CHEMICAL BIOLOGY
Nonhomologous Recombination
Thomas E. Wilson, University of Michigan Medical School, Ann Arbor, Michigan
doi: 10.1002/9780470048672.wecb396
Genetic recombination classically proceeds by the homologous recombination pathway, but direct joining of chromosome fragments lacking extensive homology also occurs and is of particular interest as a nearly ubiquitous genome alteration in cancer. At least two distinct but overlapping pathways of DNA repair are relevant to the execution of nonhomologous recombination: nonhomologous end joining (NHEJ) and microhomology mediated end joining (MMEJ). Like other DNA repair pathways, NHEJ and MMEJ seem to facilitate primarily genome maintenance, but by their nature are prone to errors, including an ability to join inappropriately paired chromosome break ends. This review considers the function of these pathways, with an emphasis on the unique and unusual biochemical properties of key enzymes, including DNA ligase IV and Pol X family DNA polymerases, and how these facilitate direct rejoining of broken chromosomes.
Genetic recombination is classically manifest as the reassortment of genes during meiosis, where extensive pairing of homologous chromosomes is associated with “crossover” between them to create new derivative hybrid chromosomes. The associated enzymatic pathway, homologous recombination (HR), is now known to execute such exchange not only during meiosis but also between homologous sequences in mitotic cells, for example between sister chromatids. Nonhomologous recombination (NHR), also called illegitimate recombination, was recognized initially and defined as genetic recombination that failed to show this canonical feature of HR, i.e., hybrid derivative chromosomes that lacked extended homology at the junction between parents. Instead, junctions often showed 1-10-bp segments of “microhomology” too short to be used by HR enzymes (Fig. 1) (1, 2). Such events seemed to defy the logic and benefits of HR in a chemically distinct alternative reaction that was, on its face, detrimental. In this review, the processes that underlie NHR are placed in context as DNA repair pathways that are in fact associated with genome preservation, but that can secondarily give rise to chromosome rearrangements and other mutations. The biochemical requirements of these reactions are discussed as well as current understanding of how the responsible enzymes have been adapted to meet these requirements.
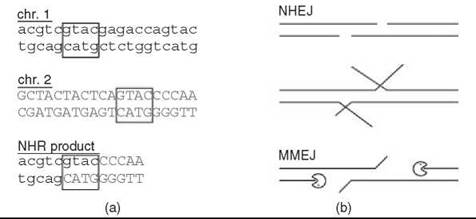
Figure 1. Microhomology use in NHR. (a) Demonstration of microhomology, which is evident as a sequence common to both chromosome substrates (boxed). Note that the position of initiating DSBs cannot be inferred from the product. (b) Possible mechanisms for generating microhomology. If DSB overhangs encompass a microhomology, it can be rejoined directly by NHEJ. Internal microhomologies must be exposed, either by unwinding, as suggested for DNA-PKcs (1), or by 5' resection, as suggested for deletion MMEJ (2).
Biological Background
As might be expected from the fact that both HR and NHR represent new joints between previously separate DNA duplexes, an intimate relationship exists between recombination and DNA double-strand breaks (DSBs) (Fig. 2) (3-5). Processes that give rise to DSBs not only underlie meiotic and mitotic HR, but also they predispose cells to NHR. Conversely, deficiency in cellular components required for both HR and NHR causes hypersensitivity to DSBs. Indeed, it may be most accurate from an evolutionary perspective to consider HR and NHR foremost as alternative mechanisms for silent repair of DSBs, with recombination as a secondary consequence. It is thus critical to consider the cell cycle-dependent interplay of homologous and nonhomologous mechanisms at a DSB as the stage on which NHR plays out.
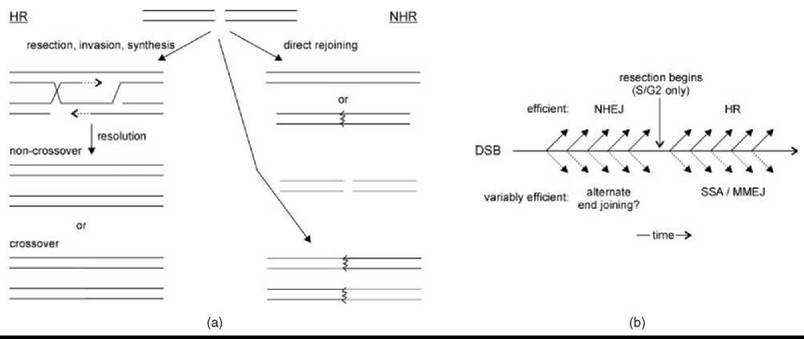
Figure 2. Interplay between HR and NHR. (a) When a DSB occurs (top), it can be resected and used to copy a donor chromosome (red) during HR (left) or rejoined directly (right), sometimes inaccurately (jagged line). HR and NHR lead to recombination by crossover resolution and engagement of a different DSB (green), respectively (bottom). (b) A timeline shows a period during which NHEJ may be iteratively attempted before DSB resection; after which, HR is attempted. Alternative pathways (below) might lead to rejoining, but these are often less efficient and uncommon when the above pathways are functioning normally.
Interplay between homologous and nonhomologous DSB repair
Cells constantly suffer DNA lesions that degrade the genome. Some DSBs are created deliberately and by specific mechanisms (see below), but many result from oxidative, radiation, or physical stresses that occur more randomly (4, 5) When acting as a DSB repair pathway, HR uses a homologous donor chromosome as a template to copy the genetic information required to bridge the broken ends without altering the template itself (Fig. 2a) (3, 4). Such a second intact copy of a chromosome is present in diploid cells, although in large, complex, and repetitive genomes, finding this potential donor is not trivial because chromosome pairs are not associated. An ideal donor exists when one of a pair of sister chromatids is broken, where the other sister is both physically associated and strictly homologous. This special relationship is strengthened by understanding that replication itself can lead to DSB formation. It is thus both predictable and observed that HR is a principal repair pathway in S or G2/M cell cycle stages after replication but before cell division (6, 7). Given that nonhomologous DSB repair processes are, by definition, not HR, it is in turn predictable and observed that such repair predominates at DSBs occurring randomly outside of replication (6, 7). But what is DSB repair in the absence of a homologous donor? Stated simply, nonhomologous repair is the direct rejoining of DSB ends (Fig. 2a) (5). The details of these mechanisms are addressed below, but simply recognizing that they are executed without an information template correctly predicts that they have a higher error rate. However, when HR is impractical or impossible because of inaccessibility of a homologous template, even error-prone rejoining of DSB ends will yield a relative preservation of the genome as compared with continuing cell division with a broken chromosome. Also, repair of simple DSBs by direct rejoining can be >99% accurate (8, 9), so that nonhomologous repair is often genetically silent.
A major difference between homologous and nonhomologous DSB repair is that HR begins with extensive resection of the 5' DSB strands, whereas NHR is comparatively duplex preserving. Mounting evidence indicates that the 5' resection is the regulated step in DSB repair pathway “choice” because it is an essentially irreversible commitment to HR (6, 8). Resection is under direct cell-cycle control, being activated by cyclin-dependent kinases, at least in yeast, and therefore largely inactive in G1 cells (6). Even in replicating cells, resection is delayed, approximately an hour in yeast, before it begins in earnest (10). The basis of these effects is poorly understood, mainly because the mechanism(s) of resection remain enigmatic. Regardless, their net effect is to provide a period during which nonhomologous repair of a DSB might be attempted, with transition to homologous repair of persistent breaks at a pace that is cell-stage dependent (Fig. 2b). The greatest opportunity for, and biologic relevance of, nonhomologous repair might thus be in nondividing cells. Multicellular organisms are composed mainly of terminally differentiated cells, where an error-prone DSB repair mechanism may be particularly tolerable. In single-cell organisms, periodic cell cycle exit is a natural part of life in the sometimes inhospitable natural environment. For example, some bacteria require nonhomologous DSB repair for resistance to desiccation (11). NHR-associated genome changes may even be desirable as an adaptive response to such stress (12).
Telomeres and breakage-fusion-bridge cycles
The ends of linear chromosomes are a type of “DSB” worth specific consideration. McClintock first recognized that specialized structures called telomeres protect chromosome ends (13, 14). A detailed description of this structure is beyond the scope of this article, but it involves a repeating DNA sequence and multiple proteins that bind to it, including DSB repair proteins (14). The key importance is that when this structure fails, because of loss of telomere proteins or degradation of the DNA repeat, chromosome ends become a DSB target for NHR mechanisms that can fuse sister chromatids by direct joining. The resulting dicentric chromosome is destined to be broken during mitosis if pulled toward opposite spindle poles, which leads to DSBs that might again lead to chromosome fusion, ongoing “breakage-fusion-bridge” cycles, and NHR before ultimate stable resolution (Fig. 3a) (13). These cycles underscore the potential complexity of the processes giving rise to observed NHR events.
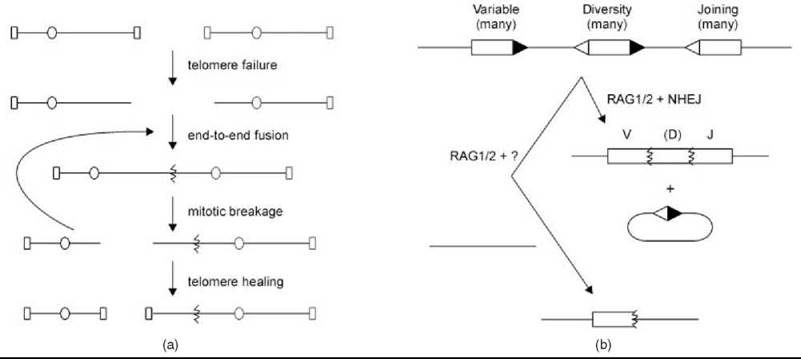
Figure 3. Special mechanisms of NHR. (a) Telomere failure unmasks chromosome ends for fusion by NHEJ, which leads to potentially iterative cycles of breakage and fusion before ultimate stable telomere resolution. Oval, centromere; rectangle, telomere. (b) Immune receptor loci contain multiple V, J, and sometimes D segments with signal sequences (triangles) that are cleaved by RAG1/2 and subjected to NHEJ to generate recombined products. Engagement of an inappropriate chromosome can lead to RAG1/2-dependent chromosomal rearrangement.
V(D)J recombination
Another important context for understanding NHR is a series of genome manipulations in the developing vertebrate immune system where nonhomologous repair mechanisms are co-opted to create deliberate mitotic chromosome rearrangements. In V(D)J recombination, specific variable, diversity, and joining sequences from a collection of such segments in the germline genome are juxtaposed in recombinant chromosomes to create functional immunoglobulin and T-cell receptor genes (Fig. 3b) (15, 16). Both by the reassortment of different coding segments and by maximizing the error-prone nature of nonhomologous junctions, a large repertoire of antigen receptor genes results. V(D)J recombination is initiated by a mechanism-specific recombinase, the RAG1/2 proteins, but it is completed by the same nonhomologous repair mechanisms used in genome preservation, which highlights the potential duplicity of these mechanisms. Although less well defined, immunoglobulin class switch recombination also seem to require nonhomologous repair (15, 16).
Chromosomal instability in cancer
Nearly all cancer cells show deranged chromosomes manifest as translocations, deletions, and amplifications. Some rearrangements likely develop as a consequence of aberrant cell growth, but others are unquestionably causative. In one pattern, which is typical of lymphoid malignancies, specific cancers are associated with recurring, usually reciprocal, translocations between nonhomologous chromosomes. Translocation t9:22 in chronic myeloid leukemia is a classic example that allows treatment with inhibitors that target the product of the resulting BCR-ABL fusion gene (17). The other main pattern, which is typical of epithelial malignancies, shows massively deranged genomes with multiple, usually nonreciprocal, rearrangements. Extensive efforts continue to identify oncogenes and tumor suppressor genes whose function is affected by these rearrangements. Junctions of rearranged chromosomes in cancer frequently show microhomology in the absence of extended homology and are therefore inferred to originate by direct rejoining/NHR. The lesions and mechanisms that yield these rearrangements are undoubtedly multiple, including both telomere-dependent and telomere-independent events. Some rearrangements represent errors of the V(D)J process because they occur in cells and loci undergoing this targeted recombination (Fig. 3b). This issue provides another caution regarding the multiplicity of NHR mechanisms, because even these errors could represent use of inappropriate loci in the V(D)J reaction, joining of RAG1/2-generated and spontaneous DSBS, or even DSB-independent transposition between chromosomes (15). Other NHR presumably originates from two independent DSBs (Fig. 2a), but the final sequence might have little to do with the configuration of the inciting damage, as elegantly demonstrated in a yeast model (18).
Chemistry of DNA End Joining
The chemical transactions of DNA end joining are predictably similar to other DNA repair pathways and even to replication in that all of these processes manipulate base-pairing and phosphodiester bonds to create intact duplex molecules. However, the direct repair of DSBs presents biochemical challenges unique to these substrates. In this section, I consider both the general and the specific chemical requirements of nonhomologous DSB repair, how they are manifest in the two main pathways of repair, and the sometimes unusual properties of key enzymes that determine their function in these pathways.
Chemical steps in strand break repair
It is important to recognize that although NHR and HR share a substrate, they are mechanistically very different. It is more useful to compare NHR with the direct repair of single-strand breaks (SSBs), which is a subset of the base-excision repair (BER) pathway (Fig. 4a) (19). Three reaction classes might be required for such repair, depending on the specific nature of the break. All DNA repair culminates in ligation of a strand nick bearing 3' OH and 5' P termini. To arrive at that point, it might first be necessary to resynthesize bases missing as a result of the DNA damage. Both polymerization and ligation might also depend on the removal or reversal of blocking nucleotide fragments residual on the break termini. It might include lyase-dependent removal of 5' deoxyribosephosphate moieties, 3' phosphatase and/or 5' kinase action, or frank nucleolysis. End trimming can be either local (i.e. confined to the damaged nucleotide), or more extensive, as in long-patch BER (19). The key point is that all of these reactions are expected to be recapitulated in direct repair of DSBs (5). The critical difference is that SSB repair occurs on a stable substrate with an immediately available information template in the intact strand. DSBs require that reactions occur either divorced from the other side of the lesion or only when the bipartite substrate is brought back together in a repair complex (Fig. 4a). It is this requirement that creates the unique challenges for nonhomologous DSB repair. Microhomology at junctions indicates that limited annealing is often exploited during rejoining, which might itself require nucleolysis to expose base-pairing potential (Fig. 1b) and which permits both local mutations caused by misalignment and NHR caused by joining of different DSBs (Fig. 2a).

Figure 4. Chemistry of strand break repair. (a) The steps of SSB and DSB rejoining are highly analogous, with an added requirement for end bridging in NHEJ. For simplicity, only select mammalian proteins are indicated. Curved line, terminal damage; red arrow, polymerization. (b) The end-bridging step is realized in unusual ways by NHEJ polymerases (Pol) and ligase (Lig). At 3' overhangs, polymerases tolerate an incomplete primer-template pair, in part using special protein motifs (cyan loop). Similarly, DNA ligase IV can ligate (carat) across from strand discontinuities.
Nonhomologous end joining
The term “nonhomologous end joining” (NHEJ) initially referred generically to direct rejoining of DSBs, but it is now most commonly used to refer to a specific and dominant pathway of nonhomologous repair defined by both DNA features and protein components (5). Regarding the DNA, NHEJ typically entails little or no nucleotide loss from DSB ends. In the strictest manifestation, which is typified by the behavior of budding yeast, NHEJ acts almost exclusively by annealing DSB overhangs (9, 20). Because such overhangs were often paired in the original duplex, overhang joining tends toward accurate repair directly analogous to BER (Fig. 4a). This restriction to overhangs is apparently relaxed in other species, but current evidence nonetheless distinguishes NHEJ and more extensive nucleotide loss (see below). Regarding proteins, NHEJ requires both specific structural proteins to bind and bridge ends and enzymes to catalyze reactions similar to BER (Fig. 4a) (5). Unlike HR, whose genes were first identified by screens in yeast and bacteria, many proteins that catalyze NHEJ were discovered in mammals by observations that their deficiency causes sensitivity to DSBs and V(D)J impairment (4, 5, 16). This observation led to the common notion that mammals and single-cell organisms predominantly use NHEJ and HR, respectively. This notion is misleading, because all eukaryotes maintain complex HR and NHEJ pathways that are highly active in the correct cell states (6, 7).
NHEJ structural proteins include Ku, which is a protein preserved in all organisms that possess NHEJ and whose utilization provides the most specific current definition of this pathway (5). Ku is a heterodimer of the Ku70 and Ku80 proteins in eukaryotes and a homodimer in prokaryotes but in all organisms forms a ring that provides initial lesion recognition by passage of the DSB end through the ring (21). Interestingly, although Ku likely binds all DSBs, it is only required for NHEJ, not for HR. The specific roles of Ku in NHEJ in higher eukaryotes center on its identity as the principal DNA binding component of the DNA-dependent protein kinase (DNA-PK), whose catalytic subunit DNA-PKcs is also required for efficient NHEJ (5). As a kinase activated by DSB ends, DNA-PKcs contributes to NHEJ in part by phosphorylating NHEJ proteins and regulating the ensuing cascade of events. However, in vitro evidence also supports a primary structural role of DNA-PKcs as part of an end bridging complex (22, 23), although cellular evidence that supports this function is needed. Indeed, one key action of activated DNA-PKcs is autophosphorylation, which in some fashion seems to release the DSB ends for access by downstream enzymes (24). Despite this central role, neither lower eukaryotes nor bacteria are known to require a DSB-dependent kinase for NHEJ, which suggests that such regulation was imposed on the reaction during evolution. Ku must therefore support other NHEJ functions, at least in organisms that lack DNA-PKcs, and indeed a potentially yeast-specific ligase contact uses a portion of Yku80 known to bind DNA-PKcs in vertebrates (25). In some yeast, an additional NHEJ requirement exists for the DSB-binding Mre11-Rad50-Xrs2 (MRX) complex (9), which may account for the bridging function in place of DNA-PKcs (26). Consistently, current best evidence does not suggest a role for the analogous Mre11-Rad50-Nbs1 (MRN) complex in vertebrate NHEJ (27), but this is poorly explored because MRN is essential for viability.
NHEJ enzymes include foremost the requisite DNA ligase. In eukaryotes, DNA ligase IV (Lig4) is both strictly required for Ku-dependent NHEJ and largely if not entirely dedicated to this pathway (5). In prokaryotes, the principal NHEJ ligase, called LigD in mycobacteria, is present in an operon with Ku, which again demonstrates a coupling of Ku with a specific NHEJ ligase (28). However, the ligase presents a point of divergence between kingdoms in that LigD is no more related to Lig4 than to other ATP-dependent ligases. Lig4 does not act alone in support of NHEJ but it interacts via tandem C-terminal BRCT domains with a coiled-coil domain of a protein called XRCC4 (29). XRCC4 itself binds DNA (30) and so is arguably a structural protein that acts as a scaffold between the ligase and the DNA but because of the strength of its association is accurately viewed as a subunit of DNA ligase IV. Strikingly, yet another protein associates with Lig4::XRCC4 called XLF/Cernunnos that is predicted to be structurally similar and evolutionarily related to XRCC4 (31).
Other NHEJ enzymes execute end processing before ligation and are generally less well defined and less strictly required. Nucleases include the protein Artemis in higher eukaryotes, which executes unique NHEJ functions in V(D)J recombination (32) and the slower repair of approximately 10% of irradiation-induced DSBs that correlate with cytotoxicity (33). However, Artemis is absent from most lower eukaryotes, perhaps because of its intimate association with DNA-PKcs and V(D)J recombination (32), and even in organisms that possess it, Artemis is unlikely to account for all end trimming. Thus, some NHEJ nucleases are yet to be described. NHEJ polymerases include select members of the Pol
X family of DNA polymerases typified by the BER enzyme Pol β, specifically Pol4 in yeast (20) and terminal deoxynucleotidyl transferase (TdT, used only in V(D)J recombination), Pol λ, and Pol μ in mammals (34). However, like nucleases, these proteins cannot account for all NHEJ polymerization, as demonstrated clearly in yeast (20). Also, deficiency of even both Pol λ and Pol μ leads to only a mild phenotype manifest as altered V(D)J joint spectra but not frank sensitivity to DSBs (35). In prokaryotes, NHEJ end processing remarkably includes two protein domains often fused to the ligase, although these can function independently (28, 36). LigD in particular is a polyfunctional protein that contains polymerase, nuclease, and ligase domains in tandem, so that mycobacterial NHEJ can be transferred with just two polypeptides (37). As with the ligase, the prokaryotic nuclease and polymerase are not particularly related to Artemis or Pol X polymerases (28, 36).
Microhomology-mediated end joining
In early experiments it was observed that DSBs were sometimes repaired by an erroneous mechanism associated with nucleotide loss from the ends (38). In much literature this repair was attributed to NHEJ in its generic sense, but more recent work has continued to refine both NHEJ and this alternative form of repair. A major clue that the latter was a distinct mode of repair was the general observation that joints formed in NHEJ mutants showed more extensive nucleotide loss. This phenomenon has been carefully explored, and data support the notion that Ku-dependent NHEJ is primarily responsible for accurate rejoining (39, 40). One name given to the alternative deletion pathway is backup NHEJ (41), but this is confusing given the more common convention of using NHEJ to describe Ku-dependent joining. The designation microhomology-mediated end joining (MMEJ) was applied to Ku-independent repair in budding yeast (42). Similar joining, which is typified by microhomology use and substantial nucleotide loss, can also be observed in insects and plants (43, 44), and until additional information suggests a better classification, MMEJ seems a suitable name for all. MMEJ is poorly explored in bacteria.
The mechanisms of MMEJ are not well established and are likely multiple. A unifying feature in all systems is that MMEJ is repressed by Ku. In mammals, developing data suggest that poly ADP-ribose polymerase and DNA ligase III might participate in one subpathway (41). Because these factors are typically associated with BER, the analogy drawn above between the direct repair of SSBs and DSBs might in fact be realized in competing repair pathways. More detailed explorations of MMEJ have been performed in yeast, where a different type of repair was revealed that is not yet possible to describe as a simple linear pathway (2, 42). In Saccharomyces cerevisiae, MMEJ shows only a partial dependence on any factor (42). Interestingly, two of these factors are the NHEJ enzymes Dnl4 and Pol4, even in the absence of Ku. Another dependence is on the 3' flap nuclease Rad1-Rad10, which is not implicated in NHEJ but does function in HR and, more relevantly, in single-strand annealing (SSA) (3, 4). SSA is a form of repair in which larger internal direct repeats are annealed. SSA is not typically described as NHR because these repeats are too big to be considered microhomologies and because it uses the HR protein Rad52 to facilitate annealing. Although Rad52 is not obviously required for MMEJ in S. cerevisiae (42), its homologue in Schizosac- charomyces pombe is, which leads to the designation of MMEJ as “micro-SSA” (2). This finding is compelling as it invokes the most consistent apparent requirement of deletion MMEJ, namely that strand resection must expose microhomologous sequences for annealing, which is probably common with HR (Fig. 1b) (2, 45). Importantly, NHEJ might sometimes use internal microhomologies close to the DSB ends through the action of proteins such as DNA-PKcs, which are distinct from 5' resection and MMEJ (Fig. 1b) (1).
A fundamental question remains whether MMEJ is a distinct mechanism of DNA repair or simply a different manifestation of previously described pathways. Yeast MMEJ is markedly inefficient and largely irrelevant to basal DNA repair (40, 43), in contrast to HR and NHEJ (6, 8). However, repair consistent with MMEJ can be very efficient in higher eukaryotes, so that although the quality changes, rates of end joining do not necessarily decrease markedly with NHEJ mutation (40, 42). Moreover, even an inefficient pathway might be of relevance to NHR, so it is important to consider which mechanisms actually catalyze rejoining of inappropriate DSB pairs. NHEJ can efficiently mediate nonhomologous integration of transformed fragments in some fungi (46). Also, it was found that some spontaneous yeast nonreciprocal rearrangements depend on NHEJ (47) and that yeast NHEJ can mediate reciprocal translocations between two induced DSBs (48). It is not known how to disable MMEJ specifically, but residual chromosome rearrangement observed in NHEJ mutants indicates that at least one other mechanism, presumably MMEJ, is also at play (47, 48). It is not yet clear how often mammalian NHEJ might execute spontaneous NHR, but it is clearly a net caretaker of the genome whose absence leads to increased chromosome rearrangements by alternative repair mechanisms (49).
DNA ligase IV
Clearly, Lig4 is specifically suited to NHEJ, but why? In part, it is because the DNA ligase IV complex can engage the NHEJ structural machinery by specific protein contacts (25). However, the enzymatic function of Lig4 might also be optimized for NHEJ, because transferring its BRCT domains to a different ligase did not transfer NHEJ capacity (50). This function is not manifest by a notably greater ability of Lig4 to ligate compatible overhangs. Instead, Lig4 has a unique ability to ligate ends that do not align to a simple nick but are incompatible or have a gap (51). Unlike most ligases, Lig4 apparently can bring the 3' OH and 5' P into position for catalysis anyway, which requires tolerance for non-duplex DNA (Fig. 4b). This activity would have the tendency to promote rejoining of complex ends and therefore chromosome stability at the expense of potentially less deleterious local mutations. Concerns are that others have not observed markedly different ligation fidelity of Lig4, but importantly this was measured nicks, not DSBs (52). Also, my laboratory has examined many NHEJ substrates in yeast genetic assays and has rarely observed outcomes consistent with promiscuous ligation (8, 20). Other NHEJ proteins might also influence the different types of outcomes. XLF/Cernunnos depletion from extract-mediated NHEJ reactions had a much greater effect on joining of complex as opposed to simple DSB substrates (53). This effect might imply a role for XLF/Cernunnos in promoting either end processing or possibly promiscuous ligation by Lig4. Again, though, Nej1, the yeast XLF homologue, is equally required for both simple and complex end configurations (our unpublished results). It is still the early days for these studies, and it is of great interest to determine how important the catalytic properties of the DNA ligase IV complex are beyond its recruitment by NHEJ structural proteins.
Pol X NHEJ polymerases
Strong evidence points to the acquisition of special enzymatic properties in the Pol X family of NHEJ polymerases that allow them to deal with the limiting substrates inherent to DSBs. The comparison with BER is particularly helpful here. Pol β is similar to Pol4, Pol λ, and Pol μ in that it fills gaps by extending a primer strand, bringing them to ligation readiness (19). However, BER and NHEJ differ markedly in the template strand, which is continuous in BER but discontinuous in NHEJ (Fig. 4). Critically, the point of template discontinuity in NHEJ depends on the polarity of the DSB (Fig. 4b). At 5' overhangs, the discontinuity is ahead of the primer-template pair, past the gap, and outside of the active site of the enzyme. At 3 overhangs, the discontinuity is behind the point of synthesis, within the active site in the position of primer-template pairing. Numerous findings indicate that Pol X NHEJ polymerases tolerate this latter substrate perturbation, whereas other polymerases cannot. Yeast Pol4 is required for synthesis only at 3', but not at 5', overhangs (20). Pol X NHEJ polymerases also show an unusual propensity toward frameshift mutations that suggest a greater flexibility in use of primer-template pairings (54). Extensive structural data also reveal that contacts with the distal 5' side of a gap are paradoxically more numerous than with the 3' (i.e., synthesis) side (55). Moreover, two protein loops unique to Pol X NHEJ polymerases make variable contacts to, or occupy the position of, the template strand (56, 57). These loops account in part for the stabilization of extrahelical bases during frameshift synthesis, and they might also contribute to stabilization of template discontinuities (Fig. 4b). One loop, in conjunction with a Pol μ/TdT histidine that bridges the dNTP and primer terminus, also facilitates template-independent synthesis and possibly template-dependent synthesis in the absence of initial primer-template base-pairing, which thus adds base-pairing potential to initially incompatible overhangs (Fig. 4b) (34, 51, 58). A picture emerges in which extended and atypical polymerase-DNA contacts overcome the instability of DSB joints caused by template strand discontinuity.
Tools and Techniques for Studying End Joining
The temporal sequence imposed in vivo on repair of single, often complex, chromatin DSB lesions is difficult if not impossible to recapitulate in vitro. It is thus advisable to take genetic data as a primary reference point for understanding NHR mechanisms. In vitro analyses nonetheless allow focused study of enzymes properties, and structural biology will continue to be irreplaceable in elucidating enzyme-substrate relationships.
Genetic analysis
A first approach to studying DSB repair is to treat cells with DSB-inducing agents such as ionizing radiation. A change in either survival or mutagenesis caused by a gene defect can be taken as evidence for its involvement in DSB repair, especially if supported by epistasis analysis to ascertain which repair pathway is affected. An alternative approach is to express in cells mega-endonucleases that create DSBs specifically at native or engineered chromosomal sites (3). In this way, repair can be tracked not only by survival but also by the status of genetic markers at the cut site. A limitation is that most joints restore the cut site and will simply be recleaved, so that repair outcomes are influenced by the method. Linking DSB formation to termination of endonuclease expression limits this issue (8), but the problem is never completely avoidable. A final approach introduces plasmids that were linearized in vitro into cells, where recircularization is required for plasmid maintenance (9, 20). Although limited by the use of naked DNA, the simplicity of this approach and the tremendous variety of available DSB ends make it common and powerful.
In vitro reconstitution
Two main approaches have been used for studying NHEJ in vitro. In the first approach, crude extracts are prepared from cells and are spiked with DNA fragments whose joining is followed by electrophoretic monitoring or PCR amplification (27, 53). Extracts have a full complement of cellular proteins in appropriate ratios and might therefore provide the best recapitulation of the in vivo situation while allowing manipulation of conditions, DSB ends, and so on. More reports reconstituted the NHEJ reaction from purified mammalian, yeast, or bacterial components (26, 37, 51). Some rejoining can be catalyzed by nothing more than a ligase, but focusing on incremental activity with combined components and complex DSBs gives confidence that results are meaningful to cellular NHEJ. Another concern is that NHEJ reconstitution has always used mass action chemistry with sometimes high concentrations of proteins and DNA, which is in marked contrast to the single pair of ends presented in vivo.
Structural analysis
Crystal structures of many NHEJ proteins have tremendously supported the discussions above. Also, electron microscopy revealed the overall shape of the large and complex DNA-PKcs structure (23). For most other domains, structural inferences can be drawn by mapping NHEJ proteins onto crystallized enzymes of the same class. However, no published structures show two DSB ends engaged together, which I argue defines the unique functions of NHEJ proteins.* Similarly, poor insight exists into the interconnected assembly of the full NHEJ repair complex. Furthermore, crystal structures are static, whereas NHEJ is dynamic with multiple different reactions.
Future Directions
Nonhomologous DSB repair has a complex relationship with genome preservation in that it can yield both silent repair and NHR. More understanding of this relationship will require improved genetic systems for monitoring the influence of nonhomologous repair pathways on genome integrity. It will also require delineation of the cell-cycle, checkpoint, and chromatin mechanisms that regulate 5' resection and the interplay of NHR and HR. Better molecular definition of the various nonhomologous repair pathways is needed, especially MMEJ, in which clear mechanism(s) are still pending. Pol X polymerases provide a conceptual framework for exploring how individual proteins engage the unstable substrates inherent to a DSB, which could be extended to the ligase and beyond. More interaction mapping and biochemical and structural analyses will be required to understand the assembly of a full NHEJ repair complex and the function of currently enigmatic contributors to it. Specific goals include single-molecule NHEJ reconstitution in vitro, structures with two-sided DSBs, and delineation of dynamic changes and regulatory switches that occur during reaction progression.
__________________________
*A recent paper has now reported a structure of ends being bridged by the polymerase domain of the bacterial LigD enzyme (Brissett NC, Pitcher RS, Juarez R, Picher AJ, Green AJ, Dafforn TR, Fox GC, Blanco L, Doherty AJ. Structure of a NHEJ polymerase-mediated DNA synaptic complex. Science 2007; 318:456-9).
References
1. Hammarsten O, DeFazio LG, Chu G. Activation of DNA-dependent protein kinase by single-stranded DNA ends. J. Biol Chem. 2000; 275:1541-1550.
2. Decottignies A. Microhomology-mediated end-joining in fission yeast is repressed by Pku70 and relies on genes involved in homologous recombination. Genetics 2007.
3. Haber JE. Transpositions and translocations induced by site-specific double-strand breaks in budding yeast. DNA Repair (Amst). 2006; 5:998-1009.
4. Cahill D, Connor B, Carney JP. Mechanisms of eukaryotic DNA double strand break repair. Front Biosci. 2006; 11:1958-1976.
5. Wilson TE. Nonhomologous end joining mechanisms, conservation, and relationship to illegitimate recombination. In: Topics in Current Genetics: Molecular Genetics of Recombination. Aguilera A, Rodney R, eds. 2007. Springer, New York. pp. 487-512.
6. Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, Haber JE, Foiani M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011-1017.
7. Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998; 17:5497-5508.
8. Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics 2002; 161:1015-1027.
9. Boulton SJ, Jackson SP. Components of the Ku-dependent nonhomologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998; 17:1819- 1828.
10. Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol. Cell 2002; 10:1189-1199.
11. Pitcher RS, Green AJ, Brzostek A, Korycka-Machala M, Dziadek J, Doherty AJ. NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair 2007; 6:1271-1276.
12. Roth JR, Kugelberg E, Reams AB, Kofoid E, Andersson DI. Origin of mutations under selection: the adaptive mutation controversy. Annu. Rev. Microbiol. 2006; 60:477-501.
13. Murnane JP. Telomeres and chromosome instability. DNA Repair 2006; 5:1082-1092.
14. Riha K, Heacock ML, Shippen DE. The role of the nonhomologous end-joining DNA double-strand break repair pathway in telomere biology. Annu. Rev. Genet. 2006; 40:237-277.
15. Lieber MR, Yu K, Raghavan SC. Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations. DNA Repair 2006; 5:1234-1245.
16. Dudley DD, Chaudhuri J, Bassing CH, Alt FW. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv Immunol. 2005; 86:43-112.
17. Savona M, Talpaz M. Chronic myeloid leukemia: changing the treatment paradigms. Oncology (Williston Park). 2006; 20:707- 711; discussion 712-704, 719, 724.
18. Narayanan V, Mieczkowski PA, Kim HM, Petes TD, Lobachev KS. The pattern of gene amplification is determined by the chromosomal location of hairpin-capped breaks. Cell 2006; 125:1283- 1296.
19. Fortini P, Dogliotti E. Base damage and single-strand break repair: mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007; 6:398-409.
20. Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of pol X family polymerase action in nonhomologous end joining. J. Biol. Chem. 2005; 280:29030-29037.
21. Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001; 412:607-614.
22. DeFazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. EMBO J. 2002; 21:3192-3200.
23. Spagnolo L, Rivera-Calzada A, Pearl LH, Llorca O. Threedimensional structure of the human DNA-PKcs/Ku70/Ku80 complex assembled on DNA and its implications for DNA DSB repair. Mol. Cell. 2006; 22:511-519.
24. Reddy YV, Ding Q, Lees-Miller SP, Meek K, Ramsden DA. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J. Biol. Chem. 2004; 279:39408-39413.
25. Palmbos PL, Daley JM, Wilson TE. Mutations of the Yku80C terminus and Xrs2 FHA domain specifically block yeast nonhomologous end joining. Mol. Cell. Biol. 2005; 25:10782-10790.
26. Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol. Cell. 2001; 8:1105-1115.
27. Di Virgilio M, Gautier J. Repair of double-strand breaks by nonhomologous end joining in the absence of Mre11. J. Cell. Biol. 2005; 171:765-771.
28. Aravind L, Koonin EV. Prokaryotic homologs of the eukaryotic DNA-end-binding protein Ku, novel domains in the Ku protein and prediction of a prokaryotic double-strand break repair system. Genome Res. 2001; 11:1365-1374.
29. Dore AS, Furnham N, Davies OR, Sibanda BL, Chirgadze DY, Jackson SP, Pellegrini L, Blundell TL. Structure of an Xrcc4-DNA ligase IV yeast ortholog complex reveals a novel BRCT interaction mode. DNA Repair 2006; 5:362-368.
30. Modesti M, Hesse JE, Gellert M. DNA binding of Xrcc4 protein is associated with V(D)J recombination but not with stimulation of DNA ligase IV activity. EMBO J. 1999; 18:2008-2018.
31. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006; 124:301-313.
32. Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002; 108:781-794.
33. Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell. 2004; 16:715-724.
34. Nick McElhinny SA, Havener JM, Garcia-Diaz M, Juarez R, Bebenek K, Kee BL, Blanco L, Kunkel TA, Ramsden DA. A gradient of template dependence defines distinct biological roles for family X polymerases in nonhomologous end joining. Mol. Cell. 2005; 19:357-366.
35. Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal de- oxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 2006; 25:31-41.
36. Pitche RS, Brissett NC, Picher AJ, Andrade P, Juarez R, Thompson D, Fox GC, Blanco L, Doherty AJ. Structure and function of a mycobacterial NHEJ DNA repair polymerase. J. Mol. Biol. 2007; 366:391-405.
37. Della M, Palmbos PL, Tseng HM, Tonkin LM, Daley JM, Topper LM, Pitcher RS, Tomkinson AE, Wilson TE, Doherty AJ. Mycobacterial Ku and ligase proteins constitute a two-component NHEJ repair machine. Science 2004; 306:683-685.
38. Roth DB, Wilson JH. Nonhomologous recombination in mammalian cells: role for short sequence homologies in the joining reaction. Mol. Cell. Biol. 1986; 6:4295-4304.
39. van Heemst D, Brugmans L, Verkaik NS, van Gent DC. Endjoining of blunt DNA double-strand breaks in mammalian fibroblasts is precise and requires DNA-PK and XRCC4. DNA Repair 2004; 3:43-50.
40. Verkaik NS, Esveldt-van Lange RE, van Heemst D, Bruggenwirth HT, Hoeijmakers JH, Zdzienicka MZ, van Gent DC. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 2002; 32:701-709.
41. Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170-6182.
42. Ma JL, Kim EM, Haber JE, Lee SE. Yeast Mre11 and Rad1 proteins define a Ku-independent mechanism to repair double-strand breaks lacking overlapping end sequences. Mol. Cell. Biol. 2003; 23:8820-8828.
43. Romeijn RJ, Gorski MM, van Schie MA, Noordermeer JN, Mullenders LH, Ferro W, Pastink A. Lig4 and rad54 are required for repair of DNA double-strand breaks induced by P-element excision in Drosophila. Genetics 2005; 169:795-806.
44. Gallego ME, Bleuyard JY, Daoudal-Cotterell S, Jallut N, White CI. Ku80 plays a role in non-homologous recombination but is not required for T-DNA integration in Arabidopsis. Plant J. 2003; 35:557-565.
45. Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-dependent single-strand DNA formation at DNA break promotes microhomology-mediated end joining. Genetics 2007; 176:2003- 2014.
46. Kegel A, Martinez P, Carter SD, Astrom SU. Genome wide distribution of illegitimate recombination events in Kluyveromyces lactis. Nucleic Acids Res. 2006; 34:1633-1645.
47. Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science 2002; 297:552-557.
48. Yu X, Gabriel A. Reciprocal translocations in Saccharomyces cerevisiae formed by nonhomologous end joining. Genetics 2004; 166:741-751.
49. Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, De-Pinho RA, Alt FW. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc. Natl. Acad. Sci U.S.A. 2000; 97:6630- 6633.
50. Teo SH, Jackson SP. Lif1p targets the DNA ligase Lig4p to sites of DNA double-strand breaks. Curr. Biol. 2000; 10:165-168.
51. Gu J, Lu H, Tippin B, Shimazaki N, Goodman MF, Lieber MR. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 2007; 26:1010-1023.
52. Wang Y, Lamarche BJ, Tsai MD. Human DNA ligase IV and the ligase IV/XRCC4 complex: analysis of nick ligation fidelity. Biochemistry 2007; 46:4962-4976.
53. Tsai CJ, Kim SA, Chu G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:7851-7856.
54. Bebenek K, Garcia-Diaz M, Patishall SR, Kunkel TA. Biochemical properties of Saccharomyces cerevisiae DNA polymerase IV. J. Biol. Chem. 2005; 280:20051-20058.
55. Garcia-Diaz M, Bebenek K, Krahn JM, Blanco L, Kunkel TA, Pedersen LC. A structural solution for the DNA polymerase lambda-dependent repair of DNA gaps with minimal homology. Mol. Cell. 2004; 13:561-572.
56. Garcia-Diaz M, Bebenek K, Krahn JM, Pedersen LC, Kunkel TA. Structural analysis of strand misalignment during DNA synthesis by a human DNA polymerase. Cell 2006; 124:331-342.
57. Juarez R, Ruiz JF, Nick McElhinny SA, Ramsden D, Blanco L. A specific loop in human DNA polymerase mu allows switching between creative and DNA-instructed synthesis. Nucleic Acids Res. 2006; 34:4572-4582.
58. Moon AF, Garcia-Diaz M, Bebenek K, Davis BJ, Zhong X, Ramsden DA, Kunkel TA, Pedersen LC. Structural insight into the substrate specificity of DNA Polymerase mu. Nat. Struct. Mol. Biol. 2007; 14:45-53.
Further Reading
Lieber MR.ed. Mechanisms of chromosomal translocation. DNA Repair. 2006; 5:997-1298.
This special issue of DNA Repair contains a series of review articles on factors that affect chromosome maintenance. Several articles are referenced herein, but the entire volume is an excellent single source for beginning a more in-depth exploration of this field.
See Also
DNA Damage
DNA Damage Repair, Chemistry of
Eukaryotic DNA Polymerases, Chemistry of
Homologous Recombination
Nucleic Acids, Enzymes that Modify