Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION IV. Structure, Function, & Replication of Informational Macromolecules
Chapter 36. RNA Synthesis, Processing, & Modification
P. Anthony Weil, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe the molecules involved and the mechanism of RNA synthesis.
Describe the molecules involved and the mechanism of RNA synthesis.
![]() Explain how eukaryotic DNA-dependent RNA polymerases, in collaboration with an array of specific accessory factors, can differentially transcribe genomic DNA to produce specific mRNA precursor molecules.
Explain how eukaryotic DNA-dependent RNA polymerases, in collaboration with an array of specific accessory factors, can differentially transcribe genomic DNA to produce specific mRNA precursor molecules.
![]() Describe the structure of eukaryotic mRNA precursors, which are highly modified at both termini.
Describe the structure of eukaryotic mRNA precursors, which are highly modified at both termini.
![]() Appreciate the fact that the majority of mammalian mRNA-encoding genes are interrupted by multiple non-protein coding sequences termed introns, which are interspersed between protein coding regions termed exons.
Appreciate the fact that the majority of mammalian mRNA-encoding genes are interrupted by multiple non-protein coding sequences termed introns, which are interspersed between protein coding regions termed exons.
![]() Explain that since intron RNA does not encode protein, the intronic RNA must be specifically and accurately removed in order to generate functional mRNAs from the mRNA precursor molecules in a series of precise molecular events termed RNA splicing.
Explain that since intron RNA does not encode protein, the intronic RNA must be specifically and accurately removed in order to generate functional mRNAs from the mRNA precursor molecules in a series of precise molecular events termed RNA splicing.
![]() Explain the steps and molecules that catalyze mRNA splicing, a process that converts the end-modified mRNA precursor molecules into mRNAs that are functional for translation.
Explain the steps and molecules that catalyze mRNA splicing, a process that converts the end-modified mRNA precursor molecules into mRNAs that are functional for translation.
BIOMEDICAL IMPORTANCE
The synthesis of an RNA molecule from DNA is a complex process involving one of the group of RNA polymerase enzymes and a number of associated proteins. The general steps required to synthesize the primary transcript are initiation, elongation, and termination. Most is known about initiation. A number of DNA regions (generally located upstream from the initiation site) and protein factors that bind to these sequences to regulate the initiation of transcription have been identified. Certain RNAs—mRNAs in particular—have very different life spans in a cell. The RNA molecules synthesized in mammalian cells are made as precursor molecules that have to be processed into mature, active RNA. It is important to understand the basic principles of messenger RNA (mRNA) synthesis and metabolism, for modulation of this process results in altered rates of protein synthesis and thus a variety of both metabolic and phenotypic changes. This is how all organisms adapt to changes of environment. It is also how differentiated cell structures and functions are established and maintained. Errors or changes in synthesis, processing, splicing, stability, or function of mRNA transcripts are a cause of disease.
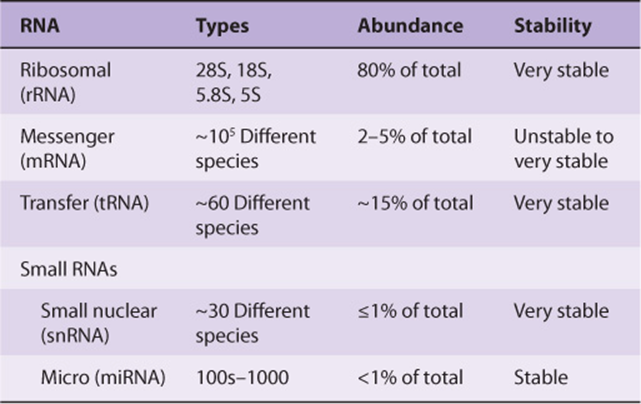
RNA EXISTS IN FOUR MAJOR CLASSES
All eukaryotic cells have four major classes of RNA (Table 36-1): ribosomal RNA (rRNA), mRNA, transfer RNA (tRNA), and small RNAs, the small nuclear RNAs and microRNAs (snRNA and miRNA). The first three are involved in protein synthesis, while the small RNAs are involved in mRNA splicing and modulation of gene expression by altering mRNA function. The various classes of RNA are different in their diversity, stability, and abundance in cells.
TABLE 36–1 Classes of Eukaryotic RNA
RNA IS SYNTHESIZED FROM A DNA TEMPLATE BY AN RNA POLYMERASE
The processes of DNA and RNA synthesis are similar in that they involve (1) the general steps of initiation, elongation, and termination with 5′-3’ polarity; (2) large, multicomponent initiation complexes; and (3) adherence to Watson-Crick base-pairing rules. However, DNA and RNA synthesis do differ in several important ways, including the following: (1) ribonucleotides are used in RNA synthesis rather than deoxyribonucleotides; (2) U replaces T as the complementary base for A in RNA; (3) a primer is not involved in RNA synthesis as RNA polymerases have the ability to initiate synthesis de novo; (4) only portions of the genome are vigorously transcribed or copied into RNA, whereas the entire genome must be copied, once and only once during DNA replication; and (5) there is no highly active, efficient proofreading function during RNA transcription.
The process of synthesizing RNA from a DNA template has been characterized best in prokaryotes. Although in mammalian cells, the regulation of RNA synthesis and the processing of the RNA transcripts are different from those in prokaryotes, the process of RNA synthesis per se is quite similar in these two classes of organisms. Therefore, the description of RNA synthesis in prokaryotes, where it is best understood, is applicable to eukaryotes even though the enzymes involved and the regulatory signals, though related, are different.
The Template Strand of DNA Is Transcribed
The sequence of ribonucleotides in an RNA molecule is complementary to the sequence of deoxyribonucleotides in one strand of the double-stranded DNA molecule (Figure 34–8). The strand that is transcribed or copied into an RNA molecule is referred to as the template strand of the DNA. The other DNA strand, the nontemplate strand, is frequently referred to as the coding strand of that gene. It is called this because, with the exception of T for U changes, it corresponds exactly to the sequence of the messenger RNA primary transcript, which encodes the (protein) product of the gene. In the case of a double-stranded DNA molecule containing many genes, the template strand for each gene will not necessarily be the same strand of the DNA double helix (Figure 36–1). Thus, a given strand of a double-stranded DNA molecule will serve as the template strand for some genes and the coding strand of other genes. Note that the nucleotide sequence of an RNA transcript will be the same (except for U replacing T) as that of the coding strand. The information in the template strand is read out in the 3′-5’ direction. Though not shown in Figure 36–1 there are instances of genes embedded within other genes.
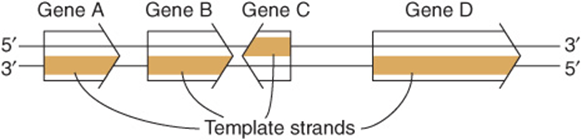
FIGURE 36–1 Genes can be transcribed off both strands of DNA. The arrowheads indicate the direction of transcription (polarity). Note that the template strand is always read in the 3′-5’ direction. The opposite strand is called the coding strand because it is identical (except for T for U changes) to the mRNA transcript (the primary transcript in eukaryotic cells) that encodes the protein product of the gene.
DNA-Dependent RNA Polymerase Initiates Transcription at a Distinct Site, the Promoter
DNA-dependent RNA polymerase is the enzyme responsible for the polymerization of ribonucleotides into a sequence complementary to the template strand of the gene (see Figures 36-2 and 36-3). The enzyme attaches at a specific site—the promoter—on the template strand. This is followed by initiation of RNA synthesis at the starting point, and the process continues until a termination sequence is reached (Figure 36–3). A transcription unit is defined as that region of DNA that includes the signals for transcription initiation, elongation, and termination. The RNA product, which is synthesized in the 5′-3’ direction, is the primary transcript. Transcription rates vary from gene to gene but can be quite high. An electron micrograph of transcription in action is presented in Figure 36–4. In prokaryotes, this can represent the product of several contiguous genes; in mammalian cells, it usually represents the product of a single gene. If a transcription unit contains only a single gene, then the 5′ termini of the primary RNA transcript and the mature cytoplasmic RNA are identical. Thus, the starting point of transcription corresponds to the 5′ nucleotide of the mRNA. This is designated position +1, as is the corresponding nucleotide in the DNA. The numbers increase as the sequence proceeds downstream from the start site. This convention makes it easy to locate particular regions, such as intron and exon boundaries. The nucleotide in the promoter adjacent to the transcription initiation site in the upstream direction is designated -1, and these negative numbers increase as the sequence proceeds upstream, away from the initiation site. This +/- numbering system provides a conventional way of defining the location of regulatory elements in the promoter.
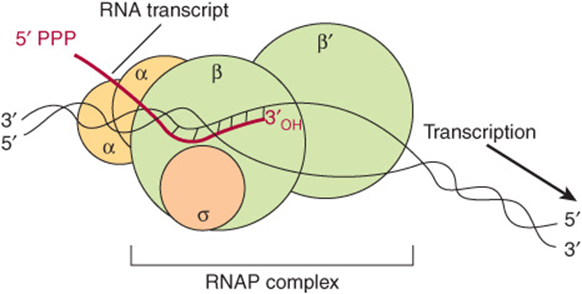
FIGURE 36–2 RNA polymerase (RNAP) catalyzes the polymerization of ribonucleotides into an RNA sequence that is complementary to the template strand of the gene. The RNA transcript has the same polarity (5’-3’) as the coding strand but contains U rather than T. E coli RNAP consists of a core complex of two α subunits and two β subunits (β and β’). The holoenzyme contains the σ subunit bound to the α2 ββ’ core assembly. The ω subunit is not shown. The transcription “bubble” is an approximately 20-bp area of melted DNA, and the entire complex covers 30-75 bp, depending on the conformation of RNAP.
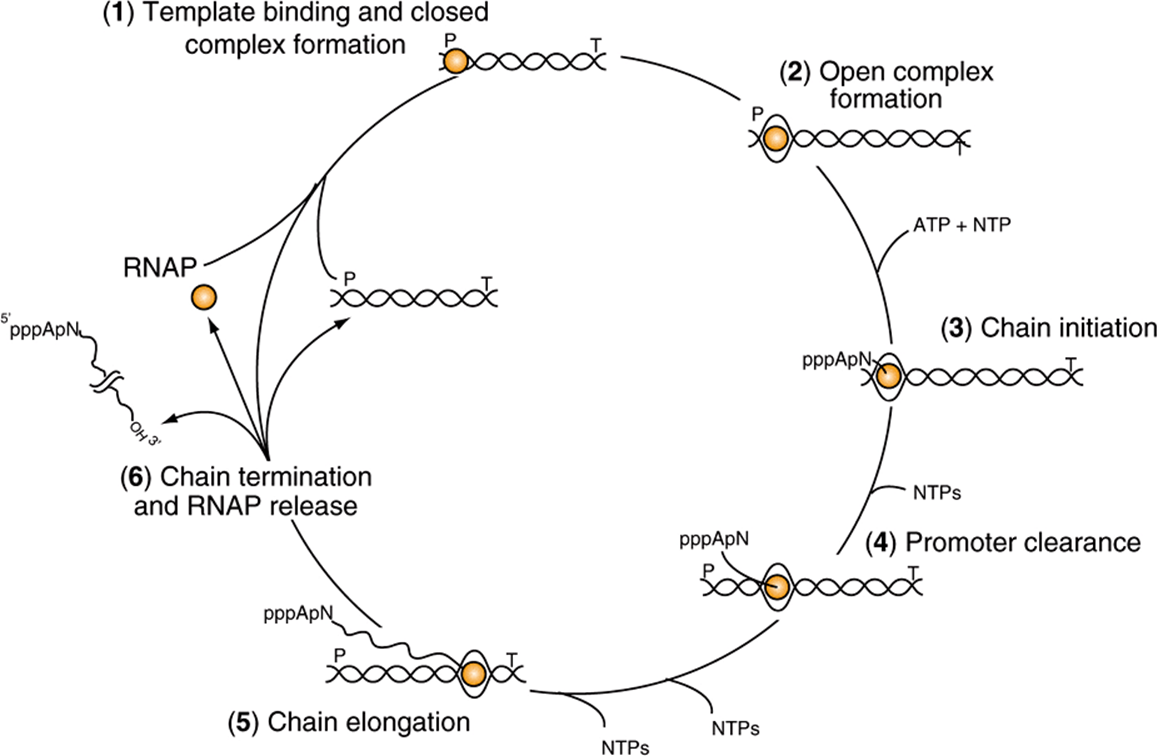
FIGURE 36–3 The transcription cycle. Transcription can be described in six steps: (1) Template binding and closed RNA polymerase-promoter complex formation: RNA polymerase (RNAP) binds to DNA and then locates a promoter (P), (2) Open promoter complex formation: once bound to the promoter, RNAP melts the two DNA strands to form an open promoter complex; this complex is also referred to as the preinitiation complex or PIC. Strand separation allows the polymerase to access the coding information in the template strand of DNA (3) Chain initiation: using the coding information of the template RNAP catalyzes the coupling of the first base (often a purine) to the second, template-directed ribonucleoside triphosphate to form a dinucleotide (in this example forming the dinucleotide 5′ pppApNOH 3′). (4) Promoter clearance: after RNA chain length reaches -10-20 nt, the polymerase undergoes a conformational change and then is able to move away from the promoter, transcribing down the transcription unit. (5) Chain elongation: Successive residues are added to the 3′-OH terminus of the nascent RNA molecule until a transcription termination signal (T) is encountered. (6) Chain termination and RNAP release: Upon encountering the transcription termination site RNAP undergoes an additional conformational change that leads to release of the completed RNA chain, the DNA template and RNAP. RNAP can rebind to DNA beginning the promoter search process and the cycle is repeated. Note that all of the steps in the transcription cycle are facilitated by additional proteins, and indeed are often subjected to regulation by positive and/or negative-acting factors.
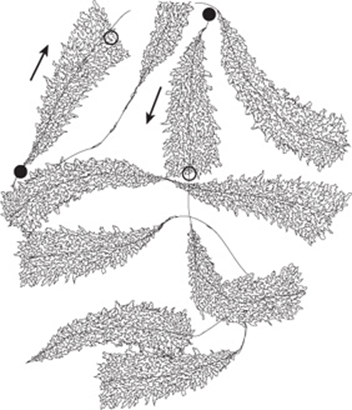
FIGURE 36–4 Schematic representation of an electron photomicrograph of multiple copies of amphibian rRNA-encoding genes in the process of being transcribed. The magnification is about 6000×. Note that the length of the transcripts increases as the RNA polymerase molecules progress along the individual rRNA genes from transcription start sites (filled circles) to transcription termination sites (open circles). RNA polymerase I (not visualized here) is at the base of the nascent rRNA transcripts. Thus, the proximal end of the transcribed gene has short transcripts attached to it, while much longer transcripts are attached to the distal end of the gene. The arrows indicate the direction (5’-3’) of transcription.
The primary transcripts generated by RNA polymerase II—one of the three distinct nuclear DNA-dependent RNA polymerases in eukaryotes—are promptly capped by 7-methyl-guanosine triphosphate caps (Figure 34–10) that persist and eventually appear on the 5’ end of mature cyto-plasmic mRNA. These caps are necessary for the subsequent processing of the primary transcript to mRNA, for the translation of the mRNA, and for protection of the mRNA against exonucleolytic attack.
Bacterial DNA-Dependent RNA Polymerase Is a Multisubunit Enzyme
The DNA-dependent RNA polymerase (RNAP) of the bacterium Escherichia coli exists as an approximately 400 kDa core complex consisting of two identical α subunits, similar but not identical β and β’ subunits, and an ω subunit. The β subunit binds Mg2+ ions and composes the catalytic subunit (Figure 36–2). The core RNA polymerase, ββ’α2ω, often termed E, associates with a specific protein factor (the sigma [σ] factor) to form holoenzyme, ββ’α2ωσ, or Eσ. The σ subunit helps the core enzyme recognize and bind to the specific deoxy-nucleotide sequence of the promoter region (Figure 36–5) to form the preinitiation complex (PIC). There are multiple, distinct σ-factor encoding genes in all bacterial species. Sigma factors have a dual role in the process of promoter recognition; σ association with core RNA polymerase decreases its affinity for nonpromoter DNA while simultaneously increasing holoenzyme affinity for promoter DNA. The multiple σ-factors compete for interaction with limiting core RNA polymerase (ie, E). Each of these unique σ-factors act as a regulatory protein that modifies the promoter recognition specificity of the resulting unique RNA polymerase holoenzyme (ie, Eσ1, Eσ2,…). The appearance of different σ-factors and their association with core RNA polymerase forming novel holoenzyme forms can be correlated temporally with various programs of gene expression in prokaryotic systems such as sporulation, growth in various poor nutrient sources, and the response to heat shock.
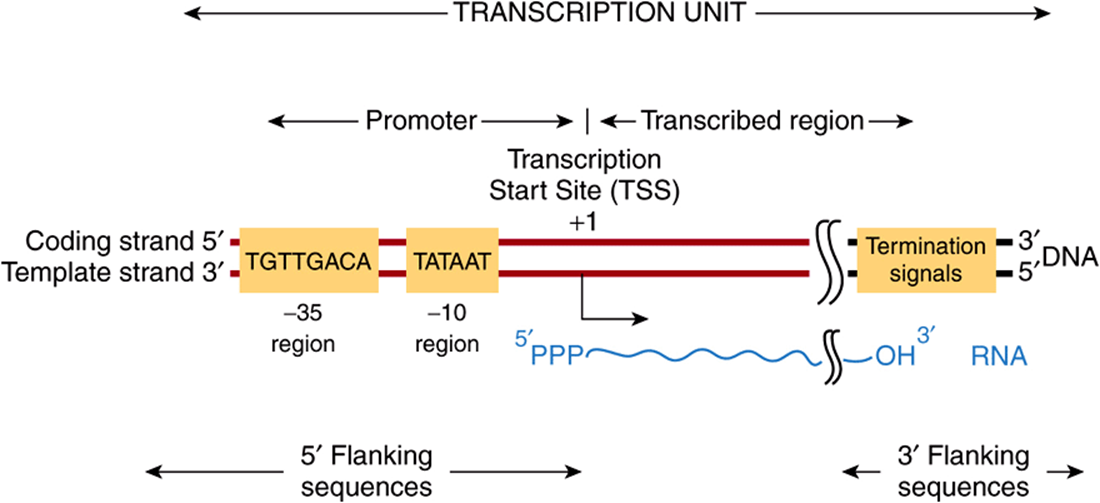
FIGURE 36–5 Bacterial promoters, such as that from E coli shown here, share two regions of highly conserved nucleotide sequence. These regions are located 35 and 10 bp upstream (in the 5′ direction of the coding strand) from the transcription start site (TSS), which is indicated as +1. By convention, all nucleotides upstream of the transcription initiation site (at +1) are numbered in a negative sense and are referred to as 5′-flanking sequences, while sequences downstream are numbered in a positive sense with the TSS as +1. Also by convention, the promoter DNA regulatory sequence elements such as the -35 and TATA box elements are described in the 5′-3’ direction and as being on the coding strand. These elements function only in double-stranded DNA, however. Other transcriptional regulatory elements, however, can often act in a direction independent fashion, and such cis-elements are drawn accordingly in any schematic (see also Figure 36–8). Note that the transcript produced from this transcription unit has the same polarity or “sense” (ie, 5′-3’ orientation) as the coding strand. Termination cis-elements reside at the end of the transcription unit (see Figure 36–6 for more detail). By convention, the sequences downstream of the site at which transcription termination occurs are termed 3′-flanking sequences.
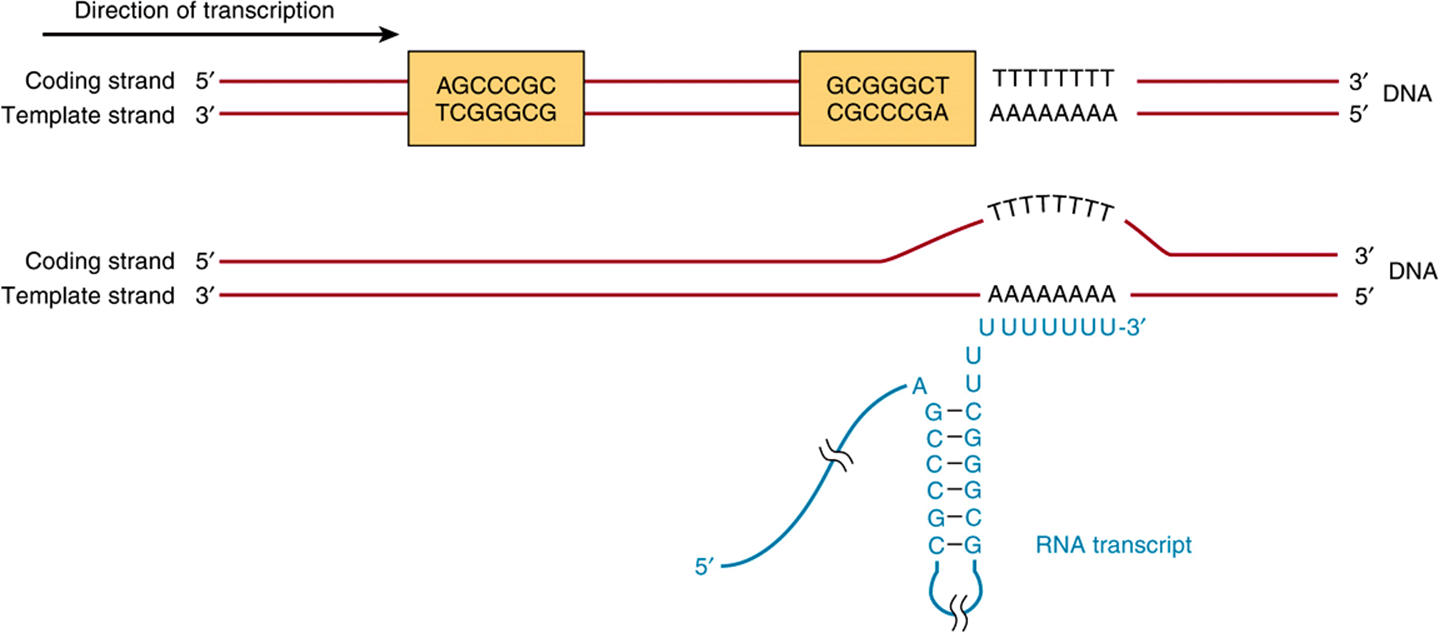
FIGURE 36–6 The predominant bacterial transcription termination signal contains an inverted, hyphenated repeat (the two boxed areas) followed by a stretch of AT base pairs (top). The inverted repeat, when transcribed into RNA, can generate the secondary structure in the RNA transcript (bottom). Formation of this RNA hairpin causes RNA polymerase to pause and subsequently the ρ (rho) termination factor interacts with the paused polymerase and induces chain termination through mechanisms not yet fully understood.
Mammalian Cells Possess Three Distinct Nuclear DNA-Dependent RNA Polymerases
The properties of mammalian nuclear polymerases are described in Table 36-2. Each of these DNA-dependent RNA polymerases is responsible for transcription of different sets of genes. The sizes of the RNA polymerases range from MW 500,000 to MW 600,000. These enzymes exhibit more complex subunit profiles than prokaryotic RNA polymerases. They all have two large subunits and a number of smaller subunits—as many as 14 in the case of RNA pol III. However, the eukaryotic RNA polymerase subunits do exhibit extensive amino acid sequence homologies with prokaryotic RNA polymerases. This homology has been shown recently to extend to the level of three-dimensional structures. The functions of each of the subunits are not yet fully understood.
A peptide toxin from the mushroom Amanita phalloides, α-amanitin, is a specific differential inhibitor of the eukaryotic nuclear DNA-dependent RNA polymerases and as such has proved to be a powerful research tool (Table 36-2). α-Amanitin blocks the translocation of RNA polymerase during phosphodiester bond formation.
TABLE 36–2 Nomenclature and Properties of Mammalian Nuclear DNA-Dependent RNA Polymerases

RNA SYNTHESIS IS A CYCLICAL PROCESS & INVOLVES RNA CHAIN INITIATION, ELONGATION, & TERMINATION
The process of RNA synthesis in bacteria—depicted in Figure 36–3—is cyclical and involves multiple steps. First RNA polymerase holoenzyme (E-σ) must bind DNA and locate a promoter (P; Figure 36–3). Once the promoter is located, the Eσ-promoter DNA complex undergoes a temperature-dependent conformational change and unwinds, or melts the DNA in and around the transcription start site (at +1). This complex is termed the preinitiation complex, or PIC. This unwinding allows the active site of the Eσ to access the template strand, which of course dictates the sequence of ribonucleotides to be polymerized into RNA. The first nucleotide (typically, though not always a purine) then associates with the nucleotide-binding site on the β subunit of the enzyme, and in the presence of the next appropriate nucleotide bound to the polymerase, RNAP catalyzes the formation of the first phosphodiester bond, and the nascent chain is now attached to the polymerization site on the β subunit of RNAP. This reaction is termed initiation. The analogy to the A and P sites on the ribosome should be noted; see Figure 37–9, below. The nascent dinucleotide retains the 5′-triphosphate of the initiating nucleotide (Figure 36–3, ATP).
RNA polymerase continues to incorporate nucleotides 3 to ~10, at which point the polymerase undergoes another conformational change and moves away from the promoter; this reaction is termed promoter clearance. The elongation phase then commences, here the nascent RNA molecule grows 5′ -3’ as consectutive NTP incorporation steps continue cyclically, antiparallel to its template. The enzyme polymerizes the ribonucleotides in the specific sequence dictated by the template strand and interpreted by Watson-Crick base-pairing rules. Pyrophosphate is released following each cycle of polymerization. As for DNA synthesis, this pyrophosphate (PPi) is rapidly degraded to 2 mol of inorganic phosphate (Pi) by ubiquitous pyrophosphatases, thereby providing irreversibility on the overall synthetic reaction. The decision, to stay at the promoter in a poised or stalled state, or transition to elongation appears to be an important regulatory step in both prokaryotic and eukaryotic mRNA gene transcription.
As the elongation complex containing RNA polymerase progresses along the DNA molecule, DNA unwinding must occur in order to provide access for the appropriate base pairing to the nucleotides of the coding strand. The extent of this transcription bubble (ie, DNA unwinding) is constant throughout transcription and has been estimated to be about 20 base pairs per polymerase molecule. Thus, it appears that the size of the unwound DNA region is dictated by the polymerase and is independent of the DNA sequence in the complex. RNA polymerase has an intrinsic “unwindase” activity that opens the DNA helix (ie, see PIC formation above). The fact that the DNA double helix must unwind, and the strands part at least transiently for transcription implies some disruption of the nucleosome structure of eukaryotic cells. Topoisomerase both precedes and follows the progressing RNA polymerase to prevent the formation of superhelical tensions that would serve to increase the energy required to unwind the template DNA ahead of RNAP.
Termination of the synthesis of the RNA molecule in bacteria is signaled by a sequence in the template strand of the DNA molecule—a signal that is recognized by a termination protein, the rho (ρ) factor. Rho is an ATP-dependent RNA-stimulated helicase that disrupts the ternary transcription elongation complex composed of RNA polymerase-nascent RNA and DNA. In some cases, bacterial RNAP can directly recognize DNA-encoded termination signals (Figure 36–3; T) without assistance by the rho factor. After termination of synthesis of the RNA, the enzyme separates from the DNA template and probably dissociates to free core enzyme and free factor. With the assistance of another σ-factor, the core enzyme then recognizes a promoter at which the synthesis of a new RNA molecule commences. In eukaryotic cells, termination is less well understood but the proteins catalyzing RNA processing, termination, and polyadenylation proteins all appear to load onto RNA polymerase II soon after initiation (see below). More than one RNA polymerase molecule may transcribe the same template strand of a gene simultaneously, but the process is phased and spaced in such a way that at any one moment each is transcribing a different portion of the DNA sequence (Figures 36-1 and 36-4).
THE FIDELITY & FREQUENCY OF TRANSCRIPTION IS CONTROLLED BY PROTEINS BOUND TO CERTAIN DNA SEQUENCES
Analysis of the DNA sequence of specific genes has allowed the recognition of a number of sequences important in gene transcription. From the large number of bacterial genes studied, it is possible to construct consensus models of transcription initiation and termination signals.
The question, “How does RNAP find the correct site to initiate transcription?” is not trivial when the complexity of the genome is considered. E coli has ![]() transcription initiation sites (ie, gene promoters) in
transcription initiation sites (ie, gene promoters) in ![]() base pairs (bp) of DNA. The situation is even more complex in humans, where as many as 105 transcription initiation sites are distributed throughout 3 × 109 bp of DNA. RNAP can bind, with low affinity, to many regions of DNA, but it scans the DNA sequence—at a rate of ≥103 bp/s—until it recognizes certain specific regions of DNA to which it binds with higher affinity. These regions are termed promoters, and it is the association of RNAP with promoters that ensures accurate initiation of transcription. The promoter recognition-utilization process is the target for regulation in both bacteria and humans.
base pairs (bp) of DNA. The situation is even more complex in humans, where as many as 105 transcription initiation sites are distributed throughout 3 × 109 bp of DNA. RNAP can bind, with low affinity, to many regions of DNA, but it scans the DNA sequence—at a rate of ≥103 bp/s—until it recognizes certain specific regions of DNA to which it binds with higher affinity. These regions are termed promoters, and it is the association of RNAP with promoters that ensures accurate initiation of transcription. The promoter recognition-utilization process is the target for regulation in both bacteria and humans.
Bacterial Promoters Are Relatively Simple
Bacterial promoters are approximately 40 nucleotides (40 bp or four turns of the DNA double helix) in length, a region small enough to be covered by an E coli RNA holopolymerase molecule. In a consensus promoter, there are two short, conserved sequence elements. Approximately 35-bp upstream of the transcription start site there is a consensus sequence of eight nucleotide pairs (consensus: 5′-TGTTGACA-3’) to which the RNAP binds to form the so-called closed complex. More proximal to the transcription start site—about 10 nucleotides upstream—is a six-nucleotide-pair A+T-rich sequence (consensus: 5′-TATAAT-3’). These conserved sequence elements together comprise the promoter, and are shown schematically in Figure 36–5. The latter sequence has a low melting temperature because of its lack of GC nucleotide pairs. Thus, the so-called TATA “box” is thought to ease the dissociation of the two DNA strands so that RNA polymerase bound to the promoter region can have access to the nucleotide sequence of its immediately downstream template strand. Once this process occurs, the combination of RNA polymerase plus promoter is called the open complex. Other bacteria have slightly different consensus sequences in their promoters, but all generally have two components to the promoter; these tend to be in the same position relative to the transcription start site, and in all cases the sequences between the two promoter elements have no similarity but still provide critical spacing functions that facilitate recognition of -35 and -10 sequences by RNA polymerase holoenzyme. Within a bacterial cell, different sets of genes are often coordinately regulated. One important way that this is accomplished is through the fact that these co-regulated genes share particular -35 and -10 promoter sequences. These unique promoters are recognized by different σ-factors bound to core RNA polymerase (ie, Eσ1, Eσ2,…).
Rho-dependent transcription termination signals in E coli also appear to have a distinct consensus sequence, as shown in Figure 36–6. The conserved consensus sequence, which is about 40 nucleotide pairs in length, can be seen to contain a hyphenated or interrupted inverted repeat followed by a series of AT base pairs. As transcription proceeds through the hyphenated, inverted repeat, the generated transcript can form the intramolecular hairpin structure, also depicted in Figure 36–6.
Transcription continues into the AT region, and with the aid of the ρ termination protein the RNA polymerase stops, dissociates from the DNA template, and releases the nascent transcript.
As discussed in detail in Chapter 38 bacterial gene transcription is controlled through the action of repressor and activator proteins. These proteins typically bind to unique and specific DNA sequences that lie adjacent to promoters. These repressors and activators affect the ability of the RNA polymerase to bind promoter DNA and/or form open complexes. The net effect is to stimulate or inhibit PIC formation and transcription initiation—consequently blocking or enhancing specific RNA synthesis.
Eukaryotic Promoters Are More Complex
It is clear that the signals in DNA that control transcription in eukaryotic cells are of several types. Two types of sequence elements are promoter-proximal. One of these defines where transcription is to commence along the DNA, and the other contributes to the mechanisms that control how frequently this event is to occur. For example, in the thymidine kinase gene of the herpes simplex virus, which utilizes transcription factors of its mammalian host for its early gene expression program, there is a single unique transcription start site, and accurate transcription from this start site depends upon a nucleotide sequence located 32 nucleotides upstream from the start site (ie, at -32) (Figure 36–7). This region has the sequence of TATAAAAG and bears remarkable similarity to the functionally related TATA box that is located about 10 bp upstream from the prokaryotic mRNA start site (Figure 36–5). Mutation or inactivation of the TATA box markedly reduces transcription of this and many other genes that contain this consensus cis-active element (see Figures 36-7 and 36-8). The TATA box is usually located 25-30 bp upstream from the transcription start site in mammalian genes that contain it. The consensus sequence for a TATA box is TATAAA, though numerous variations have been characterized. The human TATA box is bound by the 34 kDa TATA-binding protein (TBP), which is a subunit in at least two multisubunit complexes, TFIID and SAGA/P-CAF. The non-TBP subunits of TFIID are proteins called TBP-associated factors (TAFs). This complex of TBP and TAFs is referred to as TFIID. Binding of the TBP-TAF TFIID complex to the TATA box sequence is thought to represent a first step in the formation of the transcription complex on the promoter.
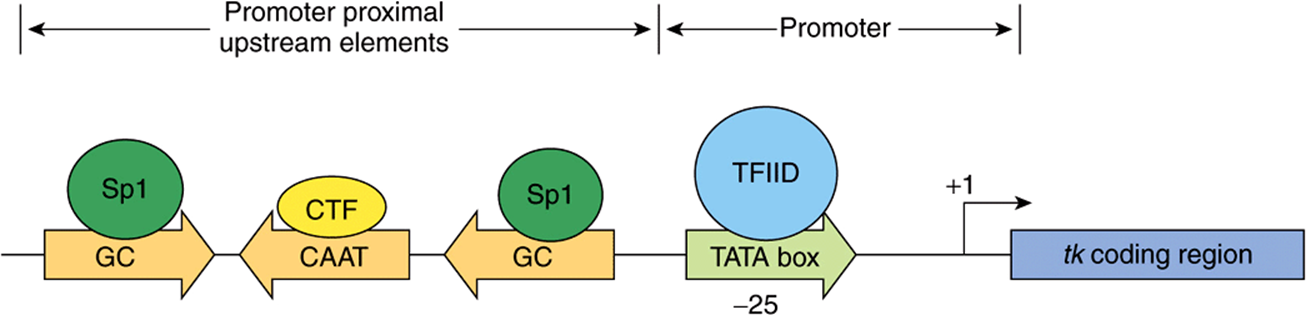
FIGURE 36–7 Transcription elements and binding factors in the herpes simplex virus thymidine kinase (tk) gene. DNA-dependent RNA polymerase II (not shown) binds to the region encompassing the TATA box (which is bound by transcription factor TFIID) and TSS at +1 (see also Figure 36–9) to form a multicomponent preinitiation complex capable of initiating transcription at a single nucleotide (+1). The frequency of this event is increased by the presence of upstream cis-acting elements (the GC and CAAT boxes) located either near to the promoter (promoter proximal) or distant from the promoter (distal elements; Figure 36–8). Proximal and distal DNA cis-elements are bound by trans-acting transcription factors, in this example Sp1 and CTF (also called C/EBP, NF1, NFY). These cis-elements can function independently of orientation (arrows).
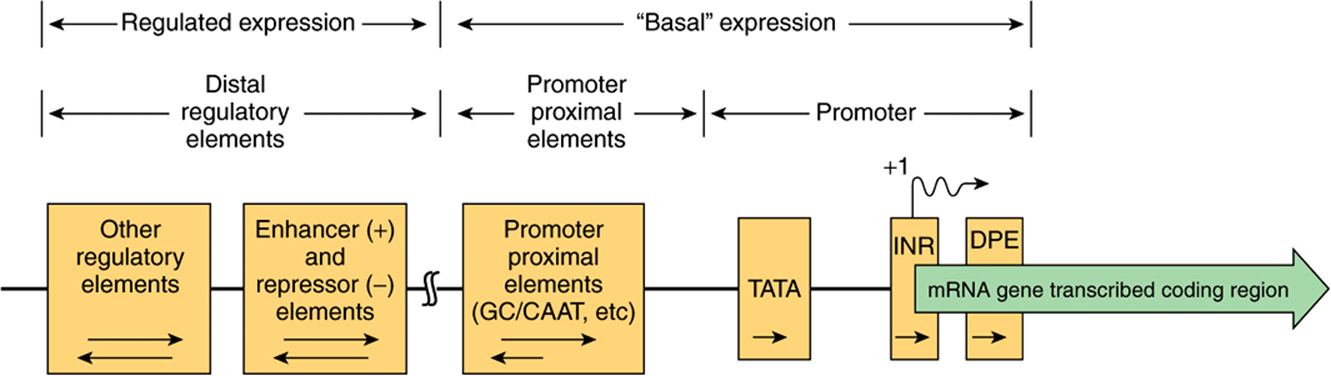
FIGURE 36–8 Schematic diagram showing the transcription control regions in a hypothetical mRNA-producing, eukaryotic gene transcribed by RNA polymerase II. Such a gene can be divided into its coding and regulatory regions, as defined by the transcription start site (arrow; +1). The coding region contains the DNA sequence that is transcribed into mRNA, which is ultimately translated into protein. The regulatory region consists of two classes of elements. One class is responsible for ensuring basal expression. The “promoter,” is often composed of the TATA box and/or Inr and/or DPE elements, directs RNA polymerase II to the correct site (fidelity). However, in certain genes that lack TATA, the so-called TATA-less promoters, an initiator (Inr) and/or DPE elements may direct the polymerase to this site. Another component, the upstream elements, specifies the frequency of initiation; such elements can either be proximal (50-200 bp) or distal (1000-105 bp) to the promoter as shown. Among the best studied of the proximal elements is the CAAT box, but several other elements (bound by the transactivator proteins Sp1, NF1, AP1, etc) may be used in various genes. The distal elements enhance or repress expression, several of which mediate the response to various signals, including hormones, heat shock, heavy metals, and chemicals. Tissue-specific expression also involves specific sequences of this sort. The orientation dependence of all the elements is indicated by the arrows within the boxes. For example, the proximal promoter elements (TATA box, INR, DPE) must be in the 5′-3’ orientation, while the proximal upstream elements often work best in the 5′-3’ orientation, but some can be reversed. The locations of some elements are not fixed with respect to the transcription start site. Indeed, some elements responsible for regulated expression can be located interspersed with the upstream elements or can be located downstream from the start site.
Some number of eukaryotic mRNA-encoding genes lack a consensus TATA box. In such instances, additional cis-elements, an initiator sequence (Inr) and/or the downstream promoter element (DPE), direct the RNA polymerase II transcription machinery to the promoter and in so doing provide basal transcription starting from the correct site. The Inr element spans the start site (from –3 to +5) and consists of the general consensus sequence TCA+1 G/T T T/C (A+1 indicates the first nucleotide transcribed). The proteins that bind to Inr in order to direct pol II binding include TFIID. Promoters that have both a TATA box and an Inr may be stronger or more vigorously transcribed than those that have just one of these elements. The DPE has the consensus sequence A/GGA/T CGTG and is localized about 25-bp downstream of the + 1 start site. Like the Inr, DPE sequences are also bound by the TAF subunits of TFIID. In a survey of thousands of eukaryotic protein coding genes, roughly 30% contained a TATA box and Inr, 25% contained Inr and DPE, 15% contained all three elements, whereas ~30% contained just the Inr.
Sequences generally, though not always, just upstream from the start site determine how frequently a transcription event occurs. Mutations in these regions reduce the frequency of transcriptional starts 10-fold to 20-fold. Typical of these DNA elements are the GC and CAAT boxes, so named because of the DNA sequences involved. As illustrated in Figure 36–7, each of these boxes binds a specific protein, Sp1 in the case of the GC box and CTF by the CAAT box; both bind through their distinct DNA-binding domains (DBDs). The frequency of transcription initiation is a consequence of these protein-DNA interactions and complex interactions between particular domains of the transcription factors (distinct from the DBD domains—so-called activation domains; ADs) of these proteins and the rest of the transcription machinery (RNA polymerase II, the basal, or general factors, GTFs, TFIIA, B, D, E, F, H and other coregulatory factors such as Mediator, chromatin remodellers and chromatin modifying factors). (See below and Figures 36-9 and 36-10.) The protein-DNA interaction at the TATA box involving RNA polymerase II and other components of the basal transcription machinery ensures the fidelity of initiation.
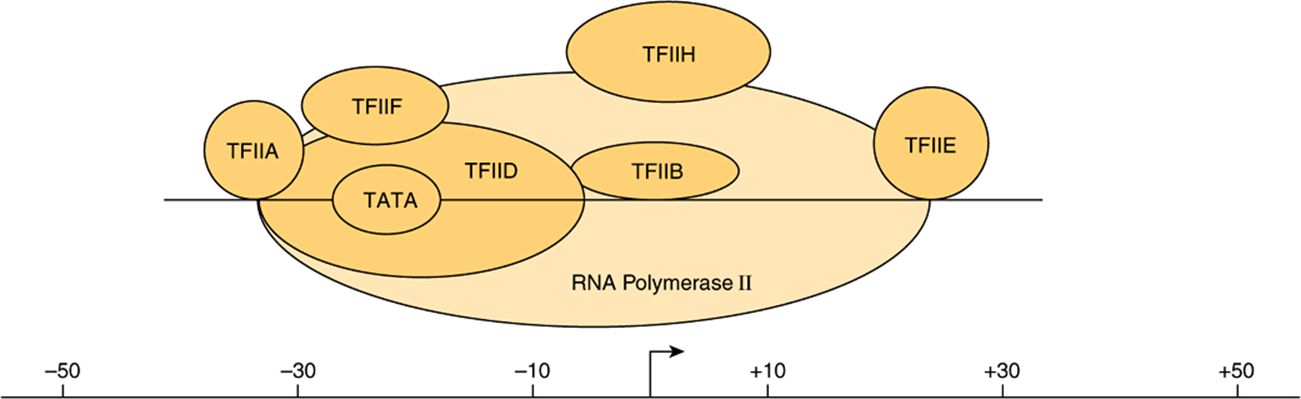
FIGURE 36–9 The eukaryotic basal transcription complex. Formation of the basal transcription complex begins when TFIID binds to the TATA box. It directs the assembly of several other components by protein-DNA and protein-protein interactions; TFIIA, B, E, F, H, and polymerase II (pol II). The entire complex spans DNA from position –30 to +30 relative to the transcription start site (TSS; +1, marked by bent arrow). The atomic level, X-ray-derived structures of RNA polymerase II alone and of the TBP subunit of TFIID bound to TATA promoter DNA in the presence of either TFIIB or TFIIA have all been solved at 3 Å resolution. The structures of TFIID and TFIIH complexes have been determined by electron microscopy at 30 Å resolution. Thus, the molecular structures of the transcription machinery are beginning to be elucidated. Much of this structural information is consistent with the models presented here.
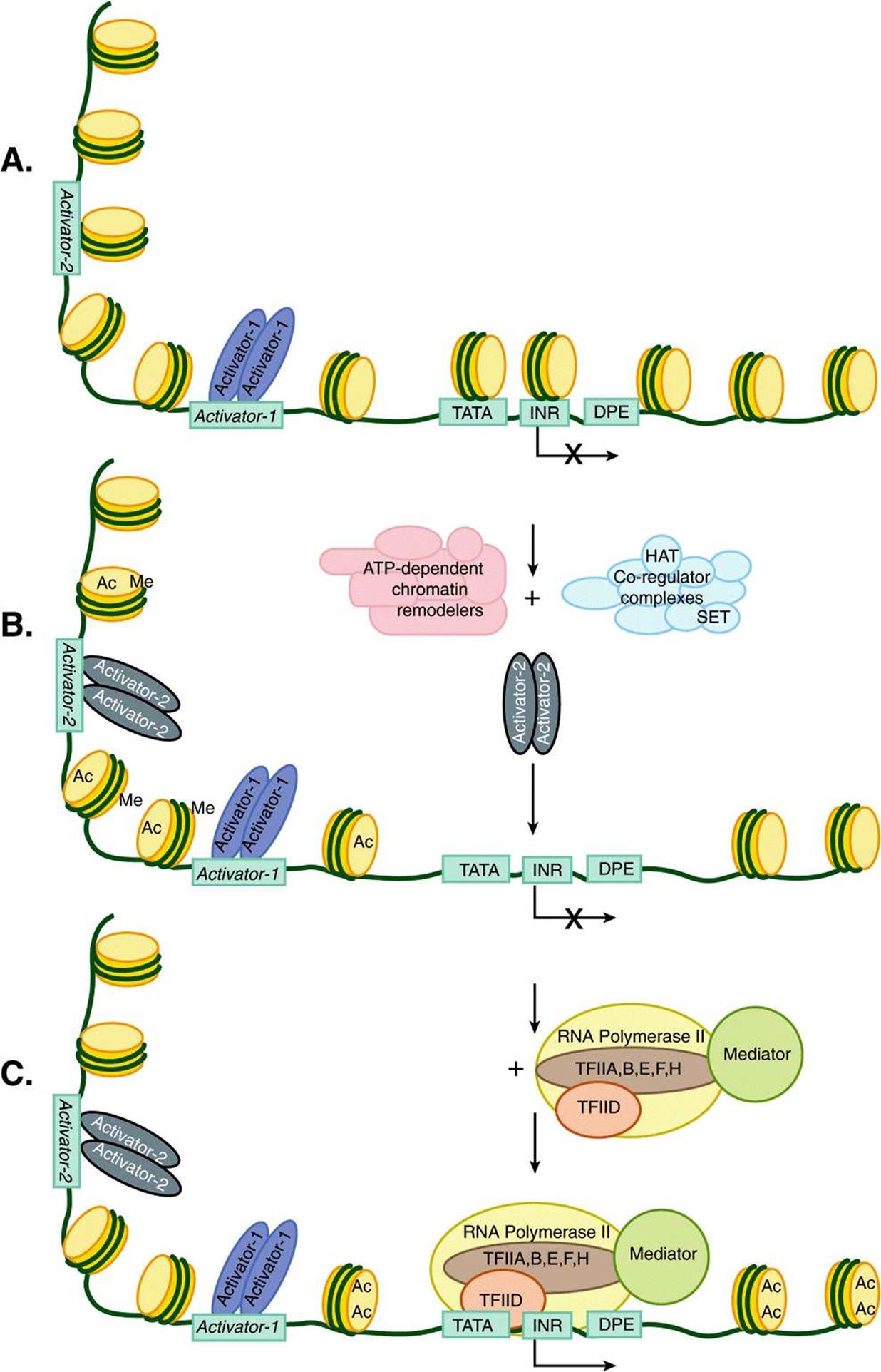
FIGURE 36–10 Nucleosome eviction by chromatin-active coregulators facilitates PIC formation and transcription. Shown in A, is an inactive mRNA encoding gene (see X over TSS) with a single dimeric transcription factor (Activator-1; violet ovals) bound to its cognate enhancer binding site (Activator-1). This particular enhancer element was nucleosome-free and hence available for interaction with its cognate activator binding protein. However, this gene is still inactive (X over TSS) due to the fact that a portion of its enhancer (in this illustration the enhancer is bipartitite and composed of Activator-1 and Activator-2, DNA-binding sites) and the entirety of the promoter are covered by nucleosomes. (B) Enhancer DNA-bound Activator-1 interacts with any of a number of distinct ATP-dependent chromatin remodelers and chromatin-modifying Co-regulator complexes. These coregulators together have the ability to both move and/or remove nucleosomes (ATP-dependent remodelers) as well as to covalently modify nucleosomal histones using intrinsic acetylases (HAT; resulting in acetylation [Ac]) and methylases (SET; resulting in methylation [Me], among other PTMs; Table 35-1), carried by subunits of these complexes. (C) The resulting changes in nucleosome position and nucleosome occupancy thus allow for the binding of the second Activator-2 dimer to Activator-2 DNA sequences, which leads to the binding of the transcription machinery (TFIIA,B,D,E,F,H; Polymerase II and Mediator) to the promoter (TATA-INR-DPE) and the formation of an active PIC, which leads to activated transcription.
Together, the promoter and promoter-proximal cis-active upstream elements confer fidelity and frequency of initiation upon a gene. The TATA box has a particularly rigid requirement for both position and orientation. As with bacterial promoters, single-base changes in any of these cis-elements can have dramatic effects on function by reducing the binding affinity of the cognate trans-factors (either TFIID/TBP or Sp1, CTF, and similar factors). The spacing of the TATA box, Inr, and DPE is also critical.
A third class of sequence elements can either increase or decrease the rate of transcription initiation of eukaryotic genes. These elements are called either enhancers or repressors (or silencers), depending on how they effect RNA synthesis. They have been found in a variety of locations both upstream and downstream of the transcription start site and even within the transcribed protein coding portions of some genes. Enhancers and silencers can exert their effects when located thousands or even tens of thousands of bases away from transcription units located on the same chromosome. Surprisingly, enhancers and silencers can function in an orientation-independent fashion. Literally, hundreds of these elements have been described. In some cases, the sequence requirements for binding are rigidly constrained; in others, considerable sequence variation is allowed. Some sequences bind only a single protein, but the majority bind several different proteins. Together, these many transfactors binding to promoter distal and proximal cis-elements regulate transcription in response to a vast array of biological signals. Such transcriptional regulatory events contribute importantly to control of gene expression.
Specific Signals Regulate Transcription Termination
The signals for the termination of transcription by eukaryotic RNA polymerase II are only poorly understood. It appears that the termination signals exist far downstream of the coding sequence of eukaryotic genes. For example, the transcription termination signal for mouse β-globin occurs at several positions 1000-2000 bases beyond the site at which the mRNA poly(A) tail will eventually be added. Less is known about the termination process or whether specific termination factors similar to the bacterial ρ factor are involved. However, it is known that formation of the mRNA 3’ terminal, which is generated posttranscriptionally, is somehow coupled to events or structures formed at the time and site of initiation. Moreover, mRNA formation, and in this case mRNA 3’-end formation depends on a special structure present on the C-terminus of the largest subunit of RNA polymerase II, the C-terminal domain, or CTD (see below), and this process appears to involve at least two steps as follows. After RNA polymerase II has traversed the region of the transcription unit encoding the 3′ end of the transcript, RNA endonucleases cleave the primary transcript at a position about 15 bases 3′ of the consensus sequence AAUAAA that serves in eukaryotic transcripts as a cleavage and polyadenylation signal. Finally, this newly formed 3’ terminal is polyadenylated in the nucleoplasm, as described below.
THE EUKARYOTIC TRANSCRIPTION COMPLEX
A complex apparatus consisting of as many as 50 unique proteins provides accurate and regulatable transcription of eukaryotic genes. The RNA polymerase enzymes (pol I, pol II, and pol III) transcribe information contained in the template strand of DNA into RNA. These polymerases must recognize a specific site in the promoter in order to initiate transcription at the proper nucleotide. In contrast to the situation in prokaryotes though, in vitro eukaryotic RNA polymerases alone are not able to discriminate between promoter sequences and other, nonpromoter regions of DNA. All eukaryotic RNA polymerase forms require other proteins known as general transcription factors or GTFs. RNA polymerase II requires TFIIA, B, D (or TBP), E, F, and H to both facilitate promoter-specific binding of the enzyme and formation of the preinitiation complex (PIC). RNA polymerases I and III require their own polymerase-specific GTFs. Moreover, RNA polymerase II and GTFs do not respond to activator proteins and can only catalyze basal or (non)-unregulated transcription in vitro. Another set of proteins—the coactivators, or coregulators—work in conjunction with DNA-binding transactivator proteins to communicate with Pol II/GTFs to regulate the rate of transcription (see below).
Formation of the Pol II Transcription Complex
In bacteria, a σ-factor-polymerase holoenzyme complex, Eσ, selectively binds to promoter DNA to form the PIC. The situation is much more complex in eukaryotic genes. mRNA-encoding genes, which are transcribed by pol II, are described as an example. In the case of pol II-transcribed genes, the function of σ-factors is assumed by a number of proteins. PIC formation requires pol II and the six GTFs (TFIIA, TFIIB, TFIID, TFIIE, TFIIF, TFIIH). These GTFs serve to promote RNA polymerase II transcription on essentially all genes. Some of these GTFs are composed of multiple subunits. TFIID, which binds to the TATA box promoter element through its TBP subunit, is the only one of these factors that is independently capable of specific, high affinity binding to promoter DNA. TFIID consists of 15 subunits, TBP and 14 TBP Associated Factors, or TAFs.
TBP binds to the TATA box in the minor groove of DNA (most transcription factors bind in the major groove) and causes an approximately 100-degree bend or kink of the DNA helix. This bending is thought to facilitate the interaction of TAFs with other components of the transcription initiation complex, the multicomponent eukaryotic promoter and possibly with factors bound to upstream elements. Although initially defined as a component solely required for transcription of pol II gene promoters, TBP, by virtue of its association with distinct, polymerase-specific sets of TAFs, is also an important component of pol I and pol III transcription initiation complexes even if they do not contain TATA boxes.
The binding of TFIID marks a specific promoter for transcription. Of several subsequent in vitro steps, the first is the binding of TFIIA, then TFIIB to the TFIID-promoter complex. This results in a stable ternary complex, which is then more precisely located and more tightly bound at the transcription initiation site. This complex then attracts and tethers the pol II-TFIIF complex to the promoter. Addition of TFIIE and TFIIH are the final steps in the assembly of the PIC. TFIIE appears to join the complex with pol II-TFIIF, and TFIIH is then recruited. Each of these binding events extends the size of the complex so that finally about 60 bp (from –30 to +30 relative to +1, the nucleotide from which transcription commences) are covered (Figure 36–9). The PIC is now complete and capable of basal transcription initiated from the correct nucleotide. In genes that lack a TATA box, the same factors are required. In such cases, the Inr or DPE serve to (see Figure 36–8) position the complex for accurate initiation of transcription.
Promoter Accessibility and Hence PIC Formation Is Often Modulated by Nucleosomes
On certain eukaryotic genes, the transcription machinery (pol II, etc) cannot access the promoter sequences (ie, TATA-INR-DPE) because these essential promoter elements are wrapped up in nucleosomes (Figures 35-2 and 35-3 and 36-10). Only after transcription factors bind to enhancer DNA upstream of the promoter and recruit chromatin remodeling and modifying coregulatory factors such as the Swi/Snf, SRC-1, p300/CBP (see Chapter 42,) or P/CAF factors, are the repressing nucleosomes removed (Figure 36–10). Once the promoter is “open” following nucleosome eviction, GTFs and RNA polymerase II can bind and initiate mRNA gene transcription. Note that the binding of transactivators and coregulators can be sensitive to, and/or directly control the covalent modification status of the histones within the nucleosomes in and around the promoter and enhancer, and thereby increase or decrease the ability of all the other components required for PIC formation to interact with a particular gene. This so-called epigenetic code of histone and protein modifications can contribute importantly to gene transcription control. Indeed, mutations in proteins that catalyze (code writers), remove (code erasers), or differentially bind (code readers) modified histones can lead to human disease.
Phosphorylation Activates Pol II
Eukaryotic pol II consists of 12 subunits. The two largest subunits (MW 150 and 190 kDa) are homologous to the bacterial β and β’ subunits. In addition to the increased number of subunits, eukaryotic pol II differs from its prokaryotic counterpart in that it has a series of heptad repeats with consensus sequence Tyr-Ser-Pro-Thr-Ser-Pro-Ser at the carboxyl terminus of the largest pol II subunit. This carboxyl terminal repeat domain (CTD) has 26 repeated units in brewers’ yeast and 52 units in mammalian cells. The CTD is a substrate for several enzymes (kinases, phosphatases, prolyl isomerases, glycosylases), Phosphorylation of the CTD was the first CTD PTM discovered. The kinase component of TFIIH can modify the CTD. Covalently modified CTD is binding site for a wide array of proteins, and it has been shown to interact with many mRNA modifying and processing enzymes and nuclear transport proteins. The association of these factors with the CTD of RNA polymerase II (and other components of the basal machinery) thus serves to couple transcription initiation with mRNA capping splicing, 3′ end formation and transport to the cytoplasm (see below). Pol II polymerization is activated when phosphorylated on the Ser and Thr residues and displays reduced activity when the CTD is dephosphorylated. CTD phosphorylation/dephosphorylation is critical for promoter clearance, elongation, termination, and even appropriate mRNA processing. Pol II lacking the CTD tail is incapable of activating transcription, and cells expressing pol II lacking the CTD are inviable. These results underscore the importance of this domain.
Pol II can associate with other proteins termed Mediator or Med proteins to form a complex sometimes referred to as the pol II holoenzyme; this complex can form on the promoter or in solution prior to PIC formation (see below). The Med proteins are essential for appropriate regulation of pol II transcription by serving myriad roles, both activating and repressing transcription. Thus Mediator, like TFIID is a transcriptional coregulator (Figure 36–11).Complex forms of RNA polymerase II holoenzyme (pol II plus Med) have been described in human cells that contain over 30 Med proteins (Med1-Med31).
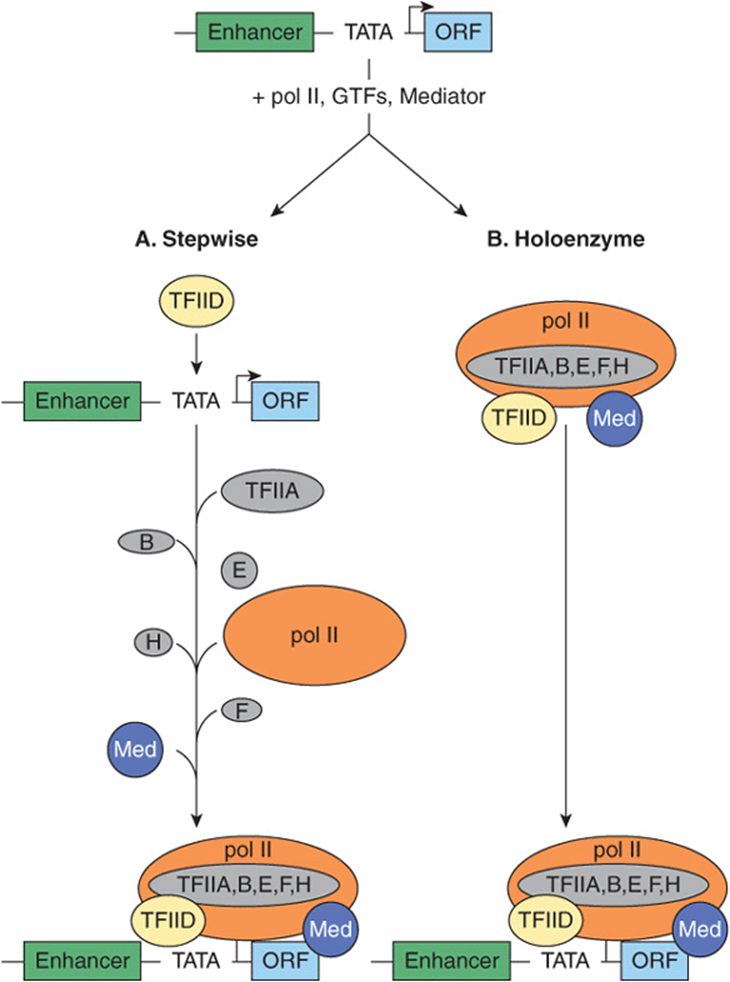
FIGURE 36–11 Models for the formation of an RNA polymerase II preinitiation complex. Shown at top is a typical mRNA encoding transcription unit: enhancer-promoter (TATA)-initiation site (bent arrow) and transcribed region (ORF; open reading frame). PICs have been shown to form by at least two distinct mechanisms: (A) the stepwise binding of GTFs, pol II, and Mediator, or (B) by the binding of a single multiprotein complex composed of pol II, Med, and the six GTFs. DNA-binding transactivator proteins specifically bind enhancers and in part facilitate PIC formation (or PIC function) by binding directly to the TFIID-TAF subunits or Med subunits of Mediator (not shown, see Figure 36–10); the molecular mechanism(s) by which such protein-protein interactions stimulate transcription remain a subject of intense investigation.
The Role of Transcription Activators & Coregulators
TFIID was originally considered to be a single protein, TBP. However, several pieces of evidence led to the important discovery that TFIID is actually a complex consisting of TBP and the 14 TAFs. The first evidence that TFIID was more complex than just the TBP molecules came from the observation that TBP binds to a 10-bp segment of DNA, immediately over the TATA box of the gene, whereas native holo-TFIID covers a 35 bp or larger region (Figure 36–9). Second, TBP has a molecular mass of 20-40 kDa (depending on the species), whereas the TFIID complex has a mass of about 1000 kDa. Finally, and perhaps most importantly, TBP supports basal transcription but not the augmented transcription provided by certain activators, for example, Sp1 bound to the GC box. TFIID, on the other hand, supports both basal and enhanced transcription by Sp1, Oct1, AP1, CTF, ATF, etc. (Table 36-3). The TAFs are essential for this activator-enhanced transcription. There are likely several forms of TFIID that differ slightly in their complement of TAFs. Thus different combinations of TAFs with TBP—or one of several recently discovered TBP-like factors (TLFs)—bind to different promoters, and recent reports suggest that this may account for the tissue or cell-selective gene activation noted in various promoters and for the different strengths of certain promoters. TAFs, since they are required for the action of activators, are often called coactivators or coregulators. There are thus three classes of transcription factors involved in the regulation of pol II genes: pol II and GTFs, coregulators, and DNA-binding activator-repressors (Table 36-4). How these classes of proteins interact to govern both the site and frequency of transcription is a question of central importance and active investigation. It is currently thought that coregulators both act as a bridge between the DNA-binding transactivators and pol II/GTFs and modify chromatin.
TABLE 36–3 Some of the Mammalian RNA Polymerase II Transcription Control Elements, Their Consensus Sequences, and the Factors That Bind to Them
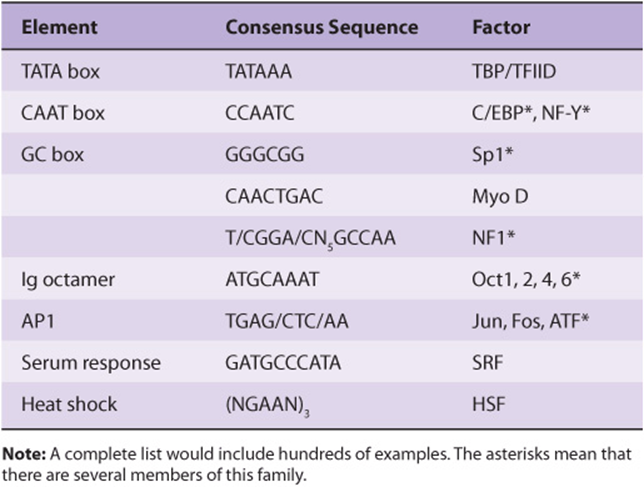
TABLE 36–4 Three Classes of Transcription Factors Involved in mRNA Gene Transcription

Two Models Can Explain the Assembly of the Preinitiation Complex
The formation of the PIC described above is based on the sequential addition of purified components as observed through in vitro experiments. An essential feature of this model is that PIC assembly takes place on a DNA template where the transcription proteins all have ready access to DNA. Accordingly, transcription activators, which have autonomous DNA binding and activation domains (see Chapter 38), are thought to function by stimulating PIC formation. Here the TAF or mediator complexes are viewed as bridging factors that communicate between the upstream-bound activators, and the GTFs and pol II. This view assumes that there is stepwise assembly of the PIC—promoted by various interactions between activators, coactivators, and PIC components, and is illustrated in panel A of Figure 36–11. This model was supported by observations that many of these proteins can indeed bind to one another in vitro.
Recent evidence suggests that there is another possible mechanism of PIC formation and thus transcription regulation. First, large preassembled complexes of GTFs and pol II are found in cell extracts, and these complexes can associate with the promoter in a single step. Second, the rate of transcription achieved when activators are added to limiting concentrations of pol II holoenzyme can be matched by increasing the concentration of the pol II holoenzyme in the absence of activators. Thus, at least in vitro, one can establish conditions where activators are not in themselves absolutely essential for PIC formation. These observations led to the “recruitment” hypothesis, which has now been tested experimentally. Simply stated, the role of activators and some coactivators may be solely to recruit a preformed holoenzyme-GTF complex to the promoter. The requirement for an activation domain is circumvented when either a component of TFIID or the pol II holoenzyme is artificially tethered, using recombinant DNA techniques, to the DBD of an activator. This anchoring, through the DBD component of the activator molecule, leads to a transcriptionally competent structure, and there is no further requirement for the activation domain of the activator. In this view, the role of activation domains is to direct preformed holoenzyme-GTF complexes to the promoter; they do not assist in PIC assembly (see panel B, Figure 36–11). In this model, the efficiency of the recruitment process directly determines the rate of transcription at a given promoter.
RNA MOLECULES ARE PROCESSED BEFORE THEY BECOME FUNCTIONAL
In prokaryotic organisms, the primary transcripts of mRNA-encoding genes begin to serve as translation templates even before their transcription has been completed. This can occur because the site of transcription is not compartmentalized into a nucleus as it is in eukaryotic organisms. Thus, transcription and translation are coupled in prokaryotic cells. Consequently, prokaryotic mRNAs are subjected to little processing prior to carrying out their intended function in protein synthesis. Indeed, appropriate regulation of some genes (eg, the Trp operon) relies upon this coupling of transcription and translation. Prokaryotic rRNA and tRNA molecules are transcribed in units considerably longer than the ultimate molecule. In fact, many of the tRNA transcription units encode more than one tRNA molecule. Thus, in prokaryotes, the processing of these rRNA and tRNA precursor molecules is required for the generation of the mature functional molecules.
Nearly all eukaryotic RNA primary transcripts undergo extensive processing between the time they are synthesized and the time at which they serve their ultimate function, whether it be as mRNA, miRNAs, or as a component of the translation machinery such as rRNA or tRNA. Processing occurs primarily within the nucleus. The processes of transcription, RNA processing, and even RNA transport from the nucleus, are highly coordinated. Indeed, a transcriptional coactivator termed SAGA in yeasts and P/CAF in human cells is thought to link transcription activation to RNA processing by recruiting a second complex termed TREX to transcription elongation, splicing, and nuclear export. TREX (transcription-export) represents a likely molecular link between transcription elongation complexes, the RNA splicing machinery, and nuclear export (see Figure 36–12). This coupling presumably dramatically increases both the fidelity and rate of processing and movement of mRNA to the cytoplasm for translation.
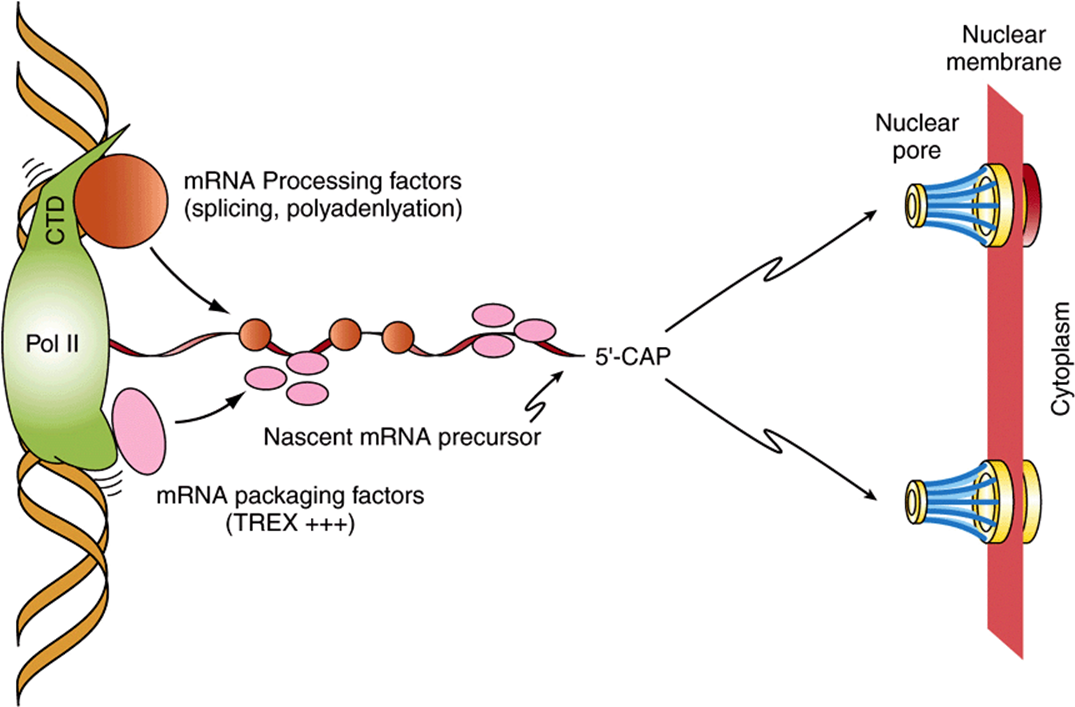
FIGURE 36–12 RNA polymerase II-mediated mRNA gene transcription is cotranscriptionally coupled to RNA processing and transport. Shown is RNA pol II actively transcribing an mRNA encoding gene (elongation top to bottom of figure). RNA processing factors (ie, SR/RNP-motif-containing splicing factors as well as polyadenylation and termination factors) interact with the C-terminal domain (CTD) of pol II, while mRNA packaging factors such as THO/TREX complex are recruited to the nascent mRNA primary transcript either through direct pol II interactions as shown or through interactions with SR/splicing factors resident on the nascent mRNA. Note that the CTD is not drawn to scale. The evolutionarily conserved CTD of the Rpb1 subunit of pol II is in reality 5-10 times the length of the polymerase due to its many prolines and consequent unstructured nature, and thus a significant docking site for RNA processing and transport proteins. In both cases, nascent mRNA chains are thought to be more rapidly and accurately processed due to the rapid recruitment of these many factors to the growing mRNA (precursor) chain. Following appropriate mRNA processing, the mature mRNA is delivered to the nuclear pores dotting the nuclear membrane, where, upon transport through the pores, the mRNAs can be engaged by ribosomes and translated into protein. (Adapted from Jensen et al: Mol Cell. 2005;11:1129-1138.)
The Coding Portions (Exons) of Most Eukaryotic Genes Are Interrupted by Introns
The RNA sequences that appear in mature RNAs are termed exons. In mRNA encoding genes, exons are often interrupted by long sequences of DNA that neither appear in mature mRNA, nor contribute to the genetic information ultimately translated into the amino acid sequence of a protein molecule (see Chapter 35). In fact, these sequences often interrupt the coding region of structural genes. These intervening sequences, or introns, exist within most but not all mRNA encoding genes of higher eukaryotes. In human mRNA encoding genes, exons average ~150 nt, while introns are much more heterogenous, ranging from 10-100 nt to 30,000 nucleotides in length. The intron RNA sequences are cleaved out of the transcript, and the exons of the transcript are appropriately spliced together in the nucleus before the resulting mRNA molecule appears in the cytoplasm for translation (Figures 36-13 and 36-14). One speculation for this exon-intron gene organization is that exons, which often encode an activity domain, or functional module of a protein, represent a convenient means of shuffling genetic information, permitting organisms to quickly test the results of combining novel protein functional domains.
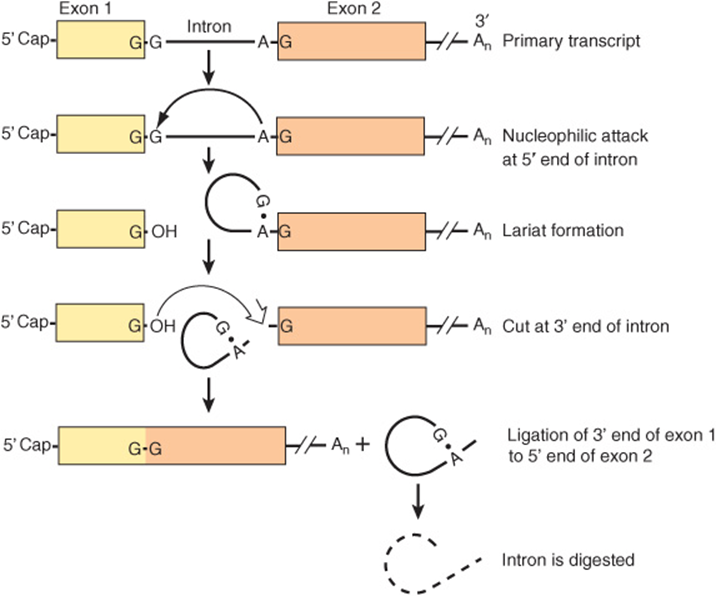
FIGURE 36–13 The processing of the primary transcript to mRNA. In this hypothetical transcript, the 5′ (left) end of the intron is cut (↓) and a lariat forms between the G at the 5′ end of the intron and an A near the 3′ end, in the consensus sequence UACUAAC. This sequence is called the branch site, and it is the 3′ most A that forms the 5′-2’ bond with the G. The 3′ (right) end of the intron is then cut (⇓). This releases the lariat, which is digested, and exon 1 is joined to exon 2 at G residues.
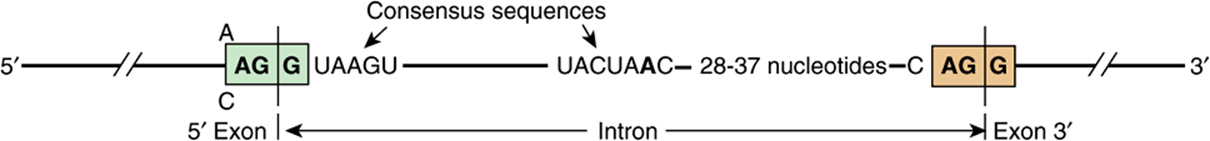
FIGURE 36–14 Consensus sequences at splice junctions. The 5′ (donor; left) and 3′ (acceptor; right) sequences are shown. Also shown is the yeast consensus sequence (UACUAAC) for the branch site. In mammalian cells, this consensus sequence is PyNPyPyPuAPy, where Py is a pyrimidine, Pu is a purine, and N is any nucleotide. The branch site is located 20-40 nucleotides upstream from the 3′ splice site.
Introns Are Removed & Exons Are Spliced Together
Several different splicing reaction mechanisms for intron removal have been described. The one most frequently used in eukaryotic cells is described below. Although the sequences of nucleotides in the introns of the various eukaryotic transcripts—and even those within a single transcript—are quite heterogeneous, there are reasonably conserved sequences at each of the two exon-intron (splice) junctions and at the branch site, which is located 20-40 nucleotides upstream from the 3′ splice site (see consensus sequences in Figure 36–14). A special multicomponent complex, the spliceosome, is involved in converting the primary transcript into mRNA. Spliceosomes consist of the primary transcript, five snRNAs (U1, U2, U4, U5, and U6) and more than 60 proteins, many of which contain conserved “RNP” and “SR” protein motifs. Collectively, the five snRNAs and RNP/SR-containing proteins form a small nuclear ribonucleoprotein termed an snRNA complex. It is likely that this penta-snRNP spliceosome forms prior to interaction with mRNA precursors. snRNPs are thought to position the exon and intron RNA segments for the necessary splicing reactions. The splicing reaction starts with a cut at the junction of the 5′-exon (donor or left) and intron (Figure 36–13). This is accomplished by a nucleophilic attack by an adenylyl residue in the branch point sequence located just upstream from the 3′ end of this intron. The free 5′ terminal then forms a loop or lariat structure that is linked by an unusual 5′-2’ phosphodiester bond to the reactive A in the PyNPyPyPuAPy branch site sequence (Figure 36–14). This adenylyl residue is typically located 20-30 nucleotides upstream from the 3′ end of the intron being removed. The branch site identifies the 3′ splice site. A second cut is made at the junction of the intron with the 3′ exon (donor on right). In this second transesterification reaction, the 3′ hydroxyl of the upstream exon attacks the 5′ phosphate at the downstream exon-intron boundary, and the lariat structure containing the intron is released and hydrolyzed. The 5′ and 3′ exons are ligated to form a continuous sequence.
The snRNAs and associated proteins are required for formation of the various structures and intermediates. U1 within the snRNP complex binds first by base pairing to the 5′ exonintron boundary. U2 within the snRNP complex then binds by base pairing to the branch site, and this exposes the nucleophilic A residue. U4/U5/U6 within the snRNP complex mediates an ATP-dependent protein-mediated unwinding that results in disruption of the base-paired U4-U6 complex with the release of U4. U6 is then able to interact first with U2, then with U1. These interactions serve to approximate the 5′ splice site, the branch point with its reactive A, and the 3′ splice site. This alignment is enhanced by U5. This process also results in the formation of the loop or lariat structure. The two ends are cleaved, probably by the U2-U6 within the snRNP complex. U6 is certainly essential, since yeasts deficient in this snRNA are not viable. It is important to note that RNA serves as the catalytic agent. This sequence of events is then repeated in genes containing multiple introns. In such cases, a definite pattern is followed for each gene, though the introns are not necessarily removed in sequence—1, then 2, then 3, etc.
Alternative Splicing Provides for Different mRNAs
The processing of mRNA molecules is a site for regulation of gene expression. Alternative patterns of mRNA splicing result from tissue-specific adaptive and developmental control mechanisms. Interestingly, recent studies suggest that alternative splicing is controlled, at least in part, through chromatin epigenetic marks (ie, Table 35-1). This form of coupling of transcription and mRNA processing may either be kinetic and/or mediated through interactions between specific histone PTMs and alternative splicing factors that can load onto nascent mRNA gene transcripts during the process of transcription (Figure 36–12).
As mentioned above, the sequence of exon-intron splicing events generally follows a hierarchical order for a given gene. The fact that very complex RNA structures are formed during splicing—and that a number of snRNAs and proteins are involved—affords numerous possibilities for a change of this order and for the generation of different mRNAs. Similarly, the use of alternative termination-cleavage polyadenylation sites also results in mRNA variability. Some schematic examples of these processes, all of which occur in nature, are shown in Figure 36–15.
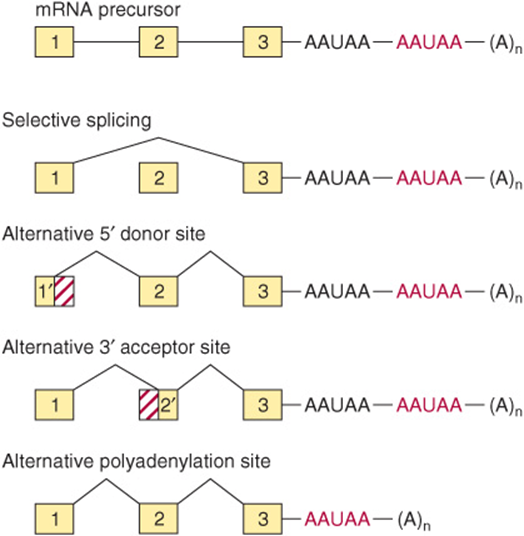
FIGURE 36–15 Mechanisms of alternative processing of mRNA precursors. This form of mRNA processing involves the selective inclusion or exclusion of exons, the use of alternative 5′ donor or 3′ acceptor sites, and the use of different polyadenylation sites.
Faulty splicing can cause disease. At least one form of β-thalassemia, a disease in which the β-globin gene of hemoglobin is severely underexpressed, appears to result from a nucleotide change at an exon-intron junction, precluding removal of the intron and therefore leading to diminished or absent synthesis of the β-chain protein. This is a consequence of the fact that the normal translation reading frame of the mRNA is disrupted by a defect in the fundamental process of RNA splicing, underscoring the accuracy that the process of RNA-RNA splicing must maintain.
Alternative Promoter Utilization Provides a Form of Regulation
Tissue-specific regulation of gene expression can be provided by alternative splicing, as noted above, by control elements in the promoter or by the use of alternative promoters. The glucokinase (GK) gene consists of 10 exons interrupted by 9 introns. The sequence of exons 2-10 is identical in liver and pancreatic β cells, the primary tissues in which GK protein is expressed. Expression of the GK gene is regulated very differently—by two different promoters—in these two tissues. The liver promoter and exon 1L are located near exons 2-10; exon 1L is ligated directly to exon 2. By contrast, the pancreatic β-cell promoter is located about 30 kbp upstream. In this case, the 3′ boundary of exon 1B is ligated to the 5′ boundary of exon 2. The liver promoter and exon 1L are excluded and removed during the splicing reaction (see Figure 36–16). The existence of multiple distinct promoters allows for cell-and tissue-specific expression patterns of a particular gene (mRNA). In the case of GK, insulin and cAMP (Chapter 42) control GK transcription in liver, while glucose controls GK expression in β cells.
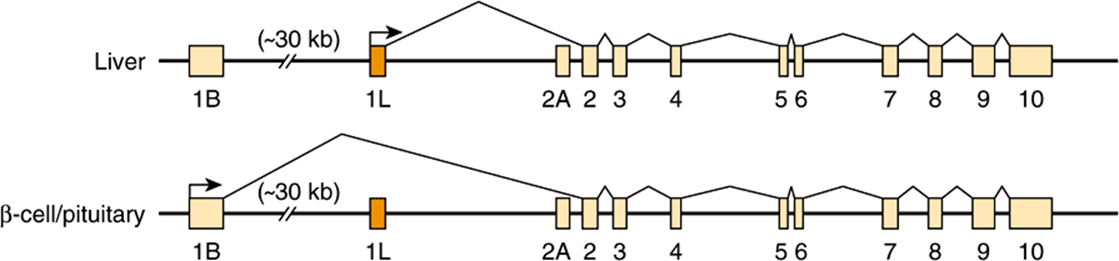
FIGURE 36–16 Alternative promoter use in the liver and pancreatic β-cell glucokinase (GK) genes. Differential regulation of the glucokinase gene is accomplished by the use of tissue-specific promoters. The β-cell GK gene promoter and exon 1B are located about 30 kbp upstream from the liver promoter and exon 1L. Each promoter has a unique structure and is regulated differently. Exons 2-10 are identical in the two genes, and the GK proteins encoded by the liver and β-cell mRNAs have identical kinetic properties.
Both Ribosomal RNAs & Most Transfer RNAs Are Processed from Larger Precursors
In mammalian cells, the three rRNA molecules (28S, 18S, 5.8S) are transcribed as part of a single large 45S precursor molecule. The precursor is subsequently processed in the nucleolus to provide these three RNA components for the ribosome subunits found in the cytoplasm. The rRNA genes are located in the nucleoli of mammalian cells. Hundreds of copies of these genes are present in every cell. This large number of genes is required to synthesize sufficient copies of each type of rRNA to form the 107 ribosomes required for each cell replication. Whereas a single mRNA molecule may be copied into 105 protein molecules, providing a large amplification, the rRNAs are end products. This lack of amplification requires both a large number of genes and a high transcription rate, typically synchronized with cell growth rate. Similarly, tRNAs are often synthesized as precursors, with extra sequences both 5′ and 3′ of the sequences comprising the mature tRNA. A small fraction of tRNAs contain introns.
RNAs CAN BE EXTENSIVELY MODIFIED
Essentially all RNAs are covalently modified after transcription. It is clear that at least some of these modifications are regulatory.
Messenger RNA Is Modified at the 5′ & 3′ Ends
As mentioned above, mammalian mRNA molecules contain a 7-methylguanosine cap structure at their 5′ terminal (Figure 34–10), and most have a poly(A) tail at the 3′ terminal. The cap structure is added to the 5′ end of the newly transcribed mRNA precursor in the nucleus prior to transport of the mRNA molecule to the cytoplasm. The 5’ cap of the RNA transcript is required both for efficient translation initiation and protection of the 5′ end of mRNA from attack by 5′ → 3′ exonucleases. The secondary methylations of mRNA molecules, those on the 2′-hydroxy and the N7 of adenylyl residues, occur after the mRNA molecule has appeared in the cytoplasm.
Poly(A) tails are added to the 3′ end of mRNA molecules in a posttranscriptional processing step. The mRNA is first cleaved about 20 nucleotides downstream from an AAUAA recognition sequence. Another enzyme, poly(A) polymerase, adds a poly(A) tail which is subsequently extended to as many as 200 A residues. The poly(A) tail appears to protect the 3′ end of mRNA from 3′ → 5′ exonuclease attack and facilitate translation. The presence or absence of the poly(A) tail does not determine whether a precursor molecule in the nucleus appears in the cytoplasm, because all poly(A)-tailed nuclear mRNA molecules do not contribute to cytoplasmic mRNA, nor do all cytoplasmic mRNA molecules contain poly(A) tails (histone mRNAs are most notable in this regard). Following nuclear transport cytoplasmic enzymes in mammalian cells can both add and remove adenylyl residues from the poly(A) tails; this process has been associated with an alteration of mRNA stability and translatability.
The size of some cytoplasmic mRNA molecules, even after the poly(A) tail is removed, is still considerably greater than the size required to code for the specific protein for which it is a template, often by a factor of 2 or 3. The extra nucleotides occur in untranslated (nonprotein coding) regions both 5′ and 3′ of the coding region; the longest untranslated sequences are usually at the 3′ end. The exact function of 5’ UTR and 3′ UTR sequences is unknown, but they have been implicated in RNA processing, transport, storage, degradation, and translation; each of these reactions potentially contributes additional levels of control of gene expression. The micro-RNAs typically target sequences within the 3′ UTR. Many of these posttranscriptional events involving mRNAs occur in cytoplasmic organelles termed P bodies (Chapter 37).
Micro-RNAs Are Derived from Large Primary Transcripts Through Specific Nucleolytic Processing
The majority of miRNAs are transcribed by RNA pol II into primary transcripts termed pri-miRNAs. pri-miRNAs are 5′-capped and 3′-polyadenylated (Figure 36–17). pri-miRNAs are synthesized from transcription units encoding one or several distinct miRNAs; these transcription units are either located independently in the genome or within the intronic DNA of other genes. miRNA-encoding genes must therefore minimally posess a distinct promoter, coding region and polyadenylation/termination signals. pri-miRNAs have extensive 2° structure, and this intramolecular structure is maintained following processing by the Drosha-DGCR8 nuclease; the portion containing the RNA hairpin is preserved, transported through the nuclear pore and once in the cytoplasm, further processed to a 21 or 22-mer by the dicer nuclease. Ultimately, one of the two strands is selected for loading into the RISC, or RNA-induced silencing complex to form a mature, functional miRNA. siRNAs are produced similarly. Once in the RISC complex, miRNAs can modulate mRNA function (see Chapter 39). Recent data suggest that regulatory miRNA-encoding genes may be linked, and hence co-evolve with their target genes.
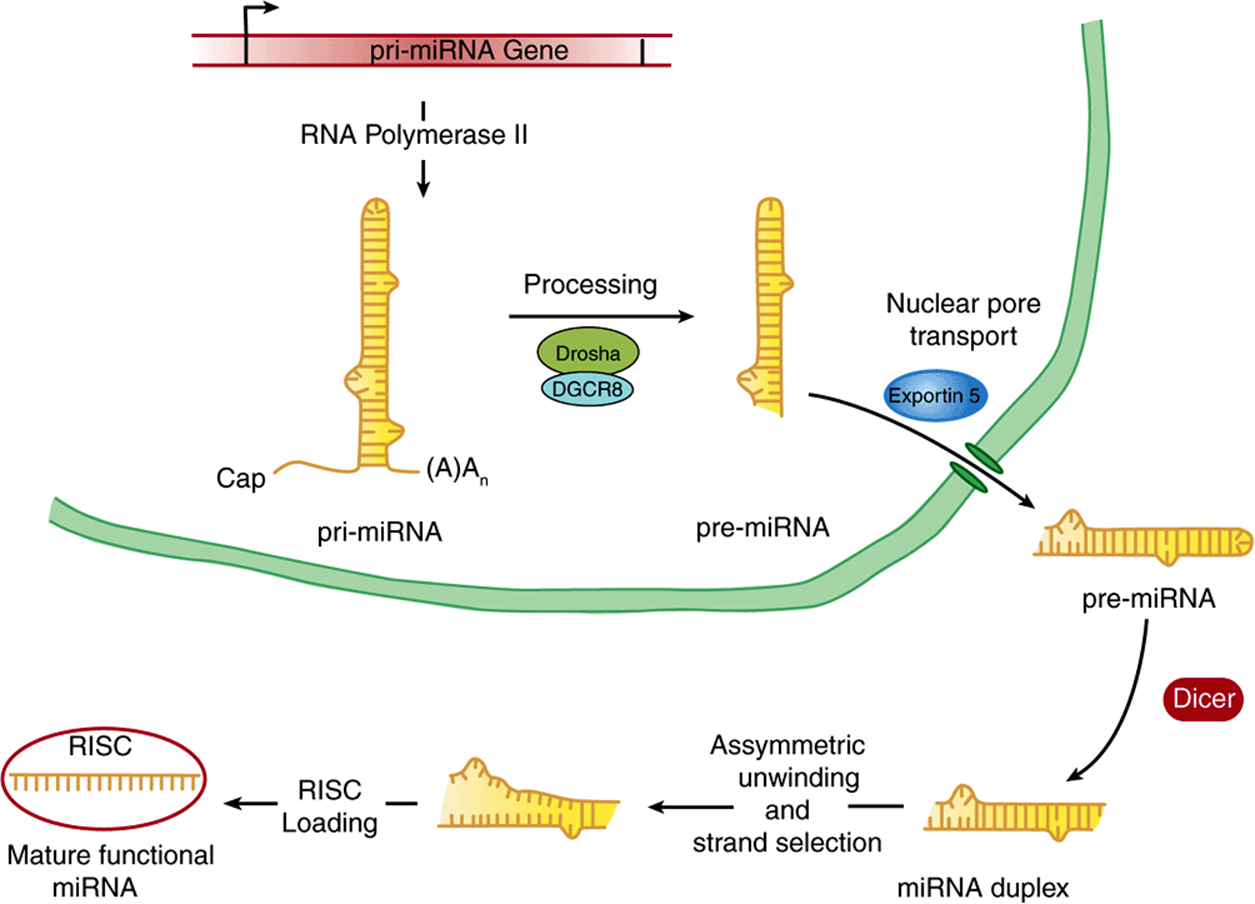
FIGURE 36–17 Biogenesis of miRNAs. miRNA encoding genes are transcribed by RNA pol II into a primary miRNA transcript (pri-miRNA), which is 5′-capped and polyadenylated as is typical of mRNA coding primary transcripts. This pri-miRNA is subjected to processing within the nucleus by the action of the Drosha-DGCR8 nuclease, which trims sequences from both 5′ and 3′ ends generating the pre-miRNA. This partially processed double-stranded RNA is transported through the nuclear pore by exportin-5. The cytoplasmic pre-miRNA is then trimmed further by the action of the multisubunit nuclease termed Dicer, to form the miRNA duplex. One of the two resulting 21-22 nucleotide-long RNA strands is selected, the duplex unwound, and the selected strand loaded into the RISC complex, thereby generating the mature, functional miRNA.
RNA Editing Changes mRNA After Transcription
The central dogma states that for a given gene and gene product there is a linear relationship between the coding sequence in DNA, the mRNA sequence, and the protein sequence (Figure 35–7). Changes in the DNA sequence should be reflected in a change in the mRNA sequence and, depending on codon usage, in protein sequence. However, exceptions to this dogma have been recently documented. Coding information can be changed at the mRNA level by RNA editing. In such cases, the coding sequence of the mRNA differs from that in the cognate DNA. An example is the apolipoprotein B (apoB) gene and mRNA. In liver, the single apoB gene is transcribed into an mRNA that directs the synthesis of a 100-kDa protein, apoB100. In the intestine, the same gene directs the synthesis of the primary transcript; however, a cytidine deaminase converts a CAA codon in the mRNA to UAA at a single specific site. Rather than encoding glutamine, this codon becomes a termination signal, and a 48-kDa protein (apoB48) is the result. ApoB100 and apoB48 have different functions in the two organs. A growing number of other examples include a glutamine to arginine change in the glutamate receptor and several changes in trypanosome mitochondrial mRNAs, generally involving the addition or deletion of uridine. The exact extent of RNA editing is unknown, but current estimates suggest that <0.01% of mRNAs are edited in this fashion. Recently, editing of miRNAs has been described suggesting that these two forms of posttranscriptional control mechanisms could cooperatively contribute to gene regulation.
Transfer RNA Is Extensively Processed & Modified
As described in Chapters 34 & 37, the tRNA molecules serve as adapter molecules for the translation of mRNA into protein sequences. The tRNAs contain many modifications of the standard bases A, U, G, and C, including methylation, reduction, deamination, and rearranged glycosidic bonds. Further posttranscriptional modification of the tRNA molecules includes nucleotide alkylations and the attachment of the characteristic CpCpAOH terminal at the 3′ end of the molecule by the enzyme nucleotidyl transferase. The 3′ OH of the A ribose is the point of attachment for the specific amino acid that is to enter into the polymerization reaction of protein synthesis. The methylation of mammalian tRNA precursors probably occurs in the nucleus, whereas the cleavage and attachment of CpCpAOH are cytoplasmic functions, since the terminals turn over more rapidly than do the tRNA molecules themselves. Enzymes within the cytoplasm of mammalian cells are required for the attachment of amino acids to the CpCpAOH residues (see Chapter 37).
RNA CAN ACT AS A CATALYST
In addition to the catalytic action served by the snRNAs in the formation of mRNA, several other enzymatic functions have been attributed to RNA. Ribozymes are RNA molecules with catalytic activity. These generally involve transesterification reactions, and most are concerned with RNA metabolism (splicing and endoribonuclease). Recently, a rRNA component has been implicated in hydrolyzing an aminoacyl ester and thus to play a central role in peptide bond function (peptidyl transferases; see Chapter 37). These observations, made using RNA molecules derived from the organelles from plants, yeast, viruses, and higher eukaryotic cells, show that RNA can act as an enzyme, and have revolutionized thinking about enzyme action and the origin of life itself.
SUMMARY
![]() RNA is synthesized from a DNA template by the enzyme RNA polymerase.
RNA is synthesized from a DNA template by the enzyme RNA polymerase.
![]() While bacteria contain but a single RNA polymerase (β,β’ α2) there are three distinct nuclear DNA-dependent RNA polymerases in mammals: RNA polymerases I, II, and III. These enzymes catalyze the transcription of rRNA(Pol I), mRNA/miRNAs (Pol II), and tRNA and 5S rRNA (Pol III) encoding genes.
While bacteria contain but a single RNA polymerase (β,β’ α2) there are three distinct nuclear DNA-dependent RNA polymerases in mammals: RNA polymerases I, II, and III. These enzymes catalyze the transcription of rRNA(Pol I), mRNA/miRNAs (Pol II), and tRNA and 5S rRNA (Pol III) encoding genes.
![]() RNA polymerases interact with unique cis-active regions of genes, termed promoters, in order to form preinitiation complexes (PICs) capable of initiation. In eukaryotes, the process of pol II PIC formation requires, in addition to polymerase, multiple general transcription factors (GTFs), TFIIA, B, D, E, F, and H.
RNA polymerases interact with unique cis-active regions of genes, termed promoters, in order to form preinitiation complexes (PICs) capable of initiation. In eukaryotes, the process of pol II PIC formation requires, in addition to polymerase, multiple general transcription factors (GTFs), TFIIA, B, D, E, F, and H.
![]() Eukaryotic PIC formation can occur on accessible promoters either step-wise—by the sequential, ordered interactions of GTFs and RNA polymerase with DNA promoters—or in one step by the recognition of the promoter by a pre-formed GTF-RNA polymerase holoenzyme complex.
Eukaryotic PIC formation can occur on accessible promoters either step-wise—by the sequential, ordered interactions of GTFs and RNA polymerase with DNA promoters—or in one step by the recognition of the promoter by a pre-formed GTF-RNA polymerase holoenzyme complex.
![]() Transcription exhibits three phases: initiation, elongation, and termination. All are dependent upon distinct DNA cis-elements and can be modulated by distinct trans-acting protein factors.
Transcription exhibits three phases: initiation, elongation, and termination. All are dependent upon distinct DNA cis-elements and can be modulated by distinct trans-acting protein factors.
![]() The presence of nucleosomes can occlude the binding of both transfactors and the transcription machinery to their cognate DNA cis-elements, thereby inhibiting transcription.
The presence of nucleosomes can occlude the binding of both transfactors and the transcription machinery to their cognate DNA cis-elements, thereby inhibiting transcription.
![]() Most eukaryotic RNAs are synthesized as precursors that contain excess sequences which are removed prior to the generation of mature, functional RNA. These processing reactions provide additional potential steps for regulation of gene expression.
Most eukaryotic RNAs are synthesized as precursors that contain excess sequences which are removed prior to the generation of mature, functional RNA. These processing reactions provide additional potential steps for regulation of gene expression.
![]() Eukaryotic mRNA synthesis results in a pre-mRNA precursor that contains extensive amounts of excess RNA (introns) that must be precisely removed by RNA splicing to generate functional, translatable mRNA composed of exonic coding and 5′ and 3′ noncoding sequences.
Eukaryotic mRNA synthesis results in a pre-mRNA precursor that contains extensive amounts of excess RNA (introns) that must be precisely removed by RNA splicing to generate functional, translatable mRNA composed of exonic coding and 5′ and 3′ noncoding sequences.
![]() All steps—from changes in DNA template, sequence, and accessibility in chromatin to RNA stability and translatability—are subject to modulation and hence are potential control sites for eukaryotic gene regulation.
All steps—from changes in DNA template, sequence, and accessibility in chromatin to RNA stability and translatability—are subject to modulation and hence are potential control sites for eukaryotic gene regulation.
REFERENCES
Bourbon H-M, Aguilera A, Ansari AZ, et al: A unified nomenclature for protein subunits of mediator complexes linking transcriptional regulators to RNA polymerase II. Mol Cell 2004;14:553.
Busby S, Ebright RH: Promoter structure, promoter recognition, and transcription activation in prokaryotes. Cell 1994;79:743.
Cramer P, Bushnell DA, Kornberg RD: Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 2001;292:1863.
Fedor MJ: Ribozymes. Curr Biol 1998;8:R441.
Gott JM, Emeson RB: Functions and mechanisms of RNA editing. Ann Rev Genet 2000;34:499.
Harel-Sharvit L, Eldad N, Haimovich G, et al: RNA polymerase II subunits link transcription and mRNA decay to translation. Cell 2010;143:552-563.
Kawauchi J, Mischo H, Braglia P, et al: Budding yeast RNA polymerases I and II employ parallel mechanisms of transcriptional termination. Genes Dev 2008;22:1082.
Keaveney M, Struhl K: Activator-mediated recruitment of the RNA polymerase machinery is the predominant mechanism for transcriptional activation in yeast. Mol Cell 1998;1:917.
Maniatis T, Reed R: An extensive network of coupling among gene expression machines. Nature 2002;416:499.
Mapendano CK, Lykke-Andersen S, Kjems J, et al: Crosstalk between mRNA 3′ end processing and transcription initiation Mol Cell 2010;40:410-422.
Nechaev S, Adelman K: Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochim Biophys Acta 2011;1809:34-45.
Orphanides G, Reinberg D: A unified theory of gene expression. Cell 2002;108:439.
Price DH: Poised polymerases: on your mark … get set … go!. Mol Cell 2008;30:7.
Reed R, Cheng H: TREX, SR proteins and export of mRNA. Curr Opin Cell Biol 2005;17:269.
Trcek T, Singer RH: The cytoplasmic fate of an mRNP is determined cotranscriptionally: exception or rule? Genes Dev 2010;24:1827-1831.
Tucker M, Parker R: Mechanisms and control of mRNA decapping in Saccharomyces cerevisiae. Ann Rev Biochem 2000;69:571.
West S, Proudfoot NJ, Dye MJ, et al: Molecular dissection of mammalian RNA polymerase II transcriptional termination. Mol Cell 2008;29:600.