Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 28
A Jewish couple of Eastern European descent presents to the clinic for prenatal counseling after their only child died early in childhood. The family could not remember the name of the disorder but said it was common in their ancestry. Their first child was normal at birth, a slightly larger than normal head circumference, an abnormal “eye finding,” and a severe progressive neurologic disease with decreased motor skills and eventually death. The results of the autopsy are consistent with Tay-Sachs disease.
![]() What type of inheritance is this disorder?
What type of inheritance is this disorder?
![]() What is the biochemical cause of the disorder?
What is the biochemical cause of the disorder?
ANSWERS TO CASE 28:
Tay-Sachs Disease
Summary: A Jewish couple of Eastern European descent presents for prenatal counseling after having a child die with a progressive neurologic disorder with loss of motor skills and abnormally large head and an abnormal eye examination. Autopsy shows Tay-Sachs disease.
• Inheritance: Autosomal recessive; 1:30 carrier rate in Ashkenazi Jews.
• Molecular basis of disorder: Lysosomal storage disorder with deficiency of hexosaminidase A enzyme resulting in GM2 gangliosides accumulating throughout the body.
CLINICAL CORRELATION
Tay-Sachs disease is a fatal genetic disorder where harmful amounts of lipids called ganglioside GM2 accumulate in the nerve cells and brains of those affected. Infants with this disorder appear normal for the first several months of life, and then as the lipids distend the nerve cells and brain cells, progressive deterioration occurs; the child becomes blind, deaf, and eventually unable to swallow. Tay-Sachs disease mainly occurs in Jewish children of Eastern European descent, and death from bronchopneumonia usually occurs by 3 to 4 years of age. A reddish spot on the retina also develops, and symptoms first appear around 6 months of age. It is a lysosomal storage disorder with insufficient activity of the enzyme hexosaminidase A, which catalyzes the biodegradation of the gangliosides. The diagnosis is made by clinical suspicion and serum hexosaminidase level. Currently, no treatment is available for this disease.
APPROACH TO:
Lysosomal Storage
OBJECTIVES
1. Describe the enzymatic defects that lead to the lysosomal storage disorders.
2. Describe the synthesis and degradation of sphingolipids.
DEFINITIONS
CERAMIDE: A component of all sphingolipids that is composed of a long-chain fatty acyl group in an amide linkage to sphingosine.
GANGLIOSIDE: Complex carbohydrate-rich glycosphingolipids containing 3 or more monosaccharides esterified to ceramide with at least one of the sugars being N-acetylneuraminic acid (sialic acid). Gangliosides are membrane components found on nerve endings.
GANGLIOSIDOSES: The build-up of gangliosides in the lysosome as a result of the deficiency of one or more enzymes involved in the degradation of gangliosides.
I-CELL DISEASE: A disease in which there is a buildup of a number of different biomolecules that are normally degraded in the lysosome as a result of a deficiency in an enzyme that modifies lysosomal enzymes so that they are targeted to the lysosome.
LYSOSOMAL STORAGE DISORDERS: Genetic diseases that are a result of a deficiency of 1 or more enzymes present in the lysosome. This leads to a buildup of the biomolecules that are normally degraded in the lysosome.
SPHINGOLIPID: Any of a number of lipid molecules that contain sphingosine as part of its molecular structure. These include ceramide, sphingomyelin, cerebrosides, sulfatides, and gangliosides.
TAY-SACHS DISEASE: A genetic disease that is a result of a deficiency in hexosaminidase A (β-N-acetylhexosaminidase), an enzyme involved in the degradation of gangliosides in the lysosome. The disease is prevalent in Jewish children of Eastern European descent and leads to a buildup of ganglioside GM2 in nerve cells of the brain and neuronal dysfunction.
DISCUSSION
Sphingolipids are a class of lipids found in biologic membranes in which the backbone of the lipid molecule is sphingosine, an 18-carbon amino alcohol. This is in contrast to the glycerol backbone found in lipids such as phospholipids. The precursor molecules for the synthesis of sphingosine are palmitoyl-CoA and serine, which condense to form 3-dehydrosphinganine (3-ketodihydrosphingosine; Figure 28-1). This is followed by a reduction reaction using NADPH to form sphinganine (dihydrosphingosine). Ceramide is formed in a 2-step reaction by the formation of an amide bond between sphinganine and a long-chain fatty acid (usually behenic acid, a saturated C-22 fatty acid). This is followed by an oxidation reaction using FAD that introduces a transdouble bond into the sphinganine backbone.
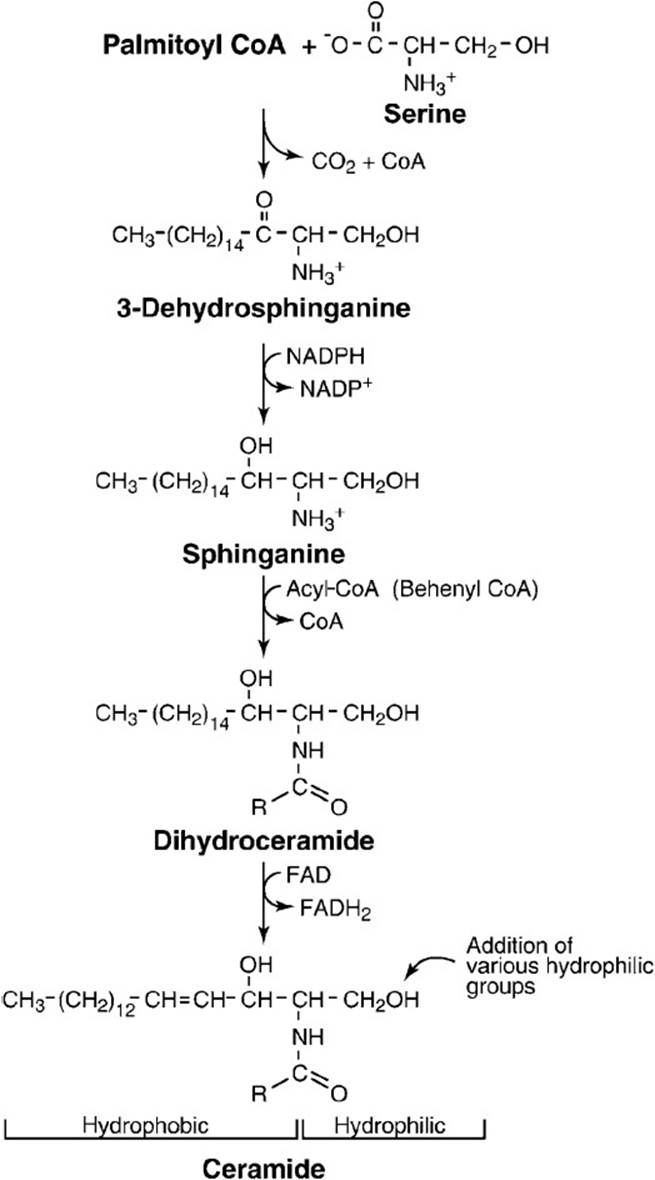
Figure 28-1. Biosynthesis of ceramide.
Ceramide is a building block for all other sphingolipids. Figure 28-2 shows the synthesis of some of these sphingolipids from ceramide. The reaction of phosphatidyl choline and ceramide (via esterification) yields sphingomyelin, a phosphate-containing subclass of sphingolipids found in the nervous tissue of higher animals. The addition of 1 or more sugar residues to ceramide yields the glycosphingolipids. The glycosphingolipid cerebroside contains one residue of glucose or galactose.
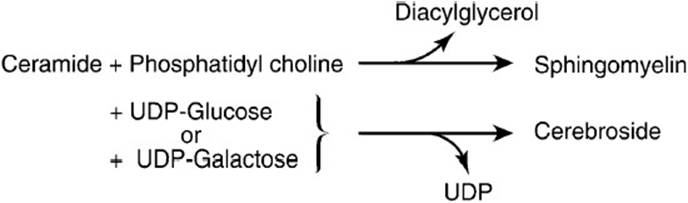
Figure 28-2. The formation of sphingomyelin and cerebrosides from ceramide.
Gangliosides are complex carbohydrate-rich sphingolipids containing 3 or more sugar residues esterified to ceramide with one of the sugars being sialic acid (N-acetylneuraminic acid). Gangliosides are synthesized by the stepwise addition of nucleoside-activated sugar residues to ceramide. Ganglioside nomenclature is a bit unusual. The ganglioside is represented by the letter G followed by a subscript letter and a number. The subscript letter indicates the number of sialic acid residues present in the molecule (M = 1, D = 2, T = 3), and the number is equal to 5 minus the number of neutral sugars. More than 60 gangliosides have been characterized, and the structure of one of these, ganglioside GM1, is schematically represented in Figure 28-3. Glycosphingolipids function in cell–cell recognition and tissue immunity. Gangliosides are found at nerve endings and may be important in nerve impulse transmission.
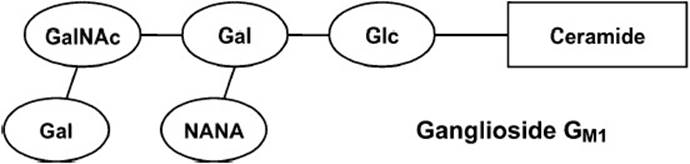
Figure 28-3. The schematic structure of ganglioside GM1. The nomenclature is as follows: G stands for ganglioside; the subscript letter indicates the number of sialic acid residues; the numeral is equal to 5 minus the number of neutral sugars. Gal = galactose, GalNAc = N-acetylgalactosamine, Glc = glucose, NANA = N-acetylneuraminic acid (sialic acid).
Sphingolipids are constantly being turned over in the lysosomes of cells by specific hydrolytic enzymes that remove the sugars in a stepwise fashion. Defects in these enzymes can occur and result in disease states resulting from the accumulation of undegraded sphingolipids. Lysosomal storage diseases are a group of approximately 40 different diseases that occur in approximately 1 in 5000 live births. Many of these diseases are characterized by a deficiency in a specific lysosomal enzyme, but some diseases can be caused by the inability of enzymes to be translocated to the lysosome (I-cell disease), defective transport of small molecules out of the lysosome (cystinosis), or a deficiency in sphingolipid activator proteins, which are small molecular weight proteins that participate in the degradation of sphingolipids. The net result in all of these diseases is a lack of the ability of the cell to breakdown sphingolipids in the lysosome. The accumulation of these macromolecules or partial derivatives of these molecules in the lysosome results in the pathologic conditions associated with the lysosomal storage diseases known as sphingolipidoses.
Proteins destined for the lysosome must contain certain carbohydrate signals. In I-cell disease, the enzyme that catalyzes the addition of a mannose-6-phosphate moiety to the protein is defective. Without the addition of this mannose residue the proteins cannot be transported to the lysosome.
Tay-Sachs disease results from a deficiency in the enzyme hexosaminidase A (β-N-acetylhexosaminidase). The human gene for this enzyme is found at chromosome position 15q23-q24. The deficiency in hexosaminidase A leads to the accumulation of ganglioside GM2 in the nerve cells of the brain. Hexosaminidase A removes a terminal N-acetylgalactosamine residue from ganglioside GM2 to form ganglioside GM3 (Figure 28-4). The inability of patients with Tay-Sachs disease to remove these sugar residues results in the accumulation of gangliosides in the lysosome. This results in swelling of the neurons containing these lipid-filled lysosomes and the disruption of neuronal function.
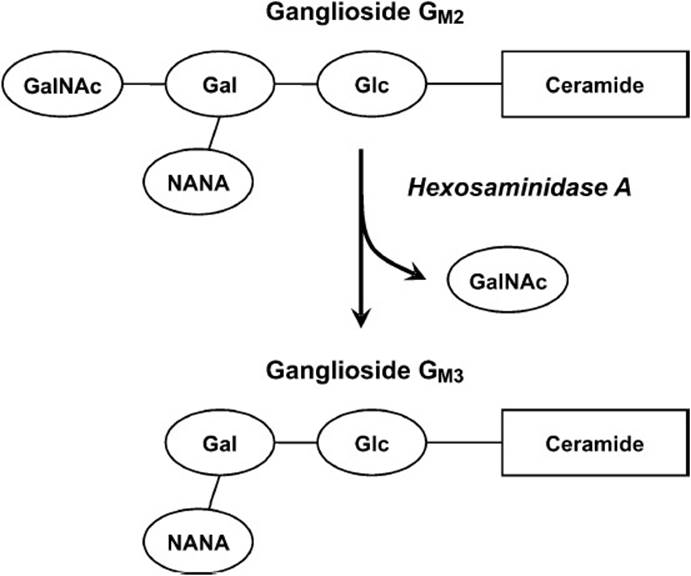
Figure 28-4. The reaction catalyzed by hexosaminidase A, the enzyme deficient in patients with Tay-Sachs disease.
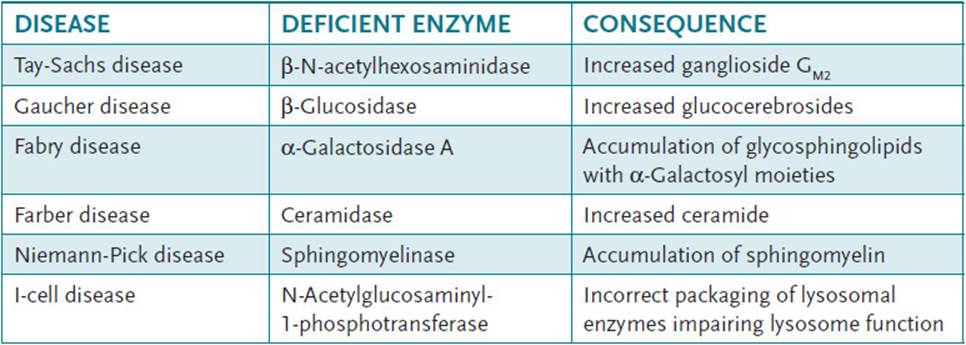
Other lysosomal storage disorders include GM1 gangliosidoses, GM2 gangliosidoses, Gaucher disease, Niemann-Pick disease, Fabry disease, fucosidosis, Schindler disease, metachromatic leukodystrophy, Krabbe disease, multiple sulfatase deficiency, Farber disease, and Wolman disease. Table 28-1 illustrates the enzyme deficiencies found in some of these disorders.
Table 28-1 • LYSOSOMAL STORAGE DISORDERS
COMPREHENSION QUESTIONS
28.1 A couple is seen in your office for genetic counseling regarding Tay-Sachs disease. They are very knowledgeable and request more information about the specific enzyme that is defective in this disease. You explain that Tay-Sachs results from the lack of an enzyme activity necessary for which of the following?
A. Removal of N-acetylgalactosamine from ganglioside GM2
B. Addition of N-acetylgalactosamine to ganglioside GM2
C. Removal of the disaccharide galactose-N-acetylgalactosamine from ganglioside GM2
D. Addition of the disaccharide galactose-N-acetylgalactosamine to ganglioside GM2
E. Removal of a galactose residue from ganglioside GM2
28.2 Tay-Sachs disease involves the metabolism of gangliosides. Gangliosides are composed of a ceramide backbone with at least which one of the following?
A. Phosphorylated sugar residue
B. Glucose residue
C. Galactose residue
D. Sialic acid residue
E. Fructose residue
28.3 The genetic disease which results from a mutation in the gene coding for the enzyme hexosaminidase (β-N-acetylhexosaminidase) is called what?
A. Huntington disease
B. Lesch-Nyhan syndrome
C. Tay-Sachs disease
D. Amyotrophic lateral sclerosis
E. Neurofibromatosis
ANSWERS
28.1 A. Tay-Sachs disease is the result of the lack of the enzyme β-N-acetylhexosaminidase. This enzyme hydrolyzes a terminal N-acetylgalactosamine from the ganglioside GM2. This ganglioside is found in high concentrations in the nervous system and is normally degraded in the lysosome by the sequential removal of terminal sugars. The lack of β-N-acetylhexosaminidase results in the accumulation of the partially degraded ganglioside in the lysosome leading to significant swelling of the lysosome. The abnormally high level of lipid in the lysosome of the neuron affects its function resulting in the disease.
28.2 D. Gangliosides are carbohydrate-rich lipids in which an oligosaccharide chain is attached to ceramide. The oligosaccharide chain must contain at least one acidic sugar such as N-acetylneuraminate or N-glycosylneuraminate. These sugars are commonly referred to as sialic acid residues. Gangliosides are synthesized by the stepwise addition of sugar residues to ceramide.
28.3 C. Tay-Sachs disease is the result of the lack of the enzyme β-N-acetylhexosaminidase. Affected infants show weakness and retarded motor skills before 1 year of age. Other abnormalities follow, and death usually occurs before 3 years of age.
BIOCHEMISTRY PEARLS
![]() Sphingolipids are a class of lipids found in biologic membranes in which the backbone of the lipid molecule is sphingosine, an 18-carbon amino alcohol.
Sphingolipids are a class of lipids found in biologic membranes in which the backbone of the lipid molecule is sphingosine, an 18-carbon amino alcohol.
![]() Sphingolipids are constantly being turned over in the lysosomes of cells by specific hydrolytic enzymes that remove the sugars in a stepwise fashion. Defects in these enzymes can result in accumulation of undegraded sphingolipids.
Sphingolipids are constantly being turned over in the lysosomes of cells by specific hydrolytic enzymes that remove the sugars in a stepwise fashion. Defects in these enzymes can result in accumulation of undegraded sphingolipids.
![]() This disease results from a deficiency in the enzyme hexosaminidase A (β-N-acetylhexosaminidase). The deficiency in hexosaminidase A leads to the accumulation of ganglioside GM2 in the nerve cells of the brain.
This disease results from a deficiency in the enzyme hexosaminidase A (β-N-acetylhexosaminidase). The deficiency in hexosaminidase A leads to the accumulation of ganglioside GM2 in the nerve cells of the brain.
![]() Other lysosomal storage disorders include GM1 gangliosidoses, GM2 gangliosidoses, Gaucher disease, Niemann-Pick disease, Fabry disease, fucosidosis, Schindler disease, metachromatic leukodystrophy, Krabbe disease, multiple sulfatase deficiency, Farber disease, and Wolman disease.
Other lysosomal storage disorders include GM1 gangliosidoses, GM2 gangliosidoses, Gaucher disease, Niemann-Pick disease, Fabry disease, fucosidosis, Schindler disease, metachromatic leukodystrophy, Krabbe disease, multiple sulfatase deficiency, Farber disease, and Wolman disease.
REFERENCES
Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th ed. New York: Freeman; 2002:721-722.
Devlin TM, ed. Textbook of Biochemistry with Clinical Correlations. 7th ed. New York: Wiley-Liss; 2010.