Lippincott’s Illustrated Reviews: Biochemistr, Sixth Edition (2014)
UNIT VI: Storage and Expression of Genetic Information
Chapter 34. Blood clotting
I. OVERVIEW
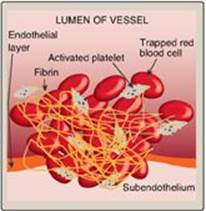
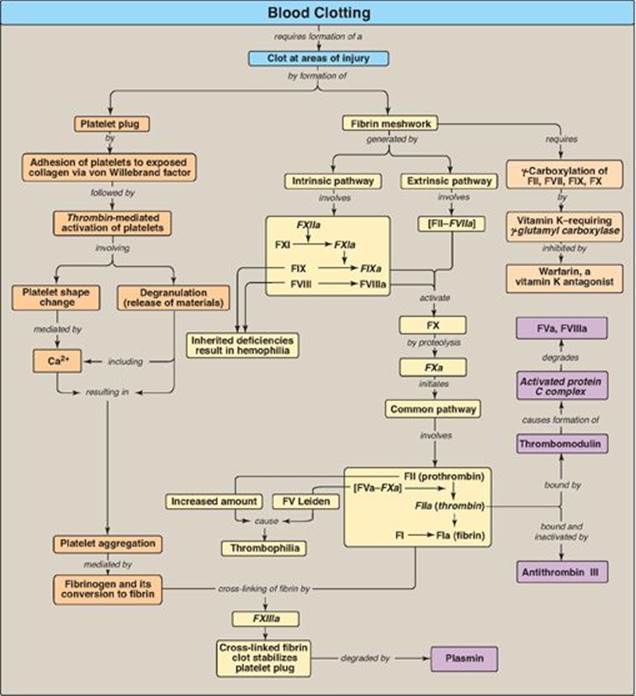
Blood clotting (coagulation) is designed to rapidly stop bleeding from a damaged blood vessel in order to maintain a constant blood volume (hemostasis). Coagulation is accomplished through vasoconstriction and the formation of a clot (thrombus) that consists of a plug of platelets and a meshwork of the protein fibrin that stabilizes the platelet plug. Clotting occurs in association with membranes on the surface of platelets and damaged blood vessels (Figure 34.1). [Note: If clotting occurs within an intact vessel such that the lumen is occluded and blood flow is impeded, a condition known as thrombosis, serious tissue damage and even death can occur. This is what happens, for example, during a myocardial infarction (MI).] Processes to limit clot formation to the area of damage and remove the clot once vessel repair is underway also play essential roles in hemostasis. [Note: Separate discussions of the formation of the platelet plug and the fibrin meshwork facilitate presentation of these multistep, multicomponent processes. However, the two work together to maintain hemostasis.]
Figure 34.1 A blood clot formed by a plug of activated platelets and a meshwork of fibrin at the site of vessel injury.
II. FIBRIN MESHWORK FORMATION
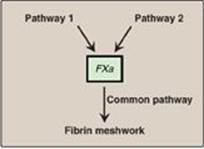
The formation of the fibrin meshwork involves two unique pathways that converge to form a common pathway (Figure 34.2). In each pathway, the major components are proteins (called factors) designated by Roman numerals. The factors are glycoproteins that are synthesized and secreted by the liver, primarily. [Note: Several factors are also denoted by alternative names. For example, active factor X (FXa), the point of pathway convergence, is also known as Stuart factor.]
Figure 34.2 Three pathways involved in formation of the fibrin meshwork.
A. Protolytic cascade
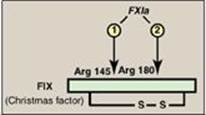
Within the pathways, a cascade is set up in which proteins are converted from an inactive form, or zymogen, to an active form by proteolytic cleavage in which the protein product of one activation reaction initiates another. The active form of a factor is denoted by a lower case “a” after the numeral. The active proteins FIIa, FVIIa, FIXa, FXa, FXIa, and FXIIa are enzymes that function as serine proteases with trypsin-like specificity and, therefore, cleave a peptide bond on the carboxyl side of an arginine or lysine residue in a polypeptide. For example, FIX (Christmas factor) is activated through cleavage at arginine 145 and arginine 180 by FXIa (Figure 34.3). The proteolytic cascade results in enormous rate acceleration, because one active protease can produce many molecules of active product each of which, in turn, can activate many molecules of the next protein in the cascade. In some cases, activation can be caused by a conformational change in the protein in the absence of proteolysis. [Note: Nonproteolytic proteins play a role as accessory proteins (cofactors) in the pathways. FIII, FV, and FVIII are the accessory proteins.]
Figure 34.3 Activation of FIX (Christmas factor) via proteolysis by the serine protease FXIa. [Note: Activation can occur by conformational change for some of the factors.] a = active; Arg = arginine.
B. Role of phosphatidylserine and calcium
The presence of the negatively charged phospholipid phosphatidylserine (PS) and positively charged calcium ions (Ca2+) accelerates the rate of some steps in the cascade.
1. Negatively charged phosphatidylserine: PS is located primarily on the intracellular (cytosolic) face of the plasma membrane. Its exposure signals injury to the endothelial cells that line blood vessels. PS is also exposed on the surface of activated platelets.
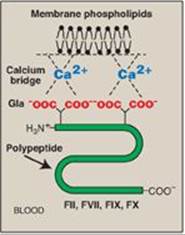
2. Calcium ions: Ca2+ binds the negatively charged γ-carboxyglutamate (Gla) residues present in certain clotting serine proteases (FII, FVII, FIX, and FX), facilitating the binding of these proteins to exposed phospholipids (Figure 34.4). The Gla residues are good chelators of Ca2+ because of their two adjacent negatively charged carboxylate groups (Figure 34.5). [Note: The use of chelating agents such as sodium citrate to bind Ca2+ in blood-collecting tubes or bags prevents the blood from clotting.]
Figure 34.4 Ca2+ facilitates the binding of γ-carboxyglutamate (Gla)- containing factors to membrane phospholipids. F = factor.
C. Formation of γ-carboxyglutamate residues
γ-Carboxylation is a posttranslational modification in which 9–12 glutamate residues (at the amino or N terminus of the target protein) get carboxylated at the γ carbon, thereby forming γ-carboxyglutamate (Gla) residues. The process occurs in the rough endoplasmic reticulum (RER) of the liver.
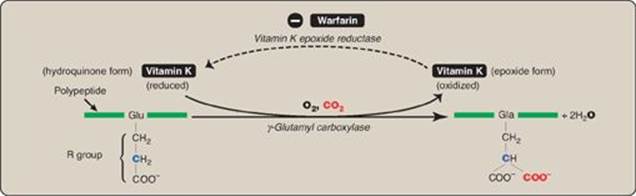
1. γ-Carboxylation: This carboxylation reaction requires a protein substrate, O2, CO2, γ-glutamyl carboxylase, and the hydroquinone form of vitamin K as a coenzyme (Figure 34.6). In the reaction, the hydroquinone form of vitamin K gets oxidized to its epoxide form as O2 is reduced to water. [Note: Vitamin K, a fat-soluble vitamin (see p. 389), is reduced from the quinone form to the hydroquinone coenzyme form by vitamin K reductase (Figure 34.7).]
2. Inhibition by warfarin: The formation of Gla residues is sensitive to inhibition by warfarin, a synthetic analog of vitamin K that inhibits the enzyme vitamin K epoxide reductase (VKOR). The reductase, an integral protein complex of the RER membrane, is required to regenerate the functional hydroquinone form of vitamin K from the epoxide form generated in the γ-carboxylation reaction. Thus, warfarin is an anticoagulant that inhibits clotting by functioning as a vitamin K antagonist. Warfarin salts are used therapeutically to limit clot formation. [Note: Warfarin is used commercially as a pest-control agent such as in rat poison. It was developed by the Wisconsin Alumni Research Foundation, hence the name.]
Figure 34.5 Gla residue.
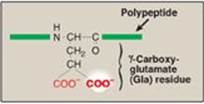
Figure 34.6 γ-Carboxylation of a glutamate (Glu) residue to γ-carboxyglutamate (Gla) by vitamin K-requiring γ-glutamyl carboxylase. The γ carbon is shown in blue.
Genetic differences (genotypes) in the gene for subunit 1 of the VKOR complex (VKORC1) influence patient response to warfarin. For example, a polymorphism in the promoter region of the gene decreases gene expression, resulting in less VKOR being made, thereby necessitating a lower dose of warfarin to achieve a therapeutic level. Polymorphisms in the cytochrome P450 enzyme (CYP2C9) that metabolizes warfarin are also known. In 2010, the U.S. Food and Drug Administration added a genotype-based dose table to the warfarin label (package insert). The influence of genetics on an individual’s response to drugs is known as pharmacogenetics.
D. Pathways
Three distinct pathways are involved in formation of the fibrin meshwork: the extrinsic pathway, the intrinsic pathway, and the common pathway. Production of FXa by the extrinsic and intrinsic pathways initiates the common pathway (see Figure 34.2).
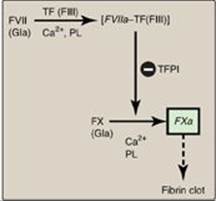
1. Extrinsic pathway: This pathway involves a protein, tissue factor (TF), that is not in the blood but becomes exposed when blood vessels get injured. TF (FIII) is a transmembrane glycoprotein abundant in vascular subendothelium. It is an extravascular accessory protein and not a protease. Any injury that exposes FIII to blood rapidly (within seconds) initiates the extrinsic (or TF) pathway. Once exposed, TF binds a circulating Gla-containing protein, FVII, activating it through conformational change. [Note: FVII can also be activated proteolytically by thrombin (see Section 3. below).] Binding of FVII to TF requires the presence of Ca2+ and phospholipids. The TF–FVIIa complex then binds and activates FX by proteolysis (Figure 34.8). Therefore, activation of FX by the extrinsic pathway occurs in association with the membrane. The extrinsic pathway is quickly inactivated by tissue factor pathway inhibitor (TFPI) that, in a FXa-dependent process, binds to the TF–FVIIa complex and prevents further production of FXa. [Note: TF and FVII are unique to the extrinsic pathway.]
Figure 34.7 The vitamin K cycle. VKOR = vitamin K epoxide reductase.
Figure 34.8 The extrinsic or tissue factor (TF) pathway. Binding of FVII to exposed TF (FIII) activates FVII. [Note: The pathway is quickly inhibited by tissue factor pathway inhibitor (TFPI).] F = factor; Gla = γ-carboxyglutamate; PL = phospholipid; a = active.
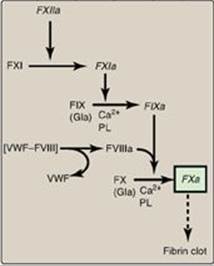
2. Intrinsic pathway: All of the protein factors involved in the intrinsic pathway are present in the blood and are, therefore, intravascular. The intrinsic pathway involves two phases: the contact phase and the FX-activation phase, each with known deficiencies.
a. Contact phase: This phase results in the activation of FXII (Hageman factor) to FXIIa by conformational change through binding to a negative surface. Deficiencies in FXII (or in the other proteins of this phase, high-molecular-weight kininogen and prekallikrein) do not result in bleeding problems, calling into question the importance of this phase in coagulation. However, the contact phase does play a role in inflammation. [Note: FXII can be activated proteolytically by thrombin (see Section 3. below)].
b. Factor X–activation phase: The sequence of events leading to the activation of Factor X to FXa by the intrinsic pathway is initiated by FXIIa (Figure 34.9). FXIIa activates FXI, and FXIa activates FIX, a Gla-containing protein. FIXa combines with FVIIIa (a bloodborne accessory [nonenzymatic] protein), and the complex activates FX, a Gla-containing serine protease. [Note: The complex containing FIXa, FVIIIa, and FX forms on exposed negatively charged membrane regions, and FX gets activated to FXa. This complex is sometimes referred to as Xase. Binding of the complex to membrane phospholipids requires Ca2+.]
c. Factor XII deficiency: A deficiency in FXII does not lead to a bleeding disorder. This is because FXI, the next protein in the cascade, can be activated proteolytically by thrombin (see Section 3. below).
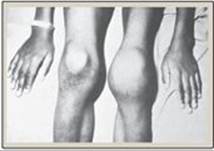
d. Hemophilia: Hemophilia is a coagulopathy, a defect in the ability to clot. Hemophilia A (the most common form of hemophilia) results from deficiency of FVIII, whereas deficiency of FIX results in hemophilia B. Each deficiency is characterized by decreased and delayed ability to clot and/or formation of abnormally friable (easily disrupted) clots. This can be manifested, for example, by bleeding into the joints (Figure 34.10). The extent of the factor deficiency determines the severity of the disease. Current treatment for hemophilia is factor replacement therapy using factors obtained from pooled human blood or from recombinant DNA technology. Gene replacement therapy is a goal. Because the genes for both proteins are on the X chromosome, hemophilia is an X-linked disorder. [Note: Deficiency of FXI results in a bleeding disorder that sometimes is referred to as hemophilia C.]
Figure 34.9 FX activation phase of the intrinsic pathway. [Note: von Willebrand factor (VWF) stabilizes FVIII in the circulation.] Gla = γ-carboxyglutamate; PL = phospholipid; a = active; F = factor.
The inactivation of the extrinsic pathway by TFPI results in dependence on the intrinsic pathway for continued production of FXa. This explains why individuals with hemophilia bleed even though they have an intact extrinsic pathway.
Figure 34.10 Acute bleeding into joint spaces (hemarthrosis) in an individual with hemophilia.
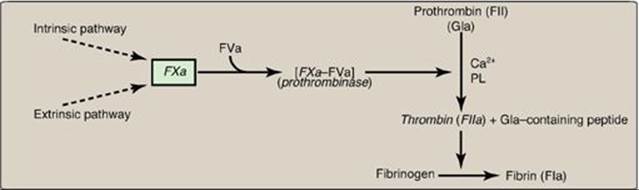
3. Common pathway: FXa produced by both the intrinsic and the extrinsic paths initiates the common pathway, a sequence of events that results in the generation of fibrin (FIa) (Figure 34.11). FXa associates with FVa (a bloodborne accessory [nonenzymic] protein) and, in the presence of Ca2+ and phospholipids, forms a membrane-bound complex referred to as prothrombinase. The complex cleaves prothrombin (FII) to thrombin (FIIa). [Note: FVa potentiates the proteolytic activity of FXa.] The binding of Ca2+ to the Gla residues in FII facilitates the binding of FII to the membrane and to the prothrombinase complex, with subsequent cleavage to thrombin. Cleavage excises the Gla-containing region, releasing thrombin from the membrane and, thereby, freeing it to activate fibrinogen (FI) in the blood. [Note: This is the only example of cleavage of a Gla protein that results in the release of a Gla-containing peptide. The peptide travels to the liver where it is thought to act as a signal for increased production of clotting proteins.] Oral, direct inhibitors of FXa have been approved for limited clinical use as anticoagulants.
A common point mutation (G20210A) in which an adenine (A) replaces a guanine (G) at nucleotide 20210 in the 3′ untranslated region of the gene for prothrombin leads to increased levels of prothrombin in the blood. This results in thrombophilia, a condition characterized by an increased tendency to clot.
Figure 34.11 Generation of fibrin by FXa and the common pathway. F = factor; Gla = γ-carboxyglutamate; PL = phospholipid; a = active.
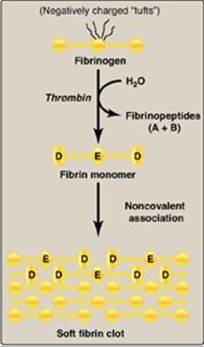
a. Conversion of fibrinogen to fibrin by thrombin: Fibrinogen is a soluble glycoprotein made by the liver. It consists of dimers of three different polypeptide chains [(Aα)2(Bβ)2(γ)2] held together at the N termini by disulfide bonds. [Note: Aα and Bβ each represent a single polypeptide.] The N termini of the Aα and Bβ chains form “tufts” on the central of three globular domains (Figure 34.12). The tufts are negatively charged and result in repulsion between fibrinogen molecules. Thrombin cleaves the charged tufts (releasing fibrinopeptides A and B), and fibrinogen becomes fibrin. As a result of the loss of charge, the fibrin monomers are able to noncovalently associate in a staggered array, and a soft (soluble) fibrin clot is formed.
Figure 34.12 Conversion of fibrinogen to fibrin and formation of the soft fibrin clot. [Note: D and E refer to domains on fibrin.]
b. Cross-linking of fibrin: The associated fibrin molecules get covalently cross-linked. This converts the soft clot to a hard (insoluble) clot. FXIIIa, a transglutaminase, covalently links the γ-carboxamide of a glutamine residue in one fibrin molecule to the ε-amino of a lysine residue in another through formation of an isopeptide bond and release of ammonia (Figure 34.13). [Note: FXIII is also activated by thrombin.]
c. Importance of thrombin: The activation of FX by the extrinsic path provides the “spark” of FXa that results in the initial activation of thrombin. Active thrombin then activates factors of the common (FV, FI, FXIII), intrinsic (FXI, FVIII), and extrinsic (FVII) pathways (Figure 34.14). It also activates FXII of the contact phase. The extrinsic pathway, then, initiates clotting by the generation of FXa, and the intrinsic pathway amplifies and sustains clotting after the extrinsic pathway has been inhibited by TFPI. [Note: Hirudin, a peptide secreted from the salivary gland of medicinal leeches, is a potent, direct, oral thrombin inhibitor. Recombinant hirudin has been approved for limited clinical use.] Additional crosstalk between the pathways of clotting is achieved by the FVIIa–TF-mediated activation of the intrinsic pathway and the FXIIa-mediated activation of the extrinsic pathway. The complete picture of physiologic blood clotting via the formation of a hard fibrin clot is shown in Figure 34.15. The factors of the clotting cascade are shown organized by function in Figure 34.16.
Figure 34.13 Cross-linking of fibrin. FXIIIa forms a covalent isopeptide bond between a lysine residue and a glutamine residue. F = factor.
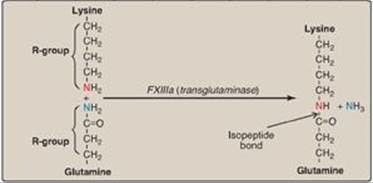
Figure 34.14 The importance of thrombin in formation of the fibrin clot. a = active; F = factor.
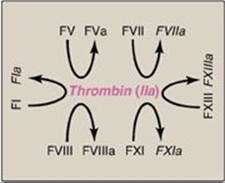
Figure 34.15 The complete picture of physiologic blood clotting via the formation of a cross-linked (hard) fibrin clot. a = active; F = factor; TF = tissue factor; TFPI = tissue factor pathway inhibitor; PL = phospholipid; Gla = γ-carboxyglutamate.
Clinical laboratory tests are available to evaluate the extrinsic through common pathways (prothrombin time [PT] using thromboplastin and expressed as the International Normalized Ratio [INR]) and the intrinsic through common pathways (activated partial thromboplastin time [aPTT]). Thromboplastin is a combination of phospholipids + FIII. A derivative, partial thromboplastin, contains just the phospholipid portion because FIII isn’t needed to activate the intrinsic pathway.
Figure 34.16 Protein factors of the clotting cascade organized by function. The activated form would be denoted by an a after the numeral. [Note: Ca2+ is IV. There is no VI. I (fibrin) is neither a protease nor an accesory protein. XIII is a transglutaminase.] Gla = γ-carboxyglutamate.

III. LIMITING CLOTTING
The ability to limit clotting to areas of damage (anticoagulation) and to remove clots once repair processes are underway (fibrinolysis) are exceedingly important aspects of hemostasis. These actions are performed by proteins that inactivate clotting factors either by binding to them and removing them from the blood or by degrading them and by proteins that degrade the fibrin meshwork.
A. Inactivating proteins
Proteins synthesized by the liver and by the blood vessels themselves balance the need to form clots at sites of vessel injury with the need to limit their formation beyond the injured area.
Figure 34.17 Inactivation of FIIa (thrombin) by binding of antithrombin III (ATIII) and transport to the liver. [Note: Heparin increases the affinity of ATIII for FIIa.] a = active; F = factor.
![]()
1. Antithrombin: Antithrombin III (ATIII), also referred to simply as antithrombin (AT), is a hepatic protein that circulates in the blood. It inactivates free thrombin by binding to it and carrying it to the liver (Figure 34.17). Thus, ATIII removes thrombin from the blood, preventing it from participating in coagulation. [Note: ATIII is a serine protease inhibitor, or “serpin.” A serpin contains a reactive loop to which a specific protease binds. Once bound, the protease cleaves a peptide bond in the serpin causing a conformational change that traps the enzyme in a covalent complex. a1-Antitrypsin (see p. 50) is also a serpin.] The affinity of ATIII for thrombin is greatly increased when ATIII is bound to heparin, an intracellular glycosaminoglycan released in response to injury by mast cells associated with blood vessels. Heparin, an anticoagulant, is used therapeutically to limit clot formation. [Note: In contrast to the anticoagulant warfarin, which has a slow onset, a long half-life, and is administered orally, heparin has a rapid onset, a short half-life, and requires intravenous administration. The two drugs are commonly used in an overlapping manner in the treatment of thrombosis.] ATIII also inactivates FXa and the other serine proteases of clotting, FIXa, FXIa, FXIIa, and the FVIIa–TF complex. [Note: AT binds to a specific pentasaccharide within the oligosaccharide form of heparin. Inhibition of FIIa requires the oligosaccharide form, whereas inhibition of FXa requires only the pentasaccharide form. Fondaparinux, a synthetic version of the pentasaccharide, is used clinically to inhibit FXa.]
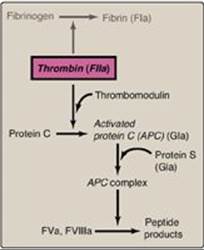
2. Protein C–protein S complex: Protein C, a circulating Gla-containing protein made in the liver, is activated by thrombin complexed with thrombomodulin. Thrombomodulin, an integral membrane glycoprotein of endothelial cells, binds thrombin, thereby decreasing thrombin’s affinity for fibrinogen and increasing its affinity for protein C. Protein C in complex with protein S, also a Gla-containing protein, forms the activated protein C (APC) complex that cleaves the accessory proteins FVa and FVIIIa that are required for maximal activity of FXa (Figure 34.18). Protein S helps anchor APC to the clot. Thrombomodulin, then, modulates the activity of thrombin, converting it from a protein of coagulation to a protein of anticoagulation, thereby limiting the extent of clotting. Factor V Leiden is a mutant form of FV (glutamine is substituted for arginine at position 506) that is resistant to APC. It is the most common inherited cause of thrombophilia in the United States, with highest frequency in the Caucasian population. Heterozygotes have a 7-fold increase in the risk for venous thrombosis, and homozygotes have up to a 50-fold increase. [Note: Women with FV Leiden are at even greater risk of thrombosis during pregnancy or when taking estrogen.]
Figure 34.18 Formation and action of the APC complex. Gla = γ-carboxyglutamate; a = active; F = factor.
Thrombophilia (hypercoagulability) can result from deficiencies of proteins C, S, and ATIII; from the presence of FV Leiden and antiphospholipid antibodies; and from excess production of prothrombin (G20210A mutation). [Note: A thrombus that forms in the deep veins of the leg (deep venous thrombosis, or DVT) can cause a pulmonary embolism (PE) if the clot (or a piece of it) breaks off, travels to the lungs, and blocks circulation.]
Figure 34.19 Fibrinolysis. TPA = tissue plasminogen activator; i = inactive; a = active; PAI = plasminogen activator inhibitor. [Note: Plasmin bound to fibrin is protected from its inhibitor.]
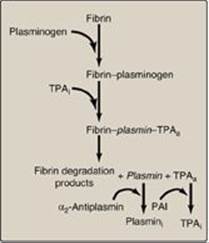
B. Fibrinolysis
Clots are temporary patches that must be removed once wound repair has begun. The fibrin clot is cleaved by the protein plasmin to fibrin-degradation products (Figure 34.19). [Note: Measurement of D-dimer, a fibrin-degradation product containing two cross-linked D domains released by the action of plasmin, can be used to assess the extent of clotting (see Figure 34.12).] Plasmin is a serine protease that is generated from plasminogen by plasminogen activators. Plasminogen, secreted by the liver into the circulation, binds to fibrin and is incorporated into clots as they form. Tissue plasminogen activator (TPA, t-PA), made by vascular endothelial cells and secreted in an inactive form in response to thrombin, becomes active when bound to fibrin–plasminogen. Bound plasmin and TPA are protected from their inhibitors, α2-antiplasmin (a serpin) and plasminogen activator inhibitors, respectively. Once the fibrin clot is dissolved, plasmin and TPA become available to their inhibitors. Therapeutic fibrinolysis in patients with an MI or an ischemic stroke can be achieved by treatment with commercially available TPA made by recombinant DNA techniques. [Note: Urokinase is a plasminogen activator (u-PA) made in a variety of tissues and originally isolated from urine. Streptokinase (from bacteria) also activates plasminogen and works on both free and fibrin-bound plasminogen.]
Plasminogen contains structural motifs known as kringle domains that mediate protein–protein interactions. Lipoprotein(a) [Lp(a)] also contains kringle domains and, thus, competes with plasminogen for binding to fibrin. The potential to inhibit fibrinolysis may be the basis for the association of elevated Lp(a) with increased risk for cardiovascular disease (see p. 236).
Figure 34.20 Size comparison of platelets, erythrocytes, and a leukocyte.
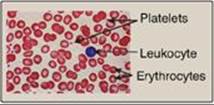
IV. PLATELET PLUG FORMATION
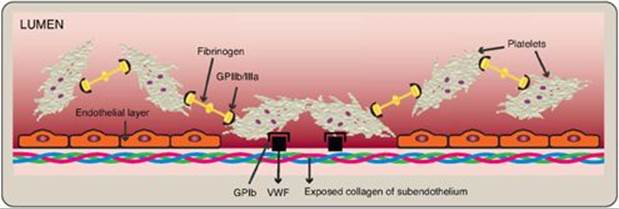
Platelets (thrombocytes) are small, anucleate fragments of megakaryocytes that adhere to exposed collagen of damaged endothelium, get activated, and aggregate to form a platelet plug (Figure 34.20). Formation of the platelet plug is referred to as primary hemostasis. In a normal adult there are 150,000–450,000 platelets per μl of blood. They have a lifespan of up to 10 days, after which they are taken up by the liver and spleen and destroyed. Clinical laboratory tests to measure platelet number and activity are available.
Figure 34.21 Binding of platelets via the receptor glycoprotein Ib (GPIb) to von Willebrand factor (VWF). VWF is bound to the exposed collagen at a site of injury.
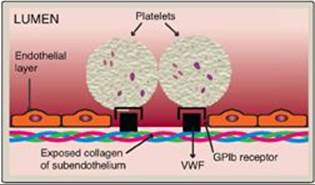
A. Adhesion
Adhesion of platelets to exposed collagen at the site of vessel injury is mediated by the protein von Willebrand factor (VWF). VWF binds to collagen, and platelets bind to VWF via glycoprotein Ib (GPIb), a component of a membrane receptor complex (GPIb–V–IX) on the platelet surface (Figure 34.21). Binding to VWF stops the forward movement of platelets. [Note: Deficiency in the receptor for VWF results in Bernard-Soulier syndrome, a disorder of decreased platelet function and number.] VWF is a glycoprotein that is released from platelets. It also is made and secreted by endothelial cells. In addition to mediating the binding of platelets to collagen, VWF also binds to and stabilizes FVIII in the blood. Deficiency of VWF results in von Willebrand disease (VWD), the most common inherited defect in the ability to clot (coagulopathy). VWD coagulopathy results from decreased binding of platelets to collagen and a deficiency in FVIII (due to increased degradation). Platelets can also bind directly to collagen via the membrane receptor GPVI. Once adhered, platelets get activated. [Note: Damage to the endothelium also exposes FIII, initiating the extrinsic pathway of blood clotting and activation of FX (see Figure 34.8).]
Figure 34.22 Platelet activation by thrombin. [Note: Protease-activated receptors are a type of G protein-coupled receptor.] PIP2 = phosphoinositol bisphosphate; DAG = diacylglycerol; IP3 = inositol trisphosphate;TXA2 = thromboxane A2; ADP = adenosine diphosphate; PDGF = platelet-derived growth factor; VWF = von Willebrand factor; F = factor.
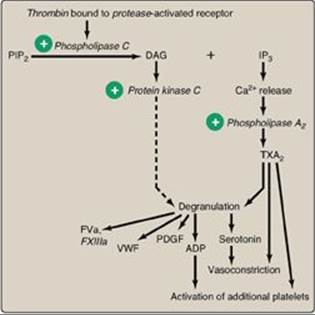
B. Activation
Once adhered to areas of injury, platelets get activated. Platelet activation involves morphologic (shape) changes and degranulation, the process by which platelets secrete the contents of their α and δ (or dense) storage granules. Activated platelets also expose PS on their surface. Thrombin is the most potent platelet activator. Thrombin binds to and activates protease-activated receptors, a type of G protein–coupled receptor (GPCR), on the surface of platelets (Figure 34.22). Thrombin is primarily associated with Gq proteins (see p. 205), resulting in activation of phospholipase C and a rise in diacylglycerol (DAG) and inositol trisphosphate (IP3). [Note: Thrombomodulin, through its binding of thrombin, decreases the availability of thrombin for platelet activation (see Figure 34.18).]
1. Degranulation: DAG activates protein kinase C, a key event for degranulation. IP3 causes the release of Ca2+ (from dense granules). The Ca2+ activates phospholipase A2, which cleaves membrane phospholipids to release arachidonic acid, the substrate for the synthesis of thromboxane A2 (TXA2) in activated platelets by cyclooxygenase-1 (COX-1) (see p. 214). TXA2 causes vasoconstriction, augments degranulation, and binds to platelet GPCRs, causing activation of additional platelets. Recall that aspirin irreversibly inhibits COX and, consequently, TXA2 synthesis and is referred to as an “antiplatelet” drug. Degranulation also results in release of serotonin and adenosine diphosphate (ADP) from dense granules. Serotonin causes vasoconstriction. ADP binds to GPCRs on the surface of platelets, activating additional platelets. [Note: Some antiplatelet drugs, such as clopidogrel, are ADP-receptor antagonists.] Platelet-derived growth factor (involved in wound healing), VWF, FV, FXIII, and fibrinogen are among other proteins released from α granules. [Note: Platelet-activating factor (PAF), an ether phospholipid (see p. 202) synthesized by a variety of cell types including endothelial cells and platelets, binds PAF receptors (GPCRs) on the surface of platelets and activates them.]
2. Shape change: The change in shape of activated platelets from discoidal to spherical with pseudopod-like processes that facilitate platelet–platelet and platelet–surface interactions (Figure 34.23) is initiated by the release of Ca2+ from dense granules. Ca2+ bound to calmodulin mediates the activation of myosin light-chain kinase that phosphorylates the myosin light chain (see p. 151), resulting in a major reorganization of the platelet cytoskeleton.
Figure 34.23 Activated platelets undergo Ca2+-initiated shape change.

C. Aggregation
Activation causes dramatic changes in platelets that lead to their aggregation. Structural changes in a surface receptor (GPIIb/IIIa) expose binding sites for fibrinogen. Bound fibrinogen molecules link activated platelets to one another (Figure 34.24), with a single fibrinogen able to bind two platelets. The fibrinogen is converted to fibrin by thrombin and then covalently cross-linked by FXIIIa coming from both the blood and the platelets. [Note: The exposure of PS on the surface of activated platelets allows formation of the Xase complex (VIIIa, IXa, X, and Ca2+) with subsequent formation of FXa and generation of thrombin.] Fibrin strengthens the platelet plug. [Note: Rare defects in the platelet receptor for fibrinogen result in Glanzmann thrombasthenia (decreased platelet function), whereas autoantibodies to this receptor are a cause of immune thrombocytopenia (decreased platelet number).]
Unnecessary activation of platelets is prevented because 1) an intact vascular wall is separated from the blood by a monolayer of endothelial cells, preventing the contact of platelets with collagen; 2) endothelial cells synthesize prostaglandin I2 (PGI2, or prostacyclin) and nitric oxide, each of which causes vasodilation; and 3) endothelial cells have a cell-surface ADPase that converts ADP to AMP.
Figure 34.24 Linking of platelets by fibrinogen via the receptor glycoprotein (GP) IIb/IIIa. [Note: The shapes in the fibrinogen molecule represent the two D and one E domains.] GPIb = glycoprotein Ib receptor; VWF = von Willebrand factor.
V. CHAPTER SUMMARY
Blood clotting (coagulation) is designed to rapidly stop bleeding from a damaged blood vessel in order to maintain a constant blood volume (hemostasis). Coagulation is accomplished through formation of a clot (thrombus) consisting of a plug of platelets (thrombocytes) and a meshwork of the protein fibrin (Figure 34.25). Wound to a tissue damages blood vessels and exposes collagen. Platelets adhere to the exposed collagen, get activated, and aggregate to form a platelet plug. Adhesion is mediated by von Willebrand Factor (VWF). VWF binds collagen, and platelets bind VWF via GPIb within a receptor complex on the platelet surface. Deficiency of VWF results in von Willebrand disease, the most common inherited coagulopathy. Once adhered, platelets get activated. Platelet activation involves changes in shape (discoidal to spherical with pseudopodia) and degranulation, the process by which platelets release the contents of their storage granules. Thrombin is the most potent activator of platelets. Thrombin binds to protease-activated G protein–coupled receptors on the surface of platelets. Activated platelets release substances that cause vasoconstriction (serotonin and thromboxane A2 [TXA2]), recruit and activate other platelets (adenosine diphosphate and TXA2) and support the formation of a fibrin clot(factor [F] V, FXIII, and fibrinogen). Activation causes changes in platelets that lead to their aggregation. Structural changes in a surface receptor (GPIIb/IIIa) expose binding sites for fibrinogen. Fibrinogen molecules link activated platelets to one another. The fibrinogen is activated to fibrin by thrombin and then cross-linked by FXIIIa, a transglutaminase coming both from the blood and from platelets. The initial loose plug of platelets (primary hemostasis) is strengthened by the fibrin meshwork (secondary hemostasis).
The formation of the fibrin meshwork involves the extrinsic and intrinsic pathways (and their associated protein factors) that converge at FXa to form the common pathway. Many of the protein factors are serineproteases with trypsin-like specificity. Ca2+ binds the negatively charged γ-carboxyglutamate (Gla) residues present in certain of the clotting proteins (FII, FVII, FIX, and FX), facilitating the binding of these proteins to exposed phosphatidylserine at the site of injury and on the surface of platelets. γ-Glutamyl carboxylase and its coenzyme, the hydroquinone form of vitamin K, are required for formation of Gla residues. In the reaction, vitamin K gets oxidized to the nonfunctional epoxide form. Warfarin, a synthetic analog of vitamin K used clinically to reduce clotting, inhibits the enzyme vitamin K epoxide reductase that regenerates the functional reduced form. The extrinsic pathway is initiated by exposure of FIII (tissue factor [TF]), an accessory protein, in vascular subendothelium. Exposed TF binds a circulating Gla-containing protein, FVII, activating it through conformational change. The TF–FVIIa complex then binds and activates FX by proteolysis. The extrinsic pathway is rapidly inhibited by tissue factor pathway inhibitor. The intrinsic pathway is initiated by FXIIa. FXIIaactivates FXI, and FXIa activates FIX. FIXa combines with FVIIIa (an accessory protein), and the complex activates FX. FVIII deficiency results in hemophilia A, whereas FIX deficiency results in the less common hemophilia B. FXa associates with FVa (an accessory protein), forming prothrombinase that cleaves prothrombin (FII) to thrombin (FIIa). Thrombin then cleaves fibrinogen to fibrin (FIa). Fibrin monomers associate, forming a soluble (soft) fibrin clot. The fibrin molecules get cross-linked by FXIIIa, forming an insoluble (hard) fibrin clot. Proteins synthesized by the liver and by blood vessels themselves balance coagulation with anticoagulation. Antithrombin III, a serine protease inhibitor, or serpin, binds to and removes thrombin from the blood. Its affinity for thrombin is increased by heparin, which is used therapeutically to limit clot formation. Protein C, a Gla-containing protein, is activated by the thrombin–thrombomodulin complex. Thrombomodulin decreases thrombin’s affinity for fibrinogen, converting it from a protein of coagulation to a protein of anticoagulation. Protein C in complex with protein S (a Gla-containing protein) forms the activated protein C (APC) complex that cleaves the accessory proteins FVa and FVIIIa. Factor V Leiden is resistant to APC. It is the most common inherited thrombophilic condition in the United States. The fibrin clot is cleaved (fibrinolysis) by the protein plasmin, a serine protease that is generated from plasminogen by plasminogen activators such as tissue plasminogen activator (TPA, t-PA). Recombinant TPA is used clinically. Disorders of platelets and coagulation proteins can result in deviations in the ability to clot. Prothrombin time and activated partial thromboplastin time are used to evalulate the clotting cascade.
Figure 34.25 Key concept map for blood clotting. a = active; F = factor.
Study Questions
Choose the ONE best answer.
For Questions 31.1–31.5, match the most appropriate protein of clotting to the description.
|
A. |
FI |
|
|
B. |
FII |
|
|
C. |
FIII |
|
|
D. |
FV |
|
|
E. |
FVII |
|
|
F. |
FVIII |
|
|
G. |
FIX |
|
|
H. |
FX |
|
|
I. |
FXI |
|
|
J. |
FXIII |
34.1 This factor activates components of the intrinsic, extrinsic, and common pathways.
34.2 This factor converts the soluble clot to an insoluble clot.
34.3 This factor initiates the common pathway.
34.4 This factor is an accessory protein that potentiates the activity of factor Xa.
34.5 This factor is a γ-carboxyglutamate–containing serine protease of the extrinsic pathway.
Correct answers = B, J, H, D, E. Thrombin (FII) is formed in the common pathway and activates components in each of the three pathways of the clotting cascade. Factor (F)XIII, a transglutaminase, covalently cross-links associated fibrin monomers, thereby converting a soluble clot to an insoluble one. The generation of FXa by the intrinsic and extrinsic pathways initiates the common pathway. FV increases the activity of FXa. It is one of three accessory (nonprotease) proteins. The others are FIII (tissue factor) and FVIII (complexes with FIX to activate FX). FVII is a γ-carboxyglutamate–containing serine protease that complexes with FIII in the extrinsic pathway.
34.6 In which patient would prothrombin time be unaffected and activated partial thromboplastin time be prolonged?
A. A patient on aspirin therapy
B. A patient with end-stage liver disease
C. A patient with hemophilia
D. A patient with thrombocytopenia
Correct answer = C. Prothrombin time (PT) measures the activity of the extrinsic through the common pathways, and activated partial thromboplastin time (aPTT) measures the activity of the intrinsic through the common pathways. Patients with hemophilia are deficient in either factor (F)VIII (hemophilia A) or FIX (hemophilia B), components of the common pathway. They have an intact extrinsic pathway. Therefore, the PT is unaffected, and the aPTT is prolonged. Patients on aspirin therapy and those with thrombocytopenia have alterations in platelet function and number, respectively, and not in the proteins of the clotting cascade. Therefore, both the PT and the aPTT are unaffected. Patients with end-stage liver disease have decreased ability to synthesize clotting proteins. They show prolonged PT and aPTT.
34.7 Which one of the following can be ruled out in a patient with thrombophilia?
A. A deficiency of antithrombin III
B. A deficiency of factor IX
C. A deficiency of protein C
D. An excess of prothrombin
E. Expression of factor V Leiden
Correct answer = B. Symptomatic deficiencies in clotting factors will present with a decreased ability to clot (coagulopathy). Thrombophilia, however, is characterized by an increased tendency to clot. Choices A, C, D, and E result in thrombophilia.
34.8 Current guidelines for the treatment of patients with acute ischemic stroke (a stroke caused by a blood clot obstructing a vessel that supplies blood to the brain) include the recommendation that tissue plasminogen activator (TPA) be used shortly after the onset of symptoms. The basis of the recommendation for TPA is that it activates:
A. antithrombin III.
B. the activated protein C complex.
C. the receptor for von Willebrand factor.
D. the serine protease that degrades fibrin.
E. thrombomodulin.
Correct answer = D. Tissue plasminogen activator (TPA) converts plasminogen to plasmin. Plasmin (a serine protease) degrades the fibrin meshwork, removing the obstruction to blood flow. Antithrombin III in association with heparin binds thrombin and carries it to the liver, decreasing thrombin’s availability in the blood. The activated protein C complex degrades the accessory proteins FV and FVIII. The platelet receptor for von Willebrand factor is not affected by TPA. Thrombomodulin binds thrombin and converts it from a protein of coagulation to one of anticoagulation by decreasing its activation of fibrinogen and increasing its activation of protein C.
34.9 The adhesion, activation, and aggregation of platelets provide the initial plug at the site of vessel injury. Which of the following statements concerning the formation of this platelet plug is correct?
A. Activated platelets undergo a shape change that decreases their surface area.
B. Formation of a platelet plug is prevented in intact vessels by the production of thromboxane A2 by endothelial cells.
C. The activation phase requires production of cyclic adenosine monophosphate.
D. The adhesion phase is mediated by the binding of platelets to von Willebrand factor via glycoprotein Ib.
E. Thrombin activates platelets by binding to a protease-activated G protein–coupled receptor and causing activation of protein kinase A.
Correct answer = D. The adhesion phase of platelet plug formation is initiated by the binding of von Willebrand factor to a receptor (glycoprotein Ib) on the surface of platelets. Shape change from discoidal to spherical with pseudopodia increases the surface area of platelets. Thromboxane A2 is made by platelets. It causes platelet activation and vasoconstriction. Adenosine diphosphate is released from activated platelets, and it itself activates platelets. Thrombin works primarily through receptors coupled to Gq proteins causing activation of phospholipase C.
34.10 Several days after having had their home treated for an infestation of rats, the parents of a 3-year-old girl become concerned that she might be ingesting the poison-containing pellets. After calling the Poison Hotline, they take her to the Emergency Department. Blood studies reveal a prolonged prothrombin and activated partial thromboplastin time and a decreased concentration of factor (F)II, FVII, FIX, and FX. Why might administration of vitamin K be a rational approach to the treatment of this patient?
Many rodent poisons are super warfarins, drugs that have a long half-life in the body. Warfarin inhibits γ-carboxylation (production of γ-carboxyglutamate, or Gla, residues), and the clotting proteins reported as decreased are the Gla-containing proteases of the clotting cascade. [Note: Proteins C and S of anticlotting are also Gla-containing proteins.] Because warfarin functions as a vitamin K antagonist, administration of vitamin K is a rational approach to treatment.