Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION III. Metabolism of Proteins & Amino Acids
Chapter 29. Catabolism of the Carbon Skeletons of Amino Acids
Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Name the principal catabolites of the carbon skeletons of the common amino acids and the major metabolic fates of these catabolites.
Name the principal catabolites of the carbon skeletons of the common amino acids and the major metabolic fates of these catabolites.
![]() Write an equation for an aminotransferase (transaminase) reaction and illustrate the role played by the coenzyme.
Write an equation for an aminotransferase (transaminase) reaction and illustrate the role played by the coenzyme.
![]() Outline the metabolic pathways for each of the common amino acids, and identify reactions associated with clinically significant metabolic disorders.
Outline the metabolic pathways for each of the common amino acids, and identify reactions associated with clinically significant metabolic disorders.
![]() Provide examples of aminoacidurias that arise from defects in glomerular tubular reabsorption, and the consequences of impaired intestinal absorption of tryptophan.
Provide examples of aminoacidurias that arise from defects in glomerular tubular reabsorption, and the consequences of impaired intestinal absorption of tryptophan.
![]() Explain why metabolic defects in different enzymes of the catabolism of a specific amino acid can be associated with similar clinical signs and symptoms.
Explain why metabolic defects in different enzymes of the catabolism of a specific amino acid can be associated with similar clinical signs and symptoms.
![]() Describe the implications of a metabolic defect in glutamate-γ-semialdehyde dehydrogenase for the catabolism of proline and of 4-hydroxyproline.
Describe the implications of a metabolic defect in glutamate-γ-semialdehyde dehydrogenase for the catabolism of proline and of 4-hydroxyproline.
![]() Explain how the α-amino nitrogen of proline and of lysine is removed by processes other than transamination.
Explain how the α-amino nitrogen of proline and of lysine is removed by processes other than transamination.
![]() Draw analogies between the reactions that participate in the catabolism of fatty acids and of the branched-chain amino acids.
Draw analogies between the reactions that participate in the catabolism of fatty acids and of the branched-chain amino acids.
![]() Identify the specific metabolic defects in hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
Identify the specific metabolic defects in hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
BIOMEDICAL IMPORTANCE
The prior chapter described the removal and metabolic fate of the nitrogen atoms of the common L-α-amino acids. This chapter will address the metabolic fates of the resulting hydrocarbon skeletons of these amino acids. Discussed are the enzymes and intermediates formed during the conversion of the carbon skeletons to amphibolic intermediates, and several metabolic diseases or “inborn errors of metabolism” associated with these processes. While most disorders of amino acid catabolism are rare, if left untreated they can result in irreversible brain damage and early mortality. Prenatal or early postnatal detection of metabolic disorders and timely initiation of treatment thus are essential. The ability to detect the activities of enzymes in cultured amniotic fluid cells facilitates prenatal diagnosis by amniocentesis. All states now conduct screening tests of newborns for as many as 30 metabolic diseases. These tests include, but are not limited to, disorders associated with defects in the catabolism of amino acids. The most reliable screening tests use tandem mass spectrometry to detect, in a few drops of neonate blood, catabolites suggestive of a given metabolic defect. The metabolites detected pinpoint the metabolic defect as the lowered or absent activity of a given enzyme. Treatment consists primarily of feeding diets low in the amino acid whose catabolism is impaired.
Mutations in the exons or in the regulatory regions of a gene that encodes an enzyme of amino acid metabolism can result in the failure to synthesize that enzyme or in the synthesis of a partially or completely nonfunctional enzyme. Mutations may have no significant effect of the activity of the encoded enzyme. By contrast, mutations that compromise the overall three-dimensional structure or the structure of catalytic or regulatory sites may be associated with adverse metabolic consequences. Low catalytic efficiency of a mutant enzyme can result from impaired positioning of residues involved in catalysis, or in binding a substrate, coenzyme, or metal ion. Mutations may also impair the ability of certain enzymes to respond appropriately to the signals that modulate their activity by altering an enzyme’s affinity for an allosteric regulator of activity. Since different mutations can have similar effects on any of the above factors, various mutations may give rise to the same clinical signs and symptoms. At a molecular level, these therefore are distinct molecular diseases. To supplement the disorders of amino acid metabolism discussed in this chapter, readers should consult major reference works on this topic such as Scriver et al 2001.
AMINO ACIDS ARE CATABOLIZED TO INTERMEDIATES FOR CARBOHYDRATE AND LIPID BIOSYNTHESIS
Nutritional studies in the period 1920-1940, reinforced and confirmed by studies using isotopically labeled amino acids conducted from 1940 to 1950, established the interconvertibility of the carbon atoms of fat, carbohydrate, and protein. These studies also revealed that all or a portion of the carbon skeleton of every amino acid is convertible either to carbohydrate (13 amino acids), fat (one amino acid), or both fat and carbohydrate (five amino acids) (Table 29-1). Figure 29–1 outlines overall aspects of these interconversions.
TABLE 29–1 Fate of the Carbon Skeletons of the Common L-α-Amino Acids
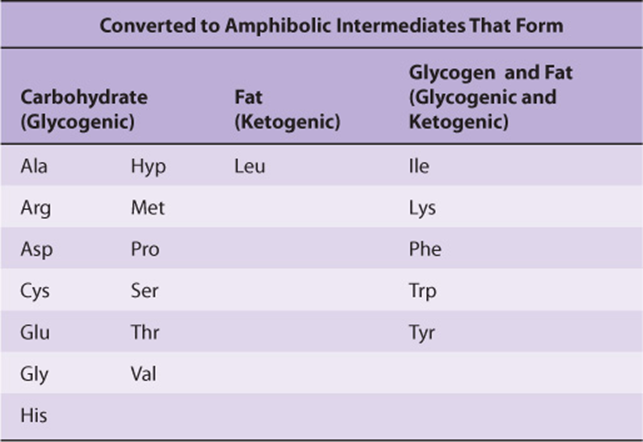
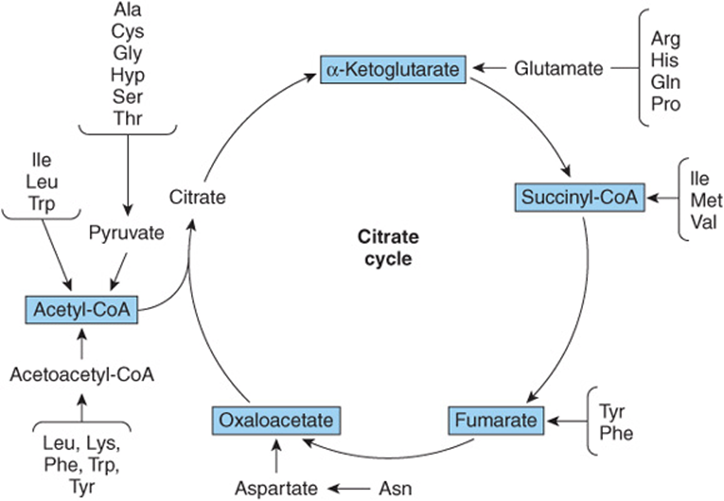
FIGURE 29–1 Overview of the amphibolic intermediates that result from catabolism of the common amino acids.
TRANSAMINATION TYPICALLY INITIATES AMINO ACID CATABOLISM
Removal of α-amino nitrogen by transamination, a reaction catalyzed by an aminotransferase or transaminase (see Figure 28–6), is the first catabolic reaction of all the common amino acids except proline, hydroxyproline, threonine, or lysine. The hydrocarbon skeleton that remains is then degraded to amphibolic intermediates as outlined in Figure 29–1.
Asparagine and Aspartate Form Oxaloacetate
All four carbons of asparagine and of aspartate form oxaloacetate via reactions catalyzed by asparaginase and a transaminase (Figure 29–2, top). Metabolic defects in transaminases, which fulfill central amphibolic functions, may be incompatible with life. Consequently, no known metabolic defect is associated with this short catabolic pathway.
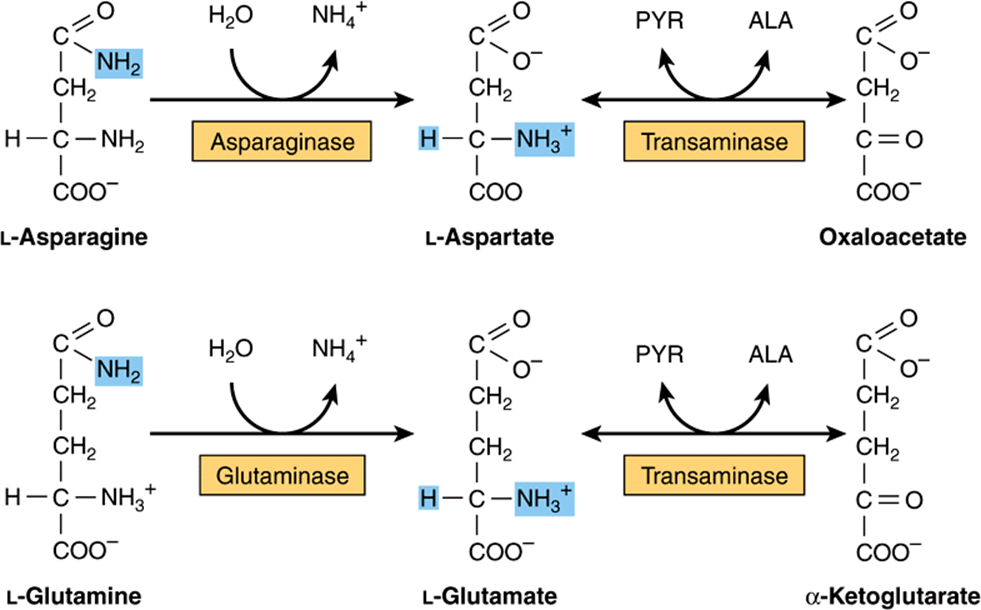
FIGURE 29–2 Catabolism to amphibolic intermediates of L-asparagine (top) and of L-glutamine (bottom). (PYR, pyruvate; ALA, L-alanine.) In this and subsequent figures, blue highlights emphasize the portions of the molecules that are undergoing chemical change.
Glutamine and Glutamate Form α-Ketoglutarate
The catabolism of glutamine and of glutamate parallels that of asparagine and aspartate in reactions catalyzed by glutaminase and transaminase that forms α-ketoglutarate (Figure 29–2, bottom). While both glutamate and aspartate are substrates for the same transaminase, deamidation of their corresponding amides is catalyzed by different enzymes: asparaginase and glutaminase. Possibly for the reason stated earlier, there are no known metabolic defects of the glutamine-glutamate catabolic pathway.
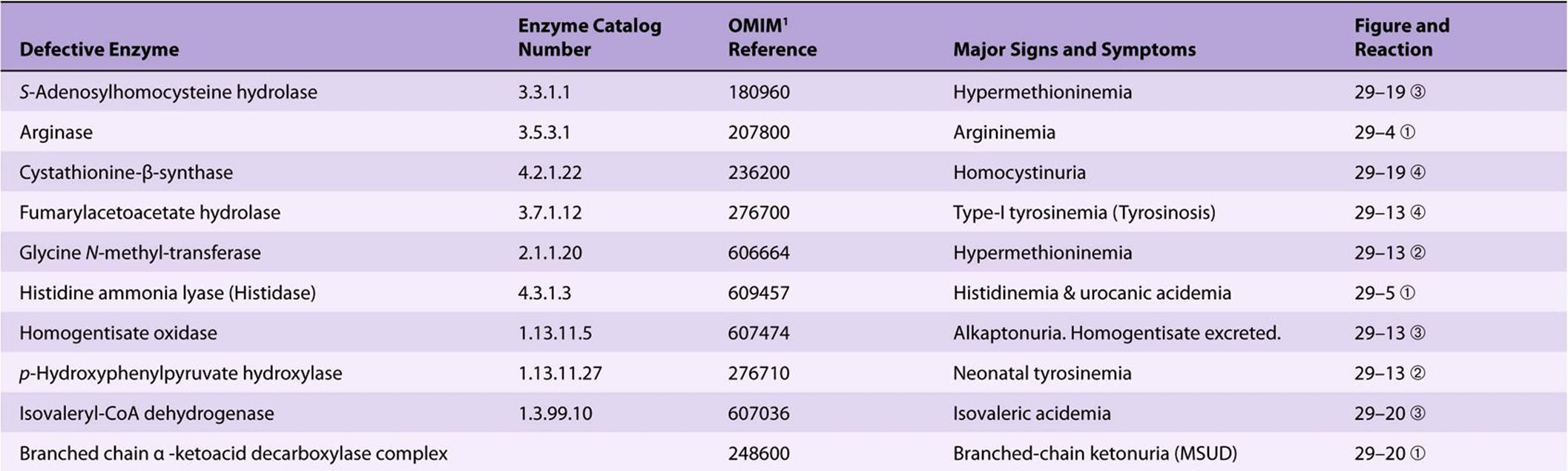
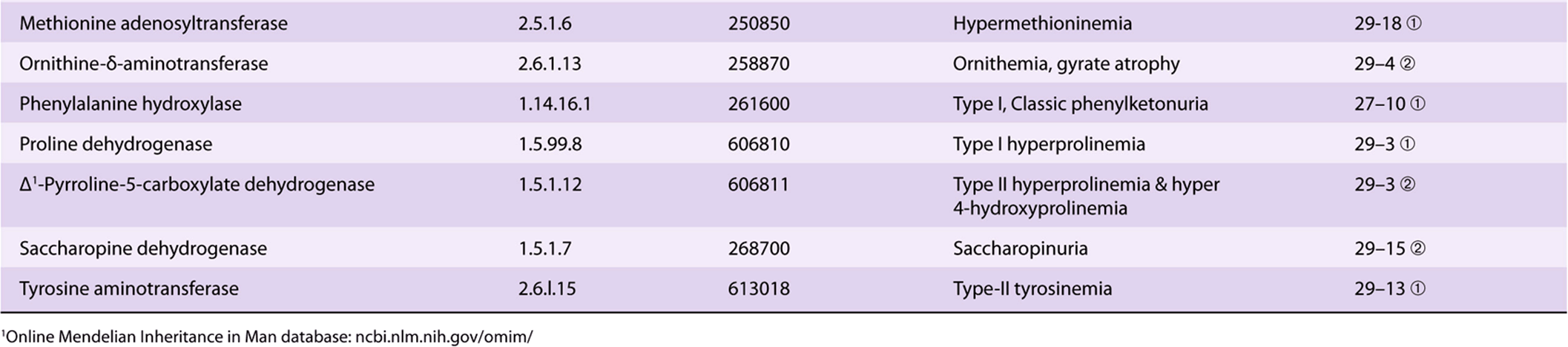
Significant metabolic disorders are, however, associated with the catabolism of many other amino acids. Discussed below under the catabolism of each amino acid, these disorders are summarized in Table 29-2. This table lists the impaired enzyme, its IUB enzyme catalog (EC) number, a cross-reference to a specific figure and numbered reaction, and a numerical link to the Online Mendelian Inheritance in Man database (OMIM).
TABLE 29–2 Metabolic Diseases of Amino Acid Metabolism
Proline
The catabolism of proline takes place in mitochondria. Since proline does not participate in transamination, the nitrogen of this imino acid is retained throughout its oxidation to Δ1-pyrrolline-5-carboxylate, ring opening to glutamate-γ-semialdehyde, and oxidation to glutamate, and is only removed during transamination of glutamate to a-ketoglutarate (Figure 29–3). There are two metabolic disorders of proline catabolism. Both are inherited as autosomal recessive traits and are consistent with a normal adult life. The metabolic block in type I hyperprolinemia is at proline dehydrogenase. There is no associated impairment of hydroxyproline catabolism. The metabolic block in type II hyperprolinemia is at glutamate-γ-semialdehyde dehydrogenase, a mitochondrial matrix enzyme that also participates in the catabolism of arginine, ornithine, and hydroxyproline (see below). Since proline and hydroxyproline catabolism are affected, both Δ1-pyrroline-5-carboxylate and Δ1-pyrroline-3-hydroxy-5-carboxylate (see Figure 29–12) are excreted.
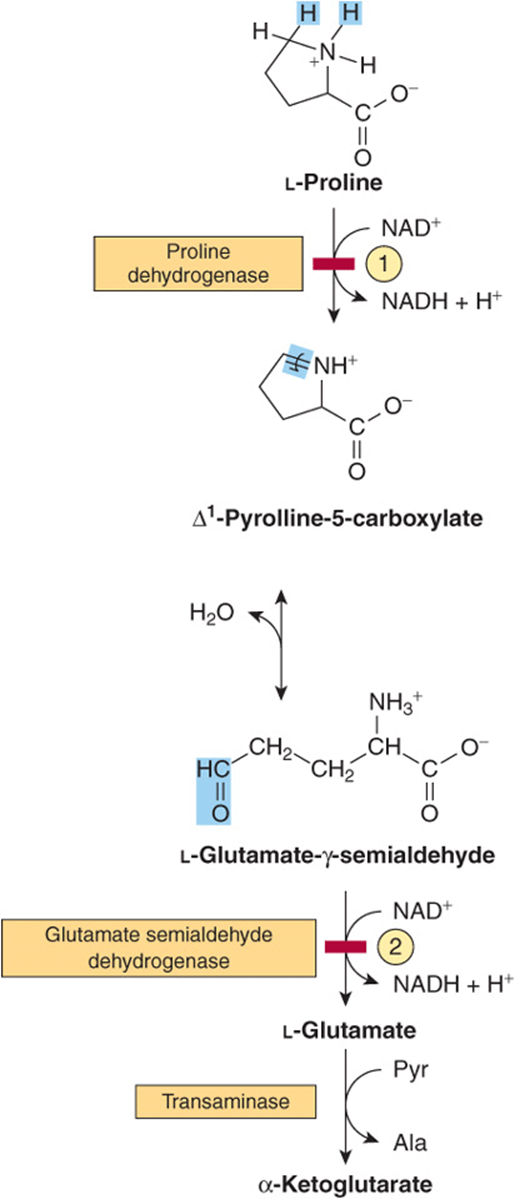
FIGURE 29–3 Catabolism of proline. Red bars and circled numerals indicate the locus of the inherited metabolic defects in ![]() type-I hyperprolinemia and
type-I hyperprolinemia and ![]() type-II hyperprolinemia.
type-II hyperprolinemia.
Arginine and Ornithine
The initial reactions in arginine catabolism are conversion to ornithine followed by transamination of ornithine to glutamate-γ-semialdehyde (Figure 29–4). Subsequent catabolism of glutamate-γ-semialdehyde to α-ketoglutarateoccurs as described for proline (see Figure 29–3). Mutations in ornithine δ-aminotransferase (ornithine transaminase) elevate plasma and urinary ornithine and are associated with gyrate atrophy of the choroid and retina.Treatment involves restricting dietary arginine. In the hyperornithinemia-hyperammonemia syndrome, a defective mitochondrial ornithine-citrulline antiporter (see Figure 28–13) impairs transport of ornithine into mitochondria for use in urea synthesis.
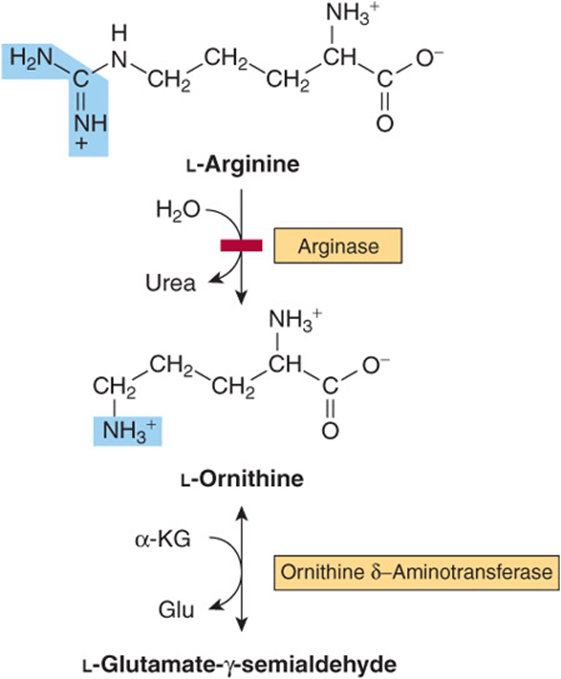
FIGURE 29–4 Catabolism of arginine. Arginase-catalyzed cleavage of L-arginine forms urea and L-ornithine. This reaction (red bar) represents the site of the inherited metabolic defect in hyperargininemia. Subsequent transamination of L-ornithine to glutamate-γ-semialdehyde is followed by conversion to α-ketoglutarate (see Figure 29–3).
Histidine
Catabolism of histidine proceeds via urocanate, 4-imida-zolone-5-propionate, and N-formiminoglutamate (Figlu). Formimino group transfer to tetrahydrofolate forms glutamate, then α-ketoglutarate (Figure 29–5). In folic acid deficiency, transfer of the formimino group is impaired, and Figlu is excreted. Excretion of Figlu following a dose of histidine thus can be used to detect folic acid deficiency. Benign disorders of histidine catabolism include histidinemia and urocanic aciduria associated with impaired histidase.
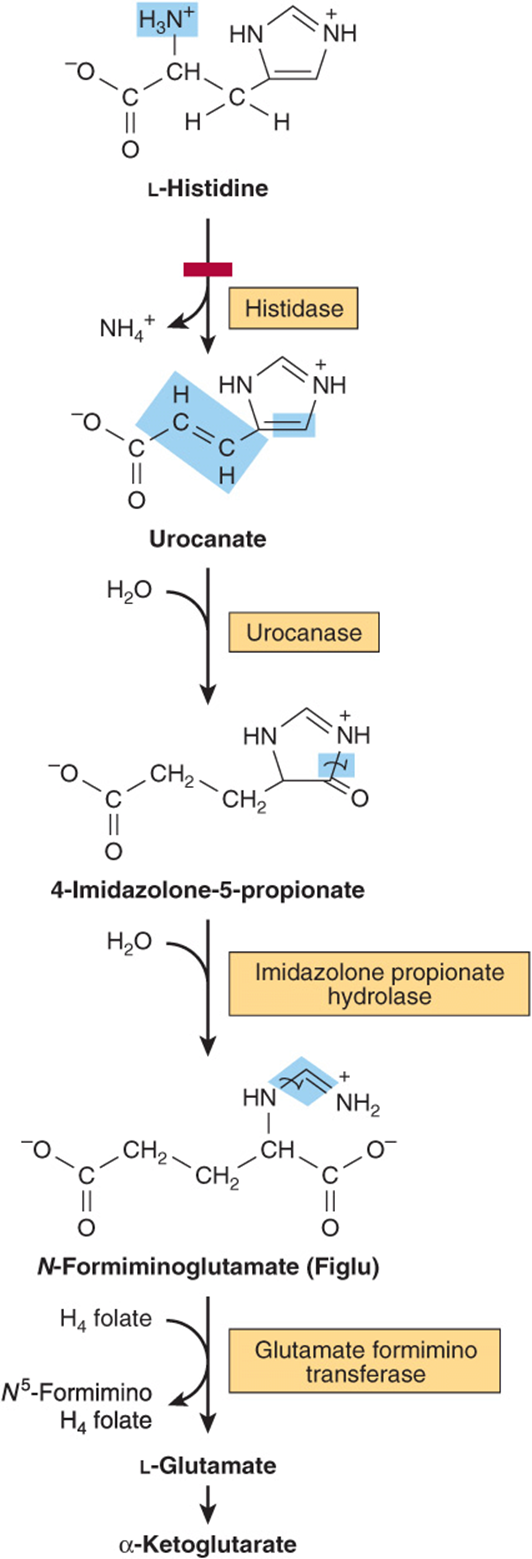
FIGURE 29–5 Catabolism of L-histidine to α-ketoglutarate. (H4 folate, tetrahydrofolate.) The red bar indicates the site of an inherited metabolic defect.
CATABOLISM OF GLYCINE, SERINE, ALANINE, CYSTEINE, THREONINE, AND 4HYDROXYPROLINE
Glycine
The glycine cleavage complex of liver mitochondria splits glycine to CO2 and NH4+ and forms N5, N10-methylene tetrahydrofolate.
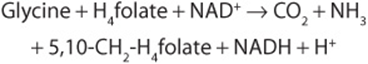
The glycine cleavage system (Figure 29–6) consists of three enzymes and an “H-protein” that has a covalently attached dihydrolipoyl moiety. Figure 29–6 also illustrates the individual reactions and intermediates in glycine cleavage. In nonketotic hyperglycinemia, a rare inborn error of glycine degradation presently known only in Finland, glycine accumulates in all body tissues including the central nervous system. The defect in primary hyperoxaluria is the failure to catabolize glyoxylate formed by the deamination of glycine. Subsequent oxidation of glyoxylate to oxalate results in uro-lithiasis, nephrocalcinosis, and early mortality from renal failure or hypertension. Glycinuria results from a defect in renal tubular reabsorption.
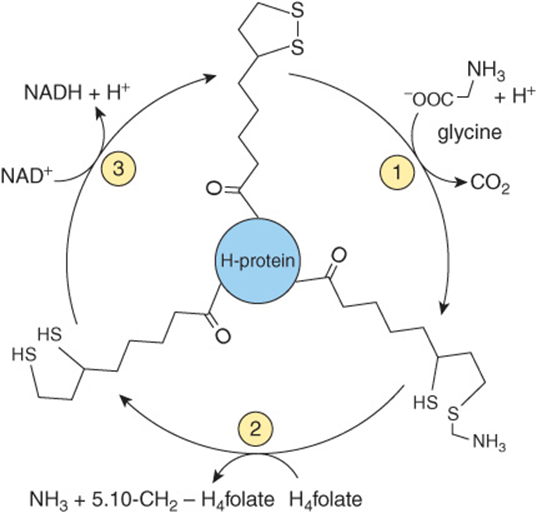
FIGURE 29–6 The glycine cleavage system of liver mitochondria. The glycine cleavage complex consists of three enzymes and an “H-protein” that has covalently attached dihyrolipoate. Catalysts for the numbered reactions are ![]() glycine dehydrogenase (decarboxylating),
glycine dehydrogenase (decarboxylating), ![]() an ammonia-forming aminomethyltransferase, and
an ammonia-forming aminomethyltransferase, and ![]() dihydrolipoamide dehydrogenase. (H4folate, tetrahydrofolate).
dihydrolipoamide dehydrogenase. (H4folate, tetrahydrofolate).
Serine
Following conversion to glycine, catalyzed by serine hydroxymethyltransferase, serine catabolism merges with that of glycine (Figure 29–7).
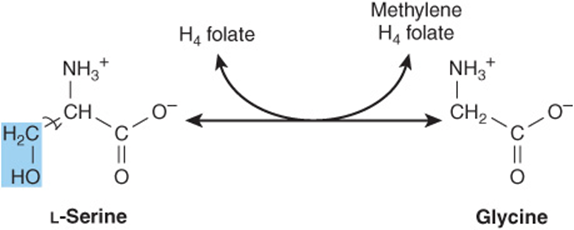
FIGURE 29–7 Interconversion of serine and glycine by serine hydroxymethyltransferase. (H4folate, tetrahydrofolate).
Alanine
Transamination of a-alanine forms pyruvate. Probably on account of its central role in metabolism there is no known metabolic defect of a-alanine catabolism.
Cystine and Cysteine
Cystine is first reduced to cysteine by cystine reductase (Figure 29–8). Two different pathways then convert cysteine to pyruvate (Figure 29–9). There are numerous abnormalities of cysteine metabolism. Cystine, lysine, arginine, and ornithine are excreted in cystine-lysinuria (cystinuria), a defect in renal reabsorption of these amino acids. Apart from cystine calculi, cystinuria is benign. The mixed disulfide of L-cysteine and L-homocysteine (Figure 29–10)excreted by cystinuric patients is more soluble than cystine and reduces formation of cystine calculi.
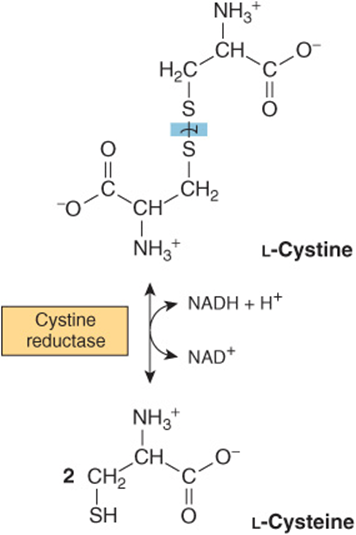
FIGURE 29–8 Reduction of cystine to cysteine in the cystine reductase reaction.
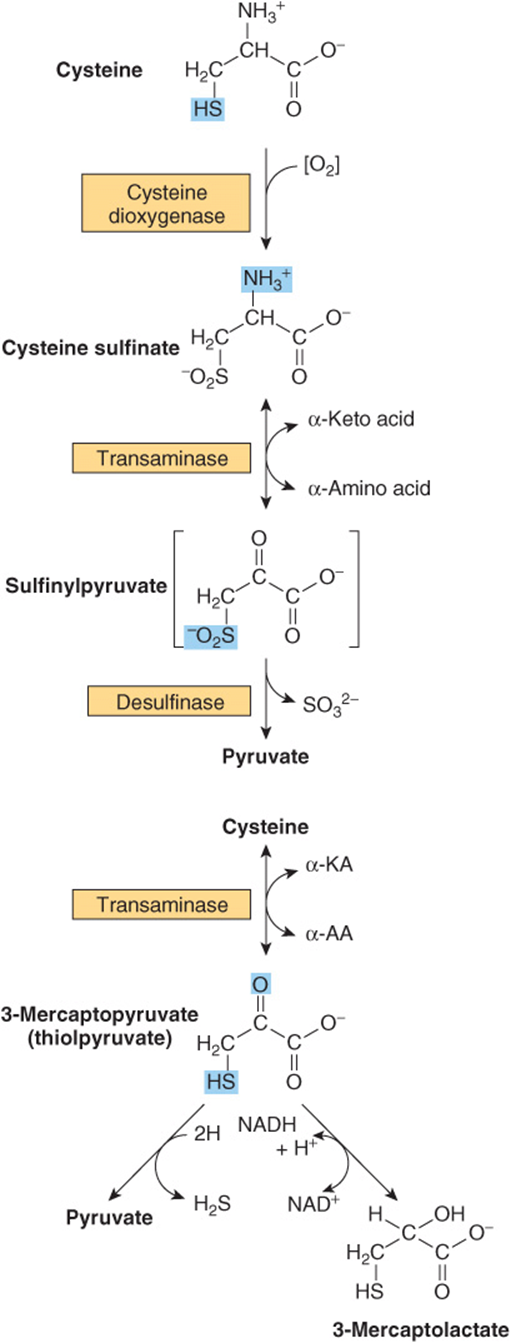
FIGURE 29–9 Two pathways catabolize L-cysteine: the cysteine sulfinate pathway (top) and the 3-mercaptopyruvate pathway (bottom).
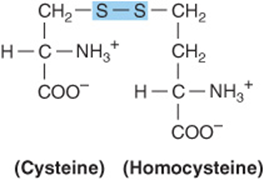
FIGURE 29–10 Structure of the mixed disulfide of cysteine and homocysteine.
Several metabolic defects result in vitamin B6-responsive or vitamin B6-unresponsive homocystinurias. These include a deficiency in the reaction catalyzed by cystathionine β-synthase:
![]()
Consequences include osteoporosis and mental retardation. Defective carrier-mediated transport of cystine results in cystinosis (cystine storage disease) with deposition of cystine crystals in tissues and early mortality from acute renal failure. Epidemiologic and other data link plasma homocysteine levels to cardiovascular risk, but the role of homocysteine as a causal cardiovascular risk factor remains controversial.
Threonine
Threonine aldolase cleaves threonine to acetaldehyde and glycine. Catabolism of glycine is discussed above. Oxidation of acetaldehyde to acetate is followed by formation of acetyl-CoA (Figure 29–11).
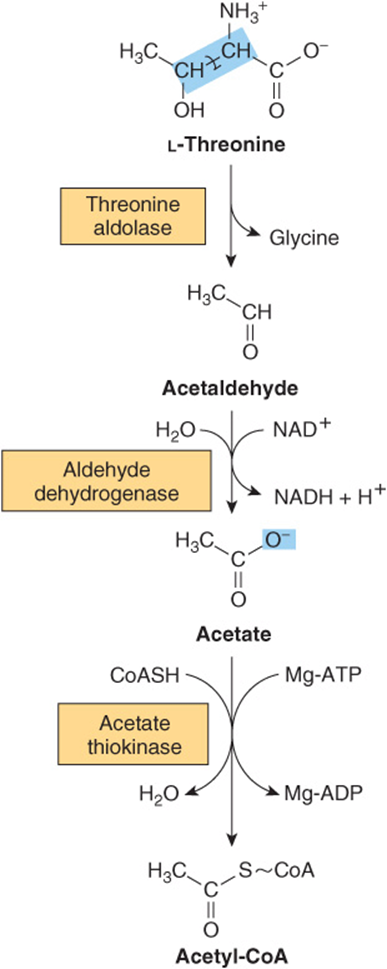
FIGURE 29–11 Intermediates in the conversion of threonine to glycine and acetyl-CoA.
4-Hydroxyproline
Catabolism of 4-hydroxy-L-proline forms, successively, L-Δ1-pyrroline-3-hydroxy-5-carboxylate, β-hydroxy-L-glutamate-β-semialdehyde, erythro-β-hydroxy-L-glutamate, and α-keto-β-hydroxyglutarate. An aldol-type cleavage then forms glyoxylate plus pyruvate (Figure 29–12). A defect in 4-hydroxyproline dehydrogenase results in hyperhy-droxyprolinemia, which is benign. There is no associated impairment of proline catabolism. As noted above under proline, a defect in glutamate-β-semialdehyde dehydrogenase is accompanied by excretion of Δ1 -pyrroline-3-hydroxy-5-carboxylate.
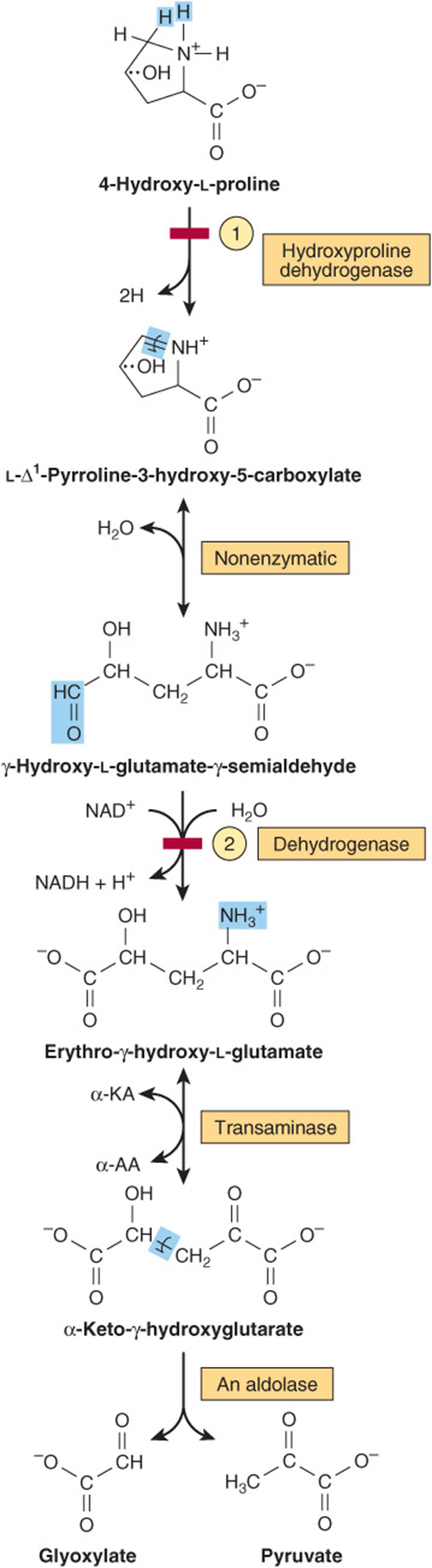
FIGURE 29–12 Intermediates in L-hydroxyproline catabolism. (α-AA, α-amino acid; α-KA, α-keto acid.) Red bars indicate the sites of the inherited metabolic defects in ![]() hyperhydroxyprolinemia and
hyperhydroxyprolinemia and ![]() type II hyperprolinemia.
type II hyperprolinemia.
ADDITIONAL AMINO ACIDS THAT FORM ACETYL-CoA
Tyrosine
Figure 29–13 illustrates the intermediates and enzymes that participate in the catabolism of tyrosine to amphibolic intermediates. Following transamination of tyrosine to p-hydroxyphenylpyruvate, successive reactions form maleylacetoacetate, fumarylacetoacetate, fumarate, acetoacetate, and ultimately acetyl-CoA and acetate.
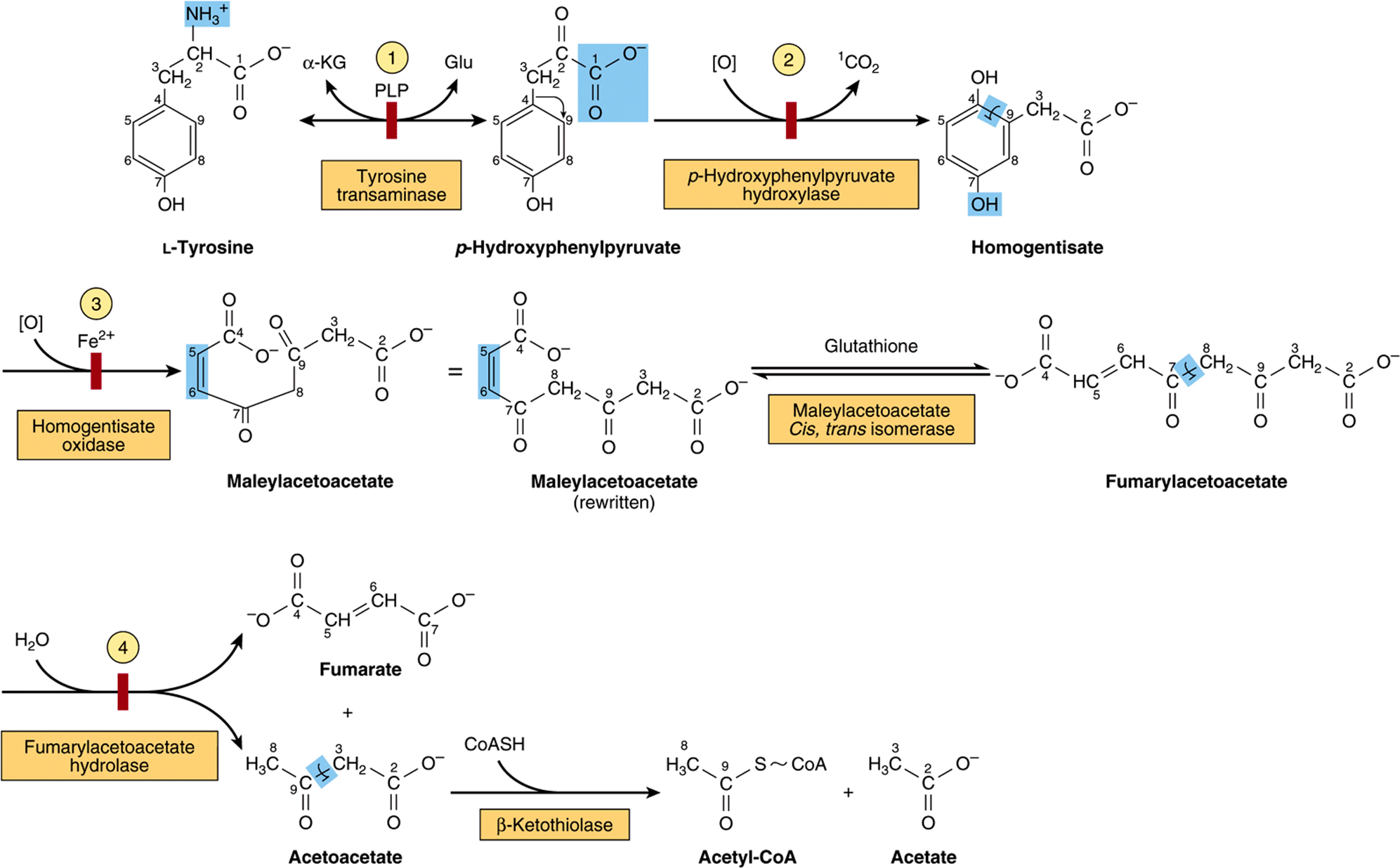
FIGURE 29–13 Intermediates in tyrosine catabolism. Carbons are numbered to emphasize their ultimate fate. (α-KG, α-ketoglutarate; Glu, glutamate; PLP, pyridoxal phosphate.) Red bars indicate the probable sites of the inherited metabolic defects in ![]() type II tyrosinemia;
type II tyrosinemia; ![]() neonatal tyrosinemia;
neonatal tyrosinemia; ![]() alkaptonuria; and
alkaptonuria; and ![]() type I tyrosinemia, or tyrosinosis.
type I tyrosinemia, or tyrosinosis.
Several metabolic disorders are associated with the tyrosine catabolic pathway. The probable metabolic defect in type I tyrosinemia (tyrosinosis) is at fumarylacetoacetate hydrolase (reaction 4, Figure 29–1). Therapy employs a diet low in tyrosine and phenylalanine. Untreated acute and chronic tyrosinosis leads to death from liver failure. Alternate metabolites of tyrosine are also excreted in type II tyrosinemia (Richner-Hanhart syndrome), a defect in tyrosine aminotransferase (reaction 1, Figure 29–13), and in neonatal tyrosinemia, due to the lowered p-hydroxyphenylpyruvate hydroxylase activity (reaction 2, Figure 29–13). Therapy employs a diet low in protein.
The metabolic defect in alkaptonuria is a defective homogentisate oxidase, the enzyme that catalyzes reaction 3 of Figure 29–13. The urine darkens on exposure to air due to oxidation of excreted homogentisate. Late in the disease, there is arthritis and connective tissue pigmentation (ochronosis) due to oxidation of homogentisate to benzoquinone acetate, which polymerizes and binds to connective tissue. First described in the sixteenth century based on the observation that the urine darkened on exposure to air, alkaptonuria provided the basis for Sir Archibald Garrod’s early twentieth century classic ideas concerning heritable metabolic disorders. Based on the presence of ochronosis and on chemical evidence, the earliest known case of alkaptonuria is, however, its 1977 detection in an Egyptian mummy dating from 1500 B.C.
Phenylalanine
Phenylalanine is first converted to tyrosine (see Figure 27–10). Subsequent reactions are those of tyrosine (Figure 29–13). Hyperphenylalaninemias arise from defects in phenylalanine hydroxylase (type I, classic phenylketonuria (PKU), frequency 1 in 10,000 births), in dihydrobiopterin reductase (types II and III), or in dihydrobiopterin biosynthesis (types IV and V) (see Figure 27–10). Alternative catabolites are excreted (Figure 29–14). A diet low in phenylalanine can prevent the mental retardation of PKU.
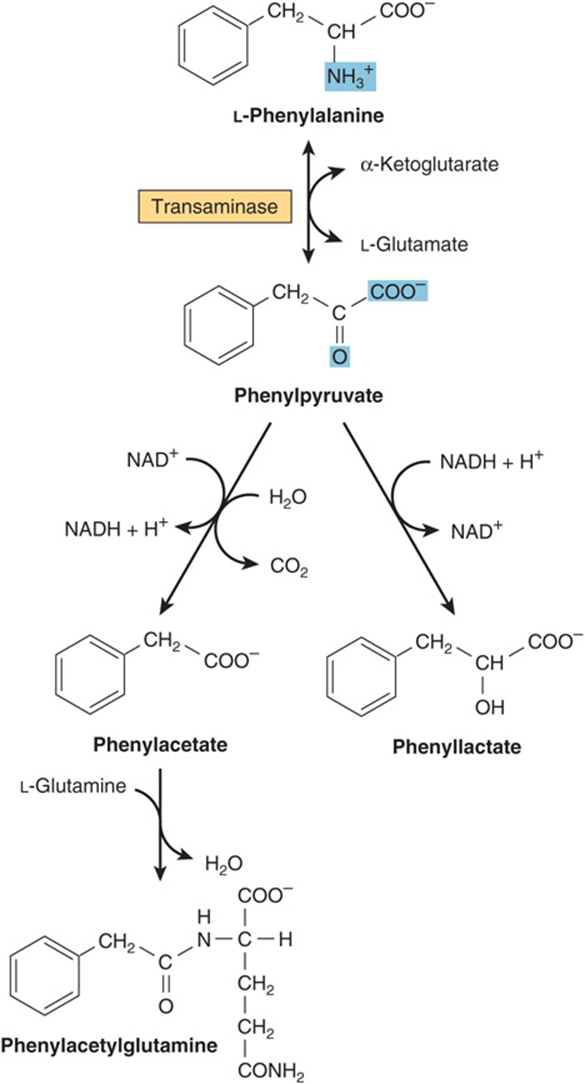
FIGURE 29–14 Alternative pathways of phenylalanine catabolism in phenylketonuria. The reactions also occur in normal liver tissue but are of minor significance.
DNA probes facilitate prenatal diagnosis of defects in phenylalanine hydroxylase or dihydrobiopterin reductase. Elevated blood phenylalanine may not be detectable until 3-4 days postpartum. False-positives in premature infants may reflect delayed maturation of enzymes of phenylalanine catabolism. An older and less reliable screening test employs FeCl3 to detect urinary phenylpyruvate. FeCl3 screening for PKU of the urine of newborn infants is compulsory in many countries, but in the United States has been largely supplanted by tandem mass spectrometry.
Lysine
The first six reactions of L-lysine catabolism in human liver form crotonyl-CoA, which is then degraded to acetyl-CoA by the reactions of fatty acid catabolism (see Figure 22–3). In what follows, circled numerals refer to the corresponding numbered reactions of Figure 29–15. Reactions 1 and 2 convert the Schiff base formed between α-ketoglutarate and the ε-amino group of lysine to L-α-aminoadipate-δ-semialdehde. Reactions 1 and 2 both are catalyzed by a single bifunctional enzyme, aminoadipate semialdehde synthase (also called lysine 2-oxoglutarate reductase-saccharopine dehydrogenase). Reduction of L-α-aminoadipate-δ-semialdehde to L-α-aminoadipate (reaction 3) is followed by transamination to a-ketoadipate (reaction 4). Conversion to the thioester glutaryl-CoA (reaction 5) is followed by the decarboxylation of glutaryl-CoA to crotonyl-CoA (reaction 6). Subsequent reactions are those of the fatty acid catabolism.
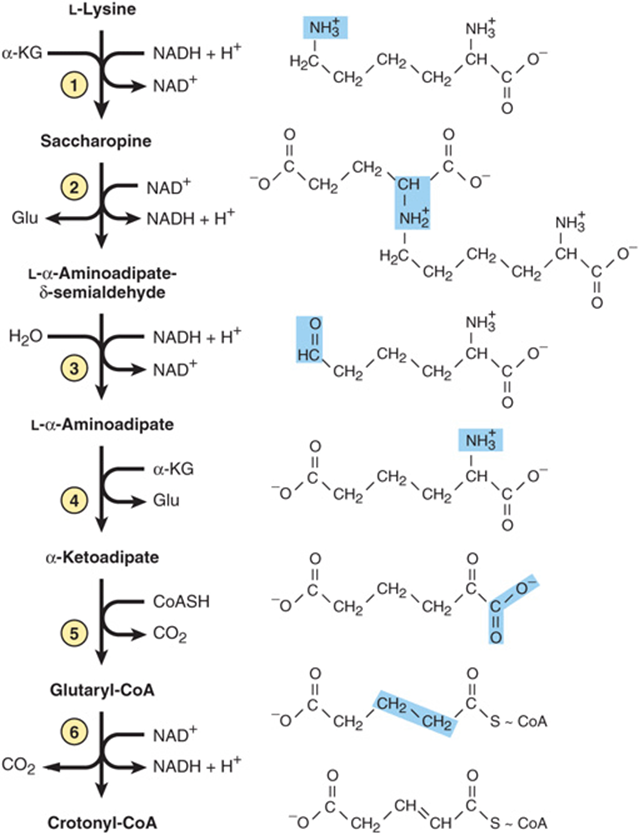
FIGURE 29–15 Reactions and intermediates in the catabolism of L-lysine.
Metabolic defects associated with reactions of the lysine catabolic pathway include hyperlysinemias. Hyperlysinemia can result from a defect in activity 1 or 2 of the bifunctional enzyme aminoadipate semialdehde synthase. Hyperlysinemia is accompanied by elevated levels of blood saccharopine only if the defect involves activity 2. A metabolic defect at reaction 6 results in an inherited metabolic disease that is associated with striatal and cortical degeneration, and is characterized by elevated concentrations of glutarate and its metabolites glutaconate and 3-hydroxyglutarate. The challenge in management of these metabolic defects is to restrict dietary intake of L-lysine without producing malnutrition.
Tryptophan
Tryptophan is degraded to amphibolic intermediates via the kynurenine-anthranilate pathway (Figure 29–16) Tryptophan oxygenase (tryptophan pyrrolase) opens the indole ring, incorporates molecular oxygen, and forms N-formylkynurenine. Tryptophan oxygenase, an iron porphyrin metalloprotein that is inducible in liver by adrenal corticosteroids and by tryptophan, is feedback inhibited by nicotinic acid derivatives, including NADPH. Hydrolytic removal of the formyl group of N-formylkynurenine, catalyzed by kynurenine formylase, produces kynurenine. Since kynureninase requires pyridoxal phosphate, excretion of xanthurenate (Figure 29–17) in response to a tryptophan load is diagnostic of vitamin B6 deficiency. Hartnup disease reflects impaired intestinal and renal transport of tryptophan and other neutral amino acids. Indole derivatives of unabsorbed tryptophan formed by intestinal bacteria are excreted. The defect limits tryptophan availability for niacin biosynthesis and accounts for the pellagra-like signs and symptoms.
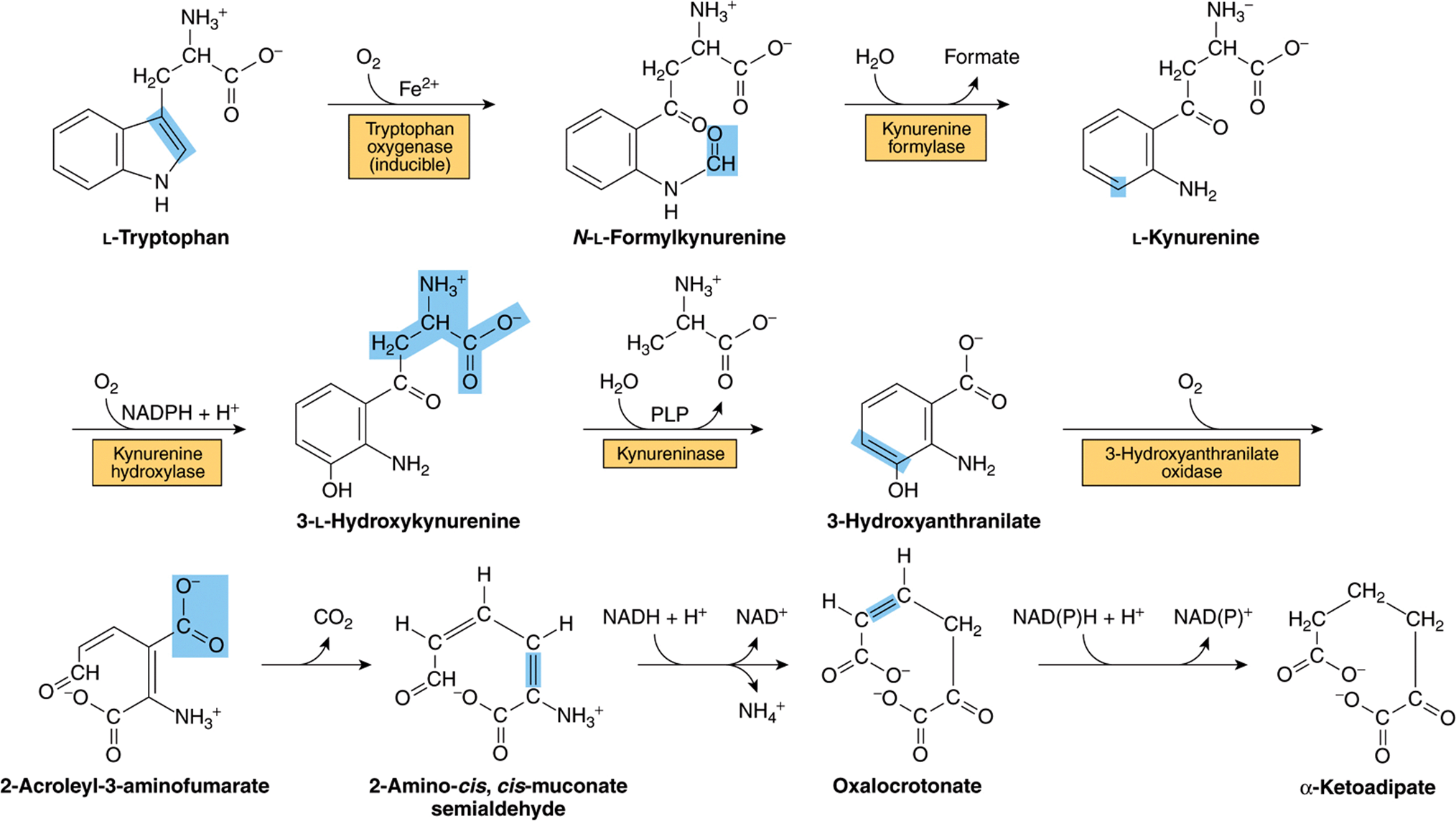
FIGURE 29–16 Reactions and intermediates in the catabolism of L-tryptophan. (PLP, pyridoxal phosphate.)
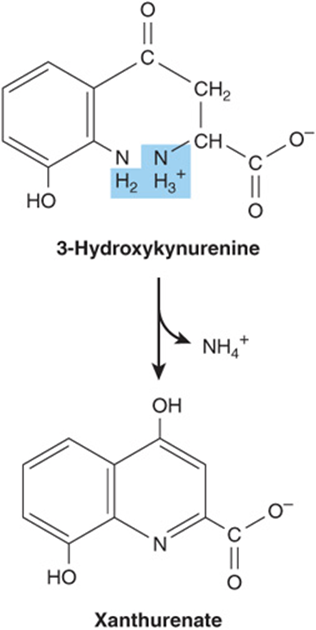
FIGURE 29–17 Formation of xanthurenate in vitamin B6 deficiency. Conversion of the tryptophan metabolite 3-hydroxykynurenine to 3-hydroxyanthranilate is impaired (see Figure 29–16). A large portion is therefore converted to xanthurenate.
Methionine
Methionine reacts with ATP forming S-adenosylmethionine, “active methionine” (Figure 29–18). Subsequent reactions form propionyl-CoA (Figure 29–19), which three subsequent reactions convert to succinyl-CoA (see Figure 20–2).
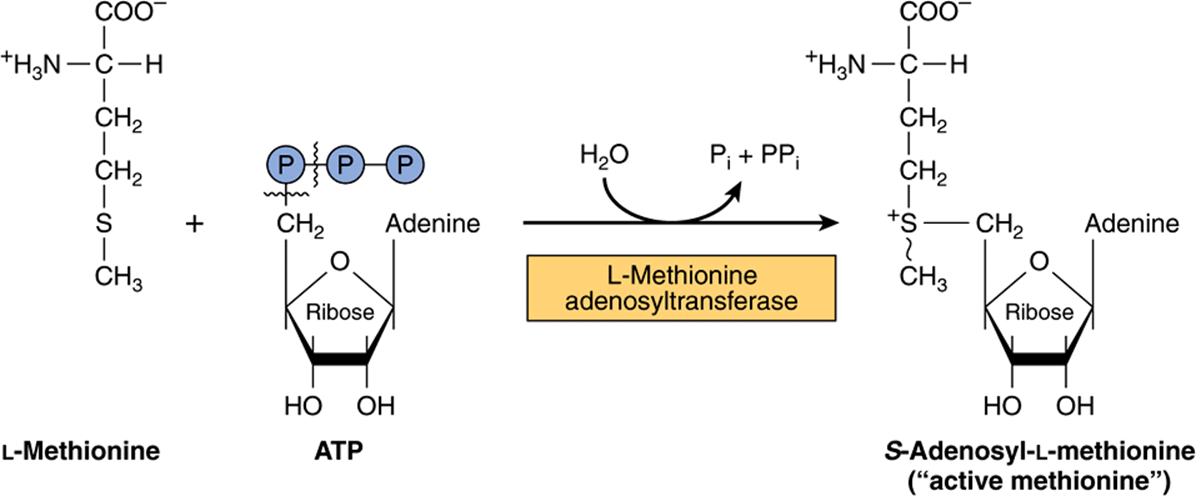
FIGURE 29–18 Formation of S-adenosylmethionine. ~ CH3 represents the high group transfer potential of “active methionine.”
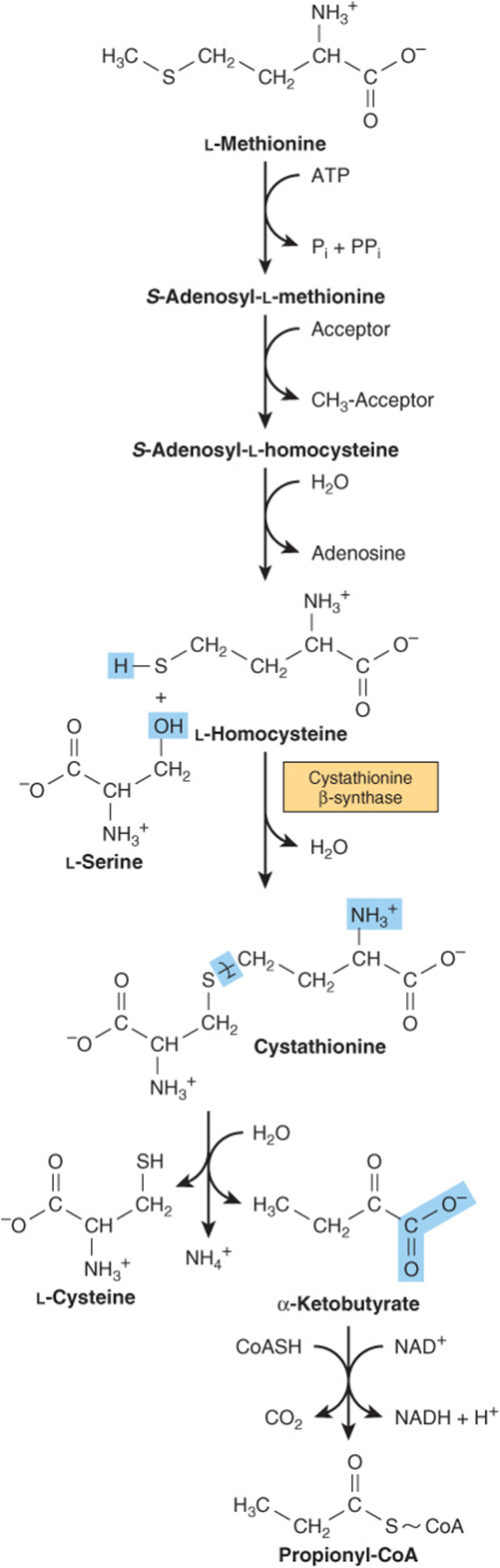
FIGURE 29–19 Conversion of methionine to propionyl-CoA.
THE INITIAL REACTIONS ARE COMMON TO ALL THREE BRANCHED-CHAIN AMINO ACIDS
The first three reactions of the catabolism of isoleucine, leucine, and valine (Figure 29–20) are analogous to reactions of fatty acid catabolism (see Figure 22–3). Following transamination (Figure 29–20, reaction 1), the carbon skeletons of the resulting a-keto acids undergo oxidative decarboxylation and conversion to coenzyme A thioesters. This multistep process is catalyzed by the mitochondrial branched-chain α-keto acid dehydrogenase complex, whose components are functionally identical to those of the pyruvate dehydrogenase complex (PDH) (see Figure 18–5). Like PDH, the branched-chain a-ketoacid dehydrogenase complex consists of five components.
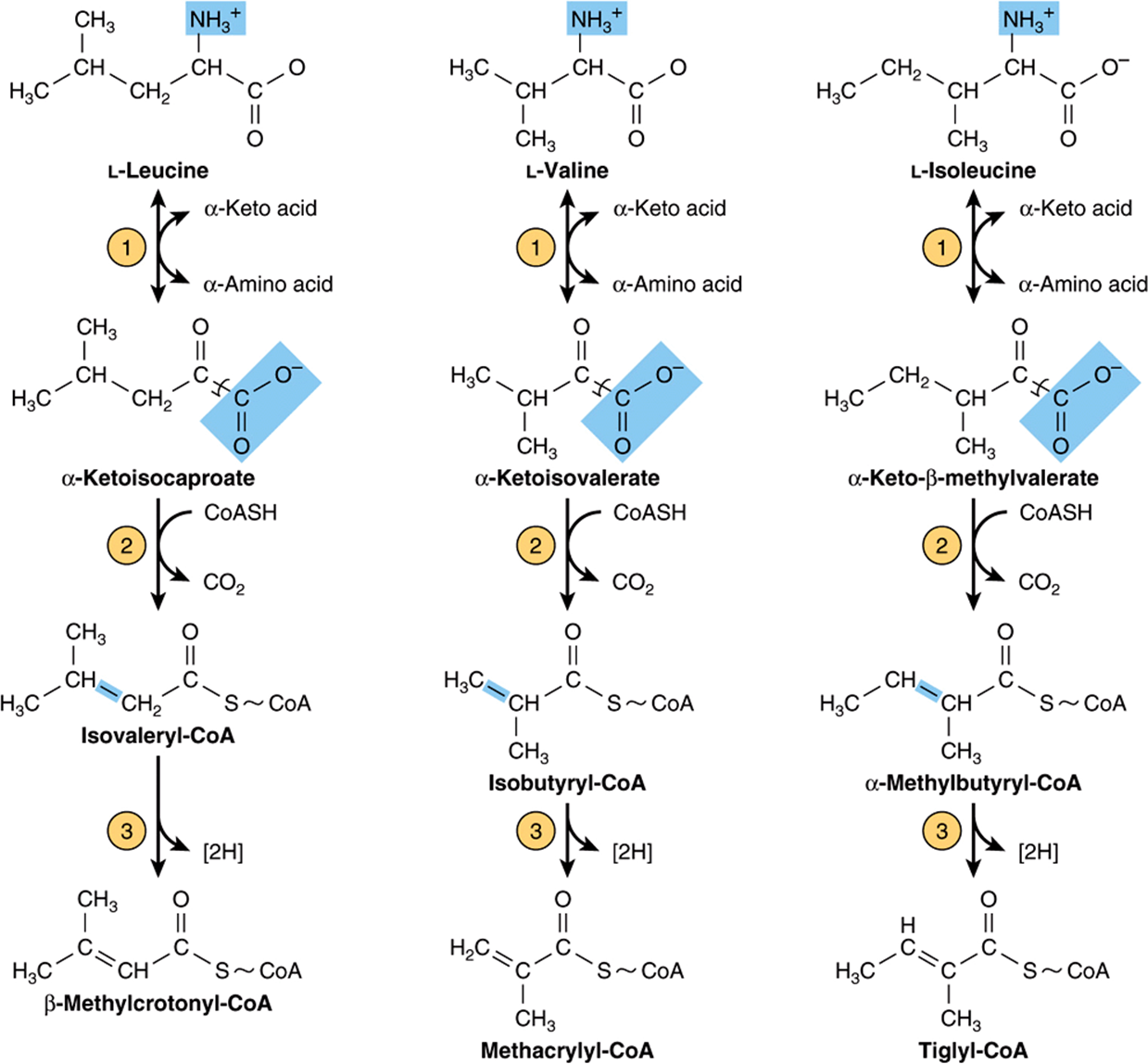
FIGURE 29–20 The first three reactions in the catabolism of leucine, valine, and isoleucine. Note the analogy of reactions 2 and 3 to reactions of the catabolism of fatty acids (see Figure 22–3). The analogy to fatty acid catabolism continues, as shown in subsequent figures.
E1: thiamin pyrophosphate (TPP)-dependent branched-chain a-ketoacid decarboxylase.
E2: dihydrolipoyl transacylase (contains lipoamide).
E3: dihydrolipoamide dehydrogenase (contains FAD).
Protein kinase.
Protein phosphatase.
As for PDH [see Figure 18–6(B)], the protein kinase and protein phosphatase regulate activity of the branched-chain a-keto acid dehydrogenase complex via phosphorylation (inactivation) and dephosphorylation (activation).
Dehydrogenation of the resulting coenzyme A thioesters (reaction 3, Figure 29–20) proceeds like the dehydrogenation of lipid-derived fatty acyl-CoA thioesters (see Figure 22–3). Subsequent reactions that are unique for each amino acid skeleton are given in Figures 29-21, 29-22, and 29-23.
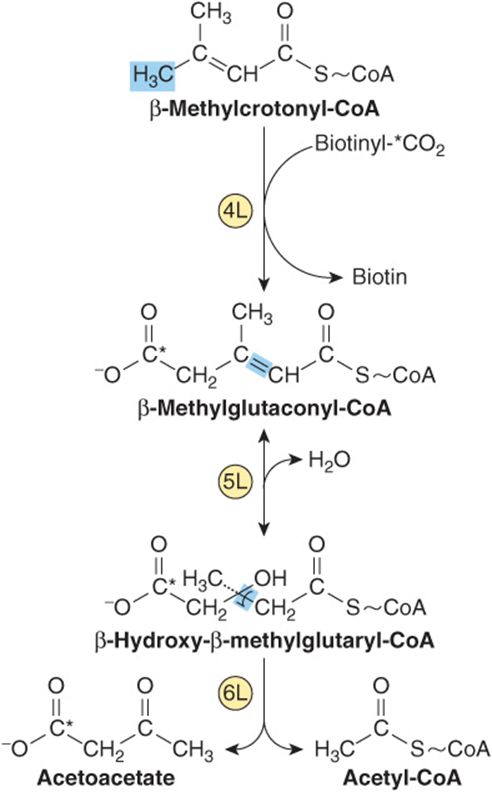
FIGURE 29–21 Catabolism of the β-methylcrotonyl-CoA formed from L -leucine. Asterisks indicate carbon atoms derived from CO2.
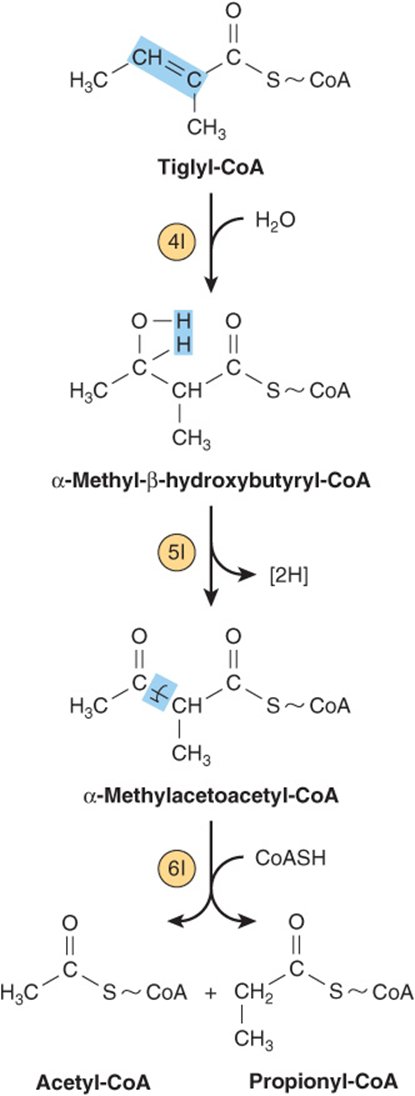
FIGURE 29–22 Subsequent catabolism of the tiglyl-CoA formed from L-isoleucine.
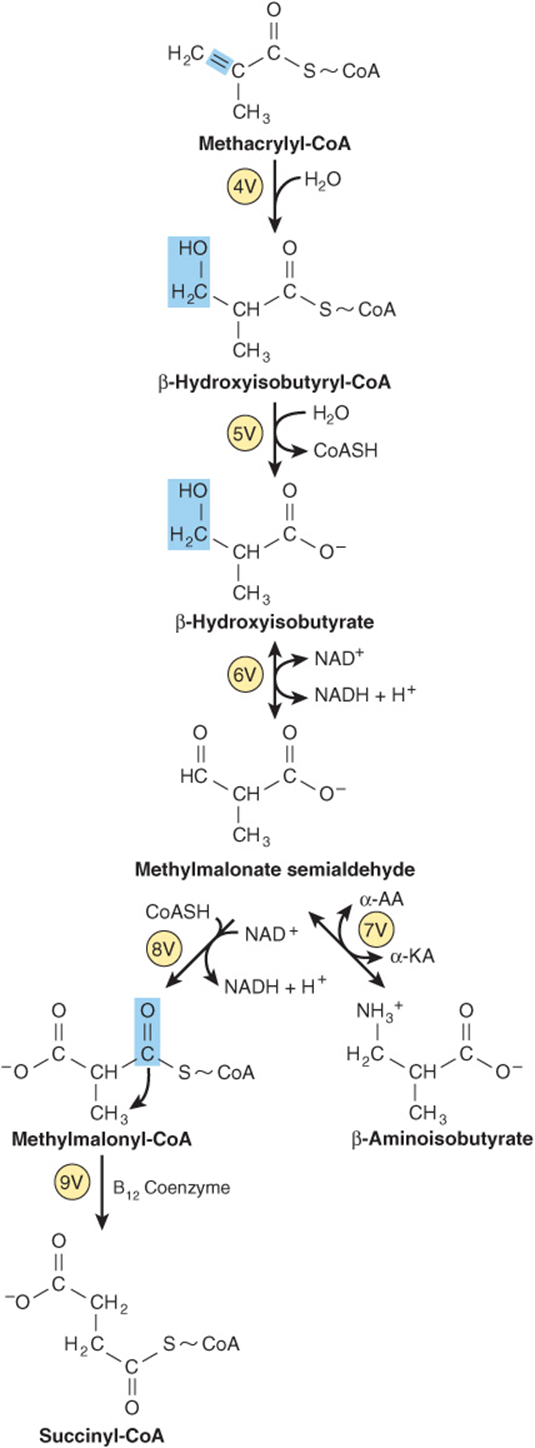
FIGURE 29–23 Subsequent catabolism of the methacrylyl-CoA formed from L-valine (see Figure 29–20). (α-AA, α-amino acid; α-KA, α-keto acid.)
METABOLIC DISORDERS OF BRANCHED-CHAIN AMINO ACID CATABOLISM
As the name implies, the odor of urine in maple syrup urine disease (branched-chain ketonuria, or MSUD) suggests maple syrup, or burnt sugar. The biochemical defect in MSUD involves the α-keto acid decarboxylase complex (reaction 2, Figure 29–20). Plasma and urinary levels of leucine, isoleucine, valine, and their a-keto acids and a-hydroxy acids (reduced a-keto acids) are elevated, but the urinary ketoacids derive principally from leucine. Signs and symptoms of MSUD include often fatal ketoacidosis, neurological derangements, mental retardation, and a maple syrup odor of urine. The mechanism of toxicity is unknown. Early diagnosis by enzymatic analysis is essential to avoid brain damage and early mortality by replacing dietary protein by an amino acid mixture that lacks leucine, isoleucine, and valine.
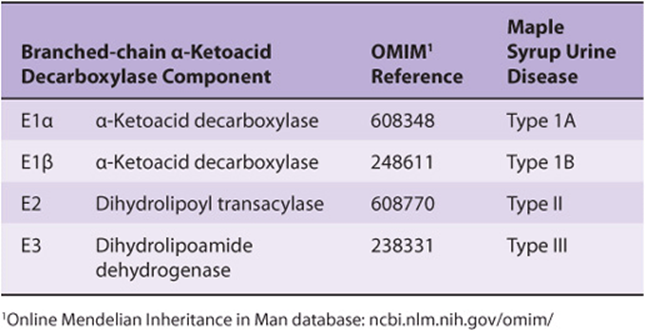
The molecular genetics of MSUD are heterogeneous. MSUD can result from mutations in the genes that encode E1α, E1β, E2, and E3. Based on the locus affected, genetic subtypes of MSUD are recognized. Type IA MSUD arises from mutations in the E1β gene, type IB in the E1β gene, type II in the E2 gene, and type III in the E3 gene (Table 29-3). In intermittent branched-chain ketonuria, the a-keto acid decarboxylase retains some activity, and symptoms occur later in life. In isovaleric acidemia, ingestion of protein-rich foods elevates isovalerate, the deacylation product of isovaleryl-CoA. The impaired enzyme in isovaleric acidemia is isovaleryl-CoA dehydrogenase(reaction 3, Figure 29–20). Vomiting, acidosis, and coma follow ingestion of excess protein. Accumulated isovaleryl-CoA is hydrolyzed to isovalerate and excreted.
TABLE 29–3 Maple Syrup Urine Disease Can Reflect Impaired Function of Various Components of the α-Ketoacid Decarboxylase Complex
SUMMARY
![]() Excess amino acids are catabolized to amphibolic intermediates that serve as sources of energy or for the biosynthesis of carbohydrates and lipids.
Excess amino acids are catabolized to amphibolic intermediates that serve as sources of energy or for the biosynthesis of carbohydrates and lipids.
![]() Transamination is the most common initial reaction of amino acid catabolism. Subsequent reactions remove any additional nitrogen and restructure hydrocarbon skeletons for conversion to oxaloacetate, a-ketoglutarate, pyruvate, and acetyl-CoA.
Transamination is the most common initial reaction of amino acid catabolism. Subsequent reactions remove any additional nitrogen and restructure hydrocarbon skeletons for conversion to oxaloacetate, a-ketoglutarate, pyruvate, and acetyl-CoA.
![]() Metabolic diseases associated with glycine catabolism include glycinuria and primary hyperoxaluria.
Metabolic diseases associated with glycine catabolism include glycinuria and primary hyperoxaluria.
![]() Two distinct pathways convert cysteine to pyruvate. Metabolic disorders of cysteine catabolism include cystine-lysinuria, cystine storage disease, and the homocystinurias.
Two distinct pathways convert cysteine to pyruvate. Metabolic disorders of cysteine catabolism include cystine-lysinuria, cystine storage disease, and the homocystinurias.
![]() Threonine catabolism merges with that of glycine after threonine aldolase cleaves threonine to glycine and acetaldehyde.
Threonine catabolism merges with that of glycine after threonine aldolase cleaves threonine to glycine and acetaldehyde.
![]() Following transamination, the carbon skeleton of tyrosine is degraded to fumarate and acetoacetate. Metabolic diseases of tyrosine catabolism include tyrosinosis, Richner-Hanhart syndrome, neonatal tyrosinemia, and alkaptonuria.
Following transamination, the carbon skeleton of tyrosine is degraded to fumarate and acetoacetate. Metabolic diseases of tyrosine catabolism include tyrosinosis, Richner-Hanhart syndrome, neonatal tyrosinemia, and alkaptonuria.
![]() Metabolic disorders of phenylalanine catabolism include PKU and several hyperphenylalaninemias.
Metabolic disorders of phenylalanine catabolism include PKU and several hyperphenylalaninemias.
![]() Neither nitrogen of lysine undergoes direct transamination. The same effect is, however, achieved by the intermediate formation of saccharopine. Metabolic diseases of lysine catabolism include periodic and persistent forms of hyperlysinemia-ammonemia.
Neither nitrogen of lysine undergoes direct transamination. The same effect is, however, achieved by the intermediate formation of saccharopine. Metabolic diseases of lysine catabolism include periodic and persistent forms of hyperlysinemia-ammonemia.
![]() The catabolism of leucine, valine, and isoleucine presents many analogies to fatty acid catabolism. Metabolic disorders of branched-chain amino acid catabolism include hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
The catabolism of leucine, valine, and isoleucine presents many analogies to fatty acid catabolism. Metabolic disorders of branched-chain amino acid catabolism include hypervalinemia, maple syrup urine disease, intermittent branched-chain ketonuria, isovaleric acidemia, and methylmalonic aciduria.
REFERENCES
Blacher J, Safar ME: Homocysteine, folic acid, B vitamins and cardiovascular risk. J Nutr Health Aging 2001;5:196.
Bliksrud YT, Brodtkorb E, Andresen PA, et al: Tyrosinemia type I, de novo mutation in liver tissue suppressing an inborn splicing defect. J Mol Med 2005;83:406.
Dobrowolski, Pey AL, Koch R, et al: Biochemical characterization of mutant phenylalanine hydroxylase enzymes and correlation with clinical presentation in hyperphenylalaninaemic patients. J Inherit Metab Dis 2009:32;10.
Flusser H, Korman SH, Sato K, et al: Mild glycine encephalopathy (NKH) in a large kindred due to a silent exonic GLDC splice mutation. Neurology 2005;64:1426.
Garg U, Dasouki M: Expanded newborn screening of inherited metabolic disorders by tandem mass spectrometry. Clinical and laboratory aspects. Clin Biochem 2006;39:315.
Gerstner B, Gratopp A, Marcinkowski M, et al: Glutaric acid and its metabolites cause apoptosis in immature oligodendrocytes: a novel mechanism of white matter degeneration in glutaryl-CoA dehydrogenase deficiency. Pediatr Res 2005;57;771.
Häussinger D, Schliess F: Glutamine metabolism and signaling in the liver. Front Biosci 2007;12:371.
Heldt K, Schwahn B, Marquardt I, et al: Diagnosis of maple syrup urine disease by newborn screening allows early intervention without extraneous detoxification. Mol Genet Metab 2005;84:313.
Moshal K, Camel CK, Kartha GK, et al: Cardiac dys-synchronization and arrhythmia in hyperhomocysteinemia. Curr Neurovasc Res 2007;4:289.
Muller E, Kolker S: Reduction of lysine intake while avoiding malnutrition: major goals and major problems in dietary treatment of glutaryl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2004;27:903.
Sacksteder KA, Biery BJ, Morrell JC, et al: Identification of the alpha-aminoadipic semialdehyde synthase gene which is defective in familial hyperlysinemia. Am J Hum Genet 2000;66:1736.
Scriver CR, Sly WS, Childs B, et al (editors): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, 2001.
Stenn FF, Milgram JW, Lee SL, et al: Biochemical identification of homogentisic acid pigment in an ochronotic Egyptian mummy. Science 1977;197:566.
Waters PJ, Scriver CR, Parniak MA: Homomeric and heteromeric interactions between wild-type and mutant phenylalanine hydroxylase subunits: evaluation of two-hybrid approaches for functional analysis of mutations causing hyperphenylalaninemia. Mol Genet Metab 2001;73:230.