Medical Microbiology
Section 1 The adversaries – microbes
7 Prions
Introduction
Prions are unusual infectious agents associated with a number of human, animal and fungal diseases. In humans they can cause degenerative changes in the brain: the transmissible spongiform encephalopathies. Kuru is the classic example of such a condition, epidemiological studies confirming human–human transmission. Prions lack a nucleic acid genome and are highly resistant to all conventional forms of disinfection processes. They are small proteinaceous particles that are modified forms of a normal cellular protein, and cause disease by converting normal protein into further abnormal forms. Prion-related conditions can arise endogenously by mutation (and be inherited) or be acquired exogenously during medical procedures or by ingestion of contaminated material. The prion diseases are part of a spectrum of neurodegenerative disorders in which soluble proteins are modified and accumulate as insoluble beta-sheet rich amyloid fibrils. The other neurogenerative disorders that include different types of dementia are not infectious but are sporadic or inherited, sharing a common pathogenesis. Endogenous sporadic Creutzfeldt–Jakob disease (CJD) has been known for some time as have Gerstmann–Sträussler–Scheinker disease, fatal familial insomnia and kuru. However, in the 1990s another form of this disease (variant CJD, vCJD) was associated with eating beef from cattle infected with the prion that causes bovine spongiform encephalopathy.
‘Rogue protein’ pathogenesis
Prions are unique infectious agents
There are a number of human and animal diseases – the spongiform encephalopathies – whose pathology is characterized by the development of large vacuoles in the CNS. These include kuru and Creutzfeldt–Jakob diseases (CJD) in humans, bovine spongiform encephalopathy (BSE) in cattle and scrapie in sheep. Sporadic CJD is the most common prion disease in humans worldwide and the incidence is approximately 1.5 per million people. For a long time, these diseases were thought to be caused by so-called unconventional slow viruses, but it is now known that the agents concerned are prions; small, proteinaceous infectious particles. Their characteristics include:
• small size (< 100 nm, therefore filterable)
• lack of a nucleic acid genome
• extreme resistance to heat, disinfectants and irradiation (but susceptible to high concentrations of phenol, periodate, sodium hydroxide, sodium hypochlorite)
• slow replication – typically diseases have a long incubation period and usually appear late in life. Incubation periods of up to 35 years have been recorded in humans, but variant CJD can produce symptoms much more rapidly
• cannot be cultured in vitro
• do not elicit immune or inflammatory responses.
Prions are host-derived molecules
Studies on scrapie gave some insight into the nature of prions and their role in disease. The infectious agent is a host-derived 30–35 kDa glycoprotein (termed PrPSc, prion protein scrapie) that is associated with the characteristic intracellular fibrils seen in diseased tissue. PrPSc is derived from a naturally occurring cellular prion protein (PrPc), expressed predominantly on the surface of nerve cells and coded by a single copy gene of unknown function (located on chromosome 20 in humans). Mice with the PrPc gene disrupted are resistant to scrapie, and they show no gross abnormalities. The two proteins have a similar sequence, but differ in structure and protease resistance; PrPSc is globular and enzyme resistant; PrPc is linear and enzyme susceptible. The association of PrPSc with PrPc results in conversion of the latter into the abnormal form, the change being largely conformational, from alpha helices to beta-pleated sheets. Affected cells produce more PrPc and the process is then repeated, the accumulating PrPSc aggregating into amyloid fibrils and plaques (Fig. 7.1). Replication can lead to very high titres of infectious particles and up to 108–109/g of brain tissue have been recorded.
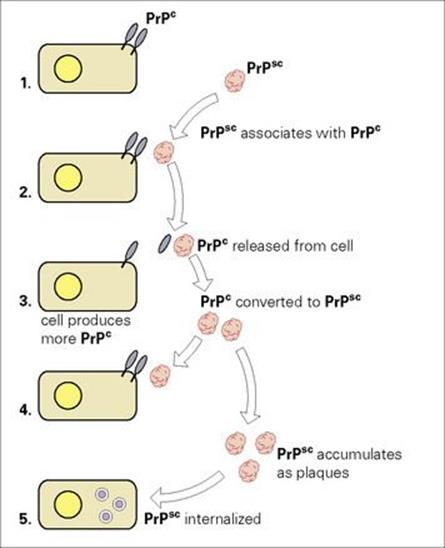
Figure 7.1 How prions may damage cells. (1) Normal cells express PrPc at the cell membrane as linear proteins. (2) PrPSc exists as a free globular glycoprotein, which can interact with PrPc. (3) PrPc is released from the cell membrane and is converted into PrPSc. (4) Cells produce more PrPcand the cycle is repeated. (5) PrPSc accumulates as plaques, and is internalized by cells.
Evidence that the interaction of PrPSc with PrPc causes these events is based on extensive experiments in sheep and mice, the main conclusions being:
• Scrapie infectivity in material co-purifies with PrPSc.
• Purified PrPSc confers greater scrapie activity.
• Mice lacking the PrPc gene do not develop disease when injected with prions.
• Introduction of a PrP transgene from a prion donor species (e.g. hamster) into a recipient species (e.g. mouse) facilitates cross-species transmission, suggesting that homology between the PrP genes of donor and recipient is the main molecular determinant of such transmission.
• In vitro, PrPSc can convert PrPc into PrPSc, with the transfer of biochemical characteristics.
The development of scrapie in sheep shows strong genetic influences, some breeds being much more resistant than others, and similar genetic effects have been shown in mice. In humans, homozygosity for methionine at codon 129 of the prion protein gene is a major determinant of susceptibility to sporadic, iatrogenic and vCJD. There is also variation in prions, different strains being described. These combinations of host and prion variation result in a spectrum of disease onset and severity.
Development, transmission and diagnosis of prion diseases
PrP is a modified host protein and the gene is located on chromosome 20. The normal form of the prion protein is referred to as PrPc. PrPSc is an abnormal isoform of PrP and accumulates in brain tissue. It only differs to PrPc by having an increased beta-sheet content, which makes it more stable. In particular, it is quite resistant to proteolysis. The normally folded protein PrPC is converted to an abnormal conformation by direct contact with the misfolded form PrPSc. If the load of the latter increases it can lead to a rapid neurodegenerative phenotype. PrPSc can be built into different structures and so these PrPSc species can result in a variety of prion diseases such as sporadic CJD and two other human prion diseases: Gerstmann–Sträussler–Scheinker syndrome and fatal familial insomnia. There is some evidence that people have a genetic predisposition for sporadic CJD. There is a naturally occurring polymorphism at codon 129 of the PrPc gene on chromosome 20 and this codes for the amino acid methionine or valine. Compared with the unaffected population, people with sporadic CJD are many times more likely to be methione homozygous at this locus.
With the exception of those cases where prions arise by mutation, transmission and spread of prion disease requires exposure to the infective agent. Ways in which this could occur include eating contaminated food material, use of contaminated medical products (blood, hormone extracts, transplants), the introduction of prions from contaminated instruments during surgical procedures, as prions bind strongly to metal surfaces, and possibly mother–fetus transmission during pregnancy (although none of the hundreds of infants born to mothers with kuru developed the disease). The disease kuru was transmitted by eating the brains of dead humans in funeral rites, and vCJD is associated with eating contaminated beef products. In these cases, prions survive digestion and are taken up across the intestinal mucosa. They are then carried in lymphoid cells, eventually being transferred into neural tissues and entering the CNS.
Prions can cross species boundaries
Although prions from one species are more effective in transmitting disease to the same species, transmission can occur between different species (Fig. 7.2). The most serious example of this is the transfer of prions from cattle infected with BSE to humans through consumption of infected meat, which has been associated with outbreaks of vCJD. BSE itself arose as a result of transfer to cattle of prions from sheep infected with scrapie, and in 1996 it became clear that human vCJD and BSE were caused by the same prion strain. Unlike CJD itself, vCJD caused disease in younger individuals (14 years and upwards) with a much shorter incubation period. The number of human infections likely to arise from the UK epidemic of BSE in cattle (thought to have affected more than 2 million animals) is still controversial, though some believe the potential to be quite small. CJD surveillance was started in the United Kingdom in 1990 in order to identify the number of human infections arising from the UK epidemic of BSE in cattle that was thought to have affected more than 3 million animals. This estimate was based on the likely number of asymptomatic animals and the clinical diagnosis of BSE made in over 180 000 cattle. vCJD was first reported in the UK in 1996 by the National CJD Surveillance Unit. Those affected had a clinical and pathological phenotype distinct from sporadic CJD and were homozygous for methionine at codon 129. Again, this demonstrated a genetic predisposition for vCJD. vCJD is the only prion disease affecting humans that can be acquired from another species and is caused by BSE. This has also been shown by animal transmission studies in which the infectious agent associated with vCJD was shown to have the same biological properties as that causing BSE. Epidemiological studies suggest that the most likely route of transmission is the oral route, the affected individual having eaten beef contaminated with the BSE agent. PrPSc has been found in the lymphoreticular system including the tonsils and spleen as well as neurological tissues and the prion may be carried in the blood by lymphocytes.
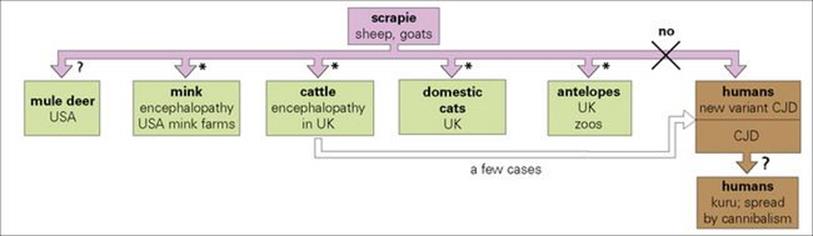
Figure 7.2 The spread of scrapie agents between species. Nearly all have been transmitted to laboratory rodents and primates.
(*Infections transferred by scrapie-infected sheep materials present in foodstuff. Most of these infectious agents have mutations at amino acid residue 129 of the prion protein, which are thought to cause conversion of the protein into the pathogenic form.)
Overall, by July 2010, 220 people had developed vCJD in 11 countries around the world, 173 of whom were diagnosed in the UK. That number was much lower than predicted by mathematical modellers in the 1990s. As the incubation period can be very long, it is unclear how many people could be at risk and asymptomatic. Issues surrounding diagnostic tests include assay sensitivity and specificity, resulting in difficulty in comparing studies. A large study was carried out in the UK investigating more than 32 000 anonymized tonsil tissues for disease related prion protein referred to as PrPCJD from people who underwent an elective tonsillectomy. Of these, 12 753 were from the 1961–1985 birth cohort that included the time most vCJD cases had arisen and 19,908 were from the 1986–95 cohort that would potentially have been exposed to BSE-contaminated meat products. PrPCJD was not detected in any samples.
Prion diseases are difficult to diagnose
Because prions cannot be cultured, and since there is no immune response, prion disease in its early stages cannot be diagnosed easily. Clinical appearances usually indicate the probable occurrence of prion disease and this can be confirmed histologically post mortem. Tonsillar tissue is a good source of PrPSc in clinical cases and these prions can be identified by immunoblotting or immunohistochemistry. Tonsillar and other tissue homogenates can also be tested for the presence of the abnormal prion protein by enzyme immunoassays. These have been used in a number of studies and the development of diagnostic tests is important not only to make a diagnosis but also from a public health standpoint to prevent infection, as transmission by blood and blood products has been reported.
Lessons from Kuru
Kuru is a condition that was identified with cannibalistic behaviour in Papua New Guinea. There were more than 2700 infections between 1957 and 2004, the incubation period of the disease being estimated at more than 50 years. The fatality rate fell from over 200 per year in the late 1950s to 6 per year in the early 1990s. This reduction followed the prohibition of cannibalistic behaviour in the 1950s. A study investigating suspected kuru cases between 1996 and 2004 identified 11 infected individuals. The minimum estimated incubation periods in this group ranged from 34 to 41 years, the range in males being from 39 to at least 56 years. Analysis of the prion protein gene (PRNP) showed that most patients with kuru were heterozygous at codon 129.
Prevention and treatment of prion diseases
Prion diseases are incurable
Although, as of 2012, there is neither an effective treatment nor vaccine, chemotherapeutic strategies involve stopping the conversion of the normal form of prion protein to the abnormal form PrPSc. Experimental studies in rodents demonstrated some protection when polyanionic and tricyclic compounds are given shortly after infection. Transgenic mouse models have helped elucidate the pathogenesis of human prion disease. Current understanding of the nature of the interactions between PrPSc and PrPc may eventually offer some hope of regulating the development of disease by reducing or destabilizing the formation of PrPSc. Immunomodulation and mucosal immunization may be potential therapeutic and preventative approaches, especially as the alimentary tract is likely to be the main route of transmission.
![]()
 Key Facts
Key Facts
• Prions are unusual infectious agents, causing diseases characterized by changes in the brain (spongiform encephalopathies) and motor disturbances.
• Prions are host-derived glycoproteins and lack a nucleic acid genome. They are extremely resistant to disinfection procedures.
• Transmission of prions is usually by ingestion of contaminated tissues, but can occur via medical procedures.
• Diseases caused by prions include kuru, Creutzfeldt–Jakob disease (CJD), variant CJD and bovine spongiform encephalopathy (BSE).
![]()
![]()
 Conflicts
Conflicts
Of all the pathogens covered in this book, prions win the human–pathogen conflict. They are resistant to almost all disinfectant procedures and elicit minimal immune responses. They are never exposed to the outside world and cannot therefore be intercepted. They have no nucleic acids and no metabolic systems, so cannot be targeted by antimicrobial drugs. Prions can arise by mutation and hijack normal protein-folding control, producing abnormal molecules that are resistant to enzymes. Prions can cross from one species to another, and have crossed from animals to humans. Infection is therefore possible from meat-based food products. The presence of prions in meat is hard to detect; once ingested, prions can travel from the intestine to lymphoid and then to nervous tissues, ultimately causing profound and usually fatal changes in the CNS. Genetic characteristics of potential hosts seem to play an important role in determining the course of disease after exposure. Examples of prion-induced diseases are Creutzfeldt–Jakob disease (CJD); variant CJD (linked to ‘mad cow disease’); and kuru. These diseases can be diagnosed but there is currently no effective treatment.
![]()