Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION II. Bioenergetics & the Metabolism of Carbohydrates & Lipids
Chapter 19. Metabolism of Glycogen
David A. Bender, PhD & Peter A. Mayes, PhD, DSc
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe the structure of glycogen and its importance as a carbohydrate reserve.
Describe the structure of glycogen and its importance as a carbohydrate reserve.
![]() Describe the synthesis and breakdown of glycogen and how the processes are regulated in response to hormone action.
Describe the synthesis and breakdown of glycogen and how the processes are regulated in response to hormone action.
![]() Describe the various types of glycogen storage diseases.
Describe the various types of glycogen storage diseases.
BIOMEDICAL IMPORTANCE
Glycogen is the major storage carbohydrate in animals, corresponding to starch in plants; it is a branched polymer of α-D-glucose (Figure 14–13). It occurs mainly in liver and muscle, with modest amounts in the brain. Although the liver content of glycogen is greater than that of muscle, because the muscle mass of the body is considerably greater than that of the liver, about three-quarters of total body glycogen is in muscle (Table 19-1).
TABLE 19–1 Storage of Carbohydrate in a 70 kg Human Being
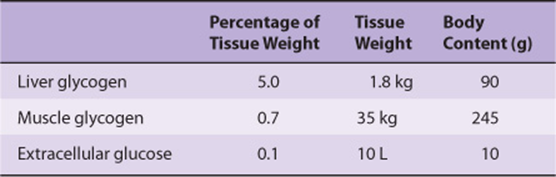
Muscle glycogen provides a readily available source of glucose 1-phosphate for glycolysis within the muscle itself. Liver glycogen functions to store and export glucose to maintain the blood glucose concentration in the fasting state. The liver concentration of glycogen is about 450 mM after a meal, falling to about 200 mM after an overnight fast; after 12-18 h of fasting, liver glycogen is almost totally depleted. Although muscle glycogen does not directly yield free glucose (because muscle lacks glucose 6-phosphatase), pyruvate formed by glycolysis in muscle can undergo transamination to alanine, which is exported from muscle and used for gluconeogenesis in the liver (Figure 20–4). Glycogen storage diseases are a group of inherited disorders characterized by deficient mobilization of glycogen or deposition of abnormal forms of glycogen, leading to liver damage and muscle weakness; some glycogen storage diseases result in early death.
The highly branched structure of glycogen (Figure 14–13) provides a large number of sites for glycogenolysis, permitting rapid release of glucose 1-phosphate for muscle activity. Endurance athletes require a slower, more sustained release of glucose 1-phosphate. The formation of branch points in glycogen is slower than the addition of glucose units to a linear chain, and some endurance athletes practice carbohydrate loading—exercise to exhaustion (when muscle glycogen in largely depleted) followed by a high-carbohydrate meal, which results in rapid glycogen synthesis, with fewer branch points than normal.
GLYCOGENESIS OCCURS MAINLY IN MUSCLE & LIVER
The Pathway of Glycogen Biosynthesis Involves a Special Nucleotide of Glucose
As in glycolysis, glucose is phosphorylated to glucose 6-phosphate, catalyzed by hexokinase in muscle and glucokinase in liver (Figure 19–1). Glucose 6-phosphate is isomerized to glucose 1-phosphate by phosphoglucomutase.The enzyme itself is phosphorylated, and the phosphate group takes part in a reversible reaction in which glucose 1,6-bisphosphate is an intermediate. Next, glucose 1-phosphate reacts with uridine triphosphate (UTP) to form the active nucleotide uridine diphosphate glucose (UDPGlc) and pyrophosphate (Figure 19–2), catalyzed by UDPGlc pyrophosphorylase. The reaction proceeds in the direction of UDPGlc formation because pyrophosphatasecatalyzes hydrolysis of pyrophosphate to 2 × phosphate, so removing one of the reaction products. UDPGlc pyrophosphorylase has a low Km for glucose 1-phosphate and is present in relatively large amounts, so that it is not a regulatory step in glycogen synthesis.
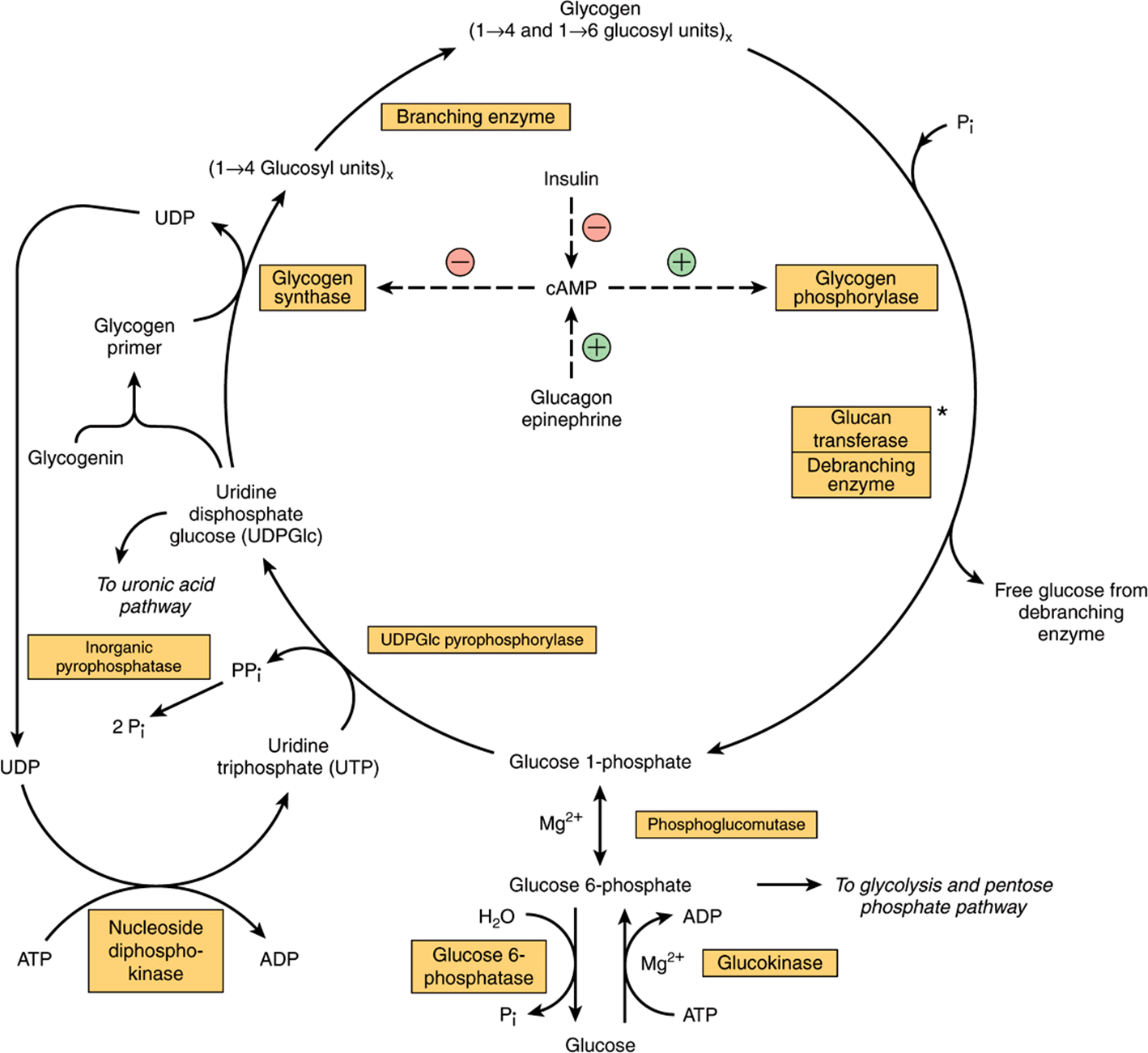
FIGURE 19–1 Pathways of glycogenesis and of glycogenolysis in the liver. ![]() , Stimulation;
, Stimulation; ![]() , inhibition.) Insulin decreases the level of cAMP only after it has been raised by glucagon or epinephrine; that is, it antagonizes their action. Glucagon is active in heart muscle but not in skeletal muscle. *Glucan transferase and debranching enzyme appear to be two separate activities of the same enzyme.
, inhibition.) Insulin decreases the level of cAMP only after it has been raised by glucagon or epinephrine; that is, it antagonizes their action. Glucagon is active in heart muscle but not in skeletal muscle. *Glucan transferase and debranching enzyme appear to be two separate activities of the same enzyme.
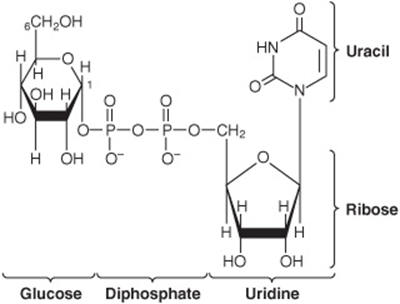
FIGURE 19–2 Uridine diphosphate glucose (UDPGlc).
The initial steps in glycogen synthesis involve the protein glycogenin, a 37 kDa protein that is glucosylated on a specific tyrosine residue by UDPGlc. Glycogenin catalyzes the transfer of a further seven glucose residues from UDPGlc, in 1 → 4 linkage, to form a glycogen primer that is the substrate for glycogen synthase. In muscle the glycogenin remains at the core of the glycogen granule (Figure 14–13), but in liver many glycogen granules do not have a central glycogenin molecule. Glycogen synthase catalyzes the formation of a glycoside bond between C-1 of the glucose of UDPGlc and C-4 of a terminal glucose residue of glycogen, liberating uridine diphosphate (UDP). The addition of a glucose residue to a preexisting glycogen chain, or “primer,” occurs at the nonreducing, outer end of the molecule, so that the branches of the glycogen molecule become elongated as successive 1 → 4 linkages are formed (Figure 19–3).
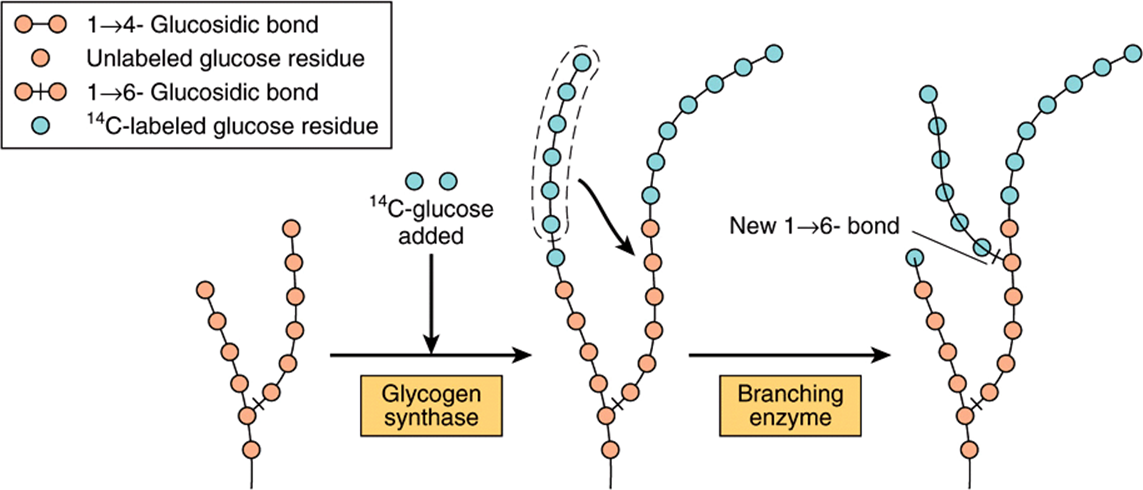
FIGURE 19–3 The biosynthesis of glycogen. The mechanism of branching as revealed by feeding 14C-labeled glucose and examining liver glycogen at intervals.
Branching Involves Detachment of Existing Glycogen Chains
When a growing chain is at least 11 glucose residues long, branching enzyme transfers a part of the 1 → 4-chain (at least six glucose residues) to a neighboring chain to form a 1 → 6 linkage, establishing a branch point. The branches grow by further additions of 1 → 4-glucosyl units and further branching.
GLYCOGENOLYSIS IS NOT THE REVERSE OF GLYCOGENESIS, BUT IS A SEPARATE PATHWAY
Glycogen phosphorylase catalyzes the rate-limiting step in glycogenolysis—the phosphorolytic cleavage (phosphorolysis; cf hydrolysis) of the 1 → 4 linkages of glycogen to yield glucose 1-phosphate (Figure 19–4). There are distinct isoenzymes of glycogen phosphorylase in liver, muscle and brain, encoded by distinct genes. Glycogen phosphorylase requires pyridoxal phosphate (see Chapter 44) as its coenzyme. Unlike the reactions of amino acid metabolism (Chapter 29), in which the aldehyde is the reactive group, in phosphorylase it is the phosphate group that it catalytically active.
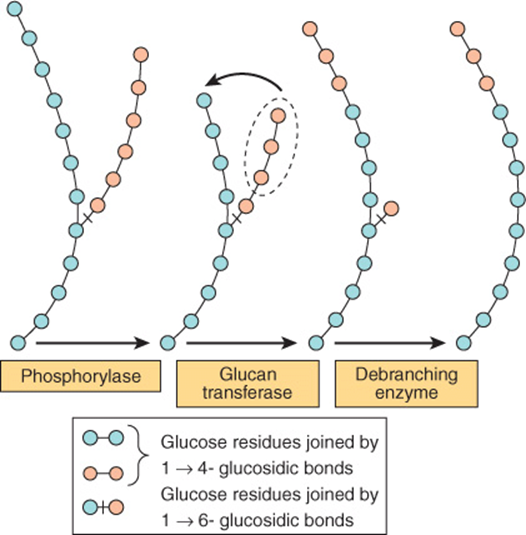
FIGURE 19–4 Steps in glycogenolysis.
The terminal glucosyl residues from the outermost chains of the glycogen molecule are removed sequentially until approximately four glucose residues remain on either side of a 1 → 6 branch (Figure 19–4). The debranching enzyme has two distinct catalytic sites in a single polypeptide chain. One is a glucan transferase that transfers a trisaccharide unit from one branch to the other, exposing the 1 → 6 branch point. The other is a 1,6-glycosidase that catalyzes hydrolysis of the 1 → 6 glycoside bond to liberate free glucose. Further phosphorylase action can then proceed. The combined action of phosphorylase and these other enzymes leads to the complete breakdown of glycogen.
The reaction catalyzed by phosphoglucomutase is reversible, so that glucose 6-phosphate can be formed from glucose 1-phosphate. In liver, but not muscle, glucose 6-phosphatase catalyzes hydrolysis of glucose 6-phosphate, yielding glucose that is exported, leading to an increase in the blood glucose concentration. Glucose 6-phosphatase is in the lumen of the smooth endoplasmic reticulum, and genetic defects of the glucose 6-phosphate transporter can cause a variant of type I glycogen storage disease (see Table 19-2).
TABLE 19–2 Glycogen Storage Diseases
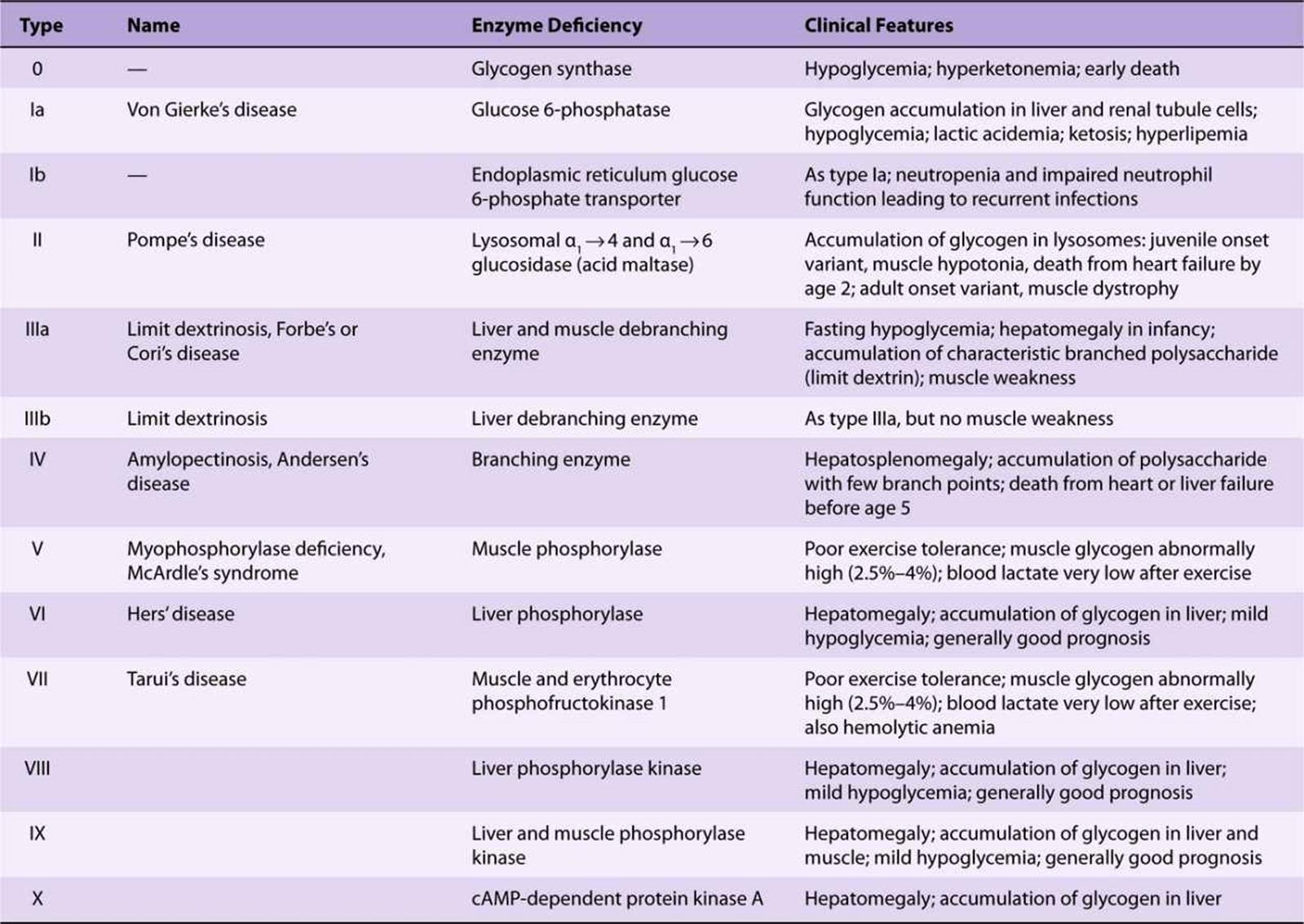
Glycogen granules can also be engulphed by lysosomes, where acid maltase catalyzes the hydrolysis of glycogen to glucose. This may be especially important in glucose homeostasis in neonates, but genetic lack of lysosomal acid maltase leads to type II glycogen storage disease (Pompe’s disease, Table 19-2). The lysosomal catabolism of glycogen is under hormonal control.
CYCLIC AMP INTEGRATES THE REGULATION OF GLYCOGENOLYSIS & GLYCOGENESIS
The principal enzymes controlling glycogen metabolism—glycogen phosphorylase and glycogen synthase—are regulated in opposite directions by allosteric mechanisms and covalent modification by reversible phosphorylation and dephosphorylation of enzyme protein in response to hormone action (Chapter 9). Phosphorylation of glycogen phosphorylase increases its activity; phosphorylation of glycogen synthase reduces its activity.
Phosphorylation is increased in response to cyclic AMP (cAMP) (Figure 19–5) formed from ATP by adenylyl cyclase at the inner surface of cell membranes in response to hormones such as epinephrine, norepinephrine, and glucagon. cAMP is hydrolyzed by phosphodiesterase, so terminating hormone action; in liver insulin increases the activity of phosphodiesterase.
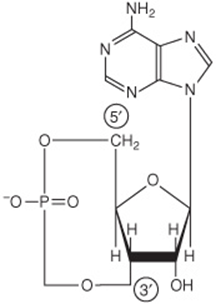
FIGURE 19–5 3′,5′-Adenylic acid (cyclic AMP; cAMP).
The Control of Glycogen Phosphorylase Differs Between Liver & Muscle
In the liver, the role of glycogen is to provide free glucose for export to maintain the blood concentration of glucose; in muscle the role of glycogen is to provide a source of glucose 6-phosphate for glycolysis in response to the need for ATP for muscle contraction. In both tissues, the enzyme is activated by phosphorylation catalyzed by phosphorylase kinase (to yield phosphorylase a) and inactivated by dephosphorylation catalyzed by phosphoprotein phosphatase (to yield phosphorylase b), in response to hormonal and other signals.
There is instantaneous overriding of this hormonal control. Active phosphorylase a in both tissues is allosterically inhibited by ATP and glucose 6-phosphate; in liver, but not muscle, free glucose is also an inhibitor. Muscle phosphorylase differs from the liver isoenzyme in having a binding site for 5′ AMP, which acts as an allosteric activator of the (inactive) dephosphorylated b-form of the enzyme. 5′ AMP acts as a potent signal of the energy state of the muscle cell; it is formed as the concentration of ADP begins to increase (indicating the need for increased substrate metabolism to permit ATP formation), as a result of the reaction of adenylate kinase: 2 × ADP ↔ ATP + 5′ AMP.
cAMP Activates Glycogen Phosphorylase
Phosphorylase kinase is activated in response to cAMP (Figure 19–6). Increasing the concentration of cAMP activates cAMP-dependent protein kinase, which catalyzes the phosphorylation by ATP of inactive phosphorylase kinase b to active phosphorylase kinase a, which in turn, phosphorylates phosphorylase b to phosphorylase a. In the liver, cAMP is formed in response to glucagon, which is secreted in response to falling blood glucose. Muscle is insensitive to glucagon; in muscle, the signal for increased cAMP formation is the action of norepinephrine, which is secreted in response to fear or fright, when there is a need for increased glycogenolysis to permit rapid muscle activity.
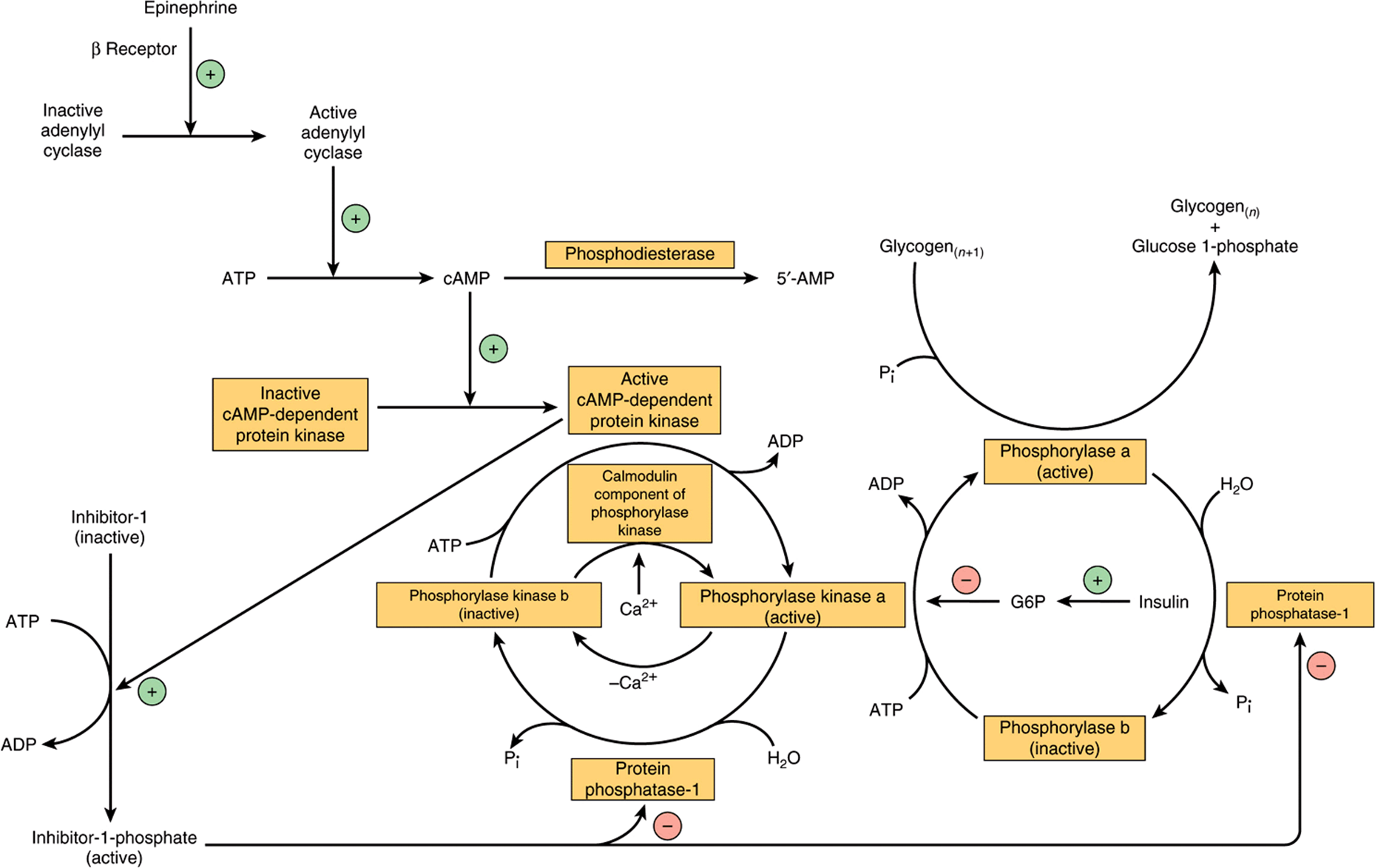
FIGURE 19–6 Control of phosphorylase in muscle. The sequence of reactions arranged as a cascade allows amplification of the hormonal signal at each step. (G6P, glucose 6-phosphate; n, number of glucose residues.)
Ca2+ Synchronizes the Activation of Glycogen Phosphorylase With Muscle Contraction
Glycogenolysis in muscle increases several 100-fold at the onset of contraction; the same signal (increased cytosolic Ca2+ ion concentration) is responsible for initiation of both contraction and glycogenolysis. Muscle phosphorylase kinase, which activates glycogen phosphorylase, is a tetramer of four different subunits, α, β, γ, and δ. The α and β subunits contain serine residues that are phosphorylated by cAMP-dependent protein kinase. The δ subunit is identical to the Ca2+-binding protein calmodulin (Chapter 42), and binds four Ca2+. The binding of Ca2+ activates the catalytic site of the γ subunit even while the enzyme is in the dephosphorylated b state; the phosphorylated a form is only fully activated in the presence of high concentrations of Ca2+.
Glycogenolysis in Liver Can Be cAMP-Independent
In the liver, there is cAMP-independent activation of glycogenolysis in response to stimulation of α1 adrenergic receptors by epinephrine and norepinephrine. This involves mobilization of Ca2+ into the cytosol, followed by the stimulation of a Ca2+/calmodulin-sensitive phosphorylase kinase. cAMP-independent glycogenolysis is also activated by vasopressin, oxytocin, and angiotensin II acting either through calcium or the phosphatidylinositol bisphosphate pathway (Figure 42–10).
Protein Phosphatase-1 Inactivates Glycogen Phosphorylase
Both phosphorylase a and phosphorylase kinase a are dephosphorylated and inactivated by protein phosphatase-1. Protein phosphatase-1 is inhibited by a protein, inhibitor-1, which is active only after it has been phosphorylated by cAMP-dependent protein kinase. Thus, cAMP controls both the activation and inactivation of phosphorylase (Figure 19–6). Insulin reinforces this effect by inhibiting the activation of phosphorylase b. It does this indirectly by increasing uptake of glucose, leading to increased formation of glucose 6-phosphate, which is an inhibitor of phosphorylase kinase.
Glycogen Synthase & Phosphorylase Activities Are Reciprocally Regulated
There are different isoenzymes of glycogen synthase in liver, muscle, and brain. Like phosphorylase, glycogen synthase exists in both phosphorylated and nonphosphorylated states, and the effect of phosphorylation is the reverse of that seen in phosphorylase (Figure 19–7). Active glycogen synthase a is dephosphorylated and inactive glycogen synthase b is phosphorylated.
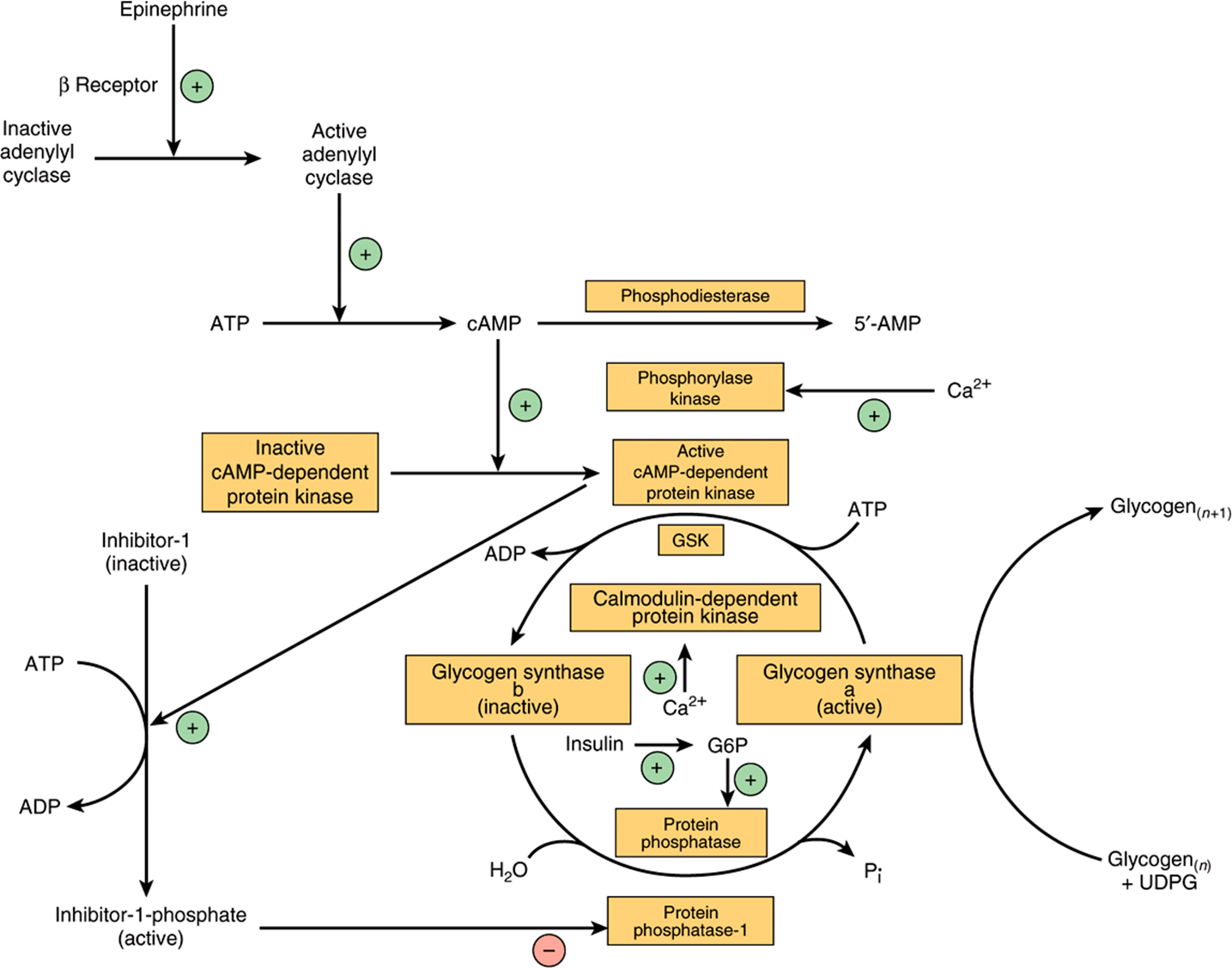
FIGURE 19–7 Control of glycogen synthase in muscle. (GSK, glycogen synthase kinase; G6P, glucose 6-phosphate; n, number of glucose residues.)
Six different protein kinases act on glycogen synthase, and there are at least 9 different serine residues in the enzyme that can be phosphorylated. Two of the protein kinases are Ca2+/calmodulin dependent (one of these is phosphory-lase kinase). Another kinase is cAMP-dependent protein kinase, which allows cAMP-mediated hormonal action to inhibit glycogen synthesis synchronously with the activation of glycogenolysis. Insulin also promotes glycogenesis in muscle at the same time as inhibiting glycogenolysis by raising glucose 6-phosphate concentrations, which stimulates the dephosphorylation and activation of glycogen synthase. Dephosphorylation of glycogen synthase b is carried out by protein phosphatase-1, which is under the control of cAMP-dependent protein kinase.
REGULATION OF GLYCOGEN METABOLISM IS EFFECTED BY A BALANCE IN ACTIVITIES BETWEEN GLYCOGEN SYNTHASE & PHOSPHORYLASE
At the same time as phosphorylase is activated by a rise in concentration of cAMP (via phosphorylase kinase), glycogen synthase is converted to the inactive form; both effects are mediated via cAMP-dependent protein kinase (Figure 19–8). Thus, inhibition of glycogenolysis enhances net glycogenesis, and inhibition of glycogenesis enhances net glycogenolysis. Also, the dephosphorylation of phosphorylase a, phosphorylase kinase, and glycogen synthase b is catalyzed by a single enzyme with broad specificity —protein phosphatase-1. In turn, protein phosphatase-1 is inhibited by cAMP-dependent protein kinase via inhibitor-1. Thus, glycogenolysis can be terminated and glycogenesis can be stimulated, or vice versa, synchronously, because both processes are dependent on the activity of cAMP-dependent protein kinase. Both phosphorylase kinase and glycogen synthase may be reversibly phosphorylated at more than one site by separate kinases and phosphatases. These secondary phosphorylations modify the sensitivity of the primary sites to phosphorylation and dephosphorylation (multisite phosphorylation).Also, they allow insulin, by way of increased glucose 6-phosphate, to have effects that act reciprocally to those of cAMP (see Figures 19-6 & 19-7).
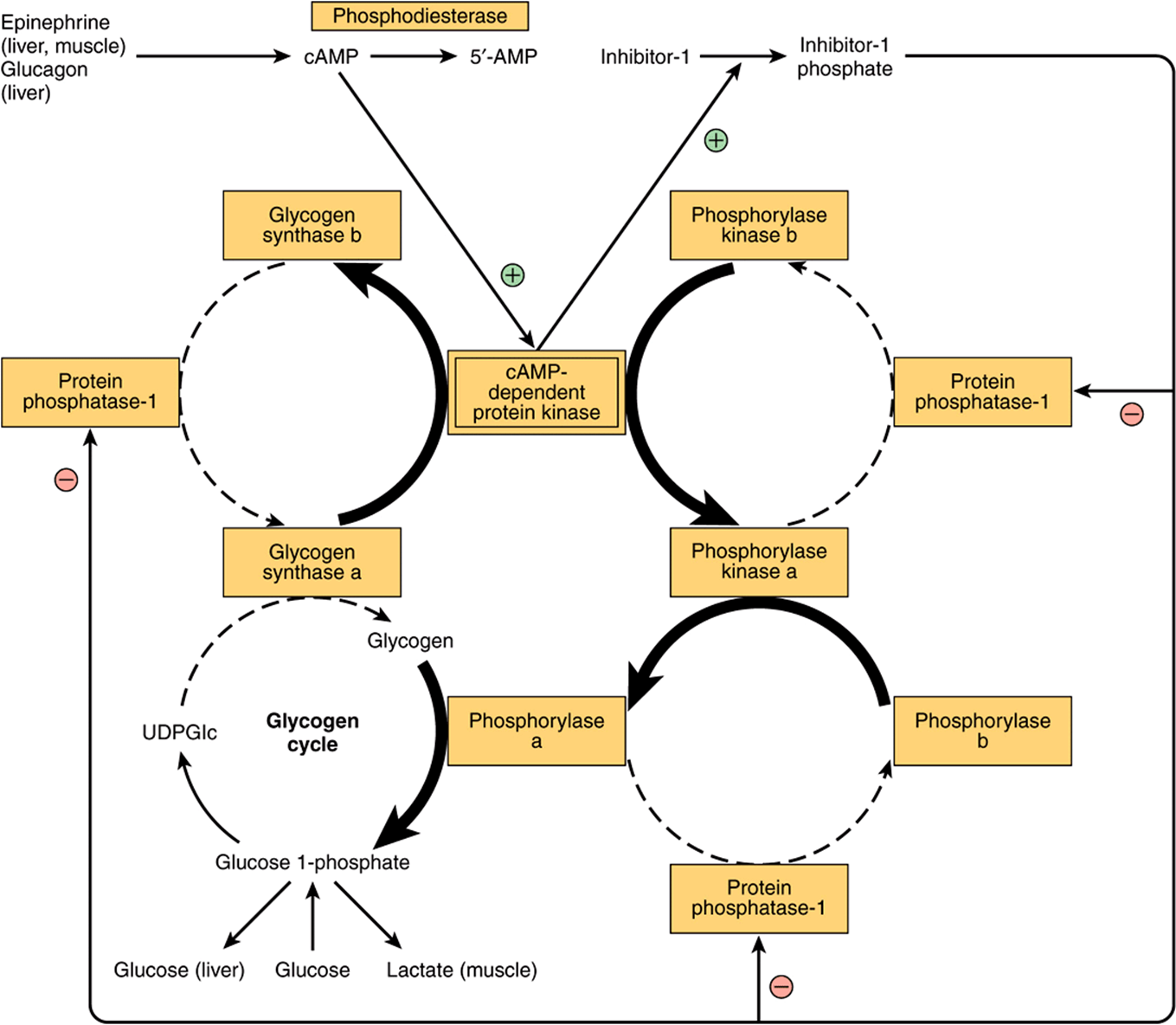
FIGURE 19–8 Coordinated control of glycogenolysis and glycogenesis by cAMP-dependent protein kinase. The reactions that lead to glycogenolysis as a result of an increase in cAMP concentrations are shown with bold arrows, and those that are inhibited by activation of protein phosphatase-1 are shown with dashed arrows. The reverse occurs when cAMP concentrations decrease as a result of phosphodiesterase activity, leading to glycogenesis.
CLINICAL ASPECTS
Glycogen Storage Diseases Are Inherited
“Glycogen storage disease” is a generic term to describe a group of inherited disorders characterized by deposition of an abnormal type or quantity of glycogen in tissues, or failure to mobilize glycogen. The principal diseases are summarized in Table 19-2.
SUMMARY
![]() Glycogen represents the principal storage carbohydrate in the body, mainly in the liver and muscle.
Glycogen represents the principal storage carbohydrate in the body, mainly in the liver and muscle.
![]() In the liver, its major function is to provide glucose for extrahepatic tissues. In muscle, it serves mainly as a ready source of metabolic fuel for use in muscle. Muscle lacks glucose 6-phosphatase and cannot release free glucose from glycogen.
In the liver, its major function is to provide glucose for extrahepatic tissues. In muscle, it serves mainly as a ready source of metabolic fuel for use in muscle. Muscle lacks glucose 6-phosphatase and cannot release free glucose from glycogen.
![]() Glycogen is synthesized from glucose by the pathway of glycogenesis. It is broken down by a separate pathway, glycogenolysis.
Glycogen is synthesized from glucose by the pathway of glycogenesis. It is broken down by a separate pathway, glycogenolysis.
![]() Cyclic AMP integrates the regulation of glycogenolysis and glycogenesis by promoting the simultaneous activation of phosphorylase and inhibition of glycogen synthase. Insulin acts reciprocally by inhibiting glycogenolysis and stimulating glycogenesis.
Cyclic AMP integrates the regulation of glycogenolysis and glycogenesis by promoting the simultaneous activation of phosphorylase and inhibition of glycogen synthase. Insulin acts reciprocally by inhibiting glycogenolysis and stimulating glycogenesis.
![]() Inherited deficiencies of enzymes of glycogen metabolism in both liver and muscle cause glycogen storage diseases.
Inherited deficiencies of enzymes of glycogen metabolism in both liver and muscle cause glycogen storage diseases.
REFERENCES
Alanso MD, Lomako J, Lomako WM, et al: A new look at the biogenesis of glycogen. FASEB J. 1995;9:1126.
Bollen M, Keppens S, Stalmans W: Specific features of glycogen metabolism in the liver. Biochem J 1998;336:19.
Ferrer JC, Favre C, Gomis RR, et al: Control of glycogen deposition. FEBS Lett 2003;546:127-132.
Forde JE, Dale TC: Glycogen synthase kinase 3: a key regulator of cellular fate. Cell Mol Life Sci 2007;64:1930.
Graham TE, Yuan Z, Hill AK, et al: The regulation of muscle glycogen: the granule and its proteins. Acta Physiol (Oxf) 2010;199:489.
Greenberg CC, Jurczak MJ, Danos AM, et al: Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab 2006;291:E1.
Jensen J, Lai YC: Regulation of muscle glycogen synthase phosphorylation and kinetic properties by insulin, exercise, adrenaline and role in insulin resistance. Arch Physiol Biochem 2009;115:13.
Jentjens R, Jeukendrup A: Determinants of post-exercise glycogen synthesis during short-term recovery. Sports Med 2003;33:117.
McGarry JD, Kuwajima M, Newgard CB, et al: From dietary glucose to liver glycogen: the full circle round. Annu Rev Nutr 1987;7:51.
Meléndez-Hevia E, Waddell TG, Shelton ED: Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem J 1993;295:477.
Ozen H: Glycogen storage diseases: new perspectives. World J Gastroenterol 2007;13:2541.
Radziuk J, Pye S: Hepatic glucose uptake, gluconeogenesis and the regulation of glycogen synthesis. Diabetes Metab Res Rev 2001;17(4):250.
Roach PJ: Glycogen and its metabolism. Curr Mol Med 2002;2(2):101.
Roden M, Bernroider E: Hepatic glucose metabolism in humans—its role in health and disease. Best Pract Res Clin Endocrinol Metab 2003;17:365.
Rybicka KK: Glycosomes—the organelles of glycogen metabolism. Tissue Cell 1996;28:254.
Shearer J, Graham TE: New perspectives on the storage and organization of muscle glycogen. Can J Appl Physiol 2002;27:179.
Shin YS: Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol 2006;13:115.
Wolfsdorf JI, Holm IA: Glycogen storage diseases. Phenotypic, genetic, and biochemical characteristics, and therapy. Endocrinol Metab Clin North Am 1999;28:801.
Yeaman SJ, Armstrong JL, Bonavaud SM, et al: Regulation of glycogen synthesis in human muscle cells. Biochem Soc Trans 2001;29:537.