CHEMICAL BIOLOGY
Origins of Life: Emergence of the RNA World
Michael P. Robertson, Center for Molecular Biology of RNA, University of California, Santa Cruz
doi: 10.1002/9780470048672.wecb424
The RNA World hypothesis refers to a period during the evolution of life on Earth, preceding the DNA/RNA/protein-based systems of modern biology, during which RNA alone directed and catalyzed the chemistry of life. The concept is an elegant solution to the problem of devolving our intertwined 3-biopolymer biochemistry toward its origins as the simplest system imaginable. In support of this idea, a scattering of molecular clues within our cells point suggestively toward an RNA-only period leading up to the ribozyme-mediated invention of protein synthesis. Indeed, the RNA World hypothesis is a very sensible scenario when considered from the perspective of trying to simplify life's complexity. The alternative perspective, however, imagining a sterile, primordial Earth and trying to assemble abiotically produced precursors into the RNA World is an extremely difficult problem. Certain steps along the logical chemical pathway to the RNA World have been demonstrated in the laboratory, but many others remain problematic. The exploration of RNA's catalytic properties has been encouraging, but complex ribozymes are extremely scarce in RNA populations. If the RNA World did once exist, it came about by a mechanism that has not yet been adequately elucidated, including the possibility that a different, unknown, biopolymer system may have preceded the RNA World.
The mechanism of the emergence of life on Earth is one of the most fundamental mysteries of the biological sciences. Despite centuries of pontification and decades of serious experimental efforts, the details remain poorly understood. The most satisfying attempts to deduce the genesis and earliest evolution of primitive life have focused on what is known of modern biology and attempted to simplify it back to the most fundamental and primitive (1, 2). This simplification exercise quickly leads to a paradox in determining which of the essential biopolymers of life, proteins or nucleic acids, would have come first. Conventional logic indicates that it could not have been protein because nucleic acids (DNA and RNA) carry the information that specifies how to make protein and are required to direct their synthesis. Conversely, it would also seem that it could not have been DNA either, which, although containing all the coded instructions to make a living cell, still requires protein enzymes and RNA to carry out those instructions. Without proteins, DNA would be just an inert, impotent molecule. A resolution to this paradox was suggested early on by a few notable investigators (3-5), but it was not until the discovery of catalytically active RNA (6, 7) demonstrated that a single biomolecule could possess both the coded instructions to make copies of itself and the functional abilities to carry out those instructions that the theory gained wide acceptance.
The assumption of a so-called ‘RNA World’ era that preceded the biological assimilation of DNA and protein, during which RNA would have been the primary facilitator of a metabolism of unknown complexity, offers an elegant and appealing solution to the challenge of inventing the system of multiple, interrelated biopolymers that exists today. Clues pointing to this bygone RNA World era are scattered throughout our biochemistry. Considered individually, they are not overwhelmingly convincing, but taken together, they make a compelling case that an RNA-based biology might have preceded what we have today.
Despite the deserved enthusiasm that the RNA World hypothesis generates, there remain many puzzling difficulties and unanswered questions. An RNA World is quite appealing from the perspective of modern biology looking backwards in time, but is extremely difficult to envision from a perspective at the formation of the Earth looking forward in time. Bits and pieces of the components of RNA can be synthesized, sometimes efficiently, in prebiotic soup-type conditions, but there is not yet a plausible scenario to assemble them into a monomer or dimer of RNA, let alone a polymer of sufficient complexity and functionality to kick-start a living system. Allowing that some unknown series of mechanisms, including the possibility of a preRNA World, led to the formation of long RNA polymers and eventually ribozymes, the creation of a self-replicating system based solely on RNA, the simplest possible RNA World scenario, is still a daunting challenge that well over a decade of focused experimental attempts have so far failed to solve. Even so, the prospects remain optimistic. Each year brings new insight into the complex characteristics and abilities of RNA, as the boundaries of its capabilities are explored further.
Life, Dogma, and the RNA World
The term ‘RNA World’ (8) refers to a hypothesized period of the early evolution of life, during which RNA, in the absence of DNA or proteins, served as the sole biopolymer of life. Such a life form would likely be unfamiliar to what we are accustomed to, and so a thoughtful, general definition for what constitutes “life” becomes important in order to identify the boundary that separates non-living from what can be considered alive. However, drafting a fundamental, general, succinct definition of the processes and characteristics that represent life has been difficult, controversial, and often unsatisfying (9). The formulation that seems to enjoy the most widespread support is the “chemical Darwinian” definition, which states, simply, that “life is a self-sustained chemical system capable of undergoing Darwinian evolution” (10). The power of this statement is that the primary natural force that has shaped the diversification and evolution of all the organismal life that we know of, namely Darwinian evolution, is also applicable at the simplest, sub-organismal, molecular stages of life. Implicit in the chemical Darwinian model is a system that can sustain itself and create progeny copies. The copies were slightly less than perfect, such that natural selection could encourage a lineage of generations, improving and diversifying, evolving eventually into the entire biosphere.
The “central dogma of molecular biology” (11, 12) describes the flow and directionality of information exchange among the three primary information-rich biopolymers of contemporary biology, namely, DNA, RNA, and proteins (Fig. 1). The biochemistry of life today is intertwined between these major, discreet components, each one dependent upon the others for its existence. However, a scenario in which two, let alone all three, of these individual systems arose simultaneously and were able to function synergistically is very difficult to imagine. In theory, a system comprised only of RNA could direct and execute the replication of its own genetic information, producing progeny via a self-templated RNA polymerization mechanism catalyzed by the RNA itself, thus satisfying the minimal definition for life with a single biopolymer.
The central component of this RNA World scenario is a replication system that can make copies of the genetic material to grow and produce progeny. The most basic scenario is a simple templated ligation of small oligomers in a cyclical replication scheme (13-16), but ultimately a catalytic entity responsible for this crucial function of replication would be necessary. The simplest example is a single self-replicating ribozyme that can copy itself. More elaborate scenarios for a mature RNA World might also involve ancillary functions such as producing the nucleotides, or building blocks, for the replicase to work with. A case can be made for numerous other functions as well, but one that seems very probable is the invention of protein synthesis. The observation that a nearly protein-free preparation of ribosomal RNA can catalyze the crucial chemical peptidyl transferase step of ribosomal protein synthesis (17), and subsequent X-ray crystal structures of the ribosome showing the arrangement of RNA around the catalytic center (18), together indicate that the cellular machine for making protein is, at its core, a ribozyme. The invention of protein synthesis marks a boundary between the RNA World and the era that persists to this day, in which proteins have become the dominant agents of catalysis across all forms of life.
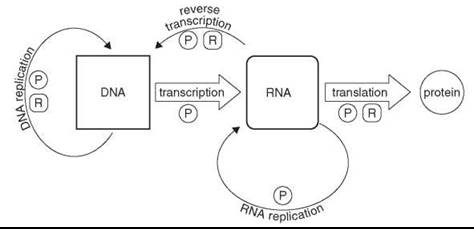
Figure 1. The central dogma of molecular biology. The central dogma describes the flow of information between the principle biopolymers of the cell. The primary exchange is shown horizontally, left to right, namely, DNA directs the synthesis of RNA, which in turn directs the synthesis of protein. Alternate observed exchanges are depicted off the horizontal with less prominent lines. The letters ''P'' and ''R'' indicate that protein or RNA, respectively, are required for particular exchanges.
Prebiotic Synthesis of RNA Components
Nucleotides are the primary building blocks necessary to assemble an RNA world. Each nucleotide is comprised of a base attached to a sugar through an N-glycosylic bond, with a different site on the sugar phosphylated (Fig. 2). The information content of a nucleic acid polymer is encoded in the identity and order of the bases-adenine, cytosine, guanine, and uracil in RNA, or thymine in place of uracil in DNA. D-ribose, the sugar moiety in RNA, bridges the backbone with 5',3'-phosphodiester linkages to adjacent nucleotides in addition to attaching to the base via the C1' position.

Figure 2. The components of RNA. (a) The primary bases of RNA, which includes the purines adenine and guanine and the pyrimidines cytosine and uracil. (b) A purine nucleoside (adenosine) and a pyrimidine nucleoside (uridine). (c) The nucleotide adenosine 5'-triphosphate. (d) An RNA strand with the sequence AGCU.
Bases
The bases are nitrogenous aromatic heterocycles, bicyclic in the case of the purines, and pyrimidines with only a single ring. Both systems are decorated with complementary arrays of hydrogen bond donors and acceptors that facilitate specific base-pairing interactions, usually between a purine and a pyrimidine, e.g. adenosine (A) pairs with uridine (U), and guano- sine (G) pairs with cytidine (C) (19) (Fig. 3). These canonical Watson-Crick pairings form the basis for the storage and copying of double-stranded DNA and the genetic transfer from DNA to RNA, although non-canonical pairings are common in nucleic acids that adopt structures more complex than a simple double helix.

Figure 3. Watson-Crick base pairs. (a) Adenosine pairs with uridine, and (b) guanosine pairs with cytidine. Hydrogen bonds are indicated with vertical grey dashes.
Purines
Demonstrations of the abiotic synthesis of the nucleic acid bases has been a relative success among the steps necessary to understand the synthesis of RNA nucleotides. Beginning in 1953, when the Miller-Urey spark discharge experiments (20) triggered a shift towards experimental testing in origins of life research, the simplest subunits of life have been favorite targets for investigation. In 1960, Oro demonstrated the synthesis of adenine in yields of less than 1% from simple aqueous solutions of ammonium cyanide (21, 22). Stoichiometrically, adenine is a pentamer of HCN, and although some uncertainty about the precise reaction pathway exists, the detection of 4-aminoimidazole-5-carboxamidine (AICA) and 4-aminoimidazole-5-carbonitrile (AICN) in the complex reaction mixture, suggests a role for either or both of these molecules as intermediates in alternate pathways to the synthesis of adenine from HCN (Fig. 4)(23, 24). Diaminomaleonitrile (DAMN), a tetramer of HCN and an abundant component of HCN polymerizations, can be converted to AICN through a photochemical rearrangement and may be an important precursor to adenine under certain conditions (25, 26). Alternatively, Ferris and coworkers have suggested that very little or no free adenine is formed in these discreet scenarios, but rather adenine is released only during the hydrolysis of the complex and insoluble cyanide polymers (27). The synthesis of guanine has also been detected under identical conditions at levels 10 to 40-fold less than adenine (27, 28). The presumed reaction pathways are similar to adenine’s but proceed through a 4-aminoimidazole-5-carboxamide intermediate.
Adenine, guanine, and other purines have also been reported in low yields in several discharge/irradiation experiments performed in methane/ammonia mixtures (29-31). Since HCN is known to form under these conditions (32), it is likely that these purine syntheses proceed through HCN-mediated mechanisms similar to the original Oro synthesis.

Figure 4. Prebiotic synthesis of adenine. This schematic shows potential pathways and intermediates in the synthesis of adenine from ammonium cyanide.
Pyrimidines
A variety of routes for the synthesis of pyrimidines have also been demonstrated. Uracil is formed in high yields by heating malic acid and urea in sulfuric (33) or polyphosphoric acid (34). Although the simple starting materials are prebiotically attractive, the extreme conditions and anhydrous requirement of this reaction make it of questionable relevance for the prebiotic Earth in anything other than highly specialized local environments. A number of other more plausible pyrimidine syntheses have also been explored, but these tend to be much less efficient. Oro reported low levels of uracil synthesis by heating aqueous acrylonitrile with urea and ammonium chloride (35). A variety of pyrimidines, including orotic acid, 5-hydroxyuracil, and 4,5-dihydroxypyrimidine have been detected at low levels in HCN polymerizations (27, 36), as has uracil (37). Schwartz and Chittenden demonstrated the synthesis of 5,6-dihydrouracil (DHU), a constituent of transfer RNA, by condensation of β-alanine and urea through a β-ureidopropionic acid intermediate, which can then cyclize to form DHU (38, 39). Subsequent photolytic dehydrogenation of DHU produces uracil in a reaction promoted by hydrated clay minerals, particularly montmorillonite. Orgel and coworkers produced reasonable amounts of cytosine and its hydrolysis product, uracil, from solutions of cyanoacetylene and cyanate or urea (40). Cyanoacetylene is an attractive prebiotic molecule in the sense that it is a reactive compound formed abundantly in spark discharge experiments (41). However, its reactivity makes cyanoacetylene short-lived in the environment, the chief byproduct being the hydrolysis product cyanoacetaldehyde. Ferris combined cyanoacetaldehyde with guanidine to form 2,4-diaminopyrimidine, which can in turn hydrolyze to make cytosine, and then uracil (42). Nearly 30 years after Orgel’s experiments with cyanoacetylene and urea, Miller, starting directly with cyanoacetaldehyde and exploiting the extreme solubility of urea, was able to produce cytosine and uracil in combined yields greater than 50% (43).
Concentration mechanisms
The investigation of prebiotic chemistry often involves a compromise between extreme conditions that favor efficient product formation on a timescale amenable to laboratory investigation and milder conditions that are more plausible on the early Earth. The most optimistic estimates for the production of reactive biological precursor molecules, while substantial in their global amounts, when diluted throughout the volume of the Earth’s hydrosphere would result in only meager concentrations (44). In some cases low reactant concentrations simply result in slower reaction rates, which may not present serious difficulties when considered in the context of a geologic time scale. In other instances, however, reaction pathways at low concentrations may differ significantly from those at higher concentrations. HCN polymerizations, for example, produce biologically interesting materials only at HCN concentrations of about 0.01 M or higher. At HCN concentrations lower than 0.01 M, hydrolysis out-competes oligomerization, resulting in the accumulation of formamide and formic acid instead of HCN oligomers (45). Concentration mechanisms are often invoked to address these difficulties and boost efficiencies, and, indeed, certain concentration processes seem likely to have played a role in nature. Miller’s rationale for using such high concentrations of urea in his synthesis of cytosine was the “drying lagoon” model of prebiotic synthesis in which periodic evaporation of bodies of water to near dryness, such as beaches, lagoons, and tide pools, concentrate the most soluble compounds to extremely high concentrations, even if they were initially very dilute (46). In the case of HCN, its volatility precludes evaporative concentration mechanisms, but freezing can be an effective means of concentration, as the HCN-water eutectic is 74.5% HCN at —23.4 °C (47). HCN polymerizations proceed effectively under these conditions, the extremely high concentrations compensating kinetically for the low temperatures (47).
Extraterrestrial synthesis of bases
Aspects of every putative prebiotic synthesis are vulnerable to critical analysis, and experiments are often open to questions of interpretation and relevance. This is especially true in origin of life research where virtually everything is unknown and will remain so, as no direct evidence survives from that time. Uncertainties about specific conditions, sources, and reaction pathways, however, in the context of the discussion of nucleic acid bases, are secondary to the likelihood that these particular fundamental building blocks of life were produced in ubiquitous abiotic processes throughout our solar system and presumably beyond. Analyses of carbonaceous meteorites have detected amino acids (48, 49) and nucleic acid bases (50-52) that are extraterrestrial in origin. Moreover, the distribution of compounds and their relative abundances are reminiscent of those observed in known prebiotic simulation experiments. These findings suggest that conditions favoring the synthesis of these fundamental biological materials, whether they be through the mechanisms reviewed above or others that have not been considered, were at work during the formation and early evolution of our solar system, and, consequently, the nucleic acid bases are very likely to have been present on the prebiotic Earth.
In addition to invoking the evidence of extraterrestrial synthesis of biomolecules as a means to corroborate that these reactions could have occurred on the prebiotic Earth, they can also be invoked directly as potential source material for the origin of life. The earliest stages of Earth’s history are characterized by a steady bombardment of extraterrestrial objects such as meteorites and comets that could have delivered large amounts of biologically useful material to the Earth. Less exciting than meteor or comet impacts, but much more significant in terms of the amounts of extraterrestrial material delivered to Earth, are interplanetary dust particles (IDPs). IDPs are small carbonaceous particles on the order of tens to hundreds of micrometers in size that are formed by comet evaporation or asteroid collisions and they “rain down” onto the Earth’s surface in amounts estimated to be at least several tons per day (53, 54). This significant source of extraterrestrially synthesized organic material is expected to have supplemented, perhaps substantially so, any endogenously produced biological precursors.
Sugars
A sugar moiety, specifically the pentose D-ribose, is attached to the base as the other half of a nucleoside. The synthesis of sugars from formaldehyde, a compound generated in spark discharge experiments and frequently invoked as a prebiotic reagent, has long been known (55), and its applicability as a prebiotic process has been well investigated. The so-called formose reaction produces a rich mixture of sugars from a solution of formaldehyde, but requires an unrealistically basic pH and improbably high concentrations of formaldehyde to seem prebiotically feasible (56). The reaction has been modified to proceed under much milder conditions at neutral pH, and can be catalyzed by certain common minerals, which is a more attractive prebiotic scenario (57, 58). However, a second problem with the formose reaction exists-its lack of synthetic selectivity. RNA nucleosides utilize a specific sugar, D-ribose, but the formose reaction generates a complex mixture of trioses through hexoses, including branched molecules, without a preference for ribose, and without enantiomeric enrichment (59). In contrast, the base-catalyzed aldolization of glycoaldehyde phosphate in the presence formaldehyde produces ribose-2,4-diphosphate as the major product (60). Further refinements to this procedure using a clay mineral catalyst allow the reaction to proceed efficiently with dilute reactants at neutral pH (61). In addition, other mechanisms to further enrich for ribose from a mixture of sugars have been demonstrated, such as selective membrane permeability (62) or selective reactivity with reagents that form adducts preferentially with ribose (63). It is also possible that unknown mechanisms or untested catalysts may exist that can produce D-ribose more efficiently, but have not yet been discovered. Alternatively, it is possible that selectivity could be achieved at the stage of nucleoside synthesis, if a catalyst for that assembly were able to selectively utilize D-ribose out of a complex mixture, but again, no evidence for such a scenario has been demonstrated.
An additional problem with prebiotic accumulation of sugars lies in the relative instability of the compounds. This is especially true in the alkaline conditions of the standard formose reaction, where sugars are made very rapidly but undergo further reactions that quickly render them unusable. Even under milder conditions, the instability of the sugars make it unlikely that ribose could persist in solution and accumulate on the prebiotic Earth on time scales that are typically assumed for the origin of life (64). However, the clay minerals that have been shown to catalyze the synthesis of ribose phosphate have also been shown to provide a protected environment to sequester those products, resulting in increased stability relative to free solution (61). An alternative approach involves creating stable adducts of ribose with other molecules. Recently, Benner and co-workers have demonstrated that certain borate minerals can substantially stabilize sugars, such as ribose, that have hydroxyl groups on adjacent carbons, sequestering the sugar in a stable complex and thereby prolonging the sugar’s availability for further assembly (65). Similarly, the product of ribose and cyanamide forms an insoluble adduct that crystallizes out of solution protecting the ribose from solution-mediated degradation.
Nucleosides
A nucleoside is the combination of a base and ribose. Their formation, in theory, is most directly achieved by a reaction involving pre-formed bases and ribose, but this approach, so far, has shown little promise. The best attempts have been the demonstrations that the dry-phase heating of purine bases and ribose leads to the formation of purine nucleosides (66, 67). Further, the yields are enhanced several-fold in the presence of certain salts, including evaporated seawater. However, the same investigators were unable to substantiate an earlier report claiming the formation of adenosine from dilute solutions of adenine and ribose exposed to ultraviolet light, and were also unable to detect any nucleoside formation in aqueous solution under a wide variety of conditions. This dry-phase synthesis, while producing nucleosides in yields of several percent, is complicated by a side reaction in which bases bearing exocyclic amines (adenine, guanine) are preferentially ribosylated on the amine group in yields of up to 74% (66). Similar reactions with hypoxanthine, which does not have an exocyclic amine group, were not affected by this side reaction, and produced inosine in yields comparable to the adenine and guanine reactions. As was the case with a lack of selectivity between synthesis of D and L enantiomers, a similar lack of selectivity exists in the formation of α and β anomers.
The problem of pyrimidine nucleoside synthesis has been even more challenging than purines. In dry-phase heating experiments, the pyrimidines do not form detectable amounts of nucleoside (66). Alternative strategies include using prebiotic reagents to build a base on a pre-formed ribose (68-70), or the converse, building a sugar on a pre-formed base. Both tactics have had degrees of success, but neither seem to be the simple and robust processes that might be expected of a convincing prebiotic synthesis, especially of such a crucial class of biomolecules. Still another approach uses other pyrimidines (not C, U, or T) (71) or pyrimidine-like bases (72) that more readily form nucleosides with ribose. In a subsequent step, these non-standard nucleosides are then either chemically converted to a standard pyrimidine or else invoked as a precursor to conventional RNA
Nucleotides
The last step to make an RNA monomer is to add phosphate. This step can be coupled with chemical activation of the nucleoside in order to make the polymerization of monomers energetically favorable. A phosphate links each nucleoside monomer together in an RNA polymer through phosphodiester bonds between the 5' and 3' hydroxyls on the riboses of adjacent nucleotides. The activated form of the nucleotide used by all RNA polymerases in contemporary biology is the nucleoside 5'-triphosphate (NTP). The energy that drives polymerization is accessed by breaking the bond linking the α and β phosphate groups, with pyrophosphate released as a byproduct. Attempts to create nucleotides by phosphorylating nucleosides under prebiotic conditions have had mixed success. The most successful approach involves the dry-phase phosphorylation of nucleosides with phosphate salts by heating a dried solution of nucleoside and inorganic phosphate. This reaction produces an assortment of different mono-phosphorylated nucleotides (NMPs) (73). The addition of urea to the reaction increases the yields substantially, but still produces a mixture of products, including 3' and 2' phosphates and 2',3'-cyclic phosphates (NMcPs), which, in many cases, are the major product (74). The relative yields of products are sensitive to reaction conditions, which can be fine tuned to favor the accumulation of 5'-NMP (75). For instance, the temperature of the reaction affects the distribution of products, particularly the formation of NMcP. The earliest experiments were carried out at temperatures of 100oC and higher and resulted in the greatest proportion of NMcP phosphorylated products. Simply lowering the temperature to 65 °C greatly reduces the amount of NMcP relative to the 5'-NMP product. However, lowering the temperature below 60 °C inhibits all phosphorylation (75).
An alternative to the dry-phase phosphorylation schemes involves the use of a condensing agent in aqueous solution. Small yields of NMPs are produced from heated solutions of nucleoside, phosphate, and certain prebiotically feasible condensing agents such as cyanogen, cyanate, and cyanoacetylene (76, 77). The yields are low because none of the tested condensing agents provide any selectivity for the nucleoside over water. Consequently, water is able to out-compete nucleoside for phosphorylation due to its substantial concentration advantage. With what little phosphorylation of the nucleoside that does occur, as with the dry-phase reactions, NMcP is found as a major product of these reactions. The observation that the unimolecular cyclization of 3'-NMP to NMcP proceeds efficiently under these conditions is consistent with this model (78).
Other possibilities include adding or building a base onto a pre-formed sugar phosphate as mentioned in the previous section (69, 70) or utilizing phosphate derived from minerals or extraterrestrial sources. For example, the phosphorous-containing mineral schreibersite, found in many meteorites, decomposes in aqueous solution generating compounds such as phosphate, pyrophosphate, and phosphite (79). The proposed degradation pathway proceeds through a phosphite radical intermediate, which could potentially be useful in producing polyphosphates. In this regard, phosphite has been suggested as an attractive prebiotic alternative to phosphate due to advantages in solubility and reactivity (80).
The mono-phosphorylation discussed above completes the nucleotide unit, but does not create the high energy activated phosphate that drives polymerization, so an additional step must be invoked to complete the activated nucleotide. This could occur either by using a nucleotide with an activated phosphate group or in the form of using an external activating agent to effect the polymerization process. Polyphosphates can be made under similar conditions (81, 82), but generally require higher temperatures to achieve, and their utilization in a prebiotic setting has been questioned (83).
Cyclic phosphates
The emphasis in the area of nucleotide synthesis is almost always focused on 5' activated phosphates, based in large measure, on nature’s (current) preference for NTPs. Alternatives to this model include 2',3'-cyclic phosphates, which have a strong case for consideration. First, they are often the major products, sometimes substantially so, of the prebiotic phosphorylation experiments discussed above (74, 78). The cyclic phosphate is an activated phosphodiester and so is ready for potential polymerization without the need for an additional activating step or condensing agent. Finally, since the natural breakdown product of any RNA polymer is NMcPs, their direct utilization in the polymerization process, whether spontaneous or catalyzed, is attractive on the basis of simplicity and efficiency. The main objections to the utilization of NMcPs are based not on their likely prebioticity, but rather on their weak performances in non-enzymatic template-directed polymerization experiments (84).
Nonenzymatic Assembly of Oligonucleotides
The prebiotic utility of nucleotides lies in their ability to form long, polymeric chains that store genetic information and form complex and functional macromolecular shapes. Any initial assembly of oligonucleotides presumably would have been non-nucleotide-templated and occurred either in solution or perhaps on a clay or mineral surface. The energy to drive polymerization could come either in the form of a pre-activated nucleotide or by utilizing an external activating agent during polymerization (discussed above). The latter approach, which bypasses the pre-activation of the nucleoside phosphate, would balance that advantage in the inefficiency of polymerization. Typical experiments yield small amounts of mostly dimers and trimers, and among these are a mixture of the “natural” 3',5' internucleotide linkages with unnatural 2',5' linkages, as well as other complex products including 5',5'-pyrophosphate linkages (85). The alternative strategy of pre-activating the nucleotide has produced the most robust laboratory results. The choice of activating group is dictated primarily by practical considerations. Nucleoside triphosphates, nature’s chosen activating group, react too slowly to be practical for the laboratory. Other activating groups have been explored, and among these, the most studied have been the phosphorimidazolides. The prevalent use of phosphorimidazolides in these types of non-enzymatic polymerization studies is based on the simplicity of their synthesis and utilization, as well as their successful results. The steps and components of their synthesis can be considered plausibly prebiotic, but unlikely to persist and accumulate to reasonable levels. Their popularity as a polymerization substrate is less an endorsement of their prebiotic relevance and more the adoption of a well-behaved model system with which to address general and, hopefully, fundamental questions about the behavior of non-enzymatic RNA polymerization.
Polymerizations of this type in solution produce predominantly shorter than 5-mer oligonucleotides with a mixed and variable composition of 2',5' and 3',5' linkages (86). The distribution of products is highly sensitive to reaction conditions, including the presence of metal ions, modification of the leaving group, and sequence context. The addition of the clay montmorillonite to otherwise similar reactions has a favorable effect on polymerization (87). The effect is likely due to the concentration of monomers within the clay’s cationic layers. These clay-catalyzed experiments have produced polynucleotides up to 55 nucleotides long with approximately 80 percent 3',5'-linked composition (88).
Template-directed synthesis
The appeal of RNA or any nucleic acid as a genetic material is due to its ability to base-pair and consequently serve as a template for the synthesis of complementary copies of itself. Eventually, random, non-templated RNA assembly would have had to give way to template-directed systems able to propagate beneficial genetic information from one generation to the next. When a template is added to prebiotic polymerization simulations, the effects can vary dramatically depending on the nucleotide composition of the template. In basic experiments with homopolymeric templates, the complementary strand is polymerized from monomers that base-pair with the template. The incorporation of purines, particularly guanosine, into the complementary strand is much more efficient than pyrimidine incorporation (89). These trends hold true for mixed sequence systems: good templates are generally those with a high percentage of cytidine, directing the incorporation of guanosine. Such a system might be a good template from which to make a reverse-complementary copy, but that copy would be an extremely poor template from which to regenerate the original oligonucleotide. The complementary nature of nucleic acid replication cannot succeed with such an extreme templating bias unless another, currently unknown, rescue mechanism is postulated.
The increased efficiency of oligomerizing dimers, trimers, or longer oligomers instead of monomers is an attempt to address some of these difficulties. The energetics of base-pairing longer stretches of nucleotides increases binding efficiency, but unless the pieces are sufficiently long, the templating peculiarities just discussed can still apply to oligomerizations and severely restrict the number of replicable sequences. The feasibility of this kind of system has been characterized using a carefully chosen palindromic system with trimers ligating on a hexamer template (14). A modified system using a manual denaturation step to separate the product strands from the template strands during each generation of copies has demonstrated exponential replication of nucleic acids (15).
Enantiomeric cross-inhibition
A previously mentioned difficulty in prebiotic synthesis, now re-surfacing for this discussion of prebiotic template-directed oligonucleotide synthesis, is the problem of sugar handedness. Unless a reasonable mechanism or process is found that leads to the accumulation of one sugar enantiomer over another, the prebiotic synthesis of sugars must be assumed to produce an equal mixture of D and L ribose, and, consequently, also in the absence of a reasonable selectivity mechanism, lead to a mixture of D and L nucleosides. Studies of uncatalyzed templated polymerizations using a mixture of nucleoside enantiomers indicate that the non-biological “L” enantiomer acts as a chain terminator when incorporated against a D enantiomer template, blocking further templated extension of the oligonucleotide (90). This is another serious difficulty without a satisfactory resolution with regard to the uncatalyzed template-directed polymerization of RNA nucleotides.
Ribozyme to Replicase
Ignoring, for the moment, the challenges associated with the abiotic synthesis of RNA polymers, the emergence of the RNA World still depends on the ability of RNA to catalyze its own replication in some fashion. An RNA-dependent RNA polymerase is the class of enzyme that modern biology would use for this purpose, and a ribozyme with this functionality is thought to be central to the beginnings and propagation of the RNA World. No such ribozyme has yet been discovered in nature, but a laboratory re-creation of a ribozyme with that functionality, though not proof of its historical existence, would show that it could once have existed, and validate a fundamental pillar of the RNA World hypothesis. The isolation of RNA ligase ribozymes from a population of random sequence RNA molecules was the start of an experimental progression that has come closest to realizing the goal of an RNA polymerase ribozyme (91). RNA ligase ribozymes catalyze the identical chemical reaction as an RNA polymerase, but instead of the triphosphate-bearing half of the reaction being an NTP, it is instead present as the 5'-triphosphate of the 5' terminus of the ribozyme itself (Fig. 5). In terms of in vitro selection targets, ligase activity appears to be relatively prevalent in RNA populations after having been isolated under different reaction conditions by several teams using different starting populations of RNA. The phrase “relatively prevalent”, in this case, means on the order of one ligase ribozyme in every 1013-1014 RNA molecules (91), only about an order of magnitude lower frequency than experiments isolating self-cleaving ribozymes from random sequence populations (92, 93). While these probabilities may seem relatively low, they correspond to finding a ~100 nucleotide ligase ribozyme in approximately every microgram of random RNA, a trivial amount of material assuming that mechanisms for the prebiotic synthesis of RNA oligomers existed at all.
A strategy to convert the RNA ligase ribozyme into a general RNA polymerase involves separating the template and substrate from the ribozyme, optimizing the enzyme to “ligate” single nucleotides as substrates, and translocating along the template to the next position. Following a symbiotic regime of rational design and in vitro evolution, Bartel and his coworkers created a polymerase ribozyme able to polymerize along a template up to 14 nucleotides, the equivalent of a full turn of RNA helix (94). Technological advances of in vitro selection methodologies have led to a modest improvement of more than 20 polymerized nucleotides, which represents the current state of the art in polymerase ribozymes (95). This lineage of experiments has generated the most sophisticated artificial ribozyme known to science, but still falls well short of the presumed efficiency that would be needed in the RNA World.
Nucleoside-2',3'-cyclic phosphates have been mentioned as candidates for activated monomers in the context of nonenzymatic polymerizations, and also deserve mention in the context of ribozyme-catalyzed polymerizations. The initial experimental verification of the possibility of a cyclic phosphate polymerase was the isolation of cyclic phosphate RNA ligase deoxyribozymes (96). Once again, in vitro selection was used to explore large populations of both DNA (97)and RNA (M.P. Robertson, J.E. Blaustein, and W.G. Scott, unpublished results) and isolate new ligases. Attempts to convert these ligases into polymerases have not been reported.
RNA has proven to be a remarkably malleable tool in the hands of biochemists, and a satisfactory RNA-dependent RNA polymerase ribozyme will probably be developed in the laboratory eventually. When that happens, it will be the culmination of at least a decade’s worth of directed efforts, using complex combinations of rational design and in vitro evolution. It will be an important demonstration that further strengthens the case that an RNA World could have existed, but it will also show that RNA polymerase ribozymes do not appear to be a simple functionality that is likely to appear de novo from a naive population of RNA molecules. The possibility of a self-replicating RNA system less sophisticated than a general RNA-dependent RNA polymerase ribozyme cannot be eliminated.
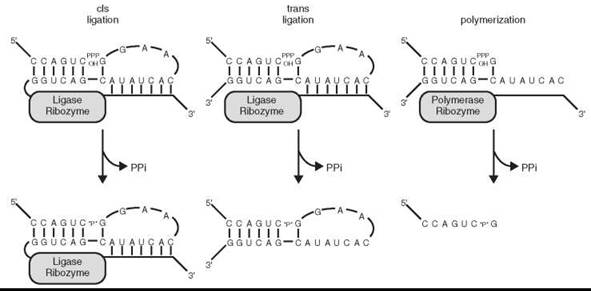
Figure 5. RNA ligation and polymerization. A schematic representation, based on the Class I RNA ligase ribozyme, illustrating that the difference between a ligase and a polymerase lies primarily in the substrates utilized and not in the chemistry of the reactions.
Ribozymic Metabolism
The minimal theoretical requirement to initiate the RNA World is a self-replicating RNA polymerase ribozyme. Realistically, supporting functionalities would soon be necessary to sustain an emerging RNA World. For example, a reliable supply of activated nucleotides would have been essential as the polymerases depleted the presumably scarce supply of abiotically produced material. To demonstrate that ribozymes are capable of catalyzing such complex chemistries, a nucleotide synthetase ribozyme was created in the laboratory (98). The ribozyme appends a base to an activated ribose to form a nucleoside. This is just one step in what can be imagined as an ever more complex network of metabolic pathways supporting the propagation of the RNA organism. But while the boundaries of the RNA World begin with an RNA polymerase, and begin to end with the invention of protein synthesis, less can be inferred about what happened in between and what level of complexity was achieved before the RNA World began to wane. Based on the defined starting and end points of the RNA World, a polymerase with a supporting metabolism to produce its activated nucleotide substrates would be necessary. Closer to the end, a ribosomal-type translation system with a supporting metabolism to produce its necessary substrates is believed to have been in place. Beyond these examples, modern biology may hold clues of extant molecular relics of RNA World functionalities. In fact, the case in favor of an RNA World is built, in part, on the ubiquity of nucleotide-based cofactors throughout biochemistry and the suggestion that they are remnants of an extensive RNA-catalyzed metabolism (99). While the range of chemical reactions that natural ribozymes are known to catalyze is very limited, in vitro selection has generated a rich collection of ribozymes with diverse functionalities indicating that, under the right conditions, RNA is capable of catalyzing a large subset of the reactions normally catalyzed by protein enzymes, though not as efficiently.
Another characteristic of living systems that is thought to have been utilized very early in the evolution of life, if not concurrently or even preceding RNA replication, is compart- mentalization. All modern cells are enclosed by membranes that act as a barrier to regulate the intake and escape of molecules from the system. This functionality is necessary to contain the byproducts of metabolism and as a mechanism to link genotype with phenotype, which would be just as important for the earliest forms of life. For instance, an RNA replicase operating free in solution without boundaries might be able to replicate RNA molecules that it encounters, but the most efficient replicases would not benefit from an evolutionary advantage unless their systems and progeny were segregated from less efficient competitors (100). In simple in vitro systems involving vesicle-encapsulated RNA, the synthesis of additional RNA resulted in spontaneous vesicle growth at the expense of empty vesicles (101). Preliminary studies with these types of protocell systems have shown that basic properties like growth, encapsulation, and division occur as a consequence of purely physical properties and do not necessarily require additional catalytic functionalities in order to be operational (101).
preRNA
In most discussions, the RNA World is synonymous with the origin of life, but this need not necessarily be the case. The difficulties associated with the prebiotic synthesis of long RNA polymers have led many to speculate that an alternative, simpler biopolymer-that can somehow eliminate the problems associated with RNA-preceded the RNA World. Most of these “preRNA” candidates retain the familiar Watson-Crick base pairing between strands, but a number of different types of alternative backbone systems have been proposed, including a non-cyclic carbohydrate (102), peptide (103), and a variety of non-ribose sugars (104, 105). But unlike RNA, no recognizable molecular artifacts have yet been found in support of any of them. In the absence of direct evidence, the enthusiasm for any particular candidate can only be judged against a number of practical factors, including the ease and likelihood of its prebiotic synthesis pathway and the ability to ultimately transfer its sequence information to an RNA polymer. With examples of the genetic transfer of sequence information from a non-RNA polymer to a strand of RNA having been demonstrated (106), the main hurdle, like RNA, is in the ease and likelihood of its prebiotic synthesis. preRNA candidates have one or more attributes that address a shortcoming of RNA, but none of the candidates are without flaws themselves. In this respect, no current preRNA candidate has distinguished itself as clearly superior to the others.
Summary
The mysteries of how life originated on this planet and came to achieve its current complexity hold a special fascination in the imagination of humankind. The earliest eras of life’s evolution are very poorly understood, but fragments of evidence exist. The RNA World hypothesis outlines a description of how the earliest molecular forms of life may have arisen. The fragmented evidence that points most convincingly toward an RNA World relies almost entirely on what is known of modern biochemistry and the plausibility of reverse engineering a pathway from now to then. The converse, however, the period of time leading up to the moment of RNA-based life, is more challenging to imagine. Difficulties and uncertainties exist at every step leading up to the emergence of the RNA World, from the inefficiencies of abiotic organic syntheses to the probability of inventing a ribozyme more complex than any that has been seen today. Because of the difficulties of creating an RNA World from scratch, many believe that a preRNA World based on a simpler, nucleic acid-like polymer, preceded the RNA World as the first living system.
References
1. Joyce GF. RNA evolution and the origins of life. Nature 1989; 338:217-224.
2. Benner SA, Ellington AD, Tauer A. Modern metabolism as a palimpsest of the RNA world. Proc. Natl. Acad. Sci. U. S. A. 1989; 86:7054-7058.
3. Woese C. The Genetic Code: The Molecular Basis for Genetic Expression. 1967. Harper & Row, New York.
4. Orgel LE. Evolution of the genetic apparatus. J. Mol. Biol. 1968; 38:381-393.
5. Crick FHC. The origin of the genetic code. J. Mol. Biol. 1968; 38:367-379.
6. Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983; 35:849-857.
7. Kruger K, Grabowski PJ, Zaug AJ, Sands J, Gottschling DE, Cech TR. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell 1982; 31:147-157.
8. Gilbert W. The RNA world. Nature 1986; 319:618.
9. Cleland CE, Chyba CF. Defining ‘life’. Orig. Life Evol. Biosph. 2002; 32:387-393.
10. Joyce GF. In: Origins of Life: The Central Concepts. Deamer DW, Fleischaker GR, eds. 1994. Jones and Bartlett Publishers, Boston, MA. pp. xi-xii.
11. Crick FHC. On protein synthesis. Symp. Soc. Exp. Biol. 1958; 12:138-163.
12. Crick F. Central dogma of molecular biology. Nature 1970; 227:561-563.
13. Zielinski WS, Orgel LE. Autocatalytic synthesis of a tetranucleotide analogue. Nature 1987; 327:346-347.
14. Sievers D, von Kiedrowski G. Self-replication of complementary nucleotide-based oligomers. Nature 1994; 369:221-224.
15. Luther A, Brandsch R, von Kiedrowski G. Surface-promoted replication and exponential amplification of DNA analogues. Nature 1998; 396:245-248.
16. James KD, Ellington AD. The fidelity of template-directed oligonucleotide ligation and the inevitability of polymerase function. Orig. Life Evol. Biosph. 1999; 29:375-390.
17. Noller HF, Hoffarth V, Zimniak L. Unusual resistance of peptidyl transferase to protein extraction procedures. Science 1992; 256:1416-1419.
18. Yusupov MM, Yusupova GZ, Baucom A, Lieberman K, Earnest TN, Cate JH, Noller HF. Crystal structure of the ribosome at 5.5 A resolution. Science 2001; 292:883-896.
19. Watson JD Crick FHC. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 1953; 171:737-738.
20. Miller SL. A production of amino acids under possible primitive earth conditions. Science. 1953; 117:528-529.
21. Oro J. Synthesis of adenine from ammonium cyanide. Biochem. Biophys. Res. Commun. 1960; 2:407-412.
22. Oro J, Kimball AP. Synthesis of purines under possible primitive earth conditions. I. Adenine from hydrogen cyanide. Arch. Biochem. Biophys. 1961; 94:217-227.
23. Oro J, Kimball AP. Synthesis of purines under possible primitive earth conditions. II. Purine intermediates from hydrogen cyanide. Arch. Biochem. Biophys. 1962; 96:293-313.
24. Roy D, Najafian K, Schleyer PV. Chemical evolution: The mechanism of the formation of adenine under prebiotic conditions. Proc. Natl. Acad. Sci. U. S. A. 2007; 104:17272-17277.
25. Ferris JP, Orgel LE. An unusual photochemical rearrangement in the synthesis of adenine from hydrogen cyanide. J. Am. Chem. Soc. 1966; 88:1074.
26. Ferris JP, Orgel LE. Studies on prebiotic synthesis. I. Aminomalononitrile and 4-amino-5-cyanoimidazole. J. Am. Chem. Soc. 1966; 88:3829-3831.
27. Ferris JP, Joshi PC, Edelson EH, Lawless JG. HCN: a plausible source of purines, pyrimidines and amino acids on the primitive earth. J. Mol. Evol. 1978; 11:293-311.
28. Levy M, Miller SL, Oro J. Production of guanine from NH(4)CN polymerizations. J. Mol. Evol. 1999; 49:165-168.
29. Ponnamperuma C, Lemmon RM, Mariner R, Calvin M. Formation of adenine by electron irradiation of methane, ammonia, and water. Proc. Natl. Acad. Sci. U. S. A. 1963; 49:737-740.
30. Yuasa S, Ishigami M. High frequency discharge experiment. I: formation of organic compounds from methane and ammonia. Orig. Life. 1975; 6:75-81.
31. Yuasa S, Flory D, Basile B, Oro J. Abiotic synthesis of purines and other heterocyclic compounds by the action of electrical discharges. J. Mol. Evol. 1984; 21:76-80.
32. Miller SL. The mechanism of synthesis of amino acids by electric discharges. Biochim. Biophys. Acta. 1957; 23:480-489.
33. Davidson D, Baudisch O. The preparation of uracil from urea. J. Am. Chem. Soc. 1926; 48:2379-2383.
34. Fox SW, Harada K. Synthesis of uracil under conditions of a thermal model of prebiological chemistry. Science 1961; 133:1923-1924.
35. Oro J. Non-enzymatic formation of purines and pyrimidines. Fed. Proc. 1963; 22:681.
36. Ferris JP, Joshi PC, Lawless JG. Chemical evolution XXIX. Pyrimidines from hydrogen cyanide. Biosystems 1977; 9:81-86.
37. Voet AB, Schwartz AW. Uracil synthesis via HCN oligomerization. Orig. Life 1982; 12:45-49.
38. Chittenden GJ, Schwartz AW. Possible pathway for prebiotic uracil synthesis by photodehydrogenation. Nature 1976; 263:350-351.
39. Schwartz AW, Chittenden GJ. Synthesis of uracil and thymine under simulated prebiotic conditions. Biosystems 1977; 9:87-92.
40. Ferris JP, Sanchez RA, Orgel LE. Studies in prebiotic synthesis III. Synthesis of pyrimidines from cyanoacetylene and cyanate. J. Mol. Biol. 1968; 33:693-704.
41. Sanchez RA, Ferris JP, Orgel LE. Cyanoacetylene in prebiotic synthesis. Science 1966; 154:784-785.
42. Ferris JP, Zamek OS, Altbuch AM, Freiman H. Chemical evolution. 18. Synthesis of pyrimidines from guanidine and cyanoacetaldehyde. J. Mol. Evol. 1974; 3:301-309.
43. Robertson MP, Miller SL. An efficient prebiotic synthesis of cytosine and uracil. Nature 1995; 375:772-774.
44. Stribling R, Miller SL. Energy yields for hydrogen cyanide and formaldehyde syntheses: the HCN and amino acid concentrations in the primitive ocean. Orig. Life Evol. Biosph. 1987; 17:261-273.
45. Sanchez RA, Ferris JP, Orgel LE. Studies in prebiotic synthesis. II. Synthesis of purine precursors and amino acids from aqueous hydrogen cyanide. J. Mol. Biol. 1967; 30:223-253.
46. Nelson KE, Robertson MP, Levy M, Miller SL. Concentration by evaporation and the prebiotic synthesis of cytosine. Orig. Life Evol. Biosph. 2001; 31:221-229.
47. Sanchez R, Ferris J, Orgel LE. Conditions for purine synthesis: did prebiotic synthesis occur at low temperatures? Science 1966; 153:72-73.
48. Kvenvolden K, Lawless J, Pering K, Peterson E, Flores J, Ponnamperuma C, Kaplan IR, Moore C. Evidence for extraterrestrial amino-acids and hydrocarbons in the Murchison meteorite. Nature 1970; 228:923-926.
49. Kvenvolden KA, Lawless JG, Ponnamperuma, C. Nonprotein amino acids in the Murchison meteorite. Proc. Natl. Acad. Sci. U. S. A. 1971; 68:486-490.
50. Hayatsu R, Studier MH, Moore LP, Anders E. Purines and triazines in the Murchison meteorite. Geochim. Cosmochim. Acta 1975; 39:471-488.
51. Stoks PG, Schwartz AW. Uracil in carbonaceous meteorites. Nature 1979; 282:709-710.
52. Stoks PG, Schwartz AW. Nitrogen-heterocyclic compounds in meteorites: significance and mechanisms of formation. Geochim. Cosmochim. Acta 1981; 45:563-569.
53. Chyba C, Sagan C. Endogenous production, exogenous delivery and impact-shock synthesis of organic molecules: an inventory for the origins of life. Nature 1992; 355:125-132.
54. Bernstein M. Prebiotic materials from on and off the early Earth. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2006; 361:1689-1700.
55. Butlerow A. Formation synthetique d’une substance sucree. C. R. Acad. Sci. 1861; 53:145-147.
56. Shapiro R. Prebiotic ribose synthesis: a critical analysis. Orig. Life Evol. Biosph. 1988; 18:71-85.
57. Gabel NW, Ponnamperuma C. Model for origin of monosaccharides. Nature 1967; 216:453-455.
58. Reid C, Orgel LE. Synthesis of sugars in potentially prebiotic conditions. Nature 1967; 216:455.
59. Decker P, Schweer H, Pohlmann R. Identification of formose sugars, presumable prebiotic metabolites, using capillary gas chromatography/gas chromatography-mass spectrometry of n-butoxime trifluoroacetates on OV-225. J. Chromatog. 1982; 244:281-291.
60. Muller D, Pitsch S, Kittaka A, Wagner E, Wintner CE, Eschenmoser A, Ohlofjgewidmet G. Chemie von a-aminonitrilen. Aldomerisierung von glycolaldehyd-phosphat zu racemischen hexose-2,4,6-triphosphaten und (in Gegenwart von Formaldehyd) racemischen pentose-2,4-diphosphaten: rac-allose-2,4,6-triphosphat und rac-ribose-2,4-diphosphat sind die Reaktionshauptprodukte. Helv. Chim. Acta 1990; 73:1410-1468.
61. Krishnamurthy R, Pitsch S, Arrhenius G. Mineral induced formation of pentose-2,4-bisphosphates. Orig. Life Evol. Biosph. 1999; 29:139-152.
62. Sacerdote, MG and Szostak, JW. Semipermeable lipid bilayers exhibit diastereoselectivity favoring ribose. Proc. Natl. Acad. Sci. U. S. A. 2005; 102:6004-6008.
63. Springsteen G, Joyce GF. Selective derivatization and sequestration of ribose from a prebiotic mix. J. Am. Chem. Soc. 2004; 126:9578-9583.
64. Larralde R, Robertson MP, Miller SL. Rates of decomposition of ribose and other sugars: implications for chemical evolution. Proc. Natl. Acad. Sci. U. S. A. 1995; 92:8158-8160.
65. Ricardo A, Carrigan MA, Olcott AN, Benner SA. Borate minerals stabilize ribose. Science 2004; 303:196.
66. Fuller WD, Sanchez RA, Orgel LE. Studies in prebiotic synthesis. VI. Synthesis of purine nucleosides. J. Mol. Biol. 1972; 67:25-33.
67. Fuller WD, Sanchez RA, Orgel LE. Studies in prebiotic synthesis: VII. Solid-state synthesis of purine nucleosides. J. Mol. Evol. 1972; 1:249-257.
68. Sanchez RA, Orgel LE. Studies in prebiotic synthesis. V. Synthesis and photoanomerization of pyrimidine nucleosides. J. Mol. Biol. 1970; 47:531-543.
69. Ingar AA, Luke RW, Hayter BR, Sutherland JD. Synthesis of cytidine ribonucleotides by stepwise assembly of the heterocycle on a sugar phosphate. ChemBioChem. 2003; 4:504-507.
70. Powner MW, Anastasi C, Crowe MA, Parkes AL, Raftery J, Sutherland JD. On the prebiotic synthesis of ribonucleotides: photoanomerisation of cytosine nucleosides and nucleotides revisited. ChemBioChem. 2007; 8:1170-1179.
71. Bean HD, Sheng Y, Collins JP, Anet FA, Leszczynski J, Hud NV. Formation of a beta-pyrimidine nucleoside by a free pyrimidine base and ribose in a plausible prebiotic reaction. J. Am. Chem. Soc. 2007; 129:9556-9557.
72. Kolb VM, Dworkin JP, Miller SL. Alternative bases in the RNA world: the prebiotic synthesis of urazole and its ribosides. J. Mol. Evol. 1994; 38:549-557.
73. Ponnamperuma C, Mack R. Nucleotide Synthesis under Possible Primitive Earth Conditions. Science 1965; 48:1221-1223.
74. Lohrmann R, Orgel LE. Urea-inorganic phosphate mixtures as prebiotic phosphorylating agents. Science 1971; 171:490-494.
75. Reimann R, Zubay G. Nucleoside phosphorylation: a feasible step in the prebiotic pathway to RNA. Orig. Life Evol. Biosph. 1999; 29:229-247.
76. Lohrmann R, Orgel LE. Prebiotic synthesis: phosphorylation in aqueous solution. Science 1968; 161:64-66.
77. Ferris JP, Goldstein G, Beaulieu DJ. Chemical evolution IV. An evaluation of cyanovinyl phosphate as a prebiotic phosphorylating agent. J. Am. Chem. Soc. 1970; 92:6598-6603.
78. Ferris JP, Yanagawa H, Hagan WJ Jr. The prebiotic chemistry of nucleotides. Orig. Life 1984; 14:99-106.
79. Pasek MA, Dworkin JP, Lauretta DS. A radical pathway for organic phosphorylation during schreibersite corrosion with implications for the origin of life. Geochim. Cosmochim. Acta 2007; 71:1721-1736.
80. Pasek MA. Rethinking early Earth phosphorus geochemistry. Proc. Natl. Acad. Sci. U. S. A. 2008; 105:853-858.
81. Osterberg R, Orgel LE. Polyphosphate and trimetaphosphate formation under potentially prebiotic conditions. J. Mol. Evol. 1972; 1:241-248.
82. Schwartz AW, van der V, Bisseling T, Chittenden GJ. Prebiotic nucleotide synthesis-demonstration of a geologically plausible pathway. Orig. Life. 1975; 6:163-168.
83. Keefe AD, Miller SL. Are polyphosphates or phosphate esters prebiotic reagents? J. Mol. Evol. 1995; 41:693-702.
84. Renz M, Lohrmann R, Orgel LE. Catalysts for the polymerization of adenosine cyclic 2',3'-phosphate on a poly (U) template. Biochim. Biophys. Acta 1971; 240:463-471.
85. Orgel LE. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol. 2004; 39:99-123.
86. Sawai H, Orgel LE. Oligonucleotide synthesis catalyzed by the Zn2+ ion. J. Am. Chem. Soc. 1975; 97:3532-3533.
87. Ferris JP, Ertem G. Oligomerization of ribonucleotides on montmorillonite: reaction of the 5'-phosphorimidazolide of adenosine. Science 1992; 257:1387-1389.
88. Ferris JP, Hill AR Jr, Liu R, Orgel LE. Synthesis of long prebiotic oligomers on mineral surfaces. Nature 1996; 381:59-61.
89. Joyce GF. Nonenzymatic template-directed synthesis of informational macromolecules. Cold Spring Harb. Symp. Quant. Biol. 1987; 52:41-51.
90. Joyce GF, Visser GM, van Boeckel CA, van Boom JH, Orgel LE, van Westrenen J. Chiral selection in poly(C)-directed synthesis of oligo(G). Nature 1984; 310:602-604.
91. Bartel DP, Szostak JW. Isolation of new ribozymes from a large pool of random sequences. Science 1993; 261:1411-1418.
92. Tang J, Breaker RR. Structural diversity of self-cleaving ribozymes. Proc. Natl. Acad. Sci. U. S. A. 2000; 97:5784-5789.
93. Salehi-Ashtiani K, Szostak JW. In vitro evolution suggests multiple origins for the hammerhead ribozyme. Nature 2001; 414: 82-84.
94. Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science 2001; 292:1319-1325.
95. Zaher HS, Unrau PJ. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA 2007; 13:1017-1026.
96. Flynn-Charlebois A, Wang Y, Prior TK, Rashid I, Hoadley KA, Coppins RL, Wolf AC, Silverman SK. Deoxyribozymes with 2'-5' RNA ligase activity. J. Am. Chem. Soc. 2003; 125:2444- 2454.
97. Coppins RL, Silverman SK. Rational modification of a selection strategy leads to deoxyribozymes that create native 3’-5’ RNA linkages. J. Am. Chem. Soc. 2004; 126:16426-16432.
98. Unrau PJ, Bartel DP. RNA-catalysed nucleotide synthesis. Nature 1998; 395:260-263.
99. White HB 3rd. Coenzymes as fossils of an earlier metabolic state. J. Mol. Evol. 1976; 7:101-104.
100. Szostak JW, Bartel DP, Luisi PL. Synthesizing life. Nature 2001; 409:387-390.
101. Chen IA, Roberts RW, Szostak JW. The emergence of competition between model protocells. Science 2004; 305:1474-1476.
102. Joyce GF, Schwartz AW, Miller SL, Orgel LE. The case for an ancestral genetic system involving simple analogues of the nucleotides. Proc. Natl. Acad. Sci. U. S. A. 1987; 84:4398-4402.
103. Nielsen PE. Peptide nucleic acids and the origin of life. Chem. Biodivers. 2007; 4:1996-2002.
104. Schoning K, Scholz P, Guntha S, Wu X, Krishnamurthy R, Eschenmoser A. Chemical etiology of nucleic acid structure: the alpha-threofuranosyl-(3'->2') oligonucleotide system. Science 2000; 290:1347-1351.
105. Beier M, Reck F, Wagner T, Krishnamurthy R, Eschenmoser A. Chemical etiology of nucleic acid structure: comparing pentopyranosyl-(2’->4’) oligonucleotides with RNA. Science 1999; 283:699-703.
106. Bohler C, Nielsen PE, Orgel LE. Template switching between PNA and RNA oligonucleotides. Nature 1995; 376:578-581.
Further Reading
Gesteland RF, Atkins JF. The RNA World. 1993. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Gesteland RF, Cech TR, Atkins JF. The RNA World. 1998. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Gesteland RF, Cech TR, Atkins JF. The RNA World. 2005. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
See Also
Origins of Life: Emergence of Nucleic Acids
In Vitro Selection and Application of Nucleic Acid Enzymes (Ribozymes and Deoxyribozymes)
Catalytic Modes in Natural Ribozymes