CHEMICAL BIOLOGY
Catalytic Antibodies: Past, Present, and Future
Andrew P. Brogan, Tobin J. Dickerson and Kim D. Brogan, The Scripps Research Institute, La Jolla, California
doi: 10.1002/9780470048672.a0000054
Catalytic antibodies have emerged as powerful tools for the chemical biologist, enabling the design and realization of specific catalysis for a wide range of chemical reactions. Catalytic antibodies are grounded upon transition state theory and envisioned as programmable mimics of enzyme catalysis. Affinity maturation of the immune response for a small-molecule hapten elicits binding site complementarity within an antibody that facilitates chemical catalysis. The evolution of hapten design strategies for chemical catalysis is presented, including the transition state analog approach, strain-induced hapten design, ''bait-and-switch,'' and ''reactive immunization.'' The range and scope of antibody catalysis is examined by reaction class, highlighting structural and mechanistic investigations to explore the roots of chemical catalysis by these designer biocatalysts. Recently, antibodies, regardless of disposition or origin, have been shown to catalyze the oxidation of water, equipping the antibody with a mechanism for antigen decomposition. These recent developments are presented along with the utilization of this pathway in the oxidative degradation of a commonly abused drug. The achievements in antibody catalysis have enriched our scientific understanding of chemical catalysis, particularly by biological molecules in aqueous systems. However, realizing more operative rate enhancements on the same order as natural enzymes remains as the ''holy grail'' of this field.
catalytic antibody, transition state, reaction mechanism, immunization, hapten
Introduction
The usual paradigm in chemical biology is that chemical means lead to biological ends; however, in the case of catalytic antibodies, the reverse is the case—eliciting catalytic antibodies through biological means provides catalysts for a chemical end. The idea that antibodies could be designed to catalyze a specific chemical transformation was first proposed by Jencks (1) and was built on the foundations of enzyme catalysis originally conceptualized by Pauling (2). In this presumption, the catalytic power of an enzyme is primarily derived from the stabilization of the high energy transition state along a given reaction coordinate (Fig. 1). Therefore, a catalyst could be generated by probing the immune response for an antibody that can bind a small molecule, or hapten, that is a transition state analog of a desired chemical reaction. The first catalytic antibodies reported in 1986 independently by Lerner et al. (3, 4) and Schultz et al. (5) employed this strategy, and since these seminal reports, more than 50 chemical reactions have been catalyzed by antibodies. The technique has been refined over the years, but the general concept remains the same and numerous reviews on catalytic antibodies have appeared in the literature (6-15). Catalytic antibodies offer unique capabilities in a range of scenarios, including stereoselective organic synthesis, therapeutic potential in the treatment of disease, the elimination of toxins, the attenuation of agents used in chemical and biological warfare, and cessation of abused and/or addictive drugs. In this article, a broad overview of antibody catalysis is presented, including modern methods for eliciting catalytic antibodies, the evolution of hapten design, and advances in antibody catalysis.

Figure 1. Relative energies of substrate (S) and product (P) along a reaction coordinate, revealing the energy differences between uncatalyzed (TSunca₮) and catalyzed (TScat₮) transition states.
Antibodies: Structure and Function
A brief introduction to the basic structure and function of an antibody is essential to understanding the catalytic power of a select few of these molecules. Immunoglobulins (Igs) are glycoproteins that can be divided into various classes and subclasses based on structure and function. Most catalytic antibodies are IgG molecules and therefore will be the focus of this review.
IgG molecules consist of four polypeptide chains, two identical light chains and two identical heavy chains, that assemble into a Y-shaped structure via a network of disulfide bonds and protein-protein interactions (Fig. 2) Globular domains within the overall IgG structure delineate the functional aspects of the molecule. The light chain consists of two domains: a variable domain (VL) and a constant domain (CL). Together the light chain makes up half of each of the two antigen binding fragments (Fab) per IgG molecule. The heavy chains are arranged in a similar fashion but have four domains, the variable (VH) and constant (CH1) domains make up the other of half of the Fab. The CH1 is fused to the crystallizable fragment (Fc) via a hinge region (H). The Fc comprises two constant domains (CH2 and CH3). The antigen-binding locus within the Fab is a compilation of the variable domains from the heavy and light chains, VH and VL, which together form the Fv (variable fragment). These domains contain the complementarity determining regions (CDRs) of high amino acid sequence variability that are directly involved in antigen recognition.
The heavy and light chains are encoded in different loci within the genome, and the source and magnitude of the immune repertoire to recognize a seemingly limitless number of antigens stems from the combinatorial integration of VH and VL. The germline diversification of IgG stems from the heavy-chain recombination of three translated genes: a VH (variable) gene, a DH (diversity) gene, and a JH (joining) gene with the light-chain gene undergoing similar recombination of VL and JL genes. This naive library of IgG molecules is further diversified by somatic mutations that occur during immune-response maturation.

Figure 2. Y-shaped structure of immunoglobulin G (IgG). Disulfide linkages and sites of oligosaccharide attachment are shown. CDRs (complementarity determining regions) are the sites of high amino acid variablility. Antigen binding site comprisesthe VH (variable heavy) and VL (variable light) domains, which together make the Fv. Fab is composed of VH, VL, CH1, and CL. H is the hinge region. Fc is composed of CH2 and CH3. There are two Fab and Fv per IgG and one Fc region per IgG.
Eliciting Catalytic Antibodies
A merging of chemistry and biology is essential to effectively probe the immune system for catalytic antibodies (Fig. 3). Haptens that are successful in eliciting catalytic antibodies are variations of the central theme that transition state stabilization in the antibody combining site will yield functional catalysts for a desired chemical reaction. The evolution of hapten design will be discussed further in subsequent sections. Once the hapten is selected and synthesized, it is attached to an immunogenic carrier protein, usually via an amide bond, for hyperimmunization. A preliminary screen for antibodies that bind the hapten using an enzyme-linked immunosorbent assay (ELISA) is followed by another screen for catalysis of the reaction for which the hapten was designed. Other screening methods have also been used, including catELISA, which screens for catalysis in the antibody pool rather than hapten binding (16, 17). There are three primary methods for eliciting catalytic antibodies currently used: polyclonal, hybridoma, and phage-display.
Polyclonal antibody production is the most primitive method and has several significant limitations (6). This method inherently yields a complex mixture of antibody molecules from the immune response after hapten hyperimmunization with no attempt to purify specific IgG molecules. Efficiency and cost-effectiveness are hallmarks of polyclonal antibody production; however, accurate characterization of this mixture is difficult. Furthermore, X-ray crystallography to examine structure-function relationships and affinity maturation to optimize the antibody is impossible with the polyclonal antibody method.
Consequently, hybridoma technology was a significant milestone enabling the isolation and production of individual antibodies (18). In this technique, antibody producing cells are isolated from the spleen after hyperimmunization with the hapten-protein conjugate. These B cells are subsequently fused with an immortal cell line, and the resultant hybrids secrete monoclonal antibodies. Monoclonal antibodies are homogeneous, can be produced in large quantities, and can be rigorously purified to remove any potential contaminants. Despite greater expense and a more time-consuming process, monoclonal antibody production is the method of choice.
Phage-display technology has many uses for catalytic antibody research (19-21). Generally, phage display involves combinatorial antibody Fab or scFv (single-chain variable fragment) libraries and their expression on phage particles. One advantage to this technology is that the hyperimmunization protocol can be avoided, thereby removing the use of animals. Naive Fab or scFv are identified in a screen for binding to a desired transition state analog. Although in theory a desired catalyst can be discovered this way, in practice this technique is more successful when an initial hyperimmunization protocol is performed followed by acquisition of the mRNA of the B cells from the spleen (19-21). The corresponding focused combinatorial antibody library is biased toward antibody fragments that recognize the hapten. Phage-display is also used as a tool in affinity maturation of a previously identified catalytic antibody. Mutagenesis of the corresponding Fab or scFv from an existing catalytic antibody can be explored using error-prone polymerase chain reaction (PCR), CDR walking, structure-guided mutagenesis, and DNA shuffling to optimize catalysis of the corresponding chemical reaction (22).

Figure 3. Flowchart illustrating the key stages in catalytic antibody generation.
Evolution of Hapten Design
The conformational changes and charge distribution along the reaction coordinate of a chemical transformation are fundamental to hapten design. Catalytic antibodies are designed to mimic the catalytic power of an enzyme, which, in part, stems from the stabilization of the high energy transition state.
Stable Transition State Analog Hapten Design
A stable chemical analog that mimics the transition state of a chemical reaction was the first approach used to elicit catalytic antibodies (Fig. 4) (3-5). Acyl transfer reactions are the most studied type of catalytic antibody reaction, and a wealth of knowledge about this reaction has been garnered through antibody acyl transferases (3-5, 23-28). Using ester hydrolysis as a representative example, nucleophilic addition of water to the carbonyl carbon results in a tetrahedral transition state followed by expulsion of the alcohol leaving group. The transition state for this reaction has a delocalized negative charge that is remarkably similar to the chemically stable phosphonate ester (29). The phosphorous (V) core, known to be an excellent mimic of the transition state in hydrolytic enzymes (30-33) and often used as a key pharmacophore in transition state analog inhibitors (29, 34-36), has been widely adopted as a central motif in catalytic antibody hapten design (7, 9, 37).
Antibody esterase 48G7 was elicited against hapten 1 and effectively catalyzed the hydrolysis of the corresponding activated ester 2 (27). The X-ray crystal structure of this catalytic antibody Fab complexed with 1 revealed the corresponding stabilization of the oxyanion by a nearby cationic ArgL96 residue (27, 38). Hydrogen bonds from the side chains of the adjacent amino acids HisH35 and TyrH33 serve to stabilize the polarized phosphoryl bonds of hapten 1 that would assist in forming the transition state of ester 2. Main-chain amide bonds from TyrL91 and TyrH100 also provide additional hydrogen-bond stabilization forces.

Figure 4. (a) Phosphonate ester as a chemically stable mimic of ester hydrolysis. (b) Transition state analog hapten 1 elicited antibody 48G7 that catalyzes the hydrolysis of ester 2. (c) Key contacts of the 48G7 Fab-1 complex.
Strain-Induced Hapten Design
A slight modihcation of the transition state analog approach to hapten design is the use of a strain-induced hapten to elicit catalytic antibodies. In this approach, a chemical modihcation of the substrate distorts the resultant hapten (Fig. 5). During hyperimmunization, the strain-induced hapten leads to altered substrate binding in the antibody combining site, facilitating the chemical reaction by lowering the energy of the transition state.
An example of this approach is demonstrated in an antibody mimic of the enzyme ferrochetalase (39). Ferrochelatase catalyzes the insertion of Fe2+ into protoporphyrin IX (3) as the last step in the heme biosynthetic pathway (40). Interestingly, N-alkylporphyrins are known to be potent inhibitors of this enzyme, because alkylation at one pyrrole nitrogen distorts the planarity of the porphyrin macrocycle (41). This hnding was used in the design of hapten 4 to catalyze the incorporation of metal ions into mesoporphyrin IX (5) by eliciting an antibody that binds the substrate in a ring-strained conformation. The lone-pair electrons on the pyrrole nitrogen of the porphyrin ring are more accessible to chelation of metal ions in the ring-strained conformation and leads to metalation of mesoporphyrin IX. Antibody 7G12 catalyzes the incorportain of Zn2+, Cu2+, Co2+, and Mn2+ into mesoporphyrin IX, whereas ferrochetalase uses Fe2+, Zn2+, Co2+, and Ni2+ as substrates in the chelation of protoporphyrin IX. X-ray crystallography of the catalytically active 5-7G12 Michaelis complex revealed that the porphyrin ring adopts a nonplanar conformation that is essential for catalysis as anticipated from the hapten design (42).

Figure 5. Strain-induced hapten 4 elicited antibody 7G12 that catalyzes metalation of mesoporphyrin IX (5).
Bait-and-Switch Hapten Design
Transition state analog hapten design has been the most universal means to elicit catalytic antibodies; however, limitations with this approach and straininduced hapten design are realized when fractional bond orders, extended bond lengths, expanded valences, distorted bond angles, and charge distributions cannot be achieved in a stable chemical analog of the transition state suitable for immunization (7, 43, 44). Furthermore, a signihcant number of haptens designed for a specihc chemical transformation have led to antibodies that bind the hapten with exquisite afhnity yet no effective catalysis was realized. This is easily explained because the somatic mutations in the IgG molecule favor tighter hapten binding, but not necessarily more effective catalysis. Additionally, the products of a reaction may have signihcant similarity to the hapten used in hyperimmunization, leading to slow release of the product and/or signihcant product inhibition. In fact, product inhibition is thought to be the major contributing factor for low efhciency hydrolytic catalytic antibodies (45, 46).
A signihcant step in the evolution of hapten design was introduced by Janda and Lerner, coined the “bait-and-switch” method (47-49). This novel advancement enables electrophilic/ nucleophilic and/or general acid/general base catalysis to be programmed into an antibody combining site. Specihcally, a point charge on the hapten in close proximity to, or in direct substitution for, a functional group to be transformed in the respective substrate is used to induce a complementary charge on an amino acid residue in the antibody combining site during hyperimmunization (Fig. 6). The substrate lacks this charge but retains a similar overall structure and the corresponding antibody binds the substrate and acts as a general acid/general base and/or as a nucleophile/electrophile in the desired chemical reaction.
The phosphodiesterase antibody MATT.F-1 is a didactic example of baitand-switch hapten design, illustrating differences from other hapten design approaches (49). The hydrolysis of a phosphodiester bond, such as those found in RNA and DNA, are catalyzed by ribonucleases (RNases) and deoxyribonucleases (DNases), respectively. RNase A is a thoroughly studied enzyme that has two catalytic histidine residues in the active site (50). The imidazole group of His12 acts as a general base by deprotonating the 2' oxygen, and the imidazolium group of His119 acts as a general acid by protonating the 5 ' phosphoryl oxygen in the classic mechanism (51).
The incorporation of a general base and a general acid in the hydrolysis of a phosphodiester was hypothesized to be elicited in an antibody combining site programmed by specific point charges designed into a bait-and-switch hapten (49). Indeed, an antibody against hapten 6 successfully catalyzed the hydrolysis of the corresponding substrate 7 (Fig. 6). In contrast to the transition state analog hapten 8 (52), which elicited the less-proficient phosphodiesterase catalytic antibody 2G12, the precise conformation of the high energy intermediate was sacrificied for charged moieties in specific locations of the hapten. The corresponding counterion charges from amino residues were elicited in the antibody combining site that led to catalytic antibodies with improved catalytic proficiency ((kcat/Km)/kuncat = 1.6 x 107 M-1 for MATT.F-1 versus ((kcat/Km)/kuncat = 1.3 x 106 M-1 for 2G12) for phosphodiesterase activity.

Figure 6. (a) The catalytic mechanism of RNase A, including the postulated transition state. (b) Bait-and-switch hapten 6 elicited antibody MATT.F-1 that catalyzes phosphodiester bond hydrolysis of substrate 7. Transition state analog hapten 8 also elicited catalytic antibodies but with slower rates.
Reactive Immunization Hapten Design
An essential concept in enzyme catalysis, partly realized through the study of catalytic antibodies, is that the proficiency of an enzyme is not solely due to the stabilization of a high energy transition state. Enzymes are not static entities, but rather they have the dynamic ability to stabilize all possible conformations of a chemical reaction along the reaction coordinate (33, 53-55). Furthermore, many enzymes can form covalent intermediates with the substrate that are essential to catalysis. The previous hapten design methods program specific complementarity in the antibody combining site; however, the hapten is a static snapshot of a dynamic chemical process resulting in catalysts limited to nonconvalent interactions that are ultimately less efficient than their enzyme counterparts. Reactive immunization is a hapten design strategy that provides a chance for catalytic antibodies to approach the catalytic efficiency of natural enzymes by using a hapten that undergoes dynamic conformational changes during hyperimmunization and traps chemical reactivity at the B-cell level (Fig. 7) (53, 56-63).
A direct comparison between reactive immunization and transition state analog hapten design was demonstrated by comparing antibody esterase activity elicited against a reactive immunization hapten, phosphonate diester 9, and a transition state analog hapten, phosphonate monoester 10 (Fig. 7) (58, 61). Hapten 9 was originally designed for the purpose of resolving a racemic mixture of naproxen esters and contains a modestly reactive diphosphonate ester that is susceptible to nucleophilic attack during hyperimmunization. Antibody 15G2, elicited against hapten 9, efficiently catalyzed the hydrolysis of 11a to S-(+)-naproxen 12a and phenol 13. Another antibody, 5A9, from the reactive immunization panel possessed turnover numbers lower than 15G2, however, kinetically resolved the hydrolysis of racemic substrate 11 to the antiinflammatory agent S-(+)-naproxen 12a in 35% yield and 90% ee. Meanwhile, antibody 6G6, raised against hapten 10, catalyzed hydrolysis of 11a with comparable turnover numbers to 15G2, but kinetically resolved the hydrolysis of racemic substrate 11 to the anti-inflammatory agent S-(+)-naproxen 12a in 50% yield and >98% ee.
This direct comparison of hapten design approaches for the same reaction revealed that antibodies generated by each method exhibit quite different catalytic behavior. The transition state analog approach provided catalytic antibodies with good turnover numbers and enanatiomeric discrimination; however, it suffered from varying degrees of product inhibition by phenol 13. Comparatively, the reactive immunization approach yielded antibodies that are ultimately better catalysts because, once an efficient catalytic mechanism evolved further complementarity did not develop, leading to broader substrate recognition with reduced product inhibition. Binding site complementarity is the selection criteria rather than chemical reactivity using the transition state analog approach and antibodies developed by this strategy are more substrate specific and yet suffer from product inhibition as a result of exquisite binding affinity.

Figure 7. Reactive immunization hapten 9 elicited antibody 15G2 that catalyzes the hydrolysis of substrate 11. Transition state analog hapten 10 elicited antibody 12C8 to catalyze the same reaction.
Advances in Antibody Catalysis
More than 50 reactions have been catalyzed by antibodies (Table 1). A brief survey of the catalytic antibody landscape is presented below, highlighting creative hapten design strategies, the breadth of reactions catalyzed, and structure-function relationships.
Antibody Cationic Cyclases
Catalytic antibody technology enables the diversification of cyclization products from polyene substrates, because the programmability of antibody catalysis is not limited to the set of naturally occurring polyenes (64-69). Accordingly, hapten 14 elicited the cationic cyclase HA5-19A4 that catalyzes the tandem cationic cyclization of polyene substrate 15 to the bridge-methylated trans-decalins 16a-c (Fig. 8) (67). Cationic cyclization reactions have three components: initiation, propagation, and termination. The zwitterionic N-oxide moiety of hapten 14 mimics the initiation step of this reaction because it is isosteric and isopolar to the first carbocation formed at the beginning of the reaction cascade. The X-ray crystal structure of the Fab fragment of HA5-19A4 complexed with hapten 14 reveals a highly complementary fit in the antibody combining site (70). The hydrophobic pocket is lined with numerous aromatic residues that stabilize the postulated reactive intermediate 17 through cation-n interactions and force it into a chair-chair conformation. Propagation of cyclization proceeds via concerted attack of the C5-C6 n-bond, avoiding accumulation of the unfavorable carbocation at C1, while forming the A ring of 16a-c through the more favorable tertiary carbocation at C5. Subsequent attack on C5 by the C9-10 n-bond forms the B ring of 16a-c. Minor by-products of the reaction include compounds with incomplete closure of the B ring, resulting from either elimination or solvolysis. Termination of the cationic cyclization was programmed into the antibody by including an epoxide group to elicit an antibody residue capable of coordinating a water molecule to facilitate quenching of the terminal carbocation by solvent or to constrain the B ring of the trans-decalin into the half-chair and facilitate proton elimination. Interestingly, termination of the HA5-19A4 catalyzed reaction occurs exclusively by proton elimination because only olefinic products 16a-c are observed.

Figure 8. (a) Hapten 14 elicited antibody HA5-19A4 that catalyzes the cationic cyclization of substrate 15. (b) Key contacts of HA5-19A4 Fab with postulated reactive intermediate 17.
Table 1. Examples of chemical reactions catalyzed by antibodies
|
Ester hydrolysis |
Aldol |
Diels-Alder |
Claisen rearragement |
|
Amide hydrolysis |
Henry reaction |
Hetero-Diels-Alder |
Oxy-Cope rearrangement |
|
Phosphodiester hydrolysis |
β-Elimination |
Aza-Diels-Alder |
Allylic rearrangement |
|
Carbamate hydrolysis |
syn Elimination |
Dipolar cycloaddition |
[2,3]-Sigmatropic rearrangement |
|
Decarboxylation |
anti Elimination |
Cationic cyclization |
Photo-Fries rearrangement |
|
Norrish reaction |
Pericyclic elimination |
Tandem cationic cyclization |
Bergmann cycloaromatization |
|
Robinson annulation |
Photodimerization |
Electrophilic cyclization |
Aromatic oxidation |
|
N-oxidation |
Metalation |
Yang cyclization |
|
For further study of the antibody catalyzed reactions see ref. 6-15.
Antibody-Catalyzed Disfavored Ring Closure
An astounding aspect of antibody catalysis is the ability of these programmable biocatalysts to preferentially form the less thermodynamically favored product (66, 71-74). The intramolecular cyclization reaction of trans-epoxyalcohol 18 is an archetypal example of antibody catalysis of a disfavored transformation, which preferentially forms the tetrahydrofuran 19 under uncatalyzed conditions due to the overwhelming stereoelectronic constraints predicted by Baldwin’s rules for ring closure (Fig. 9) (75, 76).
The N-oxide and N-methyl ammonium haptens 20 and 21 were designed to mimic the stereoelectronic features of the disfavored 6-endo transition state 22 and function as bait-and-switch haptens by programming specihc complementary charges in the antibody combining site (72, 77). Antibody 26D9, elicited against only product. Additionally, hyperimmunization using the N -methyl ammonium 20, efficiently reroutes this transformation and gives tetrahydropyran 23 as the hapten 21 produced antibody 5 C8 that also catalyzed the regio- and enantioselective epoxide opening of substrate 18 to yield the disfavored endo product 23 (77). The active sites in both antibodies contain a putative catalytic diad, as determined by X-ray crystallography, confirming bait-and-switch hapten design as a viable approach to catalyze this disfavored ring closure (77). The exact mechanism has not been established; however, the active site of Fab 5 C8 reveals plausible general acid-base catalysis occurring by AspH95 acting as a proton donor to the epoxide oxygen assisting the formation of intermediate 22 with HisL89 serving as a base enabling nucleophilic attack by the alcohol group.

Figure 9. Bait-and-switch haptens for disfavored ring closure. N-Oxide hapten 20 elicited antibody 26D9 that catalyzes the endo ring closure of substrate 18. N-Methyl ammonium hapten 21 elicited antibody 5 C8 that catalyzes the same disfavored reaction. Key contacts of 5 C8 Fab with postulated reactive intermediate 22.
Antibody Diels-Alderases
One aspect of antibody catalysis that truly ignites the imagination of the chemical biologist is that these biocatalysts are not limited to reactions that have a natural enzymatic equivalent. The Diels-Alder reaction has immense synthetic utility; however, this chemical transformation is extremely rare in nature. Furthermore, the reaction proceeds via an entropically disfavored, highly organized pericyclic transition state (78). The programmability of a catalytic antibody has enabled the catalysis of the Diels-Alder reaction previously considered beyond the realm of possibility with a protein (79-84).
A significant hurdle in the development of a Diels-Alderase catalytic antibody was minimizing product inhibition, because based on the Curtin-Hammett principle, the transition state is markedly similar to the product of a Diels-Alder reaction. A creative solution to this problem was employed in the development of antibody 1E9 (Fig. 10) (79). The endo-hexachloronorbornene hapten 24 elicited antibody 1E9 to catalyze the Diels-Alder reaction between diene tetrachlorothiophene dioxide 25 and N-ethylmaleimide 26. Product inhibition was overcome because, after the pericyclic reaction forming 27, SO2 is liberated sponateously followed by oxidation to yield the structurally dissimilar aromatic product 28; accordingly, no product inhibition was observed. X-ray crystallographic data of 1E9 Fab revealed that the antibody binding pocket is preorganized to provide signihcant shape complementarity with hapten 24 through van der Waals contacts, n-stacking with the maleimide functional group, and a hydrogen bond with AsnH35 (85). A recent study of noncovalent catalyzed Diels-Alder reactions by synthetic, protein, and nucleic acid hosts indicated that antibody 1E9 is the most effective of the noncovalent catalyst systems studied (86). The capabilities of this extraordinary catalytic antibody have been explained by theoretical calculations and the high degree of shape complementarity consistent with the X-ray crystallography data.

Figure 10. (a) Hapten 24 elicited antibody 1E9 that catalyzes the Diels-Alder reaction between diene 25 and dienophile 26. (b) Key contacts of the 1E9 Fab-24 complex.
Diels-Alderase antibody 39-A11 minimizes product inhibition while generating a more conventional Diels-Alder product (Fig. 11) (80). Bicyclo[2.2.2]octane hapten 29 was designed to mimic the proposed boat-like transition state 30 of the [4π + 2π] cycloaddition between diene 31 and dienophile 32. Product inhibition was circumvented by the structural disparity between the product cycloadduct and the pseudo-boat form of the hapten employed for immunization. X-ray crystallographic data of hapten 29 complexed with the 39-A11 Fab indicated that the diene and the dienophile are bound in a reactive conformation that reduces translational and rotational degrees of freedom (83, 87, 88). The stereoselective capabilities of this antibody are accomplished by two strategically positioned hydrogen bonds (AsnH35a, TrpH50) and π-stacking of the maleimide dienophile with TrpH50, as calculated from quantum mechanical models and docking simulations (89). This unique arrangement allows reorganization of one enantiomeric transition state, facilitating formation the chiral product 33. Interestingly, sequencing and cross-reactivity studies indicate that antibodies 39-A11 and 1E9 are structurally similar and may have the same polyspecific germline origin.

Figure 11. (a) Hapten 29 elicited antibody 39A-11 that catalyzes the Diels-Alder reaction between diene 31 and dienophile 32. (b) Key contacts of the 39A-11 Fab-29 complex.
A radical idea to generate Diels-Alderase catalytic antibodies employed a ferrocenyl moiety in hapten 34 and was designed to catalyze the reaction of diene 35 with dienophile 36 (Fig. 12) (82). Hapten 34 has two pentagonal, delocalized, n-electron ring systems stacked upon each other that were thought to be a loose transition state mimic capable of guiding the diene and dieneophile into a reactive ternary complex. Additionally, the hydrophobicity of the hapten will induce a strong immune response and generate antibodies containing hydrophobic microenvironments that sequester the reactants from aqueous solution, increasing reaction rates. The freely rotating ferrocenyl moiety may enable stereoselective catalysis of all possible diastereomers; however, the immune system must be able to bind and stabilize a single conformer of 34 to elicit an effective catalyst. Antibody 13G5 was identified to preferentially catalyze the formation of the disfavored ortho-exo-cycloadduct 37 in high regio-, diastereo, and enantioselectivity over the corresponding endo-product 38. This is finding remarkable considering the flexibility of hapten 34, because antibody 13G5 must preferentially stabilize the exo-transition state 39 over the endo-transition state 40. Quantum mechanical modeling depicts that hapten 34 resembles the van der Waals complex between the reactants more closely than the transition state, yielding antibodies that preferentially recognize the hapten rotamer that mimics 39 (90). Furthermore, the steric restraints imposed by specific hydrogen-bonding interactions revealed in the crystal structure of 13G5 Fab complexed with the inhibitor 41 (an attenuated version of hapten 34) traps 41 in one available eclipsed conformation that is a loose mimic of the early boatlike transition state for the exo Diels-Alder reaction (91).
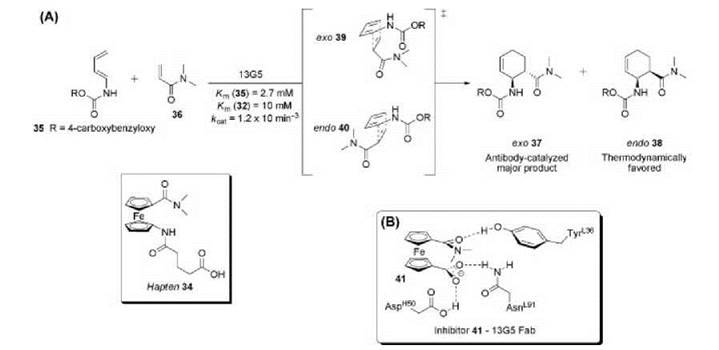
Figure 12. (a) Hapten 34 elicited antibody 13G5 that catalyzes the Diels-Alder reaction between diene 35 and dienophile 36. (b) Key contacts of 13G5 Fab with the truncated hapten analog 41.
Antibody-Catalyzed Oxy-Cope Rearrangements
The oxy-Cope rearrangement is a thoroughly studied and synthetically useful reaction in organic chemistry that proceeds through a highly organized chair-like pericyclic transition state (Fig. 13) (92, 93). The transition state analog hapten 42 elicited antibody AZ-28, which catalyzed the oxy-Cope rearrangement of substrate 43 to aldehyde 44 (94). Product inhibition was avoided by in situ chemical modification to generate the oxime. Surprisingly, the germline precursor to AZ-28 accelerated this oxy-Cope reaction 164,000-fold faster than the uncatalyzed reaction despite a much lower affinity for hapten 42. An explanation for this unprecedented discovery can be explained by X-ray crystallography of both the apo form and hapten 42-complex (95). The van der Waals and hydrogen bond interactions present in AZ-28 force the hapten into a fixed conformation that is catalytically unfavorable. In contrast, the antibody combining site of the germline precursor to AZ-28 seems to have much more flexibility, allowing dynamic changes that lead to enhanced orbital overlap and increased rate acceleration. The disparity in catalysis is further supported by molecular dynamics simulation (96). This finding highlights the discrepancy between transition state analog binding and catalysis. Affinity maturation for the transition state analog does not necessarily result in more efficient catalysts; to the contrary, flexible substrate binding by the germline precursor was a more robust catalytic antibody.

Figure 13. Hapten 42 elicited antibody AZ-28 that catalyzes the oxy-Cope rearrangement of substrate 43.
Antibody Aldolases
The aldol reaction is a fundamental C-C bond forming reaction that is ubiquitous in both chemical synthesis and nature (97). Class I aldolase enzymes possess a reactive lysine residue that forms an enamine intermediate with carbonyl substrates, enabling nucleophilic attack on the corresponding electrophile in the enzyme active site (98, 99). Aldolase catalytic antibodies with a similar reactive lysine residue (59, 60, 100-102) were generated using the reactive immunization hapten 45, which contains a moderately reactive β-1,3-diketone moiety (Fig. 14). Two highly efficient aldolase antibodies, 38 C2 and 33F12, were obtained from the catalytic screen (59, 60). The β-1,3-diketone functionality successfully trapped a lysine residue in the antibody combining site forming Schiff base 46 and ultimately the reactive enamine 47, which directly participates in the mechanism of the aldol reaction in the antibody active site. Both 38 C2 and 33F12 catalyze the aldol reaction between acetone and aldehyde 48 with enzymic catalytic proficiency ((kcat/Km)/kuncat) of nearly 109. The X-ray crystal structure of 33F12 revealed that LysH93 is essential to the catalytic mechanism, which initiates the reaction by forming a stable enamine with the ketone substrate (60). The surrounding hydrophobic residues help to stabilize the unprotonated form of the lysine є-amino group (pKa of 10 in bulk water). A significant perturbation of LysH93 must occur to maintain its uncharged status. Antibodies 38 C2 and 33F12 are actually better catalysts of the retro-aldol reaction; however, these catalysts are extremely robust and 38 C2 participated in major steps in the total synthesis of epothilones A-F (103, 104).
An attempt to improve on these antibodies employed a hybrid approach to hapten design by using a transition state mimic sulfone along with a β-1,3-diketone moiety to trap a reactive site lysine residue (Fig. 15) (62). The hybrid hapten 49 is an excellent mimic of the aldol transition state 50, eliciting two aldolase antibodies, 93F3 and 84G3. In the aldol reaction of 51 with 3-pentanone, antibody 93F3 provided syn-aldol 52 in 90% de and 90% ee, whereas antibody 38 C2 only afforded the anti-isomer in 62% de and 59% ee. These second-generation aldolase antibodies 93F3 and 84G3 showed 103-fold increase in proficiency over antibody 38 C2.
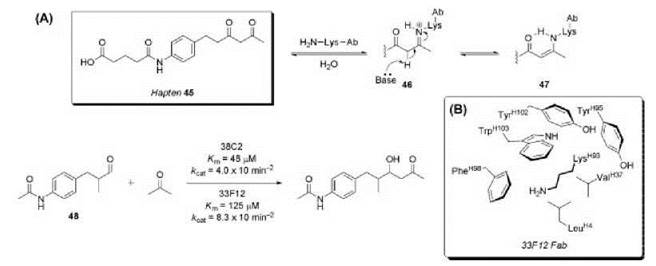
Figure 14. (a) Reactive immunization hapten 45 elicited antibodies 38 C2 and 33F12 that catalyze the aldol reaction of aldehyde 48 with acetone. (b) Hydrophobic environment surrounding LysH93 in 33F12 Fab.
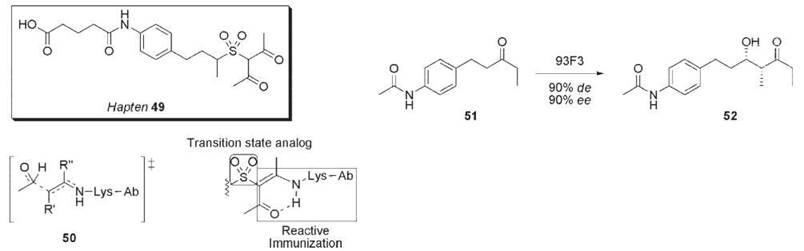
Figure 15. Hapten 49, a hybrid of transition state analog and reactive immunization, elicited antibody 93F3 that catalyzes the aldol reaction of ketone 51 with 3-pentanone.
Antibody Catalyzed Photo-Fries Rearrangement
The photo-Fries rearrangement involves the absorption of light energy by a diphenyl ether substrate resulting in homolytic C-O bond cleavage followed by radical recombination to yield biphenyl products. Multiple products can be formed in this reaction, depending on the electron withdrawing/donating characteristics of the aromatic substituents. Furthermore, the escape of free radicals from the solvent cage leads to additional phenolic products (105). For example, ultraviolet (UV) irradiation of 4-phenoxyaniline 53 forms aromatic products 54-58 (Fig. 16). The electron donating character of the amine substituent leads to preferential cleavage via path a (products 54 and 55) as opposed to path b (products 56-58) that would be favored by an electron withdrawing substituent. An antibody-mediated reaction that suppresses the escape of free radicals and primarily forms biphenyl 54 was generated against haptens 59 (antibody MT2-21C4) and 60 (antibody MT4-3G2) (106). The rotational freedom of hapten 59 was used to explore the combinatorial power of the immune response to elict catalysts that stabilize radicals in the combining site and seek either path a or path b. Hapten 60 is rigid and designed to assist the immune repertoire in the selection of a catalyst by limiting the conformations that can be accessed. Antibodies elicited against both haptens catalyzed the photo-Fries reaction revealing the dynamic ability of an antibody to stabilize a high energy surface in the catalysis of a reaction. Additional photochemical catalytic antibodies for the Norrish type II reaction have been developed (107-109), including an enantioselective Yang cyclization (108, 109).
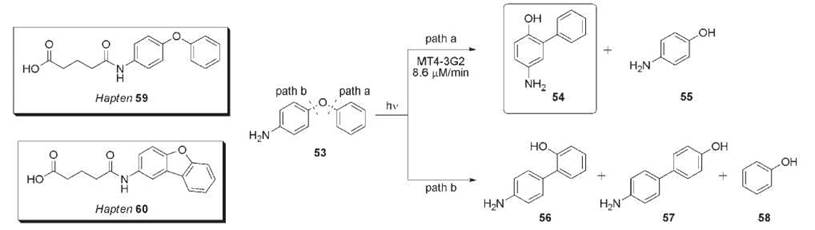
Figure 16. Haptens 59 and 60 elicited antibody MT4-3G2 that catalyzes the photo-Fries reaction of 53.
Recent Developments in Catalytic Antibodies
Since the seminal discovery of chemical catalysis by an antibody, numerous complex chemical transformations have been catalyzed by these molecules. However, in biological systems, the primary function of an antibody is to serve as a mediator between recognition of a foreign substance and its destruction. Specihcally, the variable region of an antibody has evolved to recognize an antigen and then recruit effector systems such as complement and phagocytic cells to destroy the foreign entity.
This paradigm has been challenged by the recent discovery that antibodies have the innate ability to not only recognize foreign substances but to also destroy them (110). Lerner and coworkers have found that all antibodies, regardless of source or antigenic specihcity can catalyze the oxidation of water by singlet oxygen (1O2) via a pathway that is postulated to include trioxygen species, such as dihydrogen trioxide (H2O3) and possibly ozone (O3), in the formation of hydrogen peroxide (H2O2) as the ultimate product (110-114). Further examination of this phenomenon indicated that 1O2 could be generated by either direct UV irradiation of the antibody molecule, by visible light and a triplet oxygen (3O2) sensitizer like hematoporphyrin IX, or by thermal decomposition of endoperoxides. In each case, the antibody catalyzed formation of H2O2 was triggered.
Typically, proteins are not stable under extended exposure to UV irradiation; however, antibodies efficiently form H2O2 linearly up to 40 mole equivalents before an observed decrease in rate. Furthermore, H2O2 seems to inhibit its own production and >500 equivalents of H2O2 can be generated by an antibody when H2O2 is removed by catalase. Isotopic labeling experiments suggest that water is the electron source in this oxidation pathway. Functionally, the water oxidation pathway has been postulated to play a role in a range of clinical scenarios, including bacterial killing, inflammation, and the pathogenesis of atherosclerosis (112, 115).
The manipulation of the water oxidation pathway expands the realm of possibilities with catalytic antibodies. A specific example recently reported is the catalytic oxidative degradation of nicotine (53) by antibodies TD1-10E8 and TD1-36H10 (Fig. 17) (116). Catalytic antibodies that degrade cocaine by ester hydrolysis have been previously identified, and the use of catalytic antibodies in the cessation of drug abuse is an active area of research (37, 117-121). Developing an antibody capable of degrading nicotine has been particularly challenging because its chemical structure is not amenable to decomposition by catalytic antibodies using any of the previously outlined hapten design strategies. Many oxidative degradation products of nicotine are known (122) and antibody catalysis could parallel this pathway with a sufficient 1O2 source. Riboflavin interacts with immunoglobulins, although this interaction is not completely understood (123) and is a known photosensitizer. The riboflavin-antibody interaction was exploited in the generation of reactive oxygen species to oxidatively decompose nicotine. However, the initial screen using a tight binding (K d < 10 μM) nicotine antibody panel elicited against hapten 54, riboflavin, and either UV or visible light led to no catalysis of nicotine degradation over the appropriate control reactions. Conversely, a weak binding panel of antibodies elicited against the less congruent nicotine hapten 55 (K d > 1 μM) effectively catalyzed the formation of nicotine oxidation products 56 and 57. It is important to note that, in this study, a weak binding nicotine antibody was converted to a catalytic antibody using visible light and riboflavin as a photosensitizer. The inherent utility of the water oxidation pathway is an active research area, and the antibody catalyzed oxidative degradation of nicotine is the first example of manipulating this pathway for a potential therapeutic outcome.

Figure 17. Hapten 55 elicited antibodies TD1-10E8 and TD1-36H10 that catalyze the oxidative degradation of nicotine (53) using riboflavin as a photosensitizer.
Conclusions
Caxtalytic antibodies are unparalleled as tailor-made enzyme mimics of a chemical reaction and a paramount advancement in chemical biology. The scope of antibody catalysis continues to increase although enzyme-like rates have yet to be achieved. A snapshot of the reaction coordinate using transition state analog, strain-induced, or bait-and-switch hapten design has led to numerous antibody catalyzed reactions. Enzymes and catalytic antibodies share their primary mode of action by stabilizing a high energy transition state. Enzymes have a superior evolutionary advantage, and additional mechanisms that assist enzyme catalysis, like covalent catalysis, cofactors, proximity effects, and the dynamic ability to complement an infinite number of conformations along a reaction coordinate, play a larger role than originally suspected.
Perhaps the most significant contribution of the catalytic antibody field is the realization that enzyme catalysis is not simply transition state stabilization. Reactive immunization has enabled a mimic of the dynamics involved in enzyme catalysis, and an aldolase antibody that approaches enzymatic rates has been developed using this technique (59, 60). However, only a few types of reactions have been catalyzed using this hapten design strategy. The ability to design a de novo enzyme-like catalyst for a multitude of different reactions is an intriguing lure for the chemical biologist. The advent of catalytic antibodies was essential to the current understanding of the relationship between chemical catalysis and molecular structure; however, a better understanding of this relationship is essential for the advancement of catalytic antibody research toward the ultimate goal of readily programmed catalysts with operative rates that are inherently friendly for the environment. The discovery of naturally occurring catalytic antibodies (124) and the realization that nature uses IgG molecules for chemical catalysis provides a basis to strive toward this ultimate goal.
Acknowledgment
This work was supported by The Skaggs Institute for Chemical Biology and the National Institute on Drug Abuse (DA 018507 to A.P.B. and DA 015700 to K.D.J.).
References
1. Jencks WP. Strain, distortion and conformational change. 1969. New York: McGraw-Hill.
2. Pauling L. Nature of forces between large molecules of biological interest. Nature 1948; 161(4097):707-709.
3. Tramontano A, Janda KD, Lerner RA. Chemical reactivity at an antibody binding site elicited by mechanistic design of a synthetic antigen. Proc. Natl. Acad. Sci. U.S.A. 1986; 83(18):6736-6740.
4. Tramontano A, Janda KD, Lerner RA. Catalytic antibodies. Science 1986; 234(4783):1566-1570.
5. Pollack SJ, Jacobs JW, Schultz PG. Selective chemical catalysis by an antibody. Science 1986; 234(4783):1570-1573.
6. Keinan E, ed. Catalytic antibodies. 2005. Wiley-VCH Verlag GmbH & Co KGaA, Weinheim, Germany.
7. Xu Y, Yamamoto N, Janda KD. Catalytic antibodies: hapten design strategies and screening methods. Biooig. Med. Chem. 2004; 12(20):5247-5268.
8. Schultz PG, Yin J, Lerner RA. The chemistry of the antibody molecule. Angew. Chem. Int. Ed. Engl. 2002; 41(23):4427-4437.
9. Wentworth P, Jr, Janda KD. Catalytic antibodies: structure and function. Cell Biochem. Biophys. 2001; 35(1):63-87.
10. Wentworth, P Jr, Janda KD. Catalytic antibodies. Curr. Opin. Chem. Biol. 1998; 2(1):138-144.
11. Smithrud DB, Benkovic SJ. The state of antibody catalysis. Curr. Opin. Biotechnol. 1997; 8(4):459^66.
12. Schultz PG, Lerner RA. From molecular diversity to catalysis: lessons from the immune system. Science 1995; 269(5232):1835- 1842.
13. Shokat KM, Schultz PG. Catalytic antibodies. Methods Enzymol. 1991; 203:327-351.
14. Mayforth RD, Quintans J. Designer and catalytic antibodies. N. Engl. J. Med. 1990; 323(3):173-178.
15. Lerner RA, Tramontano A. Catalytic antibodies. Sci. Am. 1988; 258(3):58-70.
16. Tawfik DS, Green BS, Chap R, Sela M, Eshhar Z. catELISA: a facile general route to catalytic antibodies. Proc. Natl. Acad. Sci. U.S.A. 1993; 90(2):373-377.
17. MacBeath G, Hilvert D. Monitoring catalytic activity by immunoassay: implications for screening. J. Am. Chem. Soc. 1994; 116(14):6101-6106.
18. Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975; 256(5517):495-497.
19. Huse WD, Sastry L, Iverson SA, Kang AS, ting-Mees M, Burton DR, Benkovic SJ, Lerner RA. Generation of a large combinatorial library of the immunoglobulin repertoire in phage lambda. Science 1989; 246(4935):1275-1281.
20. Huse WD, Stinchcombe TJ, Glaser SM, Starr L, MacLean M, Hellstrom KE, Hellstrom I, Yelton DE. Application of a filamentous phage pVIII fusion protein system suitable for efficient production, screening, and mutagenesis of F(ab) antibody fragments. J. Immunol. 1992; 149(12):3914-3920.
21. Barbas CF III, Kang AS, Lerner RA, Benkovic SJ. Assembly of combinatorial antibody libraries on phage surfaces: the gene III site. Proc. Natl. Acad. Sci. U.S.A. 1991; 88(18):7978-7982.
22. Barbas CF III, Burton DR, Scott JK, Silverman GJ, eds. Phage display of proteins and peptides: A laboratory manual. 2001. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
23. Janda KD, Schloeder D, Benkovic SJ, Lerner RA. Induction of an antibody that catalyzes the hydrolysis of an amide bond. Science 1988; 241(4870):1188-1191.
24. Zhou GW, Guo J, Huang W, Fletterick, RJ, Scanlan TS. Crystal structure of a catalytic antibody with a serine protease active site. Science 1994; 265(5175):1059-1064.
25. Guo J, Huang W, Scanlan TS. Kinetic and mechanistic characterization of an efficient hydrolytic antibody: evidence for the formation of an acyl intermediate. J. Am. Chem. Soc. 1994; 116(14):6062-6069.
26. Guo J, Huang W, Zhou GW, Fletterick RJ, Scanlan TS. Mechanistically different catalytic antibodies obtained from immunization with a single transition-state analog. Proc. Natl. Acad. Sci. U.S.A. 1995; 92(5):1694-1698.
27. Lesley SA, Patten PA, Schultz PG. A genetic approach to the generation of antibodies with enhanced catalytic activities. Proc. Natl. Acad. Sci. U.S.A. 1993; 90(4):1160-1165.
28. Charbonnier JB, Golinelli-Pimpaneau B, Gigant B, Tawfik DS, Chap R, Schindler DG, Kim SH, Green BS, Eshhar Z, Knossow M. Structural convergence in the active sites of a family of catalytic antibodies. Science 1997; 275(5303):1140-1142.
29. Collins KD, Stark GR. Aspartate transcarbamylase. Interaction with the transition state analogue N-(phosphonacetyl)-L- aspartate. J. Biol. Chem. 1971; 246(21):6599-6605.
30. Wolfenden R. Transition state analogues for enzyme catalysis. Nature 1969; 223(207):704-705.
31. Wolfenden, R. Transition state analog inhibitors and enzyme catalysis. Annu. Rev. Biophys. Bioeng. 1976; 5:271-306.
32. Wolfenden R. Conformational aspects of inhibitor design: enzymesubstrate interactions in the transition state. Bioorg. Med. Chem. 1999; 7(5):647-652.
33. Wolfenden R. Thermodynamic and extrathermodynamic requirements of enzyme catalysis. Biophys. Chem. 2003; 105(2-3):559-572.
34. Jacobsen NE, Bartlett PA. A phosphonamidate dipeptide analog as an inhibitor of carboxypeptidase A. J. Am. Chem. Soc. 1981; 103(3):654-657.
35. Bartlett PA, Marlowe CK. Phosphonamidates as transition-state analog inhibitors of thermolysin. Biochemistry 1983; 22(20):4618-4624.
36. Bartlett PA, Lamden LA. Inhibition of chymotrypsin by phosphonate and phosphonamidate peptide analogs. Bioorg. Chem. 1986; 14(4):356-377.
37. Dickerson TJ, Janda KD. Recent advances for the treatment of cocaine abuse: central nervous system immunopharmacotherapy. AAPS J. 2005; 7(3):E579-E586.
38. Wedemayer GJ, Wang LH, Patten PA, Schultz PG, Stevens RC. Crystal structures of the free and liganded form of an esterolytic catalytic antibody. J. Mol. Biol. 1997; 268(2):390-400.
39. Cochran AG, Schultz, PG. Antibody-catalyzed porphyrin metallation. Science 1990; 249(4970):781-783.
40. Dailey HA, Dailey TA, Wu CK, Medlock AE, Wang KF, Rose JP, Wang BC. Ferrochelatase at the millennium: Structures, mechanisms and [2Fe-2S] clusters. Cell Mol. Life Sci. 2000; 57(13-14):1909-1926.
41. Dailey HA, Fleming JE. Bovine ferrochelatase. Kinetic analysis of inhibition by $N$-methylprotoporphyrin, manganese, and heme. J. Biol. Chem. 1983; 258(19):11453-11459.
42. Yin J, Andryski SE, Beuscher AE, Stevens RC, Schultz PG. Structural evidence for substrate strain in antibody catalysis. Proc. Natl. Acad. Sci. U.S.A 2003; 100(3):856-861.
43. Hanson JE, Kaplan AP, Bartlett PA. Phosphonate analogues of carboxypeptidase A substrates are potent transition-state analogue inhibitors. Biochemistry 1989; 28(15):6294-6305.
44. Morgan B, Scholtz JM, Ballinger MD, Zipkin ID, Bartlett PA. Differential binding energy: a detailed evaluation of the influence of hydrogen-bonding and hydrophobic groups on the inhibition of thermolysin by phosphoruscontaining inhibitors. J. Am. Chem. Soc. 1991; 113(1):297-307.
45. Stewart JD, Benkovic SJ. Catalytic antibodies: mechanistic and practical considerations. Chem. Soc. Rev. 1993; 22(4):213-219.
46. Tramontano A, Ivanov B, Gololobov G, Paul S. Inhibition and labeling of enzymes and abzymes by phosphonate diesters. Appl. Biochem. Biotechnol. 2000; 83(1-3):233-243.
47. Janda KD, Weinhouse MI, Schloeder DM, Lerner RA, Benkovic SJ. Bait and switch strategy for obtaining catalytic antibodies with acyl-transfer capabilities. J. Am. Chem. Soc. 1990; 112(3): 1274-1275.
48. Janda KD. New strategies for the design of catalytic antibodies. Biotechnol. Prog. 1990; 6(3):178-181.
49. Wentworth P Jr, Liu Y, Wentworth AD, Fan P, Foley MJ, Janda KD. A bait and switch hapten strategy generates catalytic antibodies for phosphodiester hydrolysis. Proc. Natl. Acad. Sci. U.S.A. 1998; 95(11):5971-5975.
50. Raines RT. Ribonuclease A. Chem. Rev. 1998; 98(3):1045-1066.
51. Thompson JE, Raines RT. Value of general acid-base catalysis to ribonuclease A. J. Am. Chem. Soc. 1994; 116(12):5467-5468.
52. Weiner DP, Wiemann T, Wolfe MM, Wentworth P, Janda KD. A pentacoordinate oxorhenium(V) metallochelate elicits antibody catalysts for phosphodiester cleavage. J. Am. Chem. Soc. 1997; 119(17):4088-4089.
53. Wirsching P, Ashley JA, Lo CH, Janda KD, Lerner RA. Reactive immunization. Science 1995; 270(5243):1775-1782.
54. Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science 2003; 301(5637):1196-1202.
55. Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006; 75:519-541.
56. Lin CH, Hoffman TZ, Xie Y, Wirsching P, Janda KD. An antibody transesterase derived from reactive immunization that utilizes a wide variety of alcohol substrates. Chem. Commun. 1998; (10):1075-1076.
57. Chen DW, Kubiak RJ, Ashley JA, Janda KD. Reactive immunization elicits catalytic antibodies for polyester hydrolysis. J. Chem. Soc., Perkin Trans. 1 2001; (21):2796-2803.
58. Lo CHL, Wentworth P, Jung KW, Yoon J, Ashley JA, Janda KD. Reactive immunization strategy generates antibodies with high catalytic proficiencies. J. Am. Chem. Soc. 1997; 119(42):10251- 10252.
59. Wagner J, Lerner RA, Barbas CF III. Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995; 270(5243):1797-1800.
60. Barbas CF III, Heine A, Zhong G, Hoffmann T, Gramatikova S, Bjornestedt R, List B, Anderson J, Stura EA, Wilson IA, Lerner RA. Immune versus natural selection: antibody aldolases with enzymic rates but broader scope. Science 1997; 278(5346):2085-2092.
61. Datta A, Wentworth P, Shaw JP, Simeonov A, Janda KD. Catalytically distinct antibodies prepared by the reactive immunization versus transition state analogue hapten manifolds. J. Am. Chem. Soc. 1999; 121(45):10461-10467.
62. Zhong G, Lerner RA, Barbas CF III. Broadening the aldolase catalytic antibody repertoire by combining reactive immunization and transition state theory: new enantio- and diastereoselectivities. Angew. Chem. Int. Ed. Engl. 1999; 38(24):3738-3741.
63. Schowen RL. The elicitation of carboxylesterase activity in antibodies by reactive immunization with labile organophosphorus antigens: a role for flexibility. J. Immunol. Methods 2002; 269(1-2):59-65.
64. Li T, Janda KD, Ashley JA, Lerner RA. Antibody catalyzed cationic cyclization. Science 1994; 264(5163):1289-1293.
65. Li T, Janda KD, Lerner RA. Cationic cyclopropanation by antibody catalysis. Nature 1996; 379(6563):326-327.
66. Li T, Lerner RA, Janda KD. Antibody-catalyzed cationic reactions: rerouting of chemical reactions via antibody catalysis. Acc. Chem. Res. 1997; 30(3):115-121.
67. Hasserodt J, Janda KD, Lerner RA. Formation of bridge-methylated decalins by antibody-catalyzed tandem cationic cyclization. J. Am. Chem. Soc. 1997; 119(26):5993-5998.
68. Zhu X, Heine A, Monnat F, Houk KN, Janda KD, Wilson IA. Structural basis for antibody catalysis of a cationic cyclization reaction. J. Mol. Biol. 2003; 329(1):69-83.
69. Yamaguchi H, Tsubouchi K, Kawaguchi K, Horita E, Harada A. Peroxidase activity of cationic metalloporphyrin-antibody complexes. Chem. Eur. J. 2004; 10(23):6179-6186.
70. Paschall CM, Hasserodt J, Jones T, Lerner RA, Janda KD, Christianson DW. Convergence of catalytic antibody and terpene cyclase mechanisms: polyene cyclization directed by carbocation-p interactions. Angew. Chem. Int. Ed. Engl. 1999; 38(12):1743-1747.
71. Janda KD. Catalytic antibodies: the rerouting of chemical reactions. Biochem. Soc. Trans. 1993; 21(4):1090-1095.
72. Janda KD, Shevlin CG, Lerner RA. Antibody catalysis of a disfavored chemical transformation. Science 1993; 259(5094):490- 493.
73. Cravatt BF, Ashley JA, Janda KD, Boger DL, Lerner RA. Crossing extreme mechanistic barriers by antibody catalysis: syn elimination to a cis olefin. J. Am. Chem. Soc. 1994; 116(13):6013- 6014.
74. Yli-Kauhaluoma JT, Janda KD. Catalytic antibodies: the rerouting of chemical reactions. Towards electrophilic aromatic substitution by carbon dioxide. Ann. N.Y. Acad. Sci. 1996; 799:26-31.
75. Baldwin JE. Approach vector analysis: A stereochemical approach to reactivity. J. Chem. Soc., Chem. Commun. 1976; (18):738-741.
76. Baldwin JE. Rules for ring closure. J. Chem. Soc., Chem. Commun. 1976; (18):734-736.
77. Gruber K, Zhou B, Houk KN, Lerner RA, Shevlin CG, Wilson IA. Structural basis for antibody catalysis of a disfavored ring closure reaction. Biochemistry 1999; 38(22):7062-7074.
78. Sauer J. Diels-AIder reactions. I. New preparative aspects. Angew. Chem. Int. Ed. Engl. 1966; 5(2):211-230.
79. Hilvert D, Hill KW, Nared KD, Auditor MT. Antibody catalysis of the Diels-Alder reaction. J. Am. Chem. Soc. 1989; 111(26): 9261-9262.
80. Braisted AC, Schultz PG. An antibody-catalyzed bimolecular Diels-Alder reaction. J. Am. Chem. Soc. 1990; 112(20):7430- 7431.
81. Gouverneur, VE, Houk, KN, de Pascual-Teresa, B, Beno, B, Janda, KD, Lerner, RA. Control of the exo and endo pathways of the Diels-Alder reaction by antibody catalysis. Science 1993; 262(5131):204-208.
82. Yli-Kauhaluoma JT, Ashley JA, Lo CH, Tucker L, Wolfe MM, Janda KD. Anti-metallocene antibodies: a new approach to enantioselective catalysis of the Diels-Alder reaction. J. Am. Chem. Soc. 1995; 117(27):7041-7047.
83. Romesbeig FE, Spiller B, Schultz PG, Stevens RC. Immunological origins of binding and catalysis in a Diels-Alderase antibody. Science 1998; 279(5358):1929-1933.
84. Shi ZD, Yang BH, Wu YL, Pan YJ, Ji YY, Yeh M. First example of an antibody-catalyzed aza Diels-Alder reaction. Bioorg. Med. Chem. Lett. 2002; 12(17):2321-2324.
85. Chen J, Deng Q, Wang R, Houk K, Hilvert D. Shape complementarity, binding-site dynamics, and transition state stabilization: a theoretical study of Diels-Alder catalysis by antibody 1E9. Chembiochem. 2000; 1(4):255-261.
86. Kim SP, Leach AG, Houk KN. The origins of noncovalent catalysis of intermolecular Diels-Alder reactions by cyclodextrins, self-assembling capsules, antibodies, and RNAses. J. Org. Chem. 2002; 67(12):4250-4260.
87. Ulrich HD, Patten PA, Yang PL, Romesberg FE, Schultz PG. Expression studies of catalytic antibodies. Proc. Natl. Acad. Sci. U.S.A. 1995; 92(25):11907-119n.
88. Romesberg FE, Santarsiero BD, Spiller B, Yin J, Barnes D, Schultz PG, Stevens RC. Structural and kinetic evidence for strain in biological catalysis. Biochemistry 1998; 37(41):14404-14409.
89. Zhang X, Deng Q, Yoo SH, Houk KN. Origins and predictions of stereoselective antibody catalysis: theoretical analysis of Diels-Alder catalysis by 39A11 and its germ-line antibody. J. Org. Chem. 2002; 67(25):9043-9053.
90. Cannizzaro CE, Ashley JA, Janda KD, Houk KN. Experimental determination of the absolute enantioselectivity of an antibody-catalyzed Diels-Alder reaction and theoretical explorations of the origins of stereoselectivity. J. Am. Chem. Soc. 2003; 125(9):2489-2506.
91. Heine A, Stura EA, Yli-Kauhaluoma JT, Gao C, Deng Q, Beno BR, Houk KN, Janda KD, Wilson IA. An antibody exo Diels- Alderase inhibitor complex at 1.95 angstrom resolution. Science 1998; 279(5358):1934-1940.
92. Doering W, Roth WR. The overlap of two allyl radicals or a four-centered transition state in the Cope rearrangement. Tetrahedron 1962; 18:67-74.
93. Dewar MJS, Wade LE Jr. A study of the mechanism of the Cope rearrangement. J. Am. Chem. Soc. 1977; 99(13):4417-4424.
94. Ulrich HD, Mundorff E, Santarsiero BD, Driggers EM, Stevens RC, Schultz PG. The interplay between binding energy and catalysis in the evolution of a catalytic antibody. Nature 1997; 389(6648):271-275.
95. Mundorff EC, Hanson MA, Varvak A, Ulrich H, Schultz PG, Stevens RC. Conformational effects in biological catalysis: an antibody-catalyzed oxy-cope rearrangement. Biochemistry 2000; 39(4):627-632.
96. Asada T, Gouda H, Kollman PA. Molecular dynamics simulation study of the negative correlation in antibody AZ-28 catalyzed oxy-Cope rearrangement. J. Am. Chem. Soc. 2002; 124(42): 12535-12542.
97. Machajewski TD, Wong CH. The catalytic asymmetric aldol reaction. Angew. Chem. Int. Ed. Engl. 2000; 39(8):1352-1375.
98. Heine A, Luz JG, Wong CH, Wilson IA. Analysis of the class I aldolase binding site architecture based on the crystal structure of 2-deoxyribose-5-phosphate aldolase at 0.99A resolution. J. Mol. Biol. 2004; 343(4):1019-1034.
99. Lorentzen E, Siebers B, Hensel R, Pohl E. Structure, function and evolution of the Archaeal class I fructose-1,6-bisphosphate aldolase. Biochem. Soc. Trans. 2004; 32(2):259-263.
100. Mu YQ, Gibbs RA. Design and synthesis of chiral and racemic phosphonate-based haptens for the induction of aldolase catalytic antibodies. Bioorg. Med. Chem. 1997; 5(7):1327-1337.
101. List B, Barbas CF III, Lerner RA. Aldol sensors for the rapid generation of tunable fluorescence by antibody catalysis. Proc. Natl. Acad. Sci. U.S.A. 1998; 95(26):15351-15355.
102. List B, Lerner RA, Barbas CF III. Enantioselective aldol cyclodehydrations catalyzed by antibody 38C2. Org. Lett. 1999; 1(1):59- 61.
103. Sinha SC, Barbas CF III, Lerner, RA. The antibody catalysis route to the total synthesis of epothilones. Proc. Natl. Acad. Sci. U.S.A. 1998; 95(25):14603-14608.
104. Sinha SC, Sun J, Miller GP, Wartmann M, Lerner RA. Catalytic antibody route to the naturally occurring epothilones: total synthesis of epothilones A-F. Chem. Eur. J. 2001; 7(8):1691-1702.
105. Haga N, Takayanagi H. Mechanisms of the photochemical rearrangement of diphenyl ethers. J. Org. Chem. 1996; 61(2):735-745.
106. Dickerson TJ, Tremblay MR, Hoffman TZ, Ruiz DI, Janda KD. Catalysis of the photo-Fries reaction: antibody-mediated stabilization of high energy states. J. Am. Chem. Soc. 2003; 125(50): 15395-15401.
107. Taylor MJ, Hoffman TZ, Yli-Kauhaluoma JT, Lerner RA, Janda KD. A light-activated antibody catalyst. J. Am. Chem. Soc. 1998; 120(49):12783-12790.
108. Saphier S, Sinha SC, Keinan E. Antibody-catalyzed enantioselective Norrish type II cyclization. Angew. Chem. Int. Ed. Engl. 2003; 42(12):1378-1381.
109. Saphier S, Hu Y, Sinha SC, Houk KN, Keinan E. Origin of selectivity in the antibody 20F10-catalyzed Yang cyclization. J. Am. Chem. Soc. 2005; 127(1):132-145.
110. Wentworth AD, Jones LH, Wentworth P Jr, Janda KD, Lerner RA. Antibodies have the intrinsic capacity to destroy antigens. Proc. Natl. Acad. Sci. U.S.A. 2000; 97(20):10930-10935.
111. Wentworth P Jr, Jones LH, Wentworth AD, Zhu X, Larsen NA, Wilson IA, Xu X, Goddard WA III, Janda, KD, Eschenmoser, A, Lerner, RA. Antibody catalysis of the oxidation of water. Science 2001; 293(5536):1806-1811.
112. Wentworth P Jr, McDunn JE, Wentworth AD, Takeuchi C, Nieva J, Jones T, Bautista C, Ruedi JM, Gutierrez A, Janda KD, Babior BM, Eschenmoser A, Lerner RA. Evidence for antibody- catalyzed ozone formation in bacterial killing and inflammation. Science 2002; 298(5601):2195-2199.
113. Wentworth P Jr, Wentworth AD, Zhu X, Wilson IA, Janda KD, Eschenmoser A, Lerner RA. Evidence for the production of trioxygen species during antibody-catalyzed chemical modification of antigens. Proc. Natl. Acad. Sci. U.S.A. 2003; 100(4):1490-1493.
114. Zhu X, Wentworth P Jr, Wentworth AD, Eschenmoser A, Lerner RA, Wilson IA. Probing the antibody-catalyzed water-oxidation pathway at atomic resolution. Proc. Natl. Acad. Sci. U.S.A. 2004; 101(8):2247-2252.
115. Wentworth P Jr, Nieva J, Takeuchi C, Galve R, Wentworth AD, Dilley RB, DeLaria GA, Saven A, Babior BM, Janda KD, Eschenmoser A, Lerner RA. Evidence for ozone formation in human atherosclerotic arteries. Science2003; 302(5647):1053-1056.
116. Dickerson TJ, Yamamoto N, Janda KD. Antibody-catalyzed oxidative degradation of nicotine using riboflavin. Bioorg. Med. Chem. 2004; 12(18):4981-4987.
117. Basmadjian GP, Singh S, Sastrodjojo B, Smith BT, Avor KS, Chang F, Mills SL, Seale TW. Generation of polyclonal catalytic antibodies against cocaine using transition state analogs of cocaine conjugated to diphtheria toxoid. Chem. Pharm. Bull. 1995; 43(11):1902-1911.
118. Hagi-Pavli E, Gallacher G. Designing immunogens for the generation of antibodies that catalyse the hydrolysis of cocaine. Biochem. Soc. Trans. 1997; 25(1):83S.
119. Mets B, Winger G, Cabrera C, Seo S, Jamdar S, Yang G, Zhao K, Briscoe RJ, Almonte R, Woods JH, Landry DW. A catalytic antibody against cocaine prevents cocaine’s reinforcing and toxic effects in rats. Proc. Natl. Acad. Sci. U.S.A. 1998; 95(17):10176-10181.
120. Cashman JR, Berkman CE, Underiner GE. Catalytic antibodies that hydrolyze (—)-cocaine obtained by a high-throughput procedure. J. Pharmacol. Exp. Ther. 2000; 293(3):952-961.
121. Matsushita M, Hoffman TZ, Ashley JA, Zhou B, Wirsching P, Janda KD. Cocaine catalytic antibodies: the primary importance of linker effects. Bioorg. Med. Chem. Lett. 2001; 11(2):87-90.
122. Park TK, Kim KH, Huh TS. Studies on ozonation of nicotine. Korean Chem. Soc. 1984; 28(5):323-327.
123. Innis WS, McCormick DB, Merrill AH Jr. Variations in riboflavin binding by human plasma: identification of immunoglobulins as the major proteins responsible. Biochem. Med. 1985; 34(2):151-165.
124. Paul S, Volle DJ, Beach CM, Johnson DR, Powell MJ, Massey RJ. Catalytic hydrolysis of vasoactive intestinal peptide by human autoantibody. Science 1989; 244(4909):1158-1162.
Cross-References
antigen-antibody interactions
artificial enzymes, selective reactivities through
asymmetric synthesis of biomolecules
crystallization of proteins: overview of applications in chemical biology
enzymatic synthesis of biomolecules
enzyme catalysis, chemistry of
enzyme catalysis, roles of structural dynamics in
enzyme cofactors, chemistry of
enzyme inhibition, mechanisms of
enzyme inhibition, tools to study
enzyme kinetics, techniques to study glycomics, major techniques in
enzyme kinetics
immunological structures
noncovalent interactions in molecule recognition
organic chemistry in biology
oxidative metabolism, chemistry of
phage display
synthetic chemistry
vitamins, chemistry of
water, chemistry of