Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION I. Structures & Functions of Proteins & Enzymes
Chapter 4. Proteins: Determination of Primary Structure
Peter J. Kennelly, PhD & Victor W. Rodwell, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe multiple chromatographic methods commonly employed for the isolation of proteins from biologic materials.
Describe multiple chromatographic methods commonly employed for the isolation of proteins from biologic materials.
![]() Explain how scientists analyze the sequence or structure of a protein to extract insights into its possible physiologic function.
Explain how scientists analyze the sequence or structure of a protein to extract insights into its possible physiologic function.
![]() List several of the posttranslational alterations that proteins undergo during their lifetime and the influence of such modifications upon a protein’s function and fate.
List several of the posttranslational alterations that proteins undergo during their lifetime and the influence of such modifications upon a protein’s function and fate.
![]() Describe the chemical basis of the Edman method for determining primary structure.
Describe the chemical basis of the Edman method for determining primary structure.
![]() Give three reasons why mass spectrometry (MS) has largely supplanted chemical methods for the determination of the primary structure of proteins and the detection of posttranslational modifications.
Give three reasons why mass spectrometry (MS) has largely supplanted chemical methods for the determination of the primary structure of proteins and the detection of posttranslational modifications.
![]() Explain why MS can detect posttranslational modifications that are not detected by Edman sequencing or DNA sequencing.
Explain why MS can detect posttranslational modifications that are not detected by Edman sequencing or DNA sequencing.
![]() Describe how DNA cloning and molecular biology made the determination of the primary structures of proteins much more rapid and efficient.
Describe how DNA cloning and molecular biology made the determination of the primary structures of proteins much more rapid and efficient.
![]() Explain what is meant by “the proteome” and cite examples of its ultimate potential significance.
Explain what is meant by “the proteome” and cite examples of its ultimate potential significance.
![]() Comment on the contributions of genomics, computer algorithms, and databases to the identification of the open reading frames (ORFs) that encode a given protein.
Comment on the contributions of genomics, computer algorithms, and databases to the identification of the open reading frames (ORFs) that encode a given protein.
BIOMEDICAL IMPORTANCE
Proteins are physically and functionally complex macromolecules that perform multiple critically important roles. For example, an internal protein network, the cytoskeleton (Chapter 49) maintains cellular shape and physical integrity. Actin and myosin filaments form the contractile machinery of muscle (Chapter 49). Hemoglobin transports oxygen (Chapter 6), while circulating antibodies defend against foreign invaders (Chapter 50). Enzymes catalyze reactions that generate energy, synthesize and degrade biomolecules, replicate and transcribe genes, process mRNAs, etc (Chapter 7). Receptors enable cells to sense and respond to hormones and other environmental cues (Chapters 41 and 42). Proteins are subject to physical and functional changes that mirror the life cycle of the organisms in which they reside. A typical protein is “born” at translation (Chapter 37), matures through posttranslational processing events such as selective proteolysis (Chapters 9 and 37), alternates between working and resting states through the intervention of regulatory factors (Chapter 9), ages through oxidation, deamidation, etc (Chapter 52), and “dies” when degraded to its component amino acids (Chapter 29). An important goal of molecular medicine is to identify biomarkers such as proteins and/or modifications to proteins whose presence, absence, or deficiency is associated with specific physiologic states or diseases (Figure 4–1).

FIGURE 4–1 Diagrammatic representation of the life cycle of a hypothetical protein.. (1) The life cycle begins with the synthesis on a ribosome of a polypeptide chain, whose primary structure is dictated by an mRNA. (2) As synthesis proceeds, the polypeptide begins to fold into its native conformation (blue). (3) Folding may be accompanied by processing events such as proteolytic cleavage of an N-terminal leader sequence (Met-Asp-Phe-Gln-Val) or the formation of disulfide bonds (S—S). (4) Subsequent covalent modifications may, for example, attach a fatty acid molecule (yellow) for (5) translocation of the modified protein to a membrane. (6) Binding an allosteric effector (red) may trigger the adoption of a catalytically active conformation. (7) Over time, proteins get damaged by chemical attack, deamidation, or denaturation, and (8) may be “labeled” by the covalent attachment of several ubiquitin molecules (Ub). (9) The ubiquitinated protein is subsequently degraded to its component amino acids, which become available for the synthesis of new proteins.
PROTEINS & PEPTIDES MUST BE PURIFIED PRIOR TO ANALYSIS
Highly purified protein is essential for the detailed examination of its physical and functional properties. Cells contain thousands of different proteins, each in widely varying amounts. The isolation of a specific protein in quantities sufficient for analysis of its properties thus presents a formidable challenge that may require successive application of multiple purification techniques. Selective precipitation exploits differences in relative solubility of individual proteins as a function of pH (isoelectric precipitation), polarity (precipitation with ethanol or acetone), or salt concentration (salting out with ammonium sulfate). Chromatographic techniques separate one protein from another based upon difference in their size (size exclusion chromatography), charge (ion-exchange chromatography), hydrophobicity (hydrophobic interaction chromatography), or ability to bind a specific ligand (affinity chromatography).
Column Chromatography
In column chromatography, the stationary phase matrix consists of small beads loaded into a cylindrical container of glass, plastic, or steel called a column. Liquid-permeable frits confine the beads within this space while allowing the mobile-phase liquid to flow or percolate through the column. The stationary phase beads can be chemically derivatized to coat their surface with the acidic, basic, hydrophobic, or ligand-like groups required for ion exchange, hydrophobic interaction, or affinity chromatography. As the mobile-phase liquid emerges from the column, it is automatically collected in a series of small portions called fractions. Figure 4–2 depicts the basic arrangement of a simple bench-top chromatography system.
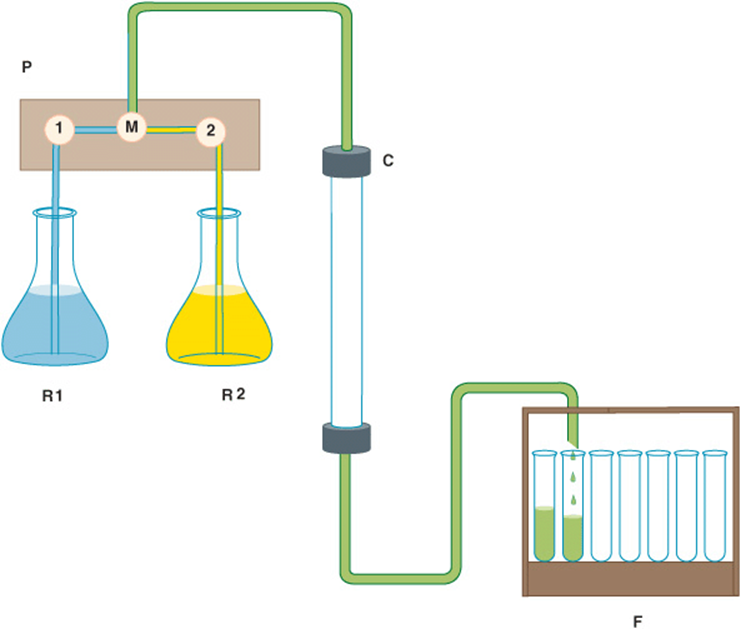
FIGURE 4–2 Components of a typical liquid chromatography apparatus. R1 and R2: Reservoirs of mobile-phase liquid. P: Programable pumping system containing two pumps, 1 and 2, and a mixing chamber, M. The system can be set to pump liquid from only one reservoir, to switch reservoirs at some predetermined point to generate a step gradient, or to mix liquids from the two reservoirs in proportions that vary over time to create a continuous gradient. C: Glass, metal, or plastic column containing stationary phase. F: Fraction collector for collecting portions, called fractions, of the eluant liquid in separate test tubes.
HPLC—High-Pressure Liquid Chromatography
First-generation column chromatography matrices consisted of long, intertwined oligosaccharide polymers shaped into spherical beads roughly a tenth of a millimeter in diameter. Unfortunately, their relatively large size perturbed mobile-phase flow and limited the available surface area. Reducing particle size offered the potential to greatly increase resolution. However, the resistance created by the more tightly packed matrix required the use of very high pressures that would crush the soft and spongy polysaccharide beads and similar materials, eg, acrylamide. Eventually, methods were developed to manufacture silicon particles of the necessary size and shape, to derivatize their surface with various functional groups, and to pack them into stainless steel columns capable of withstanding pressures of several thousand psi. Because of their greater resolving power, high-pressure liquid chromatography systems have largely displaced the once familiar glass columns in the protein purification laboratory.
Size-exclusion Chromatography
Size exclusion—or gel filtration—chromatography separates proteins based on their Stokes radius; the radius of the sphere they occupy as they tumble in solution. The Stokes radius is a function of molecular mass and shape. A tumbling elongated protein occupies a larger volume than a spherical protein of the same mass. Size-exclusion chromatography employs porous beads (Figure 4–3). The pores are analogous to indentations in a riverbank. As objects move downstream, those that enter an indentation are retarded until they drift back into the main current. Similarly, proteins with Stokes radii too large to enter the pores (excluded proteins), remain in the flowing mobile phase, and emerge before proteins that can enter the pores (included proteins). Proteins thus emerge from a gel filtration column in descending order of their Stokes radii.
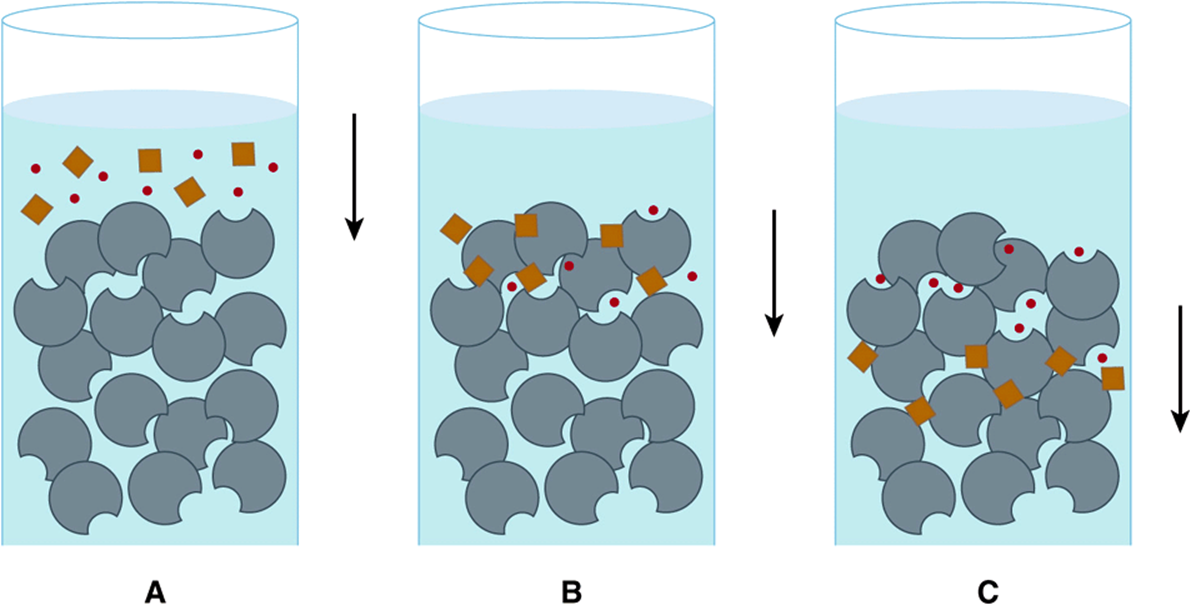
FIGURE 4–3 Size-exclusion chromatography. A: A mixture of large molecules (brown) and small molecules (red) are applied to the top of a gel filtration column. B: Upon entering the column, the small molecules enter pores in the stationary phase matrix (gray) from which the large molecules are excluded. C: As the mobile phase (blue) flows down the column, the large, excluded molecules flow with it, while the small molecules, which are temporarily sheltered from the flow when inside the pores, lag farther and farther behind.
Ion-Exchange Chromatography
In ion-exchange chromatography, proteins interact with the stationary phase by charge-charge interactions. Proteins with a net positive charge at a given pH will tightly adhere to beads with negatively charged functional groups such as carboxy-lates or sulfates (cation exchangers). Similarly, proteins with a net negative charge adhere to beads with positively charged functional groups, typically tertiary, or quaternary amines (anion exchangers). Nonadherent proteins flow through the matrix and are washed away. Bound proteins are then selectively displaced by gradually raising the ionic strength of the mobile phase, thereby weakening charge-charge interactions. Proteins elute in inverse order of the strength of their interactions with the stationary phase.
Hydrophobic Interaction Chromatography
Hydrophobic interaction chromatography separates proteins based on their tendency to associate with a stationary phase matrix coated with hydrophobic groups (eg, phenyl Sepharose, octyl Sephadex). Proteins with exposed hydrophobic surfaces adhere to the matrix via hydrophobic interactions that are enhanced by employing a mobile phase of high ionic strength. After nonadherent proteins are washed away, the polarity of the mobile phase is decreased by gradually lowering the salt concentration of the flowing mobile phase. If the interaction between protein and stationary phase is particularly strong, ethanol or glycerol may be added to the mobile phase to decrease its polarity and further weaken hydrophobic interactions.
Affinity Chromatography
Affinity chromatography exploits the high selectivity of most proteins for their ligands. Enzymes may be purified by affinity chromatography using immobilized substrates, products, coenzymes, or inhibitors. In theory, only proteins that interact with the immobilized ligand adhere. Bound proteins are then eluted either by competition with free, soluble ligand or, less selectively, by disrupting protein-ligand interactions using urea, guanidine hydrochloride, mildly acidic pH, or high salt concentrations. Commercially available stationary phase matrices contain ligands such as NAD+ or ATP analogs. Purification of recombinantly expressed proteins is frequently facilitated by modifying the cloned gene to add a new fusion domain designed to interact with a specific matrix-bound ligand (Chapter 7).
Protein Purity is Assessed by Polyacrylamide Gel Electrophoresis (PAGE)
The most widely used method for determining the purity of a protein is SDS-PAGE—polyacrylamide gel electrophoresis (PAGE) in the presence of the anionic detergent sodium dodecyl sulfate (SDS). Electrophoresis separates charged biomolecules based on the rates at which they migrate in an applied electrical field. For SDS-PAGE, acrylamide is polymerized and cross-linked to form a porous matrix. SDS binds to proteins at a ratio of one molecule of SDS per two peptide bonds, causing the polypeptide to unfold or denature. When used in conjunction with 2-mercaptoethanol or dithiothreitol to reduce and break disulfide bonds (Figure 4–4), SDS-PAGE separates the component polypeptides of multimeric proteins. The large number of anionic SDS molecules, each bearing a charge of –1, overwhelms the charge contributions of the amino acid functional groups endogenous to the polypeptides. Since the charge-to-mass ratio of each SDS-polypeptide complex is approximately equal, the physical resistance each peptide encounters as it moves through the acrylamide matrix determines the rate of migration. Since large complexes encounter greater resistance, polypeptides separate based on their relative molecular mass (Mr). Individual polypeptides trapped in the acrylamide gel after removal of the electrical field are visualized by staining with dyes such as Coomassie blue (Figure 4–5).
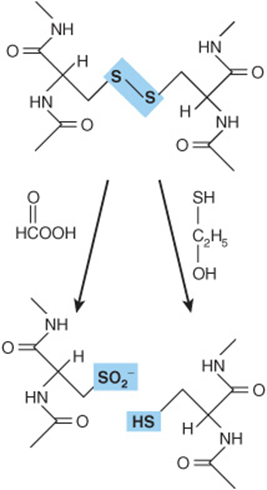
FIGURE 4–4 Oxidative cleavage of adjacent polypeptide chains linked by disulfide bonds (highlighted in blue) by performic acid (left) or reductive cleavage by β-mercaptoethanol (right) forms two peptides that contain cysteic acid residues or cysteinyl residues, respectively.
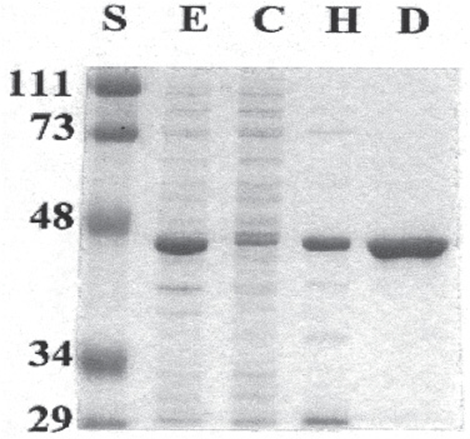
FIGURE 4–5 Use of SDS-PAGE to observe successive purification of a recombinant protein. The gel was stained with Coomassie blue. Shown are protein standards (lane S) of the indicated Mr, in kDa, crude cell extract (E), cytosol (C), high-speed supernatant liquid (H), and the DEAE-Sepharose fraction (D). The recombinant protein has a mass of about 45 kDa.
Isoelectric Focusing (IEF)
Ionic buffers called ampholytes and an applied electric field are used to generate a pH gradient within a polyacrylamide matrix. Applied proteins migrate until they reach the region of the matrix where the pH matches their isoelectric point (pI), the pH at which a molecule’s net charge is 0. IEF is used in conjunction with SDS-PAGE for two-dimensional electrophoresis, which separates polypeptides based on pI in one dimension and on Mr in the second (Figure 4–6). Two-dimensional electrophoresis is particularly well suited for separating the components of complex mixtures of proteins.
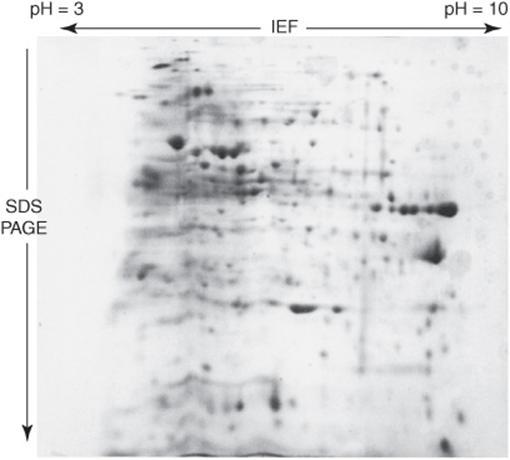
FIGURE 4–6 Two-dimensional IEF-SDS-PAGE. The gel was stained with Coomassie blue. A crude bacterial extract was first subjected to isoelectric focusing (IEF) in a pH 3–10 gradient. The IEF gel was then placed horizontally on the top of an SDS-PAGE gel, and the proteins then further resolved by SDS-PAGE. Notice the greatly improved resolution of distinct polypeptides relative to ordinary SDS-PAGE gel (Figure 4–5).
SANGER WAS THE FIRST TO DETERMINE THE SEQUENCE OF A POLYPEPTIDE
Mature insulin consists of the 21-residue A chain and the 30-residue B chain linked by disulfide bonds. Frederick Sanger reduced the disulfide bonds (Figure 4–4), separated the A and B chains, and cleaved each chain into smaller peptides using trypsin, chymotrypsin, and pepsin. The resulting peptides were then isolated and treated with acid to hydrolyze a portion of the peptide bonds and generate peptides with as few as two or three amino acids. Each peptide was reacted with 1-fluoro-2,4-dinitrobenzene (Sanger’s reagent), which derivatizes the exposed α-amino groups of the amino-terminal residues. The amino acid content of each peptide was then determined and the amino-terminal amino acid identified. The ε-amino group of lysine also reacts with Sanger’s reagent; but since an amino-terminal lysine reacts with 2 mol of Sanger’s reagent, it is readily distinguished from a lysine in the interior of a peptide. Working from di- and tripeptides up through progressively larger fragments, Sanger was able to reconstruct the complete sequence of insulin, an accomplishment for which he received a Nobel Prize, in 1958.
THE EDMAN REACTION ENABLES PEPTIDES & PROTEINS TO BE SEQUENCED
Pehr Edman introduced phenylisothiocyanate (Edman’s reagent) to selectively label the amino-terminal residue of a peptide. In contrast to Sanger’s reagent, the phenylthiohy-dantoin (PTH) derivative can be removed under mild conditions to generate a new amino-terminal residue (Figure 4–7). Successive rounds of derivatization with Edman’s reagent can therefore be used to sequence many residues of a single sample of peptide. Even so, the determination of the complete sequence of a protein by chemical methods remains a time-and labor-intensive process to this day.
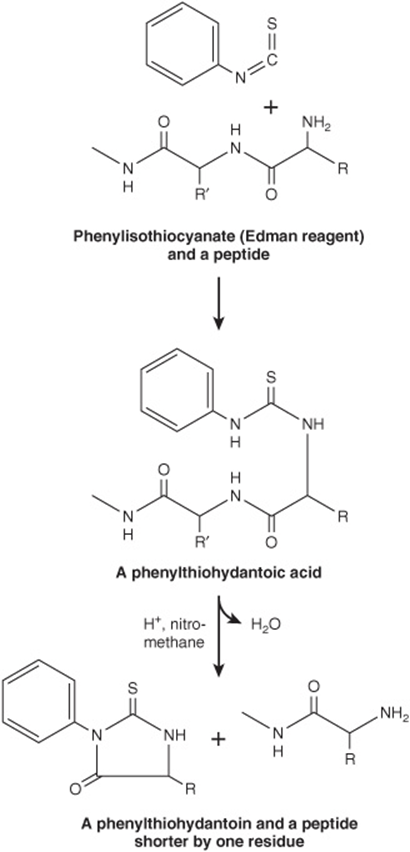
FIGURE 4–7 The Edman reaction. Phenylisothiocyanate derivatizes the amino-terminal residue of a peptide as a phenylthiohydantoic acid. Treatment with acid in a nonhydroxylic solvent releases a phenylthiohydantoin, which is subsequently identified by its chromatographic mobility, and a peptide one residue shorter. The process is then repeated.
The heterogeneous chemical properties of the amino acids meant that every step in the procedure represented a compromise between efficiency for any particular amino acid or set of amino acids and the flexibility needed to accommodate all 20. Consequently, each step in the process operates at less than 100% efficiency, which leads to the accumulation of polypeptide fragments with varying N-termini. Eventually, it becomes impossible to distinguish the correct PTH amino acid for that position in the peptide from the contaminants. As a result, the read length for Edman sequencing varies from 5 to 30 amino acid residues depending upon the quantity and purity of the peptide.
In order to determine the complete sequence of a poly-peptide several hundred residues in length, a protein must first be cleaved into smaller peptides, using either a protease or a reagent such as cyanogen bromide. Following purification by reversed phase high-pressure liquid chromatography (HPLC), these peptides are then analyzed by Edman sequencing. In order to assemble these short peptide sequences to solve the complete sequence of the intact polypeptide, it is necessary to analyze peptides whose sequences overlap one another. This is accomplished by generating multiple sets of peptides using more than one method of cleavage. The large quantities of purified protein required to test multiple protein fragmentation and peptide purification conditions constitutes the second major drawback of direct chemical protein sequencing techniques.
MOLECULAR BIOLOGY REVOLUTIONIZED THE DETERMINATION OF PRIMARY STRUCTURE
The reactions that sequentially derivatize and cleave PTH amino acids from the amino-terminal end of a peptide typically are conducted in an automated sequenator. DNA sequencing, by contrast, is both far more rapid and more economical. Recombinant techniques permit researchers to manufacture a virtually infinite supply of DNA using the original sample as template (Chapter 39). DNA sequencing methods, whose chemistry was also developed by Sanger, routinely enable polydeoxyribonucleotide sequences a few hundred residues in length to be determined in a single analysis, while automated sequencers can “read” sequences several thousand nucleotides in length. Knowledge of the genetic code enables the sequence of the encoded poly-peptide to be determined by simply translating the oligo-nucleotide sequence of its gene. Conversely, early molecular biologists designed complementary oligonucleotide probes to identify the DNA clone containing the gene of interest by reversing this process and using a segment of chemically determined amino acid sequence as template. The advent of DNA cloning thus ushered in the widespread use of a hybrid approach in which Edman chemistry was employed to sequence a small portion of the protein, then exploiting this information to determine the remaining sequence by DNA cloning and sequencing.
GENOMICS ENABLES PROTEINS TO BE IDENTIFIED FROM SMALL AMOUNTS OF SEQUENCE DATA
Today the number of organisms for which the complete DNA sequence of their genomes has been determined and made available to the scientific community numbers in the hundreds (see Chapter 10). These sequences encompass nearly all of the “model organisms” commonly employed in biomedical research laboratories: Homo sapiens, mouse, rat, Escherichia coli, Drosophila melanogaster, Caenorhabditis elegans, yeast, etc, as well as numerous pathogens. Meanwhile, across the globe, arrays of automated DNA sequenators continue to generate genome sequence data ever more rapidly and economically. Thus, for most research scientists the sequence of the protein(s) with which they are working has already been determined and lies waiting to be accessed in a database such as GenBank (Chapter 10). All that the scientist needs is to acquire sufficient amino acid sequence information from the protein, sometimes as little as five or six consecutive residues, to make an unambiguous identification. While the requisite amino acid sequence information can be obtained using the Edman technique, today mass spectrometry (MS) has emerged as the method of choice for protein identification.
MASS SPECTROMETRY CAN DETECT COVALENT MODIFICATIONS
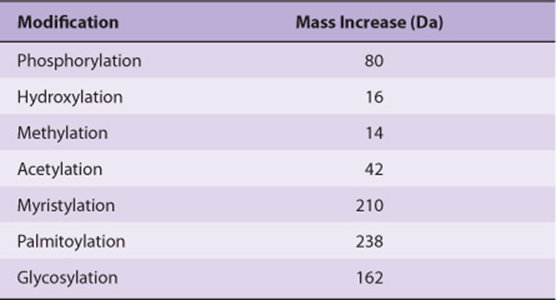
The superior sensitivity, speed, and versatility of MS have replaced the Edman technique as the principal method for determining the sequences of peptides and proteins. MS is significantly more sensitive and tolerant of variations in sample quality. Moreover, since mass and charge are common properties of a wide range of biomolecules, MS can be used to analyze metabolites, carbohydrates, and posttranslational modifications such as phosphorylation or hydroxylation that add readily identified increments of mass to a protein (Table 4–1). These modifications are difficult to detect using the Edman technique and undetectable in the DNA-derived amino acid sequence.
TABLE 4–1 Mass Increases Resulting from Common Post-Translational Modifications
MASS SPECTROMETERS COME IN VARIOUS CONFIGURATIONS
In a simple, single quadrupole mass spectrometer a sample is placed under vacuum and allowed to vaporize in the presence of a proton donor to impart a positive charge. An electrical field then propels the cations toward a curved flight tube where they encounter a magnetic field, which deflects them at a right angle to their original direction of flight (Figure 4–8). The current powering the electromagnet is gradually increased until the path of each ion is bent sufficiently to strike a detector mounted at the end of the flight tube. For ions of identical net charge, the force required to bend their path to the same extent is proportionate to their mass.
FIGURE 4–8 Basic components of a simple mass spectrometer. A mixture of molecules, represented by a red circle, green triangle, and blue diamond, is vaporized in an ionized state in the sample chamber. These molecules are then accelerated down the flight tube by an electrical potential applied to the accelerator grid (yellow). An adjustable field strength electromagnet applies a magnetic field that deflects the flight of the individual ions until they strike the detector. The greater the mass of the ion, the higher the magnetic field required to focus it onto the detector.
The flight tube for a time-of-flight (TOF) mass spectrometer is linear. Following vaporization of the sample in the presence of a proton donor, an electric field is briefly applied to accelerate the ions toward the detector at the end of the flight tube. For molecules of identical charge, the velocity to which they are accelerated—and hence the time required to reach the detector—is inversely proportional to their mass.
Quadrupole mass spectrometers generally are used to determine the masses of molecules of 4000 Da or less, whereas time-of -flight mass spectrometers are used to determine the large masses of complete proteins. Various combinations of multiple quadrupoles, or reflection of ions back down the linear flight tube of a TOF mass spectrometer, are used to create more sophisticated instruments.
Peptides Can Be Volatilized for Analysis by Electrospray Ionization or Matrix-Assisted Laser Desorption
The analysis of peptides and proteins by mass spectometry initially was hindered by difficulties in volatilizing large organic molecules. While small organic molecules could be readily vaporized by heating in a vacuum (Figure 4–9), proteins, oligonucleotides, etc, were destroyed under these conditions. Only when reliable techniques were devised for dispersing peptides, proteins, and other large biomolecules into the vapor phase was it possible to apply MS for their structural analysis and sequence determination. Dispersion into the vapor phase is accomplished by electrospray ionization and matrix-assisted laser desorption and ionization, aka MALDI. In electrospray ionization, the molecules to be analyzed are dissolved in a volatile solvent and introduced into the sample chamber in a minute stream through a capillary (Figure 4–9). As the droplet of liquid emerges into the sample chamber, the solvent rapidly disperses leaving the macromolecule suspended in the gaseous phase. The charged probe serves to ionize the sample. Electrospray ionization is frequently used to analyze peptides and proteins as they elute from an HPLC or other chromatography column already dissolved in a volatile solvent. In MALDI, the sample is mixed with a liquid matrix containing a light-absorbing dye and a source of protons. In the sample chamber, the mixture is excited using a laser, causing the surrounding matrix to disperse into the vapor phase so rapidly as to avoid heating embedded peptides or proteins (Figure 4–9).
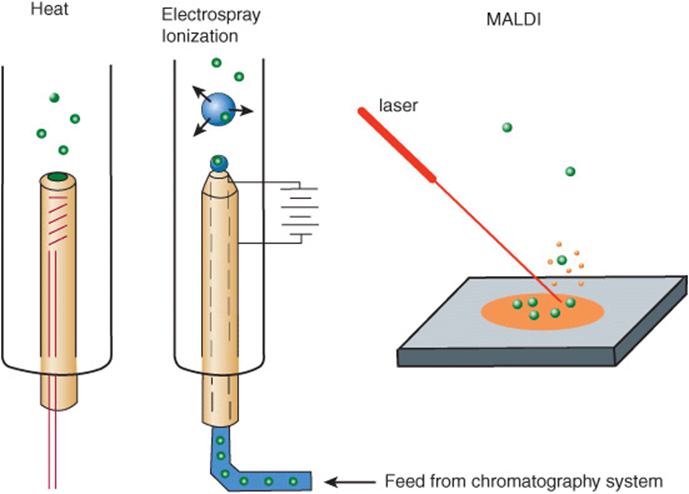
FIGURE 4–9 Three common methods for vaporizing molecules in the sample chamber of a mass spectrometer.
Peptides inside the mass spectrometer can be broken down into smaller units by collisions with neutral helium or argon atoms (collision-induced dissociation) and the masses of the individual fragments determined. Since peptide bonds are much more labile than carbon-carbon bonds, the most abundant fragments will differ from one another by units equivalent to one or two amino acids. Since—with the exceptions of (1) leucine and isoleucine and (2) glutamine and lysine—the molecular mass of each amino acid is unique, the sequence of the peptide can be reconstructed from the masses of its fragments.
Tandem Mass Spectrometry
Complex peptide mixtures can now be analyzed, without prior purification, by tandem MS, which employs the equivalent of two mass spectrometers linked in series. For this reason, such tandem instruments are often referred to as MS-MS. The first mass spectrometer separates individual peptides based upon their differences in mass. By adjusting the field strength of the first magnet, a single peptide can be directed into the second mass spectrometer, where fragments are generated and their masses determined. Alternatively, they can be held in an ion trap placed between the two quadrupoles and selectively passed to the second quadrupoles instead of being lost when the first quadrupoles is set to select ions of a different mass.
Tandem Mass Spectrometry Can Detect Metabolic Abnormalities
Tandem MS can be used to screen blood samples from new-borns for the presence and concentrations of amino acids, fatty acids, and other metabolites. Abnormalities in metabolite levels can serve as diagnostic indicators for a variety of genetic disorders, such as phenylketonuria, ethylmalonic encephalopathy, and glutaric acidemia type 1.
PROTEOMICS & THE PROTEOME
The Goal of Proteomics Is to Identify the Entire Complement of Proteins Elaborated by a Cell Under Diverse Conditions
While the sequence of the human genome is known, the picture provided by genomics alone is both static and incomplete. Proteomics aims to identify the entire complement of proteins elaborated by a cell under diverse conditions. As genes are switched on and off, proteins are synthesized in particular cell types at specific times of growth or differentiation and in response to external stimuli. Muscle cells express proteins not expressed by neural cells, and the type of subunits present in the hemoglobin tetramer undergo change pre- and postpartum. Many proteins undergo post-translational modifications during maturation into functionally competent forms or as a means of regulating their properties. Knowledge of the human genome therefore represents only the beginning of the task of describing living organisms in molecular detail and understanding the dynamics of processes such as growth, aging, and disease. As the human body contains thousands of cell types, each containing thousands of proteins, the proteome—the set of all the proteins expressed by an individual cell at a particular time—represents a moving target of formidable dimensions.
Two-Dimensional Electrophoresis & Gene Array Chips Are Used to Survey Protein Expression
One goal of proteomics is the identification of proteins whose levels of expression correlate with medically significant events. The presumption is that proteins whose appearance or disappearance is associated with a specific physiologic condition or disease are linked, either directly or indirectly, to their root causes and mechanisms. Determination of the proteomes characteristic of each cell type requires the utmost efficiency in the isolation and identification of individual proteins. The contemporary approach utilizes robotic automation to speed sample preparation and large two-dimensional gels to resolve cellular proteins. Individual polypeptides are then extracted and analyzed by Edman sequencing or mass spectroscopy. While only about 1000 proteins can be resolved on a single gel, two-dimensional electrophoresis has a major advantage, in that it examines the proteins themselves.
An alternative approach, called multidimensional protein identification technology (MudPIT) employs successive rounds of chromatography to resolve the peptides produced from the digestion of a complex biologic sample into several simpler fractions that can be analyzed separately by MS. Gene arrays, sometimes called DNA chips, in which the expression of the mRNAs that encode proteins is detected, offer a complementary approach to proteomics. While changes in the expression of the mRNA encoding a protein do not necessarily reflect comparable changes in the level of the corresponding protein, gene arrays are more sensitive than two-dimensional gels, particularly with respect to low abundance proteins, and thus can examine a wider range of gene products.
Bioinformatics Assists Identification of Protein Functions
The functions of a large proportion of the proteins encoded by the human genome are presently unknown. The development of protein arrays or chips for directly testing the potential functions of proteins on a mass scale remains in its infancy. However, recent advances in bioinformatics permit researchers to compare amino acid sequences to discover clues to potential properties, physiologic roles, and mechanisms of action of proteins. Algorithms exploit the tendency of nature to employ variations of a structural theme to perform similar functions in several proteins [eg, the Rossmann nucleotide binding fold to bind NAD(P)H, nuclear targeting sequences, and EF hands to bind Ca2+]. These domains generally are detected in the primary structure by conservation of particular amino acids at key positions. Insights into the properties and physiologic role of a newly discovered protein thus may be inferred by comparing its primary structure with that of known proteins.
SUMMARY
![]() Long amino acid polymers or polypeptides constitute the basic structural unit of proteins, and the structure of a protein provides insight into how it fulfills its functions.
Long amino acid polymers or polypeptides constitute the basic structural unit of proteins, and the structure of a protein provides insight into how it fulfills its functions.
![]() Proteins undergo post-transitional alterations during their lifetime that influence their function and determine their fate.
Proteins undergo post-transitional alterations during their lifetime that influence their function and determine their fate.
![]() The Edman method has largely been replaced by MS, a sensitive and versatile tool for determining primary structure, for identifying post-translational modifications, and for detecting metabolic abnormalities.
The Edman method has largely been replaced by MS, a sensitive and versatile tool for determining primary structure, for identifying post-translational modifications, and for detecting metabolic abnormalities.
![]() DNA cloning and molecular biology coupled with protein chemistry provide a hybrid approach that greatly increases the speed and efficiency for determination of primary structures of proteins.
DNA cloning and molecular biology coupled with protein chemistry provide a hybrid approach that greatly increases the speed and efficiency for determination of primary structures of proteins.
![]() Genomics—the analysis of the entire oligonucleotide sequence of an organism’s complete genetic material—has provided further enhancements.
Genomics—the analysis of the entire oligonucleotide sequence of an organism’s complete genetic material—has provided further enhancements.
![]() Computer algorithms facilitate identification of the ORFs that encode a given protein by using partial sequences and peptide mass profiling to search sequence databases.
Computer algorithms facilitate identification of the ORFs that encode a given protein by using partial sequences and peptide mass profiling to search sequence databases.
![]() Scientists are now trying to determine the primary sequence and functional role of every protein expressed in a living cell, known as its proteome.
Scientists are now trying to determine the primary sequence and functional role of every protein expressed in a living cell, known as its proteome.
![]() A major goal is the identification of proteins and of their post-translational modifications whose appearance or disappearance correlates with physiologic phenomena, aging, or specific diseases.
A major goal is the identification of proteins and of their post-translational modifications whose appearance or disappearance correlates with physiologic phenomena, aging, or specific diseases.
REFERENCES
Arnaud CH: Mass spec tackles proteins. Chem Eng News 2006;84:17.
Austin CP: The impact of the completed human genome sequence on the development of novel therapeutics for human disease. Annu Rev Med 2004;55:1.
Deutscher MP (editor): Guide to Protein Purification. Methods Enzymol, vol. 182, Academic Press, 1990 (Entire volume).
Gwynne P, Heebner G: Mass spectrometry in drug discovery and development: from physics to pharma. Science 2006;313:1315.
Kislinger T, Gramolini AO, MacLennan DH, et al: Multidimensional protein identification technology (MudPIT): technical overview of a profiling method optimized for the comprehensive proteomic investigation of normal and diseased heart tissue. J Am Soc Mass Spectrom 2005;16:1207.
Kolialexi A, Anagnostopoulos AK, Mavrou A, et al: Application of proteomics for diagnosis of fetal aneuploidies and pregnancy complications. J Proteomics 2009;72:731.
Levy PA: An overview of newborn screening. J Dev Behav Pediatr 2010;31:622.
Schena M, Shalon D, Davis RW, et al: Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995;270:467.
Scopes RK: Protein Purification. Principles and Practice, 3rd ed. Springer, 1994.
Semsarian C, Seidman CE: Molecular medicine in the 21st century. Intern Med J 2001;31:53.
Sharon M, Robinson CV: The role of mass spectrometry in structure elucidation of dynamic protein complexes. Annu Rev Biochem 2007;76:167.
Shendure J, Ji H: Next-generation DNA sequencing. Nature Biotechnol 2008;26:1135.
Sikaroodi M, Galachiantz Y, Baranova Al: Tumor markers: the potential of “omics” approach. Curr Mol Med 2010;10:249.
Woodage T, Broder S: The human genome and comparative genomics: understanding human evolution, biology, and medicine. J Gastroenterol 2003;15:68.