Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 47
A 3-year-old girl was brought to the emergency department (ED) after collapsing while she was playing. Her parents report that she had gone out to play in the back yard with her new dog while her mother prepared breakfast. She began to feel ill after chasing the dog and collapsed in the kitchen. Her parents immediately transported her by car to the ED, which was close by. On arrival at the ED, the child was unconscious, apneic, with sinus bradycardia (52 beats/minute). Cardiopulmonary resuscitation was begun, the girl was intubated and an intravenous line was inserted. Parents denied any recent fever, head trauma, or vomiting. The girl was born full-term without complications and reached developmental milestones. The parents report that their first child died due to sudden infant death syndrome. A bedside glucometer test revealed a blood glucose level of less than 20 mg/dL, which was later confirmed by laboratory analysis. In response, the child was infused with 10% glucose in quarter-normal saline. Laboratory analysis also reported minimal ketones in urine and serum and an elevated blood urea nitrogen level. A urinary organic acid analysis revealed elevated levels of hexanoylglycine and suberylglycine. After spending the night in the intensive care unit, the girl was successfully extubated. There were no neurologic sequelae.
![]() What is the most likely cause for the child’s hypoglycemia with low ketone bodies?
What is the most likely cause for the child’s hypoglycemia with low ketone bodies?
![]() What is the most likely molecular mechanism for this disease?
What is the most likely molecular mechanism for this disease?
![]() How is this condition treated and managed to prevent further episodes?
How is this condition treated and managed to prevent further episodes?
ANSWERS TO CASE 47:
Medium Chain Acyl-CoA Dehydrogenase Deficiency
Summary: A 3-year-old girl with bradycardia, apnea, severe hypoglycemia with low ketone production, and laboratory evidence of a defect in fatty acid oxidation.
• Most likely cause for hypoglycemia with low ketone bodies: A defect in fatty acid oxidation
• Most likely molecular mechanism: The presence of hexanoylglycine and suberylglycine (medium chain fatty acyl derivatives) suggest that there is a defect in the medium chain fatty acyl CoA dehydrogenase (MCAD), thus preventing the catabolism of fatty acyl-CoA that have fatty acyl groups of 6 to 10 carbons.
• Treatment and management: Severe episodes of hypoglycemia are resolved by intravenous administration of glucose. Long-term management of MCAD deficiency involves prevention of long-term fasting via dietary management and adequate caloric intake.
CLINICAL CORRELATION
A defect in fatty acid oxidation prevents the formation of ketone bodies, resulting in sequestering of CoA into acyl-CoA intermediates in the mitochondria. These intermediates inhibit gluconeogenesis in times of fasting, resulting in low concentrations of blood glucose. Severe episodes of hypoglycemia are resolved by intravenous administration of glucose while long-term management of MCAD deficiency involves prevention of long-term fasting via dietary management and adequate caloric intake. Infections leading to anorexia need to be aggressively managed to prevent hypoglycemia and buildup of toxic metabolites of fatty acid oxidation.
APPROACH TO:
Medium Chain Acyl-CoA Dehydrogenase Deficiency
OBJECTIVES
1. Describe how fatty acids are catabolized to yield energy for cellular processes.
2. Explain how disruption of fatty acid oxidation can lead to decreased biosynthesis of glucose and hypoglycemia.
3. Identify laboratory analyses that can be used to differentiate between mitochondrial fatty acid oxidation disorders.
4. Formulate a treatment and management plan for patients with MCAD deficiency.
DEFINITIONS
β-OXIDATION: The metabolic process by which activated fatty acids (fatty acyl-CoA derivatives) are oxidized to remove acetyl-CoA leaving a fatty acyl-CoA that is 2 carbons shorter.
ω-OXIDATION: The sequential oxidation of the terminal methyl group of a fatty acid to a carboxyl group. This oxidation occurs in the endoplasmic reticulum and results in dicarboxylic acids.
SHORT-CHAIN FATTY ACIDS: Fatty acids having 4 to 8 carbons.
MEDIUM-CHAIN FATTY ACIDS: Fatty acids having 6 to 12 carbons.
LONG-CHAIN FATTY ACIDS: Fatty acids having 10 to 16 carbons.
VERY-LONG-CHAIN FATTY ACIDS: Fatty acids having 17 to 26 carbons.
FATTY-ACYL-CoA DEHYDROGENASES: A family of mitochondrial enzymes that catalyze the first step in the β-oxidation of fatty acids; the enzymes have specificities that preferentially oxidize fatty acyl-CoA derivatives based on their chain length. Short-chain acyl-CoA dehydrogenase (SCAD) will only oxidize fatty acyl groups of 4 to 6 carbons; MCAD prefers those with 4 to 12 carbons; and long-chain acyl-CoA dehydrogenase (LCAD) prefers those with 12 to 18 carbons. Although very-long-chain fatty acids are preferentially oxidized in peroxisomes, mitochondria have a very-long-chain acyl-CoA dehydrogenase (VLCAD) that will oxidize fatty acyl CoA derivatives that have 14 to 20 carbons.
DISCUSSION
As seen in this case, severe hypoglycemia in pediatric patients can result in cardiopulmonary deficiencies and even cardiac arrest. When the hypoglycemia is accompanied by low blood ketones, it is an indication that there may be a defect in the catabolism of fatty acids that prevents the biosynthesis of glucose (gluconeogenesis) when blood glucose levels decrease.
To synthesize glucose, the gluconeogenic pathway requires a source of carbons, energy in the form of ATP and reducing equivalents in the form of NADH (see cases 21 and 22 for a discussion of gluconeogenesis and accompanying figures). The sources of carbon are lactate produced by glycolysis in muscle and red blood cells, and glycerol is released from the hydrolysis of triglycerides and the carbon skeletons of glucogenic amino acids. The end product of fatty acid oxidation, acetyl-CoA, cannot be used as a carbon source to synthesize glucose in a net fashion, but the β-oxidation of fatty acids provides the reducing equivalents in the form of FADH2 and NADH that will be used to form ATP through oxidative phosphorylation as well as the NADH that is required for the reduction step in gluconeogenesis.
Fatty acid β-oxidation is a mitochondrial process occurring in most tissues except the brain (fatty acids cannot cross the blood-brain barrier) and red blood cells (which do not have mitochondria). After being taken up by the cell, long-chain fatty acids are activated to the fatty acyl-CoA derivatives by acyl-CoA synthetase in a reaction that converts ATP to AMP and inorganic pyrophosphate. The fatty acyl-CoA can be utilized for synthesis of triglycerides, phospholipids, or cholesteryl esters; however, under conditions of low energy and low blood glucose levels, they are directed to the mitochondria for oxidation. Long-chain fatty acyl-CoA cannot cross the mitochondrial matrix, but must first be converted to carnitine derivatives by a process known as the carnitine shuttle (see Figure 36-4).
After the fatty acyl carnitine moieties enter the mitochondria and are converted back to the CoA derivatives, the CoA-activated fatty acids enter a repeating four-step oxidation process that results in the release of acetyl-CoA, which can be further oxidized to CO2 in the tricarboxylic acid (TCA) cycle or be converted to ketone bodies. This repeating 4-step process known as β-oxidation is presented in Figure 47-1.
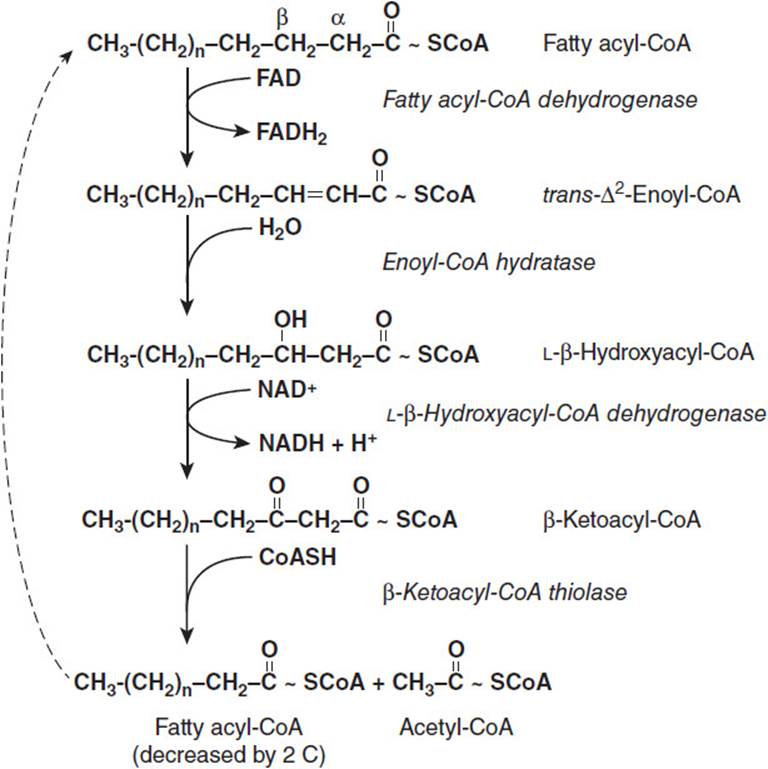
Figure 47-1. β-Oxidation pathway of fatty acyl-CoA.
The first step in the β-oxidation process is the oxidation of the fatty acyl-CoA to introduce a transdouble bond between the α- and β-carbons (carbons 2 and 3) to produce an enoyl-CoA. This reaction is catalyzed by fatty acyl-CoA dehydrogenases, a family of enzymes that have specificities based on the number of carbons in the fatty acyl chain. The electrons released by the double bond insertion are transferred to flavin adenine dinucleotide (FAD) to form FADH2. The reducing equivalents are then transferred to electron transfer flavoprotein (ETF), which feeds them into the electron transfer chain via ETF:ubiquinone oxidoreductase.
Enoyl-CoA hydratase adds a molecule of water across the trans-double bond in the second step of the β-oxidative reaction scheme. This β-hydroxyacyl-CoA product is then further oxidized by the NAD+-requiring β-hydroxyacyl-CoA dehydrogenase to produce β-ketoacyl-CoA and NADH. The final step is the cleavage of acetyl-CoA from the β-ketoacyl-CoA by β-ketothiolase. This enzymatic cleavage requires free CoA (CoASH) and results in a fatty acyl-CoA moiety that is 2 carbon units shorter than the original. This fatty acyl-CoA derivative can be further cleaved by β-oxidation until acetoacetyl-CoA is cleaved to 2 molecules of acetyl-CoA.
The fatty acyl-CoA dehydrogenases (AD) are a family of 4 enzymes that have different specificities based on the number of carbons in the fatty acyl chain. VLCAD is bound to the inner mitochondrial membrane and will oxidize fatty acyl-CoAs with chain lengths of 14-20 carbons, with its highest activity on palmitoyl-CoA (C16). The other AD enzymes are found in the mitochondrial matrix. LCAD prefers fatty acyl-CoAs with 12 to 18 carbons, which overlaps with the specificity of VLCAD. MCAD acts on CoA-activated fatty acids between 4 to 12 carbons, with SCAD only acting on those of 4 to 6 carbons. Thus, a long-chain fatty acyl-CoA such as stearoyl-CoA (C18) could be acted upon by as many as 4 fatty acyl-CoA dehydrogenases as it is degraded to 9 acetyl-CoA.
The most common genetic deficiency in the β-oxidative pathway is MCAD deficiency. MCAD deficiency is an autosomal recessive disease that mainly affects those of northwestern European heritage. More than 80% of patients diagnosed with MCAD deficiency have a missense mutation of a guanine residue for an adenine residue (c.985 A > G) that results in the substitution of a glutamate residue for a lysine in an alpha helical portion of the carboxy-terminal half of the molecule. This leads to impairment of the assembly of the tetrameric structure of the enzyme and instability.
A deficiency in MCAD results in a build-up of medium-chain fatty acyl-CoA in the mitochondria and a depletion of both free CoA and acetyl-CoA. Since CoA is tied up as medium-chain fatty-acyl CoA, this inhibits all metabolic processes that require free CoA. Thus, β-oxidation, ketone body formation, the TCA cycle, oxidative phosphorylation, and gluconeogenesis are all compromised. When blood glucose levels drop, after glycogen levels are depleted the liver is unable to make new glucose, and severe hypoglycemia can result. This explains why the affected individuals usually first present following a period of fasting.
Medium-chain fatty acyl moieties can exit the mitochondria as carnitine derivatives and can be exported to the blood. They also can be converted to dicarboxylic acids by Ω-oxidation by cytochromes P450 in the endoplasmic reticulum. These are usually excreted in the urine after N-acylation to glycine, such as suberylglycine. The presence of acylglycines in urine is suggestive of defects in β-oxidation. Specific diagnosis of MCAD deficiency can be definitively confirmed by determining the profile of medium-chain fatty acylcarnitines present in the blood by tandem mass spectrometry.
MCAD deficiency is manageable by means of careful dietary control. The goal of management is to avoid periods of fasting for 10 to 12 hours, which can lead to hypoglycemia and accumulation of toxic fatty acyl intermediates. A high-fat diet should be avoided, and meals high in carbohydrate should be given to children who are ill. L-Carnitine supplementation has been utilized by some physicians as a means of decreasing toxic medium-chain fatty acyl intermediates that accumulate during fasting or infections.
COMPREHENSION QUESTIONS
47.1 On the third day of life, a female neonate born of nonconsanguineous parents began to vomit and became lethargic and hypotonic. Laboratory analysis indicated that she was hypoglycemic, acidotic (pH 7.3), and hyperammonemic. The patient remained lethargic, hypotonic, and became unresponsive despite administration of intravenous glucose. Further laboratory analyses revealed lactic acidosis, elevated ketones, and increased levels of the organic acids butyrate, adipate, and ethylmalonate in the urine. The neonate most likely is deficient in which of the following?
A. Carnitine palmitoyltransferase I
B. Carnitine acylcarnitine translocase
C. MCAD
D. SCAD
E. Succinate dehydrogenase
47.2 A 5-year-old boy had several unexplained episodes of lethargy and coma associated with fasting, hypoglycemia with low ketones, and dicarboxylic aciduria beginning at about 18 months of age. Hospitalization following a recurrence at 5 years led to the diagnosis of a deficiency of VLCAD. Which of the following would be contraindicated in the management of this patient?
A. Diet high in carbohydrates
B. Carnitine supplementation
C. High fat, ketogenic diet
D. Frequent feeding
E. Medium-chain triglyceride supplementation
47.3 The first step in the β-oxidation of fatty acyl-CoA is catalyzed by a family of enzymes, each of which are specific for different chain length fatty acyl groups. The activity of this family of enzymes would be most negatively affected by a dietary deficiency in which of the following vitamins?
A. Ascorbic acid
B. Biotin
C. Niacin
D. Riboflavin
E. Thiamine
ANSWERS
47.1 D. The presence of the urinary organic acids butyrate, adipate, and ethylmalonate suggest a defect in the catabolism of short-chain fatty acyl groups. Patients deficient in carnitine palmitoyltransferase I usually present with hypoketotic hypoglycemia with elevated levels of plasma carnitine. Blood analysis of patients with a carnitine/acylcarnitine translocase defect would reveal elevated long-chain fatty acyl carnitines. MCAD deficiency would lead to elevations of medium-chain fatty acyl derivatives such as hexanoyl-glycine or suberyl-glycine. Succinate dehydrogenase deficiency would lead to a buildup of succinate and its derivatives. The patient most likely has a defect in SCAD.
47.2 C. With a deficiency in VLCAD, patients should avoid intake of large quantities of long-chain triglycerides, which would preclude a high fat, ketogenic diet. Patients are encouraged to eat frequent small meals high in carbohydrates to avoid hypoglycemia. Some also recommend supplementing the diet with carnitine and medium-chain triglycerides to bypass VLCAD.
47.3 D. The first step in the β-oxidation pathway of degradation of fatty acyl-CoA is catalyzed by acyl-CoA dehydrogenases. Each of these enzymes requires the participation of flavin adenine dinucleotide, which is a derivative of riboflavin. A deficiency in this enzyme would negatively affect the activity of the fatty acyl CoA dehydrogenases.
BIOCHEMISTRY PEARLS
![]() MCAD results in a build-up of medium-chain fatty acyl-CoA in the mitochondria and a depletion of both free CoA and acetyl-CoA.
MCAD results in a build-up of medium-chain fatty acyl-CoA in the mitochondria and a depletion of both free CoA and acetyl-CoA.
![]() Because CoA is tied up as medium-chain fatty-acyl CoA, it inhibits all metabolic processes that require free CoA. Thus, β-oxidation, ketone body formation, the TCA cycle, oxidative phosphorylation, and gluconeogenesis are all compromised.
Because CoA is tied up as medium-chain fatty-acyl CoA, it inhibits all metabolic processes that require free CoA. Thus, β-oxidation, ketone body formation, the TCA cycle, oxidative phosphorylation, and gluconeogenesis are all compromised.
![]() MCAD deficiency is manageable by means of careful dietary control. The goal of management is to avoid periods of fasting for 10 to 12 hours, which can lead to hypoglycemia and accumulation of toxic fatty acyl intermediates. A high-fat diet should be avoided, and meals high in carbohydrates should be given to children who are ill.
MCAD deficiency is manageable by means of careful dietary control. The goal of management is to avoid periods of fasting for 10 to 12 hours, which can lead to hypoglycemia and accumulation of toxic fatty acyl intermediates. A high-fat diet should be avoided, and meals high in carbohydrates should be given to children who are ill.
REFERENCES
Roe CR, Ding J. Mitochondrial fatty acid oxidation disorders. In: Valle D, et al, eds. The Online Metabolic & Molecular Basis of Inherited Disease. New York: McGraw-Hill, 2005. http://dx.doi.org/10.1036/ommbid.129.
Salter N, Quin G, Tracy E. Cardiac arrest in infancy: don’t forget glucose! Emerg Med J. 2010;27(9): 720-721.
Sunehag A, Haymond M, Wolfsdorf J, Hoppin A. Etiology of hypoglycemia in infants and children. Accessed March 14, 2013.