Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT VI: Storage and Expression of Genetic Information
Chapter 33. Biotechnology and Human Disease
I. OVERVIEW
In the past, efforts to understand genes and their expression have been confounded by the immense size and complexity of human deoxyribonucleic acid (DNA). The human genome contains approximately three billion (109) base pairs (bp) that encode 20,000 to 25,000 protein-coding genes located on 23 chromosomes in the haploid genome. It is now possible to determine the nucleotide sequence of long stretches of DNA, and the entire human genome has been sequenced. This effort (called the Human Genome Project and completed in 2003) was made possible by several techniques that have already contributed to our understanding of many genetic diseases (Figure 33.1). These include, first, the discovery of restriction endonucleases that permit the cleavage of huge DNA molecules into defined fragments. Second, the development of cloning techniques that provide a mechanism for amplification of specific nucleotide sequences. Finally, the ability to synthesize specific probes, which has allowed the identification and manipulation of nucleotide sequences of interest. These and other experimental approaches have permitted the identification of both normal and mutant nucleotide sequences in DNA. This knowledge has led to the development of methods for the diagnosis of genetic diseases and some successes in the treatment of patients by gene therapy. [Note: The genomes of several viruses, prokaryotes, and nonhuman eukaryotes have also been sequenced.]
Figure 33.1 Three techniques that facilitate analysis of human DNA. dsDNA = double-stranded DNA.
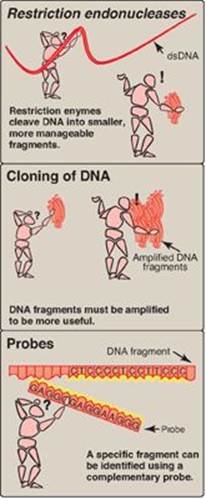
II. RESTRICTION ENDONUCLEASES
One of the major obstacles to molecular analysis of genomic DNA is the immense size of the molecules involved. The discovery of a special group of bacterial enzymes, called restriction endonucleases (restriction enzymes), which cleave double-stranded (ds) DNA into smaller, more manageable fragments, opened the way for DNA analysis. Because each enzyme cleaves DNA at a specific nucleotide sequence (restriction site), restriction enzymes are used experimentally to obtain precisely defined DNA segments called restriction fragments.
A. Specificity of restriction endonucleases
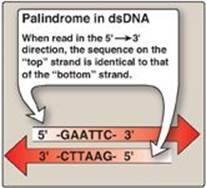
Restriction endonucleases recognize short stretches of dsDNA (four to eight bp) that contain specific nucleotide sequences. These sequences, which differ for each restriction enzyme, are palindromes, that is, they exhibit twofold rotational symmetry (Figure 33.2). This means that, within a short region of the double helix, the nucleotide sequence on the two strands is identical if each is read in the 5→3ʹ direction. Therefore, if you turn the page upside down (that is, rotate it 180° around its axis of symmetry) the sequence remains the same.
In bacteria, restriction endonucleases “restrict” the expression of nonbacterial (foreign) DNA through cleavage. Bacterial DNA is protected by methylation of bases at the restriction site.
Figure 33.2 Recognition sequence of restriction endonuclease EcoRI shows twofold rotational symmetry. dsDNA = double-stranded DNA; A = adenine; C = cytosine; G = guanine; T = thymine.
B. Nomenclature
A restriction enzyme is named according to the organism from which it was isolated. The first letter of the name is from the genus of the bacterium. The next two letters are from the name of the species. An additional letter indicates the type or strain, and a number (Roman numeral) is appended to indicate the order in which the enzyme was discovered in that particular organism. For example, HaeIII is the third restriction endonuclease isolated from the bacterium Haemophilus aegyptius.
C. “Sticky” and “blunt” ends
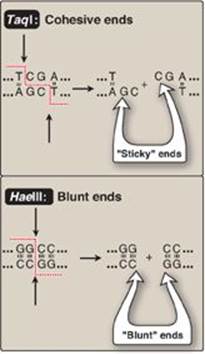
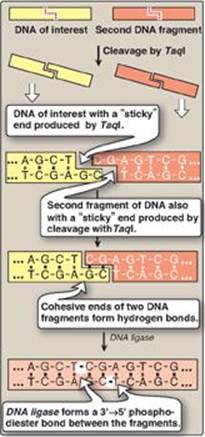
Restriction enzymes cleave dsDNA so as to produce a 3ʹ-hydroxyl group on one end and a 5ʹ-phosphate group on the other. Some restriction endonucleases, such as TaqI, form staggered cuts that produce “sticky” or cohesive ends (that is, the resulting DNA fragments have single-stranded sequences that are complementary to each other) as shown in Figure 33.3. Other restriction endonucleases, such as HaeIII, produce fragments that have “blunt” ends that are double stranded and, therefore, do not form hydrogen bonds with each other. Using the enzyme DNA ligase (see p. 406), sticky ends of a DNA fragment of interest can be covalently joined with other DNA fragments that have sticky ends produced by cleavage with the same restriction endonuclease (Figure 33.4). [Note: A ligase encoded by bacteriophage T4 can covalently join blunt-ended fragments.]
Figure 33.3 Specificity of TaqI and HaeIII restriction endonucleases; A = adenine; C = cytosine; G = guanine; T = thymine.
D. Restriction sites
A DNA sequence that is recognized and cut by a restriction enzyme is called a restriction site. Restriction endonucleases cleave dsDNA into fragments of different sizes depending upon the size of the sequence recognized. For example, an enzyme that recognizes a specific 4-bp sequence produces many cuts in the DNA molecule, one every 44 bp. In contrast, an enzyme requiring a unique sequence of 6 bp produces fewer cuts (one every 46 bp) and, therefore, longer pieces. Hundreds of these enzymes, each having different cleavage specificities (varying in both nucleotide sequences and length of recognition sites), are commercially available.
III. DNA CLONING
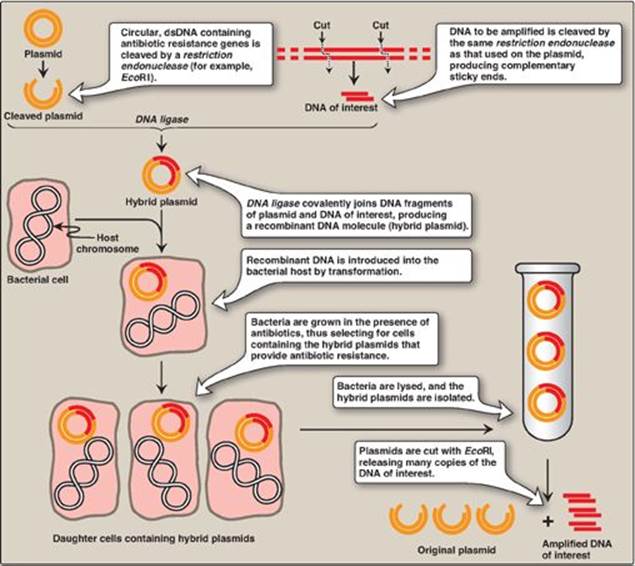
Introduction of a foreign DNA molecule into a replicating cell permits the cloning or amplification (that is, the production of many identical copies) of that DNA. [Note: Human DNA for cloning can be obtained from blood, saliva, and solid tissue.] In some cases, a single DNA fragment can be isolated and purified prior to cloning. More commonly, to clone a nucleotide sequence of interest, the total cellular DNA is first cleaved with a specific restriction enzyme, creating hundreds of thousands of fragments. Each of the resulting DNA fragments is joined to a DNA vector molecule (referred to as a cloning vector) to form a hybrid, or recombinant, DNA molecule. Each recombinant molecule carries its inserted DNA fragment into a single host cell (for example, a bacterium), where it is replicated. [Note: The process of introducing foreign DNA into a cell is called transformation for bacteria and yeast and transfection for higher eukaryotes.] As the host cell multiplies, it forms a clone in which every bacterium contains copies of the same inserted DNA fragment, hence the name “cloning.” The cloned DNA can be released from its vector by cleavage (using the appropriate restriction endonuclease) and isolated. By this mechanism, many identical copies of the DNA of interest can be produced. [Note: An alternative to amplification by biologic cloning, the polymerase chain reaction (PCR), is described on p. 479.]
Figure 33.4 Formation of recombinant DNA from restriction fragments with “sticky” ends. A = adenine; C = cytosine; G = guanine; T = thymine.
A. Vectors
A vector is a molecule of DNA to which the fragment of DNA to be cloned is joined. Essential properties of a vector include: 1) it must be capable of autonomous replication within a host cell, 2) it must contain at least one specific nucleotide sequence recognized by a restriction endonuclease, and 3) it must carry at least one gene that confers the ability to select for the vector such as an antibiotic resistance gene. Commonly used vectors include plasmids and viruses.
1. Prokaryotic plasmids: Prokaryotic organisms typically contain single, large, circular chromosomes. In addition, most species of bacteria also normally contain small, circular, extrachromosomal DNA molecules called plasmids (Figure 33.5). Plasmid DNA undergoes replication that may or may not be synchronized to chromosomal division. Plasmids may carry genes that convey antibiotic resistance to the host bacterium and may facilitate the transfer of genetic information from one bacterium to another. They can be readily isolated from bacterial cells, their circular DNA cleaved at specific sites by restriction endonucleases, and up to 10 kb (kilobases) of foreign DNA (cut with the same restriction enzyme) inserted. The recombinant plasmid can be introduced into a bacterium, producing large numbers of copies of the plasmid. The bacteria are grown in the presence of antibiotics, thus selecting for cells containing the hybrid plasmids, which provide antibiotic resistance (Figure 33.6). Artificial plasmids are routinely constructed. An example is pRB322 (Figure 33.5), which contains an origin of replication, two antibiotic resistance genes, and over 40 unique restriction sites. Use of plasmids is limited by the size of the DNA that can be inserted.
Figure 33.5 A restriction map of plasmid pBR322 indicating the positions of its antibiotic resistance genes and 6 of the over 40 unique sites recognized by specific restriction endonucleases.
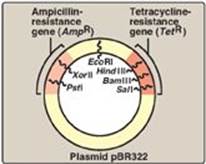
Figure 33.6 Summary of gene cloning. dsDNA = double-stranded DNA.
2. Other vectors: The development of improved vectors that can more efficiently accommodate larger DNA segments, or express the passenger genes in different cell types, has aided molecular genetics research. In addition to the prokaryotic plasmids described above, naturally occurring viruses that infect bacteria (bacteriophage l, for example) or mammalian cells (retroviruses, for example), as well as artificial constructs such as cosmids and bacterial or yeast artificial chromosomes (BACs or YACs, respectively), are currently used as cloning vectors. [Note: BACs and YACs can accept DNA inserts of 100–250 kb and 250–1000 kb, respectively.]
B. DNA libraries
A DNA library is a collection of cloned restriction fragments of the DNA of an organism. Two kinds of libraries are commonly used: genomic libraries and complementary DNA (cDNA) libraries. Genomic libraries ideally contain a copy of every DNA nucleotide sequence in the genome. In contrast, cDNA libraries contain those DNA sequences that only appear as processed messenger RNA (mRNA) molecules, and these differ from one cell type to another. [Note: cDNA lacks introns and the control regions of the genes, whereas these are present in genomic DNA.]
1. Genomic DNA libraries: A genomic library is created by digestion of the total DNA of an organism with a restriction endonuclease and subsequent ligation to an appropriate vector. The recombinant DNA molecules replicate within host bacteria. Thus, the amplified DNA fragments collectively represent the entire genome of the organism and are called a genomic library. Regardless of the restriction enzyme used, the chances are rather good that the gene of interest contains more than one restriction site recognized by that enzyme. If this is the case, and if the digestion is allowed to go to completion, the gene of interest is fragmented (that is, it is not contained in any one clone in the library). To avoid this usually undesirable result, a partial digestion is performed in which either the amount or the time of action of the enzyme is limited. This results in cleavage occurring at only a fraction of the restriction sites on any one DNA molecule, thus producing fragments of about 20 kb. Enzymes that cut very frequently (that is, those that recognize four-bp sequences) are generally used for this purpose so that the result is an almost random collection of fragments. This ensures a high degree of probability that the gene of interest is contained, intact, in some fragment.
Figure 33.7 Synthesis of cDNA from messenger RNA (mRNA) using reverse transcriptase. Additional steps (not shown) are required to clone the cDNA. [Note: Recall that DNA is resistant to alkaline hydrolysis.]
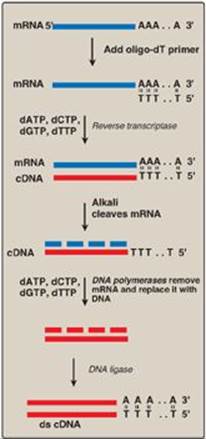
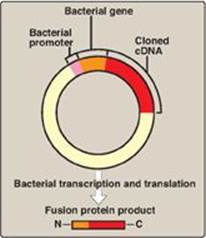
2. Complementary DNA libraries: If a protein-coding gene of interest is expressed at a high level in a particular tissue, the mRNA transcribed from that gene is likely also present at high concentrations in the cells of that tissue. For example, reticulocyte mRNA is composed largely of molecules encoding the α-globin and b-globin chains of hemoglobin. This mRNA can be used as a template to make a cDNA molecule using the enzyme reverse transcriptase (Figure 33.7). The resulting cDNA is, therefore, a double-stranded copy of mRNA. [Note: The template mRNA is isolated from transfer RNA and ribosomal RNA by the presence of its poly-A tail.] cDNA can be amplified by cloning or by PCR. It can be used as a probe to locate the gene that coded for the original mRNA (or fragments of the gene) in mixtures containing many unrelated DNA fragments. If the mRNA used as a template is a mixture of many different size species, the resulting cDNA is heterogeneous. These mixtures can be cloned to form a cDNA library. Because cDNA has no intervening sequences, it can be cloned into an expression vector for the synthesis of eukaryotic proteins by bacteria (Figure 33.8). These special plasmids contain a bacterial promoter for transcription of the cDNA and a Shine-Dalgarno (SD) sequence (see p. 438) that allows the bacterial ribosome to initiate translation of the resulting mRNA molecule. The cDNA is inserted downstream of the promoter and within a gene for a protein that is expressed in the bacterium (for example, lacZ; see p.450), such that the mRNA produced contains an SD sequence, a few codons for the bacterial protein, and all the codons for the eukaryotic protein. This allows for more efficient expression and results in the production of a fusion protein. [Note: Therapeutic human insulin is made in bacteria through this technology. However, the extensive co- and posttranslational modifications (see p. 443) required for most other human proteins (for example, blood clotting factors) necessitates the use of eukaryotic, even mammalian, hosts.]
Figure 33.8 An expression vector. The complementary DNA (cDNA) is inserted within a bacterial gene, downstream of the promoter sequence, and the sequences for the messenger RNA Shine-Dalgarno sequence, start codon, and codons for the first few amino acids of the bacterial protein. The product is a fusion protein that contains just some amino acids of the bacterial protein ![]() and all the amino acids of the cDNA-encoded protein
and all the amino acids of the cDNA-encoded protein ![]() .
.
C. Sequencing of cloned DNA fragments
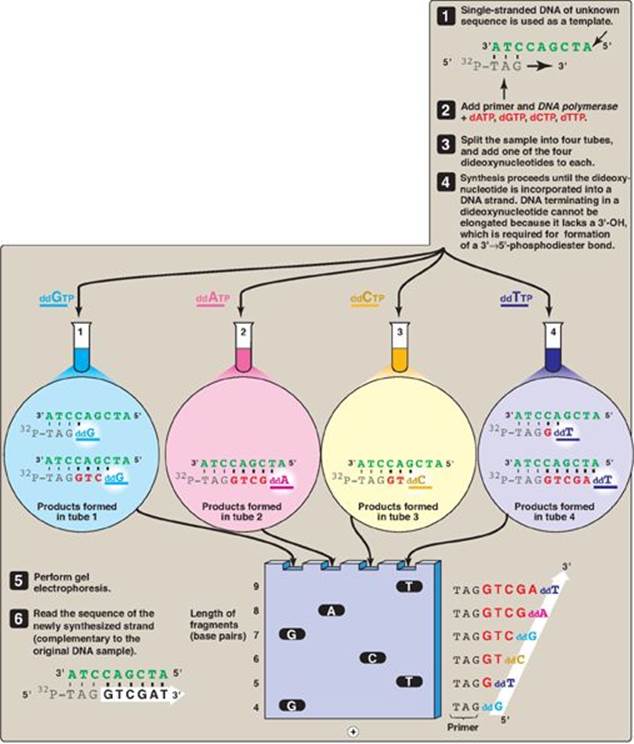
The base sequence of DNA fragments that have been cloned can be determined. The original procedure for this purpose was the Sanger dideoxy method illustrated in Figure 33.9. In this method, the single-stranded DNA (ssDNA) to be sequenced is used as the template for DNA synthesis by DNA polymerase. A radiolabeled primer complementary to the 3ʹ-end of the target DNA is added, along with the four deoxyribonucleoside triphosphates (dNTPs). The sample is divided into four reaction tubes, and a small amount of one of the four dideoxyribonucleoside triphosphates (ddNTPs) is added to each tube. Because it contains no 3ʹ-hydroxyl group, incorporation of a ddNMP terminates elongation at that point. The products of this reaction, then, consist of a mixture of DNA strands of different lengths, each terminating at a specific base. Separation of the various DNA products by size using polyacrylamide gel electrophoresis, followed by autoradiography, yields a pattern of bands from which the DNA base sequence can be read. [Note: The shorter the fragment, the farther it travels on the gel, with the shortest fragment representing that which was made first (that is, the 5ʹ-end).] In place of a labeled primer, a mixture of the four ddNTPs linked to different fluorescent dyes and in a single reaction tube is now commonly used. [Note: The Human Genome Project used highly automated variations of this technique to determine the base sequence of the human genome.] Advances in sequencing technology, so-called next generation, or deep sequencing, now allow the sequencing of longer segments in a shorter time with increased fidelity and decreased cost through the simultaneous (parallel) sequencing of many DNA pieces.
IV. PROBES
Cleavage of large DNA molecules by restriction enzymes produces a bewildering array of fragments. How can the DNA sequence of interest be picked out of a mixture of thousands or even millions of irrelevant DNA fragments? The answer lies in the use of a probe, a short piece of ssDNA or RNA, labeled with a radioisotope, such as 32P, or with a nonradioactive molecule, such as biotin or a fluorescent dye. The sequence of a probe is complementary to a sequence in the DNA of interest, called the target DNA. Probes are used to identify which band on a gel or which clone in a library contains the target DNA, a process called screening.
A. Hybridization of a probe to DNA fragments
The utility of probes hinges on the phenomenon of hybridization (or annealing) in which a probe containing a complementary sequence binds a single-stranded sequence of a target DNA. ssDNA, produced by alkaline denaturation of dsDNA, is first bound to a solid support, such as a nitrocellulose membrane. The immobilized DNA strands are prevented from self-annealing but are available for hybridization to the exogenous, radiolabeled, ssDNA probe. The extent of hybridization is measured by the retention of radioactivity on the membrane. Excess probe molecules that do not hybridize are removed by washing the membrane.
B. Synthetic oligonucleotide probes
If the sequence of all or part of the target DNA is known, short, single-stranded oligonucleotide probes can be synthesized that are complementary to a small region of the gene of interest. If the sequence of the gene is unknown, the amino acid sequence of the protein, the final gene product, may be used to construct a nucleic acid probe using the genetic code as a guide. Because of the degeneracy of the genetic code, it is necessary to synthesize several oligonucleotides. [Note: Oligonucleotides can be used to detect single-base changes in the sequence to which they are complementary. In contrast, cDNA probes contain many thousands of bases, and their binding to a target DNA with a single-base change is unaffected.]
Figure 33.9 DNA sequencing by the Sanger dideoxy method. [Note: The original method utilized a radiolabeled primer. Fluorescent dye-labeled ddNTPs are now commonly used.] A = adenine; C = cytosine; G = guanine; T = thymine; d = deoxy; dd = dideoxy.
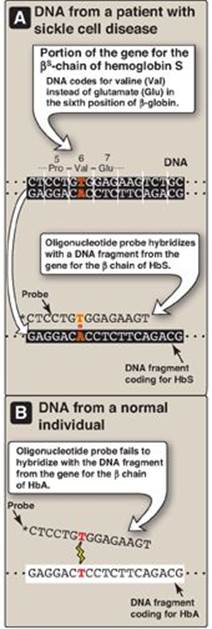
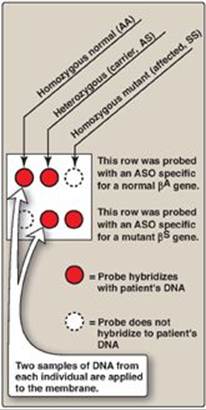
1. Detecting the βS-globin mutation: A synthetic allele-specific oligonucleotide (ASO) probe can be used to detect the presence of the sickle cell mutation in the β-globin gene (Figure 33.10). DNA, isolated from leukocytes and amplified, is denatured and applied to a membrane. A radiolabeled oligonucleotide probe, complementary to the point mutation (GAG → GTG, glutamate → valine) at codon 6 in patients with the bS gene, is applied to the membrane. DNA isolated from a heterozygous individual (sickle cell trait) or a homozygous patient (sickle cell disease) contains a sequence that is complementary to the probe, and a double-stranded hybrid forms that can be detected by electrophoresis. In contrast, DNA obtained from normal individuals is not complementary at this positon and, therefore, does not form a hybrid (see Figure 33.10). Use of a pair of such ASO probes (one specific for the normal allele and one specific for the mutant allele) allows all three possible genotypes (homozygous normal, heterozygous, and homozygous mutant) to be distinguished (Figure 33.11). [Note: ASO probes are useful only if the mutation and its location are known.]
Figure 33.10 Allele-specific oligonucleotide probe detects hemoglobin (Hb) S allele. [Note: * indicates 32P radiolabel.] A = adenine; C = cytosine; G = guanine; T = thymine; Pro = proline.
C. Biotinylated probes
Because the disposal of radioactive waste is becoming increasingly expensive, nonradiolabeled probes have been developed. One of the most successful is based on the vitamin biotin (see p. 381), which can be chemically linked to the nucleotides used to synthesize the probe. Biotin was chosen because it binds very tenaciously to avidin, a readily available protein contained in chicken egg whites. Avidin can be attached to a fluorescent dye detectable optically with great sensitivity. Thus, a DNA fragment (displayed, for example, by gel electrophoresis) that hybridizes with the biotinylated probe can be made visible by immersing the gel in a solution of dye-coupled avidin. After washing away the excess avidin, the DNA fragment that binds the probe is fluorescent. [Note: Labeled probes can allow detection and localization of DNA or RNA sequences in cell or tissue preparations, a process called in situ hybridization (ISH). If the probe is fluorescent, the technique is called FISH.]
D. Antibodies
If no amino acid sequence information is available to guide the synthesis of a probe for direct detection of the DNA of interest, a gene can be identified indirectly by cloning cDNA in an expression vector that allows the cloned cDNA to be transcribed and translated. A labeled antibody is used to identify which bacterial colony produces the protein and, therefore, contains the cDNA of interest.
V. SOUTHERN BLOTTING
Southern blotting is a technique that combines the use of restriction enzymes, electrophoresis, and DNA probes to generate, separate, and detect pieces of DNA.
A. Experimental procedure
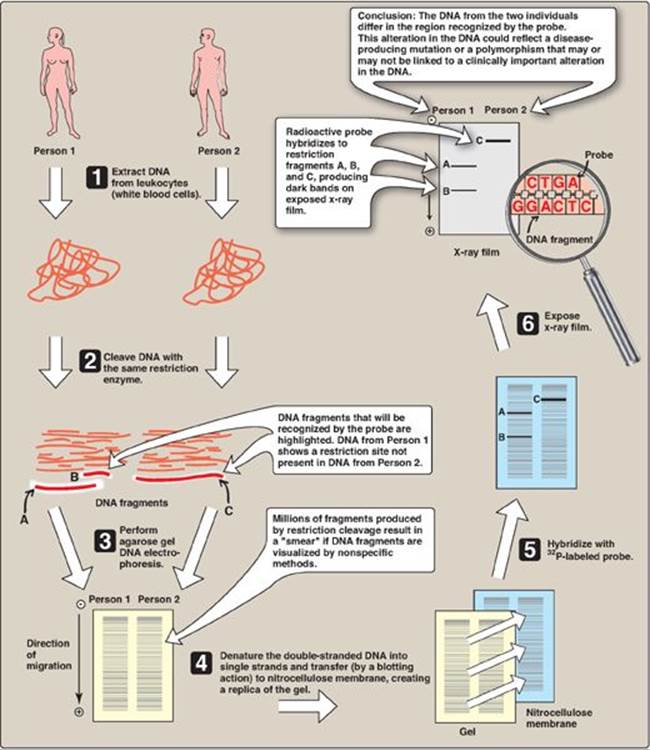
This method, named after its inventor, Edward Southern, involves the following steps (Figure 33.12). First, DNA is extracted from cells, for example, a patient’s leukocytes. Second, the DNA is cleaved into many fragments using a restriction enzyme. Third, the resulting fragments are separated on the basis of size by electrophoresis. [Note: Because the large fragments move more slowly than the smaller ones, the lengths of the fragments, usually expressed as the number of base pairs, can be calculated from comparison of the position of the band relative to standard fragments of known size.] The DNA fragments in the gel are denatured and transferred (blotted) to a nitrocellulose membrane for analysis. If the original DNA represents the individual’s entire genome, the enzymic digest contains a million or more fragments. The gene of interest is on only one (or a few if the gene itself was fragmented) of these pieces of DNA. If all the DNA segments were visualized by a nonspecific technique, they would appear as an unresolved blur of overlapping bands. To avoid this, the last step in Southern blotting uses a probe to identify the DNA fragments of interest. The patterns observed on Southern blot analysis depend both on the specific restriction endonuclease and on the probe used to visualize the restriction fragments. [Note: Variants of the Southern blot have been facetiously named “Northern” (electrophoresis of mRNA followed by hybridization with a specific probe), and “Western” (electrophoresis of protein followed by detection with an antibody directed against the protein of interest), neither of which relates to anyone’s name or to points of the compass.]
Figure 33.11 Allele-specific oligonucleotide (ASO) probes used to detect the sickle cell mutation and differentiate between sickle cell trait and disease.
B. Detection of mutations
Southern blotting can detect DNA mutations such as large insertions, deletions, or rearrangements of nucleotides. It can also detect point mutations (replacement of one nucleotide by another; see p. 433) that cause the loss or gain of restriction sites. Such mutations cause the pattern of bands to differ from those seen with a normal gene. Longer fragments are generated if a restriction site is lost. For example, in Figure 33.12, Person 2 lacks a restriction site present in Person 1. Alternatively, the point mutation may create a new cleavage site with the production of shorter fragments. [Note: Most sequence differences at restriction sites are harmless variations in the DNA.]
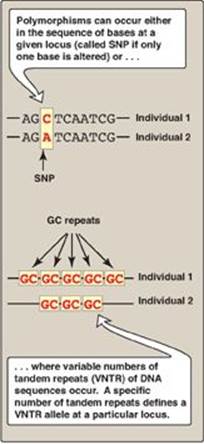
VI. RESTRICTION FRAGMENT LENGTH POLYMORPHISM
It has been estimated that the genomes of any two unrelated people are 99.5% identical. With 6 billion bp in the diploid human genome, that represents variation in about 30 million bp. These genome variations are the result of mutations that lead to polymorphisms. A polymorphism is a change in genotype that can result in no change in phenotype or a change in phenotype that is harmless; causes increased susceptibility to a disease; or, rarely, causes the disease. It is traditionally defined as a sequence variation at a given locus (allele) in more than 1% of a population. Polymorphisms primarily occur in the 98% of the genome that does not encode proteins (that is, in introns and intergenic regions). A restriction fragment length polymorphism (RFLP) is a genetic variant that can be observed by cleaving the DNA into fragments (restriction fragments) with a restriction enzyme. The length of the restriction fragments is altered if the variant alters the DNA so as to create or abolish a site of restriction endonuclease cleavage (a restriction site). RFLP can be used to detect human genetic variations, for example, in prospective parents or in fetal tissue.
Figure 33.12 Southern blotting procedure. [Note: Nonradiolabeled probes are now commonly used.]
A. DNA variations resulting in restriction fragment length polymorphism
Two types of DNA variation commonly result in RFLP: single-base changes in the DNA sequence and tandem repeats of DNA sequences.
1. Single-base changes in DNA: About 90% of human genome variation comes in the form of single nucleotide polymorphisms (SNPs, pronounced “snips”), that is, variations that involve just one base (Figure 33.13). The substitution of one nucleotide at a restriction site can render the site unrecognizable by a particular restriction endonuclease. A new restriction site can also be created by the same mechanism. In either case, cleavage with an endonuclease results in fragments of lengths differing from the normal that can be detected by DNA hybridization (see Figure 33.12). The altered restriction site can be either at the site of a disease-causing mutation (rare) or at a site some distance from the mutation. [Note: The HapMap, developed by The International Haplotype Map Project, is a catalog of common SNPs in the human genome. The data are being used in genome-wide association studies (GWAS) to identify those alleles that affect health and disease.]
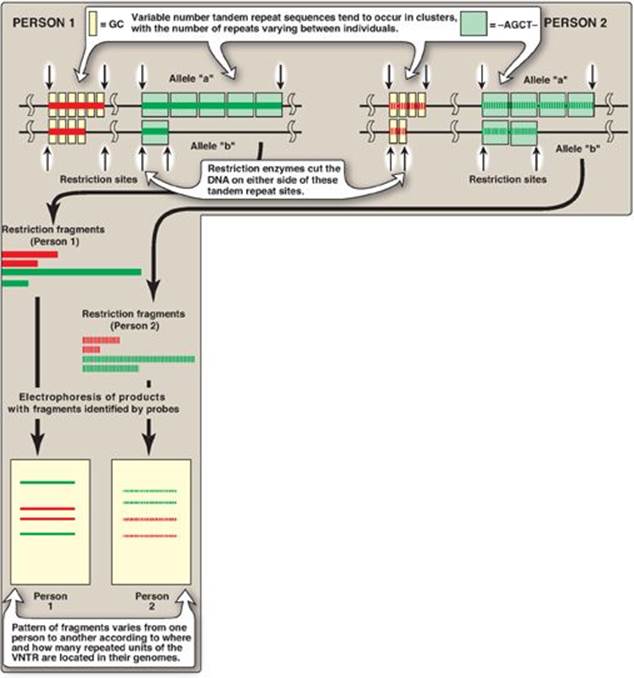
2. Tandem repeats: Polymorphism in chromosomal DNA can also arise from the presence of a variable number of tandem repeats [VNTR] see Figure 33.14). These are short sequences of DNA at scattered locations in the genome, repeated in tandem (one after another). The number of these repeat units varies from person to person but is unique for any given individual and, therefore, serves as a molecular fingerprint. Cleavage by restriction enzymes yields fragments that vary in length depending on how many repeated segments are contained in the fragment (Figure 33.14). Many different VNTR loci have been identified and are extremely useful for DNA fingerprint analysis, such as in forensic and paternity cases. It is important to emphasize that these polymorphisms, whether SNP or VNTR, are simply markers, which, in most cases, have no known effect on the structure, function, or rate of production of any particular protein.
Figure 33.13 Common forms of genetic polymorphism. SNP = single-nucleotide polymorphism. A = adenine; C = cytosine; G = guanine; T = thymine.
B. Tracing chromosomes from parent to offspring
If the DNA of an individual has gained a restriction site by base substitution, then enzymic cleavage yields at least one additional fragment. Conversely, if a mutation results in loss of a restriction site, fewer fragments are produced by enzymic cleavage. An individual who is heterozygous for a polymorphism has a sequence variation in the DNA of one chromosome and not in the homologous chromosome. In such individuals, each chromosome can be traced from parent to offspring by determining the presence or absence of the polymorphism.
Figure 33.14 Restriction fragment length polymorphism of variable number tandem repeats (VNTR). For each person, a pair of homologous chromosomes is shown.
C. Prenatal diagnosis
Families with a history of severe genetic disease, such as an affected previous child or near relative, may wish to determine the presence of the disorder in a developing fetus. Prenatal diagnosis, in association with genetic counseling, allows for an informed reproductive decision if the fetus is affected.
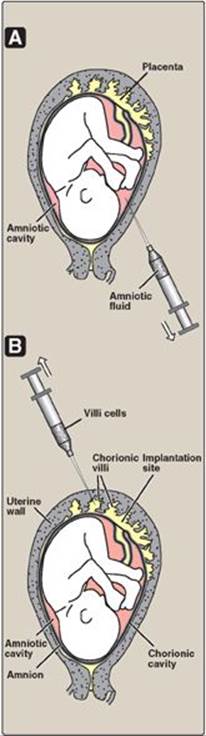
1. Methods available: The available diagnostic methods vary in sensitivity and specificity. Visualization of the fetus, for example, by ultrasound or fiberoptic devices (fetoscopy), is useful only if the genetic abnormality results in gross anatomic defects (for example, neural tube defects). The chemical composition of the amniotic fluid can also provide diagnostic clues. For example, the presence of high levels of α-fetoprotein is associated with neural tube defects. Fetal cells obtained from amniotic fluid or from biopsy of the chorionic villi can be used for karyotyping, which assesses the morphology of metaphase chromosomes. Staining and cell sorting techniques permit the rapid identification of trisomies and translocations that produce an extra chromosome or chromosomes of abnormal lengths. However, molecular analysis of fetal DNA provides the most detailed genetic picture.
2. Sources of DNA: DNA may be obtained from white blood cells, amniotic fluid, or chorionic villi (Figure 33.15). For amniotic fluid, it used to be necessary to grow cells in culture for two to three weeks in order to have sufficient DNA for analysis. The ability to amplify DNA by PCR (see p. 479) has dramatically shortened the time needed for a DNA analysis.
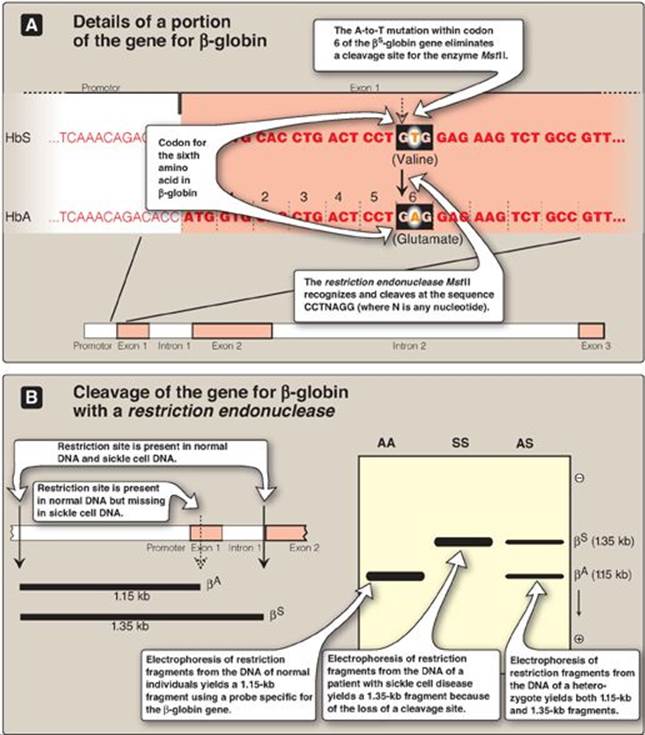
3. Direct diagnosis of sickle cell anemia using RFLP: The genetic disorders of hemoglobin (Hb) are the most common genetic diseases in humans. In the case of sickle cell anemia (Figure 33.16), the mutation that gives rise to the disease is actually one and the same mutation that gives rise to the polymorphism. Direct detection by RFLP of diseases that result from point mutations is, however, limited to only a few genetic diseases.
a. Early efforts to diagnose sickle cell anemia: In the past, prenatal diagnosis of hemoglobinopathies involved the determination of the amount and kinds of Hb synthesized in red cells obtained from fetal blood. However, the invasive procedures to obtain fetal blood have a high mortality rate (approximately 5%), and diagnosis cannot be carried out until late in the second trimester of pregnancy when HbS begins to be produced.
b. RFLP analysis: Sickle cell anemia is an example of a genetic disease caused by a point mutation (see p. 35). The sequence altered by the mutation abolishes the recognition site of the restriction endonuclease MstII: CCTNAGG (where N is any nucleotide; see Figure 33.16). Thus, the A-to-T mutation in codon 6 of the bS-globin gene eliminates a cleavage site for the enzyme. Normal DNA digested with MstII yields a 1.15-kb fragment, whereas a 1.35-kb fragment is generated from the bS gene as a result of the loss of one MstII cleavage site. Diagnostic techniques that allow analysis of fetal DNA from amniotic cells or chorionic villus sampling rather than fetal blood have proved valuable because they provide safe, early detection of sickle cell anemia as well as other genetic diseases. [Note: Genetic disorders caused by insertions or deletions between two restriction sites, rather than by the creation or loss of cleavage sites, will also display RFLP.]
Figure 33.15 Sampling of fetal cells. A. Amniotic fluid. B. Chorionic villus.
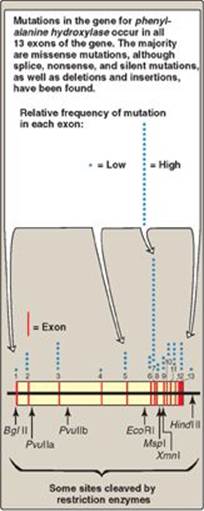
4. Indirect, prenatal diagnosis of phenylketonuria using RFLP: The gene for phenylalanine hydroxylase (PAH), deficient in phenylketonuria ([PKU] see p. 270), is located on chromosome 12. It spans about 90 kb of genomic DNA and contains 13 exons separated by introns (Figure 33.17; see p. 426 for a description of exons and introns). Mutations in PAH usually do not directly affect any restriction endonuclease recognition site. To establish a diagnostic protocol for this disease, DNA of family members of the affected individual must be analyzed. The goal is to identify genetic markers (RFLPs) that are tightly linked to the disease trait. Once these markers are identified, RFLP analysis can be used to carry out prenatal diagnosis.
a. Identification of the gene: Determinining the presence of the mutant gene by identifying the polymorphism marker can be done if two conditions are satisfied. First, if the polymorphism is closely linked to a disease-producing mutation, the defective gene can be traced by detection of the RFLP. For example, if DNA from a family carrying a disease-causing gene is examined by restriction enzyme cleavage and Southern blotting, it is sometimes possible to find an RFLP that is consistently associated with that gene (that is, they show close linkage and are coinherited). It is then possible to trace the inheritance of the gene within a family without knowledge of the nature of the genetic defect or its precise location in the genome. [Note: The polymorphism may be known from the study of other families with the disorder or may be discovered to be unique in the family under investigation.] Second, for autosomal recessive disorders, such as PKU, the presence of an affected individual in the family would aid in the diagnosis. This individual would have the mutation present on both chromosomes, allowing identification of the RFLP associated with the genetic disorder.
Figure 33.16 Detection of βS-globin mutation. kb = kilobase (1 kb = 1000 base pairs in double-stranded DNA); Hb = hemoglobin.
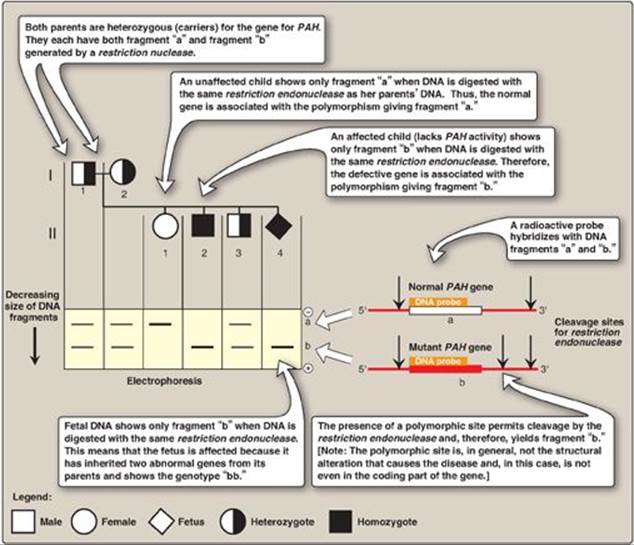
b. RFLP analysis: The presence of abnormal genes for PAH can be shown using DNA polymorphisms as markers to distinguish between normal and mutant genes. For example, Figure 33.18 shows a typical pattern obtained when DNA from the white blood cells of a family is cleaved with an appropriate restriction enzyme and subjected to electrophoresis. The vertical arrows represent the cleavage sites for the restriction enzyme used. The presence of a polymorphic site creates fragment “b” in the autoradiogram (after hybridization with a labeled PAH-cDNA probe), whereas the absence of this site yields only fragment “a.” Note that Subject II-2 demonstrates that the polymorphism, as shown by the presence of fragment “b,” is associated with the mutant gene. Therefore, in this particular family, the appearance of fragment “b” corresponds to the presence of a polymorphic site that marks the abnormal gene for PAH. The absence of fragment “b” corresponds to having only the normal gene. In Figure 33.18, examination of fetal DNA shows that the fetus inherited two abnormal genes from its parents and, therefore, has PKU.
c. Value of DNA testing: DNA-based testing is useful not only in determining if an unborn fetus is affected by PKU, but also in detecting unaffected carriers of the mutated gene to aid in family planning. [Note: PKU is treatable by dietary restriction of phenylalanine. Early diagnosis and treatment are essential in preventing severe neurologic damage in affected individuals.].
Figure 33.17 The gene for phenylalanine hydroxylase showing 13 exons, restriction sites, and some of the mutations resulting in phenylketonuria.
VII. POLYMERASE CHAIN REACTION
PCR is a test tube method for amplifying a selected DNA sequence that does not rely on the biologic cloning method described on p. 467. PCR permits the synthesis of millions of copies of a specific nucleotide sequence in a few hours. It can amplify the sequence, even when the targeted sequence makes up less than one part in a million of the total initial sample. The method can be used to amplify DNA sequences from any source, including viral, bacterial, plant, or animal. The steps in PCR are summarized in Figures 33.19 and 33.20.
A. Steps of a polymerase chain reaction
PCR uses DNA polymerase to repetitively amplify targeted portions of genomic or cDNA. Each cycle of amplification doubles the amount of DNA in the sample, leading to an exponential increase (2n, where n = cycle number) in DNA with repeated cycles of amplification. The amplified DNA products can then be separated by gel electrophoresis, detected by Southern blotting and hybridization, and sequenced.
Figure 33.18 Analysis of restriction fragment length polymorphism in a family with a child affected by phenylketonuria (PKU), an autosomal recessive disease. The molecular defect in the gene for phenylalanine hydroxylase (PAH) in the family is not known. The family wanted to know if the current pregnancy would be affected by PKU.
1. Primer construction: It is not necessary to know the nucleotide sequence of the target DNA in the PCR method. However, it is necessary to know the nucleotide sequence of short segments on each side of the target DNA. These stretches, called flanking sequences, bracket the DNA sequence of interest. The nucleotide sequences of the flanking regions are used to construct two, single-stranded oligonucleotides, usually 20–35 nucleotides long, which are complementary to the respective flanking sequences. The 3ʹ-hydroxyl end of each oligonucleotide points toward the target sequence (see Figure 33.19). These synthetic oligonucleotides function as primers in PCR reactions.
Figure 33.19 Steps in one cycle of the polymerase chain reaction.
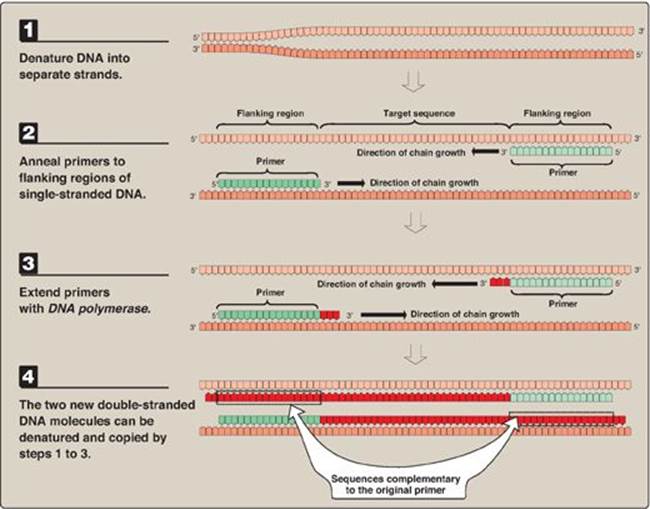
2. Denature the DNA: The DNA to be amplified is heated to separate the double-stranded target DNA into single strands.
3. Annealing of primers to single-stranded DNA: The separated strands are cooled and allowed to anneal to the two primers (one for each strand).
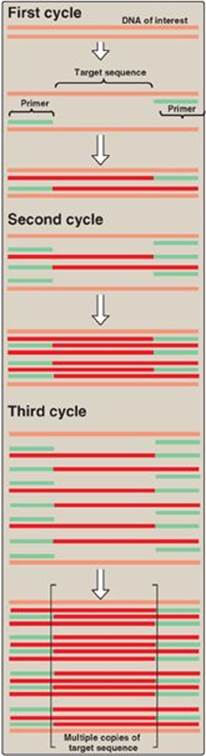
4. Chain extension: DNA polymerase and deoxyribonucleoside triphosphates (in excess) are added to the mixture to initiate the synthesis of two new strands complementary to the original DNA strands. DNA polymerase adds nucleotides to the 3ʹ-hydroxyl end of the primer, and strand growth extends across the target DNA, making complementary copies of the target. [Note: PCR products can be several thousand bp long.] At the completion of one cycle of replication, the reaction mixture is heated again to separate the strands (of which there are now four). Each strand binds a complementary primer, and the cycle of chain extension is repeated. By using a heat-stable DNA polymerase (for example, Taq polymerase from the bacterium, Thermus aquaticus that normally lives at high temperatures), the polymerase is not denatured and, therefore, does not have to be added at each successive cycle. Typically 20–30 cycles are run during this process, amplifying the DNA by a million-fold (220) to a billion-fold (230). [Note: Each extension product includes a sequence at its 5ʹ-end that is complementary to the primer (see Figure 33.19). Thus, each newly synthesized strand can act as a template for the successive cycles (see Figure 33.20). This leads to an exponential increase in the amount of target DNA with each cycle, hence, the name “polymerase chain reaction.”] Probes can be made during PCR by adding labeled nucleotides to the last few cycles.
Figure 33.20 Multiple cycles of polymerase chain reaction.
B. Advantages of polymerase chain reaction
The major advantages of PCR over biologic cloning as a mechanism for amplifying a specific DNA sequence are sensitivity and speed. DNA sequences present in only trace amounts can be amplified to become the predominant sequence. PCR is so sensitive that DNA sequences present in an individual cell can be amplified and studied. Isolating and amplifying a specific DNA sequence by PCR is faster and less technically difficult than traditional cloning methods using recombinant DNA techniques.
C. Applications
PCR has become a very common tool for a large number of applications.
1. Comparison of a normal gene with a mutant form of the gene: PCR allows the synthesis of mutant DNA in sufficient quantities for a sequencing protocol without laboriously cloning the altered DNA.
2. Detection of low-abundance nucleic acid sequences: Viruses that have a long latency period, such as human immunodeficiency virus (HIV), are difficult to detect at the early stage of infection using conventional methods. PCR offers a rapid and sensitive method for detecting viral DNA sequences even when only a small proportion of cells harbors the virus. [Note: Quantitative real time PCR (qRT-PCR) allows quantification of starting amounts of the target nucleic acid as PCR progresses (in real time) rather than at the end and is useful in determining viral load (the amount of virus).]
3. Forensic analysis of DNA samples: DNA fingerprinting by means of PCR has revolutionized the analysis of evidence from crime scenes. DNA isolated from a single human hair, a tiny spot of blood, or a sample of semen is sufficient to determine whether the sample comes from a specific individual. The DNA markers analyzed for such fingerprinting are most commonly short tandem repeat polymorphisms. These are very similar to the VNTRs described previously (see p. 475) but are smaller in size. [Note: Determination of paternity uses the same techniques.]
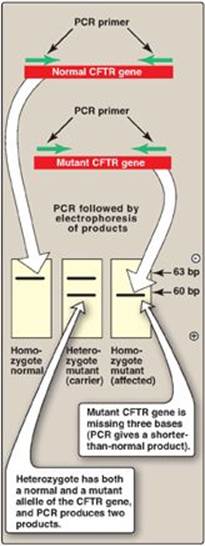
4. Prenatal diagnosis and carrier detection of cystic fibrosis: Cystic fibrosis is an autosomal recessive genetic disease resulting from mutations in the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The most common mutation is a three-base deletion that results in the loss of a phenylalanine residue from the CFTR protein (see p. 434). Because the mutant allele is three bases shorter than the normal allele, it is possible to distinguish them from each other by the size of the PCR products obtained by amplifying that portion of the DNA. Figure 33.21 illustrates how the results of such a PCR test can distinguish between homozygous normal, heterozygous (carriers), and homozygous mutant (affected) individuals.
The simultaneous amplification of multiple regions of a target DNA using multiple primer pairs is known as multiplex PCR. It allows detection of the loss of 1 or more exons in a gene with many exons such as the gene for CFTR, which has 27 exons.
Figure 33.21 Genetic testing for cystic fibrosis using the polymerase chain reaction (PCR). CFTR = cystic fibrosis transmembrane conductance regulator; bp = base pairs.
VIII. ANALYSIS OF GENE EXPRESSION
The tools of biotechnology not only allow the study of gene structure, but also provide ways of analyzing the mRNA and protein products of gene expression.
A. Determination of messenger RNA levels
mRNA levels are usually determined by the hybridization of labeled probes to either mRNA itself or to cDNA produced from mRNA. [Note: Amplification of cDNA made from mRNA by retroviral reverse transcriptase (RT) is referred to as RT-PCR.]
1. Northern blots: Northern blots are very similar to Southern blots (see Figure 33.12, p. 474), except that the original sample contains a mixture of mRNA molecules that are separated by electrophoresis, then transferred to a membrane and hybridized to a radiolabeled probe. The bands obtained by autoradiography give a measure of the amount and size of particular mRNA molecules in the sample.
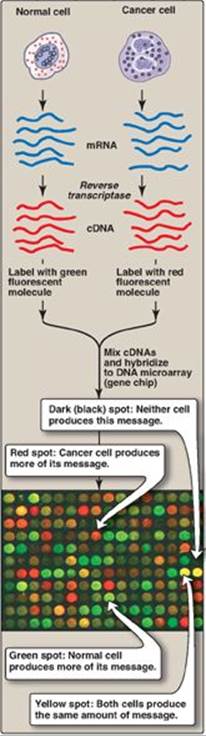
2. Microarrays: DNA microarrays contain thousands of immobilized ssDNA sequences organized in an area no larger than a microscope slide. These microarrays are used to analyze a sample for the presence of gene variations or mutations (genotyping) or to determine the patterns of mRNA production (gene expression analysis), analyzing thousands of genes at the same time. For genotyping analysis, the sample is from genomic DNA. For expression analysis, the population of mRNA molecules from a particular cell type is converted to cDNA and labeled with a fluorescent tag (Figure 33.22). This mixture is then exposed to a gene (DNA) chip, which is a glass slide or membrane containing thousands of tiny spots of DNA, each corresponding to a different gene. The amount of fluorescence bound to each spot is a measure of the amount of that particular mRNA in the sample. DNA microarrays are used to determine the differing patterns of gene expression in two different types of cell (for example, normal and cancer cells; see Figure 33.22). They can also be used to subclassify cancers, such as breast cancer, to optimize treatment. [Note: Microarrays involving proteins and the antibodies or other proteins that recognize them are being used to identify biomarkers to aid in the diagnosis, prognosis, and treatment of disease based on a patient’s protein expression profile. Protein (and DNA) microarrays are important tools in the development of personalized medicine.]
Figure 33.22 Microarray analysis of gene expression using DNA (gene) chips. [Note: Protein chips are also used.] mRNA = messenger RNA; cDNA = complementary DNA.
B. Analysis of proteins
The kinds and amounts of proteins in cells do not always directly correspond to the amounts of mRNA present. Some mRNAs are translated more efficiently than others, and some proteins undergo posttranslational modification. When analyzing the abundance and interactions of a large number of proteins, automated methods involving a variety of techniques, such as mass spectrometry and two-dimensional electrophoresis, are used. When investigating one, or a limited number of proteins, labeled antibodies are used to detect and quantify specific proteins and to determine posttranslational modifications.
1. Enzyme-linked immunosorbent assays (ELISAs): These assays are performed in the wells of a plastic microtiter dish. The antigen (protein) is bound to the plastic of the dish. The probe used consists of an antibody specific for the particular protein to be measured. The antibody is covalently bound to an enzyme, which will produce a colored product when exposed to its substrate. The amount of color produced is proportional to the amount of antibody present and, indirectly, to the amount of protein in a test sample.
2. Western blots: Western blots (also called immunoblots) are similar to Southern blots, except that protein molecules in the sample are separated by electrophoresis and blotted (transferred) to a membrane. The probe is a labeled antibody, which produces a band at the location of its antigen.
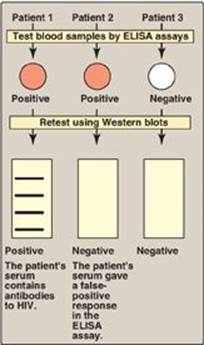
3. Detecting exposure to human immunodeficiency virus (HIV): ELISA and Western blots are commonly used to detect exposure to HIV by measuring the amount of anti-HIV antibodies present in a patient’s blood sample. ELISAs are used as the primary screening tool, because they are very sensitive. Because these assays sometimes give false positives, however, Western blots, which are more specific, are often used as a confirmatory test (Figure 33.23). [Note: ELISA and Western blots can only detect HIV exposure after anti-HIV antibodies appear in the bloodstream. PCR-based testing for HIV is more useful in the first few months after exposure.]
C. Proteomics
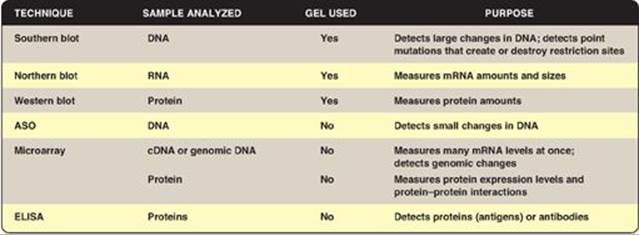
The study of the proteome or all the proteins expressed by a genome, including their relative abundance, distribution, posttranslational modifications, functions, and interactions with other macromolecules, is known as proteomics. The 20,000 to 25,000 protein-coding genes of the human genome translate into well over 100,000 proteins when posttranscriptional and posttranslational modifications are considered. Although a genome remains essentially unchanged, the amounts and types of proteins in any particular cell change dramatically as genes are turned on and off. [Note: Proteomics (and genomics) required the parallel development of bioinformatics, the computer-based organization, storage, and analysis of biologic data.] Figure 33.24 compares some of the analytic techniques discussed in this chapter.
Figure 33.23 Testing for HIV exposure by enzymelinked immunosorbent assays (ELISAs) and Western blots.
IX. GENE THERAPY
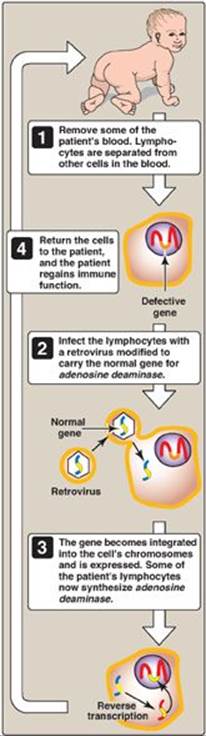
The goal of gene therapy is to treat disease through insertion of the normal, cloned DNA for a gene into the somatic cells of a patient who has a defect in that gene as a result of a disease-causing mutation. Because somatic gene therapy changes only the targeted somatic cells, the change is not passed on to the next generation. [Note: In germline gene therapy, it is the germ cells that are modified, and so the change is passed on. A long-standing moratorium on germline gene therapy is in effect world-wide.] There are two types of gene transfer: 1) ex vivo, in which cells from the patient are removed, transduced, and returned; and 2) in vivo, in which the cells are directly transduced. Both types require use of a vector (viral or nonviral) to deliver the DNA into the target cell. Challenges of gene therapy include development of vectors, achievement of long-lived expression, and prevention of side effects such as an immune response. The first successful gene therapy (1990) involved two patients with severe combined immunodeficiency disease (SCID) caused by mutations to the gene for adenosine deaminase (see p. 301). It utilized mature T lymphocytes transduced ex vivo with a viral vector (Figure 33.25). Since 1990, only a small number of patients (with a variety of disorders, such as hemophilia, cancers, and certain types of blindness) have been treated with gene therapy, with varying degrees of success.
Figure 33.24 Techniques used to analyze DNA, RNA, and proteins. ASO = allele-specific oligonucleotides. ELISA = enzymelinked immunosorbent assay; cDNA = complementary DNA; mRNA = messenger RNA.
X. TRANSGENIC ANIMALS
Transgenic animals can be produced by injecting a cloned gene into the fertilized egg. If the gene becomes successfully integrated into a chromosome, it will be present in the germline of the resulting animal and can be passed along from generation to generation. A giant mouse called “Supermouse” was produced in this way by injecting the gene for rat growth hormone into a fertilized mouse egg. [Note: Transgenic animals have been designed that produce human proteins in their milk. Antithrombin, an anticlotting protein, was produced by transgenic goats and approved for clinical use in 2009 (see online Chapter 34).] Sometimes, rather than introducing a functional gene into a mouse, a nonfunctional version is inserted. Such genetically engineered animals can be used to produce a colony of “knockout mice” that lack the product of the affected gene. Such animals can then serve as models for the study of a corresponding human disease. [Note: Knock-in mice result if the inserted gene expresses a mutated product or under- or overexpresses a product.]
Figure 33.25 Gene therapy for SCID caused by adenosine deaminase deficiency. [Note: Bone marrow stem cells and a modified retroviral vector are now used.]
XI. CHAPTER SUMMARY
Restriction endonucleases are bacterial enzymes that cleave double-stranded DNA (dsDNA) into smaller fragments. Each enzyme cleaves at a specific four to eight–base pair sequence (a restriction site), producing DNA segments called restriction fragments. The sequences that are recognized are palindromic. Restriction enzymes form either staggered cuts (sticky ends) or blunt-end cuts on the DNA. Bacterial DNA ligases can join two DNA fragments from different sources if they have been cut by the same restriction endonuclease. This hybrid combination of two fragments is called a recombinant DNA molecule. Introduction of a foreign DNA molecule into a replicating cell permits the amplification (production of many copies) of the DNA, a process called cloning. A vector is a molecule of DNA to which the fragment of DNA to be cloned is joined. Vectors must be capable of autonomous replication within the host cell, must contain at least one specific nucleotide sequence recognized by a restriction endonuclease, and must carry at least one gene that confers the ability to select for the vector such as an antibiotic resistance gene. Prokaryotic organisms normally contain small, circular, extrachromosomal DNA molecules called plasmids that can serve as vectors. They can be readily isolated from the bacterium (or artificially constructed); joined with the DNA of interest; and reintroduced into the bacterium, which will replicate, thus making multiple copies of the hybrid plasmid. A DNA library is a collection of cloned restriction fragments of the DNA of an organism. A genomic library is a collection of fragments of dsDNA obtained by digestion of the total DNA of the organism with a restriction endonuclease and subsequent ligation to an appropriate vector. It ideally contains a copy of every DNA nucleotide sequence in the genome. In contrast, complementary DNA (cDNA) libraries contain only those DNA sequences that are complementary to messenger RNA (mRNA) molecules present in a cell and differ from one cell type to another. Because cDNA has no intervening sequences, it can be cloned into an expression vector for the synthesis of human proteins by bacteria or eukaryotes. Cloned, then purified, fragments of DNA can be sequenced, for example, using the Sanger dideoxy method. A probe is a small piece of RNA or single-stranded DNA (usually labeled with a radioisotope, such as 32P, or another recognizable compound, such as biotin or a fluorescent dye) that has a nucleotide sequence complementary to the DNA molecule of interest (target DNA). Probes can be used to identify which clone of a library or which band on a gel contains the target DNA. Southern blotting is a technique that can be used to detect specific sequences present in DNA. The DNA is cleaved using a restriction endonuclease, and the pieces are separated by gel electrophoresis and are denatured and transferred (blotted) to a nitrocellulose membrane for analysis. The fragment of interest is detected using a probe. The human genome contains many thousands of polymorphisms (DNA sequence variations at a given locus). Polymorphisms can arise from single-base changes and from tandem repeats. A polymorphism can serve as a genetic marker that can be followed through families. A restriction fragment length polymorphism (RFLP) is a genetic variant that can be observed by cleaving the DNA into restriction fragments using a restriction enzyme. A base substitution in one or more nucleotides at a restriction site can render the site unrecognizable by a particular restriction endonuclease. A new restriction site also can be created by the same mechanism. In either case, cleavage with the endonuclease results in fragments of lengths differing from the normal that can be detected by DNA hybridization. This technique can be used to diagnose genetic diseases early in the gestation of a fetus. The polymerase chain reaction (PCR), another method for amplifying a selected DNA sequence, does not rely on the biologic cloning method. PCR permits the synthesis of millions of copies of a specific nucleotide sequence in a few hours. It can amplify the sequence, even when the targeted sequence makes up less than one part in a million of the total initial sample. The method can be used to amplify DNA sequences from any source. Applications of the PCR technique include: 1) efficient comparison of a normal gene with a mutant form of the gene, 2) detection of low-abundance nucleic acid sequences, 3) forensic analysis of DNA samples, and 4) prenatal diagnosis and carrier detection (for example, of cystic fibrosis). The products of gene expression (mRNA and proteins) can be measured by techniques such as the following: Northern blots are very similar to Southern blots except that the original sample contains a mixture of mRNA molecules that are separated by electrophoresis, then hybridized to a radiolabeled probe; microarrays are used to determine the differing patterns of gene expression in two different types of cells (for example, normal and cancer cells); enzyme-linked immunosorbent assays and Western blots (immunoblots) are used to detect specific proteins. Proteomics is the study of all the proteins expressed by a genome. The goal of gene therapy is the insertion of a normal cloned gene to replace a defective gene in a somatic cell. Insertion of a foreign gene into the germline of an animal creates a transgenic animal that can produce therapeutic proteins or serve as a model for human diseases.
Study Questions
Choose the ONE best answer.
33.1 HindIII is a restriction endonuclease. Which of the following is most likely to be the recognition sequence for this enzyme?
A. AAGAAG
B. AAGAGA
C. AAGCTT
D. AAGGAA
E. AAGTTC
Correct answer = C. The vast majority of restriction endonucleases recognize palindromes in double-stranded DNA, and AAGCTT is the only palindrome among the choices. Because the sequence of only one DNA strand is given, the base sequence of the complementary strand must be determined. To be a palindrome, both strands must have the same sequence when read in the 5ʹ→3ʹ direction. Thus, the complement of 5ʹ-AAGCTT-3ʹ is also 5ʹ-AAGCTT-3ʹ.
33.2 An Ashkenazi Jewish couple brings their 6-month-old son to you for evaluation of listlessness, poor head control, and a fixed gaze. You determine that he has Tay-Sachs disease, an autosomal recessive disorder. The couple also has a daughter. The family’s pedigree is shown to the right, along with Southern blots of a restriction fragment length polymorphism very closely linked to the gene for hexosaminidase A, which is defective in Tay-Sachs. Which of the statements below is most accurate with respect to the daughter?
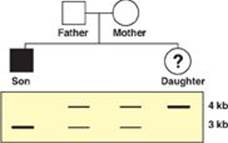
A. She has a 25% chance of having Tay-Sachs disease.
B. She has a 50% chance of having Tay-Sachs disease.
C. She has Tay-Sachs disease.
D. She is a carrier for Tay-Sachs disease.
E. She is homozygous normal.
Correct answer = E. Because they have an affected son, both the biological father and mother must be carriers for this disease. The affected son must have inherited a mutant allele from each parent. Because he shows only the 3-kilobase (kb) band on the Southern blot, the mutant allele for this disease must be linked to the 3-kb band. The normal allele must be linked to the 4-kb band, and, because the daughter inherited only the 4-kb band, she must be homozygous normal for the hexosaminidase A gene.
33.3 A physician would like to determine the global patterns of gene expression in two different types of tumor cells in order to develop the most appropriate form of chemotherapy for each patient. Which of the following techniques would be most appropriate for this purpose?
A. Enzyme-linked immunosorbent assay
B. Microarray
C. Northern blot
D. Southern blot
E. Western blot
Correct answer = B. Microarray analysis allows the determination of messenger RNA (mRNA) production (gene expression) from thousands of genes at once. A Northern blot only measures mRNA production from one gene at a time. Western blots and enzyme-linked immunosorbent assay measure protein production (also gene expression) but only from one gene at a time. Southern blots are used to analyze DNA, not DNA expression.
33.4 A 2-week-old infant is diagnosed with a urea cycle defect. Enzymic analysis showed no activity for ornithine transcarbamoylase (OTC), an enzyme of the cycle. Molecular analysis revealed that the messenger RNA (mRNA) product of the gene for OTC was identical to that of a control. Which of the techniques listed below was most likely used to analyze mRNA?
A. Dideoxy chain termination
B. Northern blot
C. Polymerase chain reaction
D. Southern blot
E. Western blot
Correct answer = B. Northern blot allows analysis of the messenger RNA present (expressed) in a particular cell or tissue. Southern blot is used for DNA analysis, whereas Western blot is used for protein analysis. Dideoxy chain termination is used to sequence DNA. Polymerase chain reaction is used to generate multiple, identical copies of a DNA sequence in vitro.
33.5 For the patient above, which phase of the central dogma was most likely affected?
Correct answer = Translation. The gene is present and is able to be expressed as evidenced by messenger RNA production. The lack of enzymic activity means that some aspect of protein synthesis is affected.