Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION II. Bioenergetics & the Metabolism of Carbohydrates & Lipids
Chapter 22. Oxidation of Fatty Acids: Ketogenesis
Kathleen M. Botham, PhD, DSc & Peter A. Mayes, PhD, DSc
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe the processes by which fatty acids are transported in the blood and activated and transported into the matrix of the mitochondria for breakdown to obtain energy.
Describe the processes by which fatty acids are transported in the blood and activated and transported into the matrix of the mitochondria for breakdown to obtain energy.
![]() Outline the β-oxidation pathway by which fatty acids are metabolized to acetyl-CoA and explain how this leads to the production of large quantities of ATP from the reducing equivalents produced during β-oxidation and further metabolism of the acetyl-CoA via the citric acid cycle.
Outline the β-oxidation pathway by which fatty acids are metabolized to acetyl-CoA and explain how this leads to the production of large quantities of ATP from the reducing equivalents produced during β-oxidation and further metabolism of the acetyl-CoA via the citric acid cycle.
![]() Identify the three compounds termed “ketone bodies” and describe the reactions by which they are formed in liver mitochondria.
Identify the three compounds termed “ketone bodies” and describe the reactions by which they are formed in liver mitochondria.
![]() Appreciate that ketone bodies are important fuels for extrahepatic tissues and indicate the conditions in which their synthesis and use are favored.
Appreciate that ketone bodies are important fuels for extrahepatic tissues and indicate the conditions in which their synthesis and use are favored.
![]() Indicate the three stages in the metabolism of fatty acids where ketogenesis is regulated.
Indicate the three stages in the metabolism of fatty acids where ketogenesis is regulated.
![]() Understand that overproduction of ketone bodies leads to ketosis and, if prolonged, ketoacidosis, and identify pathological conditions when this occurs.
Understand that overproduction of ketone bodies leads to ketosis and, if prolonged, ketoacidosis, and identify pathological conditions when this occurs.
![]() Give examples of diseases associated with impaired fatty acid oxidation.
Give examples of diseases associated with impaired fatty acid oxidation.
BIOMEDICAL IMPORTANCE
Although fatty acids are broken down by oxidation to acetyl-CoA and also synthesized from acetyl-CoA, fatty acid oxidation is not the simple reverse of fatty acid biosynthesis but an entirely different process taking place in a separate compartment of the cell. The separation of fatty acid oxidation in mitochondria from biosynthesis in the cytosol allows each process to be individually controlled and integrated with tissue requirements. Each step in fatty acid oxidation involves acyl-CoA derivatives, is catalyzed by separate enzymes, utilizes NAD+ and FAD as coenzymes, and generates ATP. It is an aerobic process, requiring the presence of oxygen.
Increased fatty acid oxidation is a characteristic of starvation and of diabetes mellitus, and leads to ketone body production by the liver (ketosis). Ketone bodies are acidic and when produced in excess over long periods, as in diabetes, cause ketoacidosis, which is ultimately fatal. Because gluconeogenesis is dependent upon fatty acid oxidation, any impairment in fatty acid oxidation leads to hypoglycemia. This occurs in various states of carnitine deficiency or deficiency of essential enzymes in fatty acid oxidation, for example, carnitine palmitoyltransferase, or inhibition of fatty acid oxidation by poisons, for example, hypoglycin.
OXIDATION OF FATTY ACIDS OCCURS IN MITOCHONDRIA
Fatty Acids Are Transported in the Blood as Free Fatty Acids
Free fatty acids (FFA)—also called unesterified (UFA) or non-esterified (NEFA) fatty acids—are fatty acids that are in the unesterified state. In plasma, longer chain FFA are combined with albumin, and in the cell they are attached to a fatty acid binding protein, so that in fact they are never really “free.” Shorter chain fatty acids are more water-soluble and exist as the unionized acid or as a fatty acid anion.
Fatty Acids Are Activated Before Being Catabolized
Fatty acids must first be converted to an active intermediate before they can be catabolized. This is the only step in the complete degradation of a fatty acid that requires energy from ATP. In the presence of ATP and coenzyme A, the enzyme acyl-CoA synthetase (thiokinase) catalyzes the conversion of a fatty acid (or FFA) to an “active fatty acid” or acyl-CoA, which uses one high-energy phosphate with the formation of AMP and PPi (Figure 22–1). The PPi. is hydrolyzed by inorganic pyrophosphatase with the loss of a further high-energy phosphate, ensuring that the overall reaction goes to completion. Acyl-CoA synthetases are found in the endoplasmic reticulum, peroxisomes, and inside and on the outer membrane of mitochondria.
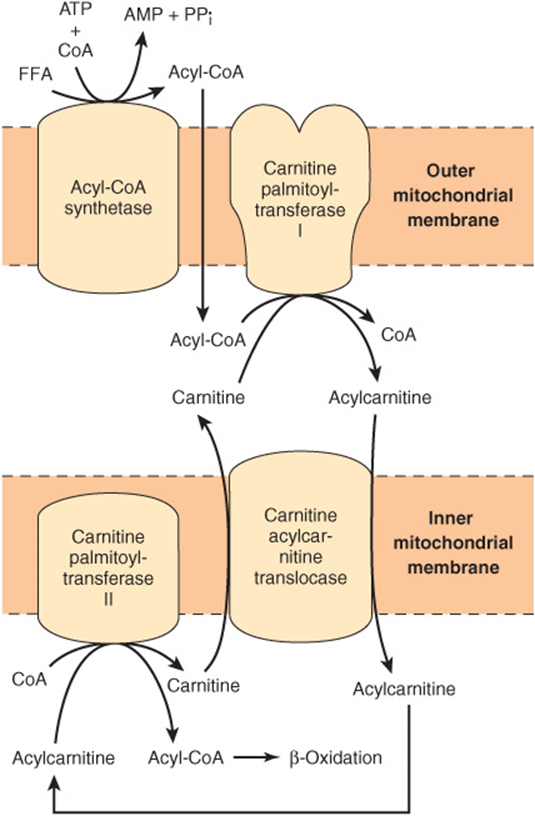
FIGURE 22–1 Role of carnitine in the transport of long-chain fatty acids through the inner mitochondrial membrane. Long-chain acyl-CoA cannot pass through the inner mitochondrial membrane, but its metabolic product, acylcarnitine, can.
Long-Chain Fatty Acids Penetrate the Inner Mitochondrial Membrane as Carnitine Derivatives
Carnitine (β-hydroxy-γ-trimethylammonium butyrate), (CH3)3N+—CH2—CH(OH)—CH2—COO–, is widely distributed and is particularly abundant in muscle. Long-chain acyl-CoA (or FFA) cannot penetrate the inner membrane of mitochondria. In the presence of carnitine, however, carnitine palmitoyltransferase-I, located in the outer mitochondrial membrane, converts long-chain acyl-CoA to acylcarnitine, which is able to penetrate the inner membrane and gain access to the β-oxidation system of enzymes (Figure 22–1). Carnitine-acylcarnitine translocase acts as an inner membrane exchange transporter. Acylcarnitine is transported in, coupled with the transport out of one molecule of carnitine. The acylcarnitine then reacts with CoA, catalyzed by carnitine palmitoyltransferase-II, located on the inside of the inner membrane, reforming acyl-CoA in the mitochondrial matrix, and carnitine is liberated.
β-OXIDATION OF FATTY ACIDS INVOLVES SUCCESSIVE CLEAVAGE WITH RELEASE OF ACETYL-CoA
In β-oxidation (Figure 22–2), two carbons at a time are cleaved from acyl-CoA molecules, starting at the carboxyl end. The chain is broken between the α(2)- and β(3)-carbon atoms—hence the name β-oxidation. The two-carbon units formed are acetyl-CoA; thus, palmitoyl-CoA forms eight acetyl-CoA molecules.
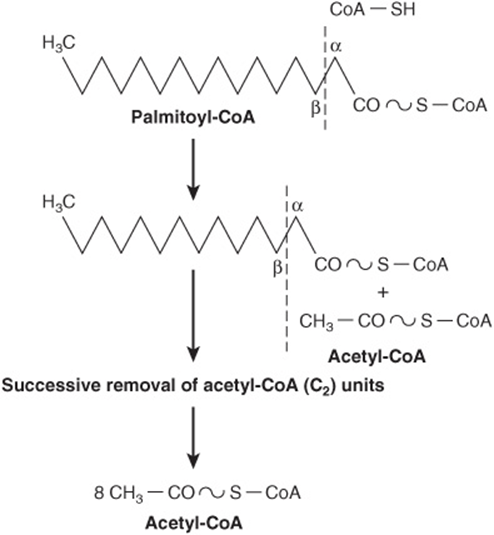
FIGURE 22–2 Overview of β-oxidation of fatty acids.
The Cyclic Reaction Sequence Generates FADH2 & NADH
Several enzymes, known collectively as “fatty acid oxidase,” are found in the mitochondrial matrix or inner membrane adjacent to the respiratory chain. These catalyze the oxidation of acyl-CoA to acetyl-CoA, the system being coupled with the phosphorylation of ADP to ATP (Figure 22–3).
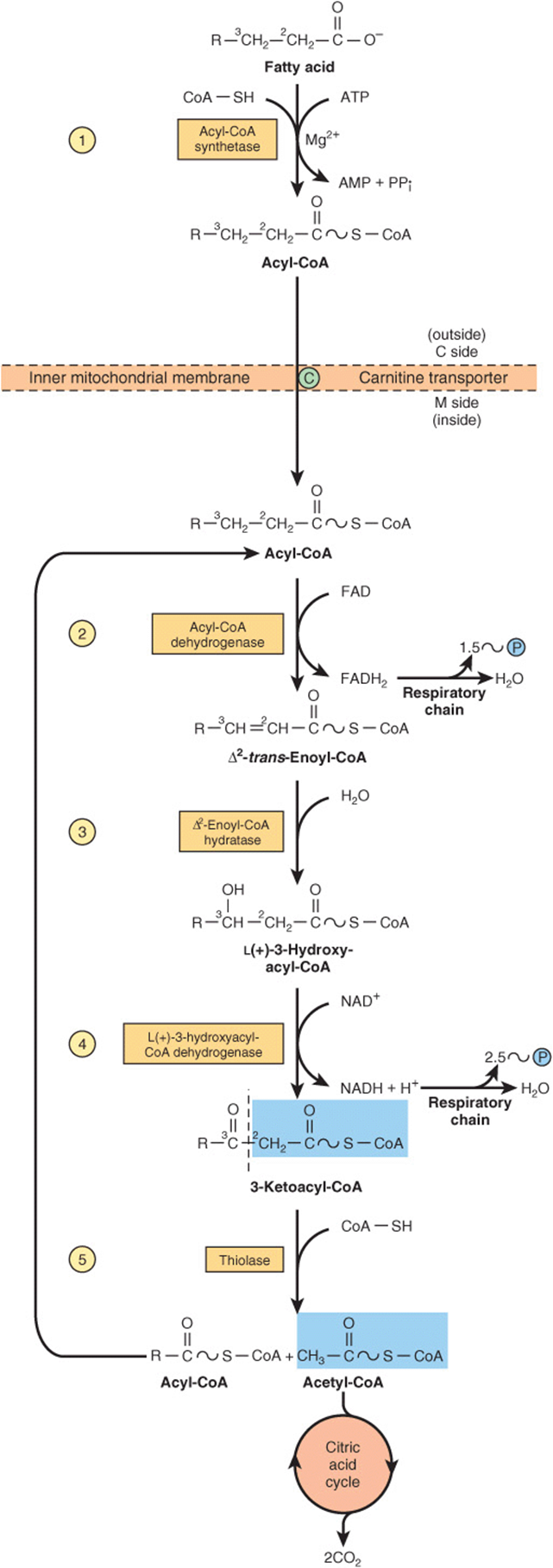
FIGURE 22–3 β-Oxidation of fatty acids. Long-chain acyl-CoA is cycled through reactions ![]() -
-![]() , acetyl-CoA being split off, each cycle, by thiolase (reaction
, acetyl-CoA being split off, each cycle, by thiolase (reaction ![]() ). When the acyl radical is only four carbon atoms in length, two acetyl-CoA molecules are formed in reaction
). When the acyl radical is only four carbon atoms in length, two acetyl-CoA molecules are formed in reaction ![]() .
.
The first step is the removal of two hydrogen atoms from the 2(α)- and 3(β)-carbon atoms, catalyzed by acyl-CoA dehydrogenase and requiring FAD. This results in the formation of Δ2-trans-enoyl-CoA and FADH2 The reoxidation of FADH2 by the respiratory chain requires the mediation of another flavoprotein, termed electron-transferring flavoprotein (Chapter 12). Water is added to saturate the double bond and form 3-hydroxyacyl-CoA, catalyzed by Δ2-enoyl-CoA hydratase. The 3-hydroxy derivative undergoes further dehydrogenation on the 3-carbon catalyzed by L(+)-3-hydroxyacyl-CoA dehydrogenase to form the corresponding 3-ketoacyl-CoA compound. In this case, NAD+ is the coenzyme involved. Finally, 3-ketoacyl-CoA is split at the 2,3-position by thiolase (3-ketoacyl-CoA-thiolase), forming acetyl-CoA and a new acyl-CoA two carbons shorter than the original acyl-CoA molecule. The acyl-CoA formed in the cleavage reaction reenters the oxidative pathway at reaction 2 (Figure 22–3). In this way, a long-chain fatty acid may be degraded completely to acetyl-CoA (C2 units). Since acetyl-CoA can be oxidized to CO2 and water via the citric acid cycle (which is also found within the mitochondria), the complete oxidation of fatty acids is achieved.
Oxidation of a Fatty Acid with an Odd Number of Carbon Atoms Yields Acetyl-CoA Plus a Molecule of Propionyl-CoA
Fatty acids with an odd number of carbon atoms are oxidized by the pathway of β-oxidation, producing acetyl-CoA, until a three-carbon (propionyl-CoA) residue remains. This compound is converted to succinyl-CoA, a constituent of the citric acid cycle (Figure 20–2). Hence, the propionyl residue from an odd-chain fatty acid is the only part of a fatty acid that is glucogenic.
Oxidation of Fatty Acids Produces a Large Quantity of ATP
Transport of electrons from FADH2 and NADH via the respiratory chain leads to the synthesis of four high-energy phosphates (Chapter 13) for each of the seven cycles needed for the breakdown of the C16 fatty acid, palmitate, to acetyl-CoA ![]() . A total of 8 mol of acetyl-CoA is formed, and each gives rise to 10 mol of ATP on oxidation in the citric acid cycle, making
. A total of 8 mol of acetyl-CoA is formed, and each gives rise to 10 mol of ATP on oxidation in the citric acid cycle, making ![]() . Two must be subtracted for the initial activation of the fatty acid, yielding a net gain of 106 mol of ATP per mole of palmitate, or
. Two must be subtracted for the initial activation of the fatty acid, yielding a net gain of 106 mol of ATP per mole of palmitate, or ![]() . This represents 68% of the free energy of combustion of palmitic acid.
. This represents 68% of the free energy of combustion of palmitic acid.
Peroxisomes Oxidize Very Long Chain Fatty Acids
A modified form of β-oxidation is found in peroxisomes and leads to the formation of acetyl-CoA and H2O2 (from the flavoprotein-linked dehydrogenase step), which is broken down by catalase (Chapter 12). Thus, this dehydrogenation in peroxisomes is not linked directly to phosphorylation and the generation of ATP. The system facilitates the oxidation of very long chain fatty acids (eg, C20, C22). These enzymes are induced by high-fat diets and in some species by hypolipidemic drugs such as clofibrate.
The enzymes in peroxisomes do not attack shorter chain fatty acids; the β-oxidation sequence ends at octanoyl-CoA. Octanoyl and acetyl groups are both further oxidized in mitochondria. Another role of peroxisomal β-oxidation is to shorten the side chain of cholesterol in bile acid formation (Chapter 26). Peroxisomes also take part in the synthesis of ether glycerolipids (Chapter 24), cholesterol, and dolichol (Figure 26–2).
OXIDATION OF UNSATURATED FATTY ACIDS OCCURS BY A MODIFIED β-OXIDATION PATHWAY
The CoA esters of unsaturated fatty acids are degraded by the enzymes normally responsible for β-oxidation until either a Δ3-cis-acyl-CoA compound or a Δ4-cis-acyl-CoA compound is formed, depending upon the position of the double bonds (Figure 22–4). The former compound is isomerized (Δ3cis → Δ2-trans-enoyl-CoA isomerase) to the corresponding Δ2-trans-CoA stage of β-oxidation for subsequent hydration and oxidation. Any Δ4-cis-acyl-CoA either remaining, as in the case of linoleic acid, or entering the pathway at this point after conversion by acyl-CoA dehydrogenase to Δ2-trans-Δ4-cis-dienoyl-CoA, is then metabolized as indicated in Figure 22–4.
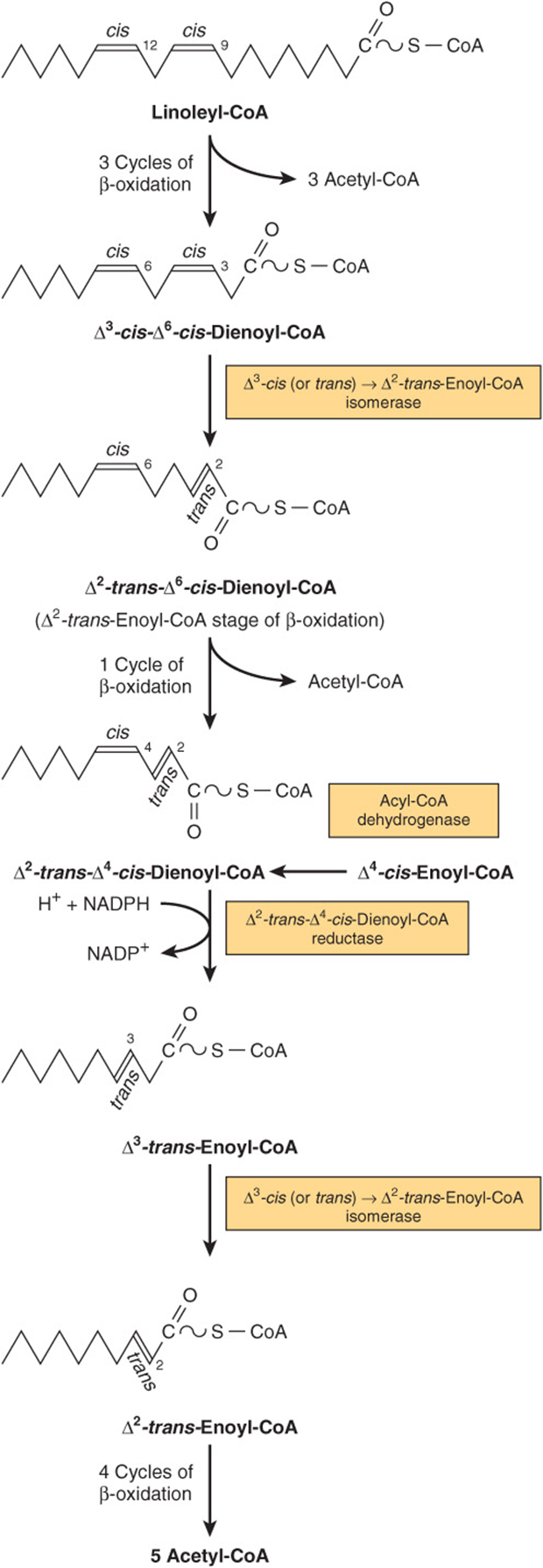
FIGURE 22–4 Sequence of reactions in the oxidation of unsaturated fatty acids, for example, linoleic acid. Δ4-cis-fatty acids or fatty acids forming Δ4-cis-enoyl-CoA enter the pathway at the position shown. NADPH for the dienoyl-CoA reductase step is supplied by intramitochondrial sources such as glutamate dehydrogenase, isocitrate dehydrogenase, and NAD(P)H transhydrogenase.
KETOGENESIS OCCURS WHEN THERE IS A HIGH RATE OF FATTY-ACID OXIDATION IN THE LIVER
Under metabolic conditions associated with a high rate of fatty acid oxidation, the liver produces considerable quantities of acetoacetate and D(-)-3-hydroxybutyrate (β-hydroxybutyrate). Acetoacetate continually undergoes spontaneous decarboxylation to yield acetone. These three substances are collectively known as the ketone bodies (also called acetone bodies or [incorrectly*] “ketones”) (Figure 22–5). Acetoacetate and 3-hydroxybutyrate are interconverted by the mitochondrial enzyme D(-)-3-hydroxybutyrate dehydrogenase; the equilibrium is controlled by the mitochondrial [NAD+]/[NADH] ratio, ie, the redox state. The concentration of total ketone bodies in the blood of well-fed mammals does not normally exceed 0.2 mmol/L except in ruminants, where 3-hydroxybutyrate is formed continuously from butyric acid (a product of ruminal fermentation) in the rumen wall. In vivo, the liver appears to be the only organ in nonruminants to add significant quantities of ketone bodies to the blood. Extrahepatic tissues utilize them as respiratory substrates. The net flow of ketone bodies from the liver to the extrahepatic tissues results from active hepatic synthesis coupled with very low utilization. The reverse situation occurs in extrahepatic tissues (Figure 22–6).
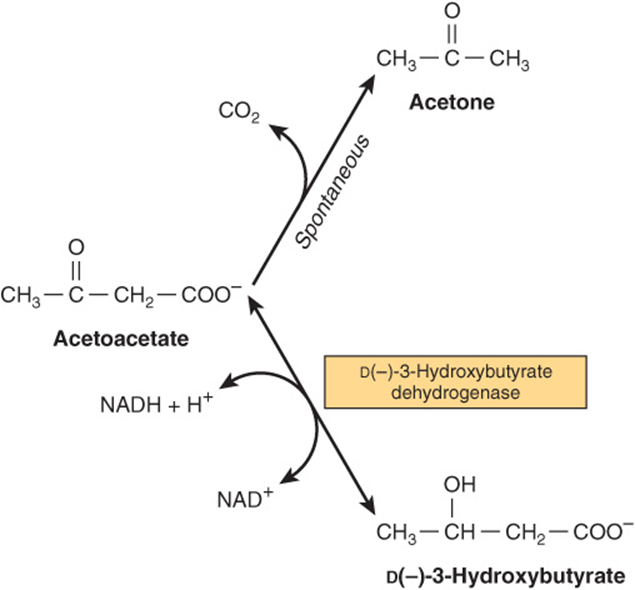
FIGURE 22–5 Interrelationships of the ketone bodies. D(-)-3-hydroxybutyrate dehydrogenase is a mitochondrial enzyme.
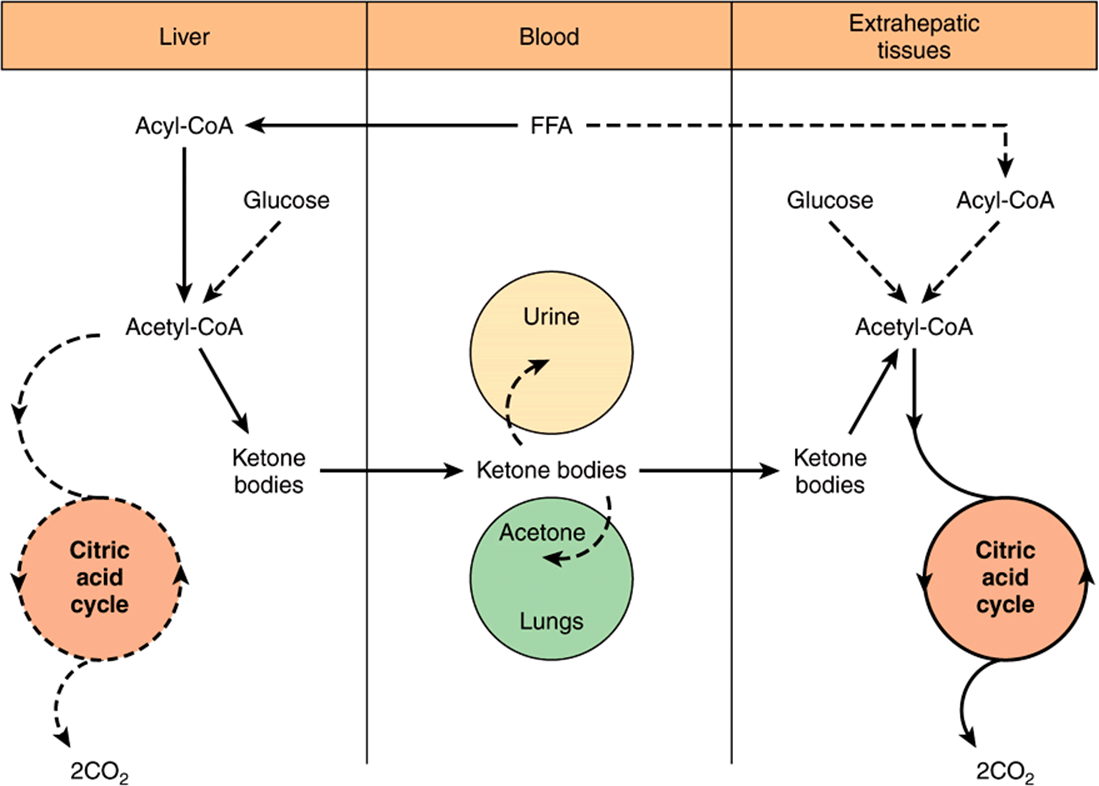
FIGURE 22–6 Formation, utilization, and excretion of ketone bodies. (The main pathway is indicated by the solid arrows.)
3-Hydroxy-3-Methylglutaryl-CoA (HMG-CoA) Is an Intermediate in the Pathway of Ketogenesis
Enzymes responsible for ketone body formation are associated mainly with the mitochondria. Two acetyl-CoA molecules formed in β-oxidation condense to form acetoacetyl-CoA by a reversal of the thiolase reaction. Acetoacetyl-CoA, which is the starting material for ketogenesis, also arises directly from the terminal four carbons of a fatty acid during β-oxidation (Figure 22–7). Condensation of acetoacetyl-CoA with another molecule of acetyl-CoA by 3-hydroxy-3-methylglutaryl-CoA synthase forms 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA). 3-Hydroxy-3-methylglutaryl-CoA lyase then causes acetyl-CoA to split off from the HMG-CoA, leaving free acetoacetate. The carbon atoms split off in the acetyl-CoA molecule are derived from the original acetoacetyl-CoA molecule. Both enzymes must be present in mitochondria for ketogenesis to take place. This occurs solely in liver and rumen epithelium. D(-)-3-Hydroxybutyrate is quantitatively the predominant ketone body present in the blood and urine in ketosis.
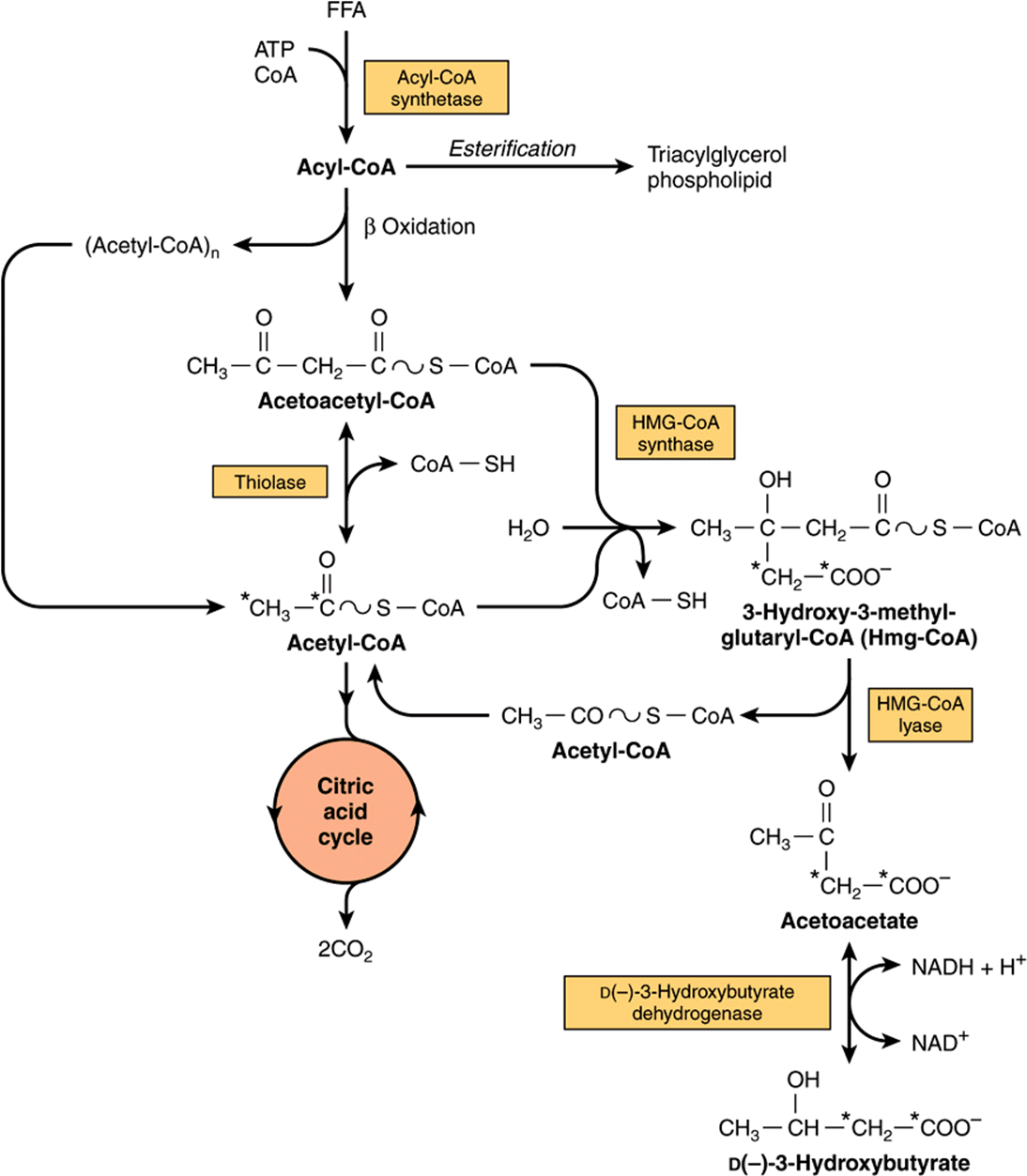
FIGURE 22–7 Pathways of ketogenesis in the liver. (FFA, free fatty acids.)
Ketone Bodies Serve as a Fuel for Extrahepatic Tissues
While an active enzymatic mechanism produces acetoacetate from acetoacetyl-CoA in the liver, acetoacetate once formed cannot be reactivated directly except in the cytosol, where it is used in a much less active pathway as a precursor in cholesterol synthesis. This accounts for the net production of ketone bodies by the liver.
In extrahepatic tissues, acetoacetate is activated to acetoacetyl-CoA by succinyl-CoA-acetoacetate CoA transferase. CoA is transferred from succinyl-CoA to form acetoacetyl-CoA (Figure 22–8). With the addition of a CoA, the acetoacetyl-CoA is split into two acetyl-CoAs by thiolase and oxidized in the citric acid cycle. If the blood level is raised, oxidation of ketone bodies increases until, at a concentration of ~12 mmol/L, the oxidative machinery is saturated. When this occurs, a large proportion of oxygen consumption may be accounted for by the oxidation of ketone bodies.
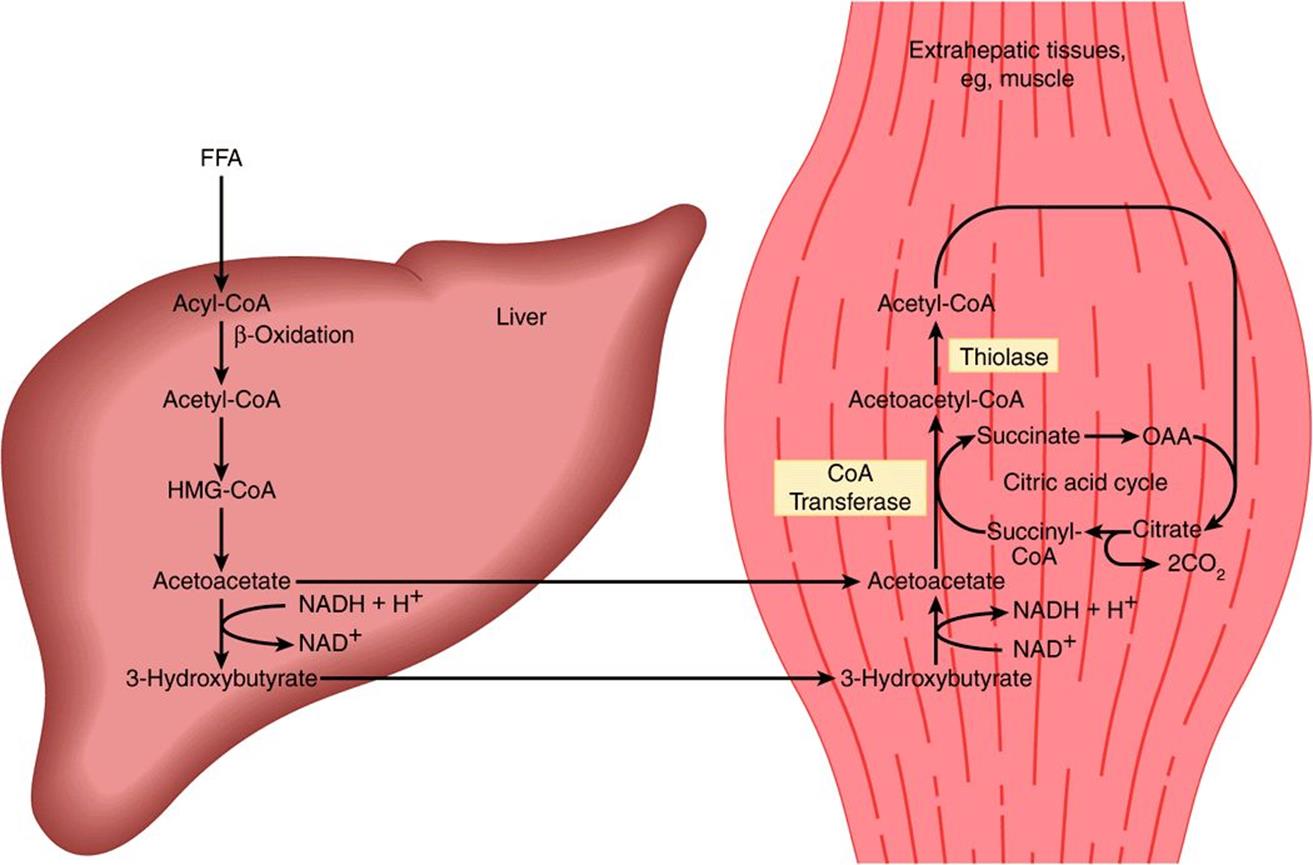
FIGURE 22–8 Transport of ketone bodies from the liver and pathways of utilization and oxidation in extrahepatic tissues.
In most cases, ketonemia is due to increased production of ketone bodies by the liver rather than to a deficiency in their utilization by extrahepatic tissues. While acetoacetate and D(-)-3-hydroxybutyrate are readily oxidized by extrahepatic tissues, acetone is difficult to oxidize in vivo and to a large extent is volatilized in the lungs.
In moderate ketonemia, the loss of ketone bodies via the urine is only a few percent of the total ketone body production and utilization. Since there are renal threshold-like effects (there is not a true threshold) that vary between species and individuals, measurement of the ketonemia, not the ketonuria, is the preferred method of assessing the severity of ketosis.
KETOGENESIS IS REGULATED AT THREE CRUCIAL STEPS
1. Ketosis does not occur in vivo unless there is an increase in the level of circulating FFA that arise from lipolysis of triacylglycerol in adipose tissue. FFA are the precursors of ketone bodies in the liver. The liver, both in fed and in fasting conditions, extracts ~30% of the FFA passing through it, so that at high concentrations the flux passing into the liver is substantial. Therefore, the factors regulating mobilization of FFA from adipose tissue are important in controlling ketogenesis (Figures 22-9 and 25-8).
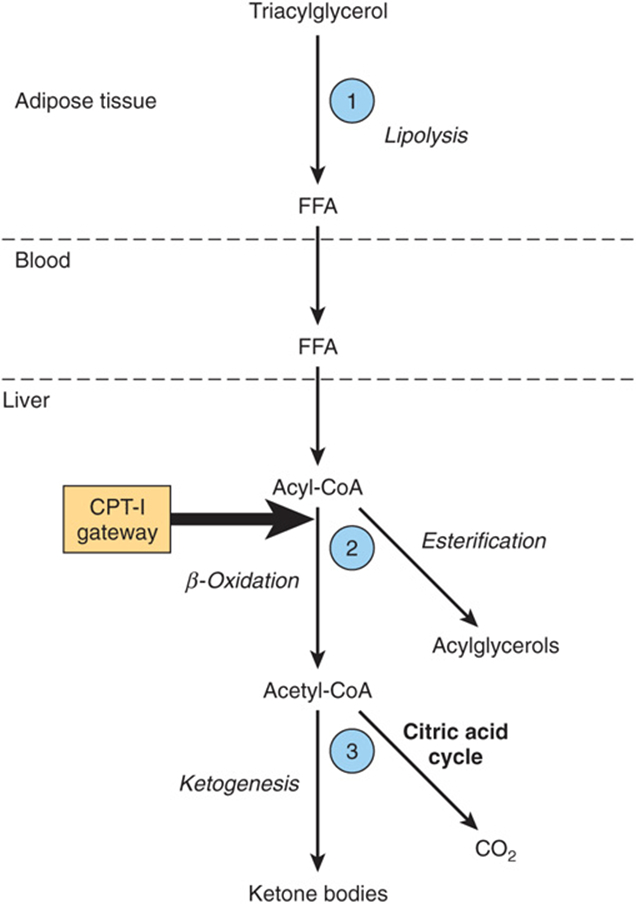
FIGURE 22–9 Regulation of ketogenesis. ![]() -
-![]() show three crucial steps in the pathway of metabolism of free fatty acids (FFA) that determine the magnitude of ketogenesis. (CPT-I, carnitine palmitoyltransferase-I.)
show three crucial steps in the pathway of metabolism of free fatty acids (FFA) that determine the magnitude of ketogenesis. (CPT-I, carnitine palmitoyltransferase-I.)
2. After uptake by the liver, FFA are either β-oxidized to CO2 or ketone bodies or esterified to triacylglycerol and phospholipid. There is regulation of entry of fatty acids into the oxidative pathway by carnitine palmitoyltransferase-I (CPT-I), and the remainder of the fatty acid taken up is esterified. CPT-I activity is low in the fed state, leading to depression of fatty acid oxidation, and high in starvation, allowing fatty acid oxidation to increase. Malonyl-CoA, the initial intermediate in fatty acid biosynthesis (Figure 23–1) formed by acetyl-CoA carboxylase in the fed state, is a potent inhibitor of CPT-I (Figure 22–10). Under these conditions, FFA enter the liver cell in low concentrations and are nearly all esterified to acylglycerols and transported out of the liver in very low density lipoproteins (VLDL). However, as the concentration of FFA increases with the onset of starvation, acetyl-CoA carboxylase is inhibited directly by acyl-CoA, and (malonyl-CoA) decreases, releasing the inhibition of CPT-I and allowing more acyl-CoA to be β-oxidized. These events are reinforced in starvation by a decrease in the (insulin)/(glucagon) ratio. Thus, β-oxidation from FFA is controlled by the CPT-I gateway into the mitochondria, and the balance of the FFA uptake not oxidized is esterified.
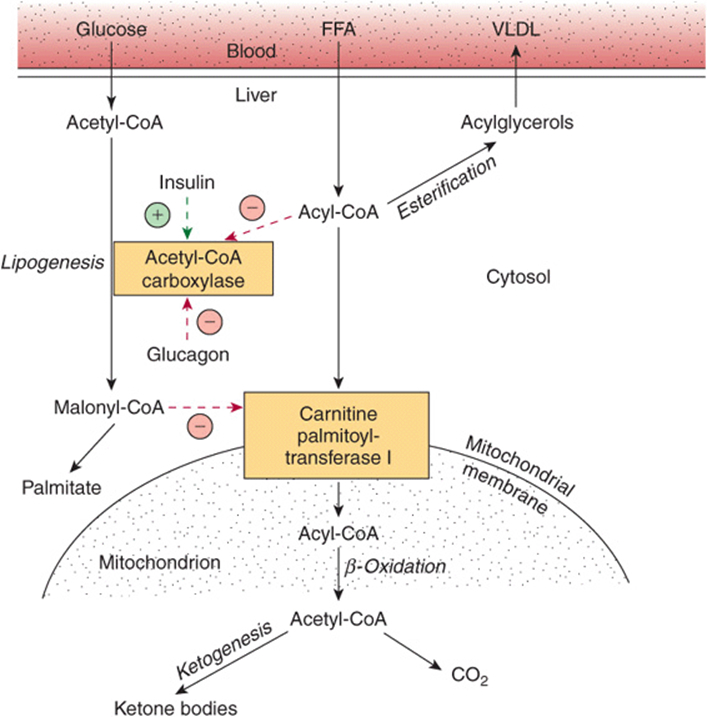
FIGURE 22–10 Regulation of long-chain fatty acid oxidation in the liver. (FFA, free fatty acids; VLDL, very low density lipoprotein.) Positive ![]() and negative
and negative ![]() regulatory effects are represented by broken arrows and substrate flow by solid arrows.
regulatory effects are represented by broken arrows and substrate flow by solid arrows.
3. In turn, the acetyl-CoA formed in β-oxidation is oxidized in the citric acid cycle, or it enters the pathway of ketogenesis to form ketone bodies. As the level of serum FFA is raised, proportionately more FFA is converted to ketone bodies and less is oxidized via the citric acid cycle to CO2. The partition of acetyl-CoA between the ketogenic pathway and the pathway of oxidation to CO2 is regulated so that the total free energy captured in ATP which results from the oxidation of FFA remains constant as their concentration in the serum changes. This may be appreciated when it is realized that complete oxidation of 1 mol of palmitate involves a net production of 106 mol of ATP via β-oxidation and CO2 production in the citric acid cycle (see above), whereas only 26 mol of ATP are produced when acetoacetate is the end product and only 21 mol when 3-hydroxybutyrate is the end product. Thus, ketogenesis may be regarded as a mechanism that allows the liver to oxidize increasing quantities of fatty acids within the constraints of a tightly coupled system of oxidative phosphorylation.
A fall in the concentration of oxaloacetate, particularly within the mitochondria, can impair the ability of the citric acid cycle to metabolize acetyl-CoA and divert fatty acid oxidation toward ketogenesis. Such a fall may occur because of an increase in the (NADH)/(NAD+) ratio caused by increased β-oxidation of fatty acids affecting the equilibrium between oxaloacetate and malate, leading to a decrease in the concentration of oxaloacetate, and when gluconeogenesis is elevated, which occurs when blood glucose levels are low. The activation of pyruvate carboxylase, which catalyzes the conversion of pyruvate to oxaloacetate, by acetyl-CoA partially alleviates this problem, but in conditions such as starvation and untreated diabetes mellitus, ketone bodies are overproduced causing ketosis.
CLINICAL ASPECTS
Impaired Oxidation of Fatty Acids Gives Rise to Diseases Often Associated with Hypoglycemia
Carnitine deficiency can occur particularly in the newborn—and especially in preterm infants—owing to inadequate biosynthesis or renal leakage. Losses can also occur in hemodialysis. This suggests a vitamin-like dietary requirement for carnitine in some individuals. Symptoms of deficiency include hypoglycemia, which is a consequence of impaired fatty acid oxidation and lipid accumulation with muscular weakness. Treatment is by oral supplementation with carnitine.
Inherited CPT-I deficiency affects only the liver, resulting in reduced fatty acid oxidation and ketogenesis, with hypoglycemia. CPT-II deficiency affects primarily skeletal muscle and, when severe, the liver.
Inherited defects in the enzymes of β-oxidation and ketogenesis also lead to nonketotic hypoglycemia, coma, and fatty liver. Defects are known in long- and short-chain 3-hydroxyacyl-CoA dehydrogenase (deficiency of the long-chain enzyme may be a cause of acute fatty liver of pregnancy). 3-Ketoacyl-CoA thiolase and HMG-CoA lyase deficiency also affect the degradation of leucine, a ketogenic amino acid (Chapter 29).
Jamaican vomiting sickness is caused by eating the unripe fruit of the akee tree, which contains the toxin hypoglycin. This inactivates medium- and short-chain acyl-CoA dehydrogenase, inhibiting β-oxidation and causing hypoglycemia. Dicarboxylic aciduria is characterized by the excretion of C6-C10 ω-dicarboxylic acids and by nonketotic hypoglycemia, and is caused by a lack of mitochondrial medium-chain acyl-CoA dehydrogenase. Refsum’s disease is a rare neurologic disorder due to a metabolic defect that results in the accumulation of phytanic acid, which is found in dairy products and ruminant fat and meat. Phytanic acid is thought to have pathological effects on membrane function, protein prenylation, and gene expression. Zellweger’s (cerebrohepatorenal) syndrome occurs in individuals with a rare inherited absence of peroxisomes in all tissues. They accumulate C26-C38polyenoic acids in brain tissue and also exhibit a generalized loss of peroxisomal functions. The disease causes severe neurological symptoms, and most patients die in the first year of life.
Ketoacidosis Results from Prolonged Ketosis
Higher than normal quantities of ketone bodies present in the blood or urine constitute ketonemia (hyperketonemia) or ketonuria, respectively. The overall condition is called ketosis. The basic form of ketosis occurs in starvationand involves depletion of available carbohydrate coupled with mobilization of FFA. This general pattern of metabolism is exaggerated to produce the pathologic states found in diabetes mellitus, the type 2 form of which is increasingly common in Western countries; twin lamb disease; and ketosis in lactating cattle. Nonpathologic forms of ketosis are found under conditions of high-fat feeding and after severe exercise in the postabsorptive state.
Acetoacetic and 3-hydroxybutyric acids are both moderately strong acids and are buffered when present in blood or other tissues. However, their continual excretion in quantity progressively depletes the alkali reserve, causing ketoacidosis. This may be fatal in uncontrolled diabetes mellitus.
SUMMARY
![]() Fatty acid oxidation in mitochondria leads to the generation of large quantities of ATP by a process called β-oxidation that cleaves acetyl-CoA units sequentially from fatty acyl chains. The acetyl-CoA is oxidized in the citric acid cycle, generating further ATP.
Fatty acid oxidation in mitochondria leads to the generation of large quantities of ATP by a process called β-oxidation that cleaves acetyl-CoA units sequentially from fatty acyl chains. The acetyl-CoA is oxidized in the citric acid cycle, generating further ATP.
![]() The ketone bodies (acetoacetate, 3-hydroxybutyrate, and acetone) are formed in hepatic mitochondria when there is a high rate of fatty acid oxidation. The pathway of ketogenesis involves synthesis and breakdown of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by two key enzymes, HMG-CoA synthase, and HMG-CoA lyase.
The ketone bodies (acetoacetate, 3-hydroxybutyrate, and acetone) are formed in hepatic mitochondria when there is a high rate of fatty acid oxidation. The pathway of ketogenesis involves synthesis and breakdown of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by two key enzymes, HMG-CoA synthase, and HMG-CoA lyase.
![]() Ketone bodies are important fuels in extrahepatic tissues.
Ketone bodies are important fuels in extrahepatic tissues.
![]() Ketogenesis is regulated at three crucial steps: (1) control of FFA mobilization from adipose tissue; (2) the activity of carnitine palmitoyltransferase-I in liver, which determines the proportion of the fatty acid flux that is oxidized rather than esterified; and (3) partition of acetyl-CoA between the pathway of ketogenesis and the citric acid cycle.
Ketogenesis is regulated at three crucial steps: (1) control of FFA mobilization from adipose tissue; (2) the activity of carnitine palmitoyltransferase-I in liver, which determines the proportion of the fatty acid flux that is oxidized rather than esterified; and (3) partition of acetyl-CoA between the pathway of ketogenesis and the citric acid cycle.
![]() Diseases associated with impairment of fatty acid oxidation lead to hypoglycemia, fatty infiltration of organs, and hypoketonemia.
Diseases associated with impairment of fatty acid oxidation lead to hypoglycemia, fatty infiltration of organs, and hypoketonemia.
![]() Ketosis is mild in starvation but severe in diabetes mellitus and ruminant ketosis.
Ketosis is mild in starvation but severe in diabetes mellitus and ruminant ketosis.
REFERENCES
Eaton S, Bartlett K, Pourfarzam M: Mammalian mitochondrial β-oxidation. Biochem J 1996;320:345.
Fukao T, Lopaschuk GD, Mitchell GA: Pathways and control of ketone body metabolism: on the fringe of lipid metabolism. Prostaglandins Leukot Essent Fatty Acids 2004;70:243.
Gurr MI, Harwood JL, Frayn K: Lipid Biochemistry. Blackwell Publishing, 2002.
Reddy JK, Mannaerts GP: Peroxisomal lipid metabolism. Annu Rev Nutr 1994;14:343.
Scriver CR, Beaudet AL, Sly WS, et al (editors): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, 2001.
Wood PA: Defects in mitochondrial beta-oxidation of fatty acids. Curr Opin Lipidol 1999;10:107.
*ΔG for the ATP reaction, as explained in Chapter 18.
*The term “ketones” should not be used because 3-hydroxybutyrate is not a ketone and there are ketones in blood that are not ketone bodies, for example, pyruvate and fructose.