Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION IV. Structure, Function, & Replication of Informational Macromolecules
Chapter 39. Molecular Genetics, Recombinant DNA, & Genomic Technology
P. Anthony Weil, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Explain the basic procedures and methods involved in recombinant DNA technology and genetic engineering.
Explain the basic procedures and methods involved in recombinant DNA technology and genetic engineering.
![]() Appreciate the rationale behind the methods used to synthesize, analyze, and sequence DNA and RNA.
Appreciate the rationale behind the methods used to synthesize, analyze, and sequence DNA and RNA.
![]() Explain how to identify and quantify individual proteins, both soluble and insoluble (ie, membrane bound or compartmentalized intracellularly) proteins, as well as proteins bound to specific sequences of genomic DNA and RNA.
Explain how to identify and quantify individual proteins, both soluble and insoluble (ie, membrane bound or compartmentalized intracellularly) proteins, as well as proteins bound to specific sequences of genomic DNA and RNA.
BIOMEDICAL IMPORTANCE*
The development of recombinant DNA, high-density DNA microarrays, high-throughput screening, low-cost genome-scale analyses, DNA sequencing and other molecular genetic methodologies has revolutionized biology and is having an increasing impact on clinical medicine. Though much has been learned about human genetic disease from pedigree analysis and study of affected proteins, in many cases where the specific genetic defect is unknown, these approaches cannot be used. The new technologies circumvent these limitations by going directly to the DNA molecule for information. Manipulation of a DNA sequence and the construction of chimeric molecules—so-called genetic engineering—provides a means of studying how a specific segment of DNA works. Novel biochemical and molecular genetic tools and direct DNA sequencing allow investigators to query and manipulate genomic sequences as well as to examine the entire complement of cellular RNA, protein profiles and protein PTM status at the molecular level.
Understanding this technology is important for several reasons: (1) it offers a rational approach to understanding the molecular basis of a number of diseases. For example, familial hypercholesterolemia, sickle-cell disease, the thalassemias, cystic fibrosis, muscular dystrophy as well as more complex multifactorial diseases like vascular and heart disease, cancer, and diabetes. (2) Human proteins can be produced in abundance for therapy (eg, insulin, growth hormone, and tissue plasminogen activator). (3) Proteins for vaccines (eg, hepatitis B) and for diagnostic testing (eg, Ebola and AIDS tests) can be obtained. (4) This technology is used both to diagnose existing diseases as well as to predict the risk of developing a given disease and individual response to pharmacological therapeutics. (5) Special techniques have led to remarkable advances in forensic medicine. (6) Gene therapy for potentially curing diseases caused by a single-gene deficiency such as sickle-cell disease, the thalassemias, adenosine deaminase deficiency, and others may be devised.
RECOMBINANT DNA TECHNOLOGY INVOLVES ISOLATION & MANIPULATION OF DNA TO MAKE CHIMERIC MOLECULES
Isolation and manipulation of DNA, including end-to-end joining of sequences from very different sources to make chimeric molecules (eg, molecules containing both human and bacterial DNA sequences in a sequence-independent fashion), is the essence of recombinant DNA research. This involves several unique techniques and reagents.
Restriction Enzymes Cleave DNA Chains at Specific Locations
Certain endonucleases—enzymes that cut DNA at specific DNA sequences within the molecule (as opposed to exonucleases, which digest from the ends of DNA molecules)—are a key tool in recombinant DNA research. These enzymes were called restriction enzymes because their presence in a given bacterium restricted the growth of certain bacterial viruses called bacteriophages. Restriction enzymes cut DNA of any source into unique, short pieces in a sequence-specific manner—in contrast to most other enzymatic, chemical, or physical methods, which break DNA randomly. These defensive enzymes (hundreds have been discovered) protect the host bacterial DNA from the DNA genome of foreign organisms (primarily infective phages) by specifically inactivating the invading phage DNA by digestion. The viral RNA-inducible interferon system (Chapter 38; Figure 38–11) provides the same sort of molecular defense against RNA viruses in mammalian cells. However, restriction endonucleases are present only in cells that also have a companion enzyme that site-specifically methylates the host DNA, rendering it an unsuitable substrate for digestion by that particular restriction enzyme. Thus, site-specific DNA methylases and restriction enzymes that target the exact same sites always exist in pairs in a bacterium.
Restriction enzymes are named after the bacterium from which they are isolated. For example, EcoRI is from Escherichia coli, and BamHI is from Bacillus amyloliquefaciens (Table 39-1). The first three letters in the restriction enzyme name consist of the first letter of the genus (E) and the first two letters of the species (co). These may be followed by a strain designation (R) and a roman numeral (I) to indicate the order of discovery (eg, EcoRIand EcoRII). Each enzyme recognizes and cleaves a specific double-stranded DNA sequence that is typically 4-7 bp long. These DNA cuts result in blunt ends (eg, HpaI) or overlapping (sticky or cohesive) ends (eg, BamHI)(Figure 39–1), depending on the mechanism used by the enzyme. Sticky ends are particularly useful in constructing hybrid or chimeric DNA molecules (see below). If the four nucleotides are distributed randomly in a given DNA molecule, one can calculate how frequently a given enzyme will cut a length of DNA. For each position in the DNA molecule, there are four possibilities (A, C, G, and T); therefore, a restriction enzyme that recognizes a 4-bp sequence cuts, on average, once every 256 bp (44), whereas another enzyme that recognizes a 6-bp sequence cuts once every 4096 bp (46). A given piece of DNA has a characteristic linear array of sites for the various enzymes dictated by the linear sequence of its bases; hence, a restriction map can be constructed. When DNA is digested with a particular enzyme, the ends of all the fragments have the same DNA sequence. The fragments produced can be isolated by electrophoresis on agarose or polyacrylamide gels (see the discussion of blot transfer, below); this is an essential step in DNA cloning as well as various DNA analyses, and a major use of these enzymes.
TABLE 39–1 Selected Restriction Endonucleases and Their Sequence Specificities
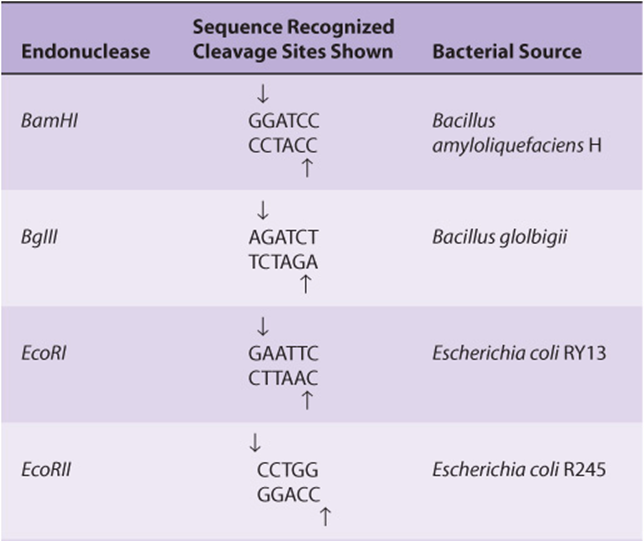
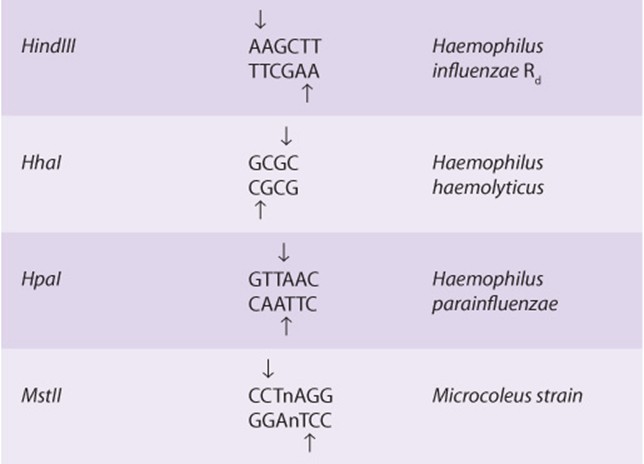

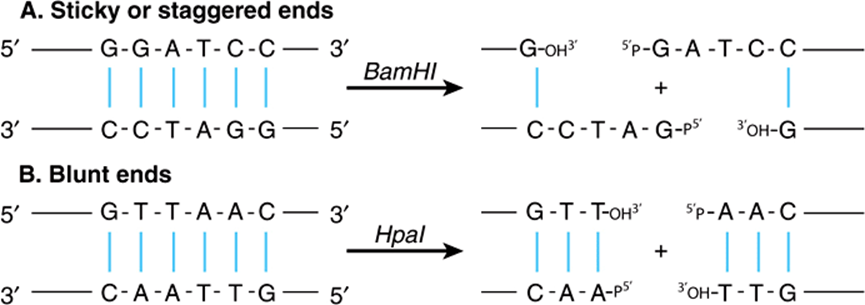
FIGURE 39–1 Results of restriction endonuclease digestion. Digestion with a restriction endonuclease can result in the formation of DNA fragments with sticky, or cohesive, ends (A) or blunt ends (B); phosphodiester backbone, black lines; interstrand hydrogen bonds between purine and pyrimidine bases, blue. This is an important consideration in devising cloning strategies.
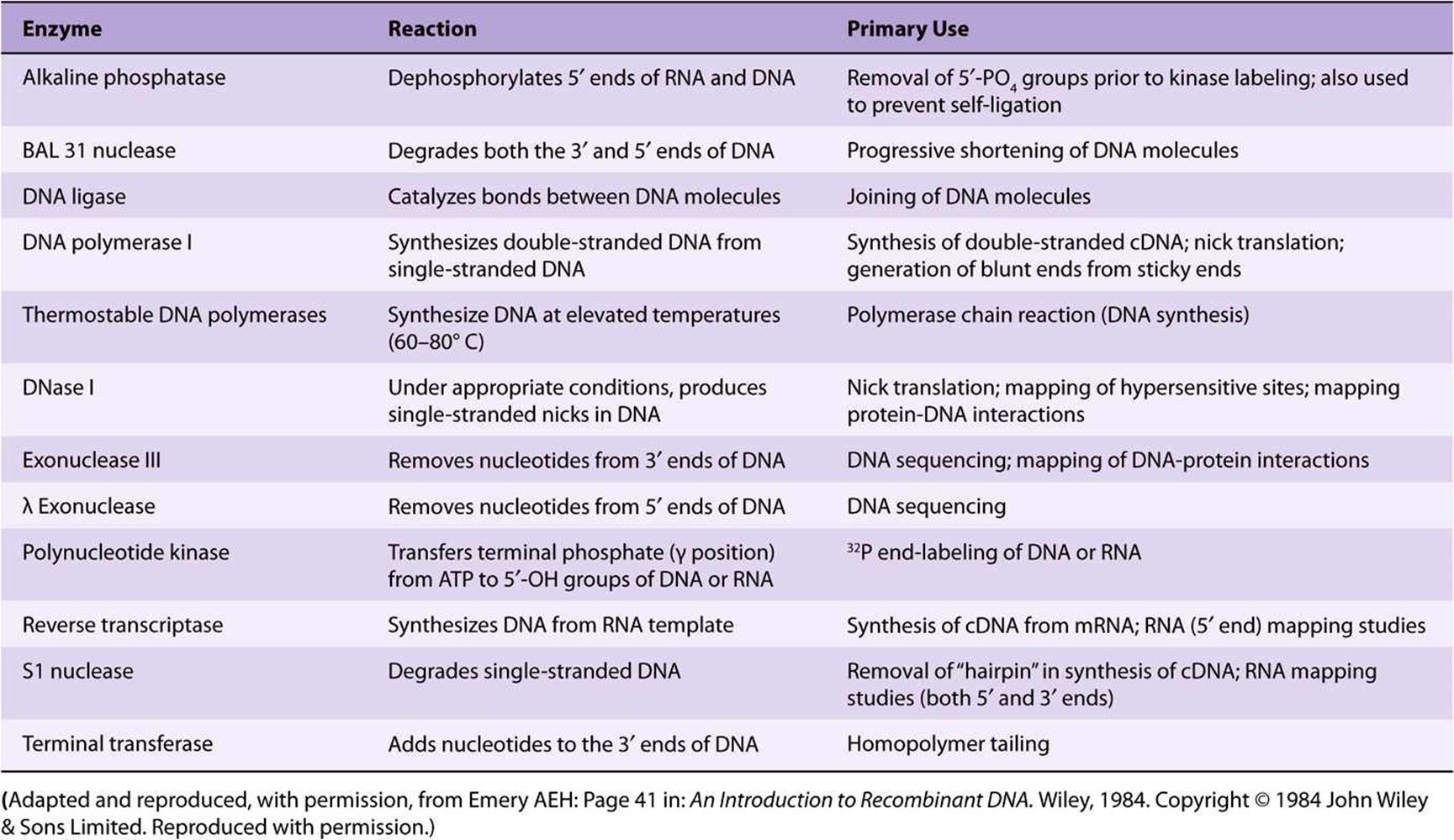
A number of other enzymes that act on DNA and RNA are an important part of recombinant DNA technology. Many of these are referred to in this and subsequent chapters (Table 39-2).
TABLE 39–2 Some of the Enzymes Used in Recombinant DNA Research
Restriction Enzymes & DNA Ligase Are Used to Prepare Chimeric DNA Molecules
Sticky, or comlementary cohesive-end ligation of DNA fragments is technically easy, but some special techniques are often required to overcome problems inherent in this approach. Sticky ends of a vector may reconnect with themselves, with no net gain of DNA. Sticky ends of fragments also anneal so that heterogeneous tandem inserts form. Also, sticky-end sites may not be available or in a convenient position. To circumvent these problems, an enzyme that generates blunt ends can be used. Blunt ends can be ligated directly; however, ligation is not directional. Two alternatives thus exist: new ends are added using the enzyme terminal transferase or synthetic sticky ends are added. If poly d(G) is added to the 3′ ends of the vector and poly d(C) is added to the 3′ ends of the foreign DNA using terminal transferase, the two molecules can only anneal to each other, thus circumventing the problems listed above. This procedure is called homopolymer tailing. Alternatively, synthetic blunt-ended duplex oligonucleotide linkers containing the recognition sequence for a convenient restriction enzyme sequence are ligated to the blunt-ended DNA. Direct blunt-end ligation is accomplished using the bacteriophage T4 enzyme DNA ligase. This technique, though less efficient than sticky-end ligation, has the advantage of joining together any pairs of ends. If blunt ends or homopolymer tailing methods are used there is no easy way to retrieve the insert. As an adjunct to the use of restriction endonucleases scientists have recently begun utilizing specific prokaryotic or eukaryotic recombinases (such as bacterial lox P sites, which are recognized by the CRE recombinase, or yeast FRT sites recognized by the Flp recombinase) to catalyze specific incorporation of two DNA fragments that carry the appropriate recognition sequences. These enzymes catalyze homologous recombination (Figure 35–9) between the relevant recognition sites.
Cloning Amplifies DNA
A clone is a large population of identical molecules, bacteria, or cells that arise from a common ancestor. Molecular cloning allows for the production of a large number of identical DNA molecules, which can then be characterized or used for other purposes. This technique is based on the fact that chimeric or hybrid DNA molecules can be constructed in cloning vectors— typically bacterial plasmids, phages, or cosmids—which then continue to replicate in a host cell under their own control systems. In this way, the chimeric DNA is amplified. The general procedure is illustrated in Figure 39–2.
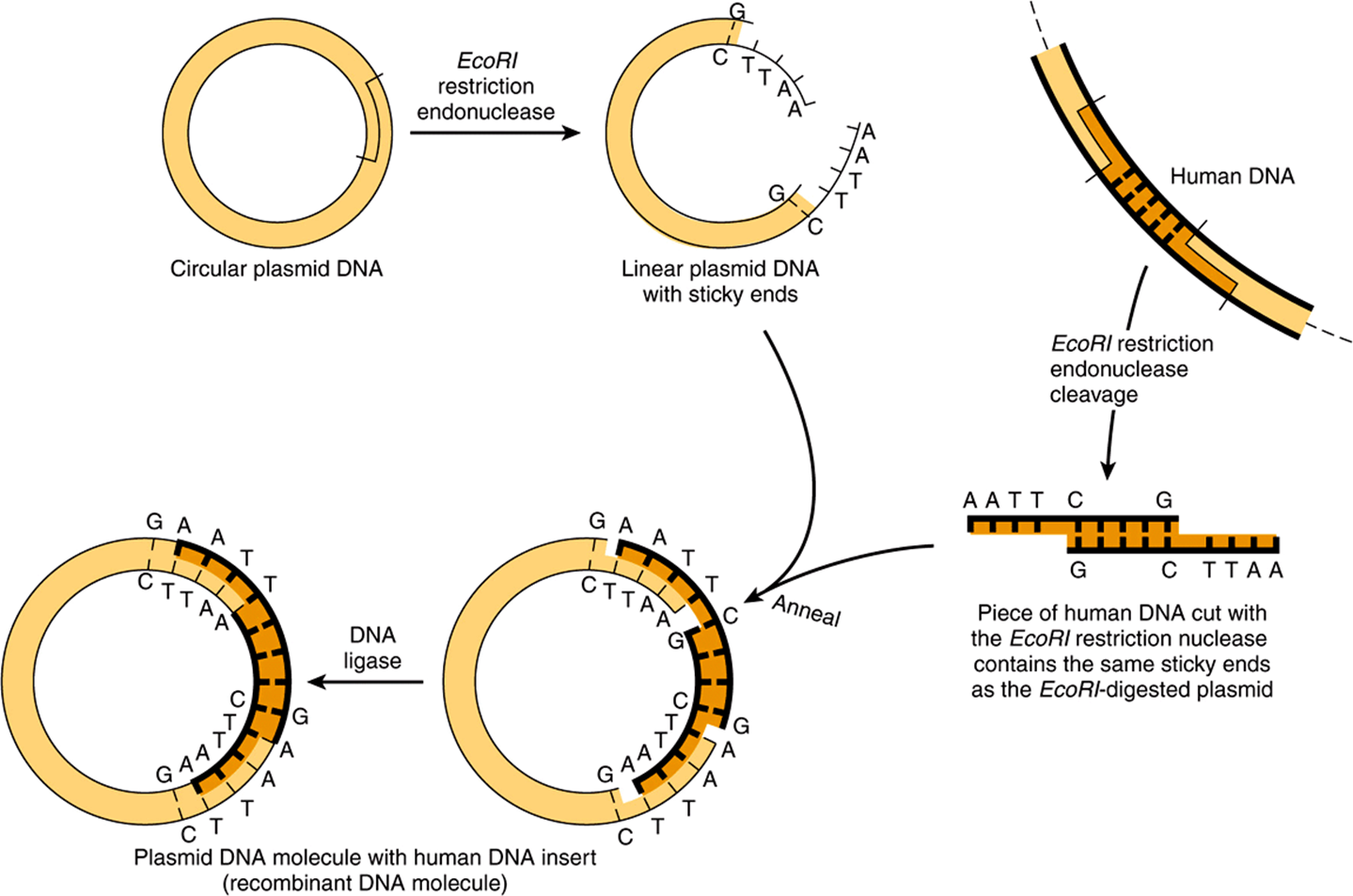
FIGURE 39–2 Use of restriction nucleases to make new recombinant or chimeric DNA molecules. When inserted back into a bacterial cell (by the process called DNA-mediated transformation), typically only a single plasmid is taken up by a single cell, and the plasmid DNA replicates not only itself but also the physically linked new DNA insert. Since recombining the sticky ends, as indicated, typically regenerates the same DNA sequence recognized by the original restriction enzyme, the cloned DNA insert can be cleanly cut back out of the recombinant plasmid circle with this endonuclease. If a mixture of all of the DNA pieces created by treatment of total human DNA with a single restriction nuclease is used as the source of human DNA, a million or so different types of recombinant DNA molecules can be obtained, each pure in its own bacterial clone. (Modified and reproduced, with permission, from Cohen SN: The manipulation of genes. Sci Am [July] 1975;233:25. Copyright © The Estate of Bunji Tagawa.)
Bacterial plasmids are small, circular, duplex DNA molecules whose natural function is to confer antibiotic resistance to the host cell. Plasmids have several properties that make them extremely useful as cloning vectors. They exist as single or multiple copies within the bacterium and replicate independently from the bacterial DNA while using primarily the host replication machinery. The complete DNA sequence of many plasmids is known; hence, the precise location of restriction enzyme cleavage sites for inserting the foreign DNA is available. Plasmids are smaller than the host chromosome and are therefore easily separated from the latter, and the desired plasmid-inserted DNA can be readily removed by cutting the plasmid with the enzyme specific for the restriction site into which the original piece of DNA was inserted.
Phages (bacterial viruses) often have linear DNA molecules into which foreign DNA can be inserted at several restriction enzyme sites. The chimeric DNA is collected after the phage proceeds through its lytic cycle and produces mature, infective phage particles. A major advantage of phage vectors is that while plasmids accept DNA pieces about 6-10 kb long, phages can accept DNA fragments 10-20 kb long, a limitation imposed by the amount of DNA that can be packed into the phage head during virus propagation.
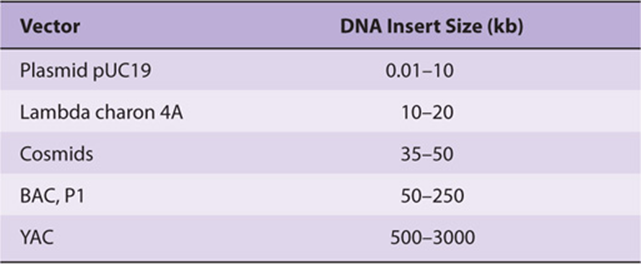
Larger fragments of DNA can be cloned in cosmids, which combine the best features of plasmids and phages. Cosmids are plasmids that contain the DNA sequences, so-called cos sites, required for packaging lambda DNA into the phage particle. These vectors grow in the plasmid form in bacteria, but since much of the unnecessary lambda DNA has been removed, more chimeric DNA can be packaged into the particle head. It is not unusual for cosmids to carry inserts of chimeric DNA that are 35-50 kb long. Even larger pieces of DNA can be incorporated into bacterial artificial chromosome (BAC), yeast artificial chromosome (YAC), or E coli bacteriophage P1-based (PAC)vectors. These vectors will accept and propagate DNA inserts of several hundred kilobases or more and have largely replaced the plasmid, phage, and cosmid vectors for some cloning and eukaryotic gene mapping applications. A comparison of these vectors is shown in Table 39-3.
TABLE 39–3 Cloning Capacities of Common Cloning Vectors
Because insertion of DNA into a functional region of the vector will interfere with the action of this region, care must be taken not to interrupt an essential function of the vector. This concept can be exploited, however, to provide a selection technique. For example, a common early plasmid vector pBR322 has both tetracycline (tet) and ampicillin (amp) resistance genes. A single PstI restriction enzyme site within the amp resistance gene is commonly used as the insertion site for a piece of foreign DNA. In addition to having sticky ends (Table 39-1 and Figure 39–1), the DNA inserted at this site disrupts the amp resistance gene and makes the bacterium carrying this plasmid amp-sensitive (Figure 39–3). Thus, cells carrying the parental plasmid, which provides resistance to both antibiotics, can be readily distinguished and separated from cells carrying the chimeric plasmid, which is resistant only to tetracycline. YACs contain selection, replication, and segregation functions that work in both bacteria and yeast cells and therefore can be propagated in either organism.
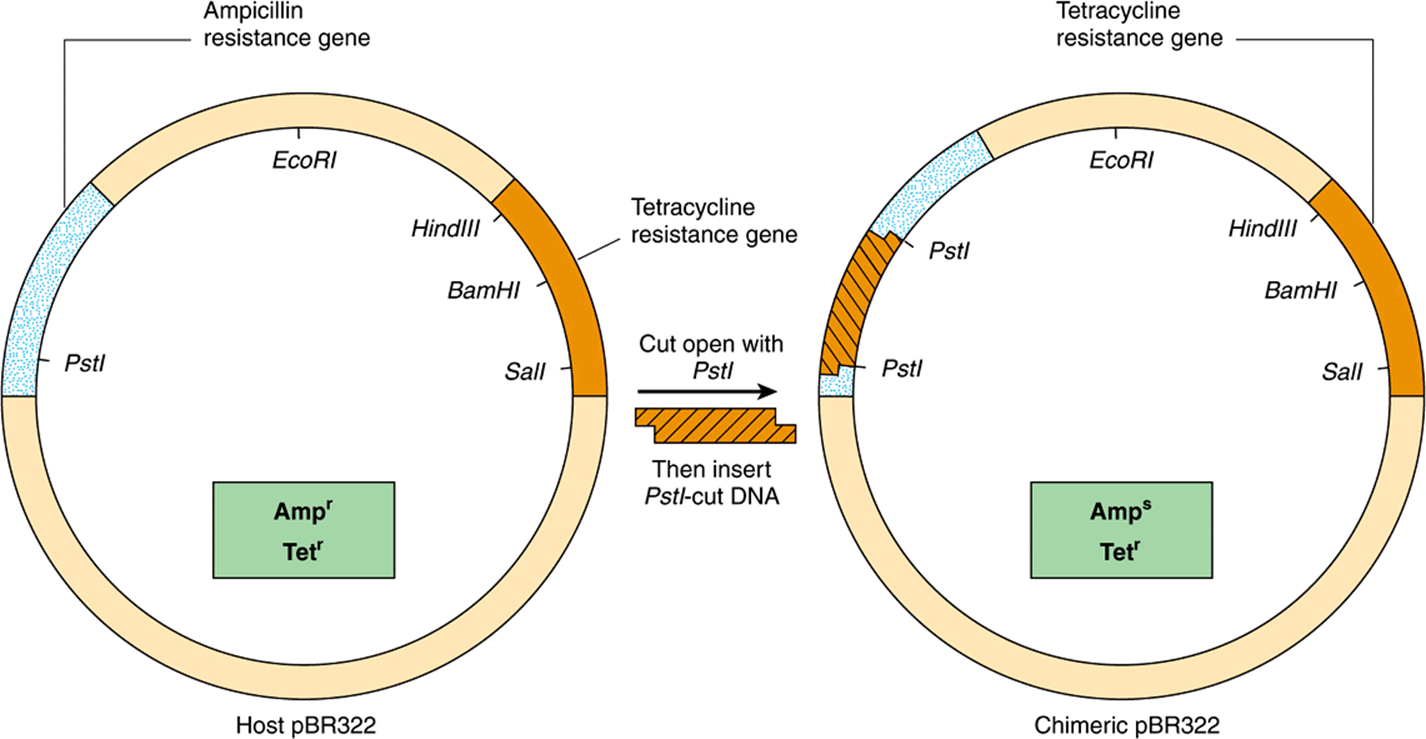
FIGURE 39–3 A method of screening recombinants for inserted DNA fragments. Using the plasmid pBR322, a piece of DNA is inserted into the unique PstI site. This insertion disrupts the gene coding for a protein that provides ampicillin resistance to the host bacterium. Hence, cells carrying the chimeric plasmid will no longer survive when plated on a substrate medium that contains this antibiotic. The differential sensitivity to tetracycline and ampicillin can therefore be used to distinguish clones of plasmid that contain an insert. A similar scheme relying upon production of an in-frame fusion of a newly inserted DNA producing a peptide fragment capable of complementing an inactive, N-terminally truncated form of the enzyme β-galactosidase, a component of the lac operon (Figure 38–2) allows for blue-white colony formation on agar plates containing a dye hydrolyzable by β-galactoside. β-Galactosidase-positive colonies are blue; such colonies contain plasmids in which a DNA was successfully inserted.
In addition to the vectors described in Table 39-3 that are designed primarily for propagation in bacterial cells, vectors for mammalian cell propagation and insert gene (cDNA)/protein expression have also been developed. These vectors are all based upon various eukaryotic viruses that are composed of RNA or DNA genomes. Notable examples of such viral vectors are those utilizing adenoviral (Ad), or adenovirus - associated viral (AAV) (DNA-based) and retroviral (RNA-based) genomes. Though somewhat limited in the size of DNA sequences that can be inserted, such mammalian viral cloning vectors make up for this shortcoming because they will efficiently infect a wide range of different cell types. For this reason, various mammalian viral vectors are being investigated for use in gene therapy and are commonly used for laboratory experiments.
A Library Is a Collection of Recombinant Clones
The combination of restriction enzymes and various cloning vectors allows the entire genome of an organism to be individually packed into a vector. A collection of these different recombinant clones is called a library. A genomic library is prepared from the total DNA of a cell line or tissue. A cDNA library comprises complementary DNA copies of the population of mRNAs in a tissue. Genomic DNA libraries are often prepared by performing partial digestion of total DNA with a restriction enzyme that cuts DNA frequently (eg, a four base cutter such as TaqI). The idea is to generate rather large fragments so that most genes will be left intact. The BAC, YAC, and P1 vectors are preferred since they can accept very large fragments of DNA and thus offer a better chance of isolating an intact eukaryotic mRNA-encoding gene on a single DNA fragment.
A vector in which the protein coded by the gene introduced by recombinant DNA technology is actually synthesized is known as an expression vector. Such vectors are now commonly used to detect specific cDNA molecules in libraries and to produce proteins by genetic engineering techniques. These vectors are specially constructed to contain very active inducible promoters, proper in-phase translation initiation codons, both transcription and translation termination signals, and appropriate protein processing signals, if needed. Some expression vectors even contain genes that code for protease inhibitors, so that the final yield of product is enhanced. Interestingly as the cost of synthetic DNA synthesis has dropped, many investigators often synthesize an entire cDNA (gene) of interest (in 100-150 nt segments) incorporating the codon preferences of the host used for expression in order to maximize protein production. New efficiencies in synthetic DNA synthesis now allow for the de novo synthesis of complete genes and even genomes. These advances usher in new and exciting possibilities in synthetic biology while concomitantly introducing potential ethical conundrums.
Probes Search Libraries or Complex Samples for Specific Genes or cDNA Molecules
A variety of molecules can be used to “probe” libraries in search of a specific gene or cDNA molecule or to define and quantitate DNA or RNA separated by electrophoresis through various gels. Probes are generally pieces of DNA or RNA labeled with a 32P-containing nucleotide—or fluorescently labeled nucleotides (more commonly now). Importantly, neither modification (32P or fluorescent-label) affects the hybridization properties of the resulting labeled nucleic acid probes. The probe must recognize a complementary sequence to be effective. A cDNA synthesized from a specific mRNA can be used to screen either a cDNA library for a longer cDNA or a genomic library for a complementary sequence in the coding region of a gene. A popular technique for finding specific genes entails taking a short amino acid sequence and, employing the codon usage for that species (see Chapter 37), making an oligonucleotide probe (or probe mixture) that will detect the corresponding DNA fragment in a genomic library. If the sequences match exactly, probes 15-20 nucleotides long will hybridize. cDNA probes are used to detect DNA fragments on Southern blot transfers and to detect and quantitate RNA on Northern blot transfers. Specific antibodies can also be used as probes provided that the vector used synthesizes protein molecules that are recognized by them.
Blotting & Hybridization Techniques Allow Visualization of Specific Fragments
Visualization of a specific DNA or RNA fragment among the many thousands of “contaminating” molecules in a complex sample requires the convergence of a number of techniques, collectively termed blot transfer. Figure 39–4illustrates the Southern (DNA), Northern (RNA), and Western (protein) blot transfer procedures. (The first is named for the person who devised the technique [Edward Southern], and the other names began as laboratory jargon but are now accepted terms.) These procedures are useful in determining how many copies of a gene are in a given tissue or whether there are any alterations in a gene (deletions, insertions, or rearrangements) because the requisite electrophoresis step separates the molecules on the basis of size. Occasionally, if a specific base is changed and a restriction site is altered, these procedures can detect a point mutation. The Northern and Western blot transfer techniques are used to size and quantitate specific RNA and protein molecules, respectively. A fourth hybridization technique, the Southwestern blot, examines protein-DNA interactions (not shown). In this method, proteins are separated by electrophoresis, blotted to a membrane, renatured, and analyzed for an interaction with a particular sequence by incubation with a specific labeled nucleic acid probe.
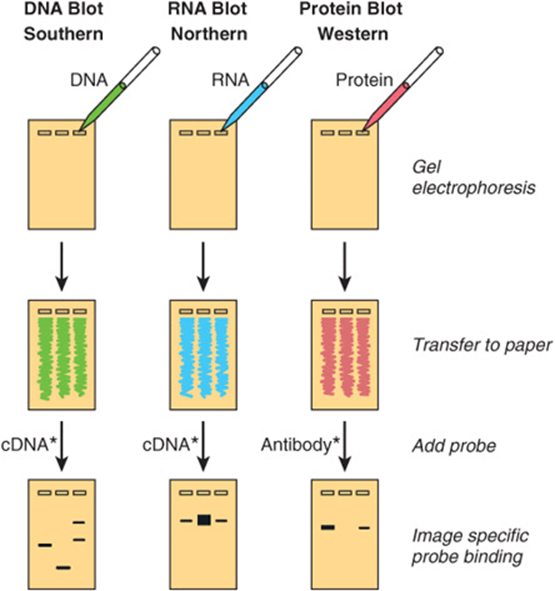
FIGURE 39–4 The blot transfer procedure. In a Southern, or DNA blot transfer, DNA isolated from a cell line or tissue is digested with one or more restriction enzymes. This mixture is pipetted into a well in an agarose or polyacrylamide gel and exposed to a direct electrical current. DNA, being negatively charged, migrates toward the anode; the smaller fragments move the most rapidly. After a suitable time, the DNA within the gel is denatured by exposure to mild alkali and transferred to nitrocellulose or nylon paper, resulting in an exact replica of the pattern on the gel, by the blotting technique devised by Southern. The DNA is bound to the paper by exposure to heat or UV, and the paper is then exposed to the labeled cDNA probe, which hybridizes to complementary strands on the filter. After thorough washing, the paper is exposed to X-ray film or an imaging screen, which is developed to reveal several specific bands corresponding to the DNA fragment that recognized the sequences in the cDNA probe. The RNA, or Northern, blot is conceptually similar. RNA is subjected to electrophoresis before blot transfer. This requires some different steps from those of DNA transfer, primarily to ensure that the RNA remains intact, and is generally somewhat more difficult. In the protein, or Western, blot, proteins are electrophoresed and transferred to special paper that avidly binds proteins and then probed with a specific antibody or other probe molecule. (Asterisks signify labeling, either radioactive or fluorescent.) In the case of Southwestern blotting (see the text; not shown), a protein blot similar to that shown above under “Western” is exposed to labeled nucleic acid, and protein-nucleic acid complexes formed are detected by autoradiography or imaging.
Colony or plaque hybridization is the method by which specific clones are identified and purified. Bacteria are grown as colonies on an agar plate and overlaid with an oriented nitrocellulose filter paper. Cells from each colony stick to the filter and are permanently fixed thereto by heat or UV, which with NaOH treatment also lyses the cells and denatures the DNA so that it is available to hybridize with the probe. A radioactive probe is added to the filter, and (after washing) the hybrid complex is localized by exposing the filter to x-ray film or imaging screen. By matching the spot on the autoradiograph (exposed and developed x-ray film) to a colony, the latter can be picked from the plate. A similar strategy is used to identify fragments in phage libraries. Successive rounds of this procedure result in a clonal isolate (bacterial colony) or individual phage plaque containing a unique DNA insert.
All of the hybridization procedures discussed in this section depend on the specific base-pairing properties of complementary nucleic acid strands described above. Perfect matches hybridize readily and withstand high temperatures in the hybridization and washing reactions. Specific complexes also form in the presence of low salt concentrations. Less than perfect matches do not tolerate such stringent conditions (ie, elevated temperatures and low salt concentrations); thus, hybridization either never occurs or is disrupted during the washing step. Gene families, in which there is some degree of homology, can be detected by varying the stringency of the hybridization and washing steps. Cross-species comparisons of a given gene can also be made using this approach. Hybridization conditions capable of detecting just a single base-pair (bp) mismatch between probe and target have been devised.
Manual & Automated Techniques Are Available to Determine the Sequence of DNA
The segments of specific DNA molecules obtained by recombinant DNA technology can be analyzed to determine their nucleotide sequence. This method depends upon having a large number of identical DNA molecules. This requirement can be satisfied by cloning the fragment of interest, using the techniques described above, or by using PCR methods (see below). The manual enzymatic method (Sanger) employs specific dideoxynucleotides that terminate DNA strand synthesis at specific nucleotides as the strand is synthesized on purified template nucleic acid. The reactions are adjusted so that a population of DNA fragments representing termination at every nucleotide is obtained. By having a radioactive label incorporated at the termination site, one can separate the fragments according to size using polyacrylamide gel electrophoresis. An autoradiograph is made, and each of the fragments produces an image (band) on an x-ray film or imaging plate. These are read in order to give the DNA sequence (Figure 39–5). Another manual method is that of Maxam and Gilbert, which employs chemical methods to cleave the DNA molecules where they contain the specific nucleotides. Techniques that do not require the use of radioisotopes are employed in automated DNA sequencing. Most commonly employed is an automated procedure in which four different fluorescent labels—one representing each nucleotide—are used. Each emits a specific signal upon excitation by a laser beam of a particular wavelength that is measured by densitive detectors, and this can be recorded by a computer. The newest DNA sequencing machines use fluorescently labeled nucleotides but detect incorporation using microscopic optics. These machines have reduced the cost of DNA sequencing dramatically, over 100X. These reductions in cost have ushered in the era of personalized genome sequencing. Indeed, using this new technology the sequence of the co-discoverer of the double helix, James Watson, was completely determined.
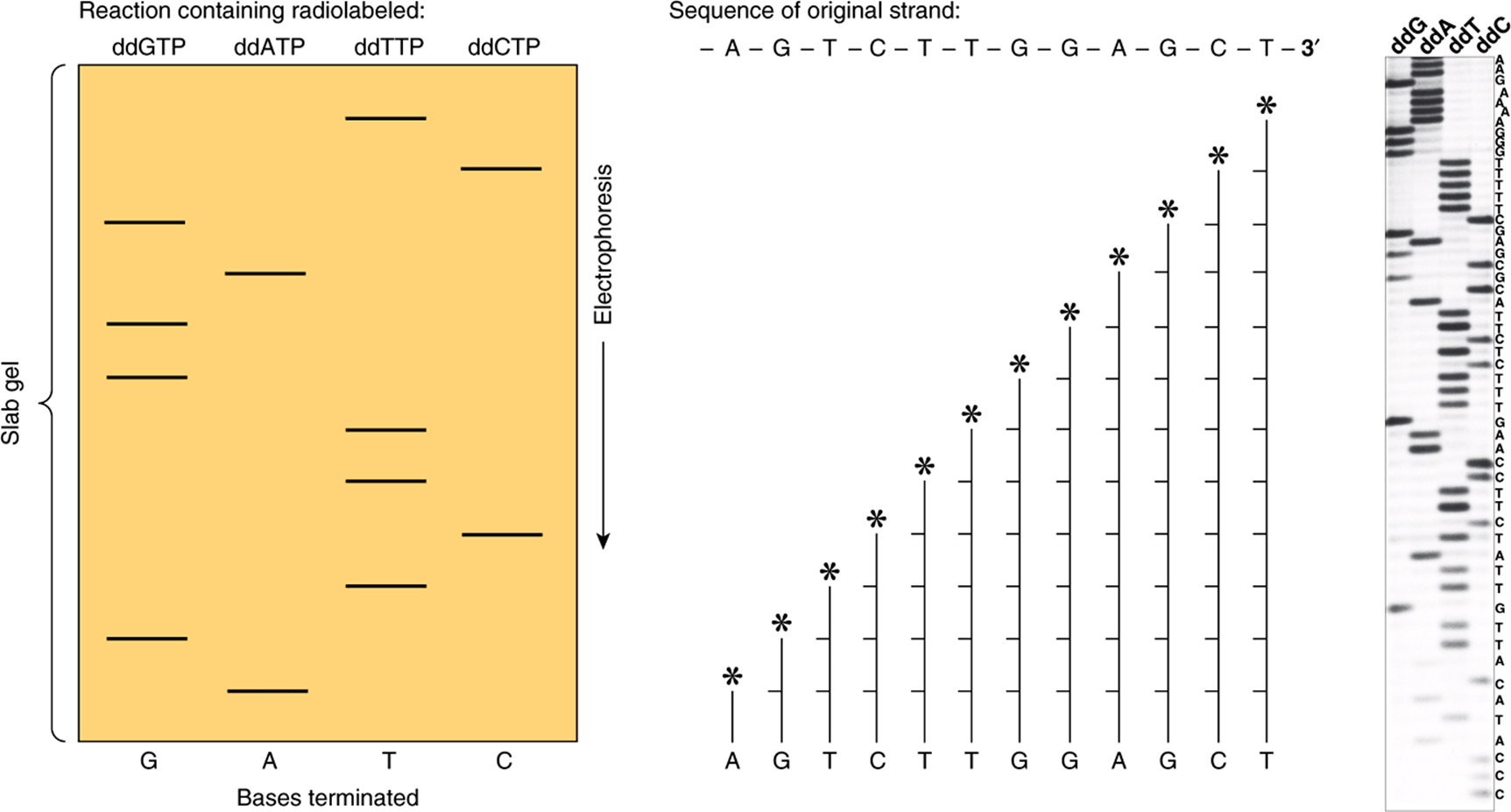
FIGURE 39–5 Sequencing of DNA by the chain termination method devised by Sanger. The ladder-like arrays represent from bottom to top all of the successively longer fragments of the original DNA strand. Knowing which specific dideoxynucleotide reaction was conducted to produce each mixture of fragments, one can determine the sequence of nucleotides from the unlabeled end toward the labeled end (*) by reading up the gel. The base-pairing rules of Watson and Crick (A-T, G-C) dictate the sequence of the other (complementary) strand. (Asterisks signify site of radiolabeling.) Shown (left, middle) are the terminated synthesis products of a hypothetical fragment of DNA, sequence shown. An autoradiogram (right) of an actual set of DNA sequencing reactions that utilized the four 32P-labeled dideoxynucleotides indicated at the top of the scanned autoradiogram (ie, dideoxy(dd)G, ddA, ddT, ddC). Electrophoresis was from top to bottom. The deduced DNA sequence is listed on the right side of the gel. Note the log-linear relationship between distance of migration (ie, top to bottom of gel) and DNA fragment length. Current state-of-the-art DNA sequencers no longer utilize gel electrophoresis for fractionation of labeled synthesis products. Moreover in the NGS sequencing platforms, synthesis is followed by monitoring incorporation of the four fluorescently labeled dXTPs.
Oligonucleotide Synthesis Is Now Routine
The automated chemical synthesis of moderately long oligonucleotides (~100 nucleotides) of precise sequence is now a routine laboratory procedure. Each synthetic cycle takes but a few minutes, so an entire molecule can be made by synthesizing relatively short segments that can then be ligated to one another. As mentioned above, the process has been miniaturized and can be significantly parallelized to allow the synthesis of 100s to 1000s of defined sequence oligonucleotides simultaneously. Oligonucleotides are now indispensable for DNA sequencing, library screening, protein-DNA binding assays, the polymerase chain reaction (PCR) (see below), site-directed mutagenesis, synthetic gene synthesis, and numerous other applications.
The Polymerase Chain Reaction (PCR) Method Amplifies DNA Sequences
The PCR is a method of amplifying a target sequence of DNA. The development of PCR has revolutionized the ways in which both DNA and RNA can be studied. PCR provides a sensitive, selective, and extremely rapid means of amplifying any desired sequence of DNA. Specificity is based on the use of two oligonucleotide primers that hybridize to complementary sequences on opposite strands of DNA and flank the target sequence (Figure 39–6). The DNA sample is first heated to separate the two strands of the template DNA containing the target sequence; the primers, added in vast excess, are allowed to anneal to the DNA; and each strand is copied by a DNA polymerase, starting at the primer sites in the presence of all four dXTPs. The two DNA strands each serve as a template for the synthesis of new DNA from the two primers. Repeated cycles of heat denaturation, annealing of the primers to their complementary sequences, and extension of the annealed primers with DNA polymerase result in the exponential amplification of DNA segments of defined length (a doubling at each cycle). Early PCR reactions used an E coliDNA polymerase that was destroyed by each heat denaturation cycle and hence needed to be re-added at the beginning of each cycle. Substitution of a heat-stable DNA polymerase from Thermus aquaticus (or the corresponding DNA polymerase from many other thermophilic bacteria), an organism that lives and replicates at 70-80°C, obviates this problem and has made possible automation of the reaction since the polymerase reactions can be run at 70°C. This has also improved the specificity and the yield of DNA.
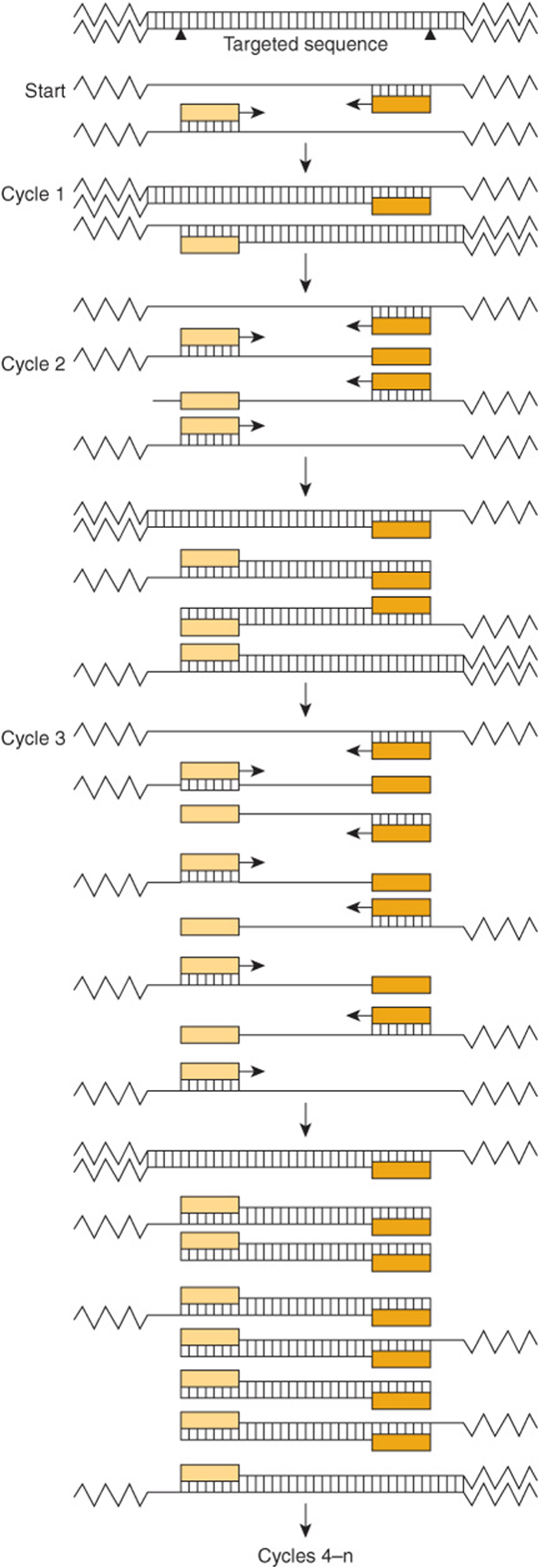
FIGURE 39–6 The polymerase chain reaction is used to amplify specific gene sequences. Double-stranded DNA is heated to separate it into individual strands. These bind two distinct primers that are directed at specific sequences on opposite strands and that define the segment to be amplified. DNA polymerase extends the primers in each direction and synthesizes two strands complementary to the original two. This cycle is repeated several times, giving an amplified product of defined length and sequence. Note that the two primers are present in vast excess.
DNA sequences as short as 50-100 bp and as long as 10 kb can be amplified. Twenty cycles provide an amplification of 106 (ie, 220) and 30 cycles, 109 (230). Each cycle takes <5-10 min so that even large DNA molecules can be amplified rapidly. The PCR allows the DNA in a single cell, hair follicle, or spermatozoon to be amplified and analyzed. Thus, the applications of PCR to forensic medicine are obvious. The PCR is also used (1) to detect infectious agents, especially latent viruses; (2) to make prenatal genetic diagnoses; (3) to detect allelic polymorphisms; (4) to establish precise tissue types for transplants; and (5) to study evolution, using DNA from archeological samples (6) for quantitative RNA analyses after RNA copying and mRNA quantitation by the so-called RTPCR method (cDNA copies of mRNA generated by a retroviral reverse transcriptase) or (7) to score in vivo protein-DNA occupancy using chromatin immunoprecipitation assays to facilitate NGS sequencing (see below). There are an equal number of applications of PCR to problems in basic science, and new uses are developed every year.
PRACTICAL APPLICATIONS OF RECOMBINANT DNA TECHNOLOGY ARE NUMEROUS
The isolation of a specific (ca. 1000 bp) mRNA-encoding gene from an entire genome requires a technique that will discriminate one part in a million. The identification of a regulatory region that may be only 10 bp in length requires a sensitivity of one part in 3 × 108; a disease such as sickle-cell anemia is caused by a single base change, or one part in 3 × 109. DNA technology is powerful enough to accomplish all these things.
Gene Mapping Localizes Specific Genes to Distinct Chromosomes
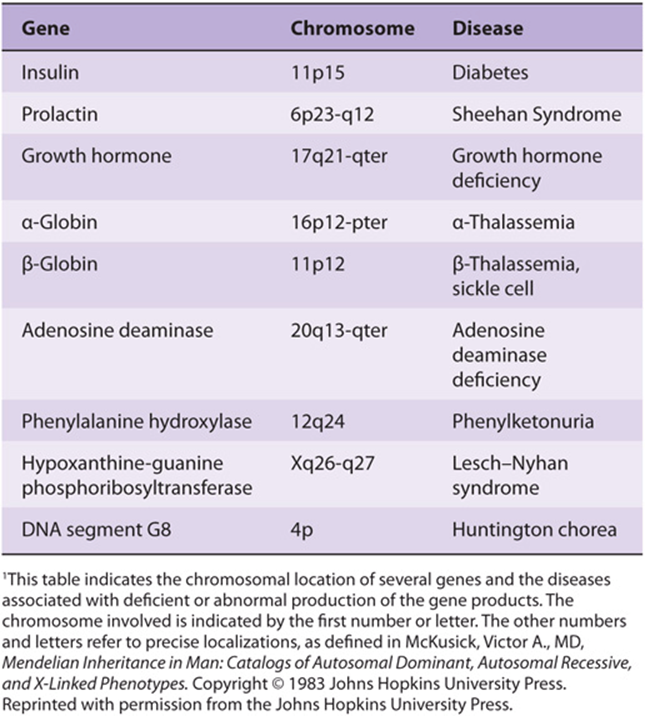
Gene localization thus can define a map of the human genome. This is already yielding useful information in the definition of human disease. Somatic cell hybridization and in situ hybridization are two techniques used to accomplish this. In in situ hybridization, the simpler and more direct procedure, a radioactive probe is added to a metaphase spread of chromosomes on a glass slide. The exact area of hybridization is localized by layering photographic emulsion over the slide and, after exposure, lining up the grains with some histologic identification of the chromosome. Fluorescence in situ hybridization (FISH), which utilizes fluorescent rather than radioactively labeled probes, is a very sensitive technique that is also used for this purpose. This often places the gene at a location on a given band or region on the chromosome. Some of the human genes localized using these techniques are listed in Table 39-4. This table represents only a sampling of mapped genes since tens of thousands of genes have been mapped as a result of the recent sequencing of the human genome. Once the defect is localized to a region of DNA that has the characteristic structure of a gene, a synthetic cDNA copy of the gene can be constructed, which contains only mRNA encoding exons, and expressed in an appropriate vector and its function can be assessed—or the putative peptide, deduced from the open reading frame in the coding region, can be synthesized. Antibodies directed against this peptide can be used to assess whether this peptide is expressed in normal persons and whether it is absent, or altered in those with the genetic syndrome.
TABLE 39–4 Localization of Human Genes1
Proteins Can Be Produced for Research, Diagnosis & Commerce
A practical goal of recombinant DNA research is the production of materials for biomedical applications. This technology has two distinct merits: (1) it can supply large amounts of material that could not be obtained by conventional purification methods (eg, interferon, tissue plasminogen activating factor, etc). (2) It can provide human material (eg, insulin and growth hormone). The advantages in both cases are obvious. Although the primary aim is to supply products—generally proteins—for treatment (insulin) and diagnosis (AIDS testing) of human and other animal diseases and for disease prevention (hepatitis B vaccine), there are other potential commercial applications, especially in agriculture. An example of the latter is the attempt to engineer plants that are more resistant to drought or temperature extremes, more efficient at fixing nitrogen, or that produce seeds containing the complete complement of essential amino acids (rice, wheat, corn, etc).
Recombinant DNA Technology Is Used in the Molecular Analysis of Disease
Normal Gene Variations
There is a normal variation of DNA sequence just as is true of more obvious aspects of human structure. Variations of DNA sequence, polymorphisms, occur approximately once in every 500-1000 nucleotides. A recent comparison of the nucleotide sequence of the genome of James Watson, the co-discoverer of DNA structure, identified about 3,300,000 single-nucleotide polymorphisms (SNPs) relative to the “standard” initially sequenced human reference genome. Interestingly, >80% of the SNPs found in Watson’s DNA had already been identified in other individuals. There are also genomic deletions and insertions of DNA (ie, copy number variations; CNV) as well as single-base substitutions. In healthy people, these alterations obviously occur in noncoding regions of DNA or at sites that cause no change in function of the encoded protein. This heritable polymorphism of DNA structure can be associated with certain diseases within a large kindred and can be used to search for the specific gene involved, as is illustrated below. It can also be used in a variety of applications in forensic medicine.
Gene Variations Causing Disease
Classic genetics taught that most genetic diseases were due to point mutations which resulted in an impaired protein. This may still be true, but if on reading previous chapters one predicted that genetic disease could result from derangement of any of the steps leading from replication to transcription to RNA processing/transport and protein synthesis, one would have made a proper assessment. This point is again nicely illustrated by examination of the β-globin gene. This gene is located in a cluster on chromosome 11 (Figure 39–7), and an expanded version of the gene is illustrated in Figure 39–8. Defective production of β-globin results in a variety of diseases and is due to many different lesions in and around the β-globin gene (Table 39-5).
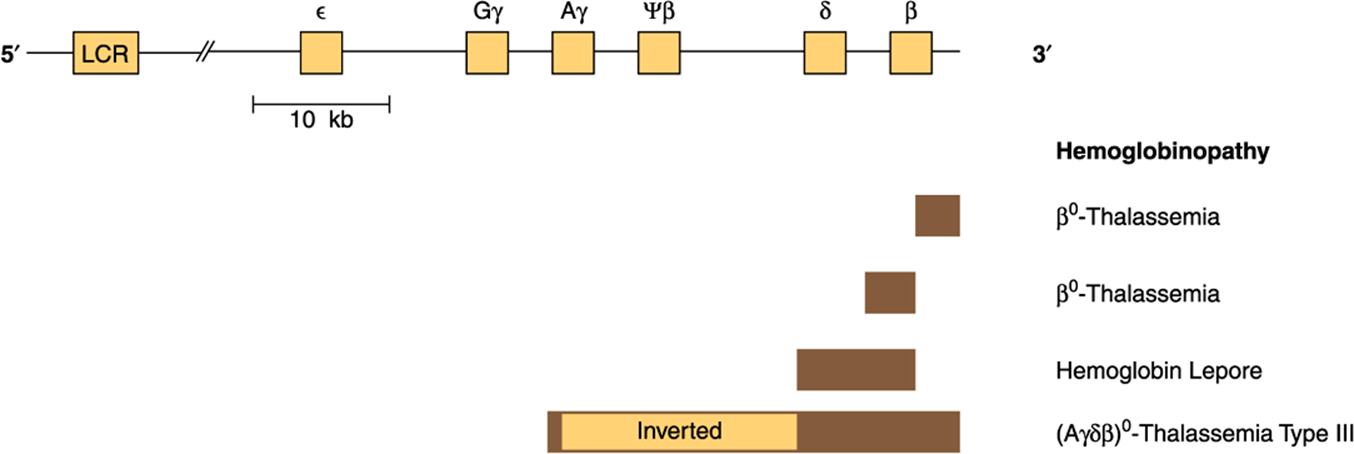
FIGURE 39–7 Schematic representation of the β-globin gene cluster and of the lesions in some genetic disorders. The β-globin gene is located on chromosome 11 in close association with the two γ-globin genes and the δ-globin gene. The β-gene family is arranged in the order 5′-ε-Gγ-Aγ-ψβ-δ-β-3’. The ε locus is expressed in early embryonic life (as α2ε2). The γ genes are expressed in fetal life, making fetal hemoglobin (HbF, α2γ2). Adult hemoglobin consists of HbA (α2β2) or HbA2(α2ψ2). The ψβ is a pseudogene that has sequence homology with β but contains mutations that prevent its expression. A locus control region (LCR), a powerful enhancer located upstream (5’) from the gene, controls the rate of transcription of the entire β-globin gene cluster. Deletions (solid bar) of the β locus cause β-thalassemia (deficiency or absence [β0] of β-globin). A deletion of δ and β causes hemoglobin Lepore (only hemoglobin is present). An inversion (Aγδβ)0 in this region (largest bar) disrupts gene function and also results in thalassemia (type III). Each type of thalassemia tends to be found in a certain group of people, eg, the (Aγδβ)0 deletion inversion occurs in persons from India. Many more deletions in this region have been mapped, and each causes some type of thalassemia.
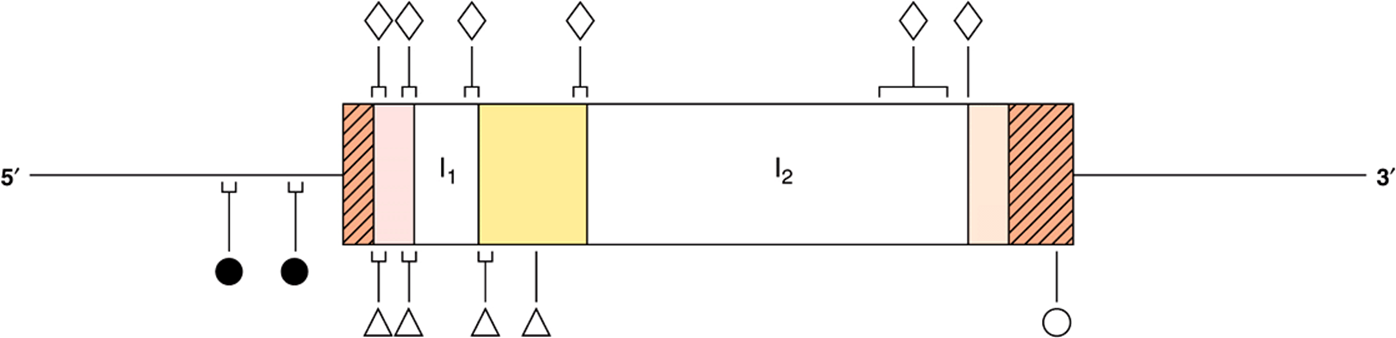
FIGURE 39–8 Mutations in the β-globin gene causing β-thalassemia. The β-globin gene is shown in the 5′ to 3′ orientation. The cross-hatched areas indicate the 5′ and 3′ nontranslated regions. Reading from the 5′ to 3′ direction, the shaded areas are exons 1-3 and the clear spaces are introns 1 (I1) and 2 (I2). Mutations that affect transcription control ![]() are located in the 5′ flanking-region DNA. Examples of nonsense mutations
are located in the 5′ flanking-region DNA. Examples of nonsense mutations ![]() , mutations in RNA processing
, mutations in RNA processing ![]() and RNA cleavage mutations
and RNA cleavage mutations ![]() have been identified and are indicated. In some regions, many distinct mutations have been found. These are indicated by the brackets.
have been identified and are indicated. In some regions, many distinct mutations have been found. These are indicated by the brackets.
TABLE 39–5 Structural Alterations of the β-Globin Gene
Point Mutations
The classic example is sickle-cell disease, which is caused by mutation of a single base out of the 3 × 109 in the genome, a T-to-A DNA substitution, which in turn results in an A-to-U change in the mRNA corresponding to the sixth codon of the β-globin gene. The altered codon specifies a different amino acid (valine rather than glutamic acid), and this causes a structural abnormality of the β-globin molecule. Other point mutations in and around the β-globin gene result in decreased or, in some instances, no production of β-globin; β-thalassemia is the result of these mutations. (The thalassemias are characterized by defects in the synthesis of hemoglobin subunits, and so β-thalassemia results when there is insufficient production of β-globin.) Figure 39–8 illustrates that point mutations affecting each of the many processes involved in generating a normal mRNA (and therefore a normal protein) have been implicated as a cause of β-thalassemia.
Deletions, Insertions, & Rearrangements of DNA
Studies of bacteria, viruses, yeasts, fruit flies, and now humans show that pieces of DNA can move from one place to another within a genome. The deletion of a critical piece of DNA, the rearrangement of DNA within a gene, or the insertion or amplification of a piece of DNA within a coding or regulatory region can all cause changes in gene expression resulting in disease. Again, a molecular analysis of thalassemias produces numerous examples of these processes—particularly deletions—as causes of disease (Figure 39–7). The globin gene clusters seem particularly prone to this lesion. Deletions in the α-globin cluster, located on chromosome 16, cause α-thalassemia. There is a strong ethnic association for many of these deletions, so that northern Europeans, Filipinos, blacks, and Mediterranean peoples have different lesions all resulting in the absence of hemoglobin A and α-thalassemia.
A similar analysis could be made for a number of other diseases. Point mutations are usually defined by sequencing the gene in question, though occasionally, if the mutation destroys or creates a restriction enzyme site, the technique of restriction fragment analysis can be used to pinpoint the lesion. Deletions or insertions of DNA larger than 50 bp can often be detected by the Southern blotting procedure while PCR-based assays can detect much smaller changes in DNA structure.
Pedigree Analysis
Sickle-cell disease again provides an excellent example of how recombinant DNA technology can be applied to the study of human disease. The substitution of T for A in the template strand of DNA in the β-globin gene changes the sequence in the region that corresponds to the sixth codon from
![]()
to

and destroys a recognition site for the restriction enzyme MstII (CCTNAGG; denoted by the small vertical arrows; Table 39-1). Other MstII sites 5′ and 3′ from this site (Figure 39–9) are not affected and so will be cut. Therefore, incubation of DNA from normal (AA), heterozygous (AS), and homozygous (SS) individuals results in three different patterns on Southern blot transfer (Figure 39–9). This illustrates how a DNA pedigree can be established using the principles discussed in this chapter. Pedigree analysis has been applied to a number of genetic diseases and is most useful in those caused by deletions and insertions or the rarer instances in which a restriction endonuclease cleavage site is affected, as in the example cited here. Such analyses are now facilitated by the PCR reaction, which can amplify and hence provide sufficient DNA for analysis from just a few nucleated cells.
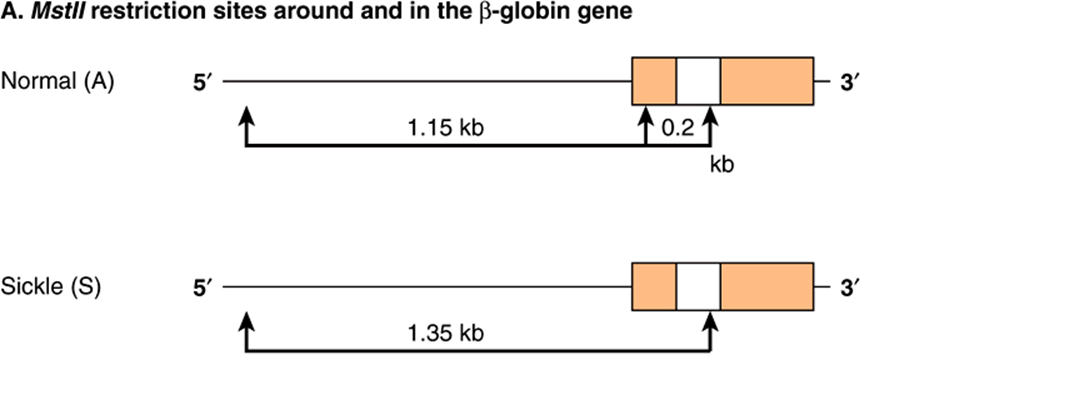
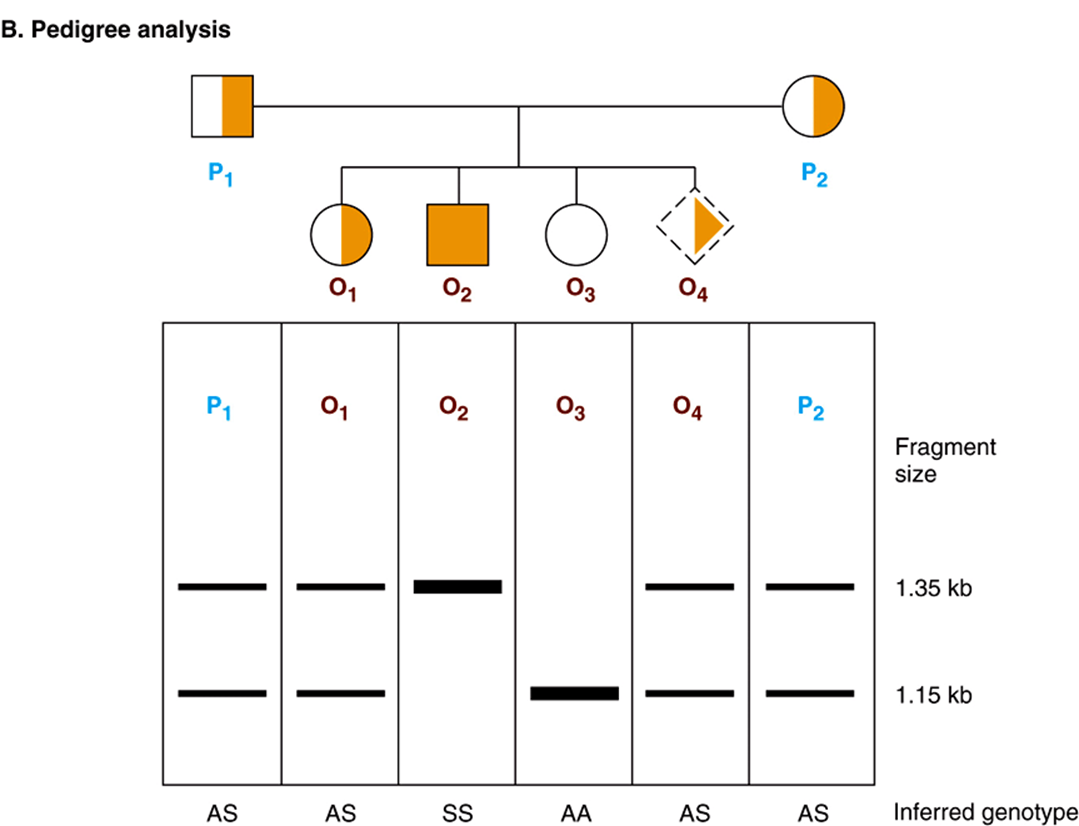
FIGURE 39–9 Pedigree analysis of sickle-cell disease. The top part of the figure (A) shows the first part of the β-globin gene and the MstII restriction enzyme sites in the normal (A) and sickle-cell (S) β-globin genes. Digestion with the restriction enzyme MstII results in DNA fragments 1.15 kb and 0.2 kb long in normal individuals. The T-to-A change in individuals with sickle-cell disease abolishes one of the three MstII sites around the β-globin gene; hence, a single restriction fragment 1.35 kb in length is generated in response to MstII. This size difference is easily detected on a Southern blot. (The 0.2-kb fragment would run off the gel in this illustration.) (B) Pedigree analysis shows three possibilities: AA = normal (open circle); AS = heterozygous (half-solid circles, half-solid square); SS = homozygous (solid square). This approach can allow for prenatal diagnosis of sickle-cell disease (dash-sided square). See the text.
Prenatal Diagnosis
If the genetic lesion is understood and a specific probe is available, prenatal diagnosis is possible. DNA from cells collected from as little as 10 mL of amniotic fluid (or by chorionic villus biopsy) can be analyzed by Southern blot transfer. A fetus with the restriction pattern AA in Figure 39–9 does not have sickle-cell disease, nor is it a carrier. A fetus with the SS pattern will develop the disease. Probes are now available for this type of analysis of many genetic diseases.
Restriction Fragment Length Polymorphism and SNPs
The differences in DNA sequence cited above can result in variations of restriction sites and thus in the length of restriction fragments. Similarly, single nucleotide polymorphisms, or SNPs, can be detected by the sensitive PCR method. An inherited difference in the pattern of restriction enzyme digestion (eg, a DNA variation occurring in more than 1% of the general population) is known as a restriction fragment length polymorphism (RFLP).Extensive RFLP and SNP maps of the human genome have been constructed. This is proving useful in the Human Genome Analysis Project and is an important component of the effort to understand various single-gene and multigenic diseases. RFLPs result from single-base changes (eg, sickle-cell disease) or from deletions or insertions (CNVs) of DNA into a restriction fragment (eg, the thalassemias) and have proved to be useful diagnostic tools. They have been found at known gene loci and in sequences that have no known function; thus, RFLPs may disrupt the function of the gene or may have no apparent biologic consequences. As mentioned above, 80% of the SNPs in the genome of a single known individual had already been mapped independently through the efforts of the SNP-mapping component of the International HapMap Project.
RFLPs and SNPs are inherited, and they segregate in a mendelian fashion. A major use of SNPs/RFLPs is in the definition of inherited diseases in which the functional deficit is unknown. SNPs/RFLPs can be used to establish linkage groups, which in turn, by the process of chromosome walking, will eventually define the disease locus. In chromosome walking (Figure 39–10), a fragment representing one end of a long piece of DNA is used to isolate another that overlaps but extends the first. The direction of extension is determined by restriction mapping, and the procedure is repeated sequentially until the desired sequence is obtained. Collections of mapped, overlapping BAC- or PAC-cloned human genomic DNAs are commercially available. The X chromosome-linked disorders are particularly amenable to the approach of chromosome walking since only a single allele is expressed. Hence, 20% of the defined RFLPs are on the X chromosome and a complete linkage map (and genomic sequence) of this chromosome have been determined. The gene for the X-linked disorder, Duchenne-type muscular dystrophy, was found using RFLPs. Similarly, the defect in Huntington disease was localized to the terminal region of the short arm of chromosome 4, and the defect that causes polycystic kidney disease is linked to the α-globin locus on chromosome 16.
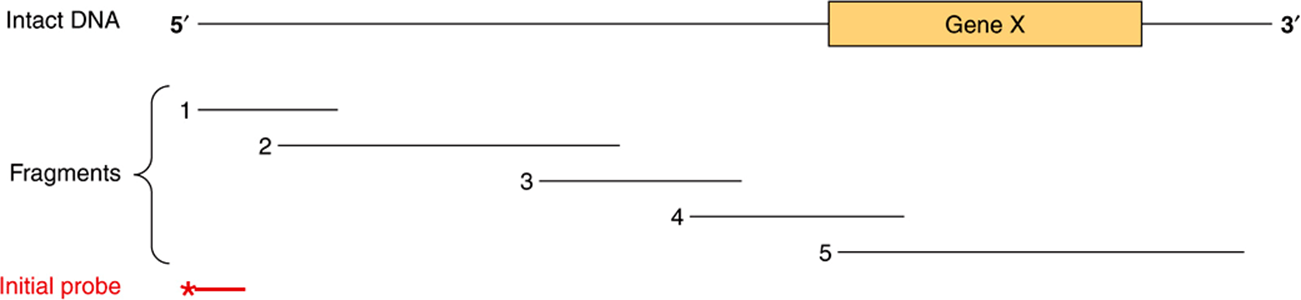
FIGURE 39–10 The technique of chromosome walking. Gene X is to be isolated from a large piece of DNA. The exact location of this gene is not known, but a probe (*——) directed against a fragment of DNA (shown at the 5′ end in this representation) is available, as is a library of clones containing a series of overlapping DNA insert fragments. For the sake of simplicity, only five of these are shown. The initial probe will hybridize only with clones containing fragment 1, which can then be isolated and used as a probe to detect fragment 2. This procedure is repeated until fragment 4 hybridizes with fragment 5, which contains the entire sequence of gene X.
Microsatellite DNA Polymorphisms
Short (2-6 bp), inherited, tandem repeat units of DNA occur about 50,000-100,000 times in the human genome (Chapter 35). Because they occur more frequently—and in view of the routine application of sensitive PCR methods—they are replacing RFLPs as the marker loci for various genome searches.
RFLPs & VNTRs in Forensic Medicine
Variable numbers of tandemly repeated (VNTR) units are one common type of “insertion” that results in an RFLP. The VNTRs can be inherited, in which case they are useful in establishing genetic association with a disease in a family or kindred; or they can be unique to an individual and thus serve as a molecular fingerprint of that person.
Direct Sequencing of Genomic DNA
As noted above, recent advances in DNA sequencing technology, the so-called next generation sequencing (NGS) platforms, have dramatically reduced the per base cost of DNA sequencing. The initial sequence of the human genome cost roughly $350,000,000. The cost of sequencing the same 3 × 109 bp diploid human genome using the new NGS platforms is estimated to be <0.03% of the original. This dramatic reduction in cost has stimulated various international initiatives to sequence the entire genomes of thousands of individuals of various racial and ethnic backgrounds in order to determine the true extent of DNA/genome polymorphisms present within the population. The resulting cornucopia of genetic information, and the ever-decreasing cost of genomic DNA sequencing is dramatically increasing our ability to diagnose and, ultimately treat human disease. Obviously, when personal genome sequencing does become commonplace, dramatic changes in the practice of medicine will result because therapies will ultimately be custom tailored to the exact genetic makeup of each individual.
Gene Therapy and Stem Cell Biology
Diseases caused by deficiency of a single gene product (Table 39-4) are all theoretically amenable to replacement therapy. The strategy is to clone a normal copy of the relevant gene (eg, the gene that codes for adenosine deaminase) into a vector that will readily be taken up and incorporated into the genome of a host cell. Bone marrow precursor cells are being investigated for this purpose because they presumably will resettle in the marrow and replicate there. The introduced gene would begin to direct the expression of its protein product, and this would correct the deficiency in the host cell.
As an alternative to “replacing” defective genes to cure human disease, many scientists are investigating the feasibility of identifying and characterizing pluripotent stem cells that have the ability to differentiate into any cell type in the body. Recent results in this field have shown that adult human somatic cells can readily be converted into apparent induced pluripotent stem cells (iPSCs) by transfection with cDNAs encoding a handful of DNA binding transcription factors. These and other new developments in the fields of gene therapy and stem cell biology promise exciting new potential therapies for curing human disease.
Transgenic Animals
The somatic cell gene replacement therapy described above would obviously not be passed on to offspring. Other strategies to alter germ cell lines have been devised but have been tested only in experimental animals. A certain percentage of genes injected into a fertilized mouse ovum will be incorporated into the genome and found in both somatic and germ cells. Hundreds of transgenic animals have been established, and these are useful for analysis of tissue-specific effects on gene expression and effects of overproduction of gene products (eg, those from the growth hormone gene or oncogenes) and in discovering genes involved in development—a process that heretofore has been difficult to study. The transgenic approach has been used to correct a genetic deficiency in mice. Fertilized ova obtained from mice with genetic hypogonadism were injected with DNA containing the coding sequence for the gonadotropin-releasing hormone (GnRH) precursor protein. This gene was expressed and regulated normally in the hypothalamus of a certain number of the resultant mice, and these animals were in all respects normal. Their offspring also showed no evidence of GnRH deficiency. This is, therefore, evidence of somatic cell expression of the transgene and of its maintenance in germ cells.
Targeted Gene Disruption or Knockout
In transgenic animals, one is adding one or more copies of a gene to the genome, and there is no way to control where that gene eventually resides. A complementary—and much more difficult—approach involves the selective removal of a gene from the genome. Gene knockout animals (usually mice) are made by creating a mutation that totally disrupts the function of a gene. This is then used to replace one of the two genes in an embryonic stem cell that can be used to create a heterozygous transgenic animal. The mating of two such animals will, by Mendelian genetics, result in a homozygous mutation in 25% of offspring. Several thousand strains of mice with knockouts of specific genes have been developed. Techniques for disrupting genes in specific cells, tissues, or organs have been developed, so-called conditional, or directed, knockouts. This can be accomplished by taking advantage of particular promoter-enhancer combinations driving expression of DNA recombinases, or alternatively expression of miRNAs, both of which inactivate gene expression. These methods are particularly useful in cases where gene ablation during early development causes embryonic lethality.
RNA and Protein Profiling, and Protein-DNA Interaction Mapping
The “-omic” revolution of the last decade has culminated in the determination of the nucleotide sequences of entire genomes, including those of budding and fission yeasts, numerous bacteria, the fruit fly, the worm Caenorhabditis elegans, plants, the mouse, rat, chicken, monkey and, most notably, humans. Additional genomes are being sequenced at an accelerating pace. The availability of all of this DNA sequence information, coupled with engineering advances, has lead to the development of several revolutionary methodologies, most of which are based upon high-density microarray technology or NGS platforms. In the case of microarrays, it is now possible to deposit thousands of specific, known, definable DNA sequences (more typically now synthetic oligonucleotides) on a glass microscope-style slide in the space of a few square centimeters. By coupling such DNA microarrays with highly sensitive detection of hybridized fluorescently labeled nucleic acid probes derived from mRNA, investigators can rapidly and accurately generate profiles of gene expression (eg, specific cellular mRNA content) from cell and tissue samples as small as 1 g or less. Thus, entire transcriptome information (the entire collection of cellular RNAs) for such cell or tissue sources can readily be obtained in only a few days. In the case of NGS sequencing, mRNAs are converted to cDNAs using reverse transcription, and these cDNAs are amplified by PCR and directly sequenced; this method is termed RNA-Seq. Methods are being developed to allow direct RNA sequencing obviating the need for the cDNA/PCR step. These methods allow for the description of the entire transcriptome. Recent methodological advances (GRO-Seq, Global Run-On sequencing, and NET-seq, native elongating transcript sequencing)allow for sequencing of RNA within elongating RNA polymerase-DNA-RNA ternary complexes, thereby allowing nucleotide-level descriptions, genome-wide, of transcription in living cells. Such transcriptome information allows one to quantitatively predict the collection of proteins that might be expressed in a particular cell, tissue, or organ in normal and disease states based upon the mRNAs present in those cells. Complementing this high-throughput, transcript-profiling method is the recent development of methods to map the location, or occupancy of specific proteins bound to discrete sites within living cells. This method, illustrated in Figure 39–11, is termed chromatin immunoprecipitation (ChIP). Proteins are crosslinked in situ in cells or tissues, chromatin isolated, sheared, and specific protein DNA complexes purified using antibodies recognizing a particular protein, or protein iso-form. DNA bound to this protein is recovered and analyzed using PCR and either gel electrophoresis, direct sequencing (ChIP-SEQ) or microarray analysis (ChIP-chip). Both ChIP-SEQ and ChIP-chip methods allow investigators to identify the entire genome-wide locations of a single protein throughout all the chromosomes; ChIP-SEQ allows mapping at nucleotide-level resolugion. Finally, methods for high-sensitivity, high-throughput mass spectrometry of metabolites (metabolomics) and complex protein samples (proteomics) have been developed. Newer mass spectrometry methods allow one to identify hundreds to thousands of proteins in samples extracted from very small numbers of cells (<1 g). Such analyses can now be used to quantitate the amounts of proteins in two samples as well as the level of certain PTMs, such as phosphorylation. This critical information tells investigators which of the many mRNAs detected in transcriptome mapping studies are actually translated into protein, generally the ultimate dictator of phenotype. New genetic means for identifying protein-protein interactions and protein function have also been devised. Systematic genome-wide gene expression knockdown, using SiRNAs (miRNAs), or synthetic lethal genetic interaction screens, have been applied to assess the contribution of individual genes to a variety of processes in model systems (yeast, worms, and flies) and mammalian cells (human and mouse). Specific network mappings of protein-protein interactions on a genome-wide basis have been identified using high-throughput variants of the two hybrid interaction test (Figure 39–12). This simple yet powerful method can be performed in bacteria, yeast, or metazoan cells, and allows for detecting specific protein-protein interactions in living cells. Reconstruction experiments indicate that protein-protein interactions with affinities of Kd ~ 1 μM or tighter can readily be detected with this method. Together, these technologies provide powerful new tools with which to dissect the intricacies of human biology.
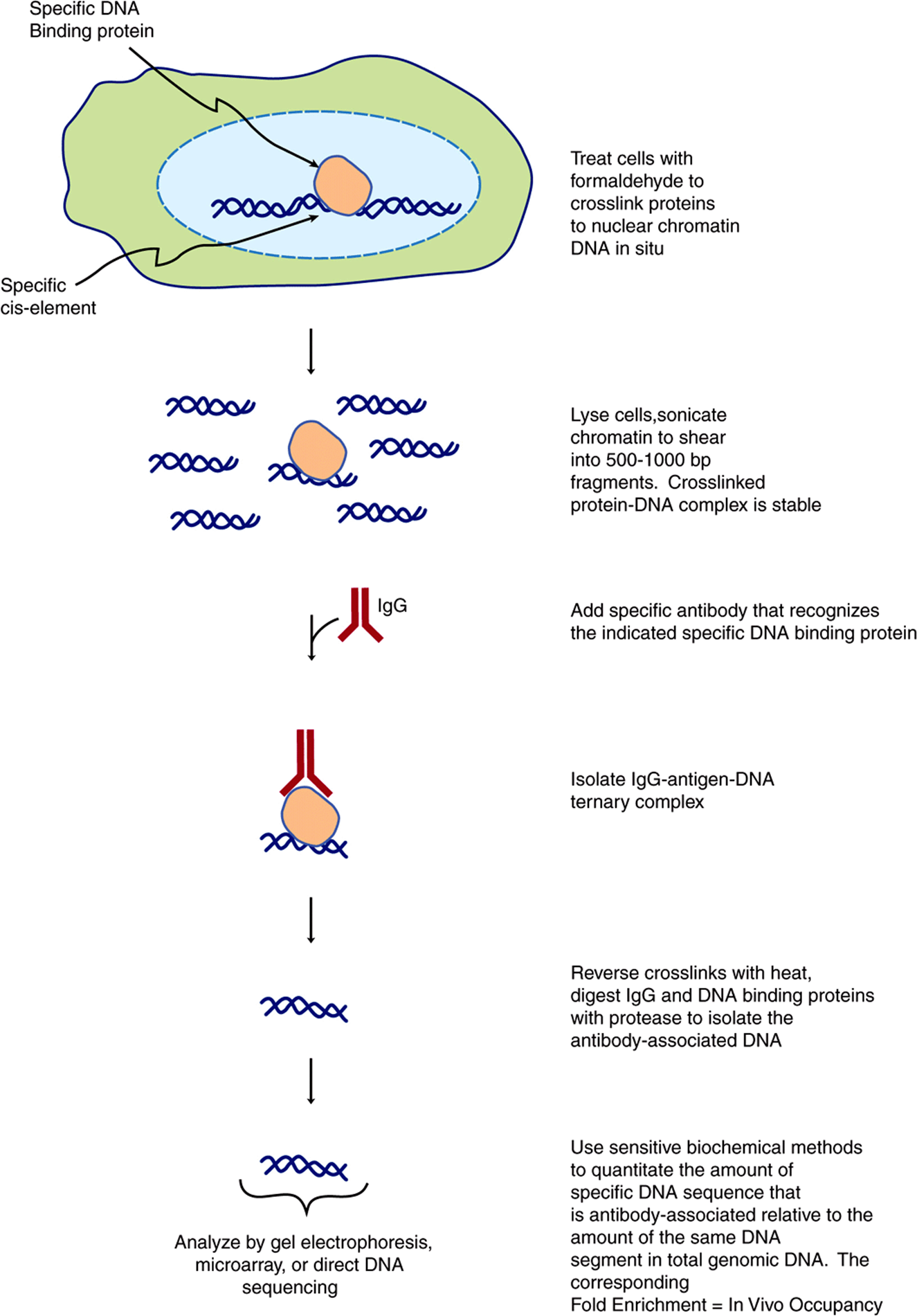
FIGURE 39–11 Outline of the chromatin immunoprecipitation (ChIP) technique. This method allows for the precise localization of a particular protein (or modified protein if an appropriate antibody is available; eg, phosphorylated or acetylated histones, transcription factors, etc) on a particular sequence element in living cells. Depending upon the method used to analyze the immunopurified DNA, quantitative or semi-quantitative information, at near nucleotide level resolution, can be obtained. Protein-DNA occupancy can be scored genome-wide in two ways. First, by ChIP-chip, a method that uses a hybridization readout. In ChIP-chip total genomic DNA is labeled with one particular fluorphore and the immunopurified DNA is labeled with a spectrally distinct fluorphore. These differentially labeled DNAs are mixed and hybridized to microarray ‘chips’ (microscope slides) that contain specific DNA fragments, or more commonly now, synthetic oligonucleotide 50-70 nucleotides long. These gene-specific oligonucleotides are deposited and covalently attached at predetermined, known X,Y coordinates on the slide. The labeled DNAs are hybridized, the slides washed and hybridization to each gene-specific oligonucleotide probe is scored using differential laser scanning and sensitive photodetection at micron resolution. The hybridization signal intensities are quantified and the ratio of IP DNA/genomic DNA signals is used to score occupancy levels. The second method, termed ChIP-seq, directly sequences immunopurified DNAs using NGS/deep sequencing methods. Both approaches rely upon efficient bioinformatic algorithms to deal with the very large datasets that are generated. ChIP-chip and ChIP-seq techniques provide a (semi-)quantitative measure of in vivo protein occupancy.
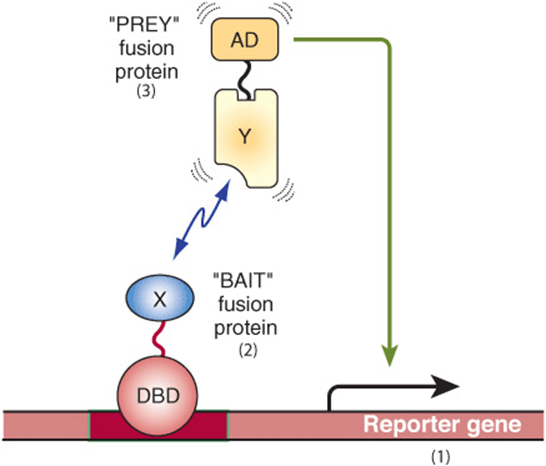
FIGURE 39–12 Overview of two hybrid system for identifying and characterizing protein-protein interactions. Shown are the basic components and operation of the two hybrid system, originally devised by Fields and Song (Nature 340:245-246 [1989]) to function in the bakers yeast system. (1) A reporter gene, either a selectable marker (ie, a gene conferring prototrophic growth on selective media, or producing an enzyme for which a colony colorimetric assay exists, such as β-galactosidase) that is expressed only when a transcription factor binds upstream to a cis-linked enhancer (dark red bar). (2) A “bait” fusion protein (DBD-X) produced from a chimeric gene expressing a modular DNA binding domain (DBD; often derived from the yeast Gal 4 protein or the bacterial Lex A protein, both high-affinity, high-specificity DNA binding proteins) fused in-frame to a protein of interest, here X. In two hybrid experiments, one is testing whether any protein can interact with protein X. Prey protein X may be fused in its entirety or often alternatively just a portion of protein X is expressed in-frame with the DBD. (3) A “prey” protein (Y-AD), which represents a fusion of a specific protein fused in-frame to a transcriptional activation domain (AD; often derived from either the Herpes simplex virus VP16 protein or the yeast Gal 4 protein). This system serves as a useful test of protein-protein interactions between proteins X and Y because in the absence of a functional transactivator binding to the indicated enhancer, no transcription of the reporter gene occurs (ie, see Figure 38–16). Thus, one observes transcription only if protein X-protein Y interaction occurs, thereby bringing a functional AD to the cis-linked transcription unit, in this case activating transcription of the reporter gene. In this scenario, protein DBD-X alone fails to activate reporter transcription because the X-domain fused to the DBD does not contain an AD. Similarly, protein Y-AD alone fails to activate reporter gene transcription because it lacks a DBD to target the Y-AD protein to the enhancer. Only when both proteins are expressed in a single cell and bind the enhancer and, via DBD-X-Y-AD protein-protein interactions, regenerate a functional transactivator binary “protein,” does reporter gene transcription result in activation and mRNA synthesis (line from AD to reporter gene).
Microarray techniques, high-throughput DNA sequencing, two-hybrid, genetic knockdown, and mass spectrometric protein and metabolite identification experiments have led to the generation of huge amounts of data. Appropriate data management and interpretation of the deluge of information forthcoming from such studies have relied upon statistical methods, and this new technology, coupled with the flood of DNA sequence information, has led to the development of the fields of bioinformatics (Chapter 11) and systems biology, new disciplines whose goals are to help manage, analyze, and integrate this flood of biologically important information. Future work at the intersection of bioinformatics, transcriptprotein/PTM profiling, and systems biology will revolutionize our understanding of physiology and medicine.
SUMMARY
![]() A variety of very sensitive techniques can now be applied to the isolation and characterization of genes and to the quantitation of gene products.
A variety of very sensitive techniques can now be applied to the isolation and characterization of genes and to the quantitation of gene products.
![]() In DNA cloning, a particular segment of DNA is removed from its normal environment using PCR or one of many restriction endonucleases. This is then ligated into a vector in which the DNA segment can be amplified and produced in abundance.
In DNA cloning, a particular segment of DNA is removed from its normal environment using PCR or one of many restriction endonucleases. This is then ligated into a vector in which the DNA segment can be amplified and produced in abundance.
![]() Cloned or in vitro synthesized DNA can be used as a probe in one of several types of hybridization reactions to detect other related or adjacent pieces of DNA, or it can be used to quantitate gene products such as mRNA.
Cloned or in vitro synthesized DNA can be used as a probe in one of several types of hybridization reactions to detect other related or adjacent pieces of DNA, or it can be used to quantitate gene products such as mRNA.
![]() Manipulation of the DNA to change its structure, so-called genetic engineering, is a key element in cloning (eg, the construction of chimeric molecules) and can also be used to study the function of a certain fragment of DNA and to analyze how genes are regulated.
Manipulation of the DNA to change its structure, so-called genetic engineering, is a key element in cloning (eg, the construction of chimeric molecules) and can also be used to study the function of a certain fragment of DNA and to analyze how genes are regulated.
![]() Chimeric DNA molecules are introduced into cells to make transfected cells or into the fertilized oocyte to make transgenic animals.
Chimeric DNA molecules are introduced into cells to make transfected cells or into the fertilized oocyte to make transgenic animals.
![]() Techniques involving cloned or synthetic DNA are used to locate genes to specific regions of chromosomes, identify the genes responsible for diseases, study how faulty gene regulation causes disease, diagnose genetic diseases, and increasingly to treat genetic diseases.
Techniques involving cloned or synthetic DNA are used to locate genes to specific regions of chromosomes, identify the genes responsible for diseases, study how faulty gene regulation causes disease, diagnose genetic diseases, and increasingly to treat genetic diseases.
REFERENCES
Brass AL, Dykxhoorn DM, Benita Y, et al: Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008;319:921.
Core LJ, Waterfall JJ, Lis JT: Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 2008;322:1845-1848.
Churchman LS, Weissman JS: Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature 2011;469:368-373.
Friedman A, Perrimon N: Genome-wide high-throughput screens in functional genomics. Curr Opin Gen Dev 2004;14:470.
Gandhi TK, Zhong J, Mathivanan S, et al: Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 2006;38:285.
Gerstein MB, Lu ZJ, Van Nostrand EL, et al: Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 2010;330:1775-1787.
Gibson DG, Glass JI, Lartigue C, et al: Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010;329:52-56.
Gilchrist DA, Fargo DC, Adelman K: Using ChIP-chip and ChIP-seq to study the regulation of gene expression: genome-wide localization studies reveal widespread regulation of transcription elongation. Methods 2009;48:398-408.
Isaacs FJ, Carr PA, Wang HH, et al: Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348-353.
Kodzius R, Kojima M, Nishiyori H, et al: CAGE: cap analysis of gene expression. Nat Meth 2006;3:211-222.
Martin JB, Gusella JF: Huntington’s disease: pathogenesis and management. N Engl J Med 1986;315:1267.
Myers RM, Stamatoyannopoulos J, Snyder M, et al: A user’s guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011;9:e1001046.
Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, 1989.
Schlabach MR, Luo J, Solimini NL, et al: Cancer proliferation gene discovery through functional genomics. Science 2008;319:620.
Suter B, Kittanakom S, Stagljar I: Interactive proteomics: what lies ahead? Biotechniques 2008;44:681.
Spector DL, Goldman RD, Leinwand LA: Cells: A Laboratory Manual. Cold Spring Harbor Laboratory Press, 1998.
Takahashi K, Tanabe K, Ohnuki M, et al: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861.
The ENCODE Project Consortium: Identification and analysis of functional in 1% of the human genome by the ENCODE Pilot Project. Nature 2007;447:799.
Watson JD, Gilman M, Witkowski JA, et al: Recombinant DNA, 2nd ed. Scientific American Books. Freeman, 1992.
Weatherall DJ: The New Genetics and Clinical Practice, 3rd ed. Oxford University Press, 1991.
Wernig M, Meissner A, Foreman R, et al: In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 2007;448:318.
Wheeler DA, Srinivasan M, Egholm M, et al: The complete genome of an individual by massively parallel DNA sequencing. Nature 2008;451:872.
Wold B, Myers RM: Sequence census methods for functional genomics. Nat Meth 2008;5:19.
GLOSSARY
ARS: Autonomously replicating sequence; the origin of replication in yeast.
Autoradiography: The detection of radioactive molecules (eg, DNA, RNA, and protein) by visualization of their effects on photographic or x-ray film.
Bacteriophage: A virus that infects a bacterium.
Blunt-ended DNA: Two strands of a DNA duplex having ends that are flush with each other.
CAGE: Cap analysis of gene expression. A method that allows the selective capture, amplification, cloning and sequencing of mRNAs via the 5′-Cap structure.
cDNA: A single-stranded DNA molecule that is complementary to an mRNA molecule and is synthesized from it by the action of reverse transcriptase.
Chimeric molecule: A molecule (eg, DNA, RNA, and protein) containing sequences derived from two different species.
ChIP, chromatin immunoprecipitation: A technique that the determination of the exact localization of a particular protein, or protein isoform, on any particular genomic location in a living cell. The method is based upon cross-linking of living cells, cell disruption, DNA fragmentation, and immunoprecipitation with specific antibodies that purify the cognate protein crosss-linked to DNA. Cross-links are reversed, associated DNA purified and specific sequences that are purified are measured using any of several different methods.
ChIP-chip, chromatin immunoprecipitation assayed via a microarray chip hybridization read-out: A hybridization-based method that uses chromatin immunoprecipitation (ChIP) techniques to map, genome-wide, the in vivo sites of binding of specific proteins within chromatin in living cells. Sequence-binding is determined by annealing fluorescently labeled DNA samples to microarrays (array).
ChIP-Seq, chromatin immunoprecipitation assayed via a NGS/deep sequencing read-out: Genomic DNA binding location in a ChIP determined by high-throughput deep\sequencing, rather than hybridization to microarrays.
Clone: A large number of organisms, cells or molecules that are identical with a single parental organism cell or molecule.
Copy number variation (CNV): Change in the copy number of specific genomic regions of DNA between two or more individuals. CNVs can be as large as 106 bp of DNA and include deletions or insertions.
Cosmid: A plasmid into which the DNA sequences from bacteriophage lambda that are necessary for the packaging of DNA (cos sites) have been inserted; this permits the plasmid DNA to be packaged in vitro.
ENCODE project: Encyclopedia of DNA elements project; an effort of multiple laboratories throughout the world to provide a detailed, biochemically informative representation of the human genome using high-throughput sequencing methods to identify and catalog the functional elements within a single restricted portion (~1% 30,000,000 bp) of one human chromosome.
Endonuclease: An enzyme that cleaves internal bonds in DNA or RNA.
Epigenetic code: The patterns of modification of chromosomal DNA (ie, cytosine methylation) and nucleosomal histone posttranslational modifications. These changes in modification status can lead to dramatic alterations in gene expression. Notably though, the actual underlying DNA sequence involved does not change.
Excinuclease: The excision nuclease involved in nucleotide exchange repair of DNA.
Exome: The nucleotide sequence of the entire complement of mRNA exons expressed in a particular cell, tissue, organ or organism. The exome differs from the transcriptome that represents the entire collection of genome transcripts; the exome represents a subset of the RNA sequences composing the transcriptome.
Exon: The sequence of a gene that is represented (expressed) as mRNA.
Exonuclease: An enzyme that cleaves nucleotides from either the 3′ or 5′ ends of DNA or RNA.
Fingerprinting: The use of RFLPs or repeat sequence DNA to establish a unique pattern of DNA fragments for an individual.
FISH: Fluorescence in situ hybidization, a method used to map the location of specific DNA sequences within fixed nuclei.
Footprinting: DNA with protein bound is resistant to digestion by DNase enzymes. When a sequencing reaction is performed using such DNA, a protected area, representing the “footprint” of the bound protein, will be detected because nucleases are unable to cleave the DNA directly bound by the protein.
GRO-Seq, global run-on sequencing: A method where nascent transcripts are specifically captured and sequenced using NGS/deep sequencing. This methods allows for the mapping of the location of active transcription complexes.
Hairpin: A double-helical stretch formed by base pairing between neighboring complementary sequences of a single strand of DNA or RNA.
Hybridization: The specific reassociation of complementary strands of nucleic acids (DNA with DNA, DNA with RNA, or RNA with RNA).
Insert: An additional length of base pairs in DNA, generally introduced by the techniques of recombinant DNA technology.
Intron: The sequence of an mRNA-encoding gene that is transcribed but excised before translation. tRNA genes can also contain introns.
Library: A collection of cloned fragments that represents, in aggregate, the entire genome. Libraries may be either genomic DNA (in which both introns and exons are represented) or cDNA (in which only exons are represented).
Ligation: The enzyme-catalyzed joining in phosphodiester linkage of two stretches of DNA or RNA into one; the respective enzymes are DNA and RNA ligases.
Lines: Long interspersed repeat sequences.
Microsatellite polymorphism: Heterozygosity of a certain microsatellite repeat in an individual.
Microsatellite repeat sequences: Dispersed or group repeat sequences of 2-5 bp repeated up to 50 times. May occur at 50-100 thousand locations in the genome.
miRNAs: MicroRNAs, 21-22 nucleotide long RNA species derived from RNA polymerase II transcription units, and 500-1500 bp in length via RNA processing. These RNAs, recently discovered, are thought to play crucial roles in gene regulation.
NET-seq, native elongating sequencing: Genome-wide analysis of eukaryotic mRNA nascent chain 3′-ends maped at nucleotide-level resolution. RNA Polymerase II elongation complexes are captured by immunopurification with anti-Pol II IgG and nascent RNAs containing a free 3′OH group are tagged via ligation with an RNA linker and subsequently amplified by PCR and subjected to deep sequencing.
Nick translation: A technique for labeling DNA based on the ability of the DNA polymerase from E coli to degrade a strand of DNA that has been nicked and then to resynthesize the strand; if a radioactive nucleoside triphosphate is employed, the rebuilt strand becomes labeled and can be used as a radioactive probe.
Northern blot: A method for transferring RNA from an agarose or polyacrylamide gel to a nitrocellulose filter, on which the RNA can be detected by a suitable probe.
Oligonucleotide: A short, defined sequence of nucleotides joined together in the typical phosphodiester linkage.
Ori: The origin of DNA replication.
PAC: A high-capacity (70-95 kb) cloning vector based upon the lytic E coli bacteriophage P1 that replicates in bacteria as an extrachromosomal element.
Palindrome: A sequence of duplex DNA that is the same when the two strands are read in opposite directions.
Plasmid: A small, extrachromosomal, circular molecule of DNA that replicates independently of the host DNA.
Polymerase chain reaction (PCR): An enzymatic method for the repeated copying (and thus amplification) of the two strands of DNA that make up a particular gene sequence.
Primosome: The mobile complex of helicase and primase that is involved in DNA replication.
Probe: A molecule used to detect the presence of a specific fragment of DNA or RNA in, for instance, a bacterial colony that is formed from a genetic library or during analysis by blot transfer techniques; common probes are cDNA molecules, synthetic oligodeoxynucleotides of defined sequence, or antibodies to specific proteins.
Proteome: The entire collection of expressed proteins in an organism.
Pseudogene: An inactive segment of DNA arising by mutation of a parental active gene; typically generated by transposition of a cDNA copy of an mRNA.
Recombinant DNA: The altered DNA that results from the insertion of a sequence of deoxynucleotides not previously present into an existing molecule of DNA by enzymatic or chemical means.
Restriction enzyme: An endodeoxynuclease that causes cleavage of both strands of DNA at highly specific sites dictated by the base sequence.
Reverse transcription: RNA-directed synthesis of DNA, catalyzed by reverse transcriptase.
RNA-Seq: A method where cellular RNA populations are converted, via linker ligation and PCR into cDNAs that are then subjected to deep sequencing to determine the complete sequence of essentially all RNAs in the preparation.
RT-PCR: A method used to quantitate mRNA levels that relies upon a first step of cDNA copying of mRNAs catalyzed by reverse transcriptase prior to PCR amplification and quantitation.
Signal: The end product observed when a specific sequence of DNA or RNA is detected by autoradiography or some other method. Hybridization with a complementary radioactive polynucleotide (eg, by Southern or Northern blotting) is commonly used to generate the signal.
Sines: Short interspersed repeat sequences.
SiRNAs: Silencing RNAs, 21-25 nt in length generated by selective nucleolytic degradation of double-stranded RNAs of cellular or viral origin. SiRNAs anneal to various specific sites within target in RNAs leading to mRNA degradation, hence gene “knockdown.”
SNP: Single nucleotide polymorphism. Refers to the fact that single nucleotide genetic variation in genome sequence exists at discrete loci throughout the chromosomes. Measurement of allelic SNP differences is useful for gene mapping studies.
snRNA: Small nuclear RNA. This family of RNAs is best known for its role in mRNA processing.
Southern blot: A method for transferring DNA from an agarose gel to nitrocellulose filter, on which the DNA can be detected by a suitable probe (eg, complementary DNA or RNA).
Southwestern blot: A method for detecting protein-DNA interactions by applying a labeled DNA probe to a transfer membrane that contains a renatured protein.
Spliceosome: The macromolecular complex responsible for precursor mRNA splicing. The spliceosome consists of at least five small nuclear RNAs (snRNA; U1, U2, U4, U5, and U6) and many proteins.
Splicing: The removal of introns from RNA accompanied by the joining of its exons.
Sticky-ended DNA: Complementary single strands of DNA that protrude from opposite ends of a DNA duplex or from the ends of different duplex molecules (see also Blunt-ended DNA, above).
Tandem: Used to describe multiple copies of the same sequence (eg, DNA) that lie adjacent to one another.
Terminal transferase: An enzyme that adds nucleotides of one type (eg, deoxyadenonucleotidyl residues) to the 3′ end of DNA strands.
Transcription: Template DNA-directed synthesis of nucleic acids, typically DNA-directed synthesis of RNA.
Transcriptome: The entire collection of expressed RNAs in a cell, tissue, organ, or organism.
Transgenic: Describing the introduction of new DNA into germ cells by its injection into the nucleus of the ovum.
Translation: Synthesis of protein using mRNA as template.
Vector: A plasmid or bacteriophage into which foreign DNA can be introduced for the purposes of cloning.
Western blot: A method for transferring protein to a nitrocellulose filter, on which the protein can be detected by a suitable probe (eg, an antibody).