Harper’s Illustrated Biochemistry, 29th Edition (2012)
SECTION VI. Special Topics
Chapter 54. The Biochemistry of Aging
Peter J. Kennelly, PhD
OBJECTIVES
After studying this chapter, you should be able to:
![]() Describe the essential features of wear and tear theories of aging.
Describe the essential features of wear and tear theories of aging.
![]() List at least four common environmental factors known to damage biological macromolecules such as proteins and DNA.
List at least four common environmental factors known to damage biological macromolecules such as proteins and DNA.
![]() Describe why nucleotide bases are especially vulnerable to damage.
Describe why nucleotide bases are especially vulnerable to damage.
![]() Describe the most physiologically important difference between mitochondrial and nuclear genomes.
Describe the most physiologically important difference between mitochondrial and nuclear genomes.
![]() Describe the oxidative theory of aging and name the primary sources of reactive oxygen species (ROS) in humans.
Describe the oxidative theory of aging and name the primary sources of reactive oxygen species (ROS) in humans.
![]() Describe three mechanisms by which cells prevent or repair damage inflicted by ROS.
Describe three mechanisms by which cells prevent or repair damage inflicted by ROS.
![]() Describe the basic tenets of metabolic theories of aging.
Describe the basic tenets of metabolic theories of aging.
![]() Describe the mechanism of the telomere “countdown clock.”
Describe the mechanism of the telomere “countdown clock.”
![]() Describe our current understanding of the genetic contribution to aging.
Describe our current understanding of the genetic contribution to aging.
![]() Explain the evolutionary implications of a genetically encoded lifespan.
Explain the evolutionary implications of a genetically encoded lifespan.
BIOMEDICAL IMPORTANCE
Consider the various stages in the lifespan of Homo sapiens. Infancy and childhood are characterized by continual growth in height and body mass. Basic motor and intellectual skills develop: walking, language, etc. Infancy and childhood also represent a period of vulnerability wherein a youngster is dependent upon adults for water, food, shelter, protection, and instruction. Adolescence witnesses a final burst of growth in the body’s skeletal framework. More importantly, a series of dramatic developmental changes occur—an accumulation of muscle mass, loss of residual “baby fat,” maturation of the gonads and brain tissue, and the emergence of secondary sex characteristics—that transform the dependent child into a strong, independent, and reproductively capable adult. Adulthood, the longest stage, is a period devoid of dramatic physical growth or developmental change. With the notable exception of pregnancy in females, it is not unusual for adults to maintain the same body weight, overall appearance, and general level of activity for two or three decades.
Barring fatal illness or injury, the onset of the final stage of life, old age, is signaled by a resurgence of physical and physiological change. Hair begins to noticeably thin, turning white or gray as it loses its pigmentation. Skin loses its suppleness and accumulates blemishes. Individuals appear to shrink as both muscle and bone mass are progressively lost. Attention span and recall decline. Eventually, inevitably, life itself comes to an end as one or more essential bodily functions ceases to operate.
Understanding the underlying causes and instigating triggers of aging and the changes that accompany it is of great biomedical importance. Hutchison-Gilford, Werner’s, and Down syndrome are three human genetic diseases whose pathologies include an acceleration of many of the physiological events associated with aging. Slowing or stopping some of the degenerative processes that cause or accompany aging can render the last stage of life much more vital, productive, and fulfilling. Co-opting the factors responsible for triggering cell death may enable physicians to selectively destroy harmful tissues and cells such as tumors, polyps, and cysts without collateral damage to healthy tissues.
LIFESPAN VERSUS LONGEVITY
From Paleolithic times through Greece’s Golden Age to Medieval times the average life expectancy for a newborn baby remained relatively constant, oscillating within the range of 25-35 years. Beginning with the Renaissance, this number has gradually increased, so that by the beginning of the twentieth century the average life expectancy of persons born in developing countries reached the mid-40s. Today, 100 years later, the current world average is 67 years, and that for developed nations is approaching 80. This has led to speculation in the popular press about how long this trend might be expected to continue. Can future generations expect to live past the century mark? Is it possible that human beings possess the potential, barring accidents and with proper care and maintenance, to live indefinitely?
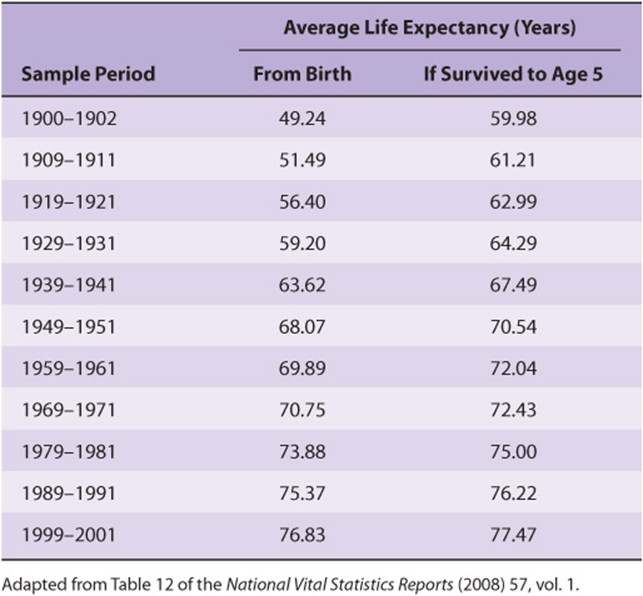
Unfortunately, this extrapolation is unlikely to be realized because it is based on a misunderstanding of the term life expectancy. Life expectancy is calculated by averaging over all births. Hence, it is dramatically influenced by infant mortality rates. While the life expectancy of a Roman child was 25 years, if one calculated the expected lifespan only for those persons who survived infancy, which we will refer to as longevity, the average nearly doubled to 48. When one factors out the dramatic decline in infant mortality rates that has taken place over the past century and a half, the apparent doubling in the human lifespan largely, but not entirely, disappears. As can be seen in Table 54-1. the predicted longevity of a 5-year-old child in the United States has increased from 70.5 in 1950 to 77.5 years in 2000. Is there some sort of upper limit to the lifespan of a properly nourished, well-maintained human being? Perhaps not.
TABLE 54–1 Average Life Expectancy by Decade, USA
AGING & MORTALITY: NONSPECIFIC OR PROGRAMMED PROCESSES?
Are aging and death nondeterminant or stochastic processes in which living creatures inevitably reach a tipping point where they succumb to a lifetime’s accumulation of damage from disease, injury, and simple wear and tear? While the human body has a certain capacity to repair and replace at the molecular and cellular levels, this capacity is variable and finite. No matter how much attention is devoted to care and maintenance, like an automobile or some other sophisticated mechanical device, sooner or later some key component of our bodies will wear out. An alternative school of thought posits that aging and death are genetically programmed processes analogous to puberty, which have evolved through a process of natural selection.
Aging and death are, in all likelihood, multifactorial processes to which numerous factors, some nondeterminant and others programmed, make important contributions. While much work remains to be done before the precise makeup of this mechanistic mosaic can be determined, a large range of potential contributors have been identified. Several of the more prominent of these are presented in the sections that follow.
WEAR & TEAR THEORIES OF AGING
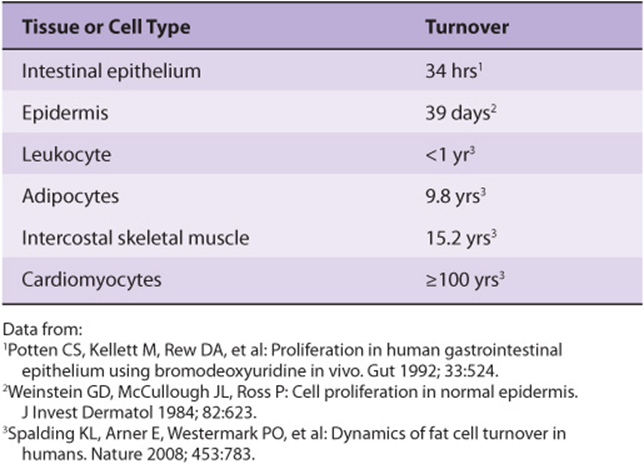
Many theories regarding aging and mortality hypothesize that the human body eventually succumbs to the accumulation of damage over time due to a variety of environmental factors that are reactive with organic biomolecules. These theories note that while repair and turnover mechanisms exist to restore or replace many classes of damaged molecules, these mechanisms are not absolutely perfect. Hence, some damage leaks through—damage that will inevitably accumulate over time, particularly in long-lived cell populations that experience little, if any, turnover (Table 54-2). Ironically, many of the agents that are most damaging to proteins, DNA, and other biomolecules are also essential for terrestrial life: water, oxygen, and sunlight.
TABLE 54–2 Time Required for All of The Average Cells of this Type to be Replaced
Hydrolytic Reactions Can Damage Proteins and Nucleotides
Water is a relatively weak nucleophile. However, because of its ubiquitous presence and high concentration (>55 M, see Chapter 2), even this weak nucleophile will react with susceptible targets inside the cell. In proteins, hydrolysis of peptide bonds leads to cleavage of the polypeptide chain. The amide bonds most frequently targeted by water are those found on the side chains of the amino acids asparagine and glutamine, presumably because they are more exposed, on average, to solvent than the amide bonds in the protein’s backbone. Hydrolysis leads to the replacement of the neutral amide group with an acidic carboxylic acid group, forming aspartate and glutamate, respectively (Figure 54–1, parts A and B). This change leads to the introduction of both a negative charge and of a potential proton donor or acceptor to the affected region of the protein. As the protein population within a living organism is subject to continual turnover, in most cases the chemically modified protein will be degraded and replaced by a newly synthesized protein.
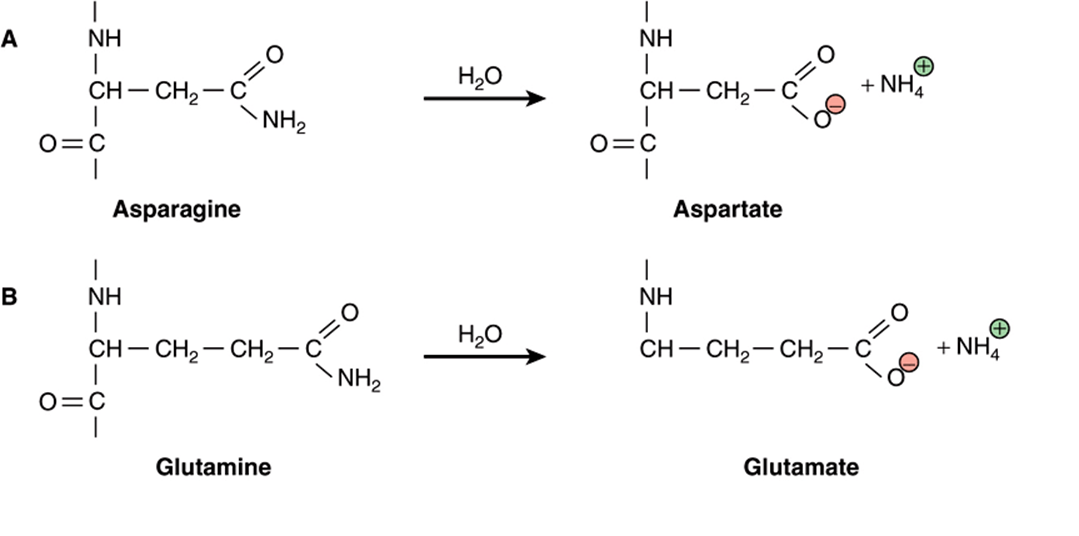
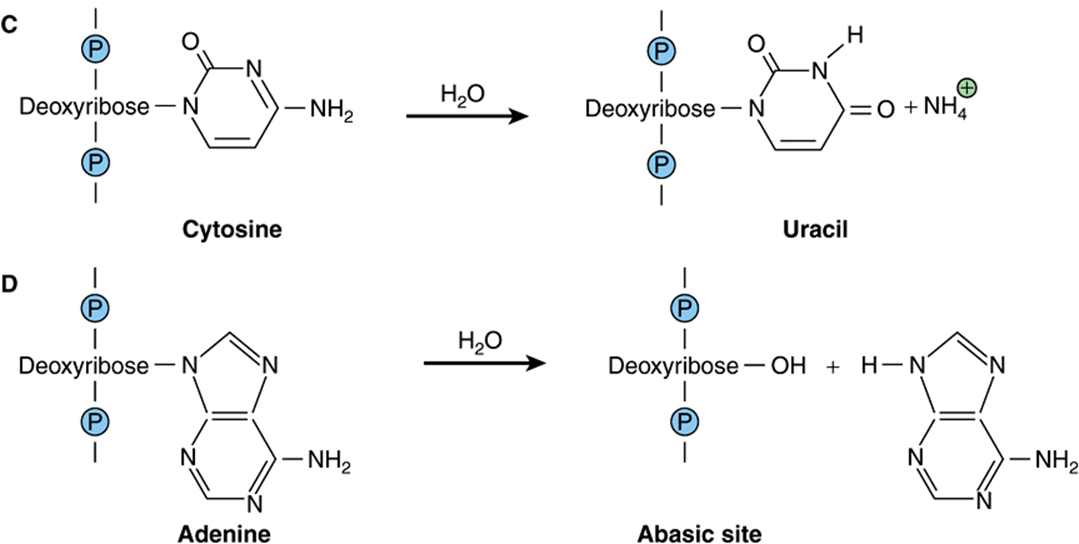
FIGURE 54–1 Examples of hydrolytic damage to biological macromolecules. Shown are a few of the ways in which water can react with and chemically alter proteins and DNA: (A) Net substitution of aspartic acid via hydrolytic deamidation of the neutral side chain of asparagine. (B) Net substitution of glutamic acid via hydrolytic deamidation of the neutral side chain of glutamine. (C) Net mutation of cytosine to uracil by water. (D) Formation of an abasic site in DNA via hydrolytic cleavage of a ribose-base bond.
Of perhaps greater potential biological consequence are the reactions of the nucleotide bases in DNA with water. The amino groups projecting from the heterocyclic aromatic rings of the nucleotide bases cytosine, adenine, and guanine are each susceptible to hydrolytic attack in which the amino group is replaced by a carbonyl to form uracil, hypoxanthine, and xanthine, respectively (Figure 54–1, part C). If the affected base is located in the cell’s DNA, the net result is a mutation that, if left unrepaired, can potentially perturb gene expression or produce a dysfunctional gene product. The bond between the nucleotide base and the deoxyribose moiety in DNA is also vulnerable to hydrolysis. In this instance the base is completely eliminated, leaving a gap in the sequence (Figure 54–1, part D) which, if left unrepaired, can lead to either a substitution or a frame-shift mutation (see Chapter 37).
Many other bonds within biological macromolecules also possess the potential to be cleaved by random chemical hydrolysis. Included in this list are the ester bonds that bind fatty acids to their cognate glycerolipids, the glucosidic bonds that link the monosaccharide units of carbohydrates, and the phosphodiester bonds that hold polynucleotides together and link the head groups of phospholipids to their diacylglycerol partners. However, these reactions appear to take place too infrequently (phosphodiester hydrolysis) or to generate insufficiently perturbing products to manifest significant biological consequences.
Respiration Generates Reactive Oxygen Species
Numerous biological processes require enzyme-catalyzed oxidation of organic molecules by molecular oxygen (O2). These processes include the hydroxylation of proline and lysine side chains in collagen (Chapter 5), the detoxification of xenobiotics by cytochrome P450 (Chapter 53), the degradation of purine nucleotides to uric acid (Chapter 33), the reoxidation of the prosthetic groups in the flavin-containing enzymes that catalyze oxidative decarboxylation (eg, the pyruvate dehydrogenase complex, Chapter 18) and other redox reactions (eg, amino acid oxidases, Chapter 28), and the generation of the chemiosmotic gradient in mitochondria by the electron transport chain (Chapter 13). Redox enzymes frequently employ prosthetic groups such as flavin nucleotides, iron-sulfur centers, or heme-bound metal ions (Chapters 12 and 13) to assist in the difficult task of generating and stabilizing the highly reactive free radical and oxyanion intermediates formed during catalysis. The electron transport chain employs specialized carriers such as ubiquinone and cytochromes to safely transport single, unpaired electrons among and within its various multiprotein complexes.
Occasionally, these highly reactive intermediates escape into the cell in the form of ROS such as superoxide and hydrogen peroxide (Figure 54–2, part A). By virtue of its structural and functional complexity and extremely high level of electron flux, “leakage” from the electron transport chain constitutes by far and away the major source of ROS in most mammalian cells. In addition, many mammalian cells synthesize and release the second messenger nitric oxide (NO.), which contains an unpaired electron, to promote vasodilation and muscle relaxation in the cardiovascular system (Chapter 49).
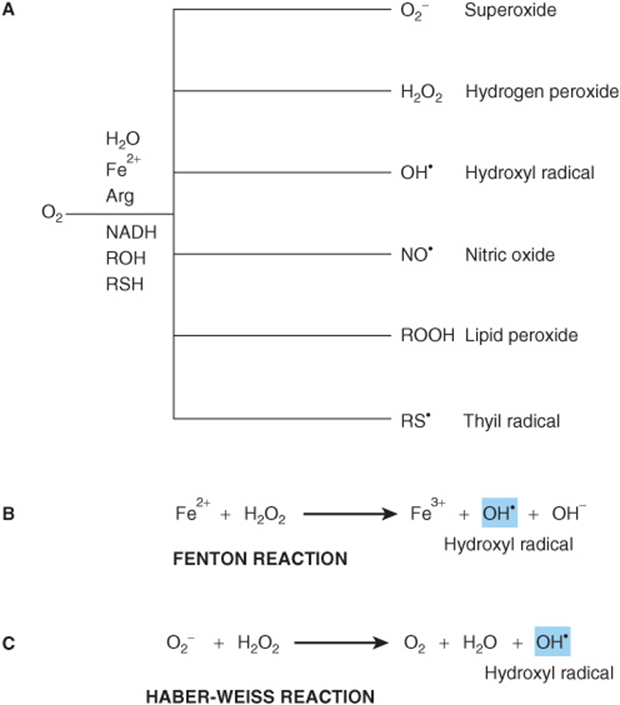
FIGURE 54–2 Reactive oxygen species (ROS) are toxic byproducts of life in an aerobic environment. (A) Many types of ROS are encountered in living cells. (B) Generation of hydroxyl radical via the Fenton reaction. (C)Generation of hydroxyl radical by the Haber-Weiss reaction.
Reactive Oxygen Species Are Chemically Prolific
The extremely high reactivity of ROS makes them extremely dangerous. ROS can react with and chemically alter virtually any organic compound, including proteins, nucleic acids, and lipids. In some cases, reaction leads to the cleavage of covalent bonds. They also display a strong tendency to form adducts, the products of the direct addition of two (or more) compounds, with nucleotide bases, polyunsaturated fatty acids, and other biological compounds possessing multiple double bonds (Figure 54–3). Adducts formed with nucleotide bases can be especially dangerous because of their potential, if left uncorrected, to cause misreads that introduce mutations into DNA.
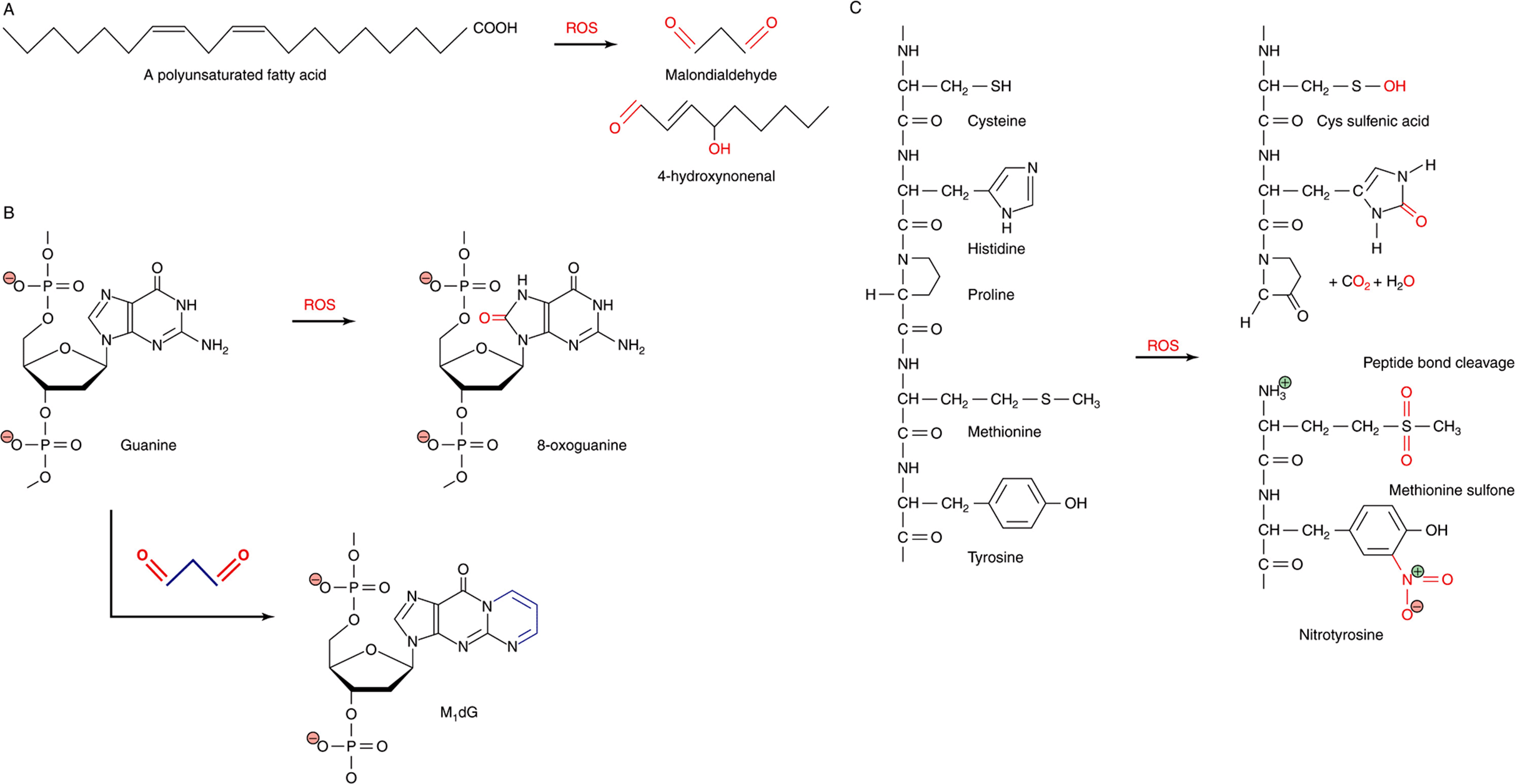
FIGURE 54–3 ROS react directly and indirectly with a wide range of biological molecules. (A) Peroxidation of unsaturated lipids generates reactive products such as malondialdehyde and 4-hydroxynonenal. (B) Guanine can be directly oxidized by ROS to produce 8-oxoguanine or form an adduct, M1dG, with the ROS product malondialdehyde. (C) Common reactions of proteins with ROS, including oxidation of amino acid side chains and cleavage of peptide bonds. Oxygen atoms derived from ROS are marked in red. Carbon atoms derived from malondialdehyde in M1dG are colored blue. The complete chemical name for M1dG is 3-(2-Deoxy-D-erythro-pentofuranosyl)pyrimido(1,2-α)purin-10(3H)-one.
The ease with which oxygen evokes the chemical changes that turn household butter rancid is a testament to the reactivity of unsaturated fats, those containing one or more carbon-carbon double bond (Chapter 23) with ROS. Lipid peroxidation can lead to the formation of cross-linked lipid-lipid and lipid-protein adducts and a loss of membrane fluidity and integrity. Loss of membrane integrity, in turn, can—in the case of the mitochondria—undermine the efficiency with which the electron transport chain converts reducing equivalents to ATP, leading to greater production of deleterious ROS. Loss of membrane integrity can also trigger apoptosis, the programmed death of a cell.
Chain Reactions Multiply the Destructiveness of ROS
The destructiveness inherent in the high reactivity of many of these ROS, particularly free radicals, is exacerbated by their capacity to participate in chain reactions in which the product of the reaction between the free radical and some biomolecule is a damaged biomolecule and another species containing a highly reactive unpaired electron. The chain will terminate when a free radical is able to acquire another lone electron to form a relatively innocuous electron pair without generating a new unpaired electron as a by-product. Such is the case when one free radical encounters another. The two “odd” electrons combine to form a pair. Alternatively, the ROS may be eliminated by one of the cell’s suite of dedicated antioxidant enzymes (Chapters 12 and 52).
The reactivity, and hence destructiveness, of individual ROS varies. Hydrogen peroxide, for example, is less reactive than superoxide, which in turn is less reactive than hydroxyl radical (OH’). Unfortunately, two pathways exist in living organisms by which highly toxic hydroxyl radical can be generated from less destructive ROS. If ferric iron is present, for example, the Fenton reaction can transform hydrogen peroxide into hydroxyl radicals (Figure 54–2, part B). The ferrous (+3) iron, in turn, can be reduced back to the ferric (+2) state by other hydrogen peroxide molecules, permitting the iron to act catalytically to produce additional hydroxyl radicals. Hydroxyl radical can also be generated when superoxide and hydrogen peroxide disproportionate, a process called the Haber-Weiss reaction (Figure 54–2, part C).
Free Radicals and the Mitochondrial Theory of Aging
In 1956, Denham Harmon proposed the so-called free radical theory of aging. It had been reported that the toxicity of hyperbaric oxygen treatment and radiation could be explained by a factor common to both, the generation of ROS. This report dovetailed nicely with Harmon’s own observation that lifespan was inversely related to metabolic rate and, by extrapolation, respiration. He therefore postulated that the cumulative damage was caused by the continual and inescapable production of ROS.
In more recent years, the proponents of the free radical theory of aging have focused attention on the mitochondria. Not only is the mitochondria host to the major source of ROS in the cell, the electron transport chain, but oxidative damage to the components of this pathway could lead to increased leakage of hydrogen peroxide, superoxide, etc, into the cytoplasm. Damage to the mitochondria would be likely to adversely affect the efficiency with which it performs its most important function, the synthesis of ATP. A significant slowing in the rate of ATP synthesis could readily lead to the types of wholesale declines in physiological function that occur in aging.
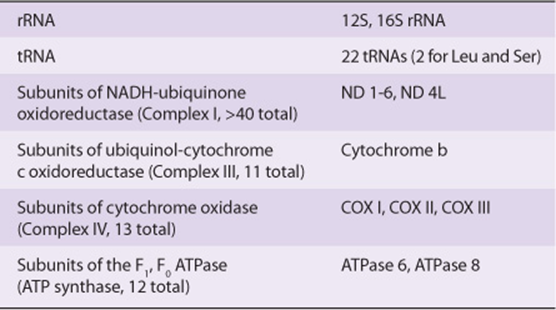
A second contributor to the proposed self-perpetuating cycle of mitochondrial redox damage is the fact that several components of the electron transport chain are encoded by the mitochondrion’s indigenous genome. The mitochondrial genome is a much reduced, vestigial remnant of the genome of the ancient bacterium that was the precursor of the current organelle. Through a process called endosymbiosis, primitive eukaryotes became dependent upon surrounding bacteria to provide certain materials, and vice versa. Eventually, the smaller bacterium was absorbed by and lived within the interior of its eukaryotic host. Over time most, but not all, of the genes contained in the bacterial genome were either eliminated as superfluous to the needs of the new fusion organism or were transferred into the host cell’s nuclear DNA. At present, the genome of the human mitochondrion encodes a small and a large ribosomal RNA, 22 tRNAs, and polypeptide subunits for complexes I, III, and IV of the electron transport chain as well as the F1, F0 ATPase (Table 54-3). The mitochondrial genome lacks the surveillance and repair mechanisms that help maintain the integrity of nuclear DNA. Hence, mutations induced by adducts or reaction with ROS, and any functional defects resulting from these mutations, become a permanent feature of each individual mitochondrion’s genome, which will continue to accumulate mutations with time.
TABLE 54–3 Genes Encoded by the Genome of Human Mitochondria
While the mitochondrial hypothesis is no longer viewed as providing a unifying explanation for all of the changes that are associated with human aging and its comorbidities, it likely is an important contributor. Powerful circumstantial evidence for this is provided by the central role played by this organelle in the sensor-response pathways that trigger apoptosis.
Mitochondria Are Key Participants in Apoptosis
Apoptosis imbues higher organisms with the ability to selectively eliminate cells that are rendered superfluous by developmental changes, such as those that take place on a continual basis during embryogenesis, or which have been damaged beyond repair. During developmental tissue remodeling, the apoptotic cell death program is triggered by receptor-mediated signals. In the case of damaged cells, any one of several interior indicators may serve as trigger: ROS, viral dsRNA, DNA damage, and heat shock. These signals trigger the opening of the permeability transition pore complex embedded in the mitochondrial outer membrane, through which molecules of the small (≈12.5 kDa) electron carrier protein cytochrome c escapes into the cytoplasm. Here, cytochrome c serves as the core protein for nucleating a multiprotein complex, called the apoptosome, that initiates a cascade of proteolytic activation events targeting the proenzyme forms of a series of cysteine proteases known as caspases. The terminal caspases, numbers 3 and 7, break down structural proteins in the cytoplasm and chromatin proteins in the nucleus; events that lead to the death of the affected cell and its elimination by phagocytosis. Needless to say, the presence of an intrinsic, receptor-mediated cell death pathway offers the hope that we can eliminate harmful cells, such as cancer, by learning how to selectively activate their apoptotic pathway.
Ultraviolet Radiation Can Be Extremely Damaging
The term ultraviolet (UV) radiation refers to those wavelengths of light that lie immediately beyond the blue or short wavelength end of the visible spectrum. While the human eye cannot detect these particular wavelengths of light, they are strongly absorbed by organic compounds possessing aromatic rings or multiple, conjugated double bonds such as the nucleotide bases of DNA and RNA; the aromatic side chains of the amino acids phenylalanine, tyrosine, and tryptophan; polyunsaturated fatty acids; heme groups; and cofactors and coenzymes such as flavins, cyanocobalamine, etc. Absorption of this short wavelength, high-energy light can cause the rupture of covalent bonds in proteins, DNA, and RNA; the formation of thymine dimers in DNA (Figure 54–4); cross-linking of proteins; and the generation of free radicals including ROS. While UV radiation does not penetrate beyond the first few layers of skin cells, the high efficiency of absorption leads to the rapid accumulation of damage to the limited population of skin cells that are impacted. Because the nucleotide bases of DNA and RNA are particularly effective at absorbing UV radiation, it is highly mutagenic. Prolonged exposure to intense sunlight can lead to the accumulation of multiple DNA lesions that can overwhelm a cell’s intrinsic repair capacity. It is thus relatively common for persons whose work or lifestyle involves prolonged exposure to sunlight to manifest aberrant skin tissue, in the form of both moles and cancerous myelomas. Many of the latter can proliferate and spread with great rapidity, necessitating careful surveillance and rapid medical intervention.
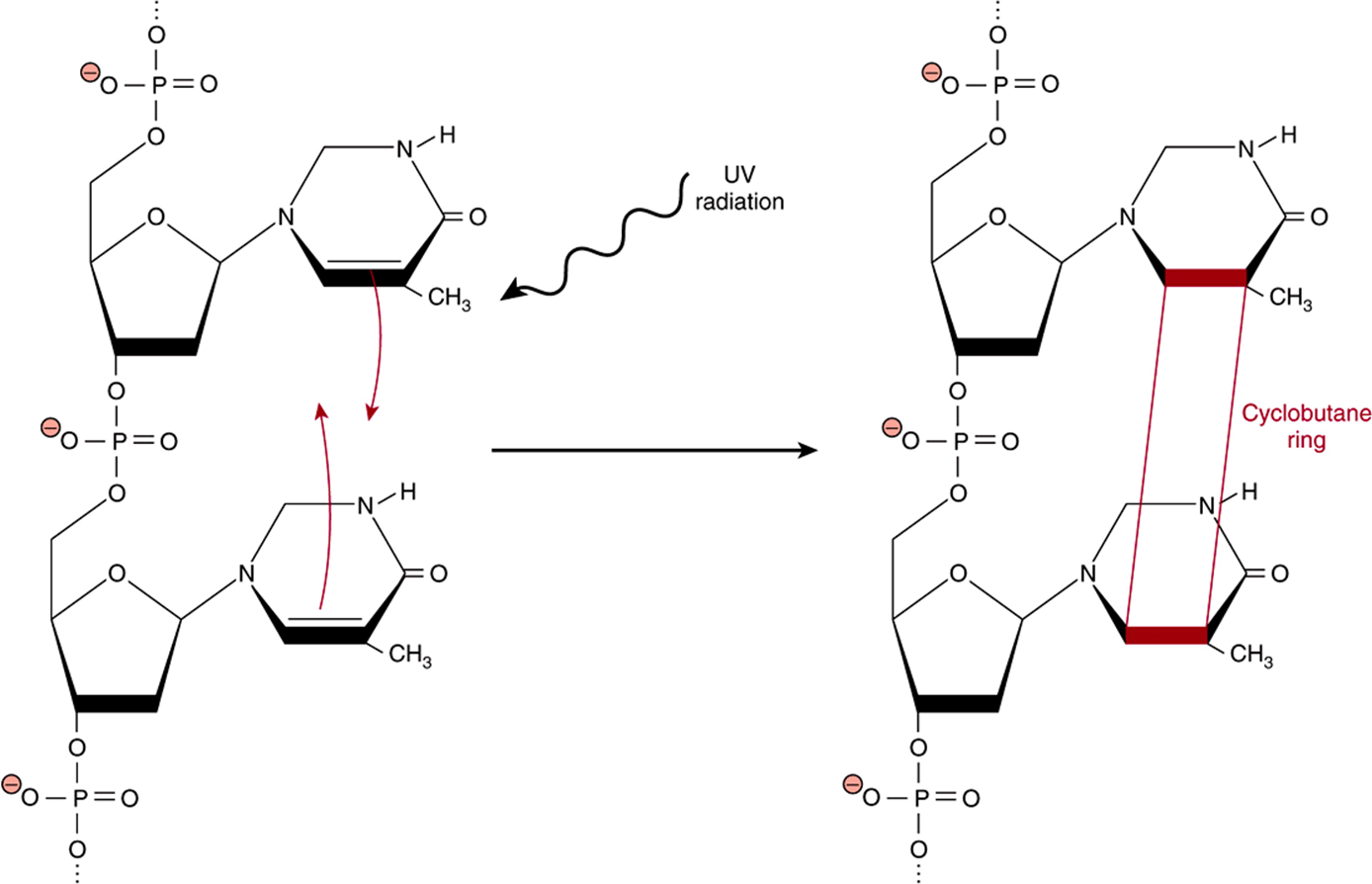
FIGURE 54–4 Formation of a thymine dimer following excitation by UV light. When consecutive thymine bases are stacked one above the other in a DNA double helix, absorption of UV light can lead to the formation of a cyclobutane ring (red, not to scale) covalently linking the two bases together to form a thymine dimer.
Protein Glycation Often Leads to the Formation of Damaging Cross-links
When amino groups such as those found on the side chain of lysine or some of the nucleotide bases are exposed to a reducing sugar such as glucose, a reversible adduct is slowly generated through the formation of a Schiff’s base between the aldehyde or ketone group of the sugar and the amine. Over time, the glycated protein undergoes a series of rearrangements to form Amadori products, which contain a conjugated carbon-carbon double bond that can react with the amino group on a neighboring protein (Figure 54–5). The net result is the formation of a covalent crosslink between two proteins or other biological macromolecules that can, in turn, undergo further glycation and crosslink to yet another macromolecule. These crosslinked aggregates are sometimes called Advanced Glycation Endproducts or AGEs.
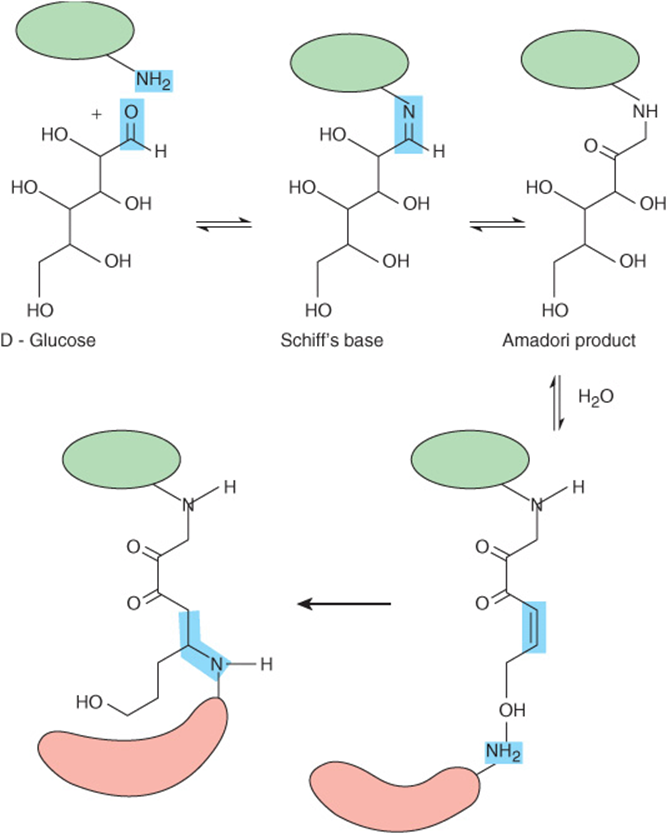
FIGURE 54–5 Protein glycation can lead to the formation of protein-protein crosslinks. Shown are the sequence of reactions that generate the Amadori product on the surface of the protein marked in green, and the subsequent formation of a protein-protein crosslink via an amino group on the surface of a second, red, protein.
The physiological impact of protein glycation can be especially pronounced when long-lived proteins such as collagen or β-crystallins are involved. Their persistence provides the opportunity for multiple glycation and crosslinking events to occur. The progressive crosslinking of the collagen network in vascular endothelial cells leads to the progressive loss of elasticity and thickening of the basement membrane in blood vessels, promoting plaque formation. The overall result is a progressive increase in the heart’s workload. In the eye, the accumulation of aggregated proteins compromises the opacity of the lens and eventually manifests itself in the form of cataracts. Impairment of glucose homeostasis renders diabetics particularly susceptible to the formation of advanced glycation end products. In fact, the glycation of hemoglobin and serum albumin are used as biomarkers for the diagnosis of diabetes and the assessment of its treatment.
MOLECULAR REPAIR MECHANISMS COMBAT WEAR & TEAR
Enzymatic and Chemical Mechanisms Intercept Damaging ROS
A corollary to the wear and tear theory of aging is that longevity reflects the effectiveness and robustness of the molecular prevention, repair, and replacement mechanisms in a given species and the individuals within it. Enzymes such as superoxide dismutase and catalase protect the cell by converting superoxide and hydrogen peroxide, respectively, to less reactive products, thereby preventing potential molecular damage before it occurs (Chapter 52). For example, fruit flies that have been genetically altered to express elevated levels of superoxide dismutase exhibit significantly extended life spans.
In the cytoplasm, the cysteine-containing tripeptide glutathione acts as a chemical redox protectant by reacting directly with ROS to generate less reactive compounds such as water. Oxidized glutathione, which consists of two tripeptides linked by an S-S bond, is then enzymatically reduced to maintain the pool of protectant (Chapter 52). Glutathione can also react directly with cysteine sulfenic acids and disulfides on proteins to restore them to their reduced state, and form adducts with toxic xenobiotics (Chapter 53). Other biomolecules such as ascorbic acid and vitamin E also possess antioxidant properties, which accounts for the fact that many “popular” diets target foods rich in these compounds in an effort to buttress the body’s ability to neutralize ROS and slow aging.
The Integrity of DNA Is Maintained by Proofreading and Repair Mechanisms
In addition to the prophylactic measures mentioned above, living organisms possess a limited capacity to replace or repair damaged macromolecules. The majority of this capacity is directed toward maintaining the integrity of the nuclear (but not the mitochondrial) genome, which is to be expected given DNA’s unique information storage function, the vulnerability of heterocyclic aromatic nucleotide bases to chemical assault and UV radiation, and the fact that—by contrast to almost every other macromolecule—each cell contains only a single unique copy of each chromosome. A somatic cell is one that forms part of the body of an organism. Maintaining the integrity of the genome begins at replication, where careful proofreading is performed to insure that the new genome formed in the process of somatic cell division faithfully replicates the template that directed it synthesis. In addition, most living organisms possess an impressive cadre of enzymes whose role is to inspect and correct aberrations that either escaped proofreading or were subsequently generated through the action of water (double strand breaks, loss of a nucleotide base, and deamidation of cytosine), UV radiation (thymine dimers and strand breaks), or exposure to chemical modifiers (adduct formation). This multilayered system is composed of mismatch repair enzymes, nucleotide excision repair enzymes, and base excision repair enzymes as well as the Ku system for repairing double-strand breaks in the phosphodiester backbone (Chapter 35). As a last resort, cells harboring damaging mutations are subject to removal by apoptosis.
Nevertheless, despite the many precautions taken to insure fidelity during replication and to repair subsequent damage listed above, some mutations inevitably slip through. Indeed, some leakage in the surveillance and repair system is necessary in order to generate the genetic variability that drives evolution. The somatic mutation theory of aging proposes that it also serves a second purpose as a driver of the aging process. Simply put, the accumulation of mutant cells over time must inevitably lead to compromised biological function that manifests itself, at least in part, as the physical changes we associate with aging.
Some Types of Protein Damage Can Be Repaired
In contrast to DNA, a cell’s capacity to repair damage to other biomolecules is relatively limited. For the most part, cells appear to rely on routine turnover, wherein the global population of a given biomolecule is degraded and replaced by new synthesis on a continuing, or constitutive, basis (Chapter 9), to remove aberrant lipids, carbohydrates, and proteins. Some proteins, particularly the fibrous proteins that contribute to the structural integrity of tendons, ligaments, bones, matrix, etc, undergo little if any turnover. These long-lived proteins tend to accumulate damage over many years, contributing to the loss of elasticity in vascular tissues and joints, loss of lens opacity, etc. The most prominent mechanisms for the repair of damaged proteins target the sulfur atoms contained in the side chains of cysteine and methionine, and the isoaspartyl groups formed by the shift of the peptide bond to side-chain carboxyl group.
The side-chain sulfhydryl group of cysteine frequently plays important catalytic, regulatory, and structural roles in proteins that are dependent upon its oxidation state. However, both its sulfhydryl group and the sulfur ether of methionine are extremely vulnerable to oxidation (Figure 54–3, part C). As is the case for many other oxidized biomolecules, the tripeptide glutathione can react directly with cysteine-disulfides, cysteine sulfenic acids, and methinonine sulfoxide to regenerate cysteine and methionine, respectively. In addition, disulfide reductases and methionine sulfoxide reductases provide an enzyme-catalyzed reduction mechanism using NADPH as electron donor. Unfortunately, the reduction potential of glutathione and NADPH is only sufficient only to reduce the lowest oxidation states of these sulfur atoms: cysteine disulfides or sulfenic acids and methionine sulfoxide. Cysteine sulfinic acid, cysteine sulfonic acid, and methinoine sulfone are refractory to reduction under physiologic conditions.
Aspartic acid residues possess the precise geometry needed to enable the side-chain carboxyl group to react with the amino group within the peptide bond formed with its alpha carboxyl group. The resulting cyclic diamide can then reopen to form either the original peptide bond or an isoaspartyl residue in which the side-chain carboxyl now forms part of the protein’s peptide backbone (Figure 54–6). Methylation of the alpha carboxyl group provides a leaving group that promotes the reformation of the cyclic diamide, which can then reopen to form the normal peptide bond linkage (Figure 54–6).
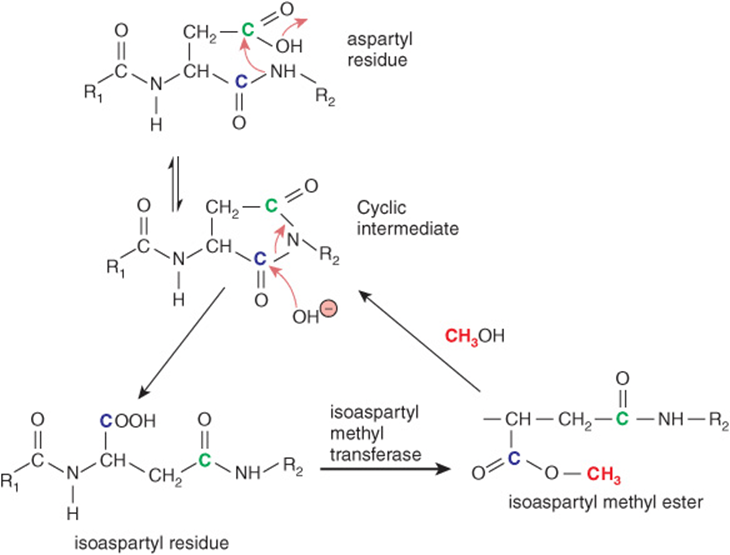
FIGURE 54–6 Formation of an isoaspartyl linkage in a polypeptide backbone and its repair via the intervention of isoaspartyl methyltransferase. Shown is the sequence of chemical and enzyme-catalyzed reactions that lead to formation of an isoaspartyl linkage and restoration of a normal peptide linkage. The carbons corresponding to the alpha and side-chain carboxylic acid groups in aspartic acid are colored blue and green, respectively. Red arrows denote the routes of nucleophilic attack during the cyclization and hydrolysis reactions. The methyl group added by isoaspartyl methyltransferase is colored pink.
Aggregated Proteins Are Highly Refractory to Degradation or Repair
Modifications to a protein’s composition or conformation that cause it to adhere to other protein molecules can lead to the formation of toxic aggregates, called amyloid. Such aggregates form the hallmark of several neurodegenerative diseases, including Parkinson’s, Alzheimer’s, Huntington’s disease, spinocerebellar ataxias, and the transmissible spongiform encephalopathies. The toxic effects of these insoluble aggregates are exacerbated by their persistence, as in this state most are generally refractory to the catalytic action of the proteases normally responsible for their turnover.
AGING AS A PREPROGRAMMED PROCESS
While molecular wear and tear undoubtedly contribute to aging, several observations suggest a role for programmed, deterministic mechanisms as well. For example, rather than “rusting” gradually, many of the physical manifestations of aging—liver spots, grayness, trembling hands, memory lapses—generally surface late in adulthood and progress at a rapid pace, as if the molecular maintenance mechanisms responsible for their repair and replacement had suddenly received a command to cease operation. Female menopause provides an unambiguous example of an age-associated physiological change that is genetically programmed and hormonally controlled. The paragraphs below describe several current theories regarding deterministic, programmed mechanisms for controlling aging and death.
Metabolic Theories of Aging: “The Brighter the Candle, the Quicker it Burns”
One of the many variants of the famous quote attributed to the ancient Chinese philosopher Lao Tzu summarizes the salient features of metabolic theories of aging. Its origins can be traced to the observation that the larger members of the animal kingdom tend to live longer than the smaller ones (Table 54-4). Reasoning that the causal basis for this correlation lay in something connected with size, rather than size itself, many scientists focused their attention on the organ most closely associated with life and vitality—the heart. In general, the resting heart rate of small animals such as hummingbirds, 250 beats per minute, tends to be higher than those of large animals such as whales, 10-30 beats per minute. Estimates of the cumulative number of times each vertebrate animal’s heart beat over the course of a lifetime exhibited an amazing convergence on 1.0 × 109 beats: one billion.
TABLE 54–4 Life Span Versus Body Mass for Several Mammals
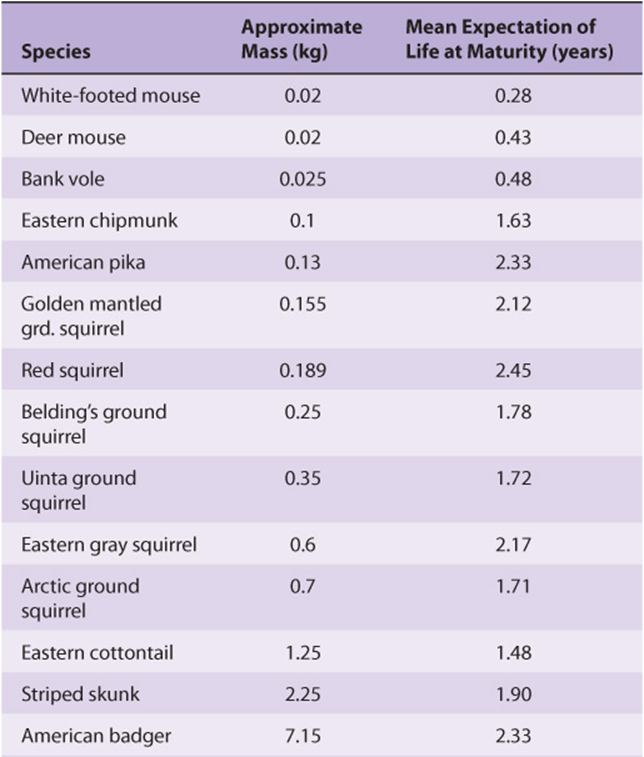
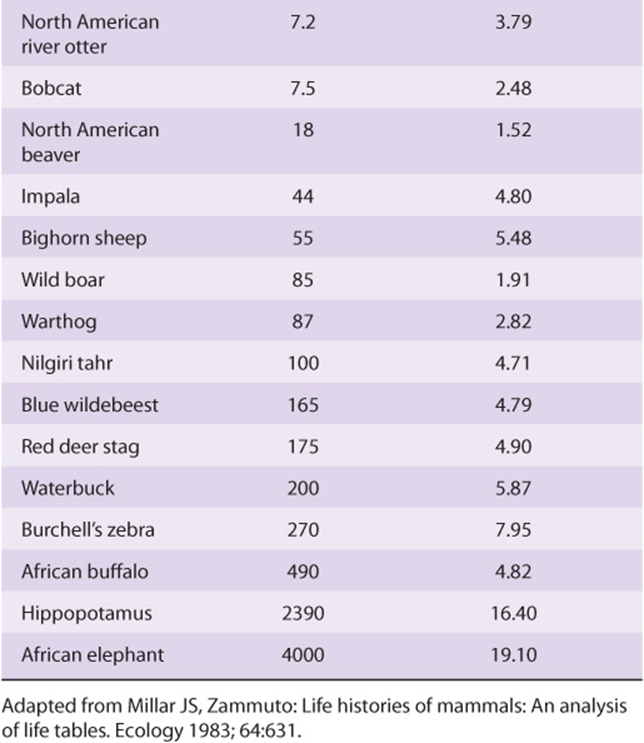
The so-called heartbeat hypothesis posited that every living creature is capable of performing only a finite number of heartbeats and/or breaths. A more nuanced variation of this basic idea, variously referred to as the metabolicor rate of living hypothesis, was put forward by Raymond Pearl in the 1920s. Pearl proposed that an individual’s lifespan was reciprocally linked to their basal metabolic rate. In other words, those who “burned the candle at both ends,” so to speak, burnt out sooner. A new round of calculations revealed that, while animals differ markedly in size, longevity, and heart rate; over their lifetime each expends a similar amount of total metabolic energy per unit body mass, 7 × 105 J/g. While intuitively appealing, a mechanistic link between lifespan and body size and energy expenditure or metabolic rate has proven elusive. Adherents of the mitochondrial theory of aging suggest that what is being “counted” is not heartbeats or energy, but the ROS that are the by-product of respiration. Over time the continued generation of energy and related consumption of O2 consumed leads to the accumulation of ROS-induced damage to DNA, proteins, and lipids until, eventually, a tipping point is reached. The rationale behind calorically restricted diets as a means to prolong life is based on the rationale that burning fewer calories will lead to a concomitant reduction in the production of damaging ROS.
Telomeres: A Molecular Countdown Clock?
A second school of thought holds that the putative countdown clock that controls aging and lifespan does not sense heartbeats, energy, or ROS. Rather, it uses telomeres to track the number of times each somatic cell divides.
Telomeres are composed of long strings of GT-rich hexanucleotide repeats that cap the ends of eukaryotic chromosomes. Unlike the closed circular DNA of bacterial genomes, the genomic DNA of eukaryotes is linear. If left unprotected, the exposed ends of these linear polynucleotides would be available to participate in potentially carcinogenic genetic recombination events. A second function of telomeres is to provide some disposable DNA to accommodate the wastage that occurs when linear DNA molecules are replicated.
This wastage is a consequence of the fact that all DNA polymerases work unidirectionally, 3′ to 5′ (Chapter 35). While in closed circular DNA this is not a problem, when trying to replicate the 5′ ends of a linear double stranded DNA via discontinuous 3′ to 5′ synthesis and ligation of small Okazaki fragments, there is simply not enough room at the end to accommodate the small RNA primer, polymerase, etc. Synthesis of the 5′ end of each strand will generally fall 100 bp or more short. Each time a cell divides, its genome will be shortened further (Figure 54–7). The telomeres provide an innocuous source of DNA whose decreasing length is of little consequence to the cell. However, once the supply of telomere DNA is exhausted, roughly 100 cell divisions for humans, all mitosis ceases and the somatic cell enters a state of replicative senescence. As more and more cells within the body enter senescence, it gradually loses the capacity to replace lost or damaged cells.
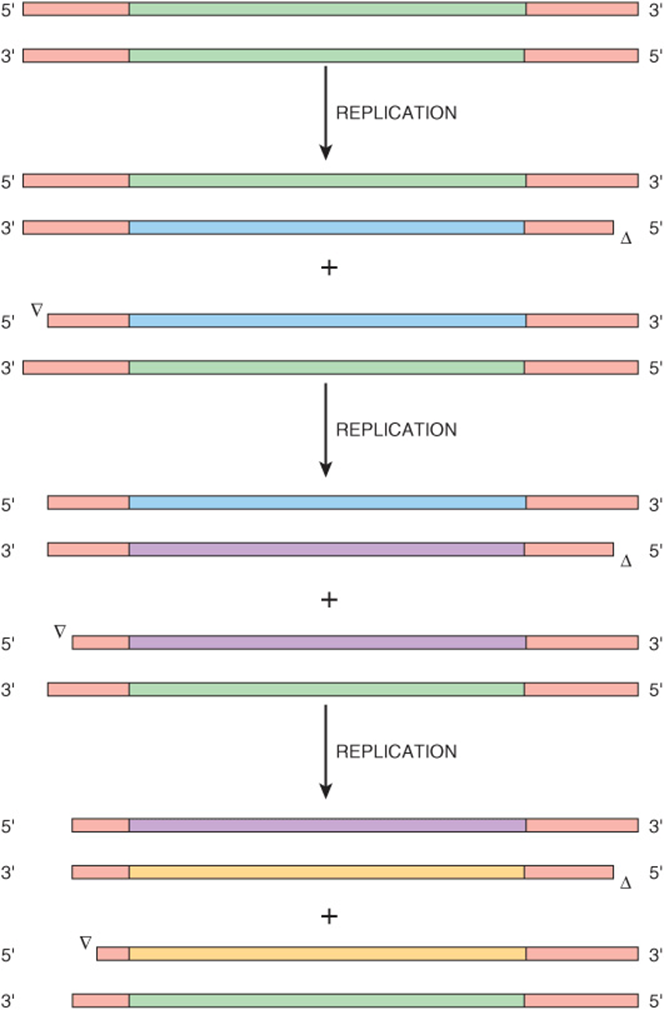
FIGURE 54–7 The telomeres at the ends of eukaryotic chromosomes progressively shorten with each cycle of replication. Shown is a schematic diagram of the linear DNA of a eukaryotic chromosome (green) containing telomeres at each end (red). During the first replication, new DNA strands are synthesized (green) using the original chromosome as template. For simplicity, the next two replication cycles (purple, yellow) show the fate of only the lower of the two nucleotide products from the preceding replicative cycle. Open arrowheads denote the site of incomplete strand synthesis. The model assumes that the single strand overhangs at the ends of each chromosome are trimmed at the completion of each cycle of cell division. Note the progressive shortening of the telomere repeats.
Organisms are able to produce progeny that contain full-length telomeres thanks to the intervention of the enzyme telomerase. Telomerase is a ribonucleoprotein that is expressed in stem cells and most cancer cells, but not in somatic cells. Using an RNA template, telomerase adds GT-rich hexanucleotide repeat sequences ranging from a few hundred (yeast) to several thousand (humans) nucleotides in length to the ends of linear DNA molecules to restore their telomeres to full length. When somatic cells are genetically engineered in the laboratory to express telomerase, they continue to divide in culture long after an unaltered control cell line stops dividing. The ability to prevent replicative senescence using an enzyme that maintains telomeres at full-length represents the most compelling evidence of the operation of a telomere clock.
Kenyon Used a Model Organism to Discover the First Aging Genes
Many advances in biomedical science are the product of research that uses a variety of so-called model organisms as their test subject. The fruit fly, Drosophila melanogaster, has yielded a rich harvest of information concerning the genes that guide cellular differentiation and organ development. Baker’s yeast and the African clawed frog Xenopus laevis, have served as the workhorses for dissecting the complex signal transduction circuitry that orchestrates the cell division cycle. A variety of cultured mammalian cell lines serve as surrogates for adipocytes, kidney cells, tumors, dendrites, etc. While at first glance it would appear that many of these model systems share little in common with humans, each possesses unique attributes that render them convenient vehicles for addressing certain problems and exploring specific systems.
Caenorhabditis elegans is a worm that has served as an important subject for the study of developmental biology. C. elegans is transparent and grows rapidly, attributes which facilitated tracing the entire developmental program for all 959 cells found in the mature adult to the fertilized egg. In the early 1990s, Cynthia Kenyon and colleagues observed that worms carrying mutations of the gene encoding an insulin receptor-like molecule, daf-2, lived 70% longer than their wild-type counterparts. Equally important, the mutant worms behaved in a manner resembling that of young wild-type C. elegans for much of this period. This is an important distinction. To qualify as an “aging gene,” its manipulation must accomplish more than merely prolonging old age by delaying the point at which life ceases. It must impact the schedule of changes associated with aging.
Investigation of further aging genes indicate they code for either one of a small set of transcription factors that include PHA-4 or DAF-16 that presumably control expression of aging critical genes, or signaling proteins such as DAF-2 that probably activate PHA-4, DAF-16, etc, in response to specific environmental signals. Much remains to be learned about the extent to which aging is controlled by genetic programming events, and how these gene products interact with the many other factors that influence vitality and longevity including nutrition, environmental stress, etc.
WHY WOULD EVOLUTION SELECT FOR LIMITED LIFESPANS?
The idea that animals would have evolved mechanisms designed specifically to limit their lifespan would appear, at first glance, to be highly counterintuitive. If the driving force behind evolution is the selection for traits that enhance fitness and survival, shouldn’t this translate into an ever-increasing life expectancy? While maximizing lifespan may represent a desirable trait from the point of view of the individual, it does not necessarily follow that this applies to a population or species as a whole. A genetically programmed limit on lifespan could benefit the group by eliminating the drain on available resources imposed by members no longer actively involved in the production, development, and training of offspring. Indeed, the current three generation lifespan can be rationalized as providing time (a) for newborns to develop into reproductively active young adults, (b) for these young adults to protect and nurture their offspring, and (c) to serve as a source of guidance and assistance to young adults facing the challenges of childbirth and childrearing.
SUMMARY
![]() Aging and longevity are controlled via the complex and largely cryptic interplay between random and deterministic factors that include genetic programming, environmental stresses, lifestyle, cellular countdown clocks, and molecular repair processes.
Aging and longevity are controlled via the complex and largely cryptic interplay between random and deterministic factors that include genetic programming, environmental stresses, lifestyle, cellular countdown clocks, and molecular repair processes.
![]() Wear and tear theories of aging hypothesize that the changes associated with old age and death itself reflect the accumulation of damage over time.
Wear and tear theories of aging hypothesize that the changes associated with old age and death itself reflect the accumulation of damage over time.
![]() The ubiquitous and life-essential environmental elements water, oxygen, and light possess an intrinsic capacity to damage biological macromolecules.
The ubiquitous and life-essential environmental elements water, oxygen, and light possess an intrinsic capacity to damage biological macromolecules.
![]() ROS such as hydroxyl radical and superoxide are particularly problematic as they are highly reactive, often participate in chain reactions that multiply their impact, and are continually generated as a by product of the complex network of redox reactions taking place in the electron transport chain.
ROS such as hydroxyl radical and superoxide are particularly problematic as they are highly reactive, often participate in chain reactions that multiply their impact, and are continually generated as a by product of the complex network of redox reactions taking place in the electron transport chain.
![]() The reactivity of their unsaturated ring systems and ability to absorb UV light render the nucleotide bases of DNA particularly vulnerable to UV or chemical damage.
The reactivity of their unsaturated ring systems and ability to absorb UV light render the nucleotide bases of DNA particularly vulnerable to UV or chemical damage.
![]() Mutations resulting from errors caused by missing or chemically modified nucleotide bases can be particularly harmful, as they may result in oncogenic transformation or render a cell vulnerable to further damage.
Mutations resulting from errors caused by missing or chemically modified nucleotide bases can be particularly harmful, as they may result in oncogenic transformation or render a cell vulnerable to further damage.
![]() Mitochondria occupy a central place in many theories of aging and death. This prominence can be attributed to several factors. Mitochondria are the site of the electron transport chain, by far the largest source of ROS in the cell.
Mitochondria occupy a central place in many theories of aging and death. This prominence can be attributed to several factors. Mitochondria are the site of the electron transport chain, by far the largest source of ROS in the cell.
![]() The efficient production of ATP is essential to cell vitality. Mitochondria play a central role in apoptosis, programmed cell death. Mitochondria lack the capacity to repair damage to their DNA.
The efficient production of ATP is essential to cell vitality. Mitochondria play a central role in apoptosis, programmed cell death. Mitochondria lack the capacity to repair damage to their DNA.
![]() In eukaryotic cells, long repeating sequences called telomeres cap the ends of their linear chromosomes. These telomeres progressively shorten each time a somatic cell divides. When a somatic cell’s telomeres become too short, it enters replicative senescence. Thus, telomeres are hypothesized to serve as a countdown clock for somatic cells.
In eukaryotic cells, long repeating sequences called telomeres cap the ends of their linear chromosomes. These telomeres progressively shorten each time a somatic cell divides. When a somatic cell’s telomeres become too short, it enters replicative senescence. Thus, telomeres are hypothesized to serve as a countdown clock for somatic cells.
![]() Animal lifespan may be genetically programmed. Mutation of the daf-2 gene in Caenorhabditis elegans yielded worms whose lifespan was 70% longer than wild type.
Animal lifespan may be genetically programmed. Mutation of the daf-2 gene in Caenorhabditis elegans yielded worms whose lifespan was 70% longer than wild type.
![]() Evolutionary selection of a limited lifespan optimizes the vitality of the species rather than that of its individual members.
Evolutionary selection of a limited lifespan optimizes the vitality of the species rather than that of its individual members.
REFERENCES
Aguzzi A, O’Connor T: Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Drug Discov 2010;9:237.
Anderson S, Bankier AT, Barrell BG, et al: Sequence and organization of the human mitochondrial genome. Nature 1981;290:457.
Arias E, Curtin LR, Wei R, et al: U.S. decennial life tables for 1999-2001, United States life tables. Natl Vital Stat Rep 2008;57:1.
Berlett BS, Stadtman ER: Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 1997;272:20313.
Clarke S: Aging as war between chemical and biochemical processes: Protein methylation and the recognition of age-damaged proteins for repair. Ageing Res Rev 2003;2:263.
Eisenberg DTA: An evolutionary overview of human telomere biology: the thrifty telomere hypothesis and notes on potential adaptive paternal effects. Am J Hum Biol 2011;23:149.
Kenyon CJ: The genetics of aging. Nature 2010;464:504.
Knight JA: The biochemistry of aging. Adv Clin Chem 2000;35:1.
Speakman JR: Body size, energy metabolism and lifespan. J Exp Biol 2005;208:1717.
Ulrich P, Cerami A: Protein glycation, diabetes, and aging. Recent Prog Horm Res 2001;56:1.