Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 37
A 45-year-old man with history of hepatitis C infection and now cirrhosis of the liver is brought to the emergency department by family members after he experienced acute changes in his mental status. The family reports that the patient has been very disoriented and confused over the last few days and has been nauseated and vomiting blood. The family first noticed disturbances in his sleep pattern followed by alterations in his personality and mood. On examination, he is disoriented with evidence of icteric sclera. His abdomen is distended with a fluid wave appreciated. He has asterixis and hyperreflexia on neurologic exam. His urine drug screen and ethyl alcohol (EtOH) screen are both negative. A blood ammonia level was noted to be elevated, and all other tests have been normal.
![]() What is the most likely cause of the patient’s symptoms?
What is the most likely cause of the patient’s symptoms?
![]() What is asterixis?
What is asterixis?
![]() What was the likely precipitating factor of the patient’s symptoms?
What was the likely precipitating factor of the patient’s symptoms?
ANSWERS TO CASE 37:
Cirrhosis
Summary: A 45-year-old man presents with cirrhosis, likely secondary to hepatitis C infection, with acute mental status change coinciding with the recent onset of hematemesis. Patient has an elevated serum ammonia level and otherwise negative workup.
• Diagnosis: Hepatic encephalopathy likely secondary to elevated ammonia levels.
• Asterixis: Nonspecific to hepatic encephalopathy. Nonrhythmic asymmetric tremor with loss of voluntary control of extremities while in a sustained position. It is also known as “liver flap.”
• Precipitating factor: Increased nitrogen load from upper gastrointestinal bleed.
CLINICAL CORRELATION
Cirrhosis is a chronic condition of the liver with diffuse parenchymal injury and regeneration leading to distortion of the liver architecture and increased resistance of blood flow through the liver. The patient usually manifests malaise, lethargy, palmar erythema, ascites, jaundice, and hepatic encephalopathy in the late stages. Toxins accumulating in the blood stream affect the patient’s mental status. The most common etiologies of cirrhosis are toxins such as alcohol, viral infections such as hepatitis B or C infection, or metabolic diseases in children (Wilson disease, hemochromatosis, or α1-antitrypsin deficiency). Treatment depends on the exact etiology, although the common therapy includes avoidance of liver toxins, salt restriction, and possibly procedures to reduce the portal pressure.
APPROACH TO:
Amino Acid Metabolism and Ammonia
OBJECTIVES
1. Describe the urea cycle.
2. Describe the strategy in amino acid metabolism.
3. Explain the biochemical means of removing excess ammonia.
DEFINITIONS
GLUTAMATE DEHYDROGENASE: A mitochondrial enzyme present in all tissues that metabolizes amino acids. It catalyzes the oxidative deamination of glutamate to α-ketoglutarate using NAD+ as the electron acceptor to also produce nicotinamide adenine dinucleotide (NADH) and ammonia. The enzyme uses the reducing equivalents of nicotinamide adenine dinucleotide phosphate (NADPH) to perform the reverse reaction.
ORNITHINE: An α-amino acid similar in structure to lysine but having one methylene group less in the side chain. It is carbamoylated to form citrulline to begin the urea cycle and is regenerated in the final step that releases urea.
TRANSAMINASE: An aminotransferase; a pyridoxal phosphate-requiring enzyme that transfers an amino group from an α-amino acid to an α-keto acid.
UREA CYCLE: The series of reactions that occur in the liver to synthesize urea for the excretion of nitrogen. The 2 nitrogen atoms present in urea arise from ammonium ion and the α-amino group of aspartate. The cycle also requires CO2 (HCO-3) and the expenditure of 4 high-energy phosphate bonds and produces fumarate.
DISCUSSION
Amino acids differ from carbohydrates and fats in that they contain nitrogen as part of their molecular structure. For the carbons in amino acids to enter into the energy generating metabolic pathways, the amino groups must first be removed so that they can be detoxified and excreted. The amino acid nitrogen is excreted predominantly as urea, but some is also excreted as free ammonia in order to buffer the urine.
The first step in the catabolism of most amino acids is the transfer of the α-amino group from the amino acid to α-ketoglutarate (α-KG). This process is catalyzed by transaminase (aminotransferase) enzymes that require pyridoxal phosphate as a cofactor. The products of this reaction are glutamate (Glu) and the α-ketoacid analog of the amino acid destined for catabolic breakdown. For example, aspartate is converted to its α-keto analog, oxaloacetate, by the action of aspartate transaminase (AST), which also produces Glu from α-KG. The transamination process is freely reversible, and the direction in which the reaction proceeds is dependent on the concentrations of the reactants and products. These reactions do not effect a net removal of amino nitrogen; the amino group is only transferred from one amino acid to another.
For net removal of amino nitrogen, a second enzymatic reaction must take place that removes the amino group from Glu for disposal. The net removal of the amino nitrogen is accomplished by the mitochondrial enzyme glutamate dehydrogenase (GDH), which catalyzes the oxidative deamination of Glu to α-KG in a reaction that uses NAD+ as the electron acceptor. The enzyme can also catalyze the reverse reaction to produce Glu, but in this case it uses the reducing equivalents from NADPH instead of NADH. The oxidative deamination reaction is allosterically activated by adenosine diphosphate (ADP) and guanosine diphosphate (GDP), whereas the reductive amination is activated by GTP and adenosine triphosphate (ATP). The overall process for net removal of the amino group from α-amino acids is summarized in Figure 37-1.
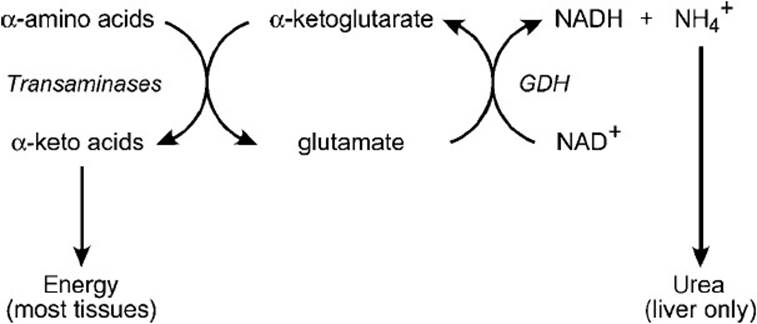
Figure 37-1. Summary of amino acid catabolism.
Ammonia is produced by almost all cells in the body; however, only the liver has the enzymatic machinery to convert it to urea. Therefore, extrahepatic ammonia must be transported to the liver. However, ammonia in the blood is toxic to cells, and therefore the nitrogen from amino acid catabolism is transported in blood either as glutamine or alanine. Glutamine is synthesized from Glu and ammonia in an ATP-requiring reaction catalyzed by glutamine synthetase. Alanine is formed from pyruvate in a transamination reaction catalyzed by alanine transaminase (ALT).
Glutamine and alanine are transported to the liver in the blood where they are taken up by cells in the periportal region. Ammonia is released by the combined action of ALT (in the case of alanine), glutaminase (in the case of glutamine), and GDH. The α-amino group of alanine is transferred to α-KG to form Glu and pyruvate. Glutaminase catalyzes the hydrolysis of the side-chain amide group releasing ammonia and Glu. Ammonia and Glu enter the mitochondria, where Glu is oxidatively deaminated by GDH. The ammonia that is released by glutaminase and GDH then enters the urea cycle (Figure 37-2), which includes enzymes that are located both in the mitochondria and the cytosol.
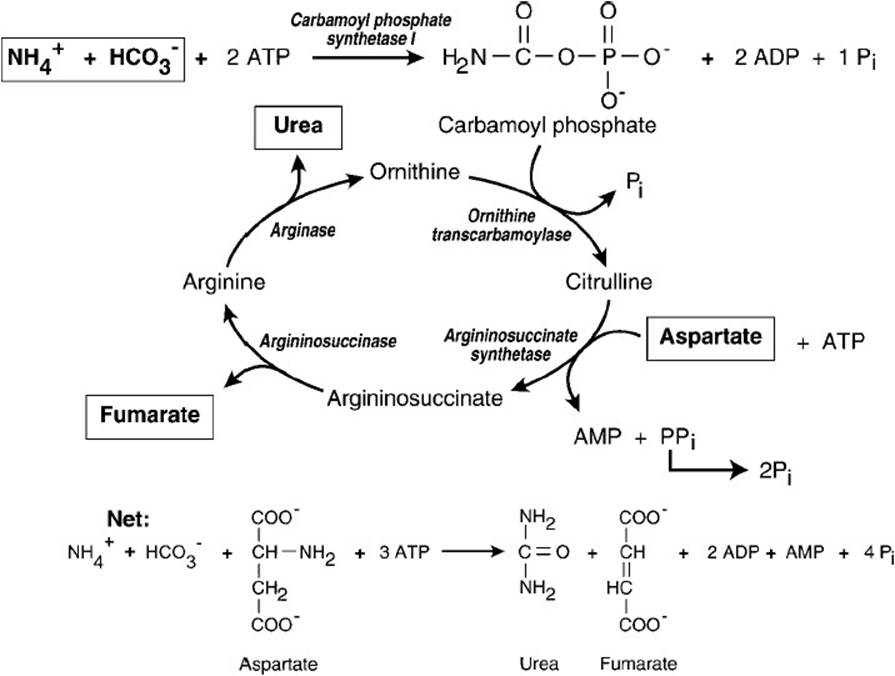
Figure 37-2. Urea cycle.
Ammonia is condensed with bicarbonate and ATP in the mitochondrion to form carbamoyl phosphate in a reaction catalyzed by carbamoyl phosphate synthetase I. Two molecules of ATP are used in this reaction; one provides the phosphate, and the other is hydrolyzed to ADP and inorganic phosphate (Pi) to provide the energy that drives the reaction to products. The activated carbamoyl group is then transferred to the amino acid ornithine by the mitochondrial enzyme ornithine transcarbamoylase to form citrulline. Citrulline then is transported out of the mitochondrion to the cytosol, where the rest of the reactions that are required to synthesize urea take place. The second nitrogen of urea comes directly from the amino acid aspartate. The side chain of citrulline condenses with the α-amino group of aspartate to form argininosuccinate in a reaction that is thermodynamically driven by the conversion of ATP to AMP and inorganic pyrophosphate (PPi). The rapid hydrolysis of PPi by pyrophosphatase releases energy and removes the PPi, thus making the formation of argininosuccinate thermodynamically irreversible. Argininosuccinate is then cleaved to arginine and fumarate by argininosuccinase (argininosuccinate lyase). Arginase then hydrolyzes the guanidino group of arginine, releasing urea and regenerating ornithine, which can then re-enter the mitochondrion and accept another carbamoyl group from carbamoyl phosphate. The urea is transported to the kidney for excretion.
Glutamine is also used by the kidney as a source of ammonia that is used to buffer the urine. Ammonia is released from glutamine by the same enzymes that are active in the liver. The free ammonia accepts a proton to form ammonium ion, thus decreasing the acidity of the urine.
Although most of the ammonia detoxified by the liver arises from the breakdown of amino acids in dietary protein or endogenous protein that is being turned over, ammonia is also produced by bacteria in the gut. This is absorbed into the portal venous blood and directly taken to the liver for conversion into urea.
When liver function is severely impaired or when collateral links between the portal and venous blood vessels arise as occurs in cirrhosis, the ability of the liver to detoxify ammonia to urea is compromised resulting in hyperammonemia. This can be exacerbated by an increase in the ammonia load, such as can occur from gastrointestinal bleeding. When blood ammonia levels rise, ammonia builds up inside the cells and drives the GDH reaction to form Glu, thus depleting α-KG stores and slowing the tricarboxylic acid (TCA) cycle. This is particularly devastating to the brain, which must have an active TCA cycle to produce the energy needed for brain function.
COMPREHENSION QUESTIONS
37.1 An 8.5-month-old infant was admitted to the hospital in a coma and a temperature of 102.9°F (39.4°C). His pulse was elevated, his liver was enlarged, and findings on electroencephalography were grossly abnormal. Because the infant could not retain milk given by gavage feeding, intravenous glucose was administered. He improved rapidly and came out of the coma within 24 hours. Analysis of his urine showed abnormally high amounts of glutamine and uracil, which suggested a high blood ammonium ion concentration. The laboratory confirmed this. Considering the data, which enzyme may be defective in this patient?
A. Arginase
B. Carbamoyl phosphate synthetase I
C. Glutamate dehydrogenase
D. Glutaminase
E. Ornithine transcarbamoylase
37.2 A newborn male infant was diagnosed as having phenylketonuria and is immediately placed on diet low in phenylalanine (Phe). Careful compliance with the diet and frequent monitoring of the patient’s plasma Phe level resulted in the level being maintained at the lower limit of the normal range. The patient appeared to be developing normally until 4 months of age, when he developed truncal hypotonia and spasticity of the limbs. Despite being on a low-phenylalanine diet, at 5 months the patient had several grand mal (epileptic) seizures. After an abnormal Phe-loading test, the patient’s urine was found to have a markedly elevated urinary biopterin concentration. Which of the following enzymes is most likely deficient in this patient?
A. Dihydropteridine reductase
B. GTP cyclohydrolase I
C. Phenylalanine hydroxylase
D. Tryptophan hydroxylase
E. Tyrosine hydroxylase
37.3 Which treatment regimen would be most beneficial to this patient in question 37.2?
A. A low-Phe diet with biopterin supplementation
B. A low-Phe diet with cobalamin (vitamin B12) supplementation
C. A low-Phe diet + L-dopa (3,4-dihydroxyphenylalanine)
D. A low-Phe diet + L-dopa and 5-hydroxytryptophan
E. A diet completely free of Phe
ANSWERS
37.1 E. The patient exhibits signs of a defect in the urea cycle. The presence of elevated uracil in addition to ammonia and glutamine points to an accumulation of carbamoyl phosphate. If ornithine transcarbamoylase is deficient, carbamoyl phosphate will accumulate in the mitochondria and leak into the cytosol, providing the starting compound for the synthesis of uracil.
37.2 A. The patient, despite being put on a low-Phe diet, exhibits neurologic problems resulting from an inability to synthesize catecholamine and indoleamine neurotransmitters. This is caused by a deficiency in dihydropteridine reductase (DHPR). DHPR regenerates tetrahydrobiopterin (BH4), which is oxidized to dihydrobiopterin by phenylalanine hydroxylase, as well as tyrosine hydroxylase and tryptophan hydroxylase (tryptophan 5-monooxygenase). If phenylalanine hydroxylase were deficient, then a diet low in Phe would alleviate the effects. Because the urinary biopterin concentration is elevated, a deficiency in GTP cyclohydrolase I is eliminated because that is an enzyme in the biosynthetic pathway of BH4. Phe hydroxylase, Tyr hydroxylase, and Trp hydroxylase activities are low because of a lack of BH4.
37.3 D. Because of the DHPR deficiency, the activities of Phe hydroxylase and Tyr hydroxylase are low, hence the synthesis of catecholamine neurotransmitters are depressed. The synthesis of the indoleamine neurotransmitter serotonin is also depressed because BH4 is required for the hydroxylation of tryptophan. The best treatment is to decrease the Phe load by a low-Phe diet and provide the precursors for the catecholamine and indoleamine neurotransmitters that occur after the enzymes affected by the deficiency of BH4, which would be L-dopa and 5-hydroxytryptophan.
BIOCHEMISTRY PEARLS
![]() Ammonia is produced by almost all cells in the body; however, only the liver has the enzymatic machinery to convert it to urea.
Ammonia is produced by almost all cells in the body; however, only the liver has the enzymatic machinery to convert it to urea.
![]() Liver disease such as cirrhosis affects the ability of the liver to detoxify ammonia to urea resulting in hyperammonemia.
Liver disease such as cirrhosis affects the ability of the liver to detoxify ammonia to urea resulting in hyperammonemia.
![]() Hyperammonemia can be exacerbated by an increase in the ammonia load, such as what can occur from gastrointestinal bleeding.
Hyperammonemia can be exacerbated by an increase in the ammonia load, such as what can occur from gastrointestinal bleeding.
REFERENCES
Coomes MW. Amino acid metabolism. In: Devlin TM, ed. Textbook of Biochemistry with Clinical Correlations. 7th ed. New York: Wiley-Liss; 2010.
Rodwell VW. Catabolism of proteins & of amino acid nitrogen. In: Murray RK, Bender DA, Botham KM, et al, eds. Harper’s Illustrated Biochemistry. 29th ed. New York: Lange Medical Books/McGraw-Hill; 2012.