Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION III. Metabolism of Proteins & Amino Acids
Chapter 28. Catabolism of Proteins & of Amino Acid Nitrogen
Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe protein turnover, indicate the mean rate of protein turnover in healthy individuals, and provide examples of human proteins that are degraded at rates greater than the mean rate.
Describe protein turnover, indicate the mean rate of protein turnover in healthy individuals, and provide examples of human proteins that are degraded at rates greater than the mean rate.
![]() Describe the events in protein turnover by both ATP-dependent and ATP-independent pathways, and the roles in protein degradation of ubiquitin, cell surface receptors, circulating asialoglycoproteins, and lysosomes.
Describe the events in protein turnover by both ATP-dependent and ATP-independent pathways, and the roles in protein degradation of ubiquitin, cell surface receptors, circulating asialoglycoproteins, and lysosomes.
![]() Indicate how the ultimate end products of nitrogen catabolism in mammals differ from those in birds and in fish.
Indicate how the ultimate end products of nitrogen catabolism in mammals differ from those in birds and in fish.
![]() Illustrate the central roles of transaminases (aminotransferases), of glutamate dehydrogenase, and of glutaminase in human nitrogen metabolism.
Illustrate the central roles of transaminases (aminotransferases), of glutamate dehydrogenase, and of glutaminase in human nitrogen metabolism.
![]() Use structural formulas to represent the reactions that convert NH3, CO2, and the amide nitrogen of aspartate into urea.
Use structural formulas to represent the reactions that convert NH3, CO2, and the amide nitrogen of aspartate into urea.
![]() Indicate the subcellular locations of the enzymes that catalyze urea biosynthesis, and the roles of allosteric regulation and of acetylglutamate in the regulation of this process.
Indicate the subcellular locations of the enzymes that catalyze urea biosynthesis, and the roles of allosteric regulation and of acetylglutamate in the regulation of this process.
![]() Explain why metabolic defects in different enzymes of urea biosynthesis, although distinct at the molecular level, present similar clinical signs and symptoms.
Explain why metabolic defects in different enzymes of urea biosynthesis, although distinct at the molecular level, present similar clinical signs and symptoms.
![]() Describe both the classical approaches and the role of tandem mass spectrometry in screening neonates for inherited metabolic diseases.
Describe both the classical approaches and the role of tandem mass spectrometry in screening neonates for inherited metabolic diseases.
BIOMEDICAL IMPORTANCE
This chapter describes how the nitrogen of amino acids is converted to urea and the rare metabolic disorders that accompany defects in urea biosynthesis. In normal adults, nitrogen intake matches nitrogen excreted. Positive nitrogen balance, an excess of ingested over excreted nitrogen, accompanies growth and pregnancy. Negative nitrogen balance, where output exceeds intake, may follow surgery, advanced cancer, and the nutritional disorders kwashiorkor, and marasmus. Ammonia, which is highly toxic, arises in humans primarily from the α-amino nitrogen of amino acids. Tissues therefore convert ammonia to the amide nitrogen of the nontoxic amino acid glutamine. Subsequent deamination of glutamine in the liver releases ammonia, which is then converted to urea, which is not toxic. If liver function is compromised, as in cirrhosis or hepatitis, elevated blood ammonia levels generate clinical signs and symptoms. Each enzyme of the urea cycle provides examples of metabolic defects and their physiologic consequences, and the cycle as a whole serves as a molecular model for the study of human metabolic defects.
PROTEIN TURNOVER OCCURS IN ALL FORMS OF LIFE
The continuous degradation and synthesis (turnover) of cellular proteins occur in all forms of life. Each day, humans turn over 1-2% of their total body protein, principally muscle protein. High rates of protein degradation occur in tissues that are undergoing structural rearrangement, for example, uterine tissue during pregnancy, skeletal muscle in starvation, and tadpole tail tissue during metamorphosis. Approximately 75% of the amino acids liberated by protein degradation are reutilized. Excess free amino acids are, however, not stored. Those not immediately incorporated into new protein are rapidly degraded. The major portion of the carbon skeletons of the amino acids is converted to amphibolic intermediates, while in humans the amino nitrogen is converted to urea and excreted in the urine.
PROTEASES & PEPTIDASES DEGRADE PROTEINS TO AMINO ACIDS
The relative susceptibility of a protein to degradation is expressed as its half-life (t1/2), the time required to lower its concentration to half the initial value. Half-lives of liver proteins range from under 30 min to over 150 h. Typical “housekeeping” enzymes have t1/2 values of over 100 h. By contrast, key regulatory enzymes may have t1/2 values as low as 0.5-2 h. PEST sequences, regions rich in proline (P), glutamate (E), serine (S), and threonine (T), target some proteins for rapid degradation. Intracellular proteases hydrolyze internal peptide bonds. The resulting peptides are then degraded to amino acids by endopeptidases that cleave internal peptide bonds, and by aminopeptidases and carboxypeptidases that remove amino acids sequentially from the amino- and carboxyl-termini, respectively.
ATP-Independent Degradation
Degradation of blood glycoproteins (see Chapter 47) follows loss of a sialic acid moiety from the nonreducing ends of their oligosaccharide chains. Asialoglycoproteins are then internalized by liver-cell asialoglycoprotein receptors and degraded by lysosomal proteases. Extracellular, membrane-associated, and long-lived intracellular proteins are degraded in lysosomes by ATP-independent processes.
ATP and Ubiquitin-Dependent Degradation
Degradation of regulatory proteins with short half-lives and of abnormal or misfolded proteins occurs in the cytosol, and requires ATP and ubiquitin. Ubiquitin, so named because it is present in all eukaryotic cells, is a small (8.5 kDa, 76 residues) polypeptide that targets many intracellular proteins for degradation. The primary structure of ubiquitin is highly conserved. Only three of 76 residues differ between yeast and human ubiquitin. Ubiquitin molecules are attached by non-α-peptide bonds formed between the carboxyl terminal of ubiquitin and the ε-amino groups of lysyl residues in the target protein (Figure 28–1). The residue present at its amino terminal affects whether a protein is ubiquitinated. Amino terminal Met or Ser retards, whereas Asp or Arg accelerates ubiquitination. Attachment of a single ubiquitin molecule to transmembrane proteins alters their subcellular localization and targets them for degradation. Soluble proteins undergo polyubiquitination, the ligase-catalyzed attachment of four or more additional ubiquitin molecules. Subsequent degradation of ubiquitin-tagged proteins takes place in the pro-teasome, a macromolecule with multiple different subunits that also is ubiquitous in eukaryotic cells (see Chapter 46). For the discovery of ubiquitin-mediated protein degradation, Aaron Ciechanover and Avram Hershko of Israel and Irwin Rose of the United States were awarded the 2004 Nobel Prize in Chemistry. Metabolic diseases associated with defects of ubiquitination include the Angelman syndrome and the von Hippel-Lindau syndrome in which there is a defect in the ubiquitin E3 ligase. For additional aspects of protein degradation and of ubiquitination, including its role in the cell cycle, see Chapters 4 and 46.
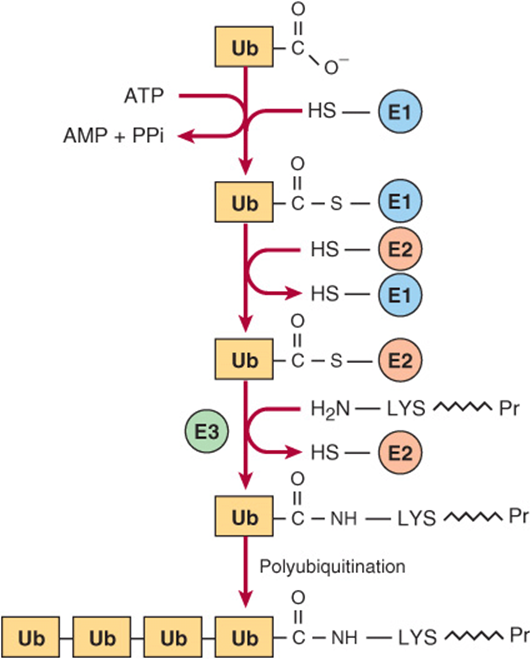
FIGURE 28–1 Reactions involved in the attachment of ubiquitin (Ub) to proteins. Three enzymes are involved. E1 is an activating enzyme, E2 a ligase, and E3 a transferase. While depicted as single entities, there are several types of E1, and over 500 types of E2. The terminal COOH of ubiquitin first forms a thioester. The coupled hydrolysis of PPi by pyrophosphatase ensures that the reaction will proceed readily. A thioester exchange reaction now transfers activated ubiquitin to E2. E3 then catalyzes the transfer of ubiquitin to the ε-amino group of a lysyl residue of the target protein. Additional rounds of ubiquitination result in subsequent polyubiquitination.
INTERORGAN EXCHANGE MAINTAINS CIRCULATING LEVELS OF AMINO ACIDS
The maintenance of steady-state concentrations of circulating plasma amino acids between meals depends on the net balance between release from endogenous protein stores and utilization by various tissues. Muscle generates over half of the total body pool of free amino acids, and liver is the site of the urea cycle enzymes necessary for disposal of excess nitrogen. Muscle and liver thus play major roles in maintaining circulating amino acid levels.
Figure 28–2 summarizes the postabsorptive state. Free amino acids, particularly alanine and glutamine, are released from muscle into the circulation. Alanine, which appears to be the vehicle of nitrogen transport in the plasma, is extracted primarily by the liver. Glutamine is extracted by the gut and the kidney, both of which convert a significant portion to alanine. Glutamine also serves as a source of ammonia for excretion by the kidney. The kidney provides a major source of serine for uptake by peripheral tissues, including liver and muscle. Branched-chain amino acids, particularly valine, are released by muscle and taken up predominantly by the brain.
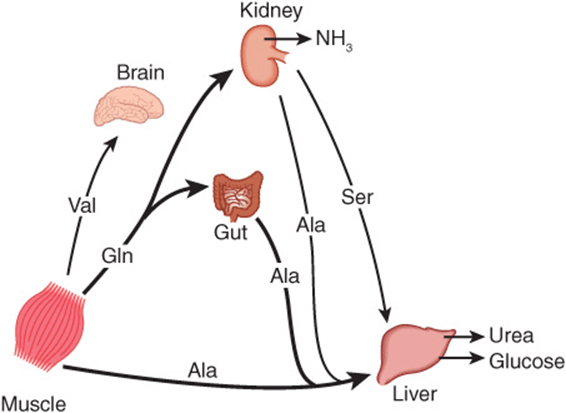
FIGURE 28–2 Interorgan amino acid exchange in normal postabsorptive humans. The key role of alanine in amino acid output from muscle and gut and uptake by the liver is shown.
Alanine is a key gluconeogenic amino acid (Figure 28–3). The rate of hepatic gluconeogenesis from alanine is far higher than from all other amino acids. The capacity of the liver for gluconeogenesis from alanine does not reach saturation until the alanine concentration reaches 20-30 times its normal physiologic level. Following a protein-rich meal, the splanchnic tissues release amino acids (Figure 28–4) while the peripheral muscles extract amino acids, in both instances predominantly branched-chain amino acids. Branched-chain amino acids thus serve a special role in nitrogen metabolism: in the fasting state, when they provide the brain with an energy source, and after feeding, when they are extracted predominantly by muscle, having been spared by the liver.
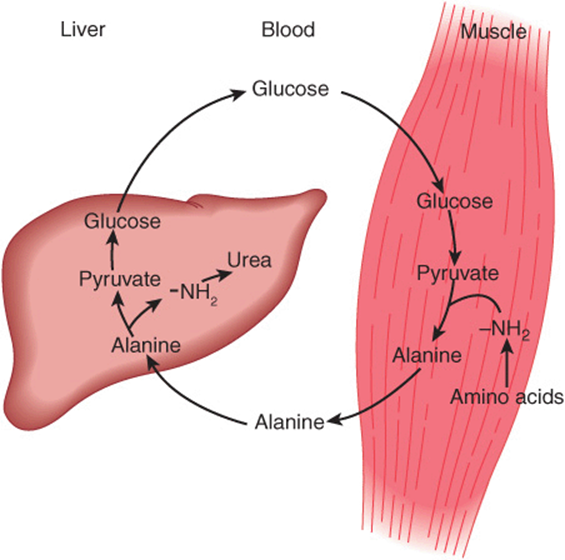
FIGURE 28–3 Theglucose-alaninecycle. Alanineissynthesized in muscle by transamination of glucose-derived pyruvate, released into the bloodstream, and taken up by the liver. In the liver, the carbon skeleton of alanine is reconverted to glucose and released into the bloodstream, where it is available for uptake by muscle and resynthesis of alanine.
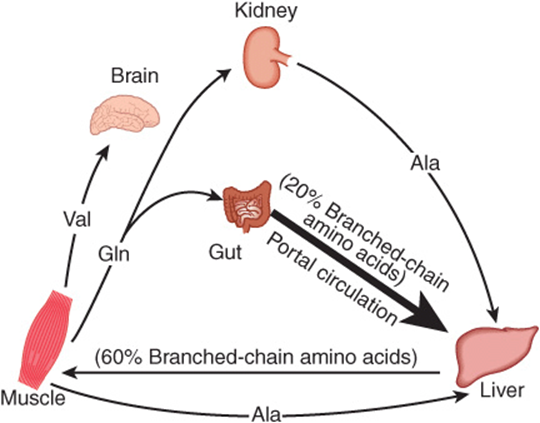
FIGURE 28–4 Summary of amino acid exchange between organs immediately after feeding.
ANIMALS CONVERT α-AMINO NITROGEN TO VARIED END PRODUCTS
Depending on their ecological niche and physiology, different animals excrete excess nitrogen as ammonia, as uric acid, or as urea. The aqueous environment of teleostean fish, which are ammonotelic (excrete ammonia), permits them to excrete water continuously to facilitate excretion of ammonia, which is highly toxic. While this approach is appropriate for an aquatic animal, birds must both conserve water and maintain low weight. Birds, which are uricotelic, address both problems by excreting nitrogen-rich uric acid (see Figure 33–11) as semisolid guano. Many land animals, including humans, are ureotelic and excrete nontoxic, highly water-soluble urea. Since urea is nontoxic to humans, high blood levels in renal disease are a consequence, not a cause, of impaired renal function.
BIOSYNTHESIS OF UREA
Urea biosynthesis occurs in four stages: (1) transamination, (2) oxidative deamination of glutamate, (3) ammonia transport, and (4) reactions of the urea cycle (Figure 28–5). The use of complementary DNA probes has shown that the expression in liver of the RNAs for all the enzymes of the urea cycle increases several fold in starvation.
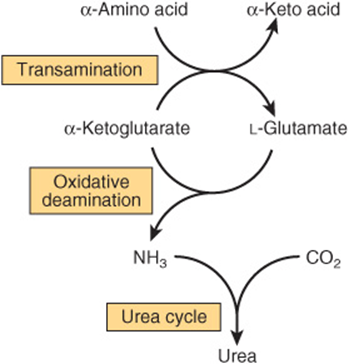
FIGURE 28–5 Overall flow of nitrogen in amino acid catabolism.
Transamination Transfers α-Amino Nitrogen to α-Ketoglutarate, Forming Glutamate
Transamination reactions interconvert pairs of α-amino acids and α-keto acids (Figure 28–6). Transamination reactions, which are freely reversible, also function in amino acid biosynthesis (see Figure 27–3). All of the common amino acids except lysine, threonine, proline, and hydroxyproline participate in transamination. Transamination is not restricted to α-amino groups. The δ-amino group of ornithine (but not the ε-amino group of lysine) readily undergoes transamination.
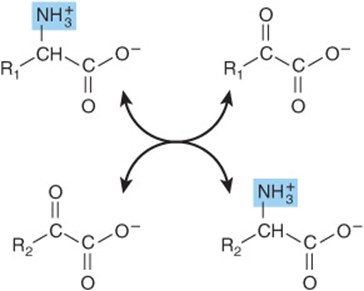
FIGURE 28–6 Transamination. The reaction is freely reversible with an equilibrium constant close to unity.
Alanine-pyruvate aminotransferase (alanine aminotransferase) and glutamate-α-ketoglutarate aminotransferase (glutamate aminotransferase) catalyze the transfer of amino groups to pyruvate (forming alanine) or to α-ketoglutarate (forming glutamate) (Figure 28–7). Each aminotransferase is specific for one pair of substrates, but nonspecific for the other pair. Since alanine is also a substrate for glutamate aminotransferase, the α-amino nitrogen from all amino acids that undergo transamination can be concentrated in glutamate. This is important because L-glutamate is the only amino acid that undergoes oxidative deamination at an appreciable rate in mammalian tissues. The formation of ammonia from α-amino groups thus occurs mainly via the α-amino nitrogen of L-glutamate.
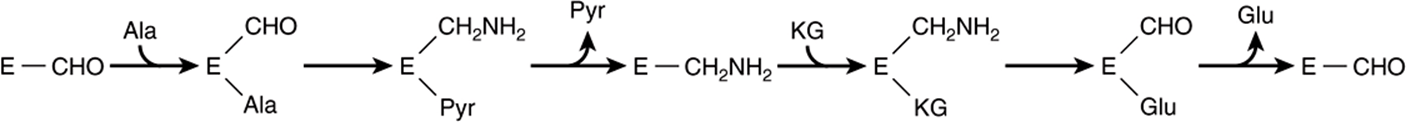
FIGURE 28–7 “Ping-pong” mechanism for transamination. E—CHO and E—CH2NH2 represent enzyme-bound pyridoxal phosphate and pyridoxamine phosphate, respectively. (Ala, alanine; Glu, glutamate; KG, α-ketoglutarate; Pyr, pyruvate.)
Transamination occurs via a “ping-pong” mechanism characterized by the alternate addition of a substrate and release of a product (Figure 28–7). Following removal of its α-amino nitrogen by transamination, the remaining carbon “skeleton” of an amino acid is degraded by pathways discussed in Chapter 29. As noted earlier, certain diseases are associated with elevated serum levels of aminotransferases (see Table 7-2).
Pyridoxal phosphate (PLP), a derivative of vitamin B6 (see Figure 44–12) is present at the catalytic site of all aminotransferases, and plays a key role in catalysis. During transamination, PLP serves as a “carrier” of amino groups. An enzyme-bound Schiff base (Figure 28–8) is formed between the oxo group of enzyme-bound PLP and the α-amino group of an α-amino acid. The Schiff base can rearrange in various ways. In transamination, rearrangement forms an α-keto acid and an enzyme-bound pyridoxamine phosphate. As noted earlier, certain diseases are associated with elevated serum levels of aminotransferases (see Table 7-2).
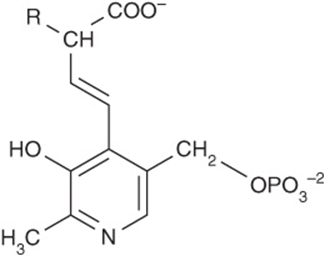
FIGURE 28–8 Structure of a Schiff base formed between pyridoxal phosphate and an amino acid.
L-GLUTAMATE DEHYDROGENASE OCCUPIES A CENTRAL POSITION IN NITROGEN METABOLISM
Transfer of amino nitrogen to α-ketoglutarate forms L-glutamate. Hepatic L-glutamate dehydrogenase (GDH), which can use either NAD+ or NADP+. releases this nitrogen as ammonia (Figure 28–9). Conversion of α-amino nitrogen to ammonia by the concerted action of glutamate aminotransferase and GDH is often termed “transdeamination.” Liver GDH activity is allosterically inhibited by ATP, GTP, and NADH, and is activated by ADP. The GDH reaction is freely reversible, and also functions in amino acid biosynthesis (see Figure 27–1).
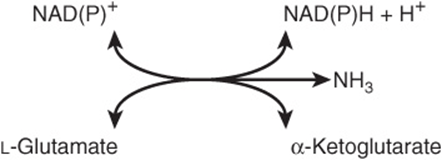
FIGURE 28–9 The L-glutamate dehydrogenase reaction. NAD(P)+ means that either NAD+ or NADP+ can serve as the oxidoreductant. The reaction is reversible, but favors glutamate formation.
Amino Acid Oxidases Remove Nitrogen as Ammonia
While their physiologic importance is uncertain, L-amino acid oxidases of liver and kidney convert an amino acid to an α-imino acid that decomposes to an α-keto acid with release of ammonium ion (Figure 28–10). The reduced flavin is reoxidized by molecular oxygen, forming hydrogen peroxide (H2O2), which then is split to O2 and H2O by catalase.
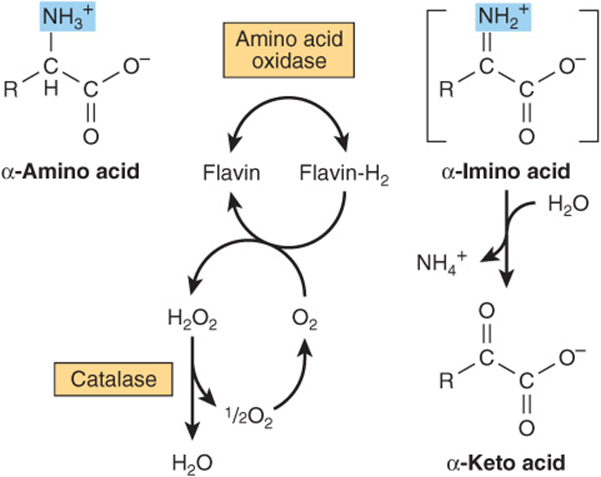
FIGURE 28–10 Oxidative deamination catalyzed by L-amino acid oxidase (L-α-amino acid: O2 oxidoreductase). The α-imino acid, shown in brackets, is not a stable intermediate.
Ammonia Intoxication Is Life Threatening
The ammonia produced by enteric bacteria and absorbed into portal venous blood and the ammonia produced by tissues are rapidly removed from circulation by the liver and converted to urea. Thus, only traces (10-20 μg/dL) normally are present in peripheral blood. This is essential, since ammonia is toxic to the central nervous system. Should portal blood bypass the liver, systemic blood ammonia levels may attain toxic levels. This occurs in severely impaired hepatic function or the development of collateral links between the portal and systemic veins in cirrhosis. Symptoms of ammonia intoxication include tremor, slurred speech, blurred vision, coma, and ultimately death. Ammonia may be toxic to the brain in part because it reacts with α-ketoglutarate to form glutamate. The resulting depletion of levels of α-ketoglutarate then impairs function of the tricarboxylic acid (TCA) cycle in neurons.
Glutamine Synthase Fixes Ammonia as Glutamine
Formation of glutamine is catalyzed by mitochondrial glutamine synthase (Figure 28–11). Since amide bond synthesis is coupled to the hydrolysis of ATP to ADP and Pi, the reaction strongly favors glutamine synthesis. During catalysis, glutamate attacks the γ-phosphoryl group of ATP, forming γ-glutamyl phosphate and ADP. Following deprotonation of NH4+, NH3 attacks γ-glutamyl phosphate, and glutamine and Pi are released. In addition to providing glutamine to serve as a carrier of nitrogen, carbon and energy between organs (Figure 28–2), glutamine synthase plays a major role in ammonia detoxification and acid-base homeostasis. A rare deficiency in neonate glutamine synthase results in severe brain damage, multiorgan failure, and death.
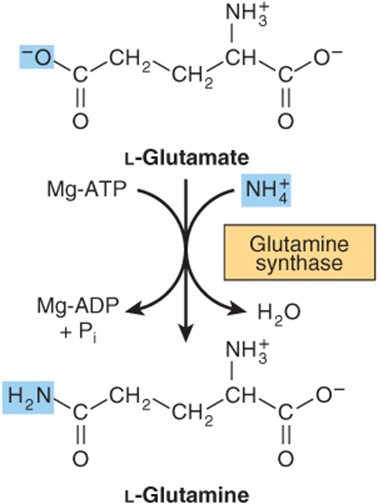
FIGURE 28–11 The glutamine synthase reaction strongly favors glutamine synthesis.
Glutaminase & Asparaginase Deamidate Glutamine & Asparagine
There are two human isoforms of mitochondrial glutaminase, termed liver-type and renal type glutaminase. Products of different genes, the glutaminases differ with respect to their structure, kinetics, and regulation. Hepatic glutaminase levels rise in response to high protein intake while renal kidney-type glutaminase increases in metabolic acidosis. Hydrolytic release of the amide nitrogen of glutamine as ammonia, catalyzed by glutaminase (Figure 28–12), strongly favors glutamate formation. An analogous reaction is catalyzed by L-asparaginase. The concerted action of glutamine synthase and glutaminase thus catalyzes the interconversion of free ammonium ion and glutamine.
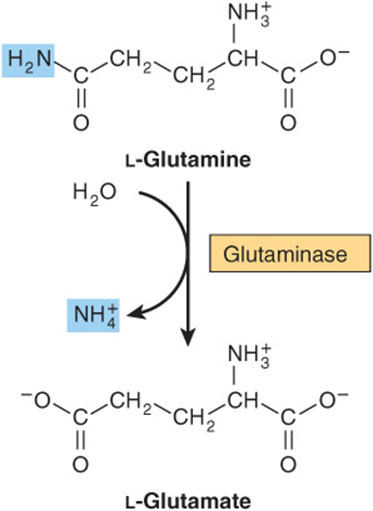
FIGURE 28–12 The glutaminase reaction proceeds essentially irreversibly in the direction of glutamate and NH4+ formation. Note that the amide nitrogen, not the α-amino nitrogen, is removed.
Formation & Secretion of Ammonia Maintains Acid-Base Balance
Excretion into urine of ammonia produced by renal tubular cells facilitates cation conservation and regulation of acid-base balance. Ammonia production from intracellular renal amino acids, especially glutamine, increases in metabolic acidosis and decreases in metabolic alkalosis.
UREA IS THE MAJOR END PRODUCT OF NITROGEN CATABOLISM IN HUMANS
Synthesis of 1 mol of urea requires 3 mol of ATP, 1 mol each of ammonium ion and of aspartate, and employs five enzymes (Figure 28–13). Of the six participating amino acids, N-acetylglutamate functions solely as an enzyme activator. The others serve as carriers of the atoms that ultimately become urea. The major metabolic role of ornithine, citrulline, and argininosuccinate in mammals is urea synthesis. Urea synthesis is a cyclic process. While ammonium ion, CO2, ATP, and aspartate are consumed, the ornithine consumed in reaction 2 is regenerated in reaction 5. There thus is no net loss or gain of ornithine, citrulline, argininosuccinate, or arginine. Some reactions of urea synthesis occur in the matrix of the mitochondrion, and other reactions in the cytosol (Figure 28–13).
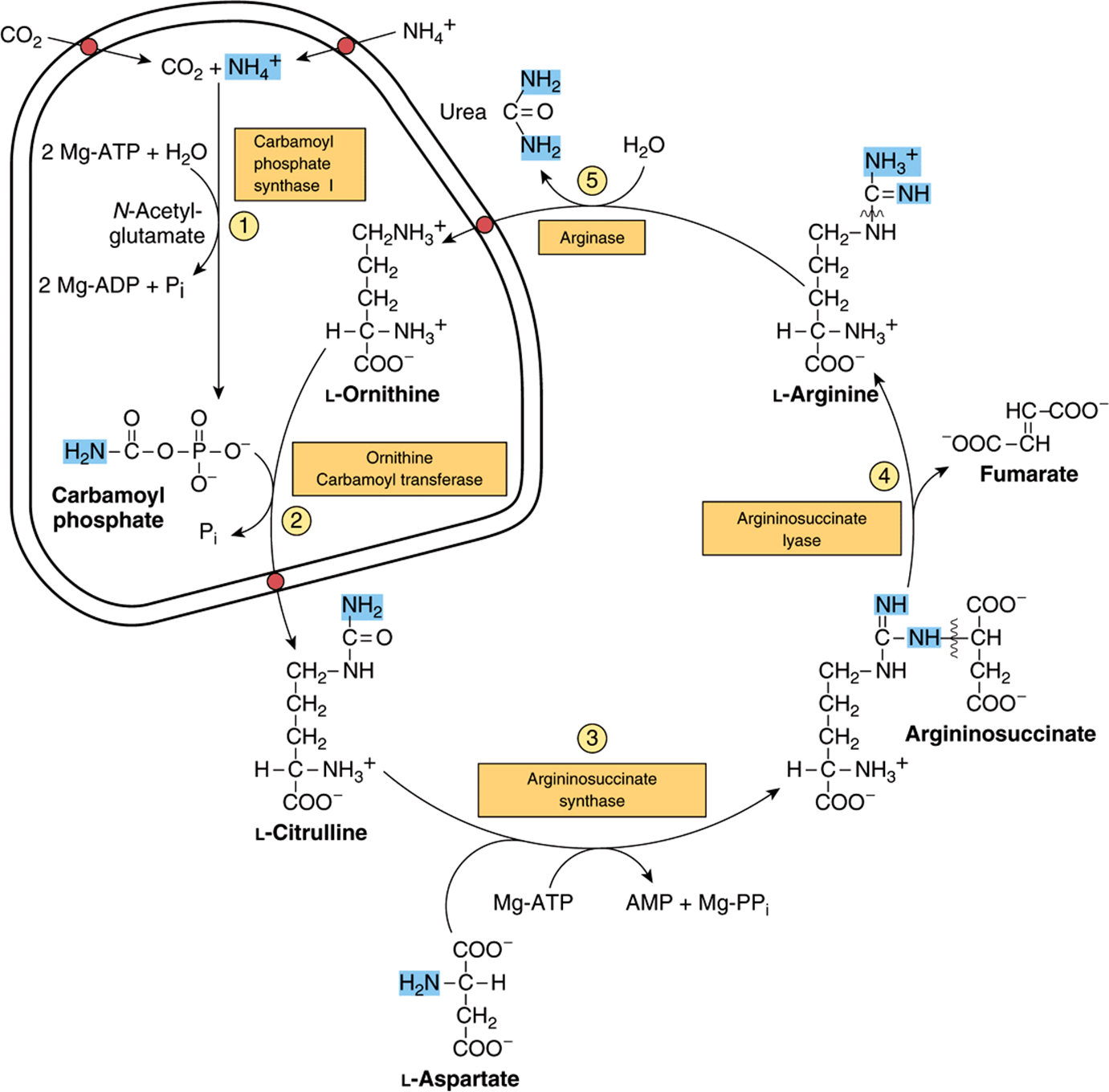
FIGURE 28–13 Reactions and intermediates of urea biosynthesis. The nitrogen-containing groups that contribute to the formation of urea are shaded. Reactions ![]() and
and ![]() occur in the matrix of liver mitochondria and reactions
occur in the matrix of liver mitochondria and reactions ![]() ,
, ![]() , and
, and ![]() in liver cytosol. CO2 (as bicarbonate), ammonium ion, ornithine, and citrulline enter the mitochondrial matrix via specific carriers (see red dots) present in the inner membrane of liver mitochondria.
in liver cytosol. CO2 (as bicarbonate), ammonium ion, ornithine, and citrulline enter the mitochondrial matrix via specific carriers (see red dots) present in the inner membrane of liver mitochondria.
Carbamoyl Phosphate Synthase I Initiates Urea Biosynthesis
Condensation of CO2, ammonia, and ATP to form carbamoyl phosphate is catalyzed by mitochondrial carbamoyl phosphate synthase I. A cytosolic form of this enzyme, carbamoyl phosphate synthase II, uses glutamine rather than ammonia as the nitrogen donor and functions in pyrimidine biosynthesis (see Figure 33–9). The concerted action of glutamate dehydrogenase and carbamoyl phosphate synthase 1 thus shuttles amino nitrogen into carbamoyl phosphate, a compound with high group transfer potential.
Carbamoyl phosphate synthase I, the rate-limiting enzyme of the urea cycle, is active only in the presence of N-acetylglutamate, an allosteric activator that enhances the affinity of the synthase for ATP. Synthesis of 1 mol of carbamoyl phosphate requires 2 mol of ATP. One ATP serves as the phosphoryl donor for formation of the mixed acid anhydride bond of carbamoyl phosphate. The second ATP provides the driving force for synthesis of the amide bond of carbamoyl phosphate. The other products are 2 mol of ADP and 1 mol of Pi (reaction 1, Figure 28–13). The reaction proceeds stepwise. Reaction of bicarbonate with ATP forms carbonyl phosphate and ADP. Ammonia then displaces ADP, forming carbamate and orthophosphate. Phosphorylation of carbamate by the second ATP then forms carbamoyl phosphate.
Carbamoyl Phosphate Plus Ornithine Forms Citrulline
L-Ornithine transcarbamoylase catalyzes transfer of the carbamoyl group of carbamoyl phosphate to ornithine, forming citrulline and orthophosphate (reaction 2, Figure 28–13). While the reaction occurs in the mitochondrial matrix, both the formation of ornithine and the subsequent metabolism of citrulline take place in the cytosol. Entry of ornithine into mitochondria and exodus of citrulline from mitochondria therefore involve mitochondrial inner membrane permeases (Figure 28–13).
Citrulline Plus Aspartate Forms Argininosuccinate
Argininosuccinate synthase links aspartate and citrulline via the amino group of aspartate (reaction 3, Figure 28–13) and provides the second nitrogen of urea. The reaction requires ATP and involves intermediate formation of citrullyl-AMP. Subsequent displacement of AMP by aspartate then forms argininosuccinate.
Cleavage of Argininosuccinate Forms Arginine & Fumarate
Cleavage of argininosuccinate is catalyzed by argininosuccinate lyase. The reaction proceeds with retention of all three nitrogens in arginine and release of the aspartate skeleton as fumarate (reaction 4, Figure 28–13). Subsequent addition of water to fumarate forms L-malate, whose subsequent NAD+-dependent oxidation forms oxaloacetate. These two reactions are analogous to reactions of the citric acid cycle (see Figure 17–3), but are catalyzed by cytosolicfumarase and malate dehydrogenase. Transamination of oxaloacetate by glutamate aminotransferase then re-forms aspartate. The carbon skeleton of aspartate-fumarate thus acts as a carrier of the nitrogen of glutamate into a precursor of urea.
Cleavage of Arginine Releases Urea & Re-Forms Ornithine
Hydrolytic cleavage of the guanidino group of arginine, catalyzed by liver arginase, releases urea (reaction 5, Figure 28–13). The other product, ornithine, reenters liver mitochondria and participates in additional rounds of urea synthesis. Ornithine and lysine are potent inhibitors of arginase, and compete with arginine. Arginine also serves as the precursor of the potent muscle relaxant nitric oxide (NO) in a Ca2+-dependent reaction catalyzed by NO synthase (see Figure 49–15).
Carbamoyl Phosphate Synthase I Is the Pacemaker Enzyme of the Urea Cycle
The activity of carbamoyl phosphate synthase I is determined by N-acetylglutamate, whose steady-state level is dictated by the balance between its rate of synthesis from acetyl-CoA and glutamate and its rate of hydrolysis to acetate and glutamate, reactions catalyzed by N-acetylglutamate synthase (NAGS) and N-acetylglutamate hydrolase, respectively.
![]()
Major changes in diet can increase the concentrations of individual urea cycle enzymes 10- to 20-fold. For example, starvation elevates enzyme levels, presumably to cope with the increased production of ammonia that accompanies enhanced starvation-induced degradation of protein.
GENERAL FEATURES OF METABOLIC DISORDERS
The comparatively rare, but well-characterized and medically devastating metabolic disorders associated with the enzymes of urea biosynthesis illustrate the following general principles of inherited metabolic diseases.
1. Similar or identical clinical signs and symptoms can characterize various genetic mutations in a gene that encodes a given enzyme or in enzymes that catalyze successive reactions in a metabolic pathway.
2. Rational therapy is based on an understanding of the relevant biochemical enzyme-catalyzed reactions in both normal and impaired individuals.
3. The identification of intermediates and of ancillary products that accumulate prior to a metabolic block provides the basis for metabolic screening tests that can implicate the reaction that is impaired.
4. Definitive diagnosis involves quantitative assay of the activity of the enzyme suspected to be defective.
5. The DNA sequence of the gene that encodes a given mutant enzyme is compared to that of the wild-type gene to identify the specific mutation(s) that cause the disease.
6. The exponential increase in DNA sequencing of human genes has identified dozens of mutations of an affected gene that are benign or are associated with symptoms of varying severity of a given metabolic disorder.
METABOLIC DISORDERS ARE ASSOCIATED WITH EACH REACTION OF THE UREA CYCLE
Defects in each enzyme of the urea cycle have been described. Many of the causative mutations have been mapped, and specific defects in the encoded enzymes have been identified. Five well-documented diseases represent defects in the biosynthesis of enzymes of the urea cycle. Molecular genetic analysis has pinpointed the loci of mutations associated with each deficiency, each of which exhibits considerable genetic and phenotypic variability (Table 28-1).
TABLE 28–1 Enzymes of Inherited Metabolic Disorders of the Urea Cycle
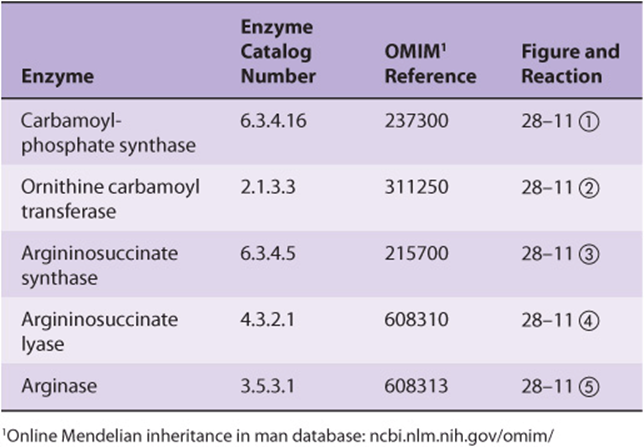
Urea cycle disorders are characterized by hyperammonemia, encephalopathy, and respiratory alkalosis. Four of the five metabolic diseases, deficiencies of carbamoyl phosphate synthase, ornithine carbamoyl transferase, argininosuccinate synthase, and argininosuccinate lyase, result in the accumulation of precursors of urea, principally ammonia and glutamine. Ammonia intoxication is most severe when the metabolic block occurs at reactions 1 or 2 (Figure 28–13), for if citrulline can be synthesized, some ammonia has already been removed by being covalently linked to an organic metabolite.
Clinical symptoms common to all urea cycle disorders include vomiting, avoidance of high-protein foods, intermittent ataxia, irritability, lethargy, and severe mental retardation. The most dramatic clinical presentation occurs in full-term infants who initially appear normal, then exhibit progressive lethargy, hypothermia, and apnea due to high plasma ammonia levels. The clinical features and treatment of all five disorders are similar. Significant improvement and minimization of brain damage can accompany a low-protein diet ingested as frequent small meals to avoid sudden increases in blood ammonia levels. The goal of dietary therapy is to provide sufficient protein, arginine, and energy to promote growth and development while simultaneously minimizing the metabolic perturbations.
Carbamoyl Phosphate Synthase I
N-Acetylglutamate is essential for the activity of carbamoyl phosphate synthase I (reaction 1, Figure 28–13). Defects in carbamoyl phosphate synthase I are responsible for the relatively rare (estimated frequency 1:62,000) metabolic disease termed “hyperammonemia type 1.”
N-Acetylglutamate Synthase
N-Acetylglutamate synthase (NAGS) catalyzes the formation from acetyl-CoA and glutamate of the N-acetylglutamate essential for carbamoyl phosphate synthase I activity.
L-glutamate + acetyl-CoA → N-acetyl-L-glutamate + CoASH
While the clinical and biochemical features of NAGS deficiency are indistinguishable from those arising from a defect in carbamoyl phosphate synthase I, a deficiency in NAGS may respond to administered N-acetylglutamate.
Ornithine Permease
The hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome (HHH syndrome) results from mutation of the ORNT1 gene that encodes the mitochondrial membrane ornithine permease. The failure to import cytosolic ornithine into the mitochondrial matrix renders the urea cycle inoperable, with consequent hyperammonemia, and hyperornithinemia due to the accompanying accumulation of cytosolic ornithine. In the absence of its normal acceptor (ornithine), mitochondrial carbamoyl phosphate carbamoylates lysine to homocitrulline, resulting in homocitrullinuria.
Ornithine Transcarbamoylase
The X-chromosome linked deficiency termed “hyperammonemia type 2” reflects a defect in ornithine transcarbamoylase (reaction 2, Figure 28–13). The mothers also exhibit hyperammonemia and an aversion to high-protein foods. Levels of glutamine are elevated in blood, cerebrospinal fluid, and urine, probably as a result of enhanced glutamine synthesis in response to elevated levels of tissue ammonia.
Argininosuccinate Synthase
In addition to patients who lack detectable argininosuccinate synthase activity (reaction 3, Figure 28–13), a 25-fold elevated Km for citrulline has been reported. In the resulting citrullinemia, plasma and cerebrospinal fluid citrulline levels are elevated, and 1-2 g of citrulline are excreted daily.
Argininosuccinate Lyase
Argininosuccinicaciduria, accompanied by elevated levels of argininosuccinate in blood, cerebrospinal fluid, and urine, is associated with friable, tufted hair (trichorrhexis nodosa). Both early- and late-onset types are known. The metabolic defect is in argininosuccinate lyase (reaction 4, Figure 28–13). Diagnosis by the measurement of erythrocyte argininosuccinate lyase activity can be performed on umbilical cord blood or amniotic fluid cells.
Arginase
Hyperargininemia is an autosomal recessive defect in the gene for arginase (reaction 5, Figure 28–13). Unlike other urea cycle disorders, the first symptoms of hyperargininemia typically do not appear until age 2 to 4 years. Blood and cerebrospinal fluid levels of arginine are elevated. The urinary amino acid pattern, which resembles that of lysine-cystinuria (see Chapter 29), may reflect competition by arginine with lysine and cysteine for reabsorption in the renal tubule.
Analysis of Neonate Blood by Tandem Mass Spectrometry Can Detect Metabolic Diseases
Metabolic diseases caused by the absence or functional impairment of metabolic enzymes can be devastating. Early dietary intervention, however, can in many instances ameliorate the otherwise inevitable dire effects. The early detection of such metabolic diseases is thus is of primary importance. Since the initiation in the United States of newborn screening programs in the 1960s, all states now conduct metabolic screening of newborns, although the scope of screen employed varies among states. The powerful and sensitive technique of tandem mass spectrometry (see Chapter 4) can in a few minutes detect over 40 analytes of significance in the detection of metabolic disorders. Most states employ tandem MS to screen newborns to detect metabolic disorders such as organic acidemias, aminoacidemias, disorders of fatty acid oxidation, and defects in the enzymes of the urea cycle. However, at present there remain significant differences in analyte coverage between states. An article in Clinical Chemistry 2006 39:315 reviews the theory of tandem MS, its application to the detection of metabolic disorders, and situations that can yield false positives, and includes a lengthy table of detectable analytes and the relevant metabolic diseases.
Can Gene Therapy Offer Promise for Correcting Defects in Urea Biosynthesis?
Gene therapy of defects in the enzymes of the urea cycle is an area of active investigation. Despite encouraging results in animal models using an adenoviral vector to treat citrullinemia, at present gene therapy provides no effective solution for human subjects.
SUMMARY
![]() Human subjects degrade 1-2% of their body protein daily at rates that vary widely between proteins and with physiologic state. Key regulatory enzymes often have short half-lives.
Human subjects degrade 1-2% of their body protein daily at rates that vary widely between proteins and with physiologic state. Key regulatory enzymes often have short half-lives.
![]() Proteins are degraded by both ATP-dependent and ATP-independent pathways. Ubiquitin targets many intracellular proteins for degradation. Liver cell surface receptors bind and internalize circulating asialoglycoproteins destined for lysosomal degradation.
Proteins are degraded by both ATP-dependent and ATP-independent pathways. Ubiquitin targets many intracellular proteins for degradation. Liver cell surface receptors bind and internalize circulating asialoglycoproteins destined for lysosomal degradation.
![]() Fish excrete highly toxic NH3 directly. Birds convert NH3 to uric acid. Higher vertebrates convert NH3 to urea.
Fish excrete highly toxic NH3 directly. Birds convert NH3 to uric acid. Higher vertebrates convert NH3 to urea.
![]() Transamination channels amino acid nitrogen into glutamate. GDH occupies a central position in nitrogen metabolism.
Transamination channels amino acid nitrogen into glutamate. GDH occupies a central position in nitrogen metabolism.
![]() Glutamine synthase converts NH3 to nontoxic glutamine. Glutaminase releases NH3 for use in urea synthesis.
Glutamine synthase converts NH3 to nontoxic glutamine. Glutaminase releases NH3 for use in urea synthesis.
![]() NH3, CO2, and the amide nitrogen of aspartate provide the atoms of urea.
NH3, CO2, and the amide nitrogen of aspartate provide the atoms of urea.
![]() Hepatic urea synthesis takes place in part in the mitochondrial matrix and in part in the cytosol.
Hepatic urea synthesis takes place in part in the mitochondrial matrix and in part in the cytosol.
![]() Changes in enzyme levels and allosteric regulation of carbamoyl phosphate synthase I by N-acetylglutamate regulate urea biosynthesis.
Changes in enzyme levels and allosteric regulation of carbamoyl phosphate synthase I by N-acetylglutamate regulate urea biosynthesis.
![]() Metabolic diseases are associated with defects in each enzyme of the urea cycle, of the membrane-associated ornithine permease, and of NAGS.
Metabolic diseases are associated with defects in each enzyme of the urea cycle, of the membrane-associated ornithine permease, and of NAGS.
![]() Tandem mass spectrometry is the technique of choice for screening neonates for inherited metabolic diseases.
Tandem mass spectrometry is the technique of choice for screening neonates for inherited metabolic diseases.
REFERENCES
Brooks P, Fuertes G, Murray RZ, et al: Subcellular localization of proteasomes and their regulatory complexes in mammalian cells. Biochem J 2000;346:155.
Caldovic L, Morizono H, Tuchman M: Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum Mutat 2007;28:754.
Crombez EA, Cederbaum SD: Hyperargininemia due to liver arginase deficiency. Mol Genet Metab 2005;84:243.
Elpeleg O, Shaag A, Ben-Shalom E, et al: N-acetylglutamate synthase deficiency and the treatment of hyperammonemic encephalopathy. Ann Neurol 2002;52:845.
Garg U, Dasouki M: Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry. Clinical and laboratory aspects. Clin Biochem 2006;39:315.
Gyato K, Wray J, Huang ZJ, et al: Metabolic and neuropsychological phenotype in women heterozygous for ornithine transcarbamylase deficiency. Ann Neurol 2004;55:80.
Häberle J, Denecke J, Schmidt E, et al: Diagnosis of N-acetylglutamate synthase deficiency by use of cultured fibroblasts and avoidance of nonsense-mediated mRNA decay. J Inherit Metab Dis 2003;26:601.
Häberle J, Görg B, Rutsch F, et al: Congenital glutamine deficiency with glutamine synthetase mutations. N Engl J Med 2005;353:1926.
Häberle J, Pauli S, Schmidt E, et al: Mild citrullinemia in caucasians is an allelic variant of argininosuccinate synthetase deficiency (citrullinemia type 1). Mol Genet Metab 2003;80:302.
Iyer R, Jenkinson CP, Vockley JG, et al: The human arginases and arginase deficiency. J Inherit Metab Dis 1998;21:86.
Pickart CM: Mechanisms underlying ubiquitination. Annu Rev Biochem 2001;70:503.
Scriver CR: Garrod’s foresight; our hindsight. J Inherit Metab Dis 2001;24:93.
Scriver CR, Sly WS, Childs B, et al (editors): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, 2001.
Yi JJ, Ehlers MD: Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol Rev 2007;59:206.