CHEMICAL BIOLOGY
Nature: A Model System for Chemists
Ronald Breslow, Department of Chemistry, Columbia University, New York, New York
doi: 10.1002/9780470048672.wecb378
Chemists have learned to imitate the principles of biologic chemistry in the quest to produce artificial enzymes, artificial receptors, and, ultimately, artificial living cells. The field of natural products chemistry is concerned chiefly with discovering the structures of the molecules produced in living systems as well as the paths by which they are produced. Biomimetic chemistry takes nature as a model and extends what exists in biology into what is possible. This process includes using hydrophobic effects in water to achieve selective reactions and making new amino acids and new nucleotides to generalize the properties of proteins and nucleic acids. Potent new catalysts are based on the chemistry performed by coenzymes using bifunctional acid-base catalysis and the water exclusion achieved by natural enzymes. Exciting new areas include molecular machines, mimics of the chemical senses, and work on the origin on earth of the homochirality observed in biomolecules. The studies using nature as a model have enriched chemistry greatly and furnished interesting insights into biochemistry.
Since the beginning of time, people have learned about the world by observing nature and by imitating aspects of what we have learned. This observation is true particularly of what we see in living creatures. For example, people observed birds and insects in flight and dreamed of being able to fly. Our most important clue was wings. We adopted this idea in the successful invention of airplanes. However, birds and insects power their flight by flapping the wings; this concept proved less useful. Nature has limitations, and one of them is that a powered propeller—and even more so a jet engine—were not practical for living creatures. Thus, we did not mimic birds slavishly; we adopted the central idea of their ability to fly and added ways to power the flight that nature could not achieve. As Philip Ball said (1): “A jumbo jet is not just a scaled up pigeon.”
Chemists have also taken many lessons and inspirations from natural chemistry; but, again, they have adapted their ideas so as not to be limited by the special requirements of natural chemistry. For example, nature designs proteins in three dimensions by folding linear polypeptide chains. This design indicates that genetic information is one dimensional, as is the sequence of nucleotides in genes. Although it might be interesting to use such a scheme in a mimic of an enzyme, chemists generally plan and then synthesize their three-dimensional structures, an option not open to nature.
Natural Products
In this brief article, I will focus on the ways we have learned from the chemistry of the life process itself. However, I must first call attention to another part of natural chemistry that has an effect on chemical thinking: the field called the Chemistry of Natural Products. Chemists have explored nature and have discovered several special types of chemicals: sugars, fats, terpenes, alkaloids, acetogenins, amino acids, heterocyclic coloring matters, macrocyclic lactones, steroid hormones, and so on. (2). These compounds have been produced by living organisms and they are secreted by or isolated from them. This field is concerned with the substances of nature and the biochemical processes by which they are formed.
Chemists have devoted much effort to exploring this natural world of chemistry as well as to determining structures; the natural world has stimulated the extension of the chemical world into models and analogs of the natural chemicals. The field of organic chemistry was influenced heavily by the types of chemical structures found in natural products; many medicinal compounds are still invented by using natural products as models for analogs. Chemists have also invented important polymers once nature showed us the natural polymeric carbohydrates, polypeptides, nucleic acids, and the polymers such as rubber that are produced from natural materials.
Biomimetic chemistry
The field concerned directly with imitating the chemistry of life processes has been called biomimetic chemistry or bio-inspired chemistry. The two terms differ in how close the mimic is to the natural process. As the author of this article, I will favor biomimetic, a word I coined (3).
Chemistry in water
A most important difference between human-made chemistry and living chemistry is that the latter occurs in water. Most chemical processes use organic solvents, which dissolve the components better; but water has special properties that are appreciated increasingly. Most significantly, the hydrophobic effect in water is the force that causes membranes, micelles, and liposomes to form, which pushes hydrocarbon chains together to diminish the high-energy hydrocarbon/water interface. The hydrophobic effect is also a major reason that hormones bind to receptors and substrates bind to enzymes, and it causes the folding of polypeptide chains into the three-dimensional structure of proteins by promoting the clustering of hydrocarbon side chains in the protein interior away from water.
Chemists now use water as a solvent for synthetic reactions, which takes advantage of the selectivities that the hydrophobic effect can induce (4). Many enzyme mimics (vide infra) also use water as the medium to promote substrate binding into the catalyst. Of course, water as a solvent also has important environmental advantages over volatile organic solvents. It remains to be seen how much impact the special properties of water as a solvent will have within chemical synthesis and manufacturing.
Molecular recognition and self-assembly
Small molecules such as hormones and allosteric effectors can bind to proteins selectively—including enzymes—and can modify their properties. Indeed, such binding is a common mechanism for the regulation of biologic effects; living cells do not simply run all processes as rapidly as possible. These natural processes have engendered a biomimetic field of molecular recognition and self-assembly, in which chemists develop such selective binding systems for analysis and as part of molecular devices. As mentioned above, medicinal chemists invent new molecules to bind to important receptors and enzymes, which imitate the natural ligands.
Proteins are assembled in living cells from amino acids, guided by the information in DNA that is passed on through RNA. Generally, chemists are inspired to generalize and extend such natural processes. A particularly interesting example is the synthesis of unnatural amino acids and the development of ways in which they can be incorporated into new kinds of proteins by modifying the biologic protein synthesis machinery (5). Some of these novel protein types promise to have new useful properties.
Nature appreciation
Another kind of generalization of natural chemistry involves viewing the natural chemistry in context by observing how well other related structures could perform the function of their natural models. In other words, why were these particular biologic molecules selected? A good example is the study of generalized forms of DNA.
In one example, some of the heterocyclic bases were modified drastically and were replaced by substituted benzene rings, and yet the base-pairing process of DNA was still observed (6). In other studies, the deoxyribose of DNA was replaced by other sugars, in some cases with an improvement in binding properties (7). As such, the 2-deoxyribose in DNA was replaced by the isomeric 3-deoxyribose to observe how the binding properties were affected (8). Interestingly, the resulting isoDNAs did not pair well with their isoDNA partners, or with the normal DNA partner that had the appropriate base sequences. This result clarified why 2-deoxyribose was selected for DNA even though the 3-deoxy isomer could be formed more easily under prebiotic conditions. However, the isoDNA base did pair well with its conjugate RNA, which furnishes insight into the difference between DNA-DNA pairing and DNA-RNA pairing (9).
The coenzyme thiamine pyrophosphate (1) plays a central role in many parts of metabolism (Fig. 1). Its mechanism of action involves the formation of a thiazolium zwitterion 2 that was stabilized by a carbene resonance form 3 (10). This discovery opened up studies of the chemistry that such “stabilized carbenes” could catalyze, as chemists realized that the otherwise impossible chemistry that thiamine pyrophosphate catalyzes in nature could be generalized and adapted for useful synthetic processes.
However, another study was an example of nature appreciation—the structure of thiamine was varied to learn what was special about the particular thiazolium derivative that was natural thiamine (11). As a chemical catalyst—ignoring the question of what effect changes would have on the ability of the coenzyme to bind to the proteins that have evolved to use it—thiamine proved to be the optimal relative to other related structures because of a balance of catalytic ability and chemical stability. The anion 4 derived from an imidazolium ring instead of a thiazolium ring was a weaker catalyst but was more stable in water (10). [The imidazolium anion and its dihydro derivative have proven to be very useful metal ion ligands, including in catalysts for olefin metathesis (12). This chemistry was developed as an outgrowth of the understanding of how nature uses thiamine in biology.]

Figure 1. Thiamine, vitamin B1, is the cofactor, as its pyrophosphate ester, for many important biologic reactions. These reactions involve the formation of an anion 2 that is stabilized by resonance with a carbene form 3. The related species 4 and derivatives have been developed as important ligands for metal ions in chemical synthesis.
Enzymatic selectivity involves geometric control
Enzymes operate by different rules than we normally use in synthetic chemistry. In nonenzymatic reactions, the intrinsic reactivity of the substrate dominates the chemistry. If we want to reduce a ketone, an aldehyde group in the same molecule will be more reactive. To obtain selectivity for the ketone group, we would need to block the aldehyde somehow. Moreover, if we want to oxidize a saturated carbon to form an alcohol, usually we would find the product alcohol oxidizes more readily, so the final product would be a ketone or aldehyde. Also, if we want to oxidize an isolated saturated carbon, a carbon-carbon double bond better not exist in the molecule or it would oxidize more readily. Generally, our synthetic procedures involve blocking such side reactions in some way. Enzymatic reactions have no such problems.
In the enzyme that oxidizes lanosterol to convert it ultimately to cholesterol (Fig. 2), three unactivated methyl groups are oxidized, whereas two double bonds and a secondary carbinol are left untouched initially. This selectivity reflects geometric control in the enzyme, and it is perhaps one of the strongest lessons that chemists have learned from nature. The enzyme binds the substrate in such a position that the oxidizing group of the enzyme, an iron oxo species, can reach the hydrogens on the inactivated methyl groups but cannot reach the more reactive double bonds or secondary carbinol (it is oxidized at some point later to permit decarboxylations, but then reduced again).
This general principle has inspired chemists to create enzyme mimics that perform biomimetic reactions that are also directed by geometric control (13). Selective attack on particular carbons of steroid substrates occurs as a function of the geometry imposed by binding, as in the enzyme. In one example, a carbon-carbon double bond was left untouched whereas a saturated inactivated carbon was oxidized, as in natural enzymes (14).
Of course, enzymes use geometry to control not only positions of reaction in a substrate but also selectivity in the formation of chiral centers and selectivity among substrates. It is fair to say that chemists were inspired to develop ways to produce optically active compounds as single enantiomers by observing how nature produces single enantiomers of amino acids and sugars, and essentially every natural compound that can exist as two enantiomers. Again, the secret in enzymatic reactions is geometric control of the processes where the chiral centers are produced because of the geometry of the enzyme-substrate complex. Such enantiomeric selectivity is the target of much modern synthesis.
A particular example from our laboratory is observed in compound 5, in which a basic amino group held rigidly on a mimic of the coenzyme pyridoxamine phosphate is able to convert ketoacids to amino acids with high enantioselectivity (15) (Fig. 3). This reaction is modeled closely to the way in which transaminase enzymes achieve the same goal.
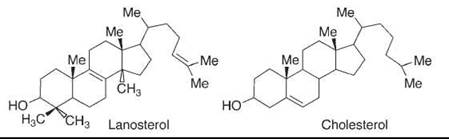
Figure 2. In the first steps of the conversion of lanosterol to cholesterol, an enzyme, cytochrome P-450, oxidizes the three methyl groups that will be removed while leaving the rest of the molecule untouched, because of geometric control that is typical in enzymes.
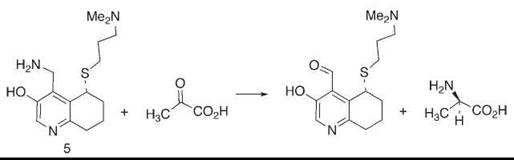
Figure 3. A mimic of the enzyme transaminase achieves high stereoselectivity in the product amino acid because of geometric control by the attached basic chain.
Enzymes can perform bifunctional acid-base catalysis
Many enzymes use coenzymes to achieve the detailed transformations they catalyze; but the enzyme proteins themselves also supply important elements of the catalysis. Enzyme proteins are the source of the entire catalytic effect when coenzymes are not involved. As one common process, acid and base groups in enzymes perform proton transfers that are critical to the catalytic mechanism. A particularly informative example is observed in the enzyme ribonuclease A, which catalyzes the cleavage of RNA (16). The catalytic process (Fig. 4) involves the imidazole ring of the amino acid histidine that removes the proton from the 2-hydroxyl of the ribose. A different protonated histidine transfers a proton to the RNA to promote the cleavage process. Studies with D2O-H2O mixtures established that the two proton transfers occur at the same time (17).
This reaction taught chemists the importance of simultaneous bifunctional catalysis, if it can be achieved. To avoid entropy problems, such bifunctional catalysts should have both catalytic groups in the same molecule, which are held in the correct position to participate in the simultaneous process. In ribonuclease A, the curling up of the peptide chain brings histidine-12 and histidine-119 close enough to let them participate in this way, even though they are separated by over 100 residues in the peptide chain. In a second step of the biologic process, the cyclic phosphate produced in the first step is hydrolyzed by the enzyme, again using the same two histidine side-chain imidazole groups.
We created a mimic of the cleavage of the cyclic phosphate by this enzyme by attaching two imidazole groups to a cyclodextrin molecule in well-defined positions (18). In water, the cyclodextrin bound a cyclic phosphate substrate—not, however, one derived from RNA—and performed the cleavage of the cyclic phosphate by using the two imidazoles as the enzyme does. That is, one functioned as a base, which delivered water to the phosphorus, and the other one, protonated, which functioned as a proton donor group in the hydrolysis reaction. The best catalyst in this process was one in which the proton was being delivered to the phosphate anion group, which formed a five-coordinate phosphorane intermediate. In a subsequent fast step, this went to the final hydrolysis product, with the proton ending up on the OH group that is formed in the hydrolysis.
We also examined the reaction rate in different mixtures of D2O and H2O, and we saw that the proton transfers were simultaneous, not sequential (19). Remarkably, the detailed data were almost identical with those observed in the enzyme process, which raised the question of whether the enzyme also used a mechanism proceeding through a five-coordinate phosphorane intermediate, rather than going to the ring-opened product directly. We examined another biomimetic system to explore this question.
We studied the reaction of a tiny fragment of RNA, uridyl-uridine 6, catalyzed by high concentrations of imidazole buffer (20) (Fig. 5). This buffer imitated the imidazole and imidazolium groups of the enzyme. In the enzyme, the effective concentration of these groups is high because of substrate binding right next to them. The products of the buffer-catalyzed reaction were uridine 7 and uridine cyclic phosphate 8, just as in an enzymatic cleavage process. However, we also saw that the buffer catalyzed an isomerization of the phosphate diester 6—originally attached to the 3-oxygen of uridine—to an isomer 9 in which it had migrated to the 2-oxygen.
The chemistry of phosphate reactions, and in particular the nonequivalence of the five groups in a phosphorane, requires that such a migration proceed through a phosphorane intermediate that undergoes pseudo-rotation to permit the migration (21). Our other kinetic studies made it clear that the cyclization process went through the same phosphorane intermediate.
We have proposed that the normal enzymatic hydrolysis process also proceeds through such a phosphorane intermediate, but isomerization does not occur because pseudo-rotation is not permitted in the enzyme (22). Complete agreement does not exist with our proposal (23). In this case, the biomimetic model system was not based only on nature’s enzyme, it pointed to possible aspects of the natural enzyme that had not been considered previously. Thus we had learned something from nature, and we returned the favor by learning something that could give insight into nature itself.

Figure 4. The enzyme ribonuclease A cleaves RNA using the imidazole group of histidine 12 as a base and the imidazolium group of histidine 119 as an acid in a simultaneous two-proton transfer process. Studies discussed in this article indicate that the process is not as simple as shown.
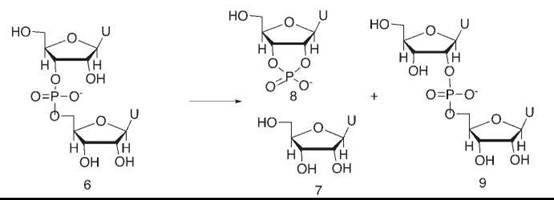
Figure 5. Imidazole buffer catalyzes both the hydrolysis of uridyl-uridine, a simple RNA fragment, and also its isomerization. This finding indicates that a phosphorane intermediate is involved, and it suggests that such a process may also be used by the enzyme ribonuclease itself.
Water exclusion by enzymes
Natural enzymes are large molecules, which raised the question of what particular advantages such large catalytic systems could have. This question stimulated Klotz and Suh to study large synthetic polyamines as enzyme mimics (24). The compounds, derived by polymerizing aziridine, are available commercially in various sizes and with different degrees of polydispersity. Klotz and Suh used a large polyaziridine with extensive cross-links, and in some cases they added alkyl chains to form a hydrophobic region. In his independent research, Suh pursued this area, examining other polymers as well such as polystyrene (25). Also, Hollfelder et al. examined this system quantitatively and concluded that there was a medium effect in the polyamines with added hydrocarbon chains (26).
Generally, their studies involved hydrolytic and fragmentation reactions. We took up the study of such systems as mimics for transaminase enzymes and showed with the polyaziridines that we achieve very large accelerations of the conversion of ketoacids to amino acids (27). This result reflected several ways in which these polyamines mimic enzymes.
First of all, transaminations involve many steps in which protons are added and subtracted from the reacting species and the reaction intermediates. In enzymes such as ribonuclease, normally imidazoles are used to perform such proton transfers. Their pKas are close to the operating pH for the enzyme, which makes them both the strongest base and the strongest acid that can exist in free form. A stronger base would be protonated by the medium and a stronger acid would be deprotonated. It can be shown that the catalytic result of increased acid or base strength does not make up for the loss of concentration of the free acid and base species.
The polyaziridines have their nitrogens so close to each other that they titrate over the range of pH 13 to pH 3, so they are half-protonated at pH 8 and have the strongest base and acid groups possible, just as in enzymes that use imidazole with a pKa near neutrality. Of course the polyaziridines achieve this not with special basic groups, just with the effect of neighboring charges that make it harder to add more positive charge.
We also saw that the potency of the polyaziridines as enzyme mimics was increased strongly when hydrophobic chains were added to some of the nitrogens (described in Refs. 24-27 and 28-30), which was evident in two ways. First of all, with the hydrophobic core that these chains produced, we achieved strong binding in water solution of ketoacids that carried hydrophobic components. The binding was much better, as reflected in a Michaelis Km, for indolepyruvic acid—that produced tryptophan on transamination—than for pyruvic acid that produced alanine, which is not surprising. Perhaps more interestingly, the hydrophobic chains increased the rate constants significantly for reaction of the complex (28), which reflects a medium effect that is invoked often for natural enzymes. Water is to some extent an enemy of rate because acids, bases, and substrates often need to lose their bound waters to react. In nature, this problem is solved by performing the reactions in the hydrophobic interior of the enzymes away from the external water. The hydrophobic chains in our enzyme mimic performed the same function, which excluded water from the reaction site.
The transformations themselves involved reactions of ketoacids with a pyridoxamine unit, either covalently attached to the polymer or reversibly bound to the hydrophobic core (29), which converted the ketoacids to amino acids, and the pyridoxamine was converted to a pyridoxal unit either covalently attached to the polymer or reversibly dissociated from the polymer. This reaction was modeled directly on the transamination process observed in natural enzymes. However, the second part of a full transamination in nature is the reaction of the pyridoxal with a different amino acid, which runs the transamination backward to form the pyridoxamine again while converting the new amino acid into its corresponding ketoacid. We found that such a process was too slow in our biomimetic system and could not compete with the rapid aldol condensation of the ketoacids with the pyridoxal.
To solve this problem, we used a mimic of a different enzyme, dialkylglycine decarboxylase (30). In this enzyme, pyridoxal phosphate reacts with an alpha-disubstituted glycine to perform an irreversible decarboxylation (Fig. 6) while converting the pyridoxal species to a pyridoxamine. We imitated this with our model transaminations using pyridoxal species that carry hydrophobic chains, and we were able to achieve as many as 100 catalytic turnovers. Thus, we could imitate one enzyme—the ordinary transaminases—by also imitating another enzyme that solved the turnover problem.
As we read more about disubstituted glycines, we learned that some such species are delivered by carbonaceous chondritic meteorites, and with partial enantioexcesses. This information offered a clue as to how biologic homochirality could have originated on earth (see below).
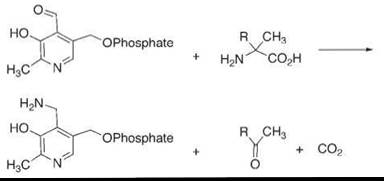
Figure 6. The mechanism used in the oxidative decarboxylation of alpha disubstituted glycines by an enzyme, which, in mimics, solved the problem of converting pyridoxal species to pyridoxamine species in biomimetic transaminase systems.
Cells as organized chemical systems
A living organism is more than just a collection of enzymes and other important species—it is an organized collection. The components of the cell are compartmentalized into organelles such as the nucleus, and the cell is structured additionally by internal and external membranes. This powerful type of system still waits to be fully mimicked.
Molecular machines
Living systems can convert chemical energy into mechanical motion. The most obvious examples are the motion in muscle contraction and the motion exhibited by flagella in bacteria and in sperm cells. These models from nature have stimulated a few chemists to try to mimic such properties with promising results (31,32). We do not yet know what practical benefits such mimics will bring, but the creation of molecular machines by imitating those in nature is an exciting adventure. It is part of the fundamental change that chemistry itself is undergoing. Instead of being concerned only with the properties of pure chemical substances, the chemistry of the future will be concerned with the properties of organized multimolecular systems. It is an exciting prospect, which is stimulated by our observation of the wonderful properties of nature’s organized multimolecular system: the living cell.
Energy from sunlight
Nature performs a process, called photosynthesis, that is of great interest to chemists and that has stimulated several attempts to model it. Many ideas about the future sources of energy on earth involve the large amount of energy that sunlight delivers. Fossil fuels—petroleum and coal—are derived from plants that stored chemical energy derived from sunlight and the animals that ate those plants. Although producing artificial photosynthesis is a fascinating challenge for chemists, it is not yet clear that it is a more practical way to obtain energy than “letting nature do it” with growing plants. However, the details of photosynthesis also inspire other approaches.
During photosynthesis, an electron of chlorophyll is pumped to a higher energy orbital by light absorption while a hole is created in the orbital that contained the electron previously. In photosynthesis itself, the excited electron produces the biologic reducing agent required for the chemistry of photosynthesis, while also generating phosphate anhydrides such as ATP. Meanwhile, the hole from a second chlorophyll—whose photoexcited electron fills the hole in the electron-deficient first chlorophyll—picks up an electron from the oxidation of water, which generates molecular oxygen. However, biologic photosynthesis requires water and carbon dioxide; water in particular is in demand for many other uses. Thus, it is more appealing for human energy generation from sunlight to use the combination of an excited electron and a hole to produce an electrical current, to use the photovoltaic effect. This process does not consume water, so it can be carried out in deserts without competing with agriculture. Many scientists are now trying to produce practical photovoltaic devices. Perhaps not always consciously modeled in nature, the processes do involve the same step as in photosynthesis—the photoexcitation of an electron.
The chemical senses
Among biologic organisms, the ability to detect specific chemicals in the environment is universal—to find food, to avoid danger, and to find receptive mates. In higher animals, this ability is lodged to some extent in the sense of taste, but it chiefly occurs in the sense of smell. One of the most baffling aspects of such chemical senses is the ability of animals to detect, selectively, substances that were created newly by chemists. How could the genetic code produce a specific receptor for every possible compound, with compounds numbering in the millions and with more continually being created? The solution to this puzzle by nature, brilliantly discovered by Axel (33) and Buck (34) in their Nobel Prize-winning work, was multiplexing. In principle, a finite group of receptors could react to a compound, but not all with the same strength of reaction. Thus a fingerprint was created, which reflects the different strengths of reaction of each receptor to the chemical. Every chemical, old or new, could elicit a different pattern of such signals and thus be perceived differently.
Eric Anslyn (35) has mimicked and adapted this principle for the detection and analysis of chemicals in a nonbiologic application. This principle promises the same ability to detect a great range of chemicals by using a multiplexed group of chemical detectors; but the different strengths of signals this group are transmitted to a computer, not to a brain.
The origin of life on earth
Nature has inspired chemists to mimic not just the details we understand, but also some important details about which we can only speculate. Chemists are making major efforts to understand how life could have begun on earth. Nature has presented us with the puzzle, in the form of biology as it now exists.
The L amino acids in proteins, the D carbohydrates, the nucleic acids, all are found to be homochiral, as observed as a single enantiomer. When the opposite enantiomers are found in biology, they are used for different functions, such as the D amino acids in some bacterial cell walls. Ordinary chemical reactions that create a new chiral center from optically inactive precursors normally produce a racemic mixture unless some chiral catalyst is present to direct the process. How did it get started before chiral catalysts such as enzymes were present?
Various theories exist, but one with considerable support takes note of the fact that some meteorites have brought to earth amino acids that are partially enantiomeric, with 3-15% enantiomeric excess of the L-amino acids (36). They are not normal amino acids, but they have a methyl group on the alpha carbon in place of the usual hydrogen so they cannot racemize. It is believed that they are formed as racemates in interstellar space, but then are deracemized partially by circularly polarized light emitted by synchrotron processes at neutron stars.
Chemists have been inspired to study how these partially enantiomeric “seeds” could induce the formation of normal biologic molecules—amino acids and carbohydrates—under credible prebiotic conditions ((36, 37), and unpublished work). Furthermore, chemists have been inspired to research how low levels of enantiopurity can be amplified into high levels—such as a 95 to 5 ratio of L-phenylalanine starting with only a 50.5 to 49.5 ratio—under credible prebiotic conditions (38, 39).
Such studies—and others attempting to understand how spontaneous chemistry in the primitive earth could have formed living cells—are not inspired directly by information about how life originated, but they are inspired by nature as we see it, which is populated as it is by life forms. The work does not aim to show how life originated, which is an historical question. However, it tries to develop the scientific evidence that can support a reasonable hypothesis of how it may have started.
Artificial life
Another important goal of modern chemistry is directly inspired by nature: the hope to produce artificial life, or at least some systems that share many of the attributes of life. The ultimate goal of biomimetic chemistry is to mimic life itself, not in the form that it now has but in alternate forms. Chemists have generalized so many types of substances—novel polymers, novel carbohydrates and amino acids, novel hormones and enzyme inhibitors—that to generalize natural chemistry is one of their most characteristic activities. Can they generalize life itself? Time will tell.
Other authors
A good fraction of modern chemistry, which includes medicinal chemistry, derives some of its inspiration from a consideration of nature as a model. It is impossible, in this brief article, to describe more than a fraction of the efforts. For additional work in the spirit of biomimetic chemistry, the following is a list of some authors not yet mentioned whose independent work should be consulted. Even here, this list is certainly not complete: Jacqueline Barton, Steven Benkovic, Albrecht Berkessel, Thomas Bruice, Jik Chin, Jean Chmielewski, E. J. Corey, Donald Cram, Peter Dervan, Francois Diederich, Mark Distefano, Jean Frechet, Samuel Gellman, John Groves, Andrew Hamilton, Donald Hilvert, Barbara Imperiali, Makoto Komiyama, Jean-Marie Lehn, Arthur Martell, Julius Rebek, Jean-Pierre Sauvage, Alanna Schepartz, Dieter Seebach, Donald Tomalia, and Steven Zimmerman.
References
1. Ball P. Stories of the Invisible: A Guided Tour of Molecules. 2001. Oxford University Press, Oxford, United Kingdom.
2. Nakanishi K, Goto T, Ito S, Natori S, Shigeo N, eds. Natural Products Chemistry. 1974. Academic Press, New York.
3. Breslow R. Biomimetic chemistry, centenary lecture. Chem. Soc. Rev. 1972; 1:553-580.
4. Breslow R. Hydrophobic effects on simple organic reactions in water. Acc. Chem. Res. 1991; 24:159-164.
5. Liu W, Brock A, Chen S, Chen S, Schultz PG. Genetic incorporation of unnatural amino acids into proteins in mammalian cells. Nature Meth. 2007; 4:239-244.
6. Krueger AT, Lu H, Lee AHF, Kool ET. Synthesis and properties of size-expanded DNAs: toward designed, functional genetic systems. Acc. Chem. Res. 2007; 40:141-150.
7. Eschenmoser A. Searching for nucleic acid alternatives. Chimia 2005; 59:836-850.
8. Dougherty JP, Rizzo CJ, Breslow R. Oligodeoxynucleotides that contain 2',5'' linkages: synthesis and hybridization properties. J. Am. Chem. Soc. 1993; 114:6254-6255.
9. Sheppard TL, Breslow RC. Selective binding of RNA, but not DNA, by complementary 2/,5//-linked DNA. J. Am. Chem. Soc. 1996; 118:9810-9811.
10. Breslow R. On the mechanism of thiamine action. IV. Evidence from studies on model systems. J. Am. Chem. Soc. 1958; 80:3719-3726.
11. Breslow R, McNelis E. Studies on model systems for thiamine action. Synthesis of reactive intermediates, and evidence on the function of the pyrimidine ring. J. Am. Chem. Soc. 1959; 81:3080-3082.
12. Vougioukalakis GC, Grubbs RH. Ruthenium olefin metathesis catalysts bearing an N-fluorophenyl-N-mesityl-substituted unsymmetrical N-heterocyclic carbene. Organometallics 2007; 26: 2469-2472.
13. Breslow R, Yang J, Yan J. Biomimetic hydroxylation of saturated carbons with artificial cytochrome P-450 enzymes—liberating chemistry from the tyranny of functional groups. Tetrahedron 2002; 58:653-659.
14. Yang J, Gabriele B, Belvedere S, Huang Y, Breslow R. Catalytic oxidations of steroid substrates by artificial cytochrome P-450 enzymes. J. Org. Chem. 2002; 67:5057-5067.
15. Zimmerman SC, Czarnik AW, Breslow R. Intramolecular general base-acid catalysis in transaminations catalyzed by pyridoxamine enzyme analogues. J. Am. Chem. Soc. 1983; 105:1694-1695.
16. Richards FM, Wycoff HW. Bovine Pancreatic Ribonuclease. The Enzymes, Volume 4. 3rd edition. 1971. Academic Press, New York. pp. 647-806.
17. Matta MS, Vo DT. Proton inventory of the second step of ribonuclease catalysis. J. Am. Chem. Soc. 1982; 87:5316-5318.
18. Anslyn E, Breslow R. Geometric evidence on the ribonuclease model mechanism. J. Am. Chem. Soc. 1989; 111:5972-5973.
19. Anslyn E, Breslow R. Proton inventory of a bifunctional ribonuclease model. J. Am. Chem. Soc. 1989; 111:8931-8932.
20. Breslow R, Xu R. Quantitative evidence for the mechanism of RNA cleavage by enzyme mimics. Cleavage and isomerization of UpU by morpholine buffers. J. Am. Chem. Soc. 1993; 115:10705-10713.
21. Breslow R. Kinetics and mechanism in RNA cleavage. Proc. Nat. Acad. Sci. U.S.A. 1993; 90:1208-1211.
22. Breslow R, Dong SD, Webb Y, Xu R. Further studies on the buffer-catalyzed cleavage and isomerization of uridyluridine. Medium and ionic strength effects on catalysis by morpholine, imidazole, and acetate buffers help clarify the mechanisms involved and their relationship to the mechanism used by the enzyme ribonuclease and by a ribonuclease mimic. J. Am. Chem. Soc. 1996; 118:6588-6600.
23. Herschlag D. Ribonuclease revisited. Catalysis via the classical general acid-base mechanism or a triester-like mechanism. J. Am. Chem. Soc. 1994; 116:11631-11635.
24. Klotz IM, Suh J. Evolution of synthetic polymers with enzyme-like catalytic activities. In: Artificial Enzymes. Breslow R, ed. 2005. Wiley-VCH. Weinheim, Germany, pp. 63-88.
25. Suh J. Synthesis of polymeric enzyme-like catalysts. Synlett 2001; 9:1343-1363.
26. Hollfelder F, Kirby AJ, Tawfik DS. Efficient catalysis of proton transfer by synzymes. J. Am. Chem. Soc. 1997; 119:9578-9579.
27. Liu L, Breslow R. Vitamin B6 enzyme models. In: Artificial Enzymes. Breslow R, ed. 2005. Wiley-VCH. Weinheim, Germany, pp. 37-62.
28. Liu L, Rozenman M, Breslow R. Hydrophobic effects on rates and substrate selectivities in polymeric transaminase mimics. J. Am. Chem. Soc. 2002; 124:12660-12661.
29. Liu L, Zhou W, Chruma JJ, Breslow R. Transamination reactions with multiple turnovers catalyzed by hydrophobic pyridoxamine cofactors in the presence of polyethylenimine polymers. J. Am. Chem. Soc. 2004; 126:8136-8137.
30. Chruma JJ, Liu L, Zhou W, Breslow R. Hydrophobic and electronic factors in the design of dialkylglycine decarboxylase mimics. Bioorg. Med. Chem. 2005; 13:5873-5883.
31. Kelly TR, Cai X, Damkaci F, Panicker SB, Tu B, Bushell SM, Cornella I, Piggott MJ, Salives R, Cavero M, Zhao Y, Jasmin S. Progress toward a rationally designed, chemically powered rotary molecular motor. J. Am. Chem. Soc. 2007; 129:376-386.
32. Pijper D, Feringa BL. Molecular transmission: controlling the twist sense of a helical polymer with a single light-driven molecular motor. Angew. Chem. Int. Ed. 2007; 46:3693-3696.
33. Axel R. Scents and sensibility: a molecular logic of olfactory perception (Nobel lecture). Angew. Chem. Int. Ed. 2005; 44:6111-6127.
34. Buck LB. Unraveling the sense of smell (Nobel lecture). Angew. Chem. Int. Ed. 2005; 44:6128-6140.
35. Anslyn EV. A marriage of supramolecular chemistry with pattern recognition. 232nd ACS National Meeting, San Francisco, CA, 2006.
36. Pizzarello S. The chemistry of life’s origin: a carbonaceous meteorite perspective. Acc. Chem. Res. 2006; 39:231-237.
37. Breslow R, Levine M. Partial transfer of enantioselective chiralities from alpha-methylated amino acids, known to be of meteoritic origin, into normal amino acids. Tetrahedron Lett. 2006; 48:1809-1812.
38. Breslow R, Levine M. Amplification of enantiomeric concentrations under credible prebiotic conditions. Proc. Nat. Acad. Sci. U.S.A. 2006; 103:12979-12980.
39. Klussman M, Iwamura H, Matthew S, Wells D, Pandya U, Armstrong A, Blackmond D. Thermodynamic control of asymmetric amplification in amino acid catalysis. Nature 2006; 441:621-623.
Further Reading
Breslow R. Studies in biomimetic chemistry. Pure & Appl. Chem. 1998; 70:267-270.
Breslow R. Imitating antibodies and enzymes. Proc. Robert A Welch Foundation 40th Conference on Chem. Res. 1997; 40:1-11.
Breslow R. Biomimetic chemistry and artificial enzymes: catalysis by design. Accts. Chem. Res. 1995; 28:146-153.
Breslow R. Binding and catalysis in water. Supramol. Chem. 1992; 411-428.
Breslow R. Hydrophobic effects on simple organic reactions in water. Accts. Chem. Res. 1991; 24:159-164.
See Also
DNA-Binding Molecular Motors
Enzyme Cofactors, Chemistry of
Life, Origins of
Organic Chemistry in Biology
Water, Properties of