CHEMICAL BIOLOGY
Natural Products: An Overview
Jon Clardy and Michael A. Fischbach, Harvard Medical School, Massachusetts
doi: 10.1002/9780470048672.wecb377
Natural products, the remarkable collection of small molecules made by living organisms in an idiosyncratic manner, have made important contributions to organic chemistry, biology, and medicine. Although natural products contain a mother lode of molecular diversity, all of their structures reflect an evolutionary history and a biosynthetic production from a limited set of starting materials and reactions. The structures of natural products differ quantifiably from the small molecules usually found in the screening libraries used in high-throughput assays. Deeper structural analysis often reveals that natural products bind important biological targets with an exquisite degree of shape and charge complementarity. In addition, natural products often employ a variety of stratagems to disguise reactive functional groups until they are needed. Biosynthetic pathways for natural products resemble those used to construct more common biomolecules; the main themes are reactions that couple monomers into larger molecules and the lavish use of redox chemistry not just to create skeletons but also to cross-link them and decorate them with oxygen-based functionality. Although the historic contributions of natural products to chemistry, biology, and medicine are clear, their current use, especially in drug discovery, seems less clear. However, efforts to explore new areas of biological diversity, mine genomes, and study the natural roles of natural products are rejuvenating natural products research and illuminating its future.
The study of natural products was a paradigm for chemical approaches to biological problems long before the field of chemical biology originated. These small molecules, especially those with pronounced biological activity, not only spurred the development of organic chemistry as early chemists tried to discover the basis of their remarkable properties, but also they connected organic chemistry to biology and medicine. Virtually all of our early medicines—and a significant fraction of today’s therapeutic agents—had their origin in natural products (1). This overview describes the distinguishing features of these small molecules, explores how this traditionally organic chemistry-based field was transformed by insights from genetics and biochemistry, and highlights some promising future directions.
What are Natural Products?
“Natural product” is such a familiar term to most chemists and biologists that it is easy to forget that is describes a group of compounds with breathtaking molecular diversity and biological activity. Two natural products easily can seem to have little, if anything, in common, so what distinguishes natural products from other small molecules? In the past, describing a natural product was fairly straightforward: It was an organic molecule obtained from a living (or formerly living) organism that had a limited distribution in nature. Biosynthetic production and sporadic occurrence were the key defining features. But as we have learned more about natural products, this seemingly clear definition has become more ambiguous as illustrated by three molecules: tryptophan, psilocin, and serotonin (Fig. 1). All prokaryotes and some eukaryotes biosynthesize tryptophan, and all organisms incorporate tryptophan into their proteins. Its widespread distribution excludes it from being a natural product, and it is usually called a primary metabolite. Psilocin, which is biosynthesized from tryptophan, is best known as the hallucinogenic principle of “magic mushrooms,” and its limited distribution in a group of small mushrooms, not to mention its role in pre-Columbian religious rituals in Mexico, make it a classic natural product (2). Serotonin, which is also biosynthesized from tryptophan, is a well-known human neurotransmitter, and its regulation is an important therapeutic target for depression and other conditions. Psilocin and serotonin bind some of the same receptors in the human brain, although to very different effect (3). But many organisms, including bacteria and plants, make serotonin for various purposes, and its classification depends on context. In humans, it is a neurotransmitter; in barnacle larvae looking for a place to settle, it is a natural product (or settling pheromone); and the difference has more to do with the scientists conducting the study than the molecule being studied (4). Is there a fundamental difference between these three molecules that have very similar structures, share large parts of their biosynthetic pathways, and bind common cellular targets? In addition to the ambiguities introduced by the limited-distribution test, our increased understanding of natural products has made even “coming from a living organism” ambiguous. Is the product of a biosynthetic pathway reconstituted in a test tube rather than a living organism a natural product? If a pathway found in nature were altered in some way, say by introducing a different enzyme for one step, then does it still produce a natural product? If enzymes from many different pathways are combined, then does this new pathway produce a natural product? It seems likely that in the future, the term “natural product” will embrace any small molecule produced by a (genetically encoded) biosynthetic pathway whether the pathway is common or rare in nature or even created in the laboratory.
Even the usual classifications of natural products by their producing organism—plant, microbial, and marine natural products are the major divisions—are breaking down under genetic insights. The important plant natural products podophyllotoxin (the basis of the anticancer agent etoposide) and camptothecin (from which the anticancer agents topotecan and irinotecan are derived) were each shown to be made by a fungus that lives inside its plant host (5-7). Similarly, patellamide, a potential anticancer agent from an ascidian, is actually made by a cyanobacterium living inside the ascidian (Fig. 1) (8). The discovery of these hidden producers, along with other developments, has greatly increased the focus of contemporary natural products research on microbial sources.
The Structures of Natural Products
At the most fundamental level, natural products differ from other small molecules because of their evolutionary history. Natural products—or, more accurately, the genes that produce them—have undergone countless rounds of alteration, selection, and amplification that have honed their biological activities to better fulfill evolutionary selection criteria. This evolutionary reworking of their structural features has created what one recent commentator described as “the extraordinary advantages of small molecule natural products as sources of agents, which interject themselves in a helpful way in various physiological processes” (9).
Synthetic molecules, especially those prepared by medicinal chemists, also have selection criteria, but they usually involve synthetic accessibility, drug-like properties, or elaborations on a successful model. Whereas natural products generate their molecular diversity from a relatively limited set of starting materials and reaction types, as will be discussed shortly, synthetic molecules can stem from much larger sets of starting materials and reactions. Several studies have compared natural products and synthetic molecules and concluded that natural products contain relatively more carbon, hydrogen, and oxygen and less nitrogen and other elements than synthetics (10). In contemporary synthetic molecules, drug-like properties (how they are absorbed, distributed, metabolized, and eliminated) are enhanced by having molecular weight cutoffs (500 daltons is popular), limited hydrogen bonding potential, and solubility criteria (11). Most natural products actually conform to these rules, but notable exceptions of clinically useful compounds that greatly exceed the molecular weight guideline and have greater water solubility than recommended can be found (Fig. 2).
For any small molecule to possess sufficient binding energy to be a potent ligand (a dissociation constant in the 10-7 to 10-9 M range), its structural features must organize the molecule’s electrostatic, hydrogen-bonding, and hydrophobic interactions to complement those of its binding partner. Successful natural and synthetic molecules achieve these conformationally restricted structures in rather different ways. Natural products often achieve this rigidification by forming macrocycles, having polycyclic structures, and minimizing gauche interactions (Fig. 2). Synthetic molecules favor possessing aromatic rings and minimizing the number of rotatable bonds. Natural products generally have more sp3-hybridized carbons and stereogenic centers (Fig. 2). Morphine, a molecule with an ancient if occasionally troubled involvement with humans, illustrates the exquisite level of three-dimensional conformational control to project hydrophobic surfaces and hydrogen bonding partners in all directions that can be achieved by natural products. In more elaborate comparisons with many molecular parameters, natural products and synthetics occupy rather different regions of “chemical space.”
A less appreciated aspect of natural products is their extraordinary repertoire of strategies employed to carry out their biological roles. They are, for example, masters of disguising reactive functionality. Sometimes the disguise can be as simple as converting a reactive aldehyde into an intramolecular hemiacetal, but it can also involve much more elaborate schemes. Some spectacular examples of this “wolf in sheep’s clothing” approach can be found in a structurally diverse family of natural products called DNA-targeting agents. These molecules must move through a biological milieu—including the cell and nuclear membranes—with many molecules that are much more reactive than DNA and then damage DNA beyond easy repair. Calicheamicin initiates a complex set of reactions to carry out this destructive mission by unmasking a thiol, which undergoes an intramolecular conjugate addition, which sets up the aromatization of the enediyne fragment, which generates an aromatic diradical, which damages both DNA strands (Fig. 2) (12). Although calicheamicin’s molecular gymnastics might seem unduly complicated, calicheamicin linked to an antibody that directs it to cancer cells has shown therapeutic efficacy as an anticancer agent in several clinical trials (13). Not all DNA-targeting agents rely on masking their reactivity. CC-1065 uses shape complementarity with specific DNA sequences to enhance its reactivity to carry out a disabling alkylation (Fig. 2) (14). It accelerates the reaction in the appropriate biological context through the exquisite alignment of the reacting partners.

Figure 1. What are natural products? Tryptophan and two molecules derived from it are shown. Psilocin is a natural product, while serotonin's categorization depends on the context in which it is studied. Podophyllotoxin and camptothecin are natural products from trees and patellamide C is from an ascidian; however, all three are actually produced by a microbial endosymbiont.

Figure 2. The structures of natural products. The antibiotic vancomycin, the analgesic morphine, the hormone estradiol, the immunosuppressant rapamycin, and the DNA damaging agents CC-1065 and calicheamicin are shown. The synthetic cholesterol-lowering drug atorvastatin is shown for comparison. The electrophilicity of the cyclopropane ring in CC-1065 (indicated by the block arrow) is activated by a change in the torsional angle of the indicated bond that occurs upon DNA binding. The activation sequence of calicheamicin is shown at the bottom; the result is a reactive aromatic diradical that damages both strands of DNA.
Biosynthetic Pathways
Biochemistry has revealed nature’s two main themes in carrying out the chemistry of life: 1) coupling reactions that join similar (or identical) small molecules into the larger molecules needed to store information, form cellular structures, and carry out catalytic functions and 2) redox reactions such as the largely oxidative reactions our bodies use to get energy from small molecules and the largely reductive reactions our bodies use to build up small molecules. Not surprisingly, the biosynthesis of natural products reprises these two reaction types. Although many natural product biosynthetic pathways employ a single theme—either coupling or redox chemistry—some use both. These two-stage pathways typically begin with the coupling-mediated assembly of a multimer followed by the redox-mediated maturation of the initially formed product. In these hybrid pathways, redox-mediated maturation reactions decorate the core structure assembled by the coupling pathway. Nature also uses redox reactions to build core structures, an approach rarely employed for the same purpose in laboratory synthesis. These general observations are expanded in the examples below.
Coupling reactions
A coupling reaction, broadly defined, joins two fragments with the accompanying loss of a small molecule (or ion). In one well-known coupling reaction, the nucleophilic amino group of one amino acid reacts with the electrophilic carboxyl carbon of a second amino acid to form an amide bond with the loss of water (Fig. 3). Proteins, RNA/DNA polymers, lipids, and polysaccharides are all produced by different monomer-joining coupling reactions. Major classes of natural products—terpenes, polysaccharides, nonribosomal peptides, and polyketides—are also produced by coupling reactions, most of which feature a phosphate in some form as the ejected fragment. The details of the coupling reaction for these different classes vary depending on the subunits to be joined and the nature of the ejected fragment. The ejected fragment is a good leaving group, meaning that the bonds being formed are stronger than those being broken, and its release provides an entropy compensation for joining the two substrates.
Terpenes are formed by coupling double-bond isomers of the same momoner, isopentenyl pyrophosphate (IPP), with loss of a pyrophosphate ion (15, 16). Three such coupling reactions join four IPPs, each with five carbon atoms, to make geranylgeranyl-pyrophosphate, the C20-precursor of diterpenes such as taxol (Fig. 3). Linear terpene precursors, which differ only in the number of IPP units joined, can be assembled by a few polyprenyl synthase enzymes, as the monomers are identical and the only variable is how many monomers to link together. In subsequent steps, these linear polyenes undergo cation-induced cyclizations to create the starting frameworks for terpenes and their derivatives.
The coupling reaction used to build polysaccharides, such as the sugar fragment in vancomycin (Fig. 2), from hexose and pentose monomers is conceptually similar to the terpene pathway, except the leaving group is a nucleoside diphosphate (a ribosylpyrophosphate) rather than inorganic pyrophosphate, and the sugars to be joined can have considerable variety. This variability in the substrates requires a larger set of coupling enzymes, or glycosyltransferases, each of which selects specific glycosyl donor and acceptor cosubstrates (17). Genomic analyses highlight the consequences of this increased specificity. Streptomyces avermitilis, a bacterium with a completely sequenced genome (18, 19), has three times as many predicted glycosyltransferases than predicted polyprenyl synthases.
The amino acid monomers of nonribosomal peptides (20) like vancomycin (21) (Fig. 2) are also linked using a phosphate-displacing coupling reaction with an adenosine monophosphate (AMP) leaving group. Unlike terpene and polysaccharide biosynthesis, the overall coupling process has two distinct reactions connected by a stable intermediate (Fig. 3). In the first reaction, the aminoacyl-adenylate is transferred to a “carrier protein” to which it is tethered through an aminoacyl thioester bond, and in the second reaction, the amine of an aminoacyl cosubstrate reacts with the thioester to form a peptide bond (22). Both reactions have good leaving groups—either AMP or a thiolate. This two-step process resembles the ribosomal assembly of peptides with the amino acid bound to a carrier protein by a thioester bond in the nonribosomal case or to a tRNA by an ester bond in the ribosomal case. Tethering substrates to intermediate carrier protein domains may be required for proper directionality of chain assembly, a role that is fulfilled by elongation factors during ribosomal peptide synthesis.
Nonribosomal peptide synthetases (NRPSs) function similarly to the ribosome in a second way: They act processively to build a peptide chain. In both cases, the amine group of the incoming aminoacyl monomer attacks the (thio)ester bond that links the nascent peptide to its carrier, which translocates the chain while elongating it by one monomer (22).
The polyketides (23) form the only major class of natural products that has monomer couplings that do not involve the loss of phosphate; like the nonribosomal peptides, the monomer couplings use a thioester intermediate. These molecules, which have biosynthetic pathways much like those of fatty acids, are assembled from the alpha-carboxylated two- and three-carbon metabolites malonyl-CoA and methylmalonyl-CoA (22). In these monomers, the alpha-carboxyl group is a caged form of carbon dioxide, and decarboxylation yields a thioester enolate that attacks a downstream thioester to form a carbon-carbon bond (Fig. 3). In this coupling reaction, decarboxylation not only provides the attacking nucleophile, but also it serves as the driving force for unidirectional assembly. Although the coupling reactions for polyketides and nonribosomal peptides differ, the enzymes that assemble them function by similar processive logic: The incoming monomer attacks the nascent chain, which translocates it while elongating it by one monomer.
Unlike many of today’s software designers, nature prioritizes compatibility across platforms, which leads to numerous molecules that are formed by hybrid pathways. Just like the lipoproteins, glycoproteins, and glycolipids from primary cellular pathways, hybrid natural products originate from variants of these chemically compatible coupling reactions in which a donor monomer of one type gets linked to an acceptor monomer of a different type. For example, vancomycin (Fig. 3) is a hybrid between a nonribosomal peptide and a polysaccharide in which the key bond is formed by a coupling enzyme that links the peptide residue 4-hydroxyphenylglycine to the hexose monomer UDP-glucose (24).

Figure 3. Biosynthetic pathways. (A) In the terpenoid coupling reaction, isomers of isopentenyl pyrophosphate are joined with the loss of pyrophosphate, leading to a linear intermediate that is cyclized to a terpenoid skeleton, as shown for the diterpene taxol. (B) In the polysaccharide coupling reaction, hexose and pentose monomers are joined with the loss of a nucleoside diphosphate, as shown for the epivancosaminyl-glucose disaccharide of vancomycin. (C) In the first step of the nonribosomal peptide coupling reaction, an aminoacyl adenylate is transferred to a carrier protein or 'thiolation' domain (denoted 'T') with loss of adenosine monophosphate. In the second step, this carrier protein-tethered aminoacyl group is coupled to the amine of an aminoacyl cosubstrate, forming a peptide bond, as shown for two residues in backbone of vancomycin. (D) In the polyketide coupling reaction, the loss of carbon dioxide from a two or three-carbon monomer yields a thioester enolate that attacks a carrier protein-tethered intermediate, forming a carbon-carbon bond as shown for the polyketone precursor of enterocin.
Redox reactions
Redox chemistry occurs in natural product biosynthesis in at least three ways: building core structures, cross-linking core structures, and adding functionality to core structures. Just as the coupling reactions in natural product pathways resemble those from macromolecular synthesis, redox transformations in natural product pathways have counterparts in primary metabolism, which indicates that the genes in natural product pathways and those in primary metabolic pathways have common ancestors. Examples to illustrate the distinctions between building cores, cross-linking cores, and decorating cores are discussed below.
Podophyllotoxin (Fig. 1) was the basis for a successful anticancer drug because of its ability to prevent tubulin polymerization, although etoposide, the successful drug developed from it, actually works by inhibiting topoisomerase II and thereby preventing DNA replication (25). Podophyllotoxin’s skeleton is assembled through a very common redox reaction, the one-electron oxidation of coniferyl alcohol and a coupling of two such radicals (26, 27) (Fig. 4). This basic structure then undergoes a reduction, aromatic and aliphatic hydroxylations, and modifications of its aromatic substituents to give podophyllotoxin (27). The initial one-electron oxidation of a phenolate anion is also used to build the skeleton of morphine (Figs. 2 and 4) (28).
An example of oxidative cross-linking takes place during vancomycin biosynthesis; three cytochrome P450 enzymes—which use the same heme cofactor as the cytochromes from the primary metabolic electron transport chain—cross-link aromatic rings in its scaffold to form its rigid cup-like shape (29, 30).
Cytochromes P450 that decorate cores with oxygen-based functionality are commonly found in biosynthetic pathways. For example, these enzymes hydroxylate the reduced polycyclic scaffold of the diterpene taxol and install functional groups that are required for target binding and increase its hydrophilicity (31).

Figure 4. Redox reactions in biosynthetic pathways. A one-electron oxidation of two coniferyl alcohol monomers is the initiating step in podophyllotoxin biosynthesis, and a similar one-electron oxidation plays a key role in the biosynthesis of the alkaloid morphine. Redox reactions are also used to crosslink nascent scaffolds, as shown for vancomycin, or to add oxygen-based functionality to reduced scaffolds, as shown for taxol.
Modifying naturally occurring pathways
The most frequently encountered natural product biosynthetic pathways—those producing polyketides, nonribosomal peptides, polysaccharides, and terpenes—are all highly “evolvable” because they have a biosynthetic logic that is highly susceptible to small changes and rearrangements. Francois Jacob, one of the fathers of molecular biology, described these changes as “molecular tinkering” to emphasize the way nature cobbles together a workable solution through small alterations and rearrangements of previously existing materials (32). The “evolvability” of natural product biosynthetic pathways takes on two forms: modularity, in the case of polyketides, nonribosomal peptides, and polysaccharides, and plasticity, in the case of terpenoids. Researchers have taken advantage of these same features to engineer new pathways that produce new products.
As mentioned above, polyketides are assembled processively by logic that resembles ribosomal peptide synthesis. But a fundamental difference is found: Whereas the ribosome is a “factory on wheels” that translocates along an mRNA template, polyketide synthases (PKSs) are multidomain enzymatic assembly lines that function both as the template and the synthesizer. Like assembly lines in factories, PKSs are modular, and each module (a set of domains) adds a single monomer to the growing polyketide chain. Researchers have exploited this modularity to alter or replace modules in the PKS that produces the backbone of erythromycin and have generated a library of “programmed” derivatives that differ at specific positions (33). A complementary approach, in which a library of artificial two-module PKSs was constructed, led to the production of new polyketide fragments and set the stage for future efforts to construct “synthetic” polyketide pathways from a standard set of biosynthetic building blocks (34). The former approach resembles the mutational component of the evolutionary approach used by nature, and efforts to introduce the component of selection are likely to improve the future prospects of “synthetic” pathway construction.
The coupling reaction described above for terpene pathways results in a linear C5n chain, whereas terpenoid natural products have a staggering array of shapes and topologies. The cyclic skeletons characteristic of terpenes are formed from these methylated precursors by enzymes called terpene cyclases (35). Terpene pathways are plastic in two ways: Relatively few mutations in these cyclase enzymes can lead to skeletons of different topology, and the redox enzymes that add oxygen-based functionality to these skeletons are often promiscuous. By exploiting the plasticity of terpene cyclases, seven mutants of the gamma-humulene synthase have been designed in which 1-5 amino acid substitutions led to the production of seven new products with a wide range of topologies (36).
The Future of Natural Products Chemistry
Although the historical contributions of natural products to the development of chemistry, biology, and medicine are clear, their role in the future development of these fields is cloudy. On the negative side, most large pharmaceutical companies have either greatly reduced or completely oxidized their natural product drug discovery programs; but on the positive side, researchers are finding new ecological niches containing dramatically new natural products, developing methods to mine genomic sequences for new natural products, and discovering new ways to connect natural products with ecology (Fig. 5).
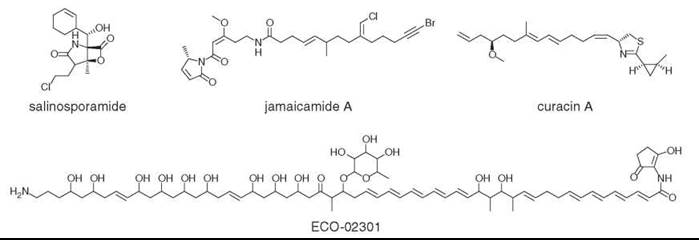
Figure 5. The future of natural products. Salinosporamide comes from a new genus of marine actinomycetes, while jamaicamide A and curacin A are produced by cyanobacteria. ECO-02301 was discovered by mining a bacterial genome.
New geographical and phylogenetic niches
Natural products chemistry has, like most things, evolved in fits and starts—periods of rapid advances followed by consolidation. The periods of rapid advance have always been associated with the opening up of new ecological niches to natural products research. In recent times, the opening up of the marine environment to natural products research spurred a few decades of truly heroic natural product discoveries. Marine natural products such as halichondrin B and ecteinascidin 743, analogs of which are in anticancer trials, were identified (37). Today, the most impressive remaining frontiers are largely microbial, and exploring the marine environment with suitably modified microbiological techniques has led to an increasing flow of molecules that are both novel and potentially useful. One example is the proteasome inhibitor salinsoporamide, which was isolated from the marine actinomycete Salinispora tropica CNB-440 (38-40). The recently sequenced genome of this organism reveals a wealth of new molecules, which highlights the potential of marine microenvironments as a source of molecular diversity and showcases the biosynthetic prowess of this new actinomycete genus (41). Likewise, the discoveries of new molecules like jamaicamide (42) and curacin (43) demonstrate the promise of marine cyanobacteria as a source of natural products (44). Some of the most impressive molecular diversity may not be found in the ocean but in the soil beneath our feet. Myxobacteria, the source of the tubulin-binding anticancer drug epothilone (45), are proving to be one of the richest sources of new natural products (46, 47).
Genome mining
The ability to connect natural products to the genes that produce them has had two important consequences. The first consequence was stimulating genetic and biochemical studies into natural product biosynthesis, which increased our knowledge of the inner workings of these pathways dramatically and set the stage for the modification of existing pathways and the construction of new pathways.
The second consequence was to create a new approach to natural product discovery based on bioinformatics. Instead of screening culture extracts to identify hits in an assay, a function-based approach, a research group scanned the genome of a bacterial strain to identify loci that encode natural product biosynthetic genes, a sequence-based search, and then isolated a new natural product from one of these loci, ECO-02301 (48). This effort was notable not only because the new molecule was identified by genomics rather than by screening but also because bioinformatics played an important role in accelerating the process of structure elucidation. Given the rapidly decreasing cost of genome sequencing and the increasingly powerful ability of bioinformatic analysis to predict natural product structure from gene sequence, genomics and bioinformatics are likely to play a more prominent role in future natural product discovery efforts.
Clues from ecology
The potential of natural products to serve as human medicines has always been the major driver of natural products research, and the relentless emphasis on their roles in treating human diseases has had the effect of largely separating natural products from the biological contexts in which they evolved and functioned. We know much more about the unnatural uses of natural products than we do about their intended uses. For example, the number of articles retrieved by searching for “taxol and cancer” (~9500) greatly exceeds the number retrieved by searching for “taxol and ecology” (three, two of which deal with the possible extinction of the taxol producer to make anticancer medications). Chemical ecology, the subfield of chemical biology that studies molecular interactions between organisms, places natural products in their evolutionary biological context, and natural products chemistry and chemical ecology have extensive overlaps. Both ecology and natural products chemistry are becoming increasingly focused on microbiology. As the great insect ecologist E. O. Wilson wrote in Naturalist, his autobiography, “if I could do it all over again and relive my vision in the twenty-first century, I would be a microbiologist” (49) As ecologists discover new types of microbes and study their roles in interspecies interactions, natural products chemists will acquire the producers of fascinating new molecules.
One example is the role of actinomycetous bacteria in maintaining an insect-fungal mutualism. Leafcutter ants, New World ants that strip foliage from plants, take the vegetation to large underground colonies where the partially chewed leaves are fed to a fungus (50). In this mutualistic relationship, the fungus eats the leaves, and the ants eat the fungus. Another fungus, which attacks the food fungus, poses a grave threat to the ant colony. Ants protect their food fungus in several ways, including carrying a symbiotic bacteria that produces a selective fungicide, a natural product that selectively kills the antagonistic but not the food fungus (51). Each of the 39 species of leafcutter ants has its own species of food fungus, its own species of antagonistic fungus, and its own symbiotic bacteria to make antifungal compounds with appropriate activity. It is likely that exploring this mutualistic relationship, and possibly other insect-bacteria mutualisms, will lead to a deeper understanding of the roles of microbes in maintaining symbioses, new natural products and their biosynthetic pathways, and possibly even new compounds with therapeutic efficacy.
References
1. Newman DJ, Cragg GM. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007; 70:461-477.
2. Hofmann A, et al. (Psilocybin, a psychotropic substance from the Mexican mushroom Psilicybe mexicana Heim.). Experientia 1958; 14:107-109.
3. Sard H, et al. SAR of psilocybin analogs: discovery of a selective 5-HT 2C agonist. Bioorg. Med. Chem. Lett. 2005; 15:4555-4559.
4. Yamamoto H, et al. Roles of dopamine and serotonin in larval attachment of the barnacle, Balanus amphitrite. J. Exp. Zool. 1999; 284:746-758.
5. Eyberger AL, Dondapati R, Porter JR. Endophyte fungal isolates from Podophyllum peltatum produce podophyllotoxin. J. Nat. Prod. 2006; 69:1121-1124.
6. Puri SC, et al. The endophytic fungus Trametes hirsuta as a novel alternative source of podophyllotoxin and related aryl tetralin lignans. J. Biotechnol. 2006; 122:494-510.
7. Puri SC, et al. An endophytic fungus from Nothapodytes foetida that produces camptothecin. J. Nat. Prod. 2005; 68:1717-1719.
8. Schmidt EW, et al. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:7315-7320.
9. Wilson RM, Danishefsky SJ. Applications of total synthesis to problems in neurodegeneration: fascinating chemistry along the way. Acc. Chem. Res. 2006; 39:539-549.
10. Feher M, Schmidt JM. Property distributions: differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003; 43:218-227.
11. Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001; 46:3-26.
12. Wolkenberg SE, Boger DL. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem. Rev. 2002; 102:2477-2495.
13. Stasi R, et al. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer Treat. Rev. 2008; 34:49-60.
14. Hurley LH, et al. Reaction of the antitumor antibiotic CC-1065 with DNA: structure of a DNA adduct with DNA sequence specificity. Science 1984; 226:843-844.
15. Cane DE. Enyzmatic formation of sesquiterpenes. Chem. Rev. 1990; 90:1089-1103.
16. Thulasiram HV, Erickson HK, Poulter CD. Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science 2007; 316:73-76.
17. Unligil UM, Rini JM. Glycosyltransferase structure and mechanism. Curr. Opin. Struct. Biol. 2000; 10:510-517.
18. Omura S, et al. Genome sequence of an industrial microorganism Streptomyces avermitilis: deducing the ability of producing secondary metabolites. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:12215-12220.
19. Ikeda H, et al. Complete genome sequence and comparative analysis of the industrial microorganism Streptomyces avermitilis. Nat. Biotechnol. 2003; 21:526-531.
20. Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides1. Annu. Rev Microbiol, 2004. 58:p.453-88.
21. Hubbard, B.K. and C.T. Walsh, Vancomycin assembly: nature’s way. Angew Chem. Int. Ed. Engl. 2003; 42:730-765.
22. Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem Rev, 2006; 106:3468-3496.
23. Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 2001; 18:380-416.
24. Lu W, et al. Characterization of a regiospecific epivancosaminyl transferase GtfA and enzymatic reconstitution of the antibiotic chloroeremomycin. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4390-4395.
25. Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. 46. Curr. Med. Chem. Anticancer Agents 2005; 5:363-372.
26. Buchanan BB, Gruissem W, Jones RL. Biochemistry & Molecular Biology of Plants. 2000. American Society of Plant Physiologists, Rockville, MD
27. Seidel V, et al. Biosynthesis of podophyllotoxin in Linum album cell cultures. Planta 2002; 215:1031-1039.
28. Zenk MH, Gerardy R, Stadler R. Phenol oxidative coupling of benzylisoquinoline alkaloids is catalysed by regio- and stereo-selective cytochrome P-450 linked plant enzymes: salutaridine and berbamunine. J. Chem. Soc. Chem. Commun. 1989: 1725-1727.
29. Woithe K, et al. Oxidative phenol coupling reactions catalyzed by OxyB: a cytochrome P450 from the vancomycin producing organism. implications for vancomycin biosynthesis. J. Am. Chem. Soc. 2007; 129:6887-6895.
30. Stegmann E, et al. Genetic analysis of the balhimycin (vancomycin-type) oxygenase genes. J. Biotechnol. 2006; 124:640-653.
31. Jennewein S, Croteau R. Taxol: biosynthesis, molecular genetics, and biotechnological applications. Appl. Microbiol. Biotechnol. 2001; 57:13-19.
32. Jacob F. Evolution and tinkering. Science 1977; 196:1161-1166.
33. McDaniel R, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc. Natl. Acad. Sci. U.S.A. 1999; 96: 1846-1851.
34. Menzella HG, et al. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat. Biotechnol. 2005; 23:1171-1176.
35. Bohlmann J, Meyer-Gauen G, Croteau R. Plant terpenoid synthases: molecular biology and phylogenetic analysis. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:4126-4133.
36. Yoshikuni Y, Ferrin TE, Keasling JD. Designed divergent evolution of enzyme function. Nature 2006; 440:1078-1082.
37. Simmons TL, et al. Marine natural products as anticancer drugs. Mol Cancer Ther, 2005; 4:333-342.
38. Fenical W, Jensen PR Developing a new resource for drug discovery: marine actinomycete bacteria. Nat. Chem. Biol. 2006; 2:666-673.
39. Jensen PR, et al. Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl. Environ. Microbiol. 2007; 73:1146-1152.
40. Feling RH, et al. Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus salinospora. Angew Chem. Int. Ed. Engl. 2003; 42:355-357.
41. Udwary DW, et al. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:10376-10381.
42. Edwards DJ, et al. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem. Biol. 2004; 11:817-833.
43. Chang Z, et al. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2004; 67:1356-1367.
44. Simmons TL, et al. Biosynthetic origin of natural products isolated from marine microorganism-invertebrate assemblages. Proc. Natl. Acad. Sci. U.S.A. 2008.
45. Bollag DM, et al. Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res. 1995; 55:2325-2533.
46. Wenzel SC, Muller R. Myxobacterial natural product assembly lines: fascinating examples of curious biochemistry. Nat. Prod. Rep. 2007; 24:1211-1224.
47. Schneiker S, et al. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat. Biotechnol. 2007; 25:1281-1289.
48. McAlpine JB, et al. Microbial genomics as a guide to drug discovery and structural elucidation: ECO-02301, a novel antifungal agent, as an example. J. Nat. Prod. 2005; 68:493-496.
49. Wilson EO, Naturalist. 1994. Washington, D.C.: Island Press. Currie CR. A community of ants, fungi, and bacteria: a multilateral approach to studying symbiosis. Annu. Rev. Microbiol. 2001; 55:357-380.
50. Currie CR, et al. Ancient tripartite coevolution in the attine ant-microbe symbiosis. Science 2003; 299:386-388.
Further Reading
Clardy J, Walsh C. Lessons from natural molecules. Nature 2004;432: 829-837.
Fischbach MA, Walsh CT. Assembly line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 2006; 106:3468-3496.
Dewick PM. Medicinal Natural Products, A Biosynthetic Approach. 2002. Chichester, UK: John Wiley & Sons Ltd.
Wyatt TD. Pheromones and Animal Behaviour. 2003. Cambridge, UK: Cambridge University Press.
See Also
Marine Natural Products, Chemical Diversity of
Chemical Diversity of Natural Products in Plants
Pharmaceuticals: Natural Products and Natural Product Models
Natural Products as Anticancer Agents
Chemical Ecology: An Overview