Case Files Biochemistry, 3rd Edition (2015)
SECTION II. Clinical Cases
CASE 2
A 1-month-old baby girl is brought to the emergency department (ED) by her mom with concerns over her baby’s decreased activity level, lack of appetite, and worsening diaper rash that has not resolved with usual antifungal treatment. The baby was born at term and mother denies any pregnancy or delivery complications. The baby was seen by the pediatrician at 2 wks and was doing okay but has had frequent upper respiratory infection symptoms. The mother denies any fever or nausea/vomiting in her newborn. On examination, the baby is afebrile but is noted to be lethargic with no lymphadenopathy. Mild hepatosplenomegaly and hypotonia are noted on physical examination. Chest radiography demonstrates an absent thymic shadow but normal heart silhouette. Laboratory values revealed elevated levels of adenosine and deoxyadenosine in the urine and blood.
![]() What is the likely diagnosis?
What is the likely diagnosis?
![]() What is the biochemical basis for this disorder?
What is the biochemical basis for this disorder?
![]() What are potential treatment options for this baby?
What are potential treatment options for this baby?
ANSWERS TO CASE 2:
Adenosine Deaminase Deficiency
Summary: A 1-month-old baby with a recent history of upper respiratory infection symptoms with an absent thymic shadow on chest radiography and elevated adenosine and deoxyadenosine levels in the urine and blood.
• Likely diagnosis: Adenosine deaminase (ADA) deficiency
• Biochemical basis for this disorder: ADA is an enzyme involved in the breakdown of purines. Specifically, it is responsible for the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine. Individuals with mutations in the ADA gene, typically inherited in an autosomal recessive manner, exhibit decreased ADA enzymatic activity together with elevations in adenosine and deoxyadenosine. Deoxyadenosine is highly cytotoxic, particularly to lymphocytes. Thus, ADA-deficient individuals exhibit a severe combined immunodeficiency and suffer from secondary complications associated with a compromised immune system.
• Potential treatment options: Haploidentical bone marrow transplantation, ADA enzyme replacement therapy, ADA gene therapy.
CLINICAL CORRELATION
ADA deficiency is a rare autosomal recessive genetic disorder that results in severe combined immunodeficiency disease (SCID) characterized by decreased levels and function of T and B lymphocytes as well as decreased levels of Natural Killer (NK) T cells. The most severe form of the disease, typically associated with near complete lack of ADA enzymatic activity, is seen in infants where secondary infections resultant from a compromised immune system appear in the first 6 months of life. However, there are less severe forms of the disease-associated mutations in ADA that still provide low levels of enzyme activity. These partially ADA-deficient individuals present later in childhood. Although the SCID associated with ADA deficiency is clearly the phenotype that leads to the most severe complications in these patients, there are other nonimmune phenotypes found in these patients. Most notable are neurologic problems and hearing loss. Other noted but less common phenotypes include lung disease, hepatocellular damage, kidney disease, and bone disorders. These nonimmune phenotypes are thought to be associated with accumulation of the ADA substrate adenosine, which is a potent cell-signaling molecule. It is clear that both the immune and nonimmune phenotypes in ADA deficiency are resultant from the accumulation of the ADA substrates deoxyadenosine (immunotoxic) and adenosine (abnormal cell signaling). The monogenetic nature of this disorder together with the relatively well-defined biochemical basis of the disorder have led to effective therapies that target the restoration of ADA enzymatic activity and the lowering of ADA substrates.
APPROACH TO:
Adenosine Deaminase Metabolism and Association With Severe Combined Immunodeficiency Disease
OBJECTIVES
1. Describe the metabolic basis of the immunodeficiency associated with ADA deficiency.
2. Explain the treatment options that are based on the ability to reverse the metabolic effects of ADA deficiency.
DEFINITIONS
ADENOSINE DEAMINASE (ADA): An important enzyme involved in the breakdown of purines. It is responsible for the deamination of adenosine and 2′-deoxyadenosine to inosine and 2′-deoxyinosine. This enzyme is found in the cytoplasm of most cells; however, it is expressed at particularly high levels in developing T lymphocytes, which accounts for the sensitivity of this cell population to loss of this enzyme.
ADENOSINE: Adenosine is a purine nucleoside that serves as a potent extracellular signal. It signals in an autocrine and paracrine manner by engaging cell surface adenosine receptors. Adenosine signaling has numerous physiologic and pathologic functions, and abnormal adenosine signaling may account for many of the nonimmune phenotypes seen in ADA deficiency.
DEOXYADENOSINE: 2′-Deoxyadenosine is generated as a breakdown product of DNA catabolism. In its further metabolism, deoxyadenosine is deaminated by ADA. If it is allowed to accumulate, as in the case of ADA deficiency, deoxyadenosine is cytotoxic to cells through a mechanism that involves its phosphorylation to deoxyadenosine triphosphate (dATP), which can promote a cell death process known as apoptosis. This is the mechanism by which lymphocytes are killed in ADA deficiency.
ADA ENZYME REPLACEMENT THERAPY: ADA enzyme replacement therapy is an effective treatment for ADA deficiency. It involves the injection of purified ADA protein that has been modified with polyethylene glycol (PEG-ADA). PEG-ADA injections effectively lower the levels of ADA substrates and alleviate the symptoms of ADA deficiency.
ADA GENE THERAPY: The monogenetic nature of this disorder lends itself to effective gene-replacement strategies. ADA deficiency was the first case of gene therapy in humans. The approach involves removal of the patient’s bone marrow followed by the stable transfer of a “good” ADA gene into the bone marrow cells that are then transferred back into patients, where they expand and populate the immune system. The metabolic disturbances are corrected, as are many of the other phenotypes seen.
DISCUSSION
Nucleotides play an essential role in a number of different metabolic processes, including RNA and DNA synthesis and transmittal of genetic information as energy stores, as structural components of coenzymes, and as signaling molecules. Because of their importance to overall metabolism, nucleotides are continuously turned over. Although the metabolism of the purine nucleotides, adenylate (AMP), 2′deoxyadenylate (dAMP), guanylate (GMP), and 2′-deoxyguanylate (dGMP) can occur throughout the body, the primary site of their degradation is the liver.
The first step in catabolism of the purine mononucleotides is the hydrolysis of the phosphate by a nucleotidase to give the corresponding nucleoside. Guanosine and deoxyguanosine are converted to the purine base, guanine, by removal of the pentose sugar by a phosphorolytic cleavage catalyzed by purine nucleoside phosphorylase. Guanine deaminase hydrolytically removes the amino group to convert guanine to xanthine. Xanthine oxidase converts xanthine to uric acid. The catabolic pathway for adenosine and deoxyadenosine is similar in strategy but has more enzymatic steps (Figure 2-1). Adenosine and deoxyadenosine are first deaminated by adenosine deaminase to yield the nucleosides inosine and deoxyinosine. Purine nucleoside phosphorylase then removes the pentose sugars from the nucleosides, releasing the purine base hypoxanthine. Hypoxanthine is sequentially converted to xanthine and then uric acid by the enzyme xanthine oxidase. Uric acid, the end product of purine catabolism, is not very soluble in blood and is excreted. If uric acid levels in blood are excessive, then sodium urate can crystallize (usually in the joints) to cause gout.
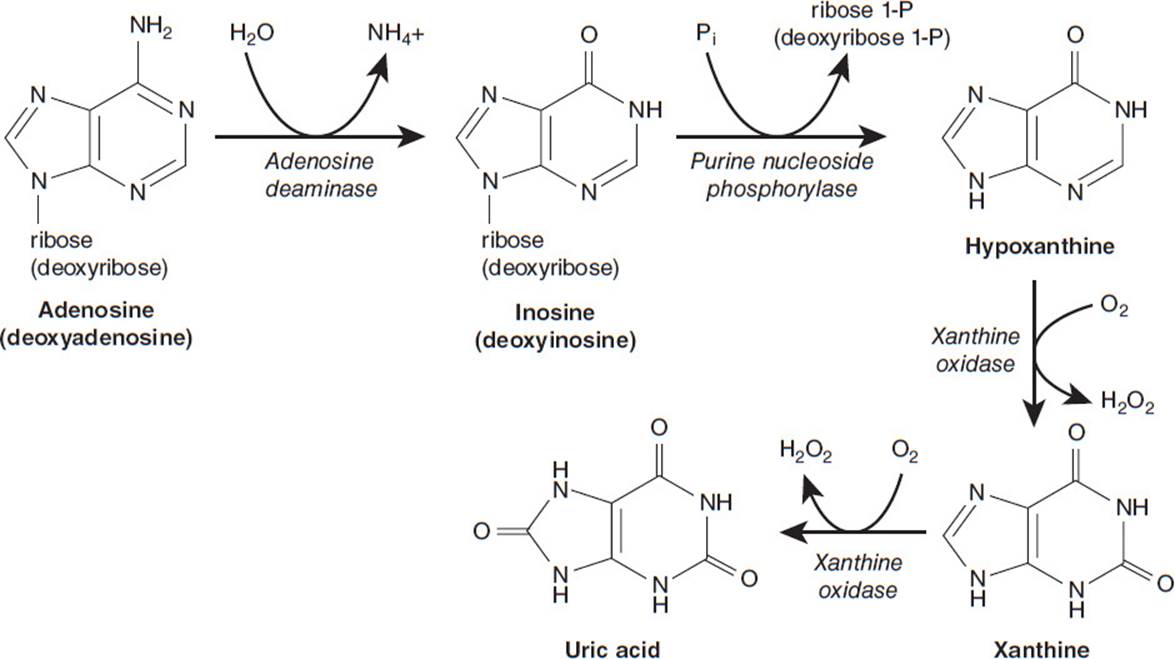
Figure 2-1. Degradation pathway of adenosine and deoxyadenosine.
ADA, the enzyme that catalyzes the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine, respectively, has been shown to be deficient in some patients with heritable immunodeficiency. Autosomal recessive mutations in ADA lead to reduction or complete loss of ADA enzymatic activity in cells (Figure 2-2, A), which in turn leads to elevations in adenosine and deoxyadenosine. Elevations in deoxyadenosine that occur in response to ADA deficiency are cytotoxic to cells by a pathway that involves the phosphorylation of deoxyadenosine to dATP. Elevations in intracellular dATP can activate pathways that lead to apoptosis, a form of cell death. This is the mechanism that accounts for the loss of lymphocytes in ADA deficiency (Figure 2-2B). Accumulation of extracellular adenosine leads to activation of adenosine receptors on numerous cells that can impact the development of non-immune phenotypes associated with ADA deficiency (Figure 2-3C). These phenotypes include neurological problems and hearing loss, as well as liver, kidney, and lung disorders.
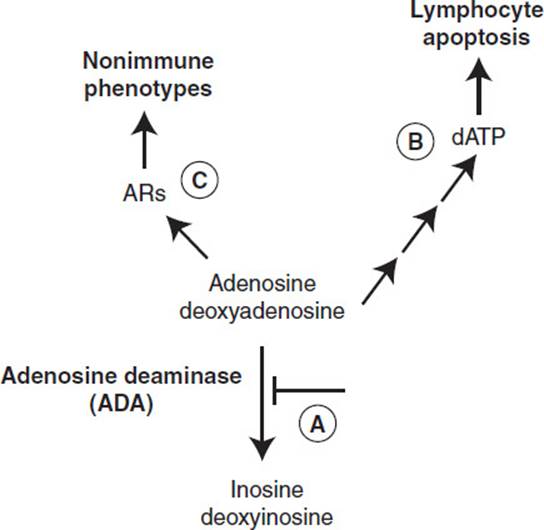
Figure 2-2. Metabolic basis of phenotypes associated with ADA deficiency.
ADA deficiency was the first immunodeficiency disease for which the molecular defect was identified, making possible both prenatal and a postnatal molecular diagnosis. In addition, understanding the metabolic basis of this disease underscored the importance of normal purine metabolism for the development of the immune system. This disorder has also allowed for the development of novel treatment strategies. PEG-ADA became the first PEG-modified protein to be used in a therapeutic setting and opened the door for the development of additional PEG-modified proteins that are in wide clinical use today. In addition, ADA deficiency was the first inherited disease to be treated with gene therapy.
COMPREHENSION QUESTIONS
2.1 A 6-month-old infant presents with an oral yeast infection and the patient history reveals that there have been persistent infections since approximately 1 month of age. Blood work reveals decreased numbers of T and B lymphocytes, and you thus suspect an immunodeficiency. Which of the following laboratory results would specifically implicate ADA deficiency as the molecular basis for the immunodeficiency seen in this patient?
A. Increased adenosine and deoxyadenosine in the blood
B. Elevated inosine and deoxyinosine in the blood
C. Decreased levels of adenosine and deoxyadenosine in the blood
D. Increased glucose levels in the blood
E. Increased creatinine levels in the blood
2.2 With a patient diagnosed with ADA-deficient SCID, which of the following treatment approaches is most likely to be successful?
A. Kidney transplantation
B. Liver transplantation together with immunoglobulin therapy
C. ADA enzyme replacement therapy
D. Immunoglobulin therapy
E. Dietary restriction of adenine
2.3 Which of the following clinical parameters should be followed in an ADA-deficient patient following the use of ADA enzyme replacement therapy to assess the efficacy of the therapy?
A. Blood urea nitrogen
B. Erythrocyte cell counts
C. Ferritin levels
D. Hemoglobin levels
E. Immunoglobulin levels
2.4 A deficiency of ADA can lead to lymphopenia and immune dysfunction. The accumulation of which substance is most likely to result in these lymphotoxic effects?
A. Adenosine
B. Deoxyadenosine
C. Hypoxanthine
D. Inosine
E. Uric acid
ANSWERS
2.1 A. ADA is responsible for deaminating adenosine and deoxyadenosine, hence, elevations in adenosine and deoxyadenosine are specifically diagnostic for ADA deficiency.
2.2 C. ADA enzyme replacement therapy is an approach that can increase ADA enzymatic activity in patients that will lead to a lowering of ADA substrates and an attenuation of the associated phenotypes.
2.3 E. Diminished immunoglobulin levels are indicative of decreased immune function and are a prominent feature of ADA deficiency. Thus, monitoring immunoglobulin levels is a way of assessing the efficacy of ADA enzyme replacement therapy. An increase in immunoglobulin levels is a positive result.
2.4 B. Deoxyadenosine is the primary lymphotoxic substance that accumulates as a result of adenosine deaminase deficiency. It can be phosphorylated to dATP, an accumulation of which leads to programmed cell death (apoptosis) of lymphoid cells. An accumulation of adenosine also occurs due to ADA deficiency, but it is not lymphotoxic. A buildup of adenosine can lead to neurologic as well as liver, kidney, and lung disorders. Inosine, hypoxanthine, and uric acid are downstream metabolites of ADA and would not be expected to accumulate with a deficiency in the enzyme.
BIOCHEMISTRY PEARLS
![]() ADA deficiency was the first immunodeficiency disease for which the molecular defect was identified.
ADA deficiency was the first immunodeficiency disease for which the molecular defect was identified.
![]() ADA catalyzes the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine, respectively.
ADA catalyzes the deamination of adenosine and deoxyadenosine to inosine and deoxyinosine, respectively.
![]() Elevations in deoxyadenosine that occur in response to ADA deficiency are cytotoxic to cells and lead to apoptosis, a form of cell death (in this case loss of lymphocytes).
Elevations in deoxyadenosine that occur in response to ADA deficiency are cytotoxic to cells and lead to apoptosis, a form of cell death (in this case loss of lymphocytes).
REFERENCE
Hershfield MS, Mitchell BS. Immunodeficiency diseases caused by adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency. In: Valle D, Vogelstein B, Kinzler KW, et al, eds. The Online Metabolic & Molecular Basis of Inherited Disease. New York: McGraw-Hill, pp 2586-2588; http://dx.doi.org/10.1036/ommbid.137.