Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 49. Muscle & the Cytoskeleton
Robert K. Murray, MD, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Understand the general biochemical features of skeletal, cardiac, and smooth muscle contraction.
Understand the general biochemical features of skeletal, cardiac, and smooth muscle contraction.
![]() Know the biologic effects of nitric oxide (NO).
Know the biologic effects of nitric oxide (NO).
![]() Indicate the different metabolic fuels required for a sprint and for the marathon.
Indicate the different metabolic fuels required for a sprint and for the marathon.
![]() Know the general structures and functions of the major components of the cytoskeleton, namely microfilaments, microtubules, and intermediate filaments.
Know the general structures and functions of the major components of the cytoskeleton, namely microfilaments, microtubules, and intermediate filaments.
![]() Understand the bases of malignant hyperthermia Duchenne and Becker muscular dystrophies, inherited cardiomyopathies, the Hutchinson-Gilford syndrome (progeria), and several skin diseases due to abnormal keratins.
Understand the bases of malignant hyperthermia Duchenne and Becker muscular dystrophies, inherited cardiomyopathies, the Hutchinson-Gilford syndrome (progeria), and several skin diseases due to abnormal keratins.
BIOMEDICAL IMPORTANCE
Proteins play an important role in movement at both the organ (eg, skeletal muscle, heart, and gut) and cellular levels. In this chapter, the roles of specific proteins and certain other key molecules (eg, Ca2+) in muscular contractionare described. A brief coverage of cytoskeletal proteins is also presented.
Knowledge of the molecular bases of a number of conditions that affect muscle has advanced greatly in recent years. Understanding of the molecular basis of Duchenne-type muscular dystrophy was greatly enhanced when it was found that it was due to mutations in the gene encoding dystrophin (see case history no. 7 in Chapter 57). Significant progress has also been made in understanding the molecular basis of malignant hyperthermia, a serious complication for some patients undergoing certain types of anesthesia. Heart failure is a very common medical condition, with a variety of causes; its rational therapy requires understanding of the biochemistry of heart muscle. One group of conditions that cause heart failure are the cardiomyopathies, some of which are genetically determined. NO has been found to be a major regulator of smooth muscle tone. Many widely used vasodilators— such as nitroglycerin, used in the treatment of angina pectoris—act by increasing the formation of NO. Muscle, partly because of its mass, plays major roles in the overall metabolism of the body.
MUSCLE TRANSDUCES CHEMICAL ENERGY INTO MECHANICAL ENERGY
Muscle is the major biochemical transducer (machine) that converts potential (chemical) energy into kinetic (mechanical) energy. Muscle, the largest single tissue in the human body, makes up somewhat less than 25% of body mass at birth, more than 40% in the young adult, and somewhat less than 30% in the aged adult. We shall discuss aspects of the three types of muscles found in vertebrates: skeletal, cardiac, and smooth. Both skeletal and cardiac muscles appear striated upon microscopic observation; smooth muscle is nonstriated. Although skeletal muscle is under voluntary nervous control, the control of both cardiac and smooth muscle is involuntary.
Sarcoplasm of Muscle Cells Contains ATP, Phosphocreatine, & Glycolytic Enzymes
Striated muscle is composed of multinucleated muscle fiber cells surrounded by an electrically excitable plasma membrane, the sarcolemma. An individual muscle fiber cell, which may extend the entire length of the muscle, contains a bundle of many myofibrils arranged in parallel, embedded in intracellular fluid termed sarcoplasm. Within this fluid is contained glycogen, the high-energy compounds ATP and phosphocreatine, and the enzymes of glycolysis.
Sarcomere Is the Functional Unit of Muscle
An overall view of voluntary muscle at several levels of organization is presented in Figure 49–1.
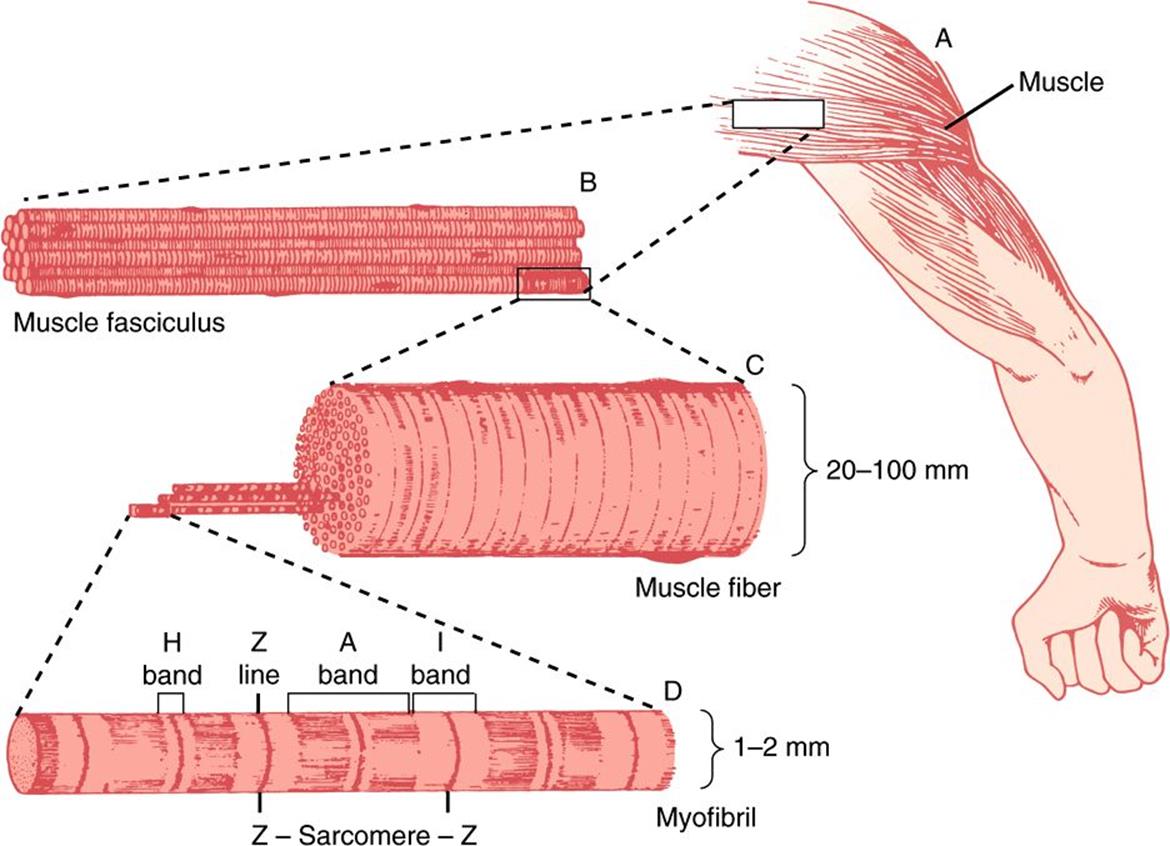
FIGURE 49–1 The structure of voluntary muscle. The sarcomere is the region between the Z lines. (Drawing by Sylvia Colard Keene. Reproduced, with permission, from Bloom W, Fawcett DW: A Textbook of Histology, 10th ed. Saunders, 1975.)
When the myofibril is examined by electron microscopy, alternating dark and light bands (anisotropic bands, meaning birefringent in polarized light, and isotropic bands, meaning not altered by polarized light) can be observed. These bands are thus referred to as A and I bands, respectively. The central region of the A band (the H band) appears less dense than the rest of the band. The I band is bisected by a very dense and narrow Z line (Figure 49–2).
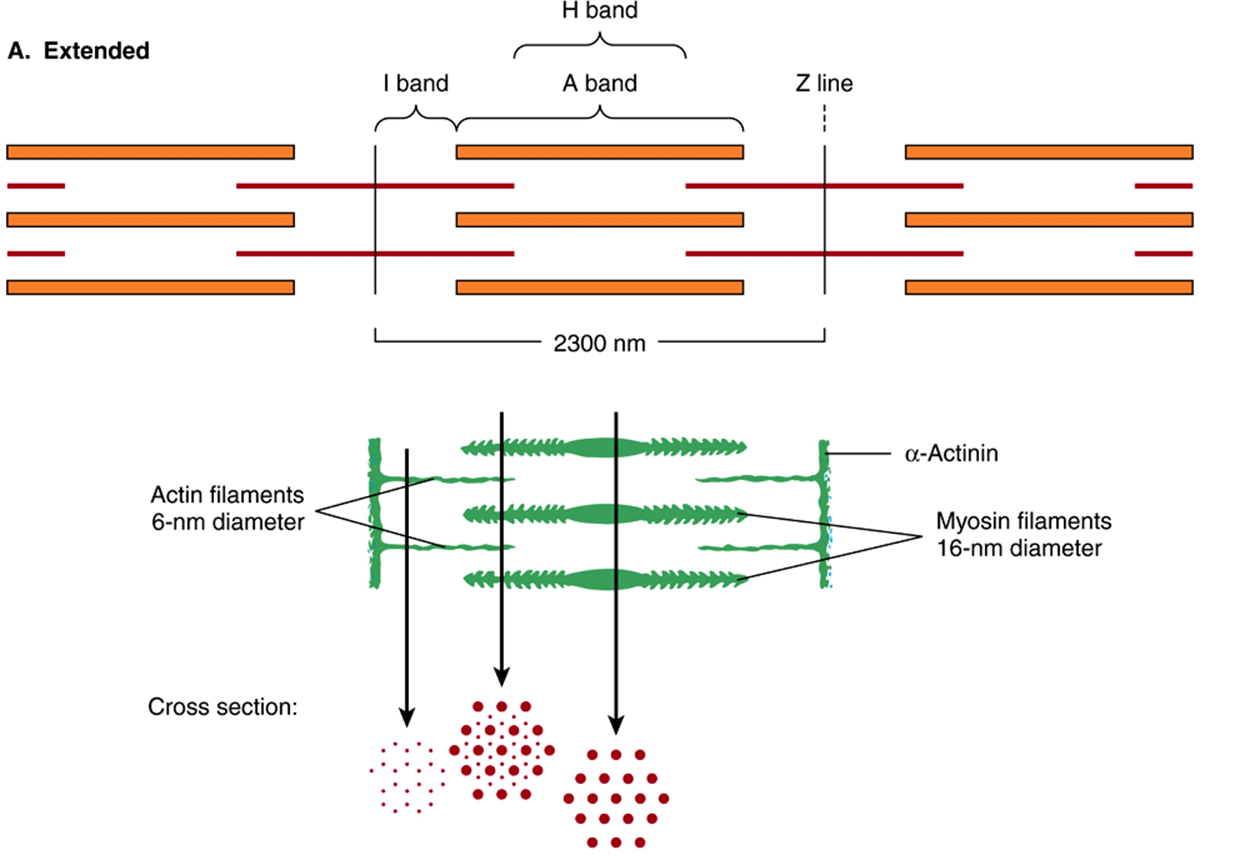
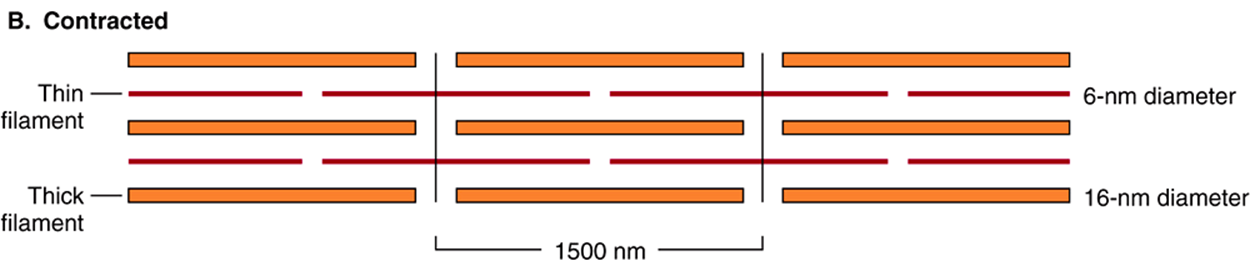
FIGURE 49–2 Arrangement of filaments in striated muscle. (A) Extended. The positions of the I, A, and H bands in the extended state are shown. The thin filaments partly overlap the ends of the thick filaments, and the thin filaments are shown anchored in the Z lines (often called Z disks). In the lower part of Figure 49–2(A), “arrowheads,” pointing in opposite directions, are shown emanating from the myosin (thick) filaments. Four actin (thin) filaments are shown attached to two Z lines via α-actinin. The central region of the three myosin filaments, free of arrowheads, is called the M band (not labeled). Cross sections through the M bands, through an area where myosin and actin filaments overlap and through an area in which solely actin filaments are present, are shown. (B) Contracted. The actin filaments are seen to have slipped along the sides of the myosin fibers toward each other. The lengths of the thick filaments (indicated by the A bands) and the thin filaments (distance between Z lines and the adjacent edges of the H bands) have not changed. However, the lengths of the sarcomeres have been reduced (from 2300 to 1500 nm), and the lengths of the H and I bands are also reduced because of the overlap between the thick and thin filaments. These morphologic observations provided part of the basis for the sliding filament model of muscle contraction.
The sarcomere is defined as the region between two Z lines (Figures 49-1 and 49-2) and is repeated along the axis of a fibril at distances of 1500-2300 nm depending upon the state of contraction.
The striated appearance of voluntary and cardiac muscle in light microscopic studies results from their high degree of organization, in which most muscle fiber cells are aligned so that their sarcomeres are in parallel register (Figure 49–1).
Thick Filaments Contain Myosin; Thin Filaments Contain Actin, Tropomyosin & Troponin
When myofibrils are examined by electron microscopy, it appears that each one is constructed of two types of longitudinal filaments. One type, the thick filament, confined to the A band, contains chiefly the protein myosin. These filaments are about 16 nm in diameter and arranged in the cross-section as a hexagonal array (Figure 49–2, center; right-hand cross-section).
The thin filament (about 7 nm in diameter) lies in the I band and extends into the A band but not into its H zone (Figure 49–2). Thin filaments contain the proteins actin, tropomyosin, and troponin (Figure 49–3). In the A band, the thin filaments are arranged around the thick (myosin) filament as a secondary hexagonal array. Each thin filament lies symmetrically between three thick filaments (Figure 49–2, center, mid cross section), and each thick filament is surrounded symmetrically by six thin filaments.
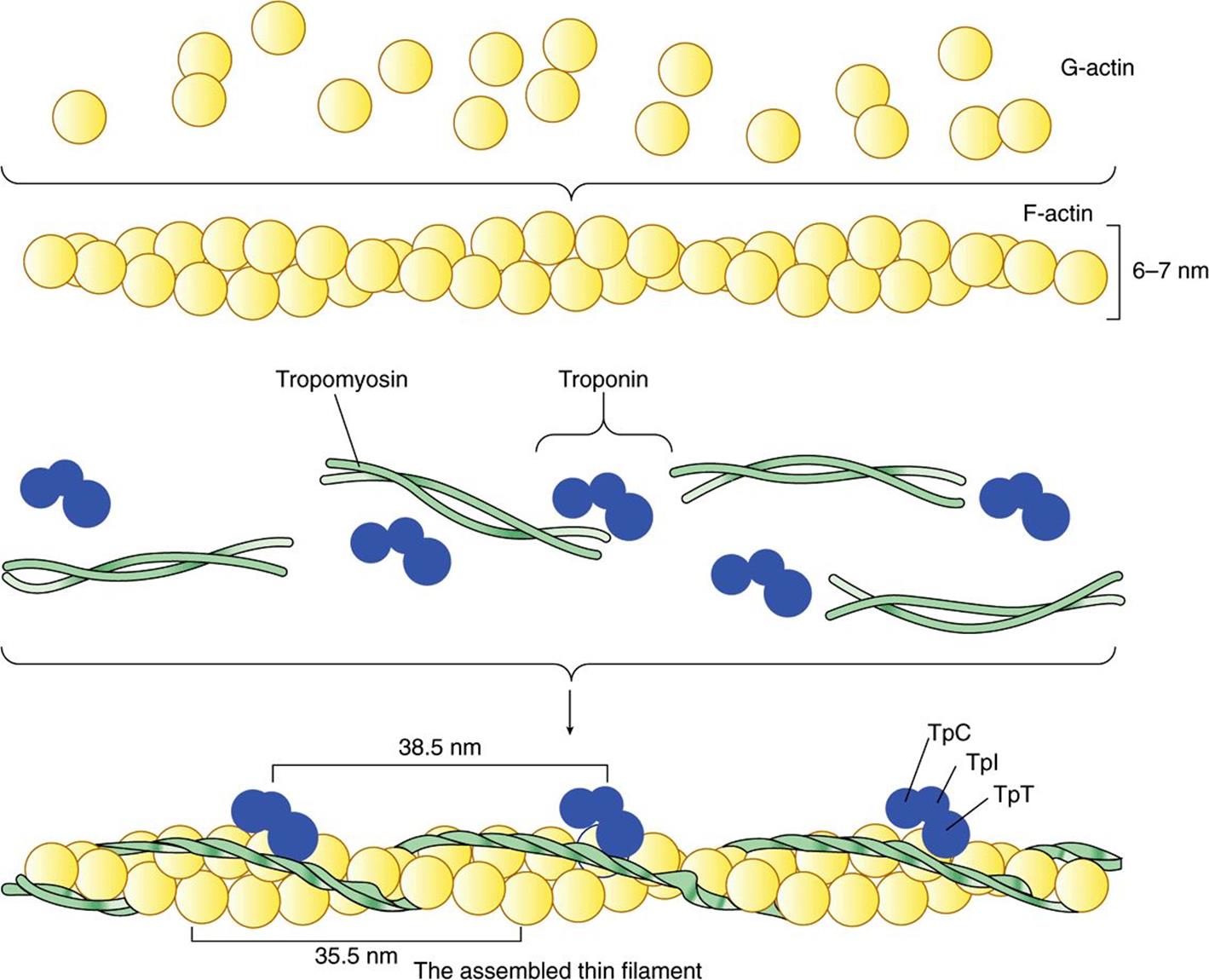
FIGURE 49–3 Schematic representation of the thin filament, showing the spatial configuration of its three major protein components: actin, myosin, and tropomyosin. The upper panel shows individual molecules of G-actin. The middle panel shows actin monomers assembled into F-actin. Individual molecules of tropomyosin (two strands wound around one another) and of troponin (made up of its three subunits) are also shown. The lower panel shows the assembled thin filament, consisting of F-actin, tropomyosin, and the three subunits of troponin (TpC, Tpl, and TpT).
The thick and thin filaments interact via cross bridges that emerge at intervals of 14 nm along the thick filaments. As depicted in Figure 49–2, the cross bridges (drawn as arrowheads at each end of the myosin filaments, but not shown extending fully across to the thin filaments) have opposite polarities at the two ends of the thick filaments. The two poles of the thick filaments are separated by a 150-nm segment (the M band, not labeled in the figure) that is free of projections.
The Sliding Filament Cross-Bridge Model Is the Foundation on Which Current Thinking About Muscle Contraction Is Built
This model was proposed independently in the 1950s by Henry Huxley and Andrew Huxley and their colleagues. It was largely based on careful morphologic observations on resting, extended, and contracting muscle. Basically, when muscle contracts, there is no change in the lengths of the thick and thin filaments, but the H zones and the I bands shorten (see legend to Figure 49–2). Thus, the arrays of inter-digitating filaments must slide past one anotherduring contraction. Cross-bridges that link thick and thin filaments at certain stages in the contraction cycle generate and sustain the tension. The tension developed during muscle contraction is proportionate to the filament overlap and to the number of cross bridges. Each cross-bridge head is connected to the thick filament via a flexible fibrous segment that can bend outward from the thick filament. This flexible segment facilitates contact of the head with the thin filament when necessary but is also sufficiently pliant to be accommodated in the interfilament spacing.
ACTIN & MYOSIN ARE THE MAJOR PROTEINS OF MUSCLE
The mass of a muscle is made up of 75% water and more than 20% protein. The two major proteins are actin and myosin.
Monomeric G-actin (43 kDa; G, globular) makes up 25% of muscle protein by weight. At physiologic ionic strength and in the presence of Mg2+, G-actin polymerizes noncovalently to form an insoluble double helical filament called F-actin (Figure 49–3). The F-actin fiber is 6-7 nm thick and has a pitch or repeating structure every 35.5 nm.
Myosins constitute a family of proteins, with at least 12 classes having been identified in the human genome. The myosin discussed in this chapter is myosin-II, and when myosin is referred to in this text, it is this species that is meant unless otherwise indicated. Myosin-I is a monomeric species that binds to cell membranes. It may serve as a linkage between microfilaments and the cell membrane in certain locations.
Myosin contributes 55% of muscle protein by weight and forms the thick filaments. It is an asymmetric hexamer with a molecular mass of approximately 460 kDa. Myosin has a fibrous tail consisting of two intertwined helices. Each helix has a globular head portion attached at one end (Figure 49–4). The hexamer consists of one pair of heavy (H) chains each of approximately 200 kDA molecular mass, and two pairs of light (L) chains each with a molecular mass of approximately 20 kDa. The L chains differ, one being called the essential light chain and the other the regulatory light chain. Skeletal muscle myosin binds actin to form actomyosin (actin-myosin), and its intrinsic ATPase activity is markedly enhanced in this complex. Isoforms of myosin exist whose amounts can vary in different anatomic, physiologic, and pathologic situations.
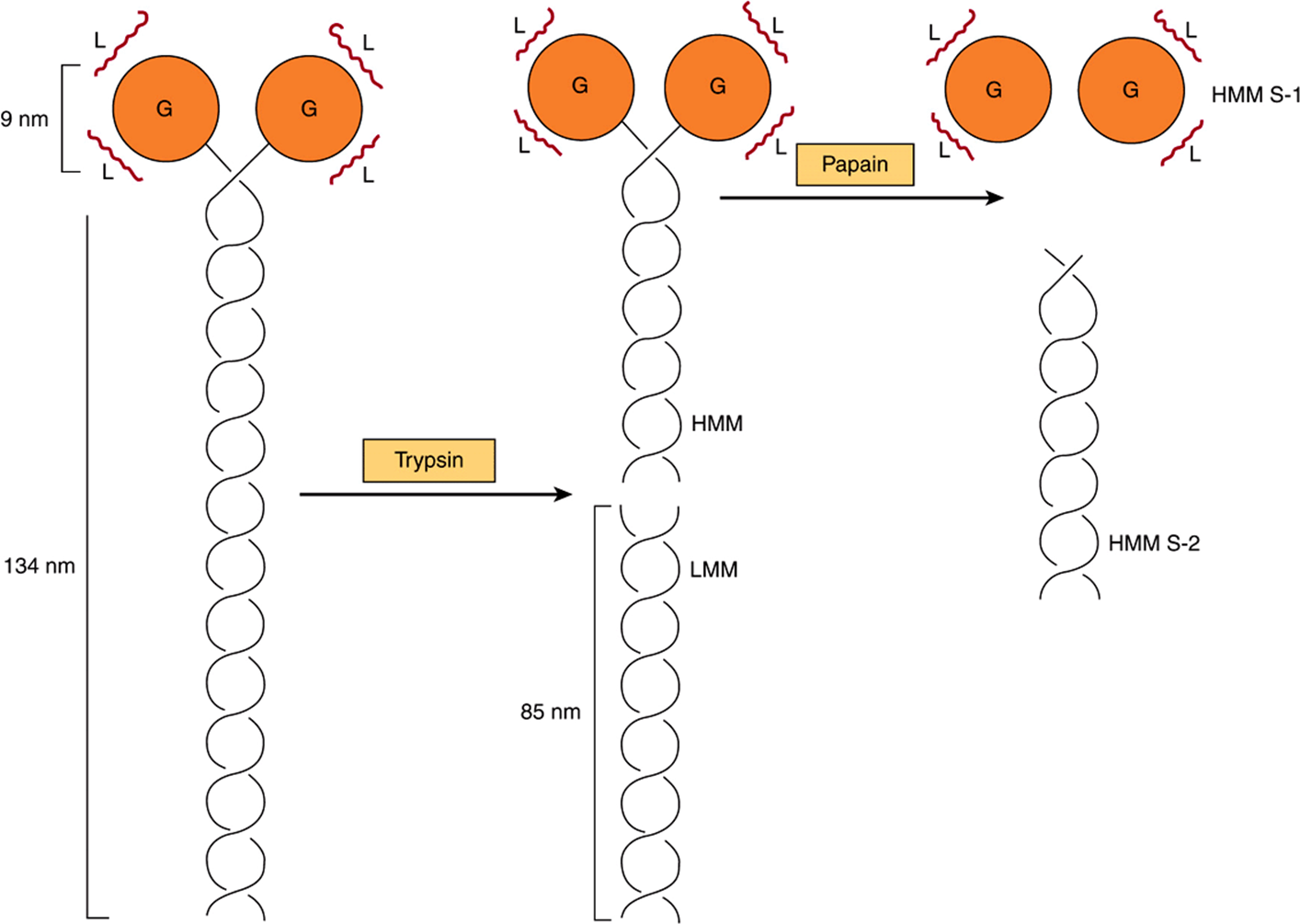
FIGURE 49–4 Diagram of a myosin molecule showing the two intertwined α-helices (fibrous portion), the globular region or head (G), the light chains (L), and the effects of proteolytic cleavage by trypsin and papain.The globular region (myosin head) contains an actin-binding site and an L chain-binding site and also attaches to the remainder of the myosin molecule.
The structures of actin and of the head of myosin have been determined by X-ray crystallography; these studies have confirmed a number of earlier findings concerning their structures and have also given rise to much new information.
Limited Digestion of Myosin with Proteases Has Helped to Elucidate Its Structure & Function
When myosin is digested with trypsin, two myosin fragments (meromyosins) are generated. Light meromyosin (LMM) consists of aggregated, insoluble α-helical fibers from the tail of myosin (Figure 49–4). LMM exhibits no ATPase activity and does not bind to F-actin.
Heavy meromyosin (HMM; molecular mass about 340 kDa) is a soluble protein that has both a fibrous portion and a globular portion (Figure 49–4). It exhibits ATPase activity and binds to F-actin. Digestion of HMM with papain generates two subfragments, S-1 and S-2. The S-2 fragment is fibrous in character, has no ATPase activity, and does not bind to F-actin.
S-1 (molecular mass approximately 115 kDa) does exhibit ATPase activity, binds L chains, and in the absence of ATP will bind to and decorate actin with “arrowheads” (Figure 49–5). Both S-1 and HMM exhibit ATPaseactivity, which is accelerated 100- to 200-fold by complexing with F-actin. As discussed below, F-actin greatly enhances the rate at which myosin ATPase releases its products, ADP and Pi. Thus, although F-actin does not affect the hydrolysis step per se, its ability to promote release of the products produced by the ATPase activity greatly accelerates the overall rate of catalysis.
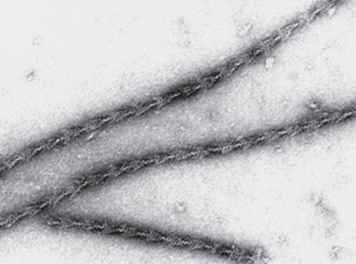
FIGURE 49–5 The decoration of actin filaments with the S-1 fragments of myosin to form “arrowheads.” (Courtesy of JA Spudich.)
CHANGES IN THE CONFORMATION OF THE HEAD OF MYOSIN DRIVE MUSCLE CONTRACTION
How can hydrolysis of ATP produce macroscopic movement? Muscle contraction essentially consists of the cyclic attachment and detachment of the S-1 head of myosin to the F-actin filaments. This process can also be referred to as the making and breaking of cross-bridges. The attachment of actin to myosin is followed by conformational changes that are of particular importance in the S-1 head and are dependent upon which nucleotide is present (ADP or ATP). These changes result in the power stroke, which drives movement of actin filaments past myosin filaments. The energy for the power stroke is ultimately supplied by ATP, which is hydrolyzed to ADP and Pi. However, the power stroke itself occurs as a result of conformational changes in the myosin head when ADP leaves it.
The major biochemical events occurring during one cycle of muscle contraction and relaxation can be represented in the five steps shown in Figure 49–6 as follows:
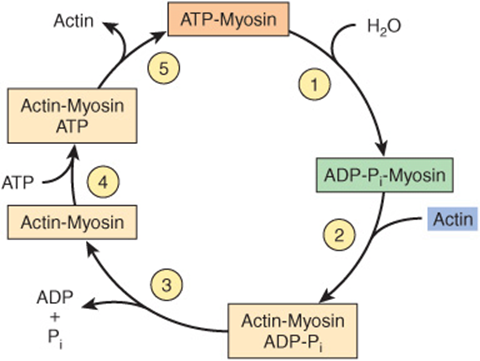
FIGURE 49–6 The hydrolysis of ATP drives the cyclic association and dissociation of actin and myosin in five reactions described in the text. (Modified, with permission, from Stryer L: Biochemistry, 2nd ed. Freeman, 1981. Copyright © 1981 by W. H. Freeman and Company.)
1. In the relaxation phase of muscle contraction, the S-1 head of myosin hydrolyzes ATP to ADP and Pi, but these products remain bound. The resultant ADP-Pi-myosin complex has been energized and is in a so-called high-energy conformation.
2. When contraction of muscle is stimulated (via events involving Ca2+, troponin, tropomyosin, and actin, which are described below), actin becomes accessible and the S-1 head of myosin finds it, binds it, and forms the actin-myosin-ADP-i complex indicated.
3. Formation of this complex promotes the release of Pi, which initiates the power stroke. This is followed by release of ADP and is accompanied by a large conformational change in the head of myosin in relation to its tail (Figure 49–7), pulling actin about 10 nm toward the center of the sarcomere. This is the power stroke. The myosin is now in a so-called low-energy state, indicated as actinmyosin.
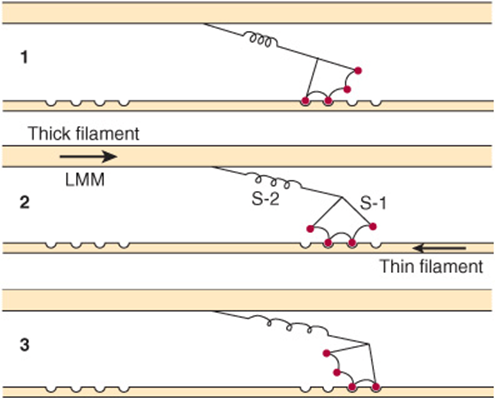
FIGURE 49–7 Representation of the active cross bridges between thick and thin filaments. This diagram was adapted by AF Huxley from HE Huxley: the mechanism of muscular contraction. Science 1969;164:1356. The latter proposed that the force involved in muscular contraction originates in a tendency for the myosin head (S-1) to rotate relative to the thin filament and is transmitted to the thick filament by the S-2 portion of the myosin molecule acting as an inextensible link. Flexible points at each end of S-2 permit S-1 to rotate and allow for variations in the separation between filaments. This figure is based on HE Huxley’s proposal, but also incorporates elastic (the coils in the S-2 portion) and stepwise-shortening elements (depicted here as four sites of interaction between the S-1 portion and the thin filament). (See Huxley AF, Simmons RM: Proposed mechanism of force generation in striated muscle. Nature [Lond] 1971;233:533.) The strengths of binding of the attached sites are higher in position 2 than in position 1 and higher in position 3 than position 2. The myosin head can be detached from position 3 with the utilization of a molecule of ATP; this is the predominant process during shortening. The myosin head is seen to vary in its position from about 90° to about 45°, as indicated in the text. (S-1, myosin head; S-2, portion of the myosin molecule; LMM) (see legend to Figure 49–4). (Reproduced from Huxley AF: Muscular contraction. J Physiol 1974;243:1. By kind permission of the author and the Journal of Physiology.)
4. Another molecule of ATP binds to the S-1 head, forming an actinmyosin-ATP complex.
5. Myosin-ATP has a low affinity for actin, and actin is thus released. This last step is a key component of relaxation and is dependent upon the binding of ATP to the actinmyosin complex.
Another cycle then commences with the hydrolysis of ATP (step 1 of Figure 49–6), re-forming the high-energy conformation.
Thus, hydrolysis of ATP is used to drive the cycle, with the actual power stroke being the conformational change in the S-1 head that occurs upon the release of ADP. The hinge regions of myosin (referred to as flexible points at each end of S-2 in the legend to Figure 49–7) permit the large range of movement of S-1 and also allow S-1 to find actin filaments.
If intracellular levels of ATP drop (eg, after death), ATP is not available to bind the S-1 head (step 4 above), actin does not dissociate, and relaxation (step 5) does not occur. This is the explanation for rigor mortis, the stiffening of the body that occurs after death.
Calculations have indicated that the efficiency of contraction is about 50%; that of the internal combustion engine is less than 20%.
Tropomyosin & the Troponin Complex Present in Thin Filaments Perform Key Functions in Striated Muscle
In striated muscle, there are two other proteins that are minor in terms of their mass but important in terms of their function. Tropomyosin is a fibrous molecule that consists of two chains, alpha and beta, that attach to F-actin in the groove between its filaments (Figure 49–3). Tropomyosin is present in all muscular and muscle-like structures. The troponin complex is unique to striated muscle and consists of three polypeptides. Troponin T (TpT) binds to tropomyosin as well as to the other two troponin components. Troponin I (TpI) inhibits the F-actin-myosin interaction and also binds to the other components of troponin. Troponin C (TpC) is a calcium-binding polypeptide that is structurally and functionally analogous to calmodulin, an important calcium-binding protein widely distributed in nature. Four molecules of calcium ion are bound per molecule of troponin C or calmodulin, and both molecules have a molecular mass of 17 kDa.
Ca2+ Plays a Central Role in Regulation of Muscle Contraction
The contraction of muscles from all sources occurs by the general mechanism described above. Muscles from different organisms and from different cells and tissues within the same organism may have different molecular mechanisms responsible for the regulation of their contraction and relaxation. In all systems, Ca2+ plays a key regulatory role. There are two general mechanisms of regulation of muscle contraction: actin-based and myosin-based.The former operates in skeletal and cardiac muscles, the latter in smooth muscle.
Actin-Based Regulation Occurs in Striated Muscle
Actin-based regulation of muscle occurs in vertebrate skeletal and cardiac muscles, both striated. In the general mechanism described above (Figure 49–6), the only potentially limiting factor in the cycle of muscle contraction might be ATP. The skeletal muscle system is inhibited at rest; this inhibition is relieved to activate contraction. The inhibitor of striated muscle is the troponin system, which is bound to tropomyosin and F-actin in the thin filament (Figure 49–3). In striated muscle, there is no control of contraction unless the tropomyosin-troponin systems are present along with the actin and myosin filaments. As described above, tropomyosin lies along the groove of F-actin, and the three components of troponin— TpT, TpI, and TpC—are bound to the F-actin-tropomyosin complex. TpI prevents binding of the myosin head to its Factin attachment site either by altering the conformation of F-actin via the tropomyosin molecules or by simply rolling tropomyosin into a position that directly blocks the sites on F-actin to which the myosin heads attach. Either way prevents activation of the myosin ATPase that is mediated by binding of the myosin head to F-actin. Hence, the TpI system blocks the contraction cycle at step 2 of Figure 49–6. This accounts for the inhibited state of relaxed striated muscle.
The Sarcoplasmic Reticulum Regulates Intracellular Levels of Ca2+ in Skeletal Muscle
In the sarcoplasm of resting muscle, the concentration of Ca2+ is 10-8 to 10-7 mol/L. The resting state is achieved because Ca2+ is pumped into the sarcoplasmic reticulum (SR) through the action of an active transport system, called the Ca2+ ATPase (Figure 49–8), initiating relaxation. The SR is a network of fine membranous sacs. Inside the SR, Ca2+ is bound to a specific Ca2+-binding protein-designated calsequestrin. The sarcomere is surrounded by an excitable membrane (the T-tubule system) composed of transverse (T) channels closely associated with the SR.
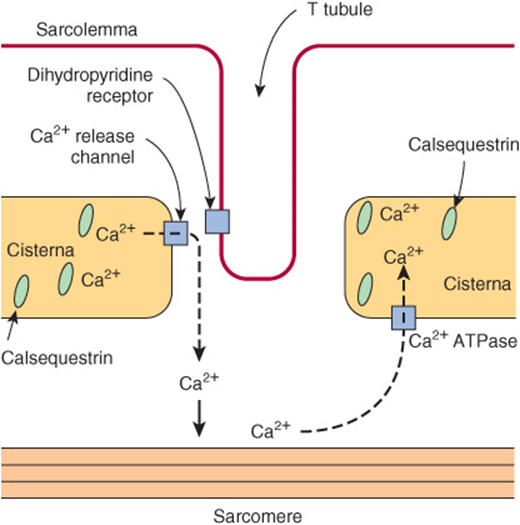
FIGURE 49–8 Diagram of the relationships among the sarcolemma (plasma membrane), a T tubule, and two cisternae of the SR of skeletal muscle (not to scale). The T tubule extends inward from the sarcolemma. A wave of depolarization, initiated by a nerve impulse, is transmitted from the sarcolemma down the T tubule. It is then conveyed to the Ca2+ release channel (RYR), perhaps by interaction between it and the dihydropyridine receptor (slow Ca2+ voltage channel), which are shown in close proximity. Release of Ca2+ from the Ca2+ release channel into the cytosol initiates contraction. Subsequently, Ca2+ is pumped back into the cisternae of the SR by the Ca2+ ATPase (Ca2+ pump) and stored there, in part bound to calsequestrin.
When the sarcolemma is excited by a nerve impulse, the signal is transmitted into the T-tubule system and a Ca2+ release channel in the nearby SR opens, releasing Ca2+ from the SR into the sarcoplasm. The concentration of Ca2+ in the sarcoplasm rises rapidly to 10-5 mol/L. The Ca2+-binding sites on TpC in the thin filament are quickly occupied by Ca2+. The TpC-4Ca2+ interacts with TpI and TpT to alter their interaction with tropomyosin. Accordingly, tropomyosin moves out of the way or alters the conformation of F-actin so that the myosin head-ADP-Pi (Figure 49–6) can interact with F-actin to start the contraction cycle.
The Ca2+ release channel is also known as the ryanodine receptor (RYR). There are two isoforms of this receptor, RYR1 and RYR2, the former being present in skeletal muscle and the latter in heart muscle and brain. Ryanodine is a plant alkaloid that binds to RYR1 and RYR2 specifically and modulates their activities. The Ca2+ release channel is a homotetramer made up of four subunits of kDa 565. It has transmembrane sequences at its carboxyl terminal, and these probably form the Ca2+ channel. The remainder of the protein protrudes into the cytosol, bridging the gap between the SR and the transverse tubular membrane. The channel is ligand-gated, Ca2+ and ATP working synergistically in vitro, although how it operates in vivo is not clear. A possible sequence of events leading to opening of the channel is shown in Figure 49–9. The channel lies very close to the dihydropyridine receptor (DHPR), a voltage-dependent calcium channel of the transverse tubule system (Figure 49–8). Experiments in vitro employing an affinity column chromatography approach have indicated that a 37-amino-acid stretch in RYR1 interacts with one specific loop of DHPR.
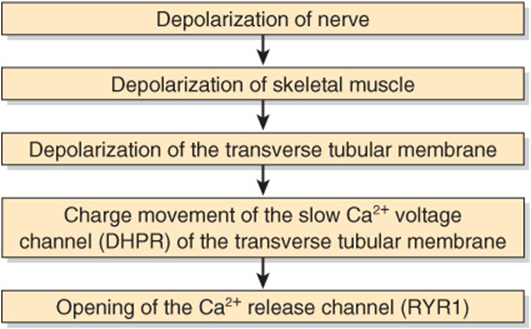
FIGURE 49–9 Possible chain of events leading to opening of the Ca2+ release channel. As indicated in the text, the Ca2+ voltage channel and the Ca2+ release channel have been shown to interact with each other in vitro via specific regions in their polypeptide chains. (DHPR, dihydropyridine receptor; RYR1, RYR 1.)
Relaxation occurs when sarcoplasmic Ca2+ falls below 10-7 mol/L owing to its resequestration into the SR by Ca2+ ATPase. TpC-4Ca2+ thus loses its Ca2+. Consequently, roponin, via interaction with tropomyosin, inhibitsfurther myosin head and F-actin interaction, and in the presence of ATP the myosin head detaches from the F-actin.
Thus, Ca2+ controls skeletal muscle contraction and relaxation by an allosteric mechanism mediated by TpC, TpI, TpT, tropomyosin, and F-actin.
A decrease in the concentration of ATP in the sarcoplasm (eg, by excessive usage during the cycle of contraction-relaxation or by diminished formation, such as might occur in ischemia) has two major effects: (1) The Ca2+ATPase (Ca2+ pump) in the SR ceases to maintain the low concentration of Ca2+ in the sarcoplasm. Thus, the interaction of the myosin heads with F-actin is promoted. (2) The ATP-dependent detachment of myosin heads from F-actin cannot occur, and rigidity (contracture) sets in. The condition of rigor mortis, following death, is an extension of these events.
Muscle contraction is a delicate dynamic balance of the attachment and detachment of myosin heads to F-actin, subject to fine regulation via the nervous system.
Table 49-1 summarizes the overall events in contraction and relaxation of skeletal muscle.
TABLE 49–1 Sequence of Events in Contraction and Relaxation of Skeletal Muscle


Mutations in the Gene Encoding the Ca2+ Release Channel Are One Cause of Human Malignant Hyperthermia
Some genetically predisposed patients experience a severe reaction, designated malignant hyperthermia (MH), on exposure to certain anesthetics (eg, halothane) and depolarizing skeletal muscle relaxants (eg, succinylcholine). The reaction consists primarily of rigidity of skeletal muscles, hypermetabolism, and high fever. A high cytosolic concentration of Ca2+ in skeletal muscle is a major factor in its causation. Unless malignant hyperthermia is recognized and treated immediately, patients may die acutely of ventricular fibrillation or survive to succumb subsequently from other serious complications. Appropriate treatment is to stop the anesthetic and administer the drug dantrolene intravenously. Dantrolene is a skeletal muscle relaxant that acts to inhibit release of Ca2+ from the SR into the cytosol, thus preventing the increase of cytosolic Ca2+ found in MH.
MH also occurs in swine. Susceptible animals homozygous for MH respond to stress with a fatal reaction (porcine stress syndrome) similar to that exhibited by humans. If the reaction occurs prior to slaughter, it affects the quality of the pork adversely, resulting in an inferior product. Both events can result in considerable economic losses for the swine industry.
The finding of a high level of cytosolic Ca2+ in muscle in MH suggested that the condition might be caused by abnormalities of the Ca2+ ATPase or of the Ca2+ release channel. No abnormalities were detected in the former, but sequencing of cDNAs for the latter protein proved insightful, particularly in swine. All cDNAs from swine with MH so far examined have shown a substitution of T for C1843, resulting in the substitution of Cys for Arg615 in the Ca2+ release channel. The mutation affects the function of the channel in that it opens more easily and remains open longer; the net result is massive release of Ca2+ into the cytosol, ultimately causing sustained muscle contraction.
The picture is more complex in humans, since MH exhibits genetic heterogeneity. Members of a number of families who suffer from malignant hyperthermia have not shown genetic linkage to the RYR1 gene. Some humans susceptible to MH have been found to exhibit the same mutation found in swine, and others have a variety of point mutations at different loci in the RYR1 gene. Certain families with MH have been found to have mutations affecting the DHPR. It is possible that mutations affecting other muscle proteins, such as calsequestrin-1, a SR Ca2+-binding protein that modulates RyR1 function may also cause MH. Figure 49–10 summarizes the probable chain of events in malignant hyperthermia. The major promise of these findings is that, once additional mutations are detected, it will be possible to screen, using suitable DNA probes, for individuals at risk of developing MH during anesthesia. Current screening tests (eg, the in vitro caffeine-halothane test) are relatively unreliable. Affected individuals could then be given alternative anesthetics, which would not endanger their lives. It should also be possible, if desired, to eliminate MH from swine populations using suitable breeding practices.
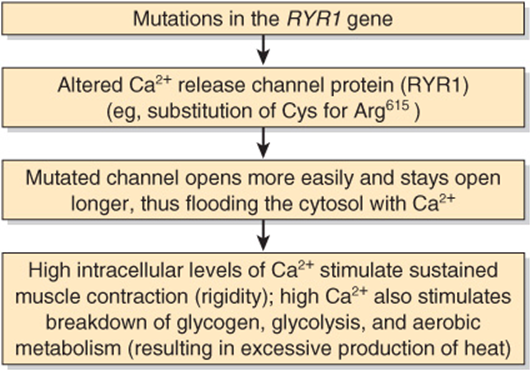
FIGURE 49–10 Simplified scheme of the causation of malignant hyperthermia (OMIM 145600). Many different point mutations have been detected in the RYR1 gene, some of which are associated with central core disease (OMIM 117000). It is estimated that at least 50% of families with members who have malignant hyperthermia are linked to the RYR1 gene. Some individuals with mutations in the gene encoding DHPR have also been detected; it is possible that mutations in other genes for proteins involved in certain aspects of muscle metabolism will also be found.
Another condition due to mutations in the RYR1 gene is central core disease. This is a rare myopathy presenting in infancy with hypotonia and proximal muscle weakness. Electron microscopy reveals an absence of mitochondria in the center of many type I (see below) muscle fibers. Damage to mitochondria induced by high intracellular levels of Ca2+ secondary to abnormal functioning of RYR1 appears to be responsible for the morphologic findings.
MUTATIONS IN THE GENE ENCODING DYSTROPHIN CAUSE DUCHENNE MUSCULAR DYSTROPHY
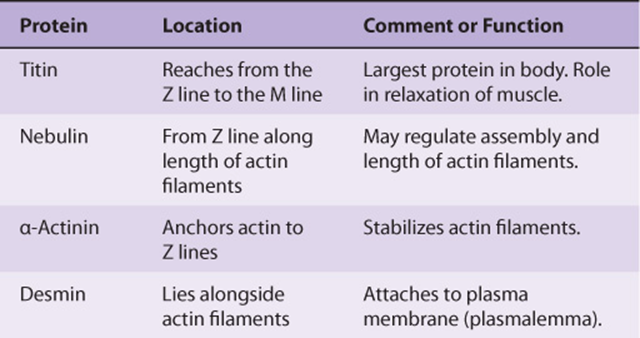
A number of additional proteins play various roles in the structure and function of muscle. They include titin (the largest protein known), nebulin, α-actinin, desmin, dystrophin, and calcineurin. Some properties of these proteins are summarized in Table 49-2.
TABLE 49–2 Some Other Important Proteins of Muscle
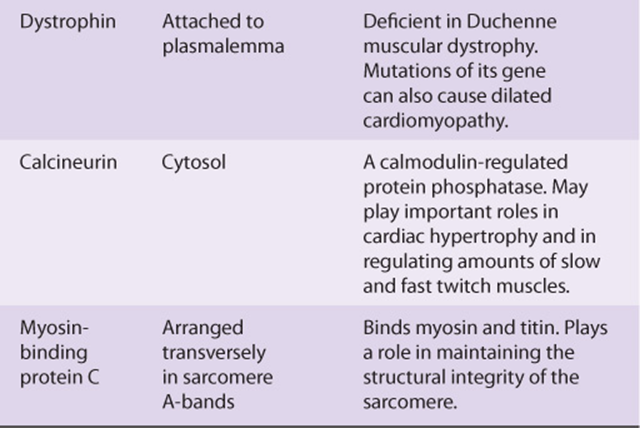
Dystrophin is of special interest. As discussed in case no. 9 of Chapter 57, mutations in the gene encoding this protein have been shown to be the cause of Duchenne muscular dystrophy and the milder Becker muscular dystrophy. They are also implicated in some cases of dilated cardiomyopathy (see below). As shown in Figure 49–11, dystrophin forms a part of a large complex of proteins that attach to or interact with the plasmalemma. Dystrophin links the actin cytoskeleton to the ECM and appears to be needed for assembly of the synaptic junction. Impairment of these processes by formation of defective dystrophin is thought to be critical in the causation of Duchenne muscular dystrophy. Mutations in the genes encoding some of the components of the sarcoglycan complex shown in Figure 49–11 are responsible for limb-girdle and certain other congenital forms of muscular dystrophy.
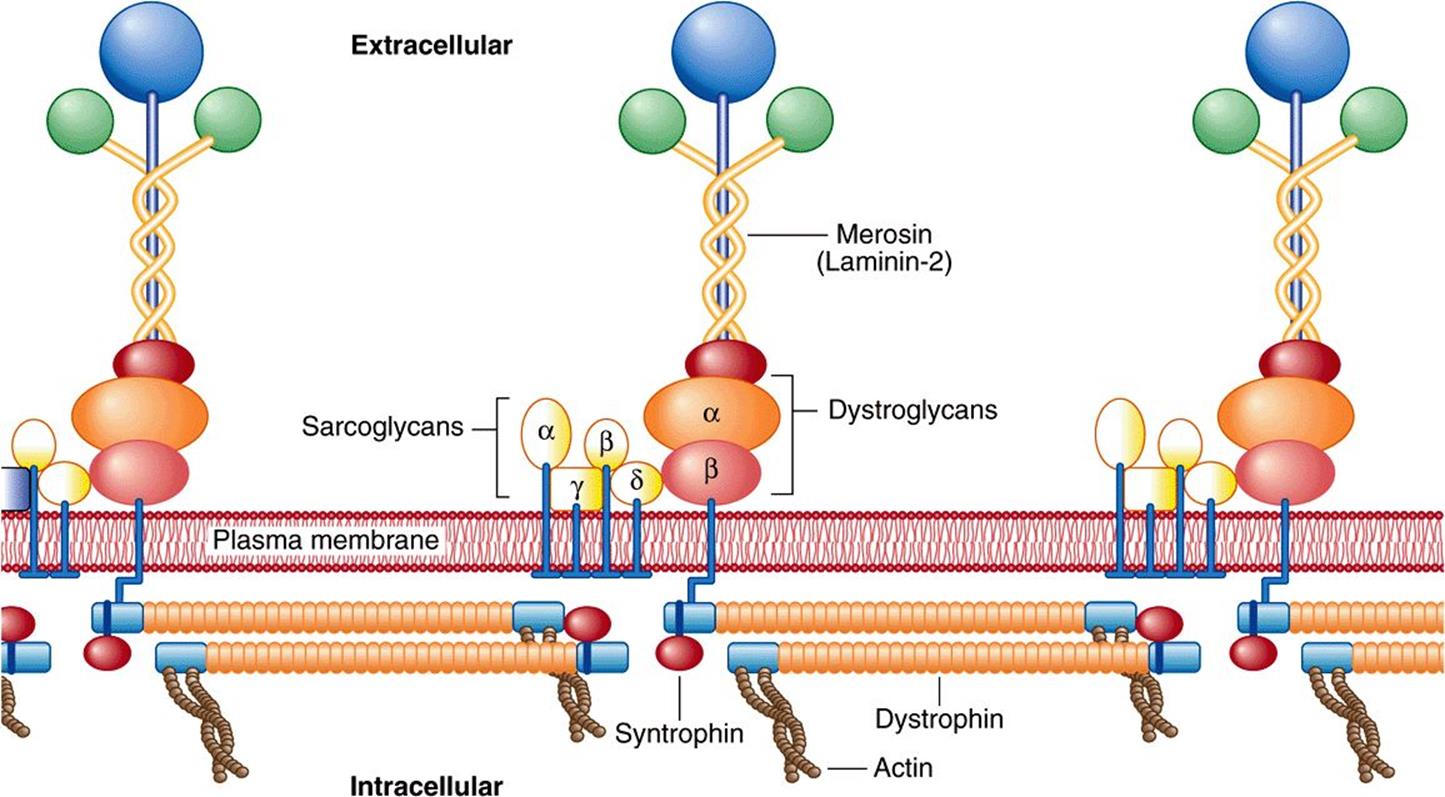
FIGURE 49–11 Organization of dystrophin and other proteins in relation to the plasma membrane of muscle cells. Dystrophin is part of a large oligomeric complex associated with several other protein complexes. The dystroglycan complex consists of α-dystroglycan, which associates with the basal lamina protein merosin (also named laminin-2, see Chapter 48), and α-dystroglycan, which binds α-dystroglycan and dystrophin. Syntrophin binds to the carboxyl terminal of dystrophin. The sarcoglycan complex consists of four transmembrane proteins: α-, β-, γ-, and δ-sarcoglycan. The function of the sarcoglycan complex and the nature of the interactions within the complex and between it and the other complexes are not clear. The sarcoglycan complex is formed only in striated muscle, and its subunits preferentially associate with each other, suggesting that the complex may function as a single unit. Mutations in the gene encoding dystrophin cause Duchenne and Becker muscular dystrophy. Mutations in the genes encoding the various sarcoglycans have been shown to be responsible for limb-girdle dystrophies (eg, OMIM 604286) and mutations in genes encoding other muscle proteins cause other types of muscular dystrophy. Mutations in genes encoding certain glycosyltransferases involved in the synthesis of the glycan chains of α-dystroglycan are responsible for certain congenital muscular dystrophies (see Chapter 47). (Reproduced, with permission, from Duggan DJ et al: Mutations in the sarcoglycan genes in patients with myopathy. N Engl J Med 1997;336:618. Copyright © 1997 Massachusetts Medical Society. All rights reserved.)
Mutations in genes encoding several glycosyltransferases involved in the synthesis of the sugar chains of α-dystroglycan have been found to be the cause of certain types of congenital muscular dystrophy (see Chapter 47).
CARDIAC MUSCLE RESEMBLES SKELETAL MUSCLE IN MANY RESPECTS
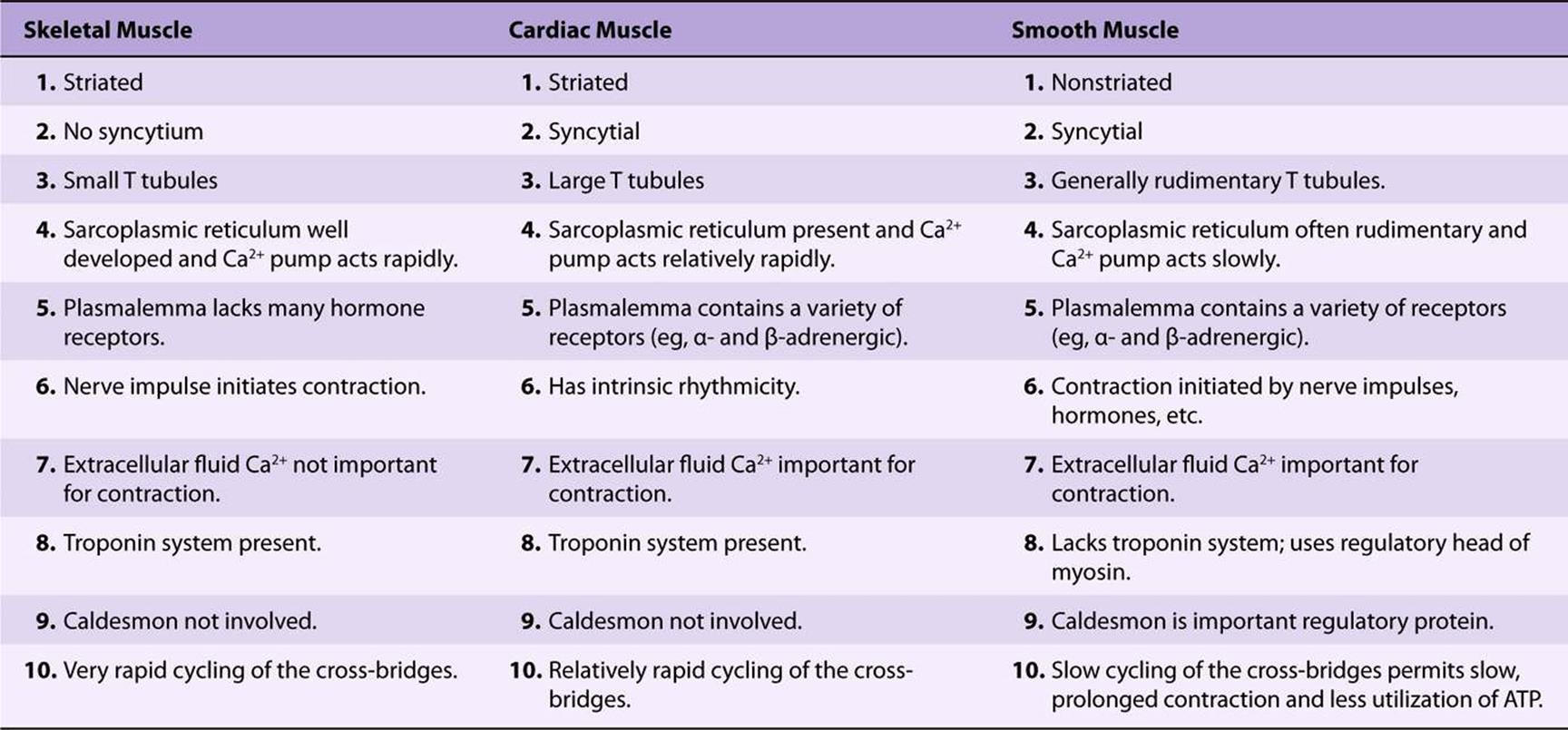
The general picture of muscle contraction in the heart resembles that of skeletal muscle. Cardiac muscle, like skeletal muscle, is striated and uses the actin-myosin-tropomyosintroponin system described above. Unlike skeletal muscle, cardiac muscle exhibits intrinsic rhythmicity, and individual myocytes communicate with each other because of its syncytial nature. The T-tubular system is more developed in cardiac muscle, whereas the SR is less extensive and consequently the intracellular supply of Ca2+ for contraction is less. Cardiac muscle thus relies on extracellular Ca2+ for contraction; if isolated cardiac muscle is deprived of Ca2+, it ceases to beat within approximately 1 min, whereas skeletal muscle can continue to contract without an extracellular source of Ca2+ for a longer period. Cyclic AMP plays a more prominent role in cardiac than in skeletal muscle. It modulates intracellular levels of Ca2+ through the activation of protein kinases; these enzymes phosphorylate various transport proteins in the sarcolemma and SR and also in the troponin-tropomyosin regulatory complex, affecting intracellular levels of Ca2+ or responses to it. There is a rough correlation between the phosphorylation of TpI and the increased contraction of cardiac muscle induced by catecholamines. This may account for the inotropic effects (increased contractility) of β-adrenergic compounds on the heart. Some differences among skeletal, cardiac, and smooth muscle are summarized in Table 49-3.
TABLE 49–3 Some Differences Among Skeletal, Cardiac, and Smooth Muscle
Ca2+ Enters Myocytes Via Ca2+ Channels & Leaves Via the Na+-Ca2+ Exchanger & the Ca2+ ATPase
As stated, extracellular Ca2+ plays an important role in contraction of cardiac muscle but not in skeletal muscle. This means that Ca2+ both enters and leaves myocytes in a regulated manner. We shall briefly consider three transmembrane proteins that play roles in this process.
Ca2+ Channels
Ca2+ enters myocytes via these channels, which allow entry only of Ca2+ ions. The major portal of entry is the L-type (long-duration current, large conductance) or slow Ca2+ channel, which is voltage-gated, opening during depolarization induced by spread of the cardiac action potential and closing when the action potential declines. These channels are equivalent to the dihydropyridine receptors of skeletal muscle (Figure 49–8). Slow Ca2+ channels are regulated by cAMP-dependent protein kinases (stimulatory) and cGMP-protein kinases (inhibitory) and are blocked by so-called calcium channel blockers (eg, verapamil). Fast (or T, transient) Ca2+ channels are also present in the plasmalemma, though in much lower numbers; they probably contribute to the early phase of increase of myoplasmic Ca2+.
The resultant increase of Ca2+ in the myoplasm acts on the Ca2+ release channel of the SR to open it. This is called Ca2+-induced Ca2+ release (CICR). It is estimated that approximately 10% of the Ca2+ involved in contraction enters the cytosol from the extracellular fluid and 90% from the SR. However, the former 10% is important, as the rate of increase of Ca2+ in the myoplasm is important, and entry via the Ca2+ channels contributes appreciably to this.
Ca2+-Na+ Exchanger
This is the principal route of exit of Ca2+ from myocytes. In resting myocytes, it helps to maintain a low level of free intracellular Ca2+ by exchanging one Ca2+ for three Na+. The energy for the uphill movement of Ca2+ out of the cell comes from the downhill movement of Na+ into the cell from the plasma. This exchange contributes to relaxation, but may run in the reverse direction during excitation. Because of the Ca2+-Na+ exchanger, anything that causes intracellular Na+ (Na+i) to rise will secondarily cause Ca2+i to rise, causing more forceful contraction. This is referred to as a positive inotropic effect. One example is when the drug digitalis is used to treat heart failure. Digitalis inhibits the sarcolemmal Na+-K+-ATPase, diminishing exit of Na+ and thus increasing Na+i. This in turn causes Ca2+ to increase, via the Ca+-Na+ exchanger. The increased Ca2+i results in increased force of cardiac contraction (see Figure 49–12), of benefit in heart failure.
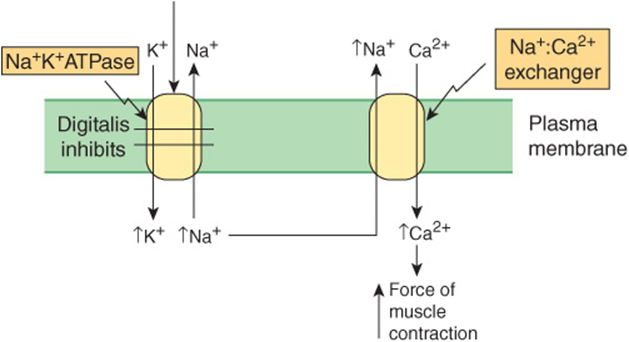
FIGURE 49–12 Scheme of how the drug digitalis (used in the treatment of certain cases of heart failure) increases cardiac contraction. Digitalis inhibits the Na+ K+ ATPase (see Chapter 40). This results in less Na+ being pumped out of the cardiac myocyte and leads to an increase of the intracellular concentration of Na+. In turn, this stimulates the Na+-Ca2+ exchanger so that more Na+ is exchanged outward, and more Ca2+ enters the myocyte. The resulting increased intracellular concentration of Ca2+ increases the force of muscular contraction.
Ca2+ ATPase
This Ca2+ pump, situated in the sarcolemma, also contributes to Ca2+ exit but is believed to play a relatively minor role as compared with the Ca2+-Na+ exchanger.
It should be noted that there are a variety of ion channels (Chapter 40) in most cells, for Na+, K+, Ca2+, etc. Many of them have been cloned in recent years and their dispositions in their respective membranes worked out (number of times each one crosses its membrane, location of the actual ion transport site in the protein, etc). They can be classified as indicated in Table 49-4. Cardiac muscle is rich in ion channels, and they are also important in skeletal muscle. Mutations in genes encoding ion channels have been shown to be responsible for a number of relatively rare conditions affecting muscle. These and other diseases due to mutations of ion channels have been termed channelopathies; some are listed in Table 49-5.
TABLE 49–4 Major Types of Ion Channels Found in Cells
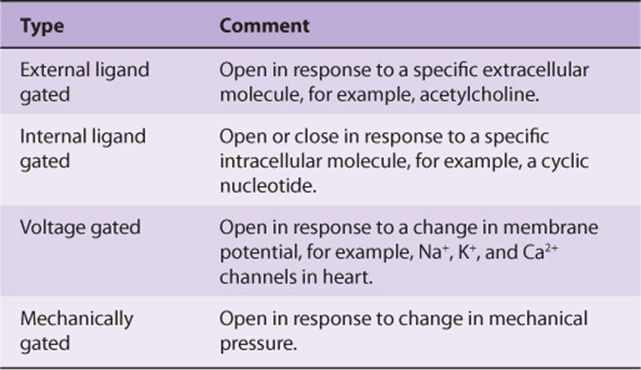
TABLE 49–5 Some Disorders (Channelopathies) Due to Mutations in Genes Encoding Polypeptide Constituents of Ion Channels
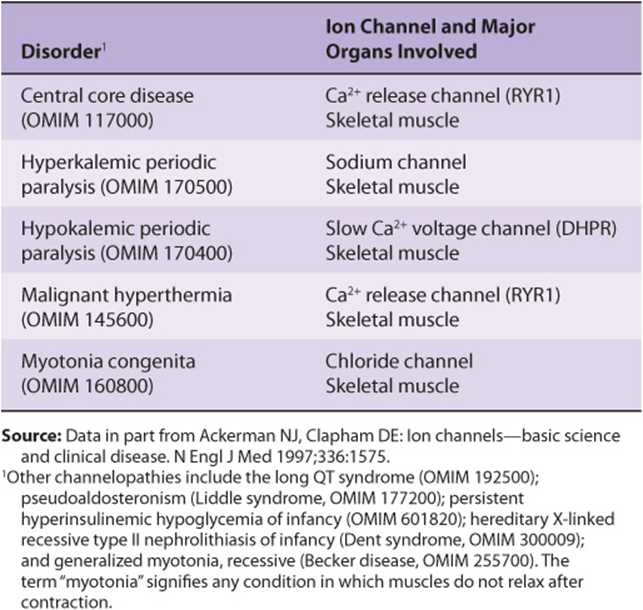
Inherited Cardiomyopathies Are Due to Disorders of Cardiac Energy Metabolism or to Abnormal Myocardial Proteins
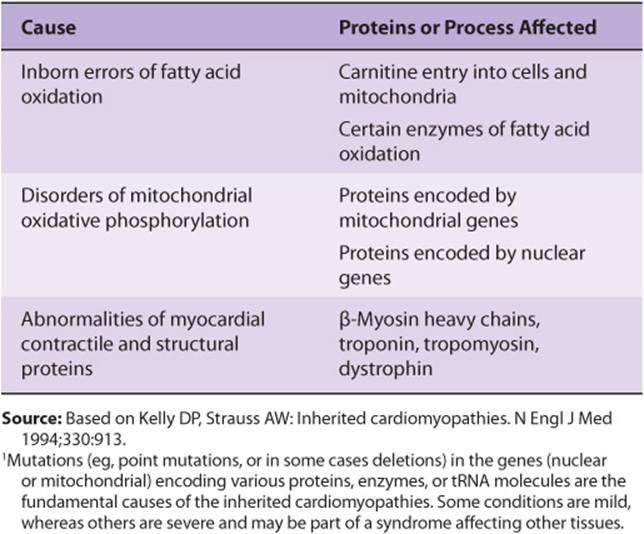
An inherited cardiomyopathy is any structural or functional abnormality of the ventricular myocardium due to an inherited cause. There are nonheritable types of cardiomyopathy, but these will not be described here. As shown in Table 49-6, the causes of inherited cardiomyopathies fall into two broad classes: (1) disorders of cardiac energy metabolism, mainly reflecting mutations in genes encoding enzymes or proteins involved in fatty acid oxidation (a major source of energy for the myocardium) and oxidative phosphorylation; (2) mutations in genes encoding proteins involved in or affecting myocardial contraction, such as myosin, tropomyosin, the troponins, and cardiac myosin-binding protein C. Mutations in the genes encoding these latter proteins cause familial hypertrophic cardiomyopathy, which will now be discussed.
TABLE 49–6 Biochemical Causes of Inherited Cardiomyopathies1
Mutations in the Cardiac β-Myosin Heavy Chain Gene Are One Cause of Familial Hypertrophic Cardiomyopathy
Familial hypertrophic cardiomyopathy is one of the most frequent hereditary cardiac diseases. Patients exhibit hypertrophy—often massive—of one or both ventricles, starting early in life, and not related to any extrinsic cause such as hypertension. Most cases are transmitted in an autosomal dominant manner; the rest are sporadic. Until recently, its cause was obscure. However, this situation changed when studies of one affected family showed that a missense mutation (ie, substitution of one amino acid by another) in the β-myosin heavy chain gene was responsible for the condition. Subsequent studies have shown a number of missense mutations in this gene, all coding for highly conserved residues. Some individuals have shown other mutations, such as formation of an α/β-myosin heavy chain hybrid gene. Patients with familial hypertrophic cardiomyopathy can show great variation in clinical picture. This in part reflects genetic heterogeneity; that is, mutation in a number of other genes (eg, those encoding cardiac actin, tropomyosin, cardiac troponins I and T, essential and regulatory myosin light chains, cardiac myosin-binding protein C, titin, and mitochondrial tRNA-glycine and tRNA-isoleucine) may also cause familial hypertrophic cardiomyopathy. In addition, mutations at different sites in the gene for β-myosin heavy chain may affect the function of the protein to a greater or lesser extent. The missense mutations are clustered in the head and head-rod regions of the myosin heavy chain. One hypothesis is that the mutant polypeptides (“poison polypeptides”) cause formation of abnormal myofibrils, eventually resulting in compensatory hypertrophy. Some mutations alter the charge of the amino acid (eg, substitution of arginine for glutamine), presumably affecting the conformation of the protein more markedly and thus affecting its function. Patients with these mutations have a significantly shorter life expectancy than patients in whom the mutation produced no alteration in charge. Thus, definition of the precise mutations involved in the genesis of FHC may prove to be of the important prognostic value; it can be accomplished by appropriate use of the polymerase chain reaction on genomic DNA obtained from one sample of blood lymphocytes. Figure 49–13 is a simplified scheme of the events causing familial hypertrophic cardiomyopathy.
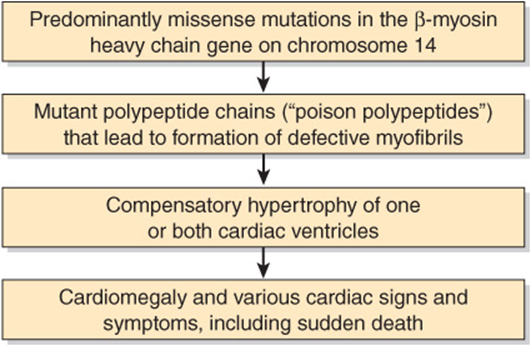
FIGURE 49–13 Simplified scheme of the causation of familial hypertrophic cardiomyopathy (OMIM 192600) due to mutations in the gene encoding β-myosin heavy chain. Mutations in genes encoding other proteins (see text) can also cause this condition.
Another type of cardiomyopathy is termed dilated cardiomyopathy. Mutations in the genes encoding dystrophin, muscle LIM protein (so-called because it was found to contain a cysteine-rich domain originally detected in three proteins: Lin-II, Isl-1, and Mec-3), the cyclic response-element binding protein (CREB), desmin, and lamin have been implicated in the causation of this condition. The first two proteins help organize the contractile apparatus of cardiac muscle cells, and CREB is involved in the regulation of a number of genes in these cells. Current research is not only elucidating the molecular causes of the cardiomyopathies but is also disclosing mutations that cause cardiac developmental disorders (eg, septal defects) and arrhythmias (eg, due to mutations affecting ion channels).
Ca2+ Also Regulates Contraction of Smooth Muscle
While all muscles contain actin, myosin, and tropomyosin, only vertebrate striated muscles contain the troponin system. Thus, the mechanisms that regulate contraction must differ in various contractile systems.
Smooth muscles have molecular structures similar to those in striated muscle, but the sarcomeres are not aligned so as to generate the striated appearance. Smooth muscles contain α-actinin and tropomyosin molecules, as do skeletal muscles. They do not have the troponin system, and the light chains of smooth muscle myosin molecules differ from those of striated muscle myosin. Regulation of smooth muscle contraction is myosin-based, unlike striated muscle, which is actin-based. However, like striated muscle, smooth muscle contraction is regulated by Ca2+.
Phosphorylation of Myosin Light Chains Initiates Contraction of Smooth Muscle
When smooth muscle myosin is bound to F-actin in the absence of other muscle proteins such as tropomyosin, there is no detectable ATPase activity, This absence of activity is quite unlike the situation described for striated muscle myosin and F-actin, which has abundant ATPase activity. Smooth muscle myosin contains light chains that prevent the binding of the myosin head to F-actin; they must be phosphorylated before they allow F-actin to activate myosin ATPase. The ATPase activity then attained hydrolyzes ATP about 10-fold more slowly than the corresponding activity in skeletal muscle. The phosphate on the myosin light chains may form a chelate with the Ca2+bound to the tropomyosin-TpC-actin complex, leading to an increased rate of formation of cross bridges between the myosin heads and actin. The phosphorylation of light chains initiates the attachment-detachment contraction cycle of smooth muscle.
Myosin Light Chain Kinase Is Activated by Calmodulin-4Ca2+ & Then Phosphorylates the Light Chains
Smooth muscle sarcoplasm contains a myosin light chain kinase that is calcium dependent. The Ca2+ activation of myosin light chain kinase requires binding of calmodulin-4Ca2+ to its kinase subunit (Figure 49–14). The calmodulin-4Ca2+-activated light chain kinase phosphorylates the light chains, which then ceases to inhibit the myosin-F-actin interaction. The contraction cycle then begins.
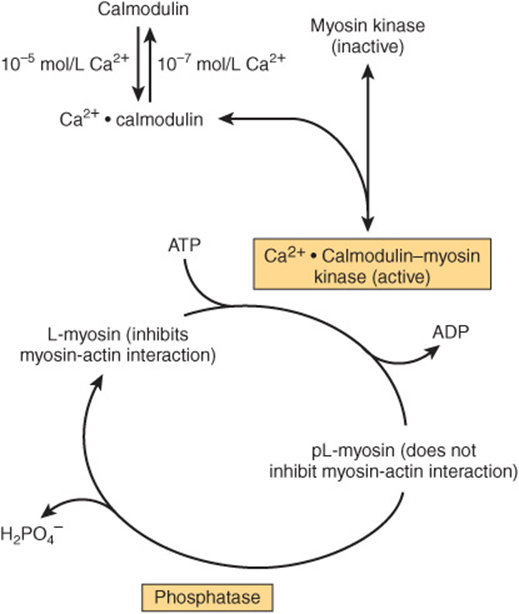
FIGURE 49–14 Regulation of smooth muscle contraction by Ca2+. The pL-myosin is the phosphorylated light chain of myosin and L-myosin is the dephosphorylated light chain. (Adapted, with permission, from Adelstein RS, Eisenberg R: Regulation and kinetics of actin-myosin ATP interaction. Annu Rev Biochem 1980;49:921. Copyright © 1980 by Annual Reviews, www.annualreviews.org.)
Another non-Ca2+-dependent pathway exists in smooth muscle for initiating contraction. This involves Rho kinase, which is activated by a variety of stimuli (not shown in Figure 49–14). This enzyme phosphorylates myosin light chain phosphatase, inhibiting it, and thus increasing the phosphorylation of the light chain. Rho kinase also directly phosphorylates the light chain of myosin. Both of these actions increase the contraction of smooth muscle.
Smooth Muscle Relaxes When the Concentration of Ca2+ Falls Below 10-7 Molar
Relaxation of smooth muscle occurs when sarcoplasmic Ca2+ falls below 10-7 mol/L. The Ca2+ dissociates from calmodulin, which in turn dissociates from the myosin light chain kinase, inactivating the kinase. No new phosphates are attached to the p-light chain, and light chain protein phosphatase, which is continually active and calcium independent, removes the existing phosphates from the light chains. Dephosphorylated myosin p-light chain then inhibits the binding of myosin heads to F-actin and the ATPase activity. The myosin head detaches from the F-actin in the presence of ATP, but it cannot reattach because of the presence of dephosphorylated p-light chain; hence, relaxation occurs.
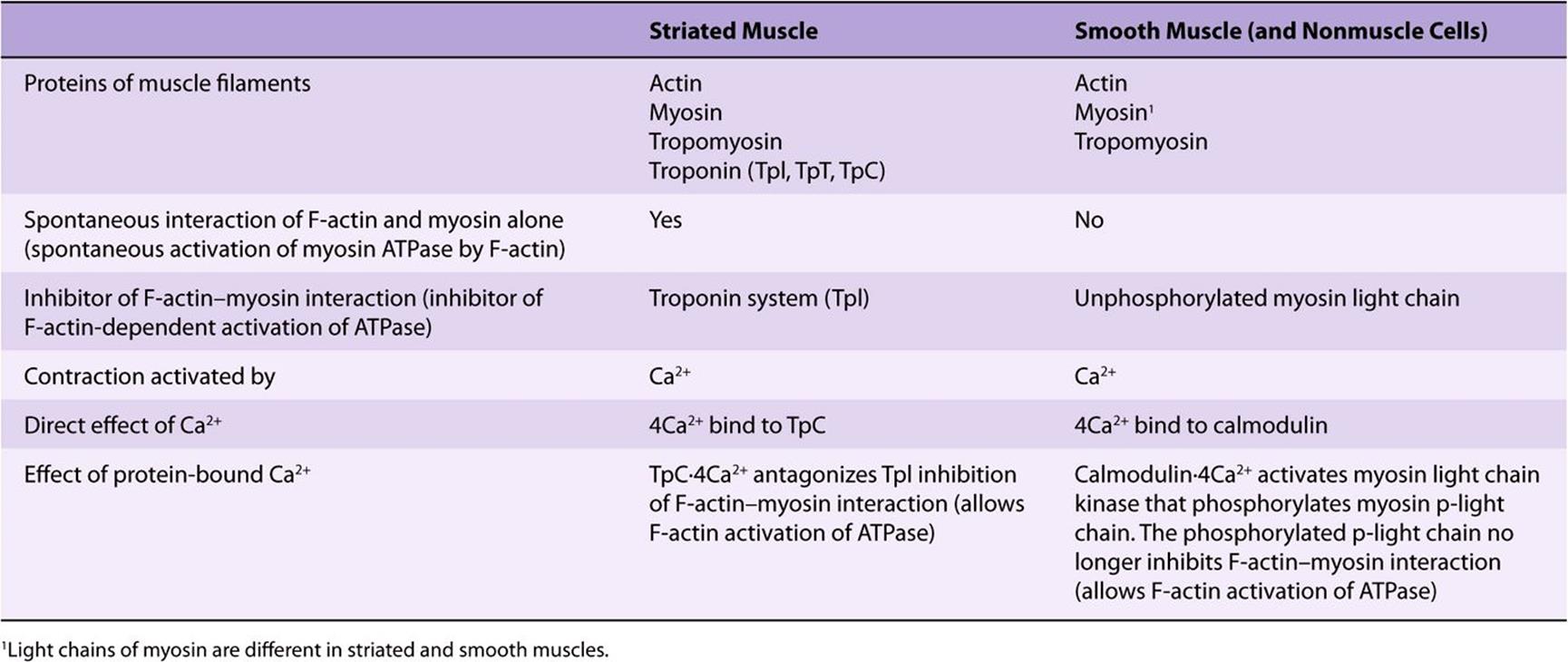
Table 49-7 summarizes and compares the regulation of actin-myosin interactions (activation of myosin ATPase) in striated and smooth muscles.
TABLE 49–7 Actin-Myosin Interactions in Striated and Smooth Muscle
The myosin light chain kinase is not directly affected or activated by cAMP. However, cAMP-activated protein kinase can phosphorylate the myosin light chain kinase (not the light chains themselves). The phosphorylated myosin light chain kinase exhibits a significantly lower affinity for calmodulin Ca2+ and thus is less sensitive to activation. Accordingly, an increase in cAMP dampens the contraction response of smooth muscle to a given elevation of sarcoplasmic Ca2+. This molecular mechanism can explain the relaxing effect of β-adrenergic stimulation on smooth muscle.
Another protein that appears to play a Ca2+-dependent role in the regulation of smooth muscle contraction is caldesmon (87 kDa). This protein is ubiquitous in smooth muscle and is also found in nonmuscle tissue. At low concentrations of Ca2+, it binds to tropomyosin and actin. This prevents interaction of actin with myosin, keeping muscle in a relaxed state. At higher concentrations of Ca2+, Ca2+-calm-odulin binds caldesmon, releasing it from actin. The latter is then free to bind to myosin, and contraction can occur. Caldesmon is also subject to phosphorylation-dephospho-rylation; when phosphorylated, it cannot bind actin, again freeing the latter to interact with myosin. Caldesmon may also participate in organizing the structure of the contractile apparatus in smooth muscle. Many of its effects have been demonstrated in vitro, and its physiologic significance is still under investigation.
As noted in Table 49-3, slow cycling of the cross-bridges permits slow prolonged contraction of smooth muscle (eg, in viscera and blood vessels) with less utilization of ATP compared with striated muscle. The ability of smooth muscle to maintain force at reduced velocities of contraction is referred to as the latch state; this is an important feature of smooth muscle, and its precise molecular bases are under study.
Nitric Oxide (NO) Relaxes the Smooth Muscle of Blood Vessels & Also Has Many Other Important Biologic Functions
Acetylcholine is a vasodilator that acts by causing relaxation of the smooth muscle of blood vessels. However, it does not act directly on smooth muscle. A key observation was that if endothelial cells were stripped away from underlying smooth muscle cells, acetylcholine no longer exerted its vasodilator effect. This finding indicated that vasodilators such as acetylcholine initially interact with the endothelial cells of small blood vessels via receptors. The receptors are coupled to the phosphoinositide cycle, leading to the intracellular release of Ca2+ through the action of inositol trisphosphate. In turn, the elevation of Ca2+ leads to the liberation of endothelium-derived relaxing factor (EDRF), which diffuses into the adjacent smooth muscle. There, it reacts with the heme moiety of a soluble guanylyl cyclase, resulting in activation of the latter, with a consequent elevation of intracellular levels of cGMP (Figure 49–15). This in turn stimulates the activities of certain cGMP-dependent protein kinases, which probably phosphorylate specific muscle proteins, causing relaxation; however, the details are still being clarified. The important coronary artery vasodilator nitroglycerin, widely used to relieve angina pectoris, acts to increase intracellular release of EDRF and thus of cGMP.
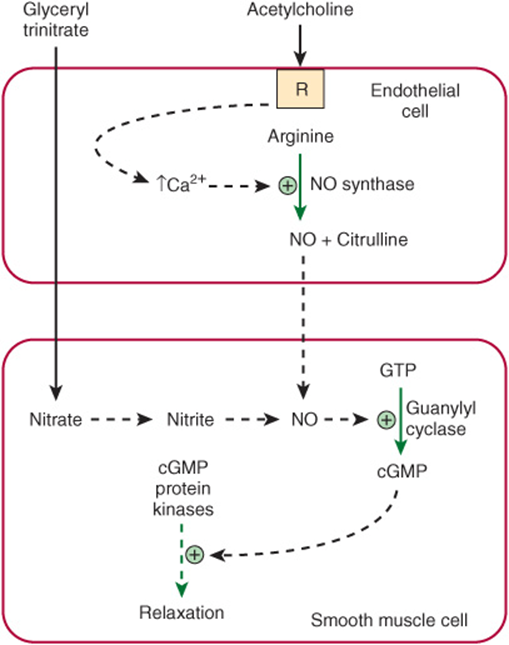
FIGURE 49–15 Diagram showing formation in an endothelial cell of nitric oxide (NO) from arginine in a reaction catalyzed by NO synthase. Interaction of an agonist (eg, acetylcholine) with a receptor (R) probably leads to intracellular release of Ca2+ via inositol trisphosphate generated by the phosphoinositide pathway, resulting in activation of NO synthase. The NO subsequently diffuses into adjacent smooth muscle, where it leads to activation of guanylyl cyclase, formation of cGMP, stimulation of cGMP protein kinases, and subsequent relaxation. The vasodilator nitroglycerin is shown entering the smooth muscle cell, where its metabolism also leads to formation of NO.
Quite unexpectedly, EDRF was found to be the gas NO. NO is formed by the action of the enzyme NO synthase, which is cytosolic. The endothelial and neuronal forms of NO synthase are activated by Ca2+ (Table 49-8). The substrate is arginine, and the products are citrulline and NO:
TABLE 49–8 Summary of the Nomenclature of the NO Synthases and of the Effects of Knockout of Their Genes in Mice

![]()
NO synthase catalyzes a five-electron oxidation of an amidine nitrogen of arginine. L-hydroxyarginine is an intermediate that remains tightly bound to the enzyme. NO synthase is a very complex enzyme, employing five redox cofactors: NADPH, FAD, FMN, heme, and tetrahydrobiopterin. NO can also be formed from nitrite, derived from vasodilators such as glyceryl trinitrate during their metabolism. NO has a very short half-life (approximately 3-4 seconds) in tissues because it reacts with oxygen and superoxide. The product of the reaction with superoxide is peroxynitrite (ONOO–), which decomposes to form the highly reactive OH· radical. NO is inhibited by hemoglobin and other heme proteins, which bind it tightly. Chemical inhibitors of NO synthase are now available that can markedly decrease formation of NO. Administration of such inhibitors to animals and humans leads to vasoconstriction and a marked elevation of blood pressure, indicating that NO is of major importance in the maintenance of blood pressure in vivo. Another important cardiovascular effect is that by increasing synthesis of cGMP, it acts as an inhibitor of platelet aggregation (Chapter 51).
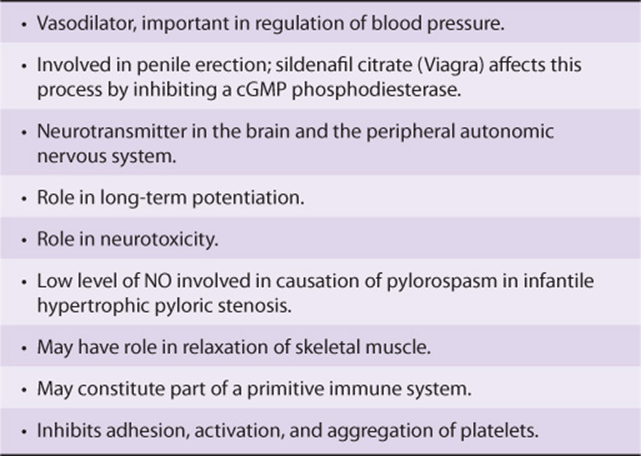
Since the discovery of the role of NO as a vasodilator, there has been intense experimental interest in this molecule. It has turned out to have a variety of physiologic roles, involving virtually every tissue of the body (Table 49-9).Three major isoforms of NO synthase have been identified, each of which has been cloned, and the chromosomal locations of their genes in humans have been determined. Gene knockout experiments have been performed on each of the three isoforms and have helped establish some of the postulated functions of NO.
TABLE 49–9 Some Physiologic Functions and Pathologic Involvements of Nitric Oxide (NO)
To summarize, research in the past decade has shown that NO plays an important role in many physiologic and pathologic processes.
SEVERAL MECHANISMS REPLENISH STORES OF ATP IN MUSCLE
The ATP required as the constant energy source for the contraction-relaxation cycle of muscle can be generated (1) by glycolysis, using blood glucose or muscle glycogen, (2) by oxidative phosphorylation, (3) from creatine phosphate, and (4) from two molecules of ADP in a reaction catalyzed by adenylyl kinase (Figure 49–16). The amount of ATP in skeletal muscle is only sufficient to provide energy for contraction for a few seconds, so that ATP must be constantly renewed from one or more of the above sources, depending upon metabolic conditions. As discussed below, there are at least two distinct types of fibers in skeletal muscle, one predominantly active in aerobicconditions and the other in anaerobic conditions; not unexpectedly, they use each of the above sources of energy to different extents.
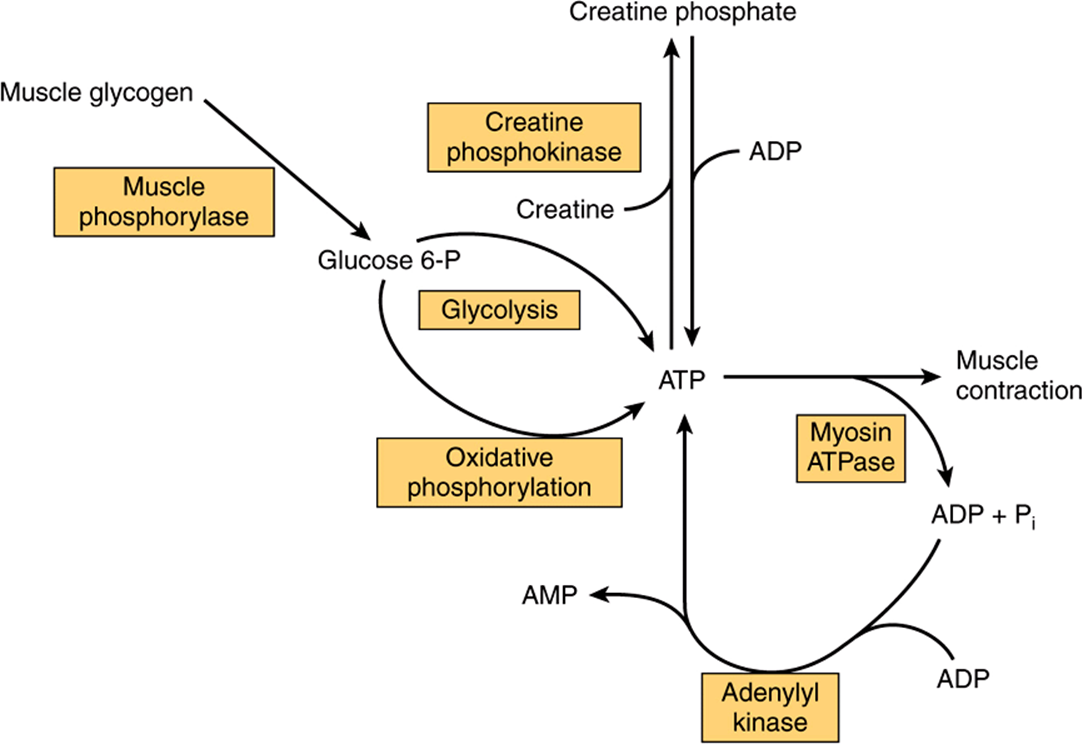
FIGURE 49–16 The multiple sources of ATP in muscle.
Skeletal Muscle Contains Large Supplies of Glycogen
The sarcoplasm of skeletal muscle contains large stores of glycogen, located in granules close to the I bands. The release of glucose from glycogen is dependent on a specific muscle glycogen phosphorylase (Chapter 19), which can be activated by Ca2+, epinephrine, and AMP. To generate glucose 6-phosphate for glycolysis in skeletal muscle, glycogen phosphorylase b must be activated to phosphorylase a via phosphorylation by phosphorylase b kinase (Chapter 19). Ca2+ promotes the activation of phosphorylase b kinase, also by phosphorylation. Thus, Ca2+ both initiates muscle contraction and activates a pathway to provide necessary energy. The hormone epinephrine also activates glycogenolysis in muscle. AMP, produced by breakdown of ADP during muscular exercise, can also activate phosphorylase b without causing phosphorylation. Muscle glycogen phosphorylase b is inactive in McArdle disease, one of the glycogen storage diseases (Chapter 19).
Under Aerobic Conditions, Muscle Generates ATP Mainly by Oxidative Phosphorylation
Synthesis of ATP via oxidative phosphorylation requires a supply of oxygen. Muscles that have a high demand for oxygen as a result of sustained contraction (eg, to maintain posture) store it attached to the heme moiety of myoglobin. Because of the heme moiety, muscles containing myoglobin are red, whereas muscles with little or no myoglobin are white. Glucose, derived from the blood glucose or from endogenous glycogen, and fatty acidsderived from the triacylglycerols of adipose tissue are the principal substrates used for aerobic metabolism in muscle.
Creatine Phosphate Constitutes a Major Energy Reserve in Muscle
Creatine phosphate prevents the rapid depletion of ATP by providing a readily available high-energy phosphate that can be used to regenerate ATP from ADP. Creatine phosphate is formed from ATP and creatine (Figure 49–16) at times when the muscle is relaxed and demands for ATP are not so great. The enzyme catalyzing the phosphorylation of creatine is creatine kinase (CK), a muscle-specific enzyme with clinical utility in the detection of acute or chronic diseases of muscle.
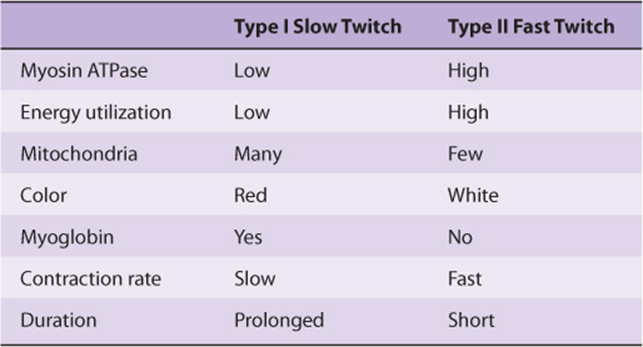
SKELETAL MUSCLE CONTAINS SLOW (RED) & FAST (WHITE) TWITCH FIBERS
Different types of fibers have been detected in skeletal muscle. One classification subdivides them into type I (slow twitch), type IIA (fast twitch-oxidative), and type IIB (fast twitch-glycolytic). For the sake of simplicity, we shall consider only two types: type I (slow twitch, oxidative) and type II (fast twitch, glycolytic) (Table 49-10). The type I fibers are red because they contain myoglobin and mitochondria; their metabolism is aerobic, and they maintain relatively sustained contractions. The type II fibers, lacking myoglobin and containing few mitochondria, are white: they derive their energy from anaerobic glycolysis and exhibit relatively short durations of contraction. The proportion of these two types of fibers varies among the muscles of the body, depending on the function (eg, whether or not a muscle is involved in sustained contraction, such as maintaining posture). The proportion also varies with training; for example, the number of type I fibers in certain leg muscles increases in athletes training for marathons, whereas the number of type II fibers increases in sprinters.
TABLE 49–10 Characteristics of Type I and Type II Fibers of Skeletal Muscle
A Sprinter Uses Creatine Phosphate & Anaerobic Glycolysis to Make ATP, Whereas a Marathon Runner Uses Oxidative Phosphorylation
In view of the two types of fibers in skeletal muscle and of the various energy sources described above, it is of interest to compare their involvement in a sprint (eg, 100 meters) and in the marathon (42.2 km; just over 26 miles) (Table 49-11).
TABLE 49–11 Types of Muscle Fibers and Major Fuel Sources Used by a Sprinter and by a Marathon Runner
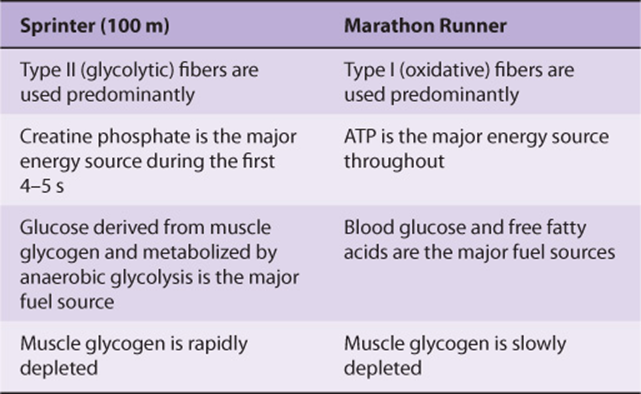
The major sources of energy in the 100-m sprint are creatine phosphate (first 4-5 sec) and then anaerobic glycolysis, using muscle glycogen as the source of glucose. The two main sites of metabolic control are at glycogen phosphorylase and at PFK-1. The former is activated by Ca2+ (released from the SR during contraction), epinephrine, and AMP. PFK-1 is activated by AMP, Pi, and NH3. Attesting to the efficiency of these processes, the flux through glycolysis can increase as much as 1000-fold during a sprint.
In contrast, in the marathon, aerobic metabolism is the principal source of ATP. The major fuel sources are blood glucose and free fatty acids, largely derived from the breakdown of triacylglycerols in adipose tissue, stimulated by epinephrine. Hepatic glycogen is degraded to maintain the level of blood glucose. Muscle glycogen is also a fuel source, but it is degraded much more gradually than in a sprint. It has been calculated that the amount of glucose in the blood, glycogen in the liver, glycogen in muscle, and triacylglycerol in adipose tissue is sufficient to supply muscle with energy during a marathon for 4, 18, 70, and approximately 4000 min, respectively. However, the rate of oxidation of fatty acids by muscle is slower than that of glucose, so that oxidation of glucose and of fatty acids is a major source of energy in the marathon.
A number of procedures have been used by athletes to counteract muscle fatigue and inadequate strength. These include carbohydrate loading, soda (sodium bicarbonate) loading, blood doping (administration of red blood cells), and ingestion of creatine and androstenedione. Their rationales and efficacies will not be discussed here.
SKELETAL MUSCLE CONSTITUTES THE MAJOR RESERVE OF PROTEIN IN THE BODY
In humans, skeletal muscle protein is the major nonfat source of stored energy. This explains very large losses of muscle mass, particularly in adults, resulting from prolonged caloric undernutrition.
The study of tissue protein breakdown in vivo is difficult, because amino acids released during intracellular breakdown of proteins can be extensively reutilized for protein synthesis within the cell, or the amino acids may be transported to other organs where they enter anabolic pathways. However, actin and myosin are methylated by a posttransla-tional reaction, forming 3-methylhistidine. During intracellular breakdown of actin and myosin, 3-methylhistidine is released and excreted into the urine. The urinary output of the methylated amino acid provides a reliable index of the rate of myofibrillar protein breakdown in the musculature of human subjects.
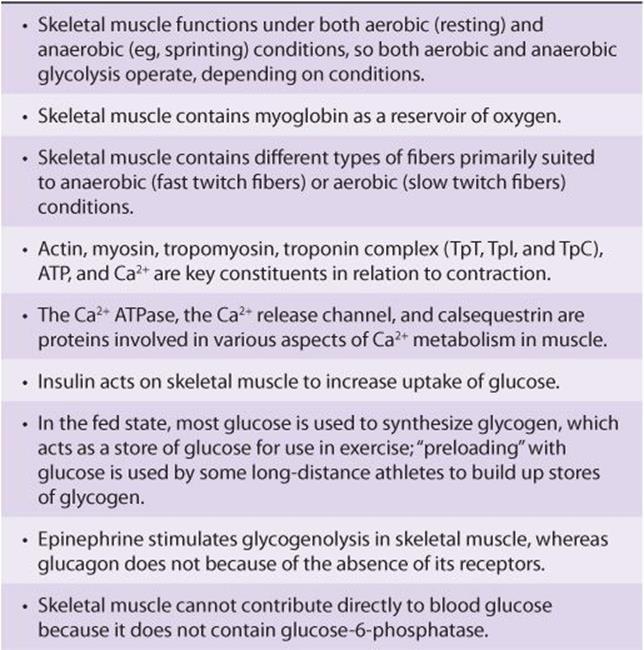
Various features of muscle metabolism, most of which are dealt with in other chapters of this text, are summarized in Table 49-12.
TABLE 49–12 Summary of Major Features of the Biochemistry of Skeletal Muscle Related to Its Metabolism1

THE CYTOSKELETON PERFORMS MULTIPLE CELLULAR FUNCTIONS
Non-muscle cells perform mechanical work, including self-propulsion, morphogenesis, cleavage, endocytosis, exocytosis, intracellular transport, and changing cell shape. These cellular functions are carried out by an extensive intracellular network of filamentous structures constituting the cytoskeleton. The cell cytoplasm is not a sac of fluid, as once thought. Essentially all eukaryotic cells contain three types of filamentous structures: actin filaments(also known as microfilaments), microtubules, and intermediate filaments. Each type of filament can be distinguished biochemically and by the electron microscope.
Some properties of these three structures are summarized in Tables 49-13 and 49-14.
TABLE 49–13 Some Properties of Microfilaments and Microtubules
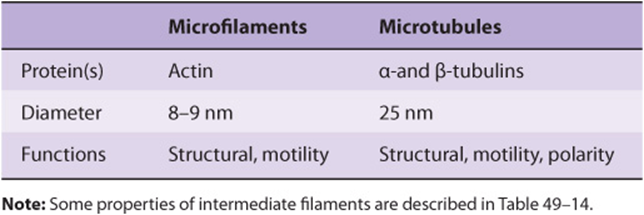
TABLE 49–14 Classes of Intermediate Filaments of Eukaryotic Cells and Their Distributions
Non-muscle Cells Contain Actin That Forms Microfilaments
G-actin is present in most if not all cells of the body. With appropriate concentrations of magnesium and potassium chloride, it spontaneously polymerizes to form double helical F-actin filaments like those seen in muscle. There are at least two types of actin in non-muscle cells: β-actin and γ-actin. Both types can coexist in the same cell and probably even copolymerize in the same filament. In the cytoplasm, F-actin forms microfilaments of 7-9.5 nm that frequently exist as bundles of a tangled-appearing meshwork. These bundles are prominent just underlying the plasma membrane of many cells and are there referred to as stress fibers. The stress fibers disappear as cell motility increases or upon malignant transformation of cells by chemicals or oncogenic viruses.
Although not organized as in muscle, actin filaments in non-muscle cells interact with myosin to cause cellular movements.
Microtubules Contain α- & β-Tubulins
Microtubules, an integral component of the cellular cytoskeleton, consist of cytoplasmic tubes 25 nm in diameter and often of extreme length (see Figure 49–17). Microtubules are necessary for the formation and function of the mitotic spindle and thus are present in all eukaryotic cells. They are also involved in the intracellular movement of endocytic and exocytic vesicles and form the major structural components of cilia and flagella. Microtubules are a major component of axons and dendrites, in which they maintain structure and participate in the axoplasmic flow of material along these neuronal processes.
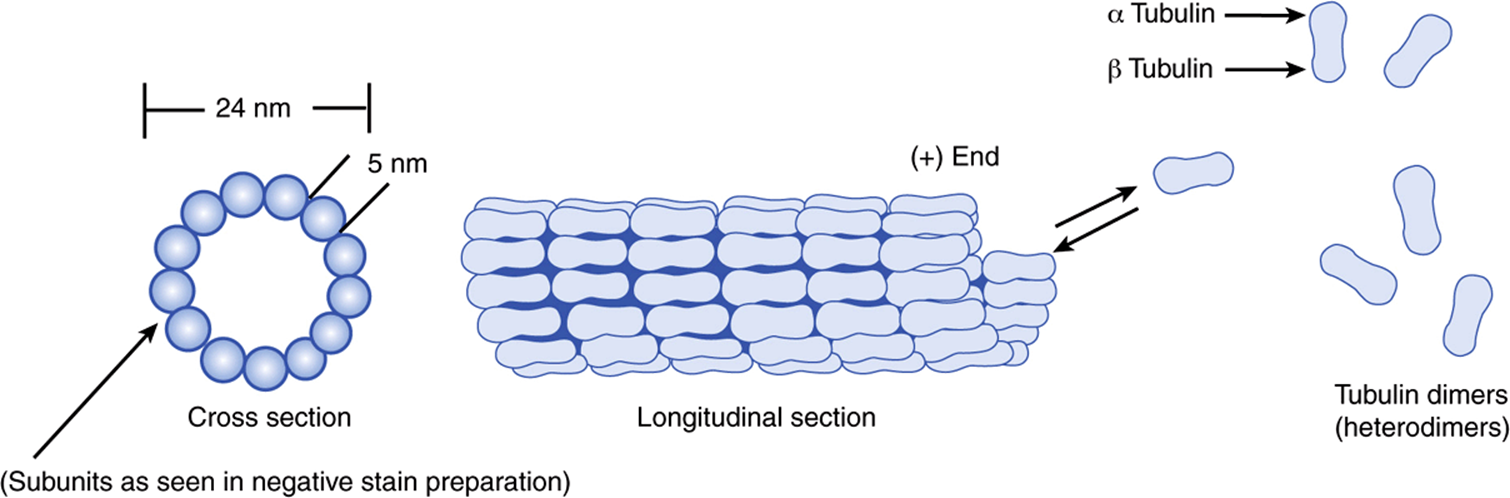
FIGURE 49–17 Schematic representation of microtubules. The upper left-hand corner shows a drawing of microtubules as seen in the electron microscope following fixation with tannic acid in glutaraldehyde. The unstained tubulin subunits are delineated by the dense tannic acid. Cross sections of tubules reveal a ring of 13 subunits of dimers arranged in a spiral. Changes in microtubule length are due to the addition or loss of individual tubulin subunits. Characteristic arrangements of microtubules (not shown here) are found in centrioles, basal bodies, cilia, and flagellae. (Reproduced, with permission, from Junqueirai LC, Carneiro J, Kelley RO: Basic Histology, 7th ed. Appleton & Lange, 1992.)
Microtubules are cylinders of 13 longitudinally arranged protofilaments, each consisting of dimers of α-tubulin and β-tubulin, closely related proteins of approximately 50 kDa molecular mass. The tubulin dimers assemble into protofilaments and subsequently into sheets and then cylinders. A microtubule-organizing center, located around a pair of centrioles, nucleates the growth of new microtubules. A third species of tubulin, γ-tubulin, appears to play an important role in this assembly. GTP is required for assembly. A variety of proteins are associated with microtubules (microtubule-associated proteins [MAPs], one of which is tau) and play important roles in microtubule assembly and stabilization. Microtubules are in a state of dynamic instability, constantly assembling and disassembling. They exhibit polarity (plus and minus ends); this is important in their growth from centrioles and in their ability to direct intracellular movement. For instance, in axonal transport, the protein kinesin, with a myosin-like ATP-ase activity, uses hydrolysis of ATP to move vesicles down the axon toward the positive end of the microtubular formation. Flow of materials in the opposite direction, toward the negative end, is powered by cytosolic dynein, another protein with ATPase activity. Similarly, axonemal dyneins power ciliary and flagellar movement. Another protein, dynamin, uses GTP and is involved in endocytosis. Kinesins, dyneins, dynamin, and myosins are referred to as molecular motors.
An absence of dynein in cilia and flagella results in immotile cilia and flagella, leading to male sterility, situs inversus and chronic respiratory infection, a condition known as Kartagener syndrome (OMIM 244400). Mutations in genes affecting the synthesis of dynein have been detected in individuals with this syndrome.
Certain drugs bind to microtubules and thus interfere with their assembly or disassembly. These include colchicine (used for treatment of acute gouty arthritis), vinblastine (a vinca alkaloid used for treating certain types of cancer), paclitaxel (Taxol) (effective against ovarian cancer), and griseofulvin (an antifungal agent).
Intermediate Filaments Differ from Microfilaments & Microtubules
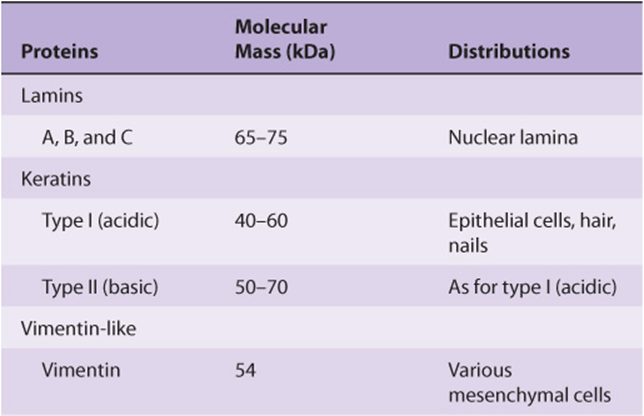
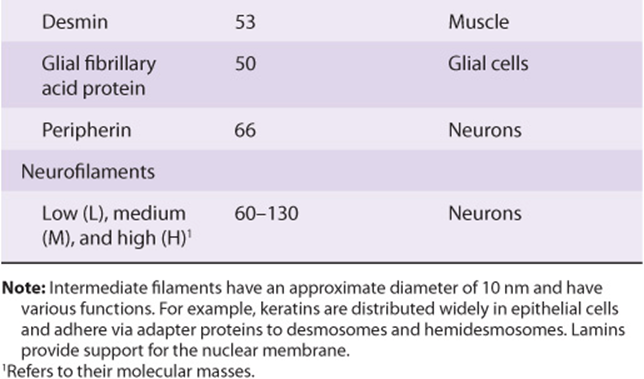
An intracellular fibrous system exists of filaments with an axial periodicity of 21 nm and a diameter of 8-10 nm that is intermediate between that of microfilaments (6 nm) and microtubules (23 nm). At least four classes of intermediate filaments are found, as indicated in Table 49-14.
They are all elongated, fibrous molecules, with a central rod domain, an amino terminal head, and a carboxyl terminal tail. They form a structure like a rope, and the mature filaments are composed of tetramers packed together in a helical manner. They are important structural components of cells, and most are relatively stable components of the cytoskeleton, not undergoing rapid assembly and disassembly and not disappearing during mitosis, as do actin and many microtubular filaments.
An important exception to this is provided by the lamins, which, subsequent to phosphorylation, disassemble at mitosis and reappear when it terminates. Lamins form a meshwork in apposition to the inner nuclear membrane.
Mutations in the gene encoding lamin A and lamin C cause the Hutchinson-Gilford progeria syndrome (progeria) [OMIM 176670], characterized by the appearance of accelerated aging and other features. A farnesylated form (see Figure 26–2 for the structure of farnesyl) of prelamin A accumulates in the condition, because the site of normal proteolytic action to cleave off the farnesylated portion of lamin A is altered by mutation. Lamin A is an important component of the structural scaffolding that maintains the integrity of the nucleus of a cell. It appears that the accumulation of the farnesylated prelamin A makes nuclei unstable, altering their shape, and somehow this predisposes to the development of signs of premature aging. Experiments in mice have indicated that administration of a farnesyltransferase inhibitor may ameliorate the development of misshapen nuclei. Children affected by this condition often die in their teens of atherosclerosis. A brief scheme of the causation of progeria is shown in Figure 49–18.
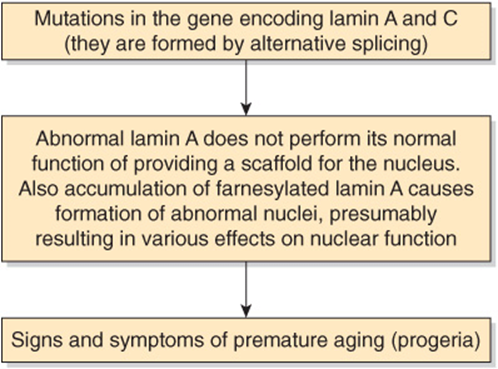
FIGURE 49–18 Scheme of the causation of progeria (Hutchinson-Gilford syndrome, OMIM 176670).
Keratins form a large family, with about 30 members being distinguished. As indicated in Table 49-14, two major types of keratins are found; all individual keratins are heterodimers made up of one member of each class.
Vimentins are widely distributed in mesodermal cells, and desmin, glial fibrillary acidic protein, and peripherin are related to them. All members of the vimentin-like family can copolymerize with each other.
Intermediate filaments are very prominent in nerve cells; neurofilaments are classified as low, medium, and high on the basis of their molecular masses. The distribution of intermediate filaments in normal and abnormal (eg, cancer) cells can be studied by the use of immunofluorescent techniques, using antibodies of appropriate specificities. These antibodies to specific intermediate filaments can also be of use to pathologists in helping to decide the origin of certain dedifferentiated malignant tumors. These tumors may still retain the type of intermediate filaments found in their cell of origin.
A number of skin diseases, mainly characterized by blistering, have been found to be due to mutations in genes encoding various keratins. Two of these disorders are epidermolysis bullosa simplex (OMIM 131800) and epidermolytic palmoplantar keratoderma (OMIM 144200). The blistering found in these disorders probably reflects a diminished capacity of various layers of the skin to resist mechanical stresses due to abnormalities in the keratin structure.
SUMMARY
![]() The myofibrils of skeletal muscle contain thick and thin filaments. The thick filaments contain myosin. The thin filaments contain actin, tropomyosin, and the troponin complex (troponins T, I, and C).
The myofibrils of skeletal muscle contain thick and thin filaments. The thick filaments contain myosin. The thin filaments contain actin, tropomyosin, and the troponin complex (troponins T, I, and C).
![]() The sliding filament cross-bridge model is the foundation of current thinking about muscle contraction. The basis of this model is that the interdigitating filaments slide past one another during contraction and cross bridges between myosin and actin generate and sustain the tension.
The sliding filament cross-bridge model is the foundation of current thinking about muscle contraction. The basis of this model is that the interdigitating filaments slide past one another during contraction and cross bridges between myosin and actin generate and sustain the tension.
![]() The hydrolysis of ATP is used to drive movement of the filaments. ATP binds to myosin heads and is hydrolyzed to ADP and Pi, by the ATPase activity of the actomyosin complex.
The hydrolysis of ATP is used to drive movement of the filaments. ATP binds to myosin heads and is hydrolyzed to ADP and Pi, by the ATPase activity of the actomyosin complex.
![]() Ca2+ plays a key role in the initiation of muscle contraction by binding to troponin C. In skeletal muscle, the SR regulates distribution of Ca2+ to the sarcomeres, whereas inflow of Ca2+ via Ca2+ channels in the sarcolemma is of major importance in cardiac and smooth muscle.
Ca2+ plays a key role in the initiation of muscle contraction by binding to troponin C. In skeletal muscle, the SR regulates distribution of Ca2+ to the sarcomeres, whereas inflow of Ca2+ via Ca2+ channels in the sarcolemma is of major importance in cardiac and smooth muscle.
![]() Many cases of malignant hyperthermia in humans are due to mutations in the gene encoding the Ca2+ release channel.
Many cases of malignant hyperthermia in humans are due to mutations in the gene encoding the Ca2+ release channel.
![]() A number of differences exist between skeletal and cardiac muscle; in particular, the latter contains a variety of receptors on its surface.
A number of differences exist between skeletal and cardiac muscle; in particular, the latter contains a variety of receptors on its surface.
![]() Some cases of familial hypertrophic cardiomyopathy are due to missense mutations in the gene coding for the β-myosin heavy chain. Mutations in genes encoding a number of other proteins have also been detected.
Some cases of familial hypertrophic cardiomyopathy are due to missense mutations in the gene coding for the β-myosin heavy chain. Mutations in genes encoding a number of other proteins have also been detected.
![]() Smooth muscle, unlike skeletal and cardiac muscle, does not contain the troponin system; instead, phosphorylation of myosin light chains initiates contraction.
Smooth muscle, unlike skeletal and cardiac muscle, does not contain the troponin system; instead, phosphorylation of myosin light chains initiates contraction.
![]() NO is a regulator of vascular smooth muscle; blockage of its formation from arginine causes an acute elevation of blood pressure, indicating that regulation of blood pressure is one of its many functions.
NO is a regulator of vascular smooth muscle; blockage of its formation from arginine causes an acute elevation of blood pressure, indicating that regulation of blood pressure is one of its many functions.
![]() Duchenne-type muscular dystrophy is due to mutations in the gene, located on the X chromosome, encoding the protein dystrophin.
Duchenne-type muscular dystrophy is due to mutations in the gene, located on the X chromosome, encoding the protein dystrophin.
![]() Two major types of muscle fibers are found in humans: white (anaerobic) and red (aerobic). The former are particularly used in sprints and the latter in prolonged aerobic exercise. During a sprint, muscle uses creatine phosphate and glycolysis as energy sources; in the marathon, oxidation of fatty acids is of major importance during the later phases.
Two major types of muscle fibers are found in humans: white (anaerobic) and red (aerobic). The former are particularly used in sprints and the latter in prolonged aerobic exercise. During a sprint, muscle uses creatine phosphate and glycolysis as energy sources; in the marathon, oxidation of fatty acids is of major importance during the later phases.
![]() Non-muscle cells perform various types of mechanical work carried out by the structures constituting the cytoskeleton. These structures include actin filaments (microfilaments), microtubules (composed primarily of α-tubulin and β-tubulin), and intermediate filaments. The latter include lamins, keratins, vimentin-like proteins, and neurofilaments. Mutations in the gene encoding lamin A cause progeria, a condition characterized by the appearance of premature aging. Mutations in genes for certain keratins cause a number of skin diseases.
Non-muscle cells perform various types of mechanical work carried out by the structures constituting the cytoskeleton. These structures include actin filaments (microfilaments), microtubules (composed primarily of α-tubulin and β-tubulin), and intermediate filaments. The latter include lamins, keratins, vimentin-like proteins, and neurofilaments. Mutations in the gene encoding lamin A cause progeria, a condition characterized by the appearance of premature aging. Mutations in genes for certain keratins cause a number of skin diseases.
REFERENCES
Alberts B, Johnson A, Lewis J, et al: Molecular Biology of the Cell, 5th ed. Garland Science, 2008. (Contains excellent coverage of muscle and the cytoskeleton).
Barrett KE, Barman SM, Boitano S, Brooks HL: Ganong’s Review of Medical Physiology, 23rd ed. McGraw-Hill Lange, 2010. (Contains excellent coverage of skeletal, cardiac and smooth muscle structure and function).
Brosnan JT, Brosnan ME: Creatine: endogenous metabolite, dietary and therapeutic supplement. Annu Rev Nutr 2007;27:241.
Cooper GM, Hausman RE: The Cell: A Molecular Approach, 5th ed. Sinauer Associates Inc., 2009. (Contains excellent coverage of muscle and the cytoskeleton).
Lodish H, Berk A, Kaiser CA, et al: Molecular Cell Biology, 6th ed. WH Freeman & Co, 2008. (Contains excellent coverage of muscle and the cytoskeleton).
Murad F: Nitric oxide and cyclic GMP in cell signalling and drug development. N Engl J Med 2006;355:2003.
Murphy RT, Starling RC: Genetics and cardiomyopathy: where are we now? Cleve Clin J Med 2005;72:465.
Neubauer S: The failing heart—an engine out of fuel. N Engl J Med 2007;356:1140.
Pollard TD, Earnshaw WC: Cell Biology, 2nd ed. Saunders, 2008. (Contains excellent coverage of muscle and the cytoskeleton).
Sanders KM: Regulation of smooth muscle excitation and contraction. Neurogastroenterol Motil 2008;20 Suppl 1:39.
Scriver CR, Beaudet AL, Valle D, et al (editors): The Metabolic and Molecular Bases of Inherited Disease, 8th ed. McGraw-Hill, 2001. (This comprehensive four-volume text and its updated online edition [see Chapter 1] contain chapters on malignant hyperthermia, channelopathies, hypertrophic cardiomyopathy, the muscular dystrophies, and disorders of intermediate filaments.)
Sweeney HL, Houdusse A: Structural and functional insights into the myosin motor mechanism. Annu Rev Biophys 2010;39:539.
Taimen P, Pfleghaar K, Shimi T, et al: A progeria mutation reveals functions for lamin A in nuclear assembly, architecture, and chromosome organization. Proc Natl Acad Sci USA 2009;106(49):20788.