Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 35
A 9-year-old boy is brought to the emergency department (ED) by his parents after 2 days of worsening nausea/vomiting and abdominal pain. The abdominal pain is located in the epigastric region and radiates to his back. He has had several episodes of similar pain in the past but none quite as severe as this one. His parents deny fever/chills and change in bowel habits. In the ED, the patient is afebrile and in moderate distress. Both the liver and spleen appear to be enlarged and he has epigastric tenderness. Several small yellow-white papules were noted on his back and buttocks. Laboratory tests reveal elevated amylase and lipase levels. On further questioning, the father reports having high triglyceride levels and several members of the mother’s family have had early heart disease. Laboratory tests performed after hospitalization revealed elevated triglyceride levels and reduced lipoprotein lipase (LPL) activity.
![]() What is etiology of the boy’s abdominal pain?
What is etiology of the boy’s abdominal pain?
![]() What is the likely underlying biochemical disorder?
What is the likely underlying biochemical disorder?
![]() What is the role of lipoprotein lipase?
What is the role of lipoprotein lipase?
ANSWERS TO CASE 35:
Hypertriglyceridemia (Lipoprotein Lipase Deficiency)
Summary: A 9-year-old boy with acute abdominal pain consistent with pancreatitis, hepatosplenomegaly, eruptive xanthomas, and a family history of hypertriglyceridemia and heart disease.
• Etiology of abdominal pain: Acute pancreatitis
• Underlying biochemical disorder: Disorder of lipoprotein metabolism
• Role of lipoprotein lipase: Hydrolysis of triglycerides from very-low-density lipoprotein (VLDL) and chylomicrons
CLINICAL CORRELATION
LPL is an enzyme found on the capillary endothelial surface of adipose tissue, heart, and skeletal muscle, and it is required, along with apoC-II, for the hydrolysis of triglycerides. ApoC-II, found on the surface of chylomicrons and VLDL, serves as an activator of LPL. A deficiency in LPL results in elevated levels of triglycerides (VLDL and chylomicrons). The cholesterol level may be normal or slightly elevated. LPL deficiency is inherited in an autosomal recessive pattern. Patients with LPL deficiency often present with recurrent episodes of pancreatitis in their childhood and may have other clinical signs of hypertriglyceridemia, including xanthomas, hepatosplenomegaly, and lipemia retinalis. Reduced serum LPL activity, following an injection of intravenous heparin, confirms the diagnosis of either LPL or apoC-II deficiency. The initial therapeutic intervention consists primarily of dietary modification (reduction of fat intake).
APPROACH TO:
Lipid Transport
OBJECTIVES
1. Describe the metabolism and transport of lipoproteins.
2. Understand the rationale for serum blood test results with the different hypertriglyceridemias.
DEFINITIONS
APOLIPOPROTEIN C-II (APOC-II): The apolipoprotein on the surface of chylomicrons and VLDLs that binds to and activates lipoprotein lipase.
APOLIPOPROTEIN E (APOE): The apolipoprotein on the surface of several lipoproteins including chylomicrons, chylomicron remnants, very-low-density lipoprotein (VLDL), VLDL remnants, and intermediate-density lipoprotein (IDL). It mediates the binding of apoE-containing lipoproteins with the LDL receptor and the chylomicron remnant (apoE) receptor.
LIPOPROTEIN LIPASE (LPL): An enzyme bound to the surface of the capillary endothelium by heparan sulfate proteoglycans. LPL catalyzes the hydrolysis of triglyceride in chylomicrons and VLDL to free fatty acids and glycerol.
HEPATIC LIPASE (HL): An enzyme bound to the surface of sinusoidal endothelial cells of liver. It catalyzes the hydrolysis of mono-, di-, and triglycerides as well as the phospholipids phosphatidyl choline and phosphatidyl ethanolamine.
DISCUSSION
Triglycerides (TGs) are safely transported in the bloodstream packaged into lipoproteins called chylomicrons or VLDLs. Chylomicrons are formed in the epithelial cells of the intestine and are responsible for the transport of dietary lipids. VLDL is synthesized in the liver and transport endogenously synthesized lipids from the liver to peripheral tissues. Both of these lipoproteins are composed of a core composed primarily of TGs enveloped by a monolayer composed of phospholipids, free cholesterol, and apolipoproteins.
Dietary triglycerides are hydrolyzed by pancreatic lipase in the lumen of the small intestine. Colipase, a protein secreted along with pancreatic lipase, binds to the TG and the pancreatic lipase and improves the hydrolytic process. TG is broken down to free fatty acids and 2-monoacylglycerols, which form micelles along with bile salts and other lipid soluble compounds such as cholesterol and fat-soluble vitamins. The free fatty acids and monoglycerides are absorbed by the microvilli of the intestinal epithelial cells. In the epithelial cell, the fatty acids and monoglycerides are reformed into triglycerides, which are packaged with phospholipids, cholesterol, and apolipoprotein B-48 into chylomicrons.
The newly synthesized chylomicrons are secreted into the lymph and enter the bloodstream via the thoracic duct. In the bloodstream, the chylomicron particles obtain proteins from high-density lipoproteins (HDLs), including apoC II and apoE, which are important for the function of the chylomicron (Figure 35-1).
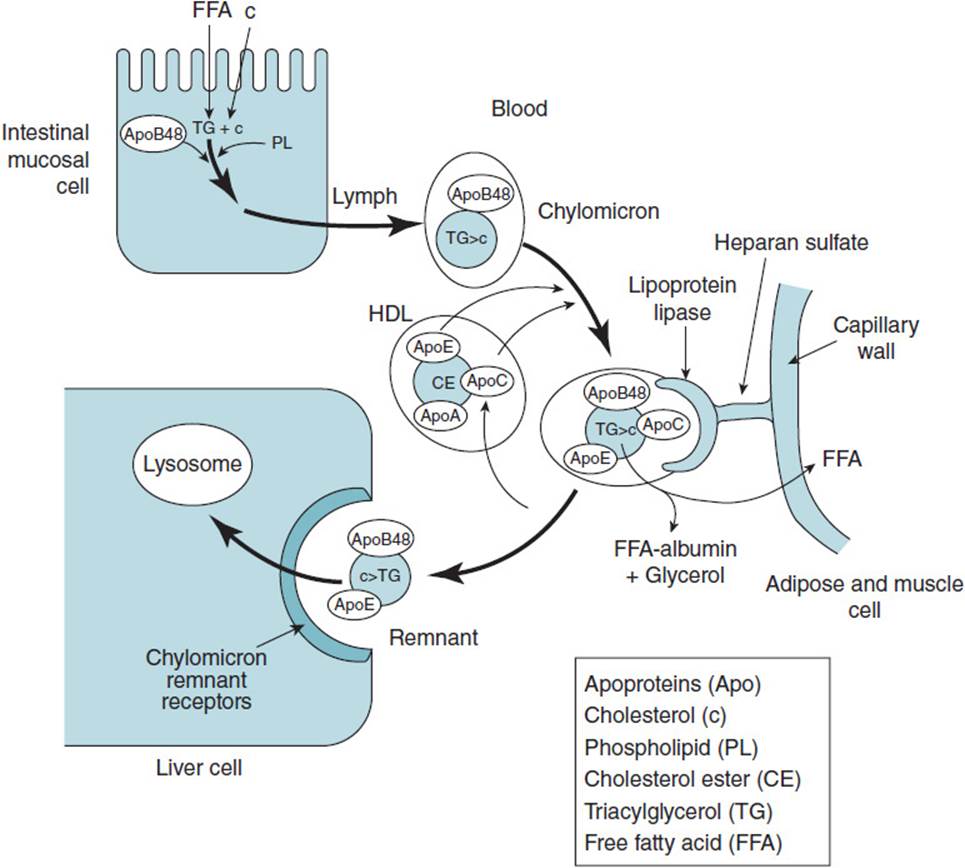
Figure 35-1. Formation and metabolism of chylomicrons. (Abbreviations used are defined in the enclosed box.)
In the capillary beds in adipose tissue, muscle tissue (especially cardiac muscle) and in lactating mammary glands, apoC II binds to and activates LPL, which is bound to the endothelial surface of the capillaries by heparan sulfate. LPL hydrolyzes the TG in the core of the chylomicron to free fatty acids and glycerol. The fatty acids are taken up by the adipose or muscle cells; glycerol is recycled back to the liver. In muscle, the fatty acids are oxidized to produce ATP and in adipose they are reformed into TG for storage. The TG-depleted chylomicron remnant remains in the blood stream until it binds to the chylomicron remnant receptor located on hepatocytes, a process mediated by apoE. The remnants are taken into the hepatocytes by endocytosis and degraded in the lysosome to fatty acids, amino acids, cholesterol, glycerol, and phosphate.
Chylomicrons appear in the blood stream shortly after consumption of a meal containing fat. However, the clearance rate for chylomicrons is fast and blood is usually free of chylomicrons following an overnight fast.
VLDLs are lipoproteins synthesized in the liver from endogenous TG, cholesterol, and phospholipids, along with several apolipoproteins including, apoB-100, apoE, and apoC-II. Following synthesis, the VLDL is secreted into the bloodstream, where it obtains more apoE and apoC II from HDL (Figure 35-2). In a process similar to that of chylomicron degradation, the VLDL apoC-II binds to and activates LPL in the capillary beds of adipose, muscle, and mammary tissue. LPL degrades the VLDL TG core, releasing free fatty acids and glycerol. About one-half of the resulting VLDL remnants bind to receptors in liver cells that recognized apoE and are taken up by endocytosis. The remaining VLDL receptors are further degraded to IDLs, which have their residual TG removed by the action of hepatic lipase to yield low-density lipoproteins (LDLs). HL is synthesized and secreted from liver and is tethered to the external surfaces of liver cells by heparan sulfate. LDL is taken up by receptor-mediated endocytosis upon binding to the LDL receptor in liver and peripheral tissues.
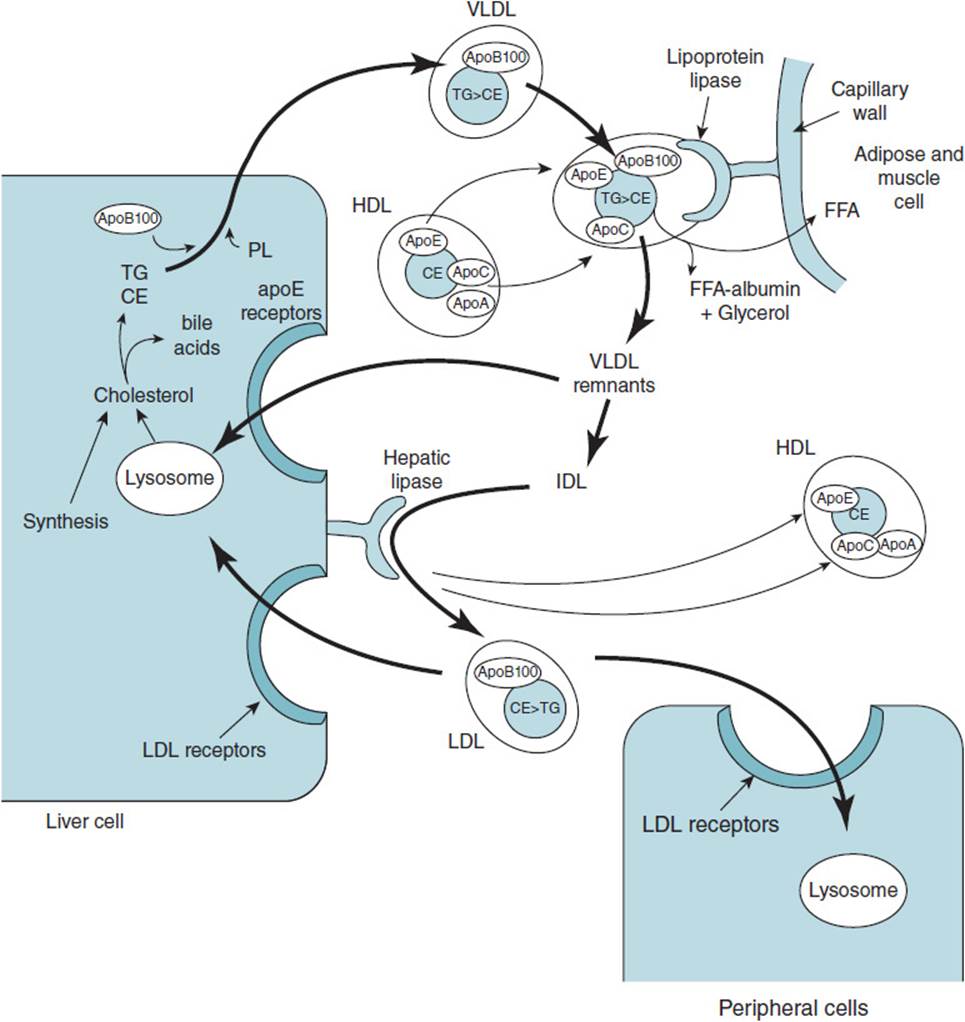
Figure 35-2. Formation of very-low-density lipoprotein and metabolism into low-density lipoprotein. (Abbreviations are as used in Figure 35-1.)
Elevated serum TG levels can occur due to a number of factors. Hypertriglyceridemia can be the result of a genetic disorder in one of the proteins involved in lipoprotein metabolism, or it can arise secondarily to a number of other disorders, including diabetes mellitus, obesity, and alcohol abuse, and as an adverse event of some medications such as β-blockers, oral estrogens, and some diuretics. Genetic deficiencies in LPL, apoC-II, or HL can give rise to elevations in circulating TG, as can cause an overproduction of apoB-100 or increased apoE2 levels. ApoE2 has a decreased affinity for hepatic receptors than apoE3 and chylomicron remnants and VLDL remnants containing apoE2 are cleared more slowly from the circulation.
The most common genetic defect leading to hypertriglyceridemia is a deficiency in LPL, which results in increased levels of both chylomicrons and VLDL. Individuals who are homozygous for the defective gene usually present with symptoms of chylomicronemia (TG levels > 2000 mg/dL, abdominal pain, pancreatitis, xanthomas, lipemia retinalis) in childhood. Clinical diagnosis of LPL deficiency requires measurement of LPL activity in plasma following intravenous injection of heparin, which displaces the LPL from its heparan sulfate tether. However, heparin also releases HL into the plasma and the postheparin plasma must be treated with antibodies specific to HL to remove it. Alternatively, LPL can also be measured in adipose tissue, which has no HL activity. A deficiency in apoC II will also show evidence of decreased postheparin plasma LPL activity. An increase in LPL activity when normal apoC II is added to the assay indicates that a defect in apoC-II is the culprit.
A deficiency in HL can also lead to elevated plasma TG levels, however, these increases in TG are usually found in VLDL remnants, LDL, and HDL, which all become more buoyant in their densities. Definitive diagnosis of HL deficiency is made by demonstrating the absence of HL activity in postheparin plasma.
COMPREHENSION QUESTIONS
35.1 A teenage boy presents with moderate to severe epigastric pain. Physical examination reveals extensive eruptive xanthomas and hepatosplenomegaly. A blood sample reveals milky plasma. Which of the following is the most likely lipoprotein to be elevated in this patient’s plasma?
A. Chylomicrons
B. Chylomicron remnants
C. HDL
D. IDL
E. LDL
35.2 Laboratory results for a patient with uncontrolled type 1 diabetes mellitus reveal hyperglycemia (634 mg/dL) and hypertriglyceridemia (498 mg/dL). The most likely cause of the hypertriglyceridemia in this patient is which of the following?
A. Deficiency in apoprotein C-II
B. Increased hepatic triglyceride synthesis
C. Decreased lipoprotein lipase activity
D. Deficiency in LDL receptors
E. Absence of hormone sensitive lipase
35.3 A 25-year-old woman was referred to a lipid research center for investigation of moderate hypertriglyceridemia because the plasma lipid and lipoprotein profiles showed abnormalities. Both HDL and LDL were more buoyant and showed elevations in TG content with the mass of TG approximately the same as that of cholesterol. A deficiency in which of the following is the most likely cause of this patient’s lipid abnormality?
A. Lecithin-cholesterol acyltransferase
B. Lipoprotein lipase
C. Apoprotein C-II
D. Hepatic lipase
E. Apoprotein B-100
ANSWERS
35.1 A. This patient presents with classical symptoms of hypertriglyceridemia: milky plasma, eruptive xanthomas, enlarged liver and spleen, and symptoms of pancreatitis. The milky plasma is due to abnormally high chylomicron levels, indicating that TG is not being removed from the transporter of dietary TG and it is accumulating in the plasma. The other lipoproteins listed have low levels of TG are much smaller and would not be expected to lead to milky plasma if elevated.
35.2 C. Decreased lipoprotein lipase activity is the result of the failure of the pancreatic β cells to produce and secrete insulin. Insulin stimulates the synthesis of lipoprotein lipase; in the absence of insulin, lipoprotein lipase activity in the capillary beds is low. While a deficiency in apoC-II would also lead to hypertriglyceridemia, this genetic defect is rare. If a patient with type I diabetes mellitus is not given insulin, then the increased glucagon–insulin ratio would stimulate gluconeogenesis and β oxidation in the liver rather than synthesis of triglycerides. Neither a deficiency of LDL receptors nor hormone-sensitive lipase would be expected to increase circulating TG levels.
35.3 D. Hepatic lipase. The abnormal buoyancies of the LDL and HDL fractions due to their increased TG content indicate that IDL is not being processed to LDL and HDL is not being remodeled. Both of these processes are accomplished by hepatic lipase. A deficiency in lecithin-cholesterol acyltransferase would lead to elevated serum cholesterol levels, almost all as free cholesterol. Deficiencies in lipoprotein lipase or apoC-II would lead to hypertriglyceridemia as increased chylomicrons and/or VLDL. An apoB-100 deficiency would lead to increased levels of LDL, but they would have the normal TG/cholesterol ratio.
BIOCHEMISTRY PEARLS
![]() TGs are safely transported in the bloodstream packaged into lipoproteins called chylomicrons or VLDLs.
TGs are safely transported in the bloodstream packaged into lipoproteins called chylomicrons or VLDLs.
![]() The most common genetic defect leading to hypertriglyceridemia is a deficiency in lipoprotein lipase, which results in increased levels of both chylomicrons and VLDL.
The most common genetic defect leading to hypertriglyceridemia is a deficiency in lipoprotein lipase, which results in increased levels of both chylomicrons and VLDL.
![]() Individuals who are homozygous for the defective gene for lipoprotein lipase usually present with symptoms of chylomicronemia (TG levels > 2000 mg/dL, abdominal pain, pancreatitis, xanthomas, lipemia retinalis) in childhood.
Individuals who are homozygous for the defective gene for lipoprotein lipase usually present with symptoms of chylomicronemia (TG levels > 2000 mg/dL, abdominal pain, pancreatitis, xanthomas, lipemia retinalis) in childhood.
REFERENCES
Brunzell JD, Deeb SS. Familial lipoprotein lipase deficiency, apo C-II deficiency, and hepatic lipase deficiency. In: Scriver CR, Beaudet AL, Sly WS, et al, eds. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw Hill; 2001.
Haymore BR, Parks JR, Oliver TG, et al. Hypertriglyceridemia. Hosp Physician. 2005;41(3):17-24.