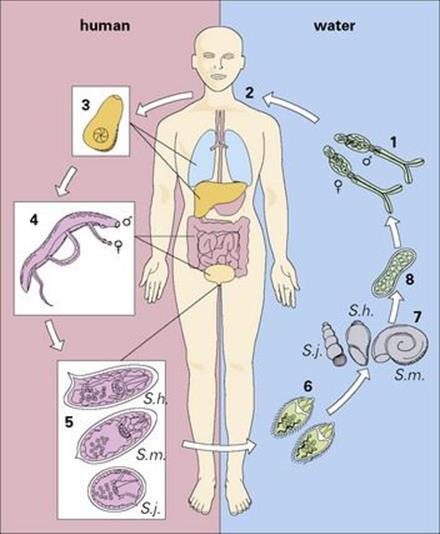
Figure 27.18 Life cycle of schistosomes. Free-swimming cercariae in water (1) penetrate unprotected skin. (2) During penetration, they lose their tails to become schistosomulae. (3) These migrate through the bloodstream via the lungs and liver to the veins of the bladder (Schistosoma haematobium) or bowel (Schistosoma mansoni, Schistosoma japonicum), where they mature (4), to produce characteristic eggs (5) within 6–12 weeks. The eggs then penetrate the bladder or colon, to be passed in the urine or the faeces (6). Eggs passed into fresh water release miracidia which penetrate snail intermediate hosts (7) where they mature into sporocysts (8). These release cercariae (1) into the water to complete the cycle.
The cycle is completed when eggs laid by the female worms move across the walls of the bladder or bowel and leave the body.
Clinical features of schistosomiasis result from allergic responses to the different life cycle stages
The stages of skin penetration, migration and egg production are each associated with pathologic changes, collectively affecting many body systems. Penetration can cause a dermatitis, which becomes more severe on repeated reinfection. The developmental stages are associated with the onset of allergic symptoms (fever, eosinophilia, lymphadenopathy, spleno- and hepatomegaly, diarrhea), but the most severe pathology arises following the onset of egg laying. The body becomes hypersensitive to antigens released by the eggs as they pass through tissues to the outside world, or become trapped in other organs after being swept away in the bloodstream.
• In urinary schistosomiasis caused by S. haematobium, movement of eggs through the bladder wall causes haemorrhage. With time, the bladder wall becomes inflamed. Infiltrated polyps develop and malignant changes may follow; nephrosis may also occur (see Ch. 20).
• Release of the eggs of S. japonicum and S. mansoni similarly causes intestinal haemorrhage and inflammation.
A more serious consequence of these infections results from the inflammatory responses to eggs that become trapped in other organs of the body, primarily the liver, but also the lung and CNS. These consequences do not develop in all patients, but if they do, severe disease may ensue (seeCh. 22). Formation of granulomas by delayed hypersensitivity reactions around eggs in the presinusoidal capillaries, interferes with blood flow and, together with extensive portal fibrosis (Symmers’ pipestem fibrosis), leads to portal hypertension. As a consequence, there is hepatosplenomegaly, collateral connections form between the hepatic vessels, and fragile oesophageal varices develop. The collateral circulation can lead to eggs being washed into the capillary bed of the lungs.
Intense inflammatory reactions are also provoked when worms killed by anthelmintic treatment are carried back from the mesenteric vessels into the liver.
Schistosomiasis is treated with praziquantel
Treatment of individuals with praziquantel removes the worms, but in advanced cases, the pathology is irreversible. Vaccine research is making progress, but a vaccine may be more useful in minimizing pathology, by reducing the numbers of eggs released, than preventing infection.
Control of infection at a population level is achieved by breaking the transmission cycle, through avoidance of infected water and improvement in sanitation. Mass treatment programmes aim to reduce morbidity but evidence is emerging that they can also reduce transmission.
Filariasis
Filarial nematodes depend upon blood-feeding arthropod vectors for transmission
The filarial nematodes parasitize the deeper tissues of the body (see Ch. 6). The most important species can be divided into those located in the lymphatics (Brugia, Wuchereria) and those in subcutaneous tissues (Onchocerca). A number of less harmful species also occur. In all species, the female worms release live microfilaria larvae, which are picked up by the vector from the blood (lymphatic species) or skin (Onchocerca). Both groups can cause severe inflammatory responses, reflected in a variety of pathologic responses in the skin and lymph nodes, but each is associated with additional and characteristic pathology. (Descriptions of the diseases caused by Onchocerca are given in Chs 25 and 26).
Lymphatic filariasis caused by Brugia and Wuchereria is transmitted by mosquitoes
The mosquitoes introduce the infective larvae into the skin as they feed. These larvae migrate to the lymphatics and develop slowly into long thin adult worms (females 80–100 mm × 0.25 mm), found in the lymph nodes and lymphatics of the limbs (usually lower) and groin. Infections become patent after about 1 year, when sheathed microfilariae appear in the blood. Infected individuals may show few clinical signs or have acute manifestations such as fever, rashes, eosinophilia, lymphangitis, lymphadenitis (Fig. 27.19) and orchitis. Later chronic obstructive changes, caused by repeated episodes of lymphangitis, may block lymphatics, leading to hydrocele and to the gross enlargement of breasts, scrotum and limbs, the latter condition being known as elephantiasis (Fig. 27.20). Secondary bacterial infection of the skin, e.g. with streptococci, is a major factor in the development and progression of filarial adenolymphangitis.
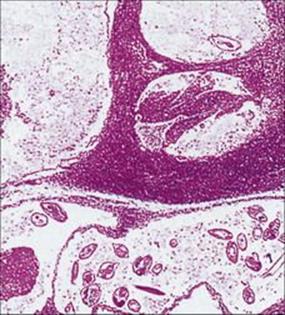
Figure 27.19 Lymph node containing adult Wuchereria, showing dilated lymphatics and tissue reaction in the vessel walls.
(Courtesy of R. Muller and J.R. Baker.)
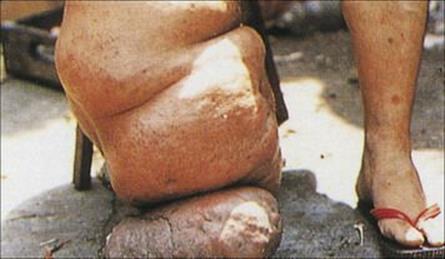
Figure 27.20 Elephantiasis of the leg, caused by Brugia malayi.
(Courtesy of A.E. Bianco.)
A feature of filarial infections in endemic regions is that not everyone exposed develops symptomatic infections. Many, although microfilaraemic, remain asymptomatic, and relatively few show gross pathology (Fig. 27.21). Some individuals develop pulmonary symptoms known as ‘tropical pulmonary eosinophilia’ (see Ch. 19).
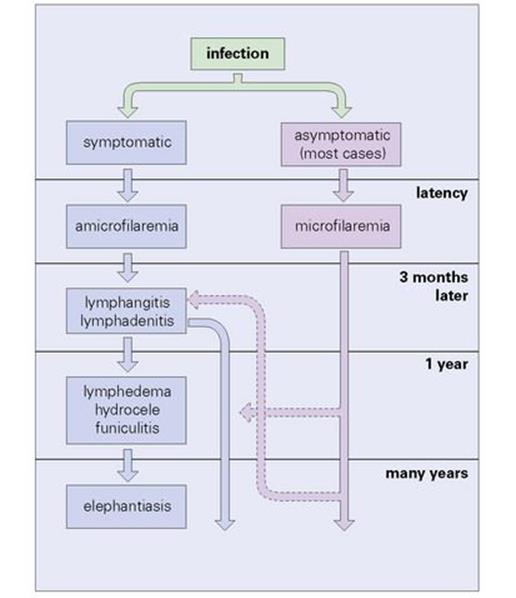
Figure 27.21 Course of lymphocytic filariasis in symptomatic cases.
(Redrawn from: Muller R., Baker J.R. Medical Parasitology. London: Gower Medical Publishing, 1990.)
Few drugs are really satisfactory for treating filariasis
Diethylcarbamazine (DEC), which primarily kills microfilariae, is no longer used for the treatment of onchocerciasis as it produces a violent allergic response when microfilariae are killed. A single low dose is, however, used in the Mazzotti test for onchocerciasis in patients whose skin snips are negative for microfilariae. Ivermectin is effective against onchocerciasis and has been used to treat lymphatic filariasis as well, in combination with albendazole. Albendazole plus DEC is used in mass drug administration programmes to eliminate lymphatic filariasis. Antibiotics, such as doxycycline, which kill the Wohlbachia symbionts, also are effective against the worms in lymphatic filariasis.
It is difficult to prevent transmission of filariasis, although this can be minimized by vector control and prevention of biting.
![]()
 Key Facts
Key Facts
• Many important infections (arboviruses, rickettsiae, Borrelia, protozoa, helminths) are transmitted by vectors – insects, ticks or snails.
• Some infections are chronic (Lyme disease, leishmaniasis, schistosomiasis) or can be lethal (malaria, viral encephalitis).
• Often they are restricted to tropical countries because of the distribution of the vector. Climate change may alter this distribution and therefore the pattern of diseases transmitted.
• Strong immune responses are mounted, often leading to immunopathologic complications. Treatment is usually by chemotherapy.
• Vector control is difficult, but can lead to disease eradication.
• With very few exceptions (yellow fever), vaccines are not available for this group of diseases.
![]()